
Home » Uncategorized (Page 106)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
TAK-733……. clinical studies for cancer treatment.

TAK-733
CAS: 1035555-63-5
Synonym: TAK-733; TAK 733; TAK733.
IUPAC/Chemical name:
(R)-3-(2,3-Dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione
Chemical Formula: C17H15F2IN4O4
Exact Mass: 504.01060
Molecular Weight: 504.23
Elemental Analysis: C, 40.49; H, 3.00; F, 7.54; I, 25.17; N, 11.11; O, 12.69
Phase I clinical studies for cancer treatment.Takeda Pharmaceutical,
Solid Tumors Therapy
Description of TAK-733: TAK-733 is an orally bioavailable small-molecule inhibitor of MEK1 and MEK2 (MEK1/2) with potential antineoplastic activity. MEK inhibitor TAK-733 selectively binds to and inhibits the activity of MEK1/2, preventing the activation of MEK1/2-dependent effector proteins and transcription factors, which may result in the inhibition of growth factor-mediated cell signaling and tumor cell proliferation. MEK1/2 (MAP2K1/K2) are dual-specificity threonine/tyrosine kinases that play key roles in the activation of the RAS/RAF/MEK/ERK pathway and are often upregulated in a variety of tumor cell types.
Current developer: Millennium Pharmaceuticals, Inc./Takeda Pharmaceutical Company Limited.
TAK-733 is being developed at Millennium Pharmaceuticals for the treatment of adult patients with advanced non-hematological malignancies. Phase I clinical trials are ongoing for the treatment of advanced metastatic melanoma. In preclinical studies, the compound has been shown to bind to and potently inhibit MEK.

………………………………….
Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer
- Takeda San Diego;10410 Science Center Drive, San Diego, CA 92121, United States
http://www.sciencedirect.com/science/article/pii/S0960894X11000941

Synthesis of compounds 26 and 27 (Route 4). Reagents and conditions: (a) 1-chloro-2,4-dinitrobenzene, K2CO3, DMF; (b) (R)-O-((2,2-dimethyl-1,3-dioxolan-4-yl)methyl)hydroxylamine or 2,2-dimethyl-1,3-dioxan-5-amine, K2CO3 or Cs2CO3, DMF; (c) HCl, THF; (d) Selectfluor, CH3CN,DMF.
TAK-733 exhibited potent enzymatic and cell activity with an IC50 of 3.2 nM against constitutively active MEK enzyme and an EC50 of 1.9 nM against ERK phosphorylation in cells. TAK-733 did not inhibit any other kinases, receptors or ion channels that were tested with inhibitor concentrations up to 10 μM. TAK-733 was found to bind plasma protein moderately (ca. 97% for human and 96% for mouse), and exhibit high permeability and high microsomal stability across species. It did not inhibit P450s up to 30 μM.
The co-crystal structure of TAK-733 in the MEK1 allosteric site has been solved (Fig. 3). As predicted, the pyridone oxygen makes a hydrogen bond with the backbone NH of Ser212. The 2-fluoro-4-iodoaniline moeity sits in the deep lipophilic pocket. The pyrimidinone oxygen makes a hydrogen bond with Lys97, and the propanediol terminal hydroxyl interacts with both Lys97 and the ADP phosphate.

-
Figure 3.
The X-ray co-crystal structure of TAK-733 in the MEK1 allosteric site.

(R)-3-(2,3-Dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione
| Molecular Weight: | 504.23 |
| TAK-733 Formula: | C17H15F2IN4O4 |
| CAS Number: | 1035555-63-5 |
TAK-733 is an orally bioavailable small-molecule inhibitor of MEK1 and MEK2 (MEK1/2) with potential antineoplastic activity. MEK inhibitor TAK-733 selectively binds to and inhibits the activity of MEK1/2, preventing the activation of MEK1/2-dependent effector proteins and transcription factors, which may result in the inhibition of growth factor-mediated cell signaling and tumor cell proliferation. MEK1/2 (MAP2K1/K2) are dual-specificity threonine/tyrosine kinases that play key roles in the activation of the RAS/RAF/MEK/ERK pathway and are often upregulated in a variety of tumor cell types.
BRAF L597 mutations in melanoma are associated with sensitivity to MEK inhibitors.
Dahlman et al. Cancer Discov. 2012 Jul 13. PMID: 22798288.Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer.
Dong et al. Bioorg Med Chem Lett. 2011 Mar 1;21(5):1315-9. PMID: 21310613.

16: 1652-1659


MEK kinases regulate the pathway that mediates proliferative and anti-apoptotic signaling factors that promote tumor growth and metastasis. TAK-733 is an MEK kinase inhibitor that entered phase I clinical trials for the treatment of cancer. A noteworthy feature of this short synthesis (25% yield overall) is the one-pot, three-step synthesis of the fluoropyridone D, in which the fluorine atom is present at the outset.
The reaction of F with the nosylate G gave a mixture of N- and O-alkylation products (8:1) from which the desired N-alkylation product was isolated by crystallization. The mixture of N-methyl pyrrolidine (NMP) and methanol used in the final deprotection step, helped to ensure formation of the desired polymorph. The nine-step discovery synthesis (3% overall yield) is also presented.
…………………………….
http://www.google.com/patents/WO2008079814A2?cl=en
Example 18: (i?)-3-(2,3-Dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8- methylpyrido[2,3-d]pyrimidine-4,7(3/f,8H)-dione

[0364] To a solution of (i?)-3-(2,3-dihydroxypropyl)-5-(2-fluoro-4-iodophenylamino)- 8-methylpyrido[2,3-d]pyrimidine-4,7(3/f,8H)-dione (example 6) (1 g, 2.06 mmol, 1 eq) in DMF (19 mL) was added dropwise a mixture of Selectfluor (801 mg, 2.26 mmol, 1.1 eq) in acetonitrile (9 niL) and DMF (5 niL) at ambient temperature while stirring under nitrogen. The reaction mixture was stirred for 10 minutes at ambient temperature and filtered. The filtrate was purified by preparatory LC/MS (30-55% CH3CN in H2O) to give (i?)-3-(2,3-dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8- methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione (example 18) as an off-white solid. The recovered starting material was subjected to the same reaction conditions to produce a second crop of product. The total yield of product was 347 mg (33% yield). 1H NMR (400 MHz, DMSO-J6) δ ppm 3.36 – 3.40 (m, 1 H) 3.43 – 3.50 (m, 1 H) 3.58 (s, 3 H) 3.61 – 3.72 (m, 1 H) 3.72 – 3.82 (m, 1 H) 4.27 – 4.37 (m, 1 H) 4.78 – 4.87 (m, 1 H) 5.14 (d, J=5.81 Hz, 1 H) 6.93 – 7.03 (m, 1 H) 7.53 (d, J=8.84 Hz, 1 H) 7.65 – 7.74 (m, 1 H) 8.52 (s, 1 H) 10.25 (d, J=LOl Hz, 1 H). [M+H] calc’d for Ci7Hi5F2IN4O4, 505; found, 505.
OTHER ISOMER
Example 19: (5)-3-(2,3-Dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8- methylpyrido[2,3-d]pyrimidine-4,7(3/f,8H)-dione
 CAREFUL PLEASE
CAREFUL PLEASE[0365] To a solution of (5)-3-(2,3-dihydroxypropyl)-5-(2-fluoro-4-iodophenylamino)- 8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione (example 5) (1.25 g, 2.58 mmol, 1 eq) in DMF (26 mL) was added a mixture of Selectfluor (1.187 g, 3.35 mmol, 1.3 eq) in acetonitrile (26 mL). The reaction mixture was irradiated with microwave at 82 0C for 10 minutes and filtered. The filtrate was purified by preparatory LC/MS (30-55% CH3CN in H2O) to give (5)-3-(2,3-dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8- methylpyrido[2,3-d]pyrimidine-4,7(3/f,8H)-dione (example 19) as an off-white solid (331 mg, 25% yield). 1H NMR (400 MHz, DMSO-J6) δ ppm 3.36 – 3.42 (m, 1 H) 3.42 – 3.51 (m, 1 H) 3.59 (s, 3 H) 3.63 – 3.72 (m, 1 H) 3.73 – 3.81 (m, 1 H) 4.32 (dd, J=13.14, 3.03 Hz, 1 H) 4.79 (br. s., 1 H) 5.10 (br. s., 1 H) 6.98 (td, J=8.46, 5.31 Hz, 1 H) 7.52 (dd, J=8.46, 1.14 Hz, 1 H) 7.68 (dd, J=10.36, 1.77 Hz, 1 H) 8.51 (s, 1 H) 10.24 (d, J=2.27 Hz, 1 H). [M+H] calc’d for Ci7Hi5F2IN4O4, 505; found, 505.
……………………………………….
http://www.google.com/patents/US8436200?cl=en

The synthetic process of the present invention allows for the preparation of (R)-3-(2,3-dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione while avoiding the use of a fluorinating reagent in the last step. That is, the present invention provides a valuable process for making (R)-3-(2,3-dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione characterized by the reaction of a compound of formula (8) with 2-fluoro-4-iodoaniline to give a compound of formula (9). Such a process avoids the use of costly and possible hazardous fluorinating regents in later steps which has significant advantages in large-scale manufacture.
Example 8.1(R)-3-(2,3-Dihydroxypropyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione
Combine (R)-3-((2,2-dimethyl-1,3-dioxolan-4-yl)methyl)-6-fluoro-5-(2-fluoro-4-iodophenylamino)-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione (24.75 g, 45.58 mmol) and ethanol (250 mL). Add aqueous 9N sulfuric acid (50 mL) over 5 minutes. Heat to 75° C. After 2 hour, cool to ambient temperature and then cool in an ice bath to give a solid. Collect the solid by filtration, rinse with ethanol (3×30 mL), and dry to give the title compound. 1H NMR (400 MHz, DMSO-d6) δ10.24 (s, 1H), 8.52 (s, 1H), 7.69 (dd, 1H, J=10.4, 1.8 Hz), 7.52 (d, 1H, J=8.6 Hz), 6.98 (m, 1H), 5.14 (brs, 1H), 4.83 (brs, 1H), 4.32 (dd, 1H, J=12.9, 2.5 Hz), 3.76 (m, 1H), 3.67 (dd, 1H, J=13.1, 12.9 Hz), 3.58 (s, 3H), 3.46 (ddd, 1H, J=10.9, 5.3, 5.1 Hz), 3.38 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ161.3 (d, J=4.0 Hz), 155.6 (d, J=22.8 Hz), 154.6 (d, J=250 Hz), 152.0, 150.6, 134.3 (d, J=231 Hz), 133.8 (d, J=7.1 Hz), 133.1 (d, J=3.0 Hz), 127.8 (d, J=10.3 Hz), 125.3 (d, J=7.0 Hz), 123.9 (d, J=21.5 Hz), 95.0 (d, J=4.0 Hz), 87.1 (d, J=7.8 Hz), 68.0, 63.8, 50.1, 28.8; 19F NMR (376 MHz, DMSO-d6) δ-124.4, −149.8; MS (M+H)+m/z calcd 505.0. found 505.0.
Google+
|
Information about this agent |
TAK-733 is currently in Phase I clinical trials and is being developed by Millennium Pharmaceuticals, Inc. (a part of Takeda Pharmaceutical Company Limited).
|
References |
1: Acquaviva J, Smith DL, Jimenez JP, Zhang C, Sequeira M, He S, Sang J, Bates RC, Proia DA. Overcoming acquired BRAF inhibitor resistance in melanoma via targeted inhibition of Hsp90 with ganetespib. Mol Cancer Ther. 2014 Feb;13(2):353-63. doi: 10.1158/1535-7163.MCT-13-0481. Epub 2014 Jan 7. PubMed PMID: 24398428.
2: Zhang Y, Xue D, Wang X, Lu M, Gao B, Qiao X. Screening of kinase inhibitors targeting BRAF for regulating autophagy based on kinase pathways. Mol Med Rep. 2014 Jan;9(1):83-90. doi: 10.3892/mmr.2013.1781. Epub 2013 Nov 7. PubMed PMID: 24213221.
3: Nakamura A, Arita T, Tsuchiya S, Donelan J, Chouitar J, Carideo E, Galvin K, Okaniwa M, Ishikawa T, Yoshida S. Antitumor activity of the selective pan-RAF inhibitor TAK-632 in BRAF inhibitor-resistant melanoma. Cancer Res. 2013 Dec 1;73(23):7043-55. doi: 10.1158/0008-5472.CAN-13-1825. Epub 2013 Oct 11. PubMed PMID: 24121489.
4: Garraway LA, Baselga J. Whole-genome sequencing and cancer therapy: is too much ever enough? Cancer Discov. 2012 Sep;2(9):766-8. doi: 10.1158/2159-8290.CD-12-0359. PubMed PMID: 22969114.
5: Dahlman KB, Xia J, Hutchinson K, Ng C, Hucks D, Jia P, Atefi M, Su Z, Branch S, Lyle PL, Hicks DJ, Bozon V, Glaspy JA, Rosen N, Solit DB, Netterville JL, Vnencak-Jones CL, Sosman JA, Ribas A, Zhao Z, Pao W. BRAF(L597) mutations in melanoma are associated with sensitivity to MEK inhibitors. Cancer Discov. 2012 Sep;2(9):791-7. Epub 2012 Jul 13. PubMed PMID: 22798288; PubMed Central PMCID: PMC3449158.
6: von Euw E, Atefi M, Attar N, Chu C, Zachariah S, Burgess BL, Mok S, Ng C, Wong DJ, Chmielowski B, Lichter DI, Koya RC, McCannel TA, Izmailova E, Ribas A. Antitumor effects of the investigational selective MEK inhibitor TAK733 against cutaneous and uveal melanoma cell lines. Mol Cancer. 2012 Apr 19;11:22. PubMed PMID: 22515704; PubMed Central PMCID: PMC3444881.
7: Dong Q, Dougan DR, Gong X, Halkowycz P, Jin B, Kanouni T, O’Connell SM, Scorah N, Shi L, Wallace MB, Zhou F. Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer. Bioorg Med Chem Lett. 2011 Mar 1;21(5):1315-9. doi: 10.1016/j.bmcl.2011.01.071. Epub 2011 Jan 22. PubMed PMID: 21310613.
| US8030317 | Dec 18, 2007 | Oct 4, 2011 | Takeda Pharmaceutical Company Limited | MAPK/ERK kinase inhibitors |
| US20080255160 | Dec 18, 2007 | Oct 16, 2008 | Qing Dong | Mapk/erk kinase inhibitors |
| WO2008000020A1 | Jun 27, 2007 | Jan 3, 2008 | Gary L Corino | Improved process |
| EP1894932A1 | Jun 10, 2005 | Mar 5, 2008 | Japan Tobacco, Inc. | 5-amino-2,4,7-trioxo-3,4,7,8-tetrahydro-2H-pyrido[2,3-d]pyrimidine derivatives and related compounds for the treatment of cancer |
| US20050222177 * | Jul 29, 2004 | Oct 6, 2005 | Irm Llc | Diseases with abnormal activation of the Abl, BCR-Abl, Bmx, CSK, TrkB, FGFR3, Fes, Lck, B-RAF, C-RAF, MKK6, alpha and beta SAPK2 kinases; antiproliferative; pyrrolo[2,3-d]pyrimidine-7-carboxylic acid [3-phenylcarbamoyl-phenyl]-amides and pyrrolo[3,2-c]pyridine analogs |
Aplaviroc, AK602, GSK-873140

Aplaviroc
4-(4-{[(3R)-1-butyl-3-[(R)-cyclohexylhydroxymethyl]-2,5-dioxo- 1,4,9-triazaspiro[5.5]undecan-9-yl]methyl}phenoxy)benzoic acid
for the treatment of HIV infection
461023-63-2 of hydrochloride
461443-59-4 (free base)
873140
AK-602
GW-873140
ONO-4128
ono…….innovator
| Ono Pharmaceutical Co., Ltd. |
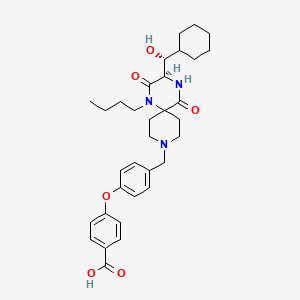
| Identifiers | |
|---|---|
| CAS number | 461023-63-2 |
| ATC code | None |
| PubChem | CID 3001322 |
| ChemSpider | 2272720 |
| UNII | 98B425P30V |
| KEGG | D06557 |
| ChEMBL | CHEMBL1255794 |
| Chemical data | |
| Formula | C33H43N3O6 |
| Mol. mass | 577.711 g/mol |
Aplaviroc (INN, codenamed AK602 and GSK-873140) is a CCR5 entry inhibitor developed for the treatment of HIV infection.[1][2] It is developed by GlaxoSmithKline.
In October 2005, all studies of aplaviroc were discontinued due to liver toxicity concerns.[3][4] Some authors have claimed that evidence of poor efficacy may have contributed to termination of the drug’s development;[5] the ASCENT study, one of the discontinued trials, showed aplaviroc to be under-effective in many patients even at high concentrations.[6]
Aplaviroc hydrochloride, an orally-effective, long-acting chemokine CCR5 receptor antagonist, had been under development by Ono and GlaxoSmithKline for the treatment of HIV infection. In early 2006, the companies discontinued development of the antagonist based on reports of elevated liver function test values from clinical studies.
Originally developed at Ono, aplaviroc was licensed to GlaxoSmithKline in 2003 for development, manufacturing and marketing. GlaxoSmithKline also obtained rights to evaluate the agent in non-HIV conditions worldwide with the exception of Japan, South Korea and Taiwan.
A low-molecular-weight compound, aplaviroc prevents HIV viral infection by blocking the binding of the virus to the CCR5 receptor
……………….
WO 2002074770
0r
http://www.google.com/patents/EP1378510A1?cl=en
Reference example 3(3)
- (3R)-1-butyl-2,5-dioxo-3-((1R)-1-hydroxy-1-cyclohexyl)-1,4,9-triazaspiro[5.5]undecane • hydrochloride
-
[0136]

TLC:Rf 0.32 (butanol:acetic acid:water = 4:2:1);
NMR (CD3OD): δ 4.16 (d, J = 2.0 Hz, 1H), 3.95 (m, 1H), 3.70 (m, 1H), 3.52 (m, 1H), 3.37 (m, 1H), 3.28 (m, 1H), 3.22-3.13 (m, 2H), 2.46-1.93 (m, 6H), 1.80-1.64 (m, 5H), 1.48-1.15 (m, 6H), 1.02-0.87 (m, 5H);
Optical rotation:[α]D +1.22 (c 1.04, methanol, 26°C).

Example 9(54)
- (3R)-1-butyl-2,5-dioxo-3-((1R)-1-hydroxy-1-cyclohexylmethyl)-9-(4-(4-carboxyphenyloxy)phenylmethyl)-1,4,9-triazaspiro[5.5]undecane • hydrochloride
-
[0359]

TLC:Rf 0.43(chloroform:methanol = 5:1);
NMR (CD3OD):δ 8.05 (d, J = 9.0 Hz, 2H), 7.61 (d, J = 9.0 Hz, 2H), 7.19 (d, J = 9.0 Hz, 2H), 7.08 (d, J = 9.0 Hz, 2H), 4.38 (s, 2H), 4.17 (d, J = 2.1 Hz, 1H), 4.02 (m, 1H), 3.78 (m, 1H), 3.60-3.40 (m, 3H), 3.30-3.10 (m, 2H), 2.56-1.86 (m, 6H), 1.82-1.60 (m, 5H), 1.52-1.16 (m, 6H), 1.06-0.82 (m, 2H), 0.97 (t, J = 7.2 Hz, 3H).

………………….
http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-9-265
Owing to the special properties of piperazines (increased solubility and H-bond acceptor capability etc.) it is often considered to be a privileged structure and therefore occurs widely, for instance in GlaxoSmithKlines investigational anti-HIV drug aplaviroc (4.37) which, despite being a promising CCR5 receptor antagonist, was discontinued due to hepatotoxicity concerns. In this compound the spirodiketopiperazine unit (4.35) was designed to mimic a type-1 β-turn (4.36) as present in G-protein coupled receptors (Figure 14) [117].
![[1860-5397-9-265-14]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-14.png?scale=2.0&max-width=1024&background=FFFFFF)
The synthesis of aplaviroc and its analogues can be accomplished via the use of an Ugi multicomponent reaction (Ugi-MCR) [118]. The procedure involved the condensation of piperidone 4.38 and butylamine (4.39) followed by reaction of the resulting imine with isocyanide 4.41 and interception of the nitrilium intermediate with the amino acid4.40 (Scheme 47) [119]. This sequence was completed by structural rearrangement and acid-mediated ring closure to produce the spirocyclic diketopiperazine 4.43. Following debenzylation this material was subjected to a reductive amination finally affording aplaviroc analogues (Scheme 47).
![[1860-5397-9-265-i47]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i47.png?scale=2.0&max-width=1024&background=FFFFFF)
- 117 Habashita, H.; Kokubo, M.; Hamano, S.; Hamanaka, N.; Toda, M.; Shibayama, S.; Tada, H.; Sagawa, K.; Fukushima, D.; Maeda, K.; Mitsuya, H. J. Med. Chem. 2006, 49, 4140–4152. doi:10.1021/jm060051s
- Dömling, A.; Huang, Y. Synthesis 2008, 2859–2883. doi:10.1055/s-0030-1257906
ref 118 - Nishizawa, R.; Nishiyama, T.; Hisaichi, K.; Matsunaga, N.; Minamoto, C.; Habashita, H.; Takaoka, Y.; Toda, M.; Shibayama, S.; Tada, H.; Sagawa, K.; Fukushima, D.; Maeda, K.; Mitsuya, H.Bioorg. Med. Chem. Lett. 2007, 17, 727–731. doi:10.1016/j.bmcl.2006.10.084
ref 119
| Patent | Submitted | Granted |
|---|---|---|
| Triazaspiro[5.5]undecane derivative and pharmaceutical composition comprising the same as active ingredient [US7262193] | 2005-09-29 | 2007-08-28 |
| Drugs containing triazaspiro[5.5]undecane derivatives as the active ingredient [US7285552] | 2004-06-03 | 2007-10-23 |
| Triazaspiro[5.5]undecane derivatives and drugs containing the same as the active ingredient [US7053090] | 2004-04-29 | 2006-05-30 |
| WO1998031364A1 * | Jan 20, 1998 | Jul 23, 1998 | Timothy Harrison | 3,3-disubstituted piperidines as modulators of chemokine receptor activity |
| WO2000014086A1 * | Jan 21, 1999 | Mar 16, 2000 | Kyowa Hakko Kogyo Kk | Chemokine receptor antagonists and methods of use therefor |
| WO2002074769A1 * | Mar 18, 2002 | Sep 26, 2002 | Kenji Maeda | Drugs containing triazaspiro[5.5]undecane derivatives as the active ingredient |
References
- Maeda, Kenji; Ogata, Hiromi; Harada, Shigeyoshi et al. (2004). “Determination of binding sites of a unique CCR5 inhibitor AK602 / ONO-4128/ GW873140 on human CCR5” (PDF). Conference on Retroviruses and Opportunistic Infections. Archived from the original on November 3, 2005.
- Nakata, Hirotomo; Maeda, Kenji; Miyakawa, Toshikazu et al. (February 2005). “Potent Anti-R5 Human Immunodeficiency Virus Type 1 Effects of a CCR5 Antagonist, AK602/ONO4128/GW873140, in a Novel Human Peripheral Blood Mononuclear Cell Nonobese Diabetic-SCID, Interleukin-2 Receptor γ-Chain-Knocked-Out AIDS Mouse Model”. Journal of Virology 79 (4): 2087–96.doi:10.1128/jvi.79.4.2087-2096.2005.
- “Aplaviroc (GSK-873,140)”. AIDSmeds.com. October 25, 2005. Retrieved September 5, 2008.[dead link]
- Nichols WG, Steel HM, Bonny T et al. (March 2008). “Hepatotoxicity Observed in Clinical Trials of Aplaviroc (GW873140)”.Antimicrobial Agents and Chemotherapy 52 (3): 858–65. doi:10.1128/aac.00821-07. PMC 2258506. PMID 18070967.
- Moyle, Graeme (December 19, 2006). “The Last Word on Aplaviroc: A CCR5 Antagonist With Poor Efficacy”. The Body.Archived from the original on 6 October 2008. Retrieved September 5, 2008.
- Currier, Judith; Lazzarin, Adriano; Sloan, Louis et al. (2008). “Antiviral activity and safety of aplaviroc with lamivudine/zidovudine in HIV-infected, therapy-naive patients: the ASCENT (CCR102881) study”. Antiviral Therapy (Lond.) 13 (2): 297–306.PMID 18505181.
Further reading
- Horster, S; Goebel, FD (April 2006). “Serious doubts on safety and efficacy of CCR5 antagonists: CCR5 antagonists teeter on a knife-edge”. Infection 34 (2): 110–13. doi:10.1007/s15010-006-6206-1. PMID 16703305.
Beta carotene may protect people with common genetic risk factor for type-2 diabetes
25 JAN 2013
STANFORD, Calif. — Stanford University School of Medicine investigators have found that for people harboring a genetic predisposition that is prevalent among Americans, beta carotene, which the body converts to a close cousin of vitamin A, may lower the risk for the most common form of diabetes, while gamma tocopherol, the major form of vitamin E in the American diet, may increase risk for the disease.
The scientists used a “big data” approach to hunt down interactions between gene variants previously associated with increased risk for type-2 diabetes and blood levels of substances previously implicated in type-2 diabetes risk. In people carrying a double dose of one such predisposing gene variant, the researchers pinpointed a highly statistically significant inverse association of beta carotene blood levels with type-2 diabetes risk, along with a suspiciously high positive association of gamma tocopherol with risk for the disease.
“Type-2 diabetes affects…
View original post 1,081 more words
New USP Requirements on Plastic Packaging Systems
DRUG REGULATORY AFFAIRS INTERNATIONAL

New USP Requirements on Plastic Packaging Systems |
| The USP describes in an article of the Pharmacopeial Forum the future requirements for plastic packaging systems. Here, the importance is laid on the selection of suitable, safe plastic materials and the verification of potential interactions. More information can be found here in the News. |
GMP News: New USP Requirements on Plastic Packaging Systems
An interesting article from the USP on the future requirements for plastic packaging systems has been published in the Pharmacopoeial Forum 39(6).
In this article, the USP’s experts group provides an overview of the already existing and also the planned General Chapters on pharmaceutical plastic packaging systems. Together both chapters aim to describe a general and chemistry-based approach for the quality and safety of packaging systems and their starting materials for the construction of these packaging systems.
Among the key topics which are discussed, you can find:
- a. The…
View original post 160 more words
SURAMIN

Suramin
A polyanionic compound with an unknown mechanism of action. It is used parenterally in the treatment of African trypanosomiasis and it has been used clinically with diethylcarbamazine to kill the adult Onchocerca. (From AMA Drug Evaluations Annual, 1992, p1643) It has also been shown to have potent antineoplastic properties.
A polyanionic compound with an unknown mechanism of action. It is used parenterally in the treatment of African trypanosomiasis and it has been used clinically with diethylcarbamazine to kill the adult Onchocerca. (From AMA Drug Evaluations Annual, 1992, p1643) It has also been shown to have potent antineoplastic properties. Suramin is manufactured by Bayer in Germany as Germanin®.
Also known as: Naphuride, Germanin, Naganol, Belganyl, Fourneau, Farma, Antrypol, Suramine, Naganin
8,8′-{Carbonylbis[imino-3,1-phenylenecarbonylimino(4-methyl-3,1-phenylene)carbonylimino]}di(1,3,5-naphthalenetrisulfonic acid) …FREE FORM
8,8′-[Ureylenebis[m-phenylenecarbonylimino(4-methyl-m-phenylene)carbonylimino]]di(1,3,5-naphthalenetrisulfonic acid) hexasodium salt
CAS 145-63-1 FREE FORM
129-46-4 of hexa sodium
LAUNCHED 1940 BAYER
| Formula | C51H40N6O23S6 |
|---|---|
| Mol. mass | 1297.29 |
The molecular formula of suramin is C51H34N6O23S6. It is a symmetric molecule in the center of which lies urea, NH-CO-NH. Suramin contains eightbenzene rings, four of which are fused in pairs (naphthalene), four amide groups in addition to the one of urea and six sulfonate groups. When given as drug it usually contains six sodium ions that form a salt with the six sulfonate groups.
Suramin is a drug developed by Oskar Dressel and Richard Kothe of Bayer, Germany in 1916, and is still sold by Bayer under the brand nameGermanin.
Suramin sodium is a heparanase inhibitor that was first launched in 1940 by Bayer under the brand name Antrypol for the treatment of helminthic infection. It was later launched by Bayer for the treatment of trypanosomiasis (African sleeping sickness).
More recently, the product has entered early clinical development at Ohio State University for the treatment of platinum-pretreated patients with stage IIIB/IV non-small cell lung cancer, in combination with docetaxel or gemcitabine.
The National Cancer Institute (NCI) is conducting phase II clinical studies for the treatment of glioblastoma multiforme and for the treatment of adrenocortical carcinoma.
According to the National Cancer Institute there are no active clinical trials (as of April 1, 2008). Completed and closed clinical trials are listed here:[1]
In addition to Germanin, the National Cancer Institute also lists the following “Foreign brand names”: 309 F or 309 Fourneau,[1] Bayer 205, Moranyl, Naganin, Naganine.

It is used for treatment of human sleeping sickness caused by trypanosomes.[2]
It has been used in the treatment of onchocerciasis.[3]
It has been investigated as treatment for prostate cancer.[4]
Also, suramin as treatment for autism is being evaluated. [5]
Suramin is administered by a single weekly intravenous injection for six weeks. The dose per injection is 1 g.
The most frequent adverse reactions are nausea and vomiting. About 90% of patients will get an urticarial rash that disappears in a few days without needing to stop treatment. There is a greater than 50% chance of adrenal cortical damage, but only a smaller proportion will require lifelongcorticosteroid replacement. It is common for patients to get a tingling or crawling sensation of the skin with suramin. Suramin will cause clouding of the urine which is harmless: patients should be warned of this to avoid them becoming alarmed.
Kidney damage and exfoliative dermatitis occur less commonly.
Suramin has been applied clinically to HIV/AIDS patients resulting in a significant number of fatal occurrences and as a result the application of this molecule was abandoned for this condition. http://www.ncbi.nlm.nih.gov/pubmed/3548350
Suramin is also used in research as a broad-spectrum antagonist of P2 receptors[6][7] and agonist of Ryanodine receptors.[8]
![ChemSpider 2D Image | 8,8'-{Carbonylbis[imino-3,1-phenylenecarbonylimino(4-methyl-3,1-phenylene)carbonylimino]}di(1,3,5-naphthalenetrisulfonic acid) | C51H40N6O23S6](https://images-blogger-opensocial.googleusercontent.com/gadgets/proxy?url=http%3A%2F%2Fwww.chemspider.com%2FImagesHandler.ashx%3Fid%3D5168%26w%3D200%26h%3D200&container=blogger&gadget=a&rewriteMime=image%2F*)
Its effect on telomerase has been investigated.[9]
It may have some activity against RNA viruses.[10]
In addition to antagonism of P2 receptors, Suramin inhibits the acitivation of heterotrimeric G proteins in a variety of other GPCRs with varying potency. It prevents the association of heteromeric G proteins and therefore the receptors Guanine exchange functionality (GEF). With this blockade the GDP will not release from the Gα subunit so it can not be replaced by a GTP and become activated. This has the effect of blocking downstream G protein mediated signaling of various GPCR proteins including Rhodopsin, the A1 Adenosine receptor, and the D2 dopamine receptor.[11]
A polyanionic compound with an unknown mechanism of action. It is used parenterally in the treatment of African trypanosomiasis and it has been used clinically with diethylcarbamazine to kill the adult Onchocerca. (From AMA Drug Evaluations Annual, 1992, p1643) It has also been shown to have potent antineoplastic properties. Suramin is manufactured by Bayer in Germany as Germanin®.
|
8-1-2012 |
InCl3-catalysed synthesis of 2-aryl quinazolin-4(3H)-ones and 5-aryl pyrazolo[4,3-d]pyrimidin-7(6H)-ones and their evaluation as potential anticancer agents. |
Bioorganic & medicinal chemistry letters |
|
9-1-2012 |
Identification of a sirtuin 3 inhibitor that displays selectivity over sirtuin 1 and 2. |
European journal of medicinal chemistry |
|
1-1-2013 |
Inhibition of the human deacylase Sirtuin 5 by the indole GW5074. |
Bioorganic & medicinal chemistry letters |
|
5-9-2013 |
Discovery of thieno[3,2-d]pyrimidine-6-carboxamides as potent inhibitors of SIRT1, SIRT2, and SIRT3. |
Journal of medicinal chemistry |
- The formula of suramin was kept secret by Bayer for commercial reasons. But it was elucidated and published in 1924 by Fourneau and his team of the Pasteur Institute, and it is only on this date that its exact chemical composition was known. (E. Fourneau, J. and Th. Tréfouël and J. Vallée (1924). “Sur une nouvelle série de médicaments trypanocides”, C. R. Séances Acad. Sci. 178: 675.)
- Darsaud A, Chevrier C, Bourdon L, Dumas M, Buguet A, Bouteille B (January 2004). “Megazol combined with suramin improves a new diagnosis index of the early meningo-encephalitic phase of experimental African trypanosomiasis”. Trop. Med. Int. Health 9 (1): 83–91.doi:10.1046/j.1365-3156.2003.01154.x. PMID 14728611.
- Anderson J, Fuglsang H (July 1978). “Further studies on the treatment of ocular onchocerciasis with diethylcarbamazine and suramin”. Br J Ophthalmol 62 (7): 450–7.doi:10.1136/bjo.62.7.450. PMC 1043255. PMID 678497.
- Ahles TA, Herndon JE, Small EJ, et al. (November 2004). “Quality of life impact of three different doses of suramin in patients with metastatic hormone-refractory prostate carcinoma: results of Intergroup O159/Cancer and Leukemia Group B 9480”. Cancer 101 (10): 2202–8.doi:10.1002/cncr.20655. PMID 15484217.
- http://medicalxpress.com/news/2013-03-drug-treatment-autism-symptoms-mouse.html
- Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, Weisman GA. (september 2006). “International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy”. Pharmacol Rev. 58 (3): 281–341.doi:10.1124/pr.58.3.3. PMID 16968944.
- Khakh BS, Burnstock G, Kennedy C, King BF, North RA, Séguéla P, Voigt M, Humphrey PP. (march 2001). “International union of pharmacology. XXIV. Current status of the nomenclature and properties of P2X receptors and their subunits”. Pharmacol Rev. 53 (1): 107–118.PMID 11171941.
- Wolner I, Kassack MU, Ullmann H, Karel A, Hohenegger M (October 2005). “Use-dependent inhibition of the skeletal muscle ryanodine receptor by the suramin analogue NF676”. Br. J. Pharmacol. 146 (4): 525–33. doi:10.1038/sj.bjp.0706359. PMC 1751178.PMID 16056233.
- Erguven M, Akev N, Ozdemir A, Karabulut E, Bilir A (August 2008). “The inhibitory effect of suramin on telomerase activity and spheroid growth of C6 glioma cells”. Med. Sci. Monit. 14(8): BR165–73. PMID 18667993.
- Mastrangelo E, Pezzullo M, Tarantino D, Petazzi R, Germani F, Kramer D, Robel I, Rohayem J, Bolognesi M, Milani M (2012) Structure-based inhibition of norovirus RNA-dependent RNA-polymerases. J Mol Biol
- Beindl W, Mitterauer T, Hohenegger M, Ijzerman AP, Nanoff C, Freissmuth M. (August 1996).“Inhibition of receptor/G protein coupling by suramin analogues”. ol. Pharmacology. 50 (2): 415–23. PMID 8700151.
- Drugs Fut 1986, 11(10): 860
- WO 2012159107
- WO 2012087336
- US 2011257109
- WO 2009022897
- WO 2009020613
- WO 2008094027
- EP 0486809
- US 5158940
- US 5173509
- WO 1993007864
- WO 1994008574

SURAMIN
- Suramin bound to proteins in the PDB
- Drug information
- Suramin, drug information by JBC Online
- Suramin in treating patients with recurrent bladder cancer
- National Cancer Institute
Enterovirus-71 (EV71) is one of the major causative reagents for hand-foot-and-mouth disease. In particular, EV71 causes severe central nervous system infections and leads to numerous dead cases. Although several inactivated whole-virus vaccines have entered in clinical trials, no antiviral agent has been provided for clinical therapy. In the present work, we screened our compound library and identified that suramin, which has been clinically used to treat variable diseases, could inhibit EV71 proliferation with an IC50 value of 40μM. We further revealed that suramin could block the attachment of EV71 to host cells to regulate the early stage of EV71 infection, as well as affected other steps of EV71 life cycle. Our results are helpful to understand the mechanism for EV71 life cycle and provide a potential for the usage of an approved drug, suramin, as the antiviral against EV71 infection.

- Suramin Hexasodium
- 129-46-4
Synonyms
- 309 F
- Antrypol
- BAY 205
- Bayer 205
- CI-1003
- EINECS 204-949-3
- Fourneau 309
- Germanin
- Moranyl
- Naganin
- Naganine
- Naganinum
- Naganol
- Naphuride sodium
- NF060
- NSC 34936
- SK 24728
- Sodium suramin
- Suramin Hexasodium
- Suramin sodium
- Suramina sodica
- Suramina sodica [INN-Spanish]
- Suramine sodique
- Suramine sodique [INN-French]
- Suramine sodium
- Suraminum natricum
- Suraminum natricum [INN-Latin]
- UNII-89521262IH
Suramin Sodium, is an anticancer agent with a wide variety of activities.
Recently suramin was shown to inhibit FSH binding to its receptor (Daugherty, R. L.; Cockett, A. T. K.; Schoen, S. R. and Sluss, P. M. “Suramin inhibits gonadotropon action in rat testis: implications for treatment of advanced prostate cancer” J. Urol. 1992, 147, 727-732).
This activity causes, at least in part, the decrease in testosterone production seen in rats and humans that were administered suramin(Danesi, R.; La Rocca, R. V.; Cooper, M. R.; Ricciardi, M. P.; Pellegrini, A.; Soldani, P.; Kragel, P. J.; Paparelli, A.; Del Tacca, M.; Myers, C. E, “Clinical and experimental evidence of inhibition of testosterone production by suramin.” J. Clin. Endocrinol. Metab. 1996, 81, 2238-2246).
Suramin is the only non-peptidic small molecule that has been reported to be an FSH receptor binding antagonist.

Suramin is 8,8′ – (carbonylbis(imino-3,1-phenylenecarbonylimino (4-methyl-3,1-phenylene) carbonylimino)) bis-1,3 ,5-naphthalenetrisulfonic acid (GB Patent No. 224849). This polyanionic compound has been used for many decades as a prophylactic and therapeutic agent for try- panosomiasis. It was subsequently shown that suramin is able to block the activity of a variety of proteins like cellular and viral enzymes and growth factors (Mitsuya, M. et al. Science 226 : 172 (1984), Hosang, M. J. Cell. Biochem. 29 : 265 (1985), De Clercq, E. Cancer Lett. 8 : 9 (1979)).

|
5-32-1977 |
Complement inhibitors |
|
|
5-25-1977 |
Aromatic amidines as antiviral agents in animals |
|
|
5-4-1977 |
Complement inhibitors |
|
|
5-4-1977 |
Complement inhibitors |
|
|
4-27-1977 |
Cyclodextrin sulfate salts as complement inhibitors |
|
|
4-20-1977 |
Ureylenebis methyl-phenylene-carbonyl-bis-dihydro-2-oxo-naphthoxazine disultonic acids |
|
|
3-30-1977 |
Water treatment for controlling the growth of algae employing biguanides |
|
|
3-2-1977 |
Isoxazole substituted nitroimidazoles |
|
|
2-16-1977 |
Amidophenyl-azo-naphthalenesulfonic complement inhibitors and method of use thereof |
|
|
2-9-1977 |
Complement inhibitors |
|
2-10-2011 |
MODULATION OF HUMAN MAST CELL ACTIVATION MODULATION OF HUMAN MAST CELL ACTIVATION |
|
|
11-18-2010 |
Admixtures for inorganic binders based on a hydrogenated disaccharide, inorganic binders containing these admixtures and process for their preparation Admixtures for inorganic binders based on a hydrogenated disaccharide, inorganic binders containing these admixtures and process for their preparation |
|
|
10-28-2010 |
THERAPEUTIC INHIBITORS OF VASCULAR SMOOTH MUSCLE CELLS |
|
|
9-9-2010 |
APPARATUS FOR USING ELECTROPORATION MEDIATED DELIVERY OF DRUGS AND GENES |
|
|
4-8-2010 |
PREPARATION AND USE OF SULFATED OLIGOSACCHARIDES |
|
|
10-29-2009 |
THERAPEUTIC INHIBITOR OF VASCULAR SMOOTH MUSCLE CELLS THERAPEUTIC INHIBITOR OF VASCULAR SMOOTH MUSCLE CELLS |
|
|
8-20-2009 |
METHOD OF MAKING MINERAL FIBRES METHOD OF MAKING MINERAL FIBRES |
|
|
6-25-2009 |
OXYGEN-FUEL BOOST REFORMER PROCESS AND APPARATUS |
|
|
4-23-2009 |
METHODS OF TREATING VASCULAR DISEASE WITH TNF ANTAGONISTS METHODS OF TREATING VASCULAR DISEASE WITH TNF ANTAGONISTS |
|
|
3-26-2009 |
COPOLYMER COMPOSITIONS FOR ORAL DELIVERY |
|
5-3-1978 |
1,3,5- Or 1,3,6-naphthalenetriyltris(sulfonylimino)aryl acids and salts |
|
|
3-22-1978 |
Nitroimidazoles |
|
|
2-15-1978 |
Treatment of rheumatoid arthritis and related diseases |
|
|
1-4-1978 |
AROMATIC AMIDINES AS ANTIVIRAL AGENTS IN ANIMALS |
|
|
1-4-1978 |
Malto-dextrin poly(H-)sulfates |
|
|
12-14-1977 |
Disazo compounds useful as complement inhibitors |
|
|
12-7-1977 |
Bis-substituted naphthalene-azo phenyleneazo-stilbene-disulfonic and naphthalene-sulfonic acid |
|
|
9-28-1977 |
UREIDOPHENYLENEBIS(CARBONYLIMINO)DINAPHTHALENETRISULFONIC ACID COMPOUNDS |
|
|
9-21-1977 |
Substituted bisnaphthylazo diphenyl ureido complement inhibitors |
|
|
9-7-1977 |
Substituted-hydroxy-naphthalenedisulfonic acid compounds |
|
1-12-1977 |
Complement inhibitors |
|
|
12-22-1976 |
Complement inhibitors |
|
|
10-13-1976 |
Complement inhibitors |
| EP0183352A2 * | Sep 27, 1985 | Jun 4, 1986 | THE UNITED STATES OF AMERICA as represented by the Secretary United States Department of Commerce | Use of suramin for clinical treatment of infection with any of the members of the family of human-t-cell leukemia (htvl) viruses including lymphadenopathy virus (lav) |
| EP0205077A2 * | Jun 3, 1986 | Dec 17, 1986 | Bayer Ag | Suramin sodium for use as an immunostimulant |
| EP0515523A1 * | Feb 13, 1991 | Dec 2, 1992 | THE UNITED STATES OF AMERICA as represented by the Secretary United States Department of Commerce | Use of suramin to treat rheumatologic diseases |
| EP0755254A1 * | Mar 24, 1995 | Jan 29, 1997 | The Trustees Of The University Of Pennsylvania | Prevention and treatment of ischemia-reperfusion and endotoxin-related injury using adenosine and purino receptor antagonists |
| EP1460087A1 * | Feb 17, 1997 | Sep 22, 2004 | The Kennedy Institute Of Rheumatology | Methods of treating vascular disease with TNF antagonists |
| EP1940376A2 * | Oct 3, 2006 | Jul 9, 2008 | Rottapharm S.P.A. | Use of neboglamine in the treatment of toxicodependency |
| EP1945204A2 * | Oct 27, 2006 | Jul 23, 2008 | Brane Discovery S.R.L. | V-atpase inhibitors for use in the treatment of septic shock |
| US5453444 * | Oct 6, 1994 | Sep 26, 1995 | Otsuka Pharmaceutical Co., Ltd. | Method to mitigate or eliminate weight loss |
| US5534539 * | Jun 12, 1995 | Jul 9, 1996 | Farmitalia Carlo Erba S.R.L. | Biologically active ureido derivatives useful as anit-metastic agenst |
| US5596105 * | Jan 13, 1995 | Jan 21, 1997 | Farmitalia Carlo Erba S.R.L. | Therapeutically active naphthalenesulfonic pyrrolecarboxamido derivatives |
| US7476693 | Mar 26, 2003 | Jan 13, 2009 | Eastern Virginia Medical School | Suramin and derivatives thereof as topical microbicide and contraceptive |
| US7608262 | Feb 16, 1996 | Oct 27, 2009 | The Kennedy Institute Of Rheumatology | Methods of preventing or treating thrombosis with tumor necrosis factor antagonists |
| US8552064 | Dec 19, 2008 | Oct 8, 2013 | Eastern Virginia Medical School | Suramin and derivatives thereof as topical microbicide and contraceptive |
| WO1994008574A1 * | Oct 12, 1993 | Apr 28, 1994 | Otsuka America Pharmaceutical | Treatment of cachexia and inhibition of il-6 activity |
| WO1994010990A1 * | Nov 12, 1993 | May 26, 1994 | British Bio Technology | Inhibition of tnf production |
| WO1997030088A2 * | Feb 17, 1997 | Aug 21, 1997 | Kennedy Inst Of Rheumatology | Methods of treating vascular disease with tnf antagonists |
| WO2004113920A1 * | Jun 18, 2004 | Dec 29, 2004 | Babon Jeff James | Screening method for substances binding to merozoite surface protein-1/42 |
| WO2008138943A2 * | May 14, 2008 | Nov 20, 2008 | Mara Galli | Prophylactic and therapeutic use of sirtuin inhibitors in tnf-alpha mediated pathologies |
| WO2009137471A2 * | May 5, 2009 | Nov 12, 2009 | University Of Miami | Azo dye related small molecule modulators of protein-protein interactions |
| WO2010016628A1 * | Jul 10, 2009 | Feb 11, 2010 | Sammy Opiyo | Conjugated suramin amino compounds for medical conditions |
| WO2012159107A1 * | May 21, 2012 | Nov 22, 2012 | Rhode Island Hospital | Inhibition of renal fibrosis |
Title: Suramin Sodium
CAS Registry Number: 129-46-4
CAS Name: 8,8¢-[Carbonylbis[imino-3,1-phenylenecarbonylimino(4-methyl-3,1-phenylene)carbonylimino]]bis-1,3,5-naphthalenetrisulfonic acid hexasodium salt
Additional Names: hexasodium sym-bis(m-aminobenzoyl-m-amino-p-methylbenzoyl-1-naphthylamino-4,6,8-trisulfonate) carbamide
Manufacturers’ Codes: Bayer 205; Fourneau 309
Trademarks: Antrypol (AstraZeneca); Germanin (Bayer); Moranyl (Specia); Naganol; Naphuride
Molecular Formula: C51H34N6Na6O23S6
Molecular Weight: 1429.17
Percent Composition: C 42.86%, H 2.40%, N 5.88%, Na 9.65%, O 25.75%, S 13.46%
Literature References: Discovered in 1917 by O. Dressel and R. Kothe: J. Dressel, J. Chem. Educ. 38, 620 (1961). Prepn: E. Fourneau et al., Compt. Rend. 178, 675 (1924); J. Trefouel, E. Fourneau, GB 224849 (1923); B. Heymann, Angew. Chem. 37, 585 (1924). Pharmacology, toxicology and clinical antiparasitic activity: F. Hawking, Adv. Pharmacol. Chemother. 15, 289-322 (1978). Inhibition of reverse transcriptase in vitro: E. De Clercq, Cancer Lett. 8, 9 (1979); vs HIV: H. Mitsuya et al., Science 226, 172 (1984). HPLC determn in plasma: R. W. Klecker, J. M. Collins, J. Liq. Chromatogr. 8, 1685 (1985). Pharmacokinetics: J. M. Collins et al., J. Clin. Pharmacol. 26, 22 (1986). Pharmacology and virustatic effect in AIDS: S. Broder et al., Lancet 2, 627 (1985); A. M. Levine et al., Ann. Intern. Med. 105, 32 (1986). Clinical trial in onchocerciasis: H. Schultz-Key et al., Trop. Med. Parasitol. 36, 244 (1985); in prostate cancer: C. Myers et al., J. Clin. Oncol. 10, 881 (1992). Review: Olenick in Antibiotics vol. 3,J. W. Corcoran, F. E. Hahn, Eds. (Springer-Verlag, New York, 1975) pp 699-703; R. La Rocca et al., Cancer Cells 2, 106-115 (1990).
Properties: White or slightly pink or cream-colored powder. Slightly bitter taste. Hygroscopic. Freely sol in water, in physiological saline; sparingly sol in 95% alcohol. Insol in benzene, ether, petr ether, chloroform. Aq solns are neutral to litmus. LD50 in mice (mg/kg): ~620 i.v. (Hawking).
Toxicity data: LD50 in mice (mg/kg): ~620 i.v. (Hawking)
Therap-Cat: Anthelmintic (Nematodes); antiprotozoal (Trypanosoma).
Therap-Cat-Vet: Antiprotozoal (Trypanosoma).
Keywords: Anthelmintic (Nematodes); Antiprotozoal (Trypanosoma); Reverse Transcriptase Inhibitor.


THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
MOBILE-+91 9323115463
web link
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
Summary of Metabolomics
Leaders in Pharmaceutical Business Intelligence Group, LLC, Doing Business As LPBI Group, Newton, MA
Summary of Metabolomics
Author and Curator: Larry H. Bernstein, MD, FCAP
This concludes the series on metabolomics, a rapidly developing science that is interconnected with a group termed – OMICS: proteomics, transcriptomics, genomics, and metabolomics. This chapter is most representative of the many important studies being done in the field, which ranges most widely because it has opened doors into nutrition and nutritional supplements, plant biochemistry, agricultural crops and breeding, animal breeding, worldwide malnutrition, diabetes, cancer, neurosciences, circulatory, respiratory, and musculosletal disorders, infectious diseases and immune system disorders. Obviously, it is not possible to cover the full range of activity, but metabolomics is most comprehensive in exploring the full range of metabolic changes that occur in health during the full age range from development to the geriatric years. It can be integrated well with gene expression, proteomics studies, and epidemiological investigations.
The subchapters are given here:
7.1 Extracellular evaluation of intracellular flux in yeast cells
View original post 1,051 more words
Flow Chemistry test facility in India

Flow Chemistry test facility in India
BOOK YOUR TRIAL
on Plantrix® Industrial system.
Contacts:

India
Vijay Kirpalani
vk@pi-inc.co
0091 9821 3420 22
More information
Stan Hoeijmakers
info@chemtrix.com
0031 (0)46 70 22 600
EVENTS
Chemtrix at CPhI India
2 – 4 December 2014
Mumbai, India
Booth H47, Hall 5
Pi Process Intensification
read at
http://hosted.verticalresponse.com/721499/f9f4fc970b/285875213/4942751fec/
|
Plantrix® Industrial Flow Chemistry Test Facility in India
|
||||||||

|
||||||||
|
|
|||||||
|
|
||||||||


Chemtrix Bv.
Urmonderbaan 22
Geleen, 6167RD
NL

Yoga back bends: feels yummy on the autonomic nervous system
Beyond Meds: Alternatives to Psychiatry
 I’m reposting this because I’ve been going through another backbend stage and I thought of this post from a while back. I like to help people see how easy yoga can be. You can start with something as simple as this and see where it takes you. Being a yogi is about listening to your body and learning from it and it really doesn’t matter if you can do really complicated poses or not. Start simple and see what happens.
I’m reposting this because I’ve been going through another backbend stage and I thought of this post from a while back. I like to help people see how easy yoga can be. You can start with something as simple as this and see where it takes you. Being a yogi is about listening to your body and learning from it and it really doesn’t matter if you can do really complicated poses or not. Start simple and see what happens.
I’ve been using yoga as a main source of rehabilitation and recovery since I was bedridden. I began doing yoga while still in bed. Now it continues to be a primary source of continued healing. Lately I’ve been doing backbends and while all the yoga I do feels like it profoundly helps my nervous system, these bends have really been making me think about my autonomic nervous system and how…
View original post 744 more words

Prefacing the e-Book Epilogue: Metabolic Genomics and Pharmaceutics
Leaders in Pharmaceutical Business Intelligence Group, LLC, Doing Business As LPBI Group, Newton, MA
Prefacing the e-Book Epilogue: Metabolic Genomics and Pharmaceutics
Author and Curator: Larry H. Bernstein, MD, FCAP
Adieu, adieu, adieu …
Sound of Music
 Snoopy – Charlie happiness
Snoopy – Charlie happiness
This work has been a coming to terms with my scientific and medical end of career balancing in a difficult time after retiring, but it has been rewarding. In the clinical laboratories, radiology, anesthesiology, and in pharmacy, there has been some significant progress in support of surgical, gynecological, developmental, medical practices, and even neuroscience directed disciplines, as well as epidemiology over a period of half a century. Even then, cancer and neurological diseases have been most difficult because the scientific basic research has either not yet uncovered a framework, or because that framework has proved to be multidimensional. In the clinical laboratory sciences, there has been enormous progress in instrumental analysis, with the recent opening of molecular methods not yet prepared for routine clinical…
View original post 3,976 more words
Cordyceps – Rare parasitic fungi could have anti-flammatory benefits
19 Nov 2012
Caterpillar fungi (Cordyceps) are rare parasites found on hibernating caterpillars in the mountains of Tibet. For centuries they have been highly prized as a traditional Chinese medicine – just a small amount can fetch hundreds of pounds.
Scientists at The University of Nottingham have been studying how this fungus could work by studying cordycepin, one of the drugs found in these mushrooms. They have already discovered that cordycepin has potential as a cancer drug. Their new work indicates that it could also have anti-inflammatory characteristics with the potential to help sufferers of asthma, rheumatoid arthritis, renal failure and stroke damage.
The research, published today in the academic journal RNA, was led by Dr Cornelia de Moor in the School of Pharmacy. It shows that cordycepin reduces inflammatory gene products in airway smooth muscle cells – the cells that contract during an asthma attack.
Several studies have suggested…
View original post 347 more words