

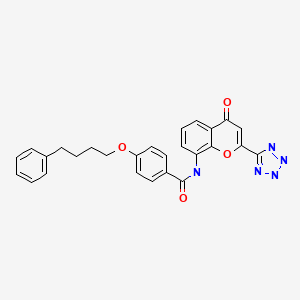
PRANLUKAST
Antiasthmatic.
-
Benzamide, N-(4-oxo-2-(1H-tetrazol-5-yl)-4H-1-benzopyran-8-yl)-4-(4-phenylbutoxy)-
-
N-(4-Oxo-2-(1H-tetrazol-5-yl)-4H-1-benzopyran-8-yl)-p-(4-phenylbutoxy)benzamide
-
4-Oxo-8-(4-(4-phenylbutoxy)benzoylamino)-2-(tetrazol-5-yl)-4H-1-benzopyran
-
N-(4-Oxo-2-(1H-tetrazol-5-yl)-4H-1-benzopyran-8-yl)-p-(4-phenylbutoxy)benzamide
hemihydrate, 103177-37-3 anhydrous, 103180-28-5 (monosodium salt)
150821-03-7, C27 H23 N5 O4 . H2O, 499.5179
Ono-1078
Ono-RS-411
RS-411
SB-205312
Ono-1070 (monosodium salt)
Ultair; Ono-1078; HY-B0290;
- Azlaire
- CCN 00401
- ONO 1078
- ONO-1078
- ONO-RS 411
- Pranlukast
- RS 411
- SB 205312
- UNII-TB8Z891092
N-[4-Oxo-2-(1H-tetrazol-5-yl)-4H-1-benzopyran-8-yl]-4-(4-phenylbutoxy)benzamide hemihydrate
Ono (Originator)Schering-Plough (Licensee)

This is described in…………
J Med Chem 1988, 31(1): 84,
WO 2010002075,
Synth Commun 1997, 27(6): 1065,
WO 1994012492
Leukotriene antagonist.
Prepn: M. Toda et al., EP 173516; eidem, US 4780469 (1986, 1988 both to Ono);
H. Nakai et al., J. Med. Chem. 31, 84 (1988).
Pharmacology: T. Obata et al., Adv. Prostaglandin Thromboxane Leukotriene Res. 15, 229 (1985); idem et al., ibid. 17,540 (1987).
Clinical evaluations in asthma: Y. Taniguchi et al., J. Allergy Clin. Immunol. 92, 507 (1993); H. Yamamoto et al. Am. J. Respir. Crit. Care Med. 150, 254 (1994).
AU 8546462; EP 0173516; JP 8650977; US 4780469; US 4939141
Pranlukast is a cysteinyl leukotriene receptor-1 antagonist. It antagonizes or reduces bronchospasm caused, principally in asthmatics, by an allergic reaction to accidentally or inadvertently encountered allergens.

Pranlukast
Pranlukast is a cysteinyl leukotriene receptor-1 antagonist. This drug works similarly to Merck & Co.‘s Singulair (montelukast). It is widely used in Japan.
Medications of this class, which go under a variety of names according to whether one looks at the American, British or European system of nomenclature, have as their primary function the antagonism of bronchospasm caused, principally in asthmatics, by an allergic reaction to accidentally or inadvertently encountered allergens.
Medications of this group are normally used as an adjunct to the standard therapy of inhaled steroids with inhaled long- and/or short-acting beta-agonists. There are several similar medications in the group; all appear to be equally effective.
Pranlukast hydrate is a leukotriene CysLT1 (LTD4) and CysLT2 (LTC4) antagonist first launched in Japan in 1995 as capsules for the oral treatment of bronchial asthma and allergic rhinitis. A dry syrup formulation of pranlukast for the treatment of asthma was approved in Japan in 1999. In April 2011, Ono filed a regulatory application in Japan seeking approval of the compound for the treatment of allergic rhinitis in pediatric patients. In December 2011, approval was obtained for this indication and launch took place immediately.
In terms of clinical development, Ono had been evaluating the drug in phase III for the treatment of sinusitis; however, in 2008 the compound was discontinued for this indication when the compound failed to demostrate the expected efficacy in the phase III studies. In March 2006, Ono discontinued development of the compound for the oral treatment of chronic obstructive pulmonary disease (COPD) based on results which suggested no evidence of efficacy. In 2000, Ono signed a license agreement with Schering-Plough to develop and market pranlukast hydrate in Latin America.
Pranlukast hemihydrate
4-Oxo-8-[(4-phenylbutoxy)benzoylamino]-2-(tetrazol-5-yl)-4H-1-benzopyran · 1/2 hydrate (common name: pranlukast, hereinafter referred to as “pranlukast” in the specification including the claims) represented by formula:
is a compound having a potential antagonistic action against leucotriene C4(LTC4) and leucotriene D4 (LTD4) and is expected as a treating agent for allergic bronchial or pulmonary diseases, allergic shock, and various allergic inflammatory diseases.

Toda synthetic complete with 3 – nitro-2 – hydroxyphenyl ko one for raw materials, ni ko with oxalic ester Claisen condensation occurs, and then heated to reflux for cyclization to construct benzo pyran ring; dehydrated by an amide synthesized ring cyano group, the cyano compound and then with sodium azide tetrazole synthesis. The nitro group on the compound in 5% Pd / C catalyzed hydrogenation of amino acid reacted with the compound Pranlukast held. This method directly using 4 – (4 – phenyl-butoxy)-benzoic acid reaction. Synthetic route is as follows:
[0006]
[0007]
[0008] ② Robert Graham and routes are routes to I-bromo-butane as a raw material, were used as a palladium catalyst, ligand compound formylation carbonylation reactions and condensation of potassium tert-butoxide, closed dehydration under acidic conditions benzopyran ring method. Synthetic route is as follows:
[0009] Robert routes:
[0010]
[0011] Graham route:
[0012]
[0013] The two synthetic routes are not disclosed in the I-Bromo butane feedstock pathway.
[0014] ③ Masayohi 2_ cyano synthetic route to a benzopyran derivative and hydrogen sulfide gas in the base-catalyzed addition reaction of 2 – thiocarbamoylbenzothiazol and pyran derivatives, and then were reacted with anhydrous hydrazine group hydrazone, with sodium nitrite under acidic conditions nitrosation reaction occurs tetrazole ring. Synthetic route is as follows:
[0015]
[0016] The materials used are not mentioned route synthesis method, it is only reflected in the improvement of the synthesis of the tetrazole ring.
[0017] ④ Giles, Hideki and Hayler are tetrazole substituent on the increase, making it easier condensation reaction, but the synthesis of substituted on the nitrogen with tetrazole difficult, and ultimately elimination reaction of lithium used tetrahydro aluminum and other hazardous reagents, is not easy to Eri industrialization. Reaction scheme is as follows:
[0018]
[0019] ⑤ Lee NK with 4_ (4_ Phenylbutoxy) benzonitrile and 2_ hydroxy _3_ iodobenzene ko 1H_4_ thiazolyl ketone and ester ko _5_ acid, concentrated sulfuric acid catalyzed cyclization iodide copper and potassium phosphate removal under the action of hydrogen iodide get Pranlukast held. Reaction scheme is as follows:
[0021] does not mention the route starting 4 – (4 – phenyl-butoxy)-benzonitrile synthesis method, while two – hydroxy – 3 – Synthesis of iodobenzene ko difficult one.

The synthesis method comprises the following steps: a. 4 – Synthesis of chlorobutanol THF was added concentrated hydrochloric acid, feeding the mass ratio of I: I. 389 ~ 5. 556,45-80 ° C was stirred for 5-18h, cooled, extracted with methylene chloride, removal of the solvent, distillation under reduced pressure to give 4 – chlorobutanol; b. 4 – phenyl butanol take benzene, aluminum chloride mixture ,0-25 ° C solution of 4 – chlorobutanol, reaction 5 -10h then poured into ice-water, a liquid, in addition to homogeneous solution U, distillation under reduced pressure, and the resulting colorless transparent liquid that is, 4 – phenyl butanol; c. I-bromo-4 – phenyl butane synthesis of 4 – phenyl butanol 40% hydrobromic acid mixture, feeding the mass ratio of I: 2. 857 ~ 11. 428, heat refluxing, cooling, liquid separation, the organic solvent divided by distillation under reduced pressure to give I-bromo-4 – phenyl butane; d. Synthesis of methyl p-hydroxybenzoate take-hydroxybenzoic acid and methanol, concentrated sulfuric acid and refluxed for 5-20h spin methanol, poured into cold water to precipitate a white solid which was filtered and dried to give the hydroxy benzoate; e. 4 – (4 – phenyl-butoxy)-benzoic acid methyl ester _ take I-bromo-4 – phenyl butane,
DMF, toluene, methyl p-hydroxybenzoate and potassium carbonate, a reflux 5 ~ 20h, cooling water, extracted with toluene, light yellow liquid rotary evaporation, recrystallization, and the resulting white solid, that is, 4 – (4 – phenyl-butoxy) – benzoic acid methyl ester; f. 4 – (4 – phenyl-butoxy yl) – benzoic acid taken 4 – (4 – phenyl-butoxy) – benzoic acid methyl ester, 15% NaOH solution was refluxed for I ~ 5h, cooled, acidified, filtered and dried to give 4 – (4 – phenylbutyrate oxy) – benzoic acid; g. sprinkle bromophenyl acetic acid ester molar ratio Preparation of I: I ~ I. 5: O. I ~ I of bromophenol, acetic anhydride, pyridine feeding, reflux 3 ~ 10h, distilled pyridine, acetic acid and excess acetic anhydride distilled under reduced pressure to give the acetic acid esters bromophenol; h. 5 – bromo-2 – Preparation of light taken acetophenone molar ratio of I: I ~ 5: I of acetic acid bromophenol esters, aluminum chloride, tetrachlorethylene for feeding, reflux O. 5 ~ 5. 5h, cooled, the reaction solution was poured into 5% hydrochloric acid and extracted with methylene chloride, the solvent evaporated under reduced pressure, to obtain a gray crystalline 5 – bromo-2 – Light acetophenone; i. 5 – bromo-3 – nitro-2 – Preparation of light acetophenone take 5 – bromo-2 – Light acetophenone, carbon tetrachloride, 50 ~ 90 ° C is added dropwise nitric acid, reflux I ~ 4h, cooled, filtered, and the resulting yellow solid which is 5 – bromo-3 – nitro-2 – hydroxyacetophenone; j. 3 – amino-2 – Light benzene ethanone Preparation of 5 – bromo-3 – nitro-2 – hydroxyacetophenone, 5% Pd / C, methylene chloride, methanol, concentrated hydrochloric acid, water, hydrogenation; the end of the reaction mixture was filtered, the filtrate was The solvent was removed, neutralized with sodium bicarbonate, and the resulting yellow solid ginger i.e., 3 – amino-2 – hydroxyacetophenone; k. 3 – [4 – (4 – phenyl-butoxy)-benzoyl amino] -2 _ light base Preparation of acetophenone 4 – (4 – phenyl-butoxy)-benzoic acid, toluene, DMF, 45 ~ 105 ° C was added dropwise SOCl2, 30min the reaction liquid droplets to the 3 – amino-2 – hydroxyphenyl toluene solution of ethyl ketone, the reaction 3 ~ 10h, cooled, neutralized with dilute hydrochloric acid, extracted with toluene, rotary evaporation, and the resulting pale yellow crystals is 3 – [4 – (4_ phenylbutoxy) benzamido] 2_-hydroxyacetophenone; I. 2 – [4 – (4 – phenyl-butoxy)-benzoyl amino] -6 – [l, 3 – dioxo-3 – ethoxycarbonyl-propyl] phenol synthetic sodium, THF, 3 – [4 – (4 – phenyl-butoxy)-benzoyl amino]-2 – hydroxyacetophenone, diethyl oxalate 4 ~ IOh After stirring the reaction was poured into dilute hydrochloric acid to precipitate the yellow solid which was filtered, and the resulting product, i.e. 2 – [4 – (4_ phenylbutoxy) benzamido] _6_ [1,3 – dioxo-3 – ethoxy propyl intended yl] phenyl discretion ·; m. 4 – oxo-8 – [4 – (4 – phenyl-butoxy)-benzoyl amino]-2 – ethoxycarbonyl-4H-benzopyran take 2 – [4 – (4 – phenyl-butoxy yl) benzoyl amino] -6 – [l, 3 – dioxo-3 – ethoxycarbonyl-propyl] phenol, THF, force mouth heat, the addition of concentrated hydrochloric acid, refluxed for 8 ~ 15h, cooled, filtered, and the resulting white solid,
that is, 4 – oxo-8 – [4 – (4 – phenyl-butoxy)-benzoyl amino]-2 – ethoxycarbonyl-4H-benzopyran; η. 4 – oxo-8 – [ 4 – (4 – phenyl-butoxy)-benzoyl amino] -2 – amino-carbonyl-4Η-benzopyran synthesis take four – oxo-8 – [4 – (4 – phenyl-butoxy)-benzoyl amino] -2 – ethoxycarbonyl-4Η-benzopyran was dissolved in DMF,
and leads to dry ammonia gas, the reaction solution changed from yellow to red, the reaction solution was poured into cold water, adjusted to acidic, and filtered to give the product 4 – oxo-8 – [4 – (4 – phenyl-butoxy)-benzoyl amino] -2 – amino-carbonyl-4Η-benzopyran; P. 4 – oxo-8 – [4 – (4 – phenylbutoxy) benzamido] -2 – cyano-4Η-benzopyran take DMF, S0C12, 4 – oxo-8 – [4 – (4 – phenyl-butoxy)-benzoic amido] _2_ aminocarbonyl-4H-benzopyran, O ~ 15 ° C under stirring for 2 ~ IOh poured into cold water, filtered, and the resulting white solid that is, 4 – oxo-8 – [4 – (4 – phenylbutoxy) benzamido] -2 – cyano-4H-benzopyran; q. Synthesis of pranlukast take four – oxo-8 – [4 – (4 – phenyl-butoxy) benzoyl amino]-2_ cyano-4H-benzopyran, ammonium chloride, sodium azide, DMF, heating I ~ 8h then poured into ice-water, dilute hydrochloric acid, filtered, and the resulting white solid that the final product Pranlukast.

The reaction of ethyl 8-nitro-4-oxo-1-benzopyran-2-carboxylate (I) with ammonia in methanol gives the corresponding amide (II), which is dehydrated with POCl3 yielding 2-cyano-8-nitro-1-benzopyran-4-one (III). The cyclization of (III) with sodium azide by means of pyridinium chloride in hot DMF affords 8-nitro-2-(tetrazol-5-yl)-1-benzopyran-4-one (IV), which is hydrogenated with H2 over Pd/C in methanol – HCl giving 8-amino-2-(tetrazol-5-yl)-1-benzopyran-4-one (V). Finally, this compound is condensed with 4-(4-phenylbutoxy)benzoic acid (VI) by means of oxalyl chloride in dichloromethane-pyridine\

………………………..
Synthetic routes
The reaction of ethyl 8-nitro-4-oxo-1-benzopyran-2-carboxylate (I) with ammonia in methanol gives the corresponding amide (II), which is dehydrated with POCl3 yielding 2-cyano-8-nitro-1- benzopyran-4-one (III). The cyclization of (III) with sodium azide by means of pyridinium chloride in hot DMF affords 8-nitro-2- (tetrazol-5-yl) -1-benzopyran-4-one (IV ), which is hydrogenated with H2 over Pd / C in methanol -. HCl giving 8-amino-2- (tetrazol-5-yl) -1-benzopyran-4-one (V) Finally, this compound is condensed with 4- (4-phenylbutoxy) benzoic acid (VI) by means of oxalyl chloride in dichloromethane-pyridine.
……………………………………
PATENT
Example 1: Synthesis of pranlukast
To 10 g of N-(4-oxo-2-(l-trityl-lH-tetrazol-5-yl)-4H-chromen-8-yl)-4-(4- phenylbutoxy) benzamide (Pharmacostech) was added 100 ml of methanol, and 10 g of a resin pre-treated with hydrochloric acid of pH 2-3 (TRILITE SCR-B gel type,
Mitsubishi Chemical Co.) was added to the reaction mixture, followed by refluxing for
5 hours. The solid components were filtered out from the reaction mixture and washed with 100 ml of methanol. The filter-in solution was subject to vacuum distillation to obtain a solid substance and the solid was dissolved in 50 ml of dimethyl acetamide (DMAC). Afterwards, 200 ml of aqueous solution was added to the DMAC solution and stirred for 1 hour at room temperature. Then, the solid formed was filtered out, dried, and left for 5 hours at room temperature to give 6.32 g (yield:
95%) of the standard compound represented by the following Formula 5: melting point, 231-2330C (decomposed); 1H-NMR (DMSOd6, 300 MHz), δ 1.9 (m, 4H), 2,7 (m,2H), 4.0 (t, 2H), 7.0 (s, 2H), 7.1 (s, IH), 7.2-7.3 (m, 5H), 7.6 (t, IH), 7.9 (t, IH), 8.0
(m, 2H), 8.3 (t, IH), 10.0 (bs, IH).

Example 2: Synthesis of pranlukastOne hundred ml of methanol was added to 10 g of N-(4-oxo-2-(l-trityl-lH- tetrazol-5-yl)-4H-chromen-8-yl)-4-(4-phenylbutoxy) benzamide (Pharmacostech), then 10 g of resin pre-treated with hydrochloric acid of pH 2-3 (TRILITE SCR-10 gel type, Mitsubishi Chemical Co.) was added to the reaction mixture, followed by refluxing for 6 hours. The solid components were filtered out from the reaction mixture and washed with 100 ml of methanol. The filter-in solution was subject to vacuum distillation to obtain a solid substance and the solid was dissolved in 50 ml of dimethyl acetamide (DMAC). Afterwards, 200 ml of aqueous solution was added to the DMAC solution and stirred for 1 hour at room temperature. Then the solid formed was filtered out, dried, and left for 5 hours at room temperature to obtain 6.18 g (yield rate: 93%) of the standard compound represent by Formula 5: melting point, 231- 233°C (decomposed); 1H-NMR (DMSOd6, 300 MHz), δ 1.9 (m, 4H), 2,7 (m, 2H), 4.0 (t, 2H), 7.0 (s, 2H), 7.1 (s, IH), 7.2-7.3 (m, 5H), 7.6 (t, IH), 7.9 (t, IH), 8.0 (m, 2H), 8.3 (t, IH), 10.0 (bs, IH).
Example 3: Synthesis of pranlukast
One hundred ml of methanol and 100 ml of methylene chloride (MC) were added to 10 g of N-(4-oxo-2-(l-trityl-lH-tetrazol-5-yl)-4H-chromen-8-yl)-4-(4- phenylbutoxy) benzamide (Pharmacostech), then 10 g of resin pre-treated with hydrochloric acid of pH 2-3 (TRILITE SCR-10 gel type) was added to the reaction mixture, followed by refluxing for 12 hours. The solid components were filtered out from the reaction mixture and washed with 100 ml of methanol. The filter-in solution was subject to vacuum distillation to obtain a solid substance and the solid was dissolved in 50 ml of dimethyl acetamide (DMAC). Afterwards, 200 ml of aqueous solution was added to the DMAC solution, and stirred for 1 hour at room temperature. Then the solid formed was filtered out, dried, and left for 5 hours at room temperature to obtain 6.18 g (yield rate: 93%) of the standard compound represent by Formula 5: melting point, 231-233°C (decomposed); 1H-NMR (DMSO-d6, 300 MHz), δ 1.9 (m, 4H), 2,7 (m, 2H), 4.0 (t, 2H), 7.0 (s, 2H), 7.1 (s, IH), 7.2-7.3 (m, 5H), 7.6 (t, IH), 7.9 (t, IH), 8.0 (m, 2H), 8.3 (t, IH), 10.0 (bs, IH).
……………………………………………………….
Pranlukast and its hydrates come into the market as a capsule of Onon® Cap. (112.5 mg pranlukast hydrates/capsule, Dong-A Pharmaceutical).
The conventional method for preparing pranlukast was disclosed in US Pat. No. 5,587,483 and pranlukart is prepared by the following reaction formula I.
Reaction Formula I
As described in the reaction formula I, the acid chloride represented by formula 11 is obtained by reacting the benzoic derivative of formula 10 with the thionyl chloride. The resulting compound is reacted with the compound represented by formula 4. The compound (n = 4) represented by formula 5 is reacted with the tetrazol derivative represented by formula 6 to introduce tetrazol group and then benzopyran ring is formed, preparing pranlukast. However, the preparation method according to the reaction formula I has quite a few problems: (a) difficult manipulation due to utilizing excess amounts of toxic thionyl chlorides around a reflux temperature when the acid chloride represented by formula 11 is obtained by reacting the benzoic derivative of formula 10 with the thionyl chloride;
(b) hard elimination of thionyl chlorides toxic in a body after terminating the reactions; (c) requirement of base in an equivalent ratio of above 4 to collect the compound represented by formula 7; (d) unsuitability of massive production in a economical area because the compound is modified into a form of natrium salt and then purified for removal of contaminants after preparing pranlukart.
On the other hand, as described in the following reaction formula II in US Pat. No. 5,874,593, nitril compounds of formula 8 are reacted with hydrazine to prepare amidrazone compounds of formula 9a and 9b, and then pranlukart is fabricated by performing a tetrazol ring reaction using nitrous acids.
Reaction Formula II
However, the preparation method according to the reaction formula II has also the following difficulties: (a) it is difficult to perform the method due to utilizing excess amounts of toxic thionyl chlorides around a reflux temperature to obtain the acid chloride derivative in the preparation of the compounds represented by formula 8; (b) it is very difficult and toxic in body to eliminate thionyl chlorides after terminating the reactions; (c) it is not easy to massively produce the compounds of interest in an industrial-scale because much hydrazine toxic in body and nitrogen oxides harmful in environment are generated and unstable nitrous acids are used during the reactions.
Likewise, US Pat. No. 5,874,593, as described in the following reaction formula III, discloses that benzoic derivatives of formula 10′ are reacted with oxalyl chlorides to isolate acid chlorides represented by formula 11′, and the resulting acid chlorides are reacted with benzopyran amine derivatives containing tetrazol of formula 12, producing various derivatives containing pranlukart.
Reaction Formula III
( I D’ ] (H ‘ )
Oxalyl chlorides are massively used because the preparation method according to the reaction formula III is very expensive cost and has highly hygroscopic characteristics. In addition, the method has to be carried out under violent conditions that the temperature is increased up to around reflux temperature using 1,2- dichloroethanol as a solvent and further reacted for 1 hr. It is also difficult to remove harmful carbon monoxide and chlorine gases massively generated in elimination of oxalyl chloride after terminating the reactions, and it is not feasible to be applied into an industrial mass-production because the reaction is carried out under conditions of anhydrous and inactive gases
EXAMPLE 1: Preparation of Pranlukart Hemihydrates 4-(4-phenylbutoxy)benzoic acid (29.1 g; 1.1 equivalent ratio; prepared according to the method disclosed in US Pat. No. 4,780,469) was dissolved in 80 ml dimethylacetamide (DMAC, Aldrich) at 00C and then thionyl chloride (14.2 g, 1.2 equivalent ratio, Aldrich) was gradually added to the solution. After the mixture solution was stirred for 10 min at 00C, the mixture of 8-amino-4-oxo-tetrazol-5-yl-4H- 1-benzopyran hydrochloride salt (26.7 g; 1 equivalent ratio; prepared according to the method disclosed in US Pat. No. 4,780,469) and triethylamine (TEA, 10.1 g, 1 equivalent ratio, Aldrich) dissolved in 80 ml dimethylacetamide (DMAC, Aldrich) was slowly added to the mixture solution, and thermally stirred for 5 hrs at 25°C.
The reaction mixture was mixed with 300 ml H2O and stirred for 1 hr at 250C. The solid material obtained by filtering the solid material produced was washed with 100 ml H2O. 200 ml 50% acetone aqueous solution was added to the solid material and then refluxed for 1 hr. After the reaction mixture was cooled to room temperature, filtered and air-dried, the mixture was kept to stand on air for 5 hrs, obtaining 47.0 g pranlukart hemihydrates (yield rate: 98%): melting point 231-233°C (decomposition); 1H-NMR (DMSO-d6, 300 MHz) δ 1.9 (m, 4H), 2,7 (m, 2H), 4.0 (t, 2H), 7.0 (s, 2H), 7.1 (s, IH), 7.2-7.3 (m, 5H), 7.6 (t, IH), 7.9 (t, IH), 8.0 (m, 2H), 8.3 (t, IH), 10.0 (bs, IH).
EXAMPLE 2: Preparation of Pranlukart Hemihydrates – Substitution of the Chlorinating Agent
4-(4-phenylbutoxy)benzoic acid (29.1 g, 1.1 equivalent ratio) was dissolved in 80 ml dimethylacetamide (DMAC) at 00C and then oxalyl chloride (15.2 g, 1.2 equivalent ratio, Aldrich) was gradually added to the solution. After the mixture solution was stirred for 10 min at 00C, the mixture of 8-amino-4-oxo-tetrazol-5-yl-4H- 1-benzopyran hydrochloride salt (26.7 g; 1 equivalent ratio) and triethylamine (TEA, 10.1 g, 1 equivalent ratio) dissolved in 80 ml dimethylacetamide (DMAC) solution was slowly added to the mixture solution, and thermally stirred for 5 hrs at 25°C.
The reaction mixture was mixed with 300 ml H2O and stirred for 1 hr at 25°C. The solid material obtained by filtering the solid material produced was washed with 100 ml H2O. 200 ml 50% acetone aqueous solution was added to the solid material and then refluxed for 1 hr. After the reaction mixture was cooled to room temperature, filtered and air-dried, the mixture was kept to stand on air for 5 hrs, obtaining 43.3 g pranlukart hemihydrates (yield rate: 92%).
EXAMPLE 3: Preparation of Pranlukart Hemihydrates – Change of Base Condition
4-(4-phenylbutoxy)benzoic acid (29.1 g, 1.1 equivalent ratio) was dissolved in 80 ml dimethylacetamide (DMAC) at 00C and then thionyl chloride (14.2 g, 1.2 equivalent ratio) was gradually added to the solution. After the mixture solution was stirred for 10 min at 00C, the mixture of 8-amino-4-oxo-tetrazol-5-yl-4H-l-benzopyran hydrochloride salt (26.7 g; 1 equivalent ratio) and pyridine (7.9 g, 1 equivalent ratio, Aldrich) dissolved in 80 ml dimethylacetamide (DMAC) solution was slowly added to the mixture solution, and thermally stirred for 5 hrs at 25°C.
The reaction mixture was mixed with 300 ml H2O and stirred for 1 hr at 25°C. The solid material obtained by filtering the solid material produced was washed with 100 ml H2O. 200 ml 50% acetone aqueous solution was added to the solid material and then refluxed for 1 hr. After the reaction mixture was cooled to room temperature, filtered and air-dried, the mixture was kept to stand on air for 5 hrs, obtaining 45.6 g pranlukart hemihydrates (yield rate: 95%).
EXAMPLE 4: Preparation of Pranlukart Hemihydrates – Change of Reaction Temperature Condition
4-(4-phenylbutoxy)benzoic acid (29.1 g, 1.1 equivalent ratio) was dissolved in 80 ml dimethylacetamide (DMAC) at 00C and then thionyl chloride (14.2 g, 1.2 equivalent ratio) was gradually added to the solution. After the mixture solution was stirred for 10 min at O0C, the mixture of 8-amino-4-oxo-tetrazol-5-yl-4H-l-benzopyran hydrochloride salt (26.7 g; 1 equivalent ratio) and triethylamine (TEZ, 10.1 g, 1 equivalent ratio) dissolved in 80 ml dimethylacetamide (DMAC) solution was slowly added to the mixture solution, and thermally stirred for 4 hrs at 500C. The reaction mixture was mixed with 300 ml H2O and stirred for 1 hr at 500C. The solid material obtained by filtering the solid material produced was washed with 100 ml H2O. 200 ml 50% acetone aqueous solution was added to the solid material and then refluxed for 1 hr. After the reaction mixture was cooled to room temperature, filtered and air-dried, the mixture was kept to stand on air for 5 hrs, obtaining 45.6 g pranlukart hemihydrates (yield rate: 95%).
EXAMPLE 5: Preparation of Pranlukart Hemihydrates – Substitution of Reaction Solvent 4-(4-phenylbutoxy)benzoic acid (29.1 g, 1.1 equivalent ratio) was dissolved in 80 ml N-methylpyrrolidine (NMP, Aldrich) at O0C and then thionyl chloride (14.2 g, 1.2 equivalent ratio) was gradually added to the solution. After the mixture solution was stirred for 10 min at 00C, the mixture of 8-amino-4-oxo-tetrazol-5-yl-4H-l-benzopyran hydrochloride salt (26.7 g; 1 equivalent ratio) and triethylamine (TEZ, 10.1 g, 1 equivalent ratio) dissolved in 80 ml N-methylpyrrolidine (NMP) solution was slowly added to the mixture solution, and thermally stirred for 4 hrs at 250C. The reaction mixture was mixed with 300 ml H2O and stirred for 1 hr at 25°C.
The solid material obtained by filtering the solid material produced was washed with 100 ml H2O. 200 ml 50% acetone aqueous solution was added to the solid material and then refluxed for 1 hr. After the reaction mixture was cooled to room temperature, filtered and air-dried, the mixture was kept to stand on air for 5 hrs, obtaining 43.3 g pranlukart hemihydrates (yield rate: 90%).
EXAMPLE 6: Preparation of Pranlukart Hemihydrates – Equivalent Ratio Change of the Chlorinating Agent
4-(4-phenylbutoxy)benzoic acid (29.1 g, 1.1 equivalent ratio) was dissolved in 80 ml dimethylacetamide (DMAC) at 00C and then thionyl chloride (14.2 g, 1 equivalent ratio) was gradually added to the solution. After the mixture solution was stirred for 10 min at 00C, the mixture of 8-amino-4-oxo-tetrazol-5-yl-4H-l-benzopyran hydrochloride salt (26.7 g, 1 equivalent ratio) and triethylamine (TEZ, 10.1 g, 1 equivalent ratio) dissolved in 80 ml dimethylacetamide (DMAC) solution was slowly added to the mixture solution, and thermally stirred for 4 hrs at 25°C.
The reaction mixture was mixed with 300 ml H2O and stirred for 1 hr at 25°C. The solid material obtained by filtering the solid material produced was washed with 100 ml H2O. 200 ml 50% acetone aqueous solution was added to the solid material and then refluxed for 1 hr. After the reaction mixture was cooled to room temperature, filtered and air-dried, the mixture was kept to stand on air for 5 hrs, obtaining 44.6 g pranlukart hemihydrates (yield rate: 93%).
……………………..
J. Med. Chem., 1988, 31 (1), pp 84–91
DOI: 10.1021/jm00396a013
…………………………………………………..
Geen, G.R.; Giles, R.G.; Grinter, T.J.; Hayler, J.D.; et al.
A direct and high yielding route to 2-(5-tetrazolyl) substituted benzopyran-4-ones: Synthesis of pranlukast
Synth Commun 1997, 27(6): 1065
A direct and high yielding route to 2-(5-tetrazolyl)benzopyran-4-ones 1, including pranlukast 1a is described. This involves the Claisen condensation reaction between the relevant hydroxyacetophenone 2 and the ethyl ester of tetrazole-2-carboxylic acid 5 to give the 1,3-diketone 6, which is then cyclised to give the desired benzopyran-4-ones 1.\
………………………………………………..
WO 1994012492

References

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

































