
Home » Preclinical drugs (Page 9)
Category Archives: Preclinical drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
GSK1904529A, GSK 4529

GSK1904529A, GSK 4529
GSK1904529A is a selective inhibitor of IGF1R with IC50 of 27 nM.
| 851.96 | |
| Formula | C44H47F2N9O5S |
| CAS Number | 1089283-49-7 |
N-(2,6-difluorophenyl)-5-[3-[2-[5-ethyl-2-methoxy-4-[4-(4-methylsulfonylpiperazin-1-yl)piperidin-1-yl]anilino]pyrimidin-4-yl]imidazo[1,2-a]pyridin-2-yl]-2-methoxybenzamide,
N-(2,6-Difluorophenyl)-5-[3-[2-[[5-ethyl-2-(methyloxy)-4-[4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl]phenyl]amino]-4-pyrimidinyl]imidazo[1,2-a]pyridin-2-yl]-2-(methyloxy)benzamide
NMR……http://www.abmole.com/download/gsk1904529a-hnmr.pdf

GSK1904529A, selectively inhibits IGF-IR and IR with IC50s of 27 and 25 nmol/L, respectively. It is a promising candidate for therapeutic use in solid and hematologic cancers. IC50s for GSK1904529A in tumor cell lines ranged from 35 nmol/L to >30 umol/L. The tumor histologic types showing the greatest sensitivity to this compound were Ewing’s sarcoma and multiple myeloma, where IC50s in three of five Ewing’s sarcoma cell lines were <100 nmol/L and IC50s in five of eight multiple myeloma cell lines were <200 nmol/L.
GSK1904529A is a small-molecule inhibitor of the insulin-like growth factor-I receptor (IGF-IR) with IC50 value of 27 nM 1.
GSK1904529A is a reversible and ATP-competitive inhibitor with Ki value of 1.6 nM. In NIH-3T3/LISN cells, GSK1904529A potently inhibited phosphorylation of IGF-IR with IC50 value of 22 nM. It also demonstrated to be a selective inhibitor since it showed poor inhibitory activity against 45 other serine/threonine and tyrosine kinases. When treated with whole-cell extracts, GSK1904529A significantly inhibited the ligand-induced phosphorylation of IGF-IR and decreased phosphorylation of downstream signaling including AKT, IRS-1 and ERK at concentrations > 0.01μM. GSK1904529A suppressed cell proliferation in a variety of tumor cells. The IC50 values for NCI-H929, TC-71, SK-N-MC, COLO 205, MCF7 and PREC are 81, 35, 43, 124, 137 and 68 nM, respectively. In COLO 205, MCF-7, and NCI-H929 cells, GSK1904529A treatment resulted in cell accumulation in G1 and decrease in S and G2-M phases. Moreover, in NIH-3T3/LISN xenograft model, once daily administration of GSK1904529A at 30 mg/kg inhibited 56% of tumor growt

…………..
Intermediates
 ,
,  ,
,  ,
, 
 ,
,

 ,
, ![]() ,
, ![]()
 ,
, 
 u can construct your synthesis
u can construct your synthesis
http://www.google.com/patents/US20080300242
Intermediate Example 2 5-[3-(2-chloro-4-pyrimidinyl)imidazo[1,2-a]pyridin-2-yl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide
Step A: Methyl 3-formyl-4-hydroxybenzoate
Methyl 4-hydroxybenzoate (3.00 g, 19.7 mmol) and magnesium chloride (2.81 g, 29.5 mmol) were stirred in 100 mL of acetonitrile. TEA (10.3 mL, 73.9 mmol) was added via syringe. Paraformaldehyde (12.0 g, 133 mmol) was added in a single portion and the reaction was heated to reflux. The reaction was stirred at reflux for 24 hours and cooled to rt. The reaction was quenched by the addition of approximately 100 mL of 1N HCl and poured into EtOAc. The layers were separated, and the organic layer was washed with brine. The combined aqueous layers were extracted with EtOAc. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. The clean fractions (by TLC) were concentrated in vacuo to afford 2.06 g (58%) of the desired product. 1H NMR (400 MHz, DMSO-d6): δ 11.54 (s, 1H), 10.27 (s, 1H), 8.21 (d, J=2.4 Hz, 1H), 8.03 (dd, J=8.8, 2.4 Hz, 1H), 7.07 (d, J=8.8 Hz, 1H), 3.79 (s, 3H).
Step B: methyl 3-formyl-4-(methyloxy)benzoate
Methyl 3-formyl-4-hydroxybenzoate (2.06 g, 11.4 mmol) and K2CO3 (2.36 g, 17.1 mmol) were stirred in 50 mL of DMF. Methyl iodide (1.42 mL, 22.8 mmol) was added via syringe, and the reaction was stirred for 6 hours at rt. The reaction was poured into H2O and diethyl ether, and the layers were separated. The organic layer was washed with brine, and the combined aqueous layers were extracted with diethyl ether. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo to afford 2.24 g of crude desired product. 1H NMR (400 MHz, DMSO-d6): δ 10.33 (s, 1H), 8.23 (d, J=2.2 Hz, 1H), 8.20 (dd, J=8.8, 2.2 Hz, 1H), 7.36 (d, J=8.8 Hz, 1H), 3.99 (s, 3H), 3.83 (s, 3H).
Step C: 2-(methyloxy)-5-[(methyloxy)carbonyl]benzoic acid
Crude methyl 3-formyl-4-(methyloxy)benzoate from the previous step was dissolved in 40 mL of dioxane with stirring. Sulfamic acid (5.87 g, 60.5 mmol) in 20 mL of H2O was added to the stirring solution. Sodium chlorite (1.68 g, 80% by weight, 18.6 mmol) in 20 mL of H2O was added dropwise via addition funnel. The reaction was stirred for 40 min and poured into EtOAc and H2O. The layers were separated, and the organic layer was washed with brine. The combined aqueous layers were extracted with EtOAc, and the combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The solid was transferred to an Erlenmeyer flask with the aid of 30-40 mL of DCM. Approximately 50 mL of hexanes was added. Air was blown over the solution to allow most of the DCM to evaporate. Diethyl ether was added (20-30 mL), and the suspension was filtered. The solid was washed with hexanes, collected, and dried to afford 1.96 g (82% over 2 steps) of the desired compound. 1H NMR (400 MHz, DMSO-d6): δ 12.92 (brs, 1H), 8.22 (d, J=2.2 Hz, 1H), 8.07 (dd, J=8.8, 2.2 Hz, 1H), 7.24 (d, J=8.8 Hz, 1H), 3.88 (s, 3H), 3.82 (s, 3H).
Step D: methyl 3-{[(2,6-difluorophenyl)amino]carbonyl}-4-(methyloxy)benzoate
2-(Methyloxy)-5-[(methyloxy)carbonyl]benzoic acid (1.96 g, 9.33 mmol) was suspended in 60 mL of DCM with stirring. DMF (0.036 mL, 0.46 mmol) was added via syringe. Oxalyl chloride (7.0 mL, 2.0M in dichloromethane, 14 mmol) was added dropwise via addition funnel. The addition funnel was rinsed with 10 mL of DCM. The reaction was stirred for 2 hours and concentrated in vacuo. The resultant solid was further dried under high vacuum pressure. The solid was dissolved in 60 mL of DCM with stirring. Pyridine (3.8 mL, 47 mmol), (4-dimethylamino)pyridine (0.0570 g, 0.467 mmol), and 2,6-difluoroaniline (3.0 mL, 28 mmol) were added to the solution. The reaction was stirred for 18 hours and poured into 1N HCl. The layers were separated, and the aqueous layer was washed once with DCM and once with diethyl ether. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. The clean fractions (by TLC) were concentrated in vacuo to afford 1.56 g (52%) of the desired product. 1H NMR (400 MHz, DMSO-d6): δ 9.81 (s, 1H), 8.31 (d, J=2.0 Hz, 1H), 8.10 (dd, J=8.8, 2.0 Hz, 1H), 7.38 (m, 1H), 7.31 (d, J=88 Hz, 1H), 7.22-7.13 (m, 2H), 3.97 (s, 3H), 3.82 (s, 3H).
Step E: 5-[(2-Chloro-4-pyrimidinyl)acetyl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide and 5-[(E)-2-(2-chloro-4-pyrimidinyl)-1-hydroxyethenyl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide
Methyl 3-{[(2,6-difluorophenyl)amino]carbonyl}-4-(methyloxy)benzoate (1.56 g, 4.86 mmol) was dissolved in 50 mL of THF with stirring and cooled to 0° C. Lithium bis(trimethylsilyl)amide (14.6 mL, 1.0M in THF, 14.6 mmol) was added slowly via syringe. 2-Chloro-4-methylpyrimidine (0.750 g, 5.83 mmol) was dissolved in 10 mL of THF and added dropwise via addition funnel. The addition funnel was rinsed with 10 mL of THF. The reaction was stirred at 0° C. for 1 hour and quenched with saturated ammonium chloride solution. The mixture was poured into H2O and EtOAc, and the layers were separated. The organic layer was washed with brine, and the combined aqueous layers were extracted with EtOAc. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. The clean fractions (by TLC) were concentrated in vacuo to afford 1.26 g (62%) of the desired product. The proton NMR is a mixture of the keto and enol tautomers (˜2:1). 1H NMR (400 MHz, DMSO-d6): δ 13.58 (s, 1H, enol), 9.83 (s, 1H, keto), 9.82 (s, 1H, enol), 8.72 (m, 1H, keto), 8.54 (m, 1H, enol), 8.34 (s, 1H, keto), 8.22 (m, 1H, both), 8.06 (m, 1H, enol), 7.56 (m, 1H, keto), 7.42-7.31 (m, 2H, both+1H, enol), 7.22-7.14 (m, 2H, both), 6.55 (s, 1H, enol), 4.66 (s, 2H, keto), 4.00 (s, 3H, keto), 3.97 (s, 3H, enol).
Step F: 5-[3-(2-chloro-4-pyrimidinyl)imidazo[1,2-a]pyridin-2-yl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide
A tautomeric mixture of 5-[(2-Chloro-4-pyrimidinyl)acetyl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide and 5-[(E)-2-(2-chloro-4-pyrimidinyl)-1-hydroxyethenyl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide (1.26 g, 3.02 mmol) was dissolved in 60 mL of DCM with stirring. NBS (0.538 g, 3.02 mmol) was added in a single portion. The reaction was stirred for 20 minutes and concentrated in vacuo. The residue was dissolved in 60 mL of dioxane with stirring, and 2-aminopyridine (0.853 g, 9.06 mmol) was added in a single portion. The reaction was heated at 60° C. with an oil bath for 24 hours and cooled to rt. The reaction was stirred at rt for an additional 40 hours. The reaction was poured into half-saturated NaHCO3 solution and EtOAc, and the layers were separated. The organic layer was washed with brine, and the combined aqueous layers were extracted twice with EtOAc. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. Impure fractions were concentrated and further purified by flash chromatography. The combined clean fractions (by TLC) from both runs were combined and concentrated in vacuo to afford 1.07 g (72%) of the desired product. 1H NMR (400 MHz, DMSO-d6): δ 9.80 (s, 1H), 9.40 (d, J=7.0 Hz, 1H), 8.57 (d, J=5.1 Hz, 1H), 8.10 (d, J=1.5 Hz, 1H), 7.84-7.77 (m, 2H), 7.57 (m, 1H), 7.39 (m, 1H), 7.33-7.26 (m, 2H), 7.24-7.14 (m, 3H), 3.99 (s, 3H).
Step A: 1,1-dimethylethyl 4-(methylsulfonyl)-1-piperazinecarboxylate
To 1,1-dimethylethyl 1-piperazinecarboxylate (568 g, 3.05 mol) in DCM (4 L) was added TEA (617 g, 6.10 mol). After stirring for 10 min at 0° C., methanesulfonyl chloride (384 g, 3.35 mol) was added via addition funnel. The mixture was stirred at rt overnight. The mixture was poured into H2O (1 L) and extracted with DCM (1 L). The organic layer was separated, washed with H2O (1 L), dried (Na2SO4), and rotovapped down to provide the title compound of step A (720 g, 2.72 mol, 90%) which was used without further purification. 1H NMR (400 MHz, CDCl3) δ 1.44 (s, 9H), 2.76 (s, 3H), 3.11-3.17 (m, 4H), 3.50-3.53 (m, 4H).
Step B: 1-(methylsulfonyl)piperazine hydrochloride
To 1,1-dimethylethyl 4-(methylsulfonyl)-1-piperazinecarboxylate (360 g, 1.36 mol) in MeOH (1 L) was added HCl (6 M in MeOH, 2 L) dropwise. The mixture was stirred at rt for 1 h. About 1 L of MeOH was rotovapped off. The resultant precipitate was filtered, washed with MeOH, and dried on high vacuum to provide the title compound of Step B (A combination of 2 batches, 570 g) which was used without further purification. 1H NMR (400 MHz, D2O) δ 2.95 (s, 3H), 3.27-3.29 (m, 4H), 3.42-3.46 (m, 4H).
Step C: 1-(methylsulfonyl)-4-(4-piperidinyl)piperazine dihydrochloride
To 1-(methylsulfonyl)piperazine hydrochloride (150 g, 632 mmol) in DCE (3.5 L) was added TEA (192 g, 1.90 mol). The mixture was stirred at rt for 1 h and then acetic acid (94.8 g, 1.58 mol) and 1,1-dimethylethyl 4-oxo-1-piperidinecarboxylate (251 g, 1.26 mol) was added. After stirring another h, the reaction was cooled with an ice water bath and NaBH(OAc)3 (294 g, 1.39 mol) was added in four portions. The mixture was stirred overnight at rt. The reaction mixture was neutralized with saturated Na2CO3 to pH 8-9. The organic phase was washed with brine and H2O, dried (Na2SO4), and rotovapped down to provide the crude Boc-protected amine (A combination of 3 batches, 720 g). This amount was split into 2 batches and used without further purification. To 1,1-dimethylethyl 4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinecarboxylate (360 g, 1.04 mol) in MeOH (1 L) was added HCl (6 M in MeOH, 2 L). The mixture was stirred at rt for 30 min. About 1 L of MeOH was rotovapped off. The resultant precipitate was filtered, washed with MeOH, and dried on high vacuum to provide the title compound of Step C (A combination of 2 batches, 600 g, 1.87 mol, 89% over 2 steps). 1H NMR (400 MHz, D2O) δ 1.87-1.91 (m, 2H), 2.33-2.36 (m, 2H), 2.97 (s, 3H), 2.99-3.05 (m, 2H), 3.45-3.59 (m, 11H).
Step A: 1-{1-[2-ethyl-5-(methyloxy)-4-nitrophenyl]-4-piperidinyl}-4-(methylsulfonyl)piperazine
A mixture of 1-ethyl-2-fluoro-4-(methyloxy)-5-nitrobenzene (Example 187, step C) (0.93 g, 4.67 mmol), 1-(methylsulfonyl)-4-(4-piperidinyl)piperazine (Example 204, step C) (1.16 g, 4.67 mmol) and K2CO3 (0.774 g, 5.60 mmol) in DMSO (20 mL) was heated at 90° C. for 48 h. The reaction had not progressed sufficiently so the reaction was then heated at 120° C. for an additional 4 h. The reaction was cooled to rt, poured into H2O and extracted with DCM. Some saturated brine solution was added and the resultant was exhaustively extracted with DCM. The combined organics were washed with H2O then dried over MgSO4. The resultant solution was concentrated onto silica and purified by flash chromatography to afford 1-{1-[2-ethyl-5-(methyloxy)-4-nitrophenyl]-4-piperidinyl}-4-(methylsulfonyl)piperazine (1.12 g, 56%). 1H NMR (400 MHz, DMSO-d6) δ ppm 7.73-7.80 (m, 1H), 6.75 (s, 1H), 3.91 (s, 3H), 3.23-3.30 (m, 1H), 3.05-3.19 (m, 3H), 2.87 (s, 2H), 2.70-2.84 (m, 2H), 2.53-2.67 (m, 5H), 1.77-1.94 (m, 2H), 1.48-1.67 (m, 2H), 1.19 (t, J=7.42 Hz, 3H).
Step B: 5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}aniline
A mixture of 1-{1-[2-ethyl-5-(methyloxy)-4-nitrophenyl]-4-piperidinyl}-4-(methylsulfonyl)piperazine (1.12 g, 2.63 mmol) and sulfided platinum on carbon (0.410 g, 0.105 mmol) in EtOAc (40 mL) was sealed in a round bottom flask with a rubber septum. The reaction mixture was purged with N2 gas and then a balloon of H2 gas was connected and the vessel was flushed with the H2 gas. The reaction was stirred at rt for 2 d. TLC analysis showed the complete consumption of the starting nitro compound so the reaction mixture was filtered through celite to remove the catalyst. The filtrate was concentrated onto silica gel and purified by flash chromatography to afford 5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}aniline (0.479 g, 46%).
1H NMR (400 MHz, DMSO-d6) δ ppm 6.60 (s, 1H), 6.46 (s, 1H), 4.35 (br. s., 2H), 3.71 (s, 3H), 3.03-3.16 (m, 4H), 2.81-2.93 (m, 5H), 2.56-2.68 (m, 6H), 2.29-2.42 (m, 1H), 1.72-1.89 (m, 2H), 1.44-1.62 (m, 2H), 1.09 (t, J=7.51 Hz, 3H).
Example 237 N-(2,6-difluorophenyl)-5-(3-{2-[(5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}phenyl)amino]-4-pyrimidinyl}imidazo[1,2-a]pyridin-2-yl)-2-(methyloxy)benzamide
A mixture of 5-[3-(2-chloro-4-pyrimidinyl)imidazo[1,2-a]pyridin-2-yl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide (Intermediate Example 2) (0.60 g, 1.22 mmol), 5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}aniline (Example 206, Step B) (0.48 g, 1.22 mmol) and HCl (4N,1,4-Dioxane, 0.61 mL, 2.44 mmol) in trifluoroethanol (15 mL) was heated at 170° C. for 40 min in the microwave. The reaction mixture was concentrated onto silica gel and purified by flash column chromatography. Recrystallization from DCM and EtOH afforded the title compound N-(2,6-difluorophenyl)-5-(3-{2-[(5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}phenyl)amino]-4-pyrimidinyl}imidazo[1,2-a]pyridin-2-yl)-2-(methyloxy)benzamide (0.61 g, 56%).
1H NMR (400 MHz, DMSO-d6)
δ ppm 9.80 (s, 1H), 9.36 (br. s., 1H), 8.50 (s, 1H), 8.26 (d, J=5.22 Hz, 1H), 8.12 (d, J=2.11 Hz, 1H), 7.80 (dd, J=8.80, 2.02 Hz, 1H), 7.71 (d, J=9.07 Hz, 1H), 7.53 (s, 1H), 7.36-7.50 (m, 2H), 7.30 (d, J=8.80 Hz, 1H), 7.14-7.25 (m, 2H), 6.91-7.00 (m, 1H), 6.83 (s, 1H), 6.58 (d, J=5.22 Hz, 1H), 4.00 (s, 3H), 3.80 (s, 3H), 3.08-3.15 (m, 4H), 3.00-3.07 (m, 2H), 2.88 (s, 3H), 2.67-2.76 (m, 2H), 2.61-2.66 (m, 4H), 2.56 (q, J=7.51 Hz, 2H), 2.38-2.46 (m, 1H), 1.80-1.91 (m, 2H), 1.50-1.68 (m, 2H), 1.11 (t, J=7.51 Hz, 3H).
MS (M+H, ES+) 852.
Separately, the Title Compound was Prepared in the Following Manner:
A mixture of 5-[3-(2-chloro-4-pyrimidinyl)imidazo[1,2-a]pyridin-2-yl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide (Intermediate Example 2) (23.0 g, 46.8 mmol), 5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}aniline (Example 206, Step B) (18.6 g, 46.8 mmol) and HCl (4N,1,4-Dioxane, 23.4 mL, 93.6 mmol) in trifluoroethanol (200 mL) was heated in a sealed vessel at 85° C. for 48 h. After cooling to rt, the reaction mixture was treated with an excess of 7N NH3 in MeOH and then subjected to filtration. The filtrate was concentrated onto silica gel and purified by flash chromatography. The chromatographed product was dissolved in DCM and treated with an excess of diethyl ether. The resultant bright yellow precipitate was collected by filtration and then recrystallized from DCM and EtOH to afford the title compound N-(2,6-difluorophenyl)-5-(3-{2-[(5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}phenyl)amino]-4-pyrimidinyl}imidazo[1,2-a]pyridin-2-yl)-2-(methyloxy)benzamide (28.2 g, 67%).
……………..
Discovery and optimization of imidazo[1,2-a]pyridine inhibitors of insulin-like growth factor-1 receptor (IGF-1R)
Bioorg Med Chem Lett 2009, 19(3): 1004……http://www.sciencedirect.com/science/article/pii/S0960894X08014376


Scheme 1.
Reagents and conditions: (a) (ClCO)2, DMF, CH2Cl2; (b) 2,6-difluoroaniline, pyridine, CH2Cl2 (84%, 2 steps); (c) LiN(SiMe3)2, THF (83%); (d) NBS, CH2Cl2, then 2-aminopyridine, dioxane, 60 °C (77%); (e) HCl or p-TSA·H2O, trifluoroethanol or isopropanol, 80–100 °C or 140–180 °C (μw) (50–90%).
References
Antitumor activity of GSK1904529A, a small-molecule inhibitor of the insulin-like growth factor-I receptor tyrosine kinase.
Sabbatini et al. Clin Cancer Res. 2009 May 1;15(9):3058-67. PMID: 19383820.
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Facebook 

 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
 LIONEL MY SON
LIONEL MY SON
/////////////GSK1904529A, IGF1R, GSK 4529, preclinical
New Parathyroid Disease Drug Etelcalcetide Seeks FDA Approval
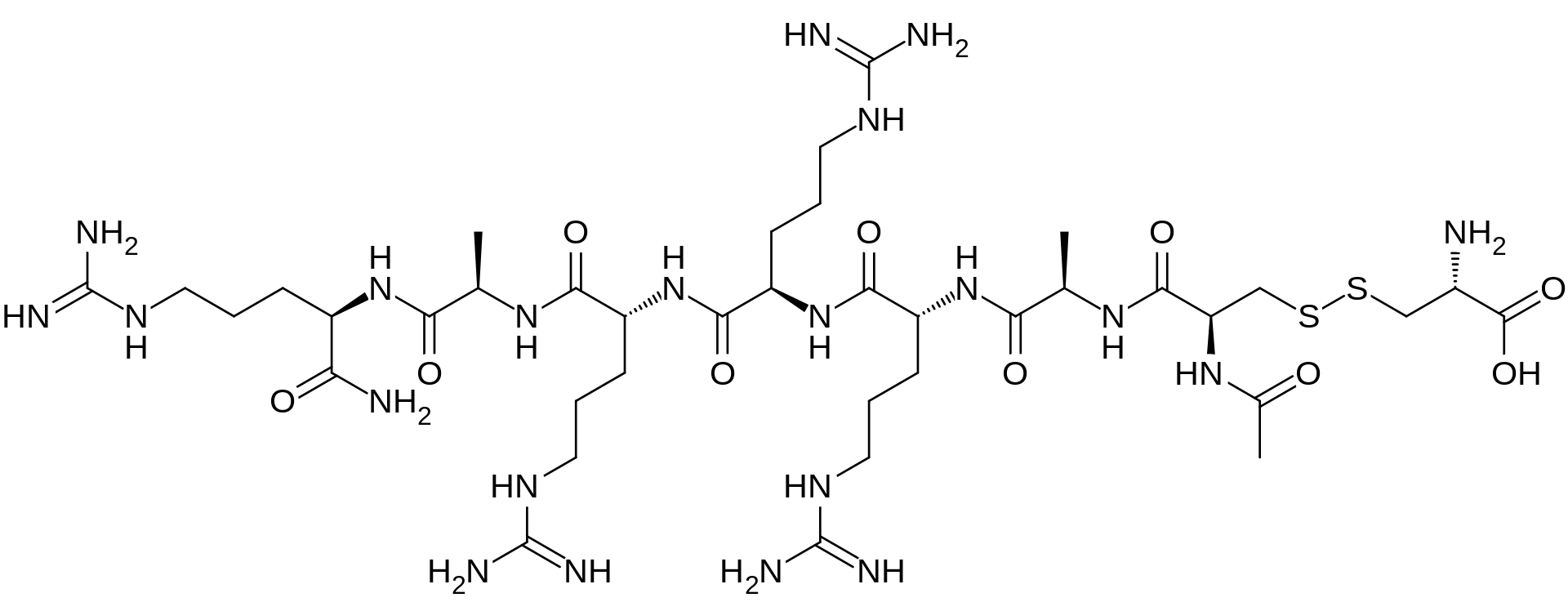
Etelcalcetide, AMG 416
AMG-416; Etelcalcetide hydrochloride; KAI-4169; KAI-4169-HCl; ONO-5163; Telcalcetide; Velcalcetide; Velcalcetide hydrochloride
D-Argininamide, N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-, disulfide with L-cysteine,
N-Acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide disulfide with L-cysteine
Secondary hyperparathyroidism
- Originator KAI Pharmaceuticals…Kai Pharmaceuticals, Inc.
- Developer Amgen; KAI Pharmaceuticals; Ono Pharmaceutical
- ClassDisulfides; Peptides
- Mechanism of ActionCalcium-sensing receptor agonists
New Parathyroid Disease Drug Seeks FDA Approval
Amgen is seeking FDA approval for etelcalcetide (AMG 461), the first calcimimetic agent administered intravenously after dialysis to treat secondary hyperparathyroidism (SHPT) in patients with chronic kidney disease (CKD).
SHPT is a common and serious condition that is often progressive among CKD patients. It usually manifests as high amounts of parathyroid hormone (PTH) associated with abnormal calcium and phosphorus levels in the body.
– See more at: http://www.pharmacytimes.com/product-news/new-parathyroid-disease-drug-seeks-fda-approval
Etelcalcetide is a D-amino peptide calcimimetic undergoing clinical evaluation for the treatment of secondary hyperparathyroidismfor patients with chronic kidney disease (CKD) on hemodialysis. Etelcalcetide is administered intravenously at the end of each dialysis session.[1][2] It exerts a pharmacological effect by binding to and activating the calcium-sensing receptor (CaSR) in theparathyroid gland, resulting in parathyroid hormone (PTH) reduction and suppression.[1] Elevated PTH is often observe in patients with CKD.[3]
On August 25, 2015 Amgen Inc. announced its submission of a New Drug Application to the Food and Drug Administration for etelcalcetide.[1]
| CAS Registry Number | 1262780-97-1 |
|---|---|
| Synonyms | Velcalcetide |
| Chemical data | |
| Formula | C38H73N21O10S2 |
| Molecular mass | 1,048.26 g·mol−1 |
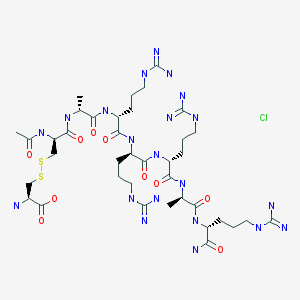
Etelcalcetide hydrochloride
RN: 1334237-71-6
UNII: 72PT5993DU
The term “AMG 416” refers to the compound having the chemical name: JV-acetyl-D- cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-arginamide disulfide with L- cysteine, which may be represented as:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2
The terms “AMG 416 hydrochloride” or “AMG 416 HQ” are interchangeable and refer to the compound having the chemical name: N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl- D-arginyl-D-alanyl-D-arginamide disulfide with L-cysteine hydrochloride, which may be represented as:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · xHCl
D-Argininamide, N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-, disulfide with L-cysteine, hydrochloride (1:?)
N-Acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide disulfide with L-cysteine hydrochloride
Amgen today announced the submission of a New Drug Application (NDA) with the United States Food and Drug Administration (FDA) for etelcalcetide (formerly AMG 416) for the treatment of secondary hyperparathyroidism (SHPT) in patients with chronic kidney disease (CKD) on hemodialysis. If approved, etelcalcetide will be the first calcimimetic agent that can be administered intravenously at the end of the dialysis session.
“Secondary hyperparathyroidism is a serious, progressive disease that can lead to significant clinical consequences and is also associated with a high pill burden for patients,” said Sean E. Harper, M.D., executive vice president of Research and Development at Amgen. “We look forward to working with regulatory authorities during the review process to bring this important treatment to market, helping to fill an unmet need for the many patients impacted by this disease.”
Etelcalcetide is a novel calcimimetic agent that suppresses the secretion of parathyroid hormone and is in clinical development for the treatment of SHPT in patients with CKD on hemodialysis. Etelcalcetide is administered intravenously three times per week at the end of each dialysis session. It acts by binding to and activating the calcium-sensing receptor on the parathyroid gland, thereby causing decreases in parathyroid hormone (PTH). Sustained elevations in PTH are known to be associated with significant clinical consequences for patients with CKD.
The submission includes data from three Phase 3 studies, all of which met the primary endpoints, including two pooled placebo-controlled trials in more than 1,000 patients and a head-to-head study evaluating etelcalcetide compared with cinacalcet.
About Secondary Hyperparathyroidism
SHPT is a common and serious condition that is often progressive among patients with CKD, and it affects many of the approximately two million people throughout the world who are receiving dialysis, including 450,000 people in the U.S. The disorder develops early in the course of CKD and usually manifests as increased levels of PTH as a result of increased production from the parathyroid glands (four small glands in the neck). Patients with end stage renal disease who require maintenance dialysis often have substantial elevations of PTH that are commonly associated with abnormal calcium and phosphorus levels and an increased risk of significant clinical consequences.
About Etelcalcetide (AMG 416)
Etelcalcetide is a novel calcimimetic agent in clinical development for the treatment of SHPT in CKD patients on hemodialysis that is administered intravenously at the end of the dialysis session. Etelcalcetide binds to and activates the calcium-sensing receptor on the parathyroid gland, thereby decreasing PTH levels.
About Sensipar® (cinacalcet)
Sensipar® (cinacalcet) is the first oral calcimimetic agent approved by the FDA for the treatment of SHPT in adult patients with CKD on dialysis. Sensipar is not indicated for use in adult patients with CKD who are not on dialysis because of an increased risk of hypocalcemia. The therapy is also approved in the U.S. for treatment of hypercalcemia in adult patients with parathyroid carcinoma and hypercalcemia in adult patients with primary HPT for whom parathyroidectomy would be indicated on the basis of serum calcium levels, but who are unable to undergo parathyroidectomy. Sensipar binds to the calcium-sensing receptor, resulting in a drop in PTH levels by inhibiting PTH synthesis and secretion. In addition, the reductions in PTH lower serum calcium and phosphorus levels.
…………………
WO 2011014707
http://www.google.com/patents/WO2011014707A2?cl=en
……………………..
WO 2014210489
http://www.google.com/patents/WO2014210489A1?cl=en
A variety of compounds having activity for lowering parathyroid hormone levels have been described. See International Publication No. WO 2011/014707. In one embodiment, the compound may be represented as follows:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2
The main chain has 7 amino acids, all in the D-configuration and the side-chain cysteine residue is in the L-configuration. The amino terminal is acetylated and the carboxyl-terminal is amidated. This compound (“AMG-416”) has utility for the treatment of secondary hyperparathyroidism (SHPT) in hemodialysis patients. A liquid formulation comprising AMG-416 may be administered to a subject intravenously. The hydrochloride salt of AMG-416 may be represented as follows:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · x(HCl)
Therapeutic peptides pose a number of challenges with respect to their formulation. Peptides in general, and particularly those that contain a disulfide bond, typically have only moderate or poor stability in aqueous solution. Peptides are prone to amide bond hydrolysis at both high and low pH. Disulfide bonds can be unstable even under quite mild conditions (close to neutral pH). In addition, disulfide containing peptides that are not cyclic are particularly prone to dimer formation. Accordingly, therapeutic peptides are often provided in lyophilized form, as a dry powder or cake, for later reconstitution. A lyophilized formulation of a therapeutic peptide has the advantage of providing stability for long periods of time, but is less convenient to use as it requires the addition of one or more diluents and there is the potential risk for errors due to the use of an improper type or amount of diluent, as well as risk of contamination. In addition, the lyophilization process is time consuming and costly.
Accordingly, there is a need for an aqueous liquid formulation comprising a peptide agonist of the calcium sensing receptor, such as AMG 416. It would be desirable for the liquid formulation to remain stable over a relevant period of time under suitable storage conditions and to be suitable for administration by intravenous or other parenteral routes.
…………………………………
Milestones
- 25 Aug 2015Preregistration for Secondary hyperparathyroidism in USA (IV)
- 29 May 2015Pooled analysis efficacy and adverse events data from two phase III trials in secondary hyperparathyroidism released by Amgen
- 21 Apr 2015Amgen plans to submit Biological License Application to USFDA and Marketing Authorisation Application to EMA for Secondary hyperparathyroidism
References
- “Amgen Submits New Drug Application For Novel Intravenous Calcimimetic Etelcalcetide (AMG 416)”
- “Velcalcetide (AMG 416), a novel peptide agonist of the calcium-sensing receptor, reduces serum parathyroid hormone and FGF23 levels in healthy male subjects
- “Evidence for Chronic Kidney Disease-Mineral and Bone Disorder Associated With Metabolic Pathway Changes”
KAI-4169, a novel calcium sensing receptor agonist, decreases serum iPTH, FGF-23 and improves serum bone markers in a phase 2 study in hemodialysis subjects with chronic kidney disease-mineral and bone disorder
49th Congr Eur Renal Assoc – Eur Dialysis Transpl Assoc (May 24-27, Paris) 2012, Abst SAO054
KAI-4169, a novel peptide agonist of the calcium sensing receptor, attenuates PTH and soft tissue calcification and restores parathyroid gland VDR levels in uremic rats
49th Congr Eur Renal Assoc – Eur Dialysis Transpl Assoc (May 24-27, Paris) 2012, Abst SAO014
Long term safety and efficacy of velcalcetide (AMG 416), a calcium-sensing receptor (CaSR) agonist, for the treatment of secondary hyperparathyroidism (SHPT) in hemodialysis (HD) patients
Kidney Week (November 5-10, Atlanta, GA) 2013, Abst SA-PO575
Preclinical PK and PD relationship for KAI-4169, a novel calcimimetic
93rd Annu Meet Endo Soc (June 4-7, Boston) 2011, Abst P1-198
KAI-4169, a novel calcimimetic for the treatment of secondary hyperparathyroidism
93rd Annu Meet Endo Soc (June 4-7, Boston) 2011, Abst P2-98
Characterization of KAI-4169, a novel peptide for the treatment of chronic kidney disease – Mineral and bone disorder, in a phase I study in healthy males
44th Annu Meet Am Soc Nephrol (ASN) (November 8-13, Philadelphia) 2011, Abst FR-PO1238
| WO2011014707A2 | Jul 29, 2010 | Feb 3, 2011 | Kai Pharmaceuticals, Inc. | Therapeutic agents for reducing parathyroid hormone levels |
////Etelcalcetide, Parathyroid Disease, Amgen Inc, AMG 416, KAI-4169; KAI-4169-HCl, ONO-5163, Telcalcetide, Velcalcetide, Velcalcetide hydrochloride
Gamendazole a novel drug candidate for male contraception.

Gamendazole
(E) 3-(1-(2,4-Dichlorobenzyl)-6-(trifluoromethyl)-1H-indazol-3-yl)acrylic Acid
trans-3-(1-Benzyl-6-(trifluoromethyl)-1H-indazol-3-yl)acrylic acid)
(E)-3-[1-[(2,4-Dichlorophenyl)methyl]-6-(trifluoromethyl)indazol-3-yl]prop-2-enoic acid
- C18H11Cl2F3N2O2
- mw415.193
- RC-MC-110
Heat Shock Protein 90 (HSP90) Inhibitors
University Of Kansas …Innovator
Gamendazole is a novel drug candidate for male contraception. It is an indazole carboxylic acid derived from lonidamine (LND). Gamendazole produced 100% antispermatogenic effects at 25 mg/kg i.p. in rats, whereas 200 mg/kg was fatal for 60% of rats tested. Since gamendazole produced 100% efficacy, it was tested orally. At a dose of 6 mg/kg, 100% of rats were infertile 4 weeks after a single administration. Complete infertility was maintained for 2 weeks, followed by complete recovery in 4 of 7 rats. The other 3 never recovered fertility. Upon dosing 6 mg/kg orally for 7 days, it produced similar infertility results, but only 2 of 7 rats recovered fertility. There were no abnormalities in rates of conception or abnormal conception in rats who recovered fertility.
In August 2004, preclinical data were presented at the 228th ACS meeting in Philadelphia, PA. Gamendazole, an indazole-3-acrylic acid derivative
Pathology reports were conducted on gamendazole treated rats. At 25 mg/kg i.p., 6 mg/kg oral, and in animals that survived 200 mg/kg i.p., there were no remarkable findings, with no evidence of inflammation, necrosis, tumors, or hemorrhage. There was also a lack of observable behavioral effects at 25 mg/kg i.p., 6 mg/kg oral, and in animals that survived 200 mg/kg i.p. Gamendazole treatment had no effect on testosterone levels, and was reported to affect Sertoli cell function, leading to decreased levels of inhibin B. Low levels of inhibin B were correlated to the infertility of the rat
Female oral contraceptive drugs are widely available in the market by several trade names, including Altravera, Brevicon, Levora, and i-pill, whereas potentially safer, more convenient, and more effective oral male contraceptives are not yet commercially available. However, there are some experimental drugs.AF-2785 1, gamendazole 2, lonidamine 3, and adjudin 4 are most promising among the experimental


Gamendazole was recently identified as an orally active antispermatogenic compound with antifertility effects. The cellular mechanism(s) through which these effects occur and the molecular target(s) of gamendazole action are currently unknown. Gamendazole was recently designed as a potent orally active antispermatogenic male contraceptive agent. Here, we report the identification of binding targets and propose a testable mechanism of action for this antispermatogenic agent. Both HSP90AB1 (previously known as HSP90beta [heat shock 90-kDa protein 1, beta]) and EEF1A1 (previously known as eEF1A [eukaryotic translation elongation factor 1 alpha 1]) were identified as binding targets by biotinylated gamendazole (BT-GMZ) affinity purification from testis, Sertoli cells, and ID8 ovarian cancer cells; identification was confirmed by matrix-assisted laser desorption/ionization-time of flight mass spectrometry and Western blot analysis. BT-GMZ bound to purified yeast HSP82 (homologue to mammalian HSP90AB1) and EEF1A1, but not to TEF3 or HBS1, and was competed by unlabeled gamendazole. However, gamendazole did not inhibit nucleotide binding by EEF1A1.
Gamendazole binding to purified Saccharomyces cerevisiae HSP82 inhibited luciferase refolding and was not competed by the HSP90 drugs geldanamycin or novobiocin analogue, KU-1. Gamendazole elicited degradation of the HSP90-dependent client proteins AKT1 and ERBB2 and had an antiproliferative effect in MCF-7 cells without inducing HSP90. These data suggest that gamendazole may represent a new class of selective HSP90AB1 and EEF1A1 inhibitors. Testis gene microarray analysis from gamendazole-treated rats showed a marked, rapid increase in three interleukin 1 genes and Nfkbia (NF-kappaB inhibitor alpha) 4 h after oral administration. A spike in II1a transcription was confirmed by RT-PCR in primary Sertoli cells 60 min after exposure to 100 nM gamendazole, demonstrating that Sertoli cells are a target. AKT1, NFKB, and interleukin 1 are known regulators of the Sertoli cell-spermatid junctional complexes. A current model for gamendazole action posits that this pathway links interaction with HSP90AB1 and EEF1A1 to the loss of spermatids and resulting infertility.
Synthesis








…………………….
2-Halo benzoic acid is converted into aroyl chloride and then to aroyl cyanide in an overall yield of 82%. Aroyl cyanides 5 are converted to 2-halophenyl glyoxylate ester 7 via ketoamide 6 in 85% yields as shown in Scheme below. Direct conversion of aroyl cyanide 5 to ester 7 is also reported[ U.S. Patent 4,596,885, 1986 .] but with lesser yields.

The 2-halophenylglyoxylate 7 esters are reacted with monosubstituted hydrazines 8 to give hydrazones 9. The monosubstituted hydrazones 9 are cyclized to give indazole esters 10. This cyclization is best conducted in the presence of DPPF · PdCl2 in 94.54% yield as shown in Scheme below.

The indazole-3-carboxylic esters 10 were reduced with sodium borohydride to alcohol 11 and were oxidized to aldehyde 12with MnO2. The aldehyde is converted to acrylic acids with malonic acid (Knoevenagel condensation) to give 88–95.6% yield of the final compounds, as shown in Schemebelow.

Preparation of (E) 3-(1-(2,4-Dichlorobenzyl)-6-(trifluoromethyl)-1H-indazol-3-yl)acrylic Acid (R = CF3) (Gamendazole) (2)
- DOI:
- 10.1080/00397911.2012.696306
Arava Veerareddya*, Gogireddy Surendrareddya & P. K. Dubeyb
pages 2236-2241
Synthetic Communications: An International Journal for Rapid Communication of Synthetic Organic Chemistry
Volume 43, Issue 16, 2013
trans 3-[l- (l^-dichlorobenzy^-ό-trifluoromethyl-lH-indazol-S-ylj-acrylic acid (RC-MC-110) is provided.

EXAMPLE 2: Synthesis of a-ll-fl^-dichlorobenzyn-ό-trifluoromethyl-lH-indazol-S-yll-acrylic acid (RC-MC-110)
Step 1 : 2-(2-nitro-4-trifluoromethylphenyl)-malonic acid dimethyl ester.
Dimethyl malonate (59.7 g, 0.44 mol) was added dropwise to a stirred solution of potassium tert-butoxide (51 g, 0.44 mol) in dry t-butanol (500 mL). To the resultant suspension, a warm solution of 2-chloro-5-trifluoromethylnitrobenzene (50 g, 0.22 mol) in t-butanol (100 mL) was added and the mixture was refluxed for 6 h (reaction monitored by TLC). After completion of the reaction, most of the t-butanol was distilled off under vacuum, and chilled water was then added to the reaction mixture. The pH was adjusted to neutral with dilute hydrochloric acid, which resulted in the precipitation of the product. The mixture was stirred for 30 minutes and the product was filtered off (68 g, 95%). This material was used without further purification in the next step. A small amount was crystallized (EtOAc/hexane, 4:6) for analysis, to yield a yellow crystalline material, mp 65-67 0C. 1H NMR (CDCl3) 8.30 (s, 1 H), 7.92 (d, J = 8.4 Hz, 1 H), 7.69 (d, J = 8.4 Hz, 1 H), 5.37 (s, 1 H), 3.80 (s, 6 H). MS (FAB) m/z: 322.1 (M+ + 1).
Step 2: (2-nitro-4-trifluoromethylphenyl)-acetic acid methyl ester.
2-(2-Nitro-4-trifluoromethylphenyl)-malonic acid dimethyl ester (68 g, 0.21 mol) was dissolved in dimethyl sulfoxide (200 mL). Sodium chloride (34 g, 0.58 mol) and water (60 mL) were added and the mixture was stirred for 16-20 h at 120 0C (reaction monitored by TLC). The reaction mixture was then cooled to room temperature and quenched into water, which caused precipitation of the product. After stirring for 30 minutes, the product (45 g, 80%) was isolated by filtration. The product was used without further purification in the next reaction. A small sample was crystallized (EtOAc/hexane, 2:8) for analysis, to yield yellow crystals, mp 104-105 0C. 1H
NMR (CDCl3) 8.3 (s, 1 H), 7.88 (d, J = 8.4 Hz, 1 H), 7.50 (d, J = 8.4 Hz, 1 H), 4.12 (s, 2 H), 3.60 (s, 3 H). MS (FAB) m/z: 275.2 (M+ + 1).
Step 3: (2-Acetylamino-4-trifluoromethylphenyl)-acetic acid methyl ester.
Hydrogenation and acetylation of (2-nitro-4-trifluoromethylphenyl)-acetic acid methyl ester (25 g, 0.095 mol) in the presence of 5% Pd-C (2.5 g, 50% wet) and acetic anhydride (38 g, 0.37 mol) in toluene (200 mL) was carried out under vigorous stirring at room temperature and atmospheric pressure for about 4-5 h (reaction monitored by TLC). The catalyst was removed by filtration and washed with toluene two times. The combined organics were evaporated in vacuo to yield the product (24.8 g, 95%), which was used without further purification in the next step. A small sample was crystallized from hexane to yield the product as a yellow solid, mp 92-94 0C. H NMR (CDCl3) 8.86 (s, 1 H), 8.21 (s, 1 H), 7.36 (d, J = 8.1 Hz, 1 H), 7.31 (d, J = 8.1 Hz, 1 H), 3.74 (s, 3 H), 3.68 (s, 2 H), 2.23 (s, 3 H). Step 4: ό-Trifluoromethyl-lH-indazole^-carboxylic acid methyl ester.
To a solution of (2-acetylamino-4-trifluoromethylphenyl)-acetic acid methyl ester (16 g, 0.058 mol) in acetic acid (50 mL) was added dropwise t-butyl nitrite (90%) (7.35 g, 0.063 mol) over a period of 20 min. at 90-95 0C. The mixture was then stirred for 0.5 h at 95 0C, poured into cold water and stirred for 1 h. The precipitates were collected by filtration and washed with water. The crude material was dissolved in ethyl acetate and dried over sodium sulfate. The solvent was removed in vacuo. This material (13.4 g, 95%) was used without further purification in the next step. A small sample was crystallized from ethyl acetate to yield a white solid, mp 240-242 0C. H NMR (DMSO-d-6) 8.25 (d, J = 8.5 Hz, 1 H), 8.04 (s, 1 H), 7.58 (d, J = 8.5 Hz, 1 H), 3.95 (s, 3 H). MS (FAB) m/z: 245.1 (M+ + 1).
Step 5: l-(2,4-Dichlorobenzyl)-6-trifluoromethyl-lH-indazole-3-carboxylic acid methyl ester.
ό-Trifluoromethyl-lH-indazole-S-carboxylic acid methyl ester (2.75 g, 0.0112 mol) was dissolved in acetonitrile (50 mL), and potassium carbonate (1O g, 0.07 mol), 2,4-dichlorobenzyl chloride (2.42 g, 0.01239 mol) and tetrabutylammonium iodide (catalytic) were added. The reaction mixture was heated to reflux and refluxed for 2 h under good stirring. The progress of the reaction was monitored by TLC. After completion of the reaction, potassium carbonate was filtered while hot and then washed with acetone. The combined solvents were distilled off under reduced pressure to afford the crude mixture of Nl and N2 benzylated products. The isomers were separated by column chromatography (silica gel, eluent started with hexane then changed to 8:2 hexane, ethyl acetate). l-(2,4-Dichlorobenzyl)-6-trifluoromethyl-lH-indazole-3-carboxylic acid methyl ester. Yield: 3.62 g (80%), white crystals mp 118-120 0C. ‘ H NMR (CDCl3) 8.39 (d, J = 8.4 Hz, 1 H) 7.74 (s, 1 H), 7.57 (d, J = 8.4 Hz, 1 H), 7.45 (d, J = 2.1 Hz, 1 H), 7.12 (dd, J = 8.4 and 2.1 Hz, 1 H), 6.78 (d, J = 8.4 Hz, 1 H), 5.82 (s, 2 H), 4.07 (s, 3 H). MS (FAB) m/z: 403 (M+ + 1).Z-^^-DichlorobenzylJ-δ-trifluoromethyl-ZH-indazole-S-carboxylic acid methyl ester. Yield: 680 mg (15%), white crystals mp 132-134 0C. ‘ H NMR (DMSO-d-6) 8.27 (s, 1 H), 8.20 (d, J = 8.7 Hz, 1 H), 7.76 (d, J = 1.8 Hz, 1 H), 7.57 (d, J = 8.7 Hz, 1 H), 7.30 (dd, J = 8.3 and 1.8 Hz, 1 H), 6.78 (d, J = 8.3 Hz, 1 H), 6.17 (s, 2 H), 3.96 (s, 3 H).
Step 6: [l-(2.4-Difluorobenzyl)-6-trifluoromethyl-lH-indazol-3-yl1-methanol.
l -(2,4-Dichlorobenzyl)-6-trifluoromethyl-lH-indazole-3-carboxylic acid methyl ester (3.0 g, 0.0075 mol) dissolved in CH2Cl2(50 mL) was cooled to -78 0C. DIBAL-H (8.18 mL, 0.00818 mol) was added slowly dropwise via a syringe under an argon blanket over a period of 15 minutes. After the complete addition of DIBAL-H, the reaction mixture was stirred at -78°C for another 2 h (reaction monitored by TLC). The reaction was quenched carefully with methanol at -78 0C. The reaction mixture was then carefully poured into water and the layers were separated. The organic layer was washed with water and dried over sodium sulfate. Removal of the solvent yielded the crude alcohol (2.6 g, 93%), which was used without purification in the next step. The alcohol was a white solid, mp 137-139 0C. 1H NMR (CDCl3) 7.97 (d, J = 8.4 Hz, 1 H), 7.66 (s, 1 H), 7.44 (d, J = 2.0 Hz, 1 H), 7.42 (d, J = 8.5 Hz, 1 H), 7.12 (dd, J = 8.3 and 2.0 Hz, 1 H), 6.93 (d, J = 8.3 Hz, 1 H), 5.65 (s, 2 H), 5.09 (s, 2 H). MS (FAB) m/z: 375 (M+ + 1).Step 7: l-(2,4-Dichlorobenzyl)-6-trifluoromethyl-lH-indazole-3-carbaldehvde.
[l-(2,4-Difluorobenzyl)-6-trifluoromethyl-lH-indazol-3-yl]-methanol (3.75 g, 0.01 mol) was dissolved in CH2Cl2 (100 mL) and manganese(IV)oxide (8.7 g, 0.1 mol) was added and stirred for 2-3 h at room temperature (reaction monitored by TLC). The solids were removed by filtration and the removal of the CH2Cl2 in vacuo yielded the crude aldehyde. The aldehyde was used without further purification in the next step. The aldehyde (3.54 g, 95%) was a white solid, mp 97-98 0C. 1H NMR (CDCl3) 10.25 (s, 1 H), 8.45 (d, J = 8.5 Hz, 1 H), 7.79 (s, 1 H), 7.60 (d, J = 8.5 Hz, 1 H), 7.48 (d, J = 2.0 Hz, 1 H), 7.20 (dd, J = 8.3 Hz and 2.0 Hz, 1 H), 6.93 (d, J = 8.3 Hz, 1 H), 5.79 (s, 2 H). MS (FAB) m/z: 373 (M+ + 1).
Step 8: 3-ri-(2,4-Dichlorobenzyl)-6-trifluoromethyl-lH-indazol-3-yll-acrylic acid ethyl ester.
l-(2,4-Dichlorobenzyl)-6-trifluoromethyl-lH-indazole-3-carbaldehyde (2.0 g, 0.00536 mol) was dissolved in CH2Cl2 (50 niL) and Wittig reagent (carbethoxymethylene) triphenylphosphorane (1.06 g, 0.0536 mol) was added to the solution. The homogeneous reaction mixture was heated to reflux in an oil bath for 12 h. The reaction progress was monitored by TLC. The reaction mixture was cooled to room temperature and worked up by quenching into water and separating the organic layer. Removal of the CH2Cl2 yielded the crude product, which was purified by column chromatography to yield the pure product (2.25 g, 95%) as a white solid, mp 186-188 0C. 1H NMR (CDCl3) 8.08 (d, J = 8.5 Hz, 1 H), 7.99 (d, J = 16.2 Hz, 1 H), 7.74 (s, 1 H), 7.52 (d, J = 8.5 Hz, 1 H), 7.47 (d, J = 2.0 Hz, 1 H), 7.16 (dd, J = 8.3 and 2.0 Hz, 1 H), 6.84 (d, J = 8.3 Hz, 1 H), 6.82 (d, J = 16.2 Hz, 1 H), 5.72 (s, 2 H), 4.32 (q, J = 7.1 Hz, 2 H), 1.38 (t, J = 7.1 Hz, 3 H). MS (FAB) m/z: 443 (M+ + 1).It will be appreciated that the acrylic acid ethyl ester can be hydrogenated using 5% Pd-C in the presence of methanol, DCM at RT and 1 atm-pressure to give the propionic acid ester derivative. For example, treatment under such conditions yields 3-[l-(2,4-dichlorobenzyl)-6- trifluoromethyl-lH-indazol-3-yl]-propionic acid ethyl ester (JWS-2-70).
Step 9: l-(2,4-Dichlorobenzyl‘)-3-r6-trifluoromethyl-lΗ-indazol-3-yll-acrvlic acid.

l-(2,4-Dichlorobenzyl)-3-[6-trifluoromethyl-lH-indazol-3-yl]-acrylic acid ethyl ester (2.0 g, 0.0045 mol) was dissolved in a mixture of tetrahydrofuran (50 mL) and methanol (25 mL). A lithium hydroxide solution (0.33 g, 0.013 mol lithium hydroxide in 7.5 mL water) was added slowly at room temperature under good stirring. The reaction mixture was then warmed to 40 0C and held at that temperature for 2 h. The reaction mixture was diluted with water and extracted with ethyl acetate in order to remove neutral impurities. The layers were separated and the aqueous layer was cooled to 0 0C and then acidified with 20% sulfuric acid to pH 2. White solids precipitated and were filtered and dried to constant weight. The crude product was recrystallized from ethyl acetate and hexane (1 :1) to afford the pure product (1.68 g, 90%) as a white solid,
REFERENCES
- 1. Corsi , G. ; Palazzo , G. ; Germani , C. ; Barcellona , P. S. ; Silvestrini , B. 1-Halobenzyl-1H-indazole-3-carboxylic acids: A new class of antispermatogenic agents . J. Med. Chem. 1976 , 19 , 778
- 2. Palazzo , G. ; Corsi , G. ; Baiocchi , L. ; Silvestrini , B. Synthesis and pharmalogical properties of 1-substituted-3-dimethylaminoalkoxy-1H-indazoles . J. Med. Chem. 1966 , 9 , 38 – 41 .
- 3. Silvestrini , B. Basic and applied research in the study of indazole carboxylic acids . Chemotherapy 1981 , 27 ( Suppl.2 ), 9 – 20 .
- 4. Silvestrini , B. ; Palazzo , G. ; De Gregorio , M. D. 3-Lonidamine and related compounds . Progr. Med. Chem. 1985 , 21 , 111 – 135 .
- 5. Cheng , C. Y. ; Silvestrini , B. ; Grima , J. ; Mo , M. Y. ; Zhu , L. J. ; Johnsson , E. ; Saso , L. ; Leone , M. G. ; Palmery , M. ; Mruk , D. Two new male contraceptives exert their effects by depleting germ cells prematurely from the testes . Biol. Reprod. 2001 , 65 , 449 – 461 .
- 6. Xia , W. ; Mruk , D. D. ; Lee , W. M. ; Ceng , C. Y. Unraveling the molecular targets pertinent to junction restructuring events during spermatogenesis using the Adjudin-induced germ cell depletion model . J. Endocrinol. 2007 , 192 , 563 – 583 .
- 7. Cheng , C. Y. ; Mruk , D. D. ; Silvestrini , B. ; Bonanomi , M. ; Wong , C. H. ; Siu , M. K. Y. ; Lee , N. P. Y. ; Mo , M. Y. AF-2364 [1-(2,4-dichlorobenzyl)-1H-indazole-3-carbohydrazide] is a potential male contraceptive: A review of recent data . Contraception2005 , 72 , 251 – 261 .
- 8. Tash , J. S. ; Attardi , B. ; Hild , S. A. ; Chakrasali , R. ; Jakkarg , S. R. ; Georg , G. I. A novel potent indazole carboxylic acid derivative blocks spermatogenesis and is contraceptive in rats after a single oral dose . Biol. Reprod. 2008 , 78 , 1127 – 1138 .
- 9. Sarkar , O. ; Mathur , P. P. Adjudin-mediated germ cell depletion alters the anti-oxidant status of adult rat testes . Mol. Reprod. Dev. 2009 , 76 , 31 – 37 .
- 10. Mok , K.-W. ; Mruk , D. D. ; Lie , P. P. Y. ; Lui , W.-Y. ; Cheng , C. Y. Adjudin, a potential male contraceptive, exerts its effects locally in the seminiferous epithelium of mammalian testes. Reproduction. 2011, 141, 571–580.
- 11. Wang , H. ; Chen , X. X. ; Wang , L.-R. ; Mao , Y.-D. ; Zhou , Z. M. ; Sha , J.-H. AF-2364 is a prospective spermicide candidate .Asian J. Androl. 2010 , 12 , 322 – 335 .
-
- “Gamendazole”. NextBio. http://www.nextbio.com. Retrieved 31 July 2011.
- Tash, Joseph (July 2008). “A Novel Potent Indazole Carboxylic Acid Derivative Blocks Spermatogenesis and Is Contraceptive in Rats after a Single Oral Dose”. Biology of Reproduction 78 (6): 1127–1138. doi:10.1095/biolreprod.106.057810. PMID 18218612.
Chakrasali, R.; Jakkaraj, S.R.; Tash, J.S.; Hild, S.A.; Attardi, B.; Georg, G.I.
Design, synthesis and in vivo evaluation of Gamendazole(R), a novel orally active male contraceptive agent
228th Am Chem Soc (ACS) Natl Meet (August 22-26, Philadelphia) 2004, Abst MEDI 305
| CHENG C.Y. ET AL: “Two New Male Contraceptives Exert Their Effects by Depleting Germ Cells Prematurely from the Testis” BIOLOGY OF REPRODUCTION, SOCIETY FOR THE STUDY OF REPRODUCTION, CHAMPAIGN, IL, US, vol. 65, no. 2, 1 August 2001 (2001-08-01), pages 449-461, XP002547492 ISSN: 0006-3363 | ||
| 2 | * | GATTA F. ET AL: “Pyrazolo[3,4-d]pyrimidines. Related to Lonidamine” JOURNAL OF HETEROCYCLIC CHEMISTRY, HETEROCORPORATION. PROVO, US, vol. 26, no. 3, 1 March 1989 (1989-03-01), pages 613-618, XP002547493 ISSN: 0022-152X |
| US3895026 * | Feb 9, 1973 | Jul 15, 1975 | Acraf | Substituted 1-benzyl-1h-indazole-3-carboxylic acids and derivatives thereof |
| WO2003097063A1 * | May 5, 2003 | Nov 27, 2003 | Bayer Ag | Derivatives of 2-(1-benzyl-1h-pyrazolo (3, 4-b)pyridine-3yl) -5-(4-pyridinyl)-4-pyrimidine amine and the use thereof as guanylate cyclase stimulators |
| WO2006015263A2 * | Jul 29, 2005 | Feb 9, 2006 | Duan Jian-Xin | Lonidamine analogs |
 |
|
 |
|
| Names | |
|---|---|
| IUPAC name
(E)-3-[1-[(2,4-Dichlorophenyl)methyl]-6-(trifluoromethyl)indazol-3-yl]prop-2-enoic acid[1]
|
|
| Other names
trans-3-(1-Benzyl-6-(trifluoromethyl)-1H-indazol-3-yl)acrylic acid)
|
|
| Identifiers | |
| 877773-32-5 |
|
| ChemSpider | 9387234 |
| Jmol-3D images | Image |
| PubChem | 11212172 |
| Properties | |
| C18H11Cl2F3N2O2 | |
| Molar mass | 415.19 g·mol−1 |
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on twitter
 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL
/////////
What is SBM-TFC-039 an SGLT Inhibitor from Sirona Biochem !!
A new “flozin” seems to me appearing on the horizon in form of SBM-TFC-039 an SGLT Inhibitor from Sirona Biochem, picked up a list from WO 2012160218, from TFChem…….see link , Sirona Biochem Announces SGLT2 Inhibitor and Skin Lightening Patent Granted, 29 Jun 2015, Patent entitled “Family of aryl, heteroaryl, o-aryl and o-heteroaryl carbasugars”
This led me to search, “Family of aryl, heteroaryl, o-aryl and o-heteroaryl carbasugars”
WO 2012160218 A1, IN 2013-DN10635, CN 103649033Tf化学公司
| Applicant | Tfchem |

List above as in http://www.google.com/patents/WO2012160218A1?cl=en
FROM THE ABOVE LIST, SBM-TFC-039 MAY BE PREDICTED/OR AS SHOWN BELOW
COMPD 16 as in/WO2012160218
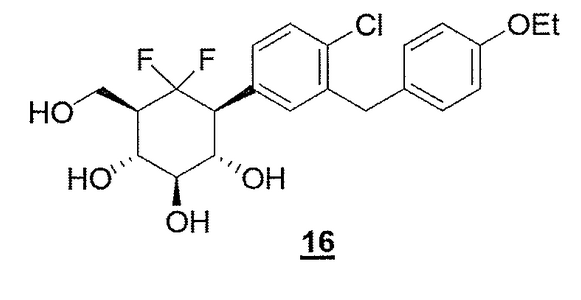
COMPD 16, PREDICTED/LIKELY SBM-TFC-039 has CAS 1413373-30-4, name D-myo-Inositol, 1-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1,2,3-trideoxy-2,2-difluoro-3-(hydroxymethyl)-
Just scrolling through the patent gave me more insight
MORE EVIDENCE….http://www.google.com/patents/WO2012160218A1?cl=en, this patent descibes compd 16 as follows
Compound 16 according to the invention has been compared to Dapaglifozin to underline the improvement of the duration of action, i.e. the longer duration of glucosuria, of the compound when the intracyclic oxygen atom of the glucose moiety is replaced by a CF2 moiety.

This assay has been carried out at a dose of 3 mg/ kg.
The results obtained are presented on Figure 5. It appears thus that 16 (3 mg/kg) triggered glucosuria that lasted beyond 24 hours compared to Dapagliflozin.
• Compound 16 according to the invention has been compared to the compound 9 of WO 2009/1076550 to underline the improvement of the duration of action of the compound when a mimic of glucose bearing a CH-OH moiety instead of the intracyclic oxygen atom is replaced by a mimic of glucose bearing a CF2 in place of the CH-OH moiet .

%7D)
| Company | Sirona Biochem Corp. |
| Description | Sodium-glucose cotransporter 2 (SGLT2) inhibitor |
| Molecular Target | Sodium-glucose cotransporter 2 (SGLT2) |
| Mechanism of Action | Sodium-glucose cotransporter 2 (SGLT2) inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Preclinical |
| Standard Indication | Diabetes |
| Indication Details | Treat Type II diabetes |
| Regulatory Designation | |
| Partner | Shanghai Fosun Pharmaceutical Group Co. Ltd. |
SBM-TFC-039
PATENT
WO 2012160218
http://www.google.com/patents/WO2012160218A1?cl=en
Examples within this first subclass include but are not limited to:

Synthesis of compound 8
C35H34O5 M = 534.64 g.mol“
Mass: (ESI ): 535.00 (M + H); 552.00 (M + H20); 785.87; 1086.67 (2M + H20)

A.

Procedure A:
To a solution of 4 (10.5g, 15.89mmol, leq) in toluene (400mL) were added 18-crown-6 (168mg, 0.64mmol, 0.04eq) and potassium carbonate (6.69g, 48.5mmol, 3.05eq.). The mixture was stirred overnight at room temperature, and then the remising insoluble material was filtered off and washed with toluene. The filtrate and the washings were combined, washed with 2N hydrochloric acid aqueous solution followed by saturated sodium hydrogencarbonate aqueous solution, dried over sodium sulphate, filtered and concentrated under reduced pressure. The residue was purified on silica gel chromatography (cyclohexane/ethyl acetate 98:2 to 80:20) to afford cyclohexenone 8 (4.07g; 48% yield) as yellowish oil.
Procedure B:
A solution of 7 (3.27g, 5.92mmol, leq) in pyridine (14mL) was cooled to 0°C before POCl3 (2.75mL, 29.6mmol, 5eq) was added dropwise. The mixture was stirred at this temperature for 10 min before the cooling bath was removed. The reaction mixture was stirred overnight at room temperature before being re-cooled to 0°C. POCI3 (2.75mL, 29.6mmol, 5eq) was added once again trying to complete the reaction. The mixture was stirred for an additional 20h at room temperature before being diluted with Et20 (20mL) and poured onto crushed ice. 1M HC1 aqueous solution (lOOmL) was added, and the mixture was extracted with Et20 (200mL & l OOmL). The combined organic extracts were washed with brine (lOOmL), dried over sodium sulphate, filtered and concentrated before being purified on silica gel chromatography (cyclohexane / ethyl acetate 98:2 to 80:20) to afford compound 8 (1.46g, 46% yield) as an orange oil. Synthesis of compound 9
C15H12BrC102 M = 339.61 g.moF1
Mass: (GC-MS): 338-340

The synthesis of this product is described in J. Med. Chem. 2008, 51, 1 145—1149.Synthesis of compound 10
C15H14B1CIO M = 325.63 g.mof1

10 The synthesis of this product is described in J. Med. Chem. 2008, 51, 1145-1 149.
Synthesis of compound 11
C50H49CIO6 M = 781.37 g.moF1
Mass: ESI+): 798.20 (M + H20)

Under inert atmosphere, Mg powder (265mg, 10.9mmol, 2.4eq) was charged into a three necked flask, followed by addition of a portion of 1/3 of a solution of the 4- bromo-l-chloro-2-(4-ethylbenzyl)benzene (2.95g, 9.1mmol; 2eq) in dry THF (25mL) and 1 ,2-dibromoethane (10 mol % of Mg; 85mg; 0.45mmol). The mixture was heated to reflux. After the reaction was initiated (exothermic and consuming of Mg), the remaining solution of 2-(4-ethylbenzyl)-4-bromo-l-chlorobenzene in dry TFIF was added dropwise. The mixture was then allowed to react for another one hour under gentle reflux until most of the Mg was consumed.
The above Grignard reagent was added dropwise into the solution of cyclohexenone 8 (2.42g, 4.53mmol, leq) in dry THF (25mL) under inert atmosphere at room temperature (about 25°C), then allowed to react for 3h. A saturated aqueous solution of ammonium chloride was added into the mixture to quench the reaction. The mixture was extracted with Et20, washed with brine, dried over sodium sulphate, filtered and concentrated. The residue was purified on silica gel chromatography (cyclohexane/ethyl acetate 100:0 to 80:20) to afford the target compound 11 as a yellow oil (3.01g, 86%).
Synthesis of compound 12
C5oH49C105 M = 765.37 g.mol“1
+): 782.13 (M + H20)

Triethylsilane (0.210mL, 1.30mmol, 3eq) and boron-trifluoride etherate (48% BF3, O. l lOmL, 0.866mmol, 2eq) were successively added into a solution of alcohol 1 1 (338mg, 0.433mmol, leq) in dichloromethane (5mL) under inert atmosphere at -20°C. After stirring for 2.5h, a saturated aqueous solution of sodium chloride was added to quench the reaction. The mixture was extracted with CH2C12 (10mLx3) and the organic layer was washed with brine, dried over Na2S04, filtrated and concentrated. The residue was purified on silica gel chromatography (cyclohexane/ethyl acetate 9.8:0.2 to 8:2) to afford the target compound 12 as a white powder (278 mg, 0.363mmol, 84%).
Synthesis of compound 13
C5oH5tC106 M = 783.39g.moF1
Mass: (ESI+): 800 (M + H20); 1581 (2M + H20)

Under inert atmosphere, borane-dimethyl sulfide complex (2M in THF, 16.7mL, 33mmol, 10.5eq) was added to a solution of 12 (2.41g; 3.15mmol, leq) in dry THF (lOOmL) cooled to 0°C. The reaction mixture was then refluxed for lh,cooled to 0°C and treated carefully with sodium hydroxide (3M in H20, 10.5mL, 31.5mmol, lOeq), followed by hydrogen peroxide (30% in H20, 3.2mL, 31.5mmol, l Oeq) at room temperature (above 30°C). The mixture was allowed to react overnight at room temperature (~25°C) before a saturated aqueous solution of ammonium chloride was added to quench the reaction. The mixture was extracted with ethyl acetate and the organic layer was washed with brine, dried over Na2S04, filtered, and concentrated. The residue was purified by silica gel chromatography (cyclohexane/ethyl acetate 97:3 to 73:27) to afford the desired compound 13 (1.05g; 43%) as a yellowish oil.
Synthesis of compound 14
C50H49CIO6 M = 781.37g.mol“1
Mass: (ESI+): 798 (M + H20); 1471; 1579 (2M + H20)

13 14
Dess-Martin periodinane (81mg; 1.91mmol; 1.5eq) was added portion wise to a solution of alcohol 13 (l .Og; 1.28mmol, leq) in anhydrous dichloromethane (20mL) at 0°C. The reaction was then stirred overnight at room temperature before being quenched with IN aqueous solution of sodium hydroxide. The organic layer was separated and the aqueous layer was extracted with dichloromethane. The combined organic layers were dried over sodium sulphate, filtered and concentrated. The residue was purified on silica gel chromatography (cyclohexane / ethyl acetate 98:2 to 82: 18), to afford the target ketone 14 (783mg, 79% yield) as a colorless oil. Synthesis of compound 15
C5oH49ClF206 M = 803.37g.moF1
19 F NMR (CDCU, 282.5MHz): -100.3 (d, J=254Hz, IF, CFF); -1 13.3 (td, Jl=254Hz, J2=29Hz, IF, CFF).
Mass: (ESI+): 820.00 (M+H20)

14 15
A solution of ketone 14 (421mg, 0.539mmol, leq) in DAST (2mL, 16.3mmol, 30eq.) was stirred under inert atmosphere at 70°C for 12h. The mixture was then cooled to room temperature and dichloromethane was added. The solution was poured on a mixture of water, ice and solid NaHC03. Agitation was maintained for 30min while reaching room temperature. The aqueous layer was extracted with dichloromethane and the organic phase was dried over Na2S04, filtered and concentrated. The crude product was purified on silica gel chromatography (cyclohexane/ethyl acetate 98:2 to 80:20) to afford the desired compound 15 as a yellowish oil ( 182mg, 42% yield).
Synthesis of compound 16
C22H25CIF2O5 M = 442.88g.mor1
19 F NMR (MeOD, 282.5MHz): -96.7 (d, J=254Hz, IF, CFF); 12.2 (td,
Jl=254Hz, J2=28Hz, IF, CFF).
Mass: (ESI+): 465.3 (M+Na)

o-Dichlorobenzene (0.320mL, 2.82mol, lOeq) followed by Pd/C 10% (0.342g, 0.32mol, l .leq) were added to a solution of 15 (228mg, 0.28mmol, leq) in a mixture of THF and MeOH (2: 1, v/v, 160mL). The reaction was placed under hydrogen atmosphere and stirred at room temperature for 2h. The reaction mixture was filtered and concentrated before being purified on silica gel chromatography (dichloromethane/methanol 100: 1 to 90: 10) to afford compound 16 (105mg, 83% yield).
Sirona Biochem’s SGLT Inhibitor Performs Better Than Johnson and Johnson’s SGLT Inhibitor, According to Study
Vancouver, British Columbia – December 7, 2012 – Sirona Biochem Corp. (TSX-V: SBM), announced its sodium glucose transporter (SGLT) inhibitor for Type 2 diabetes reduced blood glucose more effectively than Johnson and Johnson’s canagliflozin, an advanced SGLT inhibitor being considered for market approval in Europe and the U.S. Studies compared Sirona Biochem’s SGLT Inhibitor, SBM-TFC-039, with canagliflozin and were conducted on Zucker Diabetic Fatty (ZDF) rats.
In the study, SBM-TFC-039 significantly and rapidly reduced blood glucose levels at a dose of 1.0 mg/kg. Six (6) hours after administration, SBM-TFC-039 reduced blood glucose by 44% compared to canagliflozin at 26%. SBM-TFC-039 also had a longer duration of effect than canagliflozin. At 36 and 48 hours after treatment, SBM-TFC-039, at a dose of 1.0 mg/kg, was still effective at reducing blood glucose, whereas canagliflozin lost its effect after 36 hours. Studies were conducted at the Institut Universitaire de Cardiologie et de Pneumologie de Québec (IUCPQ) by Principal Investigator Dr. Denis Richard, Research Chair on Obesity and Professor, Faculty of Medicine, Department of Anatomy & Physiology at Laval University.
“SGLT Inhibitors are a ground-breaking new treatment for Type 2 diabetes and these results demonstrate that SBM-TFC-039 will be a significant competitor for other SGLT Inhibitors,” said Neil Belenkie, Chief Executive Officer of Sirona Biochem. “The first SGLT Inhibitor,Forxiga™, was approved last month by the European Commission. We believe there is tremendous market potential worldwide for SGLT Inhibitors in the treatment of diabetes.”
SBM-TFC-039 is a sodium glucose transporter (SGLT) inhibitor. SGLT inhibitors are a new class of drug candidates for the treatment of diabetes. In the kidneys, SGLT inhibitors reduce the reabsorption of glucose into the bloodstream by eliminating excess glucose into the urine.
About Sirona Biochem Corp.
Sirona Biochem is a biotechnology company developing diabetes therapeutics, skin depigmenting and anti-aging agents for cosmetic use, biological ingredients and cancer vaccine antigens. The company utilizes a proprietary chemistry technique to improve pharmaceutical properties of carbohydrate-based molecules. For more information visit www.sironabiochem.com.
![]()
Laboratory – France
TFChem
Voie de l’innovation
Pharma Parc II
Chaussée du Vexin
27100 Val de Reuil
France
Phone:+33(0)2.32.09.01.16
Fax:+33(0)2.32.25.07.64



……………………………………………………………………………….
Shanghai Fosun Pharmaceutical Group Co. Ltd.

![]()
//////
AZD 3264 an IKK2 Inhibitor from Astra Zeneca

AZD 3264
MW 441.50
CAS 1609281-86-8
Inhibition of IkB-kinase IKK2 has been identified as one of the novel pathways to treat inflammatory conditions such as asthma, chronic pulmonary obstructive disorder (COPD) and rheumatoid arthritis
……………………..
PATENT
WO 2003010158
https://www.google.com/patents/WO2003010158A1?cl=en

The synthesis began with the aromatic nucleophilic substitution reaction of 2-fluorobromobenzene (2) with (S)-N-Boc-3-pyrrolidinol 3 to give the bromo intermediate 4, which was borylated via halogen metal exchange using n-hexLi in THF followed by treatment with triisopropyl borate and acidic work-up to give the boronic acid intermediate 5. Suzuki coupling of the boronic acid 5 with bromothiophene 6(2)afforded the intermediate 7. Intermediate 7 was subjected to regioselective bromination using bromine in acetic acid. This reaction was nonregioselective and yielded 17% of the required isomer 8. The bromo compound 8 was coupled with isoxazole boronate ester 9 by another Suzuki reaction to get the title compound. The overall yield of the synthesis was <6%.


………………………..
PAPER
http://pubs.acs.org/doi/full/10.1021/op500105n

An efficient and scalable synthesis of AZD3264 is described in which the differential reactivities of various halogen atoms have been employed. The process involves five linear chemical steps with three isolated stages starting from commercially available fragments.
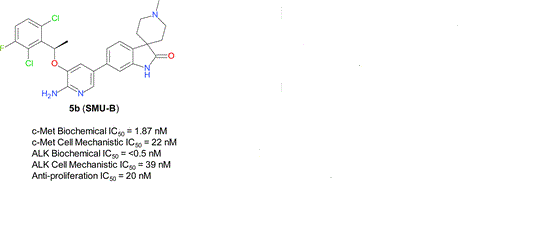
What is SMU-B?

cas 1253286-89-3
Spiro[3H-indole-3,4′-piperidin]-2(1H)-one, 5-[6-amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridinyl]-1′-methyl-
SMU-B
or is it
1253286-90-6
Spiro[3H-indole-3,4′-piperidin]-2(1H)-one, 6-[6-amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridinyl]-1′-methyl-
SMU-B

A series of novel aminopyridyl/pyrazinyl-substituted spiro[indoline-3,4′-piperidine]-2-ones were designed, synthesized, and tested in various in vitro/in vivo pharmacological and antitumor assays. 6-[6-Amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridyl]-1′-methylspiro[indoline-3,4′-piperidine]-2-one (compound 5b or SMU-B) was identified as a potent, highly selective, well-tolerated, and orally efficacious c-Met/ALK dual inhibitor, which showed pharmacodynamics effect by inhibiting c-Met phosphorylation in vivo and significant tumor growth inhibitions (>50%) in GTL-16 human gastric carcinoma xenograft models.
see..http://pubs.acs.org/doi/abs/10.1021/ml400203d
cas 1253286-90-6
6-[6-Amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridyl]-1′-methylspiro[indoline-3,4′-piperidine]-2-one (compound 5b or SMU-B)
SEE

1_4,3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] -2-nitro-approved P set

obtained in Step 1-3 (IS) -I- (2,6- dichloro-3-fluorophenyl) ethanol (2. 09g, IOmmol) was dissolved in dry THF (80 ml). Then, at room temperature under a nitrogen atmosphere, a solution of 3-hydroxy-2-nitro-pyridine (1.54g, llmmol) and triphenylphosphine (3. 409g, 13mmol), and so is completely dissolved, cooled to 0 ° C, was added Diisopropyl azodicarboxylate (DIAD, 2.63g, 13mmol), After the addition, the mixture was stirred at 0 ° C for 16 hours, the solvent was removed by rotary evaporation and the oily residue was purified by silica gel column chromatography (petroleum ether / ethyl acetate : 4/1) to give the desired product as a white solid (3. 046g, yield: 92%) o 1H-NMR (CDClySOOMHz): 8 (ppm) I. 86 (d, J = 6. 4Hz, 3H), 6 . 10 (q, J = 6. 4Hz, 1H), 7. 09 (dd, J = 7. 6,8. 8Hz, 1H), 7. 21 (dd, J = 8. 4, I. 2Hz, 1H ), 7. 31 (dd, J = 4. 8,8. 8Hz, 1H),
7. 37 (dd, J = 4. 8,8. OHz, 1H), 8. 04 (dd, J = L 6,4. 4Hz, 1H). Mass spectrum m / z:. 330 94 [M + H, 35C1,35Cl], 332. 92 [M + H, 35Cl, 37Cl].
1_5,3_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -2_ atmosphere based grant given P

to take steps 1-4 to get the 3 – [(lR) _l- (2,6_ dichloro-3-fluorophenyl) ethoxy] -2_ nitro than Li Jie (2. 649g, 8mmol) was dissolved in ethanol (15mL) was added iron powder (3. 575g, 64mmol) were mixed under nitrogen with vigorous stirring at 90 ° C oil bath, was added via syringe 0.8mL IM HCl (aq), after 10 minutes, was added 0. 8mL IMHCl (aq). Stirring was continued for 30 minutes, TLC showed the reaction. Cooled to room temperature, filtered through Celite, the filter residue washed with ethanol (3X IOmL). The combined organic phase was removed by rotary evaporation of the solvent gave the desired product as a light brown solid (2. 41g, yield: 100%) o 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 81 (d, J = 6. 8Hz, 3H ), 5. 03 (s, br, 2H), 6. 01 (q, J = 6. 8Hz, 1H), 6. 47 (dd, J = 4. 8,7. 6Hz, 1H), 6. 70 (d, J = 8. OHz, 1H), 7. 05 (t, J = 8. 8Hz, 1H), 7. 28 (dd, J = 4. 0,8. 0Hz, 1H), 7. 57 ( d, J = 5.2Hz, lH). Mass spectrum m / z:. 301 00 [M + H, 35Cl, 35Cl], 302. 77 [M + H, 35Cl, 37Cl].
l-6,5_ desert _3_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -2_ atmosphere base than Li Jie

The steps 1-5 obtained 3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] -2-yl atmosphere than Li Jie (1.506g, 5mmol) dissolved in acetonitrile (20mL) in. Then, at 0 ° C and the degree of stirring in added portionwise N- bromosuccinimide (0.908g, 5. Lmmol), After the addition, stirring was continued for 30 minutes. The solvent was removed by rotary evaporation, the crude product was obtained as a white solid was the desired product (1.045g, yield: 55%) was purified by column chromatography on silica gel. 1H-NMR (⑶Cl3,500MHz): 8 (ppm) I. 81 (d, J = 6. 8Hz, 3H), 4 85 (s, br, 2H), 6 98 (q, J = 6. 8Hz.. , 1H), 6. 82 (d, J =
2. 0Hz, 1H), 7. 08 (t, J = 8. 4Hz, 1H), 7. 31 (dd, J = 4. 8,8. 8Hz, 1H), 7. 65 (d, J = 2 . OHz, 1H). Mass spectrum m / z:… 378 84 [M + H, 35Cl, 35Cl, 79Br], 380 82 [M + H, 35Cl, 35Cl, 81Br or 35Cl, 37Cl, 79Br], 382 80 [M + H, 35Cl , 37Cl, 81Bror 37Cl, 37Cl, 79Br].
Step 2, I ‘- methyl-5- (4,4,5,5-tetramethyl -I, 3,2- dioxolane boron-2-yl) spiro [indoline Spray – 3,4 ‘- piperidin] -2_ one

2-1,5- bromo -I ‘- methyl-spiro [indoline-3,4’ – piperidin] _2_ one

[0300] 5-bromo – indol-2-one (I. 272g, 6mmol) was suspended THF (15mL) at, and cooled to -78 ° C, added dropwise with stirring IM NaN (SiMe3) THF solution of 2 (30mL, 30mmol). After the addition was stirred at _78 ° C 30 min, then 2-chloro -N- (2- chloro-ethyl) -N- methyl-ethylamine hydrochloride solid (I. 155g, 6mmol). After the addition stirring was continued for 30 minutes, then warmed to room temperature and stirred for two days. TLC showed the reaction was completed, to the pink suspension was carefully added aqueous 4M hydrochloric acid (IOmL), and then adjusted with concentrated aqueous ammonia to pH ^ 9, and extracted with DCM (3 X 80mL). The organic phases were combined, dried (Na2SO4), and concentrated to give the crude product was purified by silica gel column chromatography (7M NH3 in methanol solution / DCM: 5/95) to give the desired product (I. 38g, yield: 78%) was purified. 1H-NMR (CD3ODjOOMHz):. 8 (ppm) I. 86-1 92 (m, 2H), I 94-1 98 (m, 2H), 2 44 (s, 3H), 2 62-…. 2. 68 (m, 2H), 2. 86-2. 91 (m, 2H), 6. 76 (d, J = 7. 6Hz, 1H), 7. 33 (dd, J = I. 2,7 . 6Hz, 1H), 7. 44 (d, J = I. 6Hz, 1H), 7. 81 (s, br, 1H). Mass spectrum m / z:. 294 99 [M + H, 79Br], 296 82 [M + H, 81Br]..
2-2, V – methyl-5- (4,4,5,5-tetramethyl–1,3,2_ dioxolane Borane _2_ yl) spiro [indoline – 3,4 ‘- piperidin] -2_ one

Under nitrogen, obtained in Step 2-1 to 5-bromo -I ‘- methyl-spiro [indoline-_3,4’ – piperidin] _2_ one (147. 6mg, 0. 5mmol) , the United pinacols drop acid unitary purpose (140mg, 0. 55mmol) and acetic acid Bell (147mg, I. 5mmol) in DMSO (0. 2ml) was added in PdCl2 (dppf) • CH2Cl2 (20. 4mg, 0. 025mmol ), to the resulting solution was bubbled with nitrogen for 2 minutes, and then stirred at 80 ° C of 16 hours. LC-MS showed completion of the reaction, after cooling to room temperature, water (2mL), extracted with DCM (3X5mL). The organic phases were combined, dried (Na2SO4), and concentrated to give the desired product (170mg, yield: 100%) o MS m / z:. 342 07 [M + H], 343. 08 [M + H, 100%], 344. 11 [M + H].
Step 3,5_ [6_ atmosphere base _5_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -3_ batch P fixed base] -I ‘- A group spiro [indoline-3,4 ‘- piperidin] -2_ one
The steps 1-6 5_ desert obtained _3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] -2-yl batch atmosphere pyridine (75. 8mg , 0. 2mmol), I’- step 2_2 obtained methyl 5- (4,4,5,5-tetramethyl-l, 3,2-dioxolane Borane 2-yl) spiro [ indoline-3,4′-piperidin] -2-one (82mg, 0. 24mmol) and potassium carbonate (82. 9mg, 0. 6mmol) was dissolved in DME / water mixture solution (4 / 1,2. Oml ). Then, under nitrogen, was added Pd (PPh3) 4 (II. 6mg, 0. Olmmol), to the resulting mixture was bubbled with nitrogen for 2 minutes, and then stirred at 80 ° C of 18 hours. LC-MS showed completion of the reaction, after cooling to room temperature, water (5mL), extracted (3 X IOmL) with DCM. The organic phases were combined, dried (Na2SO4), and concentrated to give the crude product was purified by silica gel column chromatography (7M NH3 in methanol solution / DCM: 5/95) to give the desired product (88. 6mg, yield: 86%) was purified. 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 86 (d, J = 6. 4Hz, 3H), I 93-2 02 (m, 4H), 2 44 (s, 3H),…
2. 66-2. 72 (m, 2H), 2. 89-2. 93 (m, 2H), 4. 87 (s, br, 2H), 6. ll (q, J = 6. 4Hz, 1H ), 6. 88 (d, J =
8. OHz, 1H), 6. 94 (d, J = I. 2Hz, 1H), 7. 06 (t, J = 8. 4Hz, 1H), 7. 19 (dd, J = I. 2,8 . OHz, 1H),
7. 31 (m, 1H), 7. 36 (s, 1H), 7. 66 (s, br, 1H), 7. 80 (d, J = 2. OHz, 1H). Mass spectrum m / z:.. 515 05 [M + H, 35Cl, 35Cl], 517 03 [M + H, 35Cl, 37Cl].
Example 2: 6_ [6_ atmosphere base _5_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -3_ than Li Jie base] -I ‘ – methyl-spiro [indoline-3,4 ‘- piperidin] -2_ one

Step I, I ‘- methyl-6- (4,4,5,5-tetramethyl–I, 3,2- dioxolane boron-2-yl) spiro [indoline Spray – 3,4 ‘- piperidin] -2_ one
1-1,6- bromo -I ‘- methyl-spiro [indoline-3,4’ – piperidin] -2_ one

As described in Example I steps 2-1 of the method from the commercially available 6-bromo – indol-2-one was prepared, Yield: 82%. Analysis of the data obtained the desired product are = 1H-Nmr (Cd3OdJOOmHz): 8 (ppm) 1.90-1.98 (m, 4H),
2. 44 (s, 3H), 2. 64-2. 68 (m, 2H), 2. 86-2. 92 (m, 2H), 7. 05 (d, J = 2. 0Hz, 1H), 7. 16-7. 21 (m, 2H), 7. 91 (s, br, 1H). Mass spectrum m / z: 295 00 [M + H, 79Br], 296 78 [M + H, 81Br]… [0312] 1-2, 1 ‘- methyl-6- (4,4,5,5-tetramethyl-_1,3,2_ dioxolane Borane _2_ yl) spiro [indoline – 3,4 ‘- piperidin] -2_ one

In the step 1-1 of the obtained 6-bromo -I ‘- methyl-spiro [indoline-_3,4’ – piperidin] -2_ ketone and commercially available linking pinacol boronic ester material, the method of Example I was prepared in accordance with steps 2-2, Yield: 95%. Analysis of the data obtained of the target product are as follows: Mass spectrum m / z:. 342 06 [M + H], 343 04 [M + H, 100%], 344. 12 [M + H]..
Step 2,6_ [6_ atmosphere base _5_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -3 ratio Li Jie base] -I ‘- methyl-spiro [indoline-3,4 ‘- piperidin] -2_ one
Example I steps 1-6 to obtain 5-bromo -3 – [(IR) -I- (2,6- dichloro-3-fluorophenyl) ethoxy] -2-amino- pyridine, I obtained in Example 1-2 of the present embodiment in step ‘- methyl-6- (4,4,5,5-tetramethyl-l, 3,2-dioxolane-2-yl borane) spiro [indoline-_3,4 ‘- piperidin] -2-one, prepared as in Example I Step 3. Yield: 82%. 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 86 (d, J = 6. 4Hz, 3H), I. 91-1 95 (m, 2H), I 97-2 03 (m, 2H… ), 2. 45 (s, 3H), 2. 65-2. 72 (m, 2H), 2. 89-2. 95 (m, 2H), 5. 12 (s, hr, 2H),
6. 12 (q, J = 6. 4Hz, 1H), 6. 94-7. 00 (m, 3H), 7. 06 (t, J = 8. 4Hz, 1H), 7. 31 (m, 1H ), 7. 35 (d, J = 7. 2Hz, 1H), 7. 90 (d, J = 2. 0Hz, 1H), 9. 28 (s, br, 1H). Mass spectrum m / z:.. 515 05 [M + H, 35Cl, 35Cl], 517 03 [M + H, 35Cl, 37Cl].
5- [6-amino-5 – [(2,6-dichloro-3-fluorophenyl) methoxy] _3_ pyridinyl] -I’–methyl-spiro [indole: 3 [0317] Example morpholine-3,4 ‘- piperidin] -2-one
H2N N


Step I, 5_ desert _3_ (2,6_ two gas -3- integrity oxy) _2_ atmosphere based grant given P
1-1,2,6_ two gas acid gas _3_
Cl OF
Sodium hydroxide (13g, 325mmol) in water (IlOmL) was cooled to _5 ° C was added dropwise under vigorous stirring of liquid bromine (12. 5g, 78. 2mmol), added after the addition of pre-cooled to 10 ° C dioxane (75mL). The above mixture under vigorous stirring was added dropwise a pre-cooled to 5 ° C of I- (2,6- dichloro-3-fluorophenyl) ethanone (5g, 21. 2mmol) in dioxane (330mL) and water (90mL) was added. After the addition, at room temperature for 2 hours Lan Xiang, Xiang Lan then 90 C for 30 minutes. TLC was not shown with the S starting material disappeared, and was acidified with concentrated hydrochloric acid to PH~9. The resulting mixture was rotary evaporated to dryness, added water (20mL), and extracted with diethyl ether (2X80mL), the organic phases were combined, dried (Na2SO4), and concentrated to give an oily product solidified after cooling to a transparent, slightly yellow solid (3. 4g, Yield: 67%). 1H-Nmr (Cdci3AOOmHz):. 8 (ppm) 7. 21 (. Dd, J = 8. 0,8 8Hz, 1H), 7 35 (. Dd, J = 4. 4,9 2Hz, 1H), 9 . 79 (s, br, 1H). Mass spectrum m / z (ES “:. 207 11 [M_H, 35Clj35Cl], 209 10 [MH, 35Cl, 37Cl]..
1-2,2,6–dichloro-3-fluoro-benzyl alcohol
^ Coh
F
[0325] To be filled with 2,6-dichloro-3-fluoro benzoic acid (3g, 14. 35mmol) added dropwise to the flask IM BH3. THF (43mL, 43mmol), added after the mixture was stirred under reflux for 24 hours. TLC showed the reaction was complete, methanol (50mL) to destroy excess borane, and the solvent was distilled off under reduced pressure and the resulting trimethyl borate, the process is repeated twice more to give a viscous product 2. I g, yield: 75% . 1H-Nmr (Cdci3JOOmHz): 8 (ppm) 2. 09 (t, J = 6. 4Hz, 1H), 4. 97 (d, J = 6. 4Hz, 2H), 7 09 (t, J = 8. . 8Hz, 1H), 7. 32 (dd, J = 4. 8,9. 1Hz, 1H). Mass spectrum m / z (ES-):.. 193 08 [M_H, 35Cl, 35Cl], 195 12 [MH, 35Cl, 37Cl].
1-3,3_ (2,6-gas _3_ integrity oxy) _2_ nitro grant given P

Following the procedure of steps 1-4 of Example I, was prepared from 2,6-dichloro-3-fluoro-benzyl alcohol and 3-hydroxy-2-nitropyridine prepared in yield (in this example embodiment steps 1_2) : 90%. 1H-Nmr (Cdci3AOOmHz): 8 (ppm) 5. 45 (s, 2H), 7 20, 7 37 (dd, J = 4. 8. (Dd, J = 8. 0,9 2Hz, 1H.). , 9. 2Hz, 1H), 7. 59 (dd, J = 4. 4,8. 4Hz, 1H),
7. 74 (dd, J = L 2,8. 4Hz, 1H), 8. 17 (dd, J = L 6,4. 4Hz, 1H). Mass spectrum m / z:. 316 89 [M + H, 35Cl, 35Cl], 318. 89 [M + H, 35Cl, 37Cl].
1_4,3_ (2,6-gas _3_ integrity oxy) _2_ atmosphere based grant given P

The method according to Example I step 1_5 from 3- (2,6-gas -3- integrity oxy) _2_ nitro Jie ratio 唳 preparation (in this case, steps 1-3), that Yield: 95% o 1H-Nmr (Cdci3JOOmHz):. 8 (ppm) 4 65 (s, br, 2H), 5 31 (s, 2H), 6 66 (dd, J = 5. 2,8.. . 0Hz, 1H), 7. 14 (dd, J = I. 2,8. 0Hz, 1H), 7. 18 (dd, J =
8. 4,9. 2Hz, 1H), 7. 37 (dd, J = 4. 8,8. 8Hz, 1H), 7. 73 (dd, J = I. 6,5. 6Hz, 1H). Mass spectrum m / z:. 286 95 [M + H, 35Cl, 35Cl], 288 85 [M + H, 35Cl, 37Cl]..
1-5,5_ desert -3- (2,6-gas -3_ integrity oxy) ~ 2 ~ atmosphere based grant given P

Following the procedure of Example I step 1_6 embodiment, starting from 3- (2,6-gas _3_ integrity yloxy) _2_ atmosphere group given the preparation of the batch P (in the example of the present embodiment in step 1-4), Yield: 60% o 1H-Nmr (Cdci3JOOmHz):. 8 (ppm) 4 68 (s, br, 2H), 5 28 (s, 2H), 7 21 (dd, J = 8. 0,8.. . 8Hz, lH), 7. 24 (dd, J = 2. OHz, 1H), 7. 39 (dd, J = 4. 8,
9. 2Hz, 1H), 7. 78 (d, J = 2. OHz, 1H). Mass spectrum m / z:. 364 83 [M + H, 35Cl, 36Cl, 79Br], 366 77 [M + H], 368 69 [M + H]…
Step 2,5_ [6_ atmosphere base _5_ [(2,6_ two gas -3- gas) methoxy] -3_ than Li Jie base] -I-methyl-spiro [indoline _ 3,4 ‘- piperidin] -2-one
The present embodiment 5_ desert steps 1_5 obtained _3_ (2,6_ two gas _3_ integrity yloxy) pyridine ~ 2 ~ atmosphere, Examples 2-2 obtained in step I I ‘- methyl-5- (4,4,5,5-tetramethyl-borane _1,3,2- dioxolane-2-yl) spiro [indoline-_3,4’ – piperidine ] -2-one, prepared as in Example I Step 3. Yield: 85 V0o 1H-Nmr (Cdci3JOOmHz):.. 8 (ppm) I. 92-2 02 (m, 4H), 2. 43 (s, 3H), 2. 65-2 71 (m, 2H) , 2. 90-2. 91 (m, 2H), 4. 92 (s, br, 2H), 5. 52 (s, 2H), 6. 89 (d, J = 8. 4Hz, 1H), 6 . 90 (d, J = L 2Hz, 1H), 7. 06 (t, J = 8. OHz, 1H), 7. 21 (dd, J = L 2,8. OHz, 1H), 7. 31 ( m, 1H),
7. 37 (s, 1H), 7. 79 (s, br, 1H), 7. 80 (d, J = 2.0Hz, lH). MS m / z:. 501 06 [M + H, 35Cl, 35Cl], 503 04 [M + H, 35Cl, 37Cl]..
6- [6-amino-5 – [(2,6-dichloro-3-fluorophenyl) methoxy] _3_ pyridinyl] -I’- methyl-spiro [indole: 4 [0337] Example morpholine _3,4 ‘- piperidin] -2-one



H2N N
Following the procedure in Example I step of Example 3, the procedure of Example 3 to give 5-bromo-1-5 _3_ (2,6-dichloro-3-fluoro-benzyloxy) -2-amino-pyridine and Step 2 in Example I to give the embodiment 1-2 ‘- methyl-6- (4,4,5,5-tetramethyl-1,3,2-dioxolane Borane 2-yl) spiro [ indoline-3,4 ‘- piperidine] _2_ ester -one, yield:. 78 V0o 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 96-2 00 (m, 2H), 2. 01 -2. 12 (m, 2H), 2. 46 (s, 3H), 2. 66-2. 73 (m, 2H), 2. 90-2. 96 (m, 2H), 5. 30 (s , hr, 2H), 6. 94-7. 01 (m, 3H), 7. 07 (t, J =
8. 4Hz, 1H), 7. 30 (m, 1H), 7. 34 (d, J = 7. 2Hz, 1H), 7. 89 (d, J = 2. OHz, 1H), 8. 56 ( s, br, 1H). MS m / z:. 501 06 [M + H, 35Cl, 35Cl], 503 04 [M + H, 35Cl, 37Cl]..
Example 5: 5_ [5_ atmosphere base -6- [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] Batch-2-yl] -I ‘ – methyl-spiro [indoline-3,4 ‘- piperidin] -2-one
J0A = o
. | J: too
[0342] Step 1,5_ desert _2_ atmosphere base _3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] Jie than exposure
Cl 6, / ISL / Br
xy
H2N N
In at 0 ° C, NaH (80mg of NaH in mineral oil, 2mmol) force the mouth (1R) -1_ (2,6- dichloro-3-fluorophenyl) ethanol (418mg, 2mmol. See example Example I Step 1_3) in anhydrous THF (6mL) and stirred for half an hour, a solution of 2-amino-3,5-dibromo-pyrazine (506mg, 2mmol) in THF (6mL) was added. The resulting mixture was warmed to room temperature, heated under reflux for 20 hours. TLC showed the reaction was substantially complete. After cooling to room temperature, water was added (IOmL), the mixture was extracted three times with ethyl acetate (3x20mL), the organic phases were combined, dried, concentrated, and the residue to give 594mg product was purified by column chromatography (l-3Me0H inhexanes), yield: 78%. 1H-NMR (O) Cl3, 500MHz):. 8 (ppm) I. 83 (d, J = 7. 2Hz, 3H), 5. 12 (s, br, 2H), 6 73 (q, J = 6 . 8Hz, 1H), 7. 05 (t, J = 8. OHz, 1H), 7. 28 (dd, J = 4. 8,
8. 8Hz, 1H), 7. 58 (s, 1H). Mass spectrum m / z:. 379 83 [M + H, 35Cl, 35Cl, 79Br], 381. 81 [M + H, 35Cl, 35Cl, 81Br], 383 79 [M + H, 35Cl, 37Cl, 81Br]..
Step 2,5_ [5_ atmosphere base _6_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] Batch-2-yl] -I ‘- A group spiro [indoline-3,4 ‘- piperidin] -2-one
5_ bromide present embodiment obtained in step I _2_ amino _3_ [(IR) -I- (2,6_ dichloro _3_ fluorophenyl) ethoxy] pyrazine, implemented I’- methyl step 2-2 obtained in Example I-5 (4,4,5,5-tetramethyl -I, 3,2- dioxolane boron
2-yl) spiro [indoline-3,4 ‘- piperidin] -2-one, prepared as in Example I Step 3. Yield: 54%. 1H-NMR (CD3ODjOOMHz): 8 (ppm) I. 85 (d, J = 6. 8Hz, 3H), I 85-1 88 (m, 2H), I 97-2 04 (m, 2H…. ), 2. 46 (s, 3H), 2. 76-2. 82 (m, 2H), 2. 97-3. 02 (m, 2H), 6. 74 (q, J = 6. 4Hz, 1H ), 6. 85 (d, J = 8. OHz, 1H), 7. 15 (t, J = 8. 4Hz, 1H), 7. 41 (dd, J = 4. 8,9. 2Hz, lH) , 7. 54 (dd, J = I. 6,
8. OHz, 1H), 7. 69 (d, J = I. 8Hz, 1H), 7. 81 (dt, J = 2. 0,8. 0Hz, 1H), 7. 87 (s, 1H). Mass spectrum m / z:. 515 92 [M + H, 35Cl, 35Cl], 517. 90 [M + H, 35Cl, 37Cl].
Example 6: 6- [5-amino -6 – [(lR) -l_ (2,6- dichloro _3_ fluorophenyl) ethoxy] pyrazin-2-yl] -I ‘ – methyl-spiro [indoline-3,4 ‘- piperidin] -2-one

The embodiment of Example 5, 5_ bromo obtained in step I _2_ amino _3_ [(IR) -I- (2,6_ dichloro _3_ fluorophenyl) ethoxy] pyrazine, Example I’- methyl-2 obtained in steps 1-2 6- (4,4,5,5-tetramethyl–I, 3,2- dioxolane boron-2-yl) spiro [indole morpholine -3,4’_ piperidin] -2-one, prepared as in Example I Step 3. Yield: 67% 0
1H-NMR (CD3ODjOOMHz): 8 (ppm) I. 85 (d, J = 6. 8Hz, 3H), I 88-1 96 (m, 4H), 2 48 (s… , 3H), 2. 76-2. 82 (m, 2H), 2. 98-3. 05 (m, 2H), 6. 75 (q, J = 6. 4Hz, 1H), 7. 16 (t , J = 8. 8Hz, 1H), 7. 31 (d, J = 2. OHz, 1H), 7. 36-7. 43 (m, 3H), 7. 88 (s, 1H).
Mass spectrum m / z:. 515 99 [M + H, 35Clj35Cl], 517 90 [M + H, 35Cl, 37Cl]..
SEE
Bioorganic & Medicinal Chemistry Letters (2014), 24(16), 3673-3682.
School of Pharmaceutical Sciences, Southern Medical University,



 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
amcrasto@gmail.com





 Stockholm, Sweden
Stockholm, Sweden Despite the cold weather, public came and enjoyed different activities. The famous chef, Paul Svensson who works in one of the fanciest and most famous …
Despite the cold weather, public came and enjoyed different activities. The famous chef, Paul Svensson who works in one of the fanciest and most famous …
US Orphan status for Bexion’s brain tumour drug BXQ-350
SDVYCEVCEFLVKEVTKLIDNNKTEKEILDAFDKMCSKLPKSLSEECQEVVDTYGSSILSILLEEV SPELVCSMLHLCSG [SEQ ID NO: 2].
BXQ-350
Cincinnati Children’s Hospital ……..innovator
Bexion Pharmaceuticals……….under license
In February 2015, the US FDA granted saposin C Orphan designation for the treatment of glioblastoma multiforme

SAPOCIN C
Recombinant human Saposin C (SapC) bound to a liposomal formulation of the dioleoylphosphatidylserine
Bexion’s Saposin C – the active ingredient in the brain tumour therapy BXQ-350 – has been awarded Orphan Drug status by US regulators.
Read more at: http://www.pharmatimes.com/Article/15-02-17/US_Orphan_status_for_Bexion_s_brain_tumour_drug.aspx#ixzz3S3zXdHlO
Bexion Pharmaceuticals, under license from the Cincinnati Children’s Hospital, is investigating a human saposin C (SapC)/liposomal dioleoylphosphatidylserine (DOPS) conjugate, SapC-DOPS (BXQ-350), a nanovesicle-formulated pro-apoptotic sphingomyelinase activating molecular imaging agent and anticancer agent, for the potential diagnosis and treatment of cancer , . In October 2013, Bexion was planning a phase I first-in-human trial for the therapy of glioblastoma multiforme

Bexion Pharmaceuticals LLC announced today that the U.S. Food and Drug Administration (FDA) has granted the company Orphan Drug designation for Saposin C, active ingredient in its proprietary drug BXQ-350 for the potential treatment of glioblastoma multiforme.
The FDA’s Office of Orphan Drug Products Development reviews applications for Orphan Drug status to support development of medicines for underserved patient populations, or rare disorders that affect fewer than 200,000 people in the United States. The successful application submitted by Bexion and the FDA granting of Orphan Drug status entitles the company to a seven-year period of marketing exclusivity in the United States for BXQ-350, if it is approved by the FDA for the treatment of glioblastoma multiforme. Orphan Drug status also enables the company to apply for research grant funding for Phase I and II Clinical Trials, tax credits for certain research expenses, and a waiver from the FDA’s application user fee, as well as additional support from FDA and a potentially faster regulatory process.
Bexion was previously awarded a prestigious Phase II Bridge Award (Small Business Innovation Research Grant; SBIR) from the National Cancer Institute (NCI) to support the manufacture and clinical testing of BXQ-350.
“Orphan Drug status for BXQ-350 is an important milestone in the development of this new treatment modality,” stated Dr. Ray Takigiku, founder and CEO of Bexion. “Few treatment options are available for patients suffering from glioblastoma multiforme and this designation recognizes the unmet need that exists with this disease, as well as the unique attributes of BXQ-350. In addition, orphan designation allows Bexion to benefit from important financial, regulatory and commercial considerations and we have seen recently that products with orphan designation have become sought after assets.”

About Orphan Drug Designation
Orphan Drug designation is a status assigned to a medicine intended for use in rare diseases. In the U.S., the Orphan Drug Designation program confers Orphan Drug status to successful applicants for medicines intended for the safe and effective treatment, diagnosis or prevention of rare diseases or disorders that affect fewer than 200,000 people in the U.S. or that are not expected to recover the costs of developing and marketing a treatment.1
The approval of an orphan designation request does not alter the standard regulatory requirements and process for obtaining marketing approval for investigational use. Sponsors must establish safety and efficacy of a compound in the treatment of a disease through adequate and well-controlled studies. However, the FDA review process may be speedier for Orphan Drugs than those which do not receive Orphan Drug designation.
About BXQ-350
In pre-clinical studies, Bexion’s first-in-class biologic, BXQ-350 has shown promising results in selectively inducing cell death in the laboratory. BXQ-350 is a proprietary nanovesicle formulation of Saposin C (sphingolipid activator protein C, or SapC) and the phospholipid dioleoylphosphatidylserine (DOPS).
About Bexion Pharmaceuticals
Bexion Pharmaceuticals is a privately held biotech company focused on the development and commercialization of innovative cures for cancer. Initial products are based on a proprietary platform technology licensed from Cincinnati Children’s Hospital Medical Center. The technology has demonstrated potential for development as a therapeutic, diagnostic and surgical imaging reagent, and as a carrier for other pharmaceutical agents, such as oligonucleotides. For more information, visit www.bexionpharma.com or contact Margaret van Gilse atmvangilse@bexionpharma.com.
1 U.S. Food and Drug Administration web site. “Regulatory Information: Orphan Drug Act.”http://www.fda.gov/regulatoryinformation/legislation/federalfooddrugandcosmeticactfdcact/significantamendmentstothefdcact/orphandrugact/default.htm.
Margaret van Gilse859-757-1652mvangilse@bexionpharma.com
SOURCE Bexion Pharmaceuticals LLC
Glioblastoma is the most common primary CNS malignant neoplasm in adults, and accounts for nearly 75% of the cases. Although there has been steady progress in their treatment due to improvements in neuro-imaging, microsurgery, and radiation, glioblastomas remain incurable. The average life expectancy is less than one year from diagnosis, and the five-year survival rate following aggressive therapy, including gross tumor resection, is less than 10%. Glioblastomas cause death due to rapid, aggressive, and infiltrative growth in the brain. The infiltrative growth pattern is responsible for the un-resectable nature of these tumors. Glioblastomas are also relatively resistant to radiation and chemotherapy, and therefore post-treatment recurrence rates are high. In addition, the immune response to the neoplastic cells is mainly ineffective in completely eradicating residual neoplastic cells following resection and radiation therapy.
One problem in treating glioblastoma is the tumor’s protection behind the blood-brain tumor barrier (BBTB). A significant obstacle in the development of therapeutics for glioblastoma is the inability of systemic therapies to efficiently cross the BBTB. Saposin C (SapC) is a sphingolipid- activating protein that functions to catabolize glycosphingolipids. SapC-DOPS forms stable nanovesicles which can efficiently cross the blood-brain tumor barrier and fuse with GBM cells inducing cell death.
Rapamycin is a macrolide antibiotic produced by Streptomyces hygroscopicus, which was discovered first for its properties as an antifungal agent. Streptomyces hygroscopicus has also been implicated as a cancer agent.
There remains a need in the art for new therapeutics for the treatment of glioblastoma.
…………………………………………………………………..
https://www.google.com/patents/US20040229799?cl=en22
Example 1Purification of Recombinant Saposin C
[0106] Recombinant saposin C was overexpressed in E. coli cells by using the isopropyl-1-thio-β-D-galactopyranoside inducing pET system (Qi et al. (1994) J. Biol. Chem. 269:16746-16753, herein incorporated by reference in its entirety). Expressed polypeptides with a His-tag were eluted from nickel columns. After dialysis, the polypeptides were further purified by HPLC chromatography as follows. A C4 reverse phase column was equilibrated with 0.1% trifluoroacetic acid (TFA) for 10 minutes. The proteins were eluted in a linear (0-100%) gradient of 0.1% TFA in acetonitrile over 60 minutes. The major protein peak was collected and lyophilized. Protein concentration was determined as previously described (Qi et al. (1994) J. Biol. Chem. 269:16746-16753).
Example 2Bath Sonication of Sanosin C and Dioleoylphosphatidylserine
[0107] Dioleoylphosphatidylserine (DOPS) was obtained from Avanti Polar Lipids (Alabaster AL). Twenty to thirty imoles of DOPS in chloroform were dried under N2 and vacuum to lipid films. Five to ten μmoles saposin C polypeptide was added to the dried films and suspended in 50 μl McIlvanine buffer (pH 4.7). The suspension was then brought to a 1 ml volume with either cell culture medium or phosphate buffered saline (PBS) (Ausubel et al. (2002) Current Protocols in Molecular Biology. John Wiley & Sons, New York, New York, herein incorporated by reference). The mixture was sonicated in a bath sonicator for approximately 20 minutes. Ice was added as needed to prevent overheating the samples.

………………………………………………………………
http://www.google.com/patents/WO2014078522A1?cl=en
The SapC-DOPS composition comprises a phospholipid, an isolated saposin C-related polypeptide, wherein the polypeptide comprises an amino acid sequence at least 75% identical to the entire length of SEQ ID NO: 2, and a pharmaceutically acceptable carrier, wherein the phospholipid forms a nano vesicle incorporating the polypeptide. In certain embodiments, the polypeptide comprises an amino acid sequence at least 85% identical to the entire length of SEQ ID NO: 2. In certain embodiments, the polypeptide comprises an amino acid sequence at least 95% identical to the entire length of SEQ ID NO: 2. In certain embodiments, the polypeptide comprises an amino acid sequence at least 99% identical to the entire length of SEQ ID NO: 2.
The Sequence Listing, filed electronically and identified as SEQ_LIST_OSIF-2013- 102.txt, was created on November 12, 2013, is 5,548 in size, and is hereby incorporated by reference.
[0004] SEQ ID NO: 1
siy
J su c n 61y &n
*8 a 210 2iS
t n«
:?e
<H ¾■ yts ca« ¾»* **u v ΆΧ» s?s ass ¾«¾
:»o
L st S«x ri» r s
SEQ ID NO: 2

BEXION PHARMA

-
-
Russell Street, Covington, KY 41011, United States
 $2.9 Million Grant Awarded to Covington-Based Bexion for Next Step in Cancer Fight
$2.9 Million Grant Awarded to Covington-Based Bexion for Next Step in Cancer Fight 921 Spring Street Covington, Kentucky 41016 United States
921 Spring Street Covington, Kentucky 41016 United States 112 East 4th Street, Covington, KY 41011.
112 East 4th Street, Covington, KY 41011. 1182 Riverhouse Way Covington KY : 427657
1182 Riverhouse Way Covington KY : 427657
-
N-{2-[7-(Cyclohexylmethyl)-1,6-dihydro-2H-indeno[5,4-b]furan-8-yl]ethyl}acetamide

N-[2-(7-Benzyl-1,6-dihydro-2H-indeno[5,4-b]furan-8-yl)ethyl]acetamide
N-{2-[7-(Cyclohexylmethyl)-1,6-dihydro-2H-indeno[5,4-b]furan-8-yl]ethyl}acetamide
Acetamide, N-[2-[7-(cyclohexylmethyl)-1,6-dihydro-2H-indeno[5,4-b]furan-8-yl]ethyl]-
339.47, C22 H29 N O2
cas 1287785-08-3
Melatonin MT2 Agonists
Takeda……..innovator
Treatment of Sleep Disorders,
-
Melatonin (N-acetyl-5-methoxytryptamine), which is a hormone synthesized and secreted principally in the pineal gland, increases in dark circumstances and decreases in light circumstances. Melatonin exerts suppressively on pigment cells and the female gonads, and acts as a synchronous factor of biological clock while taking part in transmittance of photoperiodic code. Therefore, melatonin is expected to be used for the therapy of diseases related with melatonin activity, such as reproduction and endocrinic disorders, sleep-awake rhythm disorders, jet-lag syndrome and various disorders related to aging, etc.
-
Recently, it has been reported that the production of melatonin melatonin could reset the body’s aging clock (see Ann. N. Y. Acad. Sci., Vol. 719, pp. 456-460 (1994)). As previously reported, however, melatonin is easily metabolized by metabolic enzymes in vivo (see Clinical Examinations, Vol. 38, No. 11, pp. 282-284 (1994)). Therefore, it cannot be said that melatonin is suitable as a pharmaceutical substance.
-
Various melatonin agonists and antagonists such as those mentioned below are known.
- (1) EP-A-578620 discloses compounds of:
-
-

- (2) EP-A-420064 discloses a compound of:

- (3) EP-A-447285 discloses a compound of:

- (4) EP-A-662471 discloses a compound of:

- (5) EP-A-591057 discloses a compound of:

- (6) EP-A-527687 discloses compounds of:

X=S, 0, Y=CH
X=0, NH, Y=N - (7) EP-A-506539 discloses compounds of:

-
-
Tricyclic or more poly-cyclic compounds with a cyclic ether moiety, such as those mentioned below, are known.
- (1) Compounds of:

are disclosed in Tetrahedron Lett., Vol. 36, p. 7019 (1995).
- (2) Compounds of:


are disclosed in J. Med. Chem., Vol. 35, p. 3625 (1992).
- (3) Compounds of:

are disclosed in Tetrahedron, Vol. 48, p. 1039 (1992).
- (4) Compounds of:

are disclosed in Tetrahedron Lett., Vol. 32, p. 3345 (1991).
- (5) A compound of:

is disclosed in Bioorg. Chem., Vol. 18, p. 291 (1990).
- (6) A compound of:

is disclosed in J. Electroanal. Chem. Interfacial Electrochem., Vol. 278, p. 249 (1990).
see
- (1) Compounds of:














http://www.google.co.in/patents/EP0885210B1?cl=en
Highly Potent MT2-Selective Agonists
N-{2-[7-(Cyclohexylmethyl)-1,6-dihydro-2H-indeno[5,4-b]furan-8-yl]ethyl}acetamide (15)
MSA 100 a serotonin receptor antagonist.

(2E)-N-[2-[2-[(2S)-1-Methyl-2-piperidinyl]ethyl]phenyl]-3-phenyl-2-propenamide (1)
(S)-2′[2-1-(methyl-2-piperidyl) ethyl] cinnamanilide
(2E)-Λ/-[2-[2-[(2S)-1-Methyl-2-piperidinyl]ethyl]phenyl]-3-phenyl-2- propenamide
CAS 951155-17-2
C23H28 N2O, 348.48
It was reported that the (S)-enantiomer of 2′-[2-(1-methyl-2-piperidyl)ethyl]cinnamanilide (MSA100, 1) is an active 5-HT (5-hydroxytryptamine or serotonin) receptor antagonist; however, its (R)-isomer is totally or substantially devoid of the same activity.(1)
-
1.(a) Amer, M. S., U.S. Patent 5,780, 487, 1998.(b) Prashad, M.; Liu, Y.; Hu, B.; Girgis, M. J.; Schaefer, F., WO2007/111705, 2007.(c) Prashad, M.; Liu, Y.; Hu, B.; Girgis, M. J.; Schaefer, F., WO2007/111706, 2007.
…………………………………………………………………..
http://www.google.com/patents/WO2007111706A2?cl=en





Example 4:
Synthesis of (2E)-/V-[2-[2-[(2S)~1 -Methyl-2-piperidinyl]ethyI]phenyl]-3-phenyl-2- propenamide
(a) Free Base Generation:
A 250-mL, 4-necked, round-bottomed flask, equipped with a mechanical stirrer, digital thermometer and nitrogen inlet-outlet, heating cooling bath, and addition funnel, is charged with 6.28 g (S)-2-[2-(1-methyl-2-piperidinyl)ethyl]-benzenamine (1R,3S)-(+)-camphoric acid salt (1 :1) and 60 mL of isopropyl acetate. Stir the mixture at 20-25 0C under nitrogen and add a solution of 1.60 g of sodium hydroxide in 20 mL of water over a period of 5 min while maintaining an internal temperature at 20-25 0C. Stir the suspension efficiently until all the solid dissolves (5 min). Separate the organic layer and save. Extract the aqueous layer with 20 mL of isopropyl acetate. Combine the organic layers and wash it with 20 mL of water. Separate the organic layer and concentrate it under vacuum (20-100 mbar) at an internal temperature at 20-40 0C (external temperature 30-60 0C) to obtain ~65 mL of a solution of (S)-2-[2-(1-methyl-2-piperidinyl)ethyl]-benzenamine (containing 3.28 g of free base) in isopropyl acetate. Save this solution for the next step and store it under nitrogen.
(b) Reaction:
A 250-mL, 4-necked, round-bottomed flask, equipped with a mechanical stirrer, digital thermometer, nitrogen inlet-outlet, heating mantle, condenser, and addition funnel is charged with -65 mL of a solution of (S)-2-[2-(1-methyl-2-piperidinyl)ethyl]-benzenamine (containing 3.28 g of free base) in isopropyl acetate and 6.22 g of potassium carbonate. Stir the reaction mixture under nitrogen at an internal temperature at 23 ± 3 0C to afford a suspension. Add 3.75 g of cinnamoyl chloride over a period of 5 min while maintaining an internal temperature at 23 ± 3 0C to obtain a slurry. Heat the reaction mixture to an internal temperature at 85 ± 5 0C (external temperature 90-100 0C) over a period of 30-60 min. Stir the reaction mixture at this temperature for an additional 2 h. Cool the reaction mixture to 23 ± 3 0C over a period of 1 h. Add 50 mL of water. Stir the reaction mixture at 23 ± 3 0C for 30-60 min to obtain a bi-phasic solution. Separate the organic layer. Add 80 mL of 0.5 N HCI solution over a period of 10 min while maintaining an internal temperature at 23 ± 3 0C to afford a bi-phasic solution. Separate the aqueous layer. Add 60 mL of isopropyl acetate. Stir the reaction mixture and add a solution of 2.00 g of sodium hydroxide in 25 mL of water over a period of 10 min while maintaining an internal temperature at 23 ± 3 0C to afford a bi- phasic solution. Separate the organic layer and save. Extract the aqueous layer with 60 mL of isopropyl acetate. Combine the organic layers and wash it with 40 mL of water. Separate the organic layer and concentrate it under vacuum (20-100 mbar) at an internal temperature at 20-40 0C (external temperature 30-60 0C) to obtain 22mL (19.3 g) of a solution of (iii) in isopropyl acetate. Stir and heat the reaction mixture to an internal temperature at 85 ± 5 0C (external temperature 90-100 0C) over a period of 30-60 min. Add 96 mL of hepatane over a period of 10 min while maintaining an internal temperature at 85 ± 5 0C. Stir and Cool the reaction mixture to 23 ± 3 0C over a period of 1 h. Stir the resulting slurry at 23 ± 3 0C for an additional 2 h. Collect the solid by filtration over a polypropylene filter paper in a Buchner funnel with suction. Wash the solid with a total of 28 mL of a mixture of isopropyl acetate and heptane (1/6) in two equal portions of 14 mL each. Dry the solid at 45-50 0C under vacuum (13-40 mbar) with nitrogen bleeding to obtain a constant weight (LOD < 1%, 4 h) of 4.06 g of (2£)-A/-[2-[2-[(2S)-1-methyl-2-piperidinyl]ethyl]phenyl]-3-phenyl-2-propenamide as an off white solid.
Theoretical Yield: 5.23 g
Yield: 77.6%
Purity: 99.8% (HPLC area %).
Enantiomeric purity: (R)-(iii) was not detected by Chiral HPLC. Example 5:
Alternative synthesis of (2£)-/V-[2-[2-[(2S)-1-MethyI-2-piperidinyl]ethyl]phenyI]-3- phenyl-2-propenamide
(a) Free base generation:
In a 500 ml round bottomed flask equipped with a mechanical stirrer the resolved camphoric acid salt (IV) (20 g) in isopropyl acetate (120 g) is added at an internal temperature of 20 to 25 0C (external temperature 20 0C). Then, at an internal temperature of 25 to 30 0C (external temperature 20 0C) a solution of sodium hydroxide (38.24 g) in water (60 g) is added to the reaction mixture over a period of 5 minutes. The reaction mixture (suspension) is then stirred for a further 30 minutes. The resulting orange emulsion is then allowed to separate into a two-phase mixture and the water phase is removed. The organic phase is then subjected to a rotary evaporator and the isopropyl acetate is distilled at an internal temperature of 60 0C and under reduced pressure (250 mbar). Approximately 90 g of isopropyl acetate is distilled. Prior to distilling, the organic phase is a clear, bright orange colour and of a volume of approximately 160 ml ( 13Og),
(b) Reaction:
In a 1.5 I flask equipped with a mechanical stirrer and at an internal temperature of 35 0C (external temperature 38 0C) and under inert conditions (nitrogen) 2-butanone (160 g) and isopropyl acetate (20 g) is added to the reaction mixture of part (a). Then, at an internal temperature of 35 0C (external temperature of 38 0C) a solution of cinnamoyl chloride (8.9 g) in 2-butanone (20 g) is added drop wise. Then, the reaction mixture is treated with more 2- butanone (2 x 5 g). The resulting suspension is then stirred for 20 minutes at an internal temperature of 350C. The pH of the mixture is between 6 and 8.
(c) Resolution:
The suspension of step (b) is then cooled to an internal temperature of 25 0C (external temperature 20 0C) and at the same time a mixture of water (200 g) and isopropyl acetate (60 g) is added. The reaction mixture is then stirred for a further 15 minutes at an internal temperature of 25 0C (external temperature 20 0C). The resulting two-phase reaction mixture is then separated and the water phase removed. The resulting yellow upper layer is then treated with 2.5 mol/l hydrochloric acid (200 g). The resulting two-phase mixture is then separated and the water phase is transferred into a 750 ml flask equipped with a mechanical stirrer. The organic phase is then washed with 2.5 mol/l hydrochloric acid (200 g) and the resulting two-phase mixture is separated and the water phase is added to the first water phase. The combined water phases are then treated with acetic acid (300 g) and sodium hydroxide (150 g) is added. The reaction mixture is then stirred at an internal temperature of 25 to 30 0C (external temperature 20 0C) for 15 minutes. The resulting two-phase reaction mixture is then separated.
(d) Crystallization
The organic phase from the above reaction step (c) is reduced in volume on a rorary evaporator at an external temperature of 60 0C and at 250 mbar. Then, the reduced-volume reaction mixture is treated with isopropanol (60 g) and the resulting reaction mixture is reduced in volume on a rotary evaporator at an external temperature of 60 0C and under a vacuum of 150 mbar. Then, at an internal temperature of 50 to 55 0C (external temperature 60 0C) the reaction mixture is treated with water (20 g) and the resulting suspension is further treated with the product (iv) (10 mg) in isopropanol (0.01 g). The reaction mixture is then stirred for a further 15 minutes at an internal temperature of 50 to 55 0C (external temperature 60 0C). Then, further water is added over a period of 15 to 30 minutes and the reaction mixture is maintained at an internal temperature of 50 to 55 0C (external temperature 60 0C). Then, the resulting suspension is cooled to an internal temperature of 22 to 22 0C (external temperature 20 0C. Then, the suspension is stirred for a further 30 minutes at an internal temperature of 22 to 22 0C (external temperature 20 0C) and the resulting solid is collected by filtration and washed with a mixture of water and isopropyl acetate (2 x 20 g), where the water: isopropyl acetate ratio is of 5:1 g/g. The resulting solid may then be dried under a vacuum at a temperature of 55 0C.
Yield: 14.8 g (89.3% of theory). mp: 127.3 to 130.2 0C
Example 6: Recrystallisation of (2E)-Λ/-[2-[2-[(2S)-1-Methyl-2-piperidinyl]ethyl]phenyl]-3-phenyl-2- propenamide
(a) In a 200 ml round bottomed flask equipped with a magnetic stirrer, containing the product (iv) (15 g) is added isopropanol (25 g) and heptane (heptane fraction from petroleum having a boiling point of 65 to 100 0C) (25 g) is added. Then, the reaction mixture is heated to an internal temperature of 75 0C (external temperature 95 0C) and refluxed for approximately 30 minutes, whilst stirring. Then, the reaction mixture is filtered over a glass fibre filter at an internal temperature of 70 to 75 0C (external temperature 85 0C) in to a 350 ml flask equipped with a magnetic stirrer. Then, a mixture of isopropanol (5 g) and heptane (5 g) is added and the reaction mixture is heated to an internal temperature of 70 0C (external temperature 95 0C). Then, further heptane is added drop wise to the reaction mixture at an internal temperature of 65 to 75 0C (external temperature 75 0C).
(b) Crystallization
The solution from step (a) is then cooled to an internal temperature of 40 0C (external temperature 40 0C) over a period of 15 minutes. The, at an internal temperature of 40 0C, the solution is treated with a suspension of the recrystallized product (v) (11 mg) in heptane is added and the reaction mixture is stirred for 30 minutes at an internal temperature of 40 0C (external temperature 40 to 45 0C). Then, the reaction mixture is treated with some further heptane (15 g) at an internal temperature of 40 0C. The resulting suspension is then cooled to an internal temperature of -10 0C (external temperature -10 to -15 0C) over a period of 30 minutes and then further stirred for a further hour. The reaction mixture is then filtered at an internal temperature of -10 0C (external temperature -10 to -15 0C) and the resulting solid may be washed in a mixture of isopropanol and heptane, where the isopropanohheptane ratio is 1 :1.5. The solid may be washed twice (2 x 11.25 g). The solid may then be dried in a vacuum at a temperature of 60 0C.
Yield: 17.8 g (89% of theory) mp: 127.4 to 132.0 0C.
………………………………………………………………..

An efficient process was developed for the manufacture of MSA100, a serotonin receptor antagonist, via a five-step synthetic route furnishing a high quality of active pharmaceutical ingredient. Highlights of this synthesis include: (1) replacing carcinogenic methyl iodide with methyl p-toluenesulfonate as the methylating reagent; (2) a hydrogenation protocol with optimized temperature, pressure, and mass-transfer conditions that avoided one side product and reduced the other one effectively; (3) chemical resolution employing D-camphoric acid in a mixed-solvent system; (4) amidation under anhydrous conditions for controlling a Michael adduct impurity; and (5) plausible mechanisms for the formation of side products.
Example I PREPARATION AND CONFIRMATION OF S-MPEC


racemic-APEMEP-HI -5/1-


– 6 –
1. 2 – nitrobenzaldehyde
2. 2 – picoline
3. 2 – (o-nitrostyryl) pyridine (NSP)
4. 2 – “(o-nitrostyryl)- 1 -methylpyridinium iodide 5. RS-2- (o-aminophenethyl)-l-methylpiperidine. HI
6. S-[2-(o-aminophenethyl)- 1 -methylpiperidine-dibenzoyl-L-tartrate] (S-APEMP. DBLT OR .L-DBT)
7. S-2′- [2-(l-methyl-2-piperidyl) ethyl] cinnamanilide (S-MPEC) 7a. Cinnamoyl chloride S-MPEC CHEMICAL PROCESS
(A) 2-(O-Nitrostyryl) – 1 -Methylpyridinium Iodide fNSMP-P
To a 50 L round bottomed flask was added 2-nitrobenzaldehyde (3,500 g. 23.2 moles), 2-picoline (3.2L., 32.8 moles) and acetic anhydride. The mixture was stirred efficiently under an inert atmosphere (nitrogen or another inert gas) and heated to reflux for 27 hrs. The mixture was cooled to under 100 C, for safe handling, and quenched in a suitable vessel equipped with external cooling and efficient stirring on 10.5 Kg. of ice. The pH was adjusted to 11 with 45% aqueous sodium hydroxide at a rate to keep the temperature below 50°C. After cooling to 20-30°C, the granular solid was collected by filtration, washed well with water. Yield 6572 g. of crude 2-(o-nitrostyryl) pyridine (NSP).
This solid was transferred to a 50L, round bottomed flask, dissolved in acetone (14L.) and iodomethane (2.94L., 47.7 moles) (quaternizing methylating agent) was added. (Other such (alkylating) agents may be used, generally having the formula CH3X, X being an anion such as sulfate, methyl sulfate, halide (Cl, Br, I), etc.). The mixture was heated to reflux under an inert atmosphere (nitrogen or another inert gas) for 18 hrs. After cooling to 20°C. the precipitate was collected by filtration and washed with acetone or a 1 :1 mixture of acetone:ethyl acetate (3×3.5L.). Drying to constant weight at 50-60°C. yielded 6,839 g. (80%) of NSMP.I. (B) RS- 2-(o-Aminophenethyl)-l-Methylpiperidine. Hvdroiodide
ΓRS-APEMP.HΠ
In a 5 gallon reactor, a solution of NSNP.I (935 g., 2.5 moles) in – 7 – methanol (14L.) was reduced in a hydrogen atmosphere (Psi. 55) in the presence of Pt/C (5 or 10%, 98g.). After removal of the catalyst and evaporation of the filtrate in the usual manner, the residue was dissolved in hot methanol (2.8L.). Ethyl acetate (2.8L) was added to the hot mixture to induce crystallization, yield 516.3 g. (59%) of RS-APEMP.HI.
(C) S-[2-(o-Aminophenethv0- 1 -Methylpiperidine Dibenzoyl-L-Tartrate] (S- APEMP.DBLT)
A solution of RS-APEMP.HI (516g., 1.5 mole) ethyl acetate (5.5g.) (or other low boiling water immiscible solvent such as benzene, toluene etc.) was extracted with 5% aqueous sodium hydroxide to liberate the free base (organic phase), washing the organic phase with water, drying over a suitable drying agent (such as anhydr. sodium sulfate, magnesium sulfate, potassium carbonate etc.) After separating the solvent from the drying agent the solution was evaporated in vacuo and the residual RS-APEMP free base was dissolved in methanol (l .O.L.) and a solution of dibenzoyl-L-tartaric acid (540 g., 1.5 moles) in methanol (2.3 L.) was added. The mixture was held overnight at room temperature. The crystalline precipitate was collected and recrystallized from methanol (3.4 L.), yield 246g. of S-APEMP.DBLT. (28.6%, wt; 57.2% of the S-APEMP). (O) S-2′-r2-π-Methyl-2-Piperidvnethyll Cinnamanilide f S-MPEC) A solution of S-APEMP.DBLT (287 g, 0.5 mole) in ethyl acetate
(3.2 L.) (or other low boiling water immiscible solvent) was extracted with 7.5% aqueous sodium bicarbonate (3.2 L.) to liberate the S-APEMP. After a water wash and drying over a suitable drying agent the solvent was removed in vacuo. The oily residue, S-APEMP, was dissolved in ethyl acetate (1.0 L.) and anhydrous potassium carbonate (412 g, 3.0 moles) (or other suitable acid acceptor such as triethyl amine, pyridine etc.) was added. Cinnamoyl chloride (143 g., 0.7 mole) in 700 ml. of ethyl acetate was added slowly. After the initial reaction, the mixture was refluxed for 14 hrs. After cooling to room temperature the mixture was extracted with water (1.7 L.) and dried over a suitable drying agent. After removing the drying agent the solvent was removed in vacuo and the residue was dissolved in hot ethyl acetate (280 ml.) and allowed to slowly cool to room temperature; filtration yielded S-MPEC, (136 g., 79% yield). Analysis: Calcd. For C, H, N : C, 79.27; H, 8.10;N, 8.04. Found: C, 79.27; H, 8.06; N, 8.07. HPLC(chiral)purity: 99.5%, [oc ]D25, -46° (c=0.01,EtOH); Melting point: 128°C.
TABLE 2 CERTIFICATE OF ANALYSIS Compound Name: (-)-2′-[2-(l-Methyl-2 piperidyl)ethyl]cinnamanilide(/-MPEC,S- MPEC

……………………………….
synthesis of 2′[2-1-(methyl -2-piperidyl) ethyl] cinnamanilide (Y1), which is a compound of formula (Y) where Ra is hydrogen and R1 is methyl:

Processes for the preparation of compounds of formula (Y) are described in U.S. Pat. No. 3,931,195 which comprises the step of alkylating compounds of formula (i) (below) with an alkyl halide, such as methyl iodide for methylation. The same methylation step is described in EP0973741 for the synthesis of compound (Y1).

Thus, the processes described in the prior art involve the use of a highly toxic reagent (e.g. methyl iodide) and provides a yield of about 50%

Zifaxaban, TY-602, Zhifeishaban 知非沙班……Tianjin Institute of Pharmaceutical Research China

Zifaxaban

Zifaxaban
cas 1378266-98-8
rotation (-)
C20 H16 Cl N3 O4 S
C20H16ClN3O4 S, M = 429.87
Tianjin Institute of Pharmaceutical Research
Deep vein thrombosis; Lung embolism
Factor Xa antagonist
TY-602; zhifeishaban; zifaxaban
Chinese J Struc Chem. 2014, 33 (7), 1091-1095.
(S) -5- chloro -N- ((2- oxo _3_ (4_ (2_ oxo _2H_-1-yl) phenyl) oxazolidin-5 -1,3_ yl) methyl) thiophene-2-carboxamide
5-Chloro-N-(5S)-2-oxo-3-[4-(2-oxopyridin-1(2H)-yl)phenyl]oxazolidin-5-ylimethyllthiophene-2-carboxamide]

The title compound(zifaxaban 2, C20H16ClN3O4 S, Mr = 429.87) was synthesized and its crystal structure was determined by single-crystal X-ray diffraction. Zifaxaban crystallizes in monoclinic, space group P21 with a = 5.7900(12), b = 13.086(3), c = 12.889(3) A, β = 100.86(3)°, V = 959.1(3) A3, Z = 2, Dc = 1.489 g/cm3, F(000) = 444, μ = 0.342 mm-1, the final R = 0.0320 and wR = 0.0640 for 2717 observed reflections(I > 2σ(I)).
The absolute configuration of the stereogenic center in the title compound was confirmed to be S by single-crystal X-ray diffraction. Four existing intermolecular hydrogen bonds help to stabilize the lattice and the molecule in the lattice to adopt an L-shape conformation.
Zifaxaban was slightly more active than rivaroxaban 1 in in vitro assay against human FXa and therefore is promising as a drug candidate.
zifaxaban (first disclosed in CN102464658), useful for treating thromboembolic disorders. Zifaxaban, a factor Xa antagonist, is being developed by Tianjin Institute of Pharmaceutical Research, for treating deep vein thrombosis and pulmonary embolism (preclinical, as of November 2014). In May 2014, an IND was filed in China. In June 2014, the institute was seeking to outlicense this product.
In vivo within the cardiovascular, blood coagulation or blood analysis some have formed out of the process of forming a solid mass with the aggregation, called thrombosis, the formation of a solid mass called a thrombus blocks. Thrombosis is an abnormal flow of blood coagulation status due to platelet activation and coagulation factors are activated in accordance therewith.
The blood coagulation was originally a protective mechanism of the organism, there is a mutual antagonism in blood coagulation system and the anti-clotting system. Under physiological conditions, blood clotting factors continue to be activated to produce thrombin, fibrin formation trace, calm on the vascular endothelium, but these traces of fibrin and constantly being activated fibrinolytic system dissolution, while being activated coagulation factors are constantly mononuclear phagocyte system swallowed. The dynamics of the coagulation system and fibrinolysis system, which ensures the blood coagulation potential can also always ensure that the fluid state of the blood.
Sometimes, however, in certain factors can promote the coagulation process, breaking the above dynamic balance triggered the coagulation process, the blood can form a thrombosis or embolism, such as leading to myocardial infarction, stroke, deep vein thrombosis, pulmonary embolism and other thromboembolic disease.
Thromboembolic disease is cardiovascular disease against the most serious diseases, is the first killer of human health. In China, with the improvement and increased aging of the population’s living standards, the incidence of such diseases, mortality, morbidity is increasing every year.
The existing anti-thromboembolic diseases into anti-platelet drugs, anticoagulants and fibrinolytic drugs. Among them, the anti-clotting drugs are the main contents of antithrombotic therapy, mainly thrombin inhibitors and vitamin K antagonists. Heparin and low molecular weight heparin, represented by the presence of oral thrombin inhibitor invalid, non-selective inhibition and high risk of bleeding and other shortcomings. Although warfarin is representative of vitamin K antagonists can be administered orally, but there are narrow therapeutic index, high risk of bleeding and other shortcomings.
Studies have shown that the coagulation process is usually divided into intrinsic coagulation pathway and the extrinsic coagulation pathway. Coagulation process involves a lot of coagulation factors, coagulation factor activated are each the next inactive clotting factor precursor is converted into the activated form. Endogenous, exogenous pathway final summary, the blood coagulation factor X is converted to Xa.
Therefore, theoretically, the direct inhibition of ¾ factor activity should produce effective anti-clotting effect, without the side effects of thrombin inhibitors with. As direct inhibition) (a factor activity on normal hemostasis reaction / adjustment process produces minimal impact. For example, platelets remain low catalytic activity of thrombin on the ability to respond to, and thus does not affect the formation of platelet thrombi, so bleeding integrated minimize the risk of the levy.
research also proved this point. Recently reported a variety of compounds can selectively inhibit efficient Xa, which play a preventive and / or treatment of thromboembolic disease effect (W003000256A1; CN00818966; US2007259913A1; US2007259913A1). Among them, rivaroxaban (Rivaroxaban) was listed in 2008 for hip or knee replacement surgery prophylaxis and treatment of venous thrombosis, with oral, fixed dose and other advantages.
rivaroxaban drawback is the high price of raw materials, low yield preparation, purification of the product is difficult, high production costs. Patent CN00818966 8 reported rivaroxaban synthetic routes as follows:
4

where the first reaction (Preparation of 4- (4-morpholino-3-yl) nitrobenzene) yield of only 17.6%, and rivaroxaban difficult purification.

………………………………
Patent
http://www.google.com/patents/CN103232446A?cl=en
(S) -5- chloro -N- ((2- oxo-3- (4- (2_ oxo -2H- pyridin-1-yl) phenyl) -1, 3_ oxazolidine -5 – yl) methyl) thiophene-2-carboxamide.
[0011] Meanwhile, patent CN201110337461.4 described formula (I) Preparation of the compound:
[0012]

……………………………………..
Patent
CN102464658
http://www.google.com/patents/CN102464658B?cl=en
Example 1
[0046] (S) -5- chloro -N- ((2- oxo-3- (4_ (2_ Batch oxo _2H_ piperidinyl) phenyl) _1,3_ oxazolidin-5-yl) methyl ) thiophene-2-carboxamide (II)
[0048] A, 1- (4- amino-phenyl) -IH- pyridin _2_ -one (Compound VII) is
[0049] The reaction flask was charged with 104g of pyridine -2 (IH) – one (Compound IX), 200g of iodoaniline (compound VIII), 26gCuI, 151g of potassium carbonate, 18g8- hydroxyquinoline, 500mlDMF, nitrogen, heated to reflux, Insulation reaction was stirred 10h. Filtered hot, the filtrate evaporated under reduced pressure to make the solvent, the residue was added ethyl acetate, IL, 0 ° C incubated with stirring lh, filtered and the solid dried, 2L acetonitrile and purified to give 98g dark red solid. Refined liquor was concentrated to 500ml, the ice bath was stirred lh, filtered to give a dark red solid 19g. Total product were 117g, yield 68.9%.
[0050] 1H-NMR (DMSO-Cl6), δ (ppm):… 5 306 (s, 2H), 6 236 (d, 1H), 6 406 (d, 1H), 6 601 (d,. 2H), 6. 977 (d, 2H), 7. 459 (m, 2H).
[0051] B, (R) -2- (2- hydroxy-3- ((2-oxo–2H- pyridin-1-yl) phenyl) amino) propyl) isoindoline-1,3- -dione (Compound V) is
[0052] The reaction flask was added 40gl_ (4- aminophenyl) -IH- pyridin-2-one (Compound VII), 45g (S) _N_ glycidyl phthalimide (Compound VI), 300ml95% ethanol, heating to reflux, the gradual emergence of solid insulation mixing IOh, cooled to room temperature, filtered, and the filter cake washed with ethanol (150ml X 2), and dried to give an off-white solid 38g.
[0053] The mother liquor was taken, evaporated to dryness under reduced pressure, was added 15g (Q-N_ glycidyl phthalimide (Compound VII), 150ml95% ethanol, heated to reflux, stirred incubated 10h, concentrated under reduced pressure, cooled to room temperature , stirred at room temperature for 2h, washed with ethanol and dried to give an off-white solid 33g.
[0054] A total of an off-white solid 71g, yield of 84.8%, without purification, was used directly in the next step.
[0055] 1H-NMR (DMS0_d6), δ (ppm):… 3 053 (m, 1H), 3 194 (m, 1H), 4 644 (m, 2H), 4 020 (m, 1H). , 5. 168 (d, 1H), 5. 851 (t, 1H), 6. 230 (m, 1H), 6. 404 (d, 1H), 6. 665 (d, 2H), 7. 041 ( d, 2H), 7. 435 (m, 1H), 7. 537 (m, 1H), 7. 855 (m, 4H).
[0056] C, ⑶-2- ((2- oxo-3- (4- (2_ oxo _2H_ pyridyl) phenyl) oxazolidin _5_ -1,3_ yl) methyl ) Preparation of isoindoline-1,3-dione (Compound IV) of the
[0057] The reaction flask was charged 50g Compound V, 27gN, N’- carbonyldiimidazole (⑶I), 4_ catalytic amount of dimethylaminopyridine (DMAP), 150mlN, N- dimethylformamide (DMF), stirred for 90 temperature ° C, the reaction was kept for 8 hours to make the solvent was evaporated under reduced pressure, added to IL of water, stirred and dispersed, filtered, washed with water (150mlX “, washed with ethanol (100ml X 1), dried to give a white solid 48g, yield of 90%.
[0058] 1H-NMR (DMSo-CI6), δ (ppm):…. 3 984 (m, 3H), 4 251 (t, 1H), 4 968 (m, 1H), 6 301 (m, 1H), 6. 459 (d, 1H), 7. 423 (d, 2H), 7. 514 (m, 1H), 7. 615 (m, 3H), 7. 892 (m, 4H).
[0059] D, (S) -5- (aminomethyl) -3- (4- (2_ oxo _2H_-1-yl) phenyl) oxazolidin _2_ -1,3_ one hydrochloride (compound III) Synthesis of
[0060] The reaction flask was charged 50g compound IV, 200ml of ethanol, 60ml aqueous methylamine (40%), heated to reflux, stirred incubated 2h, cooled, evaporated under reduced pressure to make the solvent to give a sticky solid.
[0061] added to 300ml of ethanol, 20ml of hydrochloric acid, heated to reflux, stirred incubated lh, cooled to room temperature, incubated with stirring 2h, filtered, washed with ethanol, and dried to obtain;. 34 5g of white solid, yield 88.7%.
1H-NMR (DMS0_d6), δ (ppm):…. 3 240 (m, 2H), 3 980 (m, 1H), 4 255 (m, 1H), 5 028 (m, 1H) , 6. 321 (m, 1H), 6. 475 (d, 1H), 7. 504 (m, 3H), 7. 634 (m, 3H), 8. 561 (s, 1H).
Ε, (S) -5- chloro -N – ((2- oxo-3- (4- (2-oxo–2Η- pyridin-1-yl) phenyl) oxazolidin _1,3_ 5-yl) methyl) thiophene-2-carboxamide Preparation of thiophene (II) of
The reaction flask was charged 15g Compound III, 200ml of tetrahydrofuran, 40ml of water was added with stirring 6. 2g of sodium carbonate was added dropwise 10g5- chloro-thiophene-2-carbonyl chloride (Compound II-1) in tetrahydrofuran IOOml, 30~35 ° C insulation stirred 5h, point board to control the reaction was complete.
to make the solvent was distilled off under reduced pressure, 50ml of water was added, stirring was filtered, the filter cake washed with water and dried to give 18. 5g of white solid.
200ml of acetic acid and purified room temperature overnight, filtered, and the filter cake washed with ethanol and dried to give a white solid 16g, 80% yield.
Melting point: 204 8 ~205 8 ° C;
1H-NMR (DMSo-CI6), δ (ppm):…. 3 623 (t, 2H), 3 893 (m, 1H), 4 230 (t, 1H), 4 871 (m, 1H), 6. 308 (t, 1H), 6. 468 (d, 1H), 7. 193 (d, 1H), 7. 426 (m, 2H), 7. 500 (m, 1H), 7. 637 (m, 4H), 8. 967 (t, 1H);
MS (ESI): m / z = 430 (M + H);
HPLC: rt (%) = 14. 38 (99. 62);
[a] 20d = -37 6 ° (c 0. 3004, DMS0);
WO-2014183667Acetic acid solvate of oxazolidinone derivative, preparation method for the solvate, and application thereof
WO-2014183665Oxazolidinone derivative crystal form I and preparation method and use thereof
WO-2014183666Oxazolidinone derivate crystal form II, preparation method therefor, and application thereo
SEE ABAN SERIES AT…………http://organicsynthesisinternational.blogspot.in/p/aban-series.html
/////////
