
Home » Uncategorized (Page 13)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Aneratrigine


Aneratrigine
2097163-74-9
5-chloro-2-fluoro-4-[4-fluoro-2-[methyl-[2-(methylamino)ethyl]amino]anilino]-N-(1,3-thiazol-4-yl)benzenesulfonamide
5-chloro-2-fluoro-4-((4-fluoro-2-(3-(methylamino)pyridin-1-yl)phenyl)amino)-N-(thiazol-4-yl)benzenesulfonamide hydrochloride
| Benzenesulfonamide, 5-chloro-2-fluoro-4-[[4-fluoro-2-[methyl[2-(methylamino)ethyl]amino]phenyl]amino]-N-4-thiazolyl- |
C19H20ClF2N5O2S2 488.0 g/mol
UNII 6A5ZY5LT78
WHO
SYN

Assignee: Daewoong Pharmaceutical Co., Ltd.
World Intellectual Property Organization, WO2017082688
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017082688&_cid=P11-M0UEPF-95506-1
Preparation of 5-chloro-2-fluoro-4-((4-fluoro-2-(3-(methylamino)pyridin-1-yl)phenyl)amino)-N-(thiazol-4-yl)benzenesulfonamide hydrochloride
Step 1) Preparation of tert-butyl (1-(2-amino-5-fluorophenyl)pyridin-3-yl)(methyl)carbamate
2,4-Difluoro-1-nitrobenzene (2.0 g, 12.6 ng/mol) and tert-butyl methyl (pyridin-3-yl)carbamate (2.5 g, 1.0 eq.) were dissolved in DMF (20 mL), and K2C03 ( 2.6 g , 1.5 eq .) was added. The internal temperature was maintained at 60–70 ° C and the mixture was stirred for 2 hours. The completion of the reaction was confirmed by TLC when the reaction solution turned deep yellow. After cooling to room temperature, ethyl acetate (EA)/H20 was added, stirred, and the layers were separated. MgS04 was added to the separated organic layer, stirred, dried, and filtered. After concentrating the filtrate under reduced pressure, the residue was dissolved in EtOH (10 mL) and distilled water (10 mL), and then Na 2 S 2 0 4 (13.0 g, 6 eq.) was added. After stirring for 2 hours while maintaining the internal temperature at 60 to 70 ° C, the completion of the reaction was confirmed by TLC when the yellow color of the reaction solution lightened and became almost colorless. After cooling to room temperature, distilled water (50 mL) was added and extracted twice with EA (100 mL). MgS0 4 was added to the organic layer, stirred, dried, and filtered. The filtrate was concentrated under reduced pressure, and the obtained residue was separated by column chromatography (n-Hexane/EA = 3/1) to obtain the title compound (2.0 g, 51. ).
1H NMR (MeOD): 6.73(m, 1H), 6.57(t, 1H), 3.23(m, 1H), 3.10(m, 2H), 2.94(m, 1H), 2.91(s, 3H), 2.25( m, 1H), 1.99(m, 1H)
Step 2) Preparation of tert-butyl thiazol-4-ylcarbamate
Thiazole-4-carboxylic acid (5.0 g, 38.8 vol) was dissolved in t-Bu0H (100 mL), and then TEA (8.1 mL, 1.5 eq.) and DPPA (7.1 mL, 1.5 eq.) were added. The internal temperature was maintained at 90–100 ° C, and the mixture was stirred for 3 days. The completion of the reaction was confirmed by TLC. The product was concentrated under reduced pressure, distilled water (50 mL) was added, and the solution was washed with EA (100 mL).
It was extracted twice. MgSQ 4 was added to the organic layer, stirred, dried, and filtered.
After concentrating the filtrate under reduced pressure, the residue was added to a small amount of EA, slurried, and the resulting solid was filtered to obtain the white title compound (4.0 g, 51.5%).
1H NMR (MeOD): 8.73(s, 1H), 7.24(s, 1H), 1.52(s, 9H)
Step 3) Preparation of tert-butyl ((4-bromo-5-chloro-2-fluorophenyl)sulfonyl)(thiazol-4-yl)carbamate
Step 2) The tert-Butyl thiazol-4-ylcarbamate (4.0 g, 20.0 ng ol) prepared in the reaction vessel was placed in a reaction vessel and the interior was replaced with nitrogen gas. After dissolving in THF (32 mL), it was cooled to _78 ° C using dry ice— acetone. After cooling, LiHMDS (22.4 mL, 1.5 eq.) was slowly added and the reaction mass was stirred for 30 minutes. 4-Bromo-5-chloro-2-fluorobenzenesulfonyl chloride (6.0 g, 1.0 eq.) was dissolved in THF (10 mL) and slowly added to the reaction mixture. The reaction mass was stirred overnight and the completion of the reaction was confirmed by TLC. Distilled water (50 mL) was added and extracted twice with EA (100 mL). MgS0 4 was added to the organic layer, stirred, dried, and filtered. After concentrating the filtrate under reduced pressure, the residue was crystallized from THF/n-hexane to obtain the title compound (4.4 g, 59.0%).
1H NMR (MeOD): 9.00(s, 1H), 8.22(d, 1H), 7.90(d, 1H), 7.78(s, 1H), 1.35(s, 9H)
Step 4) Preparation of tert-butyl (l-(2-((4-(N-(tert-butyloxycarbonyl)-N-(thiazol-4-yl)sulfamoyl)-2-chloro-5-fluorophenyl)amino)-5-fluorophenyl)pyrlidin-3-yl)(methyl)carbamate
Tert-butyl (1-(2-amino-5-fluorophenyl)pyrlidin-3-yl)(methyl)carbamate (0.5 g, 1.1 ng ol) prepared in Step 1) and tert-butyl ((4-bromo-5-chloro-2-fluorophenyl)sulfonyl)(thiazol-4-yl)carbamate (0.9 g, 1.2 eq.) prepared in Step 3) were dissolved in 1,4-dioxane (10 mL). Pd(OAc) 2 (0.03 g, 0.1 eq), rac-BINAP (0.19 g, 0.2 eq.), Cs 2 C0 3 (1.5 g, 3.0 eq.) were added to the reaction solution. After reacting at 120 ° C for 30 minutes using a microwave initiator, the completion of the reaction was confirmed by TLC. Distilled water (50 mL) was added and extracted twice with EA (100 mL).
MgS0 4 was added to the organic layer, stirred, filtered and dried. The filtrate was concentrated under reduced pressure, and the residue was separated by column chromatography (EA/n-Hexane = 1/1). This was repeated twice to obtain the title compound (2.0 g, 88.2%).
1H NMR (MeOD): 8.95(s, 1H), 7.94(d, 1H), 7.65(s, 1H), 7.14(t, 1H), 6.70(d, 1H), 6.64(t, 1H), 6.07( d, 1H)ᅳ 3.40(m, 1H), 3.28(m, 2H), 3.16(m, 1H), 2.64(s, 3H), 2.06(m, 1H), 1.89(m, 1H), 1.41(s , 9H), 1.36(s, 9H)
Step 5) Preparation of 5-chloro-2-fluoro-4-((4-fluoro-2-(3-(methylamino)pyridin-1-yl)phenyl)amino)-N-(thiazol-4-yl)benzenesulfonamide hydrochloride
Step 4) was prepared by adding 1.25 M HCl in MeOH (15 mL) to tert-butyl (1-(2-((4-(Ν-(tert-butoxycarbonyl)-N-(thiazol-4-yl)sulfamoyl)—2-chloro-5-fluorophenyl)amino)-5-fluorophenyl)pyrlidin-3-yl) (methyl)carbamate (2.0 g, 2.9 µl). The mixture was heated to 40–50 ° C and stirred overnight, and the completion of the reaction was confirmed by TLC. The product was concentrated, and methylene chloride (15 mL) was added to the residue, which was stirred for 1 hour, and the resulting solid was filtered to obtain the title compound (0.9 g, 58.8%).
1H 證 (MeOD): 8.73(s, 1H), 7.75(d, 1H), 7.12(t, 1H), 7.00(s, 1H), 6.69(d, 1H), 6.67(t, 1H), 6.05( d, 1H), 3.73(m, 1H) , 3.54(m, 1H), 3.45(m, 1H), 3.38(m, 1H), 3.26(m, 1H), 2.63(s, 3H) , 2.31(m , 1H), 1.96(m, 1H)
PATENTS
0002705578SODIUM CHANNEL BLOCKER
20180346459Substituted benzenesulfonamides as sodium channel blockers
2018533606ナトリウムチャネル遮断剤
3375782SODIUM CHANNEL BLOCKER
108349963SODIUM CHANNEL BLOCKER
1020170056461SODIUM CHANNEL BLOCKER

////////////Aneratrigine, DAEWOONG
Zevotrelvir, EDP 235
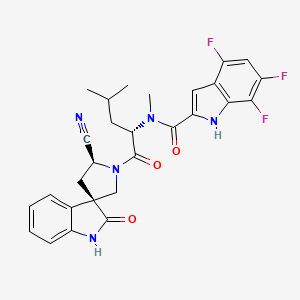
Zevotrelvir, EDP 235
cas 2773516-53-1
N-[(2S)-1-[(2′S,3R)-2′-cyano-2-oxospiro[1H-indole-3,4′-pyrrolidine]-1′-yl]-4-methyl-1-oxopentan-2-yl]-4,6,7-trifluoro-N-methyl-1H-indole-2-carboxamide
C28H26F3N5O3, 537.5
Zevotrelvir (Compound 52) is a coronavirus inhibitor with IC50 ranges of <0.1 μM and <0.1mM for 229E hCoV and SARS-CoV-23C-like (3CL) proteases, respectively. Zevotrelvir has the potential to study viral infections.
Coronaviruses are enveloped, positive-sense, single-stranded RNA viruses. The genomic RNA of CoVs has a 5′-cap structure and 3′-poly-A tail and contains at least 6 open reading frames (ORFs). The first ORF (ORF 1a/b) directly translates two polyproteins: pp1a and pp1ab. These polyproteins are processed by a 3C-Like protease (3CLpro), also known as the main protease (Mpro), into 16 non-structural proteins. These non-structural proteins engage in the production of subgenomic RNAs that encode four structural proteins, namely envelope, membrane, spike, and nucleocapsid proteins, among other accessory proteins. As a result, it is understood that 3C-Like protease has a critical role in the coronavirus life cycle.
3CLpro is a cysteine protease involved in most cleavage events within the precursor polyprotein. Active 3CLpro is a homodimer containing two protomers and features a Cys-His dyad located in between domains I and II.3CLpro is conserved among coronaviruses and several common features are shared among the substrates of 3CLpro in different coronaviruses. As there is no human homolog of 3CLpro, it is an ideal antiviral target. Although compounds have been reported to inhibit 3CLpro activity, they have not been approved as coronavirus therapies. (Refer to
WO 2004101742 A2, US 2005/0143320 Al, US 2006/0014821 Al, US 2009/0137818
Al, WO 2013049382 A2, WO 2013166319 A1, WO2018042343, WO2018023054, WO 2022013684, WO 2021252644, WO2022020711, WO 2022020242, US 11,174,231 B1, US 11,124,497 B1, WO 2005113580, and WO2006061714).
There is a need in the art for novel therapeutic agents that treat, ameliorate or prevent SARS-CoV-2 infection. The present invention provides the process of novel compounds which act in inhibiting or preventing SARS-CoV-2 viral replication and thus are used in the treatment of COVID-19 (see PCT/US21/60247).
Synthesis of substituted spirooxindole and its intermediate has been previously published (Refer to PCT/US21/60247, WO2019086142, WO 2020221811, WO2020221826, J. Med. Chem.2012, 55, 9069). However, the scale-up using previous process is very challenging due to the safety concern associated with certain intermediates, instability of certain intermediates as well as lack of purification process other than column chromatograph. Thus, there is a strong need for developing a safe and efficient processes for the large-scale preparation of these novel substituted spirooxindole derivatives.
SYNTHESIS

https://patents.google.com/patent/US11352363B1/en

PATENT
SYN
[1]. Guoqiang Wang, et al. Novel spiropyrrolidine derived antiviral drugs. Patent CN114524821A.
1.20230295175PROCESSES FOR THE PREPARATION OF SUBSTITUTED SPIROOXINDOLE DERIVATIVES
2.WO/2023/177854PROCESSES FOR THE PREPARATION OF SUBSTITUTED SPIROOXINDOLE DERIVATIVES
3.WO/2022/109363NOVEL SPIROPYRROLIDINE DERIVED ANTIVIRAL AGENTS
Enanta Pharmaceuticals, Inc.
WO2023177854




Example 15. Preparation of Preparation of (3R,5’S)-1′-(N-methyl-N-(4,6,7-trifluoro-1H-indole-2-carbonyl)-L-leucyl)-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (Compound (n))

DMF (760 kg, 8V) was added into the reaction at 0 °C (-5~5 °C) followed by compound (j) (63 kg, 1.05 eq) and N-Methylmorpholine (56 kg, 2 eq), HATU
(106 kg, 1.0 eq) and Compound (m-1) (100 kg, 1.0 eq). The reactor was rinsed with DMF (190 kg, 2V) under and warmed up to 25 °C (20~30 °C) and stirred for 5 h (3~6 h) at 25 °C (20~30 °C). After that, additional HATU (0.1 eq) was added and the reaction mixture was stirred for 16-24 h.25% Ammonium hydroxide (38 kg) was added to the reaction mixture at 25 °C (20~30 °C) and stirred for 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was then added to water (5000 kg, 50V) at 20-30°C over 2 h and the resulting slurry was stirred for 2 h (1~5 h) at 25 °C (20~30 °C). The mixture was filtered and the cake was rinsed with water (500 kg, 5 V). The cake was dissolved in ethyl acetate (1350 kg, 15 V) and washed with 10% sodium chloride solution (500 kg) for three times. The organic layer was separated to 1.5-2.5V at not more than 45℃ under vacuum. The solution was cooled to 25 °C (20~30 °C) and Dichloromethane (660 kg, 5V) was added. The mixture was stirred for 2 h (2~5 h) at 25 °C (20~30 °C) and a slurry was formed. n-Heptane (137 kg, 2V) was added dropwise over 0.5 h (0.5~2 h) at 25 °C (20~30 °C) and stirred for additional 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was filtered and the wet cake was rinsed with DCM/heptane (5/2). The wet cake was dried at 50 °C (45~55 °C) for 20 h (15~25 h) to provide Compound (n) as the white solid in 80-85% yield.
1H NMR (300 MHz, DMSO-d6) δ 12.46 (s, 1H), 10.68 (s, 1H), 7.56 (s, 1H), 7.15 – 7.00 (m, 3H), 6.91 (t, J = 4.4 Hz, 2H), 6.84 (d, J = 7.7 Hz, 1H), 6.55 (d, J = 2.8 Hz, 1H), 5.34 (t, J = 7.3 Hz, 1H), 4.63 (dd, J = 9.8, 8.0 Hz, 1H), 3.83 (q, J = 10.3 Hz, 2H), 3.45 (qd, J = 7.0, 5.1 Hz, 1H), 3.16 (s, 3H), 2.35 – 2.13 (m, 2H), 1.69 (t, J = 7.1 Hz, 2H), 1.56 (dq, J = 13.1, 6.5 Hz, 1H), 0.93 (dd, J = 12.2, 6.3 Hz, 6H).
Example 16. Preparation of (3R,5’S)-1′-(N-methyl-N-(4,6,7-trifluoro-1H-indole-2-carbonyl)-L-leucyl)-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (Compound (n))
DMF solution of Compound (m-2) (1 kg, 1.0 eq.) was added to a reactor at around 0-10oC. Compound (l) (600 g, 1.0 eq.), NMM (3.00 eq., 850 g) and HATU (1.00 eq., 1.06 kg) was added to the reactor while maintaining the temperature at 0-10oC; The reaction was warmed to 20±5oC, and stirred for at least 6 hours at 20±5oC. HATU (0.20 eq., 210 g) was added to the reactor at 20±5oC and stirred for at least 6 hours at 20±5oC.25% Ammonium hydroxide (390 g, 1.0 eq) was added to the reaction mixture at 20 °C and stirred for 2 h (1~3 h) at 20 °C. EtOAc (14.0 V) and water (14 V) was added at around 25oC over 20 minutes, and the
solution was stirred for at least 30 min. Aqueous phase was extracted with EtOAc for three times and the organic phase was combined, and washed with 10% aq. NaCl for three times at 20±5oC. The organic phase was concentrated to 6 V then EtOH (7.0 V) was charged. The EtOAc-EtOH solvent swap was repeated for three times and concentrated to 5 V before water (7.0 v) was added at 20±5oC. The mixture was cooled to 0-10oC and stirred for 1 h before being filtered. The filter cake was dissolved in ethyl acetate (15 V) and washed with 10% sodium chloride solution for three times. The organic layer was concentrated to 2-3V at not more than 45℃ under vacuum. The solution was cooled to 25 °C (20~30 °C) and Dichloromethane (5V) was added. The mixture was stirred for 2 h (2~5 h) at 25 °C (20~30 °C) and a slurry was formed. n-Heptane (2V) was added dropwise over 0.5 h (0.5~2 h) at 25 °C (20~30 °C) and stirred for additional 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was filtered and wet cake was rinsed with DCM/heptane (5/2). The wet cake was dried at 50 °C (45~55 °C) for 20 h (15~25 h) to provide Compound (n) as the white solid in about 70-75% yield over two steps.
1H NMR (300 MHz, DMSO-d6) δ 12.46 (s, 1H), 10.68 (s, 1H), 7.56 (s, 1H), 7.15 – 7.00 (m, 3H), 6.91 (t, J = 4.4 Hz, 2H), 6.84 (d, J = 7.7 Hz, 1H), 6.55 (d, J = 2.8 Hz, 1H), 5.34 (t, J = 7.3 Hz, 1H), 4.63 (dd, J = 9.8, 8.0 Hz, 1H), 3.83 (q, J = 10.3 Hz, 2H), 3.45 (qd, J = 7.0, 5.1 Hz, 1H), 3.16 (s, 3H), 2.35 – 2.13 (m, 2H), 1.69 (t, J = 7.1 Hz, 2H), 1.56 (dq, J = 13.1, 6.5 Hz, 1H), 0.93 (dd, J = 12.2, 6.3 Hz, 6H).
Example 17. Preparation of (3R,5’S)-1′-(N-methyl-N-(4,6,7-trifluoro-1H-indole-2-carbonyl)-L-leucyl)-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (Compound (n))
DMF (10.0 v) was added to a reactor at 25 °C followed by Compound (l) (4.4 kg, 1.0 eq.), NMM (3.0 eq.) Compound (m-3) (1.0 eq.) and HATU (1.0 eq) at 20-25oC. The reaction mixture was stirred for at least 12 hours at 20-25 °C. Once reaction was complete, aqueous ammonium hydroxide (1.0 eq.) was to the reaction system at 20-25 °C, then stirred for at least 2 hours at 20-25oC. The reaction mixture was then added to water (220 kg, 50V) at 20-30°C over 2 h and the resulting slurry was stirred for 2 h (1~5 h) at 25 °C (20~30 °C). The mixture was filtered and the cake was rinsed with water (22 kg, 5 V). The cake was dissolved in ethyl acetate (135 g, 15 V) and washed with 10% sodium chloride solution (22 kg) for three times. The organic layer was separated to 1.5-2.5V at not more than 45 ℃ under vacuum. The solution was cooled to 25 °C (20~30 °C) and Dichloromethane (5V) was added. The mixture was stirred for 2 h (2~5 h) at 25 °C (20~30 °C) and a slurry was formed. n-Heptane (2V) was added dropwise over 0.5 h (0.5~2 h) at 25 °C (20~30 °C) and stirred for additional 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was filtered and wet cake was rinsed with DCM/heptane (5/2). The wet cake was dried at 50 °C (45~55 °C) for 20 h (15~25 h) to provide Compound (n) as the white solid in 80-85% yield.
1H NMR (300 MHz, DMSO-d6) δ 12.46 (s, 1H), 10.68 (s, 1H), 7.56 (s, 1H), 7.15 – 7.00 (m, 3H), 6.91 (t, J = 4.4 Hz, 2H), 6.84 (d, J = 7.7 Hz, 1H), 6.55 (d, J = 2.8 Hz, 1H), 5.34 (t, J = 7.3 Hz, 1H), 4.63 (dd, J = 9.8, 8.0 Hz, 1H), 3.83 (q, J = 10.3 Hz, 2H), 3.45 (qd, J = 7.0, 5.1 Hz, 1H), 3.16 (s, 3H), 2.35 – 2.13 (m, 2H), 1.69 (t, J = 7.1 Hz, 2H), 1.56 (dq, J = 13.1, 6.5 Hz, 1H), 0.93 (dd, J = 12.2, 6.3 Hz, 6H). Example 18. Preparation of N-((S)-1-((3R,5’S)-5′-cyano-2-oxospiro[indoline-3,3′-pyrrolidin]-1′-yl)-4-methyl-1-oxopentan-2-yl)-4,6,7-trifluoro-N-methyl-1H-indole-2-carboxamide toluene solvate (Compound (I))
(I))

Ethyl acetate (630 kg, 10 V) was added into reactor (R1) followed by Compound (n) (70 kg). Make sure the water content was less than 0.20% (w/w). The reaction was cooled to 0 °C (-5 – 5°C) and then triethylamine (89.6 kg) was added followed by trifluoroacetic anhydride (92.4 kg) at 0 °C (-5 – 5°C). The reaction was stirred for 1 h (0.5~2 h) at 0 °C (-5 – 5°C). Once the reaction was complete, the reaction mixture was added slowly to 0.2 N aqueous HCl solution (700 kg) over 1 h at 0 °C (-5~5 °C). The resulting solution was stirred for 30 min at 0 °C (-5~5 °C) and the organic layer was separated.1% aqueous ammonium hydroxide (700 kg) was added to the organic layer and stirred at 20 °C for 30 min (15~25 °C). The organic layer was separated and washed with 10% brine for four times. Then the organic layer was separated and distilled to 2-3 V. Toluene-EtOAc swap was performed until precipitate was observed at 3-4 V. Then Toluene (5-6 V) was added and the slurry was stirred at 50 oC for 2 h. Then the solution was cooled down to 20 oC over 1-2 h and stirred for 10 hr (6~14 hr) at 20 °C (15~25 °C). The reaction mixture was filtered and the wet cake was rinsed with toluene (120 kg, 2V). The wet cake was then dried at 50˚C (45~55 °C) for 48 hr to provide desired compound (o) as a white solid in 80-85% yield.
1H NMR (400 MHz, Acetone-d6) δ 11.17 (s, 1H), 9.65 (s, 1H), 7.02 (dd, J = 13.7, 7.3 Hz, 2H), 6.94 (dd, J = 6.0, 3.5 Hz, 1H), 6.92 – 6.85 (m, 2H), 6.81 (t, J = 7.5 Hz, 1H), 5.56 (dd, J = 9.4, 5.6 Hz, 1H), 5.21 (t, J = 8.3 Hz, 1H), 4.25 (d, J = 10.7 Hz, 1H), 3.99 (d, J = 10.6 Hz, 1H), 3.43 (s, 3H), 2.79 – 2.61 (m, 2H), 1.93 (ddd, J = 14.4, 9.5, 5.1 Hz, 1H), 1.79 (ddd, J = 14.2, 8.7, 5.6 Hz, 1H), 1.64 (dpd, J = 8.7, 6.6, 5.1 Hz, 1H), 0.98 (dd, J = 18.5, 6.6 Hz, 6H).
US20230103494
CN114524821
SCHEME

MAIN

////////Zevotrelvir, EDP 235
O=C1[C@@]2(CN([C@@H](C2)C#N)C([C@H](CC(C)C)N(C)C(C3=CC4=C(F)C=C(F)C(F)=C4N3)=O)=O)C5=CC=CC=C5N1
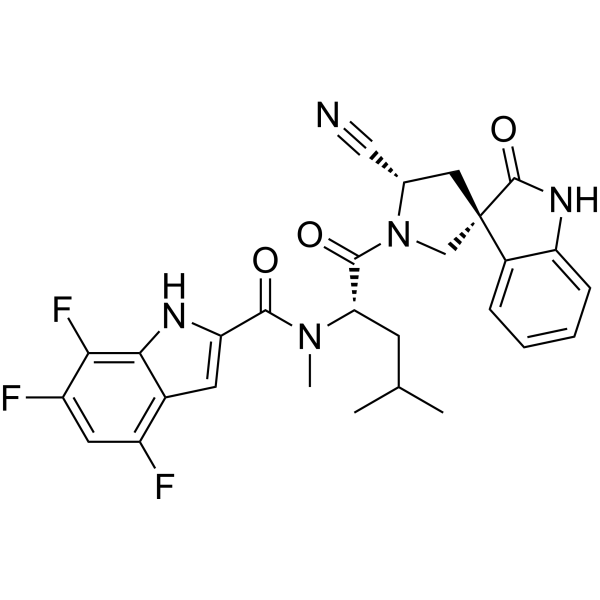
Afimetoran

Afimetoran BMS-986256, WHO 11516
cas 2171019-55-7
2-[4-[2-(7,8-dimethyl-[1,2,4]triazolo[1,5-a]pyridin-6-yl)-3-propan-2-yl-1H-indol-5-yl]piperidin-1-yl]acetamide
C26H32N6O,444.583, phase 1
Afimetoran is an immunomodulator and an antagonist of toll-like receptors 7 and 8.1,2 It is also is under investigation in clinical trial NCT04269356 (Study to Assess the Way the Body Absorbs, Distributes, Breaks Down and Eliminates Radioactive BMS-986256 in Healthy Male Participants).

Ref
WO2018005586
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018005586&_cid=P20-M0RQ0D-09010-1
The invention further pertains to pharmaceutical compositions containing at least one compound according to the invention that are useful for the treatment of conditions related to TLR modulation, such as inflammatory and autoimmune diseases, and methods of inhibiting the activity of TLRs in a mammal.
Toll/IL-1 receptor family members are important regulators of inflammation and host resistance. The Toll-like receptor family recognizes molecular patterns derived from infectious organisms including bacteria, fungi, parasites, and viruses (reviewed in Kawai, T. et al., Nature Immunol., 11:373-384 (2010)). Ligand binding to the receptor induces dimerization and recruitment of adaptor molecules to a conserved cytoplasmic motif in the receptor termed the Toll/IL-1 receptor (TIR) domain. With the exception of TLR3, all TLRs recruit the adaptor molecule MyD88. The IL-1 receptor family also contains a cytoplasmic TIR motif and recruits MyD88 upon ligand binding (reviewed in Sims, J.E. et al., Nature Rev. Immunol., 10:89-102 (2010)).
Toll-like receptors (TLRs) are a family of evolutionarily conserved, transmembrane innate immune receptors that participate in the first-line defense. As pattern recognition receptors, the TLRs protect against foreign molecules, activated by pathogen associated molecular patterns (PAMPs), or from damaged tissue, activated by danger associated molecular patterns (DAMPs). A total of 13 TLR family members have been identified, 10 in human, that span either the cell surface or the endosomal compartment. TLR7-9 are among the set that are endosomally located and respond to single-stranded RNA (TLR7and TLR8) or unmethylated single-stranded DNA containing cytosine-phosphate-guanine (CpG) motifs (TLR9).
Activation of TLR7/8/9 can initiate a variety of inflammatory responses (cytokine production, B cell activation and IgG production, Type I interferon response). In the case of autoimmune disorders, the aberrant sustained activation of TLR7/8/9 leads to worsening of disease states. Whereas overexpression of TLR7 in mice has been shown to exacerbate autoimmune disease, knockout of TLR7 in mice was found to be protective against disease in lupus-prone MRL/lpr mice. Dual knockout of TLR7 and 9 showed further enhanced protection.
As numerous conditions may benefit by treatment involving modulation of cytokines, IFN production and B cell activity, it is immediately apparent that new compounds capable of modulating TLR7 and/or TLR8 and/or TLR9 and methods of using these compounds could provide substantial therapeutic benefits to a wide variety of patients.
The present invention relates to a new class of [1,2,4]triazolo[1,5-a]pyridinyl substituted indole compounds found to be effective inhibitors of signaling through TLR7/8/9. These compounds are provided to be useful as pharmaceuticals with desirable stability, bioavailability, therapeutic index, and toxicity values that are important to their drugability.
EXAMPLE 15
2-(4-(2-(7,8-dimethyl-[1,2,4]triazolo[1,5-a]pyridin-6-yl)-3-isopropyl-1H-indol-5-yl) piperidin-1-yl)acetamide
To a reaction flask were added
6-(3-isopropyl-5-(piperidin-4-yl)-1H-indol-2-yl)-7,8-dimethyl-[1,2,4]triazolo[1,5-a]pyrid ine, 2 HCl (47.66 g, 104 mmol), DCE (220 mL), DBU (62.4 mL, 414 mmol), and 2-bromoacetamide (17.14 g, 124 mmol). The reaction flask was capped. The reaction mixture was stirred overnight at room temperature. The reaction mixture was concentrated, diluted with water, and stirred for 30 minutes then filtered. The solid was recrystallized using ethanol to afford 2-(4-(2-(7,8-dimethyl-[1,2,4]triazolo[1,5-a] pyridin-6-yl)-3-isopropyl-1H-indol-5-yl)piperidin-1-yl)acetamide (42.3 g, 93 mmol,
90% yield) as a white solid. LCMS MH+: 445. HPLC Ret. Time 1.20 min. Method QC-ACN-TFA-XB. 1HNMR (400 MHz, DMSO-d6) δ 10.97-10.86 (m, 1H), 8.78-8.69 (m, 1H), 8.54-8.40 (m, 1H), 7.64-7.49 (m, 1H), 7.30-7.21 (m, 2H), 7.17-7.09 (m, 1H), 7.06-6.93 (m, 1H), 2.99-2.82 (m, 5H), 2.62-2.54 (m, 4H), 2.24-2.12 (m, 5H), 1.92-1.72 (m, 4H), 1.37-1.29 (m, 6H).
ACS Medicinal Chemistry Letters (2022), 13(5), 812-818 83%
References
- Bristol-Myers Squibb: Investor Series [Link]
- Bristol-Myers Squibb: Investor Series [Link]
- MedKoo Biosciences: Afimetoran [Link]
//////////////Afimetoran, BMS-986256, BMS 986256, WHO 11516, phase 1
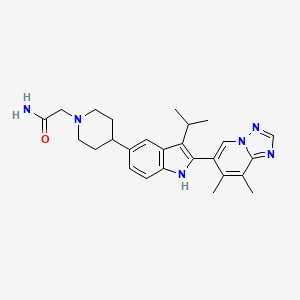
INAXAPLIN
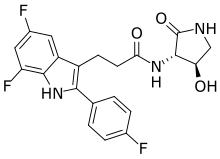
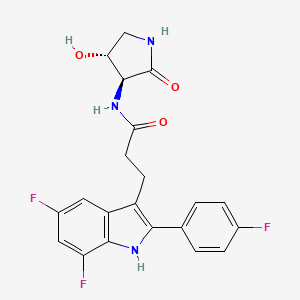
INAXAPLIN, 2446816-88-0
- 2446816-88-0
- S2SJ2RVZ6Y
- UNII-S2SJ2RVZ6Y
- C21H18F3N3O3
3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxopyrrolidin-3-yl]propanamide
Inaxaplin (VX-147) is a small-molecule apolipoprotein L1 inhibitor developed by Vertex Pharmaceuticals for APOL1-mediated kidney disease. In preliminary studies the drug has shown promise in treating people with kidney disease and multiple gain of function mutations on the APOL1 gene.[1][2]
FSGS is a disease of the podocyte (glomerular visceral epithelial cells) responsible for proteinuria and progressive decline in kidney function. NDKD is a disease characterized by hypertension and progressive decline in kidney function. Human genetics support a causal role for the G1 and G2 APOL1 variants in inducing kidney disease. Individuals with 2 APOL1 risk alleles are at increased risk of developing primary (idiopathic) FSGS, human immunodeficiency virus (HIV)-associated FSGS, and NDKD. Currently, FSGS and NDKD are managed with symptomatic treatment (including blood pressure control using blockers of the renin angiotensin system), and patients with FSGS and heavy proteinuria may be offered high dose steroids. Corticosteroids induce remission in a minority of patients and are associated with numerous side effects. These patients, in particular individuals of recent sub-Saharan African ancestry with 2 APOL1 risk alleles, experience rapid disease progression leading to end-stage renal disease (ESRD). Thus, there is an unmet medical need for treatment for FSGS and NDKD.
SCHEME
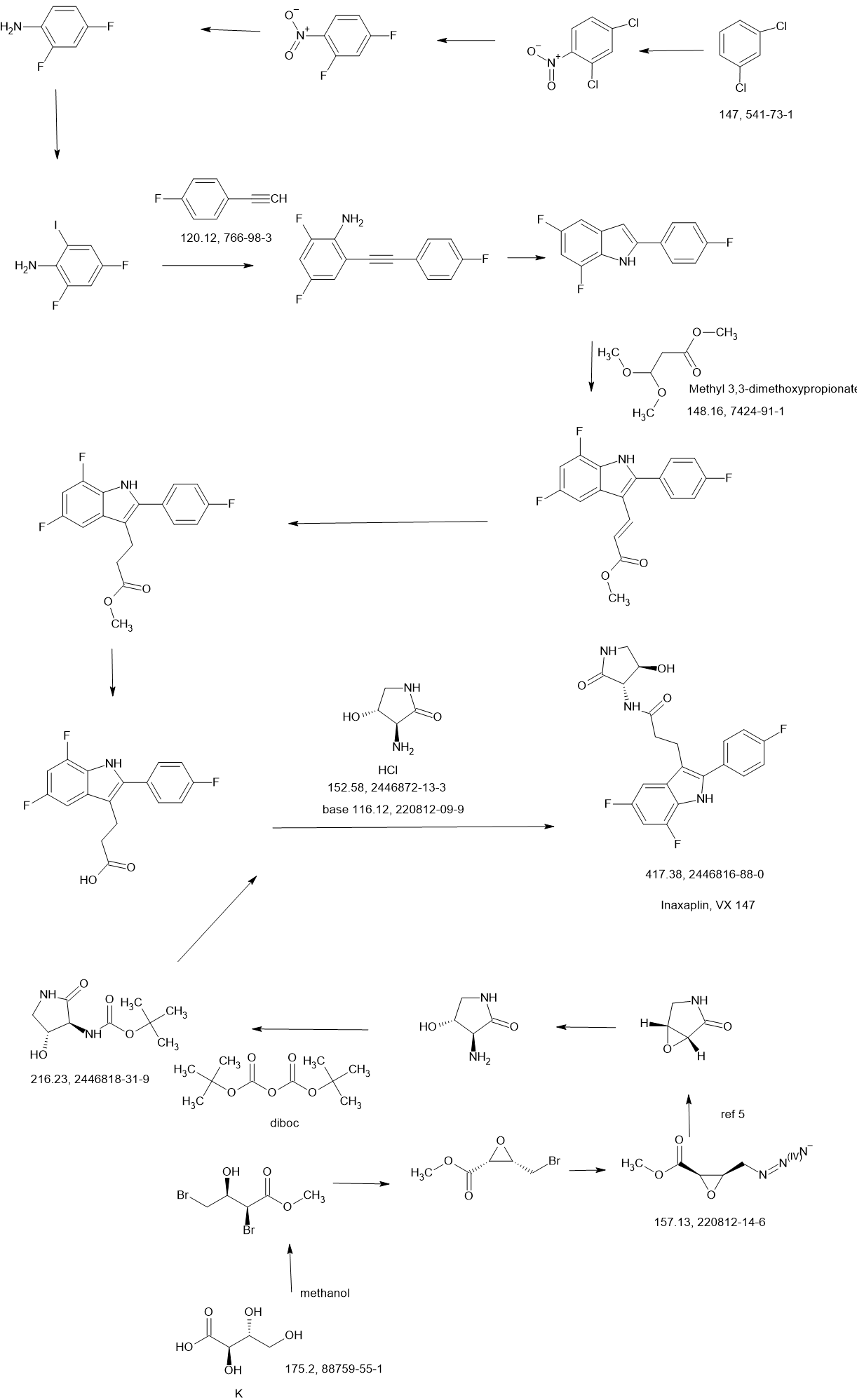
WO2021252863
US20210275496
WO2020131807
WO2021252849
PATENT
Compound 2

3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide (2)
Synthesis of 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide (2)
Alternative Procedure for Synthesis of Compound (2)
Step 1. Synthesis of Methyl (E)-3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]prop-2-enoate (C51)
Step 2. Synthesis of Methyl 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]propanoate (C52)
Step 3. Synthesis of 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]propanoic Acid (S12)
Step 4. Synthesis of 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide (2)
Re-Crystallization of Compound 2
Form A of Compound 2
Hydrate Form A of Compound 2
| 200 mg of Compound 2 was charged with 10 mL of water. The slurry was cooled to 5° C. and allowed to stir. Hydrate A was observed after 3 days of stirring. |
PATENT
PATENT

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
//////////
| Clinical data | |
|---|---|
| Other names | VX-147 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2446816-88-0 |
| PubChem CID | 147289591 |
| ChemSpider | 115009344 |
| UNII | S2SJ2RVZ6Y |
| ChEMBL | ChEMBL5083170 |
| Chemical and physical data | |
| Formula | C21H18F3N3O3 |
| Molar mass | 417.388 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
- ^ Gbadegesin, Rasheed; Lane, Brandon (August 2023). “Inaxaplin for the treatment of APOL1-associated kidney disease”. Nature Reviews Nephrology. 19 (8): 479–480. doi:10.1038/s41581-023-00721-0. PMC 10461697. PMID 37106136.
- ^ Egbuna, Ogo; Zimmerman, Brandon; Manos, George; Fortier, Anne; Chirieac, Madalina C.; Dakin, Leslie A.; Friedman, David J.; Bramham, Kate; Campbell, Kirk; Knebelmann, Bertrand; Barisoni, Laura; Falk, Ronald J.; Gipson, Debbie S.; Lipkowitz, Michael S.; Ojo, Akinlolu; Bunnage, Mark E.; Pollak, Martin R.; Altshuler, David; Chertow, Glenn M. (16 March 2023). “Inaxaplin for Proteinuric Kidney Disease in Persons with Two APOL1 Variants”. New England Journal of Medicine. 388 (11): 969–979. doi:10.1056/NEJMoa2202396. PMID 36920755. S2CID 257534251.
………///////INAXAPLIN, VX-147, VX 147
Golcadomide (CC-99282)

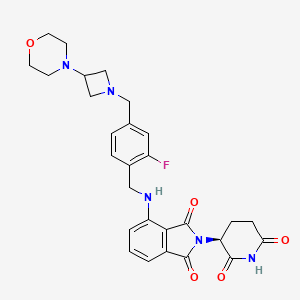
Golcadomide (CC-99282)
| Molecular Weight | 535.57 |
| Formula | C28H30FN5O5 |
| CAS No. | 2379572-34-4 |
(S)-2-(2,6-Dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline-1,3-dione
2-[(3S)-2,6-dioxopiperidin-3-yl]-4-[[2-fluoro-4-[(3-morpholin-4-ylazetidin-1-yl)methyl]phenyl]methylamino]isoindole-1,3-dione
- 2-[(3S)-2,6-Dioxo-3-piperidinyl]-4-[[[2-fluoro-4-[[3-(4-morpholinyl)-1-azetidinyl]methyl]phenyl]methyl]amino]-1H-isoindole-1,3(2H)-dione (ACI)
- (S)-2-(2,6-Dioxopiperidin-3-yl)-4-[[2-fluoro-4-[(3-morpholinoazetidin-1-yl)methyl]benzyl]amino]isoindoline-1,3-dione
- CC 1007548
- CC 99282
- CC-99282
- CC1007548
- Golcadomide
- WHO 12305
Golcadomide HCl, 2639939-36-7
Chemical Name: CC-99282 Hydrochloride; 2-[(3S)-2,6-Dioxopiperidin-3-yl]-4-[[2-fluoro-4-[(3-morpholin-4-ylazetidin-1-yl)methyl]phenyl]methylamino]isoindole-1,3-dione hydrochloride
Novel potent and orally active cereblon (CRBN) E3 ligase modulator
Ref: https://www.sciencedirect.com/science/article/abs/pii/S0268960X22000686
Golcadomide is a modulator of the E3 ubiquitin ligase complex containing cereblon (CRL4-CRBN E3 ubiquitin ligase), with potential immunomodulating and antineoplastic activities. Upon administration, golcadomide specifically binds to cereblon (CRBN), thereby affecting the ubiquitin E3 ligase activity, and targeting certain substrate proteins for ubiquitination. This induces proteasome-mediated degradation of certain transcription factors, some of which are transcriptional repressors in T-cells. This leads to modulation of the immune system, including activation of T-lymphocytes, and downregulation of the activity of other proteins, some of which play key roles in the proliferation of certain cancer cell types. CRBN, the substrate recognition component of the CRL4-CRBN E3 ubiquitin ligase complex, plays a key role in the ubiquitination of certain proteins.

SCHEME

SIDE CHAIN
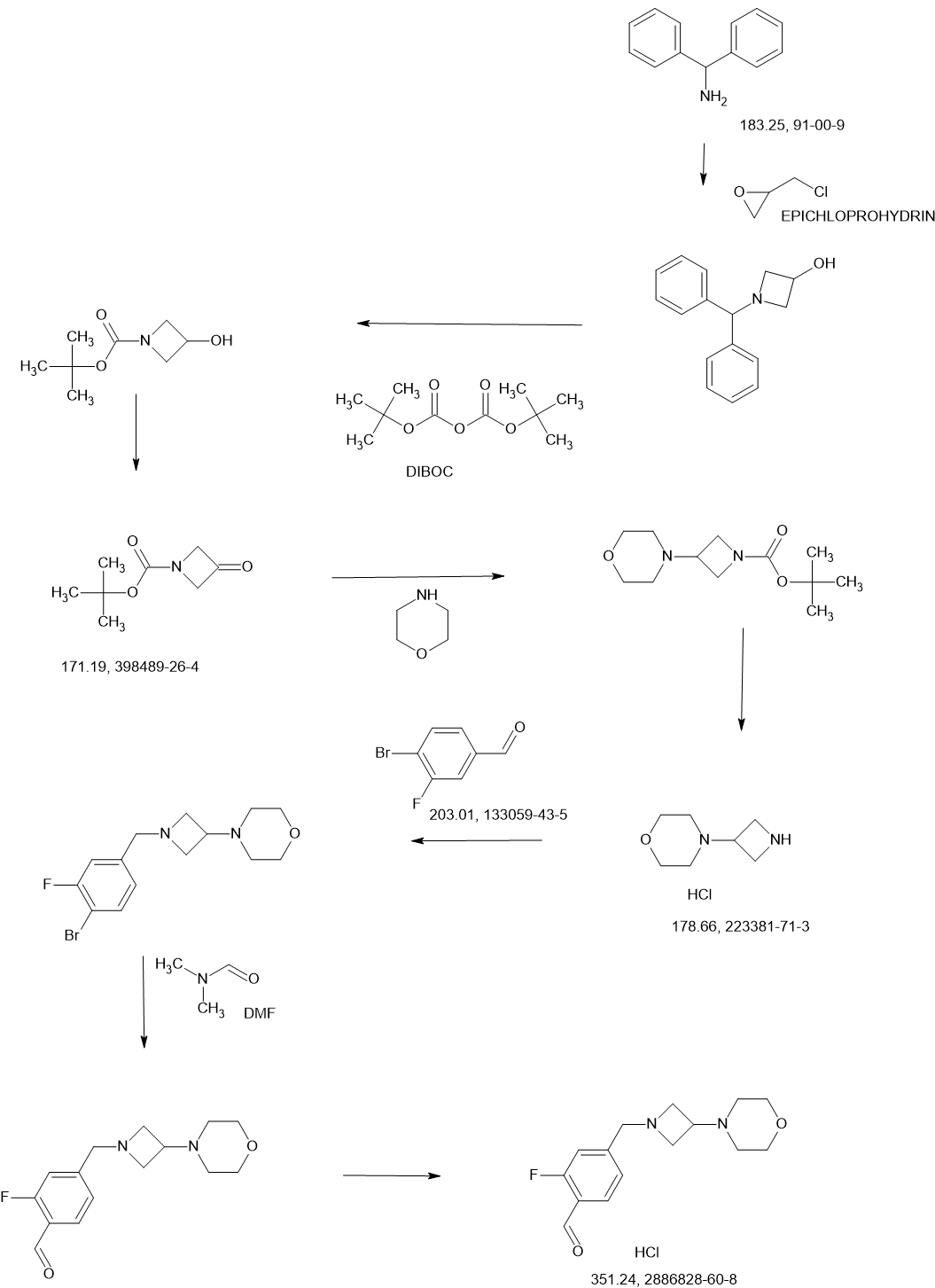
MAIN

PATENT
US20220324855
Example 1: Synthesis of (S)-2-(2,6-Dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline-1,3-dione (Compound 1)
PATENT
WO2022271557
Example 1: Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3- morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione

[00242] Synthesis of Ethyl 4-nitro-1,3-dioxo-isoindoline-2-carboxylate (Compound 10): To a solution of 4-nitroisoindoline-1,3-dione (Compound 11, 440 g, 2.29 mol) and TEA (262 g, 2.59 mol, 359 mL) in dry DMF (2.2 L) was cooled to 0 °C and ethyl chloroformate (313 g, 2.89 mol, 275 mL) was added dropwise over 5 minutes. The reaction mixture was stirred at 22 °C for 10 hours. The mixture was slowly added to chilled water (10 L) and the resulting suspension stirred for 5 minutes. The suspension was filtered and the filter cake was washed with water (1 L). The solid was dissolved with ethyl acetate (5 L) and the organic phase was washed with aqueous HCl (1 M, 1 L), water (2 L) and brine (2 L). The organic phase was dried over sodium sulfate , filtered and concentrated to give Compound 10 (360 g, 59%) as a white solid. 1 H NMR (400 MHz CDCl 3 ) δ ppm 8.24 (d, J = 7.6 Hz, 1H), 8.19 (d, J = 8.4 Hz, 1H), 8.06-8.02 (m, 1H), 4.49 (q, J = 7.2 Hz, 2H), 1.44 (t, J = 6.8 Hz, 3H).
[00243] Synthesis of tert-Butyl (4S)-5-amino-4-(4-nitro-1,3-dioxo-isoindolin-2-yl)-5-oxo-pentanoate (Compound 6): To a solution of Compound 10 (165 g, 625 mmol) and DIEA (113 g, 874 mmol, 153 mL) in dry THF (1700 mL) was added tert-butyl (4S)-4,5-diamino-5-oxo-pentanoate hydrochloride ( 149 g, 625 mmol) and heated at reflux for 10 hours. The reaction mixture was concentrated under reduced pressure. The resulting residue was diluted with methyl tert-butyl ether (5 L) and stirred at 20 °C for 1 hour. The suspension was filtered and the filter cake was dissolved with DCM (4 L). The organic phase was washed with water (1.5 L x 3), brine (1.5 L) and dried over sodium sulfate . The organic phase was filtered and concentrated under reduced pressure to give a light yellow oil. The oil was diluted with hexane / ethyl acetate (10/1, 2 L) and stirred until a light yellow suspension formed. The suspension was filtered and the filter cake was triturated and concentrated in vacuum to give Compound 6 (175 g, 74%) as a light yellow solid. 1 H NMR (400 MHz CDCl3) δ ppm 8.12 (d, J = 8.0 Hz, 2H), 7.94 (t, J = 8.0 Hz, 1H), 6.48 (s, 1H), 5.99 (s, 1H), 4.84- 4.80 (m, 1H), 2.49-2.44 (m, 2H), 2.32-2.27 (m, 2H), 1.38 (s, 9H).
[00244] Synthesis of tert-Butyl (S)-5-amino-4-(4-amino-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 5): To a suspension of Compound 6 (170.0 g, 450.5 mmol, 1.00 eq) in DMA (1.00 L) was added palladium on carbon (50.0 g, 10% purity) under nitrogen. The suspension was degassed under vacuum and purged with hydrogen gas several times. The mixture was stirred under hydrogen gas (50 psi) at 25 °C for 16 hours. The mixture was filtered and the filtrate was poured into cooled water (3.0 L). The mixture was stirred at 10 °C for 1 hour and filtered. The filter cake was washed with water (700 mL) and dissolved in DCM (1.00 L). The organic phase was dried over sodium sulfate , filtered and concentrated under reduced pressure to give Compound 5 (107 g, 68%) as a green solid. 1 H NMR (400 MHz DMSO-d 6 ) δ ppm 7.52 (s, 1H), 7.43 (dd, J = 8.4, 7.2 Hz, 1H), 7.13 (s, 1H), 6.95-6.99 (m, 2H), 6.42 (s, 2H), 5.75 (s, 1H), 4.47-4.51 (m, 1H), 2.32-2.33 (m, 1H), 2.14-2.20 (m, 3H), 1.32 (s, 9H); HPLC purity, 100.0%; SFC purity, 100.0% ee.
[00245] Synthesis of 2-fluoro-4-(hydroxymethyl)benzaldehyde (Compound 8): To a solution of 4-(((tert-butyldimethylsilyl)oxy)methyl)-2-fluorobenzaldehyde (370.0 g, 1.38 mol, 1.00 eq ) in THF (1.85 L) was added a solution of p-toluenesulfonic acid monohydrate (78.7 g, 413.6 mmol, 0.30 eq) in water (1.85 L) drop-wise at 10 °C. The mixture was stirred at 27 °C for 16 hours. TEA (80 mL) was added drop-wise and stirred for 10 minutes. The organic phase was separated and the aqueous phase was extracted with ethyl acetate (600 mL × 4). The combined
organic phase was washed with brine (1.50 L), dried over anhydrous sodium sulfate , filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give Compound 8 (137.5 g, 76%) as a yellow oil. 1 H NMR (400 MHz CDCl 3 ) δ ppm 10.34 (s, 1H), 7.86 (dd, J = 8.0, 7.2 Hz, 1H), 7.25 (s, 1H), 7.22 (d, J = 4.4 Hz, 1H) , 4.79 (d, J = 6.0 Hz, 2H), 1.91 (t, J = 6.0 Hz, 1H).
[00246] Synthesis of tert-Butyl (S)-5-amino-4-(4-((2-fluoro-4-(hydroxymethyl)benzyl)amino)-1,3-dioxoisoindolin-2-yl)-5- oxopentanoate (Compound 2-b): To a solution of Compound 5 (100.0 g, 287.9 mmol, 1.00 eq) and Compound 8 (57.7 g, 374.3 mmol, 1.30 eq) in dry DCM (1.00 L) was added TFA (164.1 g , 1.44 mol, 5.00 eq) at 0 °C. The reaction mixture was stirred at 28 °C for 2 hours. To the solution was added sodium cyanoborohydride (27.1 g, 431.8 mmol, 1.50 eq) at 0 °C. The mixture was stirred at 28 °C for 30 minutes. The reaction mixture was quenched by addition of MeOH (600 mL) and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give Compound 2-b (110.0 g, 74.0%) as a yellow solid. 1 H NMR (400 MHz, DMSO-d6) δ ppm 7.56 (s, 1H), 7.50 (dd, J = 8.4, 7.2 Hz, 1H), 7.34 (t, J = 8.0 Hz, 1H), 7.02-7.18 ( m, 4H), 6.94-7.01 (m, 2H), 4.57 (d, J = 6.0 Hz, 2H), 4.47-4.53 (m, 3H), 2.31-2.35 (m, 1H), 2.15-2.22 (m, 3H), 1.31 (s, 9H); HPLC purity, 94.0%; SFC purity, 100.0% ee.
[00247] Synthesis of tert-butyl (S)-5-amino-4-(4-((4-(chloromethyl)-2-fluorobenzyl)amino)-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 2-a): To a solution of Compound 2-b (100.0 g, 206.0 mmol, 1.00 eq) in NMP (430.0 mL) was added DIEA (79.9 g, 617.9 mmol, 3.00 eq) and MsCl (47.2 g, 411.9 mmol, 2.00 eq) at 0 °C. The ice bath was removed, and the reaction was stirred at 28°C for 10 hours. The reaction was poured into cooled water (<10°C, 2.0 L) and stirred for 10 minutes. The mixture was extracted with methyl tert-butyl ether (750 mL x 3). The combined organic layer was washed with brine (1.25 L), dried over sodium sulfate , filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give Compound 2-a (86.0 g, 81.2%) as a yellow solid.
1 H NMR (400 MHz DMSO-d 6 ) δ ppm 7.55 (s, 1H), 7.50 (dd, J = 8.4, 7.2 Hz, 1H), 7.38 (t, J = 8.0Hz, 1H), 7.31 (dd, J = 10.8, 1.6 Hz, 1H), 7.23 (dd, J = 8.0, 1.6 Hz, 1H), 7.16 (s, 1H), 7.11 (t, J = 6.4 Hz, 1H), 7.00 (d, J = 7.2 Hz, 1H), 6.95 (d, J = 8.4 Hz, 1H), 4.74 (s, 2H), 4.61 (d, J = 6.4 Hz, 2H), 4.49-4.53 (m, 1H), 2.29-2.38 (m , 1H), 2.16-2.25 (m, 3H), 1.30 (s, 9H); HPLC purity, 98.0%; SFC purity, 100.0% ee.
[00248] Synthesis of tert-butyl (S)-5-amino-4-(4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)-1,3- dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 2): To a solution of 4-(azetidin-3-yl)morpholine hydrochloride (Compound 7 HCl, 30.5 g, 170.7 mmol, 1.00 eq) and DIEA (66.2 g, 512.0 mmol, 3.00 eq) in DMSO (350.0 mL) was added to a solution of Compound 2-a (86 g, 170.65 mmol, 1.00 eq) in DMSO (350.0 mL) drop-wise at 15 °C. The reaction mixture was stirred at 28 °C for 16 hours. The reaction mixture was poured into cold half saturated brine (<10°C, 2.5 L) and extracted with ethyl acetate (1.50 L, 1.00 L, 800.0 mL). The combined organic phase was washed with saturated brine (1.50 L), dried over sodium sulfate , filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give Compound 2 (68.3 g, 65.7%) as a yellow solid. 1 H NMR (400 MHz DMSO-d 6 ) δ ppm 7.55 (s, 1H), 7.50 (dd, J = 8.4, 7.2 Hz, 1H), 7.31 (t, J = 8.0 Hz, 1H), 7.16 (s, 1H), 6.94-7.10 (m, 5H), 4.56 (d, J = 6.4 Hz, 2H), 4.49-4.52 (m, 1H), 3.54-3.55 (m, 6H) 3.31-3.32 (m, 3H), 2.81-2.88 (m, 3H), 2.29-2.38 (m, 1H), 2.15-2.25 (m, 7H), 1.30 (s, 9H); HPLC purity, 100.0%; SFC purity, 100.0% ee.
[00249] Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione (Compound 1): A solution of Compound 2 (30.0 g, 49.2 mmol, 1.00 eq) and benzenesulfonic acid (31.1 g, 196.8 mmol, 4.00 eq) in acetonitrile (480.0 mL) was stirred at reflux for 3 hours. The reaction was cooled to 20 °C, poured into cold brine:saturated sodium bicarbonate solution (1:1, <10 °C, 2.0 L) and extracted with ethyl acetate (1.0 L). The organic phase was washed with cold brine:saturated sodium bicarbonate solution (1:1, <10°C, 1.00 L) once more. The combined aqueous phase was extracted with ethyl acetate (500.0 mL x 2). The combined organic phase was washed with cold brine (<10°C, 1.0 L), dried over sodium sulfate , filtered and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography to give Compound 1 (17.5 g, 66.0%) as a yellow solid. 1 H NMR (400 MHz, DMSO-d 6 ) δ ppm 11.10 (s, 1H), 7.54 (t, J = 8.0 Hz, 1H), 7.30 (t, J = 8.0 Hz, 1H), 7.04-7.10 (m , 4H), 7.00 (d, J = 8.4 Hz, 1H), 5.07 (dd, J = 12.8, 5.2 Hz, 1H), 4.58 (d, J = 6.4 Hz, 2H), 3.53-3.55 (m, 6H) , 3.30-3.32 (m, 2H), 2.81-2.89 (m, 4H), 2.54-2.61 (m, 2H), 2.20 (m, 4H) 2.03-2.06 (m, 1H); HPLC purity, 100.0%; SFC purity, 97.2% ee; LCMS (ESI) m/z 536.1 [M+H] + .
Example 2: Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3- morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione

[00250] Synthesis of tert-butyl (S)-5-amino-4-(4-nitro-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 6): Ethyl acetate (245 mL, 5 V ), 3-nitrophthalic anhydride (49.1 g, 0.25 mol, 1 eq), and tert-butyl (S)-4,5-diamino-5-oxopentanoate hydrochloride (59.2 g, 0.25 mol,
1 eq) were charged into a reactor and cooled to 15-20°C. A premade solution of CDI (66.7 g,
0.41 mol, 1.5 eq) in DMF (245 mL, 5 V) was charged and the mixture was stirred at 20-25°C for
1 hour. The reaction was quenched with 15% (wt/wt) aqueous citric acid solution (10 V).
EtOAc (5 V) was added, the mixture was agitated and the phases split and separated. Tea
aqueous layer was extracted with EtOAc (5 V) and the combined organic layers were washed
twice with a 5% (wt/wt) aqueous citric acid solution (5 V each wash). The organic layer was
distilled at reduced pressure to 5 V and further continuously distilled at reduced pressure with the addition of iPrOH (10 V), maintaining a constant volume at 5 V. The final distillate was diluted to 13 V with iPrOH and used in the next step without further handling. 91% solution yield. [00251] Synthesis of tert-butyl (S)-5-amino-4-(4-amino-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 5): The solution of Compound 6 in iPrOH was charged to a hydrogenation reactor. 10% palladium on carbon (50% wet, 4.65g 5 wt%) was charged. The reaction mixture was stirred under 50-60 psi H2 at 40-50 o C for 16 hrs. The reaction mixture was filtered and the filter cake was washed three times with iPrOH (1 V each wash). The solution was distilled at reduced pressure to 5 V, cooled to ambient temperature and seeded (1 wt%). Water (20 V) was charged at 20-25 o C. The resulting slurry was cooled to 3-8 o C for 4-8 hrs. The solids were collected by filtration and washed three times with cold water (1.5 V each wash). The solids were dried at 35-45 o C under reduced pressure to give Compound 5 in 87% yield. 1 H NMR (500 MHz DMSO-d 6 ) δ (ppm): 7.52 (s, 1H), 7.43 (dd, J = 8.4, 7.0 Hz, 1H), 7.13 (s, 1H), 6.97 (ddd, J = 10.9, 7.7, 0.61 Hz, 2H), 6.43 (s, 2H), 4.49 (m, 1H), 2.33 (m, 1H), 2.17 (m, 3H), 1.32 (s, 9H); HPLC purity, 99.2%; Chiral purity, 99.9% ee; LCMS (ESI) m/z 348.2, [M+H] + , 292.2 [Mt-Bu+H] + . Residual IPA: 0.7 mol% by 1 H NMR.
[00252] Synthesis of 4-(1-(4-bromo-3-fluorobenzyl)azetidin-3-yl)morpholine (Compound 4): A mixture of 4-bromo-3-flurobenzaldehyde (Compound 14, 82 g, 396 mmol ) and 4-(azetidin-3-yl)-morpholine hydrochloride (Compound 7 HCl, 72 g, 396 mmol) in acetonitrile (820 ml) was agitated at 25±5°C for at least 3 hours. The mixture was cooled to 10±5°C and sodium triacetoxyborohydride (130 g, 594 mmol) was added in four portions while maintaining the temperature of the mixture below 30°C. The temperature of the mixture was adjusted to 25±5°C and stirred for at least 30 min until reaction completion. The mixture was transferred to a precooled (10-15°C) solution of aqueous citric acid (152 g in 400 ml water, 792 mmol) while maintaining the temperature below 30°C. Once the quenching process was complete, the mixture was concentrated to ~ 560 ml (7 volumes) while keeping the temperature at or below 45°C. The mixture was then washed with toluene (320 ml). To the aqueous phase was added THF and the pH was adjusted to above 12 with aqueous NaOH solution (320 ml, 10 N). The phases were separated, and the aqueous phase was removed. The organic phase was washed with brine and subsequently concentrated with addition of THF (~ 3L) until KF ≤ 0.10%. The mixture was filtered to remove any inorganics and the product Compound 4 was isolated as a solution in THF with 95% yield.
[00253] Synthesis of sodium (2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)phenyl) (hydroxy)methanesulfonate (Compound 13): A solution of Compound 4 (520 g, 1.58 mol) in
THF (380 ml) was cooled to −15 ± 5 °C. A solution of iPrMgCl . LiCl (1.3 M, 1823 ml, 2.37 mol) in THF was added over the course of at least 1 hour while maintaining the temperature below −10 °C. After addition was complete, the temperature of the reaction mixture was adjusted to 0 ± 5 °C and stirred for at least 1 hour. Once magnesiation was complete, the mixture was cooled to −15 ± 5 °C (target −15 °C to −20 °C) and a solution of DMF (245 ml g, 3.16 mol) in THF (260 ml) was added slowly over the course of at least 1 hour while maintaining the temperature below −10 °C. The temperature of the mixture was then adjusted to −15 ± 5 °C and agitated for at least 4 hours.
[00254] Upon reaction completion, the reaction mixture was charged into an aqueous 3 N HCl solution (2600 ml) over the course of at least 1 hour while maintaining the temperature below −5 °C. The temperature of the mixture was then adjusted to 5 ± 5 °C and agitation was stopped, letting the mixture settle for at least 15 minutes. The layers were separated. The lower aqueous layer containing the product was washed with 2-MeTHF (2600 ml). The aqueous layer was then charged with 2-MeTHF (2600 ml) and the temperature of the batch was adjusted to −10 ± 5 °C. To the cooled mixture, an aqueous 5 N NaOH (728 ml, 3.64 mol) solution was added while maintaining the temperature below −5 °C until the pH of the mixture was between 10 and 11. The temperature of the mixture was adjusted to 5 ± 5 °C and agitated for at least 15 minutes. The agitation of the mixture was stopped and the mixture allowed to settle for at least 15 minutes. The layers were separated, and the lower aqueous layer was back extracted two times with 2-MeTHF (2600 ml). The combined organic layer was washed with water (1040 mL) and the organic solution was evaporated to dryness, affording 372 g of crude Compound 3 freebase as an oil (yield 85%). 1 H NMR (DMSO-d 6 ) δ (ppm): 10.18 (s, 1H), 7.78 (t, J =7.7 Hz, 1H), 7.23-7.35 (m, 2H), 3.66 (s, 2H), 3.51 -3.60 (m, 4H), 3.26-3.47 (m, 2H), 2.72-2.97 (m, 3H), 2.12-2.32 (m, 4H).
[00255] The crude Compound 3 freebase (4.3 kg) was adsorbed onto silica gel (8.6 kg) with 100% DCM, loaded onto a 60 L column containing 12.9 kg silica gel (packed with 100% DCM), and eluted with DCM ( 86 L), followed successively by 1% MeOH/DCM (40 L), 3% MeOH/DCM (80 L) and 10% MeOH/DCM (40 L). The fractions were collected and concentrated at or below 38˚C to give Compound 3 as a purified oil (3.345 kg, yield 66%).
[00256] A portion of Compound 3 (1.0 kg, 3.59 mol) was dissolved in ethanol (16.0 L, 16
vol) at 20±5 °C and the mixture heated to 40 °C. A solution of Na2S2O5 (622.0 g, 3.27 mol; 0.91 eq) in water (2 L, 2 vol) was prepared at 20±5 °C and added to the freebase solution at 40 °C to obtain an off-white suspension. The batch was agitated and maintained at 40 °C for 2 hrs, then cooled to 20±5 °C and agitated for 1 to 2 hrs. The batch was filtered and washed with ethanol (2×2.0 L, 2×2 vol) to obtain an off-white solid. The wet cake was dried under vacuum at 40 °C for 18 hrs to afford about 1.88 kg of Compound 13.
[00257] Synthesis of 2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzaldehyde (Compound 3): Compound 13 (1.88 kg) was dissolved in ethyl acetate (15.0 L) at 20±5 °C . A 2 M Na 2 CO 3 solution (total 15.0 L used) was added to adjust the pH to 10.0. The batch was agitated for 1 to 1.5 hrs at 20±5 °C. After the reaction was complete, the phases were separated and the organic layer was washed with brine (2.0 L). The organic layer was concentrated to dryness at 35-38 °C to afford 852.0 g of Compound 3 as a colorless oil (yield 81%).
[00258] Synthesis of 2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzaldehyde bis-oxalic acid salt (Compound 3 bis-oxalic acid salt): A portion of the Compound 3 oil (187 g, 0.67 mol) was dissolved in isopropanol (1125 ml) and water (375 ml). A first portion (~30%) of this freebase mixture (480 ml) was slowly added over the course of at least 30 minutes to a solution of oxalic acid (125 g, 1.38 mol) in IPA (1125 ml)/water (375 ml) at 60 ± 5 °C. A second portion (~20%) of the freebase mixture (320 ml) was slowly added over the course of at least 30 minutes to the reaction mixture at 60 ± 5 °C. The reaction mixture was agitated at 60 ± 5 °C for at least 90 minutes. A third portion (~25%) of the freebase mixture (~ 400 ml) was slowly added over the course of at least 30 minutes to the reaction mixture at 60 ± 5 °C and the reaction mixture was agitated at 60 ± 5 °C for at least 90 minutes. The remaining freebase solution (400 ml) was slowly added over the course of at least 30 minutes to the reaction mixture at 60 ± 5 °C and the reaction mixture was agitated at 60 ± 5 °C for at least 90 minutes. The temperature of the mixture was adjusted to 20 ± 5 °C (target 20 °C) over the course of at least 1 hour and the mixture was agitated for at least 16 hours at 20 ± 5 °C and then filtered. The cake was washed three times with IPA (2 x 375 ml) and dried in the drying oven at ≤ 40 °C with a slow bleed of nitrogen to afford 261 g of Compound 3 bis-oxalic acid salt (yield 85%). 1 H NMR (DMSO-d 6 ) δ (ppm): 10.21 (s, 1H), 7.87 (t, J = 7.6 Hz, 1H), 7.42-7.56 (m, 2H), 4.31 (s, 2H), 3.89 -4.03 (m, 2H), 3.75-3.89 (m, 2H), 3.60 (br t, J = 4.3 Hz, 4H), 3.26 (br t, J = 6.9 Hz, 1H), 2.37 (br s, 4H) .
[00259] Synthesis of tert-butyl (S)-5-amino-4-(4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)-1,3- dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 2):
Acetonitrile (6.8 L, 8.0 X Vol) was added to a 30 L jacketed cylindrical reactor. Compound 5 (0.845 kg, 1.00 X Wt) and Compound 3 bis-oxalic acid salt (1.35 kg, 1.60 The contents of the reactor were equilibrated with agitation at 20 ± 5 °C. Trifluoroacetic acid (0.19 L, 0.22 X Vol) was added dropwise, maintaining the batch temperature at 20 ± 5 °C. The reaction mixture was stirred at 20 ± 5 °C for no less than 5 minutes and then sodium triacetoxyborohydride (0.13 kg, 015 X Wt) was added as a solid, maintaining the batch temperature at 20 ± 5 °C. The process of adding trifluoroacetic acid and then sodium triacetoxyborohydride was repeated an additional 5 times. After the last addition, the reaction mixture was sampled to determine the reaction progress. The reaction was held at 20 ± 5 °C overnight. The reaction mixture was then quenched with water (3.4 L, 4.0 X Vol), maintaining the batch temperature at 20 ± 5 °C. The mixture was then stirred at 20 ± 5 °C for no less than 30 minutes and the resulting slurry filtered through a 3 L sintered glass filter, directing the filtrates to clean containers. The reactor was rinsed with acetonitrile (0.4 L, 0.5 X Vol) and the rinse passed through the contents of the 3 L sintered glass filter, directing the filtrate to the containers containing the main batch. The contents of the containers were concentrated to ~5 X Vol under reduced pressure at a bath temperature of no more than 30 °C. The residue was transferred to a clean reactor, was rinsing with 2-MeTHF (2.5 L, 3.0 X Vol) to complete the transfer. Additional 2-MeTHF (10.1 L, 12.0 X Vol) was added to the reactor, followed by water (3.4 L, 4.0 X Vol). The mixture was agitated for no less than 15 minutes at 20 ± 5 °C, then allowed to settle for no less than 10 minutes at 20 ± 5 °C before transferring the bottom aqueous layer to new containers. An aqueous sodium bicarbonate solution (5.3 L, 6.3 X Vol, 9% wt/wt) was added to the reactor with stirring over 30 minutes, maintaining batch temperature no more than 25 °C. The mixture was agitated for no more than 15 minutes at 20 ± 5 °C, then allowed to settle for no less than 10 minutes at 20 ± 5 °C before the bottom aqueous layer was transferred to new containers. The aqueous sodium bicarbonate wash was repeated an additional 2 times to reach a pH of about 6.6 for the spent aqueous layer. A saturated aqueous solution of NaCl(0.85 L, 1.0 X Vol) was then added to reactor with agitation. The mixture was agitated for no less than 15 minutes at 20 ± 5 °C, then allowed to settle for no less than 10 minutes before the bottom aqueous layer was transferred to new containers. The remaining organics were concentrated under reduced pressure to a batch volume of ~5 X Vol at a bath temperature of about 40 °C. Acetonitrile (5.1 L, 6.0 X Vol) was added to the residual volume and the resulting solution concentrated to a batch volume of ~ 5 X Vol under reduced pressure at bath temperature of about 40 °C. The process of adding acetonitrile and concentrating under vacuum was repeated two more times to reach the distillation endpoint with a water content of about 1%. The acetonitrile solution was transferred to a clean container along with two 1.7 L (2.0 X Vol) rinses and held at 5 °C overnight. The acetonitrile solution was then filtered through a 3 L sintered glass filter, followed by a 1.7 L (2.0 X Vol) acetonitrile rinse, directing the filtrates to a clean container. The filtrate was transferred to a clean reactor and the container rinsed twice with 1.7 L (2.0 X Vol) of acetonitrile to complete the transfer. Enough acetonitrile (roughly 0.6 L) was added to adjust the total volume in the reactor to about 14 L. A solution assay of the contents of the reactor was obtained to calculate the amount of Compound 2 present for use in the next step (result = 1.3 kg = 1.00 X Wt for remainder of process).
[00260] Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione bis-besylate salt (Compound 1 bis-besylate salt): The solution of Compound 2 in acetonitrile from the previous step was diluted with acetonitrile (roughly 2 L) such that the total volume in the reactor was about 16 L. The solution was cooled with stirring to 10 ± 5 °C and held within that range for 96 hours. Benzenesulfonic acid (1.86 kg, 1.43 X Wt) was added while sparging the reaction mixture with nitrogen gas and maintaining the batch temperature at 10 ± 10 °C. The temperature of the reactor was then adjusted to 20 ± 5 °C and the mixture stirred at that temperature for 60 minutes. The total volume of reaction mixture was adjusted back to 16 L to account for solvent lost during sparging by the addition of acetonitrile (roughly 0.4 L). The reaction mixture was then heated to 55 ± 5 °C over the course of about 30 minutes and held in that range for 15 to 16 hours for reaction completion. The mixture was then cooled to 50 ± 5 °C and MTBE (3.9 L, 3.0 X Vol) was added, maintaining the batch temperature at 50 ± 5 °C. The mixture was allowed to stir at 50 ± 5 °C for about 1.5 hours to establish a self-seeded slurry. Additional MTBE (3.9 L, 3.0 X Vol) was added to the reactor over the course of about 1.75 hours at 50 ± 5 °C. The slurry was cooled to 20 ± 5 °C over the course of about 1.75 hours and held in that temperature range overnight. The slurry was filtered using a Buchner funnel. The reactor was rinsed twice with
MTBE (3.9 L each, 3.0 X Vol) and the rinse was used to wash the solids in the Buchner funnel. The solids were dried on drying trays for about 23 hours at 40 °C under reduced pressure (15-150 mbar), yielding 1.62 kg (77.9%) of Compound 1 bis-besylate salt.
[00261] Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione hydrochloride salt (Compound 1 HCl): A suspension of Compound 1 bis-besylate salt (120 g, 1 equiv.) in 2-MeTHF (25 L/kg) was added to a reactor and agitated at 10 °C. A solution of KHCO 3 (32.5 g, 2.4 equiv) in water (1.8 L, 6 L/kg) was added to the slurry over the course of 40 minutes. The mixture was stirred for an additional 30 minutes. The batch was then allowed to settle, at which point the aqueous (bottom) layer was separated and discarded. An aqueous solution of NaCl (5%, 5 L/kg, 575 ml) was added to the organic layer and the mixture was agitated for 10 minutes, after which point the temperature was raised to 20 °C. The batch was allowed to settle, at which point the aqueous (bottom) layer was discarded. The brine was repeated a second time.
Additional 2-MeTHF (500 ml) was added to dilute the organic layer, resulting in a concentration of about 20 mg product per ml. A solution of HCl (total 0.98 eq.) in 2-MeTHF was prepared and a portion (20% of total, corresponding to ~0.2 eq.) then added to the reaction mixture over the course of about 10 min. Seeds of Compound 1 hydrochloride (~5% wt) were added, but did not dissolve. The batch was held under vigorous agitation for one hour. To the slurry, the remaining portion of the HCl solution (~0.78 eq.) was added over the course of 3 hours at a constant rate. Vigorous agitation was maintained. After addition was complete, the batch was held for one hour, after which the batch was filtered, washed three times with 3 L/kg of 2-MeTHF . The filter cake was placed in a vacuum oven at 22 °C for 12 hours, at which point the temperature was raised to 40 °C. Dry cake of Compound 1 hydrochloride (58g, 75% yield) was obtained and packaged. Achiral HPLC purity: 98.91%; chiral HPLC purity: 99.68%.
Example 4: Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3- morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione

[00264] Synthesis of tert-butyl (S)-5-amino-4-(4-nitro-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 6): To a solution of 3-nitrophthalic anhydride (Compound 12,
35.15 g, 176.6 mmol, 1.00 eq) in ethyl acetate (350 mL) was added tert-butyl (4S)-4,5-diamino-5-oxo-pentanoate hydrochloride (Compound 9 HCl, 43.22 g, 181.1 mmol, 1.025 eq) ), DMF (70
mL) and 2-MeTHF (110 mL) at 25°C. 2,6-Lutidine (23.4 mL, 201 mmol, 1.14 eq) was added
slowly to maintain the temperature at or below 25°C. The mixture was aged at 25°C for 1 hour before being cooled to 5°C. CDI (4.17 g, 25.7 mmol, 0.146 eq) was added and stirred until the temperature returned to 5°C. Another portion of CDI (4.62 g, 28.5 mmol, 0.161 eq) was added and stirred until temperature returned to 5°C. CDI (8.87 g, 54.7 mmol, 0.310 eq) was added and stirred until the temperature returned to 5°C. CDI (8.91 g, 54.9 mmol, 0.311 eq) was added and stirred until the temperature returned to 5°C. The mixture was warmed to 20°C and CDI (16.4 g, 101.1 mmol, 0.573 eq) was added, and the mixture was aged at 20°C for 16 hours. The mixture was cooled to 5°C and a solution of 30 wt% citric acid and 5 wt% NaCl (350 mL) was added slowly while maintaining the temperature. The mixture was warmed to 20°C and aged for 30 minutes. The phases were split and separated. The organic phase was diluted with EtOAc (175 mL) and washed with a solution of 5 wt% citric acid (175 mL), and concentrated by distillation (75 torr, 50°C) to a volume of 175 mL EtOAc. The solvent was changed to iPrOH by constant volume distillation (75 torr, 50°C) with 350 mL iPrOH to a final volume of 175 mL. The distillate was diluted with 200 mL iPrOH to afford Compound 6 as a solution for use in the next step. 1 H NMR (500 MHz, CDCl 3 ) δ (ppm): 8.18 – 8.13 (m, 2H), 7.96 (t, J = 7.8 Hz, 1H), 6.34 (s, 1H), 5.59 (s, 1H), 4.90 (dd, J = 10.1, 4.6 Hz, 1H), 2.61 (ddt, J = 14.6, 10.1, 6.1 Hz, 1H), 2.49 (ddt, J = 14.2, 8.7, 5.2 Hz, 1H), 2.44 – 2.29 ( m, 2H), 1.44 (s, 9H).
[00265] Synthesis of tert-butyl (S)-5-amino-4-(4-amino-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 5): To a solution of Compound 6 in iPrOH (375 mL) was added 5% palladium on carbon (1.23 g, 3.5 wt%, wet). The mixture was purged with nitrogen five times and with hydrogen three times. The mixture was pressurized with hydrogen (50 psi) and aged at 50°C for 16 hours. The mixture was cooled to room temperature and purged with nitrogen three times, filtered to remove catalyst, and the filter cake was washed with iPrOH (20mL) three times. The filtrate was concentrated to 200 mL, seeded (0.454 g, 1.3 wt%) at 22 °C, and aged for 45 minutes. Water (1325 mL) was added over 3 hours at 22°C. After the addition of water, the mixture was cooled to 8°C over 2 hours and aged for 1 hour at 8°C. The slurry was filtered, and the cake was rinsed with cold water (200 mL) three times and dried under vacuum at 50°C to yield Compound 5 as a yellow solid (47.97 g, 80.6% yield, 99.62% LC purity, 103% 1 H NMR potency). 1 H NMR (500 MHz, CDCl 3 ) δ (ppm): 7.46 (dd, J = 8.3, 7.0 Hz, 1H), 7.19 (d, J = 7.2 Hz, 1H), 6.89 (d, J = 8.3 Hz, 1H), 6.28 (s, 1H), 5.41 (s, 1H), 5.28 (s, 2H), 4.83 (dd, J = 9.3, 6.0 Hz, 1H), 2.52 (p, J = 7.0 Hz, 2H), 2.36 – 2.29 (m, 2H), 1.44 (s, 9H). 13 C NMR (126 MHz, CDCl 3 ) δ (ppm): 171.80, 171.12, 169.64, 168.27, 145.70, 135.50, 132.20, 121.43, 112.98, 80.99, 53.04, 32.23, 28.02 , 24.36. LCMS (ESI): m/z 291.9 [M+H – tBu]
[00266] Synthesis of 4-(1-(4-bromo-3-fluorobenzyl)azetidin-3-yl)morpholine bis-methanesulfonic acid salt (Compound 4 bis-methanesulfonic acid salt): A mixture of 4-bromo-3- fluorobenzaldehyde (Compound 14, 102 g, 493 mmol) and 4-(azetidin-3-yl)morpholine hydrochloride (Compound 7 HCl, 90 g, 493 mmol) in acetonitrile (1000 ml) was agitated at a temperature of about 20 to 25 °C for 2 to 3 hours. The slurry was cooled to temperature of about 10 to 15 °C and sodium triacetoxyborohydride (STAB, 162 g, 739 mmol) was added in 4 portions over the course of about 45 minutes while maintaining the batch temperature at no more than 30 °C. The slurry was stirred at a temperature of about 20 to 25 °C for at least 30 minutes and then quenched by an aqueous citric acid solution (191 g, 986 mmol in 500 ml of water) at a temperature of about 40 to 45 °C over the course of 2 hours. Upon completion of the quenching process, the batch volume was reduced by vacuum distillation to about 700 ml at a temperature of no more than 45 °C. Cyclopentylmethylether (CPME, 400 ml) was added to the aqueous solution to afford a final volume of about 1100 ml. The pH was adjusted to about 8 to 9 by addition of an aqueous solution of 10 N NaOH (added volume about 430 ml). The phases were separated, and the aqueous phase discarded. The organic phase was washed with brine (100 ml) twice such that the pH was no more than 8 and the volume was adjusted to about 1000 ml with addition of extra CPME. The batch was distilled at constant volume under reduced pressure with addition of CPME until KF was no more than 0.15%. CPME was added (if needed) to adjust the batch to a volume of 1000 ml at the end of distillation. The dry CPME solution was seeded (500 to 750 mg) at ambient temperature. The seeded, dry CPME slurry was heated to a temperature of 50 to 60 °C and then charged with methanesulfonic acid in 200 ml of CPME over the course of 4 to 5 hours. The slurry was then cooled to 20 °C over the course of 4 to 5 hours and kept at 20 °C for 3 to 4 hours, filtered, rinsed with CPME and dried in a vacuum oven at 35 to 40 °C over 16 hours to give Compound 4 bis-methanesulfonic acid salt as a white solid. 1 H NMR (500 MHz DMSO-d 6 ) δ (ppm): 10.62 (br s, 1-2H), 7.85 (t, J = 7.8 Hz, 1H), 7.58 (dd, J = 9.5 Hz, 1.9 Hz, 1H), 7.34 (dd, J = 8.2 Hz, 1.8 Hz, 1H), 4.55-4.24 (m, 7H), 3.84 (br s, 4H), 3.14 (m, 4H); HPLC purity, 99.8%, LCMS (ESI) m/z 329.1 /331.1[M/M+2] + . XRPD pattern of the product is shown in FIG.10. DSC thermogram of the product is shown in FIG.11.
[00267] Preparation of 4-(1-(4-bromo-3-fluorobenzyl)azetidin-3-yl)morpholine (Compound 4): A slurry of Compound 4 bis-methanesulfonic acid salt (70 g, 134 mmol) in t -butyl methyl ether was cooled to 10±5°C. An aqueous solution of NaOH (2 N, 201 ml, 403 mmol) was added over the course of at least 30 minutes while maintaining the batch temperature at about 15 °C. After the addition of NaOH , the batch temperature was raised to 20±5°C and agitated over the course of about 20 minutes. The organic layer was separated and washed with water (210 ml) three times. The organic layer was subsequently concentrated with addition of THF (~ 1.05L) until KF ≤ 0.10%. The product Compound 4 was isolated as a solution in THF
with 95% solution yield.
[00268] Preparation of 2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzaldehyde dihydrochloride (Compound 3 di-HCl): A solution of Compound 4 (44 g, 134 mmol) in THF (total volume ~ 350 ml) was then cooled to −20 ± 5 °C. A solution of iPrMgCl . LiCl (1.3 M, 176 ml, 228 mmol) in THF was added over the course of half an hour while maintaining the temperature below −10 °C. After the addition was complete, the batch was stirred at −20 ± 5 °C for 16 to 22 hours. DMF (21 ml, 268 mmol) was then added slowly over the course of 30 minutes while maintaining the batch temperature no more than -15 °C. The batch was stirred at −20 ± 5 °C for 6 to 24 hours. 2-MeTHF (350 ml) was then added to the batch over the course of 30 minutes, followed by slow addition of 3 N HCl (235 ml, 704 mmol) while keeping the batch temperature no more than -10 °C. After the addition of aqueous HCl, the batch was warmed to 0 ± 5 °C and 2 N aqueous NaOH (154 ml, 309 mmol) was added slowly to adjust the solution pH to about 8 to 9. The batch was stirred for about 30 minutes and then warmed to 20 ± 5 °C. The organic layer was separated and washed with 15% aqueous NaCl (3 x 140 ml). The organic layer was subsequently concentrated with addition of 2-MeTHF until KF ≤ 0.10%.
[00269] A portion of the free base of 2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzaldehyde (37.4 g, 134 mmol) so obtained was dissolved in 2-MeTHF (total ~ 420 ml) , to which isopropanol (420 ml) and water (21 ml) were added at 20 ± 5 °C. The batch was then heated to 50 ± 5 °C and a solution of HCl in IPA (5 to 6 N, 28 ml, half of total HCl volume) was added over the course of 1 hour. The batch was seeded with 2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzaldehyde dihydrochloride (700 mg) and aged for 1 hour. The remaining HCl (28 ml) was then added over the course of 1 hour. The batch was agitated at 50 ± 5 °C for 4 hours and then cooled to 20 ± 5 °C for 8 hours. The slurry was filtered, washed with IPA (210 ml), and the filter cake dried under vacuum at 50 ± 5 °C to afford Compound 3 dihydrochloride salt (36 g, yield 75%). 1 H NMR (DMSO-d 6 ) δ (ppm): 12.32-12.55 (m, 1H), 10.23 (s, 1H), 7.93 (t, J =7.6 Hz, 1H), 7.66 (d, J = 10.5 Hz , 1H), 7.58 (d, J = 7.9 Hz, 1H), 4.80 (br s, 2H), 4.48-4.70 (m, 2H), 4.30 (br s, 4H), 3.78-4.00 (m, 5H), 2.93-3.15 (m, 2H). Two polymorphic forms were obtained. XRPD pattern and DSC thermogram of Form A (anhydrous) are shown in FIG.5 and FIG.6, respectively. XRPD pattern, TGA thermogram and DSC thermogram of Form B (hydrate) are shown in FIG.7, FIG.8, and FIG.9, respectively.
[00270] Synthesis of tert-butyl (S)-5-amino-4-(4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)-1,3- dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 2): A mixture of Compound 5 (12 g, 34.5 mmol, 1.0 eq) and Compound 3 dihydrochloride (14.56 g, 41.5 mmol, 1.2 eq) in MeCN (96 ml) was cooled to 0-5 °C. Trifluoroacetic acid (TFA, 2.0 ml, 26 mmol, 0.75 eq) was added, followed by sodium triacetoxyborohydride (STAB, 2.75 g, 12.95 mmol, 0.375 eq) while maintaining the internal temperature below 10 °C. The addition of TFA and STAB was repeated three additional times. After a total of four additions of TFA and STAB, the reaction was aged at 0-5°C for 1 hour. A 10% brine solution (108 ml) was then added to the reaction mixture over the course of 1 hour and partitioned with IPAc (96 ml). The mixture was warmed to 20-25 °C and aged for 30 minutes. The layers were then separated and the organic layer was washed with 2.0 M K3PO4 (114 ml). The pH of the spent aqueous layer should have a pH of about 8.5 – 9.0. The layers were separated again and the organic phase was washed with 8.5% NaHCO 3 (2 x 60 ml), with 30 minutes between each wash, followed by 24% brine (60 ml). The organic fraction was distilled to 72 ml at an internal temperature near 50°C. Toluene (72 ml) was added to bring the volume to 144 ml and distillation continued at constant volume at 50°C with feed and bleed until water content < 0.1. The mixture was heated to 50°C and acetonitrile (48 ml) was added, followed by slow addition of heptane (144 ml) while maintaining the internal temperature above 45 °C. The reaction was held at 50 °C for 2 hours. Once complete, the reaction was slowly cooled to 20-25°C over the course of 4 hours and held at 20-25°C overnight (16 hour). The yellow slurry was then filtered and the yellow cake displacement washed with 1:3:3 mixture of acetonitrile/heptane/toluene (3 x 48 ml). The final cake was then dried under reduced pressure at 50 °C under nitrogen to provide Compound 2 (87.7% isolated molar yield) with >99.0% LCAP. HPLC purity, 99.85%; Chiral purity, >99.9% ee. 1 H NMR (DMSO-d6, 500 MHz) δ (ppm) 7.55 (s, 1H), 7.51 (dd, J = 7.2, 8.4 Hz, 1H), 7.32 (t, J = 7.9 Hz, 1H), 7.16 ( s, 1H), 7.0-7.1 (m, 5H), 4.57 (d, J = 6.3 Hz, 2H), 4.5-4.5 (m, 1H), 3.5-3.6 (m, 6H), 3.3-3.4 (m, 3H), 2.8-2.9 (m, 3H), 2.3-2.4 (m, 1H), 2.1-2.3 (m, 7H), 1.31 (s, 9H); LCMS m/z 610.3 [M+H] + .
[00271] Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione bis-besylate salt (Compound 1 bis-besylate): To a suspension of Compound 2 (130 g, 1.0 equiv.) in MeCN (1.56 L, 12 L/kg) agitated at 55 °C was added a solution of benzene sulfonic acid (185 g, 5.5
equiv.) in MeCN (0.39 L, 3 L/kg) and water (0.01 L, 2.0 equiv.). The mixture was stirred at 55 °C for 16 hours. After the reaction age, crystalline seeds (1.3 g, 1 wt%) of bis-besylate salt of Compound 1 were charged into the batch, resulting in formation of a yellow slurry. The slurry was then cooled to 20 °C over the course of 90 minutes. 2-MeTHF (1.3 L, 10 L/kg) was added to the batch slowly over 2 hours at 20 °C. The batch was agitated for an additional 4 hours at 20°C. The yellow slurry was then filtered and the yellow cake re-slurried with MeTHF (1.3 L, 10 L/kg) followed by a displacement MeTHF (0.65 L, 5 L/kg) wash. The final cake was then dried under reduced pressure at 50 °C under nitrogen to give Compound 1 bis-besylate salt (160 g, 88.4% yield). HPLC purity: 98.39%; chiral HPLC purity: 100%. XRPD pattern, TGA thermogram and DSC thermogram of the product are shown in FIG.1, FIG.2, and FIG.3, respectively.
[00272] Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione hydrochloride salt (Compound 1 HCl): A suspension of Compound 1 bis-besylate salt (300 g, 1 equiv.) in EtOAc (4.68 L, 15.6 L/kg) and 2-propanol (0.12 L, 0.4 L/kg) was agitated at 15 °C. To the suspension was added a solution of KHCO 3 (82.4 g, 2.5 equiv) in water (1.8 L, 6 L/kg) over 30 minutes. The mixture was heated to 20 °C over 30-60 minutes and then agitated for 30 minutes. The batch was allowed to settle for 30 minutes, at which point the aqueous (bottom) layer was discarded. To the rich organic layer was added water (1.2 L, 4 L/kg) and the reactor contents were agitated for 30 minutes. The batch was allowed to settle for 30 minutes, at which point the aqueous (bottom) layer was discarded. To the rich organic stream was added 2-propanol (2.375 L, 7.9 L/kg) and the stream was then filtered. Water was added to the filtrate to adjust the water content to 8≤KF≤8.2. To the above agitated solution at 20 °C was added 0.2 N HCl (38 mL, 0.025 equiv prepared in EtOAC/IPA 2:1, v/v with 8wt% water) over 10 minutes. To the mixture was added crystalline seeds of Compound 1 hydrochloride salt (1.6 g, 0.5wt%) and the contents of the reactor were agitated at 20 °C for 30 minutes. To the suspension was added 0.2 N HCl (1.44 L, 0.945 equiv. prepared in EtOAC/IPA 2:1, v/v with 8 wt% water) over 4.5 hours. The slurry was agitated for 14 hours, then filtered and washed with EtOAC/IPA (750 mL, 2.5 L/kg, 2:1 v/v with 8 wt% water) followed by IPA (750 mL, 2.5 L/kg). The solids were dried under vacuum at 40 °C to afford Compound 1 hydrochloride salt (170 g, 90% yield). Achiral HPLC purity: 99.91%; chiral HPLC purity: 99.58%. XRPD analysis (FIG.4) confirmed the product (a)

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
//////////Golcadomide, CC 99282, CC 1007548, WHO 12305
C1CC(=O)NC(=O)C1N2C(=O)C3=C(C2=O)C(=CC=C3)NCC4=C(C=C(C=C4)CN5CC(C5)N6CCOCC6)F
Igermetostat
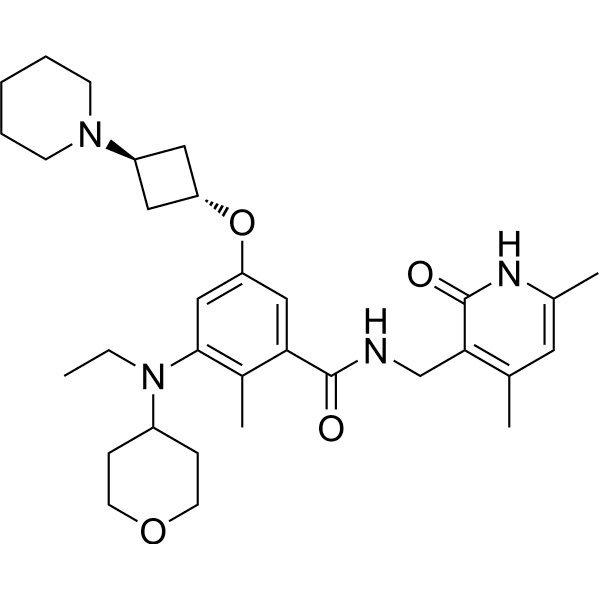

Igermetostat
C32H46N4O4. 550.73
2409538-60-7
2KES6YSH8D
INN 12655
BENZAMIDE, N-((1,2-DIHYDRO-4,6-DIMETHYL-2-OXO-3-PYRIDINYL)METHYL)-3-(ETHYL(TETRAHYDRO-2H-PYRAN-4-YL)AMINO)-2-METHYL-5-((TRANS-3-(1-PIPERIDINYL)CYCLOBUTYL)OXY)-
IGERMETOSTAT
IGERMETOSTAT [INN]
N-((1,2-DIHYDRO-4,6-DIMETHYL-2-OXO-3-PYRIDINYL)METHYL)-3-(ETHYL(TETRAHYDRO-2H-PYRAN-4-YL)AMINO)-2-METHYL-5-((TRANS-3-(1-PIPERIDINYL)CYCLOBUTYL)OXY)BENZAMIDE
N-((4,6-DIMETHYL-2-OXO-1,2-DIHYDROPYRIDIN-3-YL)METHYL)-3-(ETHYL(OXAN-4-YL)AMINO)-2-METHYL-5-(((1R,3R)-3-(PIPERIDIN-1-YL)CYCLOBUTYL)OXY)BENZAMIDE
IGERMETOSTAT is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.
SCHEME

.

PATENT
PATENT
US20230382898
The compound has been disclosed in patent WO2020020374A1. It is an enhancer of Zeste homolog 2 (EZH2) inhibitor, and can be used for preventing or treating EZH2-mediated diseases, including brain cancer, thyroid cancer, cardiac sarcoma, lung cancer, oral cancer, stomach cancer and various other cancers.
PATENT
US20210308141
Example 4: Preparation of the Compound N-((4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(trans-3-morpholinylcyclobutoxy)benzamide (4)
| The title compound was prepared by referring to Example 1 with cyclobutanol replaced by cis-3-morpholinylcyclobutyl-1-ol. The cis-3-morpholinylcyclobutyl-1-ol was prepared as follows: |
[1]. Peng, et al. Polysubstituted benzene compound and preparation method and use thereof.
World Intellectual Property Organization, WO2020020374 A1. 2020-01-30.
//////////Igermetostat
CCN(C1CCOCC1)c2cc(O[C@H]3C[C@@H](C3)N4CCCCC4)cc(C(=O)NCc5c(C)cc(C)[nH]c5=O)c2C
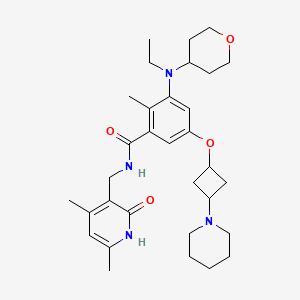
Obeldesivir

Obeldesivir,
2647441-36-7
361.35 g/mol
C16H19N5O5
[(2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxyoxolan-2-yl]methyl 2-methylpropanoate
Q55KCM7PXB; ATV006; UNII-Q55KCM7PXB
SCHEME
NEXT
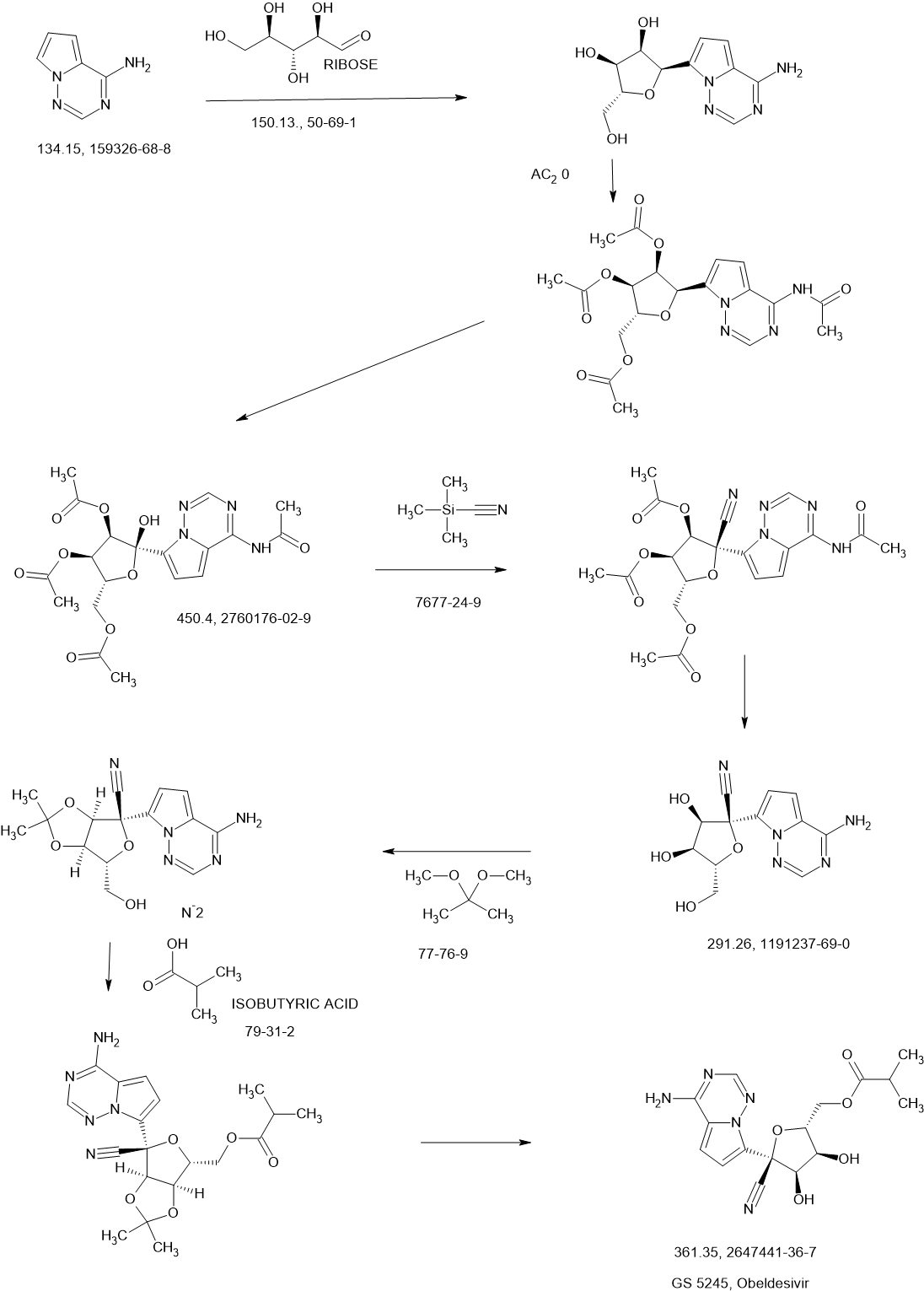
CN11687449 68%
Journal of Medicinal Chemistry (2023), 66(17), 11701-11717 50%
WO2023167938
CN115583954 67%
WO2022047065
CN113698405 49%
https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c00750
Obeldesivir (GS-5245, ATV006) is an isobutyric ester prodrug of GS-441524 made by Gilead Sciences that is currently in Phase III trials for the outpatient treatment of COVID-19 in high risk patients.[1][2] The purpose of the isobutyric ester modification on obeldesivir is to improve the oral bioavailability of the parent nucleoside, GS-441524. Obeldesivir is hydrolyzed to its parent nucleoside, GS-441524, which is in turn converted to remdesivir-triphosphate (GS-443902) by a nucleoside kinase, adenylate kinase and nucleotide diphosphate kinase.[3][4]
GS-443902 is a bioactive ATP analogue with broad-spectrum antiviral activity [5] and is the same compound formed by remdesivir, though by a different enzymatic pathway. Unlike remdesivir, which is metabolized by enzymes that are highly expressed in the liver, GS-441524 released by obeldesivir is metabolized by enzymes that are evenly expressed throughout the body. Due to their different metabolic pathways, obeldesivir can be administered orally, whereas remdesivir must be administered intravenously for COVID-19 treatment.
The pharmacokinetic properties of obeldesivir and improved was first published by Chinese researchers in May 2022. The Chinese group pursued investigation of obeldesivir independently from Gilead Sciences. Compared to IV administered GS-441524 in rats at 5 mg/kg, orally administered obeldesivir at 25 mg/kg (referred to as “ATV006”) yielded approximately 22% bioavailability.[6] Treatment with obdeldesivir reduced viral load and prevents lung pathology in KI-hACE2 and Ad5-hACE2 mouse models of SARS-CoV-2. A patent filed by Gilead Sciences with a priority date of August 27, 2020,[7] found the bioavailability of GS-441524 after oral administration of obdeldesivir (compound 15) in mice, rats, ferrets, dogs, and cynomolgus macaques to be 41%, 63.9%, 154%, 94%, and 38%, respectively. Across all species evaluated, obeldesivir showed improved oral bioavailability compared to oral administration of the parent nucleoside, GS-441524
PAPER
https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c00750
Abstract

Remdesivir 1 is an phosphoramidate prodrug that releases the monophosphate of nucleoside GS-441524 (2) into lung cells, thereby forming the bioactive triphosphate 2-NTP. 2-NTP, an analog of ATP, inhibits the SARS-CoV-2 RNA-dependent RNA polymerase replication and transcription of viral RNA. Strong clinical results for 1 have prompted interest in oral approaches to generate 2-NTP. Here, we describe the discovery of a 5′-isobutyryl ester prodrug of 2 (GS-5245, Obeldesivir, 3) that has low cellular cytotoxicity and 3–7-fold improved oral delivery of 2 in monkeys. Prodrug 3 is cleaved presystemically to provide high systemic exposures of 2 that overcome its less efficient metabolism to 2-NTP, leading to strong SARS-CoV-2 antiviral efficacy in an African green monkey infection model. Exposure-based SARS-CoV-2 efficacy relationships resulted in an estimated clinical dose of 350–400 mg twice daily. Importantly, all SARS-CoV-2 variants remain susceptible to 2, which supports development of 3 as a promising COVID-19 treatment.
Synthesis of ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl Isobutyrate (3)
To a solution of (3aR,4R,6R,6aR)-4-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carbonitrile 4 (2000 mg, 6.0 mmol) (30) and isobutyric acid (638 mg, 7.2 mmol) in DMF (5 mL), N,N′-diisopropylcarbodiimide (914 mg, 7.2 mmol) was slowly added followed by 4-(dimethylamino)pyridine (737 mg, 6.0 mmol). The reaction mixture was stirred for 4 h and then diluted with ethyl acetate, washed with water and brine, dried over sodium sulfate, and concentrated under reduced pressure. The residue was subjected to silica gel chromatography, eluting with 20% MeOH in CH2Cl2 to provide the intermediate ((3aR,4R,6R,6aR)-6-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-cyano-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl isobutyrate. LCMS m/z = 402.2 (M + 1). To a solution of the intermediate (1500 mg) in THF (10 mL), conc. HCl (2 mL) was added, and the mixture was stirred at rt for 4 h. The reaction mixture was diluted with CH2Cl2, washed with water, saturated aqueous bicarbonate, and brine, dried over sodium sulfate, concentrated, and subjected to silica gel chromatography, eluting with 30% MeOH in CH2Cl2 to afford the title compound (660 mg, 50%). 1H NMR (400 MHz, MeOH-d4) δ 7.88 (s, 1H), 6.96–6.85 (m, 2H), 4.50–4.27 (m, 4H), 4.16 (dd, J = 6.2, 5.3 Hz, 1H), 2.56 (p, J = 7.0 Hz, 1H), 1.14 (dd, J = 7.0, 3.8 Hz, 6H). LCMS m/z: 362.1 (M + 1). 13C NMR (400 MHz, CHCl3–d3) δ 175.9, 155.6, 147.9, 123.5, 110.2, 100.8, 116.9, 116.6, 81.3, 79.0, 74.0, 70.2, 62.9, 33.2, 18.7, 18.6. HRMS m/z: 362.14615, C16H20N5O5 362.14644.

PATENT
WO-2022047065-A2
PATENT
| Example 1. (3aR, 4R, 6R, 6aR)-4-(4-aminopyrrole[2,1-f][1,2,4]triazin-7-yl)-6-(hydroxymethyl- Synthesis of 2,2-dimethyltetrahydrofuran[3,4-d][1,3]dioxolyl-4-carbonitrile (compound 5) |

| Dissolve 5.62g of compound 69-0 in 30mL of acetone, then add 11.50mL of 2,2-dimethoxypropane and 1.34mL of 98% sulfuric acid, stir at 45°C for half an hour, cool to room temperature, and remove by rotary evaporation Organic solvents. Extract with 100 mL of ethyl acetate and 100 mL of saturated sodium bicarbonate solution. Repeat the extraction three times. Combine the ethyl acetate layers, add anhydrous sodium sulfate to dry, and filter to remove sodium sulfate. The organic solvent was removed by rotary evaporation, and 6.20 g of compound 5 (white solid, yield 97%) was obtained through column chromatography (eluent: petroleum ether/ethyl acetate (v/v) = 1/2). The hydrogen spectrum of the obtained compound 5 was detected, and the results are as follows: |
| 氢谱:1H NMR(400MHz,Chloroform-d)δ7.95(s,1H),7.11(d,J=4.7Hz,1H),6.69(dd,J=4.8,2.4Hz,1H),5.77(s,2H),5.42(d,J=6.6Hz,1H),5.24(dd,J=6.6,2.4Hz,1H),4.67(q,J=1.9Hz,1H),3.99(dd,J=12.5,1.9Hz,1H),3.84(dd,J=12.5,1.7Hz,1H),1.81(s,3H),1.40(s,3H)。 |
| Example 2. ((3aR,4R,6R,6aR)-6-(4-aminopyrrole[2,1-f][1,2,4]triazin-7-yl)-6-cyano-2 ,Synthesis of 2-dimethyltetrahydrofuran[3,4-d][1,3]dioxol-4-yl)methylisobutyrate (compound INT-1) |
| Dissolve 1.50g of compound 5 in 15ml of dichloromethane, then add 0.42mL of isobutyric acid and 55.40mg of 4-dimethylaminopyridine, stir for 10 minutes, add 1.02g of dicyclohexylcarbodiimide, and stir at room temperature. Stir for 24h. After column chromatography separation (eluent: petroleum ether/ethyl acetate (v/v) = 1/1), 1.71 g of compound INT-1 (white solid, yield 94%) was obtained. The hydrogen spectrum and carbon spectrum of the obtained compound INT-1 were detected, and the results are as follows: |
| Hydrogen spectrum: 1 H NMR (400MHz, CDCl 3 ,ZQF-RD01-2)δ(ppm):7.99(s,1H),6.99(d,J=4.6Hz,1H),6.62(d,J=4.6Hz,1H),5.72(br,2H),5.49(d,J=6.8Hz,1H),4.93-4.90(dd,J=6.8Hz,4.3Hz,1H),4.61-4.58(q,J=4.4Hz,1H),4.44-4.26(m,2H),2.61-2.50(m,1H),1.77(s,3H),1.42(s,3H),1.17-1.14(q,J=3.8Hz,6H)。 |
| Carbon spectrum: 13 C NMR (100MHz, CDCl 3 ,ZQF-RD01-2)δ(ppm):176.7,155.2,147.3,123.5,117.2,116.7,115.6,112.6,100.0,83.8,83.0,82.0,81.4,63.1,33.8,26.4,25.6,18.9。 |
PATENT
WO2023167938

To a solution of (3aR,4R,6R,6aR)-4-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carbonitrile, Intermediate 1 (2000 mg, 6.0 mmol) (Siegel et. al. J. Med. Chem.2017, 60, 1648-1661) in tetrahydrofuran (THF) was added N,N-dimethylaminopyridine (DMAP) (0.03 eq). To the reaction mixture, isobutyric anhydride (1.1 eq) was added slowly. After the completion of the staring material, the reaction mixture was concentrated and purified by flash chromatography using 20% methanol in DCM as an eluant to give ((3aR,4R,6R,6aR)-6-(4- aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-cyano-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4- yl)methyl isobutyrate, Intermediate 1a. LCMS: MS m/z: 402.2 (M+1).
Concentrated HCl (5 eq, 1mL) was added to a solution of Intermediate 1a (1000 mg) in acetonitrile (10 mL), which was then stirred at room temperature for 2 hours. LCMS showed the product formation. The reaction was stopped after 4 hours. The reaction mixture was diluted with ethyl acetate and quenched with saturated bicarbonate. The organic layer was separated, washed with brine, dried over sodium sulphate, and concentrated. The residue was purified by flash chromatography using 30% methanol DCM as an eluant. The collected fractions were concentrated to give Intermediate 2.1H NMR (400 MHz, Methanol-d4) δ 7.88 (s, 1H), 6.96 – 6.85 (m, 2H), 4.50 – 4.27 (m, 4H), 4.16 (dd, J = 6.2, 5.3 Hz, 1H), 2.56 (p, J = 7.0 Hz, 1H), 1.14 (dd, J = 7.0, 3.8 Hz, 6H); LCMS: MS m/z: 362.1 (M+1).
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
//////////
| Names | |
|---|---|
| IUPAC name[(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f] [1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxyoxolan-2-yl]methyl 2-methylpropanoate | |
| Identifiers | |
| CAS Number | 2647441-36-7 |
| 3D model (JSmol) | Interactive image |
| ChEMBL | ChEMBL4863829 |
| ChemSpider | 115037299 |
| PubChemCID | 162513664 |
| UNII | Q55KCM7PXB |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C16H19N5O5 |
| Molar mass | 361.358 g·mol−1 |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
References
- ^ “Clinical Trials Register”.
- ^ “Gilead touts Veklury resilience against mutated coronavirus, plots phase 3 for new COVID oral antiviral”. 19 October 2022.
- ^ Cho, A.; Saunders, O. L.; Butler, T.; Zhang, L.; Xu, J.; Vela, J. E.; Feng, J. Y.; Ray, A. S.; Kim, C. U. (2012). “Synthesis and antiviral activity of a series of 1′-substituted 4-aza-7,9-dideazaadenosine C-nucleosides”. Bioorganic & Medicinal Chemistry Letters. 22 (8): 2705–2707. doi:10.1016/j.bmcl.2012.02.105. PMC 7126871. PMID 22446091.
- ^ Yan, Victoria C.; Muller, Florian L. (2020). “Advantages of the Parent Nucleoside GS-441524 over Remdesivir for Covid-19 Treatment”. ACS Medicinal Chemistry Letters. 11 (7): 1361–1366. doi:10.1021/acsmedchemlett.0c00316. PMC 7315846. PMID 32665809.
- ^ Gordon, C. J.; Tchesnokov, E. P.; Woolner, E.; Perry, J. K.; Feng, J. Y.; Porter, D. P.; Götte, M. (2020). “Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency”. The Journal of Biological Chemistry. 295 (20): 6785–6797. doi:10.1074/jbc.RA120.013679. PMC 7242698. PMID 32284326.
- ^ Cao, Liu; et al. (2022). “The adenosine analog prodrug ATV006 is orally bioavailable and has preclinical efficacy against parental SARS-CoV-2 and variants”. Science Translational Medicine. 14 (661): eabm7621. doi:10.1126/scitranslmed.abm7621. PMC 9161374. PMID 35579533.
- ^ “Espacenet – search results”.
//////////Obeldesivir, GS-5245, ATV006
CC(C)C(OC[C@H]1O[C@@](C#N)(C2=CC=C3C(N)=NC=NN32)[C@H](O)[C@@H]1O)=O
Oditrasertib
Oditrasertib
SAR443820; DNL788
| Molecular Formula | C14H15F2N3O2 |
| Molecular Weight | 295.2846 |
- UNII-I1YQT8HC89
- I1YQT8HC89
- cas 2252271-93-3
- 4-(3,3-Difluoro-2,2-dimethylpropanoyl)-3,5-dihydro-2H-pyrido(3,4-F)(1,4)oxazepine-9-carbonitrile
PYRIDO(3,4-F)-1,4-OXAZEPINE-9-CARBONITRILE, 4-(3,3-DIFLUORO-2,2-DIMETHYL-1-OXOPROPYL)-2,3,4,5-TETRAHYDRO-
Oditrasertib (SAR443820) is an orally active, BBB-penetrable and selective reversible inhibitor of RIPK1. Oditrasertib can be used in the research of chronic inflammatory central nervous system diseases, such as amyotrophic lateral sclerosis and multiple sclerosis.
Oditrasertib is a serine/threonine kinase (STK) inhibitor.
OriginatorHarvard University
DeveloperDenali Therapeutics Inc; Sanofi
ClassAntidementias; Small molecules
Mechanism of ActionRIPK1 protein inhibitors
- 24 Feb 2025Sanofi terminates its license for DNL 788 from Denali Therapeutics
- 21 Nov 2024Chemical structure information added.
- 08 Nov 2024Discontinued – Phase-II for Multiple sclerosis (In adults) in Canada, China, France, Italy, Poland, Germany, Belgium, Spain (PO)
SCHEME
WO2018213632

PATENT
[WO2018213632A1]
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018213632

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
//////////
///////////Oditrasertib,
CC(C)(C(F)F)C(=O)N1CCOC2=C(C=NC=C2C1)C#N
- [1]. Fidalgo JDV, et al. Preparation of substituted oxazepines and related compounds as RIPK1 inhibitors and uses thereof. WO2018213632A1. 2018-11-22.[2]. Hincelin-Mery A, et al. Safety, pharmacokinetics, and target engagement of a brain penetrant RIPK1 inhibitor, SAR443820 (DNL788), in healthy adult participants. Clin Transl Sci. 2024 Jan;17(1):e13690. [Content Brief][3]. Montalban X, et al. Effect of RIPK1 inhibitor, SAR443820, on serum neurofilament light levels in patients with multiple sclerosis: a phase 2 trial design (P6-3.011)[J]. Neurology, 2023, 100(17_supplement_2): 2178.
ORZILOBEN

ORZILOBEN,
CAS 1555822-28-0
2-methyl-3-(pentyloxy)benzoic acid
| Molecular Weight | 222.28 |
|---|---|
| Formula | C13H18O3 |
Orziloben is a medium chain fatty acid (MCFA) analogue[1]
Phase IILiver disorders
OriginatorPronova BioPharma
DeveloperNorthSea Therapeutics
ClassAntifibrotics; Hepatoprotectants; Omega 3 fatty acids; Unsaturated fatty acids
Mechanism of ActionUndefined mechanism.
28 Jan 2025No recent reports of development identified for phase-I development in Liver-disorders(In volunteers) in United Kingdom (PO)- 24 Dec 2024Chemical structure information added.
- 23 Aug 2024NorthSea Therapeutics completes a phase I trial in healthy volunteers in the United Kingdom (PO) (ISRCTN12367117)
Patent
WO2020074964
The rising global epidemic of obesity and its comorbidities, e.g., type 2 diabetes mellitus and hyperlipidemia, is placing an enormous burden both on public health (mortality and morbidity) and on the available public health resources required to treat these conditions.
Current drugs that treat hyperlipidemia (e.g., statins, omega-3 fatty acids, fibrates) have mostly neutral effects on glycemic control, whilst drugs targeting glycemic control e.g., insulin, thiazolidinediones (TZDs), have adverse effects upon bodyweight and (for TZDs) other unwanted side-effects restricting their use.
In addition to hyperlipidemia and type 2 diabetes, a marked increase in the prevalence of non-alcoholic fatty liver disease (NAFLD) has occurred. NAFLD has become the most common chronic liver condition in Western populations in relation to the obesity and type 2 diabetes epidemics. The prevalence of non-alcoholic steatohepatitis (NASH), a form of NAFLD that is associated with hepatic inflammation and ballooning of hepatocytes, is expected to increase by 63% between 2015 and 2030 in the United States (Estes, Hepatology, 2018; 67(1): 123-133), where NASH is expected to become the leading cause of liver transplantation by 2020. As liver fibrosis, but not inflammation, is associated with mortality and morbidity in NASH patients, drugs which prevent progression/induce regression of fibrosis are also a focus of biomedical research.
The development of novel compounds that simultaneously target both hyperlipidemia and glycemic control, without the adverse side-effects (e.g., weight gain) typically associated with insulin sensitising drugs is thus a desirable goal. Such compounds would be even more attractive if they could additionally prevent the progression/reverse hepatic fibrosis and reduce hepatic steatosis. The present invention addresses these needs for new treatment methods, compounds, and pharmaceutical compositions.

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
//////////
/////////ORZILOBEN, 1555822-28-0, OBESITY, NST-6179, SEFA-6179, NST 6179, SEFA 6179
O=C(C1=C(C)C(OCCCCC)=CC=C1)O
Opevesostat
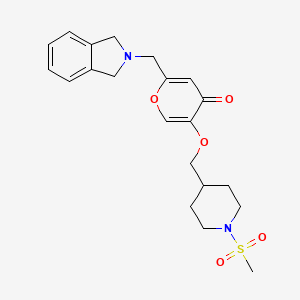


Opevesostat
- ODM208
2231294-96-3
Chemical Formula: C21H26N2O5S
MK-5684, ODM-208
Molecular Weight: 418.508
2-[(1,3-dihydro-2H-isoindol-2-yl)methyl]-5-{[1-(methanesulfonyl)piperidin-4-yl]methoxy}-4H-pyran-4-one
| Opevesostat tosylate |
4H-Pyran-4-one, 2-[(1,3-dihydro-2H-isoindol-2-yl)methyl]-5-[[1-(methylsulfonyl)-4-piperidinyl]methoxy]-, 4-methylbenzenesulfonate (1:1)
2-((1,3-DIHYDRO-2H-ISOINDOL-2-YL)METHYL)-5-((1-(METHYLSULFONYL)-4-PIPERIDINYL)METHOXY)-4H-PYRAN-4-ONE, TOSYLATE
SCHEME
SEE AT THE END OF PAGE
Opevesostat is an investigational new drug being developed for the treatment of metastatic castration-resistant prostate cancer (mCRPC).[1] It is a non-steroidal, selective inhibitor of CYP11A1 (cholesterol side-chain cleavage enzyme)[2] discovered by Orion Corporation and currently undergoing clinical development by Merck & Co., Inc. Opevesostat’s mechanism of action involves suppressing the production of steroid hormones and their precursors that may activate the androgen receptor signaling pathway, which is crucial in prostate cancer progression. As of 2024, opevesostat is being evaluated in two Phase 3 clinical trials, OMAHA1 and OMAHA2a, which are assessing its efficacy and safety in combination with hormone replacement therapy for patients with mCRPC who have failed prior treatments.[3][4][5]
useful in the treatment of a steroid receptor, in particular androgen receptor (AR), dependent conditions and diseases, and to pharmaceutical compositions containing such compounds.
Prostate cancer is worldwide the most common cancer in men. Even though the 5-year survival rate of patients with localized prostate cancer is high, the prognosis for those patients, who develop castration-resistant prostate cancer (CRPC) within that 5-year follow-up period, is poor.
The androgen receptor (AR) signalling axis is critical in all stages of prostate cancer. In the CPRC stage, disease is characterized by high AR expression, AR amplification and persistent activation of the AR signalling axis by residual tissue/tumor androgens and by other steroid hormones and intermediates of steroid biosynthesis. Thus, treatment of advanced prostate cancer involves androgen deprivation therapy (ADT) such as hormonal manipulation using gonadotropin-releasing hormone (GnRH) agonists/antagonists or surgical castration, AR antagonists or CYP17A1 inhibitors (such as abiraterone acetate in combination with prednisone).
Although therapies can initially lead to disease regression, eventually majority of the patients develop a disease that is refractory to currently available therapies. Increased progesterone levels in patients treated with abiraterone acetate has been hypothesized to be one of the resistance mechanisms. Several nonclinical and clinical studies have indicated upregulation of enzymes that catalyse steroid biosynthesis at the late stage of CRPC. Very recently it has been published that 11β-OH androstenedione can be
metabolized into 11-ketotestosterone (11-K-T) and 11-ketodehydrotestosterone (11-K-DHT) which can bind and activate AR as efficiently as testosterone and dihydrotestosterone. It has been shown that these steroids are found in high levels in plasma and tissue in prostate cancer patients, suggesting their role as AR agonists in CRPC. Furthermore, it has been addressed that prostate cancer resistance to CYP17A1 inhibition may still remain steroid dependent and responsive to therapies that can further suppress de novo intratumoral steroid synthesis upstream of CYP17A1, such as by CYP11A1 inhibition therapy (Cai, C. et al, Cancer Res., 71(20), 6503-6513, 2011).
Cytochrome P450 monooxygenase 11A1 (CYP11A1), also called cholesterol side chain cleavage enzyme, is a mitochondrial monooxygenase which catalyses the conversion of cholesterol to pregnenolone, the precursor of all steroid hormones. By inhibiting CYP11A1, the key enzyme of steroid biosynthesis upstream of CYP17A1, the total block of the whole steroid biosynthesis can be achieved. CYP11A1 inhibitors may therefore have a great potential for treating steroid hormone dependent cancers, such as prostate cancer, even in advanced stages of the disease, and especially in those patients who appear to be hormone refractory. It has been recently shown that a compound having CYP11A1 inhibitory effect significantly inhibited tumor growth in vivo in a murine CRPC xenograft model (Oksala, R. et al, Annals of Oncology, (2017) 28 (suppl.
PATENT
WO2018115591
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018115591&_cid=P20-LQXJT7-60871-1
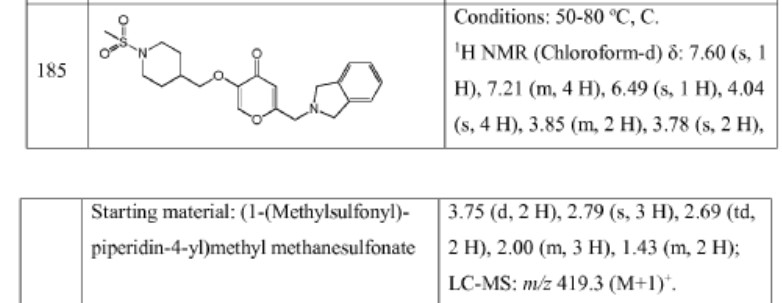

Example 4. SIMILAR
N-((4-(((6-(Isoindolin-2-ylmethyl)-4-oxo-4H-pyran-3-yl)oxy)methyl)cyclohexyl)- methyl)methanesulfonamide (Compound 173)

To a solution of 5-hydroxy-2-(isoindolin-2-ylmethyl)-4H-pyran-4-one (0.10 g, 0.41 mmol) in DMF (2 ml) were added (4-(methylsulfonamidomethyl)cyclohexyl)methyl methanesulfonate (0.14 g, 0.45 mmol) and K2CO3 (0.12 g, 0.8 mmol). The reaction mixture was heated at 80 °C for 2 h. The mixture was cooled to RT, water (10 ml) was added and the product was extracted with EtOAc. The combined extracts were washed with water, dried with Na2SO4, filtered and evaporated. The crude product was purified by column chromatography to afford the title compound (0.06 g). 1H NMR (400 MHz, Chloroform-d) δ ppm 0.92 – 1.11 (m, 4 H) 1.40 – 1.63 (m, 2 H) 1.78 – 2.00 (m, 4 H) 2.91 – 2.99 (m, 5 H) 3.65 (d, J=6.46 Hz, 2 H) 3.77 (s, 2 H) 4.03 (s, 4 H) 5.04 (br t, J=6.31 Hz, 1 H) 6.49 (s, 1 H) 7.20 (s, 4 H) 7.59 (s, 1 H).
ntermediate 58: 5-Hydroxy-2-(isoindolin-2-ylmethyl)-4H-pyran-4-one
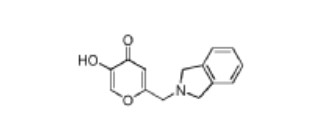
To a stirred solution of 2-(chloromethyl)-5-hydroxy-4H-pyran-4-one (2.0 g, 12.5 mmol) in acetonitrile (50 mL) were added DIPEA (3.22 mL, 25.0 mmol) and isoindoline (1.78 g, 25.0 mmol) at RT. When the reaction was complete, the precipitated solid was filtered and washed with EtOAc. The title compound was collected as pale brown solid (1.1 g). LC-MS: m/z 244.1 (M+H)+.
///////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
References
- ^ “Opevesostat – Orion”. AdisInsight.
- ^ Kim C, Jeong E, Lee YB, Kim D (July 2024). “Steroidogenic cytochrome P450 enzymes as drug target”. Toxicological Research. 40 (3): 325–333. Bibcode:2024ToxRe..40..325K. doi:10.1007/s43188-024-00237-0. PMC 11187042. PMID 38911541.
- ^ “Merck and Orion Announce Mutual Exercise of Option Providing Merck Global Exclusive Rights to Opevesostat”. StockTitan. July 2024. Retrieved 2024-11-23.
- ^ “Inside Information: Orion and MSD Announce Mutual Exercise of Option Providing MSD Global Exclusive Rights to Opevesostat”. Orion. Retrieved 2024-11-23.
- ^ “Merck and Orion Announce Mutual Exercise of Option Providing Merck Global Exclusive Rights to Opevesostat”. Merck. Retrieved 2024-11-23.
 |
|
| Clinical data | |
|---|---|
| Other names | MK-5684, ODM-208 |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C21H26N2O5S |
| Molar mass | 418.51 g·mol−1 |
| 3D model (JSmol) | |
|
show
|
|
//////////
/////////Opevesostat, ODM 208, MK-5684, ODM-208, MK 5684
O=C1C=C(CN2CC3=C(C=CC=C3)C2)OC=C1OCC4CCN(S(=O)(C)=O)CC4
SCHEME
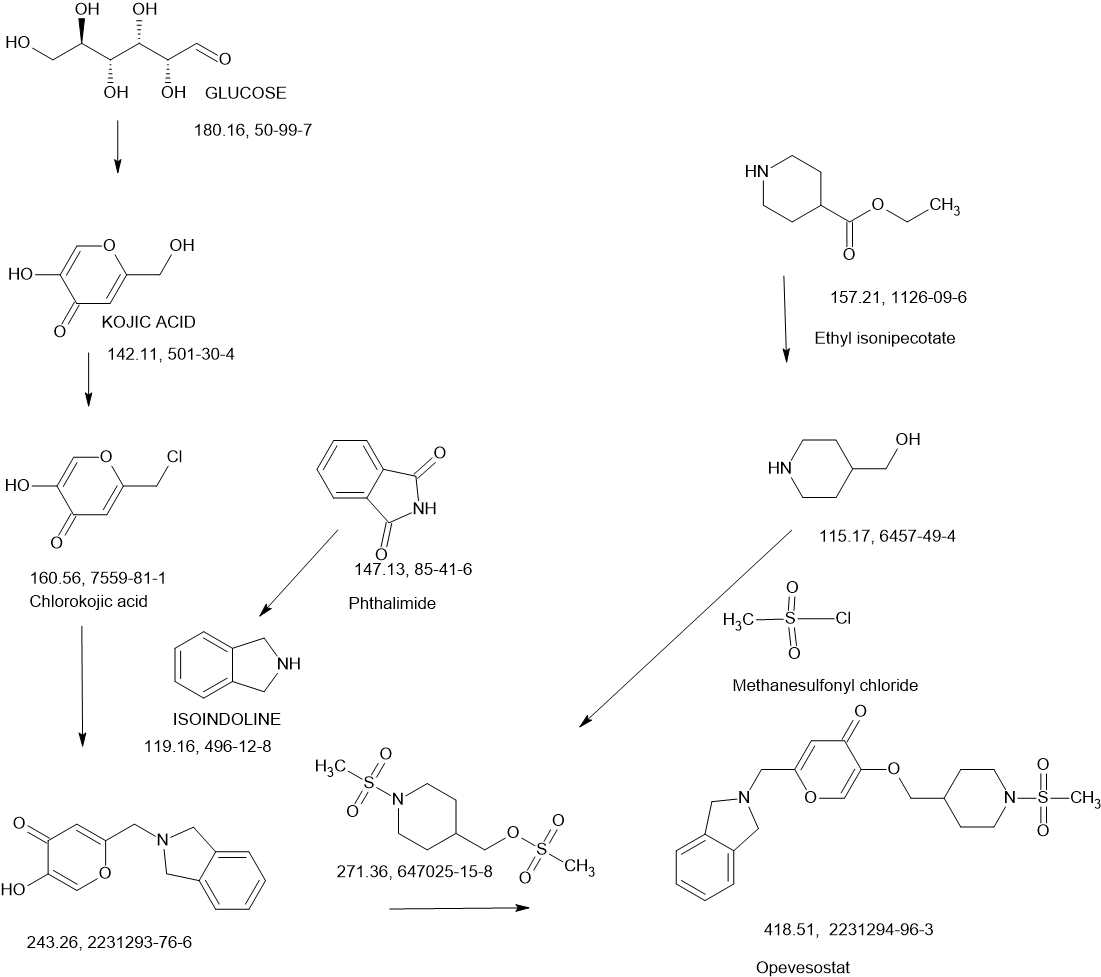
.

{kind=link}
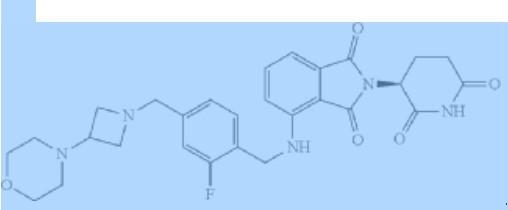{kind=link}
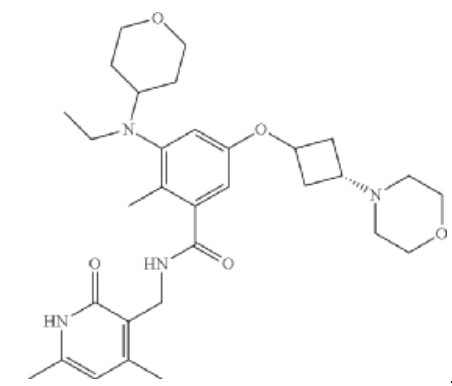{kind=link}
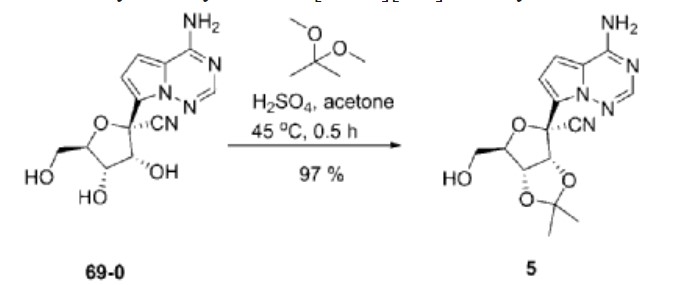{kind=link}
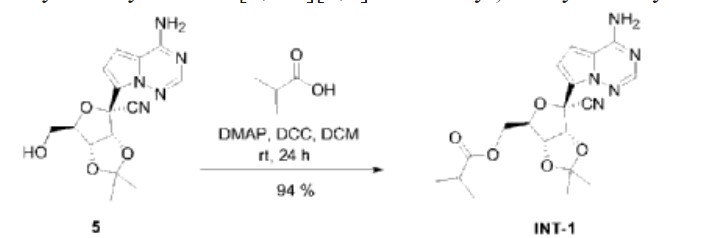{kind=link}
{kind=link}