PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Tralokinumab is a human monoclonal antibody which targets the cytokine interleukin 13,[1] and is designed for the treatment of asthma and other inflammatory diseases.[2] Tralokinumab was discovered by Cambridge Antibody Technology scientists, using Ribosome Display, as CAT-354[3] and taken through pre-clinical and early clinical development.[4] After 2007 it has been developed by MedImmune, a member of the AstraZeneca group, where it is currently in Ph3 testing for asthma and Ph2b testing for atopic dermatitis.[5][6] This makes it one of the few fully internally discovered and developed drug candidates in AstraZeneca’s late stage development pipeline.
Discovery and development
Tralokinumab (CAT-354) was discovered by Cambridge Antibody Technology scientists[7] using protein optimization based on Ribosome Display.[8] They used the extensive data sets from ribosome display to patent protect CAT-354 in a world-first of sequence-activity-relationship claims.[7] In 2004, clinical development of CAT-354 was initiated with this first study completing in 2005.[9] On 21 July 2011, MedImmune LLC initiated a Ph2b, randomized, double-blind study to evaluate the efficacy of tralokinumab in adults with asthma.[10]
In 2016, MedImmune and AstraZeneca were developing tralokinumab for asthma (Ph3) and atopic dermatitis (Ph2b) while clinical development for moderate-to-severe ulcerative colitis and idiopathic pulmonary fibrosis (IPF) have been discontinued.[9] In July of that year AstraZeneca licensed Tralokinumab to LEO Pharma for skin diseases.[11]
A phase IIb study of Tralokinumab found that treatment was associated with early and sustained improvements in atopic dermatitis symptoms and tralokinumab had an acceptable safety and tolerability profile, thereby providing evidence for targeting IL-13 in patients with atopic dermatitis.[12]
On 15 June 2017, Leo Pharma announced that they were starting phase III clinical trials with tralokinumab in atopic dermatitis.[13]
Society and culture
Legal status
On 22 April 2021, the Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Adtralza, intended for the treatment of moderate‑to‑severe atopic dermatitis.[14]
The applicant for this medicinal product is LEO Pharma A/S.
References
^ Kopf M, Bachmann MF, Marsland BJ (September 2010). “Averting inflammation by targeting the cytokine environment”. Nature Reviews. Drug Discovery. 9 (9): 703–18. doi:10.1038/nrd2805. PMID20811382. S2CID23769909.
^ Clinical trial number NCT01402986 for “A Phase 2b, Randomized, Double-blind Study to Evaluate the Efficacy of Tralokinumab in Adults With Asthma” at ClinicalTrials.gov
Immunoglobulin G4, anti-(human protein ANGPTL3 (angiopoietin-like 3)) (human monoclonal REGN1500 heavy chain), disulfide with human monoclonal REGN1500 light chain, dimer
Evinacumab is a recombinant human IgG4 monoclonal antibody targeted against angiopoietin-like protein 3 (ANGPTL3) and the first drug of its kind. The ANGPTL family of proteins serve a number of physiologic functions – including involvement in the regulation of lipid metabolism – which have made them desirable therapeutic targets in recent years.2 Loss-of-function mutations in ANGPTL3 have been noted to result in hypolipidemia and subsequent reductions in cardiovascular risk, whereas increases in function appear to be associated with cardiovascular risk, and it was these observations that provided a rationale for the development of a therapy targeted against ANGPTL3.3
In February 2021, evinacumab became the first-and-only inhibitor of ANGPTL3 to receive FDA approval after it was granted approval for the adjunctive treatment of homozygous familial hypercholesterolemia (HoFH) under the brand name “Evkeeza”.8 Evinacumab is novel in its mechanism of action compared with other lipid-lowering therapies and therefore provides a unique and synergistic therapeutic option in the treatment of HoFH.
Common side effects include nasopharyngitis (cold), influenza-like illness, dizziness, rhinorrhea (runny nose), and nausea. Serious hypersensitivity (allergic) reactions have occurred in the Evkeeza clinical trials.[2]
Evinacumab binds to the angiopoietin-like protein 3 (ANGPTL3).[2] ANGPTL3 slows the function of certain enzymes that break down fats in the body.[2] Evinacumab blocks ANGPTL3, allowing faster break down of fats that lead to high cholesterol.[2] Evinacumab was approved for medical use in the United States in February 2021.[2][3]
NAME
DOSAGE
STRENGTH
ROUTE
LABELLER
MARKETING START
MARKETING END
Evkeeza
Injection, solution, concentrate
150 mg/1mL
Intravenous
Regeneron Pharmaceuticals, Inc.
2021-02-11
Not applicable
Evkeeza
Injection, solution, concentrate
150 mg/1mL
Intravenous
Regeneron Pharmaceuticals, Inc.
2021-02-11
Not applicable
History
The effectiveness and safety of evinacumab were evaluated in a double-blind, randomized, placebo-controlled, 24-week trial enrolling 65 participants with homozygous familial hypercholesterolemia (HoFH).[2] In the trial, 43 participants received 15 mg/kg of evinacumab every four weeks and 22 participants received the placebo.[2] Participants were taking other lipid-lowering therapies as well.[2]
The primary measure of effectiveness was the percent change in low-density lipoprotein (LDL-C) from the beginning of treatment to week 24.[2] At week 24, participants receiving evinacumab had an average 47% decrease in LDL-C while participants on the placebo had an average 2% increase.[2]
The U.S. Food and Drug Administration approved Ebanga (Ansuvimab-zykl), a human monoclonal antibody, for the treatment for Zaire ebolavirus (Ebolavirus) infection in adults and children. Ebanga blocks binding of the virus to the cell receptor, preventing its entry into the cell.
Zaire ebolavirus is one of four Ebolavirus species that can cause a potentially fatal human disease. It is transmitted through blood, body fluids, and tissues of infected people or wild animals, and through surfaces and materials, such as bedding and clothing, contaminated with these fluids. Individuals who care for people with the disease, including health care workers who do not use correct infection control precautions, are at the highest risk for infection.
During an Ebola outbreak in the Democratic Republic of the Congo (DRC) in 2018-2019, Ebanga was evaluated in a clinical trial (the PALM trial). The PALM trial was led by the U.S. National Institutes of Health and the DRC’s Institut National de Recherche Biomédicale with contributions from several other international organizations and agencies.
In the PALM trial, the safety and efficacy of Ebanga was evaluated in a multi-center, open-label, randomized controlled trial. 174 participants (120 adults and 54 pediatric patients) with confirmed Ebolavirus infection received Ebanga intravenously as a single 50 mg/kg infusion and 168 participants (135 adults and 33 pediatric patients) received an investigational control. The primary efficacy endpoint was 28-day mortality. The primary analysis population was all patients who were randomized and concurrently eligible to receive either Ebanga or the investigational control during the same time period of the trial. Of the 174 patients who received Ebanga, 35.1% died after 28 days, compared to 49.4% of the 168 patients who received a control.
The most common symptoms experienced while receiving Ebanga include: fever, tachycardia (fast heart rate), diarrhea, vomiting, hypotension (low blood pressure), tachypnea (fast breathing) and chills; however, these are also common symptoms of Ebolavirus infection. Hypersensitivity, including infusion-related events, can occur in patients taking Ebanga, and treatment should be discontinued in the event of a hypersensitivity reaction.
Patients who receive Ebanga should avoid the concurrent administration of a live virus vaccine against Ebolavirus. There is the potential for Ebanga to inhibit replication of a live vaccine virus and possibly reduce the efficacy of this vaccine.
FDA granted the approval to Ridgeback Biotherapeutics, LP.
Ansuvimab, sold under the brand name Ebanga, is a monoclonal antibody medication for the treatment of Zaire ebolavirus (Ebolavirus) infection.[1][2]
The most common symptoms include fever, tachycardia (fast heart rate), diarrhea, vomiting, hypotension (low blood pressure), tachypnea (fast breathing) and chills; however, these are also common symptoms of Ebolavirus infection.[1]
Ansuvimab was approved for medical use in the United States in December 2020.[1][2]
Ansuvimab is a monoclonal antibody therapy that is infused intravenously into patients with Ebola virus disease. Ansuvimab is a neutralizing antibody,[3] meaning it binds to a protein on the surface of Ebola virus that is required to infect cells. Specifically, ansuvimab neutralizes infection by binding to a region of the Ebola virus envelope glycoprotein that, in the absence of ansuvimab, would interact with virus’s cell receptor protein, Niemann-Pick C1 (NPC1).[6][7][8] This “competition” by ansuvimab prevents Ebola virus from binding to NPC1 and “neutralizes” the virus’s ability to infect the targeted cell.[6]
Effector function
Antibodies have antigen-binding fragment (Fab) regions and constant fragment (Fc) regions. The Neutralization of virus infection occurs when the Fab regions of antibodies binds to virus antigen(s) in a manner that blocks infection. Antibodies are also able to “kill” virus particles directly and/or kill infected cells using antibody-mediated “effector functions” such as opsonization, complement-dependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity and antibody-dependent phagocytosis. These effector functions are contained in the Fc region of antibodies, but is also dependent on binding of the Fab region to antigen. Effector functions also require the use of complement proteins in serum or Fc-receptor on cell membranes. Ansuvimab has been found to be capable of killing cells by antibody-dependent cell-mediated cytotoxicity.[3] Other functional killing tests have not been performed.
Ansuvimab has also shown success with lowering the mortality rate from ~70% to about 34%. In August 2019, Congolese health authorities, the World Health Organization, and the U.S. National Institutes of Health promoted the use of ansuvimab, alongside REGN-EB3, a similar Regeneron-produced monoclonal antibody treatment, over other treatments yielding higher mortality rates, after ending clinical trials during the outbreak.[13][14]
In an experiment described in the 2016 paper, rhesus macaques were infected with Ebola virus and treated with a combination of ansuvimab and another antibody isolated from the same subject, mAb100. Three doses of the combination were given once a day starting 1 day after the animals were infected. The control animal died and the treated animals all survived.[3]
Ansuvimab monotherapy
In a second experiment described in the 2016 paper, rhesus macaques were infected with Ebola virus and only treated with ansuvimab. Three doses of ansuvimab were given once a day starting 1 day or 5 days after the animals were infected. The control animals died and the treated animals all survived.[3] Unpublished data referred to in a publication of the 2018 Phase I clinical trial results of ansuvimab, reported that a single infusion of ansuvimab provided full protection of rhesus macaques and was the basis of the dosing used for human studies.[5][4]
Experimental use in the Democratic Republic of Congo
During the 2018 Équateur province Ebola outbreak, ansuvimab was requested by the Democratic Republic of Congo (DRC) Ministry of Public Health. Ansuvimab was approved for compassionate use by the World Health OrganizationMEURI ethical protocol and at DRC ethics board. Ansuvimab was sent along with other therapeutic agents to the outbreak sites.[19][20][11] However, the outbreak came to a conclusion before any therapeutic agents were given to patients.[11]
Approximately one month following the conclusion of the Équateur province outbreak, a distinct outbreak was noted in Kivu in the DRC (2018–20 Kivu Ebola outbreak). Once again, ansuvimab received approval for compassionate use by WHO MEURI and DRC ethic boards and has been given to many patients under these protocols.[11] In November 2018, the Pamoja Tulinde Maisha (PALM [together save lives]) open-label randomized clinical control trial was begun at multiple treatment units testing ansuvimab, REGN-EB3 and remdesivir to ZMapp. Despite the difficulty of running a clinical trial in a conflict zone, investigators have enrolled 681 patients towards their goal of 725. An interim analysis by the Data Safety and Monitoring Board (DSMB) of the first 499 patient found that ansuvimab and REGN-EB3 were superior to the comparator ZMapp. Overall mortality of patients in the ZMapp and remdesivir groups were 49% and 53% compared to 34% and 29% for ansuvimab and REGN-EB3. When looking at patients who arrived early after disease symptoms appeared, survival was 89% for ansuvimab and 94% for REGN-EB3. While the study was not powered to determine whether there is any difference between REGN-EB3 and ansuvimab, the survival difference between those two therapies and ZMapp was significant. This led to the DSMB halting the study and PALM investigators dropping the remdesivir and ZMapp arms from the clinical trial. All patients in the outbreak who elect to participate in the trial will now be given either ansuvimab or REGN-EB3.[21][22][13][12]
On December 21, 2020, the US Food and Drug Administration approved Ebanga (ansuvimab-zykl) for the treatment for Zaire ebolavirus (Ebolavirus) infection in adults and children. Ebanga had been granted US Orphan Drug designation and Breakthrough Therapy designations. Ansuvimab is a human IgG1 monoclonal antibody that binds and neutralizes the virus.
The safety and efficacy of Ebanga were evaluated in the multi-center, open-label, randomized controlled PALM trial. In this study, 174 participants (120 adults and 54 pediatric patients) with confirmed Ebolavirus infection received Ebanga intravenously as a single 50 mg/kg infusion and 168 participants (135 adults and 33 pediatric patients) received an investigational control. The primary efficacy endpoint was 28-day mortality. Of the 174 patients who received Ebanga, 35.1% died after 28 days, compared to 49.4% of the 168 patients who received a control.
Ebanga is the 12th antibody therapeutic to be granted a first approval in the US or EU during 2020.
^ Jump up to:abcdef Clinical trial number NCT03478891 for “Safety and Pharmacokinetics of a Human Monoclonal Antibody, VRC-EBOMAB092-00-AB (MAb114), Administered Intravenously to Healthy Adults” at ClinicalTrials.gov
Immunoglobulin G1, anti-(calcitonin gene-related peptide) (human-oryctolagus cuniculus monoclonal ALD403 heavy chain), disulfide with human-oryctolagus cuniculus monoclonal ALD403 kappa-chain, dimer
Approved 2020 fda
ALD403, UNII-8202AY8I7H
Humanized anti-calcitonin gene-related peptide (CGRP) IgG1 antibody for the treatment of migraine.
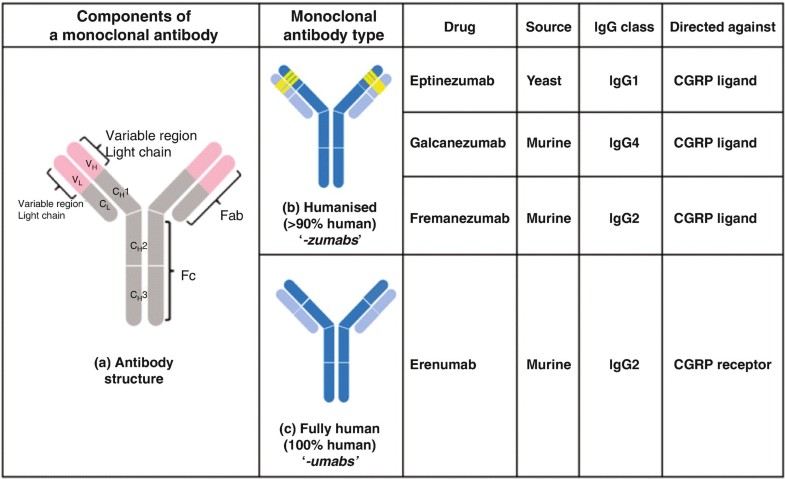
Eptinezumab, sold under the brand name Vyepti, is a medication for the preventive treatment of migraine in adults.[2] It is a monoclonal antibody that targets calcitonin gene-related peptides (CGRP) alpha and beta.[3][4] It is administered by intravenous infusion every three months.[2]
Eeptinezumab-jjmr was approved for use in the United States in February 2020.[5]
^Dodick DW, Goadsby PJ, Silberstein SD, Lipton RB, Olesen J, Ashina M, et al. (November 2014). “Safety and efficacy of ALD403, an antibody to calcitonin gene-related peptide, for the prevention of frequent episodic migraine: a randomised, double-blind, placebo-controlled, exploratory phase 2 trial”. The Lancet. Neurology. 13 (11): 1100–1107. doi:10.1016/S1474-4422(14)70209-1. PMID25297013.
Alder BioPharmaceuticals has submitted a biologics license application (BLA) for eptinezumab, a humanized IgG1 monoclonal antibody that targets calcitonin gene-related peptide (CGRP), for migraine prevention. If the US Food and Drug Administration grants approval, Alder will be on track to launch the drug in Q1 2020. The BLA included data from the PROMISE 1 and PROMISE 2 studies, which evaluated the effects of eptinezumab in episodic migraine patients (n=888) or chronic migraine patients (n=1,072), respectively. In PROMISE 1, the primary and key secondary endpoints were met, and the safety and tolerability were similar to placebo, while in PROMISE 2, the primary and all key secondary endpoints were met, and the safety and tolerability was consistent with earlier eptinezumab studies.
Alder announced one-year results from the PROMISE 1 studyin June 2018, which indicated that, following the first quarterly infusion, episodic migraine patients treated with 300 mg eptinezumab experienced 4.3 fewer monthly migraine days (MMDs) from a baseline of 8 MMDs, compared to 3.2 fewer MMDs for placebo from baseline (p= 0.0001). At one year after the third and fourth quarterly infusions, patients treated with 300 mg eptinezumab experienced further gains in efficacy, with a reduction of 5.2 fewer MMDs compared to 4.0 fewer MMDs for placebo-treated patients. In addition, ~31% of episodic migraine patients achieved, on average per month, 100% reduction of migraine days from baseline compared to ~ 21% for placebo. New 6-month results from the PROMISE 2 study were also released in June 2018. These results indicated that, after the first quarterly infusion, chronic migraine patients dosed with 300 mg of eptinezumab experienced 8.2 fewer MMDs, from a baseline of 16 MMDs, compared to 5.6 fewer MMDs for placebo from baseline (p <.0001). A further reduction in MMDs was seen following a second infusion; 8.8 fewer MMDs for patients dosed with 300 mg compared to 6.2 fewer MMDs for those with placebo. In addition, ~ 21% of chronic migraine patients achieved, on average, 100% reduction of MMDs from baseline compared to 9% for placebo after two quarterly infusions of 300 mg of eptinezumab.
Tepezza (teprotumumab-trbw) is a fully human monoclonal antibody (mAb) and a targeted inhibitor of the insulin-like growth factor 1 receptor (IGF-1R) for the treatment of active thyroid eye disease (TED).
FDA Approves Tepezza (teprotumumab-trbw) for the Treatment of Thyroid Eye Disease (TED) – January 21, 2020
Today, the U.S. Food and Drug Administration (FDA) approved Tepezza (teprotumumab-trbw) for the treatment of adults with thyroid eye disease, a rare condition where the muscles and fatty tissues behind the eye become inflamed, causing the eyes to be pushed forward and bulge outwards (proptosis). Today’s approval represents the first drug approved for the treatment of thyroid eye disease.
“Today’s approval marks an important milestone for the treatment of thyroid eye disease. Currently, there are very limited treatment options for this potentially debilitating disease. This treatment has the potential to alter the course of the disease, potentially sparing patients from needing multiple invasive surgeries by providing an alternative, non surgical treatment option,” said Wiley Chambers, M.D., deputy director of the Division of Transplant and Ophthalmology Products in the FDA’s Center for Drug Evaluation and Research. “Additionally, thyroid eye disease is a rare disease that impacts a small percentage of the population, and for a variety of reasons, treatments for rare diseases are often unavailable. This approval represents important progress in the approval of effective treatments for rare diseases, such as thyroid eye disease.”
Thyroid eye disease is associated with the outward bulging of the eye that can cause a variety of symptoms such as eye pain, double vision, light sensitivity or difficulty closing the eye. This disease impacts a relatively small number of Americans, with more women than men affected. Although this condition impacts relatively few individuals, thyroid eye disease can be incapacitating. For example, the troubling ocular symptoms can lead to the progressive inability of people with thyroid eye disease to perform important daily activities, such as driving or working.
Tepezza was approved based on the results of two studies (Study 1 and 2) consisting of a total of 170 patients with active thyroid eye disease who were randomized to either receive Tepezza or a placebo. Of the patients who were administered Tepezza, 71% in Study 1 and 83% in Study 2 demonstrated a greater than 2 millimeter reduction in proptosis (eye protrusion) as compared to 20% and 10% of subjects who received placebo, respectively.
The most common adverse reactions observed in patients treated with Tepezza are muscle spasm, nausea, alopecia (hair loss), diarrhea, fatigue, hyperglycemia (high blood sugar), hearing loss, dry skin, dysgeusia (altered sense of taste) and headache. Tepezza should not be used if pregnant, and women of child-bearing potential should have their pregnancy status verified prior to beginning treatment and should be counseled on pregnancy prevention during treatment and for 6 months following the last dose of Tepezza.
The FDA granted this application Priority Review, in addition to Fast Track and Breakthrough Therapy Designation. Additionally, Tepezza received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases or conditions. Development of this product was also in part supported by the FDA Orphan Products Grants Program, which provides grants for clinical studies on safety and efficacy of products for use in rare diseases or conditions.
The FDA granted the approval of Tepezza to Horizon Therapeutics Ireland DAC.
Teprotumumab (RG-1507), sold under the brand name Tepezza, is a medication used for the treatment of adults with thyroid eye disease, a rare condition where the muscles and fatty tissues behind the eye become inflamed, causing the eyes to be pushed forward and bulge outwards (proptosis).[1]
The most common adverse reactions observed in people treated with teprotumumab-trbw are muscle spasm, nausea, alopecia (hair loss), diarrhea, fatigue, hyperglycemia (high blood sugar), hearing loss, dry skin, dysgeusia (altered sense of taste) and headache.[1] Teprotumumab-trbw should not be used if pregnant, and women of child-bearing potential should have their pregnancy status verified prior to beginning treatment and should be counseled on pregnancy prevention during treatment and for six months following the last dose of teprotumumab-trbw.[1]
It is a human monoclonal antibody developed by Genmab and Roche. It binds to IGF-1R.
Teprotumumab was first investigated for the treatment of solid and hematologic tumors, including breast cancer, Hodgkin’s and non-Hodgkin’s lymphoma, non-small cell lung cancer and sarcoma.[2][3] Although results of phase I and early phase II trials showed promise, research for these indications were discontinued in 2009 by Roche. Phase II trials still in progress were allowed to complete, as the development was halted due to business prioritization rather than safety concerns.
Teprotumumab was subsequently licensed to River Vision Development Corporation in 2012 for research in the treatment of ophthalmic conditions. Horizon Pharma (now Horizon Therapeutics, from hereon Horizon) acquired RVDC in 2017, and will continue clinical trials.[4] It is in phase III trials for Graves’ ophthalmopathy (also known as thyroid eye disease (TED)) and phase I for diabetic macular edema.[5] It was granted Breakthrough Therapy, Orphan Drug Status and Fast Track designations by the FDA for Graves’ ophthalmopathy.[6]
In a multicenter randomized trial in patients with active Graves’ ophthalmopathy Teprotumumab was more effective than placebo in reducing the clinical activity score and proptosis.[7] In February 2019 Horizon announced results from a phase 3 confirmatory trial evaluating teprotumumab for the treatment of active thyroid eye disease (TED). The study met its primary endpoint, showing more patients treated with teprotumumab compared with placebo had a meaningful improvement in proptosis, or bulging of the eye: 82.9 percent of teprotumumab patients compared to 9.5 percent of placebo patients achieved the primary endpoint of a 2 mm or more reduction in proptosis (p<0.001). Proptosis is the main cause of morbidity in TED. All secondary endpoints were also met and the safety profile was consistent with the phase 2 study of teprotumumab in TED.[8] On 10th of July 2019 Horizon submitted a Biologics License Application (BLA) to the FDA for teprotumumab for the Treatment of Active Thyroid Eye Disease (TED). Horizon requested priority review for the application – if so granted (FDA has a 60-day review period to decide) it would result in a max. 6 month review process.[9]
Teprotumumab-trbw was approved for use in the United States in January 2020, for the treatment of adults with thyroid eye disease.[1]
Teprotumumab-trbw was approved based on the results of two studies (Study 1 and 2) consisting of a total of 170 patients with active thyroid eye disease who were randomized to either receive teprotumumab-trbw or a placebo.[1] Of the subjects who were administered Tepezza, 71% in Study 1 and 83% in Study 2 demonstrated a greater than two millimeter reduction in proptosis (eye protrusion) as compared to 20% and 10% of subjects who received placebo, respectively.[1]
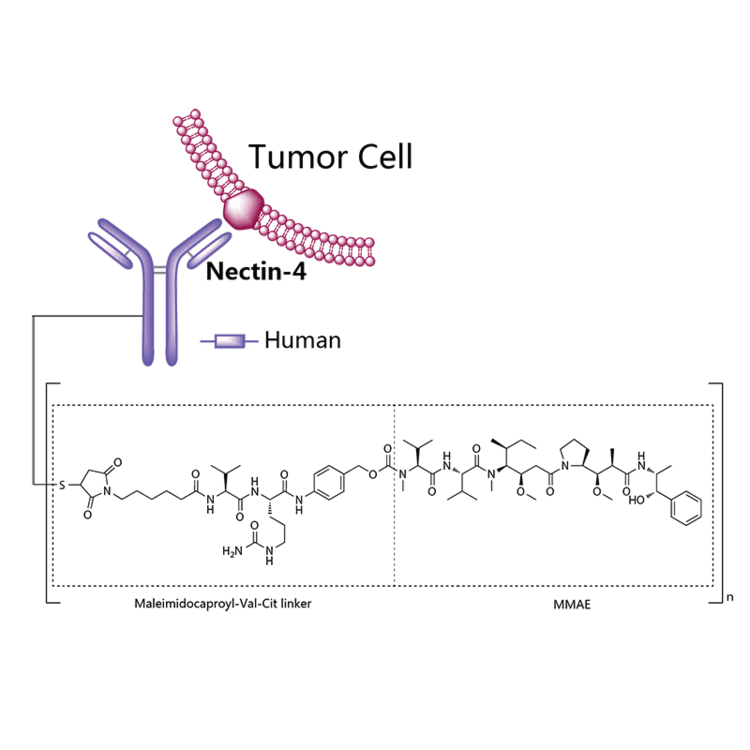
Enfortumab vedotin is an antibody-drug conjugate used in the treatment of patients with advanced, treatment-resistant urothelial cancers.3 It is comprised of a fully human monoclonal antibody targeted against Nectin-4 and a microtubule-disrupting chemotherapeutic agent, monomethyl auristatin E (MMAE), joined by a protease-cleavable link.3 It is similar to brentuximab vedotin, another antibody conjugated with MMAE that targets CD-30 instead of Nectin-4.
The clinical development of enfortumab vedotin was the result of a collaboration between Astellas Pharma and Seattle Genetics2 and it was first approved for use in the United States in December 2019 under the brand name PadcevTM.3
The most common side effects for patients taking enfortumab vedotin were fatigue, peripheral neuropathy (nerve damage resulting in tingling or numbness), decreased appetite, rash, alopecia (hair loss), nausea, altered taste, diarrhea, dry eye, pruritis (itching) and dry skin. [4]Enfortumab vedotin[1] (AGS-22M6E) is an antibody-drug conjugate[2] designed for the treatment of cancer expressing Nectin-4.[3]Enfortumab refers to the monoclonal antibody part, and vedotin refers to the payload drug (MMAE) and the linker.
The fully humanized antibody was created by scientists at Agensys (part of Astellas) using Xenomice from Amgen; the linker technology holding the antibody and the toxin together was provided by and licensed from Seattle Genetics.[5]
Results of a phase I clinical trial were reported in 2016.[2]
In December 2019, enfortumab vedotin-ejfv was approved in the United States for the treatment of adult patients with locally advanced or metastatic urothelial cancer who have previously received a programmed death receptor-1 (PD-1) or programmed death ligand 1 (PD-L1) inhibitor and a platinum-containing chemotherapy.[4]
Enfortumab vedotin was approved based on the results of a clinical trial that enrolled 125 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy.[4] The overall response rate, reflecting the percentage of patients who had a certain amount of tumor shrinkage, was 44%, with 12% having a complete response and 32% having a partial response.[4] The median duration of response was 7.6 months.[4]
Enfortumab vedotin is indicated for the treatment of adult patients with locally advanced or metastatic urothelial cancer who have previously received a programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor, and a platinum-containing chemotherapy in the neoadjuvant/adjuvant, locally advanced, or metastatic setting.3
Enfortumab vedotin is an anti-cancer agent that destroys tumor cells by inhibiting their ability to replicate.3 Patients with moderate to severe hepatic impairment should not use enfortumab vedotin – though it has not been studied in this population, other MMAE-containing antibody-drug conjugates have demonstrated increased rates of adverse effects in patients with moderate-severe hepatic impairment.3 Enfortumab vedotin may also cause significant hyperglycemia leading, in some cases, to diabetic ketoacidosis, and should not be administered to patients with a blood glucose level >250 mg/dl.3
Mechanism of action
Enfortumab vedotin is an antibody-drug conjugate comprised of multiple components.3 It contains a fully human monoclonal antibody directed against Nectin-4, an extracellular adhesion protein which is highly expressed in urothelial cancers,1 attached to a chemotherapeutic microtubule-disrupting agent, monomethyl auristatin E (MMAE). These two components are joined via a protease-cleavable linker. Enfortumab vedotin binds to cells expressing Nectin-4 and the resulting enfortumab-Nectin-4 complex is internalized into the cell. Once inside the cell, MMAE is released from enfortumab vedotin via proteolytic cleavage and goes on to disrupt the microtubule network within the cell, arresting the cell cycle and ultimately inducing apoptosis.3
PATENT
WO 2016176089
WO 2016138034
WO 2017186928
WO 2017180587
WO 2017200492
US 20170056504
PAPER
Cancer Research (2016), 76(10), 3003-3013.
General References
Hanna KS: Clinical Overview of Enfortumab Vedotin in the Management of Locally Advanced or Metastatic Urothelial Carcinoma. Drugs. 2019 Dec 10. pii: 10.1007/s40265-019-01241-7. doi: 10.1007/s40265-019-01241-7. [PubMed:31823332]
McGregor BA, Sonpavde G: Enfortumab Vedotin, a fully human monoclonal antibody against Nectin 4 conjugated to monomethyl auristatin E for metastatic urothelial Carcinoma. Expert Opin Investig Drugs. 2019 Oct;28(10):821-826. doi: 10.1080/13543784.2019.1667332. Epub 2019 Sep 17. [PubMed:31526130]
FDA Approved Drug Products: Padcev (enfortumab vedotin-ejfv) for IV injection [Link]
PADCEV™
(enfortumab vedotin-ejfv) for Injection, for Intravenous Use
DESCRIPTION
Enfortumab vedotin-ejfv is a Nectin-4 directed antibody-drug conjugate (ADC) comprised of a fully human anti-Nectin-4 IgG1 kappa monoclonal antibody (AGS-22C3) conjugated to the small molecule microtubule disrupting agent, monomethyl auristatin E (MMAE) via a protease-cleavable maleimidocaproyl valine-citrulline (vc) linker (SGD-1006). Conjugation takes place on cysteine residues that comprise the interchain disulfide bonds of the antibody to yield a product with a drug-to-antibody ratio of approximately 3.8:1. The molecular weight is approximately 152 kDa.
Figure 1: Structural Formula
Approximately 4 molecules of MMAE are attached to each antibody molecule. Enfortumab vedotin-ejfv is produced by chemical conjugation of the antibody and small molecule components. The antibody is produced by mammalian (Chinese hamster ovary) cells and the small molecule components are produced by chemical synthesis.
PADCEV (enfortumab vedotin-ejfv) for injection is provided as a sterile, preservative-free, white to off-white lyophilized powder in single-dose vials for intravenous use. PADCEV is supplied as a 20 mg per vial and a 30 mg per vial and requires reconstitution with Sterile Water for Injection, USP, (2.3 mL and 3.3 mL, respectively) resulting in a clear to slightly opalescent, colorless to slightly yellow solution with a final concentration of 10 mg/mL [see DOSAGE AND ADMINISTRATION]. After reconstitution, each vial allows the withdrawal of 2 mL (20 mg) and 3 mL (30 mg). Each mL of reconstituted solution contains 10 mg of enfortumab vedotin-ejfv, histidine (1.4 mg), histidine hydrochloride monohydrate (2.31 mg), polysorbate 20 (0.2 mg) and trehalose dihydrate (55 mg) with a pH of 6.0.
///////////////Enfortumab vedotin, AGS-22M6E, エンホルツマブベドチン (遺伝子組換え) , protein Based Therapies, Monoclonal antibody, mAb, FDA 2019
Romosozumab was originally discovered by Chiroscience,[2] which was acquired by Celltech (now owned by UCB).[3] Celltech entered in a partnership with Amgen in 2002 for the product’s development.[4]
In 2016 results from 12 months of a clinical study were reported.[5]
Some results from the FRAME[6] and ARCH clinical studies were reported on in 2017.[7]
Japan’s Ministry of Health, Labor and Welfare has granted a marketing authorization for romosozumab (EVENITY)for the treatment of osteoporosis in patients at high risk of fracture. Developed by Amgen and UCB, romosozumab is a humanized IgG2 monoclonal antibody that targets sclerostin. The approval in Japan is based on results from the Phase 3 FRAME and BRIDGE studies, which included 7,180 postmenopausal women with osteoporosis and 245 men with osteoporosis, respectively.
A biologics license application (BLA) for romosozumab as a treatment of osteoporosis in postmenopausal women at high risk for fracture was submitted to the U.S. Food and Drug Administration (FDA) in July 2016, but additional safety and efficacy data was requested in the FDA’s complete response letter, as announced by Amgen and UCB in July 2017. In July 2018, Amgen and UCB announced that the BLA had been resubmitted. In addition to data from early-stage clinical studies, the original BLA included data from the Phase 3 FRAME study. The resubmitted BLA includes results from the more recent Phase 3 ARCH study, an alendronate-active comparator trial including 4,093 postmenopausal women with osteoporosis who experienced a fracture, and the Phase 3 BRIDGE study. The FDA’s Bone, Reproductive and Urologic Drugs Advisory Committee is scheduled to review data supporting the BLA for romosozumab at a meeting on January 16, 2019.
The European Medicines Agency is also currently reviewing a marketing application for romosozumab.
Commercial production of cell culture-derived products (for example, protein-based products, such as monoclonal antibodies (mAbs)), requires optimization of cell culture parameters in order for the cells to produce enough product to meet clinical and commercial demands. However, when cell culture parameters are optimized for improving productivity of a protein product, it is also necessary to maintain desired quality specifications of the product such as glycosylation profile, aggregate levels, charge heterogeneity, and amino acid sequence integrity (Li, et al., 2010 , mAbs., 2(5):466-477).
For instance, an increase of over 20% volumetric titer results in a significant improvement in large-scale monoclonal antibody production economics. Additionally, the ability to control the glycan forms of proteins produced in cell culture is important. Glycan species have been shown to significantly influence pharmacokinetics (PK) and pharmacodynamics (PD) of therapeutic proteins such as mAbs. Moreover, the ability to modulate the relative percentage of various glycan species can have drastic results over the behavior of a protein in vivo. For example, increased mannose-5-N-acetylglycosamine-2 (“Man5”) and other high-mannose glycan species have been shown to decrease mAb in vivo half-life (Liu, 2015 , J Pharm Sci., 104(6):1866-84; Goetze et al., 2011 , Glycobiology, 21(7):949-59; and Kanda et al. 2007 , Glycobiology, 17(1):104-18). On the other hand, glycosylated mAbs with mannose-3-N-acetylglycosamine-4 (“G0”) glycan species have been shown to impact antibody dependent cellular cytotoxicity (ADCC).
Bioreactors have been successfully utilized for the cell-based production of therapeutic proteins using fed-batch, immobilized, perfusion and continuous modes. Strategies, such as the use of temperature, media formulation, including the addition of growth inhibitors, autocrine factors or cyclic mononucleotides, and hyperstimulation by osmolarity stress, have been used to enhance protein production by cells in culture. To the extent that they have worked at all, these approaches have shown only marginal success.
As such, there is a particular need for improved compositions for use in cell culture for the production of medically or industrially useful products, such as antibodies. Ideally, such compositions and methods for utilizing the same would result in higher titers, modulated (e.g. decreased) high and low molecular weight species, as well as a more favorable glycosylation profile of the derived products in cell culture.
Throughout this specification, various patents, patent applications and other types of publications (e.g., journal articles, electronic database entries, etc.) are referenced. The disclosure of all patents, patent applications, and other publications cited herein are hereby incorporated by reference in their entirety for all purposes.
Lanadelumab (INN) (alternative identifier DX-2930[1]) is a human monoclonal antibody (class IgG1 kappa)[2] that targets plasma kallikrein (pKal)[1] in order to promote prevention of angioedema in patients with hereditary angioedema.[3][4] In phase 1 clinical trialsLanadelumab was well tolerated and was reported to reduce cleavage of kininogen in the plasma of patients with hereditary angioedeman and decrease the number of patients experiencing attacks of angioedema.[1][5][6][7] As of 2017 ongoing trials for Lanadelumab include two phase 3 studies focused on investigating the utility of Lanadelumab in preventing of acute angioedema attacks in hereditary angioedema patients[8][9]
^ Jump up to:abcBanerji, Aleena; Busse, Paula; Shennak, Mustafa; Lumry, William; Davis-Lorton, Mark; Wedner, Henry J.; Jacobs, Joshua; Baker, James; Bernstein, Jonathan A. (2017-02-23). “Inhibiting Plasma Kallikrein for Hereditary Angioedema Prophylaxis”. The New England Journal of Medicine. 376 (8): 717–728. doi:10.1056/NEJMoa1605767. ISSN1533-4406. PMID28225674.
“DRUG APPROVALS INTERNATIONAL” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
The U.S. Food and Drug Administration today approved Fulphila (pegfilgrastim-jmdb) as the first biosimilar to Neulasta (pegfilgrastim) to decrease the chance of infection as suggested by febrile neutropenia (fever, often with other signs of infection, associated with an abnormally low number of infection-fighting white blood cells), in patients with non-myeloid (non-bone marrow) cancer who are receiving myelosuppressive chemotherapy that has a clinically significant incidence of febrile neutropenia.
The U.S. Food and Drug Administration today approved Fulphila (pegfilgrastim-jmdb) as the first biosimilar to Neulasta (pegfilgrastim) to decrease the chance of infection as suggested by febrile neutropenia (fever, often with other signs of infection, associated with an abnormally low number of infection-fighting white blood cells), in patients with non-myeloid (non-bone marrow) cancer who are receiving myelosuppressive chemotherapy that has a clinically significant incidence of febrile neutropenia.
“Bringing new biosimilars to patients is a top priority for the FDA, and a key part of our efforts to help promote competition that can reduce drug costs and promote access,” said FDA Commissioner Scott Gottlieb, M.D. “We’ll continue to prioritize reviews of these products to help ensure that biosimilar medications are brought to the market efficiently and through a process that makes certain that these new medicines meet the FDA’s rigorous standard for approval. This summer, we’ll release a comprehensive new plan to advance new policy efforts that promote biosimilar product development. Biologics represent some of the most clinically important, but also costliest products that patients use to promote their health. We want to make sure that the pathway for developing biosimilar versions of approved biologics is efficient and effective, so that patients benefit from competition to existing biologics once lawful intellectual property has lapsed on these products.”
Biological products are generally derived from a living organism and can come from many sources, such as humans, animals, microorganisms or yeast. A biosimilar is a biological product that is approved based on data showing that it is highly similar to a biological product already approved by the FDA (reference product) and has no clinically meaningful differences in terms of safety, purity and potency (i.e., safety and effectiveness) from the reference product, in addition to meeting other criteria specified by law.
The FDA’s approval of Fulphila is based on review of evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamic data, clinical immunogenicity data, and other clinical safety and effectiveness data that demonstrates Fulphila is biosimilar to Neulasta. Fulphila has been approved as a biosimilar, not as an interchangeable product.
The most common side effects of Fulphila are bone pain and pain in extremities. Patients with a history of serious allergic reactions to human granulocyte colony-stimulating factors such as pegfilgrastim or filgrastim products should not take Fulphila.
Serious side effects from treatment with Fulphila include rupture of the spleen, acute respiratory distress syndrome, serious allergic reactions including anaphylaxis, acute inflammation of the kidney (glomerulonephritis), an abnormally high level of white blood cells (leukocytosis), capillary leak syndrome and the potential for tumor growth. Fatal sickle cell crises have occurred.
The FDA granted approval of Fulphila to Mylan GmbH.
Crysvita (burosumab-twza) is a fibroblast growth factor 23 (FGF23) blocking antibody.
This drug is indicated for the treatment of X-linked hypophosphatemia with radiological evidence of bone disease in children of 1 year of age and older and adolescents with growing skeletons [4].
Burosumab (INN, trade name Crysvita) known as KRN23 is a human monoclonal antibody designed for the treatment of X-linked hypophosphatemia.[1][2][3] Burosumab was approved by the FDA for its intended purpose, in patients aged 1 year and older, on 17 April 2018.[4] The FDA approval fell under both the breakthrough therapy and orphan drug designations.[4]
This drug was developed by Ultragenyx and is in a collaborative license agreement with Kyowa Hakko Kirin.[5]
Burosumab (KRN23) is an entirely human monoclonal IgG1 antibody that binds excess fibroblast growth factor 23 (FGF23) and has been successfully tested in clinical trials in children with X-linked hypophosphatemic rickets [1].
The U.S. Food and Drug Administration approved Crysvita (burosumab) in April 2018. This is the first drug approved to treat adults and children ages 1 year and older with X-linked hypophosphatemia (XLH), which is a rare, inherited form of rickets. X-linked hypophosphatemia causes low circulating levels of phosphorus in the blood. It causes impaired bone growth and development in children and adolescents and issues with bone mineralization throughout a patient’s life [3].
XLH is a serious disease which affects about 3,000 children and 12,000 adults in the United States. Most children with XLH suffer from bowed or bent legs, short stature, bone pain and severe dental pain. Some adults with this condition suffer from persistent, unrelenting discomfort and complications, such as joint pain, impaired mobility, tooth abscesses and hearing loss [3]
Crysvita is specifically indicated for the treatment of X-linked hypophosphatemia (XLH) in adult and pediatric patients 1 year of age and older.
Crysvita is supplied as a subcutaneous injection. The recommended starting dose for pediatrics is 0.8 mg/kg of body weight, rounded to the nearest 10 mg, administered every two weeks. The minimum starting dose is 10 mg up to a maximum dose of 90 mg. After initiation of treatment with Crysvita, measure fasting serum phosphorus every 4 weeks for the first 3 months of treatment, and thereafter as appropriate. If serum phosphorus is above the lower limit of the reference range for age and below 5 mg/dL, continue treatment with the same dose. Follow dose adjustment schedule per the drug label. The recommended dose regimen in adults is 1 mg/kg body weight, rounded to the nearest 10 mg up to a maximum dose of 90 mg, administered every four weeks. After initiation of treatment with Crysvita, assess fasting serum phosphorus on a monthly basis, measured 2 weeks post-dose, for the first 3 months of treatment, and thereafter as appropriate. If serum phosphorus is within the normal range, continue with the same dose. See drug label for specific dose adjustments.
Mechanism of Action
Crysvita (burosumab-twza) is a fibroblast growth factor 23 (FGF23) blocking antibody. X-linked hypophosphatemia is caused by excess fibroblast growth factor 23 (FGF23) which suppresses renal tubular phosphate reabsorption and the renal production of 1,25 dihydroxy vitamin D. Burosumab-twza binds to and inhibits the biological activity of FGF23 restoring renal phosphate reabsorption and increasing the serum concentration of 1,25 dihydroxy vitamin D.
Kutilek S: Burosumab: A new drug to treat hypophosphatemic rickets. Sudan J Paediatr. 2017;17(2):71-73. doi: 10.24911/SJP.2017.2.11. [PubMed:29545670]
Kinoshita Y, Fukumoto S: X-linked hypophosphatemia and FGF23-related hypophosphatemic diseases -Prospect for new treatment. Endocr Rev. 2018 Jan 26. pii: 4825438. doi: 10.1210/er.2017-00220. [PubMed:29381780]
FDA approves first therapy for rare inherited form of rickets, x-linked hypophosphatemia [Link]
//////////////Burosumab-twza, Crysvita FDA 2018, BLA 761068, Protein Based Therapies, Monoclonal antibody, mAb, KRN 23, breakthrough therapy, orphan drug designations, Peptide, ブロスマブ

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











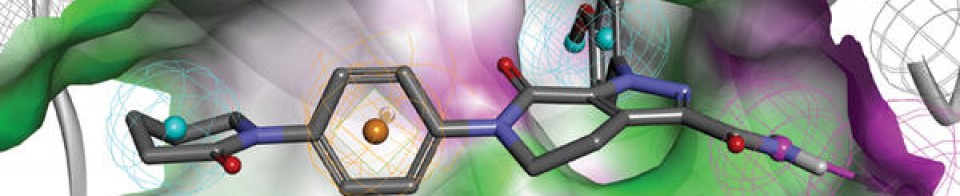




 Alder BioPharmaceuticals has submitted a biologics license application (BLA) for eptinezumab, a humanized IgG1 monoclonal antibody that targets calcitonin gene-related peptide (CGRP), for migraine prevention. If the US Food and Drug Administration grants approval, Alder will be on track to launch the drug in Q1 2020. The BLA included data from the PROMISE 1 and PROMISE 2 studies, which evaluated the effects of eptinezumab in episodic migraine patients (n=888) or chronic migraine patients (n=1,072), respectively. In PROMISE 1, the primary and key secondary endpoints were met, and the safety and tolerability were similar to placebo, while in PROMISE 2, the primary and all key secondary endpoints were met, and the safety and tolerability was consistent with earlier eptinezumab studies.
Alder BioPharmaceuticals has submitted a biologics license application (BLA) for eptinezumab, a humanized IgG1 monoclonal antibody that targets calcitonin gene-related peptide (CGRP), for migraine prevention. If the US Food and Drug Administration grants approval, Alder will be on track to launch the drug in Q1 2020. The BLA included data from the PROMISE 1 and PROMISE 2 studies, which evaluated the effects of eptinezumab in episodic migraine patients (n=888) or chronic migraine patients (n=1,072), respectively. In PROMISE 1, the primary and key secondary endpoints were met, and the safety and tolerability were similar to placebo, while in PROMISE 2, the primary and all key secondary endpoints were met, and the safety and tolerability was consistent with earlier eptinezumab studies.







{kind=link}
{kind=link}