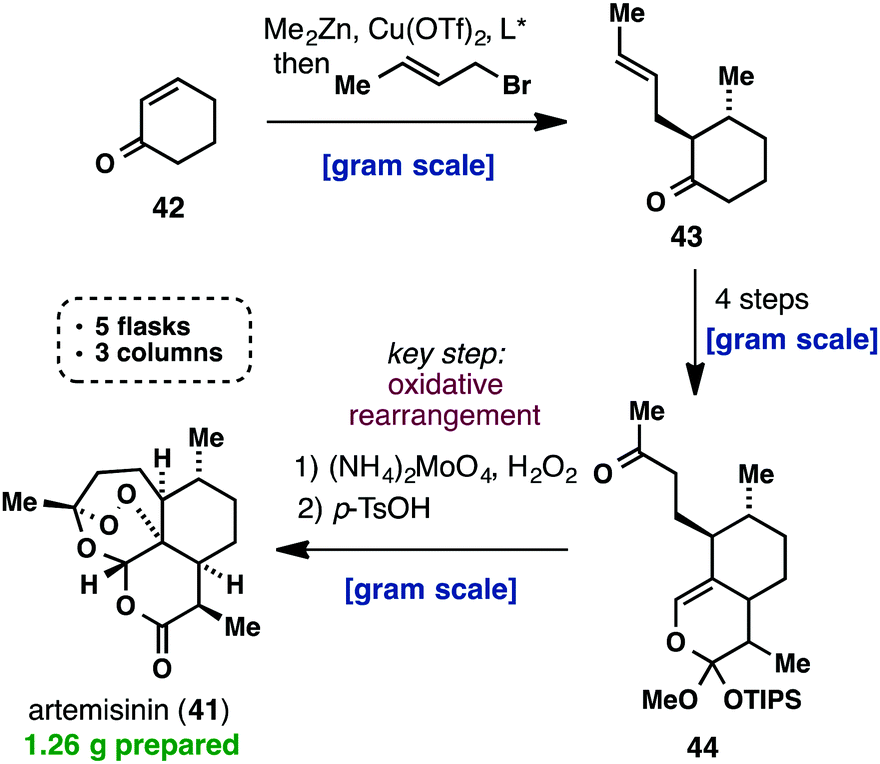
Scheme 3. Formation of acid chloride with SOCI2 in DMAc and coupling with a substituted aniline.
Scheme 4. Formation of the MW 638 impurity.
Example 4: Process 3
4-Dimethylaminocrotonoyl chloride hydrochloride and its coupling with 6-amino- 4-(3-chloro-4-(pyridin-2-ylmethoxy)phenylamino)-7-ethoxyquinoline-3-carbonitrile (procedure with thionyl chloride and DMAc).
A suspension of 4-dimethylaminocrotonic acid (17.0 g, 97.5 mmol) in DMAc (170 ml_) was cooled to -15 0C under nitrogen atmosphere. Neat thionyl chloride (12.8 g, 7.83 mmol) was added to the slurry at a rate to maintain the temperature in the reactor in the range of -18 to -14 0C (moderate exotherm). The reaction mixture was held at -17 to -15 0C for 4 hrs. A solution of the aminoquinoline (36.2 g, 81.3 mmol) in DMAc (440 ml_) was added to the reactor maintaining the temperature in the -14 to -19 0C range. The resulting mixture was held for 18 hr at approximately -15 0C. At this point HPLC analysis showed residual aniline level at 2.5%. The thick suspension of the hydrochloride salt of the coupled product was quenched with water (200 ml_) maintaining the batch temperature between -5 and -16 0C. The pH of the resulting clear solution was adjusted to 1 1 with a 13% aqueous solution of NaOH (approx. 210 ml_ of the solution was added). The suspension was further diluted with water (350 ml_) and the solids were filtered on a polypropylene cloth filter. The cake was washed with water until neutral pH of the washes and dried first in the nitrogen flow on the filter and then on a tray in vacuum at 45 to 50 0C to afford crude (.=)-/\/-(4-(3-chloro-4-(pyridin-2-ylmethoxy)phenylamino)-3-cyano-7- ethoxyquinolin-6-yl)-4-(dimethylamino)but-2-enamide (42.0 g, 91 %) as a bright-yellow crystalline solid.
………………..
WO2004066919A2
http://www.google.com/patents/WO2004066919A2?cl=en
Reaction Scheme Example 1 :
SCHEME 1
(“)
6-(4-N,N-dimethylarninocrotonyt)amido- 4-(4-benzyioxy-3-chloro)arniπo-3-cyano- 7-ethoxyquiπoline, WAY-177820 C31H3[1CIN5θ3 MW 556.07
A suspension of 4-N,N-dimethylaminocrotonic acid hydrochloride in acetonitrile and a catalytic amount of DMF is cooled to 0-10° C. Oxalyl chloride (0.95 eq) is added dropwise and the mixture warmed to 25-30° C and stirred until the chlorinating agent is completely consumed. The light yellow solution is checked for complete consumption of oxalyl chloride by HPLC then cooled to 0-10° C. A cooled solution (0-10° C) of 4-[4-benzyloxy-3-chloro]amino-6-amino-3-cyano-7- ethoxyquinoline in NMP is added dropwise and the mixture is stirred until < 2% of the starting aniline remains. The mixture is added to saturated aqueous sodium bicarbonate, the yellow precipitates are filtered and washed with water. The wet solids are heated to reflux in acetonitrile and clarified hot to remove insolubles. The solution is cooled, the precipitated product filtered and washed with cold acetonitrile. The product is dried (40-50° C, 10 mm Hg, 24 hours) to obtain the final product. Reaction Scheme Example 2:

A solution of 4-N,N-dimethylaminocrotonic acid hydrochloride in tetrahydrofuran (THF) and a catalytic amount of dimethyiformamide (DMF) is cooled to 0-5s C. Oxalyl chloride (0.95 eq) is added dropwise and the mixture warmed to 25-302C and stirred until the chlorinating agent is completely consumed. The orange solution is checked for complete consumption of oxalyl chloride by high- pressure liquid chromatography (HPLC) then cooled to 0-52 C. A solution of 4-[4-(2- pyridylmethoxy)-3-chloro]amino-6-amino-3-cyano-7-ethoxyquinoline is added dropwise and the mixture is stirred until < 0.5% of the starting aniline remains. The reaction is quenched with water and the mixture warmed to 40s C. Aqueous sodium hydroxide is added to bring the pH to 10-11. The resulting precipitates are filtered hot and washed with water. The wet solids are heated to reflux (70-759 C) in acetonitrile:THF (1 :5:1) and the solution cooled slowly to room temperature. The product is filtered and washed with acetonitrile.THF. The product is dried (50e C, 10 mm Hg, 24 hours) to 80-85% yield.
Reaction Scheme Example 3:
4-Dirnethy!amino-but-2-enoic acid |4-(3-chloro-4-fluoro-phenylamino)-3-cvano-7- ethoxy-quinolin-6-vHamide
A. 4-(dimethylamino)-2-butenoyl chloride hydrochloride
A 1 L multi-neck flask equipped with agitator, thermometer, addition funnel, and nitrogen protection is charged with acetonitrile (0.67 kg, 0.85 L) followed by adding dimethylformamide (0.00086 kg, 0.91 mL, d=0.944 g/mL). At ambient temperature, is added 4-dimethylaminocrotonic acid hydrochloride (0.0709 kg) and the mixture stirred until homogeneous. Cool the reaction mixture to (0-10° C) and add oxalyl chloride (0.0473 kg, 0.0325 L, d = 1.45 g/mL) dropwise over (20 minutes) at (0-10° C) followed by a rinse with acetonitrile (0.02 kg, 0.03 L). The temperature (0-10°C) is maintained for about (20 minutes). The temperature of the reaction mixture is adjusted to (22-26° C) over (20 minutes) and maintained over (2 hours). The temperature of reaction mixture is adjusted to (40-45° C) and held for about (5 minutes). Cool the light suspension to about (20-25° C) and check for reaction completion by high-pressure liquid chromatography (HPLC). The reaction is complete when there is < 15 % of the starting material (4-dimethylaminocrotonic acid hydrochloride) present and/or < 2 % of oxalyl chloride (detected as the dimethyl oxalate).
B. 4-Dimethy!amino-but-2-enoic acid |4-(3-chloro-4-fluoro-phenylamino)-3-cyano-7- ethoxy-quinolin-6-yll-amide (crude)
A 3 L multi-neck flask equipped with agitator, thermometer, dip tube, and nitrogen protection is charged N-methyl pyrrolidinone (0.77 kg, 0.75 L, d=1.033 g/mL). At ambient temperature is added 4-[3-chloro-4-fluorophenyl]amino-6-amino-3-cyano-7- ethoxy quinoline (0.0748 kg). The reaction mixture is heated to 40-45° C and maintained for about (15 minutes). The reaction mixture is cooled to (0-10° C) and the light suspension of 4-(dimethylamino)-2-butenoyl chloride hydrochloride in CH3CN added via dip tube and positive nitrogen pressure, over (30-45 minutes) while maintaining the temperature (0-10° C) for at least (2 hours). Reaction completion is monitored by HPLC. The reaction is complete when there is < 2 % of the starting material (4-[3-chloro-4-fluorophenyl]amino-6-amino-3-cyano-7-ethoxy quinoline) present. To a 12 L multi-neck flask equipped with agitator, thermometer, dip tube, and nitrogen protection is charged with water (2.61 kg, 2.61 L) and sodium bicarbonate (0.209 kg) with stirring until a solution is obtained followed by cooling to (20-24° C) to which is transferred the reaction mixture above which contains < 2 % of the starting material (4-[3-chloro-4-fluorophenyl]amino-6-amino-3-cyano-7-ethoxy quinoline), via dip tube and positive nitrogen pressure, to the 12 L flask over about (45-60 minutes) while maintaining the temperature at (20-24° C). The temperature is maintained at (20-24° C) for at least (1 hour). Filter the reaction mixture on a Buchner funnel, rinse with water (3 x 0.40 kg, 3 x 0.40 L), and maintain suction until dripping stops. Dry the product in a vacuum oven at about (50° C) and about (10 mm Hg) for about (28-30 hours). The yield is 78.5 g (86%) at 79.7% strength and 12.3% total impurities.
4-Dimethylamino-but-2-enoic acid r4-(3-chloro-4-fluoro-phenylamino -3-cyano-7- ethoxy-quinolin-6-vn-amide (purified small scale)
First crop: A 6 L multi-neck flask equipped with agitator, condenser, temperature probe, and nitrogen protection is charged with acetonitrile (3.14 kg, 4.00 L) followed by adding 4-dimethylamino-but-2-enoic acid [4-(3-chloro-4-fluoro-phenylamino)-3-cyano-7- ethoxy-quinolin-6-yl]-amide (0.16 kg, 0.167 moles). Heat the mixture to (75-80° C) and hold it for (1 hour). Cool the mixture to (70-75° C) and filter on a pad of diatomaceous earth to remove inorganic salts. Wash the pad with acetonitrile (2 x 0.24 kg, 2x 0.30 L), preheated to (70-75° C). Concentrate the filtrate at (20-30 mm Hg) and a maximum temperature of (40-45° C) to a volume of ( 1.2 L). To the concentrate (slurry) add prefiltered tetrahydrofuran (0.53 kg, 0.60 L). Heat to (65-70° C) to obtain a complete solution. Cool the mixture to (40-45° C) over (0.3 hours). Add seeds and continue cooling to (20-25° C) over (1 hour). Hold at (20-25° C) for a minimum of (18 hours). Collect the solid on a Buchner funnel and wash the collected solid with a prefiltered and precooled at (0-5° C) mixture of acetonitrile/tetrahydrofuran (2/1 by volume) (2 x .06 kg, 2 x 0.08 L). Dry the product in a vacuum oven at (50° C) and (10 mm Hg) for (48 hours) to a loss on drying (LOD) of less than (0.5 %). All washes and concentrates (mother liquors) are saved for further purification.
Second crop:
A 3 L multi-neck flask equipped with agitator, temperature probe, nitrogen protection, and charge with the mother liquors and washes from above. Concentrate by distillation at (20-30 mm Hg) and a maximum temperature of (40-45° C) to a volume of (0.50 L). Collect the solid on a Buchner funnel and wash the solid with prefiltered acetonitrile (0.04 kg, 0.05 L). Dry the solid product in a vacuum oven at (50° C) and (10 mm Hg) for (18 hours). A 1 L multi-neck flask equipped with agitator, condenser, temperature probe, nitrogen protection and charge with prefiltered acetonitrile (0.47 kg, 0.60 L), and the collected solid is heated as a suspension to (70-75° C) over (0.5 hours). Add prefiltered tetrahydrofuran (0.03 kg, 0.03 L) to the suspension while maintaining the temperature at (70-75° C). Cool the solution to (40-45° C) and add seed crystals. Continue cooling to (20-25° C) over (1 hour) and hold for (2 hours). Collect the resulting solid on a Buchner funnel and wash the collected solid with a prefiltered and precooled to (5° C) mixture of acetonitrile/tetrahydrofuran (20/1 by volume) (2 x 0.02 kg, 2 x 0.03 L). Dry the collected solid in a vacuum oven at (50° C) and (10 mm Hg) for (24 hours) to an LOD of less than (0.5 %). The combined yield is 27.5 g + 30.5 g (73%) in 96.2-98.4% strength and 1.5-1.7% total impurities by high pressure liquid chromatography (HPLC).
4-Dimethylamino-but-2-enoic acid f4-(3-chloro-4-fluoro-phenylamino)-3-cvano-7- ethoxy-quinolin-6-vn-amide (purified larger scale)
Acetonitrile, practical (34.0 kg) and 4-dimethylamino-but-2-enoic acid [4-(3- chloro-4-fluoro-phenylamino)-3-cyano-7-ethoxy-quinolin-6-yl]-amide (2.69 kg crude, 1.53 kg at 100% strength) are charged to a purged (100 L) reactor. Acetonitrile, practical (2.0 kg) is used as rinse for funnel and vessel walls. The brown suspension is heated at 70 to 76° C using a jacket temperature not exceeding 85° C, then held at the latter temperature for a minimum of 45 minutes, not exceeding 60 minutes. The resulting suspension is then filtered on the warm-jacketed (70-76° C) 14″ Aurora filter, while maintaining the batch temperature at 70 to 76° C. The filtrates are collected by pump into a purged (100 L) receiver, while keeping their temperature below 50° C. The diatomaceous earth pad is then washed with warm (70 to 76° C) acetonitrile, practical (3 x 2.5 kg). The filtrates and washes in (100 L) receiver are cooled to 20 to 26° C, then transferred into a stainless steel drum. Acetonitrile, practical (2.0 kg) is used as rinse. After cleaning and purging both vessels, the contents of the stainless steel drum is transferred into the (100 L) receiver. Acetonitrile, practical (2.0 kg) is used as a rinse. The batch is heated at 70 to 76° C without exceeding jacket temperature of 85° C. The batch is filtered by pump through a .0 micron single cartridge filter, while maintaining the contents at 70 to 76° C. Warm (70-76° C) acetonitrile, practical (4.0 kg) is used as rinse for vessel, filters, pump and lines. The filtrate and rinse are collected and maintained below 50° C. The batch is adjusted to 10 to 16° C, then concentrated by vacuum distillation to 28 to 33 L volume: expected distillation temperature 20 to 30° C, distillate volume 32 to 37 L. The suspension is heated to 64 to 70° C without exceeding jacket temperature of 85° C. The resulting solution is cooled to 40 to 46° C, then seeded using 4-dimethylamino-but-2~enoic acid [4-(3-chloro-4-fluoro-phenylamino)-3-cyano- 7-ethoxy-quinolin-6-yl]-amide, purified (0.5 g). The mixture is cooled to 20 to 26° C over 1 hour, then held at the latter temperature for a minimum of 2 hours. The suspension is then cooled at -3 to 3° C over 1 hour, then held for a minimum of 1 hour. The solid product is collected on a 16″ Buchner, then washed with cold (0-5° C) acetonitrile-tetrahydrofuran (20-6 v/v) mixture (2 x 2.5 kg). The wet collected solid is recrystallized once more from acetonitrile-tetrahydrofuran (20-6 v/v) to desired purity. The material is dried in a vacuum oven first at 35 to 45° C (target 40° C) for 4 hours, liquid ring pump, then 45 to 55° C (target 50° C) for 4 hours. After high vacuum is applied at the latter temperature, until LOD <0.5% (90° C, 2 hours, full vacuum) and each of acetonitrile, tetrahydrofuran and 1-methyl-2-pyrrolidinone are below 0.2%. The purified drug substance is milled (Comil), then blended. The yield is 1.10 kg (70.1 %, corrected for starting material). The strength of the material is 98.3% and a total impurities of 1.27%.
………………….
N OXIDE
http://www.google.com/patents/US20130225594
EXAMPLE 19 Formula 57-Compound 19a
19a: (E)-4-((4-((3-Chloro-4-(pyridin-2-ylmethoxy)phenyl)amino)-3-cyano-7-ethoxyquinolin-6-yl)amino)-N,N-dimethyl-4-oxobut-2-en-1-amine oxide
To a solution of compound A (200 mg, 0.36 mmol, 1.0 eq) in CH2Cl2 (20 mL) was added m-CPBA (74 mg, 0.43 mmol, 1.2 eq) and the resulting mixture was stirred at room temperature for 4 h. A saturated aqueous solution of NaHCO3 (20 mL) was then added and the organic layer was separated, dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by preparative TLC (CH2Cl2/MeOH, 10/1, v/v) to give (E)-4-((4-((3-chloro-4-(pyridin-2-ylmethoxy)phenyl)amino)-3-cyano-7-ethoxyquinolin-6-yl)amino)-N,N-dimethyl-4-oxobut-2-en-1-amine oxide (20 mg, 10%) as a yellow solid.
LC-MS (Agilent): Rt 3.03 min; m/z calculated for C30H29ClN6O4 [M+H]+ 573.19. found 573.2.
1H NMR: (400 MHz, CD3OD) δ (ppm): 8.98 (s, 1H), 8.57 (m, 1H), 8.39 (s, 1H), 7.92 (td, J=7.2, 1.6 Hz, 1H), 7.72 (d, J=8.0 Hz, 1H), 7.39 (m, 1H), 7.36 (d, J=2.4 Hz, 1H), 7.28 (s, 1H), 7.24-7.13 (m, 3H), 6.74 (d, J=15.6 Hz, 1H), 5.29 (s, 2H), 4.32 (q, J=6.8 Hz, 2H), 4.20 (d, J=7.2 Hz, 2H), 3.28 (s, 6H), 1.57 (t, J=6.8 Hz, 3H).
……………
http://www.google.fm/patents/EP1883631A1?cl=en
Scheme 2 and Scheme 3. Scheme 2
e-Acelamlno^chloro-S-cyano- 7-ethoxy quinoliπe C,4Hi2CIN2O2 +
MW 289.72
25 °C, 5 h 3-Chloro-4-(3-fluorobenzyl)oxy- anillne
C13Hi1CIFNO
MW 251.69
2 h
free base
Scheme 3
6-Acetamlno-4-chloro-3-cyanc~ 7-elhoxy qulnollne C,4H12CIN2O2 +
MW 28972
3-Chlorc-4-fluoronitrobenzene 2-Pyπdyl carblnol 3-Chloro-4-(3-pyndinylmethoxy) 3-Chloro-4-(2-pyrtdlnylmethewy)- C6H3CIFNO2 C6H7NO nitrobenzene anlllne
MW 17555 MW 109 13 C12H9CIN2O3 C12H11CIN2O d=1 1131 g/ml MW 26467 (EM 264) MW 23469
1 h
(HCI salt)
free base
maleate
Example 1
[0078] Synthesis of 3-chloro-4-(2-pyridylmethoxy)nitrobenzene
[0079] 2-pyridinyl carbinol (31.08 g, 1.05 eq) was dissolved in ACN (750 mL) and KOH flakes (85%) were added (20.6 g, 1.25 eq.). The resulting suspension was warmed to 35 °C. A solution of the 3-chloro-4-fluoronitrobenzene (50.0 g, 0.285 mol) in ACN (250 mL) was added at 35-40 °C. The mixture was held for 14 hours. The mixture was then cooled back to 20-25 °C, quenched with H2O (IL) and the resulting slurry filtered and washed with H2O (3 x 100 mL). The resulting product was isolated as a tan solid in 93% yield with a greater than 99.5% purity as determined by HPLC area. Example Ia
[0080] To accomplish the analogous synthesis of 3-chloro-4-(3-fluorobenzyloxy) nitrobenzene, 3-fluorobenzyl alcohol (0.30 kg, 2.39 mole, 1.05 eq) was dissolved in ACN (6.0 L) and to it was added potassium hydroxide flakes (85%) (0.16 kg, 1.25 eq). The resulting suspension was warmed to 35 0C. A solution of the 3-chloro-4-fluoronitrobenzene (0.40 kg, 2.28 mol) in ACN (2.0 L) was added at 35-40 °C. The mixture was held for 18 hours. The mixture was then cooled back to 20-25 °C, quenched with water (8 L) and the resulting slurry filtered and washed with water (2 x 0.40 L). The resulting product was dried at 45 °C, under 10 mm Hg pressure, for 25 hours to give 0.59 kg (92% yield). Example Ib
[0081] To prepare 4-(benzyloxy)3-chloronitrobenzene, benzyl alcohol (0.34 kg, 3.14 mole, 1.10 eq) was dissolved in acetonitrile (1.70 L) and to it was added potassium hydroxide flakes (85%) (0.24 kg, 1.50 eq). The resulting suspension was warmed to 25 0C. A solution of the 3- chloro-4-fluoronitrobenzene (0.50 kg, 2.85 mol, 1.0 eq) in acetonitrile (0.75 L) was added keeping the pot temperature < 45 0C. The mixture was held for 14 h. The mixture was then cooled back to 0-15 0C, quenched with water (2.5 L) and the resulting slurry was filtered and washed with water (2 x 0.50 L). The resulting product was dried at 50 0C, under 10 mm Hg pressure, for 24 hours to give 0.73 kg (97% yield). [0082] Experimental results for the reaction of Example 1 with different bases and solvents are shown in Table 1. The last three entries on Table 1 are large scale runs in which a 5% excess of pyridyl carbinol was used. Table 1 – Preparation of Nitroaryl Intermediate
NA = not applicable
RT = room temperature (20-25 °C)
Example 2
[0083] Preparation of 3-chloro-4-(2-pyridyhnethoxy)aniline from the nitrobenzene product of
Example 1 was accomplished with catalytic hydrogenation using platinum on carbon.
[0084] A typical hydrogenation was done using 6 volumes of THF, 2% by weight of 5%Pt/C (50% water wet), at 25 psi and at 25-30 0C for approximately 4-6 hours. The reaction is slightly exothermic and the temperature will rise to about 30-35 °C. Cooling is necessary to maintain the temperature below 30 0C.
[0085] As a specific example, a mixture of 3-chloro-4-(2-pyridylmethoxy)nitrobenzene (0.15 kg, 0.57 mole) and 2% (w/w) of 5% Pt/C (6.0 g) in tetrahydrofuran (0.90 L) was hydrogenated at 25 psi for at least 5 hours. The mixture was filtered through a celite pad and washed with tetrahydrofuran (0.60 L). The filtrate was distilled to a volume of about 0.75 L and ethanol (1.12 L) was added. Distillation was continued to a volume of about 0.75 L and ethanol (2.85 L) was added. The mixture may be used “as is” in the step of Example 3 below. Example 2 a
[0086] To accomplish an analogous synthesis of 3-chloro-4-(3-fluorobenzyloxy)aniline, zinc (0.464 kg) was added to a mixture of 3-chloro-4-(3-fluorobenzyloxy)nitrobenzene (0.40 kg, 1.42 mole) and ethanol (4.0 L). The mixture was heated to 40-50 °C. A solution of ammonium chloride (0.152 kg) in water (0.80 L) was added over 0.5 hour keeping the pot temperature at 40-50 °C. The mixture was stirred for 2 hours, filtered and washed with hot (40-50 °C) ethanol (2 x 0.40 L). The filtrate was distilled to a volume of about 0.80 L and 2- methyltetrahydrofuran (2.0 L) was added to dissolve the product. Water (0.80 L) and saturated brine (0.40 L) were added and the layers separated. The organic layer was washed with water (0.60 L), and distilled to a volume of about 0.40 L. Ethanol (2.0 L) was added and distillation continued to a volume of 1.2 L. Example 2b
[0087] To prepare 4-(benzyloxy)-3-chloroaniline, a mixture of 4-(benzyloxy)-3- chloronitrobenzene (0.325 kg, 1.23 mole, 1.0 eq) and 1% (w/w) of 5% Pt/C (3.25 g) in isopropanol (3.25 L) was hydrogenated at 25 psi for a minimum of 4.5 h. The mixture was filtered through a celite pad and washed with isopropanol (2.0 L). The filtrates were used as is in the next step.
[0088] Performing the hydrogenation in isopropyl alcohol (PA), methanol (MeOH), or ethanol
(EtOH) may result in the product being contaminated with late eluting impurity that partially precipitates out on standing in solution. It was found that performing the hydrogenation in a solvent where both the product and starting material are soluble, such as tetrahydrofuran
(THF), resulted in greater product purity and required much less solvent. Thus, THF is a preferred solvent for this step. Experimental results showing the effect of different reaction conditions are shown in Table 2. For the larger scale runs, the first aniline intermediate was not isolated (“NI”) before proceeding with the next step.
Table 2 – Hydrogenation to Form First Aniline Intermediate
* Solid impurities noted after reaction completion. ** percent by weight of starting material. Example 3
[0090] Following hydrogenation to form the first aniline intermediate, acid catalyzed coupling was performed to prepare 4~[3-chloro-4-(2-pyridylmethoxy)anilino]-3-cyano-7-ethoxy-6-N- acetylaminoquinoline, as shown below:

[0091] To perform the coupling reaction, the two reactants were heated together in alcohol at 65-78°C over 4-6 hours, yielding the product. The reaction begins as an amber slurry and thickens to a lighter beige slurry as it approaches completion. Upon scaling up from 75 g to 350 g, it proved necessary to add a catalytic amount (0.025 eq.) of methanesulfonic acid to initiate the reaction. As a specific example, 4-chloro-3-cyano-7-ethoxy-6-N- acetylaminoquinoline (0.141 kg, 0.49 mole) was added to the mixture of Example 2, followed by ethanol (0.037 L) to give a suspension. A catalytic amount of methanesulfonic acid (1.17 g) was added at 20-25 C. The resulting slurry was heated to 70-75 C and held for a minimum of 4 hours. Thickening of the slurry was evident after 1.5 hours. Following reaction completion, the mixture was cooled to room temperature and may be used “as is” in the telescoped reaction of Example 4 below. Example 3 a
[0092] To prepare 6-acetamido-4-[3-chloro-4-(3-fluorobenzyloxy)anilino]-3-cyano-7- ethoxyquinoline, ethanol (4.80 L) was added to the aniline solution followed by 4-chloro-3- cyano-7-ethoxy-6-N-acetylaminoquinoline (0.350 kg, 1.11 mole). A catalytic amount of methanesulfonic acid (2.0 ml) was added at 20-250C. The resulting suspension was heated to 70-750C and held for a minimum of 2 h. Thickening of the slurry was evident during this holding period. Following reaction completion, the mixture was used as is in the following telescoped reaction. Example 3 b
[0093] To prepare 6-acetainido-4-[4-(benzyloxy)-3-chloroanilino]-3-cyano-7-ethoxy-quinoline, isopropanol (6.75 L) was added to the aniline solution followed by 4-chloro-3-cyano-7-ethoxy- 6-N-acetylaminoquinoline (0.277 kg, 0.96 mole, 0.78 eq). A catalytic amount of methane sulfonic acid (3.50 ml) was added at 20-250C. The resulting suspension was heated to 80-850C and held for a minimum of 10 hr. Thickening of the slurry was evident during this holding period. Following reaction completion, the mixture was cooled to 25-35 0C, filtered and the cake washed with isopropanol (3 x 0.25 L). The cake was used as is in the following telescoped reaction.
[0094] As solvents EtOH, DMF or other suitable solvent may be used. Experimental results obtained using different solvents and reaction conditions are shown in Table 3. Difficulty filtering the product of this step (noted in several entries on Table 3) was circumvented by not isolating the solid at this point, but telescoping the reaction with the next step. It has been found that on the order of 20 volumes of EtOH were necessary to achieve reasonable stirring, but that the reaction can proceed in only 10 volumes of DMF, without significant loss in purity. [0095] In Table 3, where the entry is labelled NI , the intermediate product was not isolated, but carried into the next reaction step. Table 3 – Coupling Reaction
NR = no reaction, NI = not isolated; ND = not determined; NA = not available
1. Carried through to the deprotection and generation of free base to give 69.5% overall yield.
2. The overall yield after the deprotection and generation of the free base is 76.1%
3. This reaction was not filtered at all but taken as slurry to the next step.
Example 4 – Deprotection
[0096] The deprotection of the quinoline intermediate formed by the coupling reaction using
2N HCl in water is preferred as noted in Table 4 below. As in the previous Examples, the intermediate product of this step is advantageously not isolated, but carried over as a wet cake into the next step.
[0097] Preparation of 4-[3-chloro-4-(2-pyridylmethoxy)anilino]-3-cyano-7-ethoxy-6- aminoquinoline hydrochloride.
[0098] The reaction mixture from the previous step (Example 3) was taken as is and to it was added 2.7N HCl (3.3L) in H2O (16.0 L). The slurry was heated to 700C and held for 19 hours. The resulting slurry was then filtered and rinsed with 1:1 EtOHTH2O (4 x 1.0 L). The product was isolated as a wet cake and carried through to the next step. A small sample was dried at this stage and analyzed. The HCl salt had a strength of 98.9%. Example 4a
[0099] To prepare 6-amino-4-[3-chloro-4-(3-fluorobenzyloxy)anilino]-3-cyano-7- ethoxyquinoline hydrochloride, the reaction mixture from the previous step was taken as is and to it was added ethanol (1.6 L) and concentrated hydrochloric acid (1.38 L) to bring the pH to 1-3. The suspension was held at 70-75 0C for a minimum of 2 h. After 1 h, the mixture thickens and ethanol (0.80 L) was added. After 2 h, water (6.80 L) was added, the mixture stirred for 1 h and then cooled to 35-45 0C and stirred overnight (12 h). The mixture was filtered and rinsed with 1 : 1 ethanol/water (2 x 0.84 L) at 35-45 0C. The product was isolated as a wet cake and carried through to the next step. Example 4b
[00100] To prepare 6-amino-4-[4-(benzyloxy)-3-chloroanilino]-3-cyano-7- ethoxyquinoline hydrochloride, the wet cake from the previous step was taken as is and to it was added a 2 N solution of concentrated hydrochloric acid (1.16 L) in methanol (5.84 L). The suspension was heated to 63-68 0C and held for a minimum of 30 h. The mixture was cooled to 20-300C, filtered and rinsed with methanol (2 x 0.30 L). The product was isolated as a wet cake and carried through to the next step. Table 4 – Deprotection
ND = not determined (the product was used in the next step as a wet cake) NA = not available SM= starting material
Example 5 – Preparation of free base
[0100] The 4-[3-chloro-4-(2-pyridylmethoxy)anilino]-3-cyano-7-ethoxy-6-aminoquinoline HCl salt was converted to the corresponding free base by treatment with 10% potassium carbonate (1.8 L) in MeOH (2.82 L). The mixture was stirred for a minimum of 2.5 hours and the pH was 9-10. The product was filtered, washed with 1:1 methanol/water (3 x 0.19 L) and dried (at 45-50 C at a pressure of 10 mm Hg, for 24 hours) to give 0.186 kg of product with an overall yield of 86% over 4 steps.
Example 5 a
[0101] To prepare 6-amino-4-[3-chloro-4-(3-fluorobenzyloxy)anilino]-3-cyano-7- ethoxyquinoline free base, the 6-amino-4-[3-chloro-4-(3-fluorobenzyloxy)anilino]-3-cyano-7- ethoxyquinoline hydrochloride salt was converted to its corresponding free base by treatment with 10% potassium carbonate (0.22 kg in 2.27 L water) in methanol (7.21 L) until pH was 10. The mixture was stirred for a minimum of 2 h. The beige suspension was filtered, washed with 1:1 methanol/water (2 x 0.84 L) and dried (45-50 0C, 10 mm Hg, 24 h) to give 0.51 kg of product with an overall yield of 99% over 4 steps. Example 5b
[0102] To prepare 6-amino-4-[4-(benzyloxy)-3-chloroanilino]-3-cyano-7-ethoxyquinolme free base, the 6-amino-4-[4-(benzyloxy)-3-chloroamlino]-3-cyano-7-ethoxyqumoline hydrochloride salt was converted to its corresponding free base by treatment with 10% aqueous potassium carbonate (0.213 kg in 2.13 L) in methanol (6.40 L). The mixture was stirred for a minimum of 1.5 h keeping the pH at 9-10. The product was filtered, washed with water (2 x 0.50 L) and dried (50-60 0C, 10 mm Hg, 20 h) to give 0.347 kg of product with an overall yield of 82% over 4 steps.
Example 6 – Side Chain Coupling
[0103] An acid chloride of formula RV(C=O)-Cl, a mixed anhydride or an activated carboxylase R’ 2-(C=O)-LG derived from the corresponding carboxylic acid, may be used to couple a side chain at the 6 position to form a 6-amido-4-amino-3 cyanoquinoline. R’2 may be alkyl of 1-6 carbon atoms, which may be mono- or di-substituted with amino groups or cycloamino groups, or R’2 may be alkenyl of 2-6 carbon atoms which may be mono- or di- substituted with amino groups or cycloamino groups. [0104] Using the 2-step sequence shown below, an activated carboxylate is prepared in situ and coupled with the aniline. Although the acid chloride can be prepared in acetonitile, a better yield was obtained when the acid chloride was prepared in THF. In both cases, the aniline should be dissolved in NMP before amidation. It is believed that formation of product is better due to better solubility of the aniline in a THF/NMP mixture rather than in an ACN/NMP combination.

[0105] The amount of 4-N,N-dimethylaminocrotonic acid needed was 2 equivalents with respect to aniline. A slight undercharge of 1.95 eq of oxalyl chloride was added along with a catalytic amount (3 mol %) of DMF. The acid chloride was formed via the Vilsmeier intermediate. The completion test for the acid chloride reaction consists of quenching an aliquot of the reaction into ethanol and detecting by HPLC the crotonic acid ethyl ester. This method serves as a check to ensure complete consumption of oxalyl chloride. Excess oxalyl chloride will form diethyl oxalate when quenched in ethanol. [0106] The acid chloride is stable after holding for up to 5 hours at 0-10 °C, when decomposition begins. After 20 hours, complete decomposition takes place. If the acid chloride is allowed to warm, decomposition occurs and its effectiveness is diminished. [0107] The quality of the starting crotonic acid also plays a role in this coupling reaction, as commercially available crotonic acid may contain acetic acid. Acetic acid is detrimental to this reaction. 6-N-acetyl quinoline can be formed which is difficult to remove from the final product. The acetic acid can be removed by re-slurrying the crotonic acid in 4 volumes of isopropanol at room tempature, filtering and drying preferably to a level of less than 0.01%. [0108] It was found that the addition of the aniline solution in NMP to the acid chloride gave a better yield as compared to adding the acid chloride to the aniline. The addition is done keeping the temperature at 0-5 °C. The coupling reaction is slow and requires holding overnight at this temperature. It is not desirable to raise the reaction temperature as the stability of the acid chloride diminishes.
[0109] The reaction is quenched using aqueous sodium hydroxide at 40 °C and then filtered at that temperature. Quenching the reaction at 40 0C gives bigger crystals that are easily filterable. It was observed that filtration at 40 °C was faster than at room temperature. The product is recrystallized from a 1.5:1 mixture of acetonitrile:THF (15 volumes) at 70-75 0C. This in-process purification beneficially removes unreacted aniline. The recovery yields are typically greater than 85%.
[0110] To demonstrate a specific synthesis of (E)-N- {4-[3-chloro-4-(2- pyridinylmethoxy)anilino]-3-cyano-7-ethoxy-6-quinolinyl}-4-(dimethylamino)-2-butenamide, a solution of 4-N,N-dimethylaminocrotonic acid hydrochloride (186 g, 1.12 mol) in THF (1.88 L) and a catalytic amount of DMF (2 mL) was cooled to 0-5 °C. Oxalyl chloride (97 mL, 1.09 mol, 0.95 eq) was added dropwise over 45 minutes. The mixture was then warmed to 25-30 °C and stirred for 2 hours. The yellow solution was checked for complete consumption of oxalyl chloride by HPLC, then cooled to 0-5 0C.
[0111] When the reaction is deemed complete, a solution of 4-[4-(2-pyridylniethoxy)-3- chloro]amino-6-amino-3-cyano-7-ethoxyquinoline (250 g, 0.56 mol) in N-methyl-2- pyrolidinone (1.88 L) was added dropwise over 2 hours keeping the temperature at 0-5 °C. The mixture was stirred for at least 3 hours until less than about 2% of the starting aniline remains by HPLC, which takes about 3 hours.
[0112] Upon completion, the reaction was quenched with water (3.0 L), held for 30 minutes and then warmed to 40 °C. Aqueous sodium hydroxide (170 g in 1.25 L water) was added over 1.25 hours to bring the pH to 10-11. The mixture was stirred for an hour, then cooled to room temperature and held for 3 hours. The resulting precipitates were filtered and washed with water (100 mL) and heptane (100 mL). The wet solids were heated to reflux (70-75 °C) in acetonitrile:THF and the solution cooled over 3 hours to room temperature. The product was filtered and washed with cold acetonitrile:THF. The product was dried (40-50 0C, 10 mm Hg, 24 hours) to give 83% uncorrected yield. Example 6a
[0113] In an analogous synthesis of (E)-N- {4-[3-chloro-4-(3-fluorobenzyloxy)anilino]-3- cyano-7-ethoxy-6-qumolmyl}-4-(dimethylamino)-2-butenamide, a solution of 4-N5N- dimethylaminocrotonic acid hydrochloride (108 g, 0.65 mole) in tetrahydrofuran (1.13 L) and a catalytic amount of dimethylformamide (1.2 mL) was cooled to 0-5 °c. Oxalyl chloride (55 mL, 0.62 mole, 0.95 eq) was added dropwise over 50 min. The mixture was then warmed to 25-30 °c and stirred for 2 h then cooled to 0-5 °c. N-methyl-2-pyrrolidinone (0.225 L) was added over 25 min followed by a solution of 6-amino-4-[3-chloro-4-(3- fluorobenzyloxy)]anilino-3-cyano-7-ethoxy-quinoline (150 g, 0.32 mol) in N-methyl-2- pyrrolidinone (1.20 L) added dropwise over 2 hours keeping the temperature 0-5 . The mixture was stirred for at least about 3 hours, warmed to 10-15 °c and stirred for a further 12 hours. The mixture is cooled to 0-10 c, quenched by adding water (1.8 L) over 2 hours, and stirred for 30 minutes. The mixture is warmed to 40 °c. Aqueous sodium hydroxide (101 g in 0.75 L water) was added over 1 hour to bring the pH to 10-11. The mixture was stirred for an hour, filtered warm (40 °c) and washed with water (2 x 0.30 L) until the pH of the last wash was about 7. The wet solids were recrystallized by heating to reflux (70-75 °c) in 60:40 acetonitrile:tetrahydrofuran (2.25 L) and the solution cooled over 3 hours to room temperature. The product was filtered and washed with cold 60:40 acetonitrile:tetrahydrofuran (2 x 0.30 L). The product was dried (40-50 °c, 10 mm Hg, 16 h) to give 0.154 kg (83% yield). Example 6b
[0114] To prepare (E)-N- {4-[4-(benzyloxy)-3-chloroanilino]-3-cyano-7-ethoxy-6-quinolinyl}- 4-(dimethylamino)-2-butenamide free base, a solution of 4-N,N-dimethylaminocrotonic acid hydrochloride (18.6 g, 112 mmole) in acetonitrile (295 ml) and a catalytic amount of dimethylformamide (0.25 mL) was cooled to 0-5 °c. Oxalyl chloride (9.3 mL, 106 mmole, 0.95
Op eq) was added dropwise over 5 min. The mixture was then warmed to 25-30 and stirred for 1-1.5 h then cooled to 0-10 °c. A solution of 6-amino-4-[4-(benzyloxy)-3-cliloroanilino]-3- cyano-7-ethoxy-quinoline (25 g, 56 mmole) in N-methyl-2-pyrrolidinone (175 ml) was added dropwise over 30 min keeping the temperature 0-10 °c. The mixture was stirred for a minimum of 1 h at 0-10 °c. After reaction completion, the mixture was quenched by dropwise addition to a solution of sodium bicarbonate (69.7 g in 870 ml water) over 30 mins. and stirred overnight while warming to room temperature. The mixture was filtered and washed with water (3 x 25 ml). The crude product was recrystallized in refluxing (80-82 °c) acetonitrile (570 ml). The product was dried (45-50 °c, 10 mm Hg, 28 h) to give 12.81 g (41% yield). 1H NMR : δ (DMSO-d6) 9.44 (s, IH, NH), 8.97 (s, IH, Ar), 8.44 (s, IH, Ar), 7.53-7.35 (m, 7H, Ar), 7.35- 7.10 (in, 2H, Ar), 6.78 (dt, IH, -CH2CH=CH-), 6.59 (d, IH, -CH2CH=CH-), 5.21 (s, 2H, OCH2Ph), 4.30 (q, 2H, OCH2CH3), 3.07 (s, 2H, NCH2), 2.18 (s, 6H, N(CHs)2), 1-47 (t, 3H, OCH2CH3).
[0115] Results obtained with different reaction procedures at different degrees of scale-up for synthesis of the 2-pyridylmethoxy analog are shown in Table 5. Table 5 – Side Chain Coupling
* TI = total impurities
[0116] Purificatiuon of the product is conducted by recrystallization in a suitable solvent followed by reslurrying with water followed by additional recrystallization, as necessary. As noted in Table 6, in the synthesis of the 2-pyridylmethoxy analog, several trials in different solvents did not result in the isolation of a single polymorphic form of the product. Table 6
Example 7 – Formation of Salt
[0117] The free base is hygroscopic and undergoes hydrolysis readily. Forming a salt of the compound, such as a fumarate or mesylate salt, stabilizes the molecule and renders the compound more soluble. The most preferred salt is a maleate salt, which has been found to be highly crystalline and to exist substantially as a single polymorph as shown by DSC thermogram in Fig. 1.
[0118] Recrystallizing the product in the presence of an acid has been found to yield a stable salt form of the product. Experimental results achieved utilizing different solvents for the recrystallization are set forth in Table 7. As seen in Table 7, an improvement is observed when n-propanol/water is used as the solvent system. A maleate salt is the most preferred, as it exists in a single polymorphic form. Table 7 – Recrystallization
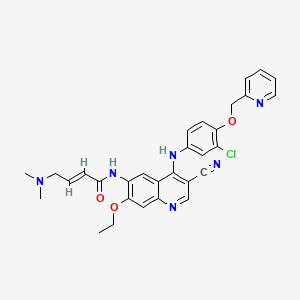
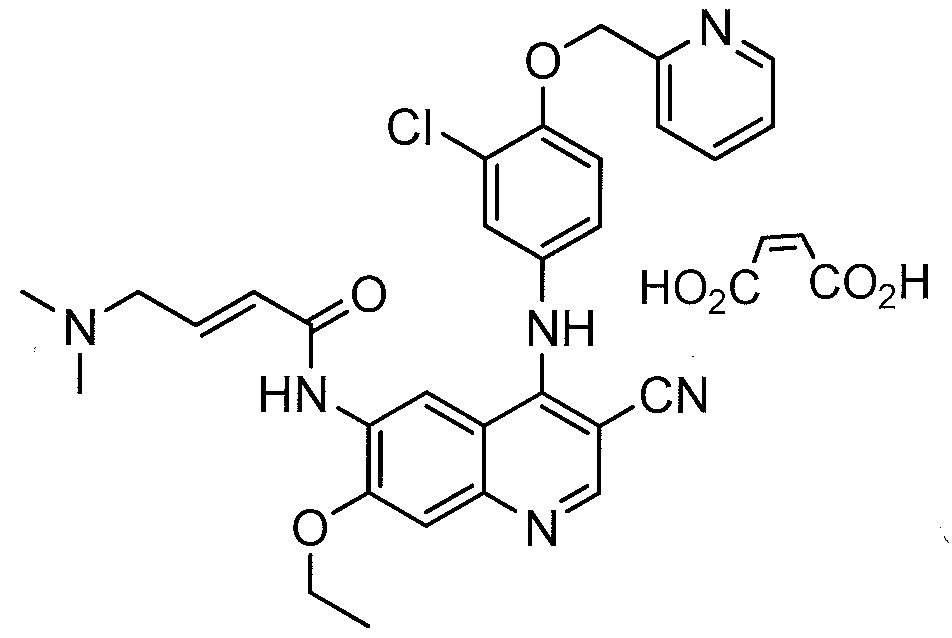
Preparation of (E)-N- {4-[3-chloro-4-(2-pyridinylmethoxy)anilino]-3-cyano-7-ethoxy-6- quinolinyl} -4-(dimethylamino)-2-butenamide maleate, WAY- 179272-B
[0120] (E)-N-{4-[3-chloro-4-(2-pyridinylmethoxy)anilino]-3-cyano-7-ethoxy-6-quinolinyl}-4- dimethylamino)-2-butenamide crude free base (0.1 kg, 0.159 mole) and maleic acid (0.019 kg, 0.164 mole) were dissolved at 40-50 in a 10% water/n-propanol mixture (1.20 L). The hot solution was clarified and cooled over 2 h to room temperature and held for 12-15 hr. The product was filtered and washed with 10% water/n-propanol (2 x 0.15 L). The product was dried (50 °c, 10 mm Hg, 24 h) to give 94.4 g (88% yield). DSC: 204 °c (single crystal form). 1H NMR : δ (DMSO-d6) 9.73 (s, IH, NH), 9.62 (s, IH, NH), 8.93 (s, IH, Ar), 8.60 (dd, IH, Ar), 8.50 (s, IH, Ar), 7.88 (dd, IH, Ar), 7.58 (d, IH, Ar), 7.40 (m, 3H, Ar), 7.24 (m, 2H, Ar), 6.75 (d, 2H, -CH=CH-), 6.03 (s, 2H, HOOC-CH=CH-COOH), 5.29 (s, 2H, OCH2PVr), 4.33 (q, 2H, OCH2CH3), 3.89 (s, 2H, NCH2), 2.76 (s, 6H, N(CH3)2), 1.47 (t, 3H, OCH2CH3). 13C NMR : δ (DMSO-d6) 168.0, 163.2, 156.9, 154.2, 153.2, 151.9, 151.3, 149.8, 148.5, 137.8, 136.5, 134.7, 133.4, 132.2, 128.0, 126.6, 124.9, 123.8, 122.3, 122.2, 117.9, 116.4, 115.1, 113.9, 109.5, 88.1, 72.0, 65.3, 57.8, 43.1, 14.9.
Example 7a
To prepare (E)-N- {4-[3-chloro-4-(3-fluorobenzyloxy)anilino]-3-cyano-7-ethoxy-6- quinolinyl}-4-(dimethylamino)-2-butenamide dimaleate,
(E)-N- {4-[3-chloro-4-(3- fluorobenzyloxy)anilino]-3-cyano-7-ethoxy-6-quinolinyl}-4-dimethylamino)-2-butenamide crude free base (0.516 kg, 0.90 mole) and maleic acid (0.214 kg, 1.84 mole) were dissolved at 40-50 °c in a 6.5% water/n-propanol mixture (12.60 L). The hot solution was clarified, rinsed with 5% water/n-propanol (0.52 L) and n-propanol (2.0 L). The mixture was held at 45 for 3 hr, cooled over 2 h to room temperature and held overnight. The mixture was further cooled to 5-10 °c. The product was filtered and washed with cold 5% water/n-propanol (0.52 L). The product was dried (45 °c, 10 mm Hg, 16-24 h) to give 0.586 kg (81% yield). DSC: 184 °c (single crystal form). 1HNMR : δ (DMSO-d6) 9.77 (s, IH, NH), 8.95 (s, IH, Ar), 8.53 (s, IH, Ar), 7.49-7.16 (m, 8H, Ar), 6.78 (m, 2H, -CH=CH-), 6.15 (s, 4H, 2 x HOOC-CH=CH-COOH), 5.26 (s, 2H, OCH2PyT), 4.33 (q, 2H, OCH2CH3), 3.97 (dd, 2H, NCH2), 2.82 (s, 6H, N(CEb)2), 1.47 (t, 3H, OCH2CH3). 13C NMR : δ (DMS0-d6) 167.0, 163.8, 162.3, 160.6, 153.6, 152.2, 151.3, 150.8, 139.5, 139.4, 133.7, 133.2, 132.2, 131.8, 130.5, 130.4, 127.4, 126.1, 124.3, 123.3, 121.7, 116.9, 115.7, 114.8, 114.5, 114.4, 114.1, 113.8, 113.1, 108.1, 87.2, 69.5, 64.6, 56.9, 42.1, 14.2. Example 7b
[0122] To prepare (E)-N- {4-[4-(benzyloxy)-3-chloroanilino]-3-cyano-7-ethoxy-6-quinolinyl}- 4-(dimethylamino)-2-butenamide maleate, (E)-N- {4-[4-(benzyloxy)-3-chloroanilino]-3-cyano- 7-ethoxy-6-quinolinyl}-4-dimethylamino)-2-butenamide crude free base (2.0 g, 3.6 mmole) and maleic acid (0.43 g, 3.7 mmole) were mixed at 40-50 c in a 10% water/n-propanol mixture (24 ml) for 2 hr. The mixture was cooled to ambient temperature, filtered and washed with 10% water/n-propanol (2 x 3 ml). The product was dried (40 °c, 10 mm Hg, 24 h) to give 0.32 g (13% yield). 1HNMR : δ (DMSO-d6) 9.75 (s, IH, NH), 8.95 (s, IH, Ar), 8.49 (s, IH, Ar), 7.49-7.37 (m, 7H, Ar), 7.23 (dd, 2H, Ar), 6.78 (s, 2H, -CH2CH=CH-), 6.06 (s, 2H, HOOC- CH=CH-COOH), 5.22 (s, 2H, OCH2Ph), 4.31 (q, 2H, OCH2CH3), 3.93 (s, 2H, NCH2), 2.79 (s, 6H, N(CH3)2), 1.46 (t, 3H, OCH2CH3).13C NMR : δ (DMSO-d6) 167.9, 163.1, 154.2, 153.3, 152.1, 151.3, 148.5, 137.3, 136.3, 134.5, 133.2, 132.3, 129.3, 129.2, 128.7, 128.3, 128.2, 128.0, 126.7, 124.9, 122.4, 117.9, 116.4, 115.2, 113.9, 109.5, 88.0, 71.1, 65.3, 57.7, 43.0, 15.0. [0123] (E)-N-{4-[4-(benzyloxy)-3-chloroanilino]-3-cyano-7-ethoxy-6-quinolinyl}-4- dimethylamino)-2-butenamide crude free base (2.0 g, 3.6 mmole) and maleic acid (0.43 g, 3.7 mmole) were mixed at 40-50 °c in a 10% water/n-propanol mixture (24 ml) for 2 hr. The mixture was cooled to ambient temperature, filtered and washed with 10% water/n-propanol (2 x 3 ml). The product was dried (40 °c, 10 mm Hg, 24 h) to give 0.32 g (13% yield). 1H NMR : δ (DMSO-d6) 9.75 (s, IH, NH), 8.95 (s, IH, Ar), 8.49 (s, IH, Ar), 7.49-7.37 (m, 7H, Ar), 7.23 (dd, 2H, Ar), 6.78 (s, 2H, -CH2CH=CH-), 6.06 (s, 2H, HOOC-CH=CH-COOH), 5.22 (s, 2H, OCH2Ph), 4.31 (q, 2H, OCH2CH3), 3.93 (s, 2H, NCH2), 2.79 (s, 6H, N(CH3)2), 1.46 (t, 3H, OCH2CH3). 13C NMR : δ (DMSO-d6) 167.9, 163.1, 154.2, 153.3, 152.1, 151.3, 148.5, 137.3, 136.3, 134.5, 133.2, 132.3, 129.3, 129.2, 128.7, 128.3, 128.2, 128.0, 126.7, 124.9, 122.4, 117.9,
116.4, 115.2, 113.9, 109.5, 88.0, 71.1, 65.3, 57.7, 43.0, 15.0.
……………….
http://www.google.com/patents/WO2009052264A2?cl=en
TABLE 1 1. STRUCTURES OF DEGRADATION PRODUCT AND PROCESS IMPURITIES
N-{4-[3-chloro-4-(2- (E)-4-({4-[3-chloro-4-(2- N -{4-[3-chloro-4-(2- pyrιdιnylmethoxy)anιlιno]-3-cyano-7- pyrιdιnylmethoxy)anιlιno]-3-cyano-7- pyrιdιnylmethoxy)anιlιno]-3-cyano-7-ethoxy- ethoxy-6-quιnolιnyl}acetamιde ethoxy-6-quιnolιnyl}amιno)-N,N,N- 6-quιnolιnyl}-N2,N2-dιmethylethanedιamιde trιmethyl-4-oxo-2-buten-1-amιnιum
Exact Mass 487 14 Exact Mass 544 16
Exact Mass 571 22
Process Impurity I Process Impurity J
SCHEME 1
The reaction of the free base and maleic acid occurs at an elevated temperature of from about 40 0C to about 60 0C, preferably between about 4O0C to about 5O0C. The ratio of watenn- propanol may vary, for example between about 1 :10 to about 1 :5, and the optimal ratio of watenn-propanol is about 1 :9. The water-alcohol solution may comprise from about 5% to about 20% by volume water and from about 80% to about 95% by volume alcohol. The alcohol may be n-propanol. In one embodiment, the water-alcohol solution comprises about 10% by volume water and about 90% by volume n-propanol. The volume of the solvent solution may be between about 8 to about 25 volumes, including about 10 to about 12 volumes. About 1.0-1.2 equivalents of maleic acid is used per equivalent of the free base, preferably about 1.03 equivalents of maleic acid per equivalent of the free base.
The resulting solution of the maleate salt may be clarified by filtration prior to cooling. The cooling step may be continued until the solution reaches a temperature of about 45°C or less, including a temperature of about 39°C or less, and more preferably to about 300C or less. In one embodiment, the solution is filtered after cooling to about room temperature, preferably from about 230C to about 25 0C. Typically, the maleate salt begins to crystallize out of solution once the temperature reaches 370C or below. The solution may be allowed to sit for at least 12 hours, preferably about 12 to about 15 hours at room temperature, and is then filtered and washed to recover the crystalline maleate salt product. The resulting filter cake may be washed with the same or a different water-alcohol solution to obtain the product. The product may be dried to obtain crystalline (E)-N-{4-[3-chloro-4-(2-pyridinylmethoxy)anilino]-3-cyano-7- ethoxy-6-quinolinyl}-4-(dimethylamino)-2-butenamide maleate. At this point, the maleate salt product recovered and isolated is typically in the form of the monohydrate form of the maleate salt.
……………
PAPTENT
http://www.google.com/patents/CN102731395A?cl=en
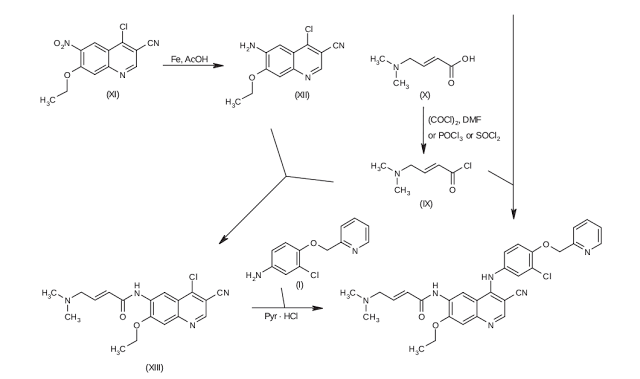
The present invention relates to a process for preparing that imatinib (neratinib, HKI-272) is a new method for its preparation and its intermediates in the preparation to the application that imatinib
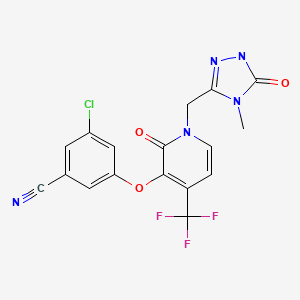
[0155] Example 14 (E)-N-(4 – (3 – chloro-4 – (2 – pyridyl) phenyl) amino] _3_ ethoxy-quinolin-6-cyano-_7_ – yl) -4 – dimethylamino-2 – butene amide
[0156]
Compound of Example 13 (20mg, 0. 037mmol) was dissolved in DMF was added potassium carbonate (10mg, 0. 07mmol), dimethylamine hydrochloride (5mg, 0. 06mmol), at room temperature for I hour, after , the reaction mixture was dropped into water, stirred for 10 minutes, filtered, washed with water and dried to give the title compound 1511 ^ 75% yield.1HNMR (300MHz, DMS0_d6): δ I. 5 (t, 3H, J = 6 · 8,13. 8), 2. 2 (br s, 6H), 3. I (d, 2H, J = 3. 8 ), 4. 3 (q, 2H, J = 7. 0,14. 2), 5. 2 (s, 2H),
6. 6 (d, 1H, J = 15. 0), 6. 8 (m, 1H), 7. 1-7. 3 (m, 2H), 7. 3-7. 4 (m, 3H), 7. 6 (d, 1H, J = 3. 9),
7. 9 (d, 1H, J = 3. 9), 8. 5 (s, 1H), 8. 6 (d, 1H, J = 3. 9), 9. 0 (s, 1H), 9. 5 (s, 1H), 9. 6 (s, 1H). ESI-MS: [M + H] + = 557. 3.
GOING BACKWARDS…………………
Example 13 (E) -4 – bromo-N-(4 – (3 – chloro-4 – (2 – pyridyl) phenyl) amino] _3_ cyano _7_ ethoxyquin -6 – yl) -2 – butene amide
Example 12 Compound (100mg, 0. 2mmol) was suspended in carbon tetrachloride was added NBS (40mg,
O. 22mmol), benzoyl peroxide (2mg, 0. Olmmol), nitrogen, refluxed for 10 hours, the reaction solution directly mixed baby gel, silica gel column chromatography to obtain the title compound isolated 60mg, yield 51%. 1HnmrgoomHz, cdci3): δ i.6 (t, 3H, J = 6. 8,13. 7), 2. 0 (d, 2H, J = 6. 9), 4. 3 (q, 2H, J = 7. 2,13. 8), 5. 3 (s, 2H), 6. I (d, 1H, J =
15. 0), 7. 0 (m, 1H), 7. 2 (m, 1H), 7. 3 (s, 1H), 7. 4 (s, 1H), 7. 6 (d, 1H, J = 8. 2), 7. 8 (d, 1H, J =
7. 6), 8. 0 (s, 1H), 8. 5 (s, 1H), 8. 6 (d, 1H, J = 4. 7), 9. 2 (s, 1H). ESI-MS: [M + H] + = 594. I.
……………
PAPER
Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity
J Med Chem 2005, 48(4): 1107
http://pubs.acs.org/doi/full/10.1021/jm040159c


 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....








































































































![VTO=\frac{\text{nominal volume of all reactors} [m^3]*\text{time per batch} [h]}{\text{output per step} [kg]}](https://i0.wp.com/upload.wikimedia.org/math/7/9/a/79aca4fd95405c01c04df9181fb3ee63.png)















 Photo by Rob Hill/MU Publications
Photo by Rob Hill/MU Publications