
Home » Uncategorized (Page 101)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
SURAMIN

Suramin
A polyanionic compound with an unknown mechanism of action. It is used parenterally in the treatment of African trypanosomiasis and it has been used clinically with diethylcarbamazine to kill the adult Onchocerca. (From AMA Drug Evaluations Annual, 1992, p1643) It has also been shown to have potent antineoplastic properties.
A polyanionic compound with an unknown mechanism of action. It is used parenterally in the treatment of African trypanosomiasis and it has been used clinically with diethylcarbamazine to kill the adult Onchocerca. (From AMA Drug Evaluations Annual, 1992, p1643) It has also been shown to have potent antineoplastic properties. Suramin is manufactured by Bayer in Germany as Germanin®.
Also known as: Naphuride, Germanin, Naganol, Belganyl, Fourneau, Farma, Antrypol, Suramine, Naganin
8,8′-{Carbonylbis[imino-3,1-phenylenecarbonylimino(4-methyl-3,1-phenylene)carbonylimino]}di(1,3,5-naphthalenetrisulfonic acid) …FREE FORM
8,8′-[Ureylenebis[m-phenylenecarbonylimino(4-methyl-m-phenylene)carbonylimino]]di(1,3,5-naphthalenetrisulfonic acid) hexasodium salt
CAS 145-63-1 FREE FORM
129-46-4 of hexa sodium
LAUNCHED 1940 BAYER
| Formula | C51H40N6O23S6 |
|---|---|
| Mol. mass | 1297.29 |
The molecular formula of suramin is C51H34N6O23S6. It is a symmetric molecule in the center of which lies urea, NH-CO-NH. Suramin contains eightbenzene rings, four of which are fused in pairs (naphthalene), four amide groups in addition to the one of urea and six sulfonate groups. When given as drug it usually contains six sodium ions that form a salt with the six sulfonate groups.
Suramin is a drug developed by Oskar Dressel and Richard Kothe of Bayer, Germany in 1916, and is still sold by Bayer under the brand nameGermanin.
Suramin sodium is a heparanase inhibitor that was first launched in 1940 by Bayer under the brand name Antrypol for the treatment of helminthic infection. It was later launched by Bayer for the treatment of trypanosomiasis (African sleeping sickness).
More recently, the product has entered early clinical development at Ohio State University for the treatment of platinum-pretreated patients with stage IIIB/IV non-small cell lung cancer, in combination with docetaxel or gemcitabine.
The National Cancer Institute (NCI) is conducting phase II clinical studies for the treatment of glioblastoma multiforme and for the treatment of adrenocortical carcinoma.
According to the National Cancer Institute there are no active clinical trials (as of April 1, 2008). Completed and closed clinical trials are listed here:[1]
In addition to Germanin, the National Cancer Institute also lists the following “Foreign brand names”: 309 F or 309 Fourneau,[1] Bayer 205, Moranyl, Naganin, Naganine.

It is used for treatment of human sleeping sickness caused by trypanosomes.[2]
It has been used in the treatment of onchocerciasis.[3]
It has been investigated as treatment for prostate cancer.[4]
Also, suramin as treatment for autism is being evaluated. [5]
Suramin is administered by a single weekly intravenous injection for six weeks. The dose per injection is 1 g.
The most frequent adverse reactions are nausea and vomiting. About 90% of patients will get an urticarial rash that disappears in a few days without needing to stop treatment. There is a greater than 50% chance of adrenal cortical damage, but only a smaller proportion will require lifelongcorticosteroid replacement. It is common for patients to get a tingling or crawling sensation of the skin with suramin. Suramin will cause clouding of the urine which is harmless: patients should be warned of this to avoid them becoming alarmed.
Kidney damage and exfoliative dermatitis occur less commonly.
Suramin has been applied clinically to HIV/AIDS patients resulting in a significant number of fatal occurrences and as a result the application of this molecule was abandoned for this condition. http://www.ncbi.nlm.nih.gov/pubmed/3548350
Suramin is also used in research as a broad-spectrum antagonist of P2 receptors[6][7] and agonist of Ryanodine receptors.[8]
![ChemSpider 2D Image | 8,8'-{Carbonylbis[imino-3,1-phenylenecarbonylimino(4-methyl-3,1-phenylene)carbonylimino]}di(1,3,5-naphthalenetrisulfonic acid) | C51H40N6O23S6](https://images-blogger-opensocial.googleusercontent.com/gadgets/proxy?url=http%3A%2F%2Fwww.chemspider.com%2FImagesHandler.ashx%3Fid%3D5168%26w%3D200%26h%3D200&container=blogger&gadget=a&rewriteMime=image%2F*)
Its effect on telomerase has been investigated.[9]
It may have some activity against RNA viruses.[10]
In addition to antagonism of P2 receptors, Suramin inhibits the acitivation of heterotrimeric G proteins in a variety of other GPCRs with varying potency. It prevents the association of heteromeric G proteins and therefore the receptors Guanine exchange functionality (GEF). With this blockade the GDP will not release from the Gα subunit so it can not be replaced by a GTP and become activated. This has the effect of blocking downstream G protein mediated signaling of various GPCR proteins including Rhodopsin, the A1 Adenosine receptor, and the D2 dopamine receptor.[11]
A polyanionic compound with an unknown mechanism of action. It is used parenterally in the treatment of African trypanosomiasis and it has been used clinically with diethylcarbamazine to kill the adult Onchocerca. (From AMA Drug Evaluations Annual, 1992, p1643) It has also been shown to have potent antineoplastic properties. Suramin is manufactured by Bayer in Germany as Germanin®.
|
8-1-2012 |
InCl3-catalysed synthesis of 2-aryl quinazolin-4(3H)-ones and 5-aryl pyrazolo[4,3-d]pyrimidin-7(6H)-ones and their evaluation as potential anticancer agents. |
Bioorganic & medicinal chemistry letters |
|
9-1-2012 |
Identification of a sirtuin 3 inhibitor that displays selectivity over sirtuin 1 and 2. |
European journal of medicinal chemistry |
|
1-1-2013 |
Inhibition of the human deacylase Sirtuin 5 by the indole GW5074. |
Bioorganic & medicinal chemistry letters |
|
5-9-2013 |
Discovery of thieno[3,2-d]pyrimidine-6-carboxamides as potent inhibitors of SIRT1, SIRT2, and SIRT3. |
Journal of medicinal chemistry |
- The formula of suramin was kept secret by Bayer for commercial reasons. But it was elucidated and published in 1924 by Fourneau and his team of the Pasteur Institute, and it is only on this date that its exact chemical composition was known. (E. Fourneau, J. and Th. Tréfouël and J. Vallée (1924). “Sur une nouvelle série de médicaments trypanocides”, C. R. Séances Acad. Sci. 178: 675.)
- Darsaud A, Chevrier C, Bourdon L, Dumas M, Buguet A, Bouteille B (January 2004). “Megazol combined with suramin improves a new diagnosis index of the early meningo-encephalitic phase of experimental African trypanosomiasis”. Trop. Med. Int. Health 9 (1): 83–91.doi:10.1046/j.1365-3156.2003.01154.x. PMID 14728611.
- Anderson J, Fuglsang H (July 1978). “Further studies on the treatment of ocular onchocerciasis with diethylcarbamazine and suramin”. Br J Ophthalmol 62 (7): 450–7.doi:10.1136/bjo.62.7.450. PMC 1043255. PMID 678497.
- Ahles TA, Herndon JE, Small EJ, et al. (November 2004). “Quality of life impact of three different doses of suramin in patients with metastatic hormone-refractory prostate carcinoma: results of Intergroup O159/Cancer and Leukemia Group B 9480”. Cancer 101 (10): 2202–8.doi:10.1002/cncr.20655. PMID 15484217.
- http://medicalxpress.com/news/2013-03-drug-treatment-autism-symptoms-mouse.html
- Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, Weisman GA. (september 2006). “International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy”. Pharmacol Rev. 58 (3): 281–341.doi:10.1124/pr.58.3.3. PMID 16968944.
- Khakh BS, Burnstock G, Kennedy C, King BF, North RA, Séguéla P, Voigt M, Humphrey PP. (march 2001). “International union of pharmacology. XXIV. Current status of the nomenclature and properties of P2X receptors and their subunits”. Pharmacol Rev. 53 (1): 107–118.PMID 11171941.
- Wolner I, Kassack MU, Ullmann H, Karel A, Hohenegger M (October 2005). “Use-dependent inhibition of the skeletal muscle ryanodine receptor by the suramin analogue NF676”. Br. J. Pharmacol. 146 (4): 525–33. doi:10.1038/sj.bjp.0706359. PMC 1751178.PMID 16056233.
- Erguven M, Akev N, Ozdemir A, Karabulut E, Bilir A (August 2008). “The inhibitory effect of suramin on telomerase activity and spheroid growth of C6 glioma cells”. Med. Sci. Monit. 14(8): BR165–73. PMID 18667993.
- Mastrangelo E, Pezzullo M, Tarantino D, Petazzi R, Germani F, Kramer D, Robel I, Rohayem J, Bolognesi M, Milani M (2012) Structure-based inhibition of norovirus RNA-dependent RNA-polymerases. J Mol Biol
- Beindl W, Mitterauer T, Hohenegger M, Ijzerman AP, Nanoff C, Freissmuth M. (August 1996).“Inhibition of receptor/G protein coupling by suramin analogues”. ol. Pharmacology. 50 (2): 415–23. PMID 8700151.
- Drugs Fut 1986, 11(10): 860
- WO 2012159107
- WO 2012087336
- US 2011257109
- WO 2009022897
- WO 2009020613
- WO 2008094027
- EP 0486809
- US 5158940
- US 5173509
- WO 1993007864
- WO 1994008574

SURAMIN
- Suramin bound to proteins in the PDB
- Drug information
- Suramin, drug information by JBC Online
- Suramin in treating patients with recurrent bladder cancer
- National Cancer Institute
Enterovirus-71 (EV71) is one of the major causative reagents for hand-foot-and-mouth disease. In particular, EV71 causes severe central nervous system infections and leads to numerous dead cases. Although several inactivated whole-virus vaccines have entered in clinical trials, no antiviral agent has been provided for clinical therapy. In the present work, we screened our compound library and identified that suramin, which has been clinically used to treat variable diseases, could inhibit EV71 proliferation with an IC50 value of 40μM. We further revealed that suramin could block the attachment of EV71 to host cells to regulate the early stage of EV71 infection, as well as affected other steps of EV71 life cycle. Our results are helpful to understand the mechanism for EV71 life cycle and provide a potential for the usage of an approved drug, suramin, as the antiviral against EV71 infection.

- Suramin Hexasodium
- 129-46-4
Synonyms
- 309 F
- Antrypol
- BAY 205
- Bayer 205
- CI-1003
- EINECS 204-949-3
- Fourneau 309
- Germanin
- Moranyl
- Naganin
- Naganine
- Naganinum
- Naganol
- Naphuride sodium
- NF060
- NSC 34936
- SK 24728
- Sodium suramin
- Suramin Hexasodium
- Suramin sodium
- Suramina sodica
- Suramina sodica [INN-Spanish]
- Suramine sodique
- Suramine sodique [INN-French]
- Suramine sodium
- Suraminum natricum
- Suraminum natricum [INN-Latin]
- UNII-89521262IH
Suramin Sodium, is an anticancer agent with a wide variety of activities.
Recently suramin was shown to inhibit FSH binding to its receptor (Daugherty, R. L.; Cockett, A. T. K.; Schoen, S. R. and Sluss, P. M. “Suramin inhibits gonadotropon action in rat testis: implications for treatment of advanced prostate cancer” J. Urol. 1992, 147, 727-732).
This activity causes, at least in part, the decrease in testosterone production seen in rats and humans that were administered suramin(Danesi, R.; La Rocca, R. V.; Cooper, M. R.; Ricciardi, M. P.; Pellegrini, A.; Soldani, P.; Kragel, P. J.; Paparelli, A.; Del Tacca, M.; Myers, C. E, “Clinical and experimental evidence of inhibition of testosterone production by suramin.” J. Clin. Endocrinol. Metab. 1996, 81, 2238-2246).
Suramin is the only non-peptidic small molecule that has been reported to be an FSH receptor binding antagonist.

Suramin is 8,8′ – (carbonylbis(imino-3,1-phenylenecarbonylimino (4-methyl-3,1-phenylene) carbonylimino)) bis-1,3 ,5-naphthalenetrisulfonic acid (GB Patent No. 224849). This polyanionic compound has been used for many decades as a prophylactic and therapeutic agent for try- panosomiasis. It was subsequently shown that suramin is able to block the activity of a variety of proteins like cellular and viral enzymes and growth factors (Mitsuya, M. et al. Science 226 : 172 (1984), Hosang, M. J. Cell. Biochem. 29 : 265 (1985), De Clercq, E. Cancer Lett. 8 : 9 (1979)).

|
5-32-1977 |
Complement inhibitors |
|
|
5-25-1977 |
Aromatic amidines as antiviral agents in animals |
|
|
5-4-1977 |
Complement inhibitors |
|
|
5-4-1977 |
Complement inhibitors |
|
|
4-27-1977 |
Cyclodextrin sulfate salts as complement inhibitors |
|
|
4-20-1977 |
Ureylenebis methyl-phenylene-carbonyl-bis-dihydro-2-oxo-naphthoxazine disultonic acids |
|
|
3-30-1977 |
Water treatment for controlling the growth of algae employing biguanides |
|
|
3-2-1977 |
Isoxazole substituted nitroimidazoles |
|
|
2-16-1977 |
Amidophenyl-azo-naphthalenesulfonic complement inhibitors and method of use thereof |
|
|
2-9-1977 |
Complement inhibitors |
|
2-10-2011 |
MODULATION OF HUMAN MAST CELL ACTIVATION MODULATION OF HUMAN MAST CELL ACTIVATION |
|
|
11-18-2010 |
Admixtures for inorganic binders based on a hydrogenated disaccharide, inorganic binders containing these admixtures and process for their preparation Admixtures for inorganic binders based on a hydrogenated disaccharide, inorganic binders containing these admixtures and process for their preparation |
|
|
10-28-2010 |
THERAPEUTIC INHIBITORS OF VASCULAR SMOOTH MUSCLE CELLS |
|
|
9-9-2010 |
APPARATUS FOR USING ELECTROPORATION MEDIATED DELIVERY OF DRUGS AND GENES |
|
|
4-8-2010 |
PREPARATION AND USE OF SULFATED OLIGOSACCHARIDES |
|
|
10-29-2009 |
THERAPEUTIC INHIBITOR OF VASCULAR SMOOTH MUSCLE CELLS THERAPEUTIC INHIBITOR OF VASCULAR SMOOTH MUSCLE CELLS |
|
|
8-20-2009 |
METHOD OF MAKING MINERAL FIBRES METHOD OF MAKING MINERAL FIBRES |
|
|
6-25-2009 |
OXYGEN-FUEL BOOST REFORMER PROCESS AND APPARATUS |
|
|
4-23-2009 |
METHODS OF TREATING VASCULAR DISEASE WITH TNF ANTAGONISTS METHODS OF TREATING VASCULAR DISEASE WITH TNF ANTAGONISTS |
|
|
3-26-2009 |
COPOLYMER COMPOSITIONS FOR ORAL DELIVERY |
|
5-3-1978 |
1,3,5- Or 1,3,6-naphthalenetriyltris(sulfonylimino)aryl acids and salts |
|
|
3-22-1978 |
Nitroimidazoles |
|
|
2-15-1978 |
Treatment of rheumatoid arthritis and related diseases |
|
|
1-4-1978 |
AROMATIC AMIDINES AS ANTIVIRAL AGENTS IN ANIMALS |
|
|
1-4-1978 |
Malto-dextrin poly(H-)sulfates |
|
|
12-14-1977 |
Disazo compounds useful as complement inhibitors |
|
|
12-7-1977 |
Bis-substituted naphthalene-azo phenyleneazo-stilbene-disulfonic and naphthalene-sulfonic acid |
|
|
9-28-1977 |
UREIDOPHENYLENEBIS(CARBONYLIMINO)DINAPHTHALENETRISULFONIC ACID COMPOUNDS |
|
|
9-21-1977 |
Substituted bisnaphthylazo diphenyl ureido complement inhibitors |
|
|
9-7-1977 |
Substituted-hydroxy-naphthalenedisulfonic acid compounds |
|
1-12-1977 |
Complement inhibitors |
|
|
12-22-1976 |
Complement inhibitors |
|
|
10-13-1976 |
Complement inhibitors |
| EP0183352A2 * | Sep 27, 1985 | Jun 4, 1986 | THE UNITED STATES OF AMERICA as represented by the Secretary United States Department of Commerce | Use of suramin for clinical treatment of infection with any of the members of the family of human-t-cell leukemia (htvl) viruses including lymphadenopathy virus (lav) |
| EP0205077A2 * | Jun 3, 1986 | Dec 17, 1986 | Bayer Ag | Suramin sodium for use as an immunostimulant |
| EP0515523A1 * | Feb 13, 1991 | Dec 2, 1992 | THE UNITED STATES OF AMERICA as represented by the Secretary United States Department of Commerce | Use of suramin to treat rheumatologic diseases |
| EP0755254A1 * | Mar 24, 1995 | Jan 29, 1997 | The Trustees Of The University Of Pennsylvania | Prevention and treatment of ischemia-reperfusion and endotoxin-related injury using adenosine and purino receptor antagonists |
| EP1460087A1 * | Feb 17, 1997 | Sep 22, 2004 | The Kennedy Institute Of Rheumatology | Methods of treating vascular disease with TNF antagonists |
| EP1940376A2 * | Oct 3, 2006 | Jul 9, 2008 | Rottapharm S.P.A. | Use of neboglamine in the treatment of toxicodependency |
| EP1945204A2 * | Oct 27, 2006 | Jul 23, 2008 | Brane Discovery S.R.L. | V-atpase inhibitors for use in the treatment of septic shock |
| US5453444 * | Oct 6, 1994 | Sep 26, 1995 | Otsuka Pharmaceutical Co., Ltd. | Method to mitigate or eliminate weight loss |
| US5534539 * | Jun 12, 1995 | Jul 9, 1996 | Farmitalia Carlo Erba S.R.L. | Biologically active ureido derivatives useful as anit-metastic agenst |
| US5596105 * | Jan 13, 1995 | Jan 21, 1997 | Farmitalia Carlo Erba S.R.L. | Therapeutically active naphthalenesulfonic pyrrolecarboxamido derivatives |
| US7476693 | Mar 26, 2003 | Jan 13, 2009 | Eastern Virginia Medical School | Suramin and derivatives thereof as topical microbicide and contraceptive |
| US7608262 | Feb 16, 1996 | Oct 27, 2009 | The Kennedy Institute Of Rheumatology | Methods of preventing or treating thrombosis with tumor necrosis factor antagonists |
| US8552064 | Dec 19, 2008 | Oct 8, 2013 | Eastern Virginia Medical School | Suramin and derivatives thereof as topical microbicide and contraceptive |
| WO1994008574A1 * | Oct 12, 1993 | Apr 28, 1994 | Otsuka America Pharmaceutical | Treatment of cachexia and inhibition of il-6 activity |
| WO1994010990A1 * | Nov 12, 1993 | May 26, 1994 | British Bio Technology | Inhibition of tnf production |
| WO1997030088A2 * | Feb 17, 1997 | Aug 21, 1997 | Kennedy Inst Of Rheumatology | Methods of treating vascular disease with tnf antagonists |
| WO2004113920A1 * | Jun 18, 2004 | Dec 29, 2004 | Babon Jeff James | Screening method for substances binding to merozoite surface protein-1/42 |
| WO2008138943A2 * | May 14, 2008 | Nov 20, 2008 | Mara Galli | Prophylactic and therapeutic use of sirtuin inhibitors in tnf-alpha mediated pathologies |
| WO2009137471A2 * | May 5, 2009 | Nov 12, 2009 | University Of Miami | Azo dye related small molecule modulators of protein-protein interactions |
| WO2010016628A1 * | Jul 10, 2009 | Feb 11, 2010 | Sammy Opiyo | Conjugated suramin amino compounds for medical conditions |
| WO2012159107A1 * | May 21, 2012 | Nov 22, 2012 | Rhode Island Hospital | Inhibition of renal fibrosis |
Title: Suramin Sodium
CAS Registry Number: 129-46-4
CAS Name: 8,8¢-[Carbonylbis[imino-3,1-phenylenecarbonylimino(4-methyl-3,1-phenylene)carbonylimino]]bis-1,3,5-naphthalenetrisulfonic acid hexasodium salt
Additional Names: hexasodium sym-bis(m-aminobenzoyl-m-amino-p-methylbenzoyl-1-naphthylamino-4,6,8-trisulfonate) carbamide
Manufacturers’ Codes: Bayer 205; Fourneau 309
Trademarks: Antrypol (AstraZeneca); Germanin (Bayer); Moranyl (Specia); Naganol; Naphuride
Molecular Formula: C51H34N6Na6O23S6
Molecular Weight: 1429.17
Percent Composition: C 42.86%, H 2.40%, N 5.88%, Na 9.65%, O 25.75%, S 13.46%
Literature References: Discovered in 1917 by O. Dressel and R. Kothe: J. Dressel, J. Chem. Educ. 38, 620 (1961). Prepn: E. Fourneau et al., Compt. Rend. 178, 675 (1924); J. Trefouel, E. Fourneau, GB 224849 (1923); B. Heymann, Angew. Chem. 37, 585 (1924). Pharmacology, toxicology and clinical antiparasitic activity: F. Hawking, Adv. Pharmacol. Chemother. 15, 289-322 (1978). Inhibition of reverse transcriptase in vitro: E. De Clercq, Cancer Lett. 8, 9 (1979); vs HIV: H. Mitsuya et al., Science 226, 172 (1984). HPLC determn in plasma: R. W. Klecker, J. M. Collins, J. Liq. Chromatogr. 8, 1685 (1985). Pharmacokinetics: J. M. Collins et al., J. Clin. Pharmacol. 26, 22 (1986). Pharmacology and virustatic effect in AIDS: S. Broder et al., Lancet 2, 627 (1985); A. M. Levine et al., Ann. Intern. Med. 105, 32 (1986). Clinical trial in onchocerciasis: H. Schultz-Key et al., Trop. Med. Parasitol. 36, 244 (1985); in prostate cancer: C. Myers et al., J. Clin. Oncol. 10, 881 (1992). Review: Olenick in Antibiotics vol. 3,J. W. Corcoran, F. E. Hahn, Eds. (Springer-Verlag, New York, 1975) pp 699-703; R. La Rocca et al., Cancer Cells 2, 106-115 (1990).
Properties: White or slightly pink or cream-colored powder. Slightly bitter taste. Hygroscopic. Freely sol in water, in physiological saline; sparingly sol in 95% alcohol. Insol in benzene, ether, petr ether, chloroform. Aq solns are neutral to litmus. LD50 in mice (mg/kg): ~620 i.v. (Hawking).
Toxicity data: LD50 in mice (mg/kg): ~620 i.v. (Hawking)
Therap-Cat: Anthelmintic (Nematodes); antiprotozoal (Trypanosoma).
Therap-Cat-Vet: Antiprotozoal (Trypanosoma).
Keywords: Anthelmintic (Nematodes); Antiprotozoal (Trypanosoma); Reverse Transcriptase Inhibitor.


THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
MOBILE-+91 9323115463
web link
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
Summary of Metabolomics
Leaders in Pharmaceutical Business Intelligence Group, LLC, Doing Business As LPBI Group, Newton, MA
Summary of Metabolomics
Author and Curator: Larry H. Bernstein, MD, FCAP
This concludes the series on metabolomics, a rapidly developing science that is interconnected with a group termed – OMICS: proteomics, transcriptomics, genomics, and metabolomics. This chapter is most representative of the many important studies being done in the field, which ranges most widely because it has opened doors into nutrition and nutritional supplements, plant biochemistry, agricultural crops and breeding, animal breeding, worldwide malnutrition, diabetes, cancer, neurosciences, circulatory, respiratory, and musculosletal disorders, infectious diseases and immune system disorders. Obviously, it is not possible to cover the full range of activity, but metabolomics is most comprehensive in exploring the full range of metabolic changes that occur in health during the full age range from development to the geriatric years. It can be integrated well with gene expression, proteomics studies, and epidemiological investigations.
The subchapters are given here:
7.1 Extracellular evaluation of intracellular flux in yeast cells
View original post 1,051 more words
Flow Chemistry test facility in India

Flow Chemistry test facility in India
BOOK YOUR TRIAL
on Plantrix® Industrial system.
Contacts:

India
Vijay Kirpalani
vk@pi-inc.co
0091 9821 3420 22
More information
Stan Hoeijmakers
info@chemtrix.com
0031 (0)46 70 22 600
EVENTS
Chemtrix at CPhI India
2 – 4 December 2014
Mumbai, India
Booth H47, Hall 5
Pi Process Intensification
read at
http://hosted.verticalresponse.com/721499/f9f4fc970b/285875213/4942751fec/
|
Plantrix® Industrial Flow Chemistry Test Facility in India
|
||||||||

|
||||||||
|
|
|||||||
|
|
||||||||


Chemtrix Bv.
Urmonderbaan 22
Geleen, 6167RD
NL

Yoga back bends: feels yummy on the autonomic nervous system
Beyond Meds: Alternatives to Psychiatry
 I’m reposting this because I’ve been going through another backbend stage and I thought of this post from a while back. I like to help people see how easy yoga can be. You can start with something as simple as this and see where it takes you. Being a yogi is about listening to your body and learning from it and it really doesn’t matter if you can do really complicated poses or not. Start simple and see what happens.
I’m reposting this because I’ve been going through another backbend stage and I thought of this post from a while back. I like to help people see how easy yoga can be. You can start with something as simple as this and see where it takes you. Being a yogi is about listening to your body and learning from it and it really doesn’t matter if you can do really complicated poses or not. Start simple and see what happens.
I’ve been using yoga as a main source of rehabilitation and recovery since I was bedridden. I began doing yoga while still in bed. Now it continues to be a primary source of continued healing. Lately I’ve been doing backbends and while all the yoga I do feels like it profoundly helps my nervous system, these bends have really been making me think about my autonomic nervous system and how…
View original post 744 more words

Prefacing the e-Book Epilogue: Metabolic Genomics and Pharmaceutics
Leaders in Pharmaceutical Business Intelligence Group, LLC, Doing Business As LPBI Group, Newton, MA
Prefacing the e-Book Epilogue: Metabolic Genomics and Pharmaceutics
Author and Curator: Larry H. Bernstein, MD, FCAP
Adieu, adieu, adieu …
Sound of Music
 Snoopy – Charlie happiness
Snoopy – Charlie happiness
This work has been a coming to terms with my scientific and medical end of career balancing in a difficult time after retiring, but it has been rewarding. In the clinical laboratories, radiology, anesthesiology, and in pharmacy, there has been some significant progress in support of surgical, gynecological, developmental, medical practices, and even neuroscience directed disciplines, as well as epidemiology over a period of half a century. Even then, cancer and neurological diseases have been most difficult because the scientific basic research has either not yet uncovered a framework, or because that framework has proved to be multidimensional. In the clinical laboratory sciences, there has been enormous progress in instrumental analysis, with the recent opening of molecular methods not yet prepared for routine clinical…
View original post 3,976 more words
Cordyceps – Rare parasitic fungi could have anti-flammatory benefits
19 Nov 2012
Caterpillar fungi (Cordyceps) are rare parasites found on hibernating caterpillars in the mountains of Tibet. For centuries they have been highly prized as a traditional Chinese medicine – just a small amount can fetch hundreds of pounds.
Scientists at The University of Nottingham have been studying how this fungus could work by studying cordycepin, one of the drugs found in these mushrooms. They have already discovered that cordycepin has potential as a cancer drug. Their new work indicates that it could also have anti-inflammatory characteristics with the potential to help sufferers of asthma, rheumatoid arthritis, renal failure and stroke damage.
The research, published today in the academic journal RNA, was led by Dr Cornelia de Moor in the School of Pharmacy. It shows that cordycepin reduces inflammatory gene products in airway smooth muscle cells – the cells that contract during an asthma attack.
Several studies have suggested…
View original post 347 more words
ROFECOXIB

ROFECOXIB
MK-966, MK-0966, Vioxx
162011-90-7
Rofecoxib /ˌrɒfɨˈkɒksɪb/ is a nonsteroidal anti-inflammatory drug (NSAID) that has now been withdrawn over safety concerns. It was marketed by Merck & Co. to treat osteoarthritis, acute pain conditions, and dysmenorrhoea. Rofecoxib was approved by the Food and Drug Administration (FDA) on May 20, 1999, and was marketed under the brand names Vioxx, Ceoxx, and Ceeoxx.
No Exclusivity found
| Patent No | Expirey Date | patent use code |
|---|---|---|
| 5474995 | Jun 24, 2013 | U-602 |
| 5474995*PED | Dec 24, 2013 | |
| 5691374 | May 18, 2015 | |
| 5691374*PED | Nov 18, 2015 | |
| 6063811 | May 6, 2017 | U-602 |
| 6063811*PED | Nov 6, 2017 | |
| 6239173 | Jun 24, 2013 | U-602 |
| 6239173*PED | Dec 24, 2013 |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 4-(4-methylsulfonylphenyl)-3-phenyl-5H-furan-2-one | |
| Clinical data | |
| Pregnancy cat. | C (AU) |
| Legal status | Prescription Only (S4) (AU)withdrawn |
| Routes | oral |
| Pharmacokinetic data | |
| Bioavailability | 93% |
| Protein binding | 87% |
| Metabolism | hepatic |
| Half-life | 17 hours |
| Excretion | biliary/renal |
| Identifiers | |
| CAS number | 162011-90-7 |
| ATC code | M01AH02 |
| PubChem | CID 5090 |
| DrugBank | DB00533 |
| ChemSpider | 4911 |
| UNII | 0QTW8Z7MCR |
| Chemical data | |
| Formula | C17H14O4S |
| Mol. mass | 314.357 g/mol |
Rofecoxib gained widespread acceptance among physicians treating patients with arthritis and other conditions causing chronic or acute pain. Worldwide, over 80 million people were prescribed rofecoxib at some time.[1]
On September 30, 2004, Merck withdrew rofecoxib from the market because of concerns about increased risk of heart attack and stroke associated with long-term, high-dosage use. Merck withdrew the drug after disclosures that it withheld information about rofecoxib’s risks from doctors and patients for over five years, resulting in between 88,000 and 140,000 cases of serious heart disease.[2] Rofecoxib was one of the most widely used drugs ever to be withdrawn from the market. In the year before withdrawal, Merck had sales revenue of US$2.5 billion from Vioxx.[3] Merck reserved $970 million to pay for its Vioxx-related legal expenses through 2007, and have set aside $4.85bn for legal claims from US citizens.
Rofecoxib was available on prescription in both tablet-form and as an oral suspension. It was available by injection for hospital use.

Mode of action
Cyclooxygenase (COX) has two well-studied isoforms, called COX-1 and COX-2. COX-1 mediates the synthesis of prostaglandinsresponsible for protection of the stomach lining, while COX-2 mediates the synthesis of prostaglandins responsible for pain and inflammation. By creating “selective” NSAIDs that inhibit COX-2, but not COX-1, the same pain relief as traditional NSAIDs is offered, but with greatly reduced risk of fatal or debilitating peptic ulcers. Rofecoxib is a selective COX-2 inhibitor, or “coxib”.
Others include Merck’s etoricoxib (Arcoxia), Pfizer’s celecoxib (Celebrex) and valdecoxib (Bextra). Interestingly, at the time of its withdrawal, rofecoxib was the only coxib with clinical evidence of its superior gastrointestinal adverse effect profile over conventional NSAIDs. This was largely based on the VIGOR (Vioxx GI Outcomes Research) study, which compared the efficacy and adverse effect profiles of rofecoxib and naproxen.[4]
Pharmacokinetics
The therapeutic recommended dosages were 12.5, 25, and 50 mg with an approximate bioavailability of 93%.[5][6][7] Rofecoxib crossed the placenta and blood–brain barrier,[5][6][8]and took 1–3 hours to reach peak plasma concentration with an effective half-life (based on steady-state levels) of approximately 17 hours.[5][7][9] The metabolic products are cis-dihydro and trans-dihydro derivatives of rofecoxib[5][9] which are primarily excreted through urine.
Fabricated efficacy studies
On March 11, 2009, Scott S. Reuben, former chief of acute pain at Baystate Medical Center, Springfield, Mass., revealed that data for 21 studies he had authored for the efficacy of the drug (along with others such as celecoxib) had been fabricated in order to augment the analgesic effects of the drugs. There is no evidence that Reuben colluded with Merck in falsifying his data. Reuben was also a former paid spokesperson for the drug company Pfizer (which owns the intellectual property rights for marketing celecoxib in the United States). The retracted studies were not submitted to either the FDA or the European Union’s regulatory agencies prior to the drug’s approval. Drug manufacturer Merckhad no comment on the disclosure.[10]
Adverse drug reactions

Aside from the reduced incidence of gastric ulceration, rofecoxib exhibits a similar adverse effect profile to other NSAIDs.
Prostaglandin is a large family of lipids. Prostaglandin I2/PGI2/prostacyclin is just one member of it. Prostaglandins other than PGI2 (such as PGE2) also play important roles in vascular tone regulation. Prostacyclin/thromboxane are produced by both COX-1 and COX-2, and rofecoxib suppresses just COX-2 enzyme, so there is no reason to believe that prostacyclin levels are significantly reduced by the drug. And there is no reason to believe that only the balance between quantities of prostacyclin and thromboxane is the determinant factor for vascular tone.[11] Indeed Merck has stated that there was no effect on prostacyclin production in blood vessels in animal testing.[12] Other researchers have speculated that the cardiotoxicity may be associated with maleic anhydride metabolites formed when rofecoxib becomes ionized under physiological conditions. (Reddy & Corey, 2005)
Adverse cardiovascular events
VIGOR study and publishing controversy
The VIGOR (Vioxx GI Outcomes Research) study, conducted by Bombardier, et al., which compared the efficacy and adverse effect profiles of rofecoxib and naproxen, had indicated a significant 4-fold increased risk of acute myocardial infarction (heart attack) in rofecoxib patients when compared with naproxen patients (0.4% vs 0.1%, RR 0.25) over the 12 month span of the study. The elevated risk began during the second month on rofecoxib. There was no significant difference in the mortality from cardiovascular events between the two groups, nor was there any significant difference in the rate of myocardial infarction between the rofecoxib and naproxen treatment groups in patients without high cardiovascular risk. The difference in overall risk was by the patients at higher risk of heart attack, i.e. those meeting the criteria for low-dose aspirin prophylaxis of secondary cardiovascular events (previous myocardial infarction, angina, cerebrovascular accident, transient ischemic attack, or coronary artery bypass).
Merck’s scientists interpreted the finding as a protective effect of naproxen, telling the FDA that the difference in heart attacks “is primarily due to” this protective effect (Targum, 2001). Some commentators have noted that naproxen would have to be three times as effective as aspirin to account for all of the difference (Michaels 2005), and some outside scientists warned Merck that this claim was implausible before VIGOR was published.[13] No evidence has since emerged for such a large cardioprotective effect of naproxen, although a number of studies have found protective effects similar in size to those of aspirin.[14][15] Though Dr. Topol’s 2004 paper criticized Merck’s naproxen hypothesis, he himself co-authored a 2001 JAMA article stating “because of the evidence for an antiplatelet effect of naproxen, it is difficult to assess whether the difference in cardiovascular event rates in VIGOR was due to a benefit from naproxen or to a prothrombotic effect from rofecoxib.” (Mukherjee, Nissen and Topol, 2001.)
The results of the VIGOR study were submitted to the United States Food and Drug Administration (FDA) in February 2001. In September 2001, the FDA sent a warning letter to the CEO of Merck, stating, “Your promotional campaign discounts the fact that in the VIGOR study, patients on Vioxx were observed to have a four to five fold increase in myocardial infarctions (MIs) compared to patients on the comparator non-steroidal anti-inflammatory drug (NSAID), Naprosyn (naproxen).”[16] This led to the introduction, in April 2002, of warnings on Vioxx labeling concerning the increased risk of cardiovascular events (heart attack and stroke).
Months after the preliminary version of VIGOR was published in the New England Journal of Medicine, the journal editors learned that certain data reported to the FDA were not included in the NEJM article. Several years later, when they were shown a Merck memo during the depositions for the first federal Vioxx trial, they realized that these data had been available to the authors months before publication. The editors wrote an editorial accusing the authors of deliberately withholding the data.[17] They released the editorial to the media on December 8, 2005, before giving the authors a chance to respond. NEJM editor Gregory Curfman explained that the quick release was due to the imminent presentation of his deposition testimony, which he feared would be misinterpreted in the media. He had earlier denied any relationship between the timing of the editorial and the trial. Although his testimony was not actually used in the December trial, Curfman had testified well before the publication of the editorial.[18]
The editors charged that “more than four months before the article was published, at least two of its authors were aware of critical data on an array of adverse cardiovascular events that were not included in the VIGOR article.” These additional data included three additional heart attacks, and raised the relative risk of Vioxx from 4.25-fold to 5-fold. All the additional heart attacks occurred in the group at low risk of heart attack (the “aspirin not indicated” group) and the editors noted that the omission “resulted in the misleading conclusion that there was a difference in the risk of myocardial infarction between the aspirin indicated and aspirin not indicated groups.” The relative risk for myocardial infarctions among the aspirin not indicated patients increased from 2.25 to 3 (although it remained statitistically insignificant). The editors also noted a statistically significant (2-fold) increase in risk for serious thromboembolic events for this group, an outcome that Merck had not reported in the NEJM, though it had disclosed that information publicly in March 2000, eight months before publication.[19]
The authors of the study, including the non-Merck authors, responded by claiming that the three additional heart attacks had occurred after the prespecified cutoff date for data collection and thus were appropriately not included. (Utilizing the prespecified cutoff date also meant that an additional stroke in the naproxen population was not reported.) Furthermore, they said that the additional data did not qualitatively change any of the conclusions of the study, and the results of the full analyses were disclosed to the FDA and reflected on the Vioxx warning label. They further noted that all of the data in the “omitted” table were printed in the text of the article. The authors stood by the original article.[20]
NEJM stood by its editorial, noting that the cutoff date was never mentioned in the article, nor did the authors report that the cutoff for cardiovascular adverse events was before that for gastrointestinal adverse events. The different cutoffs increased the reported benefits of Vioxx (reduced stomach problems) relative to the risks (increased heart attacks).[19]
Some scientists have accused the NEJM editorial board of making unfounded accusations.[21][22] Others have applauded the editorial. Renowned research cardiologist Eric Topol,[23] a prominent Merck critic, accused Merck of “manipulation of data” and said “I think now the scientific misconduct trial is really fully backed up”.[24] Phil Fontanarosa, executive editor of the prestigious Journal of the American Medical Association, welcomed the editorial, saying “this is another in the long list of recent examples that have generated real concerns about trust and confidence in industry-sponsored studies”.[25]
On May 15, 2006, the Wall Street Journal reported that a late night email, written by an outside public relations specialist and sent to Journal staffers hours before the Expression of Concern was released, predicted that “the rebuke would divert attention to Merck and induce the media to ignore the New England Journal of Medicine‘s own role in aiding Vioxx sales.”[26]
“Internal emails show the New England Journal’s expression of concern was timed to divert attention from a deposition in which Executive Editor Gregory Curfman made potentially damaging admissions about the journal’s handling of the Vioxx study. In the deposition, part of the Vioxx litigation, Dr. Curfman acknowledged that lax editing might have helped the authors make misleading claims in the article.” The Journal stated that NEJM‘s “ambiguous” language misled reporters into incorrectly believing that Merck had deleted data regarding the three additional heart attacks, rather than a blank table that contained no statistical information; “the New England Journal says it didn’t attempt to have these mistakes corrected.”[26]
Alzheimer’s studies
In 2000 and 2001, Merck conducted several studies of rofecoxib aimed at determining if the drug slowed the onset of Alzheimer’s disease. Merck has placed great emphasis on these studies on the grounds that they are relatively large (almost 3000 patients) and compared rofecoxib to a placebo rather than to another pain reliever. These studies found an elevated death rate among rofecoxib patients, although the deaths were not generally heart-related. However, they did not find any elevated cardiovascular risk due to rofecoxib.[27] Before 2004, Merck cited these studies as providing evidence, contrary to VIGOR, of rofecoxib’s safety.
APPROVe study
In 2001, Merck commenced the APPROVe (Adenomatous Polyp PRevention On Vioxx) study, a three-year trial with the primary aim of evaluating the efficacy of rofecoxib for theprophylaxis of colorectal polyps. Celecoxib had already been approved for this indication, and it was hoped to add this to the indications for rofecoxib as well. An additional aim of the study was to further evaluate the cardiovascular safety of rofecoxib.
The APPROVe study was terminated early when the preliminary data from the study showed an increased relative risk of adverse thrombotic cardiovascular events (includingheart attack and stroke), beginning after 18 months of rofecoxib therapy. In patients taking rofecoxib, versus placebo, the relative risk of these events was 1.92 (rofecoxib 1.50 events vs placebo 0.78 events per 100 patient years). The results from the first 18 months of the APPROVe study did not show an increased relative risk of adverse cardiovascular events. Moreover, overall and cardiovascular mortality rates were similar between the rofecoxib and placebo populations.[28]
In summary, the APPROVe study suggested that long-term use of rofecoxib resulted in nearly twice the risk of suffering a heart attack or stroke compared to patients receiving a placebo.
Other studies
Pre-approval Phase III clinical trials, like the APPROVe study, showed no increased relative risk of adverse cardiovascular events for the first eighteen months of rofecoxib usage (Merck, 2004). Others have pointed out that “study 090,” a pre-approval trial, showed a 3-fold increase in cardiovascular events compared to placebo, a 7-fold increase compared to nabumetone (another [NSAID]), and an 8-fold increase in heart attacks and strokes combined compared to both control groups.[29][30] Although this was a relatively small study and only the last result was statistically significant, critics have charged that this early finding should have prompted Merck to quickly conduct larger studies of rofecoxib’s cardiovascular safety. Merck notes that it had already begun VIGOR at the time Study 090 was completed. Although VIGOR was primarily designed to demonstrate new uses for rofecoxib, it also collected data on adverse cardiovascular outcomes.
Several very large observational studies have also found elevated risk of heart attack from rofecoxib. For example, a recent retrospective study of 113,000 elderly Canadians suggested a borderline statistically significant increased relative risk of heart attacks of 1.24 from Vioxx usage, with a relative risk of 1.73 for higher-dose Vioxx usage. (Levesque, 2005). Another study, using Kaiser Permanente data, found a 1.47 relative risk for low-dose Vioxx usage and 3.58 for high-dose Vioxx usage compared to current use of celecoxib, though the smaller number was not statistically significant, and relative risk compared to other populations was not statistically significant. (Graham, 2005).
Furthermore, a more recent meta-study of 114 randomized trials with a total of 116,000+ participants, published in JAMA, showed that Vioxx uniquely increased risk of renal (kidney) disease, and heart arrhythmia.[31]
Other COX-2 inhibitors
Any increased risk of renal and arrhythmia pathologies associated with the class of COX-2 inhibitors, e.g. celecoxib (Celebrex), valdecoxib (Bextra), parecoxib (Dynastat),lumiracoxib, and etoricoxib is not evident,[31] although smaller studies[32][33] had demonstrated such effects earlier with the use of celecoxib, valdecoxib and parecoxib.
Nevertheless, it is likely that trials of newer drugs in the category will be extended in order to supply additional evidence of cardiovascular safety. Examples are some more specific COX-2 inhibitors, including etoricoxib (Arcoxia) and lumiracoxib (Prexige), which are currently (circa 2005) undergoing Phase III/IV clinical trials.
Besides, regulatory authorities worldwide now require warnings about cardiovascular risk of COX-2 inhibitors still on the market. For example, in 2005, EU regulators required the following changes to the product information and/or packaging of all COX-2 inhibitors:[34]
- Contraindications stating that COX-2 inhibitors must not be used in patients with established ischaemic heart disease and/or cerebrovascular disease (stroke), and also in patients with peripheral arterial disease
- Reinforced warnings to healthcare professionals to exercise caution when prescribing COX-2 inhibitors to patients with risk factors for heart disease, such as hypertension, hyperlipidaemia (high cholesterol levels), diabetes and smoking
- Given the association between cardiovascular risk and exposure to COX-2 inhibitors, doctors are advised to use the lowest effective dose for the shortest possible duration of treatment
Other NSAIDs
Since the withdrawal of Vioxx it has come to light that there may be negative cardiovascular effects with not only other COX-2 inhibitiors, but even the majority of other NSAIDs. It is only with the recent development of drugs like Vioxx that drug companies have carried out the kind of well executed trials that could establish such effects and these sort of trials have never been carried out in older “trusted” NSAIDs such as ibuprofen, diclofenac and others. The possible exceptions may be aspirin and naproxen due to their anti-platelet aggregation properties.
Withdrawal
Due to the findings of its own APPROVe study, Merck publicly announced its voluntary withdrawal of the drug from the market worldwide on September 30, 2004.[35]
In addition to its own studies, on September 23, 2004 Merck apparently received information about new research by the FDA that supported previous findings of increased risk of heart attack among rofecoxib users (Grassley, 2004). FDA analysts estimated that Vioxx caused between 88,000 and 139,000 heart attacks, 30 to 40 percent of which were probably fatal, in the five years the drug was on the market.[36]
On November 5, the medical journal The Lancet published a meta-analysis of the available studies on the safety of rofecoxib (Jüni et al., 2004). The authors concluded that, owing to the known cardiovascular risk, rofecoxib should have been withdrawn several years earlier. The Lancet published an editorial which condemned both Merck and the FDA for the continued availability of rofecoxib from 2000 until the recall. Merck responded by issuing a rebuttal of the Jüni et al. meta-analysis that noted that Jüni omitted several studies that showed no increased cardiovascular risk. (Merck & Co., 2004).
In 2005, advisory panels in both the U.S. and Canada encouraged the return of rofecoxib to the market, stating that rofecoxib’s benefits outweighed the risks for some patients. The FDA advisory panel voted 17-15 to allow the drug to return to the market despite being found to increase heart risk. The vote in Canada was 12-1, and the Canadian panel noted that the cardiovascular risks from rofecoxib seemed to be no worse than those from ibuprofen—though the panel recommended that further study was needed for all NSAIDs to fully understand their risk profiles. Notwithstanding these recommendations, Merck has not returned rofecoxib to the market.[37]
In 2005, Merck retained Debevoise & Plimpton LLP to investigate Vioxx study results and communications conducted by Merck. Through the report, it was found that Merck’s senior management acted in good faith, and that the confusion over the clinical safety of Vioxx was due to the sales team’s overzealous behavior. The report that was filed gave a timeline of the events surrounding Vioxx and showed that Merck intended to operate honestly throughout the process. Any mistakes that were made regarding the mishandling of clinical trial results and withholding of information was the result of oversight, not malicious behavior. The Martin Report did conclude that the Merck’s marketing team exaggerated the safety of Vioxx and replaced truthful information with sales tactics.[citation needed] The report was published in February 2006, and Merck was satisfied with the findings of the report and promised to consider the recommendations contained in the Martin Report. Advisers to the US Food and Drug Administration (FDA) have voted, by a narrow margin, that it should not ban Vioxx — the painkiller withdrawn by drug-maker Merck.
They also said that Pfizer’s Celebrex and Bextra, two other members of the family of painkillers known as COX-2 inhibitors, should remain available, despite the fact that they too boost patients’ risk of heart attack and stroke. url = http://www.nature.com/drugdisc/news/articles/433790b.html The recommendations of the arthritis and drug safety advisory panel offer some measure of relief to the pharmaceutical industry, which has faced a barrage of criticism for its promotion of the painkillers. But the advice of the panel, which met near Washington DC over 16–18 February, comes with several strings attached.
For example, most panel members said that manufacturers should be required to add a prominent warning about the drugs’ risks to their labels; to stop direct-to-consumer advertising of the drugs; and to include detailed, written risk information with each prescription. The panel also unanimously stated that all three painkillers “significantly increase the risk of cardiovascular events”.
The panel voted 17 to 15 against banning Vioxx (rofecoxib) entirely; the vote on Bextra (valdecoxib) was 17 to 13 with 2 abstentions; Celebrex (celecoxib) was endorsed 31 to 1. Shares of Merck, based in Whitehouse Station, New Jersey, and New York-based Pfizer closed up 13% and 7% respectively on 18 February, 2013, the day of the votes.
The FDA is expected to act on the recommendations within weeks. Although the agency usually follows the recommendations of its outside advisers, it is not bound to do so. A top official said that, in light of the closeness of some of the votes, the agency will examine the panel members’ comments in detail before deciding what to do.
An official from Merck said during the meeting that it would consider reintroducing Vioxx, which it withdrew in September 2004. On April 7, 2005, Pfizer withdrew Bextra from the U.S. market on recommendation by the FDA. Pfizer’s other painkiller, Celebrex, is still on the market.
Litigation
As of March 2006, there had been over 10,000 cases and 190 class actions filed against Merck[citation needed] over adverse cardiovascular events associated with rofecoxib and the adequacy of Merck’s warnings. The first wrongful death trial, Rogers v. Merck, was scheduled in Alabama in the spring of 2005, but was postponed after Merck argued that the plaintiff had falsified evidence of rofecoxib use.[1]
On August 19, 2005, a jury in Texas voted 10-2 to hold Merck liable for the death of Robert Ernst, a 59-year-old man who allegedly died of a rofecoxib-induced heart attack. The plaintiffs’ lead attorney was Mark Lanier. Merck argued that the death was due to cardiac arrhythmia, which had not been shown to be associated with rofecoxib use. The jury awarded Carol Ernst, widow of Robert Ernst, $253.4 million in damages. This award will almost certainly be capped at no more than US$26.1 million because of punitive damages limits under Texas law.[2] As of March 2006, the plaintiff had yet to ask the court to enter a judgment on the verdict; Merck has stated that it will appeal.
On November 3, 2005, Merck won the second case Humeston v. Merck, a personal injury case, in Atlantic City, New Jersey. The plaintiff experienced a mild myocardial infarction and claimed that rofecoxib was responsible, after having taken it for two months. Merck argued that there was no evidence that rofecoxib was the cause of Humeston’s injury and that there is no scientific evidence linking rofecoxib to cardiac events with short durations of use. The jury ruled that Merck had adequately warned doctors and patients of the drug’s risk.[3]
The first federal trial on rofecoxib, Plunkett v. Merck, began on November 29, 2005 in Houston. The trial ended in a hung jury and a mistrial was declared on December 12, 2005. According to the Wall Street Journal, the jury hung by an eight to one majority, favoring the defense. Upon retrial in February 2006 in New Orleans, where the Vioxx multidistrict litigation (MDL) is based, a jury found Merck not liable, even though the plaintiffs had the NEJM editor testify as to his objections to the VIGOR study.
On January 30, 2006, a New Jersey state court dismissed a case brought by Edgar Lee Boyd, who blamed Vioxx for gastrointestinal bleeding that he experienced after taking the drug. The judge said that Boyd failed to prove the drug caused his stomach pain and internal bleeding.
In January 2006, Garza v. Merck began trial in Rio Grande City, Texas. The plaintiff, a 71-year-old smoker with heart disease, had a fatal heart attack three weeks after finishing a one-week sample of rofecoxib. On April 21, 2006 the jury awarded the plaintiff $7 million compensatory and $25 million punitive. The Texas state court of appeals in San Antonio later rules Garza’s fatal heart attack probably resulted from pre-existing health conditions unrelated to his taking of Vioxx, thus reversing the $32 million jury award.[4]
On April 5, 2006, the jury held Merck liable for the heart attack of 77-year-old John McDarby, and awarded Mr McDarby $4.5 million in compensatory damages based on Merck’s failure to properly warn of Vioxx safety risks. After a hearing on April 11, 2006, the jury also awarded Mr McDarby an additional $9 million in punitive damages. The same jury found Merck not liable for the heart attack of 60-year-old Thomas Cona, a second plaintiff in the trial, but was liable for fraud in the sale of the drug to Cona.
Merck has reserved $970 million to pay for its Vioxx-related legal expenses through 2007, and have set aside $4.85bn for legal claims from US citizens. Patients who claim to have suffered as a result of taking Vioxx in countries outside the US are campaigning for this to be extended.
In March 2010, an Australian class-action lawsuit against Merck ruled that Vioxx doubled the risk of heart attacks, and that Merck had breached the Trade Practices Act by selling a drug which was unfit for sale.[38]
In November 2011, Merck announced a civil settlement with the US Attorney’s Office for the District of Massachusetts, and individually with 43 US states and the District of Columbia, to resolve civil claims relating to Vioxx.[5] Under the terms of the settlement, Merck agreed to pay two-thirds of a previously recorded $950 million reserve charge in exchange for release from civil liability. Litigation with seven additional states remains outstanding. Under separate criminal proceedings, Merck plead guilty to a federal misdemeanor charge relating to the marketing of the drug across state lines, incurring a fine of $321.6 million.[6]
Other effects
Rofecoxib was shown to improve premenstrual acne vulgaris in a placebo controlled study.[39]
Synthesis

Rofecoxib synthesis.[40]
,,,,,,,,,,,,,,,,,
The oxidation of 4- (methylsulfanyl) acetophenone (X) with monoperoxyphthalic acid (MMPP) in dichloro-methane / methanol gives the corresponding sulfone (XI), which is brominated with Br2 / AlCl3 in chloroform, yielding the expected phenacyl bromide ( XII). Finally, this compound is cyclocondensed with phenylacetic acid (I) by means of 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU) and triethylamine in acetonitrile. 5) Reaction of [4- (methylsulfonyl ) phenyl] phenylacetyl-ene (XIII) with CO catalyzed by Rh4 (CO) 12 in THF at 100 C in a stainless steel autoclave at 100 Atm pressure, followed by a chromatographic separation in a silicagel column to eliminate the undesired regioisomer.

……………….
The synthesis of rofecoxib can be performed by several different ways: 1) The condensation of phenylacetic acid (I) with ethyl bromoacetate (II) by means of triethylamine in THF yields 2- (phenylacetoxy) acetic acid ethyl ester (III), which is cyclized to the hydroxyfuranone (IV) by means of potassium tert-butoxide in tert-butanol. The reaction of (IV) with triflic anhydride and diisopropylethylamine in dichloro-methane affords the corresponding triflate (V), which by reaction with LiBr in hot acetone yields the bromofuranone (VI) The condensation of (VI) with 4- (methylsulfanyl) phenylboronic acid (VII) by means of Na2CO3 and Pd (Ph3P) 4 in hot toluene gives 4- [4- (methylsulfanyl) -phenyl]. – 3-phenylfuran-2 (5H) -one (VIII), which is finally oxidized with 2KHSO5.KHSO4.K2SO4 (oxone). 2) The intermediate (VIII) can also be obtained by condensation of triflate (V) with boronic acid ( VII) by means of Na2CO3 and Pd (Ph3P) 4 in hot toluene. 3) The intermediate (VIII) can also be synthesized by the reaction of triflate (V) with tetramethylammonium chloride, giving the chlorofuranone (IX), which is then condensed with boronic acid (VII) as before.

Footnotes
- http://www.npr.org/templates/story/story.php?storyId=4054991
- “Up to 140,000 heart attacks linked to Vioxx.”. New Scientist. 2005-01-25. p. 1.
- “Merck Sees Slightly Higher 2007 Earnings”. New York Times. Reuters. 2006-12-07. p. A1.
- Bombardier, C.; Laine, L.; Reicin, A.; Shapiro, D.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M. B.; Hawkey, C. J.; Hochberg, M. C.; Kvien, T. K.; Schnitzer, T. J.; Vigor Study, G. (2000). “Comparison of Upper Gastrointestinal Toxicity of Rofecoxib and Naproxen in Patients with Rheumatoid Arthritis”. New England Journal of Medicine 343 (21): 1520–1528, 2 1528 following 1528. doi:10.1056/NEJM200011233432103. PMID 11087881. edit
- Merck & Co. VIOXX (rofecoxib tablets and oral suspension). Accessed at: http://www.merck.com/product/usa/pi_circulars/v/vioxx/vioxx_pi.pdf 01 Feb 2010
- Gold Standard Inc. Rofecoxib Vioxx Accessed at: http://www.mdconsult.com/das/pharm/body/181267313-3/946823742/full/2399 01 Feb 2010
- ^ Jump up to:a b Davies, N. M.; Teng, X. W.; Skjodt, N. M. (2003). “Pharmacokinetics of rofecoxib: a specific cyclo-oxygenase-2 inhibitor”. Clinical pharmacokinetics 42 (6): 545–556.PMID 12793839. edit
- Padi, S.; Kulkarni, S. (2004). “Differential effects of naproxen and rofecoxib on the development of hypersensitivity following nerve injury in rats”. Pharmacology, Biochemistry, and Behavior 79 (2): 349–358. doi:10.1016/j.pbb.2004.08.005. PMID 15501312. edit
- Scott, L. J.; Lamb, H. M. (1999). “Rofecoxib”. Drugs 58 (3): 499–505; discussion 506–7. doi:10.2165/00003495-199958030-00016. PMID 10493277. edit
- Winstein, Keith J. (March 11, 2009). “Top Pain Scientist Fabricated Data in Studies, Hospital Says”. The Wall Street Journal.
- Vane, J.; Bakhle, Y.; Botting, R. (1998). “Cyclooxygenases 1 and 2”. Annual review of pharmacology and toxicology 38: 97–120. doi:10.1146/annurev.pharmtox.38.1.97.PMID 9597150. edit
- sfgate.com
- Jump up^ www.saferdrugsnow.org
- Karha, J.; Topol, E. J. (2004). “The sad story of Vioxx, and what we should learn from it”. Cleveland Clinic journal of medicine 71 (12): 933–934, 936, 934–9.doi:10.3949/ccjm.71.12.933. PMID 15641522. edit
- Solomon, D. H.; Glynn, R. J.; Levin, R.; Avorn, J. (2002). “Nonsteroidal anti-inflammatory drug use and acute myocardial infarction”. Archives of Internal Medicine 162 (10): 1099–1104.doi:10.1001/archinte.162.10.1099. PMID 12020178. edit
- http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/WarningLettersandNoticeofViolationLetterstoPharmaceuticalCompanies/UCM166383.pdf
- Curfman, G.; Morrissey, S.; Drazen, J. (2005). “Expression of concern: Bombardier et al., “Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis,” N Engl J Med 2000;343:1520-8″. The New England Journal of Medicine 353 (26): 2813–2814. doi:10.1056/NEJMe058314. PMID 16339408. edit
- http://www.forbes.com/work/feeds/ap/2006/02/13/ap2523250.html. Missing or empty
|title=(help)[dead link] - Curfman, G.; Morrissey, S.; Drazen, J. (2006). “Expression of concern reaffirmed”. The New England Journal of Medicine 354 (11): 1193. doi:10.1056/NEJMe068054.PMID 16495386. edit
- Jump up^ Bombardier, C.; Laine, L.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M.; Hawkey, C.; Hochberg, M.; Kvien, T.; Schnitzer, T. J.; Weaver, A. (2006). “Response to expression of concern regarding VIGOR study”. The New England Journal of Medicine 354 (11): 1196–1199. doi:10.1056/NEJMc066096. PMID 16495387. edit
- http://pipeline.corante.com/archives/2006/02/22/nejm_vs_its_contributors_round_two.php
- http://dimer.tamu.edu/simplog/archive.php?blogid=3&pid=3293
- http://genetics.case.edu/faculty2.php?fac=ejt9
- Jump up^ http://www.medicinenet.com/script/main/art.asp?articlekey=56384&page=2
- http://www.beasleyallen.com/news/vioxx-plaintiffs-seek-mistrial-after-allegation-on-merck-study/
- David Armstrong (2006-05-15). “How the New England Journal Missed Warning Signs on Vioxx”. Wall Street Journal. p. A1.
- Konstam, M. A.; Weir, M. R.; Reicin, A.; Shapiro, D.; Sperling, R. S.; Barr, E.; Gertz, B. J. (2001). “Cardiovascular thrombotic events in controlled, clinical trials of rofecoxib”. Circulation104 (19): 2280–2288. doi:10.1161/hc4401.100078. PMID 11696466. edit
- Bresalier, R.; Sandler, R.; Quan, H.; Bolognese, J.; Oxenius, B.; Horgan, K.; Lines, C.; Riddell, R.; Morton, D.; Lanas, A.; Konstam, M. A.; Baron, J. A.; Adenomatous Polyp Prevention on Vioxx (APPROVe) Trial Investigators (2005). “Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial”. The New England Journal of Medicine 352(11): 1092–1102. doi:10.1056/NEJMoa050493. PMID 15713943. edit
- http://www.fda.gov/ohrms/dockets/ac/01/briefing/3677b2_06_cardio.pdf
- Jump up^ Wolfe, M. M. (2004). “Rofecoxib, Merck, and the FDA”. The New England Journal of Medicine 351 (27): 2875–2878; author 2878 2875–2878. doi:10.1056/NEJM200412303512719.PMID 15625749. edit
- Zhang, J.; Ding, E.; Song, Y. (2006). “Adverse effects of cyclooxygenase 2 inhibitors on renal and arrhythmia events: meta-analysis of randomized trials”. Journal of the American Medical Association 296 (13): 1619–1632. doi:10.1001/jama.296.13.jrv60015. PMID 16968832. edit
- Solomon, S.; McMurray, J.; Pfeffer, M.; Wittes, J.; Fowler, R.; Finn, P.; Anderson, W.; Zauber, A.; Hawk, E.; Bertagnolli, M.; Adenoma Prevention with Celecoxib (APC) Study Investigators (2005). “Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention”. The New England Journal of Medicine 352 (11): 1071–1080.doi:10.1056/NEJMoa050405. PMID 15713944. edit
- Nussmeier, N.; Whelton, A.; Brown, M.; Langford, R.; Hoeft, A.; Parlow, J.; Boyce, S.; Verburg, K. (2005). “Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery”. The New England Journal of Medicine 352 (11): 1081–1091. doi:10.1056/NEJMoa050330. PMID 15713945. edit
- “European Medicines Agency concludes action on COX-2 inhibitors” (pdf). European Medicines Agency. Retrieved 2008-04-16.
- “Merck Announces Voluntary Worldwide Withdrawal of VIOXX” (pdf). Retrieved 2008-04-16.
- “Congress Questions Vioxx, FDA”. PBS NewsHour. 2004-11-18. Retrieved 2013-06-03.
- “SUMMARY: Report of the Expert Advisory Panel on the Safety of Cox-2 Selective Non-steroidal Anti-Inflammatory Drugs (NSAIDs)”. Health Canada. 2005-07-06. Retrieved 2011-06-04.
- Drug unfit for sale, says judge in compo case The Age, March 6, 2010
- http://bioline.utsc.utoronto.ca/archive/00002693/01/dv04120.pdf#search=%22acne%20rofecoxib%22
- http://vioxxlawyer.org/rofecoxib-synthesis/
References
- FDA (2005). “Summary minutes for the February 16, 17 and 18, 2005, Joint meeting of the Arthritis Advisory Committee and the Drug Safety and Risk Management Advisory Committee.” Published on the internet, March 2005. Link
- Fitzgerald GA, Coxibs and Cardiovascular Disease, N Engl J Med 2004;351(17): 1709–1711. PMID 15470192.
- Grassley CE (15 Oct 2004). Grassley questions Merck about communication with the FDA on Vioxx. Press Release.
- Jüni P, Nartey L, Reichenbach S, Sterchi R, Dieppe PA, Egger M (2004). Risk of cardiovascular events and rofecoxib: cumulative meta-analysis. Lancet (published online; see also Merck response below)
- Karha J and Topol EJ. The sad story of Vioxx, and what we should learn from it Cleve Clin J Med 2004; 71(12):933-939. PMID 15641522
- Michaels, D. (June 2005) DOUBT Is Their Product, Scientific American, 292 (6).
- Merck & Co., (5 Nov 2004). Response to Article by Juni et al. Published in The Lancet on Nov. 5. Press Release.
- Merck & Co (30 Sep 2004) Merck Announces Voluntary Worldwide Withdrawal of VIOXX. Press release [7].
- D. M. Mukherjee, S. E. Nissen, and E. J. Topol, “Risk of Cardiovascular Events Associated with Selective COX-2 Inhibitors,” Journal of the American Medical Association 186 (2001): 954–959.
- Nussmeier NA, Whelton AA, Brown MT, Langford RM, Hoeft A, Parlow JL, et al. Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. N Engl J Med 2005;352(11):1081-91. PMID 15713945
- Okie, S (2005) “Raising the safety bar–the FDA’s coxib meeting.” N Engl J Med. 2005 Mar 31;352(13):1283-5. PMID 15800221.
- Leleti Rajender Reddy, Corey EJ. Facile air oxidation of the conjugate base of rofecoxib (Vioxx), a possible contributor to chronic human toxicity Tetrahedron Lett 2005, 46: 927. doi:10.1016/j.tetlet.2004.12.055
- Swan SK et al., Effect of Cyclooxygenase-2 Inhibition on Renal Function in Elderly Persons Receiving a Low-Salt Diet. Annals of Int Med 2000; 133:1–9
- Targum, SL. (1 Feb. 2001) Review of cardiovascular safety database. FDA memorandum. [8]
- Wolfe, MM et al., Gastrointestinal Toxicity of Nonsteroidal Anti-anflamattory Drugs, New England Journal of Medicine. 1999; 340; 1888-98.
External links
- National Public Radio 2004 Q&A on the case, following withdrawal announcement
- Court TV’s full coverage of the Vioxx civil trials
- Merck website on Vioxx litigation
- FDA Public Health Advisory on Vioxx
- David Michaels. Doubt is Their Product Scientific American, June 2004, p. 96-101
- JURIST, Much Pain, Much Gain: Skeptical Ruminations on the Vioxx Litigation
- Ted Frank, American Enterprise Institute, The Vioxx Litigation, Part I and Part II, December 2005
- briandeer.com – Vioxx: the UK connection
- Campaign for compensation for Vioxx victims outside the US
Literature References:
Selective cyclooxygenase-2 (COX-2) inhibitor. Prepn: Y. Ducharme et al., WO 9500501; eidem, US5474995 (both 1995 to Merck Frosst).
HPLC determn in plasma: C. M. Chavez-Eng et al., J. Chromatogr. B 748, 31 (2000).
Enzyme inhibition and clinical evaluation in dental pain: E. W. Ehrich et al., Clin. Pharmacol. Ther. 65, 336 (1999).
Evaluation of risk of gastrointestinal effects in patients with osteoarthritis: M. J. Langman et al., J. Am. Med. Assoc. 282, 1929 (1999); with rheumatoid arthritis: C. Bombardier et al., N. Engl. J. Med. 343, 1520 (2000).
Review of pharmacology and clinical experience: A. J. Matheson, D. P. Figgitt, Drugs 61, 833-865 (2001).
Keywords: Cyclooxygenase-2 Selective Inhibitor, Anti-inflammatory (Nonsteroidal), Analgesic (Non-Narcotic), rofecoxib, MK-0966, Vioxx
ABBREVIATED NEW DRUG APPLICATION (ANDA) by Anthony crasto
DRUG REGULATORY AFFAIRS INTERNATIONAL
ABBREVIATED NEW DRUG APPLICATION (ANDA) by Anthony crasto
NDA, ANDA AND IND by DR ANTHONY CRASTO
DRUG REGULATORY AFFAIRS INTERNATIONAL
NDA, ANDA AND IND by DR ANTHONY CRASTO
……….
Ibudilast

IBUDILAST, MN 166
AV-411
KC-404
MN-166
2-methyl-1-(2-propan-2-ylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one
1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)-2-methylpropan-1-one
KYORIN Kyorin Seiyaku Kk……….INNOVATOR

Ibudilast is an anti-inflammatory and neuroprotective oral agent which shows an excellent safety profile at 60 mg/day and provides significantly prolonged time-to-first relapse and attenuated brain volume shrinkage in patients with relapsing-remitting (RR) and/or secondary progressive (SP) multiple sclerosis (MS). Ibudilast is currently in development in the U.S. (codes: AV-411 or MN-166), but is approved for use as an antiinflammatory in Japan.



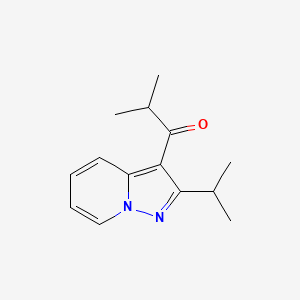
Ibudilast (development codes: AV-411 or MN-166) is an antiinflammatory drug used mainly in Japan, which acts as aphosphodiesterase inhibitor, inhibiting the PDE-4 subtype to the greatest extent,[1] but also showing significant inhibition of other PDE subtypes.[2][3]
Ibudilast has bronchodilator, vasodilator [4] and neuroprotective effects,[5][6] and is mainly used in the treatment of asthma andstroke.[7] It inhibits platelet aggregation,[8] and may also be useful in the treatment of multiple sclerosis.[9]
Ibudilast crosses the blood–brain barrier and suppresses glial cell activation. This activity has been shown to make ibudilast useful in the treatment of neuropathic pain and it not only enhances analgesia produced by opioid drugs, but also reduces the development oftolerance.[10]
It may have some use reducing methamphetamine[11] and alcohol[12] addiction.
It may have some use reducing methamphetamine addiction.[11]
Avigen has identified the potential of ibudilast (AV-411) for the treatment of neuropathic pain and other neurological indications, including opiate withdrawal. As an inhibitor of glial cells, ibudilast can deactivate these cells which produce various chemicals, including proinflammatory cytokines, in response to nerve damage or viral infection to amplify and maintain pain. Preclinical evaluation to date indicates that it reverses the painful sensory abnormality allodynia in chemotherapy- and trauma-induced neuropathic pain models.
Originator Kyorin and Banyu Pharmaceutical (now MSD KK following the merger of Banyu and Schering-Plough KK in 2010) have been developing ibudilast under a collaborative agreement. MediciNova obtained exclusive, worldwide rights outside of Japan, China, Taiwan and South Korea from Kyorin in October 2004 to develop and commercialize the compound for MS. In 2012, a codevelopment agreement was signed between MediciNova and the University of Colorado for the treatment of post-traumatic brain injury.

Sixteenth revised Japanese Pharmacopoeia chemicals, etc. IBUDILAST Ibudilast C14H18N2O: 230.31 [ 50847-11-5 ] that this product was dried when to quantify, including ibudilast (C14H18N2O) 98.5 ~ 101.0%.


http://www.google.co.in/patents/US3850941PATENT
EXAMPLE 1 Synthesis of 2-isopropyl-3-is0butyrylpyrazolo[1,5-a] pyridine (KC404) A mixture of 1-amino-Z-methylpyridinium iodide g.), isobutyric anhydride (500 g.) and K CO (81 g.) was refluxed for 8 hr. After cooling, the precipitated crystals were filtered off and water was added to the filtrate, The solution was made basic to pH 11 with K CO’ and extracted with ethyl acetate (1000 ml.). The extract’was washed with water (400 ml.), dried over Na SO and concentrated under reduced pressure. The residue was distilled to give 58 g. of colorless crystalline product, hp, 110- 175 (7.5 mm. Hg). Recrystallization from hexane gave colorless prisms, melting point 53.554.
Analysis- Calcd.: C, 73.01; H, 7.88; N, 12.17 Found: C, 72.86; H, 7.94; N, 12.09
CLIP

http://www.customsynthesis.com/ibud.html
PATENT
http://www.google.com/patents/US8119657
FIG. 6 is a synthetic reaction scheme illustrating one approach for preparing (S)-AV1013; the approach employs chiral chromatography of an N-protected form of the racemate as described in detail in Example 1.
FIG. 7 demonstrates additional reaction schemes for synthesizing (S)-AV1013.


Example 1Synthesis of (S)-2-amino-1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one hydrochloride
(S)-2-Amino-1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one hydrochloride (also referred to herein as S-AV1013.HCl) was prepared on a preparative scale using two different routes to obtain the intermediate isopropylpyrazolo[1,5-a]pyridine (IPPP). In the first approach (method 1), ibudilast was employed as the starting material to obtain IPPP; an alternate synthetic approach (method 2) employed ibudilast acid as the starting material.
Step 1Method 1Preparation of Isopropylpyrazolo[1,5-a]pyridine (IPPP) from ibudilast

A 5 L 3-neck round-bottom flask was equipped with a mechanical stirrer, thermocouple, heating mantle and a Y-adapter with a nitrogen inlet. The flask was charged with water (350 mL, USP), concentrated sulfuric acid (350 mL) and ibudilast (3-isobutyryl-2-isopropylpyrazolo[1,5-a]pyridine) (140 g, 0.608 mol). The flask was purged with nitrogen, and the mixture was stirred while it was heated to 135° C. An aliquot was removed for HPLC analysis, which showed that all starting material was consumed after 5 hours at 135° C., so the mixture was allowed to cool to room temperature overnight. The mixture was cooled in an ice bath, and water (1400 mL, USP) was added over 10 min, with the temperature maintained below 25° C. With continuous cooling in an ice bath, the mixture was neutralized by adding sodium hydroxide (50% w/w aq., 1150 mL) dropwise, with the temperature maintained below 25° C. Ethyl acetate (250 mL) was added, and the layers were separated. The aqueous layer was washed with ethyl acetate (2×300 mL). The combined ethyl acetate extracts were washed sequentially with 250 mL portions of saturated aqueous sodium bicarbonate and saturated aqueous sodium chloride, then dried over anhydrous sodium sulfate for 30 minutes. Activated carbon (20 g) and silica (60 g) were added and stirred before filtering over a pad of Celite. The filtrate was concentrated under reduced pressure to obtain 96.5 g of IPPP (2-isopropyl-pyrazolo[1,5-a]pyridine, 99% crude yield, 99.6 area % pure by HPLC) as an amber oil.
1H-NMR (CDCl3) δ 1.4 (d, 6H), 3.2 (m, 1H), 6.3 (s, 1H), 6.6 (t, 1H), 7.0 (m, 1H), 7.4 (d, 1H), 8.4 (d, 1H). HPLC: RT=9.1 min (99.6 area %).
CLIP
Ibudilast (3-isobutyryl-2-isopropylpyrazolo[l,5-α]pyridine) is a small molecule drug that has been used for many years in Japan and Korea for the treatment of bronchial asthma as well as for treatment of cerebrovascular disorders such as post-stroke dizziness. It is sold in these countries under the tradename, Ketas®. Marketed indications for ibudilast in Japan include its use as a vasodilator, for treating allergy, eye tissue regeneration, ocular disease, and treatment of allergic ophthalmic disease (Thompson Current Drug Reports). Its use in the treatment of both chronic brain infarction (ClinicalTrials.gov) and multiple sclerosis (News.Medical.Net; Pharmaceutical News, 2 Aug 2005) is currently being explored in separate, ongoing clinical trials.
The mechanisms of action of ibudilast have been widely explored. Its role as a non-selective inhibitor of cyclic nucleotide phosphodiesterase (PDE) has been described
(Fujimoto, T., et al., J. of Neuroimmunology, 95 (1999) 35-92). Additionally, ibudilast has been reported to act as an LTD4 antagonist, an anti-inflammatory, a PAF antagonist, and a vasodilatator agent (Thompson Current Drug Reports). Ibudilast is also thought to exert a neuroprotective role in the central nervous system of mammals, presumably via suppression of the activation of glial cells (Mizuno et al. (2004) Neuropharmacology 46: 404-411). New uses for ibudilast continue to be explored.http://www.google.com/patents/WO2007146087A2?cl=en
PATENT
http://www.google.com/patents/WO2007142924A1?cl=en
IBUDILAST
Ibudilast is a small molecule drug (molecular weight of 230.3) having the structure shown below.

Ibudilast is also found under ChemBank ID 3227, CAS # 50847-1 1-5, and Beilstein Handbook Reference No. 5-24-03-00396. Its molecular formula corresponds to [Ci4HIgN2O]. Ibudilast is also known by various chemical names which include 2- methyl-l-(2-(l-methylethyI)pyrazolo(l,5-a)pyridin-3-yl)l-propanone; 3-isobutyryl-2- isopropylpyrazolo(l,5-a)pyridine]; and l-(2-isopropyl-pyrazolo[l,5-a]pyridin-3-yl)-2- methyl-propan-1-one. Other synonyms for ibudilast include Ibudilastum (Latin), BRN 0656579, KC-404, and the brand name Ketas®. Ibudilast, as referred to herein, is meant to include any and all pharmaceutically acceptable salt forms thereof, prodrug forms (e.g., the corresponding ketal), and the like, as appropriate for use in its intended formulation for administration.
Ibudilast is a non-selective nucleotide phosphodiesterase (PDE) inhibitor (most active against PDE-3 and PDE-4), and has also been reported to have LTD4 and PAF antagonistic activities. Its profile appears effectively anti-inflammatory and unique in comparison to other PDE inhibitors and anti-inflammatory agents. PDEs catalyze the hydrolysis of the phosphoester bond on the 3 ‘-carbon to yield the corresponding 5′- nucleotide monophosphate. Thus, they regulate the cellular concentrations of cyclic nucleotides. Since extracellular receptors for many hormones and neurotransmitters utilize cyclic nucleotides as second messengers, the PDEs also regulate cellular responses to these extracellular signals. There are at least eight classes of PDEs: Ca2+/calmodul in-dependent PDEs (PDEl); cGMP-stimulated PDEs (PDE2); cGMP- inhibited PDEs (PDE3); cAMP-specific PDEs (PDE4); cGMP-binding PDEs (PDE5); photoreceptor PDEs (PDE6); high affinity, cAMP-specific PDEs (PDE7); and high affinity cGMP-specific PDEs (PDE9).
SYNTHESIS

DE 2315801; FR 2182914; JP 7714799, WO 0196278
By condensation of 1-amino-2-methylpyridinium iodide (I) with isobutyric anhydride (II) by means of K2CO3 at reflux temperature.
Patent
PATENT
2-methyl -l- [2- (l- methylethyl) – pyrazolo [l, 5_a] pyrimidine _3_ yl] _1_ acetone (ibudilast, generic drug name: IBUDILAST ) is an anti-allergic asthma drugs, anti-leukotrienes can twist and platelet-activating factor, promote the secretion of mucus in the respiratory tract, respiratory cilia function, enhance the role of prostacyclin, increase cerebral blood flow, improve brain metabolism. For the treatment of bronchial asthma, sequelae of cerebral embolism, cerebral arteriosclerosis.
ibudilast preparation methods are mainly the following two:
Method a: (The Jourrtal of Organic Chemistry, 1968, 33, 3766 ~3770) Synthesis Road
Lines are as follows:

The route to 2-picoline as starting material to give amino-2-methyl-pyridine iodide I-, after pyrimidine, the role of isobutyryl chloride to give the title compound. The final product obtained by this route need be purified by column chromatography, thereby increasing the difficulty of the operation, in addition to column chromatography, eluent used larger benzene toxicity, is not suitable for industrial production.
Method II: (Journal of the American Chemical Society, 2005,127, 751-760) co
A route is as follows:

The route to 2-picoline as starting material to obtain the sulfamic acid, potassium iodide I- amino-2-picoline under the action of potassium carbonate, then with isobutyric anhydride to give the title compound effect. This route of the first-stage reaction process locked, the yield is low, is not suitable for industrial production.
so there ibudilast conventional method for preparing the operational difficulties or low yield, making it impossible to achieve industrial production problems.
DETAILED DESCRIPTION IX: with a specific embodiment of the present embodiment is one of one to eight different points: in the second step of the recrystallization specific operation is as follows: First, the collected fractions was cooled to 10 ° c~25 ° C, to give a pale yellow solid, and then n-hexane was added to the pale yellow solid, and the temperature was raised to 50 ° C~68 ° C, at a temperature of 50 ° C~68 ° C incubation 5min~IOmin, then cooled to 10 ° C~ 25 ° C, and at a temperature of 10 ° C~25 ° C incubated O. 5h~Ih, and finally filtered to obtain ibudilast; the volume of the pale yellow solid quality and hexane ratio of Ig: (ImL~2mL), to obtain ibudilast.
PAPER
Bioorganic & medicinal chemistry letters (2011), 21(11), 3307-12.
Ibudilast [1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)-2-methylpropan-1-one] is a nonselective phosphodiesterase inhibitor used clinically to treat asthma. Efforts to selectively develop the PDE3- and PDE4-inhibitory activity of ibudilast led to replacement of the isopropyl ketone by a pyridazinone heterocycle. Structure–activity relationship exploration in the resulting 6-(pyrazolo[1,5-a]pyridin-3-yl)pyridazin-3(2H)-ones revealed that the pyridazinone lactam functionality is a critical determinant for PDE3-inhibitory activity, with the nitrogen preferably unsubstituted. PDE4 inhibition is strongly promoted by introduction of a hydrophobic substituent at the pyridazinone N(2) centre and a methoxy group at C-7′ in the pyrazolopyridine. Migration of the pyridazinone ring connection from the pyrazolopyridine 3′-centre to C-4′ strongly enhances PDE4 inhibition. These studies establish a basis for development of potent PDE4-selective and dual PDE3/4-selective inhibitors derived from ibudilast.

UPDATE AS ON JAN 2016
…………..MediciNova’s ibudilast gets FDA rare paediatric disease status to treat Krabbe disease
MediciNova has received rare paediatric disease status from the US Food and Drug Administration (FDA) for its MN-166 (ibudilast) to treat Type 1 Early Infantile Krabbe disease.
![]()
References
- Huang Z, Liu S, Zhang L, Salem M, Greig GM, Chan CC, Natsumeda Y, Noguchi K. Preferential inhibition of human phosphodiesterase 4 by ibudilast. Life Sciences. 2006 May 1;78(23):2663-8.
- Suzumura A, Ito A, Yoshikawa M, Sawada M. Ibudilast suppresses TNFalpha production by glial cells functioning mainly as type III phosphodiesterase inhibitor in the CNS. Brain Research. 1999 Aug 7;837(1-2):203-12.
- Gibson LC, Hastings SF, McPhee I, Clayton RA, Darroch CE, Mackenzie A, Mackenzie FL, Nagasawa M, Stevens PA, Mackenzie SJ. The inhibitory profile of Ibudilast against the human phosphodiesterase enzyme family. European Journal of Pharmacology. 2006 May 24;538(1-3):39-42.
- Kishi Y, Ohta S, Kasuya N, Sakita S, Ashikaga T, Isobe M. Ibudilast: a non-selective PDE inhibitor with multiple actions on blood cells and the vascular wall. Cardiovascular Drug Reviews. 2001 Fall;19(3):215-25.
- Mizuno T, Kurotani T, Komatsu Y, Kawanokuchi J, Kato H, Mitsuma N, Suzumura A. Neuroprotective role of phosphodiesterase inhibitor ibudilast on neuronal cell death induced by activated microglia. Neuropharmacology. 2004 Mar;46(3):404-11.
- Yoshioka M, Suda N, Mori K, Ueno K, Itoh Y, Togashi H, Matsumoto M. Effects of ibudilast on hippocampal long-term potentiation and passive avoidance responses in rats with transient cerebral ischemia. Pharmacological Research. 2002 Apr;45(4):305-11.
- Wakita H, Tomimoto H, Akiguchi I, Lin JX, Ihara M, Ohtani R, Shibata M. Ibudilast, a phosphodiesterase inhibitor, protects against white matter damage under chronic cerebral hypoperfusion in the rat. Brain Research. 2003 Nov 28;992(1):53-9.
- Rile G, Yatomi Y, Qi R, Satoh K, Ozaki Y. Potentiation of ibudilast inhibition of platelet aggregation in the presence of endothelial cells. Thrombosis Research. 2001 May 1;102(3):239-46.
- Feng J, Misu T, Fujihara K, Sakoda S, Nakatsuji Y, Fukaura H, Kikuchi S, Tashiro K, Suzumura A, Ishii N, Sugamura K, Nakashima I, Itoyama Y. Ibudilast, a nonselective phosphodiesterase inhibitor, regulates Th1/Th2 balance and NKT cell subset in multiple sclerosis. Multiple Sclerosis. 2004 Oct;10(5):494-8.
- Ledeboer A, Hutchinson MR, Watkins LR, Johnson KW. Ibudilast (AV-411). A new class therapeutic candidate for neuropathic pain and opioid withdrawal syndromes. Expert Opinion on Investigational Drugs. 2007 Jul;16(7):935-50.
- http://www.huffingtonpost.com/2013/04/03/meth-addiction-cure-ucla-ibudilast_n_2863126.html?utm_hp_ref=mostpopular#slide=more268305
- http://onlinelibrary.wiley.com/doi/10.1111/adb.12106/abstrac
SEE……Synthesis technology of ibudilast
Shandong Huagong (2014), 43, (8), 29-30. Publisher: (Shandong Huagong Bianjibu, ) CODEN:SHHUA4 ISSN:1008-021X.
Literature References:
Leukotriene D4 antagonist. Prepn: T. Irikura et al., DE 2315801; eidem, US 3850941 (1973, 1974 both to Kyorin).
Pharmacology and antiallergic activity: K. Nishino et al., Jpn. J. Pharmacol. 33, 267 (1983); H. Nagai et al., ibid. 1215.
In vitro cerebral vasodilating activity: M. Ohashi et al., Arch. Int. Pharmacodyn. 280, 216 (1986);
in vivo activity: W. M. Armstead et al., J. Pharmacol. Exp. Ther. 244, 138 (1988).
Bronchodilating activity in animals: S. Mue et al., Arch. Int. Pharmacodyn. 283,153 (1986).
Antiplatelet activity in animals: M. Ohashi et al., ibid. 321; M. Ohashi et al., Gen. Pharmacol. 17, 385 (1986).

| Patent | Submitted | Granted |
|---|---|---|
| Method for treating neuropathic pain and associated syndromes [US7534806] | 2006-07-20 | 2009-05-19 |
| Synergistic combination [US2006205806] | 2006-09-14 | |
| Pharmaceutical composition of a pde4 or pde 3/4 inhibitor and histamine receptor antagonist [US2005112069] | 2005-05-26 | |
| Methods and reagents for the treatment of immunoinflammatory disorders [US2005119160] | 2005-06-02 | |
| Methods of inducing ovulation using a non-polypeptide camp level modulator [US7507707] | 2005-07-07 | 2009-03-24 |
| Methods and reagents for the treatment of immunoinflammatory disorders [US2005192261] | 2005-09-01 | |
| Synergistic combination [US7056936] | 2004-02-19 | 2006-06-06 |
| Methods for the treatment of respiratory diseases and conditions with a selective iNOS inhibitor and a PDE inhibitor and compositions therefor [US2004087653] | 2004-05-06 | |
| Remedies for multiple sclerosis [US6395747] | 2002-05-28 | |
| Combination [US2005014762] | 2005-01-20 |
| Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US4097483 | Aug 31, 1976 | Jun 27, 1978 | Kyorin Pharmaceutical Co., Ltd. | Pyrazolo 1,5-a!pyridines |
| US7585875 * | Jun 6, 2007 | Sep 8, 2009 | Avigen, Inc. | Phosphodiesterase inhibitors; neuropathic pain, inflammation, opioid dependence or withdrawal; 2-amino-1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one for example |
| US20070015924 | Jun 15, 2006 | Jan 18, 2007 | Cardiome Pharma Corp. | stereoselective preparation of aminocyclohexyl ether compounds such as trans-(1R,2R)-aminocyclohexyl ether compounds and/or trans-(1S,2S)-aminocyclohexyl ether compounds; useful in treating arrhythmias |
| US20080070912 | Jun 6, 2007 | Mar 20, 2008 | Avigen, Inc. | Phosphodiesterase inhibitors; neuropathic pain, inflammation, opioid dependence or withdrawal; 2-amino-1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one for example |
| US20090062330 | Jul 8, 2008 | Mar 5, 2009 | Medicinova, Inc. | Treatment of progressive neurodegenerative disease with ibudilast |
| US20090318437 * | Jun 10, 2009 | Dec 24, 2009 | Gaeta Federico C A | SUBSTITUTED PYRAZOLO[1,5-a] PYRIDINE COMPOUNDS AND THEIR METHODS OF USE |
| WO2007142924A1 | May 29, 2007 | Dec 13, 2007 | Avigen Inc | Ibudilast for inhibiting macrophage migration inhibitory factor (mif) activity |
| WO2007146087A2 * | Jun 6, 2007 | Dec 21, 2007 | Avigen Inc | SUBSTITUTED PYRAZOLO [1,5-α] PYRIDINE COMPOUNDS AND THEIR METHODS OF USE |
| WO2010151551A1 * | Jun 22, 2010 | Dec 29, 2010 | Medicinova, Inc. | ENANTIOMERIC COMPOSITIONS OF 2-AMINO-1-(2-ISOPROPYLPYRAZOLO[1,5-a]PYRIDIN-3-YL)PROPAN-1-ONE AND RELATED METHODS |
| US8119657 | Jun 22, 2010 | Feb 21, 2012 | Medicinova, Inc. | Enantiomeric compositions of 2-amino-1-(2-isopropylpyrazolo[1,5-α]pyridin-3-yl)propan-1-one and related methods |
| Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2003104178A1 * | Jun 6, 2003 | Dec 18, 2003 | Cortical Pty Ltd | Napththalene derivatives which inhibit the cytokine or biological activity of macrophage migration inhibitory factor (mif) |
| WO2003104203A1 * | Jun 6, 2003 | Dec 18, 2003 | Cortical Pty Ltd | Therapeutic molecules and methods-1 |
| WO2004058713A1 * | Dec 18, 2003 | Jul 15, 2004 | Jason Chyba | Differential tumor cytotoxocity compounds and compositions |
| WO2005058304A1 * | Dec 17, 2004 | Jun 30, 2005 | Cortical Pty Ltd | Implantable device containing inhibitor of macrophage migration inhibitory factor |
| WO2006045505A1 * | Oct 19, 2005 | May 4, 2006 | Novartis Ag | Mif-inhibitors |
| WO2006108671A1 * | Apr 13, 2006 | Oct 19, 2006 | Novartis Ag | 3,4-dihydro-benzo[e][1,3]oxazin-2-ones |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-methyl-1-(2-propan-2-ylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one
|
|
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
| Identifiers | |
| CAS Number | 50847-11-5 |
| ATC code | R03DC04 |
| PubChem | CID: 3671 |
| IUPHAR/BPS | 7399 |
| DrugBank | DB05266 |
| ChemSpider | 3543 |
| UNII | M0TTH61XC5 |
| KEGG | D01385 |
| ChEMBL | CHEMBL19449 |
| Chemical data | |
| Formula | C14H18N2O |
| Molecular mass | 230.31 g/mol |
Keywords: Antiallergic; Antiasthmatic (Nonbronchodilator); Leukotriene Antagonist; Vasodilator (Cerebral).

SEE………http://apisynthesisint.blogspot.in/2016/01/ibudilast.html
////////////////////
CC(C)C1=NN2C=CC=CC2=C1C(=O)C(C)C
