
Home » Priority review (Page 8)
Category Archives: Priority review
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Pacritinib

Pacritinib
パクリチニブ;
| Formula |
C28H32N4O3
|
|---|---|
| CAS |
937272-79-2
|
| Mol weight |
472.5787
|
UPDATE FDA APPROVED 2/28/2022, Vonjo
To treat intermediate or high-risk primary or secondary myelofibrosis in adults with low platelets
A Jak2 inhibitor potentially for the treatment of acute myeloid Leukemia and myelofibrosis.
UNII-G22N65IL3O
пакритиниб
باكريتينيب
帕瑞替尼
ONX-0803; SB-1518
CAS No. 937272-79-2
472.57868 g/mol, C28H32N4O3
S*Bio Pte Ltd. and concert innovator
11-(2-pyrrolidin-1-ylethoxy)-14,19-dioxa-5,7,26-triazatetracyclo(19.3.1.1(2,6).1(8,12))heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene
| Pacritinib (SB1518) is a potent and selective inhibitor of Janus Kinase 2 (JAK2) and Fms-Like Tyrosine Kinase-3 (FLT3) with IC50s of 23 and 22 nM, respectively. | ||||||
UPDATED
Pacritinib, sold under the brand name Vonjo, is an anti-cancer medication used to treat myelofibrosis.[1][2] It is a macrocyclic Janus kinase inhibitor. It mainly inhibits Janus kinase 2 (JAK2) and Fms-like tyrosine kinase 3 (FLT3).
Common side effects include diarrhea, low platelet counts, nausea, anemia, and swelling in legs.[2]
Medical uses
Pacritinib in indicated to treat adults who have a rare form of a bone marrow disorder known as intermediate or high-risk primary or secondary myelofibrosis and who have platelet (blood clotting cells) levels below 50,000/µL.[1][2]
History
The effectiveness and safety of pacritinib were demonstrated in a study that included 63 participants with intermediate or high-risk primary or secondary myelofibrosis and low platelets who received pacritinib 200 mg twice daily or standard treatment.[2] Effectiveness was determined based upon the proportion of participants who had a 35% or greater spleen volume reduction from baseline to week 24.[2] Nine participants (29%) in the pacritinib treatment group had a 35% or greater spleen volume reduction, compared to one participant (3%) in the standard treatment group.[2]
The U.S. Food and Drug Administration (FDA) granted the application for pacritinib priority review, fast track, and orphan drug designations.[2]
Society and culture
Names
Pacritinib is the International nonproprietary name (INN).[3][4]
References
- ^ Jump up to:a b c “Enforcement Reports”. Accessdata.fda.gov. Retrieved 5 March 2022.
- ^ Jump up to:a b c d e f g h “FDA approves drug for adults with rare form of bone marrow disorder”. U.S. Food and Drug Administration. 1 March 2022. Retrieved 3 March 2022.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ World Health Organization (2010). “International nonproprietary names for pharmaceutical substances (INN). proposed INN: list 104” (PDF). WHO Drug Information. 24 (4): 386. hdl:10665/74579.
- ^ World Health Organization (2011). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 66”. WHO Drug Information. 25 (3). hdl:10665/74683.
External links
- “Pacritinib”. Drug Information Portal. U.S. National Library of Medicine.
OLD—
Pacritinib (INN[1]) is a macrocyclic Janus kinase inhibitor that is being developed for the treatment of myelofibrosis. It mainly inhibits Janus kinase 2 (JAK2). The drug is in Phase III clinical trials as of 2013.[2] The drug was discovered in Singapore at the labs of S*BIO Pte Ltd. It is a potent JAK2 inhibitor with activity of IC50 = 23 nM for the JAK2WT variant and 19 nM for JAK2V617F with very good selectivity against JAK1 and JAK3 (IC50 = 1280 and 520 nM, respectively).[3][4] The drug is acquired by Cell Therapeutics, Inc. (CTI) and Baxter international and could effectively address an unmet medical need for patients living with myelofibrosis who face treatment-emergent thrombocytopenia on marketed JAK inhibitors.[5]
Pacritinib is an orally bioavailable inhibitor of Janus kinase 2 (JAK2) and the JAK2 mutant JAK2V617F with potential antineoplastic activity. Oral JAK2 inhibitor SB1518 competes with JAK2 for ATP binding, which may result in inhibition of JAK2 activation, inhibition of the JAK-STAT signaling pathway, and so caspase-dependent apoptosis. JAK2 is the most common mutated gene in bcr-abl-negative myeloproliferative disorders; the JAK2V617F gain-of-function mutation involves a valine-to-phenylalanine modification at position 617. The JAK-STAT signaling pathway is a major mediator of cytokine activity.
Synthesis Reference
A245943 — William AD, Lee AC, Blanchard S, Poulsen A, Teo EL, Nagaraj H, Tan E, Chen D, Williams M, Sun ET, Goh KC, Ong WC, Goh SK, Hart S, Jayaraman R, Pasha MK, Ethirajulu K, Wood JM, Dymock BW: Discovery of the macrocycle 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6). 1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a potent Janus kinase 2/fms-like tyrosine kinase-3 (JAK2/FLT3) inhibitor for the treatment of myelofibrosis and lymphoma. J Med Chem. 2011 Jul 14;54(13):4638-58. doi: 10.1021/jm200326p. Epub 2011 Jun 15.
Pacritinib is an orally bioavailable inhibitor of Janus kinase 2 (JAK2) and the JAK2 mutant JAK2V617F with potential antineoplastic activity. Oral JAK2 inhibitor SB1518 competes with JAK2 for ATP binding, which may result in inhibition of JAK2 activation, inhibition of the JAK-STAT signaling pathway, and so caspase-dependent apoptosis. JAK2 is the most common mutated gene in bcr-abl-negative myeloproliferative disorders; the JAK2V617F gain-of-function mutation involves a valine-to-phenylalanine modification at position 617. The JAK-STAT signaling pathway is a major mediator of cytokine activity.


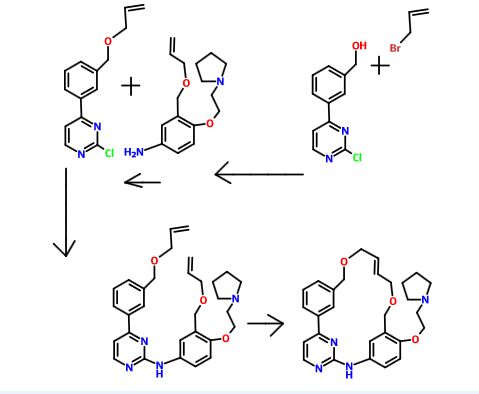
The compound 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (Compound I) was first described in PCT/SG2006/000352 and shows significant promise as a pharmaceutically active agent for the treatment of a number of medical conditions and clinical development of this compound is underway based on the activity profiles demonstrated by the compound.

-
In the development of a drug suitable for mass production and ultimately commercial use acceptable levels of drug activity against the target of interest is only one of the important variables that must be considered. For example, in the formulation of pharmaceutical compositions it is imperative that the pharmaceutically active substance be in a form that can be reliably reproduced in a commercial manufacturing process and which is robust enough to withstand the conditions to which the pharmaceutically active substance is exposed.
-
In a manufacturing sense it is important that during commercial manufacture the manufacturing process of the pharmaceutically active substance be such that the same material is reproduced when the same manufacturing conditions are used. In addition it is desirable that the pharmaceutically active substance exists in a solid form where minor changes to the manufacturing conditions do not lead to major changes in the solid form of the pharmaceutically active substance produced. For example it is important that the manufacturing process produce material having the same crystalline properties on a reliable basis and also produce material having the same level of hydration.
-
In addition it is important that the pharmaceutically active substance be stable both to degradation, hygroscopicity and subsequent changes to its solid form. This is important to facilitate the incorporation of the pharmaceutically active substance into pharmaceutical formulations. If the pharmaceutically active substance is hygroscopic (“sticky”) in the sense that it absorbs water (either slowly or over time) it is almost impossible to reliably formulate the pharmaceutically active substance into a drug as the amount of substance to be added to provide the same dosage will vary greatly depending upon the degree of hydration. Furthermore variations in hydration or solid form (“polymorphism”) can lead to changes in physico-chemical properties, such as solubility or dissolution rate, which can in turn lead to inconsistent oral absorption in a patient.
-
Accordingly, chemical stability, solid state stability, and “shelf life” of the pharmaceutically active substance are very important factors. In an ideal situation the pharmaceutically active substance and any compositions containing it, should be capable of being effectively stored over appreciable periods of time, without exhibiting a significant change in the physico-chemical characteristics of the active substance such as its activity, moisture content, solubility characteristics, solid form and the like.
-
In relation to 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene initial studies were carried out on the hydrochloride salt and indicated that polymorphism was prevalent with the compound being found to adopt more than one crystalline form depending upon the manufacturing conditions. In addition it was observed that the moisture content and ratio of the polymorphs varied from batch to batch even when the manufacturing conditions remained constant. These batch-to-batch inconsistencies and the exhibited hygroscopicity made the hydrochloride salt less desirable from a commercial viewpoint.
-
Accordingly it would be desirable to develop one or more salts of 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene which overcome or ameliorate one or more of the above identified problems.
PATENT
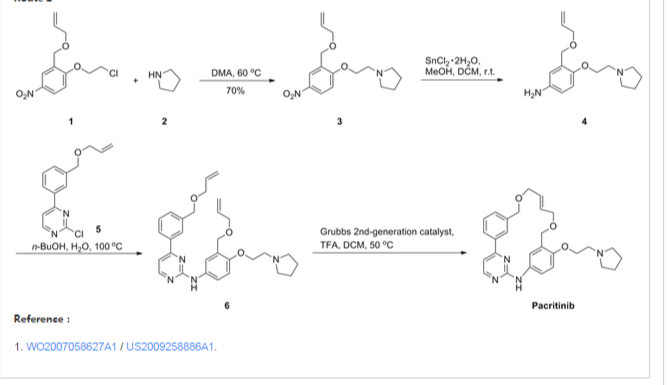
US 2011263616
http://www.google.com/patents/US20110263616
11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26triaza-tetra-cyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (Compound I) which have been found to have improved properties. In particular the present invention relates to the maleate salt of this compound. The invention also relates to pharmaceutical compositions containing this salt and methods of use of the salt in the treatment of certain medical conditions.

PATENT
http://www.google.com/patents/US8415338
Representative Procedure for the Synthesis of Compounds Type (XVIIId) [3-(2-Chloro-pyrimidin-4-yl)-phenyl]-methanol (XIIIa2)

Compound (XIIIa2) was obtained using the same procedure described for compound (XIIIa1); LC-MS (ESI positive mode) m/z 221 ([M+H]+).
4-(3-Allyloxymethyl-phenyl)-2-chloro-pyrimidine (XVa2)

Compound (XVa2) was obtained using the same procedure described for compound (XVa1); LC-MS (ESI positive mode) m/z 271 ([M+H]+).
[4-(3-Allyloxymethyl-phenyl)-pyrimidin-2-yl]-[3-allyloxymethyl-4-(2-pyrrolidin-1-yl-ethoxy)-phenyl]-amine (XVIId1)

Compound (XVIId1) was obtained using the same procedure described for compound (XVIIb1); LC-MS (ESI positive mode) m/z 501.
Macrocycle Example 3 Compound 13

Compound (13) was obtained using the same procedure described for compound (1) HPLC purity at 254 nm: 99%; LC-MS (ESI positive mode) m/z 473 ([M+H]+); 1H NMR (MeOD-d4) δ 8.79 (d, 1H), 8.46 (d, 1H), 8.34-8.31 (m, 1H), 7.98-7.96 (m, 1H), 7.62-7.49 (m, 2H), 7.35 (d, 1H), 7.15-7.10 (m, 1H), 7.07-7.02 (m, 1H), 5.98-5.75 (m, 2H, 2×=CH), 4.67 (s, 2H), 4.67 (s, 2H), 4.39-4.36 (m, 2H), 4.17 (d, 2H), 4.08 (d, 2H), 3.88-3.82 (m, 2H), 3.70 (t, 2H), 2.23-2.21 (m, 2H), 2.10-2.07 (m, 2H).
PAPER
J MC 2011, 54 4638
http://pubs.acs.org/doi/abs/10.1021/jm200326p

Discovery of the activating mutation V617F in Janus Kinase 2 (JAK2V617F), a tyrosine kinase critically involved in receptor signaling, recently ignited interest in JAK2 inhibitor therapy as a treatment for myelofibrosis (MF). Herein, we describe the design and synthesis of a series of small molecule 4-aryl-2-aminopyrimidine macrocycles and their biological evaluation against the JAK family of kinase enzymes and FLT3. The most promising leads were assessed for their in vitro ADME properties culminating in the discovery of 21c, a potent JAK2 (IC50 = 23 and 19 nM for JAK2WT and JAK2V617F, respectively) and FLT3 (IC50 = 22 nM) inhibitor with selectivity against JAK1 and JAK3 (IC50 = 1280 and 520 nM, respectively). Further profiling of 21c in preclinical species and mouse xenograft and allograft models is described. Compound 21c(SB1518) was selected as a development candidate and progressed into clinical trials where it is currently in phase 2 for MF and lymphoma.
 Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma
Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma
S*BIO Pte. Ltd., 1 Science Park Road, #05-09, The Capricorn, Singapore Science Park II, Singapore 117528
J. Med. Chem., 2011, 54 (13), pp 4638–4658
DOI: 10.1021/jm200326p
Publication Date (Web): May 23, 2011
Copyright © 2011 American Chemical Society
(21c)
The title compound was synthesized from 21a and pyrrolidine (yield, 83%; mixture of trans/cis85:15 by NMR). LC-MS (ESI positive mode) m/z 473 ([M + H]+). HRMS: theoretical C28H32N4O3MW, 472.2474; found, 473.2547. 1H NMR (MeOD-d4): δ 8.79 (d, 1H), 8.46 (d, 1H), 8.34–8.31 (m, 1H, CH), 7.98–7.96 (m, 1H), 7.62–7.49 (m, 2H), 7.35 (d, 1H), 7.15–7.10 (m, 1H), 7.07–7.02 (m, 1H), 5.98–5.75 (m, 2H), 4.67 (s, 2H), 4.67 (s, 2H), 4.39–4.36 (m, 2H), 4.17 (d, 2H), 4.08 (d, 2H), 3.88–3.82 (m, 2H), 3.70 (t, 2H), 2.23–2.21 (m, 2H), 2.10–2.07 (m, 2H); chloride content (titration) 7.7% (1.18 equivs); water content (Karl Fischer) 6.1% (1.85 equivs); Anal. Calcd. for C28H32N4O3·1.18HCl·1.85H2O: C, 61.46; H, 6.46; N, 10.24; Cl, 7.65. Found: C, 61.99; H, 6.91; N, 10.25; Cl, 7.45.
References
1 “International Nonproprietary Names for Pharmaceutical Substances (INN) List 104” (PDF). WHO Drug Information 24 (4): 386. 2010.
2“JAK-Inhibitoren: Neue Wirkstoffe für viele Indikationen”. Pharmazeutische Zeitung (in German) (21). 2013.
3William, A. D.; Lee, A. C. -H.; Blanchard, S. P.; Poulsen, A.; Teo, E. L.; Nagaraj, H.; Tan, E.; Chen, D.; Williams, M.; Sun, E. T.; Goh, K. C.; Ong, W. C.; Goh, S. K.; Hart, S.; Jayaraman, R.; Pasha, M. K.; Ethirajulu, K.; Wood, J. M.; Dymock, B. W. (2011). “Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma”. Journal of Medicinal Chemistry 54 (13): 4638–58. doi:10.1021/jm200326p. PMID 21604762.
4Poulsen, A.; William, A.; Blanchard, S. P.; Lee, A.; Nagaraj, H.; Wang, H.; Teo, E.; Tan, E.; Goh, K. C.; Dymock, B. (2012). “Structure-based design of oxygen-linked macrocyclic kinase inhibitors: Discovery of SB1518 and SB1578, potent inhibitors of Janus kinase 2 (JAK2) and Fms-like tyrosine kinase-3 (FLT3)”. Journal of Computer-Aided Molecular Design 26 (4): 437–50. doi:10.1007/s10822-012-9572-z. PMID 22527961.
5http://www.pmlive.com/pharma_news/baxter_licenses_cancer_drug_from_cti_in_$172m_deal_519143
| US8153632 * | Nov 15, 2006 | Apr 10, 2012 | S*Bio Pte Ltd. | Oxygen linked pyrimidine derivatives |
| US8415338 * | Apr 4, 2012 | Apr 9, 2013 | Cell Therapeutics, Inc. | Oxygen linked pyrimidine derivatives |
| US20110294831 * | Dec 9, 2009 | Dec 1, 2011 | S*Bio Pte Ltd. | 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene citrate salt |
| Patent | Submitted | Granted |
|---|---|---|
| OXYGEN LINKED PYRIMIDINE DERIVATIVES [US8153632] | 2009-03-19 | 2012-04-10 |
| ANTIVIRAL JAK INHIBITORS USEFUL IN TREATING OR PREVENTING RETROVIRAL AND OTHER VIRAL INFECTIONS [US2014328793] | 2012-11-30 | 2014-11-06 |
| OXYGEN LINKED PYRIMIDINE DERIVATIVES [US2013172338] | 2013-02-20 | 2013-07-04 |
| METHOD OF SELECTING THERAPEUTIC INDICATIONS [US2014170157] | 2012-06-15 | 2014-06-19 |
| CYCLODEXTRIN-BASED POLYMERS FOR THERAPEUTIC DELIVERY [US2014357557] | 2014-05-30 | 2014-12-04 |
| 11-(2-PYRROLIDIN-1-YL-ETHOXY)-14,19-DIOXA-5,7,26-TRIAZA-TETRACYCLO[19.3.1.1(2,6).1(8,12)]HEPTACOSA-1(25),2(26),3,5,8,10,12(27),16,21,23-DECAENE MALEATE SALT [US2011263616] | 2011-10-27 | |
| 11-(2-PYRROLIDIN-1-YL-ETHOXY)-14,19-DIOXA-5,7,26-TRIAZA-TETRACYCLO[19.3.1.1(2,6).1(8,12)]HEPTACOSA-1(25),2(26),3,5,8,10,12(27),16,21,23-DECAENE CITRATE SALT [US2011294831] | 2011-12-01 | |
| BIOMARKERS AND COMBINATION THERAPIES USING ONCOLYTIC VIRUS AND IMMUNOMODULATION [US2014377221] | 2013-01-25 | 2014-12-25 |
| Oxygen linked pyrimidine derivatives [US8415338] | 2012-04-04 | 2013-04-09 |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(16E)-11-[2-(1-Pyrrolidinyl)ethoxy]-14,19-dioxa-5,7,26-triazatetracyclo[19.3.1.12,6.18,12]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene
|
|
| Clinical data | |
| Legal status |
|
| Routes of administration |
Oral |
| Identifiers | |
| ATC code | None |
| PubChem | CID: 46216796 |
| ChemSpider | 28518965 |
| ChEMBL | CHEMBL2035187 |
| Synonyms | SB1518 |
| Chemical data | |
| Formula | C28H32N4O3 |
| Molecular mass | 472.58 g/mol |


S*Bio Pte Ltd
Address: 1 Science Park Rd, Singapore 117528
Phone:+65 6827 5000

S*BIO Pte Ltd. provides research and clinical development services for small molecule drugs for the treatment of cancer in Singapore. The company’s products include JAK2 inhibitors, such as SB1518 for leukemia/myelofibrosis, lymphoma, and polycythemia; and SB1578 for RA/psoriasis. The company also offers SB939, a histone deacetylases for MDS/AML+combo, prostate cancer, sarcoma, pediatric tumor, and myelofibrosis; SB2602, a mTOR inhibitor; SB2343, a mTOR/PI3K inhibitor; and SB1317, a CDK/Flt3 inhibitor. The company was founded in 2000 and is based in Singapore. S*BIO Pte Ltd. operates as a subsidiary of Chiron Corporation Limited.
SEE……..http://apisynthesisint.blogspot.in/2016/01/pacritinib.html

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
|
|
| Clinical data | |
|---|---|
| Trade names | Vonjo |
| Other names | SB1518 |
| License data |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| PDB ligand | |
| Chemical and physical data | |
| Formula | C28H32N4O3 |
| Molar mass | 472.589 g·mol−1 |
| 3D model (JSmol) | |
///////Vonjo, FDA APPTOVESD 2022, APPROVALS 2022, PACRITINIB, パクリチニブ, priority review, fast track, orphan drug, UNII-G22N65IL3O, пакритиниб , باكريتينيب , 帕瑞替尼 , SB 1518
c1cc2cc(c1)-c3ccnc(n3)Nc4ccc(c(c4)COC/C=C/COC2)OCCN5CCCC5
C1CCN(C1)CCOC2=C3COCC=CCOCC4=CC=CC(=C4)C5=NC(=NC=C5)NC(=C3)C=C2
Defibrotide


Defibrotide sodium is an oligonucleotide mixture with profibrinolytic properties. The chemical name of defibrotide sodium is polydeoxyribonucleotide, sodium salt. Defibrotide sodium is a polydisperse mixture of predominantly single-stranded (ss) polydeoxyribonucleotide sodium salts derived from porcine intestinal tissue having a mean weighted molecular weight of 13-20 kDa, and a potency of 27-39 and 28-38 biological units per mg as determined by two separate assays measuring the release of a product formed by contact between defibrotide sodium, plasmin and a plasmin substrate. The primary structure of defibrotide sodium is shown below.
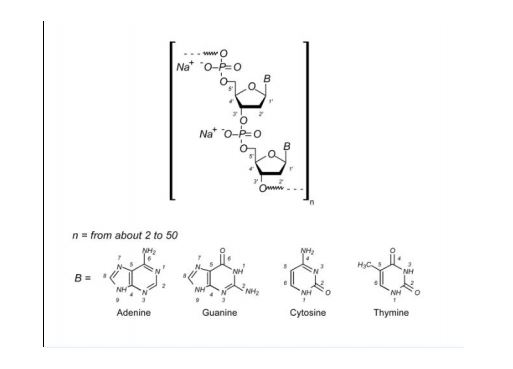
DEFITELIO (defibrotide sodium) injection is a clear, light yellow to brown, sterile, preservative-free solution in a single-patient-use vial for intravenous use. Each milliliter of the injection contains 80 mg of defibrotide sodium and 10 mg of Sodium Citrate, USP, in Water for Injection, USP. Hydrochloric Acid, NF, and/or Sodium Hydroxide, NF, may have been used to adjust pH to 6.8-7.8.
Defibrotide is the sodium salt of a mixture of single-stranded oligodeoxyribonucleotides derived from porcine mucosal DNA. It has been shown to have antithrombotic, anti-inflammatory and anti-ischemic properties (but without associated significant systemic anticoagulant effects). It is marketed under the brand names Dasovas (FM), Noravid, and Prociclide in a variety of countries, but is currently not approved in the USA. The manufacturer is Gentium.
Defibrotide is used to treat or prevent a failure of normal blood flow (occlusive venous disease, OVD) in the liver of patients who have had bone marrow transplants or received certain drugs such as oral estrogens, mercaptopurine, and many others.
In 2012, an IND was filed in Japan seeking approval of the compound for the treatment of veno-occlusive disease.
Approved 3/30/3016 US FDA, defibrotide sodium, (NDA) 208114

To treat adults and children who develop hepatic veno-occlusive disease with additional kidney or lung abnormalities after they receive a stem cell transplant from blood or bone marrow called hematopoietic stem cell transplantation
Polydeoxyribonucleotides from bovine lung or other mamalian organs with molecular weight between 15,000 and 30,000 Da
CAS 83712-60-1
Defibrotide is a polydisperse mixture of oligonucleotides produced by random, chemical cleavage (depolymerisation) of porcine DNA. It is predominantly single stranded, of varying base sequence, lengths and conformations; unfolded, folded or combined. The mean oligonucleotide length is 50 bases with a mean molecular weight of 17 ± 4 kDa. No individually defined component is at more than femtomolar concentration. The only meaningful scientific information that can be obtained about the biochemical nature of defibrotide (aside from determination of percentage of each nucleobase) is a measurement of its average length and its average percentage double stranded character. Therefore, it can be established that this active substance is of highly heterogenic nature.

Defibrotide (Defitelio, Gentium)[1] is a deoxyribonucleic acid derivative (single-stranded) derived from cow lung or porcine mucosa. It is an anticoagulant with a multiple mode of action (see below).
It has been used with antithrombin III.[2]
Jazz Pharmaceuticals plc announced that the FDA has accepted for filing with Priority Review its recently submitted New Drug Application (NDA) for defibrotide. AS ON OCT 2015
Defibrotide is an investigational agent proposed for the treatment of patients with hepatic veno-occlusive disease (VOD), also known as sinusoidal obstruction syndrome (SOS), with evidence of multi-organ dysfunction (MOD) following hematopoietic stem-cell transplantation (HSCT).
Priority Review status is designated for drugs that may offer major advances in treatment or provide a treatment where no adequate therapy exists. Based on timelines established by the Prescription Drug User Fee Act (PDUFA), FDA review of the NDA is expected to be completed by March 31, 2016.
“The FDA’s acceptance for filing and Priority Review status of the NDA for defibrotide is an important milestone for Jazz and reflects our commitment to bringing meaningful medicines to patients who have significant unmet needs,” said Karen Smith, M.D., Ph.D., Global Head of Research and Development and Chief Medical Officer of Jazz Pharmaceuticals. “We look forward to continuing to work closely with the FDA to obtain approval for defibrotide for patients with hepatic VOD with evidence of MOD in the U.S. as quickly as possible, as there are no other approved therapies for treating this rare, often fatal complication of HSCT.”
The NDA includes safety and efficacy data from three clinical studies of defibrotide for the treatment of hepatic VOD with MOD following HSCT, as well as a retrospective review of registry data from the Center for International Blood and Marrow Transplant Research. The safety database includes over 900 patients exposed to defibrotide in the clinical development program for the treatment of hepatic VOD.
The compound was originally developed under a collaboration between Sanofi and Gentium. In December 2001, Gentium entered into a license and supply agreement with Sigma-Tau Pharmaceuticals, pursuant to which the latter gained exclusive rights to distribute, market and sell the product for the treatment of VOD in the U.S. This agreement was expanded in 2005 to include all of North America, Central America and South America.
Defibrotide was granted orphan drug designations from the FDA in July 1985, May 2003 and January 2007 for the treatment of thrombotic thrombocytopenic purpura (TTP), for the treatment of VOD and for the prevention of VOD, respectively. Orphan drug was also received in the E.U. for the prevention and treatment of hepatic veno-occlusive disease (VOD) in 2004 and for the prevention of graft versus host disease (GvHD) in 2013.
Pharmacokinetics
Defibrotide is available as an oral, intravenous, and intramuscular formulation. Its oral bioavailability is in the range of 58-70% of theparenteral forms. T1/2 alpha is in the range of minutes while T1/2 beta is in the range of hours in studies with oral radiolabelleddefibrotide. These data suggest that defibrotide, in spite of its macromolecular nature, is absorbed well after oral administration. Due to the drug’s short half-life, it is necessary to give the daily dose divided in 2 to 4 doses (see below).
In 2014, Jazz Pharmaceuticals (parent of Gentium) acquired the rights of the product in U.S. and in the Americas
Mode of action
The drug appears to prevent the formation of blood clots and to help dissolve blood clots by increasing levels of prostaglandin I2, E2, and prostacyclin, altering platelet activity, increasing tissue plasminogen activator (tPA-)function, and decreasing activity of tissue plasminogen activator inhibitor. Prostaglandin I2 relaxes the smooth muscle of blood vessels and prevents platelets from adhering to each other. Prostaglandin E2 at certain concentrations also inhibits platelet aggregation. Moreover, the drug provides additional beneficial anti-inflammatory and antiischemic activities as recent studies have shown. It is yet unclear, if the latter effects can be utilized clinically (e.g., treatment of ischemic stroke).
Unlike heparin and warfarin, defibrotide appears to have a relatively mild anticoagulant activity, which may be beneficial in the treatment of patients at high risk of bleeding complications. Nevertheless, patients with known bleeding disorders (e.g., hemophilia A) or recent abnormal bleedings should be treated cautiously and under close medical supervision.
The drug was marketed under the brand names Dasovas (FM), Noravid, and Prociclide in a variety of countries. It is currently not approved in the USA. The manufacturer is Gentium.
Defibrotide also received fast track designation from the FDA for the treatment of severe VOD in recipients of stem cell transplants. In 2011, the compound was licensed to Medison Pharma by Gentium in Israel and Palestine. The license covers the management of named-patient sales program and local registration, authorization, marketing, reimbursement and medical affairs for the treatment of peripheral vascular disease.
Usual indications
Defibrotide is used to treat or prevent a failure of normal blood flow (Veno-occlusive disease, VOD) in the liver of patients having had bone marrow transplants or received certain drugs such as oral estrogens, mercaptopurine, and many others. Without intensive treatment, VOD is often a fatal condition, leading to multiorgan failure. It has repeatedly been reported that defibrotide was able to resolve the condition completely and was well tolerated.
Other indications are: peripheral obliterative arterial disease, thrombophlebitis, and Raynaud’s phenomenon. In very high doses, defibrotide is useful as treatment of acute myocardial infarction. The drug may also be used for the pre- and postoperative prophylaxis of deep venous thrombosis and can replace the heparin use during hemodialytic treatments.
It has been investigated for use in treatment of chronic venous insufficiency.[3]
Potential indications in the future
Other recent preclinical studies have demonstrated that defibrotide used in conjunction with Granulocyte Colony-Stimulating Factor (rhG-CSF) significantly increases the number of Peripheral Blood Progenitor Cells (Stem cells). The benefit of this increase in stem cells may be crucial for a variety of clinical indications, including graft engineering procedures and gene therapy programs. This would expand the clinical usefulness of defibrotide to a complete distinct area.
Very recently (since early 2006) combination therapy trials (phase I/II) with defibrotide plus melphalan, prednisone, and thalidomide in patients with multiple myeloma have been conducted. The addition of defibrotide is expected to decrease the myelosuppressive toxicity of melphalan. However, is too early for any definitive results at that stage.
Cautions and contraindications
- The efficacy of the drug has been reported to be poorer in patients with diabetes mellitus.
- Pregnancy: The drug should not be used during pregnancy, because adequate and well controlled human studies do not exist.
- Lactation: No human data is available. In order to avoid damage to the newborn, the nursing mother should discontinue either the drug or breastfeeding, taking into account the importance of treatment to the mother.
- Known Bleeding Disorders or Bleeding Tendencies having occurred recently: Defibrotide should be used cautiously. Before initiation of treatment, the usual coagulation values should be obtained as baseline and regularly controlled under treatment. The patient should be observed regularly regarding local or systemic bleeding events.
Side-effects
Increased bleeding and bruising tendency, irritation at the injection site, nausea, vomiting, heartburn, low blood pressure. Serious allergic reactions have not been observed so far.
Drug interactions
Use of heparin with defibrotide may increase the aPTT, reflecting reduced ability of the body to form a clot. Nothing is known about the concomitant application of other anticoagulants than heparin and dextran containing plasma-expanders, but it can be anticipated that the risk of serious bleeding will be increased considerably.
PATENT
WO 2001078761
G-CSF (CAS registry number 143011-2-7/Merck Index, 1996, page 4558) is a haematopoietic growth factor which is indispensable in the proliferation and differentiation of the progenitor cells of granulocytes; it is a 18-22 kDa glycoprotein normally produced in response to specific stimulation by a variety of cells, including monocytes, fibroblasts and endothelial cells. The term defibrotide (CAS registry number 83712-60-1) normally identifies a polydeoxyribonucleotide obtained by extraction (US 3,770,720 and US 3,899,481) from animal and/or vegetable tissue; this polydeoxyribonucleotide is normally used in the form of a salt of an alkali metal, generally sodium. Defibrotide is used principally for its anti- thrombotic activity (US 3,829,567) although it may be used in different applications, such as, for example, the treatment of acute renal insufficiency (US 4,694,134) and the treatment of acute myocardial ischaemia (US 4,693,995). United States patents US 4,985,552 and US 5,223,609, finally, describe a process for the production of defibrotide which enables a product to be obtained which has constant and well defined physico-chemical characteristics and is also free from any undesired side-effects
References
- “Jazz Pharma Acquiring Gentium for $1B”. Gen. Eng. Biotechnol. News (paper) 34 (2). January 15, 2014. p. 10.
- Haussmann U, Fischer J, Eber S, Scherer F, Seger R, Gungor T (June 2006). “Hepatic veno-occlusive disease in pediatric stem cell transplantation: impact of pre-emptive antithrombin III replacement and combined antithrombin III/defibrotide therapy”. Haematologica 91 (6): 795–800. PMID 16769582.
- Coccheri S, Andreozzi GM, D’Addato M, Gensini GF (June 2004). “Effects of defibrotide in patients with chronic deep insufficiency. The PROVEDIS study”. Int Angiol 23 (2): 100–7.PMID 15507885.
External links
- Palmer KJ, Goa KL. Defibrotide: a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in vascular disorders. Drugs 1993;45:259-94.
- http://www.globalrx.com/medinfo/Defibrotide.htm
- Fisher J, Holland TK, Pescador R, Porta R, Ferro L (January 1996). “Study on pharmacokinetics of radioactive labelled defibrotide after oral or intravenous administration in rats”. Thromb. Res. 81 (1): 55–63. doi:10.1016/0049-3848(95)00213-8. PMID 8747520.
- http://www.gentium.it/Defibrotide.aspx (information provided by manufacturer)
- “Melphalan: profile and news”. Archived from the original on 2007-09-28. (on cytostatic combination therapy)
- Beşişik SK, Oztürk GB, Calişkan Y, Sargin D (March 2005). “Complete resolution of transplantation-associated thrombotic microangiopathy and hepatic veno-occlusive disease by defibrotide and plasma exchange”. Turk J Gastroenterol 16 (1): 34–7. PMID 16252186.
| WO2003101468A1 * | Jun 2, 2003 | Dec 11, 2003 | Guenther Eissner | Method for the protection of endothelial and epithelial cells during chemotherapy |
| US4985552 | Jul 5, 1989 | Jan 15, 1991 | Crinos Industria Farmacobiologica S.P.A. | Process for obtaining chemically defined and reproducible polydeoxyribonucleotides |
| US5223609 | May 26, 1992 | Jun 29, 1993 | Crinos Industria Farmacobiologica S.P.A. | Process for obtaining chemically defined and reproducible polydeoxyribonucleotides |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1999026639A1 * | 24 Nov 1998 | 3 Jun 1999 | Allegheny University Of The He | Methods for mobilizing hematopoietic facilitating cells and hematopoietic stem cells into the peripheral blood |
| EP0317766A1 * | 20 Oct 1988 | 31 May 1989 | Crinos Industria Farmacobiologica S.p.A. | A method for preventing blood coaguli from being formed in the extra-body circuit of dialysis apparatus and composition useful thereof |
| EP0416678A1 * | 10 Aug 1990 | 13 Mar 1991 | Crinos Industria Farmacobiologica S.p.A. | Topical compositions containing Defibrotide |
| US5199942 * | 26 Sep 1991 | 6 Apr 1993 | Immunex Corporation | Method for improving autologous transplantation |
| US5977083 * | 5 Jun 1995 | 2 Nov 1999 | Burcoglu; Arsinur | Method for using polynucleotides, oligonucleotides and derivatives thereof to treat various disease states |
| Reference | ||
|---|---|---|
| 1 | * | CARLO-STELLA, C. (1) ET AL: “Defibrotide significantly enhances peripheral blood progenitor cell mobilization induced by recombinant human granulocyte colony – stimulating factor ( rhG – CSF.” BLOOD, ( NOVEMBER 16, 2000 ) VOL. 96, NO. 11 PART 1, PP. 553A. PRINT. MEETING INFO.: 42ND ANNUAL MEETING OF THE AMERICAN SOCIETY OF HEMATOLOGY SAN FRANCISCO, CALIFORNIA, USA DECEMBER 01-05, 2000 AMERICAN SOCIETY OF HEMATOLOGY. , XP002176349 |
| 2 | * | GURSOY A: “PREPARATION, CHARACTERIZATION AND ANTI-INFLAMMATORY EFFECT OF DEFIBROTIDE LIPOSOMES” PHARMAZIE,DD,VEB VERLAG VOLK UND GESUNDHEIT. BERLIN, vol. 48, no. 7, 1 July 1993 (1993-07-01), pages 549-550, XP000372658 ISSN: 0031-7144 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2005017160A2 * | 12 Aug 2004 | 24 Feb 2005 | Childrens Hosp Medical Center | Mobilization of hematopoietic cells |
| WO2009115465A1 * | 13 Mar 2009 | 24 Sep 2009 | Gentium Spa | Synthetic phosphodiester oligonucleotides and therapeutical uses thereof |
| EP2103689A1 * | 19 Mar 2008 | 23 Sep 2009 | Gentium S.p.A. | Synthetic phosphodiester oligonucleotides and therapeutical uses thereof |
| US7417026 | 12 Aug 2004 | 26 Aug 2008 | Children’s Hospital Medical Center | Mobilization of hematopoietic cells |
| US7915384 | 5 Jan 2009 | 29 Mar 2011 | Children’s Hospital Medical Center | Chimeric peptides for the regulation of GTPases |
| US8242246 | 28 Feb 2011 | 14 Aug 2012 | Children’s Hospital Medical Center | Chimeric peptides for the regulation of GTPases |
| US8674075 | 13 Aug 2012 | 18 Mar 2014 | Children’s Medical Center Corporation | Chimeric peptides for the regulation of GTPases |
| US8980862 | 12 Nov 2010 | 17 Mar 2015 | Gentium S.P.A. | Defibrotide for use in prophylaxis and/or treatment of Graft versus Host Disease (GVHD) |
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
oral, i.m., i.v. |
| Pharmacokinetic data | |
| Bioavailability | 58 – 70% orally (i.v. and i.m. = 100%) |
| Biological half-life | t1/2-alpha = minutes; t1/2-beta = a few hours |
| Identifiers | |
| CAS Registry Number | 83712-60-1 |
| ATC code | B01AX01 |
| DrugBank | DB04932 |
| UNII | 438HCF2X0M |
| KEGG | D07423 |
///////////Approved, 3/30/3016, US FDA, defibrotide sodium, NDA 208114, FDA 2016
Updates……….
FDA approves first treatment for rare disease in patients who receive stem cell transplant from blood or bone marrow
For Immediate Release
March 30, 2016
Release
The U.S. Food and Drug Administration today approved Defitelio (defibrotide sodium) to treat adults and children who develop hepatic veno-occlusive disease (VOD) with additional kidney or lung abnormalities after they receive a stem cell transplant from blood or bone marrow called hematopoietic stem cell transplantation (HSCT). This is the first FDA-approved therapy for treatment of severe hepatic VOD, a rare and life-threatening liver condition.
HSCT is a procedure performed in some patients to treat certain blood or bone marrow cancers. Immediately before an HSCT procedure, a patient receives chemotherapy. Hepatic VOD can occur in patients who receive chemotherapy and HSCT. Hepatic VOD is a condition in which some of the veins in the liver become blocked, causing swelling and a decrease in blood flow inside the liver, which may lead to liver damage. In the most severe form of hepatic VOD, the patient may also develop failure of the kidneys and lungs. Fewer than 2 percent of patients develop severe hepatic VOD after HSCT, but as many as 80 percent of patients who develop severe hepatic VOD do not survive.
“The approval of Defitelio fills a significant need in the transplantation community to treat this rare but frequently fatal complication in patients who receive chemotherapy and HSCT,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research.
The efficacy of Defitelio was investigated in 528 patients treated in three studies: two prospective clinical trials and an expanded access study. The patients enrolled in all three studies had a diagnosis of hepatic VOD with liver or kidney abnormalities after HSCT. The studies measured the percentage of patients who were still alive 100 days after HSCT (overall survival). In the three studies, 38 to 45 percent of patients treated with Defitelio were alive 100 days after HSCT. Based on published reports and analyses of patient-level data, the expected survival rates 100 days after HSCT would be 21 to 31 percent for patients with severe hepatic VOD who received only supportive care or interventions other than Defitelio.
The most common side effects of Defitelio include abnormally low blood pressure (hypotension), diarrhea, vomiting, nausea and nosebleeds (epistaxis). Serious potential side effects of Defitelio that were identified include bleeding (hemorrhage) and allergic reactions. Defitelio should not be used in patients who are having bleeding complications or who are taking blood thinners or other medicines that reduce the body’s ability to form clots.
The FDA granted the Defitelio application priority review status, which facilitates and expedites the development and review of certain drugs in light of their potential to benefit patients with serious or life-threatening conditions. Defitelio also received orphan drug designation, which provides incentives such as tax credits, user fee waivers and eligibility for exclusivity to assist and encourage the development of drugs for rare diseases.
Defitelio is marketed by Jazz Pharmaceuticals based in Palo Alto, California
Avatrombopag

Avatrombopag
AVATROMBOPAG; UNII-3H8GSZ4SQL; AKR-501; E5501; 570406-98-3; AS 1670542
| C29H34Cl2N6O3S2 | |
| Molecular Weight: | 649.65466 g/mol |
|---|
Elemental Analysis: C, 53.61; H, 5.28; Cl, 10.91; N, 12.94; O, 7.39; S, 9.87
1-[3-chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)-1,3-thiazol-2-yl]carbamoyl]pyridin-2-yl]piperidine-4-carboxylic acid,
1-(3-Chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)thiazol-2-yl]carbamoyl]pyridin-2-yl)piperidine-4-carboxylic acid,
1-[3-Chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)thiazol-2-yl]carbamoyl]-2-pyridyl]piperidine-4-carboxylic acid
4-Piperidinecarboxylic acid, 1-[3-chloro-5-[[[4-(4-chloro-2-thienyl)-5-(4-cyclohexyl-1-piperazinyl)-2-thiazolyl]amino]carbonyl]-2-pyridinyl]-
Phase III Clinical Trials
Drugs used in platelet disorders
Idiopathic thrombocytopenic purpura (ITP)
small-molecule thrombopoietin receptor (c-Mpl) agonist that stimulates platelet production
INNOVATOR: YAMANOUCHI PHARMACEUTICAL
DEVELOPER: Eisai

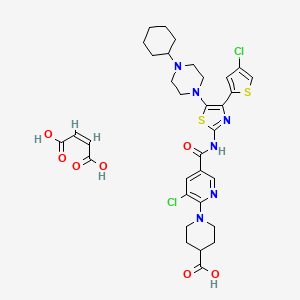
Avatrombopag maleate; UNII-GDW7M2P1IS; E5501 MALEATE; 677007-74-8; YM 477, AKR 501
| C33H38Cl2N6O7S2 | |
| Molecular Weight: | 765.72682 g/mol |
|---|
UNIIGDW7M2P1IS
(Z)-but-2-enedioic acid;1-[3-chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)-1,3-thiazol-2-yl]carbamoyl]pyridin-2-yl]piperidine-4-carboxylic acid
INTRODUCTION
Avatrombopag, also known as AKR-501, YM477, AS 1670542 or E5501, is a novel orally-active thrombopoietin (TPO) receptor agonist. AKR-501 specifically targeted the TPO receptor and stimulated megakaryocytopoiesis throughout the development and maturation of megakaryocytes just as rhTPO did. Daily oral administration of AKR-501 dose-dependently increased the number of human platelets in these mice, with significance achieved at doses of 1 mg/kg and above. The peak unbound plasma concentrations of AKR-501 after administration at 1 mg/kg in NOD/SCID mice were similar to those observed following administration of an active oral dose in human subjects. AKR-501 may be useful in the treatment of patients with thrombocytopenia. (source: Eur J Haematol. 2009 Apr;82(4):247-54).

Avatrombopag is a thrombopoietin receptor (c-Mpl) agonist in phase III clinical evaluation at Eisai for the oral treatment of chronic immune thrombocytopenia (idiopathic thrombocytopenia purpura) and for the treatment of thrombocytopenia associated with liver diseases. Phase II studies are ongoing for the treatment of thrombocytopenia during antiviral therapy (inhibition and maintenance) with Interferon for hepatitis C.
The drug candidate may hold potential in treating thrombocytopenia of diverse etiologies, including idiopathic thrombocytopenic purpura (ITP) and thrombocytopenia of myelodysplastic syndromes (MDS), in combination with or as a substitute for platelet transfusion.
AKR-501, a novel, small-molecule thrombopoietin mimetic being investigated for the treatment of thrombocytopenia. AkaRx is now a wholly-owned subsidiary of Eisai Inc. and Eisai has the exclusive worldwide rights to develop, market and manufacture AKR-501. AKR-501 is an investigational thrombopoietin receptor agonist that, based on preclinical studies, increases platelet production by stimulating megakaryocytic proliferation and differentiation. Eisai is currently conducting Phase II clinical trials of AKR-501 in the United States as a potential treatment for idiopathic thrombocytopenic purpura (ITP) and thrombocytopenia associated with liver diseases (TLD), and has confirmed proof of concept in the clinical studies for ITP. In addition, Eisai will explore the compound’s potential as a treatment for chemotherapy-induced thrombocytopenia (CIT).

E-5501 stimulates the production of thrombopoietin (TPO), a glycoprotein hormone that stimulates the production and differentiation of megakaryocytes, the bone marrow cells that fragment into large numbers of platelets. The drug candidate was originally developed at Yamanouchi, and development responsibilities were passed to AkaRx when it was formed in 2005 as a spin-off following the creation of Astellas Pharma subsequent to the merger of Yamanouchi Pharmaceutical and Fujisawa Healthcare.
In 2007, MGI Pharma was granted a license to E-5501 for the treatment of thrombocytopenia. Eisai eventually gained the rights to the product as results of its acquisition of MGI Pharma. In 2010, Eisai acquired AkaRx. AkaRx is now a wholly-owned subsidiary of Eisai Inc. and Eisai has the exclusive worldwide rights to develop, market and manufacture E-5501. In 2011, orphan drug designation was assigned by the FDA for the treatment of idiopathic thrombocytopenic purpura.
E5501 (or AKR-501 or YM477) is a small molecule agonist c-Mpl, orally available. It is in clinical trials for the treatment of chronic idiopathic thrombocytopenic purpura (ITP). It acts as an agonist of the thrombopoietin receptor active orally, mimicking its biological effect. Thrombocytopenic purpura The is the idiopathic consequence of a low number of platelets (thrombocytopenia) of unknown cause. A very low platelets can even lead to purpura (bruises), or bleeding diathesis.
February 2012: A Phase III, multicenter, randomized, double-blind, controlled against placebo, parallel group, with an open-label extension phase to assess the efficacy and safety of combined oral E5501 to standard treatment for the treatment of thrombocytopenia in adults with chronic immune thrombocytopenia, is underway.
January 2010: Eisai Inc. announced its successful acquisition of the biopharmaceutical company, AkaRx Inc. Following this acquisition, AkaRx became a wholly owned subsidiary of Eisai Inc. Eisai now owns the worldwide exclusive rights to develop , marketing and manufacture AKR-501.
October 2009: Eisai Research Institute of Boston, Inc. (established in 1987) and Eisai Medical Research Inc. (established in 2002) were merged into Eisai Inc. 2005: AkaRx was founded as a spin-out of the merger of Yamanouchi Pharmaceutical Company Ltd. and Fujisawa Pharmaceutical Company Ltd. to form Astellas Pharma Inc. AKR-501 was discovered by Yamanouchi and was licensed to AkaRx as part of the foundation of the company in 2005.
In a Phase I trial in healthy volunteers, 10 mg of AKR-501 for 14 days, increased platelet count by 50%.AKR-501 was well tolerated in both studies, mono- and multi-dose. No adverse effects were reported, even at the highest doses.
……………………
Patent
Compound A is a compound of the present invention has the following chemical structure.

That is, compounds useful as a platelet 增多 agent according to the present invention A, as well as medicaments for the Compound A as an active ingredient, in particular increasing platelets agents and Z or thrombocytopenia treating agent.


………………
PATENT

……………………
JP 2014144916/WO 2013018362
https://www.google.co.in/patents/WO2013018362A1?cl=en
1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)thiazol-2-yl]carbamoyl}pyridin-2-yl)piperidine-4-carboxylic acid as expressed by the following chemical formula (hereinafter referred to as “Compound X”) and pharmaceutically acceptable salts are known to have excellent thrombocytosis effects (patent literature 1, patent literature 2).
[Formula 1]

Patent literature 1 discloses a hydrochloride of compound X as example 16 (hereinafter referred to as “compound X hydrochloride”).
Furthermore, patent literature 2 discloses a maleic acid salt of compound X that has endothermic peaks near 198 degree C and 271 degree C in thermo gravimetric analysis (hereinafter referred to as “maleic acid salt of compound X”). However, patent literature 2 neither discloses nor suggests that the maleic acid salt of compound X exhibits crystal polymorphism.
On the other hand, compounds exhibiting crystal polymorphism demonstrate entirely different effects regardless of being the same compound, because various physical properties including physicochemical properties differ depending on the crystalline form. In pharmaceutical products in particular, if compounds that have different functional effects are expected to have the same effect, a different functional effect than expected will occur, which is thought to induce unexpected circumstances, and therefore there is demand for supply of a drug substance with constant quality. Therefore, when a compound which has crystal polymorphism is used as a medicine, one type of crystal of that compound must always be constantly provided in order to ensure constant quality and constant effects that are required of the medicine.
Under the aforementioned conditions, from the perspective of supplying a drug substance for medicines, there is a need for compound X or crystals of pharmaceutically acceptable salts thereof, which can ensure constant quality and constant effects and which can be stably supplied in mass production such as industrial production or the like, as well as for establishment of a manufacturing method thereof.
International patent publication WO 03/062233 International patent publication WO 2004/029049
The crystals of compound X maleic acid salt disclosed in patent literature 2 (hereinafter referred to as “compound X maleic acid salt A type crystals”) cannot be isolated as compound X maleic acid salt A type crystals when scaled up for mass production using the method disclosed in example 1 of patent literature 2, and therefore must be isolated in a different crystal form. (This other crystal form is referred to as “compound X maleic acid salt B type crystals”). Therefore, the compound X maleic acid salt A type crystals have a possibility that the crystal form will morph depending on the scale of production, and is clearly inappropriate as a drug substance for medicines which require constant quality and constant effects.
Preparation Example 1: Manufacture of Compound X Maleic Acid Salt B Type Crystal
310 mL of a 1 M aqueous solution of sodium hydroxide at room temperature was added to a mixture of 70.0 g of the ethyl ester of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid and 1.2 L of ethanol, the insoluble matter was filtered out, and then washed with 200 mL of ethanol. The reaction solution was stirred for 90 minutes at 60 degree C. After cooling to room temperature, 1.4 L of an aqueous solution containing 24.11 g of maleic acid was added to the solution obtained, and then the precipitate was collected by filtering.
The same operation was repeated and when combined with the previously obtained precipitate, 136.05 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid was obtained.
18.9 g of maleic acid and 2.1 L of 80% ethanol water were added to 88.90 g of the carboxylic acid obtained, and the solution was stirred for one hour at room temperature and for another hour at 100 degree C. After cooling to room temperature and further cooling with ice, the precipitated solid was filtered out to obtain 87.79 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt as a crude product.
6.84 g of maleic acid was added to 231 g of the crude product containing the crude product obtained above and those manufactured in a similar manner, dissolved in 5.5 L of 80% ethanol water, and then the precipitated solid was collected by filtering to obtain 203 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
Example 1: Manufacture of Compound X Maleic Acid Salt C Type Crystals (1)
1.52 L of ethanol, 0.38 L of water, and 15.7 g of maleic acid were added to 78.59 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid, and heated while stirring. After cooling to room temperature and further cooling with ice, the precipitated solid was collected by filtering to obtain 71.60 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt as a crude product.
296 mg of maleic acid was added to 10.0 g of the crude product obtained, dissolved in 60 mL of acetone, 60 mL of DMSO, and 30 mL of water, and then the precipitated solids were collected to obtain 8.41 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
Example 2: Manufacture of Compound X Maleic Acid Salt C Type Crystals (2)
A mixture containing 80.1 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid, 580 mL of DMSO, 580 mL of acetone, 17.2 g of maleic acid, and 290 mL of water was stirred at 69 degree C. The insoluble matter was filtered out, washed with a mixture of 32 mL of DMSO, 32 mL of acetone, and 16 mL of water, and then the filtrate was cooled and the precipitate was collected by filtering. Washing was successively performed using 150 mL of water, 80 mL of acetone, 650 mL of water, and 80 mL of acetone, followed by drying, to obtain 70.66 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
Example 3: Manufacture of Compound X Maleic Acid Salt C Type Crystals (3)
A mixture containing 20 kg of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid, 100 L of DMSO, 100 L of acetone, 4.29 kg of maleic acid, and 50 L of water is stirred at 65 degree C, and then the insoluble matter is filtered out and washed with a mixture of 8 L of DMSO, 8 L of acetone, and 4 L of water, and then the filtrate is cooled, the precipitate is collected by filtering, successively washed using 40 L of acetone, 100 L of water, and 40 L of acetone, and then dried to obtain approximately 20 kg of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
…………………………….

REFERENCES
Garabet, L.; Ghanima, W.; Lee, S.; Mowinckel, M.C.; Liebman, H.; Jonassen, C.M.; Bussel, J.; Sandset, P.M.
Thrombopoietin receptor agonists do no not cause coagulation activation: In patients with immune thrombocytopenia
25th Congr Int Soc Thromb Haemost (ISTH) (June 20-25, Toronto) 2015, Abst PO311-MON
Terrault, N.; Hassanein, T.; Joshi, S.; Lake, J.R.; Sher, L.S.; Vargas, H.E.; McIntosh, J.W.; Tang, S.; Jenkins, T.
Once-daily oral avatrombopag (E5501) prior to elective surgical or diagnostic procedures in patients with chronic liver disease and thrombocytopenia: Results from a phase 2, randomized, double-blind, placebo-controlled study (study 202)
63rd Annu Meet Am Assoc Study Liver Dis (November 9-13, Boston) 2012, Abst
Thiophenyl Triazol-3-one Derivatives As Smooth Muscle relaxers: US6613786 (2003) Priority: US20010336865P, Nov. 2, 2001 (Bristol-Myers Squibb CO, US)
Preparation Of Avatrombopag: 2-Acylaminothiazole derivative or salt thereof: EP1466912 (2004) Priority: JP20020010413, 18 Jan. 2002 (Yamanouchi Pharma Co Ltd, Japan)
Synthesis And Use Of MSE Framework-Type Molecular Sieves: US2009318696 (2009) Priority: US20080214631 20 Jun. 2008 (Exxon Mobil, US).
5,6-Dichloro-Nicotinic Acid Production By Reacting 6-Hydroxy-Nicotinic Acid With Acid Chloride Reacting With Chlorine Products, Then With Acid Chloride And Hydrolysing Products: CH664754 (1988) Priority: CH19850002692, 25 Jun. 1985 (Lonza AG, Switzerland).
David J. Kuter, New Thrombopoietic Growth Factors, Lymphoma and Myeloma Clinical Journal Volume 9, Supplement 3, S347-S356
| WO2003062233A1 | 15 Jan 2003 | 31 Jul 2003 | Yamanouchi Pharma Co Ltd | 2-acylaminothiazole derivative or salt thereof |
| WO2004029049A1 | 29 Sep 2003 | 8 Apr 2004 | Yuuji Awamura | Novel salt of 2-acylaminothiazole derivative |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| EP2764866A1 | 4 Feb 2014 | 13 Aug 2014 | IP Gesellschaft für Management mbH | Inhibitors of nedd8-activating enzyme |
| Patent | Submitted | Granted |
|---|---|---|
| CANCER TREATMENT METHOD [US2011160130] | 2011-06-30 | |
| METHOD FOR STIMULATING PLATELET PRODUCTION [US2011166112] | 2011-07-07 | |
| COMPOSITIONS AND METHODS FOR INCREASING BLOOD PLATELET LEVELS IN HUMANS [US2011224226] | 2011-09-15 | |
| Method of treating viral diseases with combinations of TPO receptor agonist and anti-viral agents [US2012020923] | 2012-01-26 |
| Patent | Submitted | Granted |
|---|---|---|
| 2-Acylaminothiazole derivative or salt thereof [US7638536] | 2005-07-14 | 2009-12-29 |
| Compositions and methods for treating thrombocytopenia [US2007203153] | 2007-08-30 | |
| Novel Combinations [US2009304634] | 2009-12-10 | |
| 2-ACYLAMINOTHIAZOLE DERIVATIVE OR SALT THEREOF [US2010222329] | 2010-09-02 | |
| 2-ACYLAMINOTHIAZOLE DERIVATIVE OR SALT THEREOF [US2010222361] | 2010-09-02 | |
| Compositions and methods for increasing blood platelet levels in humans [US2008039475] | 2008-02-14 | |
| CANCER TREATMENT METHOD [US2009022814] | 2009-01-22 | |
| Compositions and methods for treating thrombocytopenia [US2010041668] | 2010-02-18 | |
| CANCER TREATMENT METHOD [US2010075928] | 2010-03-25 |
///////E 5501, AKR 501, Phase III, eisai, Avatrombopag, y 477, orphan drug, ym 477, AS 1670542, Yamanouchi Pharma Co Ltd, Japan
UPDATE MAY 2018
Avatrombopag
https://newdrugapprovals.org/2015/08/24/avatrombopag/
The U.S. Food and Drug Administration today approved Doptelet (avatrombopag) tablets to treat low blood platelet count (thrombocytopenia) in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure. This is the first drug approved by the FDA for this use.Continue reading.
May 21, 2018
Release
The U.S. Food and Drug Administration today approved Doptelet (avatrombopag) tablets to treat low blood platelet count (thrombocytopenia) in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure. This is the first drug approved by the FDA for this use.
“Patients with chronic liver disease who have low platelet counts and require a procedure are at increased risk of bleeding,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Doptelet was demonstrated to safely increase the platelet count. This drug may decrease or eliminate the need for platelet transfusions, which are associated with risk of infection and other adverse reactions.”
Platelets (thrombocytes) are colorless cells produced in the bone marrow that help form blood clots in the vascular system and prevent bleeding. Thrombocytopenia is a condition in which there is a lower-than-normal number of circulating platelets in the blood. When patients have moderately to severely reduced platelet counts, serious or life-threatening bleeding can occur, especially during invasive procedures. Patients with significant thrombocytopenia typically receive platelet transfusions immediately prior to a procedure to increase the platelet count.
The safety and efficacy of Doptelet was studied in two trials (ADAPT-1 and ADAPT-2) involving 435 patients with chronic liver disease and severe thrombocytopenia who were scheduled to undergo a procedure that would typically require platelet transfusion. The trials investigated two dose levels of Doptelet administered orally over five days as compared to placebo (no treatment). The trial results showed that for both dose levels of Doptelet, a higher proportion of patients had increased platelet counts and did not require platelet transfusion or any rescue therapy on the day of the procedure and up to seven days following the procedure as compared to those treated with placebo.
The most common side effects reported by clinical trial participants who received Doptelet were fever, stomach (abdominal) pain, nausea, headache, fatigue and swelling in the hands or feet (edema). People with chronic liver disease and people with certain blood clotting conditions may have an increased risk of developing blood clots when taking Doptelet.
This product was granted Priority Review, under which the FDA’s goal is to take action on an application within six months where the agency determines that the drug, if approved, would significantly improve the safety or effectiveness of treating, diagnosing or preventing a serious condition.
The FDA granted this approval to AkaRx Inc.
//////////////Doptelet, avatrombopag, fda 2018, akarx, priority review,
Long-term use of AZ’ Brilinta, ticagrelor gets US priority review
An application to use AstraZeneca’s Brilinta to treat patients with a history of heart attack has been placed on a fast track regulatory pathway in the US, meaning that approval could be granted within just six months.
The US Food and Drug Administration has assigned a priority review based on Phase III data showing that Brilinta (ticagrelor), along-side low-dose aspirin, can improve long-term prevention of atherothrombotic cardiovascular events in patients with a history of myocardial infarction. The move signals the regulator’s belief that the drug could offer a benefit over existing approaches.
Read more at: http://www.pharmatimes.com/Article/15-04-29/Long-term_use_of_AZ_Brilinta_gets_US_priority_review.aspx#ixzz3YlAnNYsq
Ticagrelor, a P2Y12 (P2T) antagonist, was granted approval in the E.U. in December 2010 for the prevention of atherothrombotic events in adult patients with acute coronary syndromes (ACS).
The product was first launched in Germany and the U.K. as Brilique(TM) in January 2011. Also in 2011, the product received approval in Canada.
Ticagrelor was recommended for approval by the FDA in July 2010; however, in December 2010, a complete response letter was assigned.
In July 2011, FDA approval was granted and U.S. launch took place in August.
US priority review for Eisai cancer drug lenvatinib
![]()
US priority review for Eisai cancer drug lenvatinib
Eisai has been boosted by news that regulators in the USA have agreed to a quicker review of its anticancer agent lenvatinib.
The US Food and Drug Administration has granted a priority review to Eisai’s New Drug Application for lenvatinib as a treatment for progressive radioiodine-refractory differentiated thyroid cancer. This means that the agency has assigned a Prescription Drug User Fee Act action date of April 14 next year, eight months after the NDA was submitted.
Read more at: http://www.pharmatimes.com/Article/14-10-15/US_priority_review_for_Eisai_cancer_drug_lenvatinib.aspx#ixzz3GH3iXiDU
SEE SYNTHESIS




FDA Approves Ryanodex for the Treatment of Malignant Hyperthermia

Dantrolene sodium
1-[[[5-(4-nitrophenyl)-2-furanyl]methylene]amino]-2,4-imidazolidinedione
VIEW THIS POST AT BELOW LINK UNTIL FORMATTING IS FIXED
http://www.allfordrugs.com/2014/07/24/fda-approves-ryanodex-for
-the-treatment-of-malignant-hyperthermia/
FDA Approves Ryanodex for the Treatment of Malignant Hyperthermia
WOODCLIFF LAKE, N.J.(BUSINESS WIRE) July 23, 2014 —
Eagle Pharmaceuticals, Inc. (“Eagle” or “the Company”)
(Nasdaq:EGRX) today announced that the U. S. Food and Drug Administration (FDA)
has approved Ryanodex (dantrolene sodium) for injectable
suspension indicated for
the treatment of malignant hyperthermia (MH), along
with the appropriate supportive measures.
MH is an inherited and potentially fatal disorder triggered
by certain anesthesia agents
in genetically susceptible individuals. FDA had designated
Ryanodex as an Orphan Drug in
August 2013. Eagle has been informed by the FDA that it will learn over the next four to
six weeks if it has been granted the seven year Orphan Drug market exclusivity.
read at
http://www.drugs.com/newdrugs/fda-approves-ryanodex-malignant-
hyperthermia-4058.html?utm_source=ddc&utm_medium=email&utm_
news+summary+-+July+23%2C+2014
READ MORE AT
PATENTS, CAS NO ETC
http://www.allfordrugs.com/2014/07/24/fda-approves-ryanodex-
Purdue’s hydrocodone bitartrate tablets granted priority review designation
Hydrocodone bitartrate is morphinan-6-one, 4,5-epoxy-3-methoxy-17-methyl-, (5α)-, [R-(R*,R*)]-2,3-dihydroxybutanedioate (1:1), hydrate (2:5); also known as 4,5α-Epoxy-3-methoxy-17-methylmorphinan-6-one tartrate (1:1) hydrate (2:5); a fine white crystal or crystalline powder, which is derived from the opium alkaloid, thebaine; and may be represented by the following structural formula:
 |
Hydrocodone Bitartrate
C18H21N03•C4H606•2.5 H20
Molecular weight = 494.5
Purdue’s hydrocodone bitartrate tablets granted priority review designation
Purdue Pharma has been granted priority review designation by the US Food and Drug Administration (FDA) for its hydrocodone bitartrate extended-release tablets for treatment of chronic pain.
The once-daily, single-entity pain medication was formulated to incorporate abuse-deterrent properties designed to make the product more difficult to manipulate for misuse or abuse by various routes of administration.http://www.pharmaceutical-technology.com/news/newspurdues-hydrocodone-bitartrate-tablets-granted-priority-review-designation-4313765?WT.mc_id=DN_News
FDA Accepts Filing of NDA for IV Antibiotic Oritavancin with Priority Review

Oritavancin
(4R)-22-O-(3-Amino-2,3,6-trideoxy-3-C-methyl-alpha-L-arabinohexopyranosyl)-N3-(p-(p-chlorophenyl)benzyl)vancomycin
(3S, 6R, 7R, 22R, 23S, 26S, 36R, 38aR) -22 – (3-Amino-2 ,3,6-trideoxy-3-C-methyl-alpha-L-mannopyranosyloxy) -3 – (carbamoylmethyl ) -10,19-dichloro-44-[2-O-[3 – (4′-chlorobiphenyl-4-ylmethylamino) -2,3,6-trideoxy-3-C-methyl-alpha-L-mannopyranosyl] – beta-D-glucopyranosyloxy] –
| CAS No. | 171099-57-3 |
| CBNumber: | CB92451283 |
| Molecular Formula: | C86H97Cl3N10O26 |
| Formula Weight: | 1793.12 |
Also known as NDISACC-(4-(4-chlorophenyl)benzyl)A82846B and LY333328,N-(4-(4-chlorophenyl)benzyl)A82846B
Abbott (Supplier), Lilly (Originator), InterMune (Licensee)
The medicines company—
-
the Oritavancin Program Results.pdf
 phx.corporate-ir.net/External.File?item…t=1
phx.corporate-ir.net/External.File?item…t=1Jul 2, 2013 – Inhibits two key steps of cell wall synthesis: – Transglycosylation. – Transpeptidation. • Disrupts bacterial membrane integrity. Differentiated from …
FDA Accepts Filing of NDA for IV Antibiotic Oritavancin with Priority Review
PARSIPPANY, NJ — (Marketwired) — 02/19/14 — The Medicines Company (NASDAQ: MDCO) today announced that the U.S. Food and Drug Administration (FDA) has accepted the filing of a new drug application (NDA) for oritavancin, an investigational intravenous antibiotic, with priority review. The Medicines Company is seeking approval of oritavancin for the treatment of acute bacterial skin and skin structure infections (ABSSSI) caused by susceptible gram-positive bacteria, including methicillin-resistant Staphylococcus aureus (MRSA), administered as a single dose.
In December 2013, the FDA designated oritavancin as a Qualified Infectious Disease Product (QIDP). The QIDP designation provides oritavancin priority review, and an additional five years of exclusivity upon approval of the product for the treatment of ABSSSI. Priority review means the FDA’s goal is to take action on the application within six months, compared to 10 months under standard review. The FDA action date (PDUFA date) for oritavancin is August 6, 2014.
Oritavancin (INN, also known as LY333328) is a novel semi-synthetic glycopeptide antibiotic being developed for the treatment of serious Gram-positive infections. Originally discovered and developed by Eli Lilly, oritavancin was acquired by InterMune in 2001 and then by Targanta Therapeuticsin late 2005.[1]
In Dec 2008 the FDA declined to approve it, and an EU application was withdrawn.
In 2009 the development rights were acquired by The Medicine Co. who are running clinical trials for a possible new FDA application in 2013.[2]
Its structure is similar to vancomycin[3] It is a lipoglycopeptide
About Oritavancin
Oritavancin is an investigational intravenous antibiotic for which The Medicines Company is seeking approval in the treatment of ABSSSI caused by susceptible gram-positive bacteria, including MRSA. In clinical trials, the most frequently reported adverse events associated with oritavancin were nausea, headache, vomiting and diarrhea. Hypersensitivity reactions have been reported with the use of antibacterial agents including oritavancin.

Oritavancin shares certain properties with other members of the glycopeptide class of antibiotics, which includes vancomycin, the current standard of care for serious Gram-positive infections in the United States and Europe.[4] Data presented at the 47th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC) in September 2007 demonstrated that oritavancin possesses potent and rapid bactericidal activity in vitro against a broad spectrum of both resistant and susceptible Gram positive bacteria, including Staphylococcus aureus, methicillin-resistant Staphylococcus aureus, Enterococci, and Streptococci.[5] Two posters presented at the meeting also demonstrated that oritavancin was more active than either metronidazole or vancomycin against strains of Clostridium difficile tested.[6]
Anthrax : Research presented at the American Society for Microbiology (ASM) 107th Annual General Meeting in May 2007, suggested oritavancin’s potential utility as a therapy for exposure to Bacillus anthracis, the gram-positive bacterium that causes anthrax, having demonstrated efficacy in a mouse model both pre- and post-exposure to the bacterium[7]
 oritavancin
oritavancin
The 4′-chlorobiphenylmethyl group disrupts the cell membrane of gram positive bacteria.[8] It also acts by inhibition of transglycosylation and inhibition of transpeptidation.[9]
Results have been presented (in 2003) but possibly not yet published from two pivotal Phase 3 clinical trials testing the efficacy of daily intravenous oritavancin for the treatment of complicated skin and skin-structure infections (cSSSI) caused by Gram-positive bacteria. The primary endpoints of both studies were successfully met, with oritavancin achieving efficacy with fewer days of therapy than the comparator agents (vancomycin followed by cephalexin). In addition, oritavancin showed a significantly improved safety profile with a 19.2 percent relative reduction in the overall incidence of adverse events versus vancomycin/cephalexin (p<0.001) in the second and larger pivotal trial.[10]
A Phase 2 clinical study was planned to run until May 2008 entitled “Single or Infrequent Doses for the Treatment of Complicated Skin and Skin Structure Infections (SIMPLIFI),” evaluating the efficacy and safety of either a single dose of oritavancin or an infrequent dose of oritavancin compared to the previously studied dosing regimen of 200 mg oritavancin given once daily for 3 to 7 days.[11] Results published May 2011.[12]
Regulatory submissions
USA
On February 11, 2008, Targanta submitted a New Drug Application (NDA) to the US FDA seeking approval of oritavancin;[13] in April 2008, the FDA accepted the NDA submission for standard review.[14] On 9 Dec 2008 the FDA said insufficient data for approval of oritavancin had been provided and they requested a further phase 3 clinical study to include more patients with MRSA.[15]
Europe
June 2008, Targanta’s Marketing Authorization Application (MAA) for oritavancin was submitted and accepted for review by the European Medicines Agency (EMEA),[16] but the company later withdrew the application in Aug 2009.[17]
About The Medicines Company
The Medicines Company’s purpose is to save lives, alleviate suffering, and contribute to the economics of healthcare by focusing on 3,000 leading acute/intensive care hospitals worldwide. Its vision is to be a leading provider of solutions in three areas: acute cardiovascular care, surgery and perioperative care, and serious infectious disease care. The company operates in the Americas, Europe and the Middle East, and Asia Pacific regions with global centers today in Parsippany, NJ, USA and Zurich, Switzerland.
“We look forward to working with the FDA during the review process, and sharing the knowledge we have gained in our studies of oritavancin,” said Matthew Wikler, MD, Vice President and Medical Director, Infectious Disease Care for The Medicines Company. “We believe that upon approval, oritavancin, administered as a single dose for the treatment of ABSSSI, will offer new options for both physicians and their patients for the treatment of these infections.”
The oritavancin NDA is based on data from two Phase 3 clinical trials, SOLO I and SOLO II, which were conducted under a Special Protocol Assessment (SPA) agreement with the FDA. These Phase 3 trials evaluated the efficacy and safety of a single 1200mg dose of oritavancin compared to 7 to 10 days of twice-daily vancomycin in adults with ABSSSI, including infections caused by MRSA. The combined SOLO studies were conducted in 1,959 patients (modified intent-to -treat population, or mITT), with 405 of the patients suffering from an ABSSSI with a documented MRSA infection.
 oritavancin
oritavancin
Drug substance
Oritavancin diphosphate
CLINICAL TRIALS..http://clinicaltrials.gov/search/intervention=oritavancin
- LY 333328 diphosphate
- LY333328 diphosphate
- Oritavancin diphosphate
- UNII-VL1P93MKZN
- 192564-14-0 CAS NO
INTRODUCTION
Oritavancin
Oritavancin inhibits cell wall synthesis by complexing with the terminal D-Ala-D-Ala of a nascent peptidoglycan chain and also to the pentaglycine bridge, thus inhibiting transglyco- sylation and transpeptidation. Unlike other glycopeptides, oritavancin is able to bind to depsipeptides including D-Ala-D-Lac, which fa- cilitates its inhibition of cell wall synthesis even in organisms exhibiting VanA-type resistance. Oritavancin forms homodimers prior to binding to D-Ala-D-Ala or D-Ala-D-Lac, which increases its binding affinity for the target site.The p-chloro-phenylbenzyl side chain of oritavancin interacts with the cell membrane, exerting two beneficial effects. This binding acts to main- tain the antibacterial in a prime position for peptidoglycan interactions and it also imparts oritavancin with the ability to disrupt the bac- terial membrane potential and thus increase membrane permeability.[22,23] Oritavancin has been shown to dissipate membrane potential in both stationary and exponential phase growing bacteria, which is rare and may carry clinical implications in terms of its activity against slowly growing organisms and biofilms. The dual mechanism of action could also theoretically increase effectiveness and reduce the risk of resist- ance selection. In addition to the aforemen- tioned mechanisms, it has also been hypothesized that oritavancin inhibits RNA synthesis.


vancomycin, desmethylvancomycin, eremomycin, teicoplanin (complex of five compounds), dalbavancin, oritavancin, telavancin, and A82846B (LY264826) having structures A, B, C, D, E, F, G and H:

R = B-2-Acetylamido-glucopyraπosyl- Attorney Docket No 33746-704 602


Dalbavancin, oritavancin and telavancin are semisynthetic lipoglycopeptides that demonstrate promise for the treatment of patients with infections caused by multi-drug-resistant Gram-positive pathogens. Each of these agents contains a heptapeptide core, common to all glycopeptides, which enables them to inhibit transglycosylation and transpeptidation (cell wall synthesis). Modifications to the heptapeptide core result in different in vitro activities for the three semisynthetic lipoglycopeptides. All three lipoglycopeptides contain lipophilic side chains, which prolong their half-life, help to anchor the agents to the cell membrane and increase their activity against Gram-positive cocci. In addition to inhibiting cell wall synthesis, telavancin and oritavancin are also able to disrupt bacterial membrane integrity and increase membrane permeability; oritavancin also inhibits RNA synthesis. Enterococci exhibiting the VanA phenotype (resistance to both vancomycin and teicoplanin) are resistant to both dalbavancin and telavancin, whileoritavancin retains activity. Dalbavancin, oritavancin and telavancin exhibit activity against VanB vancomycin-resistant enterococci.
All three lipoglycopeptides demonstrate potent in vitro activity against Staphylococcus aureus and Staphylococcus epidermidis regardless of their susceptibility to meticillin, as well as Streptococcus spp. Both dalbavancin and telavancin are active against vancomycin-intermediate S. aureus (VISA), but display poor activity versus vancomycin-resistant S. aureus (VRSA). Oritavancin is active against both VISA and VRSA. Telavancin displays greater activity against Clostridium spp. than dalbavancin, oritavancin or vancomycin. The half-life of dalbavancin ranges from 147 to 258 hours, which allows for once-weekly dosing, the half-life of oritavancin of 393 hours may allow for one dose per treatment course, while telavancin requires daily administration. Dalbavancin and telavancin exhibit concentration-dependent activity and AUC/MIC (area under the concentration-time curve to minimum inhibitory concentration ratio) is the pharmacodynamic parameter that best describes their activities.
Oritavancin’s activity is also considered concentration-dependent in vitro, while in vivo its activity has been described by both concentration and time-dependent models; however, AUC/MIC is the pharmacodynamic parameter that best describes its activity. Clinical trials involving patients with complicated skin and skin structure infections (cSSSIs) have demonstrated that all three agents are as efficacious as comparators. The most common adverse effects reported with dalbavancin use included nausea, diarrhoea and constipation, while injection site reactions, fever and diarrhoea were commonly observed withoritavancin therapy. Patients administered telavancin frequently reported nausea, taste disturbance and insomnia. To date, no drug-drug interactions have been identified for dalbavancin, oritavancin or telavancin. All three of these agents are promising alternatives for the treatment of cSSSIs in cases where more economical options such as vancomycin have been ineffective, in cases of reduced vancomycin susceptibility or resistance, or where vancomycin use has been associated with adverse events.
Oritavancin diphosphate (oritavancin) is a semi-synthetic lipoglycopeptide derivative of a naturally occurring glycopeptide. Its structure confers potent antibacterial activity against gram-positive bacteria, including vancomycin-resistant enterococci (VRE), methicillin- and vancomycin-resistant staphylococci, and penicillin-resistant streptococci. The rapidity of its bactericidal activity against exponentially-growing S. aureus (≧3-log reduction within 15 minutes to 2 hours against MSSA, MRSA, and VRSA) is one of the features that distinguishes it from the prototypic glycopeptide vancomycin (McKay et al., J Antimicrob Chemother. 63(6):1191-9 (2009), Epub 2009 Apr. 15).
Oritavancin inhibits the synthesis of peptidoglycan, the major structural component of the bacterial cell wall by a mechanism that is shared with glycopeptides, such as vancomycin (Allen et al., Antimicrob Agents Chemother 41(1):66-71 (1997); Cegelski et al., J Mol Biol 357:1253-1262 (2006); Arhin et al., Poster C1-1471: Mechanisms of action of oritavancin in Staphylococcus aureus [poster]. 47th Intersci Conf Antimicro Agents Chemo, Sep. 17-20, 2007; Chicago, Ill.). Oritavancin, like vancomycin, binds to the Acyl-D-Alanyl-D-Alanine terminus of the peptidoglycan precursor, lipid-bound N-acetyl-glucosamine-N-acetyl-muramic acid-pentapeptide (Reynolds, Eur J Clin Microbiol Infect Dis 8(11):943-950 (1989); Nicas and Allen, Resistance and mechanism of action.
In: Nagarajan R, editor. Glycopeptide antibiotics. New York: Marcel Dekker 195-215 (1994); Allen et al., Antimicrob Agents Chemother 40(10):2356-2362 (1996); Allen and Nicas, FEMS Microbiology Reviews 26:511-532 (2003); Kim et al., Biochemistry 45:5235-5250 (2006)). However, oritavancin inhibits cell wall biosynthesis even when the substrate is the altered peptidoglycan precursor that is present in VRE and vancomycin-resistant S. aureus (VRSA). Thus, the spectrum of oritavancin antibacterial activity extends beyond that of vancomycin to include glycopeptide-resistant enterococci and staphylococci (Ward et al., Expert Opin Investig Drugs 15:417-429 (2006); Scheinfeld, J Drugs Dermatol 6:97-103 (2007)). Oritavancin may inhibit resistant bacteria by interacting directly with bacterial proteins in the transglycosylation step of cell wall biosynthesis (Goldman and Gange, Curr Med Chem 7(8):801-820 (2000); Halliday et al., Biochem Pharmacol 71(7):957-967 (2006); Wang et al., Poster C1-1474: Probing the mechanism of inhibition of bacterial peptidoglycan glycotransferases by glycopeptide analogs. 47th Intersci Conf Antimicro Agents Chemo, Sep. 17-20, 2007). Oritavancin also collapses transmembrane potential in gram positive bacteria, leading to rapid killing (McKay et al., Poster C1-682: Oritavancin disrupts transmembrane potential and membrane integrity concomitantly with cell killing in Staphylococcus aureus and vancomycin-resistant Enterococci. 46th Intersci Conf Antimicro Agents Chemo, San Francisco, Calif., Sep. 27-30, 2006). These multiple effects contribute to the rapid bactericidal activity of oritavancin.
Vancomycin (U.S. Patent 3,067,099); A82846A, A82846B, and A82846C (U.S. Patent 5,312,738, European Patent Publication 256,071 A1); PA-42867 factors A, C, and D (U.S. Patent4,946,941 and European Patent Publication 231,111 A2); A83850 (U.S. Patent No. 5,187,082); avoparcm (U.S. Patent 3,338,786 and U.S. Patent 4,322,343); actmoidin, also known as K288 (J. Antibiotics Series A 14:141 (1961); helevecardin (Chem. Abstracts 110:17188 (1989) and Japanese Patent Application 86/157,397); galacardin (Chem. Abstracts 110:17188 (1989) and Japanese Patent Application 89/221,320); and M47767 (European Patent Publication 339,982).
Oritavancin is in clinical development against serious gram-positive infections, where administration of the drug is via intravenous infusion using several dosages administered over a series of days. The development of alternative dosing regimens for the drug could expand treatment options available to physicians. The present invention is directed to novel dosing regimens.
Means for the preparation of the glycopeptide antibiotics, including oritavancin and analogs thereof, may be found, for example, in U.S. Pat. No. 5,840,684,
SYNTHESIS

LY-333328 was synthesized by reductocondensation of the glycopeptide antibiotic A82846B (I) with 4′-chlorobiphenyl-4-carboxaldehyde (II) by means of sodium cyanoborohydride in refluxing methanol.
J Antibiot1996, 49, (6) :575-81
(3S,6R,7R,22R,23S,26S,36R,38aR)-3-(Carbamoylmethyl)-10,19-dichloro-7,28,30,32-tetrahydroxy-6-(N-methyl-D-leucylamido)-2,5,24,38,39-pentaoxo-22-(L-vancosaminyloxy)-44-[2-O-(L-vancosaminyl)-beta-D-glucopyranosyloxy]-2,3,4,5,6,7,23,24,25,26,36,37,38,38a-tetradecahydro-1H,22H-8,11:18,21-dietheno-23,36-(iminomethano)-13,16:31,36-dimetheno-[1,6,9]oxadiazacyclohexadecino[4,5-m][10,2,16]benzoxadiazacyclotetracosine-26-carboxylic acid; A82846B (I)
4′-chloro[1,1′-biphenyl]-4-carbaldehyde (II)
LY-333328 was synthesized by reductocondensation of the glycopeptide antibiotic A82846B (I) with 4′-chlorobiphenyl-4-carboxaldehyde (II) by means of sodium cyanoborohydride in refluxing methanol.
…………………..
EXAMPLE 4
Preparation of Compound 229
A three liter 3-necked flask was fitted with a
condenser, nitrogen inlet and overhead mechanical stirring apparatus. The flask was charged with pulverized A82846B acetate salt (20.0 g, 1.21 × 10-3 mol) and methanol (1000 mL) under a nitrogen atmosphere. 4′-chlorobiphenylcarboxaldehyde (2.88 g, 1.33 × 10-2 mol, 1.1 eq.) was added to this stirred mixture, followed by methanol (500 mL). Finally, sodium cyanoborohydride (0.84 g, 1.33 × 10-2 mol, 1.1 eq.) was added followed by methanol (500 mL). The resulting mixture was heated to reflux (about 65°C).
After 1 hour at reflux, the reaction mixture attained homogeneity. After 25 hours ac reflux, the heat source was removed and the clear reaction mixture was measured with a pH meter (6.97 at 58.0°C). 1 N NaOH (22.8 mL) was added
dropwise to adjust the pH to 9.0 (at 54.7°C). The flask was equipped with a distillation head and the mixture was concentrated under partial vacuum to a weight of 322.3 grams while maintaining the pot temperature between 40-45°C.
The distillation head was replaced with an addition funnel containing 500 mL of isopropanol (IPA). The IPA was added dropwise to the room temperature solution over 1 hour. After approximately 1/3 of the IPA was added, a granular precipitate formed. The remaining IPA was added at a faster rate after precipitation had commenced. The flask was weighed and found to hold 714.4 grams of the IPA/methanol slurry.
The flask was re-equipped with a still-head and
distilled under partial vacuum to remove the remaining methanol. The resulting slurry (377.8 g) was allowed to chill in the freezer overnight. The crude product was filtered through a polypropylene pad and rinsed twice with 25 mL of cold IPA. After pulling dry on the funnel for 5 minutes, the material was placed in the vacuum oven to dry at 40°C. A light pink solid (22.87 g (theory = 22.43 g) ) was recovered. HPLC analysis versus a standard indicated 68.0% weight percent of Compound 229 (4- [4-chlorophenyl] benzyl-A82846B] in the crude solid, which translated into a
corrected crude yield of 69.3%.
The products of the reaction were analyzed by reverse-phase HPLC utilizing a Zorbax SB-C18 column with ultraviolet light (UV; 230 nm) detection. A 20 minute gradient solvent system consisting of 95% aqueous buffer/5% CH3CN at time=0 minutes to 40% aqueous buffer/60% CH3CN at time=20 minutes was used, where the aqueous buffer was TEAP (5 ml CH3CN, 3 ml phosphoric acid in 1000 ml water).
………………….
Oritavancin (also termed N-(4-(4-chlorophenyl)benzyl)A82846B and LY333328) has the following Formula III:

References
- Targanta Revives Oritavancin: Next Weapon Against cSSSI? BioWorld Today, November 26, 2007
- “Biotechs pick up slack in antibiotics development”. 17 May 2011.
- http://www.farm.ucl.ac.be/Full-texts-FARM/Domenech-2009-1.pdf “Interactions of oritavancin, a new lipoglycopeptide derived from vancomycin, with phospholipid bilayers: Effect on membrane permeability and nanoscale lipid membrane organization” 2009
- Scheinfeld, N (2007). “A comparison of available and investigational antibiotics for complicated skin infections and treatment-resistant Staphylococcus aureus and enterococcus“.J Drugs Dermatol. 6 (4): 97–103. PMID 17373167.
- 2007 ICAAC Posters: E-1612 “In Vitro Activity Profile of Oritavancin against a Broad Spectrum of Aerobic and Anaerobic Bacterial Pathogens”/E -1613 “In Vitro Activity Profile of Oritavancin (ORI) Against Organisms Demonstrating Key Resistance Profiles to Other Antimicrobial Agents”/E-1614 “In vitro Time Kill Studies of Oritavancin against Drug-resistant Isolates ofStaphylococcus aureus and Enterococci”/E-1615 “Anti-Enterococcal Activity Profile of Oritavancin, a Potent Lipoglycopeptide under Development for Use Against Gram-Positive Infections”/E-1616 “Anti-Streptococcal Activity Profile of Oritavancin, a Potent Lipoglycopeptide under Development for Use Against Gram-Positive Infections”/E-1617 “In Vitro Activity Profile of Oritavancin (ORI) Against Resistant Staphylococcal Populations From a Recent Surveillance Initiative”/E-1620 “Pharmacokinetic Concentrations of Oritavancin Kill Stationary-Phase and Biofilm Staphylococcus aureus In Vitro.” / Targanta Press Release September 19, 2007
- ICAAC 2007 Posters: “In Vitro Susceptibility of Genotypically Distinct Clostridium difficileStrains to Oritavancin” and “Activity of Metronidazole, Vancomycin and Oritavancin Against Epidemic Clostridium difficile Spores” / Targanta Press Release September 19, 2007
- ASM 2007 Poster: “Efficacy of Oritavancin in a Murine Model of Bacillus anthracis Spore Inhalation Anthrax” / Targanta Press Release May 24, 2007
- Belley; McKay, GA; Arhin, FF; Sarmiento, I; Beaulieu, S; Fadhil, I; Parr Jr, TR; Moeck, G (2010).“Oritavancin Disrupts Membrane Integrity of Staphylococcus aureus and Vancomycin-Resistant Enterococci To Effect Rapid Bacterial Killing”. Antimicrobial agents and chemotherapy 54(12): 5369–71. doi:10.1128/AAC.00760-10. PMC 2981232. PMID 20876372.
- Zhanel et al. (2012). “Oritavancin: Mechanism of Action”. Clin Infect Dis.doi:10.1093/cid/cir920.
- ICAAC 2003 Late-breaker poster: “Phase III Trial Comparing 3-7 days of Oritavancin vs. 10-14 days of Vancomycin/Cephalexin in the Treatment of Patients with Complicated Skin and Skin Structure Infections (cSSSI)” / InterMune Press Release September 15, 2003
- ClinicalTrials.gov NCT00514527
- Comparison of the Efficacy and Safety of Oritavancin Front-Loaded Dosing Regimens to Daily Dosing: An Analysis of the SIMPLIFI Trial. May 2011. doi:10.1128/AAC.00029-11.
- “Drugs.com, Targanta Submits Oritavancin New Drug Application”. Retrieved 2008-02-12.
- “FDA News, Targanta to Get FDA Decision by December”. Retrieved 2008-04-10.
- http://www.fiercebiotech.com/press-releases/fda-issues-complete-response-letter-oritavancin Dec 2008.
- “Pharmaceutical Business Review, EMEA accepts Targanta’s oritavancin MAA for review”. Retrieved 2008-06-26.
- http://www.nelm.nhs.uk/en/NeLM-Area/News/2009—August/24/European-application-for-investigational-antibiotic-oritavancin-withdrawn-/
- http://onlinelibrary.wiley.com/doi/10.1111/j.1574-6976.2003.tb00628.x/pdf
- http://www.pjps.pk/wp-content/uploads/pdfs/26/5/Paper-30.pdf
- Antimicrobial Agents and Chemotherapy, 2003 , vol. 47, 5 p. 1700 – 1706
- Antimicrobial Agents and Chemotherapy, 1999 , vol. 43, 1 p. 115 – 120
- Antimicrobial Agents and Chemotherapy, 1997 , vol. 41, 10 p. 2165 – 2172
- Tetrahedron, 2004 , vol. 60, 47 p. 10611 – 10618………… NMRhttp://www.sciencedirect.com/science/article/pii/S0040402004015108
Cooper, R.D.G.; Snyder, N.J.; Zweifel, M.J.; et al.; Reductive alkylation of glycopeptide antibiotics: Synthesis and antibacterial activity. J Antibiot 1996, 49, 6, 575-81.
Fromtling, R.A.; Castaer, J.; LY-333328. Drugs Fut 1998, 23, 1, 17.
Cooper, R.D.G.; Huff, B.E.; Nicas, T.I.; Quatroche, J.T.; Rodriguez, M.J.; Snyder, N.J.; Staszak, M.A.; Thompson, R.C.; Wilkie, S.C.; Zweifel, M.J. (Eli Lilly and Company); Glycopeptide antibiotic derivs. EP 0817797; JP 1999502534; WO 9630401 .
Cooper, R.D.G.; Huff, B.E.; Nicas, T.I.; Quatroche, J.T.; Rodriguez, M.J.; Snyder, N.J.; Staszak, M.A.; Thompson, R.C.; Wilkie, S.C.; Zweifel, M.J. (Eli Lilly and Company); Glycopeptide antibiotic derivs. EP 0667353; EP 1016670; EP 1031576 .
| EP0435503A1 * | Dec 11, 1990 | Jul 3, 1991 | Eli Lilly And Company | Improvements in or relating to gylcopeptide derivatives |
| US4639433 * | Aug 14, 1985 | Jan 27, 1987 | Eli Lilly And Company | Glycopeptide derivatives |
| US4698327 * | Apr 18, 1986 | Oct 6, 1987 | Eli Lilly And Company | Novel glycopeptide derivatives |
| US20040106590 * | Aug 29, 2003 | Jun 3, 2004 | Barry Eisenstein | Methods and reagents for treating infections of clostridium difficile and diseases associated therewith |
| US20050197333 | Dec 22, 2004 | Sep 8, 2005 | Van Duzer John H. | Rifamycin analogs and uses thereof |
| US20070014849 | Sep 20, 2006 | Jan 18, 2007 | Daniela Jabes | Use of ramoplanin to treat diseases associated with the use of antibiotics |
| US20030176327 * | Oct 18, 2002 | Sep 18, 2003 | Cassell Gail Houston | Antibiotics for treating biohazardous bacterial agents |
| US20040147441 | Aug 25, 2003 | Jul 29, 2004 | Leach Timothy S. | Methods and reagents for preventing bacteremias |
| WO1999010006A1 | Aug 18, 1998 | Mar 4, 1999 | Lilly Co Eli | Therapy for staphylococcus aureus |
| WO2000066144A2 | Apr 19, 2000 | Nov 9, 2000 | Lilly Co Eli | Monthly doses of glycopeptide antibiotics for treatment of streptococcus pneumoniae infections |
| WO2008097364A2 | Sep 24, 2007 | Aug 14, 2008 | Targanta Therapeutics Corp | Use of oritavancin for prevention and treatment of anthrax |
| WO1998052592A1 * | May 5, 1998 | Nov 26, 1998 | Lilly Co Eli | Urea and thiourea derivatives of glycopeptides |
| WO2002036612A1 * | Nov 2, 2001 | May 10, 2002 | Univ Cambridge Tech | Antibacterial agents comprising conjugates of glycopeptides and peptidic membrane-associating elements |
| WO2007138999A1 | May 25, 2007 | Dec 6, 2007 | Shionogi & Co | Glycopeptide antibiotic derivative |
| WO2009081958A1 | Dec 25, 2008 | Jul 2, 2009 | Shionogi & Co | Glycosylated glycopeptide antibiotic derivative |
| EP2314599A1 | Nov 24, 2005 | Apr 27, 2011 | National University Corporation Nagoya University | Glycopeptide antibiotic monomer derivatives |
| US5919756 * | May 1, 1997 | Jul 6, 1999 | Eli Lilly And Company | Amides |
| US5919771 * | Apr 30, 1998 | Jul 6, 1999 | Eli Lilly And Company | Urea and thiourea derivatives of glycopeptides |
| US7078380 | Nov 2, 2001 | Jul 18, 2006 | Cambridge University Technical Services Limited | Antibacterial agents comprising conjugates of glycopeptides and peptidic membrane associating elements |
| US8481696 | Dec 25, 2008 | Jul 9, 2013 | Shionogi & Co., Ltd. | Glycosylated glycopeptide antibiotic derivatives |


The US Food and Drug Administration (FDA) has granted a six-month Priority Review designation to Genzyme’s New Drug Application (NDA) for Cerdelga (eliglustat)
13 dec 2013
Genzyme’s Cerdelga NDA receives US FDA priority review designationpharmabiz.comThe US Food and Drug Administration (FDA) has granted a six-month Priority Review designation to Genzyme’s New Drug Application (NDA) for Cerdelga (eliglustat), an investigational oral therapy for adult patients with Gaucher disease
ELIGLUSTAT TARTRATE
THERAPEUTIC CLAIM Treatment of lysosomal storage disorders
CHEMICAL NAMES
1. Octanamide, N-[(1R,2R)-2-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-hydroxy-1-(1-
pyrrolidinylmethyl)ethyl]-, (2R,3R)-2,3-dihydroxybutanedioate (2:1)
2. bis{N-[(1R,2R)-2-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-hydroxy-1-(pyrrolidin-1-
ylmethyl)ethyl]octanamide} (2R,3R)-2,3-dihydroxybutanedioate
MOLECULAR FORMULA C23H36N2O4 . ½ C4H6O6
MOLECULAR WEIGHT 479.6
MANUFACTURER Genzyme Corp.
CODE DESIGNATION Genz-112638
CAS REGISTRY NUMBER 928659-70-5
old article cut paste
Genzyme Announces Positive New Data from Two Phase 3 Studies for Oral Eliglustat Tartrate for Gaucher Disease
Eliglustat tartrate (USAN)
CAS:928659-70-5
February 15, 2013
Genzyme , a Sanofi company (EURONEXT: SAN and NYSE: SNY), today announced positive new data from the Phase 3 ENGAGE and ENCORE studies of eliglustat tartrate, its investigational oral therapy for Gaucher disease type 1. The results from the ENGAGE study were presented today at the 9th Annual Lysosomal Disease Network WORLD Symposium in Orlando, Fla. In conjunction with this meeting, Genzyme also released topline data from its second Phase 3 study, ENCORE. Both studies met their primary efficacy endpoints and together will form the basis of Genzyme’s registration package for eliglustat tartrateThe data presented at this year’s WORLD symposium reinforce our confidence that eliglustat tartrate may become an important oral option for patients with Gaucher disease”The company is developing eliglustat tartrate, a capsule taken orally, to provide a convenient treatment alternative for patients with Gaucher disease type 1 and to provide a broader range of treatment options for patients and physicians. Genzyme’s clinical development program for eliglustat tartrate represents the largest clinical program ever focused on Gaucher disease type 1 with approximately 400 patients treated in 30 countries.“The data presented at this year’s WORLD symposium reinforce our confidence that eliglustat tartrate may become an important oral option for patients with Gaucher disease,” said Genzyme’s Head of Rare Diseases, Rogerio Vivaldi MD. “We are excited about this therapy’s potential and are making excellent progress in our robust development plan for bringing eliglustat tartrate to the market.”ENGAGE Study Results:In ENGAGE, a Phase 3 trial to evaluate the safety and efficacy of eliglustat tartrate in 40 treatment-naïve patients with Gaucher disease type 1, improvements were observed across all primary and secondary efficacy endpoints over the 9-month study period. Results were reported today at the WORLD Symposium by Pramod Mistry, MD, PhD, FRCP, Professor of Pediatrics & Internal Medicine at Yale University School of Medicine, and an investigator in the trial.
The randomized, double-blind, placebo-controlled study had a primary efficacy endpoint of improvement in spleen size in patients treated with eliglustat tartrate. Patients were stratified at baseline by spleen volume. In the study, a statistically significant improvement in spleen size was observed at nine months in patients treated with eliglustat tartrate compared with placebo. Spleen volume in patients treated with eliglustat tartrate decreased from baseline by a mean of 28 percent compared with a mean increase of two percent in placebo patients, for an absolute difference of 30 percent (p<0.0001).
Eliglustat tartate (Genz-112638)
What is Eliglustat?
- Eliglustat is a new investigational phase 3 compound from Genzyme Corporation that is being studied for type 1 Gaucher Disease.
- Eliglustat works as a substrate reduction therapy by reducing glucocerebroside. formation.
- This product is an oral agent (i.e. a pill) that is taken once or twice a day in contrast to an IV infusion for enzyme replacement therapy. Enzyme replacement therapy focuses on replenishing the enzyme that is deficient in Gaucher Disease and breaks down glucocerebroside that accumulates.
- The clinical trials for eliglustat tartate are sponsored by Genzyme Corporation.


Durata Therapeutics Announces FDA’s Acceptance for Priority Review of NDA for Dalvance (dalbavancin hydrochloride)

| CAS No. | 171500-79-1 |
| Chemical Name: | Dalbavancin |
| Synonyms: | MDL 63397;Dalbavancin |
| CBNumber: | CB41028737 |
| Molecular Formula: | C88H100Cl2N10O28 |
| Formula Weight: | 1816.71 |
CHICAGO, Nov 26, 2013 (GLOBE NEWSWIRE via COMTEX) — Durata Therapeutics, Inc. DRTX +6.48% today announced that the New Drug Application (NDA) for its investigational drug, Dalvance (dalbavancin hydrochloride) for injection, has been accepted for priority review by the U.S. Food and Drug Administration (FDA) with an action date of May 26, 2014. Durata is seeking FDA approval of Dalvance(TM) for the treatment of patients with acute bacterial skin and skin structure infections (ABSSSI) caused by susceptible Gram-positive microorganisms, including MRSA (methicillin resistant Staphylococcus aureushttp://www.drugs.com/nda/dalbavancin_131126.html

Dalbavancin, V-Glycopeptide, VER-001, BI-397 factor B0, BI-397
Vicuron Pharmaceuticals (Originator)
5,31-Dichloro-38-de(methoxycarbonyl)-7-demethyl-19-deoxy-56-O-[2-deoxy-2-(10-methylundecanamido)-beta-D-glucopyranuronosyl]-38-[N-[3-(dimethylamino)propyl]carbamoyl]-42-O-alpha-D-mannopyranosyl-N15-methylristomycin A aglycone; (3S,15R,18R,34R,35S,48S,50aR
Dalbavancin, which is also referred to in the scientific literature as BI397 or VER001, is a semi=synthetic glycopeptide mixture, the properties of which have been reported in U.S. Pat. Nos. 5,606,036, 5,750,509, 5,843,679, and 5,935,238.
U.S. Publication No. 20040224908;
J Antibiot1995,48,(8):869
Drugs Fut1999,24,(8):839
Dalbavancin is prepared by chemical modification of the natural glycopeptide complex A-40,926 as described in Malabarba and Donadio (1999) Drugs of the Future 24(8):839-846. The predominant component of dalbavancin is Factor Bo, which accounts for >75% of the whole complex.
[0042] The amount of each of the components present in a dalbavancin composition is dictated by a variety of factors, including, for example, the fermentation conditions employed in the preparation of the natural glycopeptide complex A-40926, which is the precursor to dalbavancin (see, e.g., U.S. Pat. No.5,843,679), the conditions employed to recover A-40926 from the fermentation broth, the chemical reactions employed to selectively esterify the caxboxyl group of the sugar moiety of A-40926, the conditions employed to amidate the peptidyl carboxyl group, the conditions employed to saponify the ester of the carboxyl group of the N-acylaminoglucuronic acid function, the conditions employed to recover dalbavancin from the synthetic mixture, and the like.
the semisynthetic glycopeptide dalbavancin was synthesized from the natural antibiotic A 40926, originally isolated from an Actinomadura culture (Malabarba et al., 1998, U.S. Pat. No. 5,750,509). Dalbavancin has shown greater efficacy against various bacterial strains than vancomycin or the antibiotic linezolid and represents a promising new treatment for skin and soft tissue infections (see, e.g., Jabés et al., 2004, Antimicrob. Agents Chemother. 48:1118-1123). According to U.S. Pat. No. 5,750,509, dalbavancin is a glycopeptide antibiotic with a monomethyl moiety at its N15 amino (see FIG. 1 for numbering), and this N15-monomethyl amino could be free (i.e. —NHCH3) or protected with an amino protecting group such as t-butoxycarbonyl, carbobenzyloxy, arylalkyl or benzyl. The method for making certain of the dalbavancin components reported in the ‘509 patent also produced N15,N15-dialkyl analogs of dalbavancin in minor-quantities, but these molecules were not characterized.
Dalbavancin (INN, trade name Zeven) is a novel second-generation lipoglycopeptideantibiotic. It belongs to the same class as vancomycin, the most widely used and one of the few treatments available to patients infected with methicillin-resistant Staphylococcus aureus (MRSA).[1]
Dalbavancin (BI397) is a novel semisynthetic lipoglycopeptide that was designed to improve upon the natural glycopeptides currently available, vancomycin and teicoplanin.[2]
It possesses in vitro activity against a variety of Gram-positive pathogens[3][4] includingMRSA and MRSE.[5] It is a once-weekly, two-dose antibiotic that Pfizer acquired when it bought Vicuron Pharmaceuticals in 2005.[6]
Dalbavancin has undergone a phase III clinical trial for adults with complicated skin infections, but in Dec 2007 the FDA said more data was needed before approval.[6] On September 9, 2008, Pfizer announced that it will withdraw all marketing applications in order to conduct another Phase 3 clinical trial.[7] Durata Therapeutics acquired the rights to dalbavancin in December 2009 and has initiated two new Phase III clinical trials for treatment of acute bacterial skin and skin structure infections.[8] Preliminary results in Dec 2012 looked good.[9]
- Vicuron Pharmaceuticals Submits New Drug Application for Dalbavancin to U.S. Food and Drug Administration
- Scheinfeld NS. (May 2006). “Dalbavancin: A review for dermatologists.”. Dermatology Online Journal 12 (6). Unknown parameter
|numero=ignored (help) PMID 17083861 - Chen AY, Zervos MJ, Vazquez JA (2007). “Dalbavancin: a novel antimicrobial”. Int. J. Clin. Pract. 61 (5): 853–63. doi:10.1111/j.1742-1241.2007.01318.x. PMC 1890846.PMID 17362476.
- Das B, Sarkar C, Biswas R, Pandey S (2008). “Review: dalbavancin-a novel lipoglycopeptide antimicrobial for gram positive pathogens”. Pak J Pharm Sci 21 (1): 78–88. PMID 18166524.
- Dalbavancin: A Novel Lipoglycopeptide Antibacterial
- UPDATE 1-Pfizer says US FDA wants more data on antibiotic. Dec 2007
- “Pfizer Will Withdraw Global Marketing Applications for Dalbavancin to Conduct a New Trial” (Press release). Pfizer Inc. 2008-09-09. Retrieved 2008-09-11.
- Durata Begins Dalbavancin Study Enrollment. Drug Discovery & Development – October 05, 2011.
- Durata Therapeutics Announces Phase 3 Clinical Trial Results for Dalbavancin in the Treatment of ABSSSI

DAPTOMYCIN

VANCOMYCIN
