
Home » Phase2 drugs (Page 5)
Category Archives: Phase2 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
ADAFOSBUVIR, адафосбувир , أدافوسبوفير ,
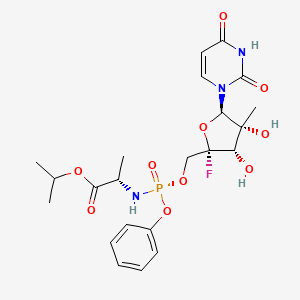


ADAFOSBUVIR
AL335; ALS-335; JNJ-64146212 , D11364
Propan-2-yl N-((P5’S)-4′-fluoro-2′-C-methyl-p-o-phenyl- 5′-uridylyl)-L-alaninate
propan-2-yl (2S)-2-{[(S)-{[(2S,3S,4R,5R)-5-(2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-yl)-2-fluoro-3,4-dihydroxy-4-methyloxolan-2-yl]methoxy}(phenoxy)phosphoryl]amino}propanoate
Isopropyl (2S)-2-{[(S)-{[(2S,3S,4R,5R)-5-(2,4-dioxo-3,4-dihydro-1(2H)-pyrimidinyl)-2-fluoro-3,4-dihydroxy-4-methyltetrahydro-2-furanyl]methoxy}(phenoxy)phosphoryl]amino}propanoate (non-preferred name
Propan-2-yl N-((P5’S)-4′-fluoro-2′-C-methyl-p-o-phenyl- 5′-uridylyl)-L-alaninate
545.5 g/mol, C22H29FN3O10P
CAS Registry Number 1613589-09-5
Adafosbuvir is under investigation in clinical trial NCT02894905 (A Study to Evaluate the Effect of Renal Impairment on the Pharmacokinetics of AL-335).
- Originator Alios BioPharma
- Developer Alios BioPharma; Janssen
- Class Antivirals; Pyrimidine nucleotides; Uracil nucleotides
- Mechanism of Action Hepatitis C virus NS 5 protein inhibitors
- Phase II Hepatitis C
- 28 Oct 2019 No recent reports of development identified for phase-I development in Hepatitis-C(In volunteers) in USA (PO)
- 28 Sep 2018 No recent reports of development identified for phase-I development in Hepatitis-C in France (PO)
- 28 Sep 2018 No recent reports of development identified for phase-I development in Hepatitis-C in Georgia (PO)
Adafosbuvir (AL 335), a monophosphate prodrug, is being developed by Alios BioPharma (a subsidiary of Johnson & Johnson) for the treatment of hepatitis C virus (HCV) infections. Adafosbuvir acts a uridine-based nucleotide analogue polymerase inhibitor. Clinical development is underway in New Zealand, Japan, the UK, the US, France, Georgia, Mauritius and Moldova.
Adafosbuvir has emerged from the company’s research programme focused on developing anti-viral nucleotides for the treatment of HCV infections , In November 2014, Alios BioPharma was acquired by Johnson & Johnson As at September 2018, no recent reports of development had been identified for phase-I development in Hepatitis-C in France (PO), Georgia (PO).
As at October 2019, no recent reports of development had been identified for phase-I development in Hepatitis-C (In volunteers) in USA (PO).
useful for the treatment of hepatitis C viral infections, assignaed to Janssen Pharmaceuticals Inc and Achillion Pharmaceuticals Inc . Janssen Pharmaceuticals, following Johnson & Johnson’s acquisition of Alios , was developing adafosbuvir, a uridine (pyrimidine) nucleotide analog, from a series of back-up compounds, that acts by inhibiting HCV NS5B polymerase, for the potential treatment of HCV infection.
As of December 2019, AL-335 dose increased from 400 to 800 mg qd in the presence of reduced simeprevir and odalasvlr doses increased ALS-022227 less than dose proportionally. However, this effect was minimal in the absence of slmeprevir [1973148]. Also, the company was also developing JNJ-4178 , a triple combination of adafosbuvir, odalasvir and simeprevir for the same indication.

McGuigan phosphoramidate nucleotide prodrugs. (a) Sofosbuvir (GS-7977) (Sp isomer), (b) BMS-986094 (Rp and Sp isomer mixture), (c) Adafosbuvir (AL-335) (Sp isomer), (d) ACH-3422*, and (e) MIV-802* (Sp isomer)

Figure 3. Clinical and preclinical 30,50-CPO prodrug. (a) GS-0938 (Rp isomer) and (b) IDX19368 (Sp isomer).

PAPER
Journal of Medicinal Chemistry (2019), 62(9), 4555-4570.
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.9b00143
We report the synthesis and biological evaluation of a series of 4′-fluoro-2′-C-substituted uridines. Triphosphates of the uridine analogues exhibited a potent inhibition of hepatitis C virus (HCV) NS5B polymerase with IC50values as low as 27 nM. In an HCV subgenomic replicon assay, the phosphoramidate prodrugs of these uridine analogues demonstrated a very potent activity with EC50 values as low as 20 nM. A lead compound AL-335(53) demonstrated high levels of the nucleoside triphosphate in vitro in primary human hepatocytes and Huh-7 cells as well as in dog liver following a single oral dose. Compound 53 was selected for the clinical development where it showed promising results in phase 1 and 2 trials.

PATENT
WO 2014209979
WO2014100505
Family members of the product case of adafosbuvir, WO2014100505 , expire in the US in December 2033.
PATENT
US 20150368286
WO 2015054465
PATENT
WO2017059147 ( US20170087174 ), claiming combination comprising simeprevir , odalasvir and AL-335
PATENT
WO-2019237297
Process for preparing AL-335 (also known as adafosbuvir) and its intermediates. AL-355 is a nucleoside inhibitor of NS3B polymerase, which plays an important role in the replication of the hepatitis C virus.



Compound 4 may be prepared in accordance with the procedures described in international patent application WO 2015/200216. Compound 4 (1.0 equiv) was then dissolved in THF (10 L/kg) and cooled down to -25℃. iPrMgCl (2M in THF) was added slowly over one hour and the resulting mixture was stirred for one hour. The Compound 3 solution previously made (see above) was then added dropwise at -25℃ and the mixture was stirred for 5h at that temperature before being warmed to -5℃ and stirred for 10 additional hours at that temperature. Once the reaction was complete, the reaction was warmed up to 5℃ and an aqueous solution of NH 4Cl (5L/kg -9 w/w%) was added slowly over 30 minutes. After phase separation, the organic layer was washed with aqueous NaHCO 3 solution (5L/kg -10 w/w%) and twice with aqueous NaCl solution (5L/kg -10 w/w%) . After solvent switch to acetonitrile, the reaction was assayed and stored under nitrogen and used as such in the next step.
/////////////ADAFOSBUVIR, AL335, ALS-335, JNJ-64146212, Alios BioPharma, Janssen, hepatitis C viral infections, D11364, адафосбувир , أدافوسبوفير , PHASE 2
CC(C)OC(=O)[C@H](C)N[P@](=O)(OC[C@@]1(F)O[C@@H](N2C=CC(=O)NC2=O)[C@](C)(O)[C@@H]1O)OC1=CC=CC=C1
Blarcamesine, ブラルカメシン ,

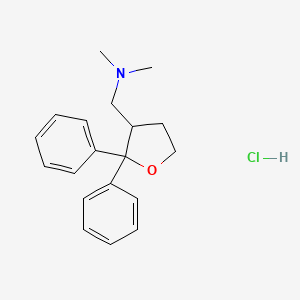
Blarcamesine
ブラルカメシン;
[(2,2-diphenyloxolan-3-yl)methyl]dimethylamine
- Anavex 2-73
- Tetrahydro-N,N-dimethyl-2,2-diphenyl-3-furanemethanamine
- THD-DP-FM
- AE-37 / AE37 / ANAVEX 2-73 FREE BASE
- UNII 9T210MMZ3F
| Formula |
C19H23NO
|
|---|---|
| Cas |
195615-83-9
195615-84-0 HCL
|
| Mol weight |
281.392
|
Treatment of Rett syndrome, Investigated for use/treatment in breast cancer.
Anti-amnesic, Muscarinic/sigma receptor agonist
- Originator Anavex Life Sciences
- Developer ABX-CRO; Anavex Life Sciences; The Michael J. Fox Foundation for Parkinsons Research
- Class Antidementias; Antidepressants; Antiepileptic drugs; Antiparkinsonians; Anxiolytics; Behavioural disorder therapies; Dimethylamines; Furans; Neuroprotectants; Neuropsychotherapeutics; Nootropics; Small molecules
- Mechanism of Action Muscarinic receptor modulators; Sigma-1 receptor agonists
- Orphan Drug Status Yes – Epilepsy; Rett syndrome
- Phase II/III Alzheimer’s disease
- Phase II Parkinson’s disease; Rett syndrome
- Preclinical Amyotrophic lateral sclerosis; Angelman syndrome; Anxiety disorders; Autistic disorder; Fragile X syndrome; Multiple sclerosis
- No development reported Cognition disorders; Epilepsy; Stroke
- 28 Oct 2019 No recent reports of development identified for phase-I development in Cognition-disorders in USA
- 09 Oct 2019 Anavex Life Sciences initiates enrolment in the long term extension ATTENTION-AD trial for Alzheimer’s disease in (country/ies)
- 02 Oct 2019 Anavex Life Sciences has patent protection covering compositions of matter and methods of treating Alzheimer’s disease for blarcamesine in USA
- Anavex Life Sciences is developing ANAVEX-2-73 and its active metabolite ANAVEX-19-144, for treating Alzheimer’s disease, epilepsy, stroke and Rett syndrome.
ANAVEX2-73 is an experimental drug is in Phase II trials for Alzheimer’s disease, phase I trials for epilepsy, and in preclinical trials for amyotrophic lateral sclerosis, Parkinson’s disease, Rett syndrome, stroke.[1][2] ANAVEX2-73 acts as a muscarinic receptor and a moderate sigma1 receptor agonist.[1] ANAVEX2-73 may function as a pro-drug for ANAVEX19-144 as well as a drug itself. ANAVEX19-144 is the active metabolite of ANAVEX 1-41, which is similar to ANAVEX2-73 but it is not as selective for sigma receptor.[2]
Properties and uses
ANAVEX2-73 has an inhibitory constant (ki) lower than 500 nM for all M1–M4 muscarinic acetylcholine receptor subtypes, demonstrating that it acts as a powerful antimuscarinic compound.[2] ANAVEX2-73 was originally tested in mice against the effect of the muscarinic receptor antagonist scopolamine, which induces learning impairment.[1] M1 receptor agonists are known to reverse the amnesia caused by scopolamine.[3] Scopolamine is used in the treatment of Parkinson’s disease and motion sickness by reducing the secretions of the stomach and intestines and can also decreases nerve signals to the stomach.[3] This is via competitive inhibition of muscarinic receptors.[3] Muscarinic receptors are involved in the formation of both short term and long term memories.[1] Experiments in mice have found that M1 and M3 receptor agonists inhibit the formation of amyloid-beta and target GSK-3B.[clarification needed]Furthermore, stimulation of the M1 receptor activates AF267B, which in turn blocks β-secretase, which cleaves the amyloid precursor protein to produce the amyloid-beta peptide. These amyloid-beta peptides aggregate together to form plaques. This enzyme[clarification needed] is involved in the formation of Tau plaques, which are common in Alzheimer’s disease.[clarification needed][4]Therefore. M1 receptor activation appears to decreases tau hyperphosphorylation and amyloid-beta accumulation.[4]
Sigma1 activation appears to be only involved in long-term memory processes. This partly explains why ANAVEX2-73 seems to be more effective in reversing scopolamine-induced long-term memory problems compared to short-term memory deficits.[1] The sigma-1 receptor is located on mitochondria-associated endoplasmic reticulum membranes and modulates the ER stress response and local calcium exchanges with the mitochondria. ANAVEX2-73 prevented Aβ25-35-induced increases in lipid peroxidation levels, Bax/Bcl-2ratio and cytochrome c release into the cytosol, which are indicative of elevated toxicity.[clarification needed] ANAVEX2-73 inhibits mitochondrial respiratory dysfunction and therefore prevents against oxidative stress and apoptosis. This drug prevented the appearance of oxidative stress. ANAVEX2-73 also exhibits anti-apoptotic and anti-oxidant activity. This is due in part because sigma-1 agonists stimulate the anti-apoptoic factor Bcl-2 due to reactive oxygen species dependent transcriptional activation of nuclear factor kB.[5] Results from Marice (2016) demonstrate that sigma1 compounds offer a protective potential, both alone and possibly with other agents like donepezil, an acetylcholinesterase inhibitor, or the memantine, a NMDA receptor antagonist.[6]
PATENT
WO9730983
PATENT
Novel crystalline forms of A2-73 (blarcamesine hydrochloride, ANAVEX2-73, AV2-73), a mixed muscarinic receptor ligand and Sig-1 R agonist useful for treating Alzheimer’s disease.
PATENT
WO2017013498
SYN
By Foscolos, George B. et alFrom Farmaco, 51(1), 19-26; 1996

References
- ^ Jump up to:a b c “ANAVEX 2-73 – AdisInsight”. Adisinsight.springer.com. Retrieved 2016-05-25.
- ^ Jump up to:a b c Malviya, M; Kumar, YC; Asha, D; Chandra, JN; Subhash, MN; Rangappa, KS (2008). “Muscarinic receptor 1 agonist activity of novel N-arylthioureas substituted 3-morpholino arecoline derivatives in Alzheimer’s presenile dementia models”. Bioorg Med Chem. 16: 7095–7101. doi:10.1016/j.bmc.2008.06.053.
- ^ Jump up to:a b Leal, NS; Schreiner, B; Pinho, CM; Filadi, R; Wiehager, B; Karlström, H; Pizzo, P; Ankarcrona, M (2016). “Mitofusin-2 knockdown increases ER-mitochondria contact and decreases amyloid β-peptide production”. J Cell Mol Med. 20: 1686–1695. doi:10.1111/jcmm.12863. PMC 4988279. PMID 27203684.
- ^ Lahmy, V; Long, R; Morin, D; Villard, V; Maurice, T (2015-09-28). “Mitochondrial protection by the mixed muscarinic/σ1 ligand ANAVEX2-73, a tetrahydrofuran derivative, in Aβ25-35 peptide-injected mice, a nontransgenic Alzheimer’s disease model”. Front Cell Neurosci. 8: 463. doi:10.3389/fncel.2014.00463. PMC 4299448. PMID 25653589.
- ^ Maurice, T (2015-09-28). “Protection by sigma-1 receptor agonists is synergic with donepezil, but not with memantine, in a mouse model of amyloid-induced memory impairments”. Behav. Brain Res. 296: 270–8. doi:10.1016/j.bbr.2015.09.020. PMID 26386305.
//////////Blarcamesine, ブラルカメシン , Orphan Drug Status, PHASE 2
CN(C)CC1CCOC1(C1=CC=CC=C1)C1=CC=CC=C1
Vidofludimus
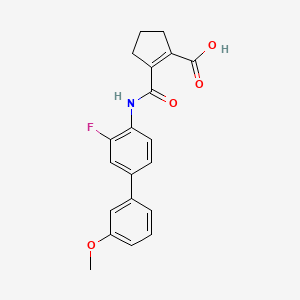
Vidofludimus
2-[[2-fluoro-4-(3-methoxyphenyl)phenyl]carbamoyl]cyclopentene-1-carboxylic acid
355.4 g/mol, C20H18FNO4
CAS 717824-30-1
4SC-101
UNII-8Y1PJ3VG81
SC12267
Vidofludimus calcium anhydrous
RN: 1354012-90-0
UNII: FW5VY7926X
IM-90838
IMU-838
Molecular Formula, 2C20-H17-F-N-O4.Ca, Molecular Weight, 748.7886
1-Cyclopentene-1-carboxylic acid, 2-(((3-fluoro-3′-methoxy(1,1′-biphenyl)-4-yl)amino)carbonyl)-, calcium salt (2:1)
Inflammatory Bowel Disease,
Immunosuppressants
Multiple Sclerosis,
Rheumatoid Arthritis,
Liver and Biliary Tract Disorders,
Antipsoriatics
Systemic Lupus Erythematosus,
Dihydroorotate Dehydrogenase (DHODH) Inhibitors
phase II clinical development at Immunic (previously Immunic AG) as an induction and maintenance therapy for patients with moderate to severe ulcerative colitis, as well as for the treatment of patients with relapsed-remitting multiple sclerosis (RRMS). Immunic is also conducting early clinical evaluation of the drug as a potential treatment for Crohn’s disease, whereas a phase II clinical trial is ongoing at the Mayo Clinic in patients suffering from primary sclerosing cholangitis.
In 2016, Immunic acquired the product from 4SC.
Vidofludimus is under investigation in clinical trial NCT03722576 (Vidofludimus Calcium for Primary Sclerosing Cholangitis).
Ca salt of vidofludimus (designated as form A) as dihydroorotate dehydrogenase (DHODH) inhibitor eg graft versus host disease, rheumatoid arthritis and multiple sclerosis
Immunic AG (a subsidiary of Immunic Inc ), following an asset acquisition from 4SC, is developing vidofludimus an orally available, small molecule DHODH inhibitor and IL-17 blocker which inhibits pyrimidine biosynthesis, for the treatment of autoimmune and inflammatory disorders including ulcerative colitis, Crohn’s disease and multiple sclerosis
PAPER
Bioorganic & Medicinal Chemistry Letters (2005), 15(21), 4854-4857
https://www.sciencedirect.com/science/article/pii/S0960894X05010127

PRODUCT PATENT
WO 03006424
SPC protection in most of the EU states until 2021 and expire in the US in January 2022 with US154 extension
PATENT
WO2019101888 claiming composition comprising vidofludimus
PATENT
WO 2012001151
WO 2016200778
WO 2018177151
PATENT
WO2012001148 claiming similar compound (assigned to 4SC Ag ) naming the inventor Daniel Vitt. Immunic AG (a subsidiary of Immunic Inc ),
Example 4: Preparation of the calcium salts
300.4 mg of Vidofludimus free acid was dissolved in 18 mL of DCM/MeOH (3:1) and sonicated for 8 minutes. 31.5 mg of calcium hydroxide was suspended in 3 mL of DCM/MeOH (3:1); this was slowly added to the Vidofludimus free acid solution. The slight suspension was stirred overnight at 25°C. The solvent was partially evaporated under nitrogen flow at 25°C. A thick light yellow suspension was observed. The solid was recovered by filtration and washed with DCM/MeOH (3:1). The material was dried for 15 min under vacuum at 25°C. The material was shown to be crystalline using the methods described in the following.
From elemental analysis, the ratio of fluorine to calcium was calculated. The elemental composition is essentially consistent with a hemi-calcium-salt.
The Raman spectrum of the newly formed compound demonstrated differences to that of the free acid (see Figure 3 for both spectra.). Note that a Raman spectrum that is not simply the superposition of the free acid, the salt former and the solvent spectra, e.g., a Raman spectrum where new peaks or shifted peaks are observed, may correspond to a salt.
However, from the Raman spectrum alone, it cannot be determined whether crystalline salt formation has occurred. Peak shifts could also be due, in principle, to complexation of the free acid and salt former as an amorphous product, to polymorphs of either the free acid or salt former, to impurities, or to degradation products. Therefore, the integrity of the molecular structure was confirmed by 1H-NMR.
In addition, the powder X-ray diffraction shown in Figure 5 show that crystalline material was obtained, however with a pattern different from that of the free acid (see Figure 6). With light microscopy the crystals were visualized (Figure 4), DSC (differential scanning calorimetry) demonstrated a melting point of about 155°C (indicating a melting of a solvate and of a non-solvated form), TG-FTIR (thermogravimetric analyzer-coupled Fourier-Transform Infrared) indicates that probably a methanol solvate and a hydrate were formed and dynamic vapor sorption revealed desolvation followed by 0.3% water uptake at about 85% r.h. and 0.4% water uptake at 95% r.h. (not reversible).
PATENT
WO-2019175396
Novel white crystalline calcium salt of vidofludimus and its solvates and hydrates (designated as polymorph A), process for its preparation, composition comprising it and its use for the treatment of rheumatoid arthritis, multiple sclerosis, inflammatory bowel disease, psoriasis, amyotrophic lateral sclerosis, lupus erythematosus, fibrosis, uveitis, rhinitis and Pneumocystis carinii are claimed. Vidofludimus is known to be an IL-17 antagonist, immunosuppressant and dihydroorotate dehydrogenase inhibitor.
Novel calcium salt polymorphs as Anti-Inflammatory, Immunomodulatory and Anti- Proliferatory Agents
Subject matter of the present invention is a white crystalline polymorph A of the Ca salt of a compound according to formula I or a solvate and/or a hydrate thereof with a molar ratio of a compound according to formula 1 or a solvate and/or a hydrate thereof to calcium which is 2±0.3. Subject matter of the present invention is in particular a compound according to formula I or a solvate and/or a hydrate thereof which is characterized by an X-ray powder diffraction pattern having characteristic peaks expressed in degrees 2theta at ±0.2 of the values shown below: 2 theta = 5.91°, 9.64°, 16.78°, 17.81°, 19.81°, 25.41° In particular the invention refers to new polymorphs of calcium salts of the Ca salt of a compound according to formula I or a solvate and/or a hydrate thereof which inhibits dihydroorotate dehydrogenase (DHODH), a process for their manufacture, pharmaceutical compositions containing them and to their use for the treatment and prevention of diseases, in particular their use in diseases where there is an advantage in inhibiting dihydroorotate dehydrogenase (DHODH). Examples of relevant diseases are given below.
Inflammatory Bowel Disease (IBD) is a group of inflammatory conditions of the colon and small intestine. With Crohn’s Disease and Ulcerative Colitis as principal types thereof. Crohn’s disease can affect the small intestine and large intestine, as well as the mouth, esophagus, stomach and the anus. Ulcerative colitis primarily affects the colon and the rectum.
Rheumatoid arthritis (RA) is a disease that is quite common especially among elder people. Its treatment with usual medications as for example non-steroid anti-inflammatory agents is not satisfactory. In view of the increasing ageing of the population, especially in the developed Western countries or in Japan the development of new medications for the treatment of RA is urgently required.
WO 2003/006425 describes certain specific compounds, which are reported to be useful for treatment and prevention of diseases where there is an advantage in inhibiting dihydroorotate dehydrogenase (DHODH). However, the specific salts according to the present invention are not disclosed. WO 2012/001148 describes the calcium salts of said compounds. However, the specific polymorphs according to the present invention are not disclosed.
WO 99/38846 and EP 0 646 578 disclose compounds which are reported to be useful for treatment of RA.
A medicament against rheumatoid arthritis with a new mechanism of action, leflunomide, was put on the market by the company Aventis under the tradename ARAVA [EP 780128, WO 97/34600]. Leflunomide has immunomodulatory as well as anti-inflammatory properties [EP 217206, DE 2524929]. The mechanism of action is based upon the inhibition of dihydroorotate dehydrogenase (DHODH), an enzyme of the pyrimidine biosynthesis.
De Julian-Ortiz (J. Med. Chem. 1999, 42, 3308-3314) describes certain potential Anti-Herpes compounds with cyclopentenoic acid moieties.
DE 33 46 814 A1 describes certain carbonic acid amide derivatives for the treatment, prevention and amelioration of diseases connected to cerebral dysfunction and symptoms caused thereby.
In the human body, DHODH catalyzes the synthesis of pyrimidines, which are in particular necessary for cellular metabolism. An inhibition of DHODH leads to block of transcription of sensitive genes in metabolically activated cells, whereas cells with normal metabolic activity obtain their required pyrimidine building blocks from the pyrimidine salvage pathway and show normal transcriptional activity. Disease relevant activated lymphocytes rely on de novo pyrimidine syntheses and react particularly sensitively to DHODH inhibition. Some substances that inhibit DHODH are important medicaments for the treatment of chronic inflammatory and auto-immune diseases.
A compound named leflunomide (ARAVA) has been the first approved inhibitor of DHODH and is used for the treatment of human diseases, in particular rheumatoid arthritis. WO 99/45926 is a further reference that discloses compounds which act as inhibitors of DHODH. Another drug which is targeting DHODH is teriflunomide (AUBAGIO®) is the metabolite of leflunomide. Teriflunomide is approved for the treatment of multiple sclerosis in some countries.
JP-A-50-121428 discloses N-substituted cyclopentene-l,2-dicarboxylic acid monoamides as herbicides and their syntheses. For example, N-(4-chlorophenyl)-l-cyclopentene-l,2-dicarboxylic acid monoamide is produced by reacting l-cyclopentene-l,2-dicarboxylic anhydride with 4- chloroaniline.
In the Journal of Med. Chemistry, 1999, Vol. 42, pages 3308-3314, virtual combinatorial syntheses and computational screening of new potential Anti-Herpes compounds are described. In Table 3 on page 3313 experimental results regarding IC50 and cytotoxicity are presented for 2-(2,3-difluorophenylcarbamoyl)-l -cyclopentene- 1 -carboxylic acid, 2-(2,6-difluorophenylcarbamoyl)-l -cyclopentene-l -carboxylic acid and 2-(2,3,4-trifluorophenyl-carbamoyl)- 1 -cyclopentene- 1 -carboxylic acid.
DE 3346814 and US 4661630 disclose carboxylic acid amides. These compounds are useful for diseases attended with cerebral dysfunction and also have anti-ulcer, anti-asthma, anti-inflammatory and hypo-cholesterol activities.
In EP 0097056, JP 55157547, DE 2851379 and DE 2921002 tetrahydrophthalamic acid derivatives are described.
It is an object of the present invention to provide effective agents, specifically in the form of certain polymorphs of their calcium salts, which can be used for the treatment of diseases which require the inhibition of DHODH.
It was also an object of the present invention to provide compounds that inhibit DHODH in a range similar to the compounds disclosed in W02003/006425 and WO 2012/001148 and at the same time show a white colour in order to facilitate double blind placebo controlled clinical studies.
It was also an object of the present invention to provide compounds and composition comprising that compounds that inhibit DHODH in a range similar to the compounds disclosed in
W02003/006425 and WO 2012/001148 and are characterized by having a THF content below
720 ppm in order to be in compliance with guidelines of the European Medicines Agency (e.g. with the version 6 December 2016 ; EMA/CHMP/ICH/82260/2006)
Particularly, it has previously been found that certain compounds of the general formula (I) shown herein below, such as 2-(3-Fluoro-3′-methoxy-biphenyl-4-ylcarbamoyl)-cyclopent-l-enecarboxylic acid (INN Vidofludimus), exhibit good anti-inflammatory activity and their usability in the oral therapy for the treatment of autoimmune diseases such as for example rheumatoid arthritis or inflammatory bowel diseases had been addressed.
Accordingly, a novel white polymorph of Calcium- vidofludimus named polymorph A with an inhibitory effect on DHODH, in particular human DHODH, was provided. Furthermore, a composition was provided comprising said white polymorph of Calcium-vidofludimus named polymorph A characterized by having a Tetrahydroduran (THF) content below 720 ppm.
[I,I ‘ – biphenyl] – 4 – yl}carbamoyl)cyclopent – 1 – ene – 1 – carboxylic acid) according to formula (I) or a solvate and/or a hydrate thereof, CAS-No 717824-30-1white crystalline calcium salt of 2 – ({3 – fluoro – 3’ – methoxy –
Thus, subject matter of the present invention is a white crystalline calcium salt of vidofludimus with a molar ratio of vidofludimus to calcium is 2±0.3 or a solvate and/or a hydrate thereof. In contrast to the pale yellow polymorph as described in EP 2588446B1, e.g. example 4, subject matter of the present invention is of white color.
White crystal can be defined as crystals with pure white color similar to the RAL color code RAL9010 that is equal or similar to the US Federal Standard 595 color code“White 506”, #27885.
A solvate for all embodiments of the invention maybe selected from the group comprising ethanol, propanol, isopropanol, butanol, ΊΊ IF, water. In a preferred embodiment for all embodiments of the invention the solvate is a hydrate. In one preferred embodiment the solvate is a calcium dihydrate for all embodiments of the invention.
In particular, subject matter of the present invention is a white crystalline polymorph A of the Ca salt of a compound according to formula I (vidofludimus) or a solvate and/or a hydrate thereof thereof which is characterized by an X-ray powder diffraction pattern having characteristic peaks expressed in degrees 2theta at ±0.2 of the values shown below:
2 theta = 5.91°, 9.64°, 16.78°, 17.81°, 19.81°, 25.41 °
PATENT
https://patents.google.com/patent/WO2012001151A1/en
/////////vidofludimus, PHASE 2, Inflammatory Bowel Disease, (IBD), Crohn’s Disease, Ulcerative Colitis, 4SC-101, UNII-8Y1PJ3VG81, SC12267, IM-90838, IMU-838, Immunic, 4SC,
COC1=CC=CC(=C1)C2=CC(=C(C=C2)NC(=O)C3=C(CCC3)C(=O)O)F
[Ca+2].COc1cccc(c1)c2ccc(NC(=O)C3=C(CCC3)C(=O)[O-])c(F)c2.COc4cccc(c4)c5ccc(NC(=O)C6=C(CCC6)C(=O)[O-])c(F)c5
CK-101
![N-[3-[2-[2,3-Difluoro-4-[4-(2-hydroxyethyl)piperazin-1-yl]anilino]quinazolin-8-yl]phenyl]prop-2-enamide.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=117909640&t=l)

CK-101, RX-518
CAS 1660963-42-7
N-[3-[2-[[2,3-Difluoro-4-[4-(2-hydroxyethyl)piperazin-1-yl]phenyl]amino]quinazolin-8-yl]phenyl]acrylamide
N-(3-(2-((2,3-Difluoro-4-(4-(2-hydroxyethyl)piperazin-1-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide
EGFR-IN-3
UNII-708TLB8J3Y
Suzhou NeuPharma (Originator)
Checkpoint Therapeutics
Non-Small Cell Lung Cancer Therapy
Solid Tumors Therapy
PHASE 2 Checkpoint Therapeutics, Cancer, lung (non-small cell) (NSCLC), solid tumour
RX518(CK-101) is an orally available third-generation and selective inhibitor of certain epidermal growth factor receptor (EGFR) activating mutations, including the resistance mutation T790M, and the L858R and exon 19 deletion (del 19) mutations, with potential antineoplastic activity.
In August 2019, Suzhou Neupharma and its licensee Checkpoint Therapeutics are developing CK-101 (phase II clinical trial), a novel third-generation, covalent, EGFR inhibitor, as a capsule formulation, for the treatment of cancers including NSCLC and other advanced solid tumors. In September 2017, the FDA granted Orphan Drug designation to this compound, for the treatment of EGFR mutation-positive NSCLC; in January 2018, the capsule was being developed as a class 1 chemical drug in China.
CK-101 (RX-518), a small-molecule inhibitor of epidermal growth factor receptor (EGFR), is in early clinical development at Checkpoint Therapeutics and Suzhou NeuPharma for the potential treatment of EGFR-mutated non-small cell lung cancer (NSCLC) and other advanced solid malignancies.
In 2015, Suzhou NeuPharma granted a global development and commercialization license to its EGFR inhibitor program, excluding certain Asian countries, to Coronado Biosciences (now Fortress Biotech). Subsequently, Coronado assigned the newly acquired program to its subsidiary Checkpoint Therapeutics.
In 2017, the product was granted orphan drug designation in the U.S. for the treatment of EGFR mutation-positive NSCLC.
There are at least 400 enzymes identified as protein kinases. These enzymes catalyze the phosphorylation of target protein substrates. The phosphorylation is usually a transfer reaction of a phosphate group from ATP to the protein substrate. The specific structure in the target substrate to which the phosphate is transferred is a tyrosine, serine or threonine residue. Since these amino acid residues are the target structures for the phosphoryl transfer, these protein kinase enzymes are commonly referred to as tyrosine kinases or serine/threonine kinases.
[0003] The phosphorylation reactions, and counteracting phosphatase reactions, at the tyrosine, serine and threonine residues are involved in countless cellular processes that underlie responses to diverse intracellular signals (typically mediated through cellular receptors), regulation of cellular functions, and activation or deactivation of cellular processes. A cascade of protein kinases often participate in intracellular signal transduction and are necessary for the realization of these cellular processes. Because of their ubiquity in these processes, the protein kinases can be found as an integral part of the plasma membrane or as cytoplasmic enzymes or localized in the nucleus, often as components of enzyme complexes. In many instances, these protein kinases are an essential element of enzyme and structural protein complexes that determine where and when a cellular process occurs within a cell.
[0004] The identification of effective small compounds which specifically inhibit signal transduction and cellular proliferation by modulating the activity of tyrosine and serine/threonine kinases to regulate and modulate abnormal or inappropriate cell proliferation, differentiation, or metabolism is therefore desirable. In particular, the identification of compounds that specifically inhibit the function of a kinase which is essential for processes leading to cancer would be beneficial.
[0005] While such compounds are often initially evaluated for their activity when dissolved in solution, solid state characteristics such as polymorphism are also important. Polymorphic forms of a drug substance, such as a kinase inhibitor, can have different physical properties, including melting point, apparent solubility, dissolution rate, optical and mechanical properties, vapor pressure, and density. These properties can have a direct effect on the ability to process or manufacture a drug substance and the drug product. Moreover, differences in these properties
can and often lead to different pharmacokinetics profiles for different polymorphic forms of a drug. Therefore, polymorphism is often an important factor under regulatory review of the ‘sameness’ of drug products from various manufacturers. For example, polymorphism has been evaluated in many multi-million dollar and even multi-billion dollar drugs, such as warfarin sodium, famotidine, and ranitidine. Polymorphism can affect the quality, safety, and/or efficacy of a drug product, such as a kinase inhibitor. Thus, there still remains a need for polymorphs of kinase inhibitors. The present disclosure addresses this need and provides related advantages as well.
PATENT
WO2015027222
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015027222


PATENT
WO-2019157225
Crystalline form II-VIII of the compound presumed to be CK-101 (first disclosed in WO2015027222 ), for treating a disorder mediated by epidermal growth factor receptor (EGFR) eg cancer.
SCHEME A
Scheme B
General Procedures
Example 1: Preparation of the compound of Formula I (N-(3-(2-((2,3-difluoro-4-(4-(2-hydroxyethyl)piperazin-l-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide)
[0253] To a solution of l,2,3-trifluoro-4-nitrobenzene (2.5 g, 14 mmol, 1.0 eq.) in DMF (20 mL) was added K2C03 (3.8 g, 28 mmol, 2.0 eq.) followed by 2-(piperazin-l-yl)ethanol (1.8 g, 14 mmol, 1.0 eq.) at 0 °C and the mixture was stirred at r.t. overnight. The mixture was poured into ice-water (200 mL), filtered and dried in vacuo to afford 2-(4-(2,3-difluoro-4-nitrophenyl)piperazin-l-yl)ethanol (2.7 g, 67.5%).
[0254] To a solution of 2-(4-(2,3-difluoro-4-nitrophenyl)piperazin-l-yl)ethanol (2.7 g, 9.0 mmol) in MeOH (30 mL) was added Pd/C (270 mg) and the resulting mixture was stirred at r.t.
overnight. The Pd/C was removed by filtration and the filtrate was concentrated to afford 2-(4-(4-amino-2,3-difluorophenyl)piperazin-l-yl)ethanol (2.39 g, 99% yield) as off-white solid.
[0255] To a solution of 8-bromo-2-chloroquinazoline (15.4 g, 63.6 mmol, 1 eq. ) and (3-aminophenyl)boronic acid (8.7 g, 63.6 mmol, 1 eq.) in dioxane/H20 (200 mL/20 mL) was added Na2C03 (13.5 g, 127.2 mmol, 2 eq.), followed by Pd(dppf)Cl2 (2.6 g, 3.2 mmol, 0.05 eq.) under N2, then the mixture was stirred at 80 °C for 12 h. Then the solution was cooled to r.t.,
concentrated and the residue was purified via column chromatography (PE/EA=3 :2, v/v) to afford 3-(2-chloroquinazolin-8-yl)aniline as yellow solid (8.7 g, 53.7% yield).
[0256] To a solution of 3-(2-chloroquinazolin-8-yl)aniline (8.7 g, 34 mmol, 1 eq.) in DCM ( 200 mL ) cooled in ice-bath was added TEA (9.5 mL, 68 mmol, 2 eq. ), followed by acryloyl chloride (4.1 mL, 51 mmol, 1.5 eq.) dropwise. The resulting mixture was stirred at r.t. for 1 h, then washed with brine, dried over anhydrous N2S04 concentrated and the residue was purified via column chromatography (PE/EA=l : 1, v:v) to afford N-(3-(2-chloroquinazolin-8-yl)phenyl)acryl amide as yellow solid(6.6 g, 65% yield).
[0257] To a suspension of 2-(4-(4-amino-2,3-difluorophenyl)piperazin-l-yl)ethanol (83 mg,
0.32 mmol, 1 eq.) and N-(3-(2-chloroquinazolin-8-yl)phenyl)acrylamide (100 mg, 0.32 mmol, 1 eq.) in n-BuOH (5 mL) was added TFA (68 mg, 0.64 mmol, 2 eq.) and the resulting mixture was stirred at 90 °C overnight. The mixture was concentrated, diluted with DCM (20 mL) , washed with Na2C03 solution (20 mL), dried over anhydrous Na2S04, concentrated and the residue was purified via column chromatography (MeOH/DCM=l/30, v:v) to afford N-(3-(2-((2,3-difluoro-4-(4-(2-hydroxyethyl)piperazin-l-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide as a yellow solid(l6.3 mg, 9.5% yield). LRMS (M+H+) m/z calculated 531.2, found 531.2. 1H NMR
(CD3OD, 400 MHz) d 9.21 (s, 1 H), 7.19-8.01 (m, 10 H), 8.90 (s, 1 H), 6.41-6.49 (m, 3 H), 5.86 (m, 1 H), 3.98-4.01 (m, 3 H), 3.70-3.76 (m, 3 H), 3.40-3.49 (m, 2 H), 3.37-3.39 (m, 4 H), 3.18 (m, 2H).
Example 2. Preparation of Form I of the compound of Formula I
[0258] Crude compound of Formula I (~30 g, 75% of weight based assay) was dissolved in ethyl acetate (3 L) at 55-65 °C under nitrogen. The resulting solution was filtered via silica gel pad and washed with ethyl acetate (3 L><2) at 55-65 °C. The filtrate was concentrated via vacuum at 30-40 °C to ~2.4 L. The mixture was heated up to 75-85 °C and maintained about 1 hour.
Then cooled down to 50-60 °C and maintained about 2 hours. The heat-cooling operation was repeated again and the mixture was then cooled down to 20-30 °C and stirred for 3 hours. The resulting mixture was filtered and washed with ethyl acetate (60 mL><2). The wet cake was dried via vacuum at 30-40 °C to get (about 16 g) of the purified Form I of the compound of Formula I.
Example 3. Preparation of Form III of the compound of Formula I
[0259] The compound of Formula I (2 g) was dissolved in EtOH (40 mL) at 75-85 °C under nitrogen. n-Heptane (40 mL) was added dropwise into reaction at 75-85 °C. The mixture was stirred at 75-85 °C for 1 hour. Then cooled down to 50-60 °C and maintained about 2 hours. The heat-cooling operation was repeated again and continued to cool the mixture down to 20-30 °C and stirred for 3 hours. The resulting mixture was filtered and washed with EtOH/n-Heptane (1/1, 5 mL><2). The wet cake was dried via vacuum at 30-40 °C to get the purified Form III of the compound of Formula I (1.7 g).
Example 4. Preparation of Form IV of the compound of Formula I The crude compound of Formula I (15 g) was dissolved in ethyl acetate (600 mL) at 75-85 °C under nitrogen and treated with anhydrous Na2S04, activated carbon, silica metal scavenger for 1 hour. The resulting mixture was filtered via neutral Al203 and washed with ethyl acetate (300 mL><2) at 75-85 °C. The filtrate was concentrated under vacuum at 30-40 °C and swapped with DCM (150 mL). n-Heptane (75 mL) was added into this DCM solution at 35-45 °C, and then the mixture was cooled down to 20-30 °C slowly. The resulting mixture was filtered and washed with DCM/n-Heptane (2/1, 10 mL><3). The wet cake was dried via vacuum at 35-40 °C to get the purified Form IV of the compound of Formula I (9.6 g).
Example 5. Preparation of Form V of the compound of Formula I
[0260] Polymorph Form III of the compound of Formula I was dried in oven at 80 °C for 2 days to obtain the polymorph Form V.
Example 6. Preparation of Form VI of the compound of Formula I
[0261] The compound of Formula I (1 g) was dissolved in IPA (20 mL) at 75-85 °C under nitrogen. n-Heptane (20 mL) was added dropwise into reaction at 75-85 °C. The mixture was stirred at 45-55 °C for 16 hours. Then heated up to 75-85 °C and maintained about 0.5 hour.
Then cooled down to 45-55 °C for 0.5 hour and continued to cool the mixture down to 20-30 °C and stirred for 3 hours. Filtered and washed with IPA/n-Heptane (1/1, 3 mL><2). The wet cake was dried via vacuum at 75-80 °C for 2 hours to get the purified Form VI of the compound of Formula I.
Example 7. Preparation of Form VIII of the compound of Formula I
[0262] The polymorph Form VI of the compound of Formula I was dried in oven at 80 °C for 2 days to obtain the polymorph Form VIII.
Example 8. X-ray powder diffraction (XRD)
[0263] X-ray powder diffraction (XRD) patterns were obtained on a Bruker D8 Advance. A CuK source (=1.54056 angstrom) operating minimally at 40 kV and 40 mA scans each sample between 4 and 40 degrees 2-theta. The step size is 0.05°C and scan speed is 0.5 second per step.
Example 9. Thermogravimetric Analyses (TGA)
[0264] Thermogravimetric analyses were carried out on a TA Instrument TGA unit (Model TGA 500). Samples were heated in platinum pans from ambient to 300 °C at 10 °C/min with a nitrogen purge of 60mL/min (sample purge) and 40mL/min (balance purge). The TGA temperature was calibrated with nickel standard, MP=354.4 °C. The weight calibration was performed with manufacturer-supplied standards and verified against sodium citrate dihydrate desolvation.
Example 10. Differential scanning calorimetry (DSC)
[0265] Differential scanning calorimetry analyses were carried out on a TA Instrument DSC unit (Model DSC 1000 or 2000). Samples were heated in non-hermetic aluminum pans from ambient to 300 °C at 10 °C/min with a nitrogen purge of 50mL/min. The DSC temperature was calibrated with indium standard, onset of l56-l58°C, enthalpy of 25-29J/g.
Example 11. Hygroscopicity (DVS)
[0266] The moisture sorption profile was generated at 25°C using a DVS Moisture Balance Flow System (Model Advantage) with the following conditions: sample size approximately 5 to 10 mg, drying 25°C for 60 minutes, adsorption range 0% to 95% RH, desorption range 95% to 0% RH, and step interval 5%. The equilibrium criterion was <0.01% weight change in 5 minutes for a maximum of 120 minutes.
Example 12: Microscopy
[0267] Microscopy was performed using a Leica DMLP polarized light microscope equipped with 2.5X, 10X and 20X objectives and a digital camera to capture images showing particle shape, size, and crystallinity. Crossed polars were used to show birefringence and crystal habit for the samples dispersed in immersion oil.
Example 13: HPLC
[0256] HPLCs were preformed using the following instrument and/or conditions.
///////////////CK-101 , CK 101 , CK101 , phase II , Suzhou Neupharma, Checkpoint Therapeutics , Orphan Drug designation, EGFR mutation-positive NSCLC, NSCLC, CANCER, SOLID TUMOUR, China, RX-518, AK543910
OCCN1CCN(CC1)c5ccc(Nc2nc3c(cccc3cn2)c4cccc(NC(=O)C=C)c4)c(F)c5F
Ritlecitinib, PF 06651600
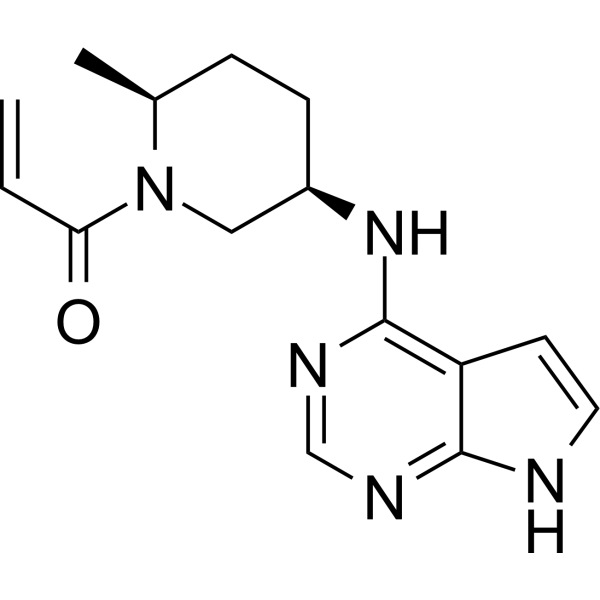


Ritlecitinib
PF-06651600
CAS 1792180-81-4
C₁₅H₁₉N₅O, 285.34, UNII-2OYE00PC25
Fda approved Litfulo, 6/23/2023, To treat severely patchy hair loss
Drug Trials Snapshot
1-((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1-yl)prop-2-en-1-one

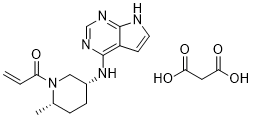
1-[(2S,5R)-2-Methyl-5-(7H-pyrrolo[2,3-d]pyrimidin-4-ylamino)-1-piperidinyl]-2-propen-1-one malonate
PF-06651600 malonate
CAS: 2140301-97-7 (malonate)
Chemical Formula: C18H23N5O5
Molecular Weight: 389.412
PHASE 2 alopecia areata, rheumatoid arthritis, Crohn’s disease, and ulcerative colitis.
Ritlecitinib, sold under the brand name Litfulo, is a medication used for the treatment of severe alopecia areata (hair loss).[6] Ritlecitinib is a kinase inhibitor which inhibits Janus kinase 3 and tyrosine kinase.[6][9][10]
The most common side effects include headache, diarrhea, acne, rashes, eczema, fever, mouth ulcers, dizziness, shingles rash, and abnormal findings in some laboratory test results.[11]
Ritlecitinib was approved for medical use in the United States in June 2023,[6][11][12] in the European Union in September 2023,[7] and in Canada in November 2023.[4]
Pfizer is developing ritlecitinib, an irreversible, covalent and selective dual JAK3/TEC inhibitor, for treating AA, RA, vitiligo and inflammatory bowel diseases, including UC and CD. In July 2021, this drug was reported to be in phase 3 clinical development.
PF-06651600 is a potent and selective JAK3 inhibitor. PF-06651600 is a potent and low clearance compound with demonstrated in vivo efficacy. The favorable efficacy and safety profile of this JAK3-specific inhibitor PF-06651600 led to its evaluation in several human clinical studies. JAK3 was among the first of the JAKs targeted for therapeutic intervention due to the strong validation provided by human SCID patients displaying JAK3 deficiencies
Pfizer has established a leading kinase research capability with multiple unique kinase inhibitors in development as potential medicines. PF-06651600 is a highly selective and orally bioavailable Janus Kinase 3 (JAK3) inhibitor that represents a potential immunomodulatory therapy. With the favorable efficacy, safety profile, and ADME properties, this JAK3-specific covalent inhibitor has been under clinical investigation for the treatment of alopecia areata, rheumatoid arthritis, Crohn’s disease, and ulcerative colitis. Supported by positive results from a Phase 2 study, 1 was granted Breakthrough Therapy designation by the FDA on Sept. 5, 2018 for treatment of alopecia areata.
SYN

PAPER
J. Med. Chem. 2017, 60 (5), 1971–1993, DOI: 10.1021/acs.jmedchem.6b01694
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.6b01694
Paper
Process Development and Scale Up of a Selective JAK3 Covalent Inhibitor PF-06651600,
Yong Tao*

A scalable process for PF-06651600 (1) has been developed through successful enabling of the first generation syntheis. The synthesis highlights include the following: (1) replacement of costly PtO2 with a less expensive 5% Rh/C catalyst for a pyridine hydrogenation, (2) identification of a diasteroemeric salt crystallization to isolate the enantiomerically pure cis-isomer directly from a racemic mixture of cis/trans isomers, (3) a high yielding amidation via Schotten–Baumann conditions, and (4) critical development of a reproducible crystallization procedure for a stable crystalline salt (1·TsOH), which is suitable for long-term storage and tablet formulation. All chromatographic purifications, including two chiral SFC chromatographic separations, were eliminated. Combined with other improvements in each step of the synthesis, the overall yield was increased from 5% to 14%. Several multikilogram batches of the API have been delivered to support clinical studies.
https://pubs.acs.org/doi/10.1021/acs.oprd.9b00198
1-((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1-yl)prop-2-en-1-one p-Toluenesulfonate (1·TsOH)
1·TsOH (4.41 kg, 9.64 mol) as a white powder in 89.6% yield (accounting for the amount of seed charged). Achiral HPLC purity: 99.6% with 0.22% of dimer 15. Chiral SFC purity: >99.7%. Mp 199 °C. Rotomers observed for NMR spectroscopies. 1H NMR (400 MHz, DMSO-d6): δ ppm 12.68 (brs, 1H), 9.22 (brs, 1H), 8.40 (s, 1H), 7.50 (d, J = 8.2 Hz, 2H), 7.45 (m, 1H), 7.12 (d, J = 8.2 Hz, 2H), 6.94 (d, J = 1.2 Hz, 1H), 6.84 (m, 1H), 6.13 (m, 1H), 5.70 (m, 1H), 4.81 (m, 0.5H), 4.54 (m, 0.5H), 4.41 (m, 0.5H), 4.12 (m, 0.5H), 3.99 (m, 1H), 3.15 (m, 0.5H), 2.82 (m, 0.5H), 2.29 (s, 3H), 1.91–1.72 (m, 4H), 1.24–1.17 (m, 3H). 13C NMR (100 MHz, DMSO-d6): δ ppm 165.52, 165.13, 150.50, 145.64, 143.06, 138.48, 129.51, 129.24, 128.67, 127.99, 127.73, 125.97, 125.02, 102.30, 49.53, 48.92, 47.27, 43.83, 42.96, 29.37, 28.41, 25.22, 21.28, 16.97, 15.51. HRMS (ESI) m/z: calculated for C15H20N5O [M + H]+286.1668; observed 286.1692.
PAPER
PATENT
WO2015083028
PATENT
WO 2015083028
https://patents.google.com/patent/WO2015083028A1
PATENT
WO 2020084435
1 -((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one has the structural formula:
The synthesis of 1 -((2S,5R)-5-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one is described in WO2015/083028, commonly assigned to the assignee of the present invention and which is incorporated herein by reference in its entirety. 1 -((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one is useful as an inhibitor of protein kinases, such as the enzyme Janus Kinase (JAK) and as such is useful therapy as an immunosuppressive agent for organ transplants, xeno transplantation, lupus, multiple sclerosis, rheumatoid arthritis, psoriasis, Type I diabetes and complications from diabetes, cancer, asthma, atopic dermatitis, autoimmune thyroid disorders, ulcerative colitis, Crohn’s disease, alopecia, vitiligo, Alzheimer’s disease, leukemia and other indications where immunosuppression would be desirable. See ACS Chem. Biol. , 2016, 11 (12), pp 3442-3451 . The present invention relates to a novel p-toluenesulfonic acid salt and crystalline solid form of the said salt of 1 -((2S,5R)-5-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one that demonstrate improved properties for use in a pharmaceutical dosage form, particularly for oral dosage forms.
Preparations
Scheme 1. Synthesis of 1
Scheme 2. Alternate Synthesis of Intermediates 7 and 10
1. K2C03, MIBK/water 1. H2, H2O
2. EtOAc, aq. NaCI Pd(OH)2/C (wet)
3. MeOH, H20 2. NaOH, MeOH
7 + 8 – ► 9 – – 10 . H2O
89% 89%
Scheme 3. First Alternate Preparation of 1
Scheme 4. Second Alternate Preparation of 1
Preparation 1
ferf-Butyl (6-methylpyridin-3-yl)carbamate (3). To a 3000L reactor was charged 2 (72.00 kg, 665.8 mol) and THF (660 kg). A solution of NH4CI (1 .07 kg , 20 mol) in water (72 kg, 4000 mol) was added. The mixture was heated to 57 °C and Di-f-butyl dicarbonate (220.0 kg, 1003 mol) was added slowly with rinse of THF (45 kg) while maintaining the temperature between 55 – 60 °C. The mixture was stirred at 55 – 60 °C for 10 h. Upon reaction completion, the slurry was cooled to 20 °C and ethyl acetate (654 kg) and water (367 kg) were added. The organic phase was separated, washed by water (2 x 360 kg) and stirred with active carbon (22 kg) for 5 h. The mixture was filtered through a layer of diatomaceous earth (22 kg) with THF rinse and the filtrates were concentrated under vacuum at <40 °C to a residual volume of ~370 L. n-Heptane (500 kg) was added slowly over 1 h and the resulting slurry was cooled to 20 °C and stirred for 2 h. The solid was collected by centrifuge with an n-heptane wash (420 kg), then dried at 45 °C under vacuum for 20 h to give 3 (131 .15 kg, 629.7 mol) as a white powder in 94.5% yield. HPLC purity: 99.9%. 1H NMR (400 MHz, DMSO-c/6): d ppm 9.42 (brs, 1 H), 8.48 (d, J = 1 .9 Hz, 1 H), 7.75 (d, J = 8.6 Hz, 1 H), 7.13 (d, J = 8.6 Hz, 1 H), 2.38 (s, 3H), 1 .49 (s, 9H). 13C NMR (100 MHz, DMSO-d6y d ppm 153.34, 151 .56, 139.75, 134.13, 126.10, 123.09, 79.87, 28.56, 23.70. HRMS (ESI) m/z: calculated for C11H17N2O2 [M + H]+ 209.1290; observed 209.1285.
Preparation 2
ferf-Butyl (6-methylpiperidin-3-yl)carbamate (rac-4). To a 3000L reactor was charged 3 (137.0 kg, 667.8 mol), ethanol (988 kg) and acetic acid (139 kg). The reactor was purged with nitrogen three times and 5 wt% Rhodium on carbon (wet, 27.4 kg, 20 wt% loading relative to 3) was added. The reactor was purged with nitrogen three times and then with hydrogen three times. The hydrogen pressure was adjusted to 0.34 – 0.38 MPa and the reactor temperature was adjusted to 47 °C. The mixture was stirred at 45 – 60 °C under hydrogen pressure at 0.34 – 0.38 MPa for 10 h. Upon reaction completion, the reactor was cooled to 20 °C and flushed with nitrogen. The mixture was filtered through a layer of diatomaceous earth (20 kg) with an ethanol rinse (1320 kg) and the filtrates were concentrated under vacuum at <50 °C to a residual volume of ~350 L. n-Heptane (571 kg) was added and the mixture was concentrated under vacuum at <50 °C to a residual volume of~350 L. This operation was repeated twice until the residual acetic acid <8.0%. Ethanol (672 kg) was added and the mixture was concentrated under vacuum at <50 °C to a residual volume of ~350 L. This operation was repeated twice until the residual n-heptane was <0.2% and water was <0.2%. Ethanol (889 kg) was added and the solution (1254 kg) was transferred to drums for use in the subsequent classical resolution step. Achiral HPLC assay indicated that the solution contained 10.8 wt% of the total reduced product (rac-4) in 96% mass recovery and chiral SFC showed that the solution contained 36.3% of the desired stereoisomer cis-4.
Preparation 3
ferf-Butyl ((3R,6S)-6-methylpiperidin-3-yl)carbamate (R)-2-(3,5-dinitrobenzamido)-2-phenylacetic acid salt (15). To a 2000L reactor (R1 ) was charged rac-4 as a 10.8 wt% solution in ethanol (620.5 kg, ~312.7 mol. of all 4 isomers). The solution was concentrated under vacuum at <45 °C to a residual volume of ~210 L and then cooled to 20 °C. To a 3000 L reactor (R2) was charged (R)-2-(3,5-dinitrobenzamido)-2-phenylacetic acid 14 (47.0 kg, 136.1 mol) and ethanol (1 125 kg). With high speed agitation, reactor R2 was heated to 70 °C, stirred at 68 – 70 °C for ~2 h to dissolve all solid 14, and then seeded with crystalline 15 (1 1 g). The solution containing 4 in reactor R1 was slowly transferred to reactor R2 over 30 min with ethanol rinse (160 kg). Reactor R2 was stirred at ~74 °C for 3 h and then cooled to 22 °C with a linear cooling rate over a period of 5 h and stirred for 16 h. The solid was collected by centrifuge with ethanol wash (2 x 200 kg). The wet cake (with 97.1 % e.e.) was charged back to reactor R2. The slurry was heated to 74 °C and the mixture was stirred for 17 h. The mixture was then cooled to 22 °C with a linear cooling rate over a period of 5 h and stirred for 4 h. The solid was collected by centrifuge with ethanol wash (2 x 200 kg) and dried at 35 °C under vacuum for 25 h to give 15 (56.05 kg, 100.2 mol) as a white powder in 30.7% yield over 2 steps. Chiral HPLC purity: 99.1 %. 1H NMR (400 MHz, DMSO-d6): d ppm 9.46 (d, J = 7.0 Hz, 1 H), 9.07 (d, J = 2.2 Hz, 2H), 8.96 (t, J = 2.2 Hz, 1 H), 7.49 (d, J = 7.3 Hz, 2H), 7.30 (t, J = 7.3 Hz, 2H), 7.23 (t, J = 7.3, 1 H), 7.1 1 (m, 1 H), 5.31 (d, J = 7.0 Hz, 1 H), 3.66 (m, 1 H), 2.98 (m, 3H), 1 .63 (m, 2H), 1 .45 (m, 2H), 1 .40 (s, 9H), 1 .1 1 (d, J = 6.7 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): d ppm 172.71 , 161 .71 , 155.42, 148.51 , 141 .27, 137.70, 128.29, 128.25, 128.02, 127.05, 121 .12, 78.49, 59.74, 50.66, 46.29, 43.34, 28.66, 26.88, 26.1 1 , 18.60.
Preparation 4
Benzyl (2S,5R)-5-amino-2-methylpiperidine-1 -carboxylate hydrochloride (7»HCI) -telescoped process. To a 2000L reactor was charged 15 (70.0 kg, 125 mol) and MTBE (500 kg). The mixture was cooled to 12 °C and 6.9 wt% aqueous NaOH solution (378 kg, 652 mol) was added slowly while maintaining the temperature between 10 – 25 °C. The mixture was stirred at 18 °C for 1 h . The organic phase was separated and washed with 3.8 wt% aqueous NaOH solution (2 x 221 kg) and then 25 wt% aqueous NaCI solution (2 x 220 kg). The organic layer (containing the free base cis-4) was concentrated under vacuum at <40 °C to a residual volume of ~300 L and then cooled to 20 °C. NaHCOs (53 kg, 632 mol) and water (200 kg) were added and the mixture was cooled to 7 °C. Benzyl chloroformate (32.30 kg, 189.3 mol) was added slowly while maintaining the temperature between 5 – 20 °C. The mixture was stirred at 17 °C for 20 h. Upon reaction completion, the mixture was cooled to 12 °C, 25 wt% aqueous ammonium hydroxide solution (79 kg, 1 160 mol) was added slowly while maintaining the temperature between 10 – 20 °C, and the mixture was stirred at 15 °C for 1 h. The organic phase was separated and washed with 25 wt% aqueous NaCI solution (3 x 90 kg). The organic layer (containing 5) was concentrated under vacuum at <45 °C to a residual volume of ~150 L. Isopropyl acetate (310 kg) was added and the mixture was concentrated under vacuum at <45 °C to a residual volume of ~150 L. This operation was repeated twice to meetthe criteria of water <0.1 % (by KF). Isopropyl acetate (130 kg) was then added and the mixture was cooled to -3 °C. 4-5N HCI in methanol (181 kg, ~730 mol) was added slowly while maintaining the temperature between -5 to 5 °C, and the mixture was stirred at 3 °C for 12 h. Upon reaction completion, the mixture was cooled to -3 °C and MTBE (940 kg) was added slowly while maintaining the temperature between -5 to 5 °C. The resulting slurry was stirred at 3 °C for 3 h. The solid was collected by centrifuge with MTBE washes (4 x 70 kg), and then dried at 45 °C under vacuum for 20 h to give 7»HCI (28.60 kg, 100.4 mol) as a white powder in 80.3% yield. Achiral HPLC purity: 100%. Chiral SFC purity: 99.8% e.e. 1H NMR (400 MHz, DMSO-d6): d ppm 8.36 (brs, 3H), 7.37 (m, 5H), 5.09 (s, 2H), 4.31 (m, 1 H), 4.16 (d, J = 8.2 Hz, 1 H), 3.00 (m, 2H), 1 .82 (m, 2H), 1 .59 (m, 2H), 1 .1 1 (d, J = 7.0 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): d ppm 154.71 , 137.24, 128.92, 128.34, 128.00, 66.89, 47.20, 45.66, 40.68, 28.16, 23.02, 15.67. HRMS (ESI) m/z. calculated for C H N O [M + H]+ 249.1603; observed 249.1598.
Preparation 5
Benzyl (2S, 5R)-5-((2-chloro-7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2 -methyl-piperidine-1 -carboxylate (9). To a 2000L reactor was charged 7»HCI (88.6 kg, 31 1 .12 mol), 8 (56.0 kg, 298 mol), K2C03 (133.0 kg, 962.3 mol), water (570 kg) and MIBK (101 kg). The mixture was heated to 90 °C and stirred at this temperature for 22 h. Upon reaction completion, the mixture was cooled to 56 °C and ethyl acetate (531 kg) was added. After cooling the mixture to 22 °C, the organic phase was separated, washed with water (570 kg) and concentrated under vacuum at <40 °C to a residual volume of ~220 L. Methanol (360 kg) was added slowly over a period of 1 h and the mixture concentrated under vacuum at <50 °C to a residual volume of ~220 L. This operation was repeated three times until residual MIBK reached <5 wt%. Methanol (270 kg) was added, followed by seeding with 9 (120 g). The mixture was stirred at 22 °C for >4 h and water (286 kg) was added slowly over 4 h. The slurry was stirred for 10 h and the solid was then collected by centrifuge. The wet cake (165.6 kg) was charged back to a clean reactor and water (896 kg) was added. The slurry was heated to 55 °C and stirred at this temperature for 7 h; and then cooled to 22 °C and stirred at this temperature for 2 h. The solid was collected by centrifuge with water wash (3 x 170 kg) and dried at 55 °C under vacuum for 20 h to give 9 (106.62 kg, 266.6 mol) as a white powder in 89.5% yield. Achiral HPLC purity: 99.7%. 1H NMR (400 MHz, DMSO-d6): d ppm 1 1 .71 (brs, 1 H), 7.72 (d, J = 7.9 Hz, 1 H), 7.38 (m, 5H), 7.10 (s, 1 H), 6.57 (d, J = 2.7 Hz, 1 H), 5.1 1 (m, 2H), 4.39 (m, 1 H), 4.17 (m, 1 H), 4.01 (m, 1 H), 3.36 (s, 2H), 2.77 (m, 1 H), 1 .73-1 .81 (m, 4H), 1 .16 (d, J = 6.6 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): d ppm 156.65, 154.74, 153.04, 151 .31 , 137.43, 128.89, 128.27, 127.96, 122.13, 101 .65, 99.51 , 66.75, 49.10, 47.32, 45.64, 42.98, 29.05, 25.08. HRMS (ESI) m/z. calculated for C20H22CIN5O2 [M + H]+ 400.1540; observed 400.1535.
Preparation 6
N-((3R,6S)-6-methylpiperidin-3-yl)-7H-pyrrolo[2,3-dlpyrimidin-4-amine monohydrate (10·H2O) To a 1600L reactor was charged water (570 kg). The reactor was purged with nitrogen three times. 10% Pd(OH)2/C (wet, 3.2 kg) and 9 (53.34 kg, 133.2 mol) were added with water rinses (2 x 55 kg). The reactor was purged with nitrogen three times and then with hydrogen three times. The hydrogen pressure was adjusted to 0.34 – 0.38 MPa and the reactor temperature was adjusted to 77 °C. The mixture was stirred at 75 – 80 °C under a hydrogen pressure of 0.34 -0.38 MPa for 10 h. Upon reaction completion, the reactor was cooled to 20 °C and purged with nitrogen. The mixture was filtered through a layer of diatomaceous earth (8 kg) with a water rinse (460 kg), and the filtrates were transferred to a 3000L reactor. Methanol (260 kg) was added, followed by slow addition of 50 wt% aqueous sodium hydroxide (12.0 kg , 150 mol) while maintaining the temperature between 15 – 25 °C. The slurry was heated to 55 °C and stirred for 2 h; then cooled to 22 °C and stirred for 10 h. The solid was collected by centrifuge with a 10:1 water/methanol wash (3 x 1 10 kg) and then dried at 55 °C under vacuum for 20 h to give 10·H2O (30.90 kg, 266.6 mol) as a white powder in 89.1 % yield. Achiral HPLC purity: 99.7%. Chiral SFC
purity: 99.8% e.e. 1H NMR (400 MHz, DMSO-d6): d ppm 1 1 .48 (brs, 1 H), 8.08 (s, 1 H), 7.07 (s, 1 H), 6.85 (d, J = 7.3 Hz, 1 H), 6.64 (s, 1 H), 4.16 (m, 1 H), 3.35 (brs, 2H), 2.96 (d, J = 12.7 Hz, 1 H), 2.82 (d, J = 12.7 Hz, 1 H), 2.67 (m, 1 H), 2.04 (brs, 1 H), 1 .92 (m, 1 H), 1 .63 (m, 1 H), 1 .44 (m, 1 H), 1 .33 (m, 1 H), 1 .03 (d, J = 6.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): d ppm 155.95, 151 .87, 150.74, 121 .20, 102.97, 99.20, 51 .27, 49.94, 44.78, 29.97, 28.69, 22.35. HRMS (ESI) m/z\ calculated for C12H17N5 [M + H]+ 232.1562; observed 232.1558.
Preparation 7
1 -((2S,5R)-5-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one (1 ). To a 100L reactor was charged water (18.0 L), 10·H2q (3.60 kg, 14.4 mol) and THF (36.0 L). The mixture was heated to 53 °C and stirred for 15 min to dissolve all the solids. The solution was then cooled to 18 °C and K3PO4 (6.38 kg, 30.1 mol) was added. The mixture was stirred at 18 °C for 10 min to dissolve all the solids, and then cooled to 10 °C. 3-Chloropropionyl chloride (2.20 kg, 17.3 mol) was added while maintaining the temperature <20 °C. The mixture was then stirred at 20 °C for 2 h. Upon reaction completion, 2 N aqueous NaOH solution (23.50 kg, 43.76 mol) was added while maintaining the temperature <25 °C. The mixture was stirred at 22 °C for >12 h until the elimination reaction was complete (11 <0.2%). KH2PO4 (10.32 kg, 75.8 mol) was added and the mixture was stirred at 20 °C for 10 min. The organic phase was separated and then washed with 23.5 wt% aqueous NaCI solution (2 x 8.5 kg). The isolated organic phase was concentrated under vacuum at <30 °C to a residual volume of ~10 L, whereupon MEK (39.6 L) was added. This operation was repeated once or twice until residual THF was <1 % and water was <2%. MgS04 (0.96 kg), Silica gel (4.90 kg) and Darco™ G-60 (0.48 kg) were added to the MEK solution, and the mixture was stirred at 20 °C for 1 h, then filtered through a layer of Diatomaceous Earth with a MEK rinse (76 L). The combined filtrates were concentrated under vacuum at <30 °C to a residual volume of ~8 L. The concentration of the residual solution was measured by qNMR, and the solution was transferred to a container with a rinse using the calculated amount of MEK to adjust the final concentration to 30 wt%. Thus, a 30 wt% solution of 1 in MEK (1 1 .09 kg, 1 1 .66 mol of 1) with 98.7% purity was obtained in 81 % yield, which was stored in a cold room (2 – 8 °C) for the next step.
Preparation 8
1 -((2S,5R)-5-((7H-pyrrolo[2,3-cdpyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one p-toluenesulfonate (1»TsOH). To a 20L reactor was charged a 30 wt% solution of 1 in MEK (9.80 kg, 10.30 mol of 1) and silica gel (0.74 kg). The mixture was stirred at 22 °C for 15 min and filtered through a 0.45 micron Teflon cartridge filter with a MEK rinse (7.89 kg, 9.8 L), collecting in a 100L reactor. Water (1 .27 L) was added, followed by a solution of p-toluenesulfonic acid monohydrate (2.18 kg, 1 1 .3 mol) in MEK (4.75 kg, 5.9 L) with a MEK rinse (3.14 kg, 3.9 L), followed by the addition of 1 »TsOH seed (188 g, 0.41 mol). The mixture was stirred at 22 °C for
4 h to form a slurry and MEK (31 .56 kg, 39.2 L) was added slowly over a period of 3 h. The slurry was stirred at 22 °C for an additional 2 h and then filtered. The cake was washed with MEK (4.02 kg, 5 L) and then dried at 50 °C under vacuum for 10 h to give 1 »TsOH (4.41 kg, 9.64 mol) as a white powder in 89.6% yield (accounting for the amount of seed charged). Achiral HPLC purity: 99.6% with 0.22% of dimer 15. Chiral SFC purity: >99.7%. m.p. 199 °C. Rotomers observed for NMR spectroscopies. Ή NMR (400 MHz, DMSO-d6): d ppm 12.68 (brs, 1 H), 9.22 (brs, 1 H), 8.40 (s, 1 H), 7.50 (d, J = 8.2 Hz, 2H), 7.45 (m, 1 H), 7.12 (d, J = 8.2 Hz, 2H), 6.94 (d, J = 1 .2 Hz, 1 H), 6.84 (m, 1 H), 6.13 (m, 1 H), 5.70 (m, 1 H), 4.81 (m, 0.5H), 4.54 (m, 0.5H), 4.41 (m, 0.5H), 4.12 (m, 0.5H), 3.99 (m, 1 H), 3.15 (m, 0.5H), 2.82 (m, 0.5H), 2.29 (s, 3H), 1 .91 -1 .72 (m, 4H), 1 .24-1 .17 (m, 3H). 13C NMR (100 MHz, DMSO-c/6): d ppm 165.52, 165.13, 150.50, 145.64, 143.06, 138.48, 129.51 , 129.24, 128.67, 127.99, 127.73, 125.97, 125.02, 102.30, 49.53, 48.92, 47.27, 43.83, 42.96, 29.37, 28.41 , 25.22, 21 .28, 16.97, 15.51 . HRMS (ESI) m/z: calculated for Ci5H2oN50 [M + H]+ 286.1668; observed 286.1692.
Comparative Example
Preparation of 1 -((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methyl-piperidin-1 -yl)prop-2-en-1 -one Malonic Acid Salt (Form 1 )
A 250 ml_ round bottom flask was charged with 1 -((2S,5R)-5-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one (4.10 g, 14.4 mmol), MEK (Methyl Ethyl Ketone (15.0 ml_/g, 687 mmol, 49.5 g, 61 .5 ml_)). To the solution, malonic acid (0.950 equiv. 13.7 mmol, 1 .42 g) was added in one portion. The mixture was heated to 50 °C and stirred at 50 °C for 15min. The heating was turned off and the slurry was stirred for 16 hours. The resulting white slurry was filtered. The filter cake was washed with MEK (2 X 5 ml_) and dried in a vacuum oven (40 °C) for 2 hours give 1 -((2S,5R)-5-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one malonic acid salt (Form 1) (4.48 g, 1 1 .5 mmol, 4.48 g, 80.1 % Yield) as white powder.
PATENT
WO-2021136482
Improved process for preparing ritlecitinib (PF06651600), useful for treating alopecia areata (AA), rheumatoid arthritis (RA), Crohn’s disease (CD) and ulcerative colitis (UC). Also claims novel intermediates of ritlecitinib and their preparation method.
PATENT
CN111732591
crystalline polymorphic form of ritlecitinib L-tartrate. Jiangsu Alicorn focuses on developing generic, innovative and first-line drugs, whose website lists an undisclosed JAK inhibitor under its product’s list.
REFERENCES
1: D’Amico F, Fiorino G, Furfaro F, Allocca M, Danese S. Janus kinase inhibitors for the treatment of inflammatory bowel diseases: developments from phase I and phase II clinical trials. Expert Opin Investig Drugs. 2018 Jul;27(7):595-599. doi: 10.1080/13543784.2018.1492547. Epub 2018 Jul 6. Review. PubMed PMID: 29938545.
2: Robinette ML, Cella M, Telliez JB, Ulland TK, Barrow AD, Capuder K, Gilfillan S, Lin LL, Notarangelo LD, Colonna M. Jak3 deficiency blocks innate lymphoid cell development. Mucosal Immunol. 2018 Jan;11(1):50-60. doi: 10.1038/mi.2017.38. Epub 2017 May 17. PubMed PMID: 28513593; PubMed Central PMCID: PMC5693788.
3: Thorarensen A, Dowty ME, Banker ME, Juba B, Jussif J, Lin T, Vincent F, Czerwinski RM, Casimiro-Garcia A, Unwalla R, Trujillo JI, Liang S, Balbo P, Che Y, Gilbert AM, Brown MF, Hayward M, Montgomery J, Leung L, Yang X, Soucy S, Hegen M, Coe J, Langille J, Vajdos F, Chrencik J, Telliez JB. Design of a Janus Kinase 3 (JAK3) Specific Inhibitor 1-((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1-yl)prop -2-en-1-one (PF-06651600) Allowing for the Interrogation of JAK3 Signaling in Humans. J Med Chem. 2017 Mar 9;60(5):1971-1993. doi: 10.1021/acs.jmedchem.6b01694. Epub 2017 Feb 16. PubMed PMID: 28139931.
4: Telliez JB, Dowty ME, Wang L, Jussif J, Lin T, Li L, Moy E, Balbo P, Li W, Zhao Y, Crouse K, Dickinson C, Symanowicz P, Hegen M, Banker ME, Vincent F, Unwalla R, Liang S, Gilbert AM, Brown MF, Hayward M, Montgomery J, Yang X, Bauman J, Trujillo JI, Casimiro-Garcia A, Vajdos FF, Leung L, Geoghegan KF, Quazi A, Xuan D, Jones L, Hett E, Wright K, Clark JD, Thorarensen A. Discovery of a JAK3-Selective Inhibitor: Functional Differentiation of JAK3-Selective Inhibition over pan-JAK or JAK1-Selective Inhibition. ACS Chem Biol. 2016 Dec 16;11(12):3442-3451. Epub 2016 Nov 10. PubMed PMID: 27791347.
5: Walker G, Croasdell G. The European League Against Rheumatism (EULAR) – 17th Annual European Congress of Rheumatology (June 8-11, 2016 – London, UK). Drugs Today (Barc). 2016 Jun;52(6):355-60. doi: 10.1358/dot.2016.52.6.2516435. PubMed PMID: 27458612.
References
- ^ Jump up to:a b “Litfulo (ritlecitinib)”. Therapeutic Goods Administration (TGA). 30 July 2024. Retrieved 12 October 2024.
- ^ https://www.tga.gov.au/resources/prescription-medicines-registrations/litfulo-pfizer-australia-pty-ltd
- ^ “Litfulo Product information”. Health Canada. 13 February 2024. Retrieved 3 March 2024.
- ^ Jump up to:a b c “Summary Basis of Decision for Litfulo”. Health Canada. 18 July 2024. Retrieved 28 August 2024.
- ^ “Regulatory Decision Summary for Litfulo”. Health Canada. 29 November 2023. Retrieved 2 April 2024.
- ^ Jump up to:a b c d e f “Litfulo- ritlecitinib capsule”. DailyMed. U.S. National Library of Medicine. 23 June 2023. Archived from the original on 29 August 2023. Retrieved 28 August 2023.
- ^ Jump up to:a b c d “Litfulo EPAR”. European Medicines Agency. 18 September 2023. Archived from the original on 19 September 2023. Retrieved 20 September 2023.
- ^ “Litfulo Product information”. Union Register of medicinal products. 18 September 2023. Archived from the original on 1 October 2023. Retrieved 1 October 2023.
- ^ “Ritlecitinib”. Inxight Drugs. Archived from the original on 25 June 2023. Retrieved 24 June 2023.
- ^ Ramírez-Marín HA, Tosti A (February 2022). “Evaluating the Therapeutic Potential of Ritlecitinib for the Treatment of Alopecia Areata”. Drug Design, Development and Therapy. 16: 363–374. doi:10.2147/DDDT.S334727. PMC 8860347. PMID 35210753.
- ^ Jump up to:a b c d e f g h “Drug Trials Snapshots: Litfulo”. U.S. Food and Drug Administration. 23 June 2023. Retrieved 29 April 2024.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “FDA Approves Pfizer’s Litfulo (Ritlecitinib) for Adults and Adolescents With Severe Alopecia Areata” (Press release). Pfizer. 23 June 2023. Archived from the original on 25 June 2023. Retrieved 24 June 2023 – via Business Wire.
- ^ Kansteiner F (26 June 2023). “Pfizer’s Litfulo enters the scene in alopecia with adolescent nod to rival Lilly’s Olumiant”. Fierce Pharma. Archived from the original on 8 July 2023. Retrieved 18 September 2023.
Further reading
- Guttman-Yassky E, Pavel AB, Diaz A, Zhang N, Del Duca E, Estrada Y, et al. (April 2022). “Ritlecitinib and brepocitinib demonstrate significant improvement in scalp alopecia areata biomarkers”. The Journal of Allergy and Clinical Immunology. 149 (4): 1318–1328. doi:10.1016/j.jaci.2021.10.036. PMID 34863853. S2CID 244824663.
- King B, Zhang X, Harcha WG, Szepietowski JC, Shapiro J, Lynde C, et al. (May 2023). “Efficacy and safety of ritlecitinib in adults and adolescents with alopecia areata: a randomised, double-blind, multicentre, phase 2b-3 trial”. Lancet. 401 (10387). London, England: 1518–1529. doi:10.1016/S0140-6736(23)00222-2. PMID 37062298. S2CID 258114404.
External links
- Clinical trial number NCT03732807 for “PF-06651600 for the Treatment of Alopecia Areata (ALLEGRO-2b/3)” at ClinicalTrials.gov
 |
|
| Clinical data | |
|---|---|
| Trade names | Litfulo |
| Other names | PF-06651600 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a624015 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
|
show
|
|
| CAS Number |
|
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank |
|
| ChemSpider | |
| UNII |
|
| KEGG |
|
| ChEBI | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C15H19N5O |
| Molar mass | 285.351 g·mol−1 |
| 3D model (JSmol) | |
|
show
|
|
|
show
|
|
////////////PF-06651600, PF 06651600, PF06651600, Breakthrough Therapy designation, PHASE 2, alopecia areata, rheumatoid arthritis, Crohn’s disease, ulcerative colitis, Ritlecitinib, Litfulo
C=CC(N1[C@@H](C)CC[C@@H](NC2=C3C(NC=C3)=NC=N2)C1)=O
LK-01, Apomorphine
LK-01
Leukos Biotech S.L.
APL-130277, H-001, Apokyn
Apomorphine hydrochloride hemihydrate
CAS 41372-20-7
Leukos Biotech (following its spin-off from Jose Carreras Leukaemia Research Institute) is developing LK-01 , a solid form of apomorphine for the sc treatment of acute myeloid leukemia (AML) and the phase II trial results were expected later in 2019.
Apomorphine (brand names Apokyn, Ixense, Spontane, Uprima) is a type of aporphine having activity as a non-selective dopamine agonist which activates both D2-like and, to a much lesser extent, D1-like receptors.[1] It also acts as an antagonist of 5-HT2 and α-adrenergic receptors with high affinity. The compound is historically a morphine decomposition product made by boiling morphine with concentrated acid, hence the –morphine suffix. Contrary to its name, apomorphine does not actually contain morphine or its skeleton, nor does it bind to opioid receptors. The apo– prefix relates to it being a morphine derivative (“[comes] from morphine”).
Historically, apomorphine has been tried for a variety of uses, including as a way to relieve anxiety and craving in alcoholics, an emetic (to induce vomiting), for treating stereotypies (repeated behaviour) in farmyard animals, and more recently in treating erectile dysfunction. Currently, apomorphine is used in the treatment of Parkinson’s disease. It is a potent emetic and should not be administered without an antiemetic such as domperidone. The emetic properties of apomorphine are exploited in veterinary medicine to induce therapeutic emesis in canines that have recently ingested toxic or foreign substances.
Apomorphine was also used as a private treatment of heroin addiction, a purpose for which it was championed by the author William S. Burroughs. Burroughs and others claimed that it was a “metabolic regulator” with a restorative dimension to a damaged or dysfunctional dopaminergic system. There is more than enough anecdotal evidence to suggest that this offers a plausible route to an abstinence-based model; however, no clinical trials have ever tested this hypothesis. A recent study indicates that apomorphine might be a suitable marker for assessing central dopamine system alterations associated with chronic heroin consumption.[2] There is, however, no clinical evidence that apomorphine is an effective and safe treatment regimen for opiate addiction.[3]
Uses
Apomorphine is used in advanced Parkinson’s disease intermittent hypomobility (“off” episodes), where a decreased response to an anti-Parkinson drug such as L-DOPA causes muscle stiffness and loss of muscle control.[4][5] While apomorphine can be used in combination with L-DOPA, the intention is usually to reduce the L-DOPA dosing, as by this stage the patient often has many of dyskinesias caused by L-DOPA and hypermobility periods.[6][7] When an episode sets in, the apomorphine is injected subcutaneously, and signs subside. It is used an average of three times a day.[6] Some people use portable mini-pumps that continuously infuse them with apomorphine, allowing them to stay in the “on” state and using apomorphine as an effective monotherapy.[7][8]
Contraindications
The main and absolute contraindication to using apomorphine is the concurrent use of adrenergic receptor antagonists; combined, they cause a severe drop in blood pressure and fainting.[6][5] Alcohol causes an increased frequency of orthostatic hypotension (a sudden drop in blood pressure when getting up), and can also increase the chances of pneumonia and heart attacks.[6] Dopamine antagonists, by their nature of competing for sites at dopamine receptors, reduce the effectiveness of the agonistic apomorphine.[6][5]
IV administration of apomorphine is highly discouraged, as it can crystallize in the veins and create a blood clot (thrombus) and block a pulmonary artery (pulmonary embolism).[6][5]
Side effects
Nausea and vomiting are common side effects when first beginning therapy with apomorphine;[9] antiemetics such as trimethobenzamide or domperidone, dopamine antagonists,[10] are often used while first starting apomorphine. Around 50% of people grow tolerant enough to apomorphine’s emetic effects that they can discontinue the antiemetic.[5][6]
Other side effects include orthostatic hypotension and resultant fainting, sleepiness, dizziness, runny nose, sweating, paleness, and flushing. More serious side effects include dyskenesias (especially when taking L-DOPA), fluid accumulation in the limbs (edema), suddenly falling asleep, confusion and hallucinations, increased heart rate and heart palpitations, and persistent erections(priaprism).[5][6][11] The priaprism is caused by apomorphine increasing arterial blood supply to the penis. This side effect has been exploited in studies attempting to treat erectile dysfunction.[12]
Pharmacology
Mechanism of action
Apomorphine’s R-enantiomer is an agonist of both D1 and D2 dopamine receptors, with higher activity at D2.[6][10] The members of the D2 subfamily, consisting of D2, D3, and D4receptors, are inhibitory G protein–coupled receptors. The D4 receptor in particular is an important target in the signaling pathway, and is connected to several neurological disorders.[13] Shortage or excess of dopamine can prevent proper function and signaling of these receptors leading to disease states.[14]
Apomorphine improves motor function by activating dopamine receptors in the nigrostriatal pathway, the limbic system, the hypothalamus, and the pituitary gland.[15] It also increases blood flow to the supplementary motor area and to the dorsolateral prefrontal cortex (stimulation of which has been found to reduce the tardive dyskinesia effects of L-DOPA).[16][17]Parkinson’s has also been found to have excess iron at the sites of neurodegeneration; both the R- and S-enantiomers of apomorphine are potent iron chelators and radical scavengers.[10][18]
Apomorphine also reduces the breakdown of dopamine in the brain (though it inhibits its synthesis as well).[19][20] It is a powerful upregulator of certain neural growth factors,[21] in particular NGF and BDNF, epigenetic downregulation of which has been associated with addictive behaviour in rats.[22][23]
Apomorphine causes vomiting by acting on dopamine receptors in the chemoreceptor trigger zone of the medulla; this activates the nearby vomiting center.[15][20][24]
Pharmacokinetics
While apomorphine has lower bioavailability when taken orally, due to not being absorbed well in the GI tract and undergoing heavy first-pass metabolism,[18][8] it has a bioavailability of 100% when given subcutaneously.[6][15] It reaches peak plasma concentration in 10–60 minutes. Ten to twenty minutes after that, it reaches its peak concentration in the cerebrospinal fluid. Its lipophilic structure allows it to cross the blood–brain barrier.[6][15]
Apomorphine possesses affinity for the following receptors (note that a higher Ki indicates a lower affinity):[25][26][27]
| Receptor | Ki (nM) | Action |
|---|---|---|
| D1 | 484 | (partial) agonista |
| D2 | 52 | partial agonist (IA = 79% at D2S; 53% at D2L) |
| D3 | 26 | partial agonist (IA = 82%) |
| D4 | 4.37 | partial agonist (IA = 45%) |
| D5 | 188.9 | (partial) agonista |
| aThough its efficacies at D1 and D5 are unclear, it is known to act as an agonist at these sites.[28] | ||
| Receptor | Ki (nM) | Action |
|---|---|---|
| 5-HT1A | 2,523 | partial agonist |
| 5-HT1B | 2,951 | no action |
| 5-HT1D | 1,230 | no action |
| 5-HT2A | 120 | antagonist |
| 5-HT2B | 132 | antagonist |
| 5-HT2C | 102 | antagonist |
| Receptor | Ki (nM) | Action |
|---|---|---|
| α1A-adrenergic | 1,995 | antagonist |
| α1B-adrenergic | 676 | antagonist |
| α1D-adrenergic | 64.6 | antagonist |
| α2A-adrenergic | 141 | antagonist |
| α2B-adrenergic | 66.1 | antagonist |
| α2C-adrenergic | 36.3 | antagonist |
It has a Ki of over 10,000 nM (and thus negligible affinity) for β-adrenergic, H1, and mACh.[1]
Apomorphine has a high clearance rate (3–5 L/kg/hr) and is mainly metabolized and excreted by the liver.[15] It is likely that while the cytochrome P450 system plays a minor role, most of apomorphine’s metabolism happens via auto-oxidation, O-glucuronidation, O-methylation, N-demethylation, and sulfation.[6][15][20] Only 3–4% of the apomorphine is excreted unchanged and into the urine. The half-life is 30–60 minutes, and the effects of the injection last for up to 90 minutes.[6][7][15]
Toxicity depends on the route of administraion; the LD50s in mice were 300 mg/kg for the oral route, 160 mg/kg for intraperitoneal, and 56 mg/kg intravenous.[29]
Chemistry
Properties
Apomorphine has a catechol structure similar to that of dopamine.[19]
Synthesis
Several techniques exist for the creation of apomorphine from morphine. In the past, morphine had been combined with hydrochloric acid at high temperatures (around 150 °C) to achieve a low yield of apomorphine, ranging anywhere from 0.6% to 46%.[30]
More recent techniques create the apomorphine in a similar fashion, by heating it in the presence of any acid that will promote the essential dehydration rearrangement of morphine-type alkaloids, such as phosphoric acid. The method then deviates by including a water scavenger, which is essential to remove the water produced by the reaction that can react with the product and lead to decreased yield. The scavenger can be any reagent that will irreversibly react with water such as phthalic anhydride or titanium chloride. The temperature required for the reaction varies based upon choice of acid and water scavenger. The yield of this reaction is much higher: at least 55%.[30]

Conversion of Morphine (I) to Apomorphine (II) in the presence of acid following the example of the morphine skeleton dehydration rearrangement, outlined by Bentley.[31]
History
The pharmacological effects of the naturally-occurring analog aporphine in the blue lotus (N. caerulea)[32] were known to the ancient Egyptians and Mayans,[33] with the plant featuring in tomb frescoes and associated with entheogenic rites. It is also observed in Egyptian erotic cartoons, suggesting that they were aware of its erectogenic properties.
The modern medical history of apomorphine begins with its synthesis by Arppe in 1845[34] from morphine and sulfuric acid, although it was named sulphomorphide at first. Matthiesen and Wright (1869) used hydrochloric acid instead of sulfuric acid in the process, naming the resulting compound apomorphine. Initial interest in the compound was as an emetic, tested and confirmed safe by London doctor Samuel Gee,[35] and for the treatment of stereotypies in farmyard animals.[36] Key to the use of apomorphine as a behavioural modifier was the research of Erich Harnack, whose experiments in rabbits (which do not vomit) demonstrated that apomorphine had powerful effects on the activity of rabbits, inducing licking, gnawing and in very high doses convulsions and death.
Treatment of alcoholism
Apomorphine was one of the earliest used pharmacotherapies for alcoholism. The Keeley Cure (1870s to 1900) contained apomorphine, among other ingredients, but the first medical reports of its use for more than pure emesis come from James Tompkins[37] and Charles Douglas.[38][39] Tompkins reported, after injection of 6.5 mg (“one tenth of a grain”):
In four minutes free emesis followed, rigidity gave way to relaxation, excitement to somnolence, and without further medication the patient, who before had been wild and delirious, went off into a quiet sleep.
Douglas saw two purposes for apomorphine:
[it can be used to treat] a paroxysm of dipsomania [an episode of intense alcoholic craving]… in minute doses it is much more rapidly efficient in stilling the dipsomaniac craving than strychnine or atropine… Four or even 3m [minim – roughly 60 microlitres] of the solution usually checks for some hours the incessant demands of the patient… when he awakes from the apomorphine sleep he may still be demanding alcohol, though he is never then so insistent as before. Accordingly it may be necessary to repeat the dose, and even to continue to give it twice or three times a day. Such repeated doses, however, do not require to be so large: 4 or even 3m is usually sufficient.
This use of small, continuous doses (1/30th of a grain, or 2.16 mg by Douglas) of apomorphine to reduce alcoholic craving comes some time before Pavlov‘s discovery and publication of the idea of the “conditioned reflex” in 1903. This method was not limited to Douglas; the Irish doctor Francis Hare, who worked in a sanatorium outside London from 1905 onwards, also used low-dose apomorphine as a treatment, describing it as “the most useful single drug in the therapeutics of inebriety”.[40] He wrote:
In (the) sanatorium it is used in three different sets of circumstances: (1) in maniacal or hysterical drunkenness: (2) during the paroxysm of dipsomania, in order to still the craving for alcohol; and (3) in essential insomnia of a special variety… [after giving apomorphine] the patient’s mental condition is entirely altered. He may be sober: he is free from the time being from any craving from alcohol. The craving may return, however, and then it is necessary to repeat the injection, it may be several times at intervals of a few hours. These succeeding injections should be quite small, 3 to 6 min. being sufficient. Doses of this size are rarely emetic. There is little facial pallor, a sensation as of the commencement of sea-sickness, perhaps a slight malaise with a sudden subsidence of the craving for alcohol, followed by a light and short doze.
He also noted there appeared to be a significant prejudice against the use of apomorphine, both from the associations of its name and doctors being reluctant to give hypodermic injections to alcoholics. In the US, the Harrison Narcotics Tax Act made working with any morphine derivatives extremely hard, despite apomorphine itself not being an opiate.
In the 1950s the neurotransmitter dopamine was discovered in the brain by Kathleen Montagu, and characterised as a neurotransmitter a year later by Arvid Carlsson, for which he would be awarded the Nobel Prize.[41] A. N. Ernst then discovered in 1965 that apomorphine was a powerful stimulant of dopamine receptors.[42] This, along with the use of sublingual apomorphine tablets, led to a renewed interest in the use of apomorphine as a treatment for alcoholism. A series of studies of non-emetic apomorphine in the treatment of alcoholism were published, with mostly positive results.[43][44][45][46][47] However, there was little clinical consequence.
Parkinson’s disease
The use of apomorphine to treat “the shakes” was first suggested by Weil in France in 1884,[48] although seemingly not pursued until 1951.[49] Its clinical use was first reported in 1970 by Cotzias et al.,[50] although its emetic properties and short half-life made oral use impractical. A later study found that combining the drug with the antiemetic domperidoneimproved results significantly.[51] The commercialization of apomorphine for Parkinson’s disease followed its successful use in patients with refractory motor fluctuations using intermittent rescue injections and continuous infusions.[52]
Aversion therapy
Aversion therapy in alcoholism had its roots in Russia in the early 1930s,[53] with early papers by Pavlov, Galant and Sluchevsky and Friken,[54] and would remain a strain in the Soviet treatment of alcoholism well into the 1980s. In the US a particularly notable devotee was Dr Voegtlin,[55] who attempted aversion therapy using apomorphine in the mid to late 1930s. However, he found apomorphine less able to induce negative feelings in his subjects than the stronger and more unpleasant emetic emetine.
In the UK, however, the publication of J Y Dent’s (who later went on to treat Burroughs) 1934 paper “Apomorphine in the treatment of Anxiety States”[56] laid out the main method by which apomorphine would be used to treat alcoholism in Britain. His method in that paper is clearly influenced by the then-novel idea of aversion:
He is given his favourite drink, and his favourite brand of that drink… He takes it stronger than is usual to him… The small dose of apomorphine, one-twentieth of a grain [3.24mg], is now given subcutaneously into his thigh, and he is told that he will be sick in a quarter of an hour. A glass of whisky and water and a bottle of whisky are left by his bedside. At six o’clock (four hours later) he is again visited and the same treatment is again administered… The nurse is told in confidence that if he does not drink, one-fortieth [1.62mg] of a grain of apomorphine should be injected during the night at nine o’clock, one o’clock, and five o’clock, but that if he drinks the injection should be given soon after the drink and may be increased to two hourly intervals. In the morning at about ten he is again given one or two glasses of whisky and water… and again one-twentieth of a grain [3.24mg] of apomorphine is injected… The next day he is allowed to eat what he likes, he may drink as much tea as he likes… He will be strong enough to get up and two days later he leaves the home.
However, even in 1934 he was suspicious of the idea that the treatment was pure conditioned reflex – “though vomiting is one of the ways that apomorphine relives the patient, I do not believe it to be its main therapeutic effect.” – and by 1948 he wrote:[3]
It is now twenty-five years since I began treating cases of anxiety and alcoholism with apomorphine, and I read my first paper before this Society fourteen years ago. Up till then I had thought, and, unfortunately, I said in my paper, that the virtue of the treatment lay in the conditioned reflex of aversion produced in the patient. This statement is not even a half truth… I have been forced to the conclusion that apomorphine has some further action than the production of a vomit.
This led to his development of lower-dose and non-aversive methods, which would inspire a positive trial of his method in Switzerland by Dr Harry Feldmann[57] and later scientific testing in the 1970s, some time after his death. However, the use of apomorphine in aversion therapy had escaped alcoholism, with its use to treat homosexuality leading to the death of a British Army Captain Billy Clegg HIll in 1962,[58] helping to cement its reputation as a dangerous drug used primarily in archaic behavioural therapies.
Opioid addiction
In his Deposition: Testimony Concerning a Sickness in the introduction to later editions of Naked Lunch (first published in 1959), William S. Burroughs wrote that apomorphine treatment was the only effective cure to opioid addiction he has encountered:
The apomorphine cure is qualitatively different from other methods of cure. I have tried them all. Short reduction, slow reduction, cortisone, antihistamines, tranquilizers, sleeping cures, tolserol, reserpine. None of these cures lasted beyond the first opportunity to relapse. I can say that I was never metabolically cured until I took the apomorphine cure… The doctor, John Yerbury Dent, explained to me that apomorphine acts on the back brain to regulate the metabolism and normalize the blood stream in such a way that the enzyme stream of addiction is destroyed over a period of four to five days. Once the back brain is regulated apomorphine can be discontinued and only used in case of relapse.
He goes on to lament the fact that as of his writing, little to no research has been done on apomorphine or variations of the drug to study its effects on curing addiction, and perhaps the possibility of retaining the positive effects while removing the side effect of vomiting.
Despite his claims throughout his life, Burroughs never really cured his addiction and was back to using opiates within years of his apomorphine “cure”.[59] However, he insisted on apomorphine’s effectiveness in several works and interviews.[citation needed]
Society and culture
- Apomorphine has a vital part in Agatha Christie‘s detective story Sad Cypress.
- The 1965 Tuli Kupferberg song “Hallucination Horrors” recommends apomorphine at the end of each verse as a cure for hallucinations brought on by a humorous variety of intoxicants; the song was recorded by The Fugs and appears on the album Virgin Fugs.
Research
There is renewed interest in the use of apomorphine to treat addiction, in both smoking cessation[60] and alcoholism.[61] As the drug is old, out of patent, and safe for use in humans, it is a viable target for repurposing.

Flow chart depicting the role of apomorphine in Alzheimer’s disease.
Apomorphine has been researched as a possible treatment for erectile dysfunction and female hypoactive sexual desire disorder, though the arousal effects were found not to be reliable enough. One large study found that only 39.4% got erections (compared to baseline 13.1); another found that apomorphine was successful 45–51% of the time, but the placebo also worked 36% of the time.[12][62] Nonetheless, it was under development as a treatment for erectile dysfunction by TAP Pharmaceuticals under the brand name Uprima. In 2000, TAP withdrew its new drug application after an FDA review panel raised questions about the drug’s safety, due to many clinical trial subjects fainting after taking the drug.[63]
Alzheimer’s disease
Apomorphine is reported to be an inhibitor of amyloid beta protein (Aβ) fiber formation, whose presence is a hallmark of Alzheimer’s disease (AD), and a potential therapeutic under the amyloid hypothesis.[64] While it promotes oligomerization of the Aβ40 group of molecules, it inhibits more advanced fibril formation; this is thought to be due to the autoxidation that occurs at the hydroxyl groups. Once this functional group was altered, the inhibitory effect could be seen to decrease, reducing either the indirect or direct interference of the fibril formation.[64]
The protective effects of apomorphine were tested in mouse models with mutations in genes related to AD, such as the amyloid precursor protein gene. Apomorphine was seen to significantly improve memory function through the increased successful completion of the Morris Water Maze. The levels of the aberrant proteins that lead to neuronal disruption were also tested in the brains of mice. Treatment was seen to decrease the intraneuronal levels of the more aggressive Aβ42 molecule when compared to the control mice. This result is consistent with the finding that another protein linked to AD, tau protein, was seen to decrease with apomorphine treatment.[65]
Veterinary use
Apomorphine is used to inducing vomiting in dogs the after ingestion of various toxins or foreign bodies. It can be given subcutaneously, intramuscularly, intravenously, or, when a tablet is crushed, in the conjunctiva of the eye.[66][67] The oral route is ineffective, as apomorphine cannot cross the blood–brain barrier fast enough, and blood levels don’t reach a high enough concentration to stimulate the chemoreceptor trigger zone.[66] It can remove around 40–60% of the contents in the stomach.[68]
One of the reasons apomorphine is a preferred drug is its reversibility:[69] in cases of prolonged vomiting, the apomorphine can be reversed with dopamine antagonists like the phenothiazines (for example, acepromazine). Giving apomorphine after giving acepromazine, however, will no longer stimulate vomiting, because apomorphine’s target receptors are already occupied.[66] An animal who undergoes severe respiratory depression due to apomorphine can be treated with naloxone.[66][67]
Apomorphine does not work in cats, who have too few dopamine receptors.[66]
PATENT
WO-2019141673
Novel crystalline forms of apomorphine or its palmitate or hydrochloride salt useful treating acute myeloid leukemia, Parkinson’s disease, sexual dysfunction and solid tumors. Also claims the process for preparing apomorphine palmitic acid cocrystal salt.
Apomorphine (APO) is a commercial available medical drug with the chemical formula C-17H-17NO2 and structure:
Apomorphine (APO) has been described for treatment of different medical indications – for instance:
– WO2015/197839A1 : leukemia such as acute myeloid leukemia (AML);
– WO2016/103262A2: Parkinson’s disease;
– WO02/39879A2: sexual dysfunction in a patient taking antidepressant medication;
– W02004/082630A2: neurological function of an individual who has a brain injury.
Apomorphine hydrochloride (HCI) is a salt present in commercially available medical products (e.g. APO-Go® PFS or Apokyn®).
A common side effect of administering apomorphine hydrochloride by e.g. subcutaneous injection is e.g. the development of subcutaneous nodules at the injection site, which can become infected, necessitating treatment or surgical involvement.
In relation to this problem – above discussed WO2016/103262A2 describes an alternative solid form of apomorphine, which is e.g. an alcohol solvate crystal of apomorphine free base, wherein the solvate forming solvent is (C-|-C8) alkanol, preferably isopropanol (IPA – i.e. a solid crystalline form of apomorphine-IPA.
Palmitic acid (hexadecanoic acid in IUPAC nomenclature) is a fatty acid found with the chemical formula CH3(CH2)14COOH.
Palmitate is the salt and ester of palmitic acid.
A herein relevant synonyms name may e.g. be palmitoate.
Beside apomorphine hydrochloride, above discussed WO2015/197839A1 and W02004/082630A2 provide a list of other possible suitable pharmaceutically acceptable salts – palmitic acid (or synonyms like palmitate or palmitoate) is not mentioned in the lists of these two WO documents.
As discussed in the review article of Schultheiss et al. (“Pharmaceutical Cocrystals and Their Physicochemical Properties”; Crystal Growth & Design, Vol. 9, No. 6, 2009, p. 2950-2967) – solid-state chemists call upon a variety of different strategies when attempting to alter the chemical and physical solid-state properties of active pharmaceutical ingredients (APIs), namely, the formation of salts, polymorphs, hydrates, solvates, and cocrystals.
Salt formation is one of the primary solid-state approaches used to modify the physical properties of APIs, and it is estimated that over half of the medicines on the market are administered as salts. However, a limitation within this approach is that the API must possess a suitable (basic or acidic)
ionizable site. In comparison, cocrystals (multicomponent assemblies held together by freely reversible, noncovalent interactions) offer a different pathway, where any API regardless of acidic, basic, or ionizable groups, could potentially be cocrystallized.
Above discussed WO02/39879A2 also provides a long list of suitable pharmaceutically acceptable salts and mentions palmitoate (see page 5, line 16).
However, in all herein relevant experimental work of this WO document was used apomorphine hydrochloride and a palmitic acid based salt is simply mentioned in a list – i.e. a palmitic acid based salt is not a preferred salt.
Alternatively expressed, by reading this WO document the skilled person has in practice no motivation to use any other solid form than apomorphine-HCI – one reason for this is that apomor-phine-HCI is used in all herein relevant experimental work of this WO document.
PATENT
WO2018130685
claiming synergistic combination comprising antimetabolite antineoplastic agent (eg cytarabine ) and type 1 serotonin receptor antagonist (5-HTR1) (eg apomorphine ), useful for treating cancer.
SYN
SYN

PAPER
- Small, L. et al.: J. Org. Chem. (JOCEAH) 5, 334 (1940)
References
- ^ Jump up to:a b Millan MJ, Maiofiss L, Cussac D, Audinot V, Boutin JA, Newman-Tancredi A (November 2002). “Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. I. A multivariate analysis of the binding profiles of 14 drugs at 21 native and cloned human receptor subtypes”. The Journal of Pharmacology and Experimental Therapeutics. 303 (2): 791–804. doi:10.1124/jpet.102.039867. PMID 12388666.
- ^ Guardia J, Casas M, Prat G, Trujols J, Segura L, Sánchez-Turet M (October 2002). “The apomorphine test: a biological marker for heroin dependence disorder?”. Addiction Biology. 7 (4): 421–6. doi:10.1080/1355621021000006206. PMID 14578019.
- ^ Jump up to:a b Dent JY (1949). “Apomorphine Treatment of Addiction”. British Journal of Addiction to Alcohol & Other Drugs. 46 (1): 15–28. doi:10.1111/j.1360-0443.1949.tb04502.x.
- ^ “Apomorphine Uses, Side Effects & Warnings”. Drugs.com. Retrieved 27 February2018.
- ^ Jump up to:a b c d e f Clayton BD, Willihnganz M (2016). Basic Pharmacology for Nurses – E-Book. Elsevier Health Sciences. pp. 210–211. ISBN 978-0-323-37697-6.
- ^ Jump up to:a b c d e f g h i j k l m “Apomorphine Hydrochloride Monograph for Professionals”. Drugs.com. Retrieved 26 February 2018.
- ^ Jump up to:a b c Chaudhuri KR, Clough C (February 1998). “Subcutaneous apomorphine in Parkinson’s disease”. BMJ. 316 (7132): 641. doi:10.1136/bmj.316.7132.641. PMC 1112674. PMID 9522772.
- ^ Jump up to:a b Schapira AH, Olanow CW (2005). Principles of Treatment in Parkinson’s Disease(illustrated ed.). Elsevier Health Sciences. p. 35. ISBN 978-0-7506-5428-9.
- ^ Dressler D (June 2005). “[Apomorphine in the treatment of Parkinson’s Disease]”. Der Nervenarzt (in German). 76 (6): 681–9. doi:10.1007/s00115-004-1830-4. PMID 15592807.
- ^ Jump up to:a b c Youdim MB, Gassen M, Gross A, Mandel S, Grünblatt E (2000). “Iron chelating, antioxidant and cytoprotective properties of dopamine receptor agonist; apomorphine”. In Mizuno Y, Calne D, Horowski R, Poewe W, Riederer P, Youdim M (eds.). Advances in Research on Neurodegeneration. 7 (illustrated ed.). Springer Science & Business Media. pp. 83–96. ISBN 978-3-211-83485-5.
- ^ “Apomorphine”. Medline Plus. US National Library of Medicine. 15 June 2017. Retrieved 26 February 2018.
- ^ Jump up to:a b Porst H, Buvat J (2008). Standard Practice in Sexual Medicine. John Wiley & Sons. p. 77. ISBN 978-1-4051-7872-3.
- ^ Ptácek R, Kuzelová H, Stefano GB (September 2011). “Dopamine D4 receptor gene DRD4 and its association with psychiatric disorders”. Medical Science Monitor. 17 (9): RA215–20. doi:10.12659/MSM.881925. PMC 3560519. PMID 21873960.
- ^ Stacy M, Silver D (2008). “Apomorphine for the acute treatment of “off” episodes in Parkinson’s disease”. Parkinsonism & Related Disorders. 14 (2): 85–92. doi:10.1016/j.parkreldis.2007.07.016. PMID 18083605.
- ^ Jump up to:a b c d e f g U.S. National Library of Medicine. “Apomorphine”. PubChem. Retrieved 26 February 2018.
- ^ Lewitt P, Oertel WH (1999). Parkinsons’s Disease: The Treatment Options. CRC Press. p. 22. ISBN 978-1-85317-379-0.
- ^ Rektorova I, Sedlackova S, Telecka S, Hlubocky A, Rektor I (2008). “Dorsolateral prefrontal cortex: a possible target for modulating dyskinesias in Parkinson’s disease by repetitive transcranial magnetic stimulation”. International Journal of Biomedical Imaging. 2008: 372125. doi:10.1155/2008/372125. PMC 2233877. PMID 18274665.
- ^ Jump up to:a b Galvez-Jimenez N (2013). Scientific Basis for the Treatment of Parkinson’s Disease, Second Edition. CRC Press. p. 195. ISBN 978-0-203-33776-9.
- ^ Jump up to:a b Iversen L (2012). Biogenic Amine Receptors. Springer Science & Business Media. p. 238. ISBN 978-1-4684-8514-1.
- ^ Jump up to:a b c Advances in Pharmacology and Chemotherapy, Volume 15. Silvio Garattini, A. Goldin, F. Hawking, Irwin J. Kopin. Academic Press. 1978. pp. 27, 93, 96. ISBN 978-0-08-058106-4.
- ^ Ohta M, Mizuta I, Ohta K, Nishimura M, Mizuta E, Hayashi K, Kuno S (May 2000). “Apomorphine up-regulates NGF and GDNF synthesis in cultured mouse astrocytes”. Biochemical and Biophysical Research Communications. 272 (1): 18–22. doi:10.1006/bbrc.2000.2732. PMID 10872797.
- ^ McGeary JE, Gurel V, Knopik VS, Spaulding J, McMichael J (October 2011). “Effects of nerve growth factor (NGF), fluoxetine, and amitriptyline on gene expression profiles in rat brain”. Neuropeptides. 45 (5): 317–22. doi:10.1016/j.npep.2011.06.002. PMID 21820738.
- ^ Heberlein A, Muschler M, Frieling H, Behr M, Eberlein C, Wilhelm J, Gröschl M, Kornhuber J, Bleich S, Hillemacher T (May 2013). “Epigenetic down regulation of nerve growth factor during alcohol withdrawal”. Addiction Biology. 18 (3): 508–10. doi:10.1111/j.1369-1600.2010.00307.x. PMID 21392176.
- ^ Riviere JE, Papich MG (2009). Veterinary Pharmacology and Therapeutics. John Wiley & Sons. p. 318. ISBN 978-0-8138-2061-3.
- ^ Roth BL, Driscol J (12 January 2011). “PDSP Ki Database”. Psychoactive Drug Screening Program (PDSP). University of North Carolina at Chapel Hill and the United States National Institute of Mental Health. Retrieved 1 July 2014. Note: Values for humans are used. If there is more than one value listed for humans, their average is used.
- ^ Newman-Tancredi A, Cussac D, Audinot V, Nicolas JP, De Ceuninck F, Boutin JA, Millan MJ (November 2002). “Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. II. Agonist and antagonist properties at subtypes of dopamine D(2)-like receptor and alpha(1)/alpha(2)-adrenoceptor”. The Journal of Pharmacology and Experimental Therapeutics. 303 (2): 805–14. doi:10.1124/jpet.102.039875. PMID 12388667.
- ^ Newman-Tancredi A, Cussac D, Quentric Y, Touzard M, Verrièle L, Carpentier N, Millan MJ (November 2002). “Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. III. Agonist and antagonist properties at serotonin, 5-HT(1) and 5-HT(2), receptor subtypes”. The Journal of Pharmacology and Experimental Therapeutics. 303 (2): 815–22. doi:10.1124/jpet.102.039883. PMID 12388668.
- ^ Hsieh GC, Hollingsworth PR, Martino B, Chang R, Terranova MA, O’Neill AB, Lynch JJ, Moreland RB, Donnelly-Roberts DL, Kolasa T, Mikusa JP, McVey JM, Marsh KC, Sullivan JP, Brioni JD (January 2004). “Central mechanisms regulating penile erection in conscious rats: the dopaminergic systems related to the proerectile effect of apomorphine”. The Journal of Pharmacology and Experimental Therapeutics. 308 (1): 330–8. doi:10.1124/jpet.103.057455. PMID 14569075.
- ^ Lewis, R.J. Sr. (2004). Sax’s Dangerous Properties of Industrial Materials (11 ed.). Wiley, John & Sons, Incorporated. p. 287. ISBN 978-0471476627.
- ^ Jump up to:a b Narayanasamy Gurusamy. “Process for making apomorphine and apocodeine”.
- ^ Bentley, K.W. (2014-04-24). The Isoquinoline Alkaloids: A Course in Organic Chemistry. Elsevier, 2014. pp. 118–120. ISBN 978-1483152233.
- ^ Poklis JL, Mulder HA, Halquist MS, Wolf CE, Poklis A, Peace MR (July 2017). “The Blue Lotus Flower (Nymphea caerulea) Resin Used in a New Type of Electronic Cigarette, the Re-Buildable Dripping Atomizer”. Journal of Psychoactive Drugs. 49 (3): 175–181. doi:10.1080/02791072.2017.1290304. PMC 5638439. PMID 28266899.
- ^ Bertol E, Fineschi V, Karch SB, Mari F, Riezzo I (February 2004). “Nymphaea cults in ancient Egypt and the New World: a lesson in empirical pharmacology”. Journal of the Royal Society of Medicine. 97 (2): 84–5. doi:10.1177/014107680409700214. PMC 1079300. PMID 14749409.
- ^ Taba P, Lees A, Stern G (2013). “Erich Harnack (1852-1915) and a short history of apomorphine”. European Neurology. 69 (6): 321–4. doi:10.1159/000346762. PMID 23549143.
- ^ Gee S (1869). “On the action of a new organic base, apomorphia”. Transactions of the Clinical Society of London. 2: 166–169.
- ^ Feser J (1873). “Die in neuester Zeit in Anwendung gekommen Arzneimittel: 1. Apomorphinum hydrochloratum”. Z Prakt Veterinairwiss: 302–306.
- ^ Tompkins J (1899). “Apomorphine in Acute Alcoholic Delirium”. Medical Record.
- ^ “Apomorphine as a hypnotic”. The Lancet. 155 (3998): 1083. 1900. doi:10.1016/s0140-6736(01)70565-x.
- ^ Douglas CJ (1899). “The withdrawal of alcohol in delirium tremens”. The New York Medical Journal: 626.
- ^ Hare, Francis (1912). On alcoholism; its clinical aspects and treatment. London: Churchill.
- ^ Benes FM (January 2001). “Carlsson and the discovery of dopamine”. Trends in Pharmacological Sciences. 22 (1): 46–7. doi:10.1016/S0165-6147(00)01607-2. PMID 11165672.
- ^ Ernst AM (May 1965). “Relation between the action of dopamine and apomorphine and their O-methylated derivatives upon the CNS”. Psychopharmacologia. 7 (6): 391–9. doi:10.1007/BF00402361. PMID 5831877.
- ^ Moynihan NH (1965). “The Treatment of Alcoholism in General Practice”. Practitioner: 223–7.
- ^ Carlsson C, Johansson PR, Gullberg B (May 1977). “A double-blind cross-over study: apomorphine/placebo in chronic alcoholics”. International Journal of Clinical Pharmacology and Biopharmacy. 15 (5): 211–3. PMID 326687.
- ^ Halvorsen KA, Martensen-Larsen O (April 1978). “Apomorphine revived: fortified, prolonged, and improved therapeutical effect”. The International Journal of the Addictions. 13 (3): 475–84. doi:10.3109/10826087809045262. PMID 352969.
- ^ Jensen SB, Christoffersen CB, Noerregaard A (December 1977). “Apomorphine in outpatient treatment of alcohol intoxication and abstinence: a double-blind study”. The British Journal of Addiction to Alcohol and Other Drugs. 72 (4): 325–30. doi:10.1111/j.1360-0443.1977.tb00699.x. PMID 341937.
- ^ Schlatter EK, Lal S (June 1972). “Treatment of alcoholism with Dent’s oral apomorphine method”. Quarterly Journal of Studies on Alcohol. 33 (2): 430–6. PMID 5033142.
- ^ Weil E (1884). “De l’apomorphine dans certain troubles nerveux”. Lyon Med (in French). 48: 411–419.
- ^ Schwab RS, Amador LV, Lettvin JY (1951). “Apomorphine in Parkinson’s disease”. Transactions of the American Neurological Association. 56: 251–3. PMID 14913646.
- ^ Cotzias GC, Papavasiliou PS, Fehling C, Kaufman B, Mena I (January 1970). “Similarities between neurologic effects of L-dopa and of apomorphine”. The New England Journal of Medicine. 282 (1): 31–3. doi:10.1056/NEJM197001012820107. PMID 4901383.
- ^ Corsini GU, Del Zompo M, Gessa GL, Mangoni A (May 1979). “Therapeutic efficacy of apomorphine combined with an extracerebral inhibitor of dopamine receptors in Parkinson’s disease”. Lancet. 1 (8123): 954–6. doi:10.1016/S0140-6736(79)91725-2. PMID 87620.
- ^ Stibe CM, Kempster P, Lees AJ & Stern GM (1988). “Subcutaneous apomorphine in parkinsonian on-off oscillations”. Lancet. 331 (8582): 403–406. doi:10.1016/S0140-6736(88)91193-2.
- ^ Ban TA (2008). Conditioning behavior and psychiatry. New Brunswick [N.J.]: AldineTransaction. ISBN 978-0-202-36235-9. OCLC 191318001.
- ^ Raikhel EA (2016). Governing habits : treating alcoholism in the post-Soviet clinic. Ithaca. ISBN 9781501703133. OCLC 965905763.
- ^ Lemere F, Voegtlin WL (June 1950). “An evaluation of the aversion treatment of alcoholism”. Quarterly Journal of Studies on Alcohol. 11 (2): 199–204. PMID 15424345.
- ^ Dent JY (1934-10-01). “Apomorphine in the Treatment of Anxiety States, with Especial Reference to Alcoholism*”. British Journal of Inebriety. 32 (2): 65–88. doi:10.1111/j.1360-0443.1934.tb05016.x. ISSN 1360-0443.
- ^ De Morsier G, Feldmann H (1952). “[Apomorphine therapy of alcoholism; report of 500 cases]”. Schweizer Archiv für Neurologie und Psychiatrie. Archives Suisses de Neurologie et de Psychiatrie. Archivio Svizzero di Neurologia e Psichiatria. 70 (2): 434–40. PMID 13075975.
- ^ “Gay injustice ‘was widespread‘“. 2009-09-12. Retrieved 2018-01-24.
- ^ Birmingham, Jed (2 November 2009). “William Burroughs and the History of Heroin”. RealityStudio.
- ^ Morales-Rosado JA, Cousin MA, Ebbert JO, Klee EW (December 2015). “A Critical Review of Repurposing Apomorphine for Smoking Cessation”. Assay and Drug Development Technologies. 13 (10): 612–22. doi:10.1089/adt.2015.680. PMID 26690764.
- ^ “Apomorphine – A forgotten treatment for alcoholism”. apomorphine.info. Retrieved 2018-01-24.
- ^ IsHak, Waguih William (2017). The Textbook of Clinical Sexual Medicine. Springer. p. 388. ISBN 978-3-319-52539-6.
- ^ “Abbott Withdraws Application for an Impotence Pill”. Bloomberg News via The New York Times. 1 July 2000.
- ^ Jump up to:a b Lashuel HA, Hartley DM, Balakhaneh D, Aggarwal A, Teichberg S, Callaway DJ (November 2002). “New class of inhibitors of amyloid-beta fibril formation. Implications for the mechanism of pathogenesis in Alzheimer’s disease”. The Journal of Biological Chemistry. 277 (45): 42881–90. doi:10.1074/jbc.M206593200. PMID 12167652.
- ^ Himeno E, Ohyagi Y, Ma L, Nakamura N, Miyoshi K, Sakae N, Motomura K, Soejima N, Yamasaki R, Hashimoto T, Tabira T, LaFerla FM, Kira J (February 2011). “Apomorphine treatment in Alzheimer mice promoting amyloid-β degradation” (PDF). Annals of Neurology. 69 (2): 248–56. doi:10.1002/ana.22319. PMID 21387370.
- ^ Jump up to:a b c d e Bill RL (2016). Clinical Pharmacology and Therapeutics for Veterinary Technicians – E-Book. Elsevier Health Sciences. p. 94. ISBN 978-0-323-44402-6.
- ^ Jump up to:a b Khan SN, Hooser SB (2012). Common Toxicologic Issues in Small Animals, an Issue of Veterinary Clinics: Small Animal Practice – E-Book. Elsevier Health Sciences. p. 310. ISBN 978-1-4557-4325-4.
- ^ Plumb, Donald C. (2011). “Apomorphine”. Plumb’s Veterinary Drug Handbook (7th ed.). Stockholm, Wisconsin: Wiley. pp. 77–79. ISBN 978-0-470-95964-0.
- ^ Peterson ME, Talcott PA (2006). Small Animal Toxicology. Elsevier Health Sciences. p. 131. ISBN 978-0-7216-0639-2.
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Apokyn |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a604020 |
| Pregnancy category |
|
| Routes of administration |
SQ |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 100% following injection |
| Protein binding | ~50% |
| Metabolism | Hepatic, phase II |
| Onset of action | 10–20 min |
| Elimination half-life | 40 minutes |
| Duration of action | 60–90 min |
| Excretion | Hepatic |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.000.327 |
| Chemical and physical data | |
| Formula | C17H17NO2 |
| Molar mass | 267.322 g/mol g·mol−1 |
| 3D model (JSmol) | |
Apomorphine
-
- ATC:N04BC07
- Use:emetic, erectile dysfunction
- Chemical name:(R)-5,6,6a,7-tetrahydro-6-methyl-4H-dibenzo[de,g]quinoline-10,11-diol
- Formula:C17H17NO2
- MW:267.33 g/mol
- CAS-RN:58-00-4
- InChI Key:VMWNQDUVQKEIOC-UHFFFAOYSA-N
- InChI:InChI=1S/C17H17NO2/c1-18-8-7-10-3-2-4-12-15(10)13(18)9-11-5-6-14(19)17(20)16(11)12/h2-6,13,19-20H,7-9H2,1H3
- EINECS:200-360-0
- LD50:56 mg/kg (M, i.v.); >100 mg/kg (M, p.o.)
///////////// LK-01, LK 01 , LK01, Apomorphine
CN1CCC2=C3C1CC4=C(C3=CC=C2)C(=C(C=C4)O)O.CN1CCC2=C3C1CC4=C(C3=CC=C2)C(=C(C=C4)O)O.O.Cl.Cl
Fluazolepali, 氟唑帕利 , Fluzoparib

Fluazolepali
CAS 2170504-09-1
Fluzoparib; SHR-3162, (HS10160)
- HS 10160
- SHR 3162
An orally available inhibitor of poly(ADP-ribose) polymerase 1 and 2 (PARP-1/2) for treatment of solid tumors (Jiangsu Hengrui Medicine Co. Ltd., Lianyungang, China)
Fluazolepali, developed by Hengrui and Howson, is intended for the treatment of recurrent ovarian cancer, triple-negative breast cancer, advanced gastric cancer and other advanced solid tumors. Currently, the drug has been introduced into China for recurrent ovarian cancer. Clinical stage.
In February 2019, a randomized, double-blind, controlled, multicenter, phase III clinical study (CTR20190294) of flazopril capsule versus placebo for maintenance of recurrent ovarian cancer was initiated in China and was sponsored by Hengrui Medicine.
Jiangsu Hansoh Pharmaceutical , in collaboration with Jiangsu Hengrui Medicine , is developing an oral capsule formulation of fluazolepali (fluzoparib; SHR-3162), a small molecule inhibitor to PARP-1 and PARP-2, for the treatment of solid tumors including epithelial ovarian, fallopian tube or primary peritoneal, breast and gastric cancer.
- Originator Jiangsu Hengrui Medicine Co.
- Class Antineoplastics
- Mechanism of Action Poly(ADP-ribose) polymerase 1 inhibitors; Poly(ADP-ribose) polymerase 2 inhibitors
- Phase II Ovarian cancer
- Phase I Breast cancer; Fallopian tube cancer; Gastric cancer; Peritoneal cancer; Solid tumours
- 09 Jul 2019 Jiangsu HengRui Medicine initiates a phase I trial in Solid tumors in China (NCT04013048) [14C]-Fluzoparib
- 01 Jul 2019 Jiangsu HengRui Medicine plans a phase I drug-drug interaction trial (In volunteers) in China (PO) (NCT04011124)
- 12 Jun 2019 Jiangsu HengRui Medicine completes a phase I trial in Gastric cancer (Combination therapy, Recurrent, Metastatic disease, Second-line therapy or greater, Late-stage disease) in China (PO) (NCT03026881)
Fluzoparib (SHR 3162) is a selective poly [ADP-ribose] polymerase 1 (PARP1) and poly [ADP-ribose] polymerase 2 inhibitor (PARP2), being developed by Jiangsu HengRui Medicine, for the treatment of cancer. PARP enzymes play a vital role in repair of DNA damage and maintaining genomic stability. Fluzoparib inhibits PARP enzymes and induces DNA-double strands breaks, G2/M arrest and apoptosis in homologous recombination repair (HR)-deficient cells. Clinical development for ovarian cancer, breast cancer, fallopian tube cancer, peritoneal cancer, gastric cancer and solid tumours is underway in China and Australia.
An orally available inhibitor of poly (ADP-ribose) polymerase (PARP) types 1 and 2, with potential antineoplastic activity. Upon oral administration, fluzoparib inhibits PARP 1 and 2 activity, which inhibits PARP-mediated repair of damaged DNA via the base excision repair (BER) pathway, enhances the accumulation of DNA strand breaks, promotes genomic instability, and leads to an induction of apoptosis. The PARP family of proteins catalyze post-translational ADP-ribosylation of nuclear proteins, which then transduce signals to recruit other proteins to repair damaged DNA. PARP inhibition may enhance the cytotoxicity of DNA-damaging agents and may reverse tumor cell chemoresistance and radioresistance. Check for active clinical trials using this agent. (NCI Thesaurus)
PATENT
WO-2019137358
Process for preparing heterocyclic compounds (presumed to be fluazolepali ) and its intermediates as PARP inhibitors useful for treating cancer.


PATENT
WO2019109938
claiming synergistic combination comprising PARP inhibitor fluazolepali and apatinib mesylate .
PATENT
WO 2018005818
WO 2018129553
WO 2018129559
WO 2018208968
WO 2018213732
WO 2018191277
WO 2018201096
WO 2018085469
WO 2018085468
WO 2019090227
WO 2019133697
WO 2019067978
WO 2019071123
WO 2019090141
///////////Fluazolepali, Jiangsu Hansoh Pharmaceutical, Jiangsu Hengrui Medicine, fluzoparib, SHR-3162, 氟唑帕利 , Phase II, Ovarian cancer, HS10160, CHINA, HS 10160
Tanzisertib
Tanzisertib
CAS 899805-25-5
trans-4-((9-((3S)-Tetrahydrofuran-3-yl)-8-((2,4,6-trifluorophenyl)amino)-9H-purin-2-yl)amino)cyclohexanol
4-[[9-[(3S)-oxolan-3-yl]-8-(2,4,6-trifluoroanilino)purin-2-yl]amino]cyclohexan-1-ol
C21-H23-F3-N6-O2, 448.4467
- CC 930
- CC-930
- Tanzisertib
- UNII-M5O06306UO
- A c-Jun amino-terminal kinase inhibitor.UNII, M5O06306UO
Treatment of Idiopathic Pulmonary Fibrosis (IPF)
- Originator Celgene Corporation
- Class Antifibrotics; Small molecules
- Mechanism of ActionJ NK mitogen-activated protein kinase inhibitors
- Orphan Drug Status Yes – Idiopathic pulmonary fibrosis
- Discontinued Discoid lupus erythematosus; Idiopathic pulmonary fibrosis
- 16 Jul 2012 Celgene Corporation terminates a phase II trial in Discoid lupus erythematosus in USA (NCT01466725)
- 23 Feb 2012 Celgene initiates enrolment in a phase II trial for Discoid lupus erythematosus in the USA (NCT01466725)
- 08 Nov 2011The Committee for Orphan Medicinal Products (COMP) recommends orphan drug designation for tanzisertib in European Union for Idiopathic pulmonary fibrosis
Tanzisertib has been granted orphan drug status by the FDA for the treatment of idiopathic pulmonary fibrosis. A positive opinion has been received from the EU Committee for Orphan Medicinal Products (COMP
Tanzisertib has been used in trials studying the treatment of Fibrosis, Discoid Lupus, Pulmonary Fibrosis, Interstitial Lung Disease, and Lung Diseases, Interstitial, among others.
PATENT
https://patents.google.com/patent/US20090048275A1/de

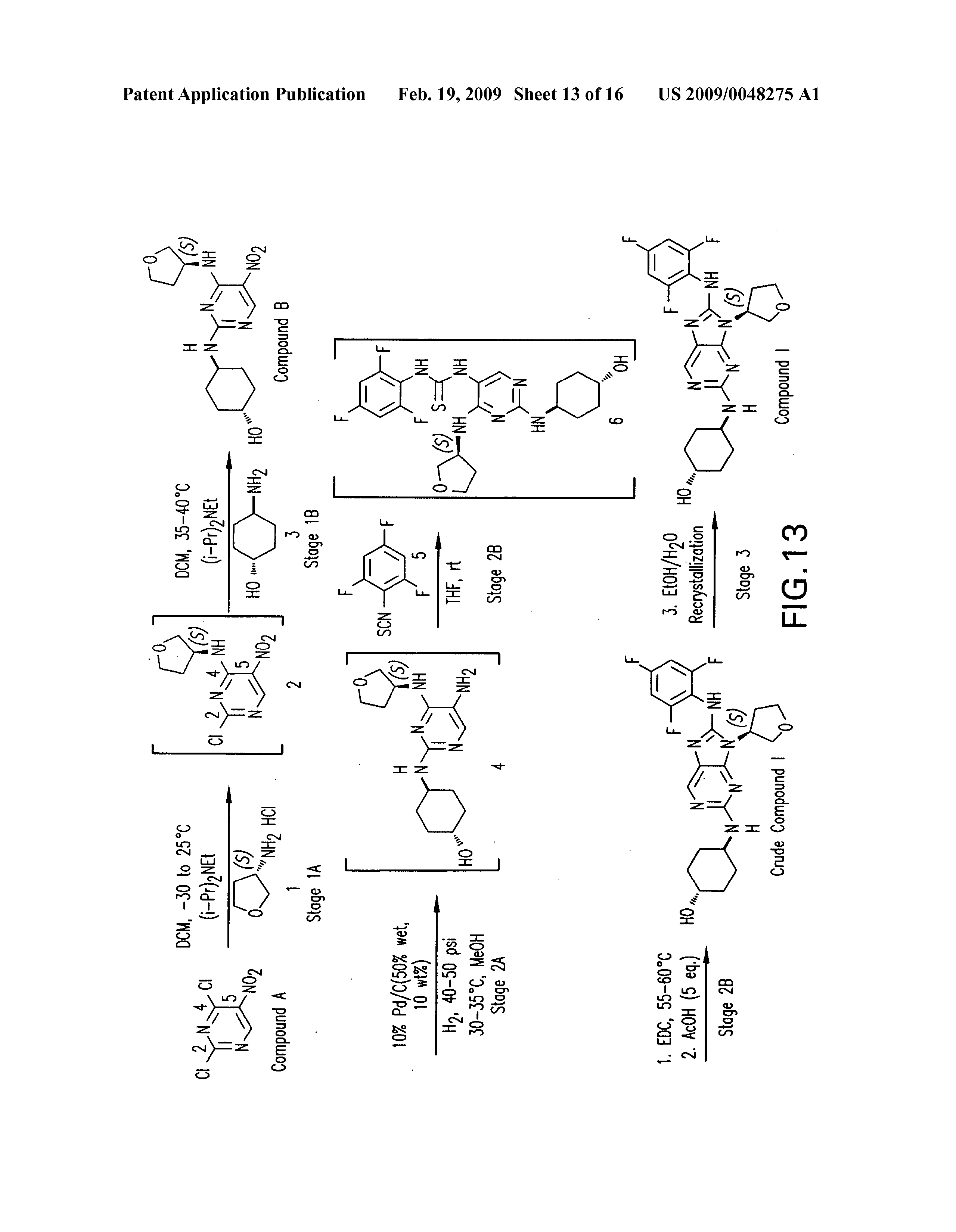

PATENT
WO 2006076595
US 20070060598
WO 2008057252
US 20080021048
US 20140094456
WO 2014055548
PATENT
WO 2015153683
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015153683
/////////Tanzisertib, CC 930, Idiopathic Pulmonary Fibrosis, Orphan Drug, phase II, CELGENE
c1c(c(c(cc1F)F)Nc2n(c3nc(ncc3n2)N[C@H]4CC[C@@H](CC4)O)[C@@H]5COCC5)F
Reldesemtiv
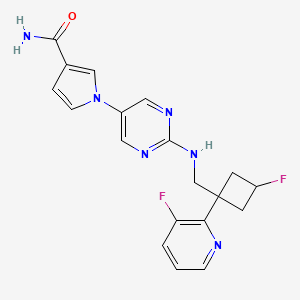
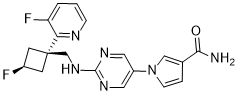
Reldesemtiv
CK-2127107
CAS 1345410-31-2
UNII-4S0HBYW6QE, 4S0HBYW6QE
MW 384.4 g/mol, MF C19H18F2N6O
1-[2-({[trans-3-fluoro-1-(3-fluoropyridin-2-yl)cyclobutyl]methyl}amino)pyrimidin-5-yl]-1H-pyrrole-3- carboxamide
1-[2-[[3-fluoro-1-(3-fluoropyridin-2-yl)cyclobutyl]methylamino]pyrimidin-5-yl]pyrrole-3-carboxamide
Reldesemtiv, also known as CK-2127107, is a skeletal muscle troponin activator (FSTA) and is a potential treatment for people living with debilitating diseases and conditions associated with neuromuscular or non-neuromuscular dysfunction, muscular weakness, and/or muscle fatigue such as SMA, COPD, and ALS.
Cytokinetics , in collaboration with Astellas , is developing reldesemtiv, the lead from a program of selective fast skeletal muscle troponin activators, in an oral suspension formulation, for the treatment of indications associated with neuromuscular dysfunction, including spinal muscular atrophy and amyotrophic lateral sclerosis.
- Originator Cytokinetics
- Developer Astellas Pharma; Cytokinetics
- Class Pyridines; Pyrimidines; Pyrroles; Small molecules
- Mechanism of Action Troponin stimulants
- Orphan Drug Status Yes – Spinal muscular atrophy
- Phase II Amyotrophic lateral sclerosis; Chronic obstructive pulmonary disease; Spinal muscular atrophy
- Suspended Muscle fatigue
- No development reported Muscular atrophy
- 05 May 2019 Safety and efficacy data from the phase II FORTITUDE-ALS trial in Amyotrophic lateral sclerosis presented at the American Academy of Neurology Annual Meeting (AAN-2019)
- 07 Mar 2019 Cytokinetics completes the phase III FORTITUDE-ALS trial for Amyotrophic lateral sclerosis in USA, Australia, Canada, Spain, Ireland and Netherlands (PO) (NCT03160898)
- 22 Jan 2019 Cytokinetics plans a phase I trial in Healthy volunteers in the first quarter of 2019
Reldesemtiv, a next-generation, orally-available, highly specific small-molecule is being developed by Cytokinetics, in collaboration with Astellas Pharma, for the improvement of skeletal muscle function associated with neuromuscular dysfunction, muscle weakness and/or muscle fatigue in spinal muscular atrophy (SMA), chronic obstructive pulmonary disease (COPD) and amyotrophic lateral sclerosis (ALS). The drug candidate is a fast skeletal muscle troponin activator (FSTA) or troponin stimulant intended to slow the rate of calcium release from the regulatory troponin complex of fast skeletal muscle fibers. Clinical development for ALS, COPD and SMA is underway in the US, Australia, Canada, Ireland, Netherlands and Spain. No recent reports of development had been identified for phase I development for muscular atrophy in the US. Due to lack of of efficacy determined at interim analysis Cytokinetics suspended phase I trial in muscle fatigue in the elderly.
The cytoskeleton of skeletal and cardiac muscle cells is unique compared to that of all other cells. It consists of a nearly crystalline array of closely packed cytoskeletal proteins called the sarcomere. The sarcomere is elegantly organized as an interdigitating array of thin and thick filaments. The thick filaments are composed of myosin, the motor protein responsible for transducing the chemical energy of ATP hydrolysis into force and directed movement. The thin filaments are composed of actin monomers arranged in a helical array. There are four regulatory proteins bound to the actin filaments, which allows the contraction to be modulated by calcium ions. An influx of intracellular calcium initiates muscle contraction; thick and thin filaments slide past each other driven by repetitive interactions of the myosin motor domains with the thin actin filaments.
[0003] Of the thirteen distinct classes of myosin in human cells, the myosin-II class is responsible for contraction of skeletal, cardiac, and smooth muscle. This class of myosin is significantly different in amino acid composition and in overall structure from myosin in the other twelve distinct classes. Myosin-II forms homo-dimers resulting in two globular head domains linked together by a long alpha-helical coiled-coiled tail to form the core of the sarcomere’s thick filament. The globular heads have a catalytic domain where the actin binding and ATPase functions of myosin take place. Once bound to an actin filament, the release of phosphate (cf. ADP-Pi to ADP) signals a change in structural conformation of the catalytic domain that in turn alters the orientation of the light-chain binding lever arm domain that extends from the globular head; this movement is termed the powerstroke. This change in orientation of the myosin head in relationship to actin causes the thick filament of which it is a part to move with respect to the thin actin filament to which it is bound. Un-binding of the globular head from the actin filament (Ca2+ regulated) coupled with return of the catalytic domain and light chain to their starting conformation/orientation completes the catalytic cycle, responsible for intracellular movement and muscle contraction.
Tropomyosin and troponin mediate the calcium effect on the interaction on actin and myosin. The troponin complex is comprised of three polypeptide chains: troponin C, which binds calcium ions; troponin I, which binds to actin; and troponin T, which binds to tropomyosin. The skeletal troponin-tropomyosin complex regulates the myosin binding sites extending over several actin units at once.
Troponin, a complex of the three polypeptides described above, is an accessory protein that is closely associated with actin filaments in vertebrate muscle. The troponin complex acts in conjunction with the muscle form of tropomyosin to mediate the
Ca2+ dependency of myosin ATPase activity and thereby regulate muscle contraction. The troponin polypeptides T, I, and C, are named for their tropomyosin binding, inhibitory, and calcium binding activities, respectively. Troponin T binds to tropomyosin and is believed to be responsible for positioning the troponin complex on the muscle thin filament. Troponin I binds to actin, and the complex formed by troponins I and T, and tropomyosin inhibits the interaction of actin and myosin. Skeletal troponin C is capable of binding up to four calcium molecules. Studies suggest that when the level of calcium in the muscle is raised, troponin C exposes a binding site for troponin I, recruiting it away from actin. This causes the tropomyosin molecule to shift its position as well, thereby exposing the myosin binding sites on actin and stimulating myosin ATPase activity.
U.S. Patent No. 8962632 discloses l-(2-((((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)amino)pyrimidin-5-yl)-lH-pyrrole-3-carboxamide, a next-generation fast skeletal muscle troponin activator (FSTA) as a potential treatment for people living with debilitating diseases and conditions associated with neuromuscular or non-neuromuscular dysfunction, muscular weakness, and/or muscle fatigue.
PATENT
WO 2011133888

PATENT
WO2016039367 ,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016039367&tab=FULLTEXT
claiming the use of a similar compound for treating stress urinary incontinence.
PATENT
WO-2019133605
Process for preparing reldesemtiv , a myosin, actin, tropomyosin, troponin C, troponin I, troponin T modulator, useful for treating neuromuscular disorders, muscle wasting, claudication and metabolic syndrome.
Scheme 1
[0091] Scheme 1 illustrates a scheme of synthesizing the compound of Formula (1C).
Scheme 2
[0092] Scheme 2 illustrates an alternative scheme of synthesizing the compound of Formula (1C).
M
TFAA DS, toluene
Et
to
2
HCI, H20
50°C
Scheme 3
[0093] Scheme 3 illustrates a scheme of converting the compound of Formula (1C) to the compound of Formula (II).
H2
Ni Raney
NH3
Scheme 4
[0094] Scheme 4 illustrates a scheme of converting the compound of Formula (II) to the compound of Formula (1).
Examples
[0095] To a flask was added N-methylpyrrolidone (30 mL), tert-butyl cyanoacetate (8.08 g) at room temperature. To a resulting solution was added potassium tert-butoxide (7.71 g), l,3-dibromo-2,2-dimethoxy propane (5.00 g) at 0 °C. To another flask, potassium iodide (158 mg), 2,6-di-tert-butyl-p-cresol (42 mg), N-methylpyrrolidone (25 mL) were added at room temperature and then resulting solution was heated to 165 °C. To this solution, previously prepared mixture was added dropwise at 140-165 °C, then stirred for 2 hours at 165 °C. To the reaction mixture, water (65 mL) was added. A resulting solution was extracted with toluene (40 mL, three times) and then combined organic layer was washed with water (20 mL, three times) and 1N NaOH aq. (20 mL). A resulting organic layer was concentrated below 50 °C under reduced pressure to give 3, 3 -dimethoxy cyclobutane- l-carbonitrile (66% yield,
GC assay) as toluene solution. 1H MR (CDCl3, 400 MHz) d 3.17 (s, 3H), 3.15 (s, 3H), 2.93-2.84 (m, 1H), 2.63-2.57 (m, 2H), 2.52-2.45 (m, 2H).
Example 2 Synthesis of methyl 3,3-dimethoxycyclobutane-l-carboxylate
[0096] A reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. MeOH (339.00 kg), 3-oxocyclobutanecarboxylic acid (85.19 kg, 746.6 mol, 1.0 eq.), Amberlyst-l5 ion exchange resin (8.90 kg, 10% w/w), and
trimethoxymethane (196.00 kg, 1847.3 mol, 2.5 eq.) were charged into the reactor and the resulting mixture was heated to 55±5°C and reacted for 6 hours to give methyl 3,3-dimethoxycyclobutane-l-carboxylate solution in MeOH. 1H NMR (CDCl3, 400 MHz) d 3.70 (s, 3H), 3.17 (s, 3H), 3.15 (s, 3H), 2.94-2.85 (m, 1H), 2.47-2.36 (m, 4H).
Example 3 Synthesis of 3, 3-dimethoxycyclobutane-l -carboxamide
[0097] The methyl 3, 3 -dimethoxy cyclobutane- l-carboxylate solution in MeOH prepared as described in Example 2 was cooled to below 25°C and centrifuged. The filter cake was washed with MeOH(7.00 kg) and the filtrate was pumped to the reactor. The solution was concentrated under vacuum below 55°C until the system had no more than 2 volumes. MeOH
(139.40 kg) was charged to the reactor and the solution was concentrated under vacuum below 55°C until the system had no more than 2 volumes. MeOH (130.00 kg) was charged to the reactor and the solution was concentrated under vacuum below 55°C until the system had no more than 2 volumes. Half of the resulting solution was diluted with MeOH (435.00 kg) and cooled to below 30°C. NH3 gas (133.80 kg) was injected into the reactor below 35°C for
24 hours. The mixture was stirred at 40±5°C for 72 hours. The resulting solution was
concentrated under vacuum below 50°C until the system had no more than 2 volumes.
MTBE(l8l.OO kg) was charged into the reactor. The resulting solution was concentrated under vacuum below 50°C until the system had no more than 2 volumes. PE (318.00 kg) was charged into the reactor. The resulting mixture was cooled to 5±5°C, stirred for 4 hours at 5±5°C, and centrifuged. The filter cake was washed with PE (42.00 kg) and the wet filter cake was put into a vacuum oven. The filter cake was dried at 30±5°C for at least 8 hours to give 3,3-dimethoxycyclobutane-l-carboxamide as off-white solid (112.63 kg, 94.7% yield). 1H NMR (CDCf, 400 MHz) d 5.76 (bs, 1H), 5.64 (bs, 1H), 3.18 (s, 3H), 3.17 (s, 3H), 2.84-2.76 (m, 1H), 2.45-2.38 (m, 4H).
[0098] A reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. Toluene (500.00 kg), 3,3-dimethoxycyclobutane-l-carboxamide (112.54kg, 706.9 mol, 1.0 eq.), and TEA (158.00 kg, 1561.3 mol, 2.20 eq) were charged into the reactor and the resulting mixture was cooled to 0+ 5°C. TFAA (164.00 kg, 781 mol, 1.10 eq.) was added dropwise at 0±5°C. The resulting mixture was stirred for 10 hours at 20±5°C and cooled below 5±5°C. H20 (110.00 kg) was charged into the reactor at below 15 °C. The resulting mixture was stirred for 30 minutes and the water phase was separated. The aqueous phase was extracted with toluene (190.00 kg) twice. The organic phases were combined and washed with H20 (111.00 kg). H20 was removed by azeotrope until the water content was no more than 0.03%. The resulting solution was cooled to below 20°C to give 3,3-dimethoxycyclobutane-l-carbonitrile solution in toluene (492.00 kg with 17.83% assay content, 87.9% yield).
Example 5 Synthesis of l-(3-fluoropyridin-2-yl)-3,3-dimethoxycyclobutane-l-carbonitrile
[0099] A reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. The 3,3-dimethoxycyclobutane-l-carbonitrile solution in toluene prepared as described in Example 4 (246.00 kg of a 17.8% solution of 3,3-dimethoxycyclobutane-l-carbonitrile in toluene, 1.05 eq.) and 2-chloro-3-fluoropyridine (39.17 kg, 297.9 mol, 1.00 eq.) were charged into the reactor. The reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. The mixture was slowly cooled to -20±5°C. NaHDMS (2M in THF) (165.71 kg, 1.20 eq) was added
dropwise at -20±5°C. The resulting mixture was stirred at -l5±5°C for 1 hour. The mixture was stirred until the content of 2-chloro-3-fluoropyridine is no more than 2% as measured by HPLC. Soft water (16.00 kg) was added dropwise at below 0°C while maintaining the reactor temperature. The resulting solution was transferred to another reactor. Aq. NH4Cl (10% w/w, 88.60 Kg) was added dropwise at below 0°C while maintaining the reactor temperature. Soft water (112.00 kg) was charged into the reactor and the aqueous phase was separated and collected. The aqueous phase was extracted with ethyl acetate (70.00 kg) and an organic phase was collected. The organic phase was washed with sat. NaCl (106.00 kg) and collected. The above steps were repeated to obtain another batch of organic phase. The two batches of organic phase were concentrated under vacuum below 70°C until the system had no more than 2 volumes. The resulting solution was cooled to below 30°C to give a l-(3-fluoropyridin-2-yl)-3, 3 -dimethoxy cyclobutane- l-carbonitrile solution. 1H NMR (CDC13, 400 MHz) d 8.42-8.38 (m, 1H), 7.50-7.45 (m, 1H), 7.38-7.33 (m, 1H), 3.28 (s, 3 H), 3.13 (s, 3H), 3.09-3.05 (m, 4H).
Example 6 Synthesis of I-(3-fluoropyridin-2-yl)-3-oxocyclohutanecarhonitrile
[0100] A reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. Water (603.00 kg) was added to the reactor and was stirred.
Concentrated HC1 (157.30 kg) was charged into the reactor at below 35°C. The l-(3-fluoropyridin-2-yl)-3, 3 -dimethoxy cyclobutane- l-carbonitrile solution prepared as described in Example 5 (206.00 kg) was charged into the reactor and the resulting mixture was heated to 50±5°C and reacted for 3 hours at 50±5°C. The mixture was reacted until the content of 1-(3 -fluoropyridin-2-yl)-3, 3 -dimethoxycyclobutane- l-carbonitrile was no more than 2.0% as measured by HPLC. The reaction mixture was cooled to below 30°C and extracted with ethyl acetate (771.00 kg). An aqueous phase was collected and extracted with ethyl acetate (770.00 kg). The organic phases were combined and the combined organic phase was washed with soft water (290.00 kg) and brine (385.30 kg). The organic phase was concentrated under vacuum at below 60°C until the system had no more than 2 volumes. Propan-2-ol (218.00 kg) was charged into the reactor. The organic phase was concentrated under vacuum at below
60°C until the system had no more than 1 volume. PE (191.00 kg) was charged into the reactor at 40±5 °C and the resulting mixture was heated to 60±5 °C and stirred for 1 hour at 60±5 °C. The mixture was then slowly cooled to 5±5 °C and stirred for 5 hours at 5±5 °C. The mixture was centrifuged and the filter cake was washed with PE (48.00 kg) and the wet filter cake was collected. Water (80.00 kg), concentrated HC1 (2.20 kg), propan-2-ol (65.00 kg), and the wet filter cake were charged in this order into a drum. The resulting mixture was stirred for 10 minutes at 20±5 °C. The mixture was centrifuged and the filter cake was washed with a mixture solution containing 18.00 kg of propan-2-ol, 22.50 kg of soft water, and 0.60 kg of concentrated HC1. The filter cake was put into a vacuum oven and dried at 30±5°C for at least 10 hours. The filter cake was dried until the weight did not change to give l-(3-fluoropyridin-2-yl)-3-oxocyclobutanecarbonitrile as off-white solid (77.15 kg, 68.0% yield). 1H NMR (CDCl3, 400 MHz) d 8.45-8.42 (m, 1H), 7.60-7.54 (m, 1H), 7.47-7.41 (m, 1H), 4.18-4.09 (m, 2H), 4.02-3.94 (m, 2H).
Example 7 Synthesis of I-(3-fhtoropyridin-2-yl)-3-hydroxycyclobulanecarbonilrile
[0101] To a solution of l-(3-fluoropyridin-2-yl)-3-oxocyclobutanecarbonitrile (231 g,
1.22 mol) in a mixture ofDCM (2 L) and MeOH (200 mL) was added NaBH4 portionwise at -78° C. The reaction mixture was stirred at -78°C. for 1 hour and quenched with a mixture of methanol and water (1 : 1). The organic layer was washed with water (500 mL><3), dried over Na2S04, and concentrated. The residue was purified on silica gel (50% EtO Ac/hexanes) to provide the title compound as an amber oil (185.8 g, 77.5%). Low Resolution Mass
Spectrometry (LRMS) (M+H) m/z 193.2.
Example 8 Synthesis of (ls,3s)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutane-l-carbonitrile
[0102] To a solution of 1 -(3 -fluoropyridin-2-yl)-3 -hydroxy cyclobutanecarbonitrile (185 g, 0.96 mol) in DCM (1 L) was added DAST portionwise at 0-10 °C. Upon the completion of addition, the reaction was refluxed for 6 hours. The reaction was cooled to rt and poured onto sat. NaHCCf solution. The mixture was separated and the organic layer was washed with water, dried over Na2S04, and concentrated. The residue was purified on silica gel (100% DCM) to provide the title compound as a brown oil (116g) in a 8: 1 transxis mixture. The above brown oil (107 g) was dissolved in toluene (110 mL) and hexanes (330mL) at 70 °C. The solution was cooled to 0 °C and stirred at 0 °C overnight. The precipitate was filtered and washed with hexanes to provide the trans isomer as a white solid (87.3 g). LRMS (M+H) m/z 195.1.
Example 9 Synthesis of ((lr,3r)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methanamine
[0103] A mixture of ( 1.v,3.v)-3-fluoro- 1 -(3-fluoropyridin-2-yl)cyclobutane- 1 -carbonitrile (71 g, 0.37 mol) and Raney nickel (~7 g) in 7N ammonia in methanol (700 mL) was charged with hydrogen (60 psi) for 2 days. The reaction was filtered through a celite pad and washed with methanol. The filtrate was concentrated under high vacuum to provide the title compound as a light green oil (70 g, 97.6%). LRMS (M+H) m/z 199.2.
Example 10 Synthesis of t-butyl 5-bromopyrimidin-2-yl((trans-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl) carbamate
[0104] A mixture of ((lr,3r)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methanamine (37.6 g, 190 mmol), 5-bromo-2-fluoropyrimidine (32.0 g, 181 mmol), DIPEA (71 mL, 407 mmol), and NMP (200 mL) was stirred at rt overnight. The reaction mixture was then diluted with EtOAc (1500 mL) and washed with saturated sodium bicarbonate (500 mL). The
organic layer was separated, dried over Na2S04, and concentrated. The resultant solid was dissolved in THF (600 mL), followed by the slow addition of DMAP (14 g, 90 mmol) and Boc20 (117.3 g, 542 mmol). The reaction was heated to 60° C. and stirred for 3 h. The reaction mixture was then concentrated and purified by silica gel chromatography
(EtO Ac/hex) to give 59.7 g oft-butyl 5-bromopyrimidin-2-yl((trans-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)carbamate as a white solid.
Example 11 Synthesis of t-butyl 5-(3-cyano- 1 H -pyrrol- 1 -yl)pyrimidin-2-yl(((lrans)-3-fhtoro-l-(3-fluoropyridin-2-yl)cyclohutyl)methyl)carhamate
[0105] To a solution oft-butyl 5-bromopyrimidin-2-yl((trans-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl) carbamate (1.0 g, 2.8 mmol) in 15 mL of toluene (degassed with nitrogen) was added copper iodide (100 mg, 0.6 mmol), potassium phosphate (1.31 g, 6.2 mmol), trans-N,N’-dimethylcyclohexane-l, 2-diamine (320 mg, 2.2 mmol), and 3-cyanopyrrole (310 mg, 3.6 mmol). The reaction was heated to 100 °C and stirred for 2 h. The reaction was then concentrated and purified by silica gel chromatography (EtOAc/hexanes) to afford 1.1 g of t-butyl 5-(3-cyano-lH-pyrrol-l-yl)pyrimidin-2-yl(((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)carbamate as a clear oil.
Example 12 Synthesis of l-(2-((((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)amino)pyrimidin-5-yl)-lH-pyrrole-3-carboxamide
[0106] To a solution oft-butyl 5-(3-cyano-lH-pyrrol-l-yl)pyrimidin-2-yl(((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)carbamate (1.1 g, 3.1 mmol) in DMSO (10 mL) was added potassium carbonate (1.3 g, 9.3 mmol). The mixture was cooled to 0 °C and hydrogen peroxide (3 mL) was slowly added. The reaction was warmed to rt and stirred for 90 min. The reaction was diluted with EtO Ac (75 mL) and washed three times with brine (50 mL). The organic layer was then dried over Na2S04, filtered, and concentrated to give a crude solid that was purified by silica gel chromatography (10% MeOH/CH2Cl2) to afford 1.07 g of a white solid compound. This compound was dissolved in 25% TFA/CH2CI2 and stirred for 1 hour. The reaction was then concentrated, dissolved in ethyl acetate (75 mL), and washed three times with saturated potassium carbonate solution. The organic layer was then dried over Na2S04, filtered, and concentrated to give a crude solid that was triturated with 75% ethyl acetate/hexanes. The resultant slurry was sonicated and filtered to give 500 mg of l-(2-((((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)amino)pyrimidin-5-yl)-lH-pyrrole-3 -carboxamide as a white solid. LRMS (M+H=385).
REFERENCES
1: Andrews JA, Miller TM, Vijayakumar V, Stoltz R, James JK, Meng L, Wolff AA, Malik FI. CK-2127107 amplifies skeletal muscle response to nerve activation in humans. Muscle Nerve. 2018 May;57(5):729-734. doi: 10.1002/mus.26017. Epub 2017 Dec 11. PubMed PMID: 29150952.
2: Gross N. The COPD Pipeline XXXII. Chronic Obstr Pulm Dis. 2016 Jul 14;3(3):688-692. doi: 10.15326/jcopdf.3.3.2016.0150. PubMed PMID: 28848893; PubMed Central PMCID: PMC5556764.
//////////////CK-2127107, CK 2127107, CK2127107, Reldesemtiv, Cytokinetics, Astellas, neuromuscular disorders, muscle wasting, claudication, metabolic syndrome, spinal muscular atrophy, amyotrophic lateral sclerosis, Orphan Drug Status, Spinal muscular atrophy, Phase II
C1C(CC1(CNC2=NC=C(C=N2)N3C=CC(=C3)C(=O)N)C4=C(C=CC=N4)F)F
SELPERCATINIB

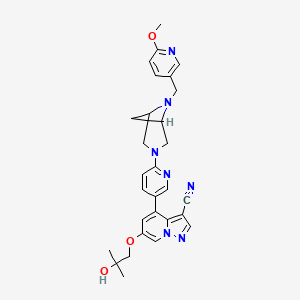
SELPERCATINIB
LOXO 292
CAS: 2152628-33-4
Chemical Formula: C29H31N7O3
Molecular Weight: 525.613
CEGM9YBNGD
UNII-CEGM9YBNGD
6-(2-hydroxy-2-methylpropoxy)-4-(6-{6-[(6-methoxypyridin- 3-yl)methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl}pyridin-3- yl)pyrazolo[1,5-a]pyridine-3-carbonitrile
Selpercatinib is a tyrosine kinase inhibitor with antineoplastic properties.
A phase I/II trial is also under way in pediatric patients and young adults with activating RET alterations and advanced solid or primary CNS tumors.
Loxo Oncology (a wholly-owned subsidiary of Eli Lilly ), under license from Array , is developing selpercatinib, a lead from a program of RET kinase inhibitors, for treating cancer, including non-small-cell lung cancer, medullary thyroid cancer, colon cancer, breast cancer, pancreatic cancer, papillary thyroid cancer, other solid tumors, infantile myofibromatosis, infantile fibrosarcoma and soft tissue sarcoma
In 2018, the compound was granted orphan drug designation in the U.S. for the treatment of pancreatic cancer and in the E.U. for the treatment of medullary thyroid carcinoma.

PATENT
WO2018071447
PATENT
US 20190106438
PATENT
WO 2019075108
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019075108&tab=PCTDESCRIPTION
Compounds of Formula I-IV, 4-(6-(4-((6-methoxypyridin-3-yl)methyl)piperazin-1-yl)pyridin-3-yl)-6-(1-methyl-1H-pyrazol-4-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile (Formula I); 6-(2-hydroxy-2-methylpropoxy)-4-(6-(6-((6-methoxypyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile (Formula II); 6-(2-hydroxy-2-methylpropoxy)-4-(6-(6-(6-methoxynicotinoyl)-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile (Formula III); and 6-(2-hydroxy-2-methylpropoxy)-4-(6-(4-hydroxy-4-(pyridin-2-ylmethyl)piperidin-1-yl)pyridin-3-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile (Formula IV) are inhibitors of RET kinase, and are useful for treating diseases such as proliferative diseases, including cancers.
[0007] Accordingly, provided herein is a compound of Formula I-IV:
and pharmaceutically acceptable salts, amorphous, and polymorph forms thereof.
PATENT
WO 2019075114
PATENT
WO-2019120194
Novel deuterated analogs of pyrazolo[1,5-a]pyrimidine compounds, particularly selpercatinib , processes for their preparation and compositions comprising them are claimed. Also claims are their use for treating pain, inflammation, cancer and certain infectious diseases.
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US10137124 | Substituted pyrazolo[1,5-a]pyridine compounds as RET kinase inhibitors | 2018-01-03 | |
| US10172851 | Substituted pyrazolo[1,5-A]pyridine compounds as RET kinase inhibitors | 2018-01-03 | |
| US10112942 | Substituted pyrazolo[1,5-A]pyridine compounds as RET kinase inhibitors | 2017-12-29 |
/////////////SELPERCATINIB, non-small-cell lung cancer, medullary thyroid cancer, colon cancer, breast cancer, pancreatic cancer, papillary thyroid cancer, other solid tumors, infantile myofibromatosis, infantile fibrosarcoma, soft tissue sarcoma, LOXO, ELI LILY, ARRAY, LOXO 292, orphan drug designation
N#CC1=C2C(C3=CC=C(N4CC(C5)N(CC6=CC=C(OC)N=C6)C5C4)N=C3)=CC(OCC(C)(O)C)=CN2N=C1