

Candidate: LHC165
Home » PHASE 1 (Page 4)
Category Archives: PHASE 1
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
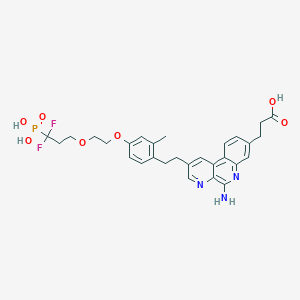
LHC 165

LHC165
3-[5-amino-2-[2-[4-[2-(3,3-difluoro-3-phosphonopropoxy)ethoxy]-2-methylphenyl]ethyl]benzo[f][1,7]naphthyridin-8-yl]propanoic acid
C29H32F2N3O7P, 603.56 g/mol
CAS 1258595-14-0
5-Amino-2-[2-[4-[2-(3,3-difluoro-3-phosphonopropoxy)ethoxy]-2-methylphenyl]ethyl]benzo[f][1,7]naphthyridine-8-propanoic acid
Benzo[f][1,7]naphthyridine-8-propanoic acid, 5-amino-2-[2-[4-[2-(3,3-difluoro-3-phosphonopropoxy)ethoxy]-2-methylphenyl]ethyl]-
- Originator Novartis
- Class Antineoplastics
- Mechanism of Action
- Undefined mechanism
- Phase I Solid tumours
- 31 Jan 2018 Phase-I clinical trials in Solid tumours (Combination therapy, Inoperable/Unresectable, Late-stage disease, Metastatic disease, Second-line therapy or greater) in USA, Belgium, Italy, Japan (Intratumoural) (NCT03301896)
- 31 Jan 2018 Phase-I clinical trials in Solid tumours (Inoperable/Unresectable, Late-stage disease, Metastatic disease, Monotherapy, Second-line therapy or greater) in USA, Japan, Italy, Belgium (Intratumoural) (NCT03301896)
- 10 Oct 2017 Novartis plans a phase I trial for Solid tumours (Monotherapy, Combination therapy, Inoperable/Unresectable, Late-stage disease, Metastatic disease, Second-line therapy or greater) in USA, Belgium, Canada, France, Germany, Italy, South Korea and Spain in November 2017 (Intratumoural) (NCT03301896)
PATENT
PATENT
US 20110053893
PATENT
WO 2011130379
PATENT
Scheme (III)
Scheme (IV)
Scheme (V)
Example 19 (Table 1: Compound 19): Synthesis of ‘3-(5-amino-2-(4-(2-(3,3-difluoro-3-phosphonopropoxy)ethoxy)-2-methylphenethyl)benzo[f][ 1, 7]naphthyridin-8-yl)propanoic acid (19)
Scheme 6
Step 1: (E)-ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)acrylate (6-3)
[517] To a solution of tert-butyl 5-bromo-2-chlorophenylcarbamate (6-1) (1.0 equiv.) in acetonitrile (0.3 M) and EtOH (0.5 M) was added K2C03 (2.0 equiv.). The reaction was degassed and flushed with N , then added (E)-ethyl 3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)acrylate (6-2) (1.2 equiv.) and Pd(PPh3)4 (0.1 equiv.). The reaction was flushed again with N2 and stirred at 100 °C overnight. After cooling to room temperature, hexane was added, and the mixture was filtered through a pad of silica, eluting with EA/Hex (1 : 1) until the product was completely eluted. The filtrate was concentrated and purified on Combiflash, eluting with 0-15% EA in Hex to give (E)-ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)acrylate (6-3) as a white solid.
Step 2: ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)propanoate (6-4)
[518] To a solution of (E)-ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)acrylate (6-3) (1.0 equiv.) in ethyl acetate/ethanol (1 : 1 , 0.3 M) was added Wilkinson’s catalyst (0.10 equiv.).
Hydrogen gas was introduced via a ballon, and the reaction was stirred at room temperature for 24 hours. The mixture was filtered through a pad of celite, washing with dichloromethane. The filtrate was concentrated in vacuo and purified by Combiflash using 0-10% ethyl acetate in hexane to give ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)propanoate (6-4) as a solid.
Step 3: ethyl 3-(3-(tert-butoxycarbonylamino)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)propanoate (6-5)
[519] A solution of ethyl 3-(3-(tert-butoxycarbonylamino)-4-chlorophenyl)propanoate (6-4) (1 .0 equiv.), 4,4,4,,4′,5,5,5′,5′-octamethyl-2,2′-bi(l ,3,2-dioxaborolane) (2.0 equiv.), tris(dibenzylideneacetone)dipalladium(0) (0.05 equiv.), 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (0.20 equiv.), and potassium acetate (2.0 equiv.) in 1 ,4-dioxane (0.2 M) was degassed and stirred at 100 °C overnight. After cooling to ambient temperature, the reaction content was concentrated in vacuo. The crude material was purified by Combiflash using 0-50% ethyl acetate in hexane to afford ethyl 3-(3-(tert-butoxycarbonylamino)-4-(4,4,5,5-tetramethyl- 1 ,3,2-dioxaborolan-2-yl)phenyl)propanoate (6-5) as a brown oil. The product was stored at -20°C and used within a month of synthesis.
Step 4: l-bromo-4-(methoxymethoxy)-2-methylbenzene (6-7)
[520] To a solution of 4-bromo-3-methylphenol (6-6) (1.0 equiv.) in DMF (0.5 M) at 0 °C was added portionwise 60% wt NaH (1.5 equiv.). The addition was controlled such that internal reaction temperature never went above 10 °C. The reaction was stirred at room temperature for 45 minutes, then a solution of chloro(methoxy)methane (1.2 equiv.) in DMF (3 M) was added dropwise via additional funnel. The reaction was stirred at room temperature for 3.5 hours, and then quenched by pouring into ice. The resulting mixture was stirred at room temperature for 1 hour. Ether was added, and the two layers were separated. The aqueous layer was extracted (lx) with ether. The combined organic layers were washed with water (2x), brine, dried over MgS04, and concentrated to give 1 -bromo-4-(methoxymethoxy)-2-methylbenzene (6-7) as a colorless oil. The crude material was used in the next step without further purification.
Step 5: triethylf (4-(methoxymethoxy)-2-methylphenyl)ethynyl)silane
[521] A solution of l -bromo-4-(methoxymethoxy)-2-methylbenzene (1.0 equiv.), triethylamine (5.0 equiv.) in DMF (0.5 M) was degassed and flushed with nitrogen. To the reaction was added TES-acetylene (1.05 equiv.), Cul (0.098 equiv.), and Pd(PPh3)2Cl2 (0.098 equiv.). The reaction was heated to 60 °C and stirred overnight. After cooling to room temperature, water and ether were added. The layers were separated, and the organic layer was washed with water (2x). The organic layer was separated and passed through a pad of silica (packed with hexane). The silica was eluted with 10% EA in Hex. The fractions were combined and concentrated to give triethyl((4-(methoxymethoxy)-2-methylphenyl)ethynyl)silane as a black oil. The crude material was used in the next step without further purification.
Step 6: l-ethynyl-4-(methoxymethoxy)-2-methylbenzene (6-8)
[522] To a solution of triethyl((4-(methoxymethoxy)-2-methylphenyl)ethynyl)silane (1.0 equiv.) at
0 °C was slowly added tetrabutylammonium fluoride (1M solution in THF, 0.20 equiv.). At this
point, the ice-bath was removed and the reaction mixture was allowed to stir at room temperature for 45 minutes. The reaction mixture was then passed through a pad of silica (packed with hexane) and eluted with 20% EtOAc in Hexanes to remove insoluble salts. The crude product was then purified by Combiflash using 0-10% EtOAc in Hexanes to give 1 -ethynyl-4-(methoxymethoxy)-2-methylbenzene (6-8) as a slightly brown liquid.
Step 7: 3-chloro-5-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)picolinonitrile (6-10)
[523] A solution of l -ethynyl-4-(methoxymethoxy)-2-methylbenzene (6-8) (1 .0 equiv.), 3,5-dichloropicolinonitrile (6-9) (0.90 equiv.), Cul (0.10 equiv.), and Pd(PPh3)2CI2 (0.10 equiv.), and triethylamine (5.0 equiv.) in DMF (0.25 M) was degassed and flushed with nitrogen. The reaction mixture was then heated to 60 °C and stirred overnight. After cooling to room temperature, water was added. The mixture was extracted with EA (2x). The combined organic layers were washed with 10% aq NH4OH (2x), brine, and concentrated. The crude material was filtered through a pad of silica (wetted with hexane). The silica was eluted with 10% EA in Hex. The fractions were combined and concentrated. The resulting solids were washed in hot ether and filtered to give a yellow solid, which was used in the next step without further purification. The filtrate was concentrated and purified by Combiflash using 0- 10% EtOAc in Hexanes to give 3-chloro-5-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)picolinonitrile (6-10) as a yellow solid.
Step 8: ethyl 3-(5-amino-2-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)-ben∑o fJfl, 7J
naphthyridin-8-yl)propanoate (6-11)
[524] A solution of 3-chloro-5-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)picolinonitrile (6-10) (1 .0 equiv.), ethyl 3-(3-(tert-butoxycarbonylamino)-4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)phenyl)propanoate (6-5) (1.25 equiv.), tris(dibenzylideneacetone)dipalladium(0) (0.10 equiv.), dicyclohexyl(2′,6′-dimethoxybiphenyl-2-yl)phosphine (0.20 equiv.), and sodium bicarbonate (3.0 equiv.) in «-butanol /H20 (5: 1 , 0.2 M) was degassed and stirred at 100 °C overnight. After cooling to ambient temperature, the reaction content was diluted with ethyl acetate and water. The two phases were separated, and the aqueous layer was extracted twice with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous MgS04, and concentrated in vacuo. The crude material was purified by flash chromatography on a COMBIFLASH® system (1SCO) using 0-40% ethyl acetate in DCM first to remove the impurity, then 0-4% MeOH in DCM to give ethyl 3-(5-amino-2-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)-benzo[f][l ,7]naphthyridin-8-yl) propanoate (6-11). Further purification was accomplished by precipitating and washing in hot ether.
Step 9: ethyl 3-(5-amino-2-(4-(methoxymethoxy)-2-methylphenethyl)benzo[fl[l ]naphthyridin-8-yl)propanoate (6-12)
[525] A solution of ethyl 3-(5-amino-2-((4-(methoxymethoxy)-2-methylphenyl)ethynyl)-benzo[f][l ,7]naphthyridin-8-yl)propanoate (6-11) (1.0 equiv.) in EtOH/THF (3: 1 , 0.16 M) was flushed with nitrogen. Then, 10% wt Pd/C (0.20 equiv. by weight) was added. The reaction was flushed with hydrogen (2x) and stirred under a hydrogen balloon. After 24 hours, the reaction was filtered through a pad of celite, washing with 5%MeOH in DCM. The filtrate was checked for the presence of starting material using LCMS. The hydrogenation reaction was repeated until no more
of the alkyne starting material or alkene intermediate was detected. The crude product was purified by Combiflash using 0-4% eOH in DCM to give ethyl 3-(5-amino-2-(4-(methoxymethoxy)-2-methylphenethyl)benzo[f][l ,7]naphthyridin-8-yl)propanoate (6-12) as a white solid.
Step 10: ethyl 3-(5-amino-2-(4-hydroxy-2-methylphenethyl)benzo[fl[l ]naphthyridin-8-yl)propanoate (6-13)
[526] Ethyl 3-(5-amino-2-(4-(methoxymethoxy)-2-methylphenethyl)benzo[fJ[l ,7]naphthyridin-8-yl)propanoate (6-12) (1 .0 equiv.) was dissolved in EtOH (0.2 M), then added a solution of 4M HC1 in dioxane (0.2 M). The product precipitated out as a yellow salt. After stirring for 3 hours, the reaction was poured into a stirring solution of ether. The mixture was stirred for 10 minutes, then filtered and washed with ether. Ethyl 3-(5-amino-2-(4-hydroxy-2-methylphenethyl)benzo[fJ[l ,7]naphthyridin-8-yl)propanoate (6-13) was obtained as a yellow solid which was dried on vacuum overnight (bis-HCl salt). Alternatively, the crude product was purified by Combiflash using 0-5% MeOH in DCM to give the free base.
Step 11: ethyl 3-(5-amino-2-(4-(2-(3-(diethoxyphosphoryl)-3,3-difluoropropoxy)ethoxy)-2-methylphenethyl)benzo[f] [1 , 7]naphthyridin-8-yl)propanoate ( 6-15)
[527] To a solution of ethyl 3-(5-amino-2-(4-hydroxy-2-methylphenethyl)benzo[fJ [ l ,7]naphthyridin-8-yl)propanoate (6-13) (1.0 equiv.) dissolved in DMF (0.14 M) was added a solution of diethyl 3-(2-bromoethoxy)-l ,l -difluoropropylphosphonate (6-14: described in Example 7 – Step 1) (1 .3 equiv.) in DMF (0.7 M) and cesium carbonate (4 equiv.). The reaction was stirred at 60 °C. After 1.5 hours (or until reaction is complete by LCMS), DCM (2 volume equivalent) was added to the reaction. The solids (inorganic) were filtered, and the filtrate was concentration. The crude product was purified by Combiflash using 0-5%MeOH in DCM to give ethyl 3-(5-amino-2-(4-(2-(3-(diethoxyphosphoryl)-3,3-difluoropropoxy)ethoxy)-2-methylphenethyl)benzo[fJ
[1 ,7]naphthyridin-8-yl)propanoate (6-15) as an oil which upon standing became a white solid.
Step 12: 3-(5-amino-2-(4-(2-(3,3-difluoro-3-phosphompropoxy)ethoxy)-2-methylphenethyl)be o[f]
[1, 7]naphthyridin-8-yl)propanoic acid (19)
[528] To a solution of ethyl 3-(5-amino-2-(4-(2-(3-(diethoxyphosphoryl)-3,3-difluoropropoxy)ethoxy)-2-methylphenethyl)benzo[f][l ,7]naphthyridin-8-yl)propanoate (6-15) (1.0 equiv.) in DCM (0.16 M) at 0 °C was added slowly TMSBr (10 equiv.). The reaction was stirred at room temperature overnight. Additional TMSBr (5.0 equiv.) was added at 0 °C, and the reaction was again stirred at room temperature overnight. The solvent was removed by evaporation and the crude orange solids dried on hi-vac briefly. The solids were suspended in EtOH (0.5 M) and added 2.5 N
NaOH (10.0 equiv.). The reaction was stirred at 80 °C for 3 hours. After cooling to room temperature, the mixture was adjusted to pH 9 to 10 and directly purified on RP-HPLC using a CI 8 column, eluting with 10-40% 95:5 (MeCN/5mM NH4OAc) in l OmM NH4OAc (pH 9) gradient. The fractions containing the product were combined and concentrated in vacuo. The resulting white gel was dissolved in refluxing 1 :1 EtOH/water (0.04 M) with the addition of a few drops of ammonium hydroxide. While hot, the mixture was slowly poured into a stirring hot solution of acetone (0.009
M) preheated at 50 °C. The acetone suspension was slowly cooled to room temperature for 15 minutes with continued stirring, and then sat in an ice bath for 10 minutes. The solids were filtered and washed successively with acetone (2x) and ether (2x). The solids were dried on hi-vac overnight to give the 3-(5-amino-2-(4-(2-(3,3-difluoro-3-phosphonopropoxy)ethoxy)-2-methylphenethyl)benzo [fj[l ,7]naphthyridin-8-yl)propanoic acid (19) as a solid. Ή NMR (Dimethylsulfoxide-d6): δ 9.02 (s, 1 H), 8.82 (s, 1H), 8.55 (d, 1H, J = 8.4 Hz), 7.58 (s, 1H), 7.48 (d, 1 H, J = 8.4 Hz), 7.07 (d, 1H, J = 8.4 Hz), 6.75 (s, 1 H), 6.68 (d, 1H, J = 8.4 Hz), 4.03-4.00 (m, 2H), 3.72-3.68 (m, 4H), 3.16-3.12 (m, 2H), 3.03-2.96 (m, 4H), 2.67-2.64 (m, 2H), 2.33-2.32 (m, 2H), 2.26 (s, 3H). LRMS [M+H] = 604.2
PATENT
US 20120237546
PATENT
WO 2012031140
PATENT
Toll-like receptors (TLRs) are pattern recognition receptors which play an essential role in the innate immunity, by recognizing invasion of microbial pathogens and initiating intracellular signal transduction pathways to trigger expression of genes, the products of which can control innate immune responses. Specifically, Toll like receptor (TLR) agonists activate innate immune cells through the TLR-MyD88-NFk and IRF3/7 pathways. TLR7, TLR8, and TLR9 belong to a subfamily of TLRs based on their genomic structure, sequence similarities, and homology. TLR7, TLR8, and TLR9 are located in intracellular endolysosomal compartments and show a unique pattern of cell type-specific expression that is thought to be responsible for different pathogen response profiles.
Small molecule agonists of TLR7 and/or TLR8 have been reported and shown to activate innate immune responses by inducing selected cytokine biosynthesis, the induction of co-stimulatory molecules, and by increased antigen-presenting capacity. Such compounds include imidazoquinoline amine derivatives (U.S. Patent No. 4689338), imidazopyridine amine derivative (U.S. Patent No. 5446153), imidazonaphthyridine derivative (U.S. Patent No.
6194425), oxazoloquinoline amine derivatives (U.S. Patent No. 61 10929); thiazoloquinoline amine derivatives (U.S. Patent No. 61 10929), selenazoloquinoline amine derivatives (U.S. Patent No. 61 10929), pyrazolopyridine derivatives (U.S. Patent No. 9145410), and
benzonaphthyridine amine derivatives (U.S. Patent Nos. 8466167 and 9045470).
The synthetic TLR7 agonist, Imiquimod (1 -(2-methylpropyl)-1 H-imidazo[ 4,5-c]quinolin-4-amine) is FDA-approved in a cream formulation for the topical treatment of cutaneous basal cell carcinoma, actinic keratosis and genital warts, and has limited activity against cutaneous melanoma and breast tumors (J. Immunol. 2014, 193(9) : 4722^1-731 ). Systemic administration of Imiquimod, and structurally similar Resiquimod, is limited by cytokine- mediated adverse effects including severe flu-like symptoms (Expert Opin. Emerging Drugs (2010), 15:544-555). Consequently, Imiquimod is used exclusively in topical applications and is not used to treat deep, non-cutaneous tumors such as melanoma or solid tumors.
An injectable lipid modified imidazoquinoline (TLR7/8 dual agonist) that forms a tissue depot with gradual, sustained release which allows for local TLR triggering activity without systemic cytokine release has been reported (J. Immunol. 2014, 193(9): 4722^731 ). However, this compound was shown to be ineffective for large tumors and in addition the serum concentration of this compound 24 hours post subcutaneous administration decreased by approximately 50% (Journal for ImmunoTherapy of Cancer, 2014, 2:12). Therefore, there remains a need for intratumor administration of a TLR7 agonist with prolonged sustained release, which may benefit the treatment of large tumors.
clip

Credit: Tien Nguyen/C&EN
Presenter: Alex Cortez, senior Investigator I at the Genomics Institute of the Novartis Research Foundation
Target: Toll-like receptor 7 (TLR7)
Disease: Solid tumors
Reporter’s notes: Cortez shared another story in the realm of immuno-oncology, although the program that yielded this compound actually started in the world of vaccines. Cortez’s team had been focusing on vaccine adjuvants, small molecules that turn on the immune system to enhance a vaccine’s effect. They developed one such class of compound that activates toll-like receptor 7 (TLR7), a protein in the immune system that recognizes dangerous-looking molecules and can trigger the release of infection-clearing proteins. After observing TLR7 agonists’ ability to induce an immune response with vaccines, the researchers wondered whether the molecules could also be effective in immuno-oncology.
They found that LHC165 adsorbed to aluminum hydroxide reduced tumor growth in mice and, intriguingly, showed signs of an abscopal effect, in which untreated tumors shrink concurrently with treated tumors. The implication is that if the immune system recognizes one tumor site, it can recognize others. As with several of the candidates presented throughout the day, LHC165 bears a phosphate group and is injected into the tumor. It’s currently in Phase I trials in patients with advanced malignancies, which means they’ve already tried second and third line therapies, as a single agent and in combination with the checkpoint inhibitor PDR001.
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9597326 | BENZONAPTHYRIDINE COMPOSITIONS AND USES THEREOF | 2011-04-13 | 2013-05-16 |
| US9950062 | COMPOUNDS AND COMPOSITIONS AS TLR ACTIVITY MODULATORS | 2010-09-01 | 2012-09-20 |
| US9517263 | BENZONAPHTHYRIDINE-CONTAINING VACCINES | 2010-06-10 | 2012-10-18 |
| US2015225432 | COMPOUNDS AND COMPOSITIONS AS TLR ACTIVITY MODULATORS | 2015-04-24 | 2015-08-13 |
| US9315530 | ADSORPTION OF IMMUNOPOTENTIATORS TO INSOLUBLE METAL SALTS | 2011-09-01 |
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2016213776 | ADSORPTION OF IMMUNOPOTENTIATORS TO INSOLUBLE METAL SALTS | 2016-04-07 | 2016-07-28 |
| US2012177681 | Formulation of immunopotentiators | 2011-09-01 | 2012-07-12 |
| US9045470 | COMPOUNDS AND COMPOSITIONS AS TLR ACTIVITY MODULATORS | 2011-03-03 | |
| US2018169204 | COMBINATION VACCINES WITH LOWER DOSES OF ANTIGEN AND/OR ADJUVANT | 2018-02-02 | |
| US9375471 | ADJUVANTED FORMULATIONS OF BOOSTER VACCINES | 2013-03-08 | 2013-09-12 |
//////LHC165, LHC 165, LHC -165, Phase I, Solid tumours, novartis
O=P(O)(O)C(F)(F)CCOCCOc4ccc(CCc1cc2c3ccc(CCC(=O)O)cc3nc(N)c2nc1)c(C)c4
CC1=C(C=CC(=C1)OCCOCCC(F)(F)P(=O)(O)O)CCC2=CN=C3C(=C2)C4=C(C=C(C=C4)CCC(=O)O)N=C3N
AB 680
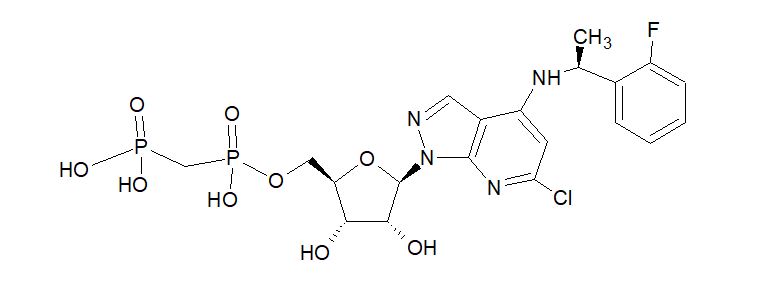
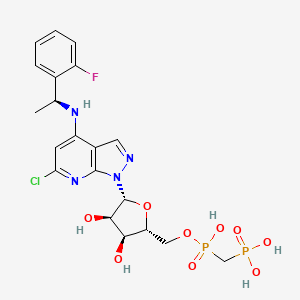
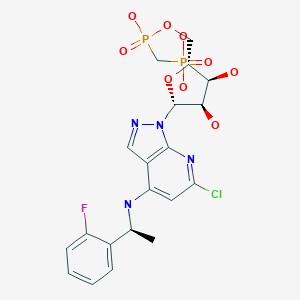

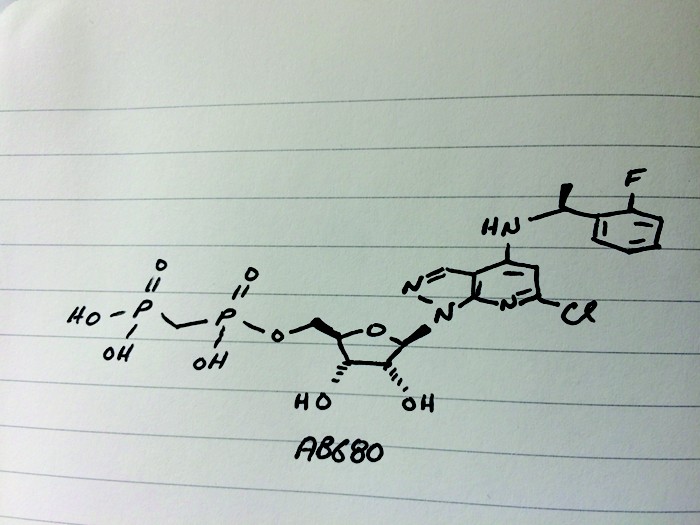
AB 680
C20H24ClFN4O9P2, 580.827 g/mol
Cas 2105904-82-1
1H-Pyrazolo[3,4-b]pyridin-4-amine, 6-chloro-N-[(1S)-1-(2-fluorophenyl)ethyl]-1-[5-O-[hydroxy(phosphonomethyl)phosphinyl]-β-D-ribofuranosyl]-
[[(2R,3S,4R,5R)-5-[6-chloro-4-[[(1S)-1-(2-fluorophenyl)ethyl]amino]pyrazolo[3,4-b]pyridin-1-yl]-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]methylphosphonic acid
[({[(2R,3S,4R,5R)-5-(6-chloro-4-{[(1S)-1-(2-fluorophenyl)ethyl]amino}-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]methoxy}(hydroxy)phosphoryl)methyl]phosphonic Acid
- Originator C
- Class Antineoplastics; Small molecules
- Mechanism of Action 5-nucleotidase inhibitors; Adenosine A2 receptor antagonists
- Phase I Cancer
- 19 Nov 2018 Arcus Biosciences plans to initiate a clinical trial in Cancer in first half of 2019
- 16 Oct 2018 Phase-I clinical trials in Cancer (In volunteers) in Australia (IV) (NCT03677973)
- 30 Sep 2018 Preclinical pharmacodynamics data in Cancer presented at 4th CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference (CRI-CIMT-EATI-AACR – 2018)
Clip
Credit: Tien Nguyen/C&EN
Presenter: Kenneth V. Lawson, senior scientist at Arcus Biosciences
Target: Ecto-5’-nucleotidase (CD73)
Disease: Cancer
Reporter’s notes: In the first talk of the day, Lawson introduced the idea of cancer drugs that target the host’s immune system. “Checkpoint inhibitors changed the way we think of treating cancer,” he said. These drugs successfully disrupt the binding interaction between a protein and a checkpoint protein that stops immune T cells from killing cancer cells. As a result, these drugs turn immune cells loose to attack tumor cells. But the drugs work only in about 30-40% of patients—an issue pharmaceutical companies like Arcus hope to address with new immunotherapies that can be taken in combination with checkpoint inhibitors.
Lawson’s team set out to inhibit an enzyme commonly found in tumors called CD73, the second of two enzymes which break down extracellular adenosine trisphosphate (ATP) to adenosine. Adenosine then binds to immunosuppressive receptors on immune cells and shuts them down. Yet developing a small molecule inhibitor of CD73 proved challenging, Lawson said. After striking out with high-throughput screening, the team turned to CD73’s natural substrate for inspiration. However, the molecule possessed more than one phosphate group, which is notoriously a liability for drug molecules because small molecules with such negative changes struggle to cross cell membranes. The team’s goal was to remove the phosphate groups, Lawson says, but things didn’t exactly go according to plan. After showing the audience a series of compounds from structure-activity relationship (SAR) studies—slides no medicinal chemistry talk would be complete without—Lawson revealed the structure of their final clinical compound AB680 as the sound of people flipping notebook sheets rippled across the room. Synthesized in 34% overall yield, the candidate ultimately included two phosphate groups—a feature that surprised audience members.
Tests revealed that AB680 can be given intravenously but the compound also showed moderate oral bioavailability. Lawson suggested a possible route for how the molecule might pass from the digestive tract to the bloodstream, a paracellular mechanism by which molecules cross the epithelium by passing through the space between cells. AB680 showed “extraordinary potency,” inhibiting CD73 in human T-cells at a concentration of 0.008 nM. The compound has a 4 day half-life, which means it could be dosed every two weeks, coinciding with the dosing schedule for patients who receive a checkpoint inhibitor. AB680 is currently in Phase 1 clinical trials with healthy patients.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US204141996&tab=PCTDESCRIPTION&maxRec=1000
|
Purinergic signaling, a type of extracellular signaling mediated by purine nucleotides and nucleosides such as ATP and adenosine, involves the activation of purinergic receptors in the cell and/or in nearby cells, resulting in the regulation of cellular functions. Most cells have the ability to release nucleotides, which generally occurs via regulated exocytosis (see Praetorius, H. A.; Leipziger, J. (1 Mar. 2010) Ann Rev Physiology 72(1): 377-393). The released nucleotides can then be hydrolyzed extracellularly by a variety of cellular membrane-bound enzymes referred to as ectonucleotidases.
|
Example 92
Synthesis of [({[(2R,3S,4R,5R)-5-(6-chloro-4-{[(1S)-1-(2-fluorophenyl)ethyl]amino}-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]methoxy}(hydroxy)phosphoryl)methyl]phosphonic Acid
PATENT

////////////////ARCUS, AB 680, AB680, AB-680, PHASE 1
https://www.arcusbio.com/wp-content/uploads/2018/04/AACR_AB680_1756_final_90x42-abstract-4886.pdf
Fc1ccccc1[C@H](C)Nc4cc(Cl)nc3c4cnn3[C@@H]2O[C@H](COP(=O)(O)CP(=O)(O)O)[C@@H](O)[C@H]2O
CC(C1=CC=CC=C1F)NC2=CC(=NC3=C2C=NN3C4C(C(C(O4)COP(=O)(CP(=O)(O)O)O)O)O)Cl
CMX-8521, CMX-521
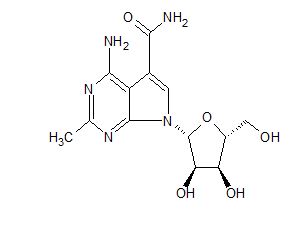
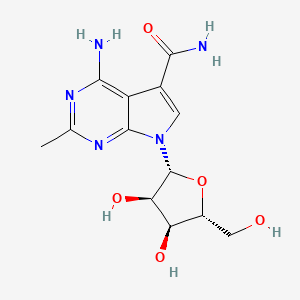
CMX-8521, CMX-521
MF C13 H17 N5 O5, MW 323.30
CAS Number 2077178-99-3
7H-Pyrrolo[2,3-d]pyrimidine-5-carboxamide, 4-amino-2-methyl-7-β-D-ribofuranosyl-
Nucleoside analogs (oral, norovirus infection), Chimerix
4-amino-7-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxamide
4-amino-7-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-2-methylpyrrolo[2,3-d]pyrimidine-5-carboxamide
CMX8521 is a nucleoside analog that inhibits the norovirus RNA polymerase. CMX8521 has in vitro activity against mouse and human norovirus.Where possible, Chimerix uses its lipid conjugate technology to build nucleoside-analog antivirals that are orally absorbed and have favorable tissue penetration.
CMX-8521 (presumed to be CMX-521) being developed by Chimerix for treating norovirus infection. In June 2018, a phase II efficacy trial was planned in 2019.
In January 2016, preclinical data were presented at the 34th Annual JP Morgan Healthcare Conference in San Francisco, CA. CMX-8521 had in vitro activity against mouse and human norovirus (EC50 = 2.1; CC50 = 114 microM). A 7-day non GLP toxicology/toxicokinetic study was completed in-life with no clinical or gross post mortem signs of toxicity. No off-target pharmacology was observed in vitro when screened against a panel of 87 receptors, transporters and enzymes associated with adverse pharmacology
PATENT
WO2017024310
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017024310
Scheme 1: General Synthesis of Compounds of the Invention

Scheme 2: General Synthesis of Compounds of the Invention

Example 7– Synthesis of Compound 1

[00315] Step 1 (Protocol #1): To a 100-L jacketed reactor were charged 4-amino-6- bromo-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (3.00 kg), (3R,4R,5R)-2-acetoxy-5- ((benzoyloxy)methyl)tetrahydrofuran-3,4-diyl dibenzoate (6.60 kg) and DCE (18.89 kg). Stirring was started and DBU (3.61) kg was added. Over a period of 03 h and 14 min, TMSOTf (8.01 kg) was added between 30.6 °C and 37.3 °C. IPC after 01 h and 30 min at approx.32 °C showed 4% of 4-amino-6-bromo-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (3.00 kg),
(3R,4R,5R)-2-acetoxy-5-((benzoyloxy)methyl)tetrahydrofuran-3,4-diyl dibenzoate remaining. IPC after 03h and 16 min at approx.32 °C showed 2% 4-amino-6-bromo-2-methyl-7H- pyrrolo[2,3-d]pyrimidine-5-carbonitrile (3.00 kg), (3R,4R,5R)-2-acetoxy-5- ((benzoyloxy)methyl)tetrahydrofuran-3,4-diyl dibenzoate remaining (spec:≤3%). The reaction mixture was diluted with DCM (39.81 kg) and quenched with potable water (15.02 kg) over an 11 min period between 9.5 °C and 15.6 °C. The extractive work-up (at approx.22 °C) was completed by a back extraction of the aqueous phase with DCM (19.90 kg), a wash with sat NaHCO3 (1.3 kg NaHCO3 in 14.9 kg potable water), a back extraction of the bicarbonate phase with DCM (19.71 kg) and a wash with brine (4.5 kg NaCl in 14.9 kg potable water). Note: the reactor was cleaned with potable water, acetone and DCM after each wash/back extraction.
[00316] The drummed organic phase containing the product was charged to the 100-L jacketed reactor through an in-line filter followed by a DCM rinse of the drum and filter with DCM (2.48 kg). The contents of the reactor were distilled to 31 L with the aid of vacuum over a period of 06 h and 04 min with a maximum temperature of 50.1 °C. At this point a thick suspension had formed. Next, over a period of 39 min, IPAc (41.88 kg) was added between 44.5 °C and 49.5 °C and the contents of the reactor were heated to 76.9 °C over a period of 01 h and 25 min. Next, the contents of the reactor were cooled to 9.9 °C over a period of 04 h and 21 min and stirred for 12 h and 26 min with a minimum temperature of 1.6 °C.
[00317] Step 1 (Protocol # 2): To a 100-L jacketed reactor were charged 4-amino-6- bromo-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (3.00 kg), (3R,4R,5R)-2-acetoxy-5- ((benzoyloxy)methyl)tetrahydrofuran-3,4-diyl dibenzoate (6.60 kg) and DCE (18.80 kg). Stirring was started and DBU (3.59) kg was added. Over a period of 01 h and 46 min, TMSOTf (7.90 kg) was added between 30.4 °C and 34.2 °C. IPC after 02 h and 49 min at approx.34 °C showed 1% of 4-amino-6-bromo-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile remaining (spec: ≤3%). The reaction mixture was diluted with DCM (40/70 kg) and quenched with potable water (14.97 kg) over an 04 min period between 9.9 °C and 18.0 °C. The extractive work-up (at approx.22 °C) was completed by a back extraction of the aqueous phase with DCM (20.34 kg), a wash with sat NaHCO3 (1.30 kg NaHCO3 in 14.90 kg potable water), a back extraction of the bicarbonate phase with DCM (20.65 kg) and a wash with brine (4.50 kg NaCl in 14.96 kg potable water). Note: the reactor was cleaned with potable water, acetone and DCM after each wash/back extraction.
[00318] The drummed organic phase containing the product was charged to the 100-L jacketed reactor through an in-line filter followed by a DCM rinse of the drum and filter with DCM (1.49 kg). The contents of the reactor were distilled to with the aid of vacuum over a period of 04 h and 49 min with a maximum temperature of 45.6 °C. At this point a thick suspension had formed. Next, over a period of 27 min, IPAc (41.70 kg) was added between 45.6 °C and 48.2 °C and the contents of the reactor were heated to 75.7 °C over a period of 01 h and 20 min. Next, the contents of the reactor were cooled to 9.4 °C over a period of 04 h and 15 min and stirred overnight with a minimum temperature of 2.3 °C.
[00319] Step 2: To the reactor were charged (2R,3R,4R,5R)-2-(4-amino-6-bromo-5- cyano-2-methyl-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-5-((benzoyloxy)methyl)tetrahydrofuran-3,4- diyl dibenzoate (10.0 kg), 10% Pd on C (Degussa, Type E101NE/W), trimethylamine (7.3 kg) and THF (44.5 kg). Hydrogen was submitted to the reactor and the mixture was stirred for 03 h and 54 min between 24.7 °C and 19.6 °C at approx.30.8 psig. IPC (HPLC) showed that
(2R,3R,4R,5R)-2-(4-amino-6-bromo-5-cyano-2-methyl-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-5- ((benzoyloxy)methyl)tetrahydrofuran-3,4-diyl dibenzoate could no longer be detected.
[00320] The reaction mixture was filtered over Celite (7.2 kg) and a polish filter and the filter residue was washed with THF (5.2 kg). The combined filtrate and wash was transferred to a 100-L jacketed reactor with the aid of a THF wash (2.12 kg). The contents of the reactor were vacuum distilled with a maximum batch temperature of 30.0 °C over a period of 05 h and 38 min to a final volume of 27 L. IPA (31.48 kg) was charged over a 40 min period to the reactor between 39.7 °C and 53.2 °C. The contents of the reactor were vacuum distilled with a maximum batch temperature of 53.2 °C over a period of 03 h and 02 min to a final volume of 33 L. IPA (48.99 kg) was charged over a 43 min period to the reactor between 53.1 °C and 57.1 °C. The contents of the reactor were heated to 60.2 °C, agitated for 12 min and cooled over a period of 04 and 28 min to 5.4 °C. Cold stirring was continued for a period of 08 h and 55 min with a minimum temperature of 1.1 °C. The slurry was filtered and washed with IPA (9.41 kg, at approx.4.5 °C). The residue was dried under vacuum with a nitrogen bleed for a period of 11 h and 44 min at a maximum temperature of 44.0 °C to provide an LOD of 0.36%. Yield: 6.58 kg (73.9 %).1H NMR confirms structure. Purity: 97.78 % (HPLC, AUC).
[00321] Step 3:

1100 g NaOH dissolved in potable water to a total volume of 1 L; 2 Diluted 500 mL conc. HCl in 2 L total with potable water [00322] A solution of (2R,3R,4R,5R)-2-(4-amino-5-cyano-2-methyl-7H-pyrrolo[2,3- d]pyrimidin-7-yl)-5-((benzoyloxy)methyl)tetrahydrofuran-3,4-diyl dibenzoate and THF was heated to 54 °C and the addition of 2.5 M NaOH was started. The initial addition gave a biphasic mixture and endothermic response (the temperature dropped to 50 °C) but as the addition continued a single phased, clear solution formed which was accompanied by a fast exotherm to 61 °C; the reaction temperature was maintained at 60 °C to 61 °C during the rest of the addition and for an additional 2 ½ h. IPC showed that no (2R,3R,4R,5R)-2-(4-amino-5-cyano-2-methyl- 7H-pyrrolo[2,3-d]pyrimidin-7-yl)-5-((benzoyloxy)methyl)tetrahydrofuran-3,4-diyl dibenzoate was left.
[00323] The reaction mixture was cooled to 21 °C and neutralized with 3 N HCl with external cooling to pH = 7.06 (Denver Instrument UB-10 pH meter equipped with a Sartorius P- P11 pH electrode, the electrode was checked with buffer solutions of pH = 4.00 and pH = 7.00); the mixture continued to cool to 8°C. The resulting neutralized mixture was distilled under vacuum with a pot temperature of 45 °C to 50 °C until the emergence of solids were observed in the pot. The suspension was cooled and stirred for 2 h at 2 °C. The beige suspension was filtered to afford a dark filtrate; the off-white residue was washed once with cold water (500 mL, 5 °C). A first LOD after 16 h gave a value of 18.73 %. HPLC) of the drying material showed the presence of 1.6% benzoate.
[00324] A brief rework study for compound 1, (containing 1.6% benzoic acid per AUC, HPLC) was executed in 10 vol of water (1 g in 10 mL):
● 3 h slurry at ambient
● 3h slurry at 50 °C
● 24 h slurry at ambient
[00325] All three experiments gave compound 1 with less than 0.1 % benzoic acid (UAC, HPLC). The slurries were fluid, were easily stirred and filtration was fast. Short term drying on the filter gave a powder-like solid indicating that a displacement wash with an organic solvent is not needed. Without wishing to be bound by theory, a loss of NMT than 1% is expected
(solubility 1 mg/mL).HPLC data for compound 1 were obtained with a method suitable for polar compounds using a Zorbax Eclipse Plus C18 column (water / ACN / TFA, 97.5 / 2.5 / 0.05). This is the same column used for steps 1 and 2.
[00326] The cold product suspension was filtered and the reactor and residue were washed with cold IPAc (approx.7.5 °C, 13.16 kg and 13.62 kg) until a colorless filtrate had been obtained. The residue was dried under vacuum and a nitrogen bleed≤ 45 °C for a period of 65 h and 19 min to an LOD of 0 %. Yield: 5.87 kg (70.7 %), 1H NMR confirmed identity; HPLC purity 98.84% (AUC). EQUIVALENTS
[0001] The disclosure can be embodied in other specific forms without departing from the spirit or essential characteristics thereof. The foregoing embodiments are therefore to be considered in all respects illustrative rather than limiting on the disclosure described herein. Scope of the disclosure is thus indicated by the appended claims rather than by the foregoing description, and all changes that come within the meaning and range of equivalency of the claims are intended to be embraced therein.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019060692&tab=PCTDESCRIPTION&maxRec=1000
Novel crystalline forms of 4-amino-7-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl) tetrahydrofuran-2-yl)-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxamide and their stable hemihydrate crystalline forms (designated as Form A-G), processes for their preparation and compositions comprising them are claimed. Also claimed is their use for treating viral infection.
Viral infections can have serious adverse effects on individuals and society as a whole. In addition to fatal viral infections such as Ebola, even non-fatal infections can have serious societal and economic consequences. For example, human noroviruses (NV) are the most common cause of epidemic acute gastroenteritis worldwide with an estimated 19-21 million cases each year in the United States including 56,000-71,000 hospitalizations and 570-800 deaths (Hall et al., Emerg.Infect.Dis. 2013 Aug; 19(8): 1198-205).
[0004] 4-amino-7-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl) tetrahydrofuran-2-yl)-2-methyl-7H-pyrrolo [2,3-d]pyrimidine-5-carboxamide (Compound 1) is an antiviral drug.
Formula 1
[0065] As used herein, “Formula I” is understood to encompass all diastereomers of 4-amino-7-(3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxamide, and pharmaceutically acceptable salts and solvates thereof. The structure of Formula I is shown below:
(Formula I).
[0066] In some embodiments, a compound of Formula I can be 4-amino-7-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxamide (“Compound 1”), or a pharmaceutically acceptable salt solvate, or isomers (e.g., enantiomers and diastereomers) thereof. The structure of Compound 1 is shown below:
| atent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9701706 | Pyrrolopyrimidine nucleosides and analogs thereof | 2016-11-22 | 2017-07-11 |
| US9708359 | PYRROLOPYRIMIDINE NUCLEOSIDES AND ANALOGS THEREOF | 2016-08-08 | |
| US2017253628 | PYRROLOPYRIMIDINE NUCLEOSIDES AND ANALOGS THEREOF | 2017-05-18 |
///////////CMX-8521, CMX 8521, CMX-521, PHASE 1
NC(=O)c2cn([C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O)c3nc(C)nc(N)c23
Epitinib
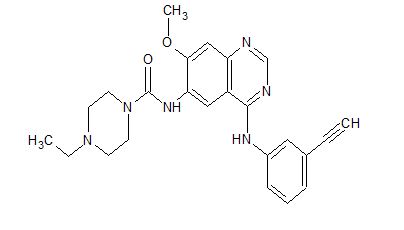
Epitinib succinate; HMPL-813; Huposuan yipitini
1203902-67-3, 430.50, C24 H26 N6 O2
1-Piperazinecarboxamide, 4-ethyl-N-[4-[(3-ethynylphenyl)amino]-7-methoxy-6-quinazolinyl]-
4-Ethyl-N-[4-[(3-ethynylphenyl)amino]-7-methoxy-6-quinazolinyl]-1-piperazinecarboxamide
Cancer; Glioblastoma; Non-small-cell lung cancer
Epitinib is in phase I clinical trials by Hutchison MediPharma for the treatment of solid tumours.
Epitinib succinate is an oral EGFR tyrosine kinase inhibitor in early clinical development at Hutchison China MediTech (Chi-Med) for the treatment of solid tumors and the treatment of glioblastoma patients with EGFR gene amplification.
- Originator Hutchison MediPharma
- Class Antineoplastics; Small molecules
- Mechanism of Action Epidermal growth factor receptor antagonists
- Phase I/II Glioblastoma; Non-small cell lung cancer
- No development reported Oesophageal cancer; Solid tumours
- 28 May 2018 No recent reports of development identified for preclinical development in Oesophageal-cancer in China (PO)
- 06 Mar 2018 Hutchison Medipharma plans a phase III pivotal study for Non-small cell lung cancer (NSCLC) patients with brain metastasis in China in 2018
- 06 Mar 2018 Phase-I/II clinical trials in Glioblastoma (Second-line therapy or greater) in China (PO)
PATENT
Binding of epidermal growth factor (EGF) to epidermal growth factor receptor (EGFR) activates tyrosine kinase activity and thereby triggers reactions that lead to cellular proliferation. Overexpression and/or overactivity of EGFR could result in uncontrolled cell division which may be a predisposition for cancer. Compounds that inhibit the overexpression and/or overactivity of EGFR are therefore candidates for treating cancer.
The relevant compound 4-ethyl-N- (4- ( (3-ethynylphenyl) amino) -7-methoxyquinazolin-6-yl) piperazine-1-carboxamide of the present invention has the effect of effectively inhibiting the overexpression and/or overactivity of EGFR. Thus, it is useful in treating diseases associated with overexpression and/or overactivity of EGFR, such as the treatment of cancer.
The phenomenon that a compound could exist in two or more crystal structures is known as polymorphism. Many compounds may exist as various polymorph crystals and also in a solid amorphous form. Until polymorphism of a compound is discovered, it is highly unpredictable (1) whether a particular compound will exhibit polymorphism, (2) how to prepare any such unknown polymorphs, and (3) how are the properties, such as stability, of any such unknown polymorphs. See, e.g., J. Bernstein “Polymorphism in Molecular Crystals” , Oxford University Press, (2002)
Since the properties of a solid material depend on the structure as well as on the nature of the compound itself, different solid forms of a compound can and often do exhibit different physical and chemical properties as well as different biopharmaceutical properties. Differences in chemical properties can be determined, analyzed and compared through a variety of analytical techniques. Those differences may ultimately be used to differentiate among different solid forms. Furthermore, differences in physical properties, such as solubility, and biopharmaceutical properties, such as bioavailability, are also of importance when describing the solid state of a pharmaceutical compound. Similarly, in the development of a pharmaceutical compound, e.g., 4-ethyl-N- (4- ( (3-ethynylphenyl) amino) -7-methoxyquinazolin-6-yl) piperazine-1-carboxamide, the new crystalline and amorphous forms of the pharmaceutical compound are also of importance.
The compound 4-ethyl-N- (4- ( (3-ethynylphenyl) amino) -7-methoxyquinazolin-6-yl) piperazine-1-carboxamide as well as the preparation thereof was described in patent CN101619043A.
pon extensive explorations and researchs, we have found that compound 4-ethyl-N- (4- ( (3-ethynylphenyl) amino) -7-methoxyquinazolin-6-yl) piperazine-1-carboxamide can be prepared into succinate salts, the chemical structure of its semisuccinate and monosuccinate being shown by Formula A. Studies have shown that, compared with its free base, the solubility of compound of Formula A is significantly increased, which is beneficial for improving the pharmacokinetic characteristics and in vivo bioavailability of the compound. We have also found that compound of Formula A can exist in different crystalline forms, and can form solvates with certain solvents. We have made extensive studies on the polymorphic forms of compound of Formula A and have finally prepared and determined the polymorphic forms which meet the requirement of pharmaceutical use. Based on these studies, the present invention provides the compound 4-ethyl-N- (4- ( (3-ethynylphenyl) amino) -7-methoxyquinazolin -6-yl) piperazine-1-carboxamide succinate and the various crystalline forms thereof, solvates and the crystalline forms thereof, which are designated as Form I, Form IV and Form V respectively.
The compound 4-ethyl-N- (4- ( (3-ethynylphenyl) amino) -7-methoxyquinazolin-6-yl) piperazine-1-carboxamide raw material used in the examples were prepared according to CN101619043A.
Example 1 Preparation of Form I of compound of Formula A
The 4-ethyl-N- (4- ( (3-ethynylphenyl) amino) -7-methoxyquinazolin-6-yl) piperazine-1-carboxamide (60g, 0.139mol) was dissolved in 150 times (volume/weight ratio) of tetrahydrofuran (9L) under refluxing. Then the obtained solution was cooled to 50℃, and succinic acid (65.8g, 0.557mol, 4 equivalents) was added in one portion. Then the obtained mixed solution was cooled naturally under stirring. The white precipitate was appeared at about 28℃. After further stirring for 18 hours, the white solid was collected by filtration, and dried at 40℃ under vacuum. A powder sample of 56.7g was obtained (yield 83%) .
1H NMR (400 MHz, cd3od) δ 8.52 (s, 1H) , 8.45 (s, 1H) , 7.93 –7.89 (m, 1H) , 7.77 –7.73 (m, 1H) , 7.35 (t, J = 7.9 Hz, 1H) , 7.24 (dd, J = 5.2, 3.8 Hz, 1H) , 7.19 (s, 1H) , 4.05 (s, 3H) , 3.69 –3.61 (m, 4H) , 3.49 (s, 1H) , 2.71 –2.64 (m, 4H) , 2.60 (q, J = 7.2 Hz, 2H) , 2.53 (s, 2H) , 1.18 (t, J = 7.2 Hz, 3H) .
The obtained powder sample is Form I of compound of Formula A, the X-ray powder diffractogram of which is shown in Figure 1. Peaks (2θ) chosen from the figure has the following values: 6.1, 7.9, 10.2, 11.6, 12.2, 13.6, 15.3, 15.9, 16.6, 17.8, 19.6, 20.4, 21.4, 21.7, 22.3, 23.5, 24.3, and 25.1 degrees, the measured 2θ values each having an error of about ± 0.2 degrees (2θ) , wherein characteristic peaks (2θ) are at 6.1, 7.9, 12.2, 15.3, 15.9, 16.6, and 20.4 degrees. DSC result is given in Figure 2, showing that the melting point range of Form I is about 193.4-197.3℃.
PATENT

PATENT
CN 108863951
PATENT
US 20100009958
PATENT
WO 2010002845
////////////Epitinib , PHASE 1, PHASE 2, Epitinib succinate, HMPL-813, Huposuan yipitini, 1203902-67-3,
CIFORADENANT
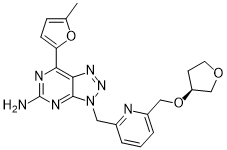

CIFORADENANT
1202402-40-1
Chemical Formula: C20H21N7O3
Molecular Weight: 407.434
CPI-444, CPI 444, CPI444, V81444, V-81444, V 81444,
UNII 8KFO2187CP
Corvus Pharmaceuticals, Inc. PHASE 1
(S)-7-(5-methylfuran-2-yl)-3-((6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methyl)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-5-amine
| 3H-1,2,3-TRIAZOLO(4,5-D)PYRIMIDIN-5-AMINE, 7-(5-METHYL-2-FURANYL)-3-((6-((((3S)-TETRAHYDRO-3-FURANYL)OXY)METHYL)-2-PYRIDINYL)METHYL)- |
(73 S)-15 -methyl-6-oxa-2(7,3)-[1,2,3]triazolo[4,5- d]pyrimidina-4(2,6)-pyridina-1(2)-furana-7(3)- oxolanaheptaphan-25 -amine adenosine receptor antagonist
Ciforadenant, also known as CPI-444 and V81444, is an orally administered antagonist of the adenosine A2A receptor. Upon oral administration, CPI-444 binds to adenosine A2A receptors expressed on the surface of immune cells, including T-lymphocytes, natural killer (NK) cells, macrophages and dendritic cells (DCs). This prevents tumor-released adenosine from interacting with the A2A receptors on these key immune surveillance cells, thereby abrogating adenosine-induced immunosuppression in the tumor microenvironment.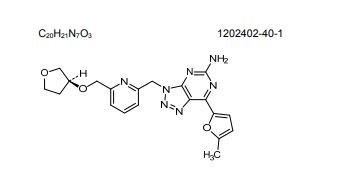
Ciforadenant is an antagonist of adenosine A2A being developed by Corvus , under license from Vernalis , for the oral treatment of advanced solid tumor; the company is also developing the drug in combination with atezolizumab , for non-small-cell lung cancer.
In 2015, Vernalis licensed the exclusive rights of the product for use of all therapeutic application to Corvus.
Synthesis
WO 2009156737

PATENT
WO 2009156737
US 8450328
WO2017112917
WO 2018175473
WO 2018009972
WO 2018049271
WO 2018022992
PATENT
PATENT
WO-2018183965
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018183965&redirectedID=true
EXAMPLES
Reaction Scheme 1
[0314] Referring to Reaction Scheme 1 , the process to manufacture triazolo[4,5]pyramidine derivatives and intermediates thereof in accordance with the present disclosure, such as the compound known as CPI-444, consists of three chemical steps and uses starting materials known as CP-55, CP-56 and CP-60. The intermediate known as CP-57 is formed at step la without isolation (telescoped) and taken to the next step to form the compound known as CP-58 at step lb. Suzuki coupling using CP-60 during step 2 generates crude CPI-444 which undergoes crystallization during step 3 to form CPI-444.
[0315] Previously described processes for making triazolo[4,5]pyramidine derivatives and intermediates thereof utilized a compound known as CP-59:
[0316] Moreover, such previously described process utilize triethylamine which takes a longer time for the layers to separate where excessive rag layer is observed during phase separation. [0317] The present inventors unexpectedly and surpisingly found that the replacement of CP-59 with CP-60 improved ease of handling and improved process efficiency. In addition, the present inventors unexpectedly and surpisingly found that the use of potassium carbonate (K2CO3) during step 2 improves the phase separation and minimizes rag layer formation upon reaction completion. Finally, Step 3 employs the use of thermocycler in order to facilitate the removal of residual solvents such as isopropyl alcohol.
[0318] Accordingly, the processes in accordance with the teachings of the present disclosure are an improvement over, and are more suitable for commercial scale-up, than processes previously described.
[0319] Starting material (C-55) is commercially available through Astatech, Inc., Keystone Business Park, 2525 Pearl Buck Road, Bristol, PA, 19007, USA; or Suven, SDE Serene Chambers, Road No.5, Avenue 7 Banjara Hills, Hyderabad, 500034, India.
[0320] CP-60 is commercially available through ARK Pharma, Inc., 3860 North Ventura Drive, Arlington Heights, IL, 60004, USA; or Boron Technology Institute, Road No. 2, Building No. 10, room No. 259, Haidian District, Beijing, China.
EXAMPLE 1. Preparation of CP-56
Reaction Scheme 1
Boc20, CbzCI
[0321] Preparation of Dimethyl pyridine-2,6-dicarboxylate:
Pyridine-2,6-dicarboxylic acid (900g, leq) is suspended in methanol(5 volume) and added H2SO4. (19g). The mixture is heated to reflux for approximately 4hr. After reaction completion, the mixture is cooled to 5- 10°C to allow the solids to precipitate. The solids are stirred for an additional hour. The solids are collected by filtration. The wet-cake is re-dissolved in DCM (3 volume) and extract in sequence with an aqueous saturated solution of NaHC03 (2 Volume) followed by with a 5% brine solution (2 Volume). The organic layer is concentrated to dryness to obtain dimethyl pyridine-2,6-dicarboxylate; 914.85g, purity 100%, yield 87.%.
[0322] Preparation of pyridine-2,6-diyldimethanol:
Dimethyl pyridine-2,6-dicarboxylate (885g, leq) is dissolved in EtOH (4425g, 5 Volume) at room temperature. The NaBH4 (341 g, 2eq) is added slowly to the reaction while keeping the internal temperature below 30°C using an ice bath. The reaction is heated to 35°C for approximately 2hrs. After reaction completion, the mixture is cooled to room temperature and adjusted with 32% HCl solution to pH value of approximately 2.5. The mixture is stirred for
2hrs to allow the solids to precipitate. The mixture is then adjusted pH value of approximately 9 using 30% NaOH solution while maintaining an internal temperature below 30°C and stirred at room temperature for about 30 min. The solids are removed by filtration. The filtrate is concentrated at 50°C. The concentrated residual is suspended with isopropanol (4160g, 8 vol)
/water (416g, 0.8 vol) and heated to 70°C for about lhr. The solution is then cooled to room
temperature and stirred for 2hr before cooling to 5-10°C for 30min. The un-dissolved solids are
removed by filtration. The filtrate is concentrated at 50°C. The concentrated residue is charged
with dichloromefhane (2700g, 5vol) and heated to 40 °C for 30min. The suspension is cooled to 5-
10°C and stirred for 30mins. The solid is collected by filtration and dried under vacuum at 40°C to obtain pyridine-2,6-diyldimethanol; 540.77g, purity 100%, yield 85.86%.
[0323] Preparation of 2,6-6 s(chloromethyl)pyridine:
2,6-bis(chloromethyl)pyridine (400g, leq) is suspended in DCM (2000g) and then cooled to 10- 15°C. Thionyl chloride (SOCb; 775g, 3eq) is charged with CH2CI2 (775g) and then added drop- wised into the reaction vessel while maintaining the internal temperature below 20 °C. The reaction is then warmed to room temperature and held for approximately 2hrs. After reaction completion, the 15% aqueous solution of a2C03 (9038g) is pre-cooled to 10-15°C before charging the reaction mixture into the carbonate solution while maintaining internal temperature below 20 °C. The mixture is stirred until gas-evolution is no longer observed. The organic layer is extracted with water (2 x 3200g) and then concentrated at 50°C to a crude product. The concentrated crude is purified by recrystallization using heptane (946g). The mixture is cooled to 5-10°C for 30min. The solid is collected by filtration and wet-cake is washed with heptane and dried at 40°C under vacuum to obtain 2,6-6zs(chloromethyl)pyridine; 442.6g, purity 100%, yield 87.0%.
[0324] Preparation of (3r,5r,7r)-l-((6-(chloromethyl)pyridin-2-yl)methyl)-l,3,5,7-tetraazaadamantan-l-ium:
2,6-to(chloromethyl)pyridine (420g, leq) is dissolved in CH2CI2 (8400g), HMTA (336g, leq) is added into the reaction vessel. The reaction is heated to approximately 40 °C for about 3hrs. Additional HMTA (168g, 0.5eq) is added into the reaction mixture and stirred overnight at room
temperature. The product is collected by filtration. The wet-cake is washed with CthCkand dried under vacuumat 50°C to obtain (3r,5r,7r)-l -((6-(chloromethyl)pyridin-2-yl)methyl)- 1 ,3, 5,7-tetraazaadamantan- 1 -ium; 730g, purity 97.01%, yield 96.58%.
[0325] Preparation of (6-(chloromethyl)pyridin-2-yl)methanamine dihydrochloride:
(3r,5r,7r)- 1 -((6-(chloromethyl)pyridin-2-yl)methyl)- 1 ,3 ,5 ,7-tetraazaadamantan- 1 -ium (730g, leq) is suspended in EtOH (4380g) before charging 37% HC1 (159g). The mixture is heated to approximately 60 °C for about lhr. After reaction completion, it is cooled to 25°C. MTBE
(1200g) is charged into the suspension. The suspension is then stirred for about 30 min and cooled to 5-10°C for about lhr. The solids are collected by filtration and washed with MTBE and dried at 50°C under vacuum to obtain (6-(chloromethyl)pyridin-2-yl)methanamine dihydrochloride; 449.56g (after assay correction), purity 98.15%, yield85.23%.
[0326] Preparation of tert-butyl ((6-(chloromethyl)pyridin-2-yl)methyl)carbamate:
(6-(chloromethyl)pyridin-2-yl)methanamine dihydrochloride [422.56g (after assay correction), leq] is dissolved in CH2CI2 (5600g) and pre-cooled to 10-15°C. K2CO3 (1632g) pre-dissolved in water (4000g) is charged into the reaction solution solution. The mixture is stirred for about lOmin and then cooled to 10-15°C. Boc-anhydride (603g) is pre-dissolved in CH2CI2 (1808g) before charging into the reactor. The mixture is warmed to room temperature and held for about an hour. After reaction completion, the organic layer is extracted with water (4000g), The organic layer is concentrated to dryness at 50 °C to obtain tert-butyl ((6-(chloromethyl)pyridin-2-yl)methyl)carbamate; 382.93g [after assay correction); purity 99.01%; yield 81%].
[0327] Preparation of tert-butyl ((6-(iodomethyl)pyridin-2-yl)methyl)carbamate:
tert-butyl ((6-(chloromethyl)pyridin-2-yl)methyl)carbamat [ 382.93g (after assay correction) , leq] is dissolved in THF (1 150) and Nal (720g) is added, the reaction is at room temperature for approximately 4hr. After reaction completion, excess Nal and NaCl are filtered off and the filtrate is concentrated at 40°C. The concentrated residue is re-dissolved in ethyl acetate (2300g) and extracted with water (2900g), the organic layer is washed with 10% aqueous solution of Na2S203 (2600g) followed by 5% brine solution (2900g). The organic layer is concentrated to a residue. The residue is re-dissolved in ethyl acetate (4200g), and then filtered. The filtrate is oncentrated and taken up in ethyl acetate (765g) and stirred at room temperature for about 2hr before slowly adding heptane (380g). The solids are filtered and dried at 50°C under vacuum to
obtain tert-butyl ((6-(iodomethyl)pyridin-2-yl)methyl)carbamate; 440g; purity 100%, Yield 85%.
[0328] Preparation of tert-butyl (S)-((6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methyl)carbamate:
A solution of t-BuOK (113g in THF (1.1 kg) is pre-cooled to 5- 10°C, before charging asolutionof (S)-tetrahydrofuran-3-ol (166g) in THF (220g). The mixture is stirred at room temperature for about lhr. A solution of tert-butyl ((6-(iodomethyl)pyridin-2-yl)methyl)carbamate (440g, leq) in THF (880g) is pre-cooled to 10-15°C before. The tetrahydrofuranyl solution is slowly charged into reaction solution while maintaining an internal temperature below 1 °C. After about 1 hour another solution of pre-cooled solution of t-BuOK (50g) and (S)-tetrahydrofuran-3-ol (66g) in THF (405g) kg) is slowly added into reaction mixture while maintaining internal temperature below 10 °C. The mixture is stirred at about 10 °C for approximately 1 hour. After reaction completion, the mixture is quenched with water (2200g) and extracted with toluene (4400g). The organic layer is washed with 5% brine (2x 2200g). The organic layer is concentrated to dryness at 50°C under vacuum to obtain tert-butyl (S)-((6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methyl)carbamate; 389g, purity 89.63%, yield 105%.
[0329] Preparation of CP-56 free base:
tert-butyl (S)-((6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methyl)carbamate (389g, leq) is dissolved in CH2CI2 (1556g) and pre-cooled to 0-5°C before charging drop-wise methanesulfonic acid ( MSA; 600g) into the reaction solution while maintaining internal temperature below 20°C. The mixture is warmed to room temperature and hold for about lhr. After reaction completion, water (389g) is added and cooled to 5-10°C. 30% NaOH is charged to adjust the reactor pH to approximately 12.5. The mixture is stirred for about 30 min before extracting with CH2CI2 (1556g). The organic layer is collected and extracted with an aqueous saturated solution of brine (584g). The organic layer is concentrated under vacuum. The residue is re-dissolved in toluene (1560g andthenconcentrated. The concentrated residue is re-dissolved in toluene (1560g) and then filtered. The filtrate is concentrated to dryness at 50°C under vacuum to obtain CP-56 free base; 221g (after assay correction), purity 91%, yield 84.23%.
[0330] Preparation of CP-56:
CP-56 free base (22 lg (after assay correction), leq) is dissolved in MeOH (260g) and EtOH (1300g) and then cooled about 15°C. Oxalic acid (47), pre-dissolved in MeOH (1 lOg is charged into reaction mixture. The reaction is at 15-20°C for 3hr. The mixture is cooled to 0-5°C and
stirred for about an Ihr. The solid is collected by filtration and the wet-cake is washed with EtOH (390g). The solid is dried under vacuum at 50°C to obtain CP-56 crude. Crude CP-56 is re-crystallized from isopropanol (865g) and H20 (lOOg). The mixture is heated to about 70°C to obtain a solution. The solution is slowly cooled to 50°C for Ihr. The mixture is cooled to 0-5°C for about another Ihr. The solid is filtered and washed with isopropanol. The wet-cake is dried at 50°C under vacuum to obtain CP-56; 164g, purity 99%, yield 95%.
[0331] Alternatively, CP-56 can be formed using the following process:
Reaction Scheme 2
7 8 9
[0332] Preparation of Dimethyl pyridine-2,6-dicarboxylate (compound 2):
Charge diacid (1; 628g) into reactor containing methanol (2Kg) and heat to reflux. After reaction completion the reaction is cooled to 30 C and stirred. The wet-cake is filtered and washed with methanol (500g). The wet-cake is dried under vacuum at about 55 °C to obtain diester (680 g, purity >99%; yield 85%).
[0333] Preparation of 6-(hydroxymethyl)picolinamide (compound 4):
Charge diester (2; 600 g) into reactor containing methanol (1.8 kg) and tetrahydrofuran (1.2 kg). Charge slowly sodium borohydride ( aBH4; about 130 g) into the reaction solution while maintaining an internal temperature below 30 °C. After reaction completion aqueous hydrochloric acid (about 350 g of 32% HC1) is charged into the reaction solution. The mixture is concentrated and then charged with dichloromethane (1.8 kg). The organic solution is extracted with water (600 g) and then concentrated to obtain the crude product (3). Crude 3 was dissolved in methanol (1.3 kg) and then charge ammonium hydroxide (20%; 1.3 kg). The solution was stirred until reaction completion before concentrating solution. The residue was taken up in water (600g) and heated to about 60 °C before cooling to 0 °C. The wet-cake was filtered, washed with water and dried in vacuum oven to obtain 6-(hydroxymethyl)picolinamide (about 220 g, >99% purity).
[0334] Preparation of 6-(chloromethyl)picolinonitrile (compound 5):
Charge 6-(hydroxymethyl)picolinamide (about 220 g) into a rector containing acetonitrile (450 g). Charge POCb (519 g and agitate at about 70 °C. After reaction completion the solution is
cooled to about 30 °C before slowly charging into a pre-cool (about 10 °C) reactor with water
(305 g). Charge toluene (1.4 kg) to extract the solution mixture. The toluene phase is washed in sequence with 20 % NaOH (600 g), saturated NaHC03 (300 g) and water (300 g). Toluene is concentrated to obtain crude Cl-nitrile, 5. Isopropyl alcohol (400 g) is charged to dissolve the wet-cake at about 45 °C before cooling to about 0 °C. The wet-cake was filtrated and washed with heptane (150 g) and dried in vacuum oven to obtain 6-(chloromethyl)picolinonitrile (180 g; > 99%.
[0335] Preparation of (S)-6-(((tetrahydrofuran-3-yl)oxy)methyl)picolinonitrile (compound 7):
Charge Cl-nitrile (180 g) into a rector containing THF (540 g). Charge Nal (185.7 g) to the reactor and stirred at 50 °C. After reaction completion, the reactor is cooled to 0 °C. In another
reactor, charge t-BuOK (145.6 g) and THF (320 g). Add (S)-tetrahydrofuran-3-ol (31 1.9 g) into the reactor while maintaining internal temperature below 50 °Cto deprotonate the alcohol. Stir
until t-BuOK dissolves. Add THF-OK / THF solution into 6-(iodomethyl)picolinonitrile solution (compound 6) while maintaining internal temperature below 10 °C. Stir at room
temperature until reaction completion. Concentrate the solution to remove THF solvent. Add
ethyl acetate (630 g) and wash by water (420 g). Extract water phase by ethyl acetate (630 g). Combine organic layer and concentrate to obtain oil crude 374 g. The residue was distilled under vaccum (P=3~4 torr, internal temperature 174 °C to 188 °C) to obtain (S)-6-
(((tetrahydrofuran-3-yl)oxy)methyl)picolinonitrile (compound 7) as an oily product (204g, >96% purity; 74% yield).
[0336] Preparation of (S)-(6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methanamine (compound 9):
Charge (S)-6-(((tetrahydrofuran-3-yl)oxy)methyl)picolinonitrile (180 g) into a rector containing MeOH (1620 g). Charge NaOMe (95.3 g) to the reactor and stirred for 30 min at 30 °C until
reaction completion. The methyl (S)-6-(((tetrahydrofuran-3-yl)oxy)methyl)picolinimidate solution (compound 8) was transferred to hydrogenation apparatus containing 50% Ni (60 g). Purge with N2 and then increase the H2 pressure. Under H2 pressure of 5 kg / cm2 and temperature of 30 °C until reaction completion. The reaction is filtered through celite. The filtrate is concentrated. Toluene is charged (1kg) and then concentrated. Then add toluene (1000 g) and filter to remove salt by-products. The filtrate was concentrated to obtain the oil residue of (S)-(6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methanamine (136 g; 85% yield, assay 80%, >91% purity).
[0337] Preparation of CP-56:
Charge (S)-(6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methanamine (170 g) into a rector containing isopropyl alcohol (600 g). Set internal temperature of 75 °C. In another reactor,
charge oxalic acid (41.1 g) and water (60 g) and heat solution. Add oxalic acid solution into
CP-56 free-base solution. Cool to 30 °C for about 4 hours and agitate. The wet-cake was filtered
and washed with isopropyl alcohol (175 g) and dried under vacuum drying with heat to obtain crude CP-56 (136.2 g). Charge CP-56 crude (123 g) into a rector containing methanol (1295 g). Stir until CP-56 was dissolved completely. Filter through celite to remove insoluble salt. The filtrate is concentrated. Charge isopropyl alcohol (500 g) and water (50 g) to dissolve CP-56 using heat. Cool to about 30 °C for about 3 hours and stir. The wet-cake was filtrated and
washed by isopropyl alcohol (165 g) and dried under vacuum drying with heat to obtain CP-56 (1 13.4 g. purity = >99 %, > 99% ee).
EXAMPLE 4. Preparation of CPI-444
CP-58 CP-60
C15H16CIN702 CPI-444
1H-17BO3
W: 361 .79 MW: 208.06 C20H21N O3
MW: 407.43
[0349] It is to be noted that other Pd coupling reagents can also be used such as Pd(PPh3)4 or Pd(PPh3)2Cl2.
[0350] A solution of CP-58 (30.0 g, 1 equiv.), CP-60 (approximately 20.8 g, 1.2 equiv.), in THF (approximately 180 mL), K2C03 (approximately 17.5 g), Pd(dtbpf)Cl2(approximately 337 mg), and water (approximately 100 mL) were stirred and heated to about 60 °C until reaction completion. The reaction was cooled to about 50 °C and the layers were allowed to separate. The aqueous layer was removed and back extracted with THF (approximately 30 mL). The THF layers were combined and water (approximately 450 ml) was added to precipitate out crude CPI-444. The slurry was cooled to about 20 °C and stirred for approximately 60 min and the slurry was filtered. The cake was washed in sequence with water (approximately 120 ml) and 2-propanol (approximately 30 ml). The wet-cake was dried in the vacuum oven to provide an off- white solid (29.74 g, 88% yield) with a purity of 98.5 %. Crude CPI-444 conforms to reference.
-444 can be prepared by the following process:
EDA and DAP are used to remove Palladium during CPI-444 formation.
[0352] The solution of CP-58 (10 g), CP-60 (6.9 g) , Pd(dtbpf)C12 (approx. 0.0015 mol eq) and K2C03 (5.8 g) in THF (6V) and H20 (3V) is heated to approximately 60 °C. The reaction is complete after approximately 30 minutes. The solution is cooled to 50 °C and aqueous layer is separated. The aqueous layer is extracted with THF (9 mL); the THF layer is added to organic solution. The organics are cooled to 40 °C, 1 ,3-diaminopropane (DAP; approximately 50 g) or ethylene diamine (EDA; approximately 45 g) is added and the mixture stirred for 1 hour. H20 (15V) is added to the organic layer over 10 min. The slurry is cooled to 20 °C for 2 hours, and stirred for an additional 1 hour. The slurry is filtered and washed with H20 (2V x 2) and z‘PrOH (IV). CPI-444 wet-cake is dried at 50 °C under full vacuum. (Yield = 90 %; purity > 99.0%).
[0353] Alternatively, CPI-444 can be prepared by the following process:
using cysteine in TNF to remove Palladium during CPI-444 formation
[0354] CP-58 (1 kg), K2C03 (0.58 kg), water (3 kg), CP-60 (0.69 kg), and THF (5.3 kg),
Pd(dtbpf)Cb (3 g). The solution is heated to 60 °C. The reaction is complete after approximately 30 minutes. Charge THF (4.5 kg) and cool to 50 °C. The aqueous layer is separated. The organic layer is charged with cysteine (0.32 kg) and water (5 kg). The mixture is agitated. NH4OH (1.1 kg) is charged to the reaction mixture and agitate for approximately 15 minutes. The layers are allowed to separate and the lower aqueous layer is separated. The organic layer is charged with cysteine (0.32 kg) and water (5 kg). The mixture is agitated. NH4OH (1.1 kg) is charged to the reaction mixture and agitate for approximately 15 minutes. The layers are allowed to separate and the lower aqueous layer is separated. THF is distilled to approximately 7 volumes under atmospheric pressure. The solution is cooled to 50 °C before charging NH4OH (0.5 kg) and agitate for 30 min. Water (14.5 kg) is charged while maintaining the internal temperature >40 °C. The reactor is cooled to 20 °C for 2 hours and hold for an additional 1 hour. CPI-444 is filtered and washed with water followed by isopropanol. CPI-444 wet-cake is dried under vacuum at 50 °C. Purity > 99%, yield 85%.
EXAMPLE 5. Removal of Residual Palladium With Biocap Filter Cartridge
[0355] A mixture of CPI-444 crude (16.00 g), THF (approximately 190 ml), L-cysteine
(approximately 8 g), and H20 (approximately 90 ml) were mixed and heated to a solution at about 60 °C for 1 hour. A solution of 28% NH OH (approximately 20 ml) was added and heated for an additional 15 minutes. The agitation was turned off to allow the layers allowed to settle. The aqueous layer was removed; the THF layer was washed with brine solution (approximately 15 ml). The combined aqueous solutions were back extracted with THF (approximately 15 ml). A 3M Biocap filter (BC0025LR55SP; available from 3M) was pretreated with THF (approximately 150 ml) at about 50 °C. The combined organic layers were recirculated through the Biocap at about 10 ml/min for approximately 3 hours and then filtered forward. The Biocap filter was rinsed with THF (approximately 130 ml) at about 50 °C. The combined filtrates were concentrated. Water
(approximately 80 ml) was added, and distilled to remove residual THF. 2-Propanol (approximately 1 10 ml) was added to the slurry, and the mixture was heated to a solution. The solution was cooled to 20 °C and water (approximately 240 ml) was added. The slurry was performed in series by heating to about 55 °C and held that that temperature for approximately 30 minutes, cooled to 20 °C over 30 minutes, and held at 20 °C for 30 minutes. This heating cycle was repeated two more. The slurry was then held at 20 °C for approximately 12 hours. The slurry was filtered, and the product was washed with water (approximately 300 ml). The wet cake (about 23 g) was dried in the vacuum oven to obtain an off white solid (13.6 g; 85% yield;99.9% purity; Pd = 25 ppm).
[0356] Reprocess of step 4. AFC-825-106
[0357] CPI-444 (16.02 g, AFC-825-48) and THF (approximately 280 ml) were charged to a flask and heated to about 50 °C for about 30 minutes to obtain a solution. A 3M Biocap filter
(BC0025LR55SP) was pretreated with THF (approximately 150 ml) at about 50 °C . The CPI-444 solution was passed through the Biocap at aboutl O ml/min. The Biocap filter was rinsed with THF (approximately 130 ml) at about 50 °C. The combined filtrates were transferred to a reactor and concentrated. Water (approximately 80 ml) was added, and distilled to remove residual THF solvent. 2-Propanol (approximately 1 10 ml) was added to the slurry and heated to about 65 °C to obtain a solution. The solution was cooled to about 20 °C before adding water (approximately 240 ml). The slurry was heated to 55 °C over 30 minutes, held at 55 °C for 30 minutes, cooled to 20 °C over 30 minutes, and held at 20 °C for 30 minutes. This heating cycle was two more times. The slurry was then held at 20 °C for 12 hours. The slurry was filtered, and the product was washed with water (approximately 300 ml). The wet cake (26.6 g) was dried in the vacuum oven overnight to obtain 15 as a white solid (95% yield; 99% purity; Pd = 5 ppm).
EXAMPLE 6. Removal of Residual Palladium With Darco KB-G
Crude CPI-444
CPI-444 Drug Substance
[0358] Crude CPI-444 (475 g, 1.17 mol, 1.00 eq), 2-MeTHF (1 1.9 L, 25.0 vol) and WFI water (2.6 L, 5.5 vol) were charged to a 19 L jacketed reactor. The mixture was mechanically agitated under a nitrogen blanket. Nitrogen was bubbled through the solution for 20 minutes. L-Cysteine (242 g, 1.99 mol, 1.71 eq) was then charged. The solution in the reactor was heated to 55±5 °C. Upon reaching 50 °C, the reaction mixture was stirred for 1 hour. 28-30% NH4OH (594 mL, 1.25 vol) was charged via addition funnel, and then the reaction mixture was stirred for 15 min. Agitation was stopped and the reaction was allowed to separate for 1 hour. The aqueous layer was removed. The organic layer was allowed to cool to ambient. The organic layer was filtered and the frit was washed with 2-MeTHF (618 mL, 1.3 vol). The organics were concentrated off by rotary evaporation. WFI water (2.42 L, 5.1 vol) and IPA (2.38 L, 5.0 vol) were used to charge the concentrated slurry to a clean 19 L jacketed reactor under N2. The mixture was heated to 65±5 °C, and then was stirred for 1 hour to obtain solution. Darco KB-G activated carbon (71.3 g, 15 wt%) was charged. The reactor was heated to 75±5 °C and stirred for 15 hours. A I L pocket filter was prepared with filter cloth and a heating jacket and heated to 70±5 °C. Reactor contents were filtered through the pocket filter using N2 pressure. The pocket filter was rinsed with a mixture of IPA/WFI water (1 : 1, 950 mL, 2 vol) followed by a mixture of IPA/WFI water (1 : 1, 1.90 L, 4 vol) and IPA/WFI water ( 1 : 1 , 1.90 L, 4 vol). Inside a 22 L three neck round bottom flask the filtrates were mechanically agitated under a N2 blanket. WFI water (7.13 L, 15 vol) was slowly added via addition funnel over 1 h at ambient temperature, and aged for 1 h. The slurry was heated to 55±5 °C and maintained the temperature for 30 min. This heating and subsequent cooling were repeated twice more. After reaching ambient
temperature the final time, the mixture was stirred for at least 2 hours. The reaction mixture was filtered and the reactor rinsed with WFI water (2.38 L, 5.0 vol, 3x). The cake was dried under N2 for 30 minutes and then transferred to a glass dish. The material was dried under full vacuum at 55±5 °C. The desired product was obtained 368.1 g (77%) as light yellow solids. This material was 99.6% pure by HPLC and had a Pd content of 3.6 ppm.
EXAMPLE 7. Removal of Residual Palladium With Polymer-Bound Thiol (SiST)
[0359] Crude CPI-444 (24.48 g, pd = 1267 ppm) and THF (244.8 mL, 10 vol) were charged to a 500 mL 4-necked flask fitted with mechanical agitation, a condenser with nitrogen balloon and a thermometer. The slurry was heated to 60 °C for 20 minutes and then slowly cooled to 45 °C. SiST (36.72 g) was added to the solution and the mixture was stirred at 42 °C for 14 h. The mixture was filtered and washed by THF (24 mL, 1 vol, twice; Pd= 13.12 ppm). H20 (120 mL, 5 vol) and IPA (120 mL, 5vol) were charged to the flask. The slurry was heated to 70 °C and maintained for 1 h (the slurry became solution). The solution was slowly cooled to room temperature and the slurry was added H20 (360 mL, 15 vol) and heated to 55 °C for 1 h. The slurry was cooled to room temperature and then heated to 55 °C for 1 h. The slurry was cooled to rt. and stirred at rt. for 2 h. The slurry was filtered and washed by H20 (100 mL, 4 vol, three times). The wet cake (28.36 g) was dried by 10 mmHg and 50 °C for overnight (14h) and the weight of CPI-444 was 19.31 g (79% recovery).
EXAMPLE 8. Removal of Residual Palladium By Recrystallization
[0360] CUNO Filter Cartridge 55 S
[0361] CPI-444 (5.0 g, Pd 14.06 ppm) and THF (50 mL, 10 vol) were charged to a 100 mL 3-necked flask fitted with stirring bar, a condenser with nitrogen balloon and a thermometer. The slurry was heated to 60 °C for 20 minutes and added CUNO 55S filter (0.75 g, 15w%). The mixture was stirred at 60 °C for 1 h. The mixture was filtered and washed by THF (5 mL, 1 vol, twice). The filtrate was concentrated. The solid, H20 (25 mL, 5 vol) and IPA (25 mL, 5vol) were charged to 250 mL 3 -necked flask fitted with stirring bar, a condenser with nitrogen balloon and a thermometer. The slurry was heated to 70 °C and maintained for 1 h (the slurry became solution). The solution was slowly cooled to rt.(40 minutes) The slurry was added H20 (75 mL, 15 vol) and then heated to 55 °C for 1 h. The slurry was cooled to rt. (30 minutes) and stirred at rt. for 2 h. The slurry was filtered and washed by H20 (20 mL, 4 vol, three times). The cake (6.355 g) was dried by 10 mmHg and 50 °C
for overnight (16 h) and the weight of CPI-444 was 4.281 g (85% recovery). Pd content(ppm) = 2.02 ppm.
[0362] Polymer-bound Thiol: SiST
[0363] CPI-444(5 g; Pd 14.06ppm) was dissolved in THF (50 mL) at 60 °C. The solution was cooled to 55 °C and SiST (7.5 g) was added to the solution. The solution was stirred at 50-55 °C for 16 h. The solution was filtered through celite and a 0.2 micron filter. The filtrate was tested for Pd content. Result: 2.43 ppm.
Catalyst
Molecular Weight: 291.6990
Molecular Weight: 337.3430
[0364] 1. A solution of S.M., CP-60, Pd(PPh3)2Cl2 and K2C03 in THF – H20 (7.9 mL, 1 : 1) was put in oil-bath at 70-75 °C.
[0365] 2. After 2 h, 0.047 g CP-60 was added to the reaction at 70-75 °C.
[0366] 3. After 1 hr, the reaction was cooled to rt. and 10 mL H20 was added to the reaction.
[0367] 4. The reaction was filtered to provide wet cake (0.812 g).
[0368] 5. The solid wet cake was dried at 45 °C and 20 mmHg for 2h to provide weight 0.499 g. (86%).
[0369] 6. The solid wet cake was stirred in 2 mL DMF for 30 mins (slurry) and then filtered. The solid was dried by 45 °C and 10 mmHg for 12h to provide weight 0.40 g; 69% yield; 98.1% purity.
//////////CIFORADENANT, CPI-444, CPI 444, CPI444, V81444, V-81444, V 81444, UNII 8KFO2187CP, Corvus Pharmaceuticals, Inc., PHASE 1,
NC1=NC2=C(N=NN2CC3=NC(CO[C@H]4CCOC4)=CC=C3)C(C5=CC=C(O5)C)=N1
THELIATINIB
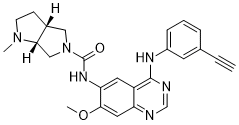
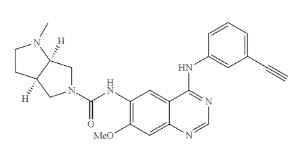
THELIATINIB
CAS: 1353644-70-8
Chemical Formula: C25H26N6O2
Molecular Weight: 442.523
HMPL-309; HMPL 309; HMPL309; Theliatinib.
- Originator Hutchison MediPharma
- Class Antineoplastics; Small molecules
- Mechanism of Action Epidermal growth factor receptor antagonists
Highest Development Phases
- Phase I Oesophageal cancer; Solid tumours
Most Recent Events
- 29 Sep 2017 Efficacy and adverse events data from a phase I trial in Oesophageal cancer released by Hutchison Pharma
- 13 Mar 2017 Phase-I clinical trials in Oesophageal cancer (First-line therapy) in China (PO) before March 2017 (Hutchison MediPharma pipeline, July 2017)
- 02 Aug 2016 Hutchison MediPharma plans a phase Ib proof-of-concept trial for Oesophageal cancer, and Head and Neck cancer in China
Theliatinib, also known as HMPL-309, is a novel small molecule, epidermal growth factor receptor tyrosine kinase inhibitor with potential antineoplastic and anti-angiogenesis activities. In vitro studies suggest that Theliatinib is a potent EGFR kinase inhibitor with good kinase selectivity and in vivo data demonstrated broad spectrum anti-tumor activity via oral dosing in multiple xerographs such as A-431, Bcap-37 and Fadu.
PRODUCT PATENT
- By Zhang, Weihan; Su, Wei-Guo; Yang, Haibin; Cui, Yumin; Ren, Yongxin; Yan, Xiaoqiang
WO2012000356 , covering quinazoline compounds as EGFR inhibitors
https://encrypted.google.com/patents/WO2012000356A1?cl=pt-PT&hl=en&output=html_text
Example 3:
(3aR,6aR)-N-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yl)-l-methyl-hexahydropyrrolo [3,4-b]pyrrole-5(lH)-carboxamide


[060] To a solution of Compound 3-a (40 g, 0.138 mol, prepared according to procedures disclosed in WO2010002845), pyridine (40 mL, 0.495 mol) and DMF (anhydrous, 22 mL) in anhydrous THF (500 mL), was added phenyl carbonochloridate 3-b (22 mL, 0.175 mol) dropwise at -10°C. The mixture was stirred at room temperature for 12 hours. The precipitates were filtered and then suspended in saturated NaHC03 solution (500 mL). The solid was filtered, washed with H20 and EtOAc, and dried in vacuum to give compound 3-c (46 g).
A mixture of compound 3-c (1 g, 2.44 mmol) and compound 3-d (369 mg, 2.92 mmol) in dioxane (30mL) was stirred at 70°C for 5 hours, and then cooled to the ambient temperature. The precipitates were filtered, washed with EtOAc, and dried in vacuum to give compound 3 (0.8 g). MS (m/e): 443.4 (M+l)+.
PATENT
https://patents.google.com/patent/WO2010002845A2/en
PATENT
US 9168253
https://patents.google.com/patent/US9168253
Example 3 (3aR,6aR)—N-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yl)-1-methyl-hexahydropyrrolo[3,4-b]pyrrole-5(1H)-carboxamide

To a solution of Compound 3-a (40 g, 0.138 mol, prepared according to procedures disclosed in WO2010002845), pyridine (40 mL, 0.495 mol) and DMF (anhydrous, 22 mL) in anhydrous THF (500 mL), was added phenyl carbonochloridate 3-b (22 mL, 0.175 mol) dropwise at −10° C. The mixture was stirred at room temperature for 12 hours. The precipitates were filtered and then suspended in saturated NaHCO3solution (500 mL). The solid was filtered, washed with H2O and EtOAc, and dried in vacuum to give compound 3-c (46 g). A mixture of compound 3-c (1 g, 2.44 mmol) and compound 3-d (369 mg, 2.92 mmol) in dioxane (30 mL) was stirred at 70° C. for 5 hours, and then cooled to the ambient temperature. The precipitates were filtered, washed with EtOAc, and dried in vacuum to give compound 3 (0.8 g). MS (m/e): 443.4 (M+1)+.
PATENT
THELIATINIB BY HUTCHISON
The present invention belongs to the field of pharmacy and provides a crystal form of a compound (3aR,6aR)-N-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yl)-1-methyl-hexahydropyrrolo[3,4-b]pyrrole-5(1H)-carboxamide, a pharmaceutical composition thereof, and a preparation method therefor and the use thereof.
(FR)La présente invention concerne le domaine de la pharmacie et fournit une forme cristalline d’un composé (3aR,6aR)-N-(4-(3-éthynylphénylamino)-7-méthoxyquinazolin-6-yl)-1-méthyl-hexahydropyrrolo[3,4-b]pyrrole-5(1H)-carboxamide, une composition pharmaceutique de celui-ci, et son procédé de préparation et son utilisation.
Novel crystalline forms of the compound presumed to be theliatinib , processes for their preparation and compositions comprising them are claimed. Also claimed is their use for treating lung cancer, colon cancer, breast cancer, ovary cancer, prostate cancer, stomach cancer, kidney cancer, liver cancer, brain cancer, esophageal cancer, bone cancer and leukemia.
Hutchison Medipharma is developing theliatinib, a small molecule EGFR tyrosine kinase and AKT cell proliferation pathway inhibitor, for treating cancer, including brain tumor, esophageal tumor and NSCLC; in September 2017, positive preliminary data were presented. Hutchison is also developing epitinib succinate , for treating cancer including glioblastoma.
Binding of epidermal growth factor (EGF) to epidermal growth factor receptor (EGFR) activates tyrosine kinase activity and triggers a response that leads to cell proliferation. Overexpression and/or overactivation of EGFR can lead to uncontrolled cell division, and uncontrolled cell division can be a cause of cancer. Therefore, compounds that inhibit the over-expression and/or over-activation of EGFR are candidates for treating tumors.
Relevant compounds of the present invention (3aR, 6aR)-N-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yl)-1-methyl-hexahydropyrrolo [3, 4-b]pyrrole-5(1H)-carboxamide, whose chemical structure is shown in Formula A, has the effect of effectively inhibiting overexpression and/or overactivation of EGFR. Therefore, it can be used for the treatment of diseases associated with overexpression and/or overactivation of EGFR, such as the treatment of cancer.
Before discovering the crystal form of a compound, it is difficult to predict (1) whether a particular compound exists in crystalline form; (2) how an unknown crystal form is made; (3) what the properties of the crystal form would be, such as stability , bioavailability and so on.
Since the properties of the solid depend on the structure and the nature of the compound itself, different solid forms of the compound often exhibit different physical and chemical properties. Differences in chemical properties can be measured, analyzed, and compared using a variety of analytical techniques that ultimately can be used to distinguish these different solid forms. Differences in physical properties, such as solubility and bioavailability, are also important in describing the solid form of the drug compound. Likewise, in the development of pharmaceutical compounds, such as compounds of Formula A, the new crystalline and amorphous forms of the pharmaceutical compounds are also important.
Patent CN102906086A discloses compound (3aR,6aR)-N-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yl)-1-methyl-hexahydropyrrolo[3 4-b]pyrrole-5(1H)-carboxamide and its preparation method.
Experimental part
The starting material of the compound of formula A used in the examples was prepared according to CN102906086A
PATENT
Example 3: (3aR, 6aR) -N- (4- (3- ethynyl-phenylamino) -7-methoxy-quinazolin-6-yl) -1-methyl-hexahydro-pyrrolo [3,4-b] pyrrol -5 (IH) – carboxamide
[0102]

[0103] at -10 ° C, to (40g, 0. 138mol, was prepared in accordance with the operation disclosed in W02010002845) Compound 3-a, pyridine (40mL, O. 495mol) and DMF (anhydrous, 22mL) in dry solution (500 mL) in THF dropwise phenyl chloroformate 3-b (22mL, O. 175mol). The mixture was stirred at room temperature for 12h. The precipitate was filtered off, and then it was suspended in saturated NaHCO3 solution (500mL). The solid was filtered off, washed with H2O and EtOAc, and dried in vacuo to give compound 3_c (46g). Compound 3-c (lg, 2. 44mmol) and the compound 3_d (369mg, 2. 92mmol) in a mixture of two anger dioxane (30mL) was stirred at 70 ° C 5 h, then cooled to ambient temperature. The precipitate was filtered off, washed with EtOAc, and dried in vacuo to give compound 3 (O. 8g). MS (m / e): 443. 4 (M + 1) +.
![]()
Theliatinib (HMPL-309)
Theliatinib (HMPL-309) is a novel small molecule, epidermal growth factor receptor tyrosine kinase inhibitor with potential antineoplastic and anti-angiogenesis activities. Theliatinib is being developed as an oral formulation for the treatment of solid tumors like non-small cell lung cancer.
Theliatinib pre-clinical studies were conducted in China. In vitro studies suggest that Theliatinib is a potent EGFR kinase inhibitor with good kinase selectivity and in vivo data demonstrated broad spectrum anti-tumor activity via oral dosing in multiple xerographs such as A-431, Bcap-37 and Fadu. Non-clinical safety studies have indicated that Theliatinib is generally well tolerated in animals.
In November 2012, HMP initiated the first-in-human clinical trials of theliatinib.
Patent Citations (4)
Publication number Priority date Publication date AssigneeTitle
CN101094840A *2004-12-292007-12-26韩美药品株式会社Quinazoline derivatives for inhibiting cancer cell growth and method for the preparation thereof
CN101619043A *2008-06-302010-01-06和记黄埔医药(上海)有限公司Quinazoline derivant and medical application thereof
CN106117182A *2016-06-202016-11-16中国药科大学Quinazoline-N-phenethyl tetrahydroisoquinoline compound and preparation method and application thereof
REFERENCES
1: Ren Y, Zheng J, Fan S, Wang L, Cheng M, Shi D, Zhang W, Tang R, Yu Y, Jiao L,
Ni J, Yang H, Cai H, Yin F, Chen Y, Zhou F, Zhang W, Qing W, Su W. Anti-tumor
efficacy of theliatinib in esophageal cancer patient-derived xenografts models
with epidermal growth factor receptor (EGFR) overexpression and gene
amplification. Oncotarget. 2017 Apr 19. doi: 10.18632/oncotarget.17243. [Epub
ahead of print] PubMed PMID: 28472779.
//////THELIATINIB, HMPL-309, HMPL 309, HMPL309, Phase I, Oesophageal cancer, Solid tumours
O=C(N1C[C@]2([H])N(C)CC[C@]2([H])C1)NC3=CC4=C(NC5=CC=CC(C#C)=C5)N=CN=C4C=C3OC
YINLITINIB



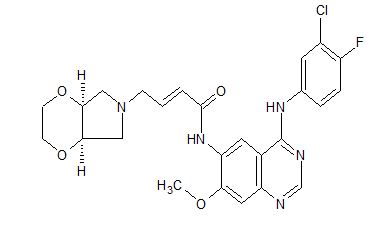
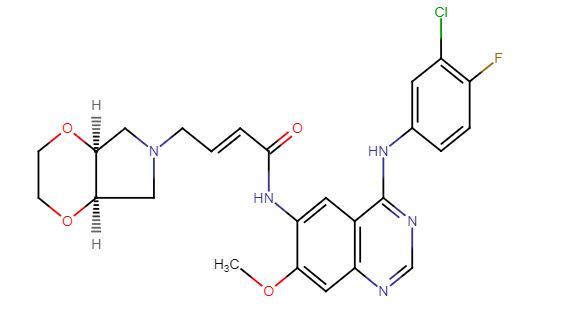
YINLITINIB
error EMAIL ME amcrasto@gmail.com
(E)-4-[(4aR,7aS)-2,3,4a,5,7,7a-hexahydro-[1,4]dioxino[2,3-c]pyrrol-6-yl]-N-[4-(3-chloro-4-fluoroanilino)-7-methoxyquinazolin-6-yl]but-2-enamide
(E)-N-(4-((3-Chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl)-4-((4aR,7aS)-tetrahydro-2H-[1,4]dioxin[2,3-c]pyrrol-6(3H)-yl)but-2-enamide
CAS 1637253-79-2
2-Butenamide, N-[4-[(3-chloro-4-fluorophenyl)amino]-7-methoxy-6-quinazolinyl]-4-[(4aR,7aS)-hexahydro-6H-1,4-dioxino[2,3-c]pyrrol-6-yl]-, (2E)-rel–
- C25 H25 Cl F N5 O4, 513.95
DNT-04110 ; yinlitinib maleate , Guangdong Hec Pharmaceutical
Use for treating proliferative diseases, atherosclerosis and pulmonary fibrosis
Phase I CHINA
NOTE AND USE YOUR JUDGMENT ON DRUG SUBSTANCE, EMAIL ME amcrasto@gmail.com
| Molecular Formula: | C25H25ClFN5O4 |
|---|---|
| Molecular Weight: | 516.973 g/mol |
Yinlitinib methoxy-d3
CAS 1637254-71-7
C25 H22 Cl D3 F N5 O4
2-Butenamide, N-[4-[(3-chloro-4-fluorophenyl)amino]-7-(methoxy-d3)-6-quinazolinyl]-4-[(4aR,7aS)-hexahydro-6H-1,4-dioxino[2,3-c]pyrrol-6-yl]-, (2E)-rel–
CN 104119350
YINLITINIB MALEATE methoxy-d3
CAS ?
EMAIL ME amcrasto@gmail.com
MAY BE DRUG COMD
| Patent ID | Patent Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9556191 | AMINOQUINAZOLINE DERIVATIVES AND THEIR SALTS AND METHODS OF USE THEREOF |
2014-04-28
|
2016-02-11
|
In March 2015, an IND was filed in China ; in February 2016, approval to conduct a clinical trial was obtained
Guangdong Hec Pharmaceutical is investigating an oral capsule formulation of yinlitinib maleate (DNT-04110), an irreversible pan-ErbB inhibitor, for the potential treatment of solid tumors . In March 2015, an IND was filed in China ; in February 2016, approval to conduct a clinical trial was obtained . In December 2016, a phase I trial was planned in China
Protein kinases (PKs) represent a large family of proteins, which play an important role in the regulation of a wide variety of cellular processes and maintaining control over cellular functions. There are two classes of protein kinases (PKs): the protein tyrosine kinases (PTKs) and the serine-threonine kinases (STKs). The protein tyrosine kinase is an enzyme that catalytically transfers the phosphate group from ATP to the tyrosine residue located at the protein substrate, and has a play in the normal cell growth. Many growth factor receptor proteins operate via the tyrosine kinase, and influence the conduction of signal passage and further regulate the cell growth by this process. However, in some circumstances, these receptors become abnormal due to either mutation or overexpression, which cause the uncontrolled cell multiplication, cause the tumor growth, and finally initiate the well-known disease, i.e., cancer. The growth factor receptor protein tyrosine kinase inhibitor, via the inhibition of the above phosphorylation process, may treat cancers and other diseases characterized by the uncontrolled or abnormal cell growth.
Epidermal growth factor receptor (EGFR), a kind of receptor tyrosine kinases, is a multifunction glycoprotein that is widely distributed on the cell membranes of the tissues of the human body, and is an oncogene analog of avian erythroblastic leukemia viral (v-erb-b). Human EGFR/HER1/ErbB-1 and HER2 (human epidermal growth factor receptor-2)/ErbB-2/Teu/p185, HER3/ErbB-3, HER4/ErbB-4 and the like are grouped into the HER/ErbB family, and belong to protein tyrosine kinases (PTKs). They are single polypeptide chains, and each is encoded respectively by genes located on different chromosomes. EGFR and the like are expressed in the epithelia-derived tumors such as squamous cell carcinoma of head and neck, mammary cancer, rectal cancer, ovarian cancer, prostate carcinoma, non-small cell lung cancer, and the like, which are associated with cell proliferation, metastasis, and the like. Pan-HER tyrosine kinase inhibitor, via the competitive binding to the kinase catalytic sites in the intracellular region against ATP, blocks the autophosphorylation of intramolecular tyrosine, blocks the tyrosine kinase activation, inhibits HER-2 family activation, and therefore inhibits cell cycle progression, accelerates cell apoptosis, and exerts the therapeutic action.
EGFR, after binding to the ligand, forms a dimer with a subgroup of HER family, and then combines with ATP to activate the tyrosine kinase activity of the EGFR itself. Therefore, the autophosphorylation occurs in several tyrosine sites of the intracellular kinase region. Pan-HER tyrosine kinase inhibitor, via simultaneity acting on EGFR and HER2/4, inhibits the activation of HER family, and plays a good role in the tumor growth inhibition.
It is indicated in the study that Pan-HER tyrosine kinase irreversible inhibitor has an inhibition effect on HER2/4, besides it effectively inhibits EGFR. The pharmaceutical drugs of this kind, having an irreversible inhibition to both of HER/ErbB families, not only increase the drug activity, but also reduce the drug resistance, and have a substantial inhibition effect on H1975 cell lines which are resistant to erlotinib.
The pharmaceutical drugs that are now commercially available include selective EGFR tyrosine kinase inhibitor gefitinb (IRESSA®, ZD1839), erlotinib (TARCEVA®, OSI-774), double EGFR/HER2 inhibitor Lapatinib (TYKERB®, GW572016), and the like. These three drugs are all reversible EGF receptor tyrosine phosphorylation kinase inhibitor. It has been found in the study that they have good therapeutic response to some tumors initially. However, several months after the treatment, the disease progression appears again and therefore a natural or secondary drug resistance forms. For example, about half of the patients administered with gefitinib or erlotinib develop resistance to gefitinib or erlotinib, which can not lead to the desired therapeutic effect. And it has been indicated by study that the development of drug resistance to selective EGFR tyrosine kinase inhibitor relates to mutations in EGFR.
The mutations of EGFR gene mostly located in the tyrosing kinase coding domain (TK, exons 18-21) are mainly deletion mutation in exon 19 and point mutation in exon 21, both of which are drug-sensitive, and few are point mutation in exon 18 and insertion mutation in exon 20. T790M mutation recognized as one of the mechanism of drug resistance is a point mutation in exon 20 of EGFR. The presence of a second-site EGFR mutation leads to the substitution of methionine for threonine at position 790 (T790M) and changes in the structure of EGFR, which hinder the binding of EGFR inhibitors to EGFR or greatly increase the affinity between EGFR and ATP, so that ATP affinity back to the level of wild-type EGFR, thus resulting in drug resistance. Further studies shows that the pre-treatment tumor samples with mutations of EGFR contain T790M mutation, which indicates that T790M mutation is not just associated with drug resistance and it may have the carcinogenic potential itself.
Irreversible inhibitor can bind to EGFR tyrosine kinase by covalent bond. Thus, the drugs can act on the entire link of epidermal growth factor signal transduction pathway, and improve efficiency of drug blocking. Many clinical studies show that some irreversible inhibitors in current development can against T790M mutation, and overcome the drug resistance caused by T790M. Meanwhile, listed drug Afatinib (BIBW 2992) and some irreversible inhibitors in clinical development (e.g., Dacomitinib, PF00299804, etc.), can inhibit multiple members of EGFR receptor family, especially to the role of EGFR and HER-2, possibly by blocking collaborative signal pathway activated by homodimer and heterodimer to enhance inhibitory effect (Oncologist, 2009, 14 (11): 1116-1130).
Upon developing the drug having an excellent antineoplastic effect, being able to reduce the drug resistance and having a good tolerance, the present inventors discover a quinazoline derivatives as tyrosine kinase inhibitors having a Pan-HER irreversible inhibition function.
PATENT
https://patents.google.com/patent/US9556191
EXAMPLES Example 1 (E)-N-(4-((3-Chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl)-4-((4aR,7aS)-tetrahydro-2H-[1,4]dioxin[2,3-c]pyrrol-6(3H)-yl)but-2-enamide
Step 1) N-(3-chloro-4-fluorophenyl)-7-methoxy-6-nitroquinazolin-4-amine
A solution of N-(3-chloro-4-fluorophenyl)-7-fluoro-6-nitroquinazolin-4-amine (10.00 g, 29.8 mmol) and sodium methanolate (2.80 g, 51.8 mmol) in methanol (150 mL) was heated to 70° C. and stirred for 4.0 hours. The reaction mixture was then cooled to 25° C. The resulting mixture was poured into ice water (500 mL), and a yellow solid precipitated out. The mixture was filtered and the filter cake was dried under vacuum to give the title compound as a yellow solid (9.00 g, 86.9%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z: 349.1 [M+1]+; and 1H NMR (400 MHz, DMSO-d6) δ: 11.60 (s, 1H), 9.55 (s, 1H), 8.08 (dd, J1=6.6 Hz, J2=2.4 Hz, 1H), 7.90 (s, 1H), 7.76-7.71 (m, 1H), 7.58 (s, 1H), 7.55 (t, J=9.4 Hz, 1H), 4.10 (s, 3H).
Step 2) N4-(3-chloro-4-fluorophenyl)-7-methoxyquinazoline-4,6-diamine
To a solution of N-(3-chloro-4-fluorophenyl)-7-methoxy-6-nitroquinazolin-4-amine (9.00 g, 25.9 mmol) in ethanol (100 mL) were added iron powder (14.50 g, 259.0 mmol) and concentrated hydrochloric acid (3.0 mL) at 25° C. The reaction mixture was heated to 90° C. and stirred for 3.0 hours. Then heating was stopped, and the resulting mixture was adjusted to pH 11 with aqueous sodium hydroxide solution (1 M) while the mixture was still at a temperature of about 60±10° C. The pH-adjusted resulting mixture was then immediately filtered hot to remove iron mud. The filtrate was concentrated in vacuo. The residue was triturated with ethanol (50 mL) and filtered. The filter cake was dried under vacuum to give the title compound as a yellow solid (6.00 g, 73.0%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z: 319.1 [M+1]+.
Step 3) (E)-4-bromobut-2-enoyl chloride
To a solution of 4-bromocrotonic acid (2.47 g, 15.0 mmol) and DMF (0.05 mL) in DCM (60 mL) was added oxalyl chloride (4.19 g, 33.0 mmol) dropwise at 0° C. The reaction mixture was stirred at 0° C. for 3.0 hours, and then concentrated in vacuo. The residue was stored in a refrigerator for the next step.
Step 4) (E)-4-bromo-N-(4-((3-chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl)but-2-enamide
To a solution of N4-(3-chloro-4-fluorophenyl)-7-methoxyquinazoline-4,6-diamine (4.00 g, 12.6 mmol) and TEA (6.0 mL, 37.8 mmol) in anhydrous tetrahydrofuran (80 mL) was added (E)-4-bromobut-2-enoyl chloride (2.74 g, 15.1 mmol) slowly at 0° C. The reaction mixture was then heated to 25° C. and stirred for 2.0 hours. The resulting mixture was poured into water (100 mL) and extracted with DCM (50 mL×3). The combined organic phases were dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was triturated with DCM (30 mL) and filtered. The filter cake was dried under vacuum to give the title compound as a brownish yellow solid (2.00 g, 34.5%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z: 465.1 [M+1]+.
Step 5) (E)-N-(4-((3-chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl)-4-((4aR,7aS)-tetrahydro-2H-[1,4]dioxin[2,3-c]pyrrol-6(3H)-yl)but-2-enamide
To a solution of (E)-4-bromo-N-(4-((3-chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl)but-2-enamide (0.50 g, 1.1 mmol) and diisopropylethylamine (0.6 mL, 3.2 mmol) in N,N-dimethylacetamide (10 mL) was added (4aR,7aS)-hexahydro-2H-[1,4]dioxino[2,3-c]pyrrole (0.42 g, 3.2 mmol) at 25° C., and the reaction mixture was then stirred at 25° C. for 5.0 hours. The resulting mixture was poured into water (70 mL) and extracted with DCM (40 mL×3). The combined organic phases were dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH (v/v)=20/1) to give the title compound as a brownish yellow solid (0.30 g, 54.5%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z: 514.1 [M+1]+; and 1H NMR (400 MHz, DMSO-d6) δ: 10.60 (s, 1H), 9.35 (s, 1H), 8.90 (s, 1H), 8.08 (dd, J1=6.6 Hz, J2=2.4 Hz, 1H), 7.76-7.70 (m, 1H), 7.58 (s, 1H), 7.55 (t, J=8.4 Hz, 1H), 6.75-6.65 (m, 1H), 6.63 (d, J=16.2 Hz, 1H), 4.10 (s, 3H), 3.78 (t, J=6.2 Hz, 4H), 3.26 (t, J=4.4 Hz, 2H), 3.20 (dd, J1=7.8 Hz, J2=2.6 Hz, 2H), 2.20 (d, J=4.6 Hz, 4H).
PATENT
WO2017067447
DIFFERENT COMPD
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017067447
claiming novel crystalline polymorphic forms of a similar EGFR, useful for treating cancer. One of these two compounds is probably yinlitinib maleate , an irreversible pan-ErbB inhibitor, being developed by Guangdong Hec Pharmaceutical , another subsidiary of HEC Pharm , for treating solid tumors; in April 2017, yinlitinib maleate was reported to be in preclinical development
Chinese patent CN 103102344 A (publication number) have disclosed the structure of 4- [ (3-chloro-4-fluorophenyl) amino] -7-methoxy-6- [3- [ (1R, 6S) -2, 5-dioxa-8-azabicyclo [4.3.0] nonan-8-yl] propoxy] quinazoline in example 6 of specification, page 57, and the structure is shown as Formula (II) . The compound of Formula (II) has a high inhibition activity against EGFR, and can be used for treating proliferative disorders.
PATENT
https://patents.google.com/patent/WO2014177038A1/en
InventorYingjun ZhangBing LiuJinlei LiuJiancun ZhangChangchun Zheng
Original AssigneeSunshine Lake Pharma Co., Ltd.
PATENT
Inventor张英俊刘兵刘金雷张健存郑常春 Original Assignee广东东阳光药业有限公司
https://patents.google.com/patent/CN104119350B/en




Example 1
[0442] (E) -N- (4- ((3- chloro-4-fluorophenyl) amino) -7-methoxy-quinazolin-6-yl) -4- ((4aR, 7aS) – tetrahydro _2H_ [1,4] dioxin burning and [2,3_c] R ratio slightly -6 (3H) – yl) butyric acid amide dilute _2_
[0443]
[0444] Synthesis Step Shu: N- (3- chloro-4-fluorophenyl) -7-methoxy-6-nitro quinazolin-4-amine
[0445] The N- (3- chloro-4-fluorophenyl) -7-fluoro-6-nitro-quinazolin-4-amine (10 • 0g, 29 • 8mmol) and sodium methoxide (2.80g, 51.8 mmol) was dissolved in methanol (150 mL), the reaction was warmed to 70 ° C 4. Oh. Was cooled to 25 ° C, the reaction mixture was poured into ice-water (500 mL), the precipitated yellow solid was filtered, the filter cake was dried in vacuo to give a yellow solid 9.00g, yield 86.9%.
[0446] MS (. ESI, pos ion) m / z: 349.1 [M + l] +;
[0447] bandit R (400MHz, DMS〇-d6) S: 11 • 60 (s, 1H), 9 • 55 (s, 1H), 8 • 08 (dd, Ji = 6 • 6Hz, J2 = 2.4Hz, lH), 7.90 (s, lH), 7.76-7.71 (m, lH), 7.58 (s, lH), 7.55 (t, J = 9.4Hz, 1H), 4.10 (s, 3H) square
[0448] Synthesis Step 2: n4- (3- chloro-4-fluorophenyl) -7-methoxy-quinazolin-4,6-diamine
[0449] The N- (3- chloro-4-fluorophenyl) -7-methoxy-6-nitro quinazolin-4-amine (9.00g, 25.9mmol) was dissolved in ethanol (100 mL), the was added reduced iron powder (14.5g, 259. Ommol) and concentrated hydrochloric acid (3mL) at 25 ° C, the reaction was warmed to 90 ° C 3.Oh. With 1M aqueous sodium hydroxide solution adjusted to pH 11, filtered hot to remove iron sludge, the mother liquor was concentrated and the residue was purified slurried with ethanol (50 mL), filtered, and the filter cake was dried in vacuo to a yellow solid 6.00g, yield 73.0%.
[0450] MS (ESI, pos ion.) M / z: 319.1 [M + l] + square
[0451] Synthesis Step 3: (E) -4- bromo-but-2-enoyl chloride
The [0452] square ° C Oxalyl chloride (4.19g, 33. Ommol) was slowly added dropwise to a solution containing 4-bromo crotonic acid (2.47g, 15. Ommol) and DMF (0.05mL) in dichloromethane (60 mL) solution of in 3. Oh reaction was stirred at 0 ° C. The reaction solution was concentrated, the residue was stored in a refrigerator until use.
[0453] Synthesis Step 4: (E) -4- bromo–N- (4- ((3- chloro-4-fluorophenyl) amino) -7-methoxy-quinazolin-6-yl) butan – 2_ dilute amide
[0454] The N4- (3- chloro-4-fluorophenyl) -7-methoxy-quinazolin-4,6-diamine (4.00g, 12.6mmol) and triethylamine (6.0mL, 37.8mmol ) was dissolved in anhydrous tetrahydro-furan in Misaki (80 mL), cooled to 0 ° C, was slowly added (E) -4- bromo-2-dilute acid chloride (2.748,15.12 dirty 〇1), warmed to 25 ° ( : 2.011 reaction the reaction mixture was poured into water (1001 ^) and extracted with methylene chloride (50mL X 3), the organic phases were combined, dried over anhydrous sodium sulfate filtered, concentrated and the residue with dichloromethane (30 mL). beating purified filtered, the filter cake was dried in vacuo 2.00g tan solid, yield 34.5%.
[0455] MS (ESI, pos ion.) M / z: 465.1 [M + l] + square
[0456] Synthesis Step 5: (E) -N- (4 _ ((3- chloro-4-fluorophenyl) amino) -7_ methoxy-quinazolin-6-yl) _4_ ((4aR, 7aS) – tetrahydro -2H- [1,4] dioxin burning and [2,3_c] P ratio slightly -6 (3H) – yl) butyric acid amide dilute _2_
[0457] The (E) -4- bromo–N- (4- ((3- chloro-4-fluorophenyl) amino) -7-methoxy-quinazolin-6-yl) but-2-ene amide (0.50g, 1.08mmol) and diisopropylethylamine (0.6mL, 3.24mmol) was dissolved in dimethylacetamide (10 mL) was added at 25 ° C (4aR, 7aS) – hexahydro–2H- [1,4] dioxane, and [2,3-c] pyrrole (0 • 42g, 3 • 24mmol) 5. Oh reaction was continued under stirring, 25 ° C. The reaction mixture was poured into water (70 mL) and extracted with methylene chloride (40mL X 3), the organic phases were combined, dried over anhydrous sodium sulfate. Filtered, concentrated and the residue purified by column chromatography (CH2Cl2 / MeOH (V / v) = 20/1), to give 0.30g tan solid, yield 54.5%.
[0458] MS (. ESI, pos ion) m / z: 514.1 [M + l] +;
[0459] XH NMR (400MHz, DMS0-d6) 8: 10.60 (s, lH), 9.35 (s, lH), 8.90 (s, lH), 8.08 (dd, Ji = 6.6Hz, J2 = 2.4Hz, 1H ), 7.76-7.70 (m, 1H), 7.58 (s, 1H), 7.55 (t, J = 8.4Hz, 1H), 6.75-6.65 (m, lH), 6.63 (d, J = 16.2Hz, lH) , 4.10 (s, 3H), 3.78 (t, J = 6.2Hz, 4H), 3.26 (t, J = 4.4Hz, 2H), 3.20 (dd, Ji = 7.8Hz, J2 = 2.6Hz, 2H), 2.20 (d, J = 4.6Hz, 4H)
PATENT
Patent applications WO 2014/177038 and CN 104119350 discloses aminoquinazoline tyrosine kinase inhibitors with irreversible inhibition effect on Pan-HER, wherein the compound (E) -N- (4- (3-chloro-4-fluorophenyl) amino) -7- (methyloxy-D3) -quinazolin-6-yl) -4- ( (4aR, 7aS) -tetra hydro-2H- [l, 4] dioxino [2, 3-c] pyrrole-6 (3H) -yl) butyl-2-enamide (i.e. compound (I) ) has an excellent antitumor effect. It can reduce the generation of drug resistance and also have good tolerance.
[0011]
EXPERIMENTAL PART
[0184]
The specific synthetic method for compound (I) (E) -N- (4- (3-chloro-4-fluorophenyl) amino) -7- (methyloxy-D3) -quinazolin-6-yl) -4- ( (4aR, 7aS) -tetra hydro-2H- [l, 4] dioxino [2, 3-c] pyrrole-6 (3H) -yl) butyl-2-enamide refers to Example 20 of Patent CN 104119350 A (Application Publication No. ) .
[0185]
EXAMPLES
[0186]
Example 1
[0187]
(E) -N- (4- (3-chloro-4-fluorophenyl) amino) -7- (methyloxy-D3) -quinazolin-6-yl) -4- ( (4aR, 7aS) -t etrahydro-2H- [l, 4] dioxino [2, 3-c] pyrrole-6 (3H) -yl) butyl-2-enamide dimesylate having crystalline form A
[0188]
1. Preparation of dimesylatesulfonate having crystalline form A
[0189]
(E) -N- (4- (3-Chloro-4-fluorophenyl) amino) -7- (methyloxy-D3) -quinazolin-6-yl) -4- ( (4a R, 7aS) -tetrahydro-2H- [l, 4] dioxino [2, 3-c] pyrrole-6 (3H) -yl) butyl-2-enamide (1.032 g, 2.0 mmol) was added to acetone (80 mL) , the mixture was heated to reflux for 30 minutes and filtered. The filtrate was refluxed, and mesylate (0.481 g, 5.0 mmol) was added. The resulting mixture was refluxed overnight. A part of solvent was evaporated under reduced pressure, then the temperature of the residue was gradually cooled to room temperature and maintained at this temperature overnight. The resulting mixture was filtered with suction. The filter cake was washed with acetone and dried at 50 ℃ for 8 hours in vacuo to give a white solid (1.15 g, 81.3%) .
PATENT
Example 6
[00221] N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-(tetrahvdro-2H-n,41dioxinor2,3-clpyrrol-6(3H -vn propoxy quinazolin-4-amine

[00222] Step Ubenzyl 3,4-dihvdroxypyrrolidine-l -carboxylate

To a solution of N- carbobenzoxy-3-pyrroline ( 1.00 g, 4.92 mmol, 1.0 eq) in acetone (20 mL) was added NMO ( 1.0 g, 7.38 mmol, 1.5 eq) followed by Os04 (cat. 10 mg in 1 mL ‘PrOH). The mixture was stirred for 3 h. To this, saturated NaHS03aqueous solution (5 mL) was added, and the mixture was stirred for another 0.5 h. The organic phase was separated from the mixture, and the water phase was extracted with EtOAc (20 mL x 3). The combined organic phases were dried over anhydrous Na2S04 and filtered. The filtrate was concentrated in vacuo and the residue was purified by a silica gel column chromatography (EtOAc) to give the compound as colorless oil (1.16 g, 100 %).
[00223] Step 2) benzyl tetrahvdro-2H-n.41dioxino[2.3-c1pyrrole-6(3H)- carboxylate

A mixture of NaOH aqueous solution (35 w/w %, 21 mL, aq.), C1CH2CH2C1 (21 mL), benzyl 3,4-dihydroxypyrrolidine-l -carboxylate (1.16 g, 4.9 mmol, 1.0 eq) and TBAB (0.31 g, 0.98 mmol, 0.2 eq) was heated at 55 °C for 48h in a round-bottom flask. The reaction mixture was cooled to room temperature and poured into water (50 mL), extracted with EtOAc (50 mL). The organic phase was separated from the mixture, and the water phase was extracted with EtOAc (20 mLx3). The combined organic phases were dried over anhydrous Na2S04 and filtered. The filtrate was concentrated in vacuo and the residue was purified with a silica gel column chromatography ( 1 : 1 (v/v) PE/EtOAc) to give the product as colorless oil (0.50 g, 39 %).
[00224] Step 3) hexahvdro-2H-n.41dioxinor2.3-clpyrrole

To a solution of benzyl tetrahydro-2H-[l ,4]dioxino[2,3-c] pyrrole-6(3H)-carboxylate (0.46 g, 1 .94 mmol) in MeOH (20 mL) was added two drops of HC02H followed by 20 % Pd(OH)2 (50mg). The reaction mixture was stirred under H2 for 4h at rt and was filtered. The filtrate was concentrated in vacuo to give the crude product, which was used for the next step without further purification.
[00225] Step 4) N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-(tetrahvdro-2H-n,41 dioxinor2,3-clpyrrol-6(3H) -yl)propoxy)quinazolin-4-amine

A mixture of hexahydro-2H-[ l ,4]dioxino[2,3-c]pyrrole (1.0 eq), N-(3-chloro-4-fluorophenyI)-6- (3-chloropropoxy)-7-methoxyquinazolin-4-amine (710 mg, 1.8 mmol, 0.95 eq), 2C03 (524 mg, 3.8 mmol, 2.0 eq) and KI (16 mg, 0.095 mmol, 0.05 eq) in DMF (12 mL) was heated at 60 °C for 3 h and cooled to room temperature. The reaction mixture was quenched with water (10 mL) and diluted with EtOAc (20 mL). The organic phase was separated from the mixture, and the water phase was extracted with EtOAc (20 mLx3). The combined organic phases were dried over anhydrous Na2S04 and concentrated in vacuo. The residue was purified by a silica gel column chromatography (20: 1 (v/v) CH2Cl2/CH3OH) to give the crude product, which was recrystallized from CH2C12/PE to afford the title compound as a grayish-white solid (230 mg, 25.00 %), HPLC:99.1 1 % . The compound was characterized by the following spectroscopic data: MS (ESI, pos. ion) m/z: 489.9 (M+1 );’H NMR (400 MHz, CDC13) δ: 2.09 (2H, m), 2.74 (4H, m), 2.99 (2H, dd, = 3.3, 10.4 Hz), 3.56 (2H, m), 3.80 (2H, m), 3.99 (3H, s), 4.12 (2H, t, J = 3.5 Hz), 4.22 (2H, t, J = 6.8 Hz), 7.14 (1 H, t, J = 8.8 Hz), 7.23 (1 H, s), 7.29 ( 1 H, d, J = 15.8 Hz), 7.60 (1 H, m), 7.89 (1 H, dd, J = 2.5, 6.5 Hz), 8.63 (1 H, s) ppm.
PATENT
Example 1
[00192] (^-N 4 (3-Chloro -fluorophenyl)amino)-7-methoxyquinazolin-6-yl)-4 (4aR,7a5)-tetrahydro-2H-[ l,4]dioxino[2,3-c]pyrrol-6(3H)

[00193] Step 1) N-(3-chloro-4-fluorophenyl)-7-methoxy-6-nitroquinazolin-4-amine
A solution of N-(3-chloro-4-fluorophenyl)-7-fluoro-6-nitroquinazolin-4-amine (10.00 g, 29.8 mmol) and sodium methanolate (2.80 g, 51.8 mmol) in methanol (150 mL) was heated to 70 °C and stirred for 4.0 hours. The reaction mixture was then cooled to 25 °C. The resulting mixture was poured into ice water (500 mL), and a yellow solid precipitated out. The mixture was filtered and the filter cake was dried under vacuum to give the title compound as a yellow solid (9.00 g, 86.9%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z : 349.1 [M+l]+; and ‘H NMR (400 MHz, DMSO-<&) δ: 11.60 (s, 1H), 9.55 (s, 1H), 8.08 (dd, Jx = 6.6 Hz, J2 = 2.4 Hz, 1H), 7.90 (s, 1H), 7.76-7.71 (m, 1H), 7.58 (s, 1H), 7.55 (t, J = 9.4 Hz, lH ), 4.10 (s, 3H).
[00194] Step 2) N4-(3-chloro-4-fluorophenyl)-7-methoxyquinazoline-4,6-diamine
To a solution of N-(3-chloro-4-fluorophenyl)-7-methoxy-6-nitroquinazolin-4-amine (9.00 g, 25.9 mmol) in ethanol (100 mL) were added iron powder (14.50 g, 259.0 mmol) and concentrated hydrochloric acid (3.0 mL) at 25 °C. The reaction mixture was heated to 90 °C and stirred for 3.0 hours. Then heating was stopped, and the resulting mixture was adjusted to pH 11 with aqueous sodium hydroxide solution (1 M) while the mixture was still at a temperature of about 60 ± 10 °C. The pH-adjusted resulting mixture was then immediately filtered hot to remove iron mud. The filtrate was concentrated in vacuo. The residue was triturated with ethanol (50 mL) and filtered. The filter cake was dried under vacuum to give the title compound as a yellow solid (6.00 g, 73.0%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z : 319.1 [M+l]+.
[00195] Step 3) (£)-4-bromobut-2-enoyl chloride
To a solution of 4-bromocrotonic acid (2.47 g, 15.0 mmol) and DMF (0.05 mL) in DCM (60 mL) was added oxalyl chloride (4.19 g, 33.0 mmol) dropwise at 0 °C. The reaction mixture was stirred at 0 °C for 3.0 hours, and then concentrated in vacuo. The residue was stored in a refrigerator for the next step.
[00196] Step 4) (ii)-4-bromo-N-(4-((3-chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl)but-2-enamide
To a solution of N4-(3-chloro-4-fluorophenyl)-7-methoxyquinazoline-4,6-diamine (4.00 g, 12.6 mmol) and TEA (6.0 mL, 37.8 mmol) in anhydrous tetrahydrofuran (80 mL) was added (E)-4-bromobut-2-enoyl chloride (2.74 g, 15.1 mmol) slowly at 0 °C. The reaction mixture was then heated to 25 °C and stirred for 2.0 hours. The resulting mixture was poured into water (100 mL) and extracted with DCM (50 mL x 3). The combined organic phases were dried over anhydrous NaaSOzi, filtered and concentrated in vacuo. The residue was triturated with DCM (30 mL) and filtered. The filter cake was dried under vacuum to give the title compound as a brownish yellow solid (2.00 g, 34.5%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z : 465.1 [M+l]+.
[00197] Step 5) (^-N 4 (3-chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl) (4aR,7aS)-tetrahydro-2H-[l,4]dioxino[2,3-c]pyrrol-6(3H)-yl)but-2-enamide
To a solution of (iT)-4-bromo-N-(4-((3-chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl)but-2-enamide (0.50 g, 1.1 mmol) and diisopropylethylamine (0.6 mL, 3.2 mmol) in N^V-dimethylacetamide (10 mL) was added (4aR,7aS)-hexahydro-2H-[l,4]dioxino[2,3-c]pyrrole (0.42 g, 3.2 mmol) at 25 °C, and the reaction mixture was then stirred at 25 °C for 5.0 hours. The resulting mixture was poured into water (70 mL) and extracted with DCM (40 mL x 3). The combined organic phases were dried over anhydrous Na2S04, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (CH2Cl2 MeOH (v/v) = 20/1) to give the title compound as a brownish yellow solid (0.30 g, 54.5%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z : 514.1 [M+l]+; and lH NMR (400 MHz, DMSO-t/tf) δ: 10.60 (s, 1H), 9.35 (s, 1H) , 8.90 (s, 1H), 8.08 (dd, Jx = 6.6 Hz, J2 = 2.4 Hz, 1H), 7.76-7.70 (m, 1H), 7.58 (s, 1H), 7.55 (t, J = 8.4 Hz, 1H ), 6.75-6.65 (m, 1H), 6.63(d, J = 16.2 Hz, 1H), 4.10 (s, 3H), 3.78 (t, J= 6.2 Hz, 4H), 3.26 (t, J = 4.4 Hz, 2H), 3.20 (dd, Jx = 7.8 Hz, J2 = 2.6 Hz, 2H), 2.20 (d, J= 4.6 Hz, 4H).
////////////DNT-04110, yinlitinib maleate , Guangdong Hec Pharmaceutical, PHASE 1, CHINA, yinlitinib
Fc1ccc(cc1Cl)Nc2ncnc3cc(OC)c(cc23)NC(=O)/C=C/CN4C[C@H]5OCCO[C@H]5C4
Fc1ccc(cc1Cl)Nc2ncnc3cc(OC([2H])([2H])[2H])c(cc23)NC(=O)/C=C/CN4C[C@H]5OCCO[C@H]5C4
SIMILAR COMPDS
|
1
|
MW: 485.9445 – |
|
|
2
|
MW: 558.8663 |
|
|
3
|
MW: 469.9455 |
|
|
4
|
MW: 485.9445 |
|
|
5
|
MW: 485.9445 |
BMS-986195

BMS-986195
- Molecular FormulaC20H23FN4O2
- Average mass370.421 Da
- CAS: 1912445-55-6
1H-Indole-7-carboxamide, 5-fluoro-2,3-dimethyl-4-[(3S)-3-[(1-oxo-2-butyn-1-yl)amino]-1-piperidinyl]-
4-[(3S)-3-(2-Butynoylamino)-1-piperidinyl]-5-fluor-2,3-dimethyl-1H-indol-7-carboxamid
(S)-4-(3-(2-Butynoylamino)piperidin-1-yl)-5-fluoro-2,3-dimethyl-1H-indole-7-carboxamide
(S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimeth -lH-indole-7-carboxamide
- Originator Bristol-Myers Squibb
- Class Anti-inflammatories; Antirheumatics
- Mechanism of Action Agammaglobulinaemia tyrosine kinase inhibitors
Highest Development Phases
- Phase I Rheumatoid arthritis
Most Recent Events
- 30 Jan 2018 Bristol-Myers Squibb completes a phase I trial in Rheumatoid arthritis (In volunteers, In adults, Combination therapy) in USA (PO) (NCT03262740)
- 10 Nov 2017 Bristol-Myers Squibb completes a phase I drug-drug interaction trial in Healthy volunteers (NCT03131973)
- 03 Nov 2017 Safety, pharmacokinetic, and pharmacodynamic data from a pharmacokinetic trial in healthy volunteers presented at the 81st American College of Rheumatology and the 52nd Association of Rheumatology Health Professionals Annual Scientific Meeting (ACR/ARHP-2017)
BMS-986195 is a potent, covalent, irreversible inhibitor of Bruton’s tyrosine kinase (BTK), a member of the Tec family of non-receptor tyrosine kinases essential in antigen-dependent B-cell signaling and function. BMS-986195 is more than 5000-fold selective for BTK over all kinases outside of the Tec family, and selectivity ranges from 9- to 1010-fold within the Tec family. BMS-986195 inactivated BTK in human whole blood with a rapid rate of inactivation (3.5×10-4 nM-1·min-1) and potently inhibited antigen-dependent interleukin-6 production, CD86 expression and proliferation in B cells (IC50 <1 nM) without effect on antigen-independent measures in the same cells.
Bristol-Myers Squibb is developing BMS-986195, an oral candidate for the treatment of rheumatoid arthritis. A phase I clinical trial in healthy adult volunteers is ongoing.
![]()

Credit: Tien Nguyen/C&EN
Presented by: Scott H. Watterson, principal scientist at Bristol-Myers Squibb
Target: Bruton’s tyrosine kinase (BTK)
Disease: Autoimmune diseases such as rheumatoid arthritis
Reporter’s notes: Completing another set of back-to-back presentations on the same target, Watterson revealed another BTK inhibitor also in Phase II clinical trials. Chemists made BMS-986195 in seven steps, and the molecule showed high levels of BTK inactivation in mice. The team aimed to develop an effective compound that required low doses and that had low metabolic degradation.
Patent
WO 2016065226
Inventor Saleem AhmadJoseph A. TinoJohn E. MacorAndrew J. TebbenHua GongQingjie LiuDouglas G. BattKhehyong NguScott Hunter WattersonWeiwei GuoBertrand Myra Beaudoin
Original Assignee Bristol-Myers Squibb Company
https://patents.google.com/patent/WO2016065226A1/en
PATENT
WO 2018045157








otein kinases, the largest family of human enzymes, encompass well over 500 proteins. Btk is a member of the Tec family of tyrosine kinases, and is a regulator of early B-cell development, as well as mature B-cell activation, signaling, and survival.
B-cell signaling through the B-cell receptor (BCR) leads to a wide range of biological outputs, which in turn depend on the developmental stage of the B-cell. The magnitude and duration of BCR signals must be precisely regulated. Aberrant BCR-mediated signaling can cause dysregulated B-cell activation and/or the formation of pathogenic auto-antibodies leading to multiple autoimmune and/or inflammatory diseases. Mutation of Btk in humans results in X-linked agammaglobulinaemia (XLA). This disease is associated with the impaired maturation of B-cells, diminished immunoglobulin production, compromised T-cell-independent immune responses and marked attenuation of the sustained calcium signal upon BCR stimulation.
Evidence for the role of Btk in allergic disorders and/or autoimmune disease and/or inflammatory disease has been established in Btk-deficient mouse models. For example, in standard murine preclinical models of systemic lupus erythematosus (SLE), Btk deficiency has been shown to result in a marked amelioration of disease progression. Moreover, Btk deficient mice are also resistant to developing collagen-induced arthritis and are less susceptible to Staphylococcus-induced arthritis.
A large body of evidence supports the role of B-cells and the humoral immune system in the pathogenesis of autoimmune and/or inflammatory diseases. Protein-based therapeutics (such as Rituxan) developed to deplete B-cells, represent an important approach to the treatment of a number of autoimmune and/or inflammatory diseases.
Because of Btk’s role in B-cell activation, inhibitors of Btk can be useful as inhibitors of B-cell mediated pathogenic activity (such as autoantibody production).
Btk is also expressed in mast cells and monocytes and has been shown to be important for the function of these cells. For example, Btk deficiency in mice is associated with impaired IgE -mediated mast cell activation (marked diminution of T F-alpha and other inflammatory cytokine release), and Btk deficiency in humans is associated with greatly reduced TNF-alpha production by activated monocytes.
Thus, inhibition of Btk activity can be useful for the treatment of allergic disorders and/or autoimmune and/or inflammatory diseases including, but not limited to: SLE, rheumatoid arthritis, multiple vasculitides, idiopathic thrombocytopenic purpura (ITP), myasthenia gravis, allergic rhinitis, multiple sclerosis (MS), transplant rejection, type I diabetes, membranous nephritis, inflammatory bowel disease, autoimmune hemolytic anemia, autoimmune thyroiditis, cold and warm agglutinin diseases, Evan’s syndrome, hemolytic uremic syndrome/thrombotic thrombocytopenic purpura (HUS/TTP), sarcoidosis, Sjogren’s syndrome, peripheral neuropathies (e.g., Guillain-Barre syndrome), pemphigus vulgaris, and asthma.
In addition, Btk has been reported to play a role in controlling B-cell survival in certain B-cell cancers. For example, Btk has been shown to be important for the survival of BCR-Abl-positive B-cell acute lymphoblastic leukemia cells. Thus inhibition of Btk activity can be useful for the treatment of B-cell lymphoma and leukemia.
In view of the numerous conditions that are contemplated to benefit by treatment involving modulation of protein kinases, it is immediately apparent that new compounds capable of modulating protein kinases such as Btk and methods of using these compounds should provide substantial therapeutic benefits to a wide variety of patients.
WO 2016/065226 discloses indole carboxamide compounds useful as Btk inhibitors, including (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide (Example 223), which has the structure:

Also disclosed is multistep synthesis process for preparing (S)-4-(3-(but-2-ynamido) piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide.
There are difficulties associated with the adaptation of the multistep synthesis disclosed in WO 2016/065226 to larger scale synthesis, such as production in a pilot plant or a manufacturing plant for commercial production. Further, there is a continuing need to find a process that has few synthesis steps, provides higher yields, and/or generates less waste.
Applicants have discovered a new synthesis process for the preparation of (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide that has fewer synthesis steps and/or provides higher yields than the process disclosed in WO 2016/065226. Furthermore, this process contains no metal-catalyzed steps, no genotoxic intermediates, and is adaptable to large scale manufacturing.
EXAMPLE 1
(S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide

Step 1 : Preparation of Methyl (S)-2-amino-4-(3-((tert-butoxycarbonyl)amino)piperidin-l-yl)-5-fluorobenz
To a 250 mL ChemGlass reactor were charged methyl 2-amino-4,5-difluoro-benzoate (11.21 g, 59.90 mmol), tert-butyl N-[(3S)-3-piperidyl]carbamate (10 g, 49.930 mmol), potassium phosphate, dibasic (10.44 g, 59.94 mmol), and dimethyl sulfoxide (100 mL, 1400 mmol). The resulting thin slurry was heated to 95 to 100 °C and agitated at this temperature for 25 hours. The mixture was cooled to 50 °C. Methanol (100 mL) was added and followed by slow addition of water (50 mL). The mixture was aged at 50 °C for 30 minutes to result in a thick white slurry. Additional water (150 mL) was slowly charged to the above mixture and agitated at 50 °C for 1 hour. The slurry was cooled to 20 °C in 1 hour and aged at this temperature for 4 hours. The slurry was filtrated. The wet cake washed with 25% MeOH in water (30 mL), water (100 mL) and dried under vacuum at 60 °C for 24 h. Methyl (S)-2-amino-4-(3-((tert-butoxycarbonyl)amino) piperidin-l-yl)-5-fluorobenzoate was obtained as a white solid (7 g, yield: 72.5%). ¾ MR (400MHz, METHANOLS) δ 7.34 (d, J=14.6 Hz, 1H), 6.27 (d, J=7.3 Hz, 1H), 3.83-3.71 (s, 3H), 3.68-3.57 (m., 1H), 3.50 -3.40 (m 1H), 3.39 -3.31 (m, 1H), 3.31-3.26 (m, 1H), 2.86-2.70 (m, 1H), 2.64 (t, J=10.0 Hz, 1H), 1.97-1.84 (m, 1H), 1.84-1.74 (m, 1H), 1.73-1.61 (m, 1H), 1.44 (s, 9H), 1.38 (m, 1H). LC-MS [M+H] 368.
Step 2: Preparation of Methyl (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate
To a reactor were charged methyl (S)-2-amino-4-(3-((tert-butoxycarbonyl)amino) piperidin-l-yl)-5-fluorobenzoate (5.0 g), DPPOH (diphenyl phosphate, 6.81 g, 2 eq) and 3-hydroxybutanone (1.2 eq, 1.44 g), followed by addition of isopropyl acetate (100 mL, 20 mL/g). The mixture was allowed to warm up to 70 to 75 °C, resulting in a yellow solution. The solution was stirred at 70 to 75 °C for 30 h to complete the cyclization.
Water (2 mL) was added and the mixture was aged at 70 °C over 24 h to remove the Boc group. The mixture was cooled to room temperature. Next, aqueous 20% K3PO4 solution (50 mL) was added and the mixture was stirred for 15 min. The organic layer was separated and washed with water (50 mL). The organic layer was then concentrated under vacuum (200 Torr) to -50 mL. The resulting slurry was stirred at 50 °C for 2 h and then heptane (100 mL) was added over 1 h. The mixture was cooled to room
temperature, stirred for 20 h, and then filtered. The cake was washed with heptane (50 mL). Methyl (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate, DPPOH salt was obtained as a light yellow solid. The wet-cake was added to a reactor. Isopropyl acetate (100 mL) was added, followed by addition of aqueous K3PO4 solution (4 g in water 50 mL). The mixture was stirred at room temperature for -half-hour, resulting in a two phase clear solution (pH >10 for aqueous). The organic layer was separated and washed with water (50 mL), and then concentrated under vacuum to a volume of 15 mL. The resulting slurry was stirred at room temperature for 4 h, then heptane (75 mL) was added over 1 h. The mixture was aged at room temperature for 24 h, then concentrated to a volume to -50 mL. The slurry was filtered. The cake was washed with heptane 20 mL and dried under vacuum at 50 °C for 24 h. Methyl (S)-4-(3- aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate was obtained as a light yellow solid (2.76 g, yield: 69%). ¾ NMR (400MHz, DMSO-d6) δ 10.64 (s, 1H), 7.33 (d, J=13.7 Hz, 1H), 3.89 (s, 3H), 3.14 (br. m., 1H), 3.07-2.90 (m, 2H), 2.84 (br. m., 1H), 2.70 (br. m., 1H), 2.35 (s, 3H), 2.33 (s, 3H), 1.87 (br. m., 1H), 1.67 (br. m., 3H). LC-MS: M+H= 320.
Alternative Preparation
Step 2: Preparation of ethyl (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate trifluoroacetic acid salt
To a reactor were charged ethyl (S)-2-amino-4-(3-((tert-butoxycarbonyl)amino) piperidin-l-yl)-5-fluorobenzoate (1.0 g, limiting reagent), DPPOH (diphenyl phosphate, 1.97 g, 3.0 eq) and 3-hydroxybutanone (1.4 eq, 0.32 g), followed by addition of toluene (20 mL, 20 mL/g). The mixture was allowed to warm up to 80-90 °C, resulting in a yellow solution. The solution was stirred at 80-90 °C for 10 h to complete the
cyclization. Water (0.4 mL, 0.4 ml/g) was added and the mixture was aged at 80-90 °C for 8 hours. The mixture was cooled to room temperature. Next, aqueous 20% K3PO4 solution (15 mL, 15 mL/g) was added and the mixture was stirred for 0.5 hour. The organic layer was separated and the aqueous layer was washed with toluene (7.5 mL, 7.5 mL/g). To combined organic layers water (10 mL, 10 mL/g) was added and the mixture was stirred for 0.5 hour. The organic layer was separated. To the organic layer water (10 mL, 10 mL/g) was added and the mixture was stirred for 0.5 hour. The organic layer was separated. The organic layer was concentrated under vacuum (100 Torr) to 8 mL (8 ml/g). Following concentration the reaction mixture was cooled to 20-25 °C and MTBE (20 mL, 20 mL/g) was added. Trifluoroacetic acid (1.2 eq., 0.36 g) was slowly added to make the salt maintaining temperature at 20-25 °C. The resulting slurry was aged for 4 hours and then filtered. The filtered solids are washed with MTBE (8 mL, 8 mL/g) and the cake
was dried under vacuum at 50 °C. (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate trifluoroacetic acid salt was obtained as a white to tan crystalline material (85% yield, 1.0 g). ¾ NMR (400 MHz, DMSO-d6) δ 10.74 (s, 1H), 8.16-7.88 (m, 2H), 7.37 (d, 7=13.6 Hz, 1H), 4.38 (q, 7=7.1 Hz, 2H), 3.18-3.01 (m, 3H), 2.96 (br s, 1H), 2.35 (s, 6H), 2.30 (s, 1H), 2.12 (br d, 7=9.3 Hz, 1H), 1.78 (br s, 2H), 1.45-1.31 (m, 4H), 1.10 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 165.1, 165.1, 158.4, 158.1, 135.4, 134.7, 134.6, 132.2, 128.8, 128.2, 126.9, 126.8, 118.7, 115.7, 110.6, 110.3,108.7, 108.6, 106.6, 106.5, 83.5, 79.8, 60.5, 54.9, 51.7, 48.7, 47.2, 28.4, 26.8, 23.6, 14.2, 11.1, 10.2
Step 3A: Preparation of (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide
A 40 mL vial was charged with methyl (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate (1.5 g, 4.70 mmol), followed by the addition of N,N-dimethylformamide (12.0 mL, 8.0 mL/g). The vial was purged with N2. Formamide (1.49 mL, 37.6 mmol) was added followed by sodium methoxide solution in methanol (35 wt%, 1.29 mL, 3.76 mmol). The resulting solution was heated at 50 °C over 8 hours. The reaction mixture was cooled down to room temperature and the reaction was quenched with water (12.0 mL, 8.0 mL/g). 2-methyltetrahydrofuran (30 mL, 20 mL/g) was added to the mixture. The mixture was shaken vigorously. The layers were separated and the aqueous layer was extracted with 2-methyltetrahydrofuran (15 mL, 10 mL/g) two more times. Organic extracts were then washed with brine and water (15 mL each, 10 mL/g). The organic layer was evaporated. Solids were dried in vacuo at 60 °C to afford (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide as a yellow solid (1.04 g, 69% yield). ¾ NMR (500MHz, DMSO-d6) δ 10.60 (br. s.,
1H), 7.91 (br. s., 1H), 7.40 (d, 7=14.0 Hz, 1H), 7.32 (br. s., 1H), 3.10 (br. s., 1H), 2.98 (br. s., 2H), 2.82 (br. s., 1H), 2.68 (br. s., 1H), 2.34 (br. s., 3H), 2.30 (br. s., 3H), 1.88 (br. s., 1H), 1.67 (br. s., 2H), 1.45 (br. s., 2H), 1.05 (br. s., 1H). LCMS [M+H] 305.24.
Step 3B: Alternative Preparation of (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide
A 100 mL Hastelloy high pressure EasyMax reactor was charged with methyl (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate (1.5 g, 4.70 mmol), followed by addition of 7 N ammonia solution in methanol (45.0 mL, 30.0 mL/g) followed by addition of l,3,4,6,7,8-hexahydro-2H-pyrimido[l,2-a]pyrimidine (1.33 g, 9.39 mmol). The reactor was sealed and purged with N2 three times. The reactor was then heated to 80 °C for 24 hrs. The reaction mixture was cooled to room temperature and the vessel contents were purged with N2 three times. Volatiles were concentrated to ~6 mL (4 mL/g) and water (24 mL, 16 mL/g) was added. The yellow precipitate was collected and filtered. The precipitate was washed with methanol/water mixture (20:80 v/v, 6 mL, 4 mL/g), and then water (18 mL, 12 mL/g). The solids were dried in vacuo at 60 °C to afford (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide as a yellow crystalline material (0.93 g, 62% yield). ¾ MR (500MHz, DMSO-de) δ 10.60 (br. s., 1H), 7.91 (br. s., 1H), 7.40 (d, J=14.0 Hz, 1H), 7.32 (br. s., 1H), 3.10 (br. s., 1H), 2.98 (br. s., 2H), 2.82 (br. s., 1H), 2.68 (br. s., 1H), 2.34 (br. s., 3H), 2.30 (br. s., 3H), 1.88 (br. s., 1H), 1.67 (br. s., 2H), 1.45 (br. s., 2H), 1.05 (br. s., 1H). LCMS [M+H] 305.24.
Alternative Preparation:
Step 3C: Preparation of (,S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide 2-butynoic acid salt
Ethyl (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxylate trifluoroacetic acid salt (1.0 g, limiting reagent) and formamide (5 mL, 5 mL/g) were added to a nitrogen inerted reactor. The temperature was maintained at 20-25 °C. To the reactor was added a solution of 20 wt% potassium t-butoxide in THF. The reaction mixture was allowed to sit for 6 hours. To reaction mixture was added Me-THF (15 mL, 15 mL/g) and 12.5 wt % aqueous NaCl (5 mL, 5 mL/g). The reaction mixture was stirred for 0.5 hour. The organic layer was separated, 5 wt% aqueous NaCl (1 mL, 1 mL/g) and 0.25 N aqueous NaOH (4 mL, 4 mL/g) were added, and then stirred for 0.5 hour. The organic layer was separated and 5 wt% aqueous NaCl (5 mL, 5 mL/g) was added, the mixture was stirred for 0.5 hour, and organic phase was separated. The rich organic phase was dried distillation at a pressure of 100 mtorr with Me-THF to obtain KF in 1.5-4wt% range at 5 mL Me-THF volume. The volume was adjusted to 15 mL Me-THF by adding Me-THF (10 mL, 10 mL/g) and EtOH (4 mL, 4 mL/g). Next, 2-butynoic acid (1.0 eq., 0.19 g) was added and the mixture was agitated for 10 hrs. The resulting slurry was filtered. The cake was washed with Me-THF (10 mL, 10 mL/g) and dried under vacuum at 75 °C to afford (,S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide 2-butynoic acid salt (0.7 g, 80% yield) as white crystalline powder. ¾ NMR (400 MHz, DMSO-d6) δ 10.68 (s, 1H), 7.98 (br s, 1H), 7.50-7.32 (m, 2H), 3.32 (br d, J=8.6 Hz, 2H), 3.21 (br t, J=10.5 Hz, 1H), 3.13-2.89 (m, 3H), 2.32 (d, J=5.1 Hz, 5H), 2.11 (br d, J=10.9 Hz, 1H), 1.81-1.67 (m, 4H), 1.55-1.28 (m, 1H).
Step 4A: Preparation of (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide
To Reactor-1 was charged N,N-dimethylformamide (DMF, 12.77 kg, 13.5 L). Reactor-1 was purged with N2 to inert. (S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide (3.0 kg, 1.0 equiv) was charged followed by 2-butynoic acid (0.854 kg, 1.04 equiv). Reactor-1 was rinsed with DMF (1.42 kg, 1.5 L). The mixture was sparged with N2 for 20 min. Triethylamine (2.99 kg, 3.0 equiv) was charged followed by a DMF rinse (1.42 kg, 1.5 L). TBTU (O-(Benzotriazol-l-yl)-N,N,N’,N’-tetramethyluronium tetrafluorob orate, 3.256 kg, 1.04 equiv) was charged followed by a DMF rinse (1.42 kg, 1.5 L). The reaction mixture was agitated for 1.5 h at 20 °C. MeTHF (46.44 kg, 60 L) was charged to the batch. The reaction was quenched with LiCl (20 wt%, 26.76 kg, 24 L) at 20 °C. The bottom aqueous layer was discharged as waste. The organic layer was washed with 2N HCl solution (24.48 kg, 24 L), 10 wt% sodium bicarbonate solution (25.44 kg, 24 L) and deionized water (24.0 kg, 24 L). THF (26.61 kg, 30 L) was charged into Reactor-1. The rich organic stream in MeTHF/TFIF was polish filtered. The stream was distilled down to 15 L at 75-100 Torn Constant volume distillation was carried out at 15 L with THF feed (39.92 kg, 45 L). The stream was heated to 60 °C for 1 hr and cooled to 50 °C. MTBE (33.30 kg, 45 L) was charged slowly over 2 h. The slurry was aged at 50 °C for 4 h and cooled to 20 °C over 2 h, and aged at 20 °C for >2 h. The 1st drop slurry was filtered and was rinsed with MTBE (8.88 kg, 12 L) twice. Wet cake was dried under vacuum 60 to 70 °C at 25 mbar overnight (>15 h). Reactor-1 was thoroughly cleaned with IPA. The dry cake was charged into Reactor-1 followed by the charge of IPA (47.10 kg, 60 L). The batch was heated to 60 °C to achieve full dissolution and cooled to 40 °C. Rich organic (24 L) was transferred to Reactor-2 for crystallization. The stream was distilled at 24 L constant volume and 100 mbar using remaining rich organic from reactor-1 as distillation feed. Following distillation completion, the batch was heated to 60 °C, aged at 60 °C for 2 h, cooled to 20 °C over 2 h, and aged at 20 °C over 2 h. The slurry was filtered. IPA (1.18 kg) was used to rinse the reactor and washed the cake. The wet cake was dried under vacuum at 70 °C and 25 mbar for >15 h. The dry cake (2.196 kg, 63.2% yield) was discharged as an off-white crystalline solid. ¾ NMR (400MHz, DMSO-d6): δ 10.62 (s, 1H), 8.48 (d, J= 7.1 Hz, 1H), 7.91 (s, 1H), 7.39 (d, J=7.4 Hz, 1H), 7.33 (s, 1H), 3.88 (m, 1H), 3.11 (t, J= 8.0 Hz, 1H), 3.0 (m, 1H), 2.96 (m, 1H), 2.78 (t, J= 10.0 Hz, 1H), 2.35 (s, 3H), 2.30 (s, 3H), 1.92 (s, 3H), 1.86 (m, 1H), 1.31 (m, 1H), 1.70 (m, 2H); 13C NMR (400 MHz, DMSO-d6): δ 168.2, 153.2, 151.9, 134.4, 133.2, 132.1, 126.5, 112.3, 108.4, 106.0, 82.3, 75.7, 56.9, 51.9, 46.3, 29.7, 24.4, 11.1, 10.2, 3.0; LC-MS: M+H= 371.2.
Step 4B: Alternative preparation of (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimeth -lH-indole-7-carboxamide
To Reactor-1 was charged N,N-dimethylformamide (DMF 4.5 mL, 4.5 mL/g). Reactor-1 was purged with N2 to inert. (,S)-4-(3-aminopiperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide 2-butynoic acid salt (1.0 g, limiting reagent) was charged followed by 2-butynoic acid (0.065g, 0.3 equiv.). The mixture was inerted with N2 for 20 min. N-methylmorpholine (0.78 g, 3.0 equiv) was charged. Next,
diphenylphosphinic chloride (0.79 g, 1.3 equiv) was charged over 0.5 h while maintaining the reaction temperature at 20-25 °C. The reaction mixture was agitated for 1.5 hour at 20 °C. Me-THF (14 mL, 14 mL/g) was charged to the reaction mixture. The reaction was quenched with the addition of aqueous NaCl (12.5 wt%, 6 mL, 6 mL/g) at 20 °C. The bottom aqueous layer was discharged as waste. Aqueous NaCl (12.5 wt%, 6 mL, 6 mL/g) at 20 °C was added to the organic layer, stirred for 0.5 hour and the bottom aqueous layer was discharged to waste. Deionized water (6 mL, 6 mL/g) was charged to the organic layer, stirred for 0.5 hour and the bottom aqueous layer was discharged to waste. THF (8 mL, 8 mL/g) was charged into Reactor-1 and the mixture was
concentrated under vacuum to remove Me-THF and water, and reconstituted in 4 L/kg of THF. The mixture was heated to 60 °C and stirred for 1 hour; the temperature was reduced to 50 °C and MTBE (12 mL, 12 mL/g) was added. The mixture was aged for 4 hours while maintaining the temperature of 50 °C and then cooled to room temperature. The solids were filtered and washed with MTBE (6.5 mL, 6.5 mL/g). The solids of crude were dried at 70 °C under vacuum for 12 hours.
Crude (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide was charged to Reactor-2, followed by THF (12 mL, 12 mL/g). The mixture was stirred for 0.5 hour. The solution was polish filtered. The solution was concentrated under vaccuum to remove THF and reconstituted in EtOH (7 mL, 7 mL/g). (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide seeds (0.01 g, 0.01 g/g) were added, the mixture was heated to 60 °C and aged for 2 hours, n-heptane (21 mL, 21 mL/g) was added slowly over 4 hours. The mixture was aged for additional 2 hours at 60 °C, followed by cooldown to room temperature. The slurry was filtered, washed with n-heptane (6 mL, 6 mL/g), and dried under vacuum at 70 °C for 12 hours. The dry cake (0.68 g, 71% yield) was discharged as an off-white crystalline solid. ¾ NMR (400MHz, DMSO-d6): δ 10.62 (s, 1H), 8.48 (d, J= 7.1 Hz, 1H), 7.91 (s, 1H), 7.39 (d, J=7.4 Hz, 1H), 7.33 (s, 1H), 3.88 (m, 1H), 3.11 (t, J= 8.0 Hz, 1H), 3.0 (m, 1H), 2.96 (m, 1H), 2.78 (t, J= 10.0 Hz, 1H), 2.35 (s, 3H), 2.30 (s, 3H), 1.92 (s, 3H), 1.86 (m, 1H), 1.31 (m, 1H), 1.70 (m, 2H); 13C MR (400 MHz, DMSO-d6): δ 168.2, 153.2, 151.9, 134.4, 133.2, 132.1, 126.5, 112.3, 108.4, 106.0, 82.3, 75.7, 56.9, 51.9, 46.3, 29.7, 24.4, 11.1, 10.2, 3.0; LC-MS: M+H= 371.2.
Applicants have discovered a new synthesis process for the preparation of (S)-4- (3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide which offers significant advantages.
The new synthesis process utilizes fewer synthesis steps (4 vs 8) than the process disclosed in WO 2016/065226.
Additionally, the process of the present invention provided (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide at an overall
yield of 22% (step 1 : 73.%, step 2: 69%, step 3 : 69%, step 4: 63%). In comparison, (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide was prepared according to the process of WO 2016/065226, which provided (S)-4-(3-(but-2-ynamido)piperidin-l-yl)-5-fluoro-2,3-dimethyl-lH-indole-7-carboxamide at an overall yield of 2.9% yield (step 1 : 91%, step 2: 71%, step 3 : 35%, step 4: 88%, step 5: 80%, step 6: 29%, step 7: 99%, step 8: 63%).
Furthermore, the process of the present invention does not include any transition metal-catalyzed steps, no genotoxic intermediates, and is adaptable to large scale manufacturing. In comparison, the process disclosed in WO 2016/065226 employed lead (Pb) in process step (8) and included a potentially genotoxic hydrazine intermediate in process step 8.
The process of the present invention has an estimated manufacturing cycle time of approximately 6 months versus a estimated manufacturing cycle time of approximately 12 months for the process disclosed in WO 2016/065226.
REFERENCE
/////////////////BMS-986195, Phase I, Rheumatoid arthritis, BMS
NC(=O)c2cc(F)c(c1c(C)c(C)nc12)N3CCC[C@@H](C3)NC(=O)C#CC
VNRX-5133 from VENATORX PHARMACEUTICALS

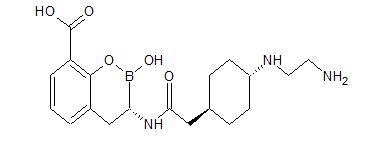
VNRX-5133
CAS: 1613268-23-7
Chemical Formula: C19H28BN3O5
Molecular Weight: 389.26
Chemical Formula: C19H28BN3O5
Molecular Weight: 389.26
3-(2-((1r,4r)-4-((2-aminoethyl)amino)cyclohexyl)acetamido)-2-hydroxy-3,4-dihydro-2H-benzo[e][1,2]oxaborinine-8-carboxylic acid
( R)-3-( 2-( trans-4-( 2-aminoethylamino)cvclohexyl)acetamido)-2-hvdroxy-3-,4-dihydro-2H-benzo[el [l,21oxaborinine-8-carboxylic acid

- Originator VenatoRx Pharmaceuticals
- Developer National Institute of Allergy and Infectious Diseases; VenatoRx Pharmaceuticals
- Class Antibacterials; Cephalosporins; Small molecules
- Mechanism of Action Beta lactamase inhibitors; Cell wall inhibitors
Highest Development Phases
- Phase I Bacterial infections
Most Recent Events
- 19 Mar 2018 VenatoRx Pharmaceuticals plans phase III pivotal trials in mid-2018
- 03 Jan 2018 VNRX 5133 receives Fast Track designation for Bacterial infections (complicated urinary tract infections and complicated intra-abdominal infections) [IV-infusion] in USA
- 03 Jan 2018 VNRX 5133 receives Qualified Infectious Disease Product status for Intra-abdominal infections in USA
- clip
- https://cen.acs.org/articles/96/web/2018/03/Drug-structures-made-public-New-Orleans.html
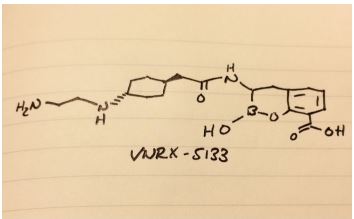 Credit: Tien Nguyen/C&EN
Credit: Tien Nguyen/C&EN
Presented by: Christopher J. Burns, president and chief executive officer of VenatoRx Pharmaceuticals
Target: β-lactamase enzymes, enzymes that inactivate β-lactam-based antibiotics enabling bacteria to resist their attacks
Disease: Gram-negative bacterial infections
Reporter’s notes: Another story with humble beginnings, this time with Burns and two colleagues sitting in a Panera Bread, with an idea. They wanted to offer a new compound in the class of β-lactam antibiotics, drugs which are “well-liked” by doctors, Burns said, and make up 60% of all antibiotic prescriptions. However, bacteria have developed defenses against these compounds in the form of β-lactamases, or as Burns dubbed them, “PAC-men.” These enzymes can chew up 1000 β-lactams per second, he said. VNRX-5133 was active against both serine-β-lactamases and metallo-β-lactamases in enzyme assays. It is being developed in combination with the antibiotic cefepime. VNRX-5133 fends off the PAC-men’s attacks, allowing cefepime to combat infection. The compound has gone through Phase I clinical trials and will be skipping ahead to Phase III later this year.
PATENT
WO 2014089365


| Applicants: | VENATORX PHARMACEUTICALS, INC [US/US]; 30 Spring Mill Drive Malvern, PA 19355 (US) |
| Inventors: | BURNS, Christopher, J.; (US). DAIGLE, Denis; (US). LIU, Bin; (US). MCGARRY, Daniel; (US). PEVEAR, Daniel C.; (US). TROUT, Robert E. Lee; (US) |
https://patents.google.com/patent/WO2014089365A1/en

Christopher J. Burns, Ph.D.
President and Chief Executive Officer
Dr. Burns is Co-Founder, President and Chief Executive Officer of VenatoRx. He brings over 25 years of corporate and R&D experience within both major (RPR/Aventis) and specialty (ViroPharma, Protez…https://www.venatorx.com/leadership/
Antibiotics are the most effective drugs for curing bacteria-infectious diseases clinically. They have a wide market due to their advantages of good antibacterial effect with limited side effects. Among them, the beta-lactam class of antibiotics (for example, penicillins,
cephalosporins, and carbapenems) are widely used because they have a strong bactericidal effect and low toxicity.
[0004] To counter the efficacy of the various beta-lactams, bacteria have evolved to produce variants of beta-lactam deactivating enzymes called beta-lactamases, and in the ability to share this tool inter- and intra-species. These beta-lactamases are categorized as “serine” or “metallo” based, respectively, on presence of a key serine or zinc in the enzyme active site. The rapid spread of this mechanism of bacterial resistance can severely limit beta-lactam treatment options in the hospital and in the community.
EXAMPLE 15 : ( R)-3-( 2-( trans-4-( 2-aminoethylamino)cvclohexyl)acetamido)-2-hvdroxy-3-,4-dihydro-2H-benzo[el [l,21oxaborinine-8-carboxylic acid

Step 1 : Synthesis of (R)-3-(2-(trans-4-(2-(tert-butoxycarbonylamino)ethylamino)cyclohexyl)acetamido)-2-hydroxy-3,4-dihydro-2H-benzo[e] [ 1 ,2]oxaborinine-8-carboxylic acid.
[00240] To (R)-3-(2-(trans-4-aminocyclohexyl)acetamido)-2-hydroxy-3,4-dihydro-2H-benzo[e][l,2]oxaborinine-8-carboxylic acid (Example 6, 15 mg) in MeOH (2 mL) was added tert-butyl 2-oxoethylcarbamate (20 mg). Pd/C (10% by weight, 10 mg) was added and the reaction mixture was stirred under ¾ balloon overnight. The reaction mixture was filtrated and the solvent was then removed under reduced pressure and the residue was carried on to the next step without further purification. ESI-MS m/z 490.1 (MH)+.
Step 2: Synthesis of (R)-3-(2-(trans-4-(2-aminoethylamino)cyclohexyl)acetamido)-2-hydroxy-3,4-dihydro-2H-benzo[e][l,2]oxaborinine-8-carboxylic acid.
[00241] To (R)-3-(2-(trans-4-(2-(tert-butoxycarbonylamino)ethylamino)cyclohexyl)acetamido)-2-hydroxy-3,4-dihydro-2H-benzo[e][l,2]oxaborinine-8-carboxylic acid (20 mg) in a flask was added 1 mL 4N HC1 in dioxane. The resulting reaction mixture was stirred at RT for 2hr. The solvent was removed in vacuo and the residue was purified by reverse phase preparative HPLC and dried using lyophilization. ESI-MS m/z 390 (MH)+.
Step 2: (R)-3-(2-(trans-4-((2-aminoethylamino)methyl)cyclohexyl)acetamido)-2-hydroxy-3,4-dihydro-2H-benzo[e] [ 1 ,2]oxaborinine-8-carboxylic acid
[00229] Prepared from 3-[2-(2-{4-[(2-tert-Butoxycarbonylamino-ethylamino)-methyl]-cyclohexyl}-acetylamino)-2-(2,9,9-trimethyl-3,5-dioxa-4-bora-tricyclo[6.1.1.02,6]dec-4-yl)-ethyl]-2-methoxy-benzoic acid tert-butyl ester and BC13 following the procedure described in Step 2 of Example 1. The crude product was purified by reverse phase preparative HPLC and dried using lyophilization. ESI-MS m/z 404 (MH)+.
/////////////////////////////VNRX-5133; VNRX5133; VNRX 5133, phase 1, VenatoRx Pharmaceuticals, BACTERIAL INFECTIONS, Christopher J. Burns
NCCN[C@@H]1CC[C@@H](CC(NC2B(O)OC(C(C(O)=O)=CC=C3)=C3C2)=O)CC1
GDC 0575
GDC 0575
GDC-0575
CAS: 1196541-47-5
C16 H20 Br N5 O, 378.27
(R)-N-(4-(3-aminopiperidin-1-yl)-5-bromo-1H-indol-3-yl)cyclopropanecarboxamide
N-[4-[(3R)-3-Amino-1-piperidinyl]-5-bromo-1H-pyrrolo[2,3-b]pyridin-3-yl]cyclopropanecarboxamide
Cyclopropanecarboxamide, N-[4-[(3R)-3-amino-1-piperidinyl]-5-bromo-1H-pyrrolo[2,3-b]pyridin-3-yl]-
- ARRY-575; GDC-0575; RG 7741; RO 6845979,
- AK 687476
- ARRY 575
- GDC 0575
- RG 7741
![]()
GDC-0575, also known as ARRY-575 and RG7741, is a potent and selective CHK1 inhibitor.
GDC-0575 is a highly selective small-molecule Chk-1 inhibitor invented by Array and licensed to Genentech. Genentech is responsible for all clinical development and commercialization activities. Array received an upfront payment of $28 million and is eligible to receive clinical and commercial milestone payments up to $380 million and up to double-digit royalties on sales.
Chk-1 is a protein kinase that regulates the tumor cell’s response to DNA damage often caused by treatment with chemotherapy. In response to DNA damage, Chk-1 blocks cell cycle progression in order to allow for repair of damaged DNA, thereby limiting the efficacy of chemotherapeutic agents. Inhibiting Chk-1 in combination with chemotherapy can enhance tumor cell death by preventing these cells from recovering from DNA damage. GDC‑0575 is designed to enhance the efficacy of some chemotherapeutic agents. GDC-0575 is currently advancing in a Phase 1 trial in patients with lymphoma or solid tumors.

- Originator Array BioPharma
- Developer Genentech
- Class Antineoplastics; Small molecules
- Mechanism of Action Checkpoint kinase 1 inhibitors
Highest Development Phases
- Phase I Lymphoma; Solid tumours
Most Recent Events
- 11 Jan 2018 Genentech completes a phase I trial in Lymphoma (Late-stage disease, Metastatic disease, Second-line therapy or greater, Combination therapy, Monotherapy) in France and USA (PO) (NCT01564251)
- 05 Dec 2017 GDC 0575 is still in phase I trials for Solid tumours and lymphoma in USA and France (Genentech pipeline, December 2017) (NCT01564251)
- 04 Nov 2017 No recent reports of development identified for phase-I development in Lymphoma in France (PO)

PATENTS
U.S. Patent, 8,841,304
U.S. Patent 8,178,131,
PAPER
Org. Process Res. Dev. 2017, 21, 664– 668
Highly Regioselective and Practical Synthesis of 5-Bromo-4-chloro-3-nitro-7-azaindole
† Department of Small Molecule Process Chemistry, Genentech, Inc., 1 DNA Way, South San Francisco, California 94080, United States
‡ Department of Pharma Technical Development, F. Hoffmann-La Roche AG, Grenzacherstrasse 124, CH-4070 Basel, Switzerland
Org. Process Res. Dev., 2017, 21 (4), pp 664–668
DOI: 10.1021/acs.oprd.7b00060
*E-mail: han.chong@gene.com.

We report an efficient and highly regiocontrolled route to prepare a functionalized 7-azaindole derivative—5-bromo-4-chloro-3-nitro-7-azaindole—from readily available parent 7-azaindole featuring a highly regioselective bromination of the 4-chloro-3-nitro-7-azaindole intermediate. In addition to the high efficiency and excellent control of regioisomeric impurities, the process is operationally simple by isolating each product via direct crystallization from the reaction mixture with no liquid–liquid extractions or distillation steps needed. We demonstrated the route on >50 kg scale and 46% overall yield to provide the target product in 97% purity by HPLC, which can serve as a useful building block for the preparation of a series of 3,4,5-substituted-7-azaindole derivatives.
https://pubs.acs.org/doi/suppl/10.1021/acs.oprd.7b00060/suppl_file/op7b00060_si_001.pdf
-Bromo-4-chloro-3-nitro-1H-pyrrolo[2,3-b]pyridine (1)(10)
Into ………….. afford 5-bromo-4-chloro-3-nitro-1H-pyrrolo[2,3-b]pyridine 1 as a tan solid (66.4 kg, 96.2 wt %, 90% yield, 96.9 A % HPLC; unreacted starting material 5: 0.99 A% HPLC; impurity 8: 0.95 A% HPLC): mp 269 °C dec; 1H NMR (300 MHz, DMSO-d6) δ 13.68 (s, 1H), 8.93 (s, 1H), 8.66 (s, 1H); 13C NMR (75 MHz, DMSO-d6) δ 146.9, 146.4, 133.9, 133.2, 12
PATENT
WO 2010118390
https://patents.google.com/patent/WO2010118390A1/und
PATENT
WO 2015027090
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015027090
PATENT
WO 2015027092
Example 1: Preparation of (i?)-5-bromo-4-(3-amino)piperidin-l-yl)-3- (cyclopropanecarboxamido)-lH-pyrrolo[2,3-&]pyridine:
[0096] Step 1 : Preparation of (i?)-5-bromo-4-(3-(/ert-butoxycarbonylamino)piperidin-l-yl)-3-nitro-lH-pyrrolo[2,3-6]pyridine:
[0097] To an inerted 10 L jacket reactor, equipped with a mechanic stirrer, a nitrogen/vacuum manifold, a thermocouple, and a condenser, were charged 2-methyl-2-butanol (3.30 L), 5-bromo-4-chloro-3-nitro-lH-pyrrolo[2,3-6]pyridine (330 g, 1.00 equiv), (R)-tert-butyl piperidin-3-ylcarbamate (456 g, 2.00 equiv), and N-methylmorpholine (115 g, 1.00 equiv). The reaction mixture was stirred at 85 °C for 48 h and cooled to 20 °C. The mixture was then washed with 15 wt % citric acid aqueous solution (3.30 kg) and water (3.30 kg). The majority of 2-methyl-2-butanol was distilled off under vacuum at 50 °C. Acetonitrile was added to bring the mixture back to its original volume. Continuous distillation was conducted until a total of 10.3 kg of acetonitrile was added. Water (3.20 kg) was slowly charged to the suspension over approximately 1 h at 55 °C. The slurry was slowly cooled to 20 °C over 4 h. The resulting solid was collected by filtration and washed with a 1 : 1 (v/v) mixture of acetonitrile and water (1.60 L). The product was dried in a vacuum oven under nitrogen at 70 °C to provide 358 g (69% yield) of (i?)-5-bromo-4-(3-(ter/-butoxycarbonylamino)piperidin-l-yl)-3-nitro-lH-pyrrolo[2,3-6]pyridine as a yellow solid. !H NMR (600 MHz, DMSO-i/6): δ 13.12 (s, 1H), 8.60 (s, 1H), 8.39 (s, 1H), 6.80 (d, J= 6.8 Hz, 1H), 3.49 (m, 1H), 3.34 (m, 2H), 3.22 (t, J = 11.2 Hz, 1H), 3.00 (t, J = 10.2 Hz, 1H), 1.88 (dd, J = 12.3, 2.8 Hz, 1H), 1.74 (m, 2H), 1.38 (m, 1H), 1.34 (s, 9H). 13C NMR (150 MHz, DMSO-<¾): δ 154.8, 148.9, 148.2, 147.9, 130.6, 128.5, 113.8, 109.6, 77.6, 54.7, 48.9, 47.3, 30.0, 28.1 (3C), 24.2. HRMS-ESI (m/z): [M + H]+ calcd for C17H23BrN504, 440.0928; found, 440.0912.
[0098] Steps 2 and 3: Preparation of (i?)-5-bromo-4-(3-(tert-butoxycarbonylamino)piperidin- 1 -yl)-3 -(cyclopropanecarboxamido)- 1 H-pyrrolo[2,3 -&]pyridine:
[0099] To an inerted 1 L pressure reactor were charged (i?)-5-bromo-4-(3-(tert-
butoxycarbonylamino)piperidin-l-yl)-3-nitro-lH-pyrrolo[2,3-6]pyridine (75.0 g, 1.00 equiv), 1% Pt + 2% V/C (11.3 g, 15 wt %), N-methylmorpholine (29.3 g, 1.70 equiv), and 2-MeTHF (750 mL). The reaction mixture was stirred at 50 °C at 5 bar of hydrogen for a minimum of 2 h. Cyclopropanecarbonyl chloride (26.7 g, 1.50 equiv) was charged into the reactor over 10 min at 15 °C. The reaction mixture was stirred at 25 °C for 1 h and filtered through Celite. The cake was washed with 2-MeTHF (150 mL). The filtrate was washed with 15 wt % aqueous ammonium chloride solution (450 mL) and water (450 mL) and then distilled in vacuo to 1/3 of it’s original volume. Toluene was added to bring the solution back to its original volume. Continuous vacuum distillation was conducted at 55 °C while adding toluene until the 2-MeTHF was below 2 wt %. The resulting solid was isolated by filtration, washed with toluene and dried in a vacuum oven at 40 °C overnight to give 69.8 g (69% corrected yield) of (i?)-5-bromo-4-(3-(tert-butoxycarbonylamino)piperidin-l-yl)-3-(cyclopropanecarboxamido)-lH-pyrrolo[2,3-6]pyridine (1 :1 toluene solvate) as an off-white solid. 1H NMR (600 MHz, THF-i 8, 4 °C): δ 10.76 (s, 1H), 9.72 (s, 1H), 8.15 (s, 1H), 7.90 (d, J = 2.4 Hz, 1H), 7.18-7.08 (m, 5H), 6.41 (d, J = 7.8 Hz, 1H), 3.82 (m, 1H), 3.60 (m, 1H), 3.44 (t, J = 10.6 Hz, 1H), 3.30 (dd, J= 10.6, 3.9 Hz, 1H), 3.03 (d, J = 10.9 Hz, 1H), 2.29 (s, 3H), 2.08 (m, 1H), 1.89 (m, 2H), 1.66 (m, 1H), 1.37 (s, 9H), 1.36 (m, 1H), 0.95-0.80 (m, 4H). 13C NMR (150 MHz, THF-ci8, 4 °C): δ 170.0, 155.8, 149.0, 147.8, 147.6, 138.4, 129.6 (2C), 128.9 (2C), 126.0, 116.6, 115.6, 111.9, 108.8, 78.5, 55.8, 50.2, 49.1, 31.8, 28.6 (3C), 26.3, 21.5, 15.8, 7.70, 7.56. HRMS-ESI (m/z): [M + H]+ calcd for C21H29BrN503, 478.1448; found, 478.1431.
[00100] Step 4: Preparation of (i?)-5-bromo-4-(3-amino)piperidin-l-yl)-3-(cyclopropanecarboxamido)- 1 H-pyrrolo [2,3 -6]pyridine :
[00101] To an inerted 1 L jacket reactor, equipped with a mechanic stirrer, a nitrogen/vacuum manifold, a thermocouple, and a condenser, were charged (i?)-5-bromo-4-(3-(tert-butoxycarbonylamino)piperidin-l-yl)-3-nitro-lH-pyrrolo[2,3-0]pyridine (1 : 1 toluene solvate) (30.0 g, 1.00 equiv), tetrahydrofuran (180 mL, 6.00 mL/g), followed by 4.5 M sulfuric acid (36.1 mL, 3.00 equiv). The reaction mixture was stirred at 50 ± 5 °C for 2 h and then cooled to 20 °C. An aqueous piperazine solution (42.4 g dissolved in 190 mL of water) was added slowly at 25 °C followed by addition of 15.0 mL of sat’d brine. The aqueous bottom layer was removed. The resulting solution was stirred at 20 °C for 5 min. Water (22.0 mL) was added. Continuous distillation was conducted at 50 °C by adjusting the feed rate of ethanol to match the distillation rate until a total of 260 mL of ethanol was added. Water (340 mL) was added at 50 °C over 1 h. The resulting solid was isolated by filtration, washed with 20% ethanol in water (2 x 60 mL) and dried in a vacuum oven at 50 °C overnight to give 16.4 g (78% corrected yield) of (i?)-5-bromo-4-(3-amino)piperidin-l-yl)-3-(cyclopropanecarboxamido)-l H-pyrrolo [2,3 -b]pyridine as a light yellow solid. (Note: The proton ( H) and carbon- 13 ( C) spectra of freebase product are very broad. Therefore, the spectra shown below are of freebase converted to a bis-HCl salt.) 1H NMR (300 MHz, DMSC ): δ 11.98 (br, 1H), 9.78 (s, 1H), 8.44 (br, 3H), 8.25 (s, 1H), 7.45 (d, J = 2.4 Hz, 1H), 3.57 (m, 1H), 3.43 (m, 1H), 3.41 (m, 1H), 3.28 (m, 1H), 3.14 (m, 1H), 2.15 (m, 1H), 1.90 (penta, J = 6.5 Hz, 1H), 1.81 (m, 1H), 1.72 (m, 1H), 1.52 (m, 1H), 0.83 (m, 4H). 13C NMR (75 MHz, DMSO- 6): 5 172.9, 149.5, 145.9, 145.1, 121.9, 114.2, 113.1, 107.8, 53.8, 51.1, 47.5, 28.6, 24.37, 14.7, 7.55, 7.45. HRMS-ESI (m/z): [M + H]+ calcd for C16H21BrN50, 378.0924; found, 378.0912.
[00102] Example 2:

[00103] Alternatively, the compound (i?)-5-bromo-4-(3-(fer/-butoxycarbonylamino)piperidin- 1 -yl)-3 -(cyclopropanecarboxamido)- 1 H-pyrrolo [2,3 -£]pyridine can be prepared from 5-bromo-4-chloro-3-nitro-lH-pyrrolo[2,3-b]pyridine and (^)-tert-butyl piperidin-3-ylcarbamate via a through process without isolating (i?)-5-bromo-4-(3-(tert-butoxycarbonylamino)piperidin-l-yl)-3-nitro-lH-pyrrolo[2,3-6]pyridine. The changes to existing procedure are shown as below: The solution of (i?)-5-bromo-4-(3-(tert-butoxycarbonylamino)piperidin- 1 -yl)-3 -nitro- 1 H-pyrrolo [2,3 -6]pyridine was hydrogenated directly in 2-methyl-2-butanol after aqueous washes with 15 wt % citric acid aqueous solution (10.0 g/g) and water (10.0 g/g). The solution concentration in 2-methyl-2-butanol was determined by HPLC weight assay.

PATENT
CHK1 is a serine/threonine kinase that regulates cell-cycle progression and is a main factor in DNA-damage response within a cell. CHK1 inhibitors have been shown to sensitize tumor cells to a variety of genotoxic agents, such as chemotherapy and radiation. U.S. Pat. No. 8,178,131 discusses a number of inhibitors of CHK1, including the compound (i?)-N-(4-(3-aminopiperidin-l-yl)-5-bromo-lH-pyrrolo[2,3-b]pyridin-3-yl)cyclopropanecarboxamide (Compound 1), which is being investigated in clinical trials for the treatment of various cancers.

Compound 1
PATENT
U.S. Patent Application, 20160200723
Example 1 Preparation of (R)-5-bromo-4-(3-amino)piperidin-1-yl)-3-(cyclopropanecarboxamido)-1H-pyrrolo[2,3-b]pyridine

Step 1: Preparation of (R)-5-bromo-4-(3-(tert-butoxycarbonylamino)piperidin-1-yl)-3-nitro-1H-pyrrolo[2,3-b]pyridine
To an inserted 10 L jacket reactor, equipped with a mechanic stirrer, a nitrogen/vacuum manifold, a thermocouple, and a condenser, were charged 2-methyl-2-butanol (3.30 L), 5-bromo-4-chloro-3-nitro-1H-pyrrolo[2,3-b]pyridine (330 g, 1.00 equiv), (R)-tert-butyl piperidin-3-ylcarbamate (456 g, 2.00 equiv), and N-methylmorpholine (115 g, 1.00 equiv). The reaction mixture was stirred at 85° C. for 48 h and cooled to 20° C. The mixture was then washed with 15 wt % citric acid aqueous solution (3.30 kg) and water (3.30 kg). The majority of 2-methyl-2-butanol was distilled off under vacuum at 50° C. Acetonitrile was added to bring the mixture back to its original volume. Continuous distillation was conducted until a total of 10.3 kg of acetonitrile was added. Water (3.20 kg) was slowly charged to the suspension over approximately 1 h at 55° C. The slurry was slowly cooled to 20° C. over 4 h. The resulting solid was collected by filtration and washed with a 1:1 (v/v) mixture of acetonitrile and water (1.60 L). The product was dried in a vacuum oven under nitrogen at 70° C. to provide 358 g (69% yield) of (R)-5-bromo-4-(3-(tert-butoxycarbonylamino)piperidin-1-yl)-3-nitro-1H-pyrrolo[2,3-b]pyridine as a yellow solid. 1H NMR (600 MHz, DMSO-d6): δ 13.12 (s, 1H), 8.60 (s, 1H), 8.39 (s, 1H), 6.80 (d, J=6.8 Hz, 1H), 3.49 (m, 1H), 3.34 (m, 2H), 3.22 (t, J=11.2 Hz, 1H), 3.00 (t, J=10.2 Hz, 1H), 1.88 (dd, J=12.3, 2.8 Hz, 1H), 1.74 (m, 2H), 1.38 (m, 1H), 1.34 (s, 9H). 13C NMR (150 MHz, DMSO-d6): δ 154.8, 148.9, 148.2, 147.9, 130.6, 128.5, 113.8, 109.6, 77.6, 54.7, 48.9, 47.3, 30.0, 28.1 (3C), 24.2. HRMS-ESI (m/z): [M+H]+ calcd for C17H23BrN5O4, 440.0928. found, 440.091
Steps 2 and 3: Preparation of (R)-5-bromo-4-(3-(tert-butoxycarbonylamino)piperidin-1-yl)-3-(cyclopropanecarboxamido)-1H-pyrrolo[2,3-b]pyridine
To an inserted 1 L pressure reactor were charged (R)-5-bromo-4-(3-(tert-butoxycarbonylamino)piperidin-1-yl)-3-nitro-1H-pyrrolo[2,3-b]pyridine (75.0 g, 1.00 equiv), 1% Pt+2% V/C (11.3 g, 15 wt %), N-methylmorpholine (29.3 g, 1.70 equiv), and 2-MeTHF (750 mL). The reaction mixture was stirred at 50° C. at 5 bar of hydrogen for a minimum of 2 h. Cyclopropanecarbonyl chloride (26.7 g, 1.50 equiv) was charged into the reactor over 10 min at 15° C. The reaction mixture was stirred at 25° C. for 1 h and filtered through Celite. The cake was washed with 2-MeTHF (150 mL). The filtrate was washed with 15 wt % aqueous ammonium chloride solution (450 mL) and water (450 mL) and then distilled in vacuo to ⅓ of it’s original volume. Toluene was added to bring the solution back to its original volume. Continuous vacuum distillation was conducted at 55° C. while adding toluene until the 2-MeTHF was below 2 wt %. The resulting solid was isolated by filtration, washed with toluene and dried in a vacuum oven at 40° C. overnight to give 69.8 g (69% corrected yield) of (R)-5-bromo-4-(3-(tert-butoxycarbonylamino)piperidin-1-yl)-3-(cyclopropanecarboxamido)-1H-pyrrolo[2,3-b]pyridine (1:1 toluene solvate) as an off-white solid. 1H NMR (600 MHz, THF-d8, 4° C.): δ 10.76 (s, 1H), 9.72 (s, 1H), 8.15 (s, 1H), 7.90 (d, J=2.4 Hz, 1H), 7.18-7.08 (m, 5H), 6.41 (d, J=7.8 Hz, 1H), 3.82 (m, 1H), 3.60 (m, 1H), 3.44 (t, J=10.6 Hz, 1H), 3.30 (dd, J=10.6, 3.9 Hz, 1H), 3.03 (d, J=10.9 Hz, 1H), 2.29 (s, 3H), 2.08 (m, 1H), 1.89 (m, 2H), 1.66 (m, 1H), 1.37 (s, 9H), 1.36 (m, 1H), 0.95-0.80 (m, 4H). 13C NMR (150 MHz, THF-d8, 4° C.): δ 170.0, 155.8, 149.0, 147.8, 147.6, 138.4, 129.6 (2C), 128.9 (2C), 126.0, 116.6, 115.6, 111.9, 108.8, 78.5, 55.8, 50.2, 49.1, 31.8, 28.6 (3C), 26.3, 21.5, 15.8, 7.70, 7.56. HRMS-ESI (m/z): [M+H]+ calcd for C21H29BrN5O3, 478.1448. found, 478.1431.
Step 4: Preparation of (R)-5-bromo-4-(3-amino)piperidin-1-yl)-3-(cyclopropanecarboxamido)-1H-pyrrolo[2,3-b]pyridine
To an inserted 1 L jacket reactor, equipped with a mechanic stirrer, a nitrogen/vacuum manifold, a thermocouple, and a condenser, were charged (R)-5-bromo-4-(3-(tert-butoxycarbonylamino)piperidin-1-yl)-3-nitro-1H-pyrrolo[2,3-b]pyridine (1:1 toluene solvate) (30.0 g, 1.00 equiv), tetrahydrofuran (180 mL, 6.00 mL/g), followed by 4.5 M sulfuric acid (36.1 mL, 3.00 equiv). The reaction mixture was stirred at 50±5° C. for 2 h and then cooled to 20° C. An aqueous piperazine solution (42.4 g dissolved in 190 mL of water) was added slowly at 25° C. followed by addition of 15.0 mL of sat′d brine. The aqueous bottom layer was removed. The resulting solution was stirred at 20° C. for 5 min. Water (22.0 mL) was added. Continuous distillation was conducted at 50° C. by adjusting the feed rate of ethanol to match the distillation rate until a total of 260 mL of ethanol was added. Water (340 mL) was added at 50° C. over 1 h. The resulting solid was isolated by filtration, washed with 20% ethanol in water (2×60 mL) and dried in a vacuum oven at 50° C. overnight to give 16.4 g (78% corrected yield) of (R)-5-bromo-4-(3-amino)piperidin-1-yl)-3-(cyclopropanecarboxamido)-1H-pyrrolo[2,3-b]pyridine as a light yellow solid. (Note: The proton (1H) and carbon-13 (13C) spectra of freebase product are very broad. Therefore, the spectra shown below are of freebase converted to a bis-HCl salt.)1H NMR (300 MHz, DMSO-d6): δ 11.98 (br, 1H), 9.78 (s, 1H), 8.44 (br, 3H), 8.25 (s, 1H), 7.45 (d, J=2.4 Hz, 1H), 3.57 (m, 1H), 3.43 (m, 1H), 3.41 (m, 1H), 3.28 (m, 1H), 3.14 (m, 1H), 2.15 (m, 1H), 1.90 (penta, J=6.5 Hz, 1H), 1.81 (m, 1H), 1.72 (m, 1H), 1.52 (m, 1H), 0.83 (m, 4H). 13C NMR (75 MHz, DMSO-d6): δ 172.9, 149.5, 145.9, 145.1, 121.9, 114.2, 113.1, 107.8, 53.8, 51.1, 47.5, 28.6, 24.37, 14.7, 7.55, 7.45. HRMS-ESI (m/z): [M+H]+ calcd for C16H21BrN5O, 378.0924. found, 378.0912.
Example 2

Alternatively, the compound (R)-5-bromo-4-(3-(tert-butoxycarbonylamino)piperidin-1-yl)-3-(cyclopropanecarboxamido)-1H-pyrrolo[2,3-b]pyridine can be prepared from 5-bromo-4-chloro-3-nitro-1H-pyrrolo[2,3-b]pyridine and (R)-tert-butyl piperidin-3-ylcarbamate via a through process without isolating (R)-5-bromo-4-(3-(tert-butoxycarbonylamino)piperidin-1-yl)-3-nitro-1H-pyrrolo[2,3-b]pyridine. The changes to existing procedure are shown as below: The solution of (R)-5-bromo-4-(3-(tert-butoxycarbonylamino)piperidin-1-yl)-3-nitro-1H-pyrrolo[2,3-b]pyridine was hydrogenated directly in 2-methyl-2-butanol after aqueous washes with 15 wt % citric acid aqueous solution (10.0 g/g) and water (10.0 g/g). The solution concentration in 2-methyl-2-butanol was determined by HPLC weight assay.
PAPER
An Efficient Through-Process for Chk1 Kinase Inhibitor GDC-0575
Chong Han*†  , Keena Green†, Kathrin Oehring‡, Arthur Meili‡, Eugen Pfeifer‡, Michelangelo Scalone‡, and Francis Gosselin†
, Keena Green†, Kathrin Oehring‡, Arthur Meili‡, Eugen Pfeifer‡, Michelangelo Scalone‡, and Francis Gosselin†
, Keena Green†, Kathrin Oehring‡, Arthur Meili‡, Eugen Pfeifer‡, Michelangelo Scalone‡, and Francis Gosselin† † Department of Small Molecule Process Chemistry, Genentech, Inc., 1 DNA Way, South San Francisco, California 94080, United States
‡ Department of Pharma Technical Development, F. Hoffmann-La Roche AG, Grenzacherstrasse 124, CH-4070 Basel, Switzerland
Abstract

We report an efficient route to prepare Chk1 kinase inhibitor GDC-0575 from 5-bromo-4-chloro-3-nitro-7-azaindole featuring a sequence of nucleophilic aromatic substitution, hydrogenative nitro-reduction, and a robust, high-yielding end-game involving deprotection–crystallization steps. The developed route was demonstrated on 10 kg scale in 30% overall yield to provide the target API in >99.8 A % HPLC purity.
(R)-5-Bromo-4-(3-amino)piperidin-1-yl)-3-(cyclopropanecarboxamido)-1H-pyrrolo[2,3-b]pyridine (GDC-0575)
To ………….. to give (R)-5-bromo-4-(3-amino)piperidin-1-yl)-3-(cyclopropanecarboxamido)-1H-pyrrolo[2,3-b]pyridine as a light yellow solid (5.1 kg, 76% yield, 99.9 A % by HPLC analysis).
Both 1H and 13C spectra of GDC-0575 freebase are very broad.
Therefore, the spectra shown below are of freebase converted to a bis-HCl salt: mp = 267 °C;
1H NMR (300 MHz, DMSO-d6): δ 11.98 (br, 1H), 9.78 (s, 1H), 8.44 (br, 3H), 8.25 (s, 1H), 7.45 (d, J = 2.4 Hz, 1H), 3.57 (m, 1H), 3.43 (m, 1H), 3.41 (m, 1H), 3.28 (m, 1H), 3.14 (m, 1H), 2.15 (m, 1H), 1.90 (penta, J = 6.5 Hz, 1H), 1.81 (m, 1H), 1.72 (m, 1H), 1.52 (m, 1H), 0.83 (m, 4H);
13C NMR (75 MHz, DMSO-d6): δ 172.9, 149.5, 145.9, 145.1, 121.9, 114.2, 113.1, 107.8, 53.8, 51.1, 47.5, 28.6, 24.37, 14.7, 7.55, 7.45;
HRMS–ESI (m/z): [M + H]+ calcd for C16H21BrN5O, 378.0924; found, 378.0912.


REFERENCES
1: Duan W, Gao L, Aguila B, Kalvala A, Otterson GA, Villalona-Calero MA. Fanconi
anemia repair pathway dysfunction, a potential therapeutic target in lung cancer.
Front Oncol. 2014 Dec 19;4:368. doi: 10.3389/fonc.2014.00368. eCollection 2014.
PubMed PMID: 25566506; PubMed Central PMCID: PMC4271581.
Publications
GDC-0575 / Cancer
07/01/2011
Oncology Research Featuring Preclinical and Clinical Cancer Therapeutics
Single-Agent Inhibition of Chk1 Is Antiproliferative in Human Cancer Cell Lines In Vitro and Inhibits Tumor Xenograft Growth In Vivo
GDC-0575 / Cancer
04/05/2011
American Association for Cancer Research Annual Meeting
Chk1 inhibition and Wee1 inhibition combine synergistically to inhibit cellular proliferation
GDC-0575 / Cancer
03/11/2011
International Symposium on Targeted Anticancer Therapies
Preclinical characterization of ARRY-575: A potent, selective, and orally bio-available small molecule inhibitor of Chk1
///////// GDC0575, GDC 0575, ARRY-575, GDC-0575, RG 7741, RO 6845979, AK 687476, ARRY 575, GDC 0575, RG 7741, PHASE 1
O=C(Nc1cnc2ncc(Br)c(c12)N3CCC[C@@H](N)C3)C4CC4
AVOID CONFUSING
 WRONG COMPD 2097938-64-0
WRONG COMPD 2097938-64-0
N ATOM MISSING IN RING
