









(2S)-N-(1-Cyanocyclopropyl)-4-fluoro-4-methyl-2-({(1S)-2,2,2-trifluoro-1-[4′-(methylsulfonyl)biphenyl-4-yl]ethyl}amino)pentanamide
Home » Phase3 drugs (Page 17)
Category Archives: Phase3 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Provectus Phase III Melanoma Trial Results Earlier Than Planned?


Rose Bengal disodium
4 ,5,6,7-Tetrachloro-2′,4′,5′,7′-tetraiodo-3-oxo-3H-spiro[2-benzofuran-1,9′-xanthene]-3′,6′-diolate disodium salt
| cas | 632-69-9 |
C20 H2 Cl4 I4 O5 . 2 Na, mw 1017.36, PH 10
| innovator | Provectus |
![]()
The FDA has granted PV-10 Orphan Drug Status for the treatment of highly lethal metastatic melanoma and metastatic liver cancer. It has a successful and expanding Compassionate Use Program in operation and successfully completed trials on metastatic cancer of the breast, liver and melanoma, with positive results in all three. Positive effects in this context is that, if you inject PV-10 into a solid tumor, it kills cancer cells, usually within a week and doesn’t harm normal tissue. Many injected tumors actually disappear while others shrink and stop growing. The dual action of the drug is that the destruction of the cancer by direct injection of PV-10 serves to sensitize the patient’s immune system to seek out and kill similar cancer throughout the body. There is convincing evidence that untreated cancer distant from the treated cancer is attacked by the patient’s immune system after treatment.
PROVECTUS COMPANY OVERVIEW
Provectus (PVCT) is a clinical stage bio-pharmaceutical drug development company. There are 3 key scientific managers running the business along with the CFO, who is also the Chief Operating Officer. They preside over a stable of expert and specialized consultants. The company has two lead drug candidates: PH-10 for significant, often severe, and common skin disorders and PV-10, a dual action, local ablation and immunological anti-cancer drug. PH-10 is currently the subject of post-Phase II trial research into mode of action. PV-10 has successfully completed Phase II trials for malignant melanoma, is currently the subject of independent research on mode of actioRose Bengal disodium is in early clinical trials at Provectus for the topical treatment of psoriasis and atopic dermatitis. An intralesional injectable formulation is also in early clinical development as breast cancer, liver cancer and melanoma therapy. Development for the treatment of actinic keratosis had been ongoing; however, no recent development for this indication has been reported. The company is seeking approval in the U.S. to begin clinical evaluation of this formulation for the treatment of liver and prostate cancer. A compassionate use program is under way for Rose Bengal disodium for the treatment of non-visceral cancers.
The drug’s mechanism of action is believed to be characterized by the creation of free radicals upon activation, which eliminate diseased cells. The compound concentrates in tumors at cytotoxic levels while quickly dissipating from healthy tissue. Simultaneously, the drug triggers an immune response that can eliminate metastatic tumor tissue.
In 2007, orphan drug designation was assigned to Rose Bengal disodium by the FDA for the treatment of metastatic melanoma. This designation was also assigned to the compound in the U.S. in 2011 for the treatment of hepatocellular carcinoma.n and efficacy in conjunction with radiation, and it will have a Phase III pivotal trial starting shortly.
SUMMARY
1. If PV-10 and the Chemotherapies act as the prior data indicate, an NDA for melanoma may be submitted by Provectus in the first half of 2015.
2. If this occurs, the FDA denial of the Breakthrough Therapy designation will not have slowed PV-10’s progress to commercialization.
3. Given the relative safety and efficacy of the different drugs, if the trial is not stopped very early for humanitarian reasons, the planned Interim Analysis is likely to result in the cancellation of the trial, prior to the end of 2015.
4. Given PV-10’s superior safety and lack of significant side effects, if it is only as good as Chemotherapy, it will deserve FDA approval.
The Phase III pivotal trial will demonstrate the safety and efficacy of PV-10 to the market and to prospective acquirers a lot earlier than many have presumed.
Rose bengal (4,5,6,7-tetrachloro-2′,4′,5′,7′-tetraiodofluorescein) is a stain. Its sodium salt is commonly used in eye drops to stain damaged conjunctival and corneal cells and thereby identify damage to the eye. The stain is also used in the preparation of Foraminifera for microscopic analysis, allowing the distinction between forms that were alive or dead at the time of collection.
A form of Rose Bengal is also being studied as a treatment for certain cancers and skin conditions. The cancer formulation of the drug, known as PV-10, is currently undergoing clinical trials for melanoma and breast cancer. The company also has formulated a drug based on Rose Bengal for the treatment of eczema and psoriasis; this drug, PH-10, is currently in clinical trials as well.
| Rose bengal | |
|---|---|
 |
|
| Identifiers | |
| CAS number | 11121-48-5 |
| ATC code | S01 |
| Jmol-3D images | Image 1 |
| Properties | |
| Molecular formula | C20H4Cl4I4O5 |
| Molar mass | 973.67 g mol−1 |
Chemical applications

Light microscopy image of the undescribed species of Spinoloricus from Loricifera stained with Rose Bengal.
Rose Bengal is also used in synthetic chemistry to generate singlet oxygen from triplet oxygen. The singlet oxygen can then undergo a variety of useful reactions, particularly [2 + 2] cycloadditions with alkenes and similar systems.
Rose Bengal can be used to form many derivatives that have important medical functions. One such derivative was created so to be sonosensative but photoinsensative, so that with a high intensity focused ultrasound, it could be used in the treatment of cancer. The derivative was formed by amidation of Rose Bengal, which turned off the fluorescent and photosensitive properties of Rose Bengal, leading to a usable compound, named in the study as RB2.[1]
Salts of Rose Bengal can also be formed, with the molecular formula C20 H4 Cl4 I4 O5 . 2 Na, molecular weight of 1017.64 g/mol and CAS # 632-69-9. Known as Rose Bengal Sodium Salt, this compound has its own unique uses and properties, but also functions as a dye.[2]
Biological applications
PV-10 was found to cause an observable response in 60 percent of tumors treated, according to researchers in a phase II melanoma study. Locoregional disease control was observed in 75 percent of patients. Also confirmed was a “bystander effect”, previously observed in the phase I trial, whereby untreated lesions responded to treatment as well, potentially due to immune system response. These data were based on the interim results of the first 40 patients treated in an 80 patient study.[3] Rose Bengal has been shown to not just prevent the growth and spread of ovarian cancer, but also to cause apoptotic cell death of the cancer cells. This has been proven in vitro, in order to prove that Rose Bengal is still a possible option in the treatment of cancer, and further research should be done.[4]
Rose Bengal is also used in animal models of ischemic stroke (photothrombotic stroke models) in biomedical research. A bolus of the compound is injected into the venous system. Then the region of interest (e.g., the cerebral cortex) is exposed and illuminated by LASER light of 561 nm. A thrombus is formed in the illuminated blood vessels, causing a stroke in the dependent brain tissue.[5][6]
Rose bengal has been used for 50 years to diagnose liver and eye cancer. It has also been used as an insecticide.[7][8]
Rose Bengal is able to stain cells whenever the surface epithelium is not being properly protected by the preocular tear film, because Rose Bengal has been proven to not be able to stain cells because of the protective functioning of these preocular tear films.[9] This is why Rose Bengal is often useful as a stain in diagnosing certain medical issues, such as conjunctival and lid disorders.[10]
Rose Bengal has been used for ocular surface staining to study the efficacy of punctal plugs in the treatment of keratoconjunctivitis sicca. [11]
Rose Bengal is being researched as an agent in creating nano sutures.[12] Wounds are painted on both sides with it and then illuminated with an intense light. This links the tiny collagen fibers together sealing the wound.[13][14][15] Healing is faster and the seal reduces chances of infection.[16][17]
Rose Bengal is used in several microbiological media, including Cooke’s Rose Bengal agar, to suppress bacterial growth.
Rose Bengal has been used as a protoplasm stain to discriminate between living and dead micro-organisms, particularly Foraminifera, since the 1950s when Bill Walton developed the technique.[18]
Electronic applications
Rose Bengal demonstrates at least six distinct electronic properties[19] which are otherwise hidden in the molecule. Rose Bengal is a double planar molecule and relative rotation of the planes generate unique electronics. Therefore, Rose Bengal is a suitable candidate for molecular electronics.
History
Rose Bengal was originally prepared in 1884 by Gnehm, as an analogue of fluorescein.[20] The name is due to its similarity to alta, a dye that women in Bengal have used for centuries to colour their feet red during weddings and festivals.
References
- Kim, Y; Valentina Rubio, Jianjun Qi, Rongmin Xia, Zheng-Zheng Shi, Leif Peterson, Ching-Hsuan Tung, and Brian E. O’Neill (2012). “Cancer treatment using an optically inert Rose Bengal derivative combined with pulsed focused ultrasound”. AIP Conference Proceedings 1481: 175.
- “Rose Bengal Sodium Salt”. Sigma-Aldrich. Sigma Aldrich Co. Retrieved 12 November 2013.
- Metastatic Melanoma PV-10 Trial Results Encouraging Says Drug Company, Medical News Today, 09 Jun 2009
- Koevary, S (2012). “Selective toxicity of rose bengal to ovarian cancer cells in vitro”. International Journal of Physiology, Pathophysiology and Pharmacology 4: 99–107.
- Salber D, et al. (2006). “Differential uptake of [18F]FET and [3H]l-methionine in focal cortical ischemia”. Nuclear Medicine and Biology 33 (8): 1029–1035. doi:10.1016/j.nucmedbio.2006.09.004. PMID 17127177.
- Watson BD, Dietrich WD, Busto R, Wachtel MS, Ginsberg MD (1985). “Induction of reproducible brain infarction by photochemically initiated thrombosis”. Ann Neurol 17 (5): 497–504. doi:10.1002/ana.410170513. PMID 4004172.
- Capinera, John L.; Squitier, Jason M. (2000). “Insecticidal Activity of Photoactive Dyes to American and Migratory Grasshoppers (Orthoptera: Acrididae)”. Journal of Economic Entomology 93 (3): 662–666. doi:10.1603/0022-0493-93.3.662. PMID 10902313.
- Martin, Phyllis; Mischke, Sue; Schroder, Robert (1998). “Compatibility of Photoactive Dyes with Insect Biocontrol Agents”. Biocontrol Science and Technology 8 (4): 501–508. doi:10.1080/09583159830018.
- Feenstra, R; Tseng, S (July 1992). “What is actually stained by rose bengal?”. Arch Ophthalmol 110: 984–993. doi:10.1001/archopht.1992.01080190090035.
- Yokoi, Norihiko (2012). “Vital staining for disorders of conjunctiva and lids”. Atarashii Ganka 29: 1599–1605.
- Ervin AM, Wojciechowski R, Schein O (2010). “Punctal occlusion for dry eye syndrome”. Cochrane Database Syst Rev 9: CD006775. doi:10.1002/14651858.CD006775.pub2. PMID 20824852.
- Chan, B; Chan, O; So, K (2008). “Effects of photochemical crosslinking on the microstructure of collagen and a feasibility study on controlled protein release”. Acta Biomaterialia 4 (6): 1627–1636. doi:10.1016/j.actbio.2008.06.007. PMID 18640085.
- O’Neill A.C., Winograd J.M, Zeballos J.M., Johnson T.S., Randolph M.A., Bujold K.E., Kochevar I.E., Redmond R.W. (2007). “Microvascular anastomosis using a photochemical tissue bonding technique”. Lasers in Surgery and Medicine 39 (9): 716–722. doi:10.1002/lsm.20548. PMID 17960755.
- Mulroy L., Kim J., Wu I., Scharper P., Melki S.A., Azar D.A., Redmond R.W., Kochevar I.E. (2000). “Photochemical keratodesmos for repair of lamellar corneal incisions”. Invest Ophthalmol Vis Sci 41 (11): 3335–3340. PMID 11006222.
- Proano C.E., Mulroy L., Erika Jones E., Azar D.A., Redmond R.W., Kochevar I.E. (2004). Invest Ophthalmol Vis Sci: 2177–2181.
- Laser Show in the Surgical Suite, Technology Review, March/April 2009
- Laser Show in the Surgical Suite, Technology Review, 02.11.2009
- Walton, W. (1952), Techniques for recognition of living foraminifera, Contrib. Cushman Found. Foraminiferal Res., 3, 56 – 60
- A new approach to extract multiple distinct conformers and co-existing distinct electronic properties of a single molecule by point-contact method Anirban Bandyopadhyay, Satyajit Sahu, Daisuke Fujita and Yutaka Wakayama, Phys. Chem. Chem. Phys., 2010 view highlights in Royal Society of Chemistry,
- Alexander, Walter (2010). “American Society of Clinical Oncology, 2010 Annual Meeting and Rose Bengal: From a Wool Dye to a Cancer Therapy”. Pharmacy and Therapeutics 35 (8): 469–474. PMC 2935646. Retrieved 5 November 2013.
- US 2010021566
- WO 2011050164
- US 2011250296
External links
- Rose Bengal at the US National Library of Medicine Medical Subject Headings (MeSH)
- Absorption and extinction data
ScinoPharm to Provide Active Pharmaceutical Ingredient 英文名称 Burixafor to F*TaiGen for Novel Stem Cell Drug

英文名称Burixafor
TG-0054
(2-{4-[6-amino-2-({[(1r,4r)-4-({[3-(cyclohexylamino)propyl]amino}methyl)cyclohexyl]methyl}amino)pyrimidin-4-yl]piperazin-1-yl}ethyl)phosphonic acid
[2-[4-[6-Amino-2-[[[trans-4-[[[3-(cyclohexylamino)propyl]amino]methyl]cyclohexyl]methyl]amino]pyrimidin-4-yl]piperazin-1-yl]ethyl]phosphonic acid
1191448-17-5
C27H51N8O3P, 566.7194
chemokine CXCR 4 receptor antagonist;
| Taigen Biotechnology Co., Ltd. |

ScinoPharm to Provide Active Pharmaceutical Ingredient to F*TaiGen for Novel Stem Cell Drug
MarketWatch
The drug has received a Clinical Trial Application from China’s FDA for the initiation of … In addition, six products have entered Phase III clinical trials.
read at

TAINAN, June 8, 2014 — ScinoPharm Taiwan, Ltd. (twse:1789) specializing in the development and manufacture of active pharmaceutical ingredients, and TaiGen Biotechnology (4157.TW; F*TaiGen) jointly announced today the signing of a manufacturing contract for the clinical supply of the API of Burixafor, a new chemical entity discovered and developed by TaiGen. The API will be manufactured in ScinoPharm’s plant in Changshu, China. This cooperation not only demonstrates Taiwan’s international competitive strength in new drug development, but also sees the beginning of a domestic pharmaceutical specialization and cooperation mechanisms, thus establishing a groundbreaking milestone for Taiwan’s pharmaceutical industry.
Dr. Jo Shen, President and CEO of ScinoPharm said, “This cooperation with TaiGen is of representative significance in the domestic pharmaceutical companies’ upstream and downstream cooperation and self-development of new drugs, and indicates the Taiwanese pharmaceutical industry’s cumulative research and development momentum is paving the way forward.” Dr. Jo Shen emphasized, “ScinoPharm’s Changshu Plant provides high-quality API R&D and manufacturing services through its fast, flexible, reliable competitive advantages, effectively assisting clients of new drugs in gaining entry into China, Europe, the United States, and other international markets.”
According to Dr. Ming-Chu Hsu, Chairman and CEO of TaiGen, “R&D is the foundation of the pharmaceutical industry. Once a drug is successfully developed, players at all levels of the value chain could reap the benefit. Burixafor is a 100% in-house developed product that can be used in the treatment of various intractable diseases. The cooperation between TaiGen and ScinoPharm will not only be a win-win for both sides, but will also provide high-quality novel dug for patients from around the world.”
Burixafor is a novel stem cell mobilizer that can efficiently mobilize bone marrow stem cells and tissue precursor cells to the peripheral blood. It can be used in hematopoietic stem cell transplantation, chemotherapy sensitization and other ischemic diseases. The results of the ongoing Phase II clinical trial in the United States are very impressive. The drug has received a Clinical Trial Application from China’s FDA for the initiation of a Phase II clinical trial in chemotherapy sensitization under the 1.1 category. According to the pharmaceutical consultancy company JSB, with only stem cell transplant and chemotherapy sensitizer as the indicator, Burixafor’s annual sales are estimated at USD1.1 billion.
ScinoPharm currently has accepted over 80 new drug API process research and development plans, of which five new drugs have been launched in the market. In addition, six products have entered Phase III clinical trials. Through the Changshu Plant’s operation in line with the latest international cGMP plant equipment and quality management standards, the company provides customers with one stop shopping services in professional R&D, manufacturing, and outsourcing, thereby shortening the customer development cycle of customers’ products and accelerating the launch of new products to the market.
TaiGen’s focus is on the research and development of novel drugs. Besides Burixafor, the products also include anti-infective, Taigexyn®, and an anti-hepatitis C drug, TG-2349. Taigexyn® is the first in-house developed novel drug that received new drug application approval from Taiwan’s FDA. TG-2349 is intended for the 160 million global patients with hepatitis C with huge market potential. TaiGen hopes to file one IND with the US FDA every 3-4 years to expand TaiGen’s product line.
About ScinoPharm
ScinoPharm Taiwan, Ltd. is a leading process R&D and API manufacturing service provider to the global pharmaceutical industry. With research and manufacturing facilities in both Taiwan and China, ScinoPharm offers a wide portfolio of services ranging from custom synthesis for early phase pharmaceutical activities to contract services for brand companies as well as APIs for the generic industry. For more information, please visit the Company’s website at http://www.scinopharm.com
About TaiGen Biotechnology
TaiGen Biotechnology is a leading research-based and product-driven biotechnology company in Taiwan with a wholly-owned subsidiary in Beijing, China. The company’s first product, Taigexyn®, have already received NDA approval from Taiwan’s FDA. In addition to Taigexyn®, TaiGen has two other in-house discovered NCEs in clinical development under IND with US FDA: TG-0054, a chemokine receptor antagonist for stem cell transplantation and chemosensitization, in Phase 2 and TG-2349, a HCV protease inhibitor for treatment of chronic hepatitis infection, in Phase 2. Both TG-0054 and TG-2349 are currently in clinical trials in patients in the US.
SOURCE ScinoPharm Taiwan Ltd.
TG-0054 is a potent and selective chemokine CXCR4 (SDF-1) antagonist in phase II clinical studies at TaiGen Biotechnology for use in stem cell transplantation in cancer patients. Specifically, the compound is being developed for the treatment of stem cell transplantation in multiple myeloma, non-Hodgkin’s lymphoma, Hodgkin’s lymphoma and myocardial ischemia.
Preclinical studies had also been undertaken for the treatment of diabetic retinopathy, critical limb ischemia (CLI) and age-related macular degeneration. In a mouse model, TG-0054 efficiently mobilizes stem cells (CD34+) and endothelial progenitor cells (CD133+) from bone marrow into peripheral circulation.
BACKGROUND
Chemokines are a family of cytokines that regulate the adhesion and transendothelial migration of leukocytes during an immune or inflammatory reaction (Mackay C.R., Nat. Immunol, 2001, 2:95; Olson et al, Am. J. Physiol. Regul. Integr. Comp. Physiol, 2002, 283 :R7). Chemokines also regulate T cells and B cells trafficking and homing, and contribute to the development of lymphopoietic and hematopoietic systems (Ajuebor et al, Biochem. Pharmacol, 2002, 63:1191). Approximately 50 chemokines have been identified in humans. They can be classified into 4 subfamilies, i.e., CXC, CX3C, CC, and C chemokines, based on the positions of the conserved cysteine residues at the N-terminal (Onuffer et al, Trends Pharmacol ScI, 2002, 23:459). The biological functions of chemokines are mediated by their binding and activation of G protein-coupled receptors (GPCRs) on the cell surface.
Stromal-derived factor- 1 (SDF-I) is a member of CXC chemokines. It is originally cloned from bone marrow stromal cell lines and found to act as a growth factor for progenitor B cells (Nishikawa et al, Eur. J. Immunol, 1988, 18:1767). SDF-I plays key roles in homing and mobilization of hematopoietic stem cells and endothelial progenitor cells (Bleul et al, J. Exp. Med., 1996, 184:1101; and Gazzit et al, Stem Cells, 2004, 22:65-73). The physiological function of SDF-I is mediated by CXCR4 receptor. Mice lacking SDF-I or CXCR4 receptor show lethal abnormality in bone marrow myelopoiesis, B cell lymphopoiesis, and cerebellar development (Nagasawa et al, Nature, 1996, 382:635; Ma et al, Proc. Natl. Acad. ScI, 1998, 95:9448; Zou et al, Nature, 1998, 393:595; Lu et al, Proc. Natl. Acad. ScI, 2002, 99:7090). CXCR4 receptor is expressed broadly in a variety of tissues, particularly in immune and central nervous systems, and has been described as the major co-receptor for HIV- 1/2 on T lymphocytes. Although initial interest in CXCR4 antagonism focused on its potential application to AIDS treatment (Bleul et al, Nature, 1996, 382:829), it is now becoming clear that CXCR4 receptor and SDF-I are also involved in other pathological conditions such as rheumatoid arthritis, asthma, and tumor metastases (Buckley et al., J. Immunol., 2000, 165:3423). Recently, it has been reported that a CXCR4 antagonist and an anticancer drug act synergistically in inhibiting cancer such as acute promuelocutic leukemia (Liesveld et al., Leukemia
Research 2007, 31 : 1553). Further, the CXCR4/SDF-1 pathway has been shown to be critically involved in the regeneration of several tissue injury models. Specifically, it has been found that the SDF-I level is elevated at an injured site and CXCR4-positive cells actively participate in the tissue regenerating process.
………………………………………………………………………..
http://www.google.com/patents/WO2009131598A1?cl=en


Compound 52
Example 1 : Preparation of Compounds 1

1-1 1-Ii 1-m
^ ^–\\ Λ xCUNN H ‘ ‘22.. P rdu/’C^ ^. , Λ>\V>v
Et3N, TFAA , H_, r [ Y I RRaanneeyy–NNiicckkeell u H f [ Y | NH2
CH2CI2, -10 0C Boc^ ‘NNA/ 11,,44–ddιιooxxaannee B Boocer”1^”–^^ LiOH, H2O, 50 0C
1-IV 1-V

Water (10.0 L) and (BoC)2O (3.33 kgg, 15.3 mol) were added to a solution of trans-4-aminomethyl-cyclohexanecarboxylic acid (compound 1-1, 2.0 kg, 12.7 mol) and sodium bicarbonate (2.67 kg, 31.8 mol). The reaction mixture was stirred at ambient temperature for 18 hours. The aqueous layer was acidified with concentrated hydrochloric acid (2.95 L, pH = 2) and then filtered. The resultant solid was collected, washed three times with water (15 L), and dried in a hot box (60 0C) to give trα/?5-4-(tert-butoxycarbonylamino-methyl)-cyclo-hexanecarboxylic acid (Compound l-II, 3.17 kg, 97%) as a white solid. Rf = 0.58 (EtOAc). LC-MS m/e 280 (M+Na+). 1H NMR (300 MHz, CDCl3) δ 4.58 (brs, IH), 2.98 (t, J= 6.3 Hz, 2H), 2.25 (td, J = 12, 3.3 Hz, IH), 2.04 (d, J= 11.1 Hz, 2H), 1.83 (d, J= 11.1 Hz, 2H), 1.44 (s, 9H), 1.35-1.50 (m, 3H), 0.89-1.03 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 181.31, 156.08, 79.12, 46.41, 42.99, 37.57, 29.47, 28.29, 27.96. M.p. 134.8-135.0 0C. A suspension of compound l-II (1.0 kg, 3.89 mol) in THF (5 L) was cooled at
-10 0C and triethyl amine (1.076 L, 7.78 mol) and ethyl chloroformate (0.441 L, 4.47 mol) were added below -10 0C. The reaction mixture was stirred at ambient temperature for 3 hours. The reaction mixture was then cooled at -10 0C again and NH4OH (3.6 L, 23.34 mol) was added below -10 0C. The reaction mixture was stirred at ambient temperature for 18 hours and filtered. The solid was collected and washed three times with water (10 L) and dried in a hot box (6O0C) to give trans-4- (tert-butoxycarbonyl-amino-methyl)-cyclohexanecarboxylic acid amide (Compound l-III, 0.8 kg, 80%) as a white solid. Rf= 0.23 (EtOAc). LC-MS m/e 279, M+Na+. 1H NMR (300 MHz, CD3OD) δ 6.63 (brs, IH), 2.89 (t, J= 6.3 Hz, 2H), 2.16 (td, J = 12.2, 3.3 Hz, IH), 1.80-1.89 (m, 4H), 1.43 (s, 9H), 1.37-1.51 (m, 3H), 0.90-1.05 (m, 2H). 13C NMR (75 MHz, CD3OD) δ 182.26, 158.85, 79.97, 47.65, 46.02, 39.28, 31.11, 30.41, 28.93. M.p. 221.6-222.0 0C.
A suspension of compound l-III (1.2 kg, 4.68 mol) in CH2Cl2 (8 L) was cooled at -1O0C and triethyl amine (1.3 L, 9.36 mol) and trifluoroacetic anhydride (0.717 L, 5.16 mol) were added below -10 0C. The reaction mixture was stirred for 3 hours. After water (2.0 L) was added, the organic layer was separated and washed with water (3.0 L) twice. The organic layer was then passed through silica gel and concentrated. The resultant oil was crystallized by methylene chloride. The crystals were washed with hexane to give £rαns-(4-cyano-cyclohexylmethyl)-carbamic acid tert-butyl ester (Compound 1-IV, 0.95 kg, 85%) as a white crystal. Rf = 0.78 (EtOAc). LC-MS m/e 261, M+Na+. 1H NMR (300 MHz, CDCl3) δ 4.58 (brs, IH), 2.96 (t, J = 6.3 Hz, 2H), 2.36 (td, J= 12, 3.3 Hz, IH), 2.12 (dd, J= 13.3, 3.3 Hz, 2H), 1.83 (dd, J = 13.8, 2.7 Hz, 2H), 1.42 (s, 9H), 1.47-1.63 (m, 3H), 0.88-1.02 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 155.96, 122.41, 79.09, 45.89, 36.92, 29.06, 28.80, 28.25, 28.00. M.p. 100.4~100.6°C.
Compound 1-IV (1.0 kg, 4.196 mol) was dissolved in a mixture of 1 ,4-dioxane (8.0 L) and water (2.0 L). To the reaction mixture were added lithium hydroxide monohydrate (0.314 kg, 4.191), Raney-nickel (0.4 kg, 2.334 mol), and 10% palladium on carbon (0.46 kg, 0.216 mol) as a 50% suspension in water. The reaction mixture was stirred under hydrogen atmosphere at 5O0C for 20 hours. After the catalysts were removed by filtration and the solvents were removed in vacuum, a mixture of water (1.0 L) and CH2Cl2 (0.3 L) was added. After phase separation, the organic phase was washed with water (1.0 L) and concentrated to give £rα/?s-(4-aminomethyl- cyclohexylmethyl)-carbamic acid tert- butyl ester (compound 1-V, 0.97 kg, 95%) as pale yellow thick oil. Rf = 0.20 (MeOH/EtOAc = 9/1). LC-MS m/e 243, M+H+. 1H NMR (300 MHz, CDCl3) δ 4.67 (brs, IH), 2.93 (t, J= 6.3 Hz, 2H), 2.48 (d, J= 6.3 Hz, 2H), 1.73-1.78 (m, 4H), 1.40 (s, 9H), 1.35 (brs, 3H), 1.19-1.21 (m, IH), 0.77-0.97 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 155.85, 78.33, 48.27, 46.38, 40.80, 38.19, 29.87, 29.76, 28.07. A solution of compound 1-V (806 g) and Et3N (1010 g, 3 eq) in 1-pentanol
(2.7 L) was treated with compound 1-VI, 540 g, 1 eq) at 900C for 15 hours. TLC showed that the reaction was completed. Ethyl acetate (1.5 L) was added to the reaction mixture at 25°C. The solution was stirred for 1 hour. The Et3NHCl salt was filtered. The filtrate was then concentrated to 1.5 L (1/6 of original volume) by vacuum at 500C. Then, diethyl ether (2.5 L) was added to the concentrated solution to afford the desired product 1-VII (841 g, 68% yield) after filtration at 250C .
A solution of intermediate 1-VII (841 g) was treated with 4 N HCl/dioxane (2.7 L) in MeOH (8.1 L) and stirred at 25°C for 15 hours. TLC showed that the reaction was completed. The mixture was concentrated to 1.5 L (1/7 of original volume) by vacuum at 500C. Then, diethyl ether (5 L) was added to the solution slowly, and HCl salt of 1-VIII (774 g) was formed, filtered, and dried under vacuum (<10 torr). For neutralization, K2CO3 (2.5 kg, 8 eq) was added to the solution of HCl salt of 1-VIII in MeOH (17 L) at 25°C. The mixture was stirred at the same temperature for 3 hours (pH > 12) and filtered (estimated amount of 1-VIII in the filtrate is 504 g). Aldehyde 1-IX (581 g, 1.0 eq based on mole of 1-VII) was added to the filtrate of 1-VIII at 0-100C. The reaction was stirred at 0-100C for 3 hours. TLC showed that the reaction was completed. Then, NaBH4 (81 g, 1.0 eq based on mole of 1-VII) was added at less than 100C and the solution was stirred at 10-150C for Ih. The solution was concentrated to get a residue, which then treated with CH2Cl2 (15 L). The mixture was washed with saturated aq. NH4Cl solution (300 mL) diluted with H2O (1.2 L). The CH2Cl2 layer was concentrated and the residue was purified by chromatography on silica gel (short column, EtOAc as mobile phase for removing other components; MeOH/28% NH4OH = 97/3 as mobile phase for collecting 1-X) afforded crude 1-X (841 g). Then Et3N (167 g, leq) and BoC2O (360 g, leq) were added to the solution of
1-X (841 g) in CH2Cl2 (8.4 L) at 25°C. The mixture was stirred at 25°C for 15 hours. After the reaction was completed as evidenced by TLC, the solution was concentrated and EtOAc (5 L) was added to the resultant residue. The solution was concentrated to 3L (1/2 of the original volume) under low pressure at 500C. Then, n-hexane (3 L) was added to the concentrated solution. The solid product formed at 500C by seeding to afford the desired crude product 1-XI (600 g, 60% yield) after filtration and evaporation. To compound 1-XI (120.0 g) and piperazine (1-XII, 50.0 g, 3 eq) in 1- pentanol (360 niL) was added Et3N (60.0 g, 3.0 eq) at 25°C. The mixture was stirred at 1200C for 8 hours. Ethyl acetate (480 mL) was added to the reaction mixture at 25°C. The solution was stirred for Ih. The Et3NHCl salt was filtered and the solution was concentrated and purified by silica gel (EtOAc/MeOH = 2:8) to afforded 1-XIII (96 g) in a 74% yield.
A solution of intermediate 1-XIII (100 mg) was treated with 4 N HCl/dioxane (2 mL) in CH2Cl2 (1 mL) and stirred at 25°C for 15 hours. The mixture was concentrated to give hydrochloride salt of compound 1 (51 mg). CI-MS (M+ + 1): 459.4
Example 2: Preparation of Compound 2

Compound 2 Intermediate 1-XIII was prepared as described in Example 1.
To a solution of 1-XIII (120 g) in MeOH (2.4 L) were added diethyl vinyl phosphonate (2-1, 45 g, 1.5 eq) at 25°C. The mixture was stirred under 65°C for 24 hours. TLC and HPLC showed that the reaction was completed. The solution was concentrated and purified by silica gel (MeOH/CH2Cl2 = 8/92) to get 87 g of 2-11 (53% yield, purity > 98%, each single impurity <1%) after analyzing the purity of the product by HPLC.
A solution of 20% TFA/CH2C12 (36 mL) was added to a solution of intermediate 2-11 (1.8 g) in CH2Cl2 (5 mL). The reaction mixture was stirred for 15 hours at room temperature and concentrated by removing the solvent to afford trifluoracetic acid salt of compound 2 (1.3 g). CI-MS (M+ + 1): 623.1
Example 3 : Preparation of Compound 3
TMSBr H H


s U
Intermediate 2-11 was prepared as described in Example 2. To a solution of 2-11 (300 g) in CH2Cl2 (1800 mL) was added TMSBr (450 g, 8 eq) at 10-150C for 1 hour. The mixture was stirred at 25°C for 15 hours. The solution was concentrated to remove TMSBr and solvent under vacuum at 400C.
CH2Cl2 was added to the mixture to dissolve the residue. TMSBr and solvent were removed under vacuum again to obtain 36O g crude solid after drying under vacuum (<1 torr) for 3 hours. Then, the crude solid was washed with 7.5 L IPA/MeOH (9/1) to afford compound 3 (280 g) after filtration and drying at 25°C under vacuum (<1 torr) for 3 hours. Crystallization by EtOH gave hydrobromide salt of compound 3 (19Og). CI-MS (M+ + 1): 567.0.
The hydrobromide salt of compound 3 (5.27 g) was dissolved in 20 mL water and treated with concentrated aqueous ammonia (pH=9-10), and the mixture was evaporated in vacuo. The residue in water (30 mL) was applied onto a column (100 mL, 4.5×8 cm) of Dowex 50WX8 (H+ form, 100-200 mesh) and eluted (elution rate, 6 mL/min). Elution was performed with water (2000 mL) and then with 0.2 M aqueous ammonia. The UV-absorbing ammonia eluate was evaporated to dryness to afford ammonia salt of compound 3 (2.41 g). CI-MS (M+ + 1): 567.3.
The ammonia salt of compound 3 (1.5 g) was dissolved in water (8 mL) and alkalified with concentrated aqueous ammonia (pH=l 1), and the mixture solution was applied onto a column (75 mL, 3×14 cm) of Dowex 1X2 (acetate form, 100-200 mesh) and eluted (elution rate, 3 mL/min). Elution was performed with water (900 mL) and then with 0.1 M acetic acid. The UV-absorbing acetic acid eluate was evaporated, and the residue was codistilled with water (5×50 mL) to afford compound 3 (1.44 g). CI-MS (M+ + 1): 567.4. Example 4: Preparation of Compound 4

Compound 4
Intermediate 1-XIII was obtained during the preparation of compound 1. To a solution of diethyl vinyl phosphonate (4-1, 4 g) in CH2Cl2 (120 mL) was added oxalyl chloride (15.5 g, 5 eq) and the mixture was stirred at 300C for 36 hours. The mixture were concentrated under vacuum on a rotatory evaporated to give quantitatively the corresponding phosphochloridate, which was added to a mixture of cyclohexyl amine (4-II, 5.3 g, 2.2 eq), CH2Cl2 (40 mL), and Et3N (6.2 g, 2.5 eq). The mixture was stirred at 35°C for 36 hours, and then was washed with water. The organic layer was dried (MgSO4), filtered, and evaporated to afford 4-III (4.7 g, 85% yield) as brown oil.
Compound 4-III (505 mg) was added to a solution of intermediate 1-XIII (500 mg) in MeOH (4 mL). The solution was stirred at 45°C for 24 hours. The solution was concentrated and the residue was purified by column chromatography on silica gel (EtOAc/ MeOH = 4: 1) to afford intermediate 4-IV (420 mg) in a 63% yield.
A solution of HCl in ether (5 mL) was added to a solution of intermediate 4- IV (420 mg) in CH2Cl2 (1.0 mL). The reaction mixture was stirred for 12 hours at room temperature and concentrated by removing the solvent. The resultant residue was washed with ether to afford hydrochloride salt of compound 4 (214 mg). CI-MS (M+ + 1): 595.1
Preparation of compound 51

TMSBr

Intermediate l-II was prepared as described in Example 1. To a suspension of the intermediate l-II (31.9 g) in toluene (150 mL) were added phosphorazidic acid diphenyl ester (51-1, 32.4 g) and Et3N (11.9 g) at 25°C for 1 hour. The reaction mixture was stirred at 800C for 3 hours and then cooled to 25°C. After benzyl alcohol (51-11, 20 g) was added, the reaction mixture was stirred at 800C for additional 3 hours and then warmed to 1200C overnight. It was then concentrated and dissolved again in EtOAc and H2O. The organic layer was collected. The aqueous layer was extracted with EtOAc. The combined organic layers were washed with 2.5 N HCl, saturated aqueous NaHCO3 and brine, dried over anhydrous MgSO4, filtered, and concentrated. The residue thus obtained was purified by column chromatography on silica gel (EtOAc/Hexane = 1 :2) to give Intermediate 51-111 (35 g) in a 79% yield. A solution of intermediate 51-111 (35 g) treated with 4 N HCl/dioxane (210 rnL) in MeOH (350 mL) was stirred at room temperature overnight. After ether (700 mL) was added, the solution was filtered. The solid was dried under vacuum. K2CO3 was added to a suspension of this solid in CH3CN and ώo-propanol at room temperature for 10 minutes. After water was added, the reaction mixture was stirred at room temperature for 2 hours, filtered, dried over anhydrous MgSO4, and concentrated. The resultant residue was purified by column chromatography on silica gel (using CH2Cl2 and MeOH as an eluant) to give intermediate 51-IV (19 g) in a 76% yield. Intermediate 1-IX (21 g) was added to a solution of intermediate 51-IV (19 g) in CH2Cl2 (570 mL). The mixture was stirred at 25°C for 2 hours. NaBH(OAc)3 (23 g) was then added at 25°C overnight. After the solution was concentrated, a saturated aqueous NaHCO3 solution was added to the resultant residue. The mixture was then extracted with CH2Cl2. The solution was concentrated and the residue was purified by column chromatography on silica gel (using EtOAc and MeOH as an eluant) to afford intermediate 51-V (23.9 g) in a 66% yield.
A solution of intermediate 51-V (23.9 g) and BoC2O (11.4 g) in CH2Cl2 (200 mL) was added to Et3N (5.8 mL) at 25°C for overnight. The solution was then concentrated and the resultant residue was purified by column chromatography on silica gel (using EtOAc and Hexane as an eluant) to give intermediate 51-VI (22 g) in a 77% yield.
10% Pd/C (2.2 g) was added to a suspension of intermediate 51-VI (22 g) in MeOH (44 mL). The mixture was stirred at ambient temperature under hydrogen atmosphere overnight, filtered, and concentrated. The residue thus obtained was purified by column chromatography on silica gel (using EtOAc and MeOH as an eluant) to afford intermediate 51-VII (16.5 g) in a 97% yield.
Intermediate 51-VII (16.5 g) and Et3N (4.4 mL) in 1-pentanol (75 mL) was allowed to react with 2,4-dichloro-6-aminopyrimidine (1-VI, 21 g) at 1200C overnight. The solvent was then removed and the residue was purified by column chromatography on silica gel (using EtOAc and hexane as an eluant) to afford intermediate 51-VIII (16.2 g) in a 77% yield.
A solution of intermediate 51-VIII (16.2 g) and piperazine (1-XII, 11.7 g) in 1-pentanol (32 mL) was added to Et3N (3.3 mL) at 1200C overnight. After the solution was concentrated, the residue was treated with water and extracted with CH2Cl2. The organic layer was collected and concentrated. The residue thus obtained was purified by column chromatography on silica gel (using EtOAc/ MeOH to 28% NH40H/Me0H as an eluant) to afford Intermediate 51-IX (13.2 g) in a 75% yield. Diethyl vinyl phosphonate (2-1) was treated with 51-IX as described in
Example 3 to afford hydrobromide salt of compound 51. CI-MS (M+ + 1): 553.3


………………………………….
Preparation of Compound 1


Water (10.0 L) and (Boc)2O (3.33 kgg, 15.3 mol) were added to a solution of trans-4-aminomethyl-cyclohexanecarboxylic acid (compound 1-I, 2.0 kg, 12.7 mol) and sodium bicarbonate (2.67 kg, 31.8 mol). The reaction mixture was stirred at ambient temperature for 18 hours. The aqueous layer was acidified with concentrated hydrochloric acid (2.95 L, pH=2) and then filtered. The resultant solid was collected, washed three times with water (15 L), and dried in a hot box (60° C.) to give trans-4-(tert-butoxycarbonylamino-methyl)-cyclo-hexanecarboxylic acid (Compound 1-II, 3.17 kg, 97%) as a white solid. Rf=0.58 (EtOAc). LC-MS m/e 280 (M+Na+). 1H NMR (300 MHz, CDCl3) δ 4.58 (brs, 1H), 2.98 (t, J=6.3 Hz, 2H), 2.25 (td, J=12, 3.3 Hz, 1H), 2.04 (d, J=11.1 Hz, 2H), 1.83 (d, J=11.1 Hz, 2H), 1.44 (s, 9H), 1.35˜1.50 (m, 3H), 0.89˜1.03 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 181.31, 156.08, 79.12, 46.41, 42.99, 37.57, 29.47, 28.29, 27.96. M.p. 134.8˜135.0° C.
A suspension of compound 1-II (1.0 kg, 3.89 mol) in THF (5 L) was cooled at 10° C. and triethyl amine (1.076 L, 7.78 mol) and ethyl chloroformate (0.441 L, 4.47 mol) were added below 10° C. The reaction mixture was stirred at ambient temperature for 3 hours. The reaction mixture was then cooled at 10° C. again and NH4OH (3.6 L, 23.34 mol) was added below 10° C. The reaction mixture was stirred at ambient temperature for 18 hours and filtered. The solid was collected and washed three times with water (10 L) and dried in a hot box (60° C.) to give trans-4-(tert-butoxycarbonyl-amino-methyl)-cyclohexanecarboxylic acid amide (Compound 1-III, 0.8 kg, 80%) as a white solid. Rf=0.23 (EtOAc). LC-MS m/e 279, M+Na+. 1H NMR (300 MHz, CD3OD) δ 6.63 (brs, 1H), 2.89 (t, J=6.3 Hz, 2H), 2.16 (td, J=12.2, 3.3 Hz, 1H), 1.80˜1.89 (m, 4H), 1.43 (s, 9H), 1.37˜1.51 (m, 3H), 0.90˜1.05 (m, 2H). 13C NMR (75 MHz, CD3OD) δ 182.26, 158.85, 79.97, 47.65, 46.02, 39.28, 31.11, 30.41, 28.93. M.p. 221.6˜222.0° C.
A suspension of compound 1-III (1.2 kg, 4.68 mol) in CH2Cl2 (8 L) was cooled at 10° C. and triethyl amine (1.3 L, 9.36 mol) and trifluoroacetic anhydride (0.717 L, 5.16 mol) were added below 10° C. The reaction mixture was stirred for 3 hours. After water (2.0 L) was added, the organic layer was separated and washed with water (3.0 L) twice. The organic layer was then passed through silica gel and concentrated. The resultant oil was crystallized by methylene chloride. The crystals were washed with hexane to give trans-(4-cyano-cyclohexylmethyl)-carbamic acid tent-butyl ester (Compound 1-IV, 0.95 kg, 85%) as a white crystal. Rf=0.78 (EtOAc). LC-MS m/e 261, M+Na+. 1H NMR (300 MHz, CDCl3) δ 4.58 (brs, 1H), 2.96 (t, J=6.3 Hz, 2H), 2.36 (td, J=12, 3.3 Hz, 1H), 2.12 (dd, J=13.3, 3.3 Hz, 2H), 1.83 (dd, J=13.8, 2.7 Hz, 2H), 1.42 (s, 9H), 1.47˜1.63 (m, 3H), 0.88˜1.02 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 155.96, 122.41, 79.09, 45.89, 36.92, 29.06, 28.80, 28.25, 28.00. M.p. 100.4˜100.6° C.
Compound 1-IV (1.0 kg, 4.196 mol) was dissolved in a mixture of 1,4-dioxane (8.0 L) and water (2.0 L). To the reaction mixture were added lithium hydroxide monohydrate (0.314 kg, 4.191), Raney-nickel (0.4 kg, 2.334 mol), and 10% palladium on carbon (0.46 kg, 0.216 mol) as a 50% suspension in water. The reaction mixture was stirred under hydrogen atmosphere at 50° C. for 20 hours. After the catalysts were removed by filtration and the solvents were removed in vacuum, a mixture of water (1.0 L) and CH2Cl2 (0.3 L) was added. After phase separation, the organic phase was washed with water (1.0 L) and concentrated to give trans-(4-aminomethyl-cyclohexylmethyl)-carbamic acid tert-butyl ester (compound 1-V, 0.97 kg, 95%) as pale yellow thick oil. Rf=0.20 (MeOH/EtOAc=9/1). LC-MS m/e 243, M+H+. 1H NMR (300 MHz, CDCl3) δ 4.67 (brs, 1H), 2.93 (t, J=6.3 Hz, 2H), 2.48 (d, J=6.3 Hz, 2H), 1.73˜1.78 (m, 4H), 1.40 (s, 9H), 1.35 (brs, 3H), 1.19˜1.21 (m, 1H), 0.77˜0.97 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 155.85, 78.33, 48.27, 46.38, 40.80, 38.19, 29.87, 29.76, 28.07.
A solution of compound 1-V (806 g) and Et3N (1010 g, 3 eq) in 1-pentanol (2.7 L) was treated with compound 1-VI, 540 g, 1 eq) at 90° C. for 15 hours. TLC showed that the reaction was completed.
Ethyl acetate (1.5 L) was added to the reaction mixture at 25° C. The solution was stirred for 1 hour. The Et3NHCl salt was filtered. The filtrate was then concentrated to 1.5 L (1/6 of original volume) by vacuum at 50° C. Then, diethyl ether (2.5 L) was added to the concentrated solution to afford the desired product 1-VII (841 g, 68% yield) after filtration at 25° C.
A solution of intermediate 1-VII (841 g) was treated with 4 N HCl/dioxane (2.7 L) in MeOH (8.1 L) and stirred at 25° C. for 15 hours. TLC showed that the reaction was completed. The mixture was concentrated to 1.5 L (1/7 of original volume) by vacuum at 50° C. Then, diethyl ether (5 L) was added to the solution slowly, and HCl salt of 1-VIII (774 g) was formed, filtered, and dried under vacuum (<10 ton). For neutralization, K2CO3 (2.5 kg, 8 eq) was added to the solution of HCl salt of 1-VIII in MeOH (17 L) at 25° C. The mixture was stirred at the same temperature for 3 hours (pH>12) and filtered (estimated amount of 1-VIII in the filtrate is 504 g).
Aldehyde 1-IX (581 g, 1.0 eq based on mole of 1-VII) was added to the filtrate of 1-VIII at 0-10° C. The reaction was stirred at 0-10° C. for 3 hours. TLC showed that the reaction was completed. Then, NaBH4 (81 g, 1.0 eq based on mole of 1-VII) was added at less than 10° C. and the solution was stirred at 10-15° C. for 1 h. The solution was concentrated to get a residue, which then treated with CH2Cl2 (15 L). The mixture was washed with saturated aq. NH4Cl solution (300 mL) diluted with H2O (1.2 L). The CH2Cl2 layer was concentrated and the residue was purified by chromatography on silica gel (short column, EtOAc as mobile phase for removing other components; MeOH/28% NH4OH=97/3 as mobile phase for collecting 1-X) afforded crude 1-X (841 g).
Then Et3N (167 g, 1 eq) and Boc2O (360 g, 1 eq) were added to the solution of 1-X (841 g) in CH2Cl2 (8.4 L) at 25° C. The mixture was stirred at 25° C. for 15 hours. After the reaction was completed as evidenced by TLC, the solution was concentrated and EtOAc (5 L) was added to the resultant residue. The solution was concentrated to 3 L (1/2 of the original volume) under low pressure at 50° C. Then, n-hexane (3 L) was added to the concentrated solution. The solid product formed at 50° C. by seeding to afford the desired crude product 1-XI (600 g, 60% yield) after filtration and evaporation.
To compound 1-XI (120.0 g) and piperazine (1-XII, 50.0 g, 3 eq) in 1-pentanol (360 mL) was added Et3N (60.0 g, 3.0 eq) at 25° C. The mixture was stirred at 120° C. for 8 hours. Ethyl acetate (480 mL) was added to the reaction mixture at 25° C. The solution was stirred for 1 h. The Et3NHCl salt was filtered and the solution was concentrated and purified by silica gel (EtOAc/MeOH=2:8) to afforded 1-XIII (96 g) in a 74% yield.
To a solution of 1-XIII (120 g) in MeOH (2.4 L) were added diethyl vinyl phosphonate (1-XIV, 45 g, 1.5 eq) at 25° C. The mixture was stirred under 65° C. for 24 hours. TLC and HPLC showed that the reaction was completed. The solution was concentrated and purified by silica gel (MeOH/CH2Cl2=8/92) to get 87 g of 1-XV (53% yield, purity>98%, each single impurity<1%) after analyzing the purity of the product by HPLC.
A solution of 20% TFA/CH2Cl2 (36 mL) was added to a solution of intermediate 1-XV (1.8 g) in CH2Cl2 (5 mL). The reaction mixture was stirred for 15 hours at room temperature and concentrated by removing the solvent to afford trifluoracetic acid salt of compound 1 (1.3 g).
CI-MS (M++1): 623.1.
(2) Preparation of Compound 2

Intermediate 1-XV was prepared as described in Example 1.
To a solution of 1-XV (300 g) in CH2Cl2 (1800 mL) was added TMSBr (450 g, 8 eq) at 10-15° C. for 1 hour. The mixture was stirred at 25° C. for 15 hours. The solution was concentrated to remove TMSBr and solvent under vacuum at 40° C. CH2Cl2 was added to the mixture to dissolve the residue. TMSBr and solvent were removed under vacuum again to obtain 360 g crude solid after drying under vacuum (<1 torr) for 3 hours. Then, the crude solid was washed with 7.5 L IPA/MeOH (9/1) to afford compound 2 (280 g) after filtration and drying at 25° C. under vacuum (<1 ton) for 3 hours. Crystallization by EtOH gave hydrobromide salt of compound 2 (190 g). CI-MS (M++1): 567.0.
The hydrobromide salt of compound 2 (5.27 g) was dissolved in 20 mL water and treated with concentrated aqueous ammonia (pH=9-10), and the mixture was evaporated in vacuo. The residue in water (30 mL) was applied onto a column (100 mL, 4.5×8 cm) of Dowex 50WX8 (H+ form, 100-200 mesh) and eluted (elution rate, 6 mL/min). Elution was performed with water (2000 mL) and then with 0.2 M aqueous ammonia. The UV-absorbing ammonia eluate was evaporated to dryness to afford ammonia salt of compound 2 (2.41 g). CI-MS (M++1): 567.3.
The ammonia salt of compound 2 (1.5 g) was dissolved in water (8 mL) and alkalified with concentrated aqueous ammonia (pH=11), and the mixture solution was applied onto a column (75 mL, 3×14 cm) of Dowex 1×2 (acetate form, 100-200 mesh) and eluted (elution rate, 3 mL/min). Elution was performed with water (900 mL) and then with 0.1 M acetic acid. The UV-absorbing acetic acid eluate was evaporated, and the residue was codistilled with water (5×50 mL) to afford compound 2 (1.44 g). CI-MS (M++1): 567.4.
(3) Preparation of Compound 3

Intermediate 1-XIII was obtained during the preparation of compound 1.
To a solution of diethyl vinyl phosphonate (3-I, 4 g) in CH2Cl2 (120 mL) was added oxalyl chloride (15.5 g, 5 eq) and the mixture was stirred at 30° C. for 36 hours. The mixture were concentrated under vacuum on a rotatory evaporated to give quantitatively the corresponding phosphochloridate, which was added to a mixture of cyclohexyl amine (3-II, 5.3 g, 2.2 eq), CH2Cl2 (40 mL), and Et3N (6.2 g, 2.5 eq). The mixture was stirred at 35° C. for 36 hours, and then was washed with water. The organic layer was dried (MgSO4), filtered, and evaporated to afford 3-III (4.7 g, 85% yield) as brown oil.
Compound 3-III (505 mg) was added to a solution of intermediate 1-XIII (500 mg) in MeOH (4 mL). The solution was stirred at 45° C. for 24 hours. The solution was concentrated and the residue was purified by column chromatography on silica gel (EtOAc/MeOH=4:1) to afford intermediate 3-IV (420 mg) in a 63% yield.
A solution of HCl in ether (5 mL) was added to a solution of intermediate 3-IV (420 mg) in CH2Cl2 (1.0 mL). The reaction mixture was stirred for 12 hours at room temperature and concentrated by removing the solvent. The resultant residue was washed with ether to afford hydrochloride salt of compound 3 (214 mg).
CI-MS (M++1): 595.1.
(4) Preparation of Compound 4

Compound 4 was prepared in the same manner as that described in Example 2 except that sodium 2-bromoethanesulfonate in the presence of Et3N in DMF at 45° C. was used instead of diethyl vinyl phosphonate. Deportations of amino-protecting group by hydrochloride to afford hydrochloride salt of compound 4.
CI-MS (M++1): 567.3
(5) Preparation of Compound 5

Compound 5 was prepared in the same manner as that described in Example 2 except that diethyl-1-bromopropylphosphonate in the presence of K2CO3 in CH3CN was used instead of diethyl vinyl phosphonate.
CI-MS (M++1): 581.4
(6) Preparation of Compound 6

Compound 6 was prepared in the same manner as that described in Example 5 except that 1,4-diaza-spiro[5.5]undecane dihydrochloride was used instead of piperazine.
CI-MS (M++1): 649.5
(7) Preparation of Compound 7


Intermediate 1-II was prepared as described in Example 1.
To a suspension of the intermediate 1-II (31.9 g) in toluene (150 mL) were added phosphorazidic acid diphenyl ester (7-I, 32.4 g) and Et3N (11.9 g) at 25° C. for 1 hour. The reaction mixture was stirred at 80° C. for 3 hours and then cooled to 25° C. After benzyl alcohol (7-II, 20 g) was added, the reaction mixture was stirred at 80° C. for additional 3 hours and then warmed to 120° C. overnight. It was then concentrated and dissolved again in EtOAc and H2O. The organic layer was collected. The aqueous layer was extracted with EtOAc. The combined organic layers were washed with 2.5 N HCl, saturated aqueous NaHCO3 and brine, dried over anhydrous MgSO4, filtered, and concentrated. The residue thus obtained was purified by column chromatography on silica gel (EtOAc/Hexane=1:2) to give Intermediate 7-III (35 g) in a 79% yield.
A solution of intermediate 7-III (35 g) treated with 4 N HCl/dioxane (210 mL) in MeOH (350 mL) was stirred at room temperature overnight. After ether (700 mL) was added, the solution was filtered. The solid was dried under vacuum. K2CO3 was added to a suspension of this solid in CH3CN and iso-propanol at room temperature for 10 minutes. After water was added, the reaction mixture was stirred at room temperature for 2 hours, filtered, dried over anhydrous MgSO4, and concentrated. The resultant residue was purified by column chromatography on silica gel (using CH2Cl2 and MeOH as an eluant) to give intermediate 7-IV (19 g) in a 76% yield.
Intermediate 1-IX (21 g) was added to a solution of intermediate 7-IV (19 g) in CH2Cl2 (570 mL). The mixture was stirred at 25° C. for 2 hours. NaBH(OAc)3 (23 g) was then added at 25° C. overnight. After the solution was concentrated, a saturated aqueous NaHCO3 solution was added to the resultant residue. The mixture was then extracted with CH2Cl2. The solution was concentrated and the residue was purified by column chromatography on silica gel (using EtOAc and MeOH as an eluant) to afford intermediate 7-V (23.9 g) in a 66% yield.
A solution of intermediate 7-V (23.9 g) and Boc2O (11.4 g) in CH2Cl2 (200 mL) was added to Et3N (5.8 mL) at 25° C. for overnight. The solution was then concentrated and the resultant residue was purified by column chromatography on silica gel (using EtOAc and Hexane as an eluant) to give intermediate 7-VI (22 g) in a 77% yield. 10% Pd/C (2.2 g) was added to a suspension of intermediate 7-VI (22 g) in MeOH (44 mL). The mixture was stirred at ambient temperature under hydrogen atmosphere overnight, filtered, and concentrated. The residue thus obtained was purified by column chromatography on silica gel (using EtOAc and MeOH as an eluant) to afford intermediate 7-VII (16.5 g) in a 97% yield.
Intermediate 7-VII (16.5 g) and Et3N (4.4 mL) in 1-pentanol (75 mL) was allowed to react with 2,4-dichloro-6-aminopyrimidine (1-VI, 21 g) at 120° C. overnight. The solvent was then removed and the residue was purified by column chromatography on silica gel (using EtOAc and hexane as an eluant) to afford intermediate 7-VIII (16.2 g) in a 77% yield.
A solution of intermediate 7-VIII (16.2 g) and piperazine (1-XII, 11.7 g) in 1-pentanol (32 mL) was added to Et3N (3.3 mL) at 120° C. overnight. After the solution was concentrated, the residue was treated with water and extracted with CH2Cl2. The organic layer was collected and concentrated. The residue thus obtained was purified by column chromatography on silica gel (using EtOAc/MeOH to 28% NH4OH/MeOH as an eluant) to afford Intermediate 7-IX (13.2 g) in a 75% yield.
Diethyl vinyl phosphonate (2-I) was treated with 7-IX as described in Example 3 to afford hydrobromide salt of compound 7.
CI-MS (M++1): 553.3
(8) Preparation of Compound 8


Cis-1,4-cyclohexanedicarboxylic acid (8-I, 10 g) in THF (100 ml) was added oxalyl chloride (8-II, 15.5 g) at 0° C. and then DMF (few drops). The mixture was stirred at room temperature for 15 hours. The solution was concentrated and the residue was dissolved in THF (100 ml). The mixture solution was added to ammonium hydroxide (80 ml) and stirred for 1 hour. The solution was concentrated and filtration to afford crude product 8-III (7.7 g).
Compound 8-III (7.7 g) in THF (200 ml) was slowly added to LiAlH4 (8.6 g) in THF (200 ml) solution at 0° C. The mixture solution was stirred at 65° C. for 15 hours. NaSO4.10H2O was added at room temperature and stirred for 1 hours. The resultant mixture was filtered to get filtrate and concentrated. The residue was dissolved in CH2Cl2 (100 ml). Et3N (27 g) and (Boc)2O (10 g) were added at room temperature. The solution was stirred for 15 h, and then concentrated to get resultant residue. Ether was added to the resultant residue. Filtration and drying under vacuum afforded solid crude product 8-IV (8.8 g).
A solution of compound 8-IV (1.1 g) and Et3N (1.7 g) in 1-pentanol (10 ml) was reacted with 2,4-dichloro-6-aminopyrimidine (1-VI, 910 mg) at 90° C. for 15 hours. TLC showed that the reaction was completed. Ethyl acetate (10 mL) was added to the reaction mixture at 25° C. The solution was stirred for 1 hour. The Et3NHCl salt was removed. The filtrate was concentrated and purified by silica gel (EtOAc/Hex=1:2) to afford the desired product 8-V (1.1 g, 65% yield).
A solution of intermediate 8-V (1.1 g) was treated with 4 N HCl/dioxane (10 ml) in MeOH (10 ml) and stirred at 25° C. for 15 hours. TLC showed that the reaction was completed. The mixture was concentrated, filtered, and dried under vacuum (<10 ton). For neutralization, K2CO3 (3.2 g) was added to the solution of HCl salt in MeOH (20 ml) at 25° C. The mixture was stirred at the same temperature for 3 hours (pH>12) and filtered. Aldehyde 1-IX (759 mg) was added to the filtrate at 0-10° C. The reaction was stirred at 0-10° C. for 3 hours. TLC showed that the reaction was completed. Then, NaBH4 (112 mg) was added at less than 10° C. and the solution was stirred at 10-15° C. for 1 hour. The solution was concentrated to get a residue, which was then treated with CH2Cl2 (10 mL). The mixture was washed with saturated NH4Cl (aq) solution. The CH2Cl2 layer was concentrated and the residue was purified by chromatography on silica gel (MeOH/28% NH4OH=97/3) to afford intermediate 8-VI (1.0 g, 66% yield).
Et3N (600 mg) and Boc2O (428 mg) were added to the solution of 8-VI (1.0 g) in CH2Cl2 (10 ml) at 25° C. The mixture was stirred at 25° C. for 15 hours. TLC showed that the reaction was completed. The solution was concentrated and purified by chromatography on silica gel (EtOAc/Hex=1:1) to afford intermediate 8-VII (720 mg, 60% yield).
To a solution compound 8-VII (720 mg) and piperazine (1-XII, 1.22 g) in 1-pentanol (10 mL) was added Et3N (1.43 g) at 25° C. The mixture was stirred at 120° C. for 24 hours. TLC showed that the reaction was completed. Ethyl acetate (20 mL) was added at 25° C. The solution was stirred for 1 hour. The Et3NHCl salt was removed and the solution was concentrated and purified by silica gel (EtOAc/MeOH=2:8) to afford 8-VIII (537 mg) in 69% yield.
To a solution of 8-VIII (537 mg) in MeOH (11 ml) was added diethyl vinyl phosphonate (2-I, 201 mg) at 25° C. The mixture was stirred under 65° C. for 24 hours. TLC and HPLC showed that the reaction was completed. The solution was concentrated and purified by silica gel (MeOH/CH2Cl2=1:9) to get 8-IX (380 mg) in a 57% yield.
To a solution of 8-IX (210 mg) in CH2Cl2 (5 ml) was added TMSBr (312 mg) at 10-15° C. for 1 hour. The mixture was stirred at 25° C. for 15 hours. The solution was concentrated to remove TMSBr and solvent under vacuum at 40° C., then, CH2Cl2 was added to dissolve the residue. Then TMSBr and solvent were further removed under vacuum and CH2Cl2 was added for four times repeatedly. The solution was concentrated to get hydrobromide salt of compound 8 (190 mg).
CI-MS (M++1): 566.9

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
amcrasto@gmail.com
MOBILE-+91 9323115463
GLENMARK SCIENTIST , INDIA
web link
web link
http://anthonycrasto.jimdo.com/
blogs are

Congratulations! Your presentation titled “Anthony Crasto Glenmark scientist, helping millions with websites” has just crossed MILLION views.
アンソニー 安东尼 Энтони 안토니 أنتوني
join my process development group on google
you can post articles and will be administered by me on the google group which is very popular across the world
LinkedIn group
blogs are
MY BLOG ON MED CHEM
New Drug Approvals
ALL ABOUT DRUGS
WORLD DRUG TRACKER
MEDICINAL CHEM INTERNATIONAL
DRUG SYN INTERNATIONAL
SCALEUP OF DRUGS
ALL FOR DRUGS ON WEB
http://scholar.google.co.uk/citations?user=bxm3kYkAAAAJ
New Drug Approvals
ALL ABOUT DRUGS
WORLD DRUG TRACKER
MEDICINAL CHEM INTERNATIONAL
DRUG SYN INTERNATIONAL
SCALEUP OF DRUGS
ALL FOR DRUGS ON WEB
http://scholar.google.co.uk/citations?user=bxm3kYkAAAAJ
MY CHINA AND JAPAN BLOGS
VIETNAM
http://me.zing.vn/u/amcrasto
ICELAND
http://amcrasto.bland.is/
RUSSIA
http://www.100zakladok.ru/amcrasto/
http://bobrdobr.ru/people/amcrasto/
GSK launches huge Phase III trial for heart drug losmapimod (GW856553)

losmapimod
Losmapimod is a p38 mitogen-activated protein kinase inhibitor.
Smithkline Beecham Corporation
6-[5-(cyclopropylcarbamoyl)-3-fluoro-2-methylphenyl]-N-(2,2-dimethylpropyl)pyridine-3-carboxamide
GW856553X, 585543-15-3, Losmapimod (USAN/INN), UNII-F2DQF16BXE, AGN-PC-00BFXU,
Molecular Formula: C22H26FN3O2
Molecular Weight: 383.459143
cas 585543-15-3
Synonym: Losmapimod; GW856553; GW-856553; GW 856553)
IUPAC/Chemical name:
6-[5-(cyclopropylcarbamoyl)-3-fluoro-2-methylphenyl]-N-(2,2-dimethylpropyl)pyridine-3-carboxamide
GlaxoSmithKline has begun a Phase III study cardiovascular outcomes study of its investigational compound losmapimod in patients with acute coronary syndrome.
The trial will assess whether losmapimod can reduce the risk of a subsequent cardiac event when administered orally twice a day for three months immediately after presentation with an ACS, such as heart attack. GSK says that some 25,500 patients will be enrolled over the study period across 39 countries.
Read more at: http://www.pharmatimes.com/Article/14-06-06/GSK_launches_huge_Phase_III_trial_for_heart_drug_losmapimod.aspx#ixzz33vFHAK14
Losmapimod, also know as GW856553 or GW856553X, is a drug developed by GlaxoSmithKline which acts as a selective inhibitor of the enzyme family known as p38 mitogen-activated protein kinases. p38 mitogen-activated protein kinases are mediators of inflammation. A Phase II human clinical trial for the treatment of COPD (chronic obstructive pulmonary disease) is underway. Inhibiting these enzymes has been shown to produce antidepressant and antipsychotic effects in animal studies, with the mechanism thought to involve increased neurogenesis probably related to BDNF release. Losmapimod has completed Phase II human clinical trials for the treatment of depression although its safety and efficacy have yet to be proven in further trials. Losmapimod is also being studied for cardiovascular disease. A Phase II trial to study its effects in myocardial infarction (heart attack) is ongoing.
Losmapimod (GW856553X) is a drug developed by GlaxoSmithKline which acts as a selective inhibitor of the enzyme family known as p38 mitogen-activated protein kinases.[1]
p38 mitogen-activated protein kinases are mediators of inflammation. A Phase II human clinical trial for the treatment of COPD(chronic obstructive pulmonary disease)[2] is underway. Inhibiting these enzymes has been shown to produce antidepressant andantipsychotic effects in animal studies, with the mechanism thought to involve increased neurogenesis[3] probably related to BDNFrelease. Losmapimod has completed Phase II human clinical trials for the treatment of depression although its safety and efficacy have yet to be proven in further trials.[4]
Losmapimod is also being studied for cardiovascular disease.[5] A Phase II trial to study its effects in myocardial infarction (heart attack) is ongoing.[6]
………………………
http://www.google.com/patents/US8252818

| Example 36 6-(5- Cyclopropylcarbamoyl- 3-fluoro-2-methyl- phenyl)-N-(2,2- dimethylpropyl)- nicotinamide |
 |
6-Chloro-N-(2,2- dimethylpropyl))nicotin- amide (Intermediate 24) | 384 | 3.01 |
……………….
https://www.google.com/patents/US7514456
General Method A
6-Bromonicotinic acid (100 mg, 0.5 mmol) was heated at 95° C. in thionyl chloride (0.63 ml) for 2 hours. The excess thionyl chloride was evaporated under vacuum and the residue dissolved in DCM (2 ml). To this solution, amine (0.5 mmol) and sodium carbonate (100 mg) were added and the reaction was stirred at room temperature for 2 hours. The reaction was filtered and the residue washed with DCM. The combined filtrate and washings were reduced to dryness to give the desired 6-chloronicotinamide.
| Retention time | |||
| Compound | Amine | MH+ | (minutes) |
| Intermediate 22: 6-Chloro-N-(3- | 3-methylbutylamine | 227 | 2.92 |
| methylbutyl)nicotinamide | |||
| Intermediate 23: 6-Chloro-N-(1- | 1-cyclopropylethylamine | 225 | 2.65 |
| cyclopropylethyl)nicotinamide | |||
| Intermediate 24: 6-Chloro-N-(2,2- | 2,2-dimethylpropylamine | 227 | 2.82 |
| dimethylpropyl))nicotinamide | |||
| Intermediate 25: 6-Chloro-N-(2,2- | 2,2- | 225 | 2.67 |
|
8-29-2012
|
Nicotinamide derivatives useful as P38 inhibitors
|
|
|
8-3-2011
|
Use of a p38 Kinase Inhibitor for Treating Psychiatric Disorders
|
|
|
11-24-2010
|
3-Aminocarbonyl, 6-phenyl substituted pyridine-1-oxides as p38 kinase inhibitors
|
|
|
8-27-2010
|
NICOTINAMIDE DERIVATES USEFUL AS P38 INHIBITORS
|
|
|
5-5-2010
|
Nicotinamide Derivatives Useful as p38 Inhibitors
|
|
|
4-8-2009
|
Nicotinamide Derivatives Useful as p38 Inhibitors
|
|
|
10-25-2006
|
Nicotinamide derivatives useful as p38 inhibitors.
|
References
- Aston N, Bamborough P, Buckton J, Edwards C, Holmes D, Jones K, Patel V, Smee P, Somers D, Vitulli G, Walker A. p38α Mitogen-Activated Protein Kinase Inhibitors: Optimization of a Series of Biphenylamides to Give a Molecule Suitable for Clinical Progression.Journal of Medicinal Chemistry 2009, 52(20), 6257. doi:10.1021/jm9004779
- Randomised, Double-Blind, Placebo-Controlled, Parallel-Group, Multi-centre, Dose Ranging Study to Evaluate the Efficacy and Safety of Losmapimod Tablets Administered Twice Daily Compared With Placebo for 24 Weeks in Adult Subjects With Chronic Obstructive Pulmonary Disease (COPD)
- Noh JS, Kang HJ, Kim YE, Sohn S, Chung YK, Kim SU, Gwag BJ. Haloperidol-Induced Neuronal Apoptosis: role of p38 and c-Jun-NH(2)-terminal protein kinase. Journal of Neurochemistry 2000, 75(6), 2327. PMID 11080184 doi:10.1046/j.1471-4159.2000.0752327.x
- A Study of GW856553X For the Treatment of Depression
- Cheriyan et al., Circulation 2011, 123(5), 515-523. Inhibition of p38 Mitogen-Activated Protein Kinase Improves Nitric Oxide–Mediated Vasodilatation and Reduces Inflammation in Hypercholesterolemia doi:10.1161/CIRCULATIONAHA.110.971986
- A Study to Evaluate the Safety of 12 Weeks of Dosing With GW856553 and Its Effects on Inflammatory Markers, Infarct Size, and Cardiac Function in Subjects With Myocardial Infarction Without ST-segment Elevation (Solstice)
|
more References |
1: Yang S, Beerahee M. Losmapimod concentration-QT relationship in healthy volunteers: meta-analysis of data from six clinical trials. Eur J Clin Pharmacol. 2013 Jun;69(6):1261-7. doi: 10.1007/s00228-012-1469-1. Epub 2013 Jan 17. PubMed PMID: 23325437.
2: Yang S, Lukey P, Beerahee M, Hoke F. Population pharmacokinetics of losmapimod in healthy subjects and patients with rheumatoid arthritis and chronic obstructive pulmonary diseases. Clin Pharmacokinet. 2013 Mar;52(3):187-98. doi: 10.1007/s40262-012-0025-6. PubMed PMID: 23254770.
3: Dewenter M, Vettel C, El-Armouche A. [Losmapimod: a novel drug against cardiovascular diseases?]. Dtsch Med Wochenschr. 2013 Jan;138(1-2):39-42. doi: 10.1055/s-0032-1327368. Epub 2012 Dec 18. Review. German. PubMed PMID: 23250695.
4: Ostenfeld T, Krishen A, Lai RY, Bullman J, Baines AJ, Green J, Anand P, Kelly M. Analgesic efficacy and safety of the novel p38 MAP kinase inhibitor, losmapimod, in patients with neuropathic pain following peripheral nerve injury: a double-blind, placebo-controlled study. Eur J Pain. 2013 Jul;17(6):844-57. doi: 10.1002/j.1532-2149.2012.00256.x. Epub 2012 Dec 14. PubMed PMID: 23239139.
5: Barbour AM, Sarov-Blat L, Cai G, Fossler MJ, Sprecher DL, Graggaber J, McGeoch AT, Maison J, Cheriyan J. Safety, tolerability, pharmacokinetics and pharmacodynamics of losmapimod following a single intravenous or oral dose in healthy volunteers. Br J Clin Pharmacol. 2013 Jul;76(1):99-106. doi: 10.1111/bcp.12063. PubMed PMID: 23215699; PubMed Central PMCID: PMC3703232.
6: Melloni C, Sprecher DL, Sarov-Blat L, Patel MR, Heitner JF, Hamm CW, Aylward P, Tanguay JF, DeWinter RJ, Marber MS, Lerman A, Hasselblad V, Granger CB, Newby LK. The study of LoSmapimod treatment on inflammation and InfarCtSizE (SOLSTICE): design and rationale. Am Heart J. 2012 Nov;164(5):646-653.e3. doi: 10.1016/j.ahj.2012.07.030. Epub 2012 Oct 16. PubMed PMID: 23137494.
7: Elkhawad M, Rudd JH, Sarov-Blat L, Cai G, Wells R, Davies LC, Collier DJ, Marber MS, Choudhury RP, Fayad ZA, Tawakol A, Gleeson FV, Lepore JJ, Davis B, Willette RN, Wilkinson IB, Sprecher DL, Cheriyan J. Effects of p38 mitogen-activated protein kinase inhibition on vascular and systemic inflammation in patients with atherosclerosis. JACC Cardiovasc Imaging. 2012 Sep;5(9):911-22. doi: 10.1016/j.jcmg.2012.02.016. PubMed PMID: 22974804.
8: Lomas DA, Lipson DA, Miller BE, Willits L, Keene O, Barnacle H, Barnes NC, Tal-Singer R; Losmapimod Study Investigators. An oral inhibitor of p38 MAP kinase reduces plasma fibrinogen in patients with chronic obstructive pulmonary disease. J Clin Pharmacol. 2012 Mar;52(3):416-24. doi: 10.1177/0091270010397050. Epub 2011 Nov 16. PubMed PMID: 22090363.
9: Cheriyan J, Webb AJ, Sarov-Blat L, Elkhawad M, Wallace SM, Mäki-Petäjä KM, Collier DJ, Morgan J, Fang Z, Willette RN, Lepore JJ, Cockcroft JR, Sprecher DL, Wilkinson IB. Inhibition of p38 mitogen-activated protein kinase improves nitric oxide-mediated vasodilatation and reduces inflammation in hypercholesterolemia. Circulation. 2011 Feb 8;123(5):515-23. doi: 10.1161/CIRCULATIONAHA.110.971986. Epub 2011 Jan 24. PubMed PMID: 21262998.
10: Welchman R. Advances and Progress in Drug Design – SMi’s ninth annual meeting. IDrugs. 2010 Apr;13(4):239-42. PubMed PMID: 20373252.
11: Willette RN, Eybye ME, Olzinski AR, Behm DJ, Aiyar N, Maniscalco K, Bentley RG, Coatney RW, Zhao S, Westfall TD, Doe CP. Differential effects of p38 mitogen-activated protein kinase and cyclooxygenase 2 inhibitors in a model of cardiovascular disease. J Pharmacol Exp Ther. 2009 Sep;330(3):964-70. doi: 10.1124/jpet.109.154443. Epub 2009 Jun 25. PubMed PMID: 19556450.
GSK Announces Phase III Cardiovascular Outcomes Study with Losmapimod in Patients with Acute Coronary Syndrome
GlaxoSmithKline plc Thursday 5 June 2014, London UK (LSE/NYSE: GSK) today announced the start of a pivotal phase III study, LATITUDE-TIMI 60, to evaluate the effects of losmapimod in patients presenting with acute coronary syndrome. The global, phase III study will assess whether losmapimod can reduce the risk of a subsequent cardiac event when administered…
AbbVie’S Investigational Oncology Compound ABT-199/GDC-0199, Venetoclax

ABT 199, RG 7601, GDC 0199
Venetoclax
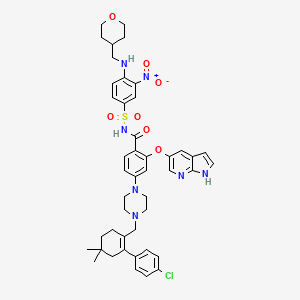
4-(4-{[2-(4-Chlorophenyl)-4,4-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
SYNTHESIS UPDATED BELOW …………..

CAS 1257044-40-8 [RN]
2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)-4-(4-((2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-enyl)methyl)piperazin-1-yl)-N-(3-nitro-4-((tetrahydro-2H-pyran-4-yl)methylamino)phenylsulfonyl)benzamide
4-[4-[[2-(4-chlorophenyl)-4,4-dimethylcyclohexen-1-yl]methyl]piperazin-1-yl]-N-[3-nitro-4-(oxan-4-ylmethylamino)phenyl]sulfonyl-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
ABT 199
- Molecular Formula: C45H50ClN7O7S
- Average mass: 868.439209 Da
- Monoisotopic mass: 867.318115 Da
-
4-(4-{[2-(4-Chlorophenyl)-4,4-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
NORTH CHICAGO, Ill., May 31, 2014/NEWS.GNOM.ES/ — AbbVie (NYSE: ABBV) released interim results from a Phase Ib clinical trial of ABT-199/GDC-0199, an investigational B-cell lymphoma 2 (BCL-2) selective inhibitor, in combination with rituximab (Abstract 7013). Results showed anoverall response rate (ORR) of 84 percent, in patients with relapsed/refractory chronic lymphocytic leukemia(CLL), the most common leukemia in the UnitedStates. These results were presented at the 50thAnnual Meeting of the American Society of ClinicalOncology (ASCO), May 30 – June 3 in Chicago.
ABT-199 is a so-called BH3-mimetic drug, which is designed to block the function of the protein Bcl 2. In 1988, it was discovered that Bcl-2 allowed leukaemia cells to become long-lived, a discovery made at the Walter and Eliza Hall Institute by Professors David Vaux, Suzanne Cory and Jerry Adams. Subsequent research led by them and other institute scientists, including Professors Andreas Strasser, David Huang, Peter Colman and Keith Watson, has explained much about how Bcl-2 and related molecules function to determine if a cell lives or dies. These discoveries have contributed to the development of a new class of drugs called BH3-mimetics that kill, and thereby rapidly remove, leukaemic cells by blocking Bcl-2. (source:http://www.wehi.edu.au).
|
Highlights of recent research using this agent |
GDC-0199 (RG7601) is a novel small molecule Bcl-2 selective inhibitor designed to restore apoptosis, also known as programmed cell death, by blocking the function of a pro-survival Bcl-2 family protein. The Bcl-2 family proteins, which are expressed at high levels in many tumors, play a central role in regulating apoptosis and, consequently, are thought to impact tumor formation, tumor growth and resistance.
Venetoclax (previously: GDC-0199, ABT-199, RG7601 )[1] is a small molecule oral drug being investigated to treat chronic lymphocytic leukemia (CLL).[2][3]
In 2015, the FDA granted Breakthrough Therapy Designation to venetoclax for CLL in previously treated (relapsed/refractory) patients with the 17p deletion genetic mutation.[3]
Mechanism of action
Venetoclax (a BH3-mimetic[4]) acts as a Bcl-2 inhibitor, ie. it blocks the anti-apoptotic B-cell lymphoma-2 (BCL2) protein, leading toprogrammed cell death in CLL cells.[2]
Clinical trials
A phase 1 trial established a dose of 400mg/day.[2]
A trial of venetoclax in combination with rituximab had an encouraging complete response rate.[5]
A phase 2 trial met its primary endpoint which was overall response rate.[3] Interim results from a Phase 2b trial are encouraging, especially in patients with the 17p deletion.[2]
A phase 3 trial (NCT02005471)[1] has started.[3]
NOW IN PHASE 3 UPDATED…………
4-(4-{[2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-en-1-yl]methyl}piperazin-1-yl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide (hereafter, “Compound 1”) is a potent and selective Bcl-2 inhibitor having, inter alia, antitumor activity as an apoptosis-inducing agent. Compound 1 has the formula:

Compound 1 is currently the subject of ongoing clinical trials for the treatment of chronic lymphocytic leukemia. U.S. Patent Publication No. 2010/0305122 describes Compound 1, and other compounds which exhibit potent binding to a Bcl-2 family protein, and pharmaceutically acceptable salts thereof. U.S. Patent Publication Nos. 2012/0108590 and 2012/0277210 describe pharmaceutical compositions comprising such compounds, and methods for the treatment of neoplastic, immune or autoimmune diseases comprising these compounds. U.S. Patent Publication No. 2012/0157470 describes pharmaceutically acceptable salts and crystalline forms of Compound 1. The disclosures of U.S. 2010/0305122; 2012/0108590; 2012/0157470 and 2012/0277210 are hereby incorporated by reference in their entireties.



PATENT
US 2015183783
http://www.google.com/patents/US20150183783
PATENT
CN 104370905
http://www.google.com/patents/CN104370905A?cl=en

ABT-199 is developed AbbVie Bel-2 inhibitors, I trial (NCT01328626) enrolled 84 patients with relapsed type / refractory CLL / SLL patients and 44 cases of relapsing / refractory non-Hodgkin lymphoma patients. ABT-199 treatment response CLL / SLL rate of 79% (complete response rate of 22%), median duration of response time was 20.5 months; ABT-199 treatment of non-Hodgkin’s lymphoma response rate of 48% (complete response rate was 7.5%). The efficacy of ABT-199 is capable of obinutuzumab, idelalisib, ibrutinib rival, is expected to become the first listed Bcl_2 inhibitors, ABT-199 is currently ongoing Phase III clinical study.
ABT-199 compound CAS number 1257044-40-8, the compound is structured as follows:

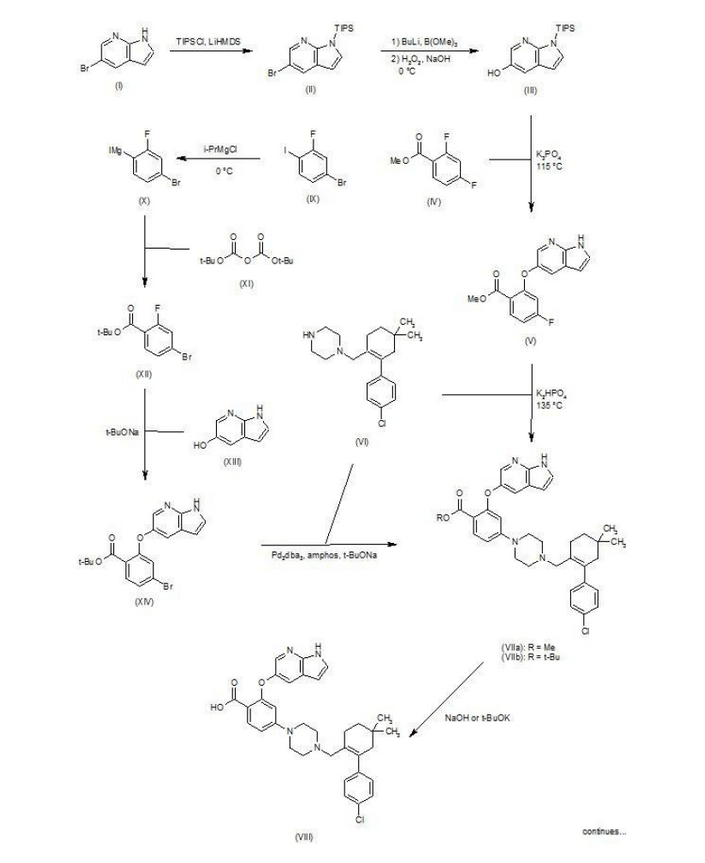
Patent W02012058392, W02012071336, W02010138588 et al. Discloses the preparation of ABT-199 in order to -IH- 5-bromo-pyrrolo [2, 3-b] pyridine as raw material to protect hydroxylation, after replacing the compound 5, and reaction of compound 6, hydrolysis to give compound 9, compound 10 and compound 9 obtained by condensation of ABT-199, a specific line as follows:

use of 2-fluoro-4-nitrobenzoate (A) as a raw material, and substituted 5-hydroxy-7-aza-indole (B), reduction to produce compound ( D), the compound (D) with the compound by cyclization after (H) substitution, hydrolysis to yield compound (J), and then with the compound (K) to afford ABT-199.

Preparation of a compound of Example (F) of the
Example

First step: Synthesis of Compound (C)
2-fluoro-4-nitrobenzoate in IL three-necked flask 50. 0g, dissolved with dimethylformamide N’N- 250ml, was added successively 5-hydroxy-7-aza-indole indole 33. 6g, potassium carbonate 34. 7g, the reaction was heated to 50 degrees under nitrogen gas protection for 2 hours, poured into 2L of ice water was added and extracted three times with ethyl acetate, the organic phase was dried with saturated sodium chloride spin dry to give Compound (C) crude 82. 0g, crude without purification in the next reaction direct investment.
Step two: Synthesis of Compound (D)
The compound of the previous step (C) of the crude product was dissolved in methanol 400ml, was added 10% palladium on carbon 4. 0g, through the reaction of hydrogen at atmospheric pressure, after the end of the reaction by TLC spin solvent to give compound (D) The crude product 73. 2g, crude without purification in the next reaction direct investment.
The third step: Synthesis of compound (F)
Take the previous step the compound (D) crude 20. 0g, t-butanol were added 150ml, compound (E) 10. g, potassium carbonate 9. 7g, completion of the addition the reaction was refluxed for 48 hours the reaction solution was cooled, added acetic acid ethyl ester was diluted, washed with water three times, the combined aqueous phases extracted once with ethyl acetate, the combined ethyl acetate phases twice, dried over anhydrous sodium sulfate and the solvent was spin, the crude product obtained was purified by silica gel column chromatography to give 13. 9g, three-step overall yield of 57.4%.
Preparation Example II Compound (H),

[0029] Take compound (G) (prepared according to W02012058392 method) 5. 0g, dissolved with 50ml of dichloromethane, was added triethylamine 5. 6ml, the reaction solution was cooled to 0-5 ° with stirring, was added dropwise methanesulfonyl chloride 2. 7g, the addition was complete the reaction was warmed to room temperature overnight, after the end of the reaction by TLC the reaction was quenched with water, the organic phase was dried over anhydrous sodium sulfate and the solvent was spin, purified by silica gel column chromatography to give compound (H) 6. 5g , a yield of 99%.
Three ABT-199 Preparation of Example

First step: Synthesis of Compound (I)
In IOOml three-necked flask were added the compound (F) 2. 5g, compound ⑶2. 3g, potassium carbonate I. 9g, Ν ‘was added and reacted at 50 degrees N- dimethylformamide 15ml, nitrogen atmosphere, TLC detection After the reaction, the reaction solution was poured into ice-water, extracted with ethyl acetate twice added ethyl acetate phase was dried over anhydrous sodium sulfate spin, and purified by silica gel column chromatography to give compound (I) 3. 6g, yield 88 %.
Step two: Synthesis of Compound (J)
In IOml single jar Compound (I) I. 0g, followed by adding water 5ml, ethanol 5ml, tetrahydrofuran 5ml, sodium hydroxide 136mg, the reaction was stirred at room temperature the reaction, ethyl acetate was added after dilution of the reaction by TLC, adjusted with IN hydrochloric acid PH4-5, extracted three times with ethyl acetate, dried over anhydrous sodium sulfate and spin dried to give compound (J) 907mg, 93% yield.
Step two: Synthesis of ABT-199
In a 25ml single neck flask was added the compound (J) 100mg, EDCI67mg, dichloromethane 10ml, the reaction was stirred for 30 minutes, was added the compound (K) (prepared in accordance with W02012058392) 55mg, finally added a catalytic amount of DMAP, the force After opening the reaction was stirred overnight, after the end of the reaction by TLC the solvent was spin, HPLC purified preparation obtained by pure ABT-199 ^ 9811, 65% yield.
PATENT
WO 2014165044
http://www.google.com/patents/WO2014165044A1?cl=en
PATENT
US 2014275540
http://www.google.com/patents/US20140275540

-

-
An exemplary reaction according to Scheme 2 is shown below.
 Scheme 3 below. Compound (E) is commercially available or may be prepared by techniques known in the art, e.g., as shown in U.S. Pat. No. 3,813,443 and Proceedings of the Chemical Society, London, 1907, 22, 302.
Scheme 3 below. Compound (E) is commercially available or may be prepared by techniques known in the art, e.g., as shown in U.S. Pat. No. 3,813,443 and Proceedings of the Chemical Society, London, 1907, 22, 302. -
 Scheme 4 below. Compound (M) is commercially available or may be prepared by techniques known in the art, e.g., as shown in GB 585940 and J. Am. Chem. Soc., 1950, 72, 1215-1218.
Scheme 4 below. Compound (M) is commercially available or may be prepared by techniques known in the art, e.g., as shown in GB 585940 and J. Am. Chem. Soc., 1950, 72, 1215-1218. -

-
In another embodiment, the compound of formula (1) is prepared from compound (D) and compound (I) as shown in Scheme 5 below. Compound (J) may be prepared by techniques known in the art, e.g., as shown in WO 2009/117626 and Organometallics, 2008, 27 (21), 5605-5611.
-

 Example 1 Synthesis of tert-butyl 4-bromo-2-fluorobenzoate (Compound (C))To a 100 ml jacketed reactor equipped with a mechanical stirrer was charged 4-bromo-2-fluoro1-iodobenzene, “Compound (A)” (5 g, 1.0 eq) and THF (25 ml). The solution was cooled to −5° C. 2 M isopropyl magnesium chloride in THF (10.8 ml, 1.3 eq) was slowly added maintaining the internal temperature below 0° C. The mixture was stirred at 0° C. for 1 h. Di-tert-butyl dicarbonate (5.44 g, 1.5 eq) in THF (10 ml) was added. After 1 h, the solution was quenched with 10% citric acid (10 ml), and then diluted with 25% NaCl (10 ml). The layers were separated and the organic layer was concentrated to near dryness and chased with THF (3×10 ml). The crude oil was diluted with THF (5 ml), filtered to remove inorganics, and concentrated to dryness. The crude oil (6.1 g, potency=67%, potency adjusted yield=88%) was taken to the next step without further purification. 1H NMR (DMSO-d6): δ 1.53 (s, 9H), 7.50-7.56 (m, 1H), 7.68 (dd, J=10.5, 1.9 Hz, 1H), 7.74 (t, J=8.2 Hz, 1H).
Example 1 Synthesis of tert-butyl 4-bromo-2-fluorobenzoate (Compound (C))To a 100 ml jacketed reactor equipped with a mechanical stirrer was charged 4-bromo-2-fluoro1-iodobenzene, “Compound (A)” (5 g, 1.0 eq) and THF (25 ml). The solution was cooled to −5° C. 2 M isopropyl magnesium chloride in THF (10.8 ml, 1.3 eq) was slowly added maintaining the internal temperature below 0° C. The mixture was stirred at 0° C. for 1 h. Di-tert-butyl dicarbonate (5.44 g, 1.5 eq) in THF (10 ml) was added. After 1 h, the solution was quenched with 10% citric acid (10 ml), and then diluted with 25% NaCl (10 ml). The layers were separated and the organic layer was concentrated to near dryness and chased with THF (3×10 ml). The crude oil was diluted with THF (5 ml), filtered to remove inorganics, and concentrated to dryness. The crude oil (6.1 g, potency=67%, potency adjusted yield=88%) was taken to the next step without further purification. 1H NMR (DMSO-d6): δ 1.53 (s, 9H), 7.50-7.56 (m, 1H), 7.68 (dd, J=10.5, 1.9 Hz, 1H), 7.74 (t, J=8.2 Hz, 1H). -
Example 2 Synthesis of tert-butyl 2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-bromobenzoate (Compound (D))
-
To a 3 L three-neck Morton flask were charged 1H-pyrrolo[2,3-b]pyridin-5-ol (80.0 g, 1.00 eq.), tert-butyl 4-bromo-2-fluorobenzoate (193 g, 1.15 eq.), and anhydrous DMF (800 mL). The mixture was stirred at 20° C. for 15 min. The resulting solution was cooled to about zero to 5° C. A solution of sodium tert-butoxide (62.0 g) in DMF (420 mL) was added slowly over 30 min while maintaining the internal temperature at NMT 10° C., and rinsed with DMF (30 mL). The reaction mixture was stirred at 10° C. for 1 hour (an off-white slurry) and adjusted the internal temperature to ˜45° C. over 30 min. The reaction mixture was stirred at 45-50° C. for 7 hr and the reaction progress monitored by HPLC (IP samples: 92% conversion % by HPLC). The solution was cooled to ˜20° C. The solution was stirred at 20° C. overnight.
-
Water (1200 mL) was added slowly to the reaction mixture at <30° C. over 1 hour (slightly exothermic). The product slurry was adjusted to ˜20° C., and mixed for NLT 2 hours. The crude product was collected by filtration, and washed with water (400 mL). The wet-cake was washed with heptane (400 mL) and dried under vacuum at 50° C. overnight to give the crude product (236.7 g).
-
Re-crystallization or Re-slurry: 230.7 g of the crude product, (potency adjusted: 200.7 g) was charged back to a 3 L three-neck Morton flask. Ethyl acetate (700 mL) was added, and the slurry heated slowly to refluxing temperature over 1 hr (small amount of solids left). Heptane (1400 mL) was added slowly, and the mixture adjusted to refluxing temperature (78° C.). The slurry was mixed at refluxing temperature for 30 min., and cooled down slowly to down to ˜−10° C. at a rate of approximate 10° C./hour), and mixed for 2 hr. The product was collected by filtration, and rinsed with heptane (200 ml).
-
The solid was dried under vacuum at ˜50° C. overnight to give 194.8 g, 86% isolated yield of the product as an off-white solid. MS-ESI 389.0 (M+1); mp: 190-191° C. (uncorrected). 1H NMR (DMSO-d6): δ 1.40 (s, 9H), 6.41 (dd, J=3.4, 1.7 Hz, 1H), 7.06 (d, J=1.8 Hz, 1H), 7.40 (dd, J=8.3, 1.8 Hz, 1H), 7.51 (t, J=3.4 Hz, 1H), 7.58 (d, J=2.6 Hz, 1H), 7.66 (d, J=8.3 Hz, 1H), 8.03 (d, J=2.7 Hz, 1H), 11.72 (s, 1H, NH).
-
Example 3 Synthesis of 2-chloro-4,4-dimethylcyclohexanecarbaldehyde (Compound (F))
-
To a 500 mL RB flask were charged anhydrous DMF (33.4 g, 0.456 mol) and CH2Cl2 (80 mL). The solution was cooled down <−5° C., and POCl3 (64.7 g, 0.422 mol) added slowly over 20 min @<20° C. (exothermic), rinsed with CH2Cl2 (6 mL). The slightly brown solution was adjusted to 20° C. over 30 min, and mixed at 20° C. for 1 hour. The solution was cooled back to <5° C. 3,3-Dimethylcyclohexanone (41.0 g, 90%, ˜0.292 mol) was added, and rinsed with in CH2Cl2 (10 mL) (slightly exothermic) at <20° C. The solution was heated to refluxing temperature, and mixed overnight (21 hours).
-
To a 1000 mL three neck RB flask provided with a mechanical stirrer were charged 130 g of 13.6 wt % sodium acetate trihydrate aqueous solution, 130 g of 12% brine, and 130 mL of CH2Cl2. The mixture was stirred and cooled down to <5° C. The above reaction mixture (clear and brown) was transferred, quenched into it slowly while maintaining the internal temperature <10° C. The reaction vessel was rinsed with CH2Cl2 (10 mL). The quenched reaction mixture was stirred at <10° C. for 15 min. and allowed to rise to 20° C. The mixture was stirred 20° C. for 15 min and allowed to settle for 30 min. (some emulsion). The lower organic phase was separated. The upper aq. phase was back extracted with CH2Cl2 (50 mL). The combined organic was washed with a mixture of 12% brine (150 g)-20% K3PO4 aq. solution (40 g). The organic was dried over MgSO4, filtered and rinsed with CH2Cl2 (30 ml). The filtrate was concentrated to dryness under vacuum to give a brown oil (57.0 g, potency=90.9 wt % by qNMR, ˜100%). 1H NMR (CDCl3): δ 0.98 (s, 6H), 1.43 (t, J=6.4 Hz, 2H), 2.31 (tt, J=6.4, 2.2 Hz, 2H), 2.36 (t, J=2.2 Hz, 2H), 10.19 (s, 1H).
-
Example 4 Synthesis of 2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-enecarbaldehyde (Compound (G))
-
To a 250 mL pressure bottle were charged 2-chloro-4,4-dimethylcyclohex-1-enecarbaldehyde (10.00 g), tetrabutylammonium bromide (18.67 g), and acetonitrile (10 mL). The mixture was stirred at 20° C. for 5 min. 21.0 wt % K2CO3 aq. solution (76.0 g) was added. The mixture was stirred at room temperature (rt) for NLT 5 min. followed by addition of 4-chlorophenylboronic acid (9.53 g) all at once. The mixture was evacuated and purged with N2 for three times. Palladium acetate (66 mg, 0.5 mol %) was added all at once under N2. The reaction mixture was evacuated and purged with N2 for three times (an orange colored mixture). The bottle was back filled with N2 and heated to ˜35° C. in an oil bath (bath temp ˜35° C.). The mixture was stirred at 30° C. overnight (15 hours). The reaction mixture was cooled to RT, and pulled IP sample from the upper organic phase for reaction completion, typically starting material <2% (orange colored mixture). Toluene (100 mL) and 5% NaHCO3-2% L-Cysteine aq. solution (100 mL) were added. The mixture was stirred at 20° C. for 60 min. The mixture was filtered through a pad of Celite to remove black solid, rinsing the flask and pad with toluene (10 mL). The upper organic phase was washed with 5% NaHCO3 aq. solution-2% L-Cysteine (100 mL) once more. The upper organic phase was washed with 25% brine (100 mL). The organic layer (105.0 g) was assayed (118.8 mg/g, 12.47 g product assayed, 87% assayed yield), and concentrated to ˜1/3 volume (˜35 mL). The product solution was directly used in the next step without isolation. However, an analytical sample was obtained by removal of solvent to give a brown oil. 1HNMR (CDCl3): δ 1.00 (s, 6H), 1.49 (t, J=6.6 Hz, 2H), 2.28 (t, J=2.1 Hz, 2H), 2.38 (m, 2H), 7.13 (m, 2H), 7.34 (m, 2H), 9.47 (s, 1H).
-
Example 5 Synthesis of tert-butyl 4-((4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)piperazine-1-carboxylate (Compound (H))
-
To a 2 L three neck RB flask provided with a mechanical stirrer were charged a solution of 4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-carbaldehyde (50.0 g) in toluene (250 mL), BOC-piperazine (48.2 g) and anhydrous THF (250 mL). The yellow solution was stirred at 20° C. for 5 min. Sodium triacetoxyborohydride (52.7 g) was added in portion (note: the internal temperature rose to ˜29.5° C. in 15 min cooling may be needed). The yellow mixture was stirred at ˜25° C. for NLT 4 hrs. A conversion of starting material to product of 99.5% was observed by HPLC after a 3 hour reaction time.
-
12.5 wt % brine (500 g) was added slowly to quench the reaction. The mixture was stirred at 20° C. for NLT 30 min and allowed to settle for NLT 15 min. The lower aq. phase (˜560 mL) was separated (note: leave any emulsion in the upper organic phase). The organic phase was washed with 10% citric acid solution (500 g×2). 500 g of 5% NaHCO3 aq. solution was charged slowly into the flask. The mixture was stirred at 20° C. for NLT 30 min., and allowed to settle for NLT 15 min. The upper organic phase was separated. 500 g of 25% brine aq. solution was charged. The mixture was stirred at 20° C. for NLT 15 min., and allowed to settle for NLT 15 min. The upper organic phase was concentrated to ˜200 mL volume under vacuum. The solution was adjusted to −30° C., and filtered off the inorganic salt. Toluene (50 mL) was used as a rinse. The combined filtrate was concentrated to ˜100 mL volume. Acetonitrile (400 mL) was added, and the mixture heated to ˜80° C. to achieve a clear solution. The solution was cooled down slowly to 20° C. slowly at rate 10° C./hour, and mixed at 20° C. overnight (the product is crystallized out at ˜45-50° C., if needed, seed material may be added at 50° C.). The slurry was continued to cool down slowly to ˜−10° C. at rate of 10° C./hours. The slurry was mixed at ˜−10° C. for NLT 6 hours. The product was collected by filtration, and rinsed with pre-cooled acetonitrile (100 mL). The solid was dried under vacuum at 50° C. overnight (72.0 g, 85%). MS-ESI: 419 (M+1); mp: 109-110° C. (uncorrected); 1H NMR (CDCl3): δ 1.00 (s, 6H), 1.46 (s, 9H), 1.48 (t, J=6.5 Hz, 2H), 2.07 (s, br, 2H), 2.18 (m, 4H), 2.24 (t, J=6.4 Hz, 2H), 2.80 (s, 2H), 3.38 (m, 4H), 6.98 (m, 2H), 7.29 (m, 2H).
-
Example 6 Synthesis of 1-((4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)piperazine dihydrochloride (Compound (I))
-
To a 2.0 L three-neck RB flask equipped with a mechanical stirrer were charged the Boc reductive amination product (Compound (H), 72.0 g) and IPA (720 mL). The mixture was stirred at rt for 5 min, and 59.3 g of concentrated hydrochloride aq. solution added to the slurry. The reaction mixture was adjusted to an internal temperature of ˜65° C. (a clear and colorless solution achieved). The reaction mixture was agitated at ˜65° C. for NLT 12 hours.
-
The product slurry was cooled down to −5° C. slowly (10° C./hour). The product slurry was mixed at ˜−5° C. for NLT 2 hours, collected by filtration. The wet cake was washed with IPA (72 mL) and dried at 50° C. under vacuum overnight to give 73.8 g (95%) of the desired product as a bis-hydrochloride IPA solvate (purity >99.5% peak area at 210 nm). MS-ESI: 319 (M+1); 1HNMR (CDCl3): δ 0.86 (s, 6H), 1.05 (d, J=6.0 Hz, 6H, IPA), 1.42 (t, J=6.1 Hz, 2H), 2.02 (s, br, 2H), 2.12 (m, 2H), 3.23 (m, 4H), 3.4 (s, br, 4H), 3.68 (s, 2H), 3.89 (septet, J=6.0 Hz, 1H, IPA), 7.11 (d, J=8.1 Hz, 2H), 7.41 (d, J=8.1 Hz, 2H).
-
Example 7 Synthesis of 3-nitro-4-(((tetrahydro-2H-pyran-4-yl)methyl)amino)-benzenesulfonamide (Compound (N))
-
To a 500 mL three-neck RB flask equipped with a mechanical stirrer were charged the 4-chloro-3-nitrobenzenesulfonamide, Compound M (10.0 g), diisopropylethylamine (17.5 g), (tetrahydro-2H-pyran-4-yl)methanamine (7.0 g) and acetonitrile (150 mL). The reaction mixture was adjusted to an internal temperature of 80° C. and agitated for no less than 12 hours.
-
The product solution was cooled down to 40° C. and agitated for no less than 1 hour until precipitation observed. The product slurry was further cooled to 20° C. Water (75 mL) was slowly charged over no less than 1 hour, and the mixture cooled to 10° C. and agitated for no less than 2 hours before collected by filtration. The wet cake was washed with 1:1 mix of acetonitrile:water (40 mL). The wet cake was then reslurried in water (80 mL) at 40° C. for no less than 1 hour before collected by filtration. The wet cake was rinsed with water (20 mL), and dried at 75° C. under vacuum to give 12.7 g of the desired product in 99.9% purity and in 91% weight-adjusted yield. 1H NMR (DMSO-d6): δ 1.25 (m, 2H), 1.60 (m, 2H), 1.89 (m, 1H), 3.25 (m, 2H), 3.33 (m, 2H), 3.83 (m, 2H), 7.27 (d, J=9.3 Hz, 1H), 7.32 (s, NH2, 2H), 7.81 (dd, J=9.1, 2.3 Hz, 1H), 8.45 (d, J=2.2 Hz, 1H), 8.54 (t, J=5.9 Hz, 1H, NH).
-
Example 8 Synthesis of tert-butyl 2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-(4-((4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)piperazin-1-yl)benzoate (Compound (K))
-
General Considerations:
-
this chemistry is considered air and moisture sensitive. While the catalyst precursors in their solid, dry form can be handled and stored in air without special precautions, contact with even small amounts of solvent may render them susceptible to decomposition. As a result, traces of oxygen or other competent oxidants (e.g., solvent peroxides) must be removed prior to combination of the catalyst precursors with solvent and care must be used to prevent ingress of oxygen during the reaction. Also, care must be taken to use dry equipment, solvents, and reagents to prevent formation of undesirable byproducts. The sodium t-butoxide used in this reaction is hygroscopic and it should be properly handled and stored prior to or during use.
-
To a 2.0 L three-neck RB flask equipped with a mechanical stirrer were charged the bis-hydrochloride salt (Compound (I), 42.5 g) and toluene (285 ml). 20% K3PO4 (285 ml) was added and the biphasic mixture was stirred for 30 min. The layers were separated and the organic layer was washed with 25% NaCl (145 ml). The organic layer concentrated to 120 g and used in the coupling reaction without further purification.
-
NaOtBu (45.2 g) and Compound (I) in toluene solution (120 g solution −30 g potency adjusted) were combined in THF (180 ml) in a suitable reactor and sparged with nitrogen for NLT 45 min. Pd2dba3 (0.646 g), Compound (J) (0.399 g), and Compound (D) (40.3 g) were combined in a second suitable reactor and purged with nitrogen until oxygen level was NMT 40 ppm. Using nitrogen pressure, the solution containing Compound (I) and NaOtBu in toluene/THF was added through a 0.45 μm inline filter to the second reactor (catalyst, Compound (J) and Compound (D)) and rinsed with nitrogen sparged THF (30 ml).
-
The resulting mixture was heated to 55° C. with stirring for NLT 16 h, then cooled to 22° C. The mixture was diluted with 12% NaCl (300 g) followed by THF (300 ml). The layers were separated.
-
The organic layer was stirred with a freshly prepared solution of L-cysteine (15 g), NaHCO3 (23 g), and water (262 ml). After 1 h the layers were separated.
-
The organic layer was stirred with a second freshly prepared solution of L-cysteine (15 g), NaHCO3 (23 g), and water (262 ml). After 1 h the layers were separated. The organic layer was washed with 12% NaCl (300 g), then filtered through a 0.45 μm inline filter. The filtered solution was concentrated in vacuo to ˜300 mL, and chased three times with heptane (600 mL each) to remove THF.
-
The crude mixture was concentrated to 6 volumes and diluted with cyclohexane (720 ml). The mixture was heated to 75° C., held for 15 min, and then cooled to 65° C. over NLT 15 min. Seed material was charged and the mixture was held at 65° C. for 4 hours. The suspension was cooled to 25° C. over NLT 8 h, then held at 25° C. for 4 hours. The solids were filtered and washed with cyclohexane (90 ml) and dried at 50° C. under vacuum.
-
Isolated 52.5 g (88.9% yield) as a white solid. Melting point (uncorrected) 154-155° C. 1H NMR (DMSO-d6): δ 0.93 (s, 6H), 1.27 (s, 9H), 1.38 (t, J=6.4 Hz, 2H), 1.94 (s, 2H), 2.08-2.28 (m, 6H), 2.74 (s, 2H), 3.02-3.19 (m, 4H), 6.33 (dd, J=3.4, 1.9 Hz, 1H), 6.38 (d, J=2.4 Hz, 1H), 6.72 (dd, J=9.0, 2.4 Hz, 1H), 6.99-7.06 (m, 2H), 7.29 (d, J=2.7 Hz, 1H), 7.30-7.36 (m, 2H), 7.41-7.44 (m, 1H), 7.64 (t, J=6.7 Hz, 1H), 7.94 (d, J=2.7 Hz, 1H), 11.53 (s, 1H).
-
Example 9 Synthesis of 2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-(4-((4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)piperazin-1-yl)benzoic acid (Compound (L))
-
Solution preparation: 10% KH2PO4 (aq): KH2PO4 (6 g) in water (56 g); 2:1 heptane/2-MeTHF:heptane (16 mL) in 2-MeTHF (8 mL).
-
Compound (K) (5.79 g), potassium tert-butoxide (4.89 g), 2-methyltetrahydrofuran (87 mL), and water (0.45 mL) were combined in a suitable reactor under nitrogen and heated to 55° C. until reaction completion. The reaction mixture was cooled to 22° C., washed with the 10% KH2PO4 solution (31 g) twice. The organic layer was then washed with water (30 g).
-
After removal of the aqueous layer, the organic layer was concentrated to 4 volumes (˜19 mL) and heated to no less than 50° C. Heptane (23 ml) was slowly added. The resulting suspension was cooled to 10° C. Solids were then collected by vacuum filtration with recirculation of the liquors and the filter cake washed with 2:1 heptane/2-MeTHF (24 ml). Drying of the solids at 80° C. under vacuum yielded 4.0 g of Compound (L) in approximately 85% weight-adjusted yield. 1H NMR (DMSO-d6): δ 0.91 (s, 6H), 1.37 (t, J=6.4 Hz, 2H), 1.94 (s, br, 2H), 2.15 (m, 6H), 2.71 (s, br, 2H), 3.09 (m, 4H), 6.31 (d, J=2.3 Hz, 1H), 6.34 (dd, J=3.4, 1.9 Hz, 1H), 6.7 (dd, J=9.0, 2.4 Hz, 1H), 7.02 (m, 2H), 7.32 (m, 2H), 7.37 (d, J=2.6 Hz, 1H), 7.44 (t, J=3.0 Hz, 1H), 7.72 (d, J=9.0 Hz, 1H), 7.96 (d, J=2.7 Hz, 1H) & 11.59 (m, 1H).
-
Example 10 Synthesis of 4-(4-{[2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-en-1-yl]methyl}piperazin-1-yl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide (Compound (I))
-
Solution preparation prior to reaction: 10% Acetic Acid:Acetic Acid (37 mL) in water (333 g); 5% NaHCO3:NaHCO3 (9 g) in water (176 g); 5% NaCl:NaCl (9 g) in water (176 g).
-
Compound (N) (13.5 g), DMAP (10.5 g), EDAC (10.7 g) and dichloromethane (300 mL) were combined in a suitable reactor and agitated at 25° C. In a second suitable reactor was charged the Acid (Compound (L), 25 g), Et3N (8.7 g) and dichloromethane (120 mL). The resulting Acid (Compound (L)) solution was slowly charged to the initial suspension of Compound (N) and agitated until reaction completion.


-
N,N-dimethylethylenediamine (9.4 g) was then charged to the reaction mixture with continued agitation. The reaction mixture was warmed to 35° C. and washed with 10% Acetic acid solution (185 mL) twice. The lower organic layer was diluted with more dichloromethane (75 mL) and methanol (12.5 mL). The organic, product layer was then washed with 5% NaHCO3 solution (185 mL) and then washed with 5% NaCl solution (185 mL) at 35° C. The lower, organic layer was separated and then concentrated to 8 vol (˜256 mL) diluted with methanol (26 mL) and warmed to 38° C. Ethyl Acetate (230 mL) was slowly charged. The resulting suspension was slowly cooled to 10° C. and then filtered. The wet cake was washed twice with a 1:1 mix of dichloromethane and ethyl acetate (˜2 vol, 64 mL). After drying the wet cake at 90° C., 32 g (84%) of Compound (I) was isolated.
-
1H NMR (DMSO-d6): δ 0.90 (s, 6H), 1.24 (m, 2H), 1.36 (t, J=6.4 Hz, 2H), 1.60 (m, 2H), 1.87 (m, 1H), 1.93 (s, br, 2H), 2.12 (m, 2H), 2.19 (m, 4H), 2.74 (s, br, 2H), 3.06 (m, 4H), 3.26 (m, 4H), 3.83 (m, 2H), 6.17 (d, J=2.1 Hz, 1H), 6.37 (dd, J=3.4, 1.9 Hz, 1H), 6.66 (dd, J=9.1, 2.2 Hz, 1H), 7.01 (m, 2H), 7.31 (m, 2H), 7.48 (m, 3H), 7.78 (dd, J=9.3, 2.3 Hz, 1H), 8.02 (d, J=2.61 Hz, 1H), 8.54 (d, J=2.33 Hz, 1H), 8.58 (t, J=5.9 Hz, 1H, NH), 11.65 (m, 1H).
PATENT

| Patent | Submitted | Granted |
|---|---|---|
| APOPTOSIS-INDUCING AGENTS FOR THE TREATMENT OF CANCER AND IMMUNE AND AUTOIMMUNE DISEASES [US2014275082] | 2014-02-10 | 2014-09-18 |
| Processes For The Preparation Of An Apoptosis-Inducing Agent [US2014275540] | 2014-03-12 | 2014-09-18 |
| APOPTOSIS INDUCING AGENTS FOR THE TREATMENT OF CANCER AND IMMUNE AND AUTOIMMUNE DISEASES [US2010305122] | 2010-12-02 | |
| Panel of micrornas that silence the MCL-1 gene and sensitize cancer cells to ABT-263 [US8742083] | 2010-12-23 | 2014-06-03 |
| Treatment Of Cancers Using PI3 Kinase Isoform Modulators [US2014377258] | 2014-05-30 | 2014-12-25 |
| METHODS OF TREATMENT USING SELECTIVE BCL-2 INHIBITORS [US2012129853] | 2011-11-22 | 2012-05-24 |
| INHIBITION OF MCL-1 AND/OR BFL-1/A1 [US2015051249] | 2013-03-14 | 2015-02-19 |
| COMBINATION THERAPY OF A TYPE II ANTI-CD20 ANTIBODY WITH A SELECTIVE BCL-2 INHIBITOR [US2014248262] | 2013-09-06 | 2014-09-04 |
References
- New Drugs Online Report for venetoclax
- Hard-to-Treat CLL Yields to Investigational Drug. ASH Dec 2015 refs: Roberts AW, et al “Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia” N Engl J Med 2015; DOI: 10.1056/NEJMoa1513257.
- Phase 2 Study of Venetoclax in Patients with Relapsed/Refractory Chronic Lymphocytic Leukemia with 17p Deletion Meets Primary Endpoint
- ABT-199 BH-3 Mimetic Enters Phase Ia Trial For Chronic Lymphocytic Leukemia. 2011
- For Refractory CLL, Venetoclax’s Complete Response Rate Is Tops. 2015
External links
- ABT-199 inc formula and structure
|
References |
1: Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH, Elmore SW. ABT-199, a potent and selective BCL-2
inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013 Jan 6. doi: 10.1038/nm.3048. [Epub ahead of print] PubMed PMID: 23291630.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-(4-{[2-(4-Chlorophenyl)-4,4-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
|
|
| Identifiers | |
| CAS Number | 1257044-40-8 |
| PubChem | CID: 49846579 |
| ChemSpider | 29315017 |
| Chemical data | |
| Formula | C45H50ClN7O7S |
| Molecular mass | 868.44 g/mol |
/////////
CC1(CCC(=C(C1)c2ccc(cc2)Cl)CN3CCN(CC3)c4ccc(c(c4)Oc5cc6cc[nH]c6nc5)C(=O)NS(=O)(=O)c7ccc(c(c7)[N+](=O)[O-])NCC8CCOCC8)C
OR
CC1(CCC(=C(C1)C2=CC=C(C=C2)Cl)CN3CCN(CC3)C4=CC(=C(C=C4)C(=O)NS(=O)(=O)C5=CC(=C(C=C5)NCC6CCOCC6)[N+](=O)[O-])OC7=CN=C8C(=C7)C=CN8)C
Saracatinib, AZD0530 in phase 3 for Ovary Cancer,

Saracatinib
NCGC00241099, cas 379231-04-6
893428-71-2 (trihydrate)
N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methyl-1-piperazinyl)ethoxy]-5-[(tetrahydro-2H-pyran-4-yl)oxy]-4-quinazolinamine
N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydropyran-4-yloxy)quinazolin-4-amine
4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methyIpiperazin-l-yI)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline
4-(6-chloro-2,3-methylenedioxyanilino)- 7-[2-(4-methylpiperazin-l -yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline
AZD0530
C27H32ClN5O5
542.03
AstraZeneca Pharmaceuticals LP
| Astrazeneca Ab, Astrazeneca Uk Ltd, |
Saracatinib (AZD0530) is a highly selective, orally available, dual-specific Src/Abl kinase inhibitor with IC50 of 2.7 and 30 nM for c-Src and Abl kinase, respectively.Saracatinib (AZD0530) demonstrated potent antimigratory and antiinvasive effects in vitro, and inhibited metastasis in a murine model of bladder cancer. Antiproliferative activity of AZD0530 in vitro varied between cell lines (IC50=0.2 ~10 mM).
c-Src, Bcr–Abl, Yes1, Lck.target
AZD0530 is orally available 5-, 7-substituted anilinoquinazoline with anti-invasive and anti-tumor activities. AZD0530 is a dual-specific inhibitor of Src and Abl, protein tyrosine kinases that are overexpressed in chronic myeloid leukemia cells. This agent binds to and inhibits these tyrosine kinases and their effects on cell motility, cell migration, adhesion, invasion, proliferation, differentiation, and survival. Specifically, AZD0530 inhibits Src kinase-mediated osteoclast bone resorption.
AZD-0530 is a highly selective, dual-specific small molecule Src/Abl kinase inhibitor currently in phase II/III clinical trials at AstraZeneca for the treatment of ovarian cancer. Phase II clinical trials are also under way at the company for the treatment of solid tumors and hematological neoplasms. The Mayo Clinic is developing AZD-0530 in phase II clinical studies for the treatment of metastatic pancreas cancer.
Additional phase II trials are under way at the National Cancer Institute (NCI) for the treatment of colorectal cancer, prostate cancer, breast cancer, lung cancer, stomach cancer, soft tissue sarcoma, stage II or IV melanoma and thymic malignancies. A phase II trial for pancreatic cancer has been suspended. Src and Abl kinase are highly expressed in various human tumor types. No recent development has been reported for research into the treatment of head and neck cancer.
Phase II study of Saracatinib (AZD0530) for for the treatment of patients with hormone receptor-negative metastatic breast cancer : Nine patients were treated on study. After a median of 2 cycles (range 1-3), no patient had achieved CR, PR, or SD >6 months. The median time to treatment failure was 82 days (12-109 days).The majority (89%) of patients discontinued saracatinib because of disease progression. One patient acquired potentially treatment-related grade 4 hypoxia with interstitial infiltrates and was removed from the study. Common adverse events included fatigue, elevated liver enzymes, nausea, hyponatremia, dyspnea, cough, and adrenal insufficiency. CONCLUSIONS: These efficacy results were not sufficiently promising to justify continued accrual to this study. Based on this series, saracatinib does not appear to have significant single-agent activity for the treatment of patients with ER(-)/PR(-) MBC. (source: Clin Breast Cancer. 2011 Oct;11(5):306-11.)
Phase II study of Saracatinib (AZD0530) in patients with metastatic or locally advanced gastric or gastro esophageal junction (GEJ) adenocarcinoma: Saracatinib has insufficient activity as a single agent in patients with advanced gastric adenocarcinoma to warrant further investigation. Further development in gastric cancer would require rational drug combinations or identification of a tumor phenotype sensitive to Src inhibition. (source: Invest New Drugs. 2011 Mar 12. [Epub ahead of print]).
Phase II study of saracatinib (AZD0530) for patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC). Nine patients were enrolled. All patients had received prior radiotherapy and six patients had received prior chemotherapy for recurrent or metastatic disease. The most common adverse event was fatigue. Eight patients had progression of disease by response evaluation criteria in solid tumors (RECIST) within the first eight-week cycle and one patient was removed from the study after 11 days due to clinical decline with stable disease according to the RECIST criteria. Median overall survival was six months. The study was closed early due to lack of efficacy according to the early stopping rule. CONCLUSION: Single-agent saracatinib does not merit further study in recurrent or metastatic HNSCC. (source: Anticancer Res. 2011 Jan;31(1):249-53.)
893428-72-3 Saracatinib difumarate
893428-73-4 also
Saracatinib (AZD0530) is a Src inhibitor for c-Src with IC50 of 2.7 nM.
…………………………….

http://www.google.com/patents/WO2001094341A9?cl=en

………………….
WO 2006064217
http://www.google.fm/patents/EP1871769A2?cl=en
4-(6-chloro-2,3-methylenedioxyanilino)- 7-[2-(4-methylpiperazin-l -yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline which compound is disclosed as Compound No. 73 within the Table in Example 14 of International Patent Application WO 01/94341. That compound is described herein by way of the Formula I

and as AZD0530, the code number by which the compound is known.
AZD0530 is an inhibitor of the Src family of non-receptor tyrosine kinase enzymes and, thereby, is a selective inhibitor of the motility of tumour cells and a selective inhibitor of the dissemination and invasiveness of mammalian cancer cells leading to inhibition of metastatic tumour growth. In particular, the compound AZD0530 is an inhibitor of c-Src non-receptor tyrosine kinase and should be of value as an anti-invasive agent for use in the containment and/or treatment of solid tumour disease in the human or animal body. The route for preparing the compound of the Formula I that is disclosed in International Patent Application WO 01/94341 involves the reaction of the compound 4-(6-chloro-2,3-methylenedioxyanilino)-7-hydroxy-5-tetrahydropyran-4-yloxyquinazoline with an alkylating agent to form the 2-(4-methylpiperazin-l-yl)ethoxy side-chain at the 7-position. The product of the reaction is disclosed in WO 01/94341 in the form of a dihydrochloride salt and in the form of a free base.
Example 14 4-(6-chloro-2,3-methylenedioxyaniIino)-7-[2-(4-methyIpiperazin-l-yI)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline (route 4)
Under an atmosphere of nitrogen gas, l-(2-hydroxyethyl)-4-methylpiperazine (13.93 g) was added to a stirred mixture of 4-(6-chloro-2,3-methylenedioxyanilino)-7-fluoro- 5-tetrahydropyran-4-yloxyquinazoline (12.9 g), sodium te/t-pentoxide (9.87 g) and 1 ,2-diethoxyethane (37.5 ml). Water (1.34 g) and 1,2-diethoxyethane (25 ml) were added and the resultant reaction mixture was stirred and heated to 86°C for 18 hours. The reaction mixture was cooled to 5O0C and, under vacuum distillation at approximately 60 millibar pressure, approximately 50 ml of reaction solvent was distilled off. The reaction mixture was neutralised to pH 7.0 to 7.6 by the addition of a mixture of concentrated aqueous hydrochloric acid (36%, 10 ml) and water (84 ml) at a rate that kept the temperature of the reaction mixture at a maximum of 6O0C. With the temperature of the reaction mixture being kept at 6O0C, the reaction mixture was extracted with ethyl acetate (225 ml). The organic solution was washed with water (50 ml). Water (25 ml) was added and, with the temperature being kept at 6O0C, the mixture was stirred for 10 minutes, then allowed to stand for 30 minutes and the aqueous layer was separated. The organic layer was concentrated to a volume of about 100 ml by distillation of solvent at about 9O0C under atmospheric pressure. The residual mixture was cooled during 1 hour to 450C and held at that temperature for 2 hours to allow crystallisation of product. The mixture was warmed briefly to 550C and then cooled during 4 hours to 180C and held at that temperature for 1 hour. The crystalline precipitate was isolated by filtration and washed in turn with water (17 ml) and with tø’t-butyl methyl ether (17 ml). There was thus obtained 4-(6-chloro-2,3-πiethylenedioxyanilino)-7-[2-(4-methylpiperazin-l-yl)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline as a trihydrate (11 g; 88% purity by HPLC using Method B, retention time 7.3 minutes); NMR Spectrum: (CDCl3) 1.65 (br s, 3H), 1.9-2.05 (m, 2H), 2.2-2.3 (m, 2H), 2.31 (s, 3H), 2.4-2.8 (m, 8H), 2.9 (m, 2H), 3.6-3.7 (m, 2H), 3.95-4.05 (m, 2H), 4.2-4.25 (m, 2H), 4.8 (m,lH), 6.05 (s, 2H), 6.55 (s, IH), 6.75 (d, IH), 6.85 (s, IH), 7.0 (d, IH), 8.55 (s, IH), 9.25 (s, IH).
A portion (10 g) of the material so obtained was placed on a filter and dried at ambient temperature in a stream of dry nitrogen gas. The resultant material was dissolved at 6O0C in dry isopropanol (140 ml) whilst maintaining a dry nitrogen atmosphere. The solution was allowed to cool to ambient temperature and to stand under a dry nitrogen atmosphere for 2 days. The resultant crystalline solid was isolated by filtration under a dry nitrogen atmosphere. The material (8 g) so obtained was a crystalline anhydrous form of 4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methylpiperazin-l -yl)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline, m.p. 142 to 1440C.
Example 15
4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methylpiperazin-l-yl)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline difumarate salt
A mixture of 4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methylpiperazin- l-yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline trihydrate (27.1 g), isopropanol (200 ml) and water (10 ml) was heated to 75°C. A mixture of fumaric acid (12.8 g), isopropanol (200 ml) and water (40 ml) was heated to 😯0C. A portion (80 ml) of the warmed solution of the quinazoline compound was added to the fumaric acid solution whilst the temperature was maintained at 750C. The resultant mixture was stirred at 750C for 75 minutes. The remainder of the quinazoline compound solution was added during 1 hour whilst the temperature was maintained at 750C. Isopropanol (50 ml) was added and the resultant mixture was stirred at 750C for 7 hours. The mixture was cooled slowly over at least 25 minutes to 5O0C and was stirred at that temperature for 6 hours. The mixture was cooled slowly over at least 20 minutes to 2O0C and was stirred at that temperature for 18.5 hours. The crystalline solid was isolated by filtration, washed twice with a 10:1 mixture of isopropanol and water (50 ml and 100 ml respectively) and dried in vacuo at 450C to constant weight. There was thus obtained 4-(6-chloro- 2,3-methylenedioxyanilino)-7-[2-(4-methylρiperazin-l-yl)ethoxy]-5-tetrahydropyran- 4-yloxyquinazoline difumarate salt (37.0 g); m.p. 233-2370C; NMR Spectrum: (DMSOd6) 1.76-1.88 (m, 2H), 2.1-2.17 (m, 2H)5 2.33 (s, 3H), 2.6 (br s, 8H), 2.78 (t, 2H), 3.51-3.6 (m, 2H)3 3.83-3.9 (m, 2H), 4.24 (t, 2H)5 4.98-5.07 (m, IH), 6.07 (s, 2H)3 6.6 (s, 4H)5 6.83 (d5 IH)3 6.84 (d, IH)5 6.91 (d3 IH)5 7.05 (d, IH)3 8.33 (s, IH)3 9.18 (s, IH).
Example 16
4-(6-chloro-2,3-methyIenedioxyaniIino)-7-[2-(4-methyIpiperazin-l-yl)ethoxy]- 5-tetrahydropyran-4-yIoxyquinazolme difumarate salt
A mixture of 4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methylpiperazin- l-yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline trihydrate (27.1 g), isopropanol (210 ml) and water (30 ml) was heated to 4O0C and the mixture was filtered. The filter was washed with isopropanol (20 ml) and the washings were added to the warm filtrate. The resultant solution was warmed to 75°C.
A mixture of fumaric acid (12.8 g), isopropanol (200 ml) and water (20 ml) was heated to 700C and the resultant mixture was filtered. A portion (110 ml) of the fumaric acid solution was added to the warmed solution of 4-(6-chloro-2,3-methylenedioxyanilino)- 7-[2-(4-methylpiperazin- 1 -yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline whilst the temperature was maintained at 75°C. Seed crystals of 4-(6-chloro-
253-methylenedioxyanilino)-7-[2-(4-methylpiperazin-l-yl)ethoxy]-5-tetrahydropyran- 4-yloxyquinazoline difumarate salt (0.02 g) were added and the resultant mixture was stirred at 750C for 1 hour. The remainder of the fumaric acid solution was added during 1 hour whilst the temperature was maintained at 750C and the resultant mixture was stirred at 750C for 14 hours.
The mixture was cooled slowly over at least 2 hours to 200C and was stirred at that temperature for 1 hour. The crystalline solid was isolated by filtration, washed twice with a 10:1 mixture of isopropanol and water (50 ml and 100 ml respectively) and dried in vacuo at 450C to constant weight. There was thus obtained 4-(6-chloro-253-methylenedioxyanilino)- 7-[2-(4-methylpiperazin-l-yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline difumarate salt (35.8 g); m.p. 234-237°C; NMR Spectrum: (DMSOd6) 1.76-1.88 (m, 2H)5 2.1-2.17 (m5 2H)5 2.33 (s5 3H)5 2.6 (br s, 8H), 2.78 (t, 2H), 3.51-3.6 (m, 2H), 3.83-3.9 (m, 2H), 4.24 (t, 2H)5 4.98-5.07 (m, IH), 6.07 (s, 2H)5 6.6 (s, 4H), 6.83 (d, IH)5 6.84 (d, IH)5 6.91 (d, IH)5 7.05 (d, IH)5 8.33 (s5 IH)5 9.18 (s5 IH).
Example 17 4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methyIpiperazin-l-yI)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline sesquifumarate salt
A mixture of 4-(6-chloro-253-methylenedioxyanilino)-7-[2-(4-methylpiperazin- l-yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline difurnarate (0.15 g) and water (20 ml) was warmed using a heat gun to obtain a solution. The sample was allowed to evaporate slowly at ambient temperature to a volume of about 3 ml under a flow of air for 24 hours whereupon a precipitate had started to form. The mixture was placed in a refridgerator at 4°C for 2 days. The resultant precipitate was isolated by filtration and washed with water. There was thus obtained 4-(6-chloro-253-methylenedioxyanilino)-7-[2-(4-methylpiperazin-l-yl)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline as a sesquifumarate tetrahydrate salt (0.084 g) which was characterised using XRPD5 DSC5 TGA5 FTIR and solution NMR techniques.
………………..
A simplified process for the manufacture of AZD0530, a potent SRC kinase inhibitor
Org Process Res Dev 2011, 15(3): 688
http://pubs.acs.org/doi/abs/10.1021/op100161y

Process research and development of a synthetic route towards a novel SRC kinase inhibitor is described. The Medicinal Chemistry route was very long and suffered from extensive use of chlorinated solvents and chromatography. A number of steps in the Medicinal Chemistry route were also unattractive for large-scale use for a variety of reasons. The route was modified to produce a shorter synthetic scheme that started from more readily available materials. By using the modified route, the title compound was manufactured on kilogram scale without recourse to chromatography and in significantly fewer steps. The scaled synthesis required two Mitsunobu couplings, which were developed and scaled successfully. An interesting hydrazine impurity was identified in the second Mitsunobu coupling; a mechanism for its formation is proposed, and a method for its control is described. The formation and control of some other interesting impurities are also described.
N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine Difumarate (AZD0530 Difumarate)
To a slurry of 30 (2.139 kg at 92% w/w, 4.73 mol) and DTAD (2.771 kg, 12.03 mol) in THF (31 L) at ambient temperature in a 100 L vessel was added a solution of triphenylphosphine (3.057 kg, 11.66 mol) in THF (8 L) over 15 min. A THF (2 L) line wash was applied, and the mixture was stirred for 10 min. The reaction mixture was cooled to 15 °C, and a filtered (to remove undissolved particulates) solution of 31 (1.050 L, 1.049 kg, 7.27 mol) in THF …DELETED…………………………….The mixture was filtered and the cake washed with IPA (7 L as a slurry wash and 7 L as a displacement wash) before drying to constant weight under reduced pressure at 50 °C to give AZD0530 difumarate (3.546 kg at 89% w/w, 4.08 mol, 86% yield).
Final Purification of N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine Difumarate (AZD0530 Difumarate)
AZD0530 difumarate (4.234 kg at 89% w/w, 4.87 mol) was refluxed in a mixture of IPA (10.L) and water (10.L, Fresenius). A solution was not obtained, so further IPA (450 mL) and water (450 mL, Fresenius) were added, and the mixture was refluxed. The resulting solution was cooled to 68 °C and screened over 3.5 min through a 20 μm in-line filter into a vessel preheated to 65 °C. IPA(20.4 L) at 65 °C was added via the first vessel and in-line filter, and the resulting solution was stirred at 65 °C for 2 h. Crystallisation was evident after 20 min. The mixture was allowed to self-cool to ambient temperature overnight before filtering and washing the cake with a mixture (prescreened through a 20 μm membrane) of water (640 mL) and IPA (5.76 L). The cake was washed with IPA (6.4 L, prescreened) and MTBE (6.4 L, prescreened) and dried to constant weight under reduced pressure at 50 °C to give AZD0530 difumarate (2.865 kg, at 95.2% w/w, 3.52 mol, 72% yield). Spectroscopic analysis was in agreement with the reported data…………Ford, J. G.; McCabe, J. F.; O’Kearney-McMullan, A.; O’Keefe, P.; Pointon, S. M.; Powell, L.; Purdie, M.; Withnall, J. WO/2006/064217, 2006.
……………………….
SEE…………N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor
J Med Chem 2006, 49(22): 6465

…………..
1: Hannon RA, Finkelman RD, Clack G, Iacona RB, Rimmer M, Gossiel F, Baselga J, Eastell R. Effects of Src kinase inhibition by saracatinib (AZD0530) on bone turnover in advanced malignancy in a Phase I study. Bone. 2012 Jan 8. [Epub ahead of print] PubMed PMID: 22245630.
2: Gucalp A, Sparano JA, Caravelli J, Santamauro J, Patil S, Abbruzzi A, Pellegrino C, Bromberg J, Dang C, Theodoulou M, Massague J, Norton L, Hudis C, Traina TA. Phase II trial of saracatinib (AZD0530), an oral SRC-inhibitor for the treatment of patients with hormone receptor-negative metastatic breast cancer. Clin Breast Cancer. 2011 Oct;11(5):306-11. Epub 2011 May 3. PubMed PMID: 21729667; PubMed Central PMCID: PMC3222913.
3: Mackay HJ, Au HJ, McWhirter E, Alcindor T, Jarvi A, Macalpine K, Wang L, Wright JJ, Oza AM. A phase II trial of the Src kinase inhibitor saracatinib (AZD0530) in patients with metastatic or locally advanced gastric or gastro esophageal junction (GEJ) adenocarcinoma: a trial of the PMH phase II consortium. Invest New Drugs. 2011 Mar 12. [Epub ahead of print] PubMed PMID: 21400081.
4: Fury MG, Baxi S, Shen R, Kelly KW, Lipson BL, Carlson D, Stambuk H, Haque S, Pfister DG. Phase II study of saracatinib (AZD0530) for patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC). Anticancer Res. 2011 Jan;31(1):249-53. PubMed PMID: 21273606.
5: Renouf DJ, Moore MJ, Hedley D, Gill S, Jonker D, Chen E, Walde D, Goel R, Southwood B, Gauthier I, Walsh W, McIntosh L, Seymour L. A phase I/II study of the Src inhibitor saracatinib (AZD0530) in combination with gemcitabine in advanced pancreatic cancer. Invest New Drugs. 2010 Dec 18. [Epub ahead of print] PubMed PMID: 21170669.
6: Dalton RN, Chetty R, Stuart M, Iacona RB, Swaisland A. Effects of the Src inhibitor saracatinib (AZD0530) on renal function in healthy subjects. Anticancer Res. 2010 Jul;30(7):2935-42. PubMed PMID: 20683035.
7: Arcaroli JJ, Touban BM, Tan AC, Varella-Garcia M, Powell RW, Eckhardt SG, Elvin P, Gao D, Messersmith WA. Gene array and fluorescence in situ hybridization biomarkers of activity of saracatinib (AZD0530), a Src inhibitor, in a preclinical model of colorectal cancer. Clin Cancer Res. 2010 Aug 15;16(16):4165-77. Epub 2010 Aug 3. PubMed PMID: 20682712.
8: Morrow CJ, Ghattas M, Smith C, Bönisch H, Bryce RA, Hickinson DM, Green TP, Dive C. Src family kinase inhibitor Saracatinib (AZD0530) impairs oxaliplatin uptake in colorectal cancer cells and blocks organic cation transporters. Cancer Res. 2010 Jul 15;70(14):5931-41. Epub 2010 Jun 15. PubMed PMID: 20551056; PubMed Central PMCID: PMC2906706.
9: Hannon RA, Clack G, Rimmer M, Swaisland A, Lockton JA, Finkelman RD, Eastell R. Effects of the Src kinase inhibitor saracatinib (AZD0530) on bone turnover in healthy men: a randomized, double-blind, placebo-controlled, multiple-ascending-dose phase I trial. J Bone Miner Res. 2010 Mar;25(3):463-71. PubMed PMID: 19775203.
10: Rajeshkumar NV, Tan AC, De Oliveira E, Womack C, Wombwell H, Morgan S, Warren MV, Walker J, Green TP, Jimeno A, Messersmith WA, Hidalgo M. Antitumor effects and biomarkers of activity of AZD0530, a Src inhibitor, in pancreatic cancer. Clin Cancer Res. 2009 Jun 15;15(12):4138-46. Epub 2009 Jun 9. PubMed PMID: 19509160.
11: Chen Y, Guggisberg N, Jorda M, Gonzalez-Angulo A, Hennessy B, Mills GB, Tan CK, Slingerland JM. Combined Src and aromatase inhibition impairs human breast cancer growth in vivo and bypass pathways are activated in AZD0530-resistant tumors. Clin Cancer Res. 2009 May 15;15(10):3396-405. PubMed PMID: 19451593.
12: Lara PN Jr, Longmate J, Evans CP, Quinn DI, Twardowski P, Chatta G, Posadas E, Stadler W, Gandara DR. A phase II trial of the Src-kinase inhibitor AZD0530 in patients with advanced castration-resistant prostate cancer: a California Cancer Consortium study. Anticancer Drugs. 2009 Mar;20(3):179-84. PubMed PMID: 19396016; PubMed Central PMCID: PMC3225398.
13: Green TP, Fennell M, Whittaker R, Curwen J, Jacobs V, Allen J, Logie A, Hargreaves J, Hickinson DM, Wilkinson RW, Elvin P, Boyer B, Carragher N, Plé PA, Bermingham A, Holdgate GA, Ward WH, Hennequin LF, Davies BR, Costello GF. Preclinical anticancer activity of the potent, oral Src inhibitor AZD0530. Mol Oncol. 2009 Jun;3(3):248-61. Epub 2009 Feb 7. PubMed PMID: 19393585.
14: de Vries TJ, Mullender MG, van Duin MA, Semeins CM, James N, Green TP, Everts V, Klein-Nulend J. The Src inhibitor AZD0530 reversibly inhibits the formation and activity of human osteoclasts. Mol Cancer Res. 2009 Apr;7(4):476-88. PubMed PMID: 19372577.
15: Schweppe RE, Kerege AA, French JD, Sharma V, Grzywa RL, Haugen BR. Inhibition of Src with AZD0530 reveals the Src-Focal Adhesion kinase complex as a novel therapeutic target in papillary and anaplastic thyroid cancer. J Clin Endocrinol Metab. 2009 Jun;94(6):2199-203. Epub 2009 Mar 17. PubMed PMID: 19293266; PubMed Central PMCID: PMC2690419.
16: Purnell PR, Mack PC, Tepper CG, Evans CP, Green TP, Gumerlock PH, Lara PN, Gandara DR, Kung HJ, Gautschi O. The Src inhibitor AZD0530 blocks invasion and may act as a radiosensitizer in lung cancer cells. J Thorac Oncol. 2009 Apr;4(4):448-54. PubMed PMID: 19240653; PubMed Central PMCID: PMC2716757.
17: Gwanmesia PM, Romanski A, Schwarz K, Bacic B, Ruthardt M, Ottmann OG. The effect of the dual Src/Abl kinase inhibitor AZD0530 on Philadelphia positive leukaemia cell lines. BMC Cancer. 2009 Feb 13;9:53. PubMed PMID: 19216789; PubMed Central PMCID: PMC2654659.
18: Chang YM, Bai L, Liu S, Yang JC, Kung HJ, Evans CP. Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530. Oncogene. 2008 Oct 23;27(49):6365-75. Epub 2008 Aug 4. PubMed PMID: 18679417.
Src inhibition with saracatinib reverses fulvestrant resistance in ER-positive ovarian cancer models in vitro and in vivo.
Simpkins et al. Clin Cancer Res. 2012 Aug 15. PMID: 22896656.
Saracatinib (AZD0530) is a potent modulator of ABCB1-mediated multidrug resistance in vitro and in vivo.
Liu et al. Int J Cancer. 2012 May 24. PMID: 22623106.
Common PIK3CA mutants and a novel 3′ UTR mutation are associated with increased sensitivity to saracatinib.
Arcaroli et al. Clin Cancer Res. 2012 May 1;18(9):2704-14. PMID: 22553375.
Phase I study of saracatinib (AZD0530) in combination with paclitaxel and/or carboplatin in patients with solid tumours.
Kaye et al. Br J Cancer. 2012 May 22;106(11):1728-34. PMID: 22531637.
Taiho’s Colon Cancer Drug Ups OS in Phase 3

TAS-102 (nonproprietary names: trifluridine and tipiracil hydrochloride)
Taiho’s Colon Cancer Drug Ups OS in Phase 3
Taiho Pharmaceutical Co. Ltd. announced results from its global Phase 3 RECOURSE trial on its oral combination anticancer drug TAS-102 in refractory metastatic colorectal cancer (mCRC). Read more…
FULL STORY
TAS-102 is an anti-cancer drug under development for colorectal cancer.[1]
Clinical trials
A phase II trial reported in 2011[2] and a phase III trial is due to end in 2014.[1][3]
Mechanism
TAS-102 consists of the cytotoxin trifluridine and the thymidine phosphorylase inhibitor (TPI) tipiracil.[4] Trifluridine is incorporated into DNA during DNA synthesis and inhibits tumor cell growth. Tipiracil protects trifluridine from being broken down when taken orally.[1]
References
- “New Drug for Colorectal Cancer Shows Promise in Phase II Trial”. 28 Aug 2012.
- “Novel Drug TAS-102 Makes Headway in Refractory Colorectal Cancer”. 4 Oct 2011.
- “Phase II study of TAS-102 for pretreated metastatic colorectal cancer”. 29 Aug 2012.
- “A novel combination antimetabolite, TAS-102, exhibits antitumor activity in FU-resistant human cancer cells through a mechanism involving FTD incorporation in DNA.”. Sep 2004.

Trifluridine
Trifluridine (also called trifluorothymidine or TFT) is an anti-herpesvirus antiviral drug, used primarily on the eye. It was sold under the trade name, Viroptic, by Glaxo Wellcome, now merged into GlaxoSmithKline. The brand is now owned by Monarch Pharmaceuticals, which is wholly owned by King Pharmaceuticals.
It is a nucleoside analogue, a modified form of deoxyuridine, similar enough to be incorporated into viral DNA replication, but the -CF3 group added to the uracil component blocks base pairing.
It is a component of the experimental anti-cancer drug TAS-102.
A Cochrane Systematic Review showed that trifluridine was a more effective treatment than idoxuridine or vidarabine, significantly increasing the relative number of successfully healed eyes in 14 days.[1]
References
- Wilhelmus KR (2010). “Antiviral treatment and other therapeutic interventions for herpes simplex virus epithelial keratitis”. Cochrane Database Syst Rev 12: CD002898. doi:10.1002/14651858.CD002898.pub4. PMID 21154352.
External links
- Costin D, Dogaru M, Popa A, Cijevschi I (2004). “Trifluridine therapy in herpetic in keratitis”. Rev Med Chir Soc Med Nat Iasi 108 (2): 409–12. PMID 15688823.
- Kuster P, Taravella M, Gelinas M, Stepp P (1998). “Delivery of trifluridine to human cornea and aqueous using collagen shields.”. CLAO J 24 (2): 122–4. PMID 9571274.
- O’Brien W, Taylor J (1991). “Therapeutic response of herpes simplex virus-induced corneal edema to trifluridine in combination with immunosuppressive agents.”. Invest Ophthalmol Vis Sci 32 (9): 2455–61. PMID 1907950.
- “Trifluridine Ophthalmic Solution, 1%” (PDF). Retrieved 2007-03-24.


Fig 4. Open Babel bond-line chemical structure with annotated hydrogens. Click to toggle size.
Spectrum Plot

Fig 5. 1H NMR spectrum of C10H11F3N2O5 in CDCL3 at 400 MHz
Trifluridine
CAS Registry Number: 70-00-8
CAS Name: a,a,a-Trifluorothymidine
Additional Names: 2¢-deoxy-5-(trifluoromethyl)uridine; 5-(trifluoromethyl)-2¢-deoxyuridine; F3TDR
Manufacturers’ Codes: NSC-75520
Trademarks: TFT Thilo (Alcon-Thilo); Virophta (Dulcis); Viroptic (Burroughs Wellcome)
Molecular Formula: C10H11F3N2O5
Molecular Weight: 296.20
Percent Composition: C 40.55%, H 3.74%, F 19.24%, N 9.46%, O 27.01%
Literature References: Prepn: C. Heidelberger et al., J. Am. Chem. Soc. 84, 3597 (1962); eidem, J. Med. Chem. 7, 1 (1964); C. Heidelberger, US 3201387 (1965 to U.S. Dept. HEW). Crystal structure: A. H. Tench, Diss. Abstr. Int. B 33, 3587 (1973). NMR study: R. J. Cushley et al., J. Am. Chem. Soc. 90, 709 (1968). Metabolism: D. L. Dexter et al., Cancer Res. 32, 247 (1972); W. J. O’Brien, H. F. Edelhauser, Invest. Ophthalmol. Visual Sci. 16, 1093 (1977). Pharmacodynamics: B. L. Wigdahl, J. R. Parkhurst,Antimicrob. Agents Chemother. 14, 470 (1978); G. J. Smith et al., Biochem. Biophys. Res. Commun. 83, 1538 (1978). Teratogenicity study: M. Itoi et al., Arch. Ophthalmol. 93, 46 (1975). Cytotoxicity and mutagenicity study: E. Huberman, C. Heidelberger, Mutat. Res. 14, 130 (1972). Clinical studies: H. E. Kaufman, Invest. Ophthalmol. Visual Sci. 17, 941 (1978); R. A. Hyndiuk et al., Arch. Ophthalmol. 96, 1839 (1978). Review of mechanism of antiviral activity: C. Heidelberger, Ann. N.Y. Acad. Sci. 255, 317 (1975). Review of pharmacology and therapeutic use: A. A. Carmine et al., Drugs 23, 329-353 (1982).
Properties: Cryst from ethyl acetate, mp 186-189°. uv max (0.1N HCl): 260 nm (e 9960); (0.1N NaOH): 260 nm (e 6590).
Melting point: mp 186-189°
Absorption maximum: uv max (0.1N HCl): 260 nm (e 9960); (0.1N NaOH): 260 nm (e 6590)
Therap-Cat: Antiviral (ophthalmic).
Keywords: Antiviral; Purines/Pyrimidinones.
………………………………………………………….

Tipiralacil, also known as TPI, is a thymidine phosphorylase inhibitor (TPI). Tipiracil is one of the active components in TAS-102, which is an anticancer drug candidate currently in clinical trials. TAS-102 consists of the cytotoxin Trifluridine and the thymidine phosphorylase inhibitor (TPI) tipiracil. Trifluridine is incorporated into DNA during DNA synthesis and inhibits tumor cell growth. Tipiracil protects trifluridine from being broken down when taken orally.
183204-72-0 (Tipiracil -HCl); 183204-74-2(Tipiracil ).

|
References |
1: Peters GJ, Bijnsdorp IV. TAS-102: more than an antimetabolite. Lancet Oncol. 2012 Dec;13(12):e518-9. doi: 10.1016/S1470-2045(12)70426-6. PubMed PMID: 23182191.
2: Yoshino T, Mizunuma N, Yamazaki K, Nishina T, Komatsu Y, Baba H, Tsuji A, Yamaguchi K, Muro K, Sugimoto N, Tsuji Y, Moriwaki T, Esaki T, Hamada C, Tanase T, Ohtsu A. TAS-102 monotherapy for pretreated metastatic colorectal cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2012 Oct;13(10):993-1001. doi: 10.1016/S1470-2045(12)70345-5. Epub 2012 Aug 28. PubMed PMID: 22951287.
3: Sobrero A. TAS-102 in refractory colorectal cancer: caution is needed. Lancet Oncol. 2012 Oct;13(10):959-61. doi: 10.1016/S1470-2045(12)70376-5. Epub 2012 Aug 28. PubMed PMID: 22951286.
4: Doi T, Ohtsu A, Yoshino T, Boku N, Onozawa Y, Fukutomi A, Hironaka S, Koizumi W, Sasaki T. Phase I study of TAS-102 treatment in Japanese patients with advanced solid tumours. Br J Cancer. 2012 Jul 24;107(3):429-34. doi: 10.1038/bjc.2012.274. Epub 2012 Jun 26. PubMed PMID: 22735906; PubMed Central PMCID: PMC3405214.
5: Suzuki N, Nakagawa F, Nukatsuka M, Fukushima M. Trifluorothymidine exhibits potent antitumor activity via the induction of DNA double-strand breaks. Exp Ther Med. 2011 May;2(3):393-397. Epub 2011 Mar 21. PubMed PMID: 22977515; PubMed Central PMCID: PMC3440718.
6: Shintani M, Urano M, Takakuwa Y, Kuroda M, Kamoshida S. Immunohistochemical characterization of pyrimidine synthetic enzymes, thymidine kinase-1 and thymidylate synthase, in various types of cancer. Oncol Rep. 2010 May;23(5):1345-50. PubMed PMID: 20372850.
7: Temmink OH, Bijnsdorp IV, Prins HJ, Losekoot N, Adema AD, Smid K, Honeywell RJ, Ylstra B, Eijk PP, Fukushima M, Peters GJ. Trifluorothymidine resistance is associated with decreased thymidine kinase and equilibrative nucleoside transporter expression or increased secretory phospholipase A2. Mol Cancer Ther. 2010 Apr;9(4):1047-57. doi: 10.1158/1535-7163.MCT-09-0932. Epub 2010 Apr 6. PubMed PMID: 20371715.
8: Bijnsdorp IV, Kruyt FA, Fukushima M, Smid K, Gokoel S, Peters GJ. Molecular mechanism underlying the synergistic interaction between trifluorothymidine and the epidermal growth factor receptor inhibitor erlotinib in human colorectal cancer cell lines. Cancer Sci. 2010 Feb;101(2):440-7. doi: 10.1111/j.1349-7006.2009.01375.x. Epub 2009 Sep 29. PubMed PMID: 19886911.
9: Bijnsdorp IV, Peters GJ, Temmink OH, Fukushima M, Kruyt FA. Differential activation of cell death and autophagy results in an increased cytotoxic potential for trifluorothymidine compared to 5-fluorouracil in colon cancer cells. Int J Cancer. 2010 May 15;126(10):2457-68. doi: 10.1002/ijc.24943. PubMed PMID: 19816940.
10: Bijnsdorp IV, Kruyt FA, Gokoel S, Fukushima M, Peters GJ. Synergistic interaction between trifluorothymidine and docetaxel is sequence dependent. Cancer Sci. 2008 Nov;99(11):2302-8. doi: 10.1111/j.1349-7006.2008.00963.x. Epub 2008 Oct 18. PubMed PMID: 18957056.
11: Overman MJ, Kopetz S, Varadhachary G, Fukushima M, Kuwata K, Mita A, Wolff RA, Hoff P, Xiong H, Abbruzzese JL. Phase I clinical study of three times a day oral administration of TAS-102 in patients with solid tumors. Cancer Invest. 2008 Oct;26(8):794-9. doi: 10.1080/07357900802087242. PubMed PMID: 18798063.
12: Overman MJ, Varadhachary G, Kopetz S, Thomas MB, Fukushima M, Kuwata K, Mita A, Wolff RA, Hoff PM, Xiong H, Abbruzzese JL. Phase 1 study of TAS-102 administered once daily on a 5-day-per-week schedule in patients with solid tumors. Invest New Drugs. 2008 Oct;26(5):445-54. doi: 10.1007/s10637-008-9142-3. Epub 2008 Jun 5. PubMed PMID: 18528634.
13: Temmink OH, Emura T, de Bruin M, Fukushima M, Peters GJ. Therapeutic potential of the dual-targeted TAS-102 formulation in the treatment of gastrointestinal malignancies. Cancer Sci. 2007 Jun;98(6):779-89. Epub 2007 Apr 18. Review. PubMed PMID: 17441963.
14: Temmink OH, Hoebe EK, van der Born K, Ackland SP, Fukushima M, Peters GJ. Mechanism of trifluorothymidine potentiation of oxaliplatin-induced cytotoxicity to colorectal cancer cells. Br J Cancer. 2007 Jan 29;96(2):231-40. PubMed PMID: 17242697; PubMed Central PMCID: PMC2360012.
Dovitinib in phase 3 for Cancer, bladder (urothelial carcinoma)

Dovitinib
(3E)-4-amino-5-fluoro-3-[5-(4-methylpiperazin-1-yl)-1,3-dihydrobenzimidazol-2-ylidene]quinolin-2-one, is one kind of white crystalline powder, odorless, little bitter taste.
4-Amino-5-fluoro-3-[6-(4-methyl-1-piperazinyl)-1H-benzimidazol-2-yl]-2(1H)-quinolinone 2-hydroxypropanoate
4-Amino-5-fluoro-3-[6-(4-methyl-1-piperazinyl)-1H-benzimidazol-2-yl]-2(1H)-quinolinone 2-hydroxypropanoate hydrate (1:1:1);
| CAS No. | 405169-16-6 (free base), 915769-50-5, 804551-71-1 of lactate | ||
| TKI-258; CHIR-258. | |||
| Formula | C21H21FN6O.C3H6O3.H2O | ||
| Molecular Weight | 500.53 |
for treatment of cancer
Novartis Ag, innovator
Dovitinib lactate is the orally bioavailable lactate salt of a benzimidazole-quinolinone compound with potential antineoplastic activity. Dovitinib strongly binds to fibroblast growth factor receptor 3 (FGFR3) and inhibits its phosphorylation, which may result in the inhibition of tumor cell proliferation and the induction of tumor cell death. In addition, this agent may inhibit other members of the RTK superfamily, including the vascular endothelial growth factor receptor; fibroblast growth factor receptor 1; platelet-derived growth factor receptor type 3; FMS-like tyrosine kinase 3; stem cell factor receptor (c-KIT); and colony-stimulating factor receptor 1; this may result in an additional reduction in cellular proliferation and angiogenesis, and the induction of tumor cell apoptosis. The activation of FGFR3 is associated with cell proliferation and survival in certain cancer cell types
Dovitinib (TKI258) is a highly potent, novel multitargeted growth factor receptor kinase inhibitor with IC50 of 1, 2, 10, 8, 27, 36 nM for FLT3, c-KIT, VEGFR1/2/3, PDGFRß and CSF-1R, respectively. It shows both antitumor and antiangiogenic activities in vivo. [1] It potently inhibits FGFR3 with an inhibitory concentration of 50% (IC50) of 5 nM in in vitro kinase assays and selectively inhibited the growth of B9 cells and human myeloma cell lines expressing wild-type (WT) or activated mutant FGFR3. Antiproliferative activity of Dovitinib (TKI258) against MV4;11 was ~24-fold greater compared with RS4;11, indicating more potent inhibition against cells with constitutively activated FLT3 ITD. [2][3]
References on Dovitinib (TKI258)
- [1] Clin Cancer Res 2005;11:3633-3641
- [2] Blood 2005;105: 2941-2948
- [3] Clin Cancer Res 2005;11:5281-5291
Dovitinib lactate is an angiogenesis inhibitor in phase III clinical trials at Novartis for the treatment of refractory advanced/metastatic renal cell cancer. Early clinical trials are also under way at the company for the oral treatment of several types of solid tumors, multiple myeloma and glioblastoma multiforme. Phase II trials are ongoing for the treatment of castration-resistant prostate cancer and for the treatment of Von-Hippel Lindau disease, for the treatment of non-small cell lung cancer (NSCLC) and for the treatment of colorectal cancer. Novartis and Seoul National University Hospital are conducting phase II clinical studies for the treatment of adenoid cystic carcinoma. Additional phase II clinical trials are ongoing at Asan Medical Center for the treatment of metastatic or advanced gastrointestinal stromal tumors (GIST). The University of Pennsylvania is conducting phase II clinical trials for the treatment of advanced malignant pheochromocytoma or paraganglioma. Phase II clinical studies are ongoing by Novartis for the treatment of advanced malignant pleural mesothelioma which has progressed following prior platinum-antifolate chemotherapy (DOVE-M) and for the oral treatment of hepatocellular carcinoma.
In 2009, Novartis discontinued development of dovitinib lactate for the treatment of acute myeloid leukemia (AML) based on the observation of time dependent drug accumulation. A phase I trial was also stopped for the same reason.
The drug candidate has been shown to inhibit multiple growth factor tyrosine kinases, including vascular endothelial growth factor receptor (VEGFR) tyrosine kinases VEGFR1 and VEGFR2, fibroblast growth factor receptor (FGFR) and platelet-derived growth factor receptor (PDGFR) tyrosine kinases. In previous studies, the benzimidazole-quinoline inhibited VEGF-mediated human microvascular endothelial cell (HMVEC) proliferation and demonstrated concentration-dependent antiangiogenic activity in in vitro assays, as well as potent antiproliferative activity against a subset of cancer cell lines.
In 2013, an orphan drug designation was assigned in the U.S. for the treatment of adenoid cystic carcinoma.
“Molecularly Targeted Agents for Renal Cell Carcinoma: The Next Generation”, C. Lance Cowey and Thomas E. Hutson -Clinical Advances in Hematology & Oncology, 2010, 8, 357.
Lee S. H.; Lopes de Menezes, D. Vora, J. Harris, A.; Ye, H. Nordahl, L.; Garrett, E.; Samara, E.; Aukerman, S. L.; Gelb, A. B. Heise, C. In Vivo Target Modulation and Biological Activity of CHIR-258, a Multitargeted Growth Factor Receptor Kinase Inhibitor, in Colon Cancer Models. Clin. Cancer Res. 2005, 11 (10), 3633–3641.
Lopes de Menezes, D. E.; Peng, J.; Garrett, E. N.; Louie, S. G.; Lee, S. H.; Wiesmann, M.; Tang, Y.; Shephard, L.; Goldbeck, C.; Oei, Y.; Ye, H.; Aukerman, S. L.; Heise, C. CHIR-258: A Potent Inhibitor of FLT3 Kinase in Experimental Tumor Xenograft Models of Human Acute Myelogenous Leukemia. Clin. Cancer Res. 2005, 11 (14), 5281–5291.
Trudel, S.; Li, Z. H.; Wei, E.; Wiesmann, M.; Chang, H.; Chen, C.; Reece, D.; Heise, C.; Stewart, A. K. CHIR-258, a novel, multitargeted tyrosine kinase inhibitor for the potential treatment of t(4;14) multiple myeloma. Blood 2005, 105 (7), 2941–2948.
Synthesis of Dovitinib
Tetrahedron Letters 47 (2006) 657–660
LHMDS mediated tandem acylation–cyclization of 2-aminobenzenecarbonitriles with 2-benzymidazol-2-yl acetates: a short and efficient route to the synthesis of 4-amino-3-benzimidazol-2-ylhydroquinolin-2-ones
William R. Antonios-McCrea, Kelly A. Frazier, Elisa M. Jazan, Timothy D. Machajewski, Christopher M. McBride, Sabina Pecchi, Paul A. Renhowe, Cynthia M. Shafer and Clarke Taylor


cas 852433-84-2
…………………………..
852433-84-2
…………………………..
WO 2002022598 or https://www.google.com/patents/EP1317442A1?cl=en


………………………………
WO 2003087095 or http://www.google.fm/patents/US20030028018?cl=un

…………………..
WO 2005046589 or http://www.google.com/patents/EP1692085A2?cl=en
lactate salt of the compound of
Structure I or the tautomer thereof is administered to the subject and/or is used to prepare the medicament. [0062] In some embodiments, the compound of Structure I has the following formula

……………………
WO 2006125130
http://www.google.com/patents/WO2006125130A1?cl=en
formula IHB

HIB
Scheme 1



Example 4
Synthesis of 4-Amino-5-fluoro-3-[6-(4-rnethyl-piperazin-1 -yl)-1 H-benzimidazol- 2-yl]-1 H-quinolin-2-one Procedure A

[0149] [6-(4-Methyl-piperazin-1-yl)-1 H-benzimidazol-2-yl]-acetic acid ethyl ester (250 g, 820 mmol) (dried with ethanol as described above) was dissolved in THF (3800 ml_) in a 5000 ml_ flask fitted with a condenser, mechanical stirrer, temperature probe, and purged with argon. 2-Amino-6-fluoro-benzonitrile (95.3 g, 700 mmol) was added to the solution, and the internal temperature was raised to 40°C. When all the solids had dissolved and the solution temperature had reached 4O0C, solid KHMDS (376.2 g, 1890 mmol) was added over a period of 5 minutes. When addition of the potassium base was complete, a heterogeneous yellow solution was obtained, and the internal temperature had risen to 62°C. After a period of 60 minutes, the internal temperature decreased back to 40°C, and the reaction was determined to be complete by HPLC (no starting material or uncyclized intermediate was present). The thick reaction mixture was then quenched by pouring it into H2O (6000 ml_) and stirring the resulting mixture until it had reached room temperature. The mixture was then filtered, and the filter pad was washed with water (1000 ml_ 2X). The bright yellow solid was placed in a drying tray and dried in a vacuum oven at 50°C overnight providing 155.3 g (47.9%) of the desired 4-amino-5-fluoro-3-[6-(4-methyl-piperazin-1-yl)-1 H-benzimidazol-2- yl]-1 H-quinolin-2-one.
Procedure B
[0150] A 5000 mL 4-neck jacketed flask was equipped with a distillation apparatus, a temperature probe, a N2 gas inlet, an addition funnel, and a mechanical stirrer. [6-(4-Methyl-piperazin-1-yl)-1 H-benzimidazol-2-yl]-acetic acid ethyl ester (173.0 g, 570 mmol) was charged into the reactor, and the reactor was purged with N2 for 15 minutes. Dry THF (2600 mL) was then charged into the flask with stirring. After all the solid had dissolved, solvent was removed by distillation (vacuum or atmospheric (the higher temperature helps to remove the water) using heat as necessary. After 1000 mL of solvent had been removed, distillation was stopped and the reaction was purged with N2. 1000 mL of dry THF was then added to the reaction vessel, and when all solid was dissolved, distillation (vacuum or atmospheric) was again conducted until another 1000 mL of solvent had been removed. This process of adding dry THF and solvent removal was repeated at least 4 times (on the 4thdistillation, 60% of the solvent is removed instead of just 40% as in the first 3 distillations) after which a 1 mL sample was removed for Karl Fischer analysis to determine water content. If the analysis showed that the sample contained less than 0.20% water, then reaction was continued as described in the next paragraph. However, if the analysis showed more than 0.20% water, then the drying process described above was continued until a water content of less than 0.20% was achieved.
[0151] After a water content of less than or about 0.20% was achieved using the procedure described in the previous paragraph, the distillation apparatus was replaced with a reflux condenser, and the reaction was charged with 2-amino-6- fluoro-benzonitrile (66.2 g, 470 mmol)( in some procedures 0.95 equivalents is used). The reaction was then heated to an internal temperature of 38-420C. When the internal temperature had reached 38-420C, KHMDS solution (1313 g, 1.32 mol, 20% KHMDS in THF) was added to the reaction via the addition funnel over a period of 5 minutes maintaining the internal temperature at about 38-50°C during the addition. When addition of the potassium base was complete, the reaction was stirred for 3.5 to 4.5 hours (in some examples it was stirred for 30 to 60 minutes and the reaction may be complete within that time) while maintaining the internal temperature at from 38-420C. A sample of the reaction was then removed and analyzed by HPLC. If the reaction was not complete, additional KHMDS solution was added to the flask over a period of 5 minutes and the reaction was stirred at 38-420C for 45-60 minutes (the amount of KHMDS solution added was determined by the following: If the IPC ratio is < 3.50, then 125 ml_ was added; if 10.0 >IPC ratio >3.50, then 56 mL was added; if 20.0 ≥IPC ratio >10, then 30 mL was added. The IPC ratio is equal to the area corresponding to 4-amino-5-fluoro-3-[6- (4-methyl-piperazin-1 -yl)-1 H-benzimidazol-2-yl]-1 H-quinolin-2-one) divided by the area corresponding to the uncyclized intermediate). Once the reaction was complete (IPC ratio > 20), the reactor was cooled to an internal temperature of 25- 300C, and water (350 mL) was charged into the reactor over a period of 15 minutes while maintaining the internal temperature at 25-35°C (in one alternative, the reaction is conducted at 400C and water is added within 5 minutes. The quicker quench reduces the amount of impurity that forms over time). The reflux condenser was then replaced with a distillation apparatus and solvent was removed by distillation (vacuum or atmospheric) using heat as required. After 1500 mL of solvent had been removed, distillation was discontinued and the reaction was purged with N2. Water (1660 mL) was then added to the reaction flask while maintaining the internal temperature at 20-300C. The reaction mixture was then stirred at 20-300C for 30 minutes before cooling it to an internal temperature of 5- 100C and then stirring for 1 hour. The resulting suspension was filtered, and the flask and filter cake were washed with water (3 x 650 mL). The solid thus obtained was dried to a constant weight under vacuum at 5O0C in a vacuum oven to provide 103.9 g (42.6% yield) of 4-amino-5-fluoro-3-[6-(4-methyl-piperazin-1-yl)-1H- benzimidazol-2-yl]-1H-quinolin-2-one as a yellow powder.
Procedure C

[0152] [6-(4-Methyl-piperazin-1-yl)-1 H-benzimidazol-2-yl]-acetic acid ethyl ester (608 g, 2.01 mol) (dried) and 2-amino-6-fluoro-benzonitrile (274 g, 2.01 mol) were charged into a 4-neck 12 L flask seated on a heating mantle and fitted with a condenser, mechanical stirrer, gas inlet, and temperature probe. The reaction vessel was purged with N2, and toluene (7.7 L) was charged into the reaction mixture while it was stirred. The reaction vessel was again purged with N2 and maintained under N2. The internal temperature of the mixture was raised until a temperature of 630C (+/- 3°C) was achieved. The internal temperature of the mixture was maintained at 63°C (+/- 30C) while approximately 2.6 L of toluene was distilled from the flask under reduced pressure (380 +/- 10 torr, distilling head t = 40°C (+/- 1O0C) (Karl Fischer analysis was used to check the water content in the mixture. If the water content was greater than 0.03%, then another 2.6 L of toluene was added and distillation was repeated. This process was repeated until a water content of less than 0.03% was achieved). After a water content of less than 0.03% was reached, heating was discontinued, and the reaction was cooled under N2 to an internal temperature of 17-19°C. Potassium t-butoxide in THF (20% in THF; 3.39 kg, 6.04 moles potassium t-butoxide) was then added to the reaction under N2 at a rate such that the internal temperature of the reaction was kept below 20°C. After addition of the potassium t-butoxide was complete, the reaction was stirred at an internal temperature of less than 2O0C for 30 minutes. The temperature was then raised to 25°C, and the reaction was stirred for at least 1 hour. The temperature was then raised to 30°C, and the reaction was stirred for at least 30 minutes. The reaction was then monitored for completion using HPLC to check for consumption of the starting materials (typically in 2-3 hours, both starting materials were consumed (less than 0.5% by area % HPLC)). If the reaction was not complete after 2 hours, another 0.05 equivalents of potassium t-butoxide was added at a time, and the process was completed until HPLC showed that the reaction was complete. After the reaction was complete, 650 mL of water was added to the stirred reaction mixture. The reaction was then warmed to an internal temperature of 50°C and the THF was distilled away (about 3 L by volume) under reduced pressure from the reaction mixture. Water (2.6 L) was then added drop wise to the reaction mixture using an addition funnel. The mixture was then cooled to room temperature and stirred for at least 1 hour. The mixture was then filtered, and the filter cake was washed with water (1.2 L), with 70% ethanol (1.2 L), and with 95% ethanol (1.2 L). The bright yellow solid was placed in a drying tray and dried in a vacuum oven at 50°C until a constant weight was obtained providing 674 g (85.4%) of the desired 4-amino-5-fluoro-3-[6-(4-methyl-piperazin-1-yl)-1 H- benzimidazol-2-yl]-1 H-quinolin-2-one.
Preparation of Lactic Acid salt of 4-Amino-5-fluoro-3-[6-(4-methyl-piperazin-1- yl)-1 H-benzimidazol-2-yl]-1 H-quinolin-2-one

D,L-Lactic Acid

[0154] A 3000 ml_ 4-necked jacketed flask was fitted with a condenser, a temperature probe, a N2 gas inlet, and a mechanical stirrer. The reaction vessel was purged with N2 for at least 15 minutes and then charged with 4-amino-5-fluoro- 3-[6-(4-methyl-piperazin-1-yl)-1 H-benzimidazol-2-yl]-1 H-quinoiin-2-one (484 g, 1.23 mol). A solution of D,L-Lactic acid (243.3 g, 1.72 mol of monomer-see the following paragraph), water (339 mL), and ethanol (1211 mL) was prepared and then charged to the reaction flask. Stirring was initiated at a medium rate, and the reaction was heated to an internal temperature of 68-720C. The internal temperature of the reaction was maintained at 68-72°C for 15-45 minutes and then heating was discontinued. The resulting mixture was filtered through a 10-20 micron frit collecting the filtrate in a 12 L flask. The 12 L flask was equipped with an internal temperature probe, a reflux condenser, an addition funnel, a gas inlet an outlet, and an overhead stirrer. The filtrate was then stirred at a medium rate and heated to reflux (internal temperature of about 780C). While maintaining a gentle reflux, ethanol (3,596 mL) was charged to the flask over a period of about 20 minutes. The reaction flask was then cooled to an internal temperature ranging from about 64-700C within 15-25 minutes and this temperature was maintained for a period of about 30 minutes. The reactor was inspected for crystals. If no crystals were present, then crystals of the lactic acid salt of 4-amino-5-fluoro-3-[6-(4-methyl- piperazin-1-yl)-1 H-benzimidazol-2-yl]-1 H-quinolin-2-one (484 mg, 0.1 mole %) were added to the flask, and the reaction was stirred at 64-7O0C for 30 minutes before again inspecting the flask for crystals.
[0155] Once crystals were present, stirring was reduced to a low rate and the reaction was stirred at 64-700C for an additional 90 minutes. The reaction was then cooled to about 00C over a period of about 2 hours, and the resulting mixture was filtered through a 25-50 micron fritted filter. The reactor was washed with ethanol (484 ml_) and stirred until the internal temperature was about 00C. The cold ethanol was used to wash the filter cake, and this procedure was repeated 2 more times. The collected solid was dried to a constant weight at 500C under vacuum in a vacuum oven yielding 510.7 g (85.7%) of the crystalline yellow lactic acid salt of 4-amino-5-fluoro-3-[6-(4-methyl-piperazin-1 -yl)-1 H-benzimidazol-2-yl]-1 H-quinolin- 2-one. A rubber dam or inert conditions were typically used during the filtration process. While the dry solid did not appear to be very hygroscopic, the wet filter cake tends to pick up water and become sticky. Precautions were taken to avoid prolonged exposure of the wet filter cake to the atmosphere.
[0156] Commercial lactic acid generally contains about 8-12% w/w water, and contains dimers and trimers in addition to the monomeric lactic acid. The mole ratio of lactic acid dimer to monomer is generally about 1.0:4.7. Commercial grade lactic acid may be used in the process described in the preceding paragraph as the monolactate salt preferentially precipitates from the reaction mixture.
[0157] It should be understood that the organic compounds according to the invention may exhibit the phenomenon of tautomerism. As the chemical structures within this specification can only represent one of the possible tautomeric forms at a time, it should be understood that the invention encompasses any tautomeric form of the drawn structure. For example, the compound having the formula NIB is shown below with one tautomer, Tautomer INBa:

HIB

Tautomer HIBa
Other tautomers of the compound having the formula NIB, Tautomer INlBb and Tautomer IHBc, are shown below:

Tautomer IIIBb

Tautomer IIIBc
……………………….
WO 2006127926
……………………..
Design, structure-activity relationships and in vivo characterization of 4-amino-3-benzimidazol-2-ylhydroquinolin-2-ones: A novel class of receptor tyrosine kinase inhibitors
J Med Chem 2009, 52(2): 278
………………………………
WO 2003087095
…………………..
WO 2005046589
……………………
WO 2006125130
……………………….
WO 2006127926
……………………..
Design, structure-activity relationships and in vivo characterization of 4-amino-3-benzimidazol-2-ylhydroquinolin-2-ones: A novel class of receptor tyrosine kinase inhibitors
J Med Chem 2009, 52(2): 278
Ezatiostat……….designed to stimulate the production of blood cells in the bone marrow

Ezatiostat
168682-53-9 (Ezatiostat); 286942-97-0 (Ezatiostat HCl salt)
gamma-Glu-S-BzCys-PhGly diethyl ester

Ezatiostat hydrochloride
Target: glutathione S-transferase P1-1 (GSTP1-1) inhibitor
Pathway: hsa00480 Glutathione metabolism
Activity: Treatment of disorders of bone marrow cellular growth and differentiation
see http://www.ncbi.nlm.nih.gov/pubmed?term=TLK-199&cmd=search
| Telik, Inc. |
innovator
TLK199; TLK-199; TLK 199; Brand name: TELINTRA®。ethyl (2R)-[(4S)-4-amino-5-ethoxy-5-oxopentanoyl]-S-benzyl-L-cysteinyl-2- phenylglycinate.
ethyl (2S)-2-amino-5-[[(2R)-3-benzylsulfanyl-1-[[(1R)-2-ethoxy-2-oxo-1-phenylethyl]amino]-1-oxopropan-2-yl]amino]-5-oxopentanoate.
IUPAC/Chemical name:
(S)-ethyl 2-amino-5-(((R)-3-(benzylthio)-1-(((S)-2-ethoxy-2-oxo-1-phenylethyl)amino)-1-oxopropan-2-yl)amino)-5-oxopentanoate
C27H35N3O6S
Exact Mass: 529.2246
nmr.http://www.medkoo.com/Product-Data/Ezatiostat/ezatiostat-QC-CRB40225web.pdf
Telintra is a small molecule product candidate designed to stimulate the production of blood cells in the bone marrow. Many conditions are characterized by depleted bone marrow, including myelodysplastic syndrome, a form of pre-leukemia in which the bone marrow produces insufficient levels of one or more of the 3 major blood elements (white blood cells, red blood cells and platelets). A reduction in blood cell levels is also a common, toxic effect of many standard chemotherapeutic drugs.
Ezatiostat is a liposomal small-molecule glutathione analog inhibitor of glutathione S-transferase (GST) P1-1 with hematopoiesis-stimulating activity. After intracellular de-esterification, the active form of ezatiostat binds to and inhibits GST P1-1, thereby restoring Jun kinase and MAPK pathway activities and promoting MAPK-mediated cellular proliferation and differentiation pathways. This agent promotes the proliferation and maturation of hematopoietic precursor cells, granulocytes, monocytes, erythrocytes and platelets
Phase II trial myelodysplastic syndrome (MDS): Cancer. 2012 Apr 15;118(8):2138-47.
Phase I trial myelodysplastic syndrome (MDS): J Hematol Oncol. 2012 Apr 30;5:18. doi: 10.1186/1756-8722-5-18; Blood. 2009 Jun 25;113(26):6533-40; J Hematol Oncol. 2009 May 13;2:20.
Ezatiostat hydrochloride is the hydrochloride acid addition salt of ezatiostat. Ezatiostat, also known as TLK199 or TER 199, is a compound of the formula:

Ezatiostat has been shown to induce the differentiation of HL-60 promyelocyte leukemia cells in vitro, to potentiate the activity of cytotoxic agents both in vitro and in vivo, and to stimulate colony formation of all three lineages of hematopoietic progenitor cells in normal human peripheral blood.
In preclinical testing, ezatiostat has been shown to increase white blood cell production in normal animals, as well as in animals in which white blood cells were depleted by treatment with cisplatin or fluorouracil. Similar effects may provide a new approach to treating myelodysplastic syndrome (MDS).
Many conditions, including MDS, a form of pre-leukemia in which the bone marrow produces insufficient levels of one or more of the three major blood elements (white blood cells, red blood cells, and platelets), are characterized by depleted bone marrow. Myelosuppression, which is characterized by a reduction in blood cell levels and in a reduction of new blood cell generation in the bone marrow, is also a common, toxic effect of many standard chemotherapeutic drugs.
Ezatiostat hydrochloride in a liposomal injectable formulation was studied in a clinical trial for the treatment of MDS, and results from this trial, reported by Raza et al., J Hem. One, 2:20 (published online 13 May 2009), demonstrated that administration of TLK199 was well tolerated and resulted in multi-lineage hematologic improvement.
Ezatiostat hydrochloride in a tablet formulation has been evaluated in a clinical trial for the treatment of MDS, as reported by Raza et al., Blood, 113:6533-6540 (prepublished online 27 April 2009) and a single-patient report by Quddus et al., J Hem. One, 3:16 (published online 23 April 2010), and is currently being evaluated in clinical trials for the treatment of MDS and for severe chronic idiopathic neutropenia.
When used for treating humans, it is important that a crystalline therapeutic agent like ezatiostat hydrochloride retains its polymorphic and chemical stability, solubility, and other physicochemical properties over time and among various manufactured batches of the agent. If the physicochemical properties vary with time and among batches, the administration of a therapeutically effective dose becomes problematic and may lead to toxic side effects or to ineffective therapy, particularly if a given polymorph decomposes prior to use, to a less active, inactive, or toxic compound.
Therefore, it is important to choose a form of the crystalline agent that is stable, is manufactured reproducibly, and has physicochemical properties favorable for its use as a therapeutic agent.
Ezatiostat hydrochloride (USAN) has the molecular weight of 566.1, the trademark of Telintra®, and the CAS registry number of 286942-97-0. Ezatiostat hydrochloride has been evaluated for the treatment of myelodysplastic syndrome (MDS), in a Phase I-IIa study using a liposomal formulation (U.S. Pat. No. 7,029,695), as reported at the 2005 Annual Meeting of the American Society for Hematology (Abstract #2250) and by Raza et al. in Journal of Hematology & Oncology, 2:20 (published online on 13 May 2009); and in a Phase I study using a tablet formulation, as reported at the 2007 Annual Meeting of the American Society for Hematology (Abstract #1454) and by Raza et al. in Blood, 113:6533-6540 (prepublished online on 27 Apr. 2009), and in a single patient case report by Quddus et al. in Journal of Hematology & Oncology, 3:16 (published online on 23 Apr. 2010).
…………………………………………………………………
http://www.google.com/patents/US20110301376
Preparation of Ezatiostat Hydrochloride
In another aspect, this invention provides a process comprising the steps of contacting a compound of formula:

or a salt thereof with a compound of formula:

or a salt thereof and an activating agent under conditions which provide a compound of formula:

In one embodiment, the process further comprises deprotecting the compound of formula:

under conditions which provide a compound of formula:

or a salt thereof. In another embodiment, the compound provided is ezatiostat hydrochloride.
In another aspect, this invention provides a process comprising contacting a compound of formula:

or a salt thereof with an ethylating agent under conditions which provide a compound of formula:

In another embodiment, the process further comprises debenzylating the compound of formula:

under conditions which provide a compound of formula:

or a salt thereof.
In another aspect, this invention provides a process comprising the steps of contacting a compound of formula:

or a salt thereof having a t-butoxycarbonyl group with an activating agent and a compound of formula:

or a salt thereof under conditions which provide a compound of formula:

In another embodiment, the process further comprises deprotecting the tertiarybutyloxycarboyl (Boc) group under conditions to provide a compound of formula:

or a salt thereof.
Certain preferred embodiments of this invention are illustrated in the reaction scheme and described below. In the peptide coupling the amino acid reagents are used generally at a 1:1 molar ratio, and the activating reagent (isobutyl chloroformate) and the base (NMM) are used in slight excess over the amino acid reagents; while in the esterification of the N-BOC-L-glutamic acid γ-benzyl ester the esterifying agent (diethyl sulfate) and base are used in about 1.4-fold excess.

EXAMPLESAs relevant and unless otherwise noted, all operations were conducted under nitrogen purge and with stirring. Water was osmosis purified, and solvents were filtered. Unless otherwise stated, all temperatures are in degrees Celcius (° C.) and the following abbreviations have the following definitions:Et EthylHCl(g) HCl gasN-BOC or N-Boc N-tertiarybutyloxycarbonylL LiterKg KilogramNMM N-methylmorpholineMol Molew/w weight by weightExample 1Preparation of S-benzyl-L-cysteinyl-D-phenylglycine ethyl ester hydrochloride (3)Without stirring, 45.1 Kg N-BOC-S-benzyl-L-cysteine (1) was added to a 600 L jacketed glass-lined reactor, followed by 45 L ethyl acetate. Stirring was started and the temperature was reduced to 13° C. NMM, 15.3 Kg, was added over 50 minutes, and rinsed in with 6 L ethyl acetate, and stirring stopped. Ethyl acetate, 315 L, was added to an 800 L cooled jacketed glass-lined reactor, followed by 20.7 Kg isobutyl chloroformate, rinsed in with 11 L ethyl acetate, and the mixture cooled to −10° C. The N-BOC-S-benzyl-L-cysteine NMM salt solution was added to the 800 L reactor over 5 hours, its reactor rinsed with 11 L ethyl acetate, and the rinse solution added to the 800 L reactor, while maintaining the temperature at (−10˜−7)° C. D-Phenylglycine ethyl ester hydrochloride, 31.2 Kg, was added in 8 portions over 50 minutes, followed by 15.3 Kg NMM in 8 portions over 1.3 hours, rinsed in with 2×5 L portions of ethyl acetate, allowing the mixture to warm to −1° C. by the end of the addition. The mixture was gradually warmed to 1° C. for 30 minutes, then to 20° C. over 2 hours, and maintained at (20˜25)° C. for 5 hours. The reaction mixture was washed twice with water: the first time adding 66 L water, stirring at room temperature for 40 minutes, allowing the phases to separate for 30 minutes, then removing the aqueous phase; the second time adding 68 L water, bringing the pH to 1.9 with the addition of 0.45 L 36% hydrochloric acid, stirring at room temperature for 35 minutes, allowing the phases to separate for 1 hour, then removing the aqueous phase. The organic phase was then heated to 38° C., and the pressure reduced to about 0.25 bar until no further gas was released, then to about (0.07-0.1) bar and solvents removed by distillation until 266 L of distillate had been removed. Four cycles of addition of 45 L ethyl acetate and removal of 45 L solvent by distillation were performed, and the water content of the remaining mixture was checked to ensure that it was below 0.1%. With the mixture at 36° C., 194 L heptanes was added, maintaining the temperature about 36° C., and held at that temperature for 2.3 hours. A further 194 L heptanes was added, allowing the temperature to cool to 30° C., and the temperature then reduced to −1° C. over 2.3 hours and then to −5° C. over 1 hour, and N-BOC-S-benzyl-L-cysteinyl-D-phenylglycine ethyl ester recovered by filtration, washing twice with 30 L each of heptanes at −5° C., giving 85 Kg (63 Kg dry basis) N-BOC-S-benzyl-L-cysteinyl-D-phenylglycine ethyl ester. Without stirring, the damp N-BOC-S-benzyl-L-cysteinyl-D-phenylglycine ethyl ester was loaded into an 800 L jacketed glass-lined reactor, followed by 257 L ethyl acetate. Stirring was started and the temperature brought to 22° C., then the nitrogen purge stopped and 12.2 Kg hydrogen chloride gas was added through an immersion tube over 1.8 hours, allowing the temperature to increase to 38° C. The temperature was increased to 41° C., and the mixture held at that temperature for 9 hours. About 280 L of solvents were removed by distillation at that temperature and a pressure of (0.2˜0.1) bar over about 2 hours. Two cycles of addition of ethyl acetate and removal of solvent by distillation were performed, using 52 L in the first cycle and 77 L in the second cycle, and the viscous solution of S-benzyl-L-cysteinyl-D-phenylglycine ethyl ester hydrochloride (3) in ethyl acetate, 148 Kg, was cooled to room temperature and filtered into a storage drum.Example 2Preparation of N-BOC-L-glutamic acid α-ethyl ester (6)Without stirring, 41 Kg N-BOC-L-glutamic acid γ-benzyl ester (4) was added to an 800 L jacketed glass-lined reactor, followed by 2.5 L water and 123 L ethyl acetate. The mixture was then stirred until the N-BOC-L-glutamic acid γ-benzyl ester completely dissolved, keeping the temperature below 15° C. Potassium carbonate fine powder, 23.4 Kg, was added in five batches, and the mixture then heated to 55° C. and maintained at that temperature for 40 minutes, giving a heterogeneous and completely fluid mixture. Diethyl sulfate, 26.2 Kg, was added over 2 hours, and rinsed in with 5 L ethyl acetate, with the temperature remaining at about 52° C. The nitrogen purge was stopped and a solution of 20 Kg ammonium chloride in 73 L water at room temperature added over 2 hours to the mixture, maintaining the temperature near 50° C., then rinsing in with 10 L water. Nitrogen purging was resumed, and the mixture was maintained at about 50° C. for 3 hours, then lowered to about 45° C., the stirring stopped, the phases allowed to separate for 30 minutes, and the lower, aqueous, phase removed. The organic phase, containing N-BOC-L-glutamic acid γ-benzyl α-ethyl ester (5), was washed three times with water, each time adding 41 L water, stirring at room temperature for 30 minutes, allowing the phases to separate for 30 minutes, then removing the aqueous phase. The organic phase was heated to 35° C., and the pressure reduced, starting at about 0.2 bar and reducing as necessary until 82 Kg solvent had been removed by distillation, leaving about (70˜80) L of slightly opalescent solution. This solution was heated to 53° C., and 102 L heptanes was added, maintaining the same temperature. The solution was then filtered, rinsing with a further 13 L heptanes, then cooled to 32° C. to cause crystallization and maintained at that temperature for 1 hour. A further 66 L heptanes was added, and the mixture cooled to 22° C. and held for 1 hour, then cooled to −5° C. and held for another 1 hour. The mixture was then filtered to isolate the N-BOC-L-glutamic acid γ-benzyl α-ethyl ester (5), which was washed twice, each time with 25 L heptanes cooled to (−5˜0)° C., and dried under vacuum at 40° C., giving 39.3 Kg N-BOC-L-glutamic acid γ-benzyl α-ethyl ester (5).A 4000 L hydrogenator was purged with nitrogen, then under nitrogen sweep and no stirring loaded with 39.2 Kg N-BOC-L-glutamic acid γ-benzyl α-ethyl ester (5), 2.0 Kg 5% palladium on carbon, and 432 L ethyl acetate, and purged (3 bar) and decompressed (0.2 bar) twice with nitrogen and twice with hydrogen. Stirring was begun and the mixture heated to (37±2)° C., hydrogenated at that temperature under 2.8 bar hydrogen pressure until no further hydrogen absorption occurred, then held under 2.8 bar hydrogen pressure for 12 hours. Completion of hydrogenation was confirmed by thin-layer chromatography of a sample. The mixture was cooled to 28° C., the hydrogen purged from the hydrogenator, and the hydrogenator purged (2 bar) and decompressed (0.2 bar) twice with nitrogen. The mixture was filtered through a filter precoated with 10 Kg powdered cellulose in 200 L ethyl acetate, then the filter washed with the ethyl acetate used to form the precoat, giving a total of 626 Kg of a dilute ethyl acetate solution containing 29.5 Kg N-BOC-L-glutamic acid α-ethyl ester (6). This was distilled at (35˜40)° C. and (0.16˜0.18) bar to give 67 L of concentrated solution, then 29 L of ethyl acetate added and the solution redistilled to again give 67 L of concentrated solution.Example 3Preparation of Ezatiostat HydrochlorideThe concentrated solution of N-BOC-L-glutamic acid α-ethyl ester (6), 61.2 Kg (containing 27.8 Kg N-BOC-L-glutamic acid α-ethyl ester), was added to a 600 L jacketed glass-lined reactor, rinsed in with 5 L ethyl acetate, then cooled to 14° C. NMM, 10.8 Kg, was added over 50 minutes and rinsed in with 5 L ethyl acetate, then stirring stopped, giving an ethyl acetate solution of N-BOC-L-glutamic acid α-ethyl ester NMM salt. Ethyl acetate, 475 L, was added to a 1300 L cooled jacketed glass-lined reactor, followed by 14.5 Kg isobutyl chloroformate, rinsed in with 2×10 L ethyl acetate, and the mixture cooled to −11° C. The N-BOC-L-glutamic acid α-ethyl ester NMM salt solution was added to the 1300 L reactor over 1.3 hours, its reactor rinsed with 10 L ethyl acetate, and the rinse solution added to the 1300 L reactor, then stirred for an additional 30 minutes, while maintaining the temperature at about −13° C.S-benzyl-L-cysteinyl-D-phenylglycine ethyl ester hydrochloride (3) in ethyl acetate, 112 Kg (containing 41.3 Kg S-benzyl-L-cysteinyl-D-phenylglycine ethyl ester hydrochloride) was added in 4 portions over 45 minutes, and rinsed in with 5 L ethyl acetate, followed by 10.8 Kg NMM in 8 portions over 1.3 hours, rinsed in with 2×5 L portions of ethyl acetate, allowing the mixture to warm to −4° C. by the end of the addition. The mixture was gradually warmed to 30° C. over 2 hours, and maintained at (30˜35)° C. for 2 hours. The reaction mixture was washed twice with water: the first time adding 100 L water, heating to 41° C., allowing the phases to separate for 30 minutes, then removing the aqueous phase; the second time adding 100 L water, bringing the pH to 2.0 with the addition of 0.8 L 36% hydrochloric acid, stirring at 43° C. for 30 minutes, allowing the phases to separate for 1 hour, then removing the aqueous phase. The organic phase was then heated to 42° C., and the pressure reduced to about 0.25 bar until no further gas was released and solvents removed by distillation until 495 L of distillate had been removed. Four cycles of addition of 120 L ethyl acetate and removal of 120 L solvent by distillation were performed, and the water content of the remaining mixture was checked to ensure that it was below 0.1%. With the mixture at 42° C., 610 L of ethyl acetate was added, maintaining the temperature about 41° C., then heating to 58° C. to ensure dissolution. The solution was filtered, rinsing the filter with 18 L ethyl acetate, and the solution allowed to cool to 22° C. The nitrogen purge was stopped and 22.2 Kg hydrogen chloride gas was added through an immersion tube over 2 hours, then the mixture held at that temperature for 2 hours. The mixture was heated to 31° C. over 1.5 hours, and held at about that temperature for 15.5 hours. Solvents were removed by distillation at 33° C. and a pressure of about 0.13 bar over about 1.5 hours to give a volume of concentrated solution of about 630 L. Ethyl acetate, 100 L, was added, and the mixture cooled to 25° C. and held at that temperature for 30 minutes. The crude ezatiostat hydrochloride was recovered by filtration and washed with 30 L ethyl acetate, giving 113 Kg damp crude ezatiostat hydrochloride, which was dried at 40° C. under vacuum for 24 hours to give 52.8 Kg dry crude ezatiostat hydrochloride.Example 4Crystallization of Ezatiostat Hydrochloride to Form Pure Crystalline Ezatiostat Hydrochloride Ansolvate Form D61.5 Kg crude ezatiostat hydrochloride was added to a reactor at room temperature, followed by 399 liter (L) ethanol, and this mixture was heated to 68° C. to completely dissolve the ezatiostat hydrochloride, filtered, then allowed to cool to 65° C. and checked for clarity and the absence of crystallization. About 1.3 Kg of ezatiostat hydrochloride ansolvate form D was suspended in 9 L of ethyl acetate, and about one-half of this suspension was added to the ethanol solution. The mixture was cooled to 63° C. and the second half of the suspension added to the mixture. The resulting mixture was cooled gradually to 45° C., 928 L ethyl acetate was added, and the mixture was cooled to 26° C. and held at about that temperature for about 5 hours, then cooled to −2° C. The mixture, containing crystalline ezatiostat hydrochloride ansolvate, was filtered, and the residue washed twice with 65 L of chilled (0-5° C.) ethyl acetate. The crystalline ezatiostat hydrochloride ansolvate was dried at 30° C. for 48 hours, then cooled to room temperature and sieved. Analysis of the material by DSC and XRPD confirmed its identity as crystalline ezatiostat hydrochloride ansolvate, and Karl Fischer analysis showed a water content of 0.1%.Example 5Purifying Ezatiostat Hydrochloride Crystals to Form Pure Crystalline Ezatiostat Hydrochloride Ansolvate Form DCrude ezatiostat hydrochloride, 51.4 Kg, was added to a 600 L jacketed glass-lined reactor at room temperature, followed by 334 L of ethanol. The mixture was heated to 68° C. to completely dissolve the ezatiostat hydrochloride. The resulting solution was filtered into a 1300 L jacketed glass-lined reactor, and an additional 27 L ethanol warmed to 66° C. used to rinse the first reactor into the second reactor through the filter. The resulting solution in the second reactor was cooled to 63° C. and checked for complete dissolution; then 4 L of a seeding suspension of crystalline ezatiostat hydrochloride ansolvate in ethyl acetate was added, and the mixture cooled to 60° C. The remaining 4 L of the seeding suspension was added, and the mixture cooled to 47° C. over 2 hours. The solids in the mixture were shown by DSC to contain more than one form of ezatiostat hydrochloride, so the stages of heating to dissolution, cooling, and adding seeding suspension (this time 2×2 L), were repeated, then the mixture cooled to 41° C. This time the solids in the mixture were confirmed by DSC to be crystalline ezatiostat hydrochloride ansolvate. Ethyl acetate, 776 L, was added, and the mixture was cooled to 25° C. over 1.3 hours and further to 20° C. over an additional 5 hours, then cooled to −3° C. The mixture, containing crystalline ezatiostat hydrochloride ansolvate, was filtered and the solids washed twice with 54 L each of chilled (−5˜0)° C. ethyl acetate. The damp solids of crystalline ezatiostat hydrochloride ansolvate, 70 Kg, were dried in a vacuum oven at 25° C. for 16 hours, 35° C. for 7 hours, then at room temperature for 1 hour, then sieved. The crystalline ezatiostat hydrochloride ansolvate, 44.2 Kg, had a loss on drying at 40° C. under vacuum for 2 hours of 0.09%, and a water content by Karl Fischer analysis of 0.09%.
……………………………………………………………
U.S. Pat. No. 5,763,570
https://www.google.com/patents/US5763570
……………………………..
http://www.google.com/patents/WO2011156025A1?cl=en
Example 1. Preparation Of Ezatiostat Hydrochloride Ansolvate By Slurrying
[0082] Ezatiostat hydrochloride monohydrate was added to methyl tert-butyl ether at room temperature in excess, so that undissolved solids were present. The mixture was then agitated in a sealed vial at room temperature for 4 days, and the solids were then isolated by suction filtration. XRPD analysis of the solids established that the isolated solids were ezatiostat hydrochloride ansolvate.
[0083] Ezatiostat hydrochloride monohydrate was added to hexanes at 60 °C in excess, so that undissolved solids were present. The mixture was then agitated in a sealed vial at 60 °C for 4 days, and the solids were then isolated by suction filtration. XRPD analysis of the solids established that the isolated solids were ezatiostat hydrochloride ansolvate.
Example 2. Preparation Of Crystalline Ezatiostat Hydrochloride Ansolvate By Heating
[0084] DSC of crystalline ezatiostat hydrochloride monohydrate showed the pattern in FIG. 1, as discussed in paragraph above. Hot stage microscopy showed an initial melt followed by a recrystallization at 153 °C and a final melt at 166 °C. VT-XRPD, where XRPD patterns were obtained at 28 °C, 90 °C, and 160 °C during heating, and 28 °C after cooling of the formerly heated material, showed the presence of ezatiostat hydrochloride monohydrate at 28 °C and 90 °C during heating and of crystalline ezatiostat hydrochloride ansolvate at 160 °C and 28 °C after cooling of the formerly heated material. This confirmed that the transition at around 153/156 °C was a conversion of ezatiostat hydrochloride monohydrate form A to crystalline ezatiostat hydrochloride ansolvate form D and that the final DSC endothermic peak at about 177 °C (166 °C in the hot stage microscopy) was due to the melting of crystalline ezatiostat hydrochloride ansolvate. This was further confirmed by XRPD of the TG-IR material, where XRPD patterns obtained at room temperature both before and after heating to about 160 °C showed that the material before heating was form A and that the material after heating was form D ansolvate. DSC of crystalline ezatiostat hydrochloride ansolvate prepared by recrystallization showed the pattern in FIG. 5, with only the endothermic peak at about
177 °C followed by a broad endotherm at about (205 – 215) °C. Accordingly, the presence of the DSC endothermic peak at about 177 °C, for example at (177±2) °C, when measured under the conditions described above, is considered characteristic of crystalline ezatiostat hydrochloride ansolvate, and the substantial absence of thermal events at temperatures below this is considered indicative of the absence of other forms of ezatiostat hydrochloride
………………………………..
http://www.google.com/patents/WO2013082462A1?cl=en
Ezatiostat, also known as TLK199 or TER 199, is a compound of the formula:

[0003] Ezatiostat has been shown to induce the differentiation of HL-60 promyelocytic leukemia cells in vitro, to potentiate the activity of cytotoxic agents both in vitro and in vivo, and to stimulate colony formation of all three lineages of hematopoietic progenitor cells in normal human peripheral blood. In preclinical testing, ezatiostat has been shown to increase white blood cell production in normal animals, as well as in animals in which white blood cells were depleted by treatment with cisplatin or fluorouracil. Similar effects may provide a new approach to treating myelodysplasia syndrome (MDS).
[0004] Many conditions, including MDS, a form of pre-leukemia in which the bone marrow produces insufficient levels of one or more of the three major blood elements (white blood cells, red blood cells, and platelets), are characterized by depleted bone marrow.
Myelosuppression, which is characterized by a reduction in blood cell levels and in a reduction of new blood cell generation in the bone marrow, is also a common, toxic effect of many standard chemotherapeutic drugs.
[0005] Ezatiostat hydrochloride is the hydrochloride acid addition salt of ezatiostat.
Ezatiostat hydrochloride in a liposomal injectable formulation was studied in a clinical trial for the treatment of MDS, and results from this trial, reported by Raza et al., J. Hem. One, 2:20 (published online 13 May 2009), demonstrated that administration of TLK199 was well tolerated and resulted in multi-lineage hematologic improvement. Ezatiostat hydrochloride in a tablet formulation has been evaluated in a clinical trial for the treatment of MDS, as reported by Raza et al, Blood, 113:6533-6540 (prepublished online 27 April 2009) and a single-patient report by Quddus et al, J. Hem. One, 3:16 (published online 23 April 2010), and is currently being evaluated in clinical trials for the treatment of MDS and for severe chronic idiopathic neutropenia.
…………………………………..
http://www.google.com/patents/WO2013082462A1?cl=en
Example 1
[0048] 80 mg of crystalline ezatiostat hydrochloride was placed in a round bottom flask and dissolved in 25 mL of methanol. The solvent was then evaporated on a rotary evaporation apparatus under reduced pressure at 30 °C. After 30 minutes, the solid sample was removed from the round bottom flask and stored in a sealed vial at 2 °C in a refrigerator. Analysis of this sample was carried out within 24 hours of removing it from the rotary evaporation apparatus.
[0049] The resulting amorphous material was analyzed by 1H NMR, 13C NMR, DSC, and X-Ray powder diffraction experiments. The DSC conditions were 30 to 300 °C at 10 °C/ min using 7 mg of the amorphous material. The X-Ray powder diffraction was taken at 0-60 of 2theta. The crystalline ezatiostat hydrochloride was also analyzed.
Phase II trials: Ezatiostat is the first GSTP1-1 inhibitor shown to cause clinically significant and sustained reduction in RBC transfusions, transfusion independence, and multilineage responses in MDS patients. The tolerability and activity profile of ezatiostat may offer a new treatment option for patients with MDS. (source: Cancer. 2012 Apr 15;118(8):2138-47.)
|
References |
1: Galili N, Tamayo P, Botvinnik OB, Mesirov JP, Brooks MR, Brown G, Raza A. Prediction of response to therapy with ezatiostat in lower risk myelodysplastic syndrome. J Hematol Oncol. 2012 May 6;5:20. doi: 10.1186/1756-8722-5-20. PubMed PMID: 22559819; PubMed Central PMCID: PMC3407785.
2: Raza A, Galili N, Mulford D, Smith SE, Brown GL, Steensma DP, Lyons RM, Boccia R, Sekeres MA, Garcia-Manero G, Mesa RA. Phase 1 dose-ranging study of ezatiostat hydrochloride in combination with lenalidomide in patients with non-deletion (5q) low to intermediate-1 risk myelodysplastic syndrome (MDS). J Hematol Oncol. 2012 Apr 30;5:18. doi: 10.1186/1756-8722-5-18. PubMed PMID: 22546242; PubMed Central PMCID: PMC3416694.
3: Lyons RM, Wilks ST, Young S, Brown GL. Oral ezatiostat HCl (Telintra®, TLK199) and idiopathic chronic neutropenia (ICN): a case report of complete response of a patient with G-CSF resistant ICN following treatment with ezatiostat, a glutathione S-transferase P1-1 (GSTP1-1) inhibitor. J Hematol Oncol. 2011 Nov 2;4:43. doi: 10.1186/1756-8722-4-43. PubMed PMID: 22047626; PubMed Central PMCID: PMC3235963.
4: Raza A, Galili N, Smith SE, Godwin J, Boccia RV, Myint H, Mahadevan D, Mulford D, Rarick M, Brown GL, Schaar D, Faderl S, Komrokji RS, List AF, Sekeres M. A phase 2 randomized multicenter study of 2 extended dosing schedules of oral ezatiostat in low to intermediate-1 risk myelodysplastic syndrome. Cancer. 2012 Apr 15;118(8):2138-47. doi: 10.1002/cncr.26469. Epub 2011 Sep 1. PubMed PMID: 21887679.
5: Quddus F, Clima J, Seedham H, Sajjad G, Galili N, Raza A. Oral Ezatiostat HCl (TLK199) and Myelodysplastic syndrome: a case report of sustained hematologic response following an abbreviated exposure. J Hematol Oncol. 2010 Apr 23;3:16. doi: 10.1186/1756-8722-3-16. PubMed PMID: 20416051; PubMed Central PMCID: PMC2873355.
6: Steensma DP. Novel therapies for myelodysplastic syndromes. Hematol Oncol Clin North Am. 2010 Apr;24(2):423-41. doi: 10.1016/j.hoc.2010.02.010. Review. PubMed PMID: 20359635.
7: D’Alò F, Greco M, Criscuolo M, Voso MT. New treatments for myelodysplastic syndromes. Mediterr J Hematol Infect Dis. 2010 Aug 11;2(2):e2010021. doi: 10.4084/MJHID.2010.021. PubMed PMID: 21415972; PubMed Central PMCID: PMC3033133.
8: Raza A, Galili N, Callander N, Ochoa L, Piro L, Emanuel P, Williams S, Burris H 3rd, Faderl S, Estrov Z, Curtin P, Larson RA, Keck JG, Jones M, Meng L, Brown GL. Phase 1-2a multicenter dose-escalation study of ezatiostat hydrochloride liposomes for injection (Telintra, TLK199), a novel glutathione analog prodrug in patients with myelodysplastic syndrome. J Hematol Oncol. 2009 May 13;2:20. doi: 10.1186/1756-8722-2-20. PubMed PMID: 19439093; PubMed Central PMCID: PMC2694211.
9: Raza A, Galili N, Smith S, Godwin J, Lancet J, Melchert M, Jones M, Keck JG, Meng L, Brown GL, List A. Phase 1 multicenter dose-escalation study of ezatiostat hydrochloride (TLK199 tablets), a novel glutathione analog prodrug, in patients with myelodysplastic syndrome. Blood. 2009 Jun 25;113(26):6533-40. doi: 10.1182/blood-2009-01-176032. Epub 2009 Apr 27. PubMed PMID: 19398716.
| WO2013082462A1 * | Nov 30, 2012 | Jun 6, 2013 | Telik, Inc. | Amorphous ezatiostat ansolvate |
| US20120251496 * | Mar 20, 2012 | Oct 4, 2012 | Telik, Inc. | Ezatiostat for treating multiple myeloma |
……….

Figure 1.
Known Yes1 kinase inhibitors, dasatinib and saracatinib.
Table 1. Select Yes1 kinase inhibitors from a HTS and their corresponding clinical phase, known targets, and IC50 values
| Compound name and NCGC ID | Structure | Clinical phase | Known targets | Yes1 IC50 (nM) |
|---|---|---|---|---|
| Dasatinib (1) NCGC00181129 |
 |
Approved | Lyn, PDGFR, KIT, Lck, BTK, Bcr–Abl, Fyn, Yes1, c-Src |
0.5 (<1.0)a |
| Saracatinib (2) NCGC00241099 |
|
Phase II/III | c-Src, Bcr–Abl, Yes1, Lck | 6.2 (0.70)a |
| AEE-788 (3) NCGC00263149 |
 |
Phase I/II | EGFR, HER-2, VEGFR-2 | 17.5 (13.1)a |
| Dovitinib (4) NCGC00249685 |
|
Phase III | FGFR, EGFR, PDGFR, VEGFR-1,2 | 31 (1.4)a |
| DCC-2036 (5) NCGC00263172 |
 |
Phase I/II | Bcr–Abl, Tie-2, Lyn, FLT3, VEGFR-2 | 2.5 (1.5)a |
| SGI-1776 (6) NCGC00263186 |
 |
Discontinued | Pim-1, FLT3 | 2670 (240)a |
| AMG-Tie-2-1 (7) NCGC00263199 |
 |
Preclinical | Tie-2 | 8.7 (22.0)a |
| AZ-23 (8) NCGC00250381 |
 |
Preclinical | Trk | 39.1 (3.0)a |
| Dorsomorphin (9) NCGC00165869 |
 |
Preclinical | AMPK, BMPR, TGFβ Receptor | 195.9 (29.8)a |
| AZ-628 (10) NCGC00250380 |
 |
Preclinical | Raf Kinase B,C | 348.3 (51.2)a |
- http://www.sciencedirect.com/science/article/pii/S0960894X13006677
- Data in parentheses were gathered by Reaction Biology Corp. using a [γ-33P]-ATP radiolabeled enzyme activity assay at an ATP concentration of 10 μM (www.reactionbiology.com).
SEE COMPILATION ON SIMILAR COMPOUNDS AT …………..http://drugsynthesisint.blogspot.in/p/nostat-series.html
Amgen-AstraZeneca Psoriasis Drug Brodalumab (AMG 827) Hits Phase 3 Endpoints

AstraZeneca and Amgen announced that the Phase 3 AMAGINE-1TM study evaluating brodalumab in patients with moderate-to-severe plaque psoriasis met all primary and secondary endpoints for both evaluated doses.
Brodalumab is a human monoclonal antibody designed for the treatment of inflammatory diseases.[1] It is being tested for the treatment of moderate to severe psoriasis[2] in Phase III clinical trials as of November 2013.[3][4]
Brodalumab was developed by Amgen, Inc.

Mechanism of action
Brodalumab binds to the interleukin-17 receptor and so prevents interleukin 17 (IL-17) from activating the receptor. This mechanism is similar to that of another anti-psoriasis antibody, ixekizumab, which however binds to IL-17 itself.[2]
At present, brodalumab is the only experimental drug in development that inhibits the IL-17 receptor, thus inhibiting several of the IL-17 ligands at once from transmitting signals to the body. Other agents currently in development seek to target the individual IL-17 ligands. By inhibiting the attachment of these ligands with the receptor, brodalumab stops the body from receiving signals that may otherwise cause inflammation and other ailments.
Researchers are currently investigating brodalumab for the treatment of psoriasis (Phase II and planned Phase III), asthma (Phase II), and psoriatic arthritis (Phase II).
Psoriasis is a chronic disease of the immune system that causes the skin cells to grow at a faster rate. Worldwide, the condition affects around 125 million individuals. Even though several types of psoriasis exist, around 80% of sufferers have plaque psoriasis. Plaque psoriasis can cause painful and itchy red, scaly patches to appear on the skin.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Interleukin 17 receptor A |
| Clinical data | |
| Legal status | Investigational |
| Identifiers | |
| CAS number | 1174395-19-7 |
| ATC code | None |
| KEGG | D10061 |
| Chemical data | |
| Formula | C6372H9840N1712O1988S52 |
| Mol. mass | 144.06 kDa |
About Brodalumab (AMG 827)
Brodalumab is a novel human monoclonal antibody that binds to the interleukin-17 (IL-17) receptor and inhibits inflammatory signaling by blocking the binding of several IL-17 ligands to the receptor. By stopping IL-17 ligands from activating the receptor, brodalumab prevents the body from receiving signals that may lead to inflammation. The IL-17 pathway plays a central role in inducing and promoting inflammatory disease processes. In addition to moderate-to-severe plaque psoriasis (Phase 3), brodalumab is currently being investigated for the treatment of psoriatic arthritis (Phase 3) and asthma (Phase 2).
About the Amgen and AstraZeneca Collaboration
In April 2012, Amgen and AstraZeneca formed a collaboration to jointly develop and commercialize five monoclonal antibodies from Amgen’s clinical inflammation portfolio. With oversight from joint governing bodies, Amgen leads clinical development and commercialization for brodalumab (Phase 3 for moderate-to-severe plaque psoriasis and psoriatic arthritis, Phase 2 for asthma) and AMG 557/MEDI5872 (Phase 1b for autoimmune diseases such as systemic lupus erythematosus). AstraZeneca, through its biologics arm MedImmune, leads clinical development and commercialization for MEDI7183/AMG 181 (Phase 2 for ulcerative colitis and Crohn’s disease), MEDI2070/AMG 139 (Phase 2 for Crohn’s disease) and MEDI9929/AMG 157 (Phase 2 for asthma).
About Amgen
Amgen is committed to unlocking the potential of biology for patients suffering from serious illnesses by discovering, developing, manufacturing and delivering innovative human therapeutics. This approach begins by using tools like advanced human genetics to unravel the complexities of disease and understand the fundamentals of human biology.
Amgen focuses on areas of high unmet medical need and leverages its biologics manufacturing expertise to strive for solutions that improve health outcomes and dramatically improve people’s lives. A biotechnology pioneer since 1980, Amgen has grown to be the world’s largest independent biotechnology company, has reached millions of patients around the world and is developing a pipeline of medicines with breakaway potential.
For more information, visit www.amgen.com and follow us on www.twitter.com/amgen.

About AstraZeneca
AstraZeneca is a global, innovation-driven biopharmaceutical business that focuses on the discovery, development and commercialisation of prescription medicines, primarily for the treatment of cardiovascular, metabolic, respiratory, inflammation, autoimmune, oncology, infection and neuroscience diseases. AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. For more information please visit: www.astrazeneca.com.
References
- “Statement On A Nonproprietary Name Adopted By The USAN Council: Brodalumab”. American Medical Association.
- “Neue Antikörper in der Pipeline”. Pharmazeutische Zeitung (in German) (12). 2012.
- ClinicalTrials.gov NCT01708590 Study of Efficacy, Safety, and Withdrawal and Retreatment With Brodalumab in Moderate to Severe Plaque Psoriasis Subjects (AMAGINE-1)
- ClinicalTrials.gov NCT01708629 Study of Efficacy and Safety of Brodalumab Compared With Placebo and Ustekinumab in Moderate to Severe Plaque Psoriasis Subjects (AMAGINE-3)
http://ksclinic.exblog.jp/18270693/
学術面で最初の講演は、米国のJames Krueger教授による「Th1細胞,Th17細胞,Th22細胞が複雑なサイトカインネットワークによって、細胞レベル、分子レベルで乾癬を引き起こす」でした。その要約を示すスライドを幾枚か失敬します(Krueger先生、ごめんなさい)。
 乾 癬の原因究明、病態(病気の起こり方)解明の主役となった免疫学的研究の最先端を行くKrueger先生の、最新情報がコンパクトにまとまった素晴らしい 講演でした。生物学的製剤の治療根拠となるサイトカインネットワークは、現在TipDC – Th17経路によって、きわめて明快に説明されるようになり、Th17細胞が放出するIL17が表皮細胞(ケラチノサイト)の乾癬化を起こします。現在使 用されている抗TNFα製剤、抗IL12/23製剤が、より上流(免疫反応の根っこ)で免疫反応を抑制するのに比べ、IL17はより末梢における乾癬の原 因サイトカインであることから、IL17の抑制は、より乾癬をピンポイントで、そして副作用もミニマムにすることが期待される。
乾 癬の原因究明、病態(病気の起こり方)解明の主役となった免疫学的研究の最先端を行くKrueger先生の、最新情報がコンパクトにまとまった素晴らしい 講演でした。生物学的製剤の治療根拠となるサイトカインネットワークは、現在TipDC – Th17経路によって、きわめて明快に説明されるようになり、Th17細胞が放出するIL17が表皮細胞(ケラチノサイト)の乾癬化を起こします。現在使 用されている抗TNFα製剤、抗IL12/23製剤が、より上流(免疫反応の根っこ)で免疫反応を抑制するのに比べ、IL17はより末梢における乾癬の原 因サイトカインであることから、IL17の抑制は、より乾癬をピンポイントで、そして副作用もミニマムにすることが期待される。
 現在、3種類のIL17抑制薬剤が開発され、治療研究が進められている。
現在、3種類のIL17抑制薬剤が開発され、治療研究が進められている。
①IL17A抗体(Secukinumab Novartis社)
②IL17A抗体(Ixekizumab Lilly社)
③IL17A受容体抗体(Brodalumab Amgen社)


その一つ、Secukinumabの効果(PASI75)=すごく乾癬がよくなる)では、たった3回の注射で90%以上の患者がPASI75を達成する。


PASI90(=乾癬がほとんどなくなる)でみても、60%の患者で達成されている。

Secukinumabの臨床効果。上の段は「プラセボ(偽薬)」、下の段がSecukinumab。

Ixekizumabの効果(PASI90)。約80%の患者で達成されている。驚異的である。

Brodalumabの臨床効果
印象深かった講演をもう一つ、詳細に紹介いたします。
米 国のAnne Bowcock教授の”The genetics of psoriasis: Old risks, novel loci (乾癬の遺伝子研究:昔から言われていた異常、新しく見つかった場所)です。Bowcock教授は、乾癬の原因遺伝子について世界で最初に報告した研究者 です。ここでも少し講演スライドを拝借(Bowcock先生、ごめんなさい)。
 Bowcock 教授は1999年、乾癬家系の詳細な遺伝子調査から第17染色体に乾癬と関わり深い遺伝子異常があることをみつけ、科学雑誌Scienceに報告した。 21世紀を迎える直前のことであり、遠からず乾癬の原因遺伝子が確定し、完治治療を開発することも夢ではないと、当時期待したものでした。
Bowcock 教授は1999年、乾癬家系の詳細な遺伝子調査から第17染色体に乾癬と関わり深い遺伝子異常があることをみつけ、科学雑誌Scienceに報告した。 21世紀を迎える直前のことであり、遠からず乾癬の原因遺伝子が確定し、完治治療を開発することも夢ではないと、当時期待したものでした。
ところが、次々と関連遺伝子はみつけられるものの(現在は30種類以上)、肝心の原因遺伝子、特定のタンパク、メカニズムは不明のままでした。
 Bowcock 教授の息の長い研究は、第17染色体上にあるCARD14と呼ばれるタンパクの、その異常が直接乾癬を起こすことを説き明かしました。CARD14は細胞 膜上にあるタンパクで、細胞外で起こる炎症から生じる様々な刺激物質を、細胞の膜から細胞の中へ伝える役割を果たしています。その伝達経路はNFκBを介 しています(乾癬ではこの経路が活発に動いていることが、高知大学の佐野教授により解明されました)。
Bowcock 教授の息の長い研究は、第17染色体上にあるCARD14と呼ばれるタンパクの、その異常が直接乾癬を起こすことを説き明かしました。CARD14は細胞 膜上にあるタンパクで、細胞外で起こる炎症から生じる様々な刺激物質を、細胞の膜から細胞の中へ伝える役割を果たしています。その伝達経路はNFκBを介 しています(乾癬ではこの経路が活発に動いていることが、高知大学の佐野教授により解明されました)。

遺伝性膿疱性乾癬患者では、このCARD14遺伝子に点突然変異が起こっていることを発見しました。この点突然変異だけで、特殊タイプではありますが、乾癬の原因が特定されたのです。

点突然変異だけではなく、CARD14遺伝子に起こりやすい変異も、ほかの遺伝子異常(PSORS1、MHC遺伝子)、あるいは環境変化が加わると乾癬を引き起こすことも証明しました。
大変感銘深い講演でした。
会議の模様、IFPA代表者会議の報告は、また後日掲載いたします(『2012年9月教室抄録』をご覧ください)。
ブログ「PHOTO & ESSAY」もご覧ください。
MK-0822; Odanacatib 奥当卡替……….has been identified as a potent and selective inhibitor of Cathepsin K.

MK-0822; Odanacatib.
603139-19-1
Formula: C25H27F4N3O3S
Mass: 525.17093
Merck Frosst Canada Ltd. phase 3
(S)-N-(1-cyanocyclopropyl)-4-fluoro-4-methyl-2-(((S)-2,2,2-trifluoro-1-(4′-(methylsulfonyl)-[1,1′-biphenyl]-4-yl)ethyl)amino)pentanamide
N1-(1-Cyanocyclopropyl)-4-fluoro-N2-[2,2,2-trifluoro-1(S)-[4′-(methylsulfonyl)biphenyl-4-yl]ethyl]-L-leucinamide
Odanacatib (pINN; codenamed MK-0822) is an investigational treatment for osteoporosis and bone metastasis. It is an inhibitor of cathepsin K, an enzyme involved in bone resorption. It is being developed by Merck & Co. As of November 2009, Merck is conducting phase III clinical trials.
Odanacatib, also known as MK-0822, is an inhibitor of cathepsin K with potential anti-osteoporotic activity. Odanacatib selectively binds to and inhibits the activity of cathepsin K, which may result in a reduction in bone resorption, improvement of bone mineral density, and a reversal in osteoporotic changes. Cathepsin K, a tissue-specific cysteine protease that catalyzes degradation of bone matrix proteins such as collagen I/II, elastin, and osteonectin plays an important role in osteoclast function and bone resorption
Osteoporosis is a disease characterized by excessive bone loss causing skeletal fragility and an increased risk of fracture. One in two women and one in eight men over the age of 50 will have an osteoporotic fracture. Cathepsin K is a recently discovered member of the papain superfamily of cysteine proteases that is abundantly expressed in osteoclasts, the cells responsible for bone resorption.
MK-0822 is in phase III clinical trials at Merck & Co. for the treatment of postmenopausal osteoporosis. Several phase II trials had been ongoing for the treatment of cancer, specifically for the treatment of women with breast cancer and metastatic bone disease and also for the treatment of osteoarthritis in the knee and for the treatment of arthritis; however, no recent development has been reported for these indications. MSD KK (formed in 2010 following the merger of Banyu and Schering-Plough KK) is developing the compound for the treatment of osteoporosis in Japan.
Bone is a living tissue that is remodeled every five to seven years in a dynamic process governed by the balance between bone formation and resorption in which osteoblasts and osteoclasts play a pivotal role. The abundant and selective expression of Cathepsin K in osteoclasts has made it an attractive therapeutic target for the treatment of osteoporosis.
Odanacatib (MK-0822) 1 has been identified as a potent and selective inhibitor of Cathepsin K.
A variety of disorders in humans and other mammals involve or are associated with abnormal bone resorption. Such disorders include, but are not limited to, osteoporosis, glucocorticoid induced osteoporosis, Paget’s disease, abnormally increased bone turnover, periodontal disease, tooth loss, bone fractures, rheumatoid arthritis, osteoarthritis, periprosthetic osteolysis, osteogenesis imperfecta, metastatic bone disease, hypercalcemia of malignancy, and multiple myeloma. One of the most common of these disorders is osteoporosis, which in its most frequent manifestation occurs in postmenopausal women. Osteoporosis is a systemic skeletal disease characterized by a low bone mass and microarchitectural deterioration of bone tissue, with a consequent increase in bone fragility and susceptibility to fracture. Osteoporotic fractures are a major cause of morbidity and mortality in the elderly population. As many as 50% of women and a third of men will experience an osteoporotic fracture. A large segment of the older population already has low bone density and a high risk of fractures. There is a significant need to both prevent and treat osteoporosis and other conditions associated with bone resorption. Because osteoporosis, as well as other disorders associated with bone loss, are generally chronic conditions, it is believed that appropriate therapy will typically require chronic treatment.
Osteoporosis is characterized by progressive loss of bone architecture and mineralization leading to the loss in bone strength and an increased fracture rate. The skeleton is constantly being remodeled by a balance between osteoblasts that lay down new bone and osteoclasts that breakdown, or resorb, bone. In some disease conditions and advancing age the balance between bone formation and resorption is disrupted; bone is removed at a faster rate. Such a prolonged imbalance of resorption over formation leads to weaker bone structure and a higher risk of fractures. Bone resorption is primarily performed by osteoclasts, which are multinuclear giant cells. Osteoclasts resorb bone by forming an initial cellular attachment to bone tissue, followed by the formation of an extracellular compartment or lacunae. The lacunae are maintained at a low pH by a proton-ATP pump. The acidified environment in the lacunae allows for initial demineralization of bone followed by the degradation of bone proteins or collagen by proteases such as cysteine proteases. See Delaisse, J. M. et al, 1980, Biochem J 192:365-368; Delaisse, J. et ah, 1984, Biochem Biophys Res Commun:44l-447; Delaisse, J. M. et α/., 1987, Bone 8^305-313, which are hereby incorporated by reference in their entirety. Collagen constitutes 95 % of the organic matrix of bone. Therefore, proteases involved in collagen degradation are an essential component of bone turnover, and as a consequence, the development and progression of osteoporosis.
Cathepsins belong to the papain superfamily of cysteine proteases. These proteases function in the normal physiological as well as pathological degradation of connective tissue. Cathepsins play a major role in intracellular protein degradation and turnover and remodeling. To date, a number of cathepsin have been identified and sequenced from a number of sources. These cathepsins are naturally found in a wide variety of tissues. For example, cathepsin B, F, H, L, K, S, W, and Z have been cloned. Cathepsin K (which is also known by the abbreviation cat K) is also known as cathepsin O and cathepsin O2. See PCT Application WO 96/13523, Khepri Pharmaceuticals, Inc., published May 9, 1996, which is hereby incorporated by reference in its entirety. Cathepsin L is implicated in normal lysosomal proteolysis as well as several diseases states, including, but not limited to, metastasis of melanomas. Cathepsin S is implicated in Alzheimer’s disease and certain autoimmune disorders, including, but not limited to juvenile onset diabetes, multiple sclerosis, pemphigus vulgaris, Graves’ disease, myasthenia gravis, systemic lupus erythemotasus, rheumatoid arthritis and Hashimoto’s thyroiditis; allergic disorders, including, but not limited to asthma; and allogenic immunbe responses, including, but not limited to, rejection of organ transplants or tissue grafts. Increased Cathepsin B levels and redistribution of the enzyme are found in tumors, suggesting a role in tumor invasion and matastasis. In addition, aberrant Cathpsin B activity is implicated in such disease states as rheumatoid arthritis, osteoarthritis, pneumocystisis carinii, acute pancreatitis, inflammatory airway disease and bone and joint disorders.
Cysteine protease inhibitors such as E-64 (trαns-epoxysuccinyl-L- leucylamide-(4-guanidino) butane) are known to be effective in inhibiting bone resorption. See Delaisse, J. M. et al., 1987, Bone 8:305-313, which is hereby incorporated by reference in its entirety. Recently, cathepsin K was cloned and found specifically expressed in osteoclasts See Tezuka, K. et al., 1994, J Biol Chem 269:1106-1109; Shi, G. P. et αZ.,1995, EEES Lett 357: 129-134; Bromme, D. and Okamoto, K., 1995, Biol Chem Hoppe Seyler 376:379-384; Bromme, D. et al, 1996, J Biol Chem 271:2126-2132: Drake, F. H. et al, 1996, J Biol Chem 271:12511- 12516, which are hereby incorporated by reference in their entirety. Concurrent to the cloning, the autosomal recessive disorder, pycnodysostosis, characterized by an osteopetrotic phenotype with a decrease in bone resorption, was mapped to mutations present in the cathepsin K gene. To date, all mutations identified in the cathepsin K gene are known to result in inactive protein. See Gelb, B. D. et al., 1996, Science 273:1236-1238; Johnson, M. R. et al., 1996, Genome Res 6:1050-1055, which are hereby incorporated by reference in their entirety. Therefore, it appears that cathepsin K is involved in osteoclast mediated bone resorption.
Cathepsin K is synthesized as a 37 kDa pre-pro enzyme, which is localized to the lysosomal compartment and where it is presumably autoactivated to the mature 27 kDa enzyme at low pH. See McQueney, M. S. et al., 1997, J Biol Chem 272:13955-13960; Littlewood-Evans, A. et al, 1997, Bone 20:81-86, which are hereby incorporated by reference in their entirety. Cathepsin K is most closely related to cathepsin S having 56 % sequence identity at the amino acid level. The S2P2 substrate specificity of cathepsin K is similar to that of cathepsin S with a preference in the PI and P2 positions for a positively charged residue such as arginine, and a hydrophobic residue such as phenylalanine or leucine, respectively. See Bromme, D. et al., 1996, J Biol Chem 271: 2126-2132; Bossard, M. J. et al, 1996, J Biol Chem 271:12517-12524, which are hereby incorporated by reference in their entirety. Cathepsin K is active at a broad pH range with significant activity between pH 4-8, thus allowing for good catalytic activity in the resorption lacunae of osteoclasts where the pH is about 4-5.
Human type I collagen, the major collagen in bone is a good substrate for cathepsin K. See Kafienah, W., et al, 1998, Biochem J 331:727-732, which is hereby incorporated by reference in its entirety. In vitro experiments using antisense oligonucleotides to cathepsin K, have shown diminished bone resorption in vitro, which is probably due to a reduction in translation of cathepsin K mRNA. See Inui, T., et al, 1997, Biol Chem 272:8109-8112, which is hereby incorporated by reference in its entirety. The crystal structure of cathepsin K has been resolved. See McGrath, M. E., et al, 1997, Nat Struct Biol 4:105-109; Zhao, B., et al, 1997, Nat Struct Biol 4: 109-11, which are hereby incorporated by reference in their entirety. Also, selective peptide based inhibitors of cathepsin K have been developed See Bromme, D., et al, 1996, Biochem 315:85-89; Thompson, S. K., et al, 1997, Proc Natl Acad Sci U S A 94: 14249-14254, which are hereby incorporated by reference in their entirety. Accordingly, inhibitors of Cathepsin K can reduce bone resorption. Such inhibitors would be useful in treating disorders involving bone resorption, such as osteoporosis.
……………….
The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K
Bioorg Med Chem Lett 2008, 18(3): 923
http://www.sciencedirect.com/science/article/pii/S0960894X07015065


Scheme 2.
Reagents and conditions: (a) ClCOOiBu, NMM, NaBH4, DME, 85%; (b) Ts2O, pyr, dichloroethane, 83%; (c) MeMgBr, toluene/THF, 85%; (d) DAST, CH2Cl2, 60%; (e) Ba(OH)2, EtOH/H2O, 100%; (f) TBSCl, Et3N; (g) CF3C(OH)OEt, PhH, 88% (two steps); (h) BrPhLi, THF; (i) TBAF, THF, 75% (two steps); (j) H5IO6, CrO3, CH3CN, 60%; (k) 1-amino-1-cyanocyclopropane hydrochloride, i-Pr2NEt, HATU, DMF, 80%; (l) MeSPhB(OH)2, PdCl2dppf, Na2CO3, DMF, 70%; (m) H2O2, Na2WO42H2O, Bu4NHSO4, EtOAc, 97%. see Supplementary data.
………………
WO 2003075836 or http://www.google.com/patents/EP1482924A2?cl=en
EXAMPLE 10
Synthesis of N l-cyanocyclopropyl)-N2{(lS)-2,2,2-trifluoro-l-[4′-(m^
1 , 1 -biphenyl-4-yl]ethyl ) -L-leucinamide

To a mixture of Ν-{(lS)-2,2,2-trifluoro-l-[4′-(methylsulfonyl)-l,l’- biphenyl-4-yl]ethyl} -L-leucine from Example 8 (0.83 g), O-(7-azabenzotriazol-l-yl)- N, N, N\ N’-tetramethyluronium hexafluorophosphate (0.78 g), cyclopropylamine hydrochloride (0.466 g) in DMF (18 mL) at 0 °C was added triethylamine (0.9 mL). The mixture was kept at room temperature for 48 hours and then poured into dilute aqueous ammonium cholride and diethyl ether. The ether layer was separated and the aqueous further extracted with diethylether. The combined ether extracts were washed with brine, dried with magnesium sulfate and the solvent was removed in vacuo. The residue was purified in SiO2 using ethyl acetate and hexanes (1:1) as eluant, followed by a swish in diethyl ether to yield the title compound.
H NMR (CD3COCD3) δ 8.15(1H, bs), 8.05(2H, d), 8.0(2H, d), 7.8(2H, d), 7.65(2H, d), 4.35-4.45(lH, m), 3.35-3.45(lH, m), 3.2(3H, s), 2.65-2.7(lH, m), 1.85-1.95(1H, m), 1.3-1.6(5H, m), 1.05-1.15(1H, m), 0.85-0.95(6H, m).
……….
WO 2008119176

http://www.google.com/patents/WO2008119176A1?cl=en
EXAMPLE 1
4-FLUORO-iV- {(1 S)-2,2,2-TRIFLUORO- 1 -[4′-(METHYLSULFONYL)BIPHENYL^- YL]ETHYL}-L-LEUCINE DICYCLOHEXYLAMINE SALT

Biphenyl acid (20.74 g) was dissolved in 2-propanol (186 mL) / water (20.7 mL). A solution of iV,jV-dicyclohexylamine (9.82 mL) in 2-propanol (21 mL) / water (2 mL) was added (-10% of volume) and the solution was seeded with DCHA salt (10 mg). A heavy seed bed formed and the slurry was let stir at rt for 30 min. Addition of DCHA was continued over 20-30 min. The slurry was let stir at rt overnight and filtered. The filter cake was washed with 2-propanol / water (2 x 30 mL, 10:1) and MTBE (2 x 30 mL). DCHA salt was obtained as a white solid, 24.4 g, 84% yield. 1H NMR (CD3OD) δ 8.07 (d, 2H, J- 8.0), 7.94 (d, 2H, J= 8.0), 7.75 (d, 2H, J= 8.0), 7.61 (d, 2H, J= 8.0), 4.31 (m, IH), 3.46 (bq, IH, J= 4), 3.22 (m, 2H), 3.19 (s, 3H), 2.11 (bm, 5H), 1.91 (bm, 5H), 1.75 (bm, 2H), 1.49 (d, 3H, J= 21.6), 1.48 (d, 3H, J= 21.6), 1.35 (m, 9H); 19F NMR (CD3OD) δ – 72.9, – 129.4; mp 209-211°C, [α]D 20 + 18.7 (c = 0.29, MeOH).
EXAMPLE 2
N-(I -CYANOCYCLOPROPYL)-4-FLUORO-N2– {(1 S)-2,2,2-TRIFLUORO- 1 -[4′- (METHYLSULFOΝYL)BIPHEΝYL-4- YL]ETHYL}-L-LEUCINAMIDE

Acid (1.9 g) was dissolved in DMAc (10 mL) and cooled to 0°C. 1 –
Aminocyclopropane carbonitrile hydrochloride (0.57 g) and HATU (1.85 g) were added. The resulting slurry was stirred for 15 min and DIEA (2.12 mL) was added over 1.5 h. The reaction was aged for 1 h. Water (11.2 mL) was added via dropping funnel over 70 min and the slurry was aged for Ih at 2O0C. The mixture was filtered and the filter cake was washed with a solution of DMAc:water (9.4 mL, 1 : 1.2), water (18.7 mL), 2-propanol (9.3 mL) The batch was dried to yield 1.67 g, 79% yield of the corresponding amide.
Amide (2.56 g), was dissolved in THF (30.7 mL) at 30°C. Water (19 mL) was added via dropping funnel. The batch was seeded and aged for Ih at 2O0C. Additional water (40.9 mL) was added over 1.5 h and the batch was aged for 16 h. The batch was filtered and washed with water (15 mL). The solids were dried to a constant weight to yield 2.50 g, 97% yield of pure amide. 1H NMR (CD3OD) δ 8.17 (bs, IH), 8.05 (d, 2H, J= 8.5), 7.96 (d, 2H, J= 8.5), 7.80 (d, 2H, J= 8.0), 7.64 (d, 2H, J= 8.0), 4.43 (m, IH), 3.55 (ddd, IH, J= 5.0, 8.5, 8.0), 3.18 (s, 3H), 2.84 (bm, IH), 2.02 (m, 2H), 1.46 (d, 3H, J= 21.5), 1.43 (d, 3H, J= 22.0), 1.36 (m, 2H), 1.07 (m, IH), 0.94 (m, IH); 13C NMR (CD3OD) δ; 19F NMR (CD3OD) δ -73.2, -136.8; IR (cm“1) 3331, 2244, 1687, 1304, 1152; mp 223-224 0C, [α]D 20 + 23.3 (c = 0.53, MeOH).
EXAMPLE 3
N-(l-CYANOCYCLOPROPYL)-4-FLUORO-iV2-{(l1S)-2,2,2-TRrFLUORO-l-[4′- (METHYLSULFONYL)BIPHENYL^-YL]ETHYL) -L-LEUCINAMIDE

A round-bottom flask was charged with biphenyl acid’DCHA salt (76.6 g, 99.2% ee, diastereomeric ratio 342:1) and DMF (590 g). Solid aminocyclopropane carbonitrile-HCl (15.2 g), HOBt-H2O (17.9 g), and EDCΗC1 (29.1 g) were all charged forming a white slurry. The batch was then heated to 38-42°C and aged for 5 hours. The batch was then cooled to 20- 250C and held overnight. HPLC analysis showed 99.4% conversion. The batch was heated to 38-42°C and water (375 g) was charged to batch over 2 hours. The batch remained as a slurry throughout the water addition. The batch was then heated to 58-620C and aged for 1 hour. Following age, water (375 g) was charged over 3 hours, at a rate of 2.1 g/min. The batch was then cooled to 15-25°C and aged overnight. The batch was filtered and washed with 39% DMF in water (2 x 300 g) and 2-propanol (180 g). The solids were dried in the filter at 40-600C for 24 hours. The desired crude product was isolated as a white solid (57g, 92% yield, 99.4 wt%). A round-bottom flask was charged with crude solid (57 g) and acetone/water solution (324 g, 88/12). The slurry was then heated to 400C, at which point the batch was in solution, and aged for an hour. Water (46 g) was then charged over 30 minutes. The batch was then seeded (1.7 g, 3.0 wt%), and the batch was aged at 40°C for an hour prior to proceeding with the crystallization. Water (255 g) was charged over 4.5 h. The batch was then cooled to 230C over 1.5 h, aged for 4 h and filtered. The solids were washed with acetone/water (158 g, 45/55) and water (176 g). The filter cake was dried with nitrogen sweep / vacuum at 55°C. The desired product (57.2 g , 99.9wt%, 99.8A% (enantiomer ND), was obtained in 94.9% yield. 1H NMR (CD3OD) δ 8.17 (bs, IH), 8.05 (d, 2H, J= 8.5), 7.96 (d, 2H, J= 8.5), 7.80 (d, 2H, J= 8.0), 7.64 (d, 2H, J= 8.0), 4.43 (m, IH), 3.55 (ddd, IH, J= 5.0, 8.5, 8.0), 3.18 (s, 3H), 2.84 (bm, IH), 2.02 (m, 2H), 1.46 (d, 3H, J= 21.5), 1.43 (d, 3H, J= 22.0), 1.36 (m, 2H), 1.07 (m, IH), 0.94 (m, IH); 13C NMR
(CD3OD) δ; 19F NMR (CD3OD) δ -73.2, -136.8; IR (cm“1) 3331, 2244, 1687, 1304, 1152; mp 223-224 0C, [α]D 20 + 23.3 (c = 0.53, MeOH).
……………..
J. Org. Chem., 2009, 74 (4), pp 1605–1610
DOI: 10.1021/jo802031
http://pubs.acs.org/doi/abs/10.1021/jo8020314

An enantioselective synthesis of the Cathepsin K inhibitor odanacatib (MK-0822) 1 is described. The key step involves the novel stereospecific SN2 triflate displacement of a chiral α-trifluoromethylbenzyl triflate 9a with (S)-γ-fluoroleucine ethyl ester 3 to generate the required α-trifluoromethylbenzyl amino stereocenter. The triflate displacement is achieved in high yield (95%) and minimal loss of stereochemistry. The overall synthesis of 1 is completed in 6 steps in 61% overall yield.
(2S)-N-(1-Cyanocyclopropyl)-4-fluoro-4-methyl-2-({(1S)-2,2,2-trifluoro-1-[4′-(methylsulfonyl)biphenyl-4-yl]ethyl}amino)pentanamide (1)
To a visually clean 5-necked 50-L round-bottomed flask equipped with a mechanical stirrer, a thermocouple, a dropping funnel, and a nitrogen inlet was added biaryl acid 12a (1.87 kg, 4.0 mol) and DMAc (9.3 L)…………………………………….deleted……………………………………………………. and dried under vacuum at 35 °C to yield 1 as a white solid (2.50 kg, 97% yield, 99.7 area %, 99.9% de by HPLC):
mp 223−224 °C;
1H NMR (CD3OD) δ 8.17 (br s, 1H), 8.05 (d, 2H, J = 8.5 Hz), 7.96 (d, 2H, J = 8.5 Hz), 7.80 (d, 2H, J = 8.0 Hz), 7.64 (d, 2H, J = 8.0 Hz), 4.43 (m, 1H), 3.55 (ddd, 1H, J = 5.0, 8.5, 8.0 Hz), 3.18 (s, 3H), 2.84 (br m, 1H), 2.02 (m, 2H), 1.46 (d, 3H, J = 21.5 Hz), 1.43 (d, 3H, J = 22.0 Hz), 1.36 (m, 2H), 1.07 (m, 1H), 0.94 (m, 1H); 13C NMR (125 MHz, acetone-d6) δ 175.2, 146.0, 141.2, 140.6, 136.1, 130.3, 128.9 (q, J = 282.8 Hz), 128.7, 128.6, 128.4, 120.9, 95.9 (d, J = 164.3 Hz), 63.5 (q, J = 30.0 Hz), 59.2 (d, J = 3.5 Hz), 44.8 (d, J = 23.1 Hz), 44.3, 27.5 (d, J = 23.9 Hz), 27.1 (d, J = 24.9 Hz), 20.7, 16.5;
19F NMR (CD3OD) δ −73.2, −136.8; IR (cm−1) 3331, 2244, 1687, 1304, 1152; [α]20D + 23.3 (c 0.53, MeOH);
HRMS calcd for C25H28F4N3O3S [MH]+ 526.1782; found 526.1781;
HPLC Phenomenex Spherisorb 4.6 mm × 25 cm column; eluants (A) 0.1% aqueous H3PO4 and (B) acetonitrile; 1 mL/min; gradient A/B 60:40 to 30:70 over 30 min; λ = 265 nm; temperature 45 °C; tR(1 (major diastereoisomer)) = 15.8 min, tR(1 (minor diastereoisomer)) = 16.4 min; HPLC (chiral) Chiralpak AD 4.6 mm × 15 cm column; eluants (A) hexanes, (B) ethanol, and (C) methanol; 1 mL/min; isocratic A/B/C 80:10:10 for 60 min; λ = 265 nm; temperature 40 °C; tR((S,S)-1) = 14.5 min, tR((R,S)-1) = 11.9 min, tR((S,R)-1) = 18.2 min, tR((R,R)-1) = 25.3 min, >99.5% (S,S).
…………..
| In vitro protocol: | XXX |
| In vivo protocol: | bone marrow of CatK(-/-) mice: Bone. 2011 Oct;49(4):623-35Pharmacokinetics and metabolism in rats, dogs, and monkeys: Drug Metab Dispos. 2011 Jun;39(6):1079-87. |
in Ovariectomized Rabbits. Calcif Tissue Int. 2013 Oct 2. [Epub ahead of print]Clinical study:Int J Clin Pharmacol Ther. 2013 Aug;51(8):688-92.J Clin Endocrinol Metab. 2013 Feb;98(2):571-80.
Br J Clin Pharmacol. 2013 May;75(5):1240-54.
J Bone Miner Res. 2010 May;25(5):937-47.
Clin Pharmacol Ther. 2009 Aug;86(2):175-82.Review papers:Clin Interv Aging. 2012;7:235-47.Clin Calcium. 2011 Jan;21(1):59-62.
IDrugs. 2009 Dec;12(12):799-809.
Ther Adv Musculoskelet Dis. 2013 Aug;5(4):199-209.
|
References |
1: Chapurlat RD. Treatment of postmenopausal osteoporosis with odanacatib. Expert Opin Pharmacother. 2014 Jan 24. [Epub ahead of print] PubMed PMID: 24456412.
2: Odanacatib, a cathepsin K inhibitor, superior to alendronate. Bonekey Rep. 2013 Sep 4;2:426. doi: 10.1038/bonekey.2013.160. eCollection 2013. PubMed PMID: 24422127; PubMed Central PMCID: PMC3789222.
3: Retraction notice: Odanacatib for the treatment of postmenopausal osteoporosis. Expert Opin Pharmacother. 2014 Jan;15(1):151. doi: 10.1517/14656566.2014.868399. Epub 2013 Nov 30. PubMed PMID: 24289716.
4: Anderson MS, Gendrano IN, Liu C, Jeffers S, Mahon C, Mehta A, Mostoller K, Zajic S, Morris D, Lee J, Stoch SA. Odanacatib, a selective cathepsin K inhibitor, demonstrates comparable pharmacodynamics and pharmacokinetics in older men and postmenopausal women. J Clin Endocrinol Metab. 2013 Dec 20:jc20131688. [Epub ahead of print] PubMed PMID: 24276460.
5: Chapurlat RD. Odanacatib for the treatment of postmenopausal osteoporosis. Expert Opin Pharmacother. 2014 Jan;15(1):97-102. doi: 10.1517/14656566.2014.853038. Epub 2013 Oct 25. Retraction in: Expert Opin Pharmacother. 2014 Jan;15(1):151. PubMed PMID: 24156249.
6: Stoch SA, Witter R, Hrenuik D, Liu C, Zajic S, Mehta A, Chandler P, Morris D, Xue H, Denker A, Wagner JA. Odanacatib does not influence the single dose pharmacokinetics and pharmacodynamics of warfarin. J Popul Ther Clin Pharmacol. 2013;20(3):e312-20. Epub 2013 Oct 2. PubMed PMID: 24142206.
7: Jensen PR, Andersen TL, Pennypacker BL, Duong le T, Delaissé JM. The bone resorption inhibitors odanacatib and alendronate affect post-osteoclastic events differently in ovariectomized rabbits. Calcif Tissue Int. 2014 Feb;94(2):212-22. doi: 10.1007/s00223-013-9800-0. Epub 2013 Oct 2. PubMed PMID: 24085265.
8: Bonnick S, De Villiers T, Odio A, Palacios S, Chapurlat R, Dasilva C, Scott BB, Le Bailly De Tilleghem C, Leung AT, Gurner D. Effects of Odanacatib on BMD and Safety in the Treatment of Osteoporosis in Postmenopausal Women Previously Treated With Alendronate: A Randomized Placebo-Controlled Trial. J Clin Endocrinol Metab. 2013 Dec;98(12):4727-35. doi: 10.1210/jc.2013-2020. Epub 2013 Sep 24. PubMed PMID: 24064689.
9: Zerbini CA, McClung MR. Odanacatib in postmenopausal women with low bone mineral density: a review of current clinical evidence. Ther Adv Musculoskelet Dis. 2013 Aug;5(4):199-209. doi: 10.1177/1759720X13490860. PubMed PMID: 23904864; PubMed Central PMCID: PMC3728981.
10: Williams DS, McCracken PJ, Purcell M, Pickarski M, Mathers PD, Savitz AT, Szumiloski J, Jayakar RY, Somayajula S, Krause S, Brown K, Winkelmann CT, Scott BB, Cook L, Motzel SL, Hargreaves R, Evelhoch JL, Cabal A, Dardzinski BJ, Hangartner TN, Duong le T. Effect of odanacatib on bone turnover markers, bone density and geometry of the spine and hip of ovariectomized monkeys: a head-to-head comparison with alendronate. Bone. 2013 Oct;56(2):489-96. doi: 10.1016/j.bone.2013.06.008. Epub 2013 Jun 24. PubMed PMID: 23806798.
11: Cabal A, Jayakar RY, Sardesai S, Phillips EA, Szumiloski J, Posavec DJ, Mathers PD, Savitz AT, Scott BB, Winkelmann CT, Motzel S, Cook L, Hargreaves R, Evelhoch JL, Dardzinski BJ, Hangartner TN, McCracken PJ, Duong le T, Williams DS. High-resolution peripheral quantitative computed tomography and finite element analysis of bone strength at the distal radius in ovariectomized adult rhesus monkey demonstrate efficacy of odanacatib and differentiation from alendronate. Bone. 2013 Oct;56(2):497-505. doi: 10.1016/j.bone.2013.06.011. Epub 2013 Jun 20. PubMed PMID: 23791777.
12: Stoch SA, Witter R, Hreniuk D, Liu C, Zajic S, Mehta A, Brandquist C, Dempsey C, Degroot B, Stypinski D, Denker A, Wagner JA. Absence of clinically relevant drug-drug interaction between odanacatib and digoxin after concomitant administration. Int J Clin Pharmacol Ther. 2013 Aug;51(8):688-92. doi: 10.5414/CP201864. PubMed PMID: 23782582.
13: Nakamura T, Shiraki M, Fukunaga M, Tomomitsu T, Santora AC, Tsai R, Fujimoto G, Nakagomi M, Tsubouchi H, Rosenberg E, Uchida S. Effect of the cathepsin K inhibitor odanacatib administered once weekly on bone mineral density in Japanese patients with osteoporosis-a double-blind, randomized, dose-finding study. Osteoporos Int. 2014 Jan;25(1):367-76. doi: 10.1007/s00198-013-2398-2. Epub 2013 May 29. PubMed PMID: 23716037.
14: Brixen K, Chapurlat R, Cheung AM, Keaveny TM, Fuerst T, Engelke K, Recker R, Dardzinski B, Verbruggen N, Ather S, Rosenberg E, de Papp AE. Bone density, turnover, and estimated strength in postmenopausal women treated with odanacatib: a randomized trial. J Clin Endocrinol Metab. 2013 Feb;98(2):571-80. doi: 10.1210/jc.2012-2972. Epub 2013 Jan 21. PubMed PMID: 23337728.
15: Fratzl-Zelman N, Roschger P, Fisher JE, Duong le T, Klaushofer K. Effects of Odanacatib on bone mineralization density distribution in thoracic spine and femora of ovariectomized adult rhesus monkeys: a quantitative backscattered electron imaging study. Calcif Tissue Int. 2013 Mar;92(3):261-9. doi: 10.1007/s00223-012-9673-7. Epub 2012 Nov 23. PubMed PMID: 23179105.
16: Stoch SA, Zajic S, Stone JA, Miller DL, van Bortel L, Lasseter KC, Pramanik B, Cilissen C, Liu Q, Liu L, Scott BB, Panebianco D, Ding Y, Gottesdiener K, Wagner JA. Odanacatib, a selective cathepsin K inhibitor to treat osteoporosis: safety, tolerability, pharmacokinetics and pharmacodynamics–results from single oral dose studies in healthy volunteers. Br J Clin Pharmacol. 2013 May;75(5):1240-54. doi: 10.1111/j.1365-2125.2012.04471.x. PubMed PMID: 23013236; PubMed Central PMCID: PMC3635595.
17: Ng KW. Potential role of odanacatib in the treatment of osteoporosis. Clin Interv Aging. 2012;7:235-47. doi: 10.2147/CIA.S26729. Epub 2012 Jul 12. Review. PubMed PMID: 22866001; PubMed Central PMCID: PMC3410681.
18: Langdahl B, Binkley N, Bone H, Gilchrist N, Resch H, Rodriguez Portales J, Denker A, Lombardi A, Le Bailly De Tilleghem C, Dasilva C, Rosenberg E, Leung A. Odanacatib in the treatment of postmenopausal women with low bone mineral density: five years of continued therapy in a phase 2 study. J Bone Miner Res. 2012 Nov;27(11):2251-8. doi: 10.1002/jbmr.1695. PubMed PMID: 22777865.
19: Jayakar RY, Cabal A, Szumiloski J, Sardesai S, Phillips EA, Laib A, Scott BB, Pickarski M, Duong le T, Winkelmann CT, McCracken PJ, Hargreaves R, Hangartner TN, Williams DS. Evaluation of high-resolution peripheral quantitative computed tomography, finite element analysis and biomechanical testing in a pre-clinical model of osteoporosis: a study with odanacatib treatment in the ovariectomized adult rhesus monkey. Bone. 2012 Jun;50(6):1379-88. doi: 10.1016/j.bone.2012.03.017. Epub 2012 Mar 24. PubMed PMID: 22469953.
20: Khosla S. Odanacatib: location and timing are everything. J Bone Miner Res. 2012 Mar;27(3):506-8. doi: 10.1002/jbmr.1541. PubMed PMID: 22354850.
21 nmr……..http://www.medkoo.com/Product-Data/Odanacatib/Odanacatib-QC-BBC20130906Web.pdf
http://www.medkoo.com/Product-Data/Odanacatib/JOC2009p1605-NMR-Data.pdf

| WO2003075836A2 | Feb 28, 2003 | Sep 18, 2003 | Axys Pharm Inc | Cathepsin cysteine protease inhibitors |
| WO2005019161A1 | Aug 19, 2004 | Mar 3, 2005 | Merck Frosst Canada Inc | Cathepsin cysteine protease inhibitors |
| WO2005021487A1 | Aug 23, 2004 | Mar 10, 2005 | Christopher Bayly | Cathepsin inhibitors |
| WO2006034004A2 | Sep 16, 2005 | Mar 30, 2006 | Axys Pharm Inc | Processes and intermediates for preparing cysteine protease inhibitors |
| CA2477657A1 * | Feb 28, 2003 | Sep 18, 2003 | Axys Pharmaceuticals, Inc. | Cathepsin cysteine protease inhibitors |
Lems, Willem F.; Geusens, Piet. Established and forthcoming drugs for the treatment of osteoporosis. Current Opinion in Rheumatology (2014), 26(3), 245-251.
Schwarz, Peter; Jorgensen, Niklas Rye; Abrahamsen, Bo. Status of drug development for the prevention and treatment of osteoporosis. Expert Opinion on Drug Discovery (2014), 9(3), 245-253.
Anderson, Matt S.; Gendrano, Isaias Noel; Liu, Chengcheng; Jeffers, Steven; Mahon, Chantal; Mehta, Anish; Mostoller, Kate; Zajic, Stefan; Morris, Denise; Lee, Jessie; et al. Odanacatib, a selective cathepsin K inhibitor, demonstrates comparable pharmacodynamics and pharmacokinetics in older men and postmenopausal women.Journal of Clinical Endocrinology and Metabolism (2014), 99(2), 552-560.
Nelo Rivera, Yadagiri R. Pendri, Sreenivas MENDE, Bramhananda N. REDDY. Process for preparing fluoroleucine alkyl esters. PCT Int. Appl., WO2013148554 A1,Oct 3, 2013
Humphrey, Guy and Yong, Kelvin. Improved amidation process for the preparation of dipeptide nitriles from fluorinated amino acids in the absence of HOBt coupling agent. PCT Int. Appl., WO2012148555, 01 Nov 2012
Kassahun, Kelem et al. Cathepsin cysteine protease inhibitors. PCT Int. Appl., WO2012112363, 23 Aug 2012
Qiu, Xiao-Long; Yue, Xuyi; Qing, Feng-Ling. Fluorine-containing chiral drugs. Chiral Drugs (2011), 195-251.
O’Shea, Paul D. et al. A Practical Enantioselective Synthesis of Odanacatib, a Potent Cathepsin K Inhibitor, via Triflate Displacement of an α-Trifluoromethylbenzyl Triflate. Journal of Organic Chemistry, 74(4), 1605-1610; 2009
O’Shea, Paul and Gosselin, Francis. Amidation process for the preparation of cathepsin K inhibitor 4-fluoro-N-[(S)-2,2,2-trifluoro-1-[4′-(methylsulfonyl)-1,1′-biphenyl-4-yl]ethyl]-L-leucine 1-cyanocyclopropylamide. PCT Int. Appl., WO2008119176, 09 Oct 2008
Gauthier, Jacques Yves et al. The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorganic & Medicinal Chemistry Letters, 18(3), 923-928; 2008
Sarah J. Dolman, Francis Gosselin, Paul D. O’Shea, Ian W. Davies. Selective metal-halogen exchange of 4,4′-dibromobiphenyl mediated by lithium tributylmagnesiate. Tetrahedron, 2006, 62, 5092–5098
Gauthier, Jacques Yves and Truong, Vouy Linh. Preparation of amino acid derivatives as cathepsin cysteine protease inhibitors. PCT Int. Appl., WO2005019161, 03 Mar 2005
Bayly, Christopher et al. Preparation of amino acid derivatives as cathepsin inhibitors. PCT Int. Appl., WO2005021487, 10 Mar 2005
Limanto, John et al. An Efficient Chemoenzymatic Approach to (S)-γ-Fluoroleucine Ethyl Ester. Journal of Organic Chemistry, 70(6), 2372-2375; 2005
Devine, Paul et al. Process for preparing fluoroleucine alkyl esters. U.S. Pat. Appl. Publ., US20050234128, 20 Oct 2005
Bayly, Christopher I. et al. Cathepsin cysteine protease inhibitors and their therapeutic use. PCT Int. Appl., WO2003075836, 18 Sep 2003
updated


{kind=link}