
Home » PROCESS (Page 4)
Category Archives: PROCESS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Minisci reactions: Versatile CH-functionalizations for medicinal chemists
Minisci reactions: Versatile CH-functionalizations for medicinal chemists
Matthew A. J. Duncton† *
Renovis, Inc. (a wholly-owned subsidiary of Evotec AG), Two Corporate Drive, South San Francisco, CA 94080, United States. E-mail: mattduncton@yahoo.com; Tel: +1 917-345-3183
First published on the web 22nd August 2011
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
The addition of a radical to a heteroaromatic base is commonly referred to as a Minsici reaction. Such reactions constitute a broad-set of selective CH-functionalization processes. This review describes some of the major applications of Minisci reactions and related processes to medicinal or biological chemistry, and highlights some potential developments within this area.
Introduction
The aim of this review is to summarize the use of Minisci reactions within medicinal chemistry, and to highlight some future opportunities to continue progression of this chemistry. As such, it is not an aim that detailed mechanistic information, or a comprehensive list of examples be described. For this, the reader is directed to excellent articles from Minisci, Harrowven and Bowman.1–3 Rather, the review is written to show that Minisci reactions are extremely valuable CH-functionalization processes within medicinal chemistry. However, their use has been somewhat under-utilized when compared with other well-known selective transformations (e.g. palladium-catalysed cross-couplings). Therefore, it is hoped that in the future, Minisci chemistry will continue to develop, such that the reactions become a staple-set of methods for medicinal and biological chemists alike.
To aid discussion, the review is divided in to several sections. First, some historical perspective is given. This is followed by a discussion of scope and limitations. The main-body of the review describes some specific examples of Minisci reactions and related processes, with a focus on their use within medicinal, or biological chemistry. Finally, brief mention is given to potential future applications, some of which may be beneficial in providing ‘high-content’ diverse libraries for screening.
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
http://pubs.rsc.org/en/content/articlehtml/2011/md/c1md00134e
…………………….
WIKI
The Minisci reaction is a named reaction in organic chemistry. It is a radical substitution to an aromatic compound, in particular to a heteroaromatic base, that introduces an alkyl group. The reaction was published about in 1971 by F. Minisci.[1] The aromatic compound is generally electron-deficient and with N-aromatic compounds the nitrogen atom is protonated.[2] A typical reaction is that between pyridine and pivalic acid to 2-tert-butylpyridine with silver nitrate, sulfuric acid and ammonium persulfate. The reaction resembles Friedel-Crafts alkylation but with opposite reactivity and selectivity.[3]
The Minisci reaction proceeds regioselectively and enables the introduction of a wide range of alkyl groups.[4] A side-reaction is acylation.[5] The ratio between alkylation and acylation depends on the substrate and the reaction conditions. Due to the simple raw materials and the simple reaction conditions the reaction has many applications in heterocyclic chemistry.[6][7]

Mechanism
A free radical is formed from the carboxylic acid in an oxidative decarboxylation with silver salts and an oxidizing agent. The oxidizing agent reoxidizes the silver salt. The radical then reacts with the aromatic compound. The ultimate product is formed by rearomatisation. The acylated product is formed from the acyl radical.[4][5]

References
- F. Minisci, R. Bernardi, F. Bertini, R. Galli, M. Perchinummo: Nucleophilic character of alkyl radicals—VI : A new convenient selective alkylation of heteroaromatic bases, in: Tetrahedron 1971, 27, 3575–3579.
- Minisci reaction Jie Jack Li in Name Reactions 2009, 361-362, doi:10.1007/978-3-642-01053-8_163
- Strategic applications of named reactions in organic synthesis: background and detailed mechanisms László Kürti, Barbara Czakó 2005
- F. Fontana, F. Minisci, M. C. N. Barbosa, E. Vismara: Homolytic acylation of protonated pyridines and pyrazines with α-keto acids: the problem of monoacylation, in: J. Org. Chem. 1991, 56, 2866–2869; doi:10.1021/jo00008a050.
- M.-L. Bennasar, T. Roca, R. Griera, J. Bosch: Generation and Intermolecular Reactions of 2-Indolylacyl Radicals, in: Org. Lett. 2001, 3, 1697–1700; doi:10.1021/ol0100576.
- P. B. Palde, B. R. McNaughton, N. T. Ross, P. C. Gareiss, C. R. Mace, R. C. Spitale, B. L. Miller: Single-Step Synthesis of Functional Organic Receptors via a Tridirectional Minisci Reaction, in: Synthesis 2007, 15, 2287–2290; doi:10.1055/s-2007-983792.
- J. A. Joules, K. Mills: Heterocyclic Chemistry, 5. Auflage, S. 125–141, Blackwell Publishing, Chichester, 2010, ISBN 978-1-4051-9365-8.
New e-book: Case Studies in Sample Storage
New e-book: Case Studies in Sample Storage
Learn how lab professionals solved their sample storage problems at leading research organizations. Case studies include adapting sample storage for changing demands in compound management and incorporating sample libraries from acquired companies.
DOWNLOAD E-BOOK
Latin American Active Pharmaceutical Ingredients Industry Catches Up on the US

Latin American Active Pharmaceutical Ingredients Industry Catches Up on the US
The market for active pharmaceutical ingredient in the Americas shows a clear north–south divide: 88% percent for the US and Canada, the rest for South and Central American companies. but as the economy in Latin America booms and prosperity growths, these markets are just about to catch up on the US new figures indicate….http://www.process-worldwide.com/management/markets_industries/articles/374702/
DRUG PROCESS CHEMISTRY

Process chemistry is the arm of pharmaceutical chemistry concerned with the development and optimization of a synthetic scheme and pilot plant procedure to manufacture compounds for the drug development phase. Process chemistry is distinguished from medicinal chemistry, which is the arm of pharmaceutical chemistry tasked with designing and synthesizing molecules on small scale in the early drug discovery phase.

Medicinal chemists are largely concerned with synthesizing a large number of compounds as quickly as possible from easily tunable chemical building blocks (usually for SAR studies). In general, the repertoire of reactions utilized in discovery chemistry is somewhat narrow (for example, the Buchwald-Hartwig amination, Suzuki coupling and reductive amination are commonplace reactions).[1] In contrast, process chemists are tasked with identifying a chemical process that is safe, cost and labor efficient, “green,” and reproducible, among other considerations.
Oftentimes, in searching for the shortest, most efficient synthetic route, process chemists must devise creative synthetic solutions that eliminate costly functional group manipulations and oxidation/reduction steps.
This article will focus exclusively on the chemical and manufacturing processes associated with the production of small molecule drugs. Biological medical products (more commonly called “biologics”) represent a growing proportion of approved therapies, but the manufacturing processes of these products are beyond the scope of this article.
Additionally, the many complex factors associated with chemical plant engineering (for example, heat transfer and reactor design) and drug formulation will be treated cursorily.

Process Chemistry Considerations
Cost efficiency is of paramount importance in process chemistry and, consequently, is a focus in the consideration of pilot plant synthetic routes. The drug substance that is manufactured, prior to formulation, is commonly referred to as the active pharmaceutical ingradient (API) and will be referred to as such herein.
API production cost can be broken into two components: the “material cost” and the “conversion cost.”[2] The ecological and environmental impact of a synthetic process should also be evaluated by an appropriate metric (e.g. the EcoScale).
An ideal process chemical route will score well in each of these metrics, but inevitably tradeoffs are to be expected. Most large pharmaceutical process chemistry and manufacturing divisions have devised weighted quantitative schemes to measure the overall attractiveness of a given synthetic route over another. As cost is a major driver, material cost and volume-time output are typically weighted heavily.
The chemical and processing industries (CPI) provide the building blocks for many products. By using large amounts of heat and energy to physically or chemically transform materials, these industries help meet the world’s most fundamental needs for food, shelter and health, as well as products that are vital to such advanced technologies as computing, telecommunications and biotechnology.
These industries face major challenges to meet the needs of the present without compromising the needs of the future generations in the face of increasing industrial competitiveness. This translates into the need to make processes much more energy efficient, safer and more flexible, and to reduce emissions to meet the many competitive challenges within a global economy.
The chemical and processing industries refer to processes where materials undergo chemical conversion during their production into finished products, as well as – or instead of – the physical conversions common to industry in general.
In the chemical process industry the products differ chemically from the raw materials as a result of undergoing one or more chemical reactions during the manufacturing process.
The chemical process industries broadly include the traditional chemical industries, both organic and inorganic; the petroleum industry; the petrochemical industry, which produces the majority of plastics, synthetic fibers, and synthetic rubber from petroleum and natural-gas raw materials; and a series of allied industries in which chemical processing plays a substantial part.
While the chemical process industries are primarily the realm of the chemical engineer and the chemist, they also involve a wide range of other scientific, engineering, and economic specialists.

Material Cost
The material cost of a chemical process is the sum of the costs of all raw materials, intermediates, reagents, solvents and catalysts procured from external vendors. Material costs may influence the selection of one synthetic route over another or the decision to outsource production of an intermediate.
Conversion Cost
The conversion cost of a chemical process is a factor of that procedure’s overall efficiency, both in materials and time, and its reproducibility. The efficiency of a chemical process can be quantified by its atom economy, yield, volume-time output, and environmental factor (E-factor), and its reproducibility can be evaluated by the Quality Service Level (QSL) and Process Excellence Index (PEI) metrics.

An illustrative example of atom economy using the Claisen rearrangement and Wittig reaction.
Atom Economy
The atom economy of a reaction is defined as the number of atoms from the starting materials that are incorporated into the final product. Atom economy can be viewed as an indicator of the “efficiency” of a given synthetic route.[3]

For example, the Claisen rearrangement and the Diels-Alder cycloaddition are examples of reaction that are 100 percent atom economical. On the other hand, a prototypical Wittig reaction has especially poor atom economy (merely 20 percent in the example shown).
Process synthetic routes should be designed such that atom economy is maximized for the entire synthetic scheme. Consequently, “costly” reagents such as protecting groups and high molecular weight leaving groups should be avoided where possible. An atom economy value in the range of 70 to 90 percent for an API synthesis is ideal, but it may be impractical or impossible to access certain complex targets within this range. Nevertheless, atom economy is a good metric to compare two routes to the same molecule.
Yield
Yield is defined as the amount of product obtained in a chemical reaction. According to Vogel’s Textbook of Practical Organic Chemistry, yields around 100% are called quantitative, yields above 90% are excellent, yields above 80% are very good, yields above 70% are good, yields above 50% are fair, and yields below 40% are poor. The yield that has practical significance in a process chemistry setting is the isolated yield, referring to the yield of the isolated product after all extraction and purification steps. In a final API synthesis, isolated yields of 80 percent or above for each synthetic step are expected.

An illustrative example of convergent synthesis.
There are several strategies that are employed in the design of a process route to ensure adequate overall yield of the pharmaceutical product. The first is the concept of convergent synthesis. Assuming a very good to excellent yield in each synthetic step, the overall yield of a multistep reaction can be maximized by combining several key intermediates at a late stage that are prepared independently from each other.
Another strategy to maximize isolated yield (as well as time efficiency) is the concept of telescoping synthesis (also called one-pot synthesis). This approach describes the process of eliminating workup and purification steps from a reaction sequence, typically by simply adding reagents sequentially to a reactor. In this way, unnecessary losses from these steps can be avoided.
Finally, to minimize overall cost, synthetic steps involving expensive reagents, solvents or catalysts should be designed into the process route as late stage as possible, to minimize the amount of reagent used.
In a pilot plant or manufacturing plant setting, yield can have a profound effect on the material cost of an API synthesis, so the careful planning of a robust route and the fine-tuning of reaction conditions are crucially important. After a synthetic route has been selected, process chemists will subject each step to exhaustive optimization in order to maximize overall yield. Low yields are typically indicative of unwanted side product formation, which can raise red flags in the regulatory process as well as pose challenges for reactor cleaning operations.
Volume-Time Output
The volume-time output (VTO) of a chemical process represents the cost of occupancy of a chemical reactor for a particular process or API synthesis. For example, a high VTO indicates that a particular synthetic step is costly in terms of “reactor hours” used for a given output. Mathematically, the VTO for a particular process is calculated by the total volume of all reactors (m3) that are occupied times the hours per batch divided by the output for that batch of API or intermediate (measured in kg).
![VTO=\frac{\text{nominal volume of all reactors} [m^3]*\text{time per batch} [h]}{\text{output per step} [kg]}](https://i0.wp.com/upload.wikimedia.org/math/7/9/a/79aca4fd95405c01c04df9181fb3ee63.png)
The process chemistry group at Boehringer-Ingelheim, for example, targets a VTO of less than 1 for any given synthetic step or chemical process.
Additionally, the raw conversion cost of an API synthesis (in dollars per batch) can be calculated from the VTO, given the operating cost and usable capacity of a particular reactor. Oftentimes, for large-volume APIs, it is economical to build a dedicated production plant rather than to use space in general pilot plants or manufacturing plants.
Environmental Factor (E-factor) and Process Mass Intensity (PMI)
Both of these measures, which capture the environmental impact of a synthetic reaction, intend to capture the significant and rising cost of waste disposal in the manufacturing process. The E-factor for an entire API process is computed by the ratio of the total mass of waste generated in the synthetic scheme to the mass of product isolated.

A similar measure, the process mass intensity (PMI) calculates the ratio of the total mass of materials to the mass of the isolated product.

For both metrics, all materials used in all synthetic steps, including reaction and workup solvents, reagents and catalysts, are counted, even if solvents or catalysts are recycled in practice. Inconsistencies in E-factor or PMI computations may arise when choosing to consider the waste associated with the synthesis of outsourced intermediates or common reagents. Additionally, the environmental impact of the generated waste is ignored in this calculation; therefore, the environmental quotient (EQ) metric was devised, which multiplies the E-factor by an “unfriendliness quotient” associated with various waste streams. A reasonable target for the E-factor or PMI of a single synthetic step is any value between 10 and 40.
Quality Service Level (QSL)
The final two “conversion cost” considerations involve the reproducibility of a given reaction or API synthesis route. The quality service level (QSL) is a measure of the reproducibility of the quality of the isolated intermediate or final API. While the details of computing this value are slightly nuanced and unimportant for the purposes of this article, in essence, the calculation involves the ratio of satisfactory quality batches to the total number of batches. A reasonable QSL target is 98 to 100 percent.
Process Excellence Index (PEI)
Like the QSL, the process excellence index (PEI) is a measure of process reproducibility. Here, however, the robustness of the procedure is evaluated in terms of yield and cycle time of various operations. The PEI yield is defined as follows:

In practice, if a process is high-yielding and has a narrow distribution of yield outcomes, then the PEI should be very high. Processes that are not easily reproducible may have a higher aspiration level yield and a lower average yield, lowering the PEI yield.
Similarly, a PEI cycle time may be defined as follows:

For this expression, the terms are inverted to reflect the desirability of shorter cycle times (as opposed to higher yields). The reproducibility of cycle times for critical processes such as reaction, centrifugation or drying may be critical if these operations are rate-limiting in the manufacturing plant setting. For example, if an isolation step is particularly difficult or slow, it could become the bottleneck for an API synthesis, in which case the reproducibility and optimization of that operation become critical.
For an API manufacturing process, all PEI metrics (yield and cycle times) should be targeted at 98 to 100 percent.
EcoScale
In 2006, Van Aken, et al.[4] developed a quantitative framework to evaluate the safety and ecological impact of a chemical process, as well as minor weighting of practical and economical considerations. Others have modified this EcoScale by adding, subtracting and adjusting the weighting of various metrics. Among other factors, the EcoScale takes into account the toxicity, flammability and explosive stability of reagents used, any nonstandard or potentially hazardous reaction conditions (for example, elevated pressure or inert atmosphere), and reaction temperature. Some EcoScale criteria are redundant with previously considered criteria (e.g. E-factor).
Synthetic Case Studies
Boehringer Ingelheim HCV Protease Inhibitor (BI 201302)
Macrocyclization is a recurrent challenge for process chemists, and large pharmaceutical companies have necessarily developed creative strategies to overcome these inherent limitations. An interesting case study in this area involves the development of novel NS3 protease inhibitors to treat Hepatitis C patients by scientists at Boehringer-Ingelheim.[5] The process chemistry team at BI was tasked with developing a cheaper and more efficient route to the active NS3 inhibitor BI 201302, a close analog of BILN 2061. Two significant shortcomings were immediately identified with the initial scale-up route to BILN 2061, depicted in the scheme below.[6] The macrocyclization step posed four challenges inherent to the cross-metathesis reaction.
- High dilution is typically necessary to prevent unwanted dimerization and oligomerization of the diene starting material. In a pilot plant setting, however, a high dilution factor translates into lower throughput, higher solvent costs and higher waste costs.
- High catalyst loading was found to be necessary to drive the RCM reaction to completion. Because of high licencing costs of the ruthenium catalyst that was used (1st generation Hoveyda catalyst), a high catalyst loading was financially prohibitive. Recycling of the catalyst was explored, but proved impractical.
- Long reaction times were necessary for reaction completion, due to slow kinetics of the reaction using the selected catalyst. It was hypothesized that this limitation could be overcome using a more active catalyst. However, while the second-generation Hoveyda and Grubbs catalysts were kinetically more active than the first-generation catalyst, reactions using these catalysts formed large amounts of dimeric and oligomeric products.
- An epimerization risk under the cross-methathesis reaction conditions. The process chemistry group at Boehringer-Ingelheim performed extensive mecahnistic studies showing that epimerization most likely occurs through a ruthenacyclopentene intermediate.[7] Furthermore, the Hoveyda catalyst employed in this scheme minimizes epimerization risk compared with the alalogous Grubbs catalyst.
Additionally, the final double SN2 sequence to install the quinoline heterocycle was identified as a secondary inefficiency in the synthetic route.

Analysis of the cross-methathesis reaction revealed that the conformation of the acyclic precursor had a profound impact on the formation of dimers and oligomers in the reaction mixture. By installing a Boc protecting group at the C-4 amide nitrogen, the Boehringer-Ingelheim chemists were able to shift the site of initiation from the vinylcyclopropane moiety to the nonenoic acid moiety, improving the rate of the intramolecular reaction and decreasing the risk of epimerization. Additionally, the catalyst employed was switched from the expensive 1st generation Hoveyda catalyst to the more reactiveless expensive Grela catalyst.[8] These modifications allowed the process chemists to run the reaction at a standard reaction dilution of 0.1-0.2 M, given that the rates of competing dimerization and oligomerization reactions was so dramatically reduced.
Additionally, the process chemistry team envisioned a SNAr strategy to install the quinoline heterocycle, instead of the SN2 strategy that they had employed for the synthesis of BILN 2061. This modification prevented the need for inefficient double inversion by proceeding through retention of stereochemistry at the C-4 position of the hydroxyproline moiety.[9]

It is interesting to examine this case study from a VTO perspective. For the unoptimized cross-metathesis reaction using the Grela catalyst at 0.01 M diene, the reaction yield was determined to be 82 percent after a reaction and workup time of 48 hours. A 6-cubic meter reactor filled to 80% capacity afforded 35 kg of desired product. For the unoptimized reaction:
This VTO value was considered prohibitively high and a steep investment in a dedicated plant would have been necessary even before launching Phase III trials with this API, given its large projected annual demand. But after reaction development and optimization, the process team was able to improve the reaction yield to 93 percent after just 1 hour (plus 12 hours for workup and reactor cleaning time) at a diene concentration of 0.2 M. With these modifications, a 6-cubic meter reactor filled to 80% capacity afforded 799 kg of desired product. For this optimized reaction:
Thus, after optimization, this synthetic step became less costly in terms of equipment and time and more practical to perform in a standard manufacturing facility, eliminating the need for a costly investment in a new dedicated plant.
Simvastatin, originally developed by Merck, is the most frequently prescribed statin today, with more nearly 100 million prescriptions filled in 2010, according to IMS Health. The traditional synthesis of the drug entailed a multi-step chemical process starting from Lovastatin. The chemical process was using large amounts of hazardous reagents as well as large quantities of solvents.
Professor Yi Tang, at UCLA conceived an initial synthesis that used an engineered enzyme. Codexis Inc. licensed the intellectual property from UCLA, optimized the initial enzyme and developed the new process for commercial use as shown in Figure 1. Following the quantitative hydrolysis of lovastatin to monacolin J acid, Codexis developed a novel, non-natural acyl donor enzyme to regioselectively acylate the C8 position and effect cyclization to simvastatin. This mild bioenzymatic process reduces the 4 steps chemical synthesis to only two steps. The Codexis process is significantly more efficient, cost effective and environmentally friendly.

This is the reaction scheme for producing the drug Simvastatin. The process was an award-winner at last month’s Green Chemistry Challenge Awards held by the Environmental Protection Agency
“We started working on Simvastatin in 2008 and completed the planning process in 2010,” Huisman said. “Then, we started the commercialization process, which takes time because you need regulatory approval of the new process we were working on. We licensed some technology from Yi Tang and UCLA and were then able to continue.”
Codexis took the three-step process used to make Simvastatin and cut out two of the steps, Huisman said.
“From the starting material, it (Simvastatin) has three reactive groups, or hydroxy groups, and what we need to do is convert two of the three groups,” Huisman explained. “We took out a protective step and a de-protective step. We took out two of the steps, and it was intense chemical processing. We then were able to accomplish everything in one step. We also circumvented the use of several nasty chemicals, as well.”
By cutting out two steps, “the overall yield goes up tremendously, about 35 percent,” Huisman added. “And we’re generating 25 times less waste than we did in the old process.”
Huisman said the new process doesn’t change the drug’s effects at all, and that scientists have been trying to do this type of work on commercial drugs for decades.
“In order for this to be a commercial process, the enzyme needs to be improved,” he said. “We needed to speed up the enzyme 1,000-fold to make this process workable; it took a team of scientists about nine months to optimize the enzymes and speed it up.”

Codexis 2 step enzymatic process versus the 4 step chemical synthesis
AZIDES
A popular procedure for making 5-substituted tetrazoles is the reaction of sodium azide with a nitrile, often in the presence of an ammonium salt. The example shown below is from Organic Syntheses (Novartis Process R&D and Ley’s group at Cambridge), providing the useful enantiocatalyst shown on an 80 mmol scale. The excess sodium azide was destroyed with sodium nitrite and sulfuric acid, which converts hydrazoic acid into nitrogen and nitrous oxide gases.

While the above procedure may be popular, any time you use sodium azide you should be thinking, “hydrazoic acid can be generated, it’s explosive and toxic, and I need to take the appropriate safety precautions.” That’s precisely what happened during some recent process R&D work at Merck Frosst on the steroyl-CoA desaturase inhibitor MK-8245. The discovery chemistry route used NaN3/pyridinium chloride as shown below, but the process group felt that the potential for significant amounts of hydrazoic acid generation was too high.
Armed with the ability to detect hydrazoic acid in the headspace above the reaction mixture using online IR, the Merck Frosst researchers surveyed alternatives. Sharpless’s zinc bromide procedure, proposed to minimize hydrazoic acid formation by control of the pH, led to a reading of 2000 ppm of HN3 in the headspace, which is below the detonation threshold of 15,000 ppm but was still felt to be undesirable. In their own survey of conditions, the Merck Frosst scientists found something quite new and significant: Reaction with sodium azide in the presence of a catalytic amount of zinc oxide in aqueous THF (pH 8) proceeded efficiently, and most notably, with only 2 ppm of HN3 in the headspace! They were able to make 7 kg of the tetrazole in one run in nearly quantitative yield. Nice!

I’d be remiss if I didn’t mention Bu3SnN3 and Me3SiN3/Cu(I) as sodium azide surrogates, sometimes used on large scale. Shown below is an application to valsartan (see here and here) with recycling of the tin by-products. The intermediate stannyl tetrazole and leftover Bu3SnN3 were converted with HCl to Bu3SnCl, which was then converted to the fluoride, which was removed by filtration and recycled to Bu3SnCl.

Additional Topics
Transition-Metal Catalysis and Organocatalysis
Biocatalysis and Enzymatic Engineering
Recently, large pharmaceutical process chemists have relied heavily on the development of enzymatic reactions to produce important chiral building blocks for API synthesis. Many varied classes of naturally occurring enzymes have been co-opted and engineered for process pharmaceutical chemistry applications. The widest range of applications come from ketoreductases and transaminases, but there are isolated examples from hydrolases, aldolases, oxidative enzymes, esterases and dehalogenases, among others.[10]
One of the most prominent uses of biocatalysis in process chemistry today is in the synthesis of Januvia®, a DPP-4 inhibitor developed by Merck for the management of type II diabetes. The traditional process synthetic route involved a late-stage enamine formation followed by rhodium-catalyzed asymmetric hydrogenation to afford the API sitagliptin. This process suffered from a number of limitations, including the need to run the reaction under a high-pressure hydrogen environment, the high cost of a transition-metal catalyst, the difficult process of carbon treatment to remove trace amounts of catalyst and insufficient stereoselectivity, requiring a subsequent recrystallization step before final salt formation.[11][12]

Merck’s process chemistry department contracted Codexis, a medium-sized biocatalysis firm, to develop a large-scale biocatalytic reductive amination for the final step of its sitagliptin synthesis. Codexis engineered a transaminase enzyme from the bacteria Arthrobacter through 11 rounds of directed evolution. The engineered transaminase contained 27 individual point mutations and displayed activity four orders of magnitude greater than the parent enzyme. Additionally, the enzyme was engineered to handle high substrate concentrations (100 g/L) and to tolerate the organic solvents, reagents and byproducts of the transamination reaction. This biocatalytic route successfully avoided the limitations of the chemocatalyzed hydrogenation route: the requirements to run the reaction under high pressure, to remove excess catalyst by carbon treatment and to recrystallize the product due to insufficient enantioselectivity were obviated by the use of a biocatalyst. Merck and Codexis were awarded the Presidential Green Chemistry Challenge Award in 2010 for the development of this biocatalytic route toward Januvia®.[13]

ATORVASTATIN
Biocatalytic process development firm Codexis was recognized with the award in the greener reaction conditions category for developing a “green-by-design” enzymatic process to replace a chemical process for making ethyl (R)-4-cyano-3-hydroxybutyrate. This chemical, also known as hydroxynitrile, is the key chiral building block used to make atorvastatin, the active ingredient in Pfizer‘s cholesterol-lowering drug Lipitor.
The new process is helping to lower atorvastatin’s long-term production costs, according to John H. Grate, senior vice president of R&D and chief technology officer at Codexis. The savings could be financially significant for Pfizer and future generics manufacturers given that Lipitor is the world’s top pharmaceutical, with annual sales of about $13 billion.

Hydroxynitrile is used in the early stages of atorvastatin synthesis to build the chiral dihydroxy acid side chain that’s essential to the drug’s activity, Grate told C&EN. Demand for the intermediate is about 200 metric tons per year, and it’s currently being made by several fine chemicals producers. The competition to supply the intermediate to Pfizer has spurred several firms to chase after a better way to prepare hydroxynitrile (Angew. Chem. Int. Ed. 2005, 44, 362).

Chemical engineering professor Galen J. Suppes of the University of Missouri, Columbia, was honored with the academic award for his group’s work to create a low-cost catalytic process to convert the glycerol by-product from biodiesel production into propylene glycol–turning 1,2,3-propanetriol into 1,2-propanediol. At first glance, this achievement may not sound that exciting. But the repercussions of Suppes’s accomplishment are expected to have a major impact on the future use of biodiesel fuel, the world glycerol market, and the environmental health and safety of antifreeze and deicing chemicals.
 Photo by Rob Hill/MU Publications Photo by Rob Hill/MU Publications |
| GREEN SOLUTION Suppes and his group uncovered ideal reaction conditions for the catalytic conversion of by-product glycerol to useful propylene glycol. |
Biodiesel is a mixture of fatty acid methyl esters made by esterifying soybean oil or other vegetable oil or animal fat. The triglycerides in the oil consist of three long fatty acid chains connected to a propyl headgroup. Sodium hydroxide is used to cleave the chains, which in turn are reacted with methanol to form methyl esters, leaving the residual glycerol headgroup as a by-product. About 1 kg of crude glycerol is formed for every 9 kg of biodiesel produced.
Millions of gallons of glycerol are flooding the world market as biodiesel production is ramping up in the U.S. and Europe, Suppes explained. The fallout from this glycerol glut is that chemical companies have shuttered some glycerol production plants and are considering glycerol as a starting material to make a host of feedstock chemicals (C&EN, Feb. 6, page 7).
Suppes entered the picture about four years ago when he realized that an inexpensive method to convert glycerol to propylene glycol could be valuable, he said. Utilizing the glycerol not only would help offset the cost of biodiesel production, but the inexpensive propylene glycol could be used as a low-toxicity replacement for ethylene glycol in automotive antifreeze.
Suppes’s system involves low-pressure hydrogenolysis of glycerol using a copper chromite catalyst, CuO•Cr2O3 (Appl. Catal. A 2005, 281, 225). In the two-step process, glycerol is first dehydrated to form acetol (1-hydroxy-2-propanone), which is then hydrogenated to form propylene glycol.
 |
| GREEN LEFTOVERS Glycerol by-product from biodiesel production can be used as a feedstock in Suppes’ process to produce acetol or propylene glycol from renewable resources. |
Copper chromite hydrogenolysis catalysts aren’t new, but the success of the Missouri process is in achieving high selectivity for propylene glycol by controlling the temperature and hydrogen pressure of the reaction, Suppes noted. In the past, researchers tended to use reaction temperatures that were too high, leading to a higher percentage of by-products. Thus, they “missed the window of opportunity to achieve high selectivity,” Suppes said. Tinkering with temperature, pressure, and several different catalysts, Suppes and his colleagues optimized the system to operate at about 220 °C and less than 10 bar versus about 260 °C and more than 150 bar for other systems.
Another key part of the synthesis is the ability to isolate the acetol intermediate, Suppes added. Acetol is a synthetic starting material used to make polyols. But when made from petroleum, it costs about $5.00 per lb, discouraging its widespread use. Suppes envisions that producing acetol from biomass-based glycerol using his process could lower the cost to 50 cents per lb, “opening up even more potential applications and markets for products made from glycerol.”
Suppes’s propylene glycol process has been patented and is being licensed through the Missouri Soybean Merchandising Council, which provided partial funding for the research. The first commercial facility, with an annual capacity of 11.5 million gal, is being built in an undisclosed location in the U.S. by Senergy Chemical. It’s expected to be in operation by the end of this year.

E7398, INN eribulin mesylate
The most awe-inspiring example of a positive tangible outcome from the combination of basic research into the synthesis of a system, and a correctly weighted assessment of ‘scalability’, is Halaven® (2, E7398, INN eribulin mesylate). Most chemists in industry and academia alike would have considered using total synthesis to support clinical development and commercialization of this compound a ‘fool’s errand,’ but the Kishi group and Eisai Inc. did not. The fact is that this compound solves a major clinical problem, so taking on the issues (length of synthesis, stability limitations, stereochemical problems, etc.) had a big payoff (reducing the relative weighting or importance of these factors in assessing the viability of a commercial chemical synthesis). As depicted below, a highly convergent approach, combined with powerful methodology for stitching together key fragments 5 and 6 (Nozaki–Hiyama–Kishi (NHK) coupling) and a strategy of targeting crystalline intermediates were all key elements that culminated in this landmark accomplishment

The commercial synthesis of Halaven® (2), a landmark achievement in process chemistry
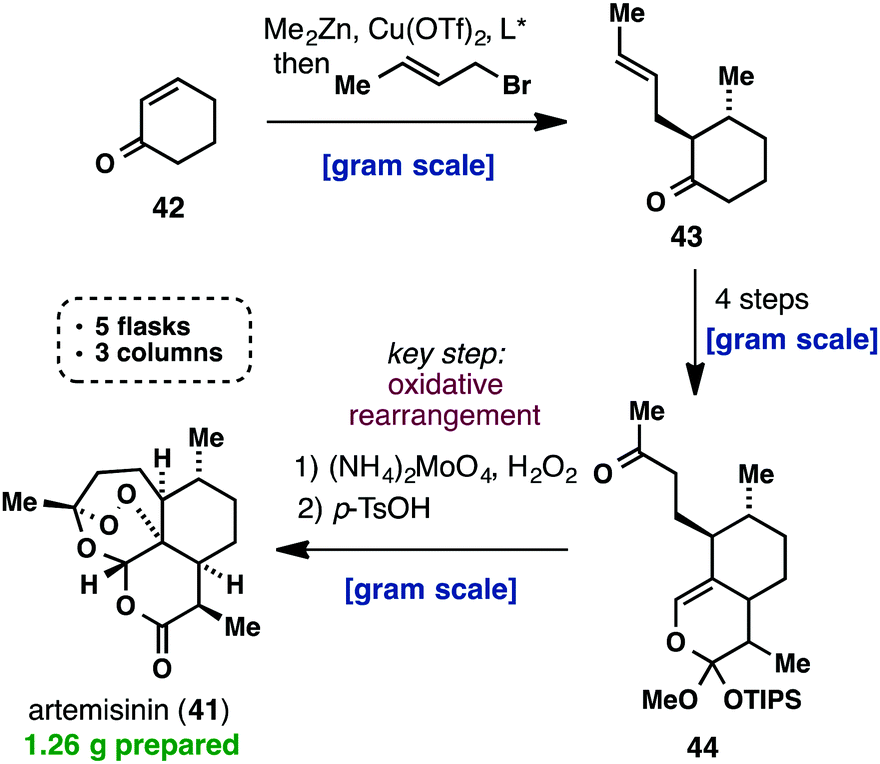
Artemisinin (Cook, 2012).
(+)-Artemisinin (41) is currently the most effective drug against Plasmodium falciparum malaria as part of an artemisinin-based combination therapy (ACT). Although it can be isolated on an industrial scale from Artemisia annua, the market price of artemisinin (41) has fluctuated widely and traditional extraction does not provide enough material to meet the worldwide demand. Interestingly, recent efforts towards a cheaper and more efficient production of artemisinin (41) have mainly taken place in the areas of synthetic biology, semisynthesis and plant engineering, while there has been a lack of practical approaches using a straightforward total synthesis. Despite the fact that all the total syntheses of artemisinin, until 2010, were impressive from a feasibility point of view, none of them provided a solution for the low-cost synthesis of 41. This changed when Cook’s group recently published a scalable synthesis of artemisinin (41), which provides a blueprint for the cost-effective production of 41 and its derivatives below Key to their successful strategy was the use of reaction cascades that rapidly built complexity, starting from the cheap feedstock chemical, cyclohexenone (42). The latter was first subjected to a one-pot conjugate addition/alkylation sequence, to give ketone 43. A three-step sequence consisting of formylation, cycloaddition and a Wacker-type oxidation, yielded 9.4 g of methyl ketone 44. The challenging formation of the unusual peroxide bridge was initially met with failure, but was eventually realized by a reaction with singlet oxygen to give 41 amongst other oxidized intermediates. The entire synthetic sequence was conducted on a gram scale, required only three chromatographic purifications and was carried out in only five flasks. Considering the low cost of the commodity chemicals used and the conciseness of Cook’s synthesis, it is certainly worth being further investigated.
 |
||
| Cook’s scalable route to (+)-artemisinin (41). | ||
Continuous/Flow Manufacturing
In recent years, much progress has been made in the development and optimization of flow reactors for small-scale chemical synthesis (the Jamison Group at MIT and Ley Group at Cambridge University, among others, have pioneered efforts in this field). The pharmaceutical industry, however, has been slow to adopt this technology for large-scale synthetic operations. For certain reactions, however, continuous processing may possess distinct advantages over batch processing in terms of safety, quality and throughput.
A case study of particular interest involves the development of a fully continuous process by the process chemistry group at Eli Lilly and Company for an asymmetric hydrogenation to access a key intermediate in the synthesis of LY500307,[14] a potent ERβ agonist that is entering clinical trials for the treatment of patients with schizophrenia, in addition to a regimen of standard antipsychotic medications. In this key synthetic step, a chiral rhodium-catalyst is used for the enantioselective reduction of a tetrasubstituted olefin. After extensive optimization, it was found that in order to reduce the catalyst loading to a commercially practical level, the reaction required hydrogen pressure up to 70 atm. The pressure limit of a standard chemical reactor is about 10 atm, although high-pressure batch reactors may be acquired at significant capital cost for reactions up to 100 atm. Especially for an API in the early stages of chemical development, such an investment clearly bears a large risk.
An additional concern was that the hydrogenation product has an unfavorable eutectic point, so it was impossible to isolate the crude intermediate in more than 94 percent ee by batch process. Because of this limitation, the process chemistry route toward LY500307 necessarily involved a kinetically controlled crystallization step after the hydrogenation to upgrade the enantiopurity of this penultimate intermediate to >99 percent ee.

The process chemistry team at Eli Lilly successfully developed a fully continuous process to this penultimate intermediate, including reaction, workup and kinetically controlled crystallization modules (the engineering considerations implicit in these efforts are beyond the scope of this article). An advantage of flow reactors is that high-pressure tubing can be utilized for hydrogenation and other hyperbaric reactions. Because the head space of a batch reactor is eliminated, however, many of the safety concerns associated with running high-pressure reactions are obviated by the use of a continuous process reactor. Additionally, a two-stage mixed suspension-mixed product removal (MSMPR) module was designed for the scalable, continuous, kinetically controlled crystallization of the product, so it was possible to isolate in >99 percent ee, eliminating the need for an additional batch crystallization step.
This continuous process afforded 144 kg of the key intermediate in 86 percent yield, comparable with a 90 percent isolated yield using the batch process. This 73-liter pilot-scale flow reactor (occupying less than 0.5 m3 space) achieved the same weekly throughput as theoretical batch processing in a 400-liter reactor. Therefore, the continuous flow process demonstrates advantages in safety, efficiency (eliminates the need for batch crystallization) and throughput, compared with a theoretical batch process.
US scientists have found a way to stop solid byproducts clogging channels in continuous flow reactors, a problem that has hampered their progress for use in manufacturing pharmaceuticals.
Klavs Jensen, Stephen Buchwald and their team at the Massachusetts Institute of Technology believe that flow methods will become increasingly important in the future of pharmaceuticals and chemical manufacturing. ‘One of the biggest hurdles is handling solids,’ says group member Timothy Noël. ‘Precipitates can form during the reactions, which usually lead to irreversible clogging of microchannels in the reactors.’ Previous methods suggested to overcome this problem include introducing another solvent to dissolve the solids, but this can reduce the overall efficiency of the reactions. Now, the team have used an ultrasound bath to break up the byproducts to prevent clogging.
Traditionally, pharmaceutical manufacture is done in a batch-based system, but the process suffers from interruptions and the need to transport material between batch reactors. Performing these reactions in a continuous flow system would speed up the process and reduce chemical waste.

|
Reagents were introduced into a tube, which was then placed in an ultrasonic bath heated to 60 degrees Celsius. When the reagents exited the reactor, the reaction was mixed with a quench of water and ethyl acetate in a larger tube, allowing plenty of time for salt byproducts to dissolve
|
The team tested the method on palladium-catalysed C-N cross-coupling reactions, making amines that are common in biologically active molecules. The reactions couple aryl halides to nitrogen nucleophiles and form byproducts – inorganic salts – that are insoluble in the solvents used.
As a result, says Noël, they were able to obtain diarylamine products with reaction times ranging from 20 seconds to 10 minutes. At very short residence times (time in the reactor under reaction conditions) they observed a significantly higher rate for the reaction in flow compared to the equivalent batch experiments. With high conversions in short reaction times, they were able to reduce the catalyst loading in flow to just 0.1 mol per cent. ‘Extremely low catalyst loadings such as these are of particular interest to the pharmaceutical industry,’ says Noël.
Noël believes that in the future microfluidics will be used to construct increasingly complex molecules. Different devices will automate and integrate many synthetic steps that are currently performed using the more traditional and time-consuming batch-based practices.
Oliver Kappe, from the Christian Doppler Laboratory for Microwave Chemistry, Institute of Chemistry, Karl-Franzens-University Graz says: ‘Jensen and Buchwald clearly demonstrate that immersing a flow device into an ultrasound bath can prevent clogging problems that unfortunately are all too familiar to the flow/microreactor community.’
Direct Fluorination and Microreactor Technology
Elemental fluorine has long been considered to be too reactive and uncontrollable for use as a reagent in organic synthesis and this perception still predominates. Prof. Poliakoff’s comments on the popular Periodic Table video series (www.PeriodicVideos.com), ‘It was much more exciting than I thought …you see the flames,’ and general comments in standard advanced organic chemistry textbooks (J. March, Advanced Organic Chemistry, ‘Direct fluorination of aromatic rings with F2 is not feasible at room temperature because of the extreme reactivity of F2….not yet of preparative significance) are typical.
Despite this background, research into the use of elemental fluorine for organic synthesis at Durham has overcome the many problems of using fluorine gas for the safe synthesis of fine chemicals, in particular, by use of dilute fluorine gas in nitrogen, appropriate solvent choice (high dielectric constant media such as formic acid, sulfuric acid or acetonitrile), reactor vessel design, gas flow regulator systems and stainless steel/monel fluorine gas handling lines have developed over the years to allow selective direct fluorination of a range of aliphatic, dicarbonyl, aromatic, heteroaromatic, heterocyclic, steroid and carbohydrate derivatives to be established and the mechanism (regiochemistry, stereochemistry, selectivity, etc.) of these processes to be assessed. Indeed, direct fluorination of aromatic rings is feasible at room temperature ! Research expanding the use of fluorine gas continues to develop new selective fluorination methodology for the synthesis of a range of aromatic, heterocylic and aliphatic systems.2,3

In particular, a process for the synthesis of a fluoroketoester first carried out in Durham was developed by our industrial collaborators, F2 Chemicals Ltd (UK), for the Pfizer company and forms a key starting material in the multi step synthesis of the widely used anti-fungal agent V-Fend (Voriconazole) throughout the clinical trial, launch and commercialization periods. In the period from January 2008 to March 2011 approximately 17 tonnes of the fluoroketoester were manufactured for Pfizer by F2 Chemicals Ltd. Global sales of V-Fend in the 2008-2010 period total $2.4 billion (Pfizer annual financial reports) and in 2010 was 17th position in Pfizer’s best selling products and it is one of the global top 100 best selling pharmaceutical products.

Further reaction control in selective fluorination reactions was achieved by the design, fabrication and commissioning of single and multi-channel continuous flow reactor systems, establishing the use of convenient, inexpensive flow reactors for gas – liquid processes using flow regimes in the laboratory. Techniques for the supply of individual gas and liquid reagents from single sources to a parallel array of many flow channels at the same flow rate and pressure whilst maintaining laminar flow within the reactor channels and telescoped gas – liquid / liquid – liquid processes involving fluorination and ring formation in one continuous flow process have been developed.

References
- Roughley, S. D.; Jordan, A. M. (2011). “The medicinal chemist’s toolbox: an analysis of reactions used in the pursuit of drug candidates”. J. Med. Chem. 54: 3451.
- Dach, R.; Song, J. J.; Roschangar, F.; Samstag, W.; Senanayake, C. H. (2012). “The eight criteria defining a good chemical manufacturing process”. Org. Process Res. Dev. 16: 1697.
- Trost, B. M. (1991). “The atom economy – a search for synthetic efficiency”. Science 254: 1471.
- Van Aken, K.; Strekowski, L.; Patiny, L. (2006). “EcoScale, a semi-quantitative tool to select an organic preparation based on economical and ecological parameters”. Beilstein J. Org. Chem. 2 (No. 3).
- Faucher, A-M.; Bailey, M. D.; Beaulieu, P. L.; Brochu, C.; Duceppe, J-S.; Ferland, J-M.; Ghiro, E.; Gorys, V.; Halmos, T.; Kawai, S. H.; Poirier, M.; Simoneau, B.; Tsantrizos, Y. S.; Llinas-Brunet, M. (2004). “Synthesis of BILN 2061, an HCV NS3 protease inhibitor with proven antiviral effect in humans”. Org. Lett. 6: 2901.
- Yee, N. K.; Farina, V.; Houpis, I. N.; Haddad, N.; Frutos, R. P.; Gallou, F.; Wang, X-J.; Wei, X.; Simpson, R. D.; Feng, X.; Fuchs, V.; Xu, Y.; Tan, J.; Zhang, L.; Xu, J.; Smith-Keenan, L. L.; Vitous, J.; Ridges, M. D.; Spinelli, E. M.; Johnson, M. (2006). “Efficient large-scale synthesis of BILN 2061, a potent HCV protease inhibitor, by a convergent approach based on ring-closing metathesis”. J. Org. Chem. 71: 7133.
- Zeng, X.; Wei, X.; Farina, V.; Napolitano, E.; Xu, Y.; Zhang, L.; Haddad, N.; Yee, N. K.; Grinberg, N.; Shen, S.; Senanayake, C. H. (2006). “Epimerization reaction of a substituted vinylcyclopropane catalyzed by ruthenium carbenes: mechanistic analysis”. J. Org. Chem. 71: 8864.
- Grela, K.; Harutyunyan, S.; Michrowska, A. (2002). “A highly efficient ruthenium catalyst for metathesis reactions”. Angew. Chem. Int. Ed. 41: 4038.
- Wei, X.; Shu, C.; Haddad, N.; Zeng, X.; Patel, N. D.; Tan, Z.; Liu, J.; Lee, H.; Shen, S.; Campbell, S.; Varsolona, R. J.; Busacca, C. A.; Hossain, A.; Yee, N. K.; Senanayake, C. H. (2013). “A highly convergent and efficient synthesis of a macrocyclic hepatitis C virus protease inhibitor BI 201302”. Org. Lett. 15: 1016.
- Bornscheuer, U. T.; Huisman, G. W.; Kazlauskas, R. J.; Lutz, S.; Moore, J. C.; Robins, K. (2012). “Engineering the third wave of biocatalysis”. Nature 485: 185.
- Savile, C. K.; Janey, J. M.; Mundorff, E. C.; Moore, J. C.; Tam, S.; Jarvis, W. R.; Colback, J. C.; Krebber, A.; Fleitz, F. J.; Brands, J.; Devine, P. N.; Huisman, G. W.; Hughes, G. J. (2010). “Biocatalytic asymmetric synthesis of chiral amines applied to sitagliptin manufacture”. Science 329: 305.
- Desai, A. A. (2011). “Sitagliptin manufacture: a compelling tale of green chemistry, process intensification, and industrial asymmetric catalysis”. Angew. Chem. Int. Ed. 50: 1974.
- Busacca, C. A.; Fandrick, D. R.; Song, J. J.; Sananayake, C. H. (2011). “The growing impact of catalysis in the pharmaceutical industry”. Adv. Synth. Catal. 353: 1825.
- Johnson, M. D.; May, S. A.; Calvin, J. R.; Remacle, J.; Stout, J. R.; Dieroad, W. D.; Zaborenko, N.; Haeberle, B. D.; Sun, W-M.; Miller, M. T.; Brannan, J. (2012). “Development and scale-up of a continuous, high-pressure, asymmetric hydrogenation reaction, workup, and isolation”. Org. Process Res. Rev. 16: 1017.
How to Handle Drug Polymorphs… Case Study of Trelagliptin Succinate

PART 1………..http://drugsynthesisint.blogspot.in/p/gliptin-series.html
PART 2 ……http://organicsynthesisinternational.blogspot.in/p/gliptin-series-22.html


If you’re involved in late drug discovery, API manufacture, drug product formulation, clinical material production, or manufacture of final dosage form, a basic understanding and awareness of solid form issues could save you a great deal of difficulty, time, and money during drug development.


Investigational new drug, writing an application for clinical trial authorization, permission marketing …The control of polymorphism in drug candidates is now ubiquitous.
READ………….An Overview of Solid Form Screening During Drug Development, http://www.icdd.com/ppxrd/10/presentations/PPXRD-10_Ann_Newman.pdf
ANN NEWMAN
When addressing the subject of polymorphism, the first reference that comes to mind is that of the occurred during the manufacture of ritonavir incident. Abbott molecule inhibitor of HIV protease marketed as Norvir, is a cautionary example of the challenges of polymorphism.
Indeed, during the production of ritonavir in 1997, a new polymorph unmarked emerged. Its precipitation and unexpected outbreak led to the cessation of the production of Norvir and seriously compromise the process. The incident has deeply marked the pharmaceutical industry.
It is ironic that the process used to discover pharmaceutical drug targets is the same one that decreases the actual efficacy of those drugs once ingested. If you remember from basic chemistry, there are compounds that exist in highly ordered crystalline states and those that remain in amorphous form.
The discovery of drug targets has often been accomplished through X-ray crystallography, which requires a sample (for example, of a defective enzyme linked to cancer or high cholesterol) to be crystallized so that the diffraction patterns can be made sense of. Scientists may spend years trying to crystallize one molecule or compound so that they can identify regions that, for example, may be blocked by pharmaceuticals.
However, when it comes to the molecular arrangement of those pharmaceuticals, crystallization actually decreases their bioavailability and solubility. Thus, it may be better for these drugs to be in amorphous form. Pierric Marchand, general manager of the company Holodiag, dedicated to the study and characterization of solid state, summarizes that ” today, it is not reasonable to not worry about the problem of polymorphism ” .
” In recent years, manufacturers have realized the essential side of expertise , “says Jean-Rémi David, commercial director Calytherm. The services company specializing in the field of physico-chemical analysis, based in Herault, has just relocated last year in supporting pharmaceutical development to meet demand. ” This is a concern for all deal with potential impacts on the effectiveness or the formulation , “says Stéphane Suchet, quality manager in the group of fine and specialty chemicals Axyntis.
Polymorphic forms are the amorphous and crystalline forms such as hydrate or solvate forms. When a molecule of interest exists in polymorphic forms, it is called polymorphism, according to the definition of the FDA (Food and Drug Administration).
Polymorphism is present at all stages of development of a drug from research to marketing. ” Keep in mind that organic molecule loves to polymorphism , “says Marchand Pierric. However, for a marketing authorization for example, must learn the criteria for the polymorphism of the molecule. ” In terms of the formulation, for example we can check whether the selected polymorphism is unchanged , “explains Pierric Marchand.

A significant influence on several levels Because the consequences of polymorphism are multiple. ” They are at three levels: bioequivalence, manufacturability and stability “lists Fabienne Lacoulonche, founder and scientific director of Calytherm.In terms of bioequivalence, different polymorphic forms may have different properties of solubility and dissolution rate … ” For poorly soluble active ingredients, you can have much more bioavailable than other crystalline forms , “Fabienne Lacoulonche information.
In terms of manufacturability, some parameters such as temperature, moisture can lead to changes in the crystalline form. ” The complexity is to anticipate changes polymorphism, both at laboratory scale, pilot and industrial , “adds the founder of Calytherm. Finally, polymorphism plays on stability. Active ingredient or finished product, are subjected to stability studies in this direction. ” When the molecule is identified, we try to highlight the existence of several forms of polymorphism, explains Stéphane Suchet (Axyntis) .
When developing a new substance, the assessment is systematic . ” Isolation of crystals from a screening is carried out in different solvents by various analytical techniques. Ideally, it will be concluded the absence of polymorphism. ” But if different polymorphic forms are present, we rework the terms of our crystallization process to control the formation of the same polymorph reproducibly ideally form the thermodynamically more stable , “says Stéphane Suchet. X-ray diffraction and other thermal analysis ”
The ICH guidelines provide decision trees to guide the industry in controlling polymorphism says Fabienne Lacoulonche (Calytherm.) We use it for writing the CTD (Common Technical Document) .
“Polymorphism is a phenomenon” complex and difficult to control, because the crystallization is dependent on many parameters , she develops. must understand the maximum . ” For this, several analytical methods are available to industry. The main technique is the X-ray diffraction ” It is a robust, rapid, which allows to characterize the different polymorphs , “summarizes Pierric Marchand (Holodiag).Non-destructive, it can work both on small quantities on large samples. Temperature and atmosphere are controlled, and analytical capabilities are broad.

But if this technique indispensable allows for routine and development, it is not sufficient in itself. Just to add a battery of additional tests, thermal analysis. ” It takes coupling methods “ confirms Fabienne Lacoulonche (Calytherm). The X-ray diffraction is a method of choice, but sometimes it is not sufficient.
The coupling with a thermal analysis method (technical ATG, or DSC thermal analysis, differential scanning calorimetry or thermomicroscopique) allows to distinguish between two polymorphic whose RX diffractograms obtained are comparable.
TGA can be coupled with IR or mass spectrometry, DSC with RX. Raman spectroscopy is also part of complementary methods. ” The difficulty increases when we want to characterize the shape of the active ingredient in the finished product , says Fabienne Lacoulonche. example, by X-ray diffraction, the peaks related to the active ingredient in the diffractogram of the finished product may be masked by those excipients: it is then necessary to use other methods, such as Raman microscopy. “In general, a single method of analysis is not sufficient to characterize the polymorphism of an active substance in the active substance or finished product: the complementarity of different methods that will conclude precisely on the polymorphism of a crystalline substance.
In addition, ” the diffractometer remains an expensive device, which requires installation in an air-conditioned and a cooling room , “says Marchand Pierric (Holodiag). To this is added the need to have expertise and qualified personnel to carry out the analyzes. ” We must master these techniques and the ability to interpret the results , “says Jean-Rémi David (Calytherm). However, polymorphism is a “problem well under control , “said Stéphane Suchet (Axyntis),” systematically evaluated although it is however not always immune to miss a polymorphic form, knowing that the screening performed in the development can never be completely comprehensive … ”

FDA
FDA may refuse to approve an ANDA referencing a listed drug if the application contains insufficient information to show that the drug substance is the “same” as that of the reference listed drug. A drug substance in a generic drug product is generally considered to be the same as the drug substance in the reference listed drug if it meets the same standards for identity.
In most cases, the standards for identity are described in the USPalthough FDA may prescribe additional standards when necessary. Because drug product performance depends on the product formulation, the drug substance in a proposed generic drug product need not have the same physical form (particle size, shape, or polymorph form) as the drug substance in the reference listed drug. An ANDA applicant is required to demonstrate that the proposed product meets the standards for identity, exhibits sufficient stability and is bioequivalent to the reference listed drug.
FDA PRESENTATION……polymorphs and co-crystals – ICDD Regulatory Considerations on Pharmaceutical Solids: Polymorphs/Salts and Co-Crystals.. THIS IS A MUST READ ITEM
Over the years FDA has approved many generic drug products based upon a drug substance with different physical form from that of the drug substance in the respective reference listed drug (e.g., warfarin sodium, famotidine, and ranitidine). Also many ANDAs have been approved in which the drug substances differed from those in the corresponding reference listed drugs with respect to solvation or hydration state (e.g., terazosin hydrochloride, ampicillin, and cefadroxil). Several regulatory documents and literature reports (67-69) address issues relevant to the regulation of polymorphism.
The concepts and principles outlined in these are applicable for an ANDA. However, certain additional considerations may be applicable in case of ANDAs. Often at the time FDA receives an ANDA a monograph defining certain key attributes of the drug substance and drug product may be available in the Unites States Pharmacopoeia (USP). These public standards play a significant role in the ANDA regulatory review process and in case of polymorphism, when some differences are noted, lead to additional requirements and considerations.
This commentary is intended to provide a perspective on polymorphism in pharmaceutical solid in the context of ANDAs. It highlights major considerations for monitoring and controlling drug substance polymorphs and describes a framework for regulatory decisions regarding drug substance “sameness” considering the role and impact of polymorphism in pharmaceutical solids.

Since polymorphs exhibit certain differences in physical (e.g., powder flow and compactability, apparent solubility and dissolution rate) and solid state chemistry (reactivity) attributes that relate to stability and bioavailability it is essential that the product development and the FDA review process pay close attention to this issue.
This scrutiny is essential to ensure that polymorphic differences (when present) are addressed via design and control of formulation and process conditions to physical and chemical stability of the product over the intended shelf-life, and bioavailability/bioequivalence.
Most pharmaceuticals are distributed as solid doseages. In order to take action, they must dissolve in the gut and be absorbed into the blood stream. In many cases, the rate at which the drug dissolves can limit its effectiveness. Pharmaceutical compounds can be packed into more than one arrangement in the solid states known as polymorphs. Rapid and efficient methods of polymorph formation can be used to increase drug efficacy and shelf life.
Regulatory agencies worldwide require that, as part of any significant filing, a company has to demonstrate that it has made a reasonable effort to identify the polymorphs of their drug substance and has checked for polymorph interconversions. Due to the unpredictable behaviour of polymorphs and their respective differences in physicochemical properties, companies also have to demonstrate consistency in manufacturing between batches of the same product. Proper understanding of the polymorph landscape and nature of the polymorphs will contribute to manufacturing consistency.
POLYMORPHISM AND PATENTS http://www.collegio.unibo.it/uploads/ideas/joelbernstein.pdf A MUST CLICK FOR PHARMA CHEMISTS

READ………..High-throughput crystallization: polymorphs, salts, co-crystalsand solvates of pharmaceutical solidshttp://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.85.5397&rep=rep1&type=pdf

Definitions
“Crystalline”, as the term is used herein, refers to a material, which may be hydrated and/or solvated, and has sufficient ordering of the chemical moiety to exhibit a discernable diffraction pattern by XRPD or other diffraction techniques. Often, a crystalline material that is obtained by direct crystallization of a compound dissolved in a solution or by interconversion of crystals obtained under different crystallization conditions, will have crystals that contain the solvent used in the crystallization, termed a crystalline solvate. Also, the specific solvent system and physical embodiment in which the crystallization is performed, collectively termed crystallization conditions, may result in the crystalline material having physical and chemical properties that are unique to the crystallization conditions, generally due to the orientation of the chemical moieties of the compound with respect to each other within the crystal and/or the predominance of a specific polymorphic form of the compound in the crystalline material.
Depending upon the polymorphic form(s) of the compound that are present in a composition, various amounts of the compound in an amorphous solid state may also be present, either as a side product of the initial crystallization, and/or a product of degradation of the crystals comprising the crystalline material. Thus, crystalline, as the term is used herein, contemplates that the composition may include amorphous content; the presence of the crystalline material among the amorphous material being detectably among other methods by the composition having a discernable diffraction pattern.
The amorphous content of a crystalline material may be increased by grinding or pulverizing the material, which is evidenced by broadening of diffraction and other spectral lines relative to the crystalline material prior to grinding. Sufficient grinding and/or pulverizing may broaden the lines relative to the crystalline material prior to grinding to the extent that the XRPD or other crystal specific spectrum may become undiscernable, making the material substantially amorphous or quasi-amorphous. Continued grinding would be expected to increase the amorphous content and further broaden the XRPD pattern with the limit of the XRPD pattern being so broadened that it can no longer be discerned above noise. When the XRPD pattern is broadened to the limit of being indiscernible, the material may be considered no longer a crystalline material, but instead be wholly amorphous. For material having increased amorphous content and wholly amorphous material, no peaks should be observed that would indicate grinding produces another form.
“Amorphous“, as the term is used herein, refers to a composition comprising a compound that contains too little crystalline content of the compound to yield a discernable pattern by XRPD or other diffraction techniques. Glassy materials are a type of amorphous material. Glassy materials do not have a true crystal lattice, and technically resembling very viscous non-crystalline liquids. Rather than being true solids, glasses may better be described as quasi-solid amorphous material. “Broad” or “broadened”, as the term is used herein to describe spectral lines, including XRPD, NMR and IR spectroscopy, and Raman spectroscopy lines, is a relative term that relates to the line width of a baseline spectrum. The baseline spectrum is often that of an unmanipulated crystalline form of a specific compound as obtained directly from a given set of physical and chemical conditions, including solvent composition and properties such as temperature and pressure.
For example, broadened can be used to describe the spectral lines of a XRPD spectrum of ground or pulverized material comprising a crystalline compound relative to the material prior to grinding. In materials where the constituent molecules, ions or atoms, as solvated or hydrated, are not tumbling rapidly, line broadening is indicative of increased randomness in the orientation of the chemical moieties of the compound, thus indicative of an increased amorphous content. When comparisons are made between crystalline materials obtained via different crystallization conditions, broader spectral lines indicate that the material producing the relatively broader spectral lines has a higher level of amorphous material.
“About” as the term is used herein, refers to an estimate that the actual value falls within ±5% of the value cited. “Forked” as the term is used herein to describe DSC endotherms and exotherms, refers to overlapping endotherms or exotherms having distinguishable peak positions
. 
Classes of multicomponent pharmaceutical materials. (a) Schematic of crystalline materials showing neutral and charged species. The red box indicates polymorphs are possible for all the multicomponent crystals contained within the box (adapted from Reference 7). (b) Schematic of amorphous solid dispersions showing binary, ternary, and quaternary possibilities for polymers and surfactants. Other solubilization techniques using cyclodextrins and phospholipids are included for completeness but have a different mechanism for solubilization when compared to polymer and surfactant systems.
The red box indicates that properties can change with water or solvent content. General methods for precipitating and crystallizing a compound may be applied to prepare the various polymorphs described herein. These general methods are known to those skilled in the art of synthetic organic chemistry and pharmaceutical formulation, and are described, for example, by J. March, “Advanced Organic Chemistry: Reactions, Mechanisms and Structure ” 4th Ed. (New York: Wiley-Interscience, 1992).
In general, a given polymorph of a compound may be obtained by direct crystallization of the compound or by crystallization of the compound followed by inter-conversion from another polymorphic form or from an amorphous form. Depending on the method by which a compound is crystallized, the resulting composition may contain different amounts of the compound in crystalline form as opposed to as an amorphous material.
Also, the resulting composition may contain differing mixtures of different polymorphic forms of the compound. Compositions comprising a higher percentage of crystalline content {e.g., forming crystals having fewer lattice defects and proportionately less glassy material) are generally prepared when using conditions that favor slower crystal formation, including slow solvent evaporation and those affecting kinetics.
Crystallization conditions may be appropriately adjusted to obtain higher quality crystalline material as necessary. Thus, for example, if poor crystals are formed under an initial set of crystallization conditions, the solvent temperature may be reduced and ambient pressure above the solution may be increased relative to the initial set of crystallization conditions in order to slow down crystallization.  Precipitation of a compound from solution, often affected by rapid evaporation of solvent, is known to favor the compound forming an amorphous solid as opposed to crystals. A compound in an amorphous state may be produced by rapidly evaporating solvent from a solvated compound, or by grinding, pulverizing or otherwise physically pressurizing or abrading the compound while in a crystalline state.
Precipitation of a compound from solution, often affected by rapid evaporation of solvent, is known to favor the compound forming an amorphous solid as opposed to crystals. A compound in an amorphous state may be produced by rapidly evaporating solvent from a solvated compound, or by grinding, pulverizing or otherwise physically pressurizing or abrading the compound while in a crystalline state.
Seven crystalline forms and one amorphous solid were identified by conducting a polymorph screen (Example 3). Described herein are Form A, Form B, Form C, Form D, Form E, Form F, Form G, and Amorphous Form of Compound I. Where possible, the results of each test for each different polymorph are provided. Forms A, C, D and E were prepared as pure forms. Forms B, F, and G were prepared as mixtures with Form A.
Various tests were performed in order to physically characterize the polymorphs of Compound I including X-ray powder diffraction (XRPD), differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), hot stage microscopy, Fourier transform infrared spectroscopy (FT-IR), Fourier transform Raman spectrometry, linked thermogravimetric-infrared spectroscopy (TG-IR), solution proton nuclear magnetic resonance (1H-NMR), solid state 13carbon nuclear magnetic resonance (13C-NMR), and moisture sorption and desorption analysis (M S/Des).

Salt screening
Physicochemical properties of drug substances, such as solubility, dissolution rate, and physicochemical stability can be altered significantly by salt formation. Consequently, important properties of the drug product such as bioavailability or shelve life can be radically influenced. Crystallics’ technology platform for crystallization screening accommodates salt screening studies using only minimal amounts of drug substance while still performing a large number of experiments. High-throughput salt screening is used for both early phase salt selection studies and broad patent protection.
Salt selection – A powerful strategy for crystal form optimization
Pharmaceutical developers have focused efforts on finding and formulating a thermodynamically stable crystalline form with acceptable physical properties for a given compound. This is reasonable, given the need to avoid cascading from a meta-stable form to a more stable one in unpredictable fashion.
Occasionally certain physical properties, such as low aqueous solubility, are limiting to performance of the compound, leading to poor oral bioavailability or insufficient solubility for an injection formulation. One of the main strategies used to affect physical performance of a compound and one that is often employed by pharmaceutical scientists is the practice of salt selection (23). At least half of compounds in marketed products are in the form of a salt for one reason or another.
This fact alone speaks to the versatility of the salt selection approach. Salt forms of a pharmaceutical can have many benefits, such as improved stability characteristics, optimal bioavailability and aqueous solubility for an injectable formulation. Salts, like all other crystalline forms, are subject to polymorphism and solvate formation, thus requiring the same form identification studies as are needed for a neutral compound.
A remarkable example of co-optimization of properties is indinavir (HIV protease inhibitor), which is marketed as the sulfate salt ethanol solvate (24,25) The crystalline free base has variable oral bioavailability in dogs (26,27) and humans (28). While acidic solutions of the base compound showed good oral pharmacokinetics, the stability of the drug in acidic solution is not consistent with a product (26). Therefore, the discovery of the salt form ensured both shelf stability and robust bioavailability performance. The salt selection strategy is limited in two ways.
First, salt formation relies on the presence of one or more ionizable functional groups in the molecule; many drugs and development compounds lack this feature.
Second, our ability to predict a priori whether a given compound will form a crystalline salt (or salts) is non-existent. The ability to actively identify crystalline salt forms has been confined to manual empirical evaluation using multiple salt formers for a given acid or base. Recently advances have been made in the area of high-throughput salt selection and crystal engineering strategies associated with salt formation (14,29-32).
In one case, we have advocated the simultaneous assessment of polymorphism as a way to help rank the developability of different crystalline salts (14). While salt forms will continue to have a prominent place in pharmaceutical science, the need for enhanced productivity dictates that every advantage must be sought to aid the design of an appropriate crystalline form of an active molecule.
Specifically, the ability to design scaffolds into crystalline forms will enhance our capacity to convert interesting molecules into effective drugs. Crystal engineering offers some additional tools in this regard. 
CASE STUDY FORM A ONLY US8084605
TRELAGLIPTIN SUCCINATE
Form A may be prepared by crystallization from the various solvents and under the various crystallization conditions used during the polymorph screen (e.g., fast and slow evaporation, cooling of saturated solutions, slurries, and solvent/antisolvent additions). Tables B and C of Example 3 summarize the procedures by which Form A was prepared.
For example, Form A was obtained by room temperature slurry of an excess amount of Compound I in acetone, acetonitrile, dichloromethane, 1,4-dioxane, diethyl ether, hexane, methanol, isopropanol, water, ethylacetate, tetrahydrofuran, toluene, or other like solvents on a rotating wheel for approximately 5 or 7 days.
The solids were collected by vacuum filtration, and air dried in the hood. Also, Form A was precipitated from a methanol solution of Compound I by slow evaporation (SE). Form A was characterized by XRPD, TGA, hot stage microscopy, IR, Raman spectroscopy, solution 1H-NMR, and solid state 13C-NMR. Figure 1 shows a characteristic XRPD spectrum (CuKa, λ=1.5418A) of Form A. The XRPD pattern confirmed that Form A was crystalline. Major X-Ray diffraction lines expressed in °2Θ and their relative intensities are summarized in Table 1. Table 1. Characteristic XRPD Peaks (CuKa) of Form A


The above set of XRPD peak positions or a subset thereof can be used to identify Form A. One subset comprises peaks at about 11.31, 11.91, 12.86, 14.54, 15.81, 16.83, 17.59, 19.26, 19.52, 21.04, 22.32, 26.63, and 29.87 °2Θ. Another subset comprises peaks at about 11.31, 11.91, 19.26, 21.04, and 22.32 °2Θ; the peaks of this subset show no shoulder peaks or peak split greater than 0.2 °2Θ. Another subset comprises peaks at about 11.31, 11.91 and 22.32 °2Θ.  Figure 2 is a TGA thermogram of Form A. TGA analysis showed that Form A exhibited insignificant weight loss when heated from 25 0C to 165 0C; this result is indicative that Form A was an anhydrous solid.
Figure 2 is a TGA thermogram of Form A. TGA analysis showed that Form A exhibited insignificant weight loss when heated from 25 0C to 165 0C; this result is indicative that Form A was an anhydrous solid.  Figure 3 shows a characteristic DSC thermogram of Form A. DSC analysis showed a single endothermic event occurred at approximately 195 0C (peak maximum). This endothermic event was confirmed by hot stage microscopy which showed the melting of Form A, which onset around 177 0C and the melting point estimated to be at approximately 184 0C.
Figure 3 shows a characteristic DSC thermogram of Form A. DSC analysis showed a single endothermic event occurred at approximately 195 0C (peak maximum). This endothermic event was confirmed by hot stage microscopy which showed the melting of Form A, which onset around 177 0C and the melting point estimated to be at approximately 184 0C. 
Figure 4 (A-D) shows a characteristic FT-IR spectrum of Form A. The major bands expressed in reciprocal wavelengths (wavenumber in cm”1) are positioned at about 3815, 3736, 3675, 3460, 3402, 3141, 3098, 3068, 3049, 2953, 2934, 2854, 2760, 2625, 2536, 2481, 2266, 2225, 2176, 1990, 1890, 1699, 1657, 1638, 1626, 1609, 1586, 1553, 1517, 1492, 1478, 1450, 1419, 1409, 1380, 1351, 1327, 1289, 1271, 1236, 1206, 1180, 1158, 1115, 1087, 1085, 1064, 1037, 1027, 971, 960, 951, 926, 902, 886, 870, 831, 820, 806, 780, 760, 740, 728, 701, 685, 668, 637, 608, 594, 567, 558, and 516 cm”1 (values rounded to the nearest whole number). This unique set of IR absorption bands or a subset thereof can be used to identify Form A.
One such subset comprises absorption bands at about 3141, 3098, 3068, 3049, 2953, 2934, 2854, 2266, 2225, 1699, 1657, 1609, 1586, 1553, 1517, 1492, 1478, 1450, 1380, 1351, 1327, 1236, 1206, 1115, 1063, 902, 886, 870, 820, 780, 760, 685, 608, 594, and 516 cm 1. Another subset comprises absorption bands at about 3141, 2953, 2934, 2854, 2266, 2225, 1699, 1657, 1450, 1206, 886, 760, 685, 594, and 516 cm 1. Yet another subset comprises absorption bands at about 3141, 2953, 2934, 2266, 1699, 1657, 1450, and 1206 cm 1.
Aprepitant case study FTIR.. READING MATERIAL http://alpha.chem.umb.edu/chemistry/ch361/spring%2005/ftir%20polymorph.pdf
Figure 5 (A-D) shows a characteristic Raman spectrum of Form A. The major Raman bands expressed in reciprocal wavelengths (wavenumber in cm”1) are positioned at about 3100, 3068, 3049, 2977, 2954, 2935, 2875, 2855, 2787, 2263, 2225, 2174, 1698, 1659, 1626, 1607, 1586, 1492, 1478, 1451, 1439, 1409, 1400, 1382, 1351, 1328, 1290, 1281, 1271, 1237, 1223, 1213, 1180, 1155, 1134, 1115, 1084, 1063, 1035, 971, 952, 926901, 868, 805, 780, 759, 740, 727, 701, 686, 669, 609, 594, 566, 558, 516, 487, 479, 433, 418, 409, 294, 274, 241, 218, 191 and 138 cm”1 (values rounded to the nearest whole number). This unique set of Raman bands or a subset thereof may be used to identify Form A.
One such subset comprises Raman bands at about 2954, 2935, 2225, 1698, 1659, and 1607 cm”1. Another subset comprises Raman bands at about 3068, 2954, 2935, 2225, 1698, 1659, 1607, 1586, 1223, 1180, 901, 780, 759, 669, and 516 cm”1. Yet another subset comprises Raman bands at about 3100, 3068, 2225, 1698, 1659, 1607, 1586, 1351, 1237, 1223, 1180, 1155, 1134, 1115, 1063, 952, 926, 901, 868, 805, 780, 759, 740, 669, 609, and 516 cm”1.
Form A was further characterized by solution 1H NMR and solid-state 13carbon NMR. The spectra are reported in Figures 6 and 7, respectively. Chemical assignments were not performed; however, the spectra are consistent with the known chemical structure of Compound I. US8084605

 Example 11. Characterization of Form A Material prepared by the procedure of Example 1 was designated as Form A. The material was characterized by XRPD, TGA, DSC, hot stage microscopy, FT-IR, FT- Raman, 1H NMR, and 13C NMR. The analyses were conducted according to the procedures outlined in Section B of Example 3.
Example 11. Characterization of Form A Material prepared by the procedure of Example 1 was designated as Form A. The material was characterized by XRPD, TGA, DSC, hot stage microscopy, FT-IR, FT- Raman, 1H NMR, and 13C NMR. The analyses were conducted according to the procedures outlined in Section B of Example 3.
The characteristic spectra and thermograms for Form A are reported in Figures 1-7. The characterization data are summarized in Table D. Table D. Characterization Data of Form A of Compound I US8084605

Amorphous solid dispersion screening
Using the amorphous form of a drug substance offers several advantages with respect to dissolution rate and solubility of the substance. However, reduced chemical stability, increased hygroscopicity and, most important, physical instability are the major drawbacks of using the amorphous phase in the final drug product. These drawbacks can be overcome by stabilizing the amorphous phase of the API in a polymer matrix, e.q. an amorphous solid dispersion. Amorphous phases dissolve more rapidly than crystalline forms, and can significantly increase bioavailability of poorly water soluble drugs substances. However, the use of amorphous materials requires confidence that crystallization will not occur during the product lifespan. For a material that has never been obtained in a crystalline form, focus should be put on attempting to crystallize it. Crystallics has extensive experience of obtaining crystalline phases from amorphous materials.
Dispersions of a drug substance onto a polymeric matrix has received increased attention in recent years. A successful dispersion results in an amorphous solid material and will show improved dissolution rates and higher apparent solubility characteristics, as well as, sufficient resistance to chemical degradation and should be physically stable e.q. sufficient high glass transition temperature avoiding crystallization of the API.
A variety of factors contribute to the formation of a suitable Amorphous Solid Dispersion (ASD), including the nature of the polymer, the drug polymer ratio, the impact of surfactants and the solvent used in the process. Crystallics has developed high-throughput solid dispersion screening technology in order to find the optimal combination of these factors.
Example 10. Preparation of Amorphous Form US8084605
A sample of Compound I (40 mg) was dissolved in 1000 μl of water. The solution was filtered through a 0.2 μm nylon filter into a clean vial then frozen in a dry ice/acetone bath. The vials were covered with a Kimwipe then placed on a lyophilizer overnight. The resulting solids yielded Amorphous Form. 8. Amorphous Form The Amorphous Form of Compound I was prepared by lyophilization of an aqueous solution of Compound I (Example 10). The residue material was characterized by XRPD and the resulting XRPD spectrum displayed in Figure 26. The XRPD spectrum shows a broad halo with no specific peaks present, which confirms that the material is amorphous. The material was further characterized by TGA, DSC, hot stage microscopy, and moisture sorption analysis. 
TGA analysis (Figure 27) showed a 1.8% weight loss from 25 0C to 95 0C, which was likely due to loss of residual solvent. 
DSC analysis (Figure 28) showed a slightly concave baseline up to an exotherm at 130 0C (recrystallization), followed by an endotherm at 194 0C, which results from the melting of Form A. Hot stage microscopy confirmed these recrystallization and melting events (micrographs not included). An approximate glass transition was observed (Figure29) at an onset temperature of 82 0
C. 
Moisture sorption/desorption data (Figure 30 and Example 25) showed a 1.0% weight loss on equilibration at 5% relative humidity. Approximately 8% of weight was gained up to 65% relative humidity. Approximately 7% of weight was lost at 75% relative humidity. This is likely due to the recrystallization of the amorphous material to a crystalline solid. A 4.4% weight gain was observed on sorption from 75% to 95% relative humidity. Approximately 4.7% weight was lost on desorption from 95% to 5% relative humidity. 
The solid material remaining after the moisture sorption analysis was determined to be Form A by XRPD (Figure 31). Table H. Characterization Data of Amorphous Form US8084605

T=temperature, RH=relative humidity, MB = moisture sorption/desorption analysis Example 19: Relative Humidity Stressing Experiments

Table B. Crystallization Experiments of Compound I from Solvents




a) FE = fast evaporation; SE = slow evaporation; RT = room temperature; SC = slow cool; CC = crash cool, MB = moisture sorption/desorption analysis b) qty = quantity; PO = preferred orientation Table C. Crystallization Experiments of Compound I in Various Solvent/Antisolvent

a precipitated by evaporation of solvent Table A. Approximate Solubilities of Compound I US8084605


a) Approximate solubilities are calculated based on the total solvent used to give a solution; actual solubilities may be greater because of the volume of the solvent portions utilized or a slow rate of dissolution. Solubilities are reported to the nearest mg/mL.
Example 3.
Polymorph Screen Compound I as prepared by the method described in Example 1 was used as the starting material for the polymorph screen. Solvents and other reagents were of ACS or HPLC grade and were used as received. A. Sample Generation. Solids for form identification were prepared via the following methods from Compound I.
1. Fast Evaporation (FE) A solution of Compound I was prepared in test solvents. The sample was placed in the hood, uncovered, to evaporate under ambient conditions. The solids were analyzed by XRPD for form identification.
2. Slow Evaporation (SE) A solution of Compound I was prepared in test solvents. The sample was placed in the hood, covered with foil rendered with pinholes, to evaporate under ambient conditions. The solids were analyzed by XRPD for form identification.
3. Room Temperature (RT) Slurries An excess amount of Compound I was slurried in test solvent on a rotating wheel for approximately 5 or 7 days. The solids were typically collected by vacuum filtration, air dried in the hood, and analyzed by XRPD for form identification.
4. Elevated Temperature Slurries Excess Compound I was slurried in test solvents at 47 0C on a shaker block for approximately 5 days. The solids were collected by vacuum filtering, dried in the hood, and then analyzed by XRPD for form identification.
5. Slow Cooling Crystallization (SC)
A saturated or near saturated solution of Compound I was prepared at elevated temperature. The samples were filtered through warmed 0.2 μm filters into warmed vials. The heat source was turned off and the samples slowly cooled to ambient temperature. If precipitation did not occur within a day the samples were placed in the refrigerator. The samples were transferred to a freezer if precipitation did not occur within several days. The solids were collected by decanting the solvent or vacuum filtration, dried in the hood and analyzed by XRPD for form identification.
6. Crash Cooling Crystallization (CC) A saturated or near saturated solution of Compound I was prepared at elevated temperature. The samples were filtered through warmed 0.2 μm filters into warmed vials then rapidly cooled in an acetone/dry ice or ice bath. If precipitation did not occur after several minutes the samples were placed in the refrigerator or freezer. Solids were collected by decanting solvent or vacuum filtration, dried in the hood, and then analyzed by XRPD. Samples that did not precipitate under subambient conditions after several days were evaporated in the hood and analyzed by XRPD for form identification.
7. Solvent/Antisolvent Crystallization (S/AS) A solution of Compound I was prepared in test solvent. A miscible antisolvent was added with a disposable pipette. Precipitate was collected by vacuum filtration or decanting solvent. The samples were stored under subambient conditions if precipitation did not occur. If solids were not observed after several days the samples were evaporated in the hood. Collected solids were analyzed by XRPD for form identification.
8. Relative Humidity (RH) Stressing Experiments Samples of Compound I were placed uncovered in approximately 58%, 88%, and 97% relative humidity jars. The samples were stored in the jars for approximately 8 days. The solids were collected and analyzed by XRPD for form identification.
9. Lyophilization Compound I was dissolved in water in a glass vial. The solution was frozen by swirling the vial in an acetone/dry ice bath. The frozen sample was placed on the lyophilizer until all of the frozen solvent was removed. The solids were collected and analyzed by XRPD for form identification.
10. Grinding Experiments Aliquots of Compound I were ground manually with a mortar and pestle as a dry solid and a wet paste in water. The samples were ground for approximately three minutes. The solids were collected and analyzed by XRPD for form identification.
11. Dehydration Experiments Hydrated samples of Compound I were dehydrated at ambient conditions (2 days) and in an ambient temperature vacuum oven (1 day). The solids were collected and analyzed by XRPD for form identification.
12. Vapor Stress Experiments Amorphous Compound I was placed in acetone, ethanol, and water vapor chambers for up to eight days. The solids were collected and analyzed by XRPD for form identification.
STABILITY STUDY Stability studies are commonly performed for new drug entities with chemical stability and impurity formation being investigated. It is also important to monitor the physical stability under these same conditions to anticipate any form changes that may occur. As an example, many hydrates will dehydrate to a lower hydrate or anhydrous form at elevated temperatures. Anhydrous materials can also undergo form transformations to other anhydrous forms upon heating.
These types of changes can be monitored using heating studies in an oven with subsequent XRPD analysis or in-situ variable temperature XRPD can be used to look for changes. In other cases, anhydrates will convert to hydrates or the API in an amorphous solid dispersion may crystallize under elevated relative humidity (RH) conditions.
Equilibration in RH chambers with subsequent analysis by XRPD or in-situ variable RH XRPD experiments can be used to readily identify these form changes. Once the effect of temperature and RH on form changes is understood, this can be factored into other processes such as drying, formulation, storage, and packaging B. Sample Characterization. The following analytical techniques and combination thereof were used determine the physical properties of the solid phases prepared.
1. X-Ray Powder Diffraction (XRPD)
XRPD is commonly used as the initial method of analysis for form screens. For polymorph, salt, and co-crystal screens XRPD is used to determine if a new form has been produced by comparing the powder pattern to all known forms of the API and the counterion/guest. If a new form is found by XRPD, additional characterization by other methods is in order. For amorphous solid dispersion screens, XRPD is used to confirm a lack of crystallinity indicated by an amorphous halo in the powder pattern.
The halos will move depending on the concentration and interactions of the API and polymer. Computational methods have also been used with XRPD data to establish miscibility of amorphous solid dispersions X-ray powder diffraction is a front line technique in solid form screening and selection based on its ability to give a fingerprint of the solid-state structure of a pharmaceutical material. Understanding the solid forms of a pharmaceutical compound provides a road map to help direct a variety of development activities ranging from crystallization, formulation, packaging, storage, and performance.
Different screening and selection strategies are warranted in early and late development because different information is needed at the various stages. Solid form selection and formulation approaches need to be investigated together and tailored to the situation. It is important to include solid form selection and possible changes in form as part of the risk management strategy throughout the drug development process.
X-ray powder diffraction (XRPD) analyses were performed using an Inel XRG- 3000 diffractometer equipped with a CPS (Curved Position Sensitive) detector with a 2Θ (2Θ) range of 120°. Real time data were collected using Cu-Ka radiation starting at approximately 4 °2Θ at a resolution of 0.03 °2Θ. The tube voltage and amperage were set to 40 kV and 30 mA, respectively. The pattern is displayed from 2.5 to 40 °2Θ. Samples were prepared for analysis by packing them into thin- walled glass capillaries. Each capillary was mounted onto a goniometer head that is motorized to permit spinning of the capillary during data acquisition. The samples were analyzed for approximately 5 minutes.
Instrument calibration was performed using a silicon reference standard. Peak picking was performed using the automatic peak picking in the Shimadzu XRD-6000 Basic Process version 2.6. The files were converted to Shimadzu format before performing the peak picking analysis. Default parameters were used to select the peaks.
2. Thermogravimetric Analysis (TGA)
Thermogravimetric (TG) analyses were performed using a TA Instruments 2950 thermogravimetric analyzer. Each sample was placed in an aluminum sample pan and inserted into the TG furnace. The furnace was first equilibrated at 25 0C, then heated under nitrogen at a rate of 10 °C/min, up to a final temperature of 350 0C. Nickel and Alumel™ were used as the calibration standards.
3. Differential Scanning Calorimetry (DSC)
Differential scanning calorimetry (DSC) was performed using a TA Instruments differential scanning calorimeter 2920. The sample was placed into an aluminum DSC pan, and the weight accurately recorded. The pan was covered with a lid and then crimped. The sample cell was equilibrated at 25 0C and heated under a nitrogen purge at a rate of 10 °C/min, up to a final temperature of 350 0C. Indium metal was used as the calibration standard. Reported temperatures are at the transition maxima. For studies of the glass transition temperature (Tg) of the amorphous material, the sample cell was equilibrated at ambient temperature, then heated under nitrogen at a rate of 20 °C/min, up to 100 0C. The sample cell was then allowed to cool and equilibrate at -20 0C. It was again heated at a rate of 20 °C/min up to 100 0C and then cooled and equilibrated at -20 0C. The sample cell was then heated at 20 °C/min up to a final temperature of 350 0C. The Tg is reported from the onset point of the transition.
4. Hot Stage Microscopy.
Hot stage microscopy was performed using a Linkam hot stage (model FTIR 600) mounted on a Leica DM LP microscope. The samples were prepared between two cover glasses and observed using a 20χ objective with crossed polarizers and first order compensator. Each sample was visually observed as the stage was heated. Images were captured using a SPOT Insight™ color digital camera with SPOT Software v. 3.5.8. The hot stage was calibrated using USP melting point standards.
5. Thermogravimetric-Infrared (TG-IR)
Thermogravimetric infrared (TG-IR) analyses were acquired on a TA Instruments thermogravimetric (TG) analyzer model 2050 interfaced to a Magna 560® Fourier transform infrared (FT-IR) spectrophotometer (Thermo Nicolet) equipped with an Ever-Glo mid/far IR source, a potassium bromide (KBr) beamsplitter, and a deuterated triglycine sulfate (DTGS) detector. The TG instrument was operated under a flow of helium at 90 and 10 cc/min for the purge and balance, respectively. Each sample was placed in a platinum sample pan, inserted into the TG furnace, accurately weighed by the instrument, and the furnace was heated from ambient temperature to 250 0C at a rate of 20 °C/min.
The TG instrument was started first, immediately followed by the FT-IR instrument. Each IR spectrum represents 32 co-added scans collected at a spectral resolution of 4 cm“1. A background scan was collected before the beginning of the experiment. Wavelength calibration was performed using polystyrene. The TG calibration standards were nickel and Alumel™. Volatiles were identified from a search of the High Resolution Nicolet TGA Vapor Phase spectral library.
6. Fourier Transform Infrared Spectroscopy (FT-IR)
Infrared spectra were acquired on a Magna-IR 560® or 860® Fourier transform infrared (FT-IR) spectrophotometer (Thermo Nicolet) equipped with an Ever-Glo mid/far IR source, an extended range potassium bromide (KBr) beamsplitter, and a deuterated triglycine sulfate (DTGS) detector. A diffuse reflectance accessory (the Collector™, Thermo Spectra-Tech) was used for sampling. Each spectrum represents 256 co-added scans collected at a spectral resolution of 4 cm“1. Sample preparation consisted of physically mixing the sample with KBr and placing the sample into a 13 -mm diameter cup. A background data set was acquired on a sample of KBr. A Log 1/R (R = reflectance) spectrum was acquired by taking a ratio of these two data sets against each other. Wavelength calibration was performed using polystyrene. Automatic peak picking was performed using Omnic version 7.2.
7. Fourier Transform Raman Spectroscopy (FT-Raman)
FT-Raman spectra were acquired on a Raman accessory module interfaced to a Magna 860® Fourier transform infrared (FT-IR) spectrophotometer (Thermo Nicolet). This module uses an excitation wavelength of 1064 nm and an indium gallium arsenide (InGaAs) detector. Approximately 0.5 W of Nd)YVO4 laser power was used to irradiate the sample. The samples were prepared for analysis by placing the material in a glass tube and positioning the tube in a gold-coated tube holder in the accessory. A total of 256 sample scans were collected from at a spectral resolution of 4 cm“1, using Happ-Genzel apodization. Wavelength calibration was performed using sulfur and cyclohexane. Automatic peak picking was performed using Omnic version 7.2.
8. Solid State Nuclear Magnetic Resonance Spectroscopy (13C-NMR)
The solid-state 13C cross polarization magic angle spinning (CP/MAS) NMR spectrum was acquired at ambient temperature on a Varian UN1TYINOVA-400 spectrometer (Larmor frequencies: 13C = 100.542 MHz, 1H = 399.799 MHz). The sample was packed into a 4 mm PENCIL type zirconia rotor and rotated at 12 kHz at the magic angle. The spectrum was acquired with phase modulated (SPINAL-64) high power 1H decoupling during the acquisition time using a 1H pulse width of 2.2 μs (90°), a ramped amplitude cross polarization contact time of 5 ms, a 30 ms acquisition time, a 10 second delay between scans, a spectral width of 45 kHz with 2700 data points, and 100 co-added scans. The free induction decay (FID) was processed using Varian VNMR 6.1C software with 32768 points and an exponential line broadening factor of 10 Hz to improve the signal-to- noise ratio. The first three data points of the FID were back predicted using the VNMR linear prediction algorithm to produce a flat baseline. The chemical shifts of the spectral peaks were externally referenced to the carbonyl carbon resonance of glycine at 176.5 ppm. 9. Solution Nuclear Magnetic Resonance Spectroscopy (1H-NMR) The solution 1H NMR spectrum was acquired at ambient temperature with a
spectrometer at a 1H Larmor frequency of 399.803 MHz. The sample was dissolved in methanol. The spectrum was acquired with a 1H pulse width of 8.4 μs, a 2.50 second acquisition time, a 5 second delay between scans, a spectral width of 6400 Hz with 32000 data points, and 40 co-added scans. The free induction decay (FID) was processed using Varian VNMR 6.1C software with 65536 points and an exponential line broadening factor of 0.2 Hz to improve the signal-to-noise ratio. The spectrum was referenced to internal tetramethylsilane (TMS) at 0.0 ppm. 10.
Moisture Sorption/Desorption Analysis Moisture sorption/desorption data were collected on a VTI SGA-100 Vapor Sorption Analyzer. Sorption and desorption data were collected over a range of 5% to 95% relative humidity (RH) at 10% RH intervals under a nitrogen purge. Samples were not dried prior to analysis. Equilibrium criteria used for analysis were less than 0.0100% weight change in 5 minutes, with a maximum equilibration time of 3 hours if the weight criterion was not met. Data were not corrected for the initial moisture content of the samples. NaCl and PVP were used as calibration standards
Does solid form matter?
Sometimes the properties of two solid forms of a drug are quite similar. In other cases, the physical and chemical properties can vary dramatically and have great impact on pharmacokinetics, ease of manufacturing, and dosage form stability. Properties that can differ among solid forms of a substance include color, solubility, crystal shape, water sorption and desorption properties, particle size, hardness, drying characteristics, flow and filterability, compressibility, and density.
Different solid forms can have different melting points, spectral properties, and thermodynamic stability. In a drug substance, these variations in properties can lead to differences in dissolution rate, oral absorption, bioavailability, levels of gastric irritation, toxicology results, and clinical trial results. Ultimately both safety and efficacy are impacted by properties that vary among different solid forms. Stability presents a special concern, since it’s easy to inadvertently generate the wrong form at any point in the development process.
Because energy differences between forms are usually relatively small, form interconversion is common and can occur during routine API manufacturing operations and during drug product formulation, storage, and use. The stakes are high. Encountering a new solid form during late stages of development can delay filing. A new form appearing in drug product can cause product withdrawal.
When should a search for solid forms begin?
The key to speed in the drug development process is to do it right the first time. For solid pharmaceuticals, that means:
- identify the optimum solid form early in drug development
- make the same form for clinical material and commercial API
- develop a crystallization process that assures control of solid form
- produce a drug product with solid form stability through expiration
scientists strongly recommend that investigation of possible solid forms of a new chemical entity be carried out as early in the development process as drug supply will allow. The best approach has three stages. The first stage, more relevant to some development processes than to others, is a milligram-scale abbreviated screen on efficacious compounds prior to final IND candidate selection. This early information can be used to guide selection of salts and solid forms for scale-up and toxicology studies. The second stage is full polymorph screening and selection of optimum solid form. This stage is important to all development processes and should certainly occur before the first GMP material is produced. In the case of ionic drugs, various salts should be prepared and screened for polymorphs and hydrates. The third stage, an exhaustive screen carried out before drug launch, is an effort to find and patent all of the forms of a high-potential drug. Staging the screening in this way optimizes the balance among the factors of early knowledge of options, probability of commercial success, and judicious investment of R&D money.
 Delay in understanding solid form issues results in problems like different batches of clinical material having different solid forms. Another common and preventable dilemma arises when clinical trials are carried out with one form while commercial production generates another. In this case, bridging studies are required to demonstrate to regulatory agencies that the clinicals are relevant. ICH guidelines require a search for solid forms, comparison of properties that might affect product efficacy, and, if appropriate, setting of solid form specifications.
Delay in understanding solid form issues results in problems like different batches of clinical material having different solid forms. Another common and preventable dilemma arises when clinical trials are carried out with one form while commercial production generates another. In this case, bridging studies are required to demonstrate to regulatory agencies that the clinicals are relevant. ICH guidelines require a search for solid forms, comparison of properties that might affect product efficacy, and, if appropriate, setting of solid form specifications.
How is solid form controlled in API manufacture?
It is important to control solid form during API synthesis in order to demonstrate complete process control to regulatory agencies. Different solid forms can have different solubilities and can affect recovery of API. Purification efficiencies can vary due to differential exclusion of impurities. Filtration and transfer characteristics often differ between forms. Ease of drying can vary due to different abilities to bind solvent and water in the crystal lattice. A prevalent but incorrect belief is that solid form is determined primarily by choice of crystallization solvent. In fact, it is well established that parameters like temperature, supersaturation level, rate of concentration or cooling, seeding, and ripening can have an overriding effect. These variables must be controlled to ensure consistency of solid form in API.
Can solid form problems arise in drug products, too?
The potential for solid form variation does not end at API production. Solid form issues remain through formulation, manufacture, storage, and use of drug product. It is common to observe form transformation during standard manufacturing operations like wet granulation and milling. Excipient interactions and compaction can induce form changes. Changes can occur in the final dosage form over time. Suspensions, including those in transdermal patches, are particularly vulnerable because they provide a low-energy pathway (dissolution/recrystallization) for form interconversion. Lyophile cakes are normally amorphous, but can crystallize on storage leading to difficulty in reconstitution. Even products containing drug in solution, such as filled gel caps, can be affected if the solution is or becomes supersaturated with respect to one of the possible solid forms of the drug.
How can you tell when you have a solid form problem?
Whenever there is a specification failure in drug product or drug substance, solid form changes should be considered in the search for causes. Particularly symptomatic is failure to meet melting point or dissolution specifications. Changes in humidity, crystallization conditions, or crystallization solvent can produce unwanted forms. Solvents known to readily produce solvates include water, alcohols, chlorinated hydrocarbons, cyclic ethers, ketones, nitriles, and amides. Changes in the appearance of gel caps or cracking of tablet coatings can indicate solid form problems. Various solid-state analytical techniques can be used to identify solid form in API. Some techniques can even determine solid form of API in intact final dosage form. Among the most useful techniques for solid-state characterization are melting point, DSC, TGA, hot stage and optical microscopy, solid-state NMR, IR and Raman spectroscopy, and X-ray powder diffraction.
Is there any good news about polymorphism?
Polymorphism presents opportunities as well as challenges. Investigation of the properties of different forms of a commercial drug can lead to new products with improved onset time, greater bioavailability, sustained release properties, or other therapeutic enhancements. New forms can bring improvements in manufacturing costs or API purity. These improvements are patentable and can provide a competitive advantage. An underutilized potential of polymorphism is to solve formulation problems that cause the abandonment of potentially useful drugs in which much investment has already been made. 
SOLUBILITY
Solubility is an important parameter for new molecules especially with the emergence of many poorly soluble compounds in the drug discovery and development pipeline. Polymorphic forms can exhibit solubility differences that vary within a factor of 1-5, amorphous solid dispersions show an improvement one or two orders of magnitude higher, and salts and co-crystals fall between these extremes . A comparison of solubility values of pure forms will provide important information when deciding on a solid form or dosage form. X-ray powder diffraction will allow identification of pure forms for these types of measurements.
However, form changes during solubility and dissolution experiments are also possible and need to be investigated. Solids remaining at the end of solubility and dissolution experiments should always be analyzed initially by XRPD to determine if a form transformation has occurred under these conditions. If a form change has occurred, XRPD patterns can be compared to known forms (polymorphs, hydrates, salts, free acid/base) in order to identify the solids remaining. If a pattern is obtained that does not correspond to known forms, complementary methods will be needed to determine properties such as hydration state or a change in stoichiometry as would be observed from breaking a salt and forming the free acid/base or the formation of salts in buffered solutions.
FORMULATION
Formulators are charged with the responsibility to formulate a product which is physically and chemically stable, manufacturable, and bioavailable. Most drugs exhibit structural polymorphism, and it is preferable to develop the most thermodynamically stable polymorph of the drug to assure reproducible bioavailability of the product over its shelf life under a variety of real-world storage conditions. There are occasional situations in which the development of a metastable crystalline or amorphous form is justified because a medical benefit is achieved. Such situations include those in which a faster dissolution rate or higher concentration are desired, in order to achieve rapid absorption and efficacy, or to achieve acceptable systemic exposure for a low-solubility drug.
Another such situation is one in which the drug remains amorphous despite extensive efforts to crystallize it. If there is no particular medical benefit, there is less justification for accepting the risks of intentional development of a metastable crystalline or amorphous form. Whether or not there is medical benefit, the risks associated with development of a metastable form must be mitigated by laboratory work which provides assurance that (a) the largest possible form change will have no substantive effect on product quality or bioavailability, and/or (b) a change will not occur under all reasonable real-world storage conditions, and/or (c) analytical methodology and sampling procedures are in place which assure that a problem will be detected before dosage forms which have compromised quality or bioavailability can reach patients.
Crystal engineering and co-crystals
Crystal engineering is generally considered to be the design and growth of crystalline molecular solids with the aim of impacting material properties. A principal tool is the hydrogen bond, which is responsible for the majority of directed intermolecular interactions in molecular solids. Co-crystals are multi-component crystals based on hydrogen bonding interactions without the transfer of hydrogen ions to form salts – this is an important feature, since Brønsted acid-base chemistry is not a requirement for the formation of a co-crystal.
Co-crystallization is a manifestation of directed self-assembly of different components. Co-crystals have been described of various organic substances over the years (33,34) and given various names, such as addition compounds (35,36) molecular complexes (37,38) and heteromolecular co-crystals (39). Regardless of naming convention, the essential meaning is that of a multi-component crystal where no covalent chemical modification of the constituents occurs as a result of the crystal formation. Pharmaceuticals co-crystals have only recently been discussed as useful materials for drug products.
Pharmaceutical co-crystals
Pharmaceutical co-crystals can be defined as crystalline materials comprised of an active pharmaceutical ingredient (API) and one or more unique co-crystal formers, which are solids at room temperature. Co-crystals can be constructed through several types of interaction, including hydrogen bonding, p-stacking, and van der Waals forces. Solvates and hydrates of the API are not considered to be co-crystals by this definition. However, co-crystals may include one or more solvent/water molecules in the crystal lattice. An example of putative design, a construction and preparation process is shown in Figure 2 for the 5-fluororuracil:urea 1:1 co-crystal(40).
This real example neatly illustrates the opportunity and challenge that exists currently with designing pharmaceutical co-crystals. Firstly, the ‘design’ is challenging because we have no ability to predict the exact crystal structure that may result from a crystallization attempt. By analogy to the challenge of deriving protein structure from first principles, the primary sequence (chemical structure in our case) is known and elements of secondary structure (the 2-D tape construction in Figure 2) are somewhat discernible from primary information. Prediction of the actual 3-D folded conformation (tertiary structure or obtained by self-assembly) is not possible. In other words, while we currently have the ability to project which things associate in what approximate manner on the secondary level, crystal structure prediction is essentially an intractable proposition.
By extension, and just as the exact function of a protein and quantitative parameters of activity are not predictable from primary and secondary structure, the prediction of crystal properties is not possible in the absence of structural information and measurements. There is early evidence that practitioners were aware that apparent co-crystallization of drugs could lead to useful preparations (41). In fact, a ‘chemical compound’ composed of sulfathiazole and proflavin dubbed flavazole was used to treat bacterial infection during the Second World War (42).
The case of flavazole reveals insight into how two different molecules might interact in a putative co-crystal:“… flavazole is definitely a chemical compound containing equimolar proportions of sulphathiazole and proflavin base. It is believed that combination occurs through the acidic sulphonamide group (SO2NH) of the sulphathiazole and the basic centres of the proflavin. Perhaps the most realistic expression of the formula would be to place proflavin and sulphathiazole side by side with a comma between them.” (42) In the second half of the 20th century, interest in co-crystals evolved into the directed study of intermolecular interactions in crystalline solids (43-45). The technical development of routine single-crystal structure determination led to a watershed of data, now largely accessible through the Cambridge Structural Database (CSD) (46,47).
The structural data have become useful for understanding the intermolecular interactions in co-crystals in atomic level detail (48). Using insight gained from analysis of the CSD and directed experimentation, scientists attempt design of co-crystals with specific properties, such as color or non-linear optical response, by selecting starting components with appropriate molecular properties likely to exhibit specific intermolecular interactions in a crystal (49-52).
However, even when chemically compatible functional groups are present it is not possible to accurately predict if a co-crystal, a eutectic mixture or simply a physical mixture will result from any given experiment. As a result of these complexities, attention has been directed at the identification and characterization of intermolecular packing motifs with the goal of developing principles for co-crystal materials (53).

Figure 2. Steps involved in crystal engineering of a pharmaceutical phase, exemplified by the real example of co-crystallization of 5-fluorouracil and urea. Scientists in India have reported a rare example of synthon polymorphism in co-crystals of 4,4′-bipyridine and 4-hydroxybenzoic acid.

Polymorphism is defined as the ability of a material to exist in more than one form or crystal structure. It has important implications for the properties of such materials; for example in pharmaceuticals, the dissolution rate of a drug can be dependent on the polymorphic form. While this is a common phenomenon in single crystals it is much less common in co-crystals, systems where the structure has at least two distinct components. Gautam Desiraju from the Indian Institute of Science, found that when 4,4′-bipyridine and 4-hydroxybenzoic acid were dissolved together in a solvent such as methanol they would co-crystallise to form two different polymorphs. They noticed that a third form, a pseudopolymorph, was also present.
PROSPECTS FOR CRYSTAL ENGINEERING AND PHARMACEUTICAL CO-CRYSTALS
At the beginning of the 21st century, the field of crystal engineering has experienced significant development. Importantly, crystal engineering principles are now being actively considered for application to pharmaceuticals to modulate the properties of these valuable materials (54).
Because the physical properties that influence the performance of pharmaceutical solids are reasonably well appreciated, there is a unique opportunity to apply crystal engineering techniques and the appropriate follow-up studies to solve real world problems, such as poor physical and chemical stability or inadequate dissolution for appropriate biopharmaceutical performance of an oral drug. As structures and series of pharmaceutical co-crystals have begun to appear, we again find that properties cannot be predicted from the structures. Nevertheless, occasional trends have been suggested.
For example, insoluble drug compounds co-crystallized with highly water soluble complements tend to achieve kinetic solubilities in aqueous media several times greater than the pure form (55,56).
There are also more possible phases for each given active compound to consider, thus there will arguably be a greater opportunities for property enhancement. In terms of stability enhancement and solubilization, the example of the series of itraconazole co-crystals with pharmaceutically acceptable 1,4-diacids (55) suggests a strategy alternative to amorphous drug formulation. The co-crystal options presented retain the stability inherent in a crystalline state, while allowing for solubilization that significantly exceeds that of crystalline itraconazole base and rivals the performance of the engineered amorphous bead formulation (Sporanox®).
Where are we now? From recent literature it appears that knowledge gained over the past century and increasingly sophisticated screening techniques developed within the last decade are paving the way towards design of co-crystals with potentially improved pharmaceutical properties (55-58) In terms of the application to pharmaceutical systems, the field of crystal engineering is developing the retro-synthetic understanding of crystal structure using reasoning that is analogous to that applied by organic chemists. For example, the retro-synthetic approach in covalent synthesis operates on the level of a single molecule, while the analogous effort in crystal engineering focuses on the “supermolecule”:
 piracetam
piracetam
The assemblies that define the crystalline arrangement of the molecules as they self-organize into the solid-state. The parallels between the development of crystal engineering and synthetic organic chemistry run still deeper. Methodologies for carrying out these crystallizations are being developed alongside the development of new robust motifs (6,53,55,57,60). The importance of the solubility and dissolution relationships of the components of a putative co-crystal is becoming a matter of significant investigation (56,60). The same can be said for the roles of additives in templating novel forms.
Mechanical milling of materials has also been documented as a means to make co-crystals, and a recent example of polymorphic forms of caffeine:glutaric acid illustrates the opportunities of this type of processing to influence crystal form (61). With an increase in the understanding of the modes of self-assembly, one can start to address the design aspect towards making pharmaceutical co-crystals.
There remain several limitations to the application of what is currently known to the design of useful materials. As mentioned earlier, it remains intractable to reliably predict crystal structure. Multi-component crystals are well out of reach for prediction due in part to complex energetic landscapes, lack of appropriate charge density models and a large number of degrees of freedom, making computation unfeasible. Moreover, there is only a qualitative understanding of the interplay between intermolecular interactions and materials performance, especially for properties relevant to pharmaceuticals such as solubility, dissolution profile, hygroscopicity and melting point.
But the saving grace of the co-crystal approach comes in two guises: Complementarity and diversity. On the topic of complementarity, it is possible, by way of CSD database mining for instance, to identify trends of hetero-synthon occurrence in model systems. As for the diversity aspect, the space of possible co-crystal formers is large, limited only by pharmaceutical acceptability. Coupled with parameters such as stoichiometry variation and increase in the number of components (binary systems can be expanded into ternary ones, etc.), the opportunities appear vast.
THE FUTURE OF CRYSTAL ENGINEERING IN PHARMACEUTICAL SCIENCE
READ
Novel Challenges in Crystal Engineering: Polymorphs and New …
Where are we going? At this point, we have only just scratched the surface of materials science-driven pharmaceutical product design. In the 21st century, practitioners of pharmaceutical chemistry need to enumerate and exploit the opportunities of crystal form design that nature affords us, and thus gain increasing ability to design the materials we need from the molecules that we seek to convert into pharmaceuticals.
Learning will be facilitated by advances in crystallization automation (6,62), microscopy-spectroscopy techniques (Raman and IR microscopy) and new techniques such as terahertz spectroscopy and AFM, along with increasingly sophisticated X-ray diffraction lab instrumentation. In addition, further enhancements in the data mining tools associated with the CSD operating on an ever increasing number of high-quality crystal structures will undoubtedly lead to new knowledge and principles of interaction. 
The challenge placed before pharmaceutical scientists, now and in the future, is the following: (i) to understand the requirement of a particular compound in terms of materials structure and properties, and (ii) to creatively integrate crystal engineering within the limits of pharmaceutical acceptability of components to obtain new forms of active ingredients with desirable properties for formulation and delivery. It should become the collective mantra of medicinal chemists, process engineers and pharmaceutical scientists to “design and make the material we need.” This mantra can form the common aspiration for an industry that is in significant need of innovation and productivity enhancement.
Applications and Advantages
Applications
- Drug companies can use this technology to protect themselves against others generating and patenting polymorphs
Advantages
- Having more than one solid form of a drug allows optimization of drug dissolution behavior and shelf life

IP AND POLYMORPHS


REFERENCES
| [1] | E. L Paul, H.-H. Tung, and M. Midler. Organic crystallization processes. Powder Technology, 150:133-143, 2005. |
| [2] | C. R.Gardner,, C. T. Walsh and Ö. Almarsson. Drugs as materials: Valuing physical form in drug discovery, Nature Reviews Drug Disc, 926-934, 2004. |
| [3] | W. Ostwald. Studien uber die Bildung und Umwandlung fester Korper. Z Phys Chem, 22:289, 1897. |
| [4] | L. Kofler and A. Kofler. Thermo-mikro-methoden zur Kenneichnung organischer Stoffe und stoffgemische. Innsbruck, Wagner, 1954. |
| [5] | Bernstein J., Polymorphism in Molecular Crystals. Oxford University Press: New York, New York, 2002. |
| [6] | S. L. Morissette, Ö. Almarsson, M. L. Peterson, J. Remenar, M. Read, A. Lemmo, S. Ellis, M. J. Cima and C. R .Gardner. High-throughput Crystallization: Polymorphs, Salts, Co-crystals and Solvates of Pharmaceutical Solids. Adv Drug Deliv RevB, 275-300, 2004. |
| [7] | D. Sharmistha and D. J.Grant. Crystal Structures of Drugs: Advances in Determination, Prediction and Engineering. Nature Reviews Drug Discovery,3: 42-57, 2004. |
| [8] | D. Singhal and W. Curatolo. Drug Polymorphism and dosage form design: a practical perspective. Adv Drug Deliv Rev, 56: 335-347, 2004. |
| [9] | N. Lewis. Shedding Some Light on Crystallization Issues: Lecture Transcript for the First international Symposium on Aspects of Polymorphism and Crystallization – Chemical Development Issues. Org Proc Res Dev, 4: 407-412, 2000. |
| [10] | J. Bauer, S. Spanton, R. Henry, J. Quick, W. Dziki, W. Porter and J. Morris. Ritonavir: An Extraordinary Example of Conformational Polymorphism. Pharm Res,18: 859-866, 2001. |
| [11] | A. T. Hulme, S. L. Price and D. A. Tocher. A New Polymorph of 5-Fluorouracil Found Following Computational Crystal Structure Predictions. J Am Chem Soc, 127:1116-1117, 2005. |
| [12] | J. Lucas and P. Burgess. When Form Equals Substance: The Value of Form Screening in Product Life-Cycle Management. Pharma Voice, 2004. |
| [13] | J. Lucas and P. Burgess. The Paxil Patent: Four Simple Words, One Complex Claim Construction Case. Pharmaceutical Law & Industry, 2:2004. |
| [14] | J. F. Remenar, J. M. MacPhee, B. K. Larson, V. A. Tyagi, J. Ho, D. A. McIlroy, M. B. Hickey, P. B. Shaw and Ö. Almarsson.. Salt selection and simultaneous polymorphism assessment via high-throughput crystallization: the case of sertraline, Org Proc Res Dev, 7:990-996, 2003. |
| [15] | S. R. Byrn, R. R. Pfeiffer, M. Ganey, C. Hoiberg, and G. Poochikian. Pharmaceutical Solids: A strategic approach to regulatory considerations. Pharmaceutical Research, 12:945, 1995. |
| [16] | ICH Steering Committee. 11-10-2000. Good Manufacturing Practice Guide For Active Pharmaceutical Ingredients Q7a, ICH Harmonized Tripartite Guideline. |
| [17] | Ö. Almarsson and C. R. Gardner. Novel approaches the issues of developability. Current Drug Discovery, January:21-26, 2003. |
| [18] | B. C. Hancock and G.Zografi. Characteristics and significance of the amorphous state in pharmaceutical systems. J Pharm Sci, 86:1-12, 1997. |
| [19] | A. R. M. Serajuddin. Solid Dispersion of Poorly Water-Soluble Drugs: Early Promises, Subsequent Problems, and Recent Breakthroughs. J Pharm Sci, 88:1058- 1066, 1999. |
| [20] | L.Yu. Amorphous pharmaceutical solids: preparation, characterization and stabilization. Adv Drug Deliv Rev, 48:27-42, 2001. |
| [21] | See www.tricor.com |
| [22] | Y. Wu, A. Loper, E. Landis, I. Hettrick, L. Novak, K. Lynn, C. Chen, K. Thompson, R. Higgins, S. Shelukar, G. Kwei and D. Storey. The role of biopharmaceutics in the development of a clinical nanoparticle formulation of MK-0869: a Beagle dog model predicts improved bioavailability and diminished food effect on absorption in human. Int J Pharm, 285:135-46, 2004. |
| [23] | P. H. Stahl and M. Nakano Pharmaceutical Aspects of the Drug Salt Form. Handbook of Pharmaceutical Salts: Properties, Selection, and Use. New York: Wiley-VCH/VCHA, 2002. |
| [24] | J. H. Lin, D. Ostovic and J. P. Vacca, The Integration of Medicinal Chemistry, Drug Metabolism, and Pharmaceutical Research and Development in Drug Discovery and Development, chapter 11: The Story of Crixivan®, and HIV Protease Inhibitor, in Integration of Pharmaceutical Discovery and Development (ed. Borchardt et al.), Plenum Press, New York 1998. |
| [25] | B. D. Johnson, A. Howard, R. Varsolona, J. McCauley and D. K. Ellison. Indinavir Sulfate, in Harry G. Brittain (ed),Analytical Profiles of Drug Substances and Excipients, Academic Press: San Diego, 1999; V26, pp. 319-357. |
| [26] | G.Y. Kwei, L. B. Novak, L. A. Hettrick, E. R. Reiss, D. Ostovic, A. E. Loper, C. Y. Lui, R. J. Higgins, I. W. Chen, J. H. and Lin. , Rediospecific Intestinal Absorption of HIV protease inhibitor L-735,524 in beagle dogs. Pharm Res, 12:884, 1995. |
| [27] | J. H. Lin, I.-W. Chen, K. J. Vastag, and D. Ostovic. pH-dependent oral absorption of L-735,524, a potent HIV protease inhibitor, in rats and dogs. Drug Metab Disp 23:730-735, 1995. |
| [28] | K. C. Yeh, P. J. Deutsch, H. Haddix, M. Hesney, V. Hoagland, W. D. Ju, S. J. Justice, B. Osborne, A. T. Sterrett, J. A. Stone, E. Woolf and S. Waldman. Single-Dose Pharmacokinetics of Indinavir and the Effect of Food. Antimicrob. Agents Chemother, 42:332, 1998. |
| [29] | P J. Desrosiers. The potential of preform. Modern Drug Discovery, 7:40-43, 2004. |
| [30] | E A. Collier. A crystallization / crystal engineering approach to aid salt selection – anions. UMIST – Institute of Science and Technology, Dept. of Chem. Eng. 2004. |
| [31] | O. Félix, M. W. Hosseini, A. De Cian and J. Fischer. Crystal engineering of 2-D hydrogen bonded molecular networks based on the self-assembly of anionic and cationic modules. Chem Commun, 281-282, 2000. |
| [32] | C. B. Aakeroy and M. Niewenhuzen. Hydrogen-bonded layers of hydrogen malate anions – a framework for crystal engineering. J Am Chem Soc, 116:10983-10991, 1994. |
| [33] | For example, d-glucose:sodium chloride monohydrate is described in F. v. Kobell and J. F. Prakt Chemie, 28:489, 1843. |
| [34] | Quinhydrone was described in F. Wöhler. Untersuchungen über das Chinon. Annalen, 51:153, 1844. |
| [35] | For example, A. Buguet. Cryoscopy of Organic Mixtures and Addition Compounds. Compt Rend, 149:857-8, 1910. |
| [36] | H. Grossmann. Thiourea. Chemiker-Zeitung,31:1195-6, 1908. |
| [37] | For example see, A. Damiani, P. De Santis, E. Giglio, A. M. Liquori, R. Puliti and A. Ripamonti. The crystal structure of the 1:1 molecular complex between 1,3,7,9-tetramethyluric acid and pyrene. Acta Crystallogr,19:340-8, 1965. |
| [38] | J. N. Van Niekerk and D. H. Saunder. The crystal structure of the molecular complex of 4,4′-dinitrobiphenyl with biphenyl. Acta Crystallogr,1:44, 1948. |
| [39] | S. Pekker, E. Kovats, G. Oszlanyi, G. Benyei, G. Klupp, G. Bortel, I. Jalsovszky, E. Jakab, F. Borondics, K. Kamaras, M. Bokor, G. Kriza, K. Tompa and G. Faigel. Rotor-stator molecular crystals of fullerenes with cubane. Nature Materials, 4:764-767, 2005. |
| [40] | 5-Fluorouracil:urea. C4H3N2O2F.CH4N2O; C2/c; a = 9.461(3) Å, b = 10.487(3) Å, c = 15.808(4) Å, β = 99.89(7)º; Z = 4; T = 100(2) K; GOF = 1.023, R2 = 0.0663; wR2 = 0.1753. |
| [41] | C. G. Santesson. Addition Combinations. Archiv fuer Experimentelle Pathologie und Pharmakologie, 118:313-24, 1926. |
| [42] | J. McIntosh, R. H. M. Robinson, F. R. Selbie, J. P. Reidy, H. Elliot Blake and L. Guttmann. Lancet, 249:97-99, 1945. |
| [43] | K. Hoogsteen. The crystal and molecular structure of a hydrogen-bonded complex between 1-methylthymine and 9-methyladenine. Acta Crystallogr, 16:907-16, 1963. |
| [44] | F. S. Mathews,and A. Rich. The molecular structure of a hydrogen-bonded complex of N-ethyladenine and N-methyluracil. J Mol Bio, 8(1):89-95, 1964. |
| [45] | H. M. Sobell, K. Tomita and A. Rich. The crystal structure of an intermolecular complex containing a guanine and a cytosine derivative. Proc Natl Acad Sci USA, 49:885-92, 1963. |
| [46] | F. H. Allen. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr, B58:380-388, 2002. |
| [47] | I. J. Bruno,; J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor. New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallogr, B58:389-397, 2002. |
| [48] | F. H. Allen, O. Kennard and R. Taylor. Systematic analysis of structural data as a research technique in organic chemistry Acc Chem Res, 16:146-153, 1983. |
| [49] | M. C. Etter. A new role for hydrogen-bond acceptors in influencing packing patterns of carboxylic acids and amides. J Am Chem Soc, 104:1095-6, 1982. |
| [50] | M. C. Etter and G. M. Frankenbach. Hydrogen-bond directed cocrystallization as a tool for designing acentric organic solids. Chem Mater, 1:10-12, 1989. |
| [51] | Desiraju, G. R., Crystal Engineering. The Design of Organic Solids, Materials Science Monographs 54, Elsevier, Amsterdam, 1989. |
| [52] | J. A. R. P. Sarma and G. R. Desiraju. Crystal engineering via donor-acceptor interactions. X-ray crystal structure and solid state reactivity of the 1:1 complex, 3,4-dimethoxycinnamic acid-2,4-dinitrocinnamic acid. J Chem Soc, Chem Commun, :45-46, 1983. |
| [53] | For example, C. B. Åakeroy, J. Desper and B. A. Helfrich. CrystEngComm, 6:19-24, 2004. |
| [54] | Ö. Almarsson and M. J. Zaworotko. Crystal Engineering of the Composition of Pharmaceutical Phases. Do Pharmaceutical Co-crystals Represent a New Path to Improved Medicines? Chem Commun, :1889 – 1896, 2004. |
| [55] | J. F. Remenar, S. L. Morissette, M. L. Peterson, B. Moulton, J. M. MacPhee,H. Guzmán and Ö. Almarsson. Crystal engineering of novel cocrystals of a triazole drug with 1,4-dicarboxylic acids. J Am Chem Soc, 125:8456-8457, 2003. |
| [56] | S. L. Childs, L. J. Chyall, J. T. Dunlap, V. N. Smolenskaya, B. C. Stahly and G. P. Stahly. Crystal Engineering Approach To Forming Cocrystals of Amine Hydrochlorides with Organic Acids. Molecular Complexes of Fluoxetine Hydrochloride with Benzoic, Succinic, and Fumaric Acids. J Am Chem Soc,126:13335-13342, 2004. |
| [57] | S. Fleischman, L. Morales; B. Moulton, N. Rodríguez-Hornedo, R. Bailey Walsh and M. J. Zaworotko. Crystal engineering of the composition of pharmaceutical phases.Chem Commun,186, 2003. |
| [58] | A. V. Trask, W. D. S. Motherwell and W. Jones. Solvent-drop Grinding: Green Polymorph Control of Cocrystallisation. Chem Commun, 890-891, 2004. |
| [59] | N. Variankaval, R. Wenslow, J. Murry, R. Hartman, R. Helmy, E. Kwong, S.-D. Clas, C. Dalton and I. Santos. Preparation and Solid-State Characterization of Nonstoichiometric Cocrystals of a Phosphodiesterase-IV Inhibitor and L-Tartaric Acid. Cryst Growth Des, 6:690-700 2006. |
| [60] | S. J. Hehm, B. Rodriguez-Spong and N Rodriguez-Hornedo. Phase Solubility Diagrams of Cocrystal Are Explained by Solubility Product and Solution Complexation: Cryst Growth Des, 6:592-600, 2006. |
| [61] | A. V. Trask, N. Shan, W. D. S. Motherwell, W. Jones, S. Feng, R. B. H. Tan and K. J. Carpenter. Selective Polymorph Transformation via Solvent-drop Grinding. Chem Commun,, 880-882, 2005. |
| [62] | A. V. Trask, N. Shan, W. D. S. Motherwell, W. Jones, S. Feng, R. B. H. Tan and K. J. Carpenter. Selective Polymorph Transformation via Solvent-drop Grinding. Chem Commun,, 880-882, 2005. |
63……..POLYMORPHISM AND PATENTS http://www.collegio.unibo.it/uploads/ideas/joelbernstein.pdf 64…Aprepitant case study FTIR.. READING MATERIALhttp://alpha.chem.umb.edu/chemistry/ch361/spring%2005/ftir%20polymorph.pdf 65…..READ………….An Overview of Solid Form Screening During Drug Development, http://www.icdd.com/ppxrd/10/presentations/PPXRD-10_Ann_Newman.pdf 66…..CRYSTALLIZATION..http://www.intechopen.com/books/advanced-topics-on-crystal-growth/crystallization-from-the-conformer-to-the-crystal
67International Conference on Harmonization Q6A Guideline: Specifications for New Drug Substances and Products: Chemical Substances, October 1999.
68Center for Drug Evaluation and Research Guidance: Submitting Supporting Documentation in Drug Applications for the Manufacture of Drug Substances, February 1987.
69S. Byrn, R. Pfeiffer, M. Ganey, C. Hoiberg, and G. Poochikian. Pharmaceutical solids: A strategic approach to regulatory considerations. Pharm. Res. 12:945-954 (1995). 70
- H. Brittain. Methods for the characterization of polymorphs and solvates. In H. G. Brittain (ed.) Polymorphism in Pharmaceutical Solids. Marcel Dekker, Inc., New York, 1999, pp. 227-278.
- L. X. Yu and G. L. Amidon GL. Analytical Solutions to Mass Transfer. In: G. L. Amidon, P. I. Lee, and E. M. Topp (eds.) Transport Processes in Pharmaceutical Systems. Marcel Dekker, Inc., 1999, p. 23-54.
- G. L. Amidon, H. Lennernas, V. P. Shah, and J. R. Crison. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 12:413-420 (1995).
- L. X. Yu, G. L. Amidon, J. E. Polli, H. Zhao, M. Mehta, D. P. Conner, V. P. Shah, L. J. Lesko, M.-L. Chen, V. H. L. Lee, and A. S. Hussain. Biopharmaceutics Classification System: The scientific basis for biowaiver extension. Pharm. Res. 19:921-925 (2002).
- S. R. Byrn, R. R. Pfeiffer, and J. G. Stowell. Solid-State Chemistry of Drugs. 2nd Edition, SSCI, Inc., West Lafayette, Indiana, pp. 259-366.
- H. G. Brittain and E. F. Fiese. Effect of pharmaceutical processing on drug polymorphs and solvates. In H. G. Brittain (ed.) Polymorphism in Pharmaceutical Solids. Marcel Dekker, Inc., New York, 1999, pp. 331-362.
71 CRYSTALS POLYMORPHS IN PHARMAhttp://www.fcfar.unesp.br/arquivos/475753.pdf
72 API………….POLYMORPHISM pharmaceutical ingredients (APIs).http://www.ncbi.nlm.nih.gov/pubmed/19275600
73 polymorphs and co-crystals – ICDD POWER POINT PRESENTATION
74Thermodynamic stability and transformation of pharmaceutical … http://pac.iupac.org/publications/pac/pdf/2005/pdf/7703×0581.pdf
75http://www.imc.cas.cz/nmr/projekt/ws/springer.pdf
76 High-throughput crystallization: polymorphs, salts, co-crystalsand solvates of pharmaceutical solidshttp://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.85.5397&rep=rep1&type=pdf
77 Crystalline Solid – University of Utah College of Pharmacy Homepharmacy.utah.edu/pharmaceutics/pdf/Crystalline.pdfForm – a term encompassing all solids – polymorphs, solvates, amorphous … inPolymorphism in Pharmaceutical Solids
78..related to xrd
- Bish, DL and Post, JE, editors. 1989. Modern Powder Diffraction. Reviews in Mineralogy, Vol. 20. Mineralogical Society of America
- Cullity, B. D. 1978. Elements of X-ray diffraction. 2nd ed. Addison-Wesley, Reading, Mass
- I. Ivanisevic, R. B. McClurg and P. J. Schields: Uses of X-Ray Powder Diffraction in the Pharmaceutical Industry, Pharmaceutical Sciences Encyclopedia: Drug Discovery, Development and Manufacturing, Ed. by Shayne C. Gad., p. 1-42 (2010).
- S. R. Byrn, S. Bates and I. Ivanisevic: Regulatory Aspects of X-ray Powder Diffraction, Part 1, American Pharmaceutical Review, p.55-59 (2005)
- D. Beckers: The power of X-ray analysis, Pharmaceutical Technology Europe (2010), p.29,30.
- M. Sakata, S. Aoyagi, T. Ogura & E. Nishibori (2007): Advanced Structural Analyses by Third Generation Synchrotron Radiation Powder Diffraction, AIP Conference Proceedings, Vol. 879, pp. 1829-1832 (2007).
- Bergamaschi, A.; Cervellino, A.; Dinapoli, R.; Gozzo, F.; Henrich, B.; Johnson, I.; Kraft, P.; Mozzanica, A.; Schmitt, B.; Shi, X.: The MYTHEN detector for X-ray powder diffraction experiments at the Swiss Light Source. J. Synchrotron Rad. 17 (2010) 653–668.
- R.B. Von Dreele, P.W. Stephens, G.D. Smith, and R.H. Blessing: The First Protein Crystal Structure Determined from X-ray Powder Diffraction Data: a Variant of T3R3 Human Insulin Zinc Complex Produced by Grinding,Acta Cryst. D 56, 1549-53 (2000).
- Margiolaki, I., Wright, J. P., Fitch, A. N., Fox, G. C. & Von Dreele, R. B.: Synchrotron X-ray powder diffraction study of hexagonal turkey egg-white lysozyme, Acta Cryst. D61, 423–432 (2005). See also: Margiolaki, I. & Wright, J. P.: Powder crystallography on macromolecules, Acta Cryst. A64, 169–180 (2008).
- Fundamentals of Crystallography,C. Giacovazzo Ed., International Union of Crystallography, Oxford Science Publications, Third Edition (2011).
- The basics of Crystallography and Diffraction (Third edition, 2010), Christopher Hammond, IUCr, Oxford Science Publications; ISBN 978-0-19-954645-9. See also: X-Ray Structure Determination, A practical Guide, George H. Stout and Lyle H. Jensen, Wiley Interscience.
- Bruni G, Gozzo F, Capsoni D, Bini M, Macchi P, Simoncic P, Berbenni V., Milanese C., Girella A., Ferrari S. and Marini A., Thermal, Spectroscopic, and Ab Initio Structural Characterization of Carprofen Polymorphs, J. Pharm. Sciences 100(6), 2321 (2011).
- Brunelli, M., Wright, J. P., Vaughan, G. B. M., Mora, A. J. & Fitch, A. N.: Solving Larger Molecular Crystal Structures from Powder Diffraction Data by Exploiting Anisotropic Thermal Expansion. Angew. Chem. (2003) 115, 2075–2078.
- T. Wessels, Ch. Baerlocher and L.B. McCusker: Single-crystal-like diffraction data from polycrystalline materials, Science (1999), 284, 477-479.
- Shankland, K., David, W. I. F., Csoka, T. & McBride, L.: Structure solution of Ibuprofen from powder diffraction data by the application of a genetic algorithm combined with prior conformational analysis, Intl. J. Pharmaceut. (1998) 165, 117–126.
- A. Altomare, C. Cuocci, C. Giacovazzo, A. Moliterni and R. Rizzi: The dual-space resolution bias correction algorithm: applications to powder data, J. Appl. Cryst. (2010). 43, 798-804.
- G.Oszlanyi and A. Suto: The charge flipping algorithm, Acta Cryst. (2008). A64, 123–134 and references herein.
- Boccaleri, E., Carniato, F., Croce, G., Viterbo, D., van Beek, W., Emerich H. and Milanesio, M.,In-situ simultaneous Raman/high-resolution X-ray powder diffraction study of transformations occurring in materials at non-ambient conditions, J. Appl. Cryst., 2007, 40, 684-693.
- Scarlett N.V.Y. and Madsen I. C., Quantification of phases with partial or no known crystal structures, Powder Diffraction (2006) 21, 278-284
- Giannini C., Guagliardi A. and Millini R., Quantitative phase analysis by combining the Rietveld and the whole-pattern decomposition methods, J. Appl. Cryst., 2002, 35, 481-490.
- Scardi, P.; Leoni, M.: Line profile analysis: pattern modeling versus profile fitting, J. Appl. Cryst. 39 (2006) 24–31. Scardi, P.; Leoni, M.: Whole Powder Pattern Modelling, Acta Crystall. A58 (2002) 190–200
- Local structure from total scattering and atomic pair distribution function (PDF) analysis, In Powder diffraction: theory and practice, (Royal Society of Chemistry, London England, 2008), Robert E. Dinnebier and Simon J. L. Billinge, Eds., pp. 464 – 493.
- Neder R. B. And Korsunskiy V. I., Structure of nanoparticles from powder diffraction data using the pair distribution function, 2005 J. Phys.: Condens. Matter 17 S125
- A. Cervellino, C. Giannini, A. Guagliardi, Determination of nanoparticle, size distribution and lattice parameter from x-ray data for monoatomic materials with f.c.c. cubic unit cell, J. Appl. Cryst. 36, 1148-1158 (2003).
- P.W. Stephens, D.E. Cox, and A.N. Fitch, Synchrotron Radiation Powder Diffraction in Structure Determination by Powder Diffraction, pp. 49-87, edited by W.I.F. David, K. Shankland, L.B. McCusker, and C. Baerlocher, (Oxford University Press, 2002)
- Joel Bernstein: Polymorphism in Molecular Crystals, IUCr Monographs on Crystallography (2002), Oxford Science Publications.
- Polymorphism in Pharmaceutical Solids, Ed. By Harry G. Brittain, Drugs and The Pharmaceutical Sciences, Vol. 192
- Tremayne, M.: The impact of powder diffraction on the structural study of organic materials., Philosophical Transactions of the Royal Society of London Series A: Chemistry and Life Sciences Triennial Issue, 362: 2691 (2004).
- Law D, Schmitt EA, Marsh KC, Everitt EA,Wang W, Fort JJ, Krill SL, Qiu Y. Ritonavir-PEG 8000 amorphous solid dispersions: in vitro and in vivo evaluations. J. Pharm. Sci. 2004; 93 (3): 563–567.
- Bruni G., Berbenni V., Milanese C., Girella A., Cardini A., Lanfranconi S. and Marini A.,Determination of the nateglinide polymorphic purity through DSC, J. Pharm. and Biomed. Anal. 54 (2011) 1196-1199.
- Aaltonen J., Alleso M., Mirza S., Koradia V., Gordon K.C and Rantanen J., Solid form screening – A review. European Journal of Pharmaceutics and Biopharmaceutics 71 (2009), 23-37.
- Srivastava D., The Food and Drug Administration and Patent Law at a Crossroads: The Listing of Polymorph Patents as a Barrier to Generic Drug Entry. Food and Drug Law Journal, Vol. 59, No.2 (2004), 339-354.
- Grabowski H. G., Kyle M., Mortimer R., Long G. & Kirson N., Evolving Brand-name And Generic Drug Competition May Warrant A Revision Of The Hatch-Waxman Act. Health Affairs, 30, no.11 (2011):2157-2166.
- Rakowski W. A and Mazzochi D. M., The case of disappearing polymorph: ‘Inherent anticipation’ and the impact of Smithkline Beecham Corp. v Apotex Corp. (Paxil®) on patent validity and infringement by inevitable conversion’. Journal of Generic Medicine, Vol.1, No.2 (2006):131-139.
- Cabri W., Ghetti P., Pozzi G. and Alpegiani M., Polymorphisms and Patent, Market, and Legal Battles: Cefdinir Case Study. Organic Process Research & Development 2007, 11, 64-72.
- Bauer J, Spanton S, Henry R, Quick J, Dziki W, Porter W, Morris J., Ritonavir: an extraordinary example of conformational polymorphism, Pharm Res. 2001 Jun;18(6):859-66.
79………..related to estimation
- McCrone, W.C. in Physics and Chemistry of the Organic Solid State, Vol. 2, (Eds.: D. Fox, M.M. Labes, A. Weissberger), Interscience, New York, 1965, pp. 725-767.
- Bernstein, J., Davey, R.L., and Henck, JO, Concomitant Polymorphism, Angew.Chem.Int. Ed. 1999,38, 3440-3461.
- Polymorphism in Pharmaceutical Solids, Second Edition (Drugs and the Pharmaceutical Sciences) Harry G. Brittain (Editor) Informa HealthCare; 2nd edition (July 27, 2009)
- Otsuka, M., Kato, F. and Matsuda, Y., Determination of indomethacin polymorphic contents by chemometric near-infrared spectroscopy and conventional powder X-ray diffractometry, Analyst, 2001, 126, 1578 – 1582.
- Patel, A.D., Luner, P. E. and Kemper, M. S., Low-level Determination of Polymorph composition in Physical Mixtures by Near-Infrared Reflectance Spectroscopy, J Pharm Sci 2001, 90, 360-370.
- Agatonovic-Kustrin S, Wu V, Rades, T, Saville, D, Tucker, I G, Powder diffractometric assay of two polymorphic forms of ranitidine hydrochloride Int J Pharm 1999, 184, 107.
- Hurst, V.J., Schroeder, P.A., and Styron, R.W. Accurate quantification of quartz and other phases by powder X-ray diffractometry. Analytica Chimica. Acta, 1997, 337, 233-252
- Campbell Roberts, S.N., Williams A.C., Grimsey, I.M., and Booth S.W., Quantitative analysis of mannitol polymorphs. X-ray powder diffractometry—exploring preferred orientation effects J. Pharm. Biomed. Anal., 2002, 28, 1149-1159.
SEE
PART 1………..http://drugsynthesisint.blogspot.in/p/gliptin-series.html
PART 2 ……http://organicsynthesisinternational.blogspot.in/p/gliptin-series-22.html
Characterization of the “hygroscopic” properties of active pharmaceutical ingredients
Characterization of the “hygroscopic” properties of active pharmaceutical ingredients.
Source
SSCI, Inc., West Lafayette, IN, USA. ann.newman@aptuit.com
http://www.ncbi.nlm.nih.gov/pubmed/17630643
Abstract
The amount of water vapor taken up by an active pharmaceutical ingredient (API) as a function of relative humidity is routinely evaluated to characterize and monitor its “hygroscopicity” throughout the drug development process. In this minireview we address the necessity of going beyond the measurement of water vapor sorption isotherms to establish the various mechanisms by which solids interact with water and the important role played by the crystalline or amorphous form of the solid. Practical approaches for choosing experimental conditions under which water vapor sorption should be measured, including the pre-treatment of samples and the time allowed to reach an equilibrium state are presented. With the assistance of a flowchart, we provide a basis for the systematic examination of samples to establish the likely mechanisms of sorption and the indicators pointing toward future problems with physical and chemical instabilities. Finally, we present strategies for managing materials that might be susceptible to the detrimental effects of water vapor sorption.
(Copyright) 2008 Wiley-Liss, Inc.
