

| Patent ID
|
Patent Title
|
Submitted Date
|
Granted Date
|
|---|---|---|---|
| US2016108123 | ANTIBODY MOLECULES TO PD-L1 AND USES THEREOF |
2015-10-13
|
2016-04-21
|
| US2014343086 | COMPOUNDS AND COMPOSITIONS FOR INHIBITING THE ACTIVITY OF ABL1, ABL2 AND BCR-ABL1 |
2014-07-31
|
2014-11-20
|
| US8829195 | Compounds and compositions for inhibiting the activity of ABL1, ABL2 and BCR-ABL1 |
2013-05-13
|
2014-09-09
|
Home » fda 2021 (Page 5)
Category Archives: fda 2021
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
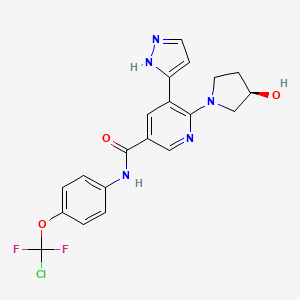
ABL 001, Asciminib
ABL001 / Asciminib
Cas 1492952-76-7
Chemical Formula: C20H18ClF2N5O3
Molecular Weight: 449.8428
Elemental Analysis: C, 53.40; H, 4.03; Cl, 7.88; F, 8.45; N, 15.57; O, 10.67
N-[4-[Chloro(difluoro)methoxy]phenyl]-6-[(3R)-3-hydroxypyrrolidin-1-yl]-5-(1H-pyrazol-5-yl)pyridine-3-carboxamide
3-Pyridinecarboxamide, N-[4-(chlorodifluoromethoxy)phenyl]-6-[(3R)-3-hydroxy-1-pyrrolidinyl]-5-(1H-pyrazol-3-yl)-
PHASE 3, Chronic Myeloid Leukemia, NOVARTIS
UPDATE FDA APPROVED 10/29/2021,
| Scemblix |
To treat Philadelphia chromosome-positive chronic myeloid leukemia with disease that meets certain criteria
Asciminib, sold under the brand name Scemblix, is a medication used to treat Philadelphia chromosome-positive chronic myeloid leukemia (Ph+ CML).[1][2][3] Asciminib is a protein kinase inhibitor.[1]
The most common adverse reactions include upper respiratory tract infections, musculoskeletal pain, fatigue, nausea, rash, and diarrhea.[2]
Asciminib was approved for medical use in the United States in October 2021.[1][4][5]
The U.S. Food and Drug Administration (FDA) granted the application for asciminib priority review, fast track, orphan drug, and breakthrough therapy designations.[2][6][7]
Asciminib is an orally bioavailable, allosteric Bcr-Abl tyrosine kinase inhibitor with potential antineoplastic activity. Designed to overcome resistance, ABL001 binds to the Abl portion of the Bcr-Abl fusion protein at a location that is distinct from the ATP-binding domain. This binding results in the inhibition of Bcr-Abl-mediated proliferation and enhanced apoptosis of Philadelphia chromosome-positive (Ph+) hematological malignancies. The Bcr-Abl fusion protein tyrosine kinase is an abnormal enzyme produced by leukemia cells that contain the Philadelphia chromosome.
ABL001 has been used in trials studying the health services research of Chronic Myelogenous Leukemia and Philadelphia Chromosome-positive Acute Lymphoblastic Leukemia.

- Originator Novartis
- Developer Novartis; Novartis Oncology
- Class Antineoplastics; Pyrazoles; Pyrrolidines; Small molecules
- Mechanism of Action Bcr-abl tyrosine kinase inhibitors
Highest Development Phases
- Phase III Chronic myeloid leukaemia
- No development reported Precursor cell lymphoblastic leukaemia-lymphoma
Most Recent Events
- 04 Nov 2017 No recent reports of development identified for phase-I development in Acute-lymphoblastic-leukaemia(Second-line therapy or greater) in Australia (PO)
- 04 Nov 2017 No recent reports of development identified for phase-I development in Acute-lymphoblastic-leukaemia(Second-line therapy or greater) in France (PO)
- 04 Nov 2017 No recent reports of development identified for phase-I development in Acute-lymphoblastic-leukaemia(Second-line therapy or greater) in Germany (PO)
- The tyrosine kinase activity of the ABLl protein is normally tightly regulated, with the N-terminal cap region of the SH3 domain playing an important role. One regulatory mechanism involves the N-terminal cap glycine-2 residue being myristoylated and then interacting with a myristate binding site within the SHI catalytic domain. A hallmark of chronic myeloid leukemia (CML) is the Philadelphia chromosome (Ph), formed by the t(9,22) reciprocal chromosome translocation in a haematopoietic stem cell. This chromosome carries the BCR-ABL1 oncogene which encodes the chimeric BCR-ABL1 protein, that lacks the N-terminal cap and has a constitutively active tyrosine kinase domain.Although drugs that inhibit the tyrosine kinase activity of BCR-ABL1 via an ATP-competitive mechanism, such as Gleevec® / Glivec® (imatinib), Tasigna® (nilotinib) and Sprycel® (dasatinib), are effective in the treatment of CML, some patients relapse due to the emergence of drug-resistant clones, in which mutations in the SHI domain compromise inhibitor binding. Although Tasigna® and Sprycel® maintain efficacy towards many Gleevec-resistant mutant forms of BCR-ABLl, the mutation in which the threonine-315 residue is replaced by an isoleucine (T315I) remains insensitive to all three drugs and can result in CML patients developing resistance to therapy. Therefore, inhibiting BCR-ABLl mutations, such as T315I, remains an unmet medical need. In addition to CML, BCR-ABLl fusion proteins are causative in a percentage of acute lymphocytic leukemias, and drugs targeting ABL kinase activity also have utility in this indication.Agents targeting the myristoyl binding site (so-called allosteric inhibitors) have potential for the treatment of BCR-ABLl disorders (J. Zhang, F. J. Adrian, W. Jahnke, S. W. Cowan- Jacob, A. G. Li, R. E. Iacob4, T. Sim, J. Powers, C. Dierks, F. Sun, G.-R. Guo, Q. Ding, B. Okram, Y. Choi, A. Wojciechowski, X. Deng, G. Liu, G. Fendrich, A. Strauss, N. Vajpai, S. Grzesiek, T. Tuntland, Y. Liu, B. Bursulaya, M. Azam, P. W. Manley, J. R. Engen, G. Q. Daley, M. Warmuth., N. S. Gray. Targeting BCR-ABL by combining allosteric with ATP -binding-site inhibitors. Nature 2010;463:501-6). To prevent the emergence of drug resistance from ATP inhibitor and/or allosteric inhibitor use, a combination treatment using both types of inhibitor can be developed for the treatment of BCR-ABLl related disorders. In particular, the need exists for small molecules, or combinations thereof, that inhibit the activity of BCR-ABLl and BCR-ABLl mutations via the ATP binding site, the myristoyl binding site or a combination of both sites.Further, inhibitors of ABL 1 kinase activity have the potential to be used as therapies for the treatment of metastatic invasive carcinomas and viral infections such as pox and Ebola viruses.The compounds from the present invention also have the potential to treat or prevent diseases or disorders associated with abnormally activated kinase activity of wild-type ABL1, including non-malignant diseases or disorders, such as CNS diseases in particular neurodegenerative diseases (for example Alzheimer’s, Parkinson’s diseases), motoneuroneuron diseases (amyotophic lateral sclerosis), muscular dystrophies, autoimmune and inflammatory diseases (diabetes and pulmonary fibrosis), viral infections, prion diseases.
Asciminib is an allosteric inhibitor of BCR-ABL kinase in phase III clinical development at Novartis for the treatment of patients with chronic myelogenous leukemia (CML) in chronic phase who have been previously treated with ATP-binding site tyrosine kinase inhibitors. Early clinical trials are also under way in patients with Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) and as first-line threapy of CML.
PATENT
To illustrate tautomerism with the following specific examples, (R)-N-(4- (chlorodifluoromethoxy)phenyl)-6-(3-hydroxypyrrolidin-l-yl)-5-(lH-pyrazol-5-yl)nicotinamide
(right structure, below) is a tautomer of (R)-N-(4-(chlorodifluoromethoxy)phenyl)-6-(3-hydroxypyrrolidin-l-yl)-5-(lH-pyrazol-3-yl)nicotinamide (left structure, below) and vice versa:

[0045] Where the plural form (e.g. compounds, salts) is used, this includes the singular
Example 9
(R)-N-(4-(Chlorodifluoromethoxy)phenyl)-6-(3-hvdroxypyrrolidin-l-yl)-5-(lH-pyrazol-5- vDnicotinamide

[00365] A mixture of (R)-5-Bromo-N-(4-(chlorodifluoromethoxy)phenyl)-6-(3-hydroxypyrrolidin-l-yl)nicotinamide (Stage 9.2, 100 mg, 0.216 mmol) and 5-(4 ,4,5,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 -((2-(trimethylsilyl)ethoxy)methyl)- IH-pyrazole (215 mg, 0.663 mmol), Pd(PPh3)2Cl2 (17 mg, 0.024 mmol), Na2C03 (115 mg, 1.081 mmol), DME (917 μί), water (262 μΕ) and EtOH (131 μί) in a MW vial was sealed, evacuated / purged 3 times with argon and subjected to MW irradiation at 125°C for 20 min. The RM was diluted with 2 mL
of DME, stirred with Si-Thiol (Silicycle 1.44 mmol/g, 90 mg, 0.130 mmol) for 3 h. The mixture was centrifuged and the supernatant was filtered through a 0.45 μηι PTFE filter and the solvent was evaporated off under reduced pressure. The crude product was purified by flash
chromatography (RediSep® Silica gel column, 12 g, cyclohexane / EtOAc from 40% to 100% EtOAc) to afford the protected intermediate as a colorless oil. Ethylene diamine (96 μί, 1.428 mmol) and TBAF 1 M in THF (1.428 mL, 1.428 mmol) were then added and the RM was stirred at 80-85°C for 5 days. The solvent was evaporated off under reduced pressure and the residue was dissolved in EtOAc (40 mL), washed 3 times with sat. aq. NaHCC and brine, dried over Na2S04 and The solvent was evaporated off under reduced pressure to give a residue which was purified by preparative SFC (Column DEAP, from 25% to 30% in 6 min) to yield the title compound as a white solid.
[00366] Alternatively, Example 9 was prepared by adding TFA (168 mL, 2182 mmol) to a solution of N-(4-(chlorodifluoromethoxy)phenyl)-6-((R)-3-hydroxypyrrolidin-l-yl)-5-(l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazol-5-yl)nicotinamide (Stage 9.1, 31.3 g, 54.6 mmol) in DCM (600 mL). The mixture was stirred at RT for 2.5 h. The solvent was evaporated off under reduced pressure and the residue was dissolved in EtOAc (1.5 L),washed with a sat. solution of NaHC03 (3 x 500 mL) and brine (500 mL), dried over Na2S04 and the solvent was evaporated off under reduced pressure to give a residue which was suspended in DCM (300 mL), stirred at RT for 15 min, filtered, washed with DCM (200 mL), dried and purified by chromatography (Silica gel, 1 kg, DCM / MeOH 95:5). The residue was dissolved in MeOH (500 mL) and treated with Si-Thiol (Biotage, 5.0 g , 6.5 mmol) for 16 h at 25°C. The resin was filtered off, the solvent was evaporated off under reduced pressure and the residue was crystallized from MeCN to afford the title compound as a white crystalline solid.
[00367] Alternatively, Example 9 was prepared by the dropwise addition of aqueous HC1
(7.7 mL of 6M) to a solution of N-(4-(chlorodifluoromethoxy)phenyl)-6-((R)-3-hydroxypyrrolidin- 1 -yl)-5-( 1 -(tetrahydro-2H-pyran-2-yl)- 1 H-pyrazol-5-yl)nicotinamide (Stage 9.1, 3.8 g, 7.12 mmol) in MeOH (20 mL) and THF (10 mL) with cooling (below 35°C). The mixture was stirred at 22°C for 2 h and then added to cooled (10°C) 1.2 M NaOH (22 mL).
Throughout the addition the temperature was kept below 30°C and pH was kept in the range of 9-10. The RM was then stirred for 30 min at 30°C. The solvent was evaporated off under reduced pressure, until the desired compound precipitated. The precipitate was filtered and dried to give the title compound as a yellow solid.
[00368] Analytical data for Example 9: HPLC (Condition 5) tR = 5.54 min, HPLC Chiral
(CHIRALCEL® OD-H, 250 x 4.6 mm, eluent : n-heptane/EtOH/MeOH (85: 10:5), 1 mL/min, UV 210 nm) tR = 10.17 min, UPLC-MS (condition 3) tR = 0.93 min, m/z = 450.3 [M+H]+, m/z = 494.1 [M+formic acid-H]“; XH-NMR (400 MHz, DMSO-d6) δ ppm 1.65 – 1.76 (m, 1 H) 1.76 – 1.87 (m, 1 H) 2.93 (d, J=l 1.73 Hz, 1 H) 3.19 – 3.29 (m, 2 H) 3.35 – 3.51 (m, 1 H) 4.10 – 4.25 (m, 1 H) 4.89 (br. s, 1 H) 6.41 (br. s, 1 H) 7.33 (d, J=8.50 Hz, 2 H) 7.57/7.83 (br. s, 1 H) 7.90 (d, J=8.50 Hz, 2 H) 8.07 (br. s, 1 H) 8.77 (br. s, 1 H) 10.23 (s, 1 H) 12.97/13.15 (br. s, 1 H).
[00369] Stage 9.1 : N-(4-(Chlorodifluoromethoxy)phenyl)-6-((R)-3-hydroxypyrrolidin- 1 -yl)-5-( 1 -(tetrahydro-2H-pyran-2- l)- 1 H-pyrazol-5-yl)nicotinamide

[00370] l-(Tetrahydro-2H-pyran-2-yl)-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (29.6 g, 102 mmol), K3P04 (51.6 g, 236 mmol) and Pd(PPh3)4 (4.55 g, 3.93 mmol) were added to a suspension of (R)-5-bromo-N-(4-(chlorodifluoromethoxy)phenyl)-6-(3-hydroxypyrrolidin-l-yl)nicotinamide (Stage 9.2, 36.4 g, 79 mmol) in toluene (360 mL) under an argon atmosphere and the mixture was stirred at 110°C for 4 h. The RM was poured into brine (500 mL) and extracted with EtOAc (2 x 1 L). The combined extracts were washed with brine (500 mL), dried over Na2S04, and the solvent was evaporated off under reduced pressure to give a residue which was purified by chromatography (Silica gel column, 1.5 kg, DCM / MeOH 95:5) to afford a dark yellow foam, that was dissolved in MeOH / DCM (1 L of 3: l) and treated with Si-Thiol (Biotage, 35 g , 45.5 mmol) for 17 h at 30°C. The resin was filtered off, and solvent was evaporated off under reduced pressure, until the desired compound crystallized. The product was filtered washed with MeOH and dried to afford the title compound.
[00371] Alternatively, Stage 9.1 was prepared by adding 4-(chlorodifluoromethoxy)aniline
(16.6 g, 84.9 mmol), NMM (21.7 g, 212.1 mmol), hydroxybenzotriazole hydrate (HOBt H20, 11.9 g, 77.77 mmol) and l-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCIHCl, 20.9 g, 109.0 mmol) to a solution of 6-((R)-3-hydroxypyrrolidin-l-yl)-5-(l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazol-5-yl)nicotinic acid (Stage 9.4, 29.83 g, 70.7 mmol) in THF (271 mL). The mixture was stirred for 1.5 h at 25°C and then at 65°C for 16 h. After cooling the RM to 35 °C, further EDCIHCl (13.3 g, 69.4 mmol) was added and the RM was stirred for 1.5 h at 35°C then again at 65°C for 16 h. After cooling the RM to 35°C, water (150 mL) was added, the THF was removed under reduced pressure, EtOAc (180 mL) was added and the mixture was stirred for at 35 °C fori h. The two layers were separated and the aq. phase was then extracted with EtOAc (60 mL). The combined organic layers were washed with water (90 mL), brine (90 mL). The solvent was evaporated off under reduced pressure to give a brown solid which was purified by column chromatography (Silica gel, DCM / MeOH 40: 1 to 20: 1) to afford the title compound as a yellow solid.
[00372] Analytical data for Stage 9.1: HPLC (Condition 5) tR = 6.12 min, UPLC-MS
(Condition 3) tR = 1.06 min, m/z = 533.2 [M+H]+; XH-NMR (400 MHz, DMSO-d6) δ ppm 1.36 -2.02 (m, 7 H) 2.23 – 2.38 (m, 1 H) 3.08 – 3.29 (m, 2 H) 3.32 – 3.52 (m, 2 H) 3.73 – 3.93 (m, 1 H) 4.13 – 4.25 (m, 1 H) 4.80 – 4.90 (m, 1 H) 4.95 – 5.17 (m, 1 H) 6.33 – 6.50 (m, 1 H) 7.33 (d, J=8.99 Hz, 2 H) 7.61 (d, J=1.56 Hz, 1 H) 7.86 (d, J=8.99 Hz, 2 H) 7.97 – 8.11 (m, 1 H) 8.82 (s, 1 H) 10.13 – 10.25 (m, 1 H).
[00373] Stage 9.2: (R)-5-Bromo-N-(4-(chlorodifluoromethoxy)phenyl)-6-(3-hydroxypyrrolidin- 1 -yl)nicotinamide

[00374] (R)-Pyrrolidin-3-ol (9.55 g, 109.6 mmol) and DIPEA (35.1 ml, 201.3 mmol) were added to a suspension of 5-bromo-6-chloro-N-(4-(chlorodifluoromethoxy)phenyl)nicotinamide (Stage 9.3, 37.7 g, 91.5 mmol) in iPrOH (65 mL) and stirred at 140°C for 1 h. EtOAc (700 mL) was added and the solution was washed IN HC1 (2 x 200 mL), sat. NaHCC (200 mL) and brine (2 x 200 mL), dried over Na2S04, and the solution was concentrated under reduced pressure until crystallization commenced. n-Heptane (1 L) were added and the mixture was stirred at RT for 30 min, filtered and washed with ΪΡΓ20 (500 mL) to afford the title compound as a white crystalline solid. HPLC (Condition 5) tR = 6.68 min, UPLC-MS (Condition 3) tR = 1.10 min, m/z =
462.2/464.2 [M+H]+; XH-NMR (400 MHz, DMSO-d6) δ ppm 1.78 – 2.01 (m, 2 H) 3.55 (d, J=l 1.34 Hz, 1 H) 3.66 – 3.75 (m, 1 H) 3.79 – 3.93 (m, 2 H) 4.34 (br. s, 1 H) 4.98 (d, =3.13 Hz, 1 H) 7.32 (d, J=8.99 Hz, 2 H) 7.84 (d, J=8.99 Hz, 2 H) 8.33 (d, J=1.96 Hz, 1 H) 8.66 (d, J=1.96 Hz, 1 H) 10.21 (s, 1 H).
[00375] Stage 9.3: 5-Bromo-6-chloro-N- 4-(chlorodifluoromethoxy)phenyl)nicotinamide

[00376] DMF (2.55 mL, 33.0 mmol) and SOCl2 (24.08 ml, 330 mmol) were added to a suspension of 5-bromo-6-chloro-nicotinic acid (26 g, 110 mmol) in toluene (220 mL) and the RM was stirred at 80°C for 1 h. The solvent was evaporated off under reduced pressure and the residue was dissolved in THF (220 mL) and cooled to -16°C. DIPEA (38.4 mL, 220 mmol) was added, followed by dropwise addition of a solution of 4-(chlorodifluoromethoxy)aniline (22.35 g, 115 mmol) in THF (220 mL) over 15 min. The suspension was stirred for 1 h at RT. The solvent was evaporated off under reduced pressure and the residue was dissolved in TBME (700 mL), washed with IN HC1 (2 x 200 mL), sat. NaHC03 (200 mL) and brine (2 x 200 mL), dried over Na2S04, and the solvent was evaporated off under reduced pressure to give the product which was crystallized from EtOAc – n-heptane to afford the title compound as a white crystalline solid. HPLC (Condition 5) tR = 7.77 min, UPLC-MS (Condition 3) tR = 1.24 min, m/z =
409.1/411.1/413.1 [M+H]+; XH-NMR (400 MHz, DMSO-d6) δ ppm 7.38 (d, =8.99 Hz, 2 H) 7.85 (d, =8.99 Hz, 2 H) 8.72 (br. s, 1 H) 8.92 (br. s, 1 H) 10.68 (s, 1 H).
[00377] Stage 9.4: 6-((R)-3-Hydroxypyrrolidin-l-yl)-5-(l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazol-5-yl)nicotinic acid

[00378] Aq. NaOH (180 niL of 2.6 M) was added to a solution of methyl 6-((R)-3-hydroxypyrrolidin- 1 -yl)-5-(l -(tetrahydro-2H-pyran-2-yl)- 1 H-pyrazol-5-yl)nicotinate (Stage 9.5, 11 lg, 299 mmol) in MeOH (270 mL) and the RM was stirred at RT for 14 h. The MeOH was evaporated off under reduced pressure and the aq. residue was treated with brine (90 mL), extracted with MeTHF twice (540 mL + 360 mL) and the combined organic layers were washed with water (90 mL). MeTHF was added to the combined aq. layers, the biphasic mixture was cooled to 0 °C and acidified (pH = 4-4.5) with aq. HC1 solution (18%) and extracted with
MeTHF. The combined organic extracts were washed with brine and the solvent was evaporated off under reduced pressure to give a residue which was recrystallized from a EtOAc / TBME (1 : 1) to afford the title compound as a white solid. HPLC (Condition 7) tR = 4.74 min, LC-MS
(Condition 8) tR = 3.37 min, m/z = 359.0 [M+H]+; XH-NMR (400 MHz, DMSO-d6) δ ppm 1.44 (br. s, 2 H), 1.51 (d, J=11.54 Hz, 2 H), 1.64 – 1.86 (m, 4 H), 1.90 (br. s, 1 H), 2.31 (d, J=9.29 Hz, 1 H), 2.77 (br. s, 1 H), 3.10 (br. s, 1 H), 3.21 (d, J=8.78 Hz, 2 H), 3.27 – 3.51 (m, 4 H), 3.87 (d, J=11.54 Hz, 1 H), 4.16 (br. s, 1 H), 4.75 – 4.93 (m, 1 H), 5.04 (br. s, 1 H), 6.35 (d, J=17.32 Hz, 1 H), 7.51 – 7.64 (m, 1 H), 7.64 – 7.82 (m, 1 H), 8.67 (d, J=2.26 Hz, 1 H), 12.58 (br. s, 1 H).
[00379] Stage 9.5: Methyl 6-((R)-3-hydroxypyrrolidin-l-yl)-5-(l-(tetrahydro-2H-pyran-2-yl)- 1 H-pyrazol-5-yl)nicotinate

[00380] A mixture of (R)-methyl 5-bromo-6-(3-hydroxypyrrolidin-l-yl)nicotinate (Stage
9.6, 90 g, 299 mmol), l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazole-5-boronic acid pinacol ester (103.9 g, 373.6 mmol), K3P04 (126.9 g, 597.7 mmol), Pd(PPh3)2Cl2 (6.29 g, 8.97 mmol) in toluene (900 mL) was stirred at 92°C and for 16 h. After cooling the mixture to RT, the solution was washed with water (450 mL), 5% NaHCC solution (430 mL) and the solvent was evaporated off under reduced pressure to give a residue which was used without further purifications in the next step. HPLC (Condition 7) tR = 6.929 min, LC-MS (Condition 8) tR = 4.30 min, m/z = 373.0 [M+H ; XH-NMR (400 MHz, DMSO-d6) δ ppm 1.19 – 1.28 (m, 1 H), 1.35 – 1.63 (m, 4 H), 1.63 -1.86 (m, 3 H), 1.89 (br. s, 1 H), 2.12 – 2.39 (m, 1 H), 3.11 (br. s, 1 H), 3.18 – 3.48 (m, 4 H), 3.78 (s, 4 H), 3.88 (d, J=11.54 Hz, 1 H), 4.08 – 4.24 (m, 1 H), 4.86 (dd, J=18.20, 2.89 Hz, 1 H), 5.02 (d, J=8.28 Hz, 1 H), 6.39 (br. s, 1 H), 7.58 (d, J=1.25 Hz, 1 H), 7.78 (br. s, 1 H), 8.69 (t, J=2.01 Hz, 1 H).
[00381] Stage 9.6: (R)-methyl 5-bromo-6-(3-hydroxypyrrolidin-l-yl)nicotinate

[00382] DIPEA (105.3 g, 142.2 mL, 814.4 mmol) was added to a solution of methyl-5-bromo-6-chroronicotinate (85 g, 339.5 mmol) and (R)-pyrrolidin-3-ol (54.2 g, 441.2 mmol) in isopropyl acetate and the RM was stirred at 70°C for 14 h . The solvent was evaporated off under reduced pressure to give a the residue which was dissolved in toluene (850 mL), washed with water (127 mL) and brine (127 mL)and concentrated under reduced pressure until precipitation commenced. n-Heptane (340 mL) was slowly added to the stirred mixture at 22 °C, which was then cooled to 0 °C and the product was filtered, washed with a toluene / n-heptane mixture
(1 : 1.5) and dried to give the title compound as a yellow solid. HPLC (Condition 7) tR = 8.54 min, LC-MS (Condition 8) tR = 4.62 min, m/z = 300.9/302.9 [M+H]+; XH-NMR (400 MHz, DMSO-d6) δ ρριη 1.77 – 1.99 (m, 2 H), 3.57 (d, J=11.54 Hz, 1 H), 3.72 (ddd, J=l 1.11, 7.97, 3.26 Hz, 1 H), 3.78 (s, 3 H), 3.81 -3.90 (m, 2 H), 4.26 – 4.39 (m, 1 H), 4.99 (br. s, 1 H), 8.11 (d, J=2.01 Hz, 1 H), 8.56 (d, J=1.76 Hz, 1 H).
PAPER
- By Wylie, Andrew A.; Schoepfer, Joseph; Jahnke, Wolfgang; Cowan-Jacob, Sandra W.; Loo, Alice; Furet, Pascal; Marzinzik, Andreas L.; Pelle, Xavier; Donovan, Jerry; Zhu, Wenjing; et al
- From Nature (London, United Kingdom) (2017), 543(7647), 733-737.
By Wylie, Andrew A. et alFrom Nature (London, United Kingdom), 543(7647), 733-737; 2017
PAPER
- By Molica, Matteo; Massaro, Fulvio; Breccia, Massimo
- From Expert Opinion on Pharmacotherapy (2017), 18(1), 57-65.
PATENT
US 20170216289
PAPER
- By El Rashedy, Ahmed A.; Olotu, Fisayo A.; Soliman, Mahmoud E. S.
- From Chemistry & Biodiversity (2018), 15(3), n/a.

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
 |
|
| Clinical data | |
|---|---|
| Trade names | Scemblix |
| Other names | ABL001 |
| Routes of administration |
By mouth |
| Drug class | Tyrosine kinase inhibitor |
| ATC code |
|
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII |
|
| KEGG | |
| ChEMBL |
|
| PDB ligand | |
| Chemical and physical data | |
| Formula | C20H18ClF2N5O3 |
| Molar mass | 449.84 g·mol−1 |
| 3D model (JSmol) | |
References
- ^ Jump up to:a b c d “Scemblix- asciminib tablet, film coated”. DailyMed. Retrieved 4 November 2021.
- ^ Jump up to:a b c “FDA approves asciminib for Philadelphia chromosome-positive chronic myeloid leukemia”. U.S. Food and Drug Administration (FDA) (Press release). 29 October 2021. Retrieved 4 November 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Breccia M, Colafigli G, Scalzulli E, Martelli M (August 2021). “Asciminib: an investigational agent for the treatment of chronic myeloid leukemia”. Expert Opinion on Investigational Drugs. 30 (8): 803–811. doi:10.1080/13543784.2021.1941863. PMID 34130563.
- ^ “Scemblix: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 29 October 2021.
- ^ “FDA approves Novartis Scemblix (asciminib), with novel mechanism of action for the treatment of chronic myeloid leukemia”. Novartis (Press release). Retrieved 29 October 2021.
- ^ “Asciminib Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 27 February 2017. Retrieved 29 October 2021.
- ^ “Novartis receives FDA Breakthrough Therapy designations for investigational STAMP inhibitor asciminib (ABL001) in chronic myeloid leukemia”. Novartis (Press release). 8 February 2020. Retrieved 29 October 2021.
External links
- “Asciminib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02081378 for “A Phase I Study of Oral ABL001 in Patients With CML or Ph+ ALL” at ClinicalTrials.gov
- Clinical trial number NCT03106779 for “Study of Efficacy of CML-CP Patients Treated With ABL001 Versus Bosutinib, Previously Treated With 2 or More TKIs” at ClinicalTrials.gov
////////////////ABL001, Asciminib, ABL 001, ABL-001, PHASE 3, Chronic Myeloid Leukemia, NOVARTIS
O=C(NC1=CC=C(OC(F)(Cl)F)C=C1)C2=CN=C(N3C[C@H](O)CC3)C(C4=CC=NN4)=C2

NEW DRUG APPROVALS
one time
$10.00
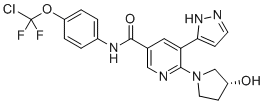
TRILACICLIB, G1T28
Trilaciclib
update 2021/2/12 US FDA APPROVED COSELA
- Molecular FormulaC24H30N8O
- Average mass446.548 Da
- G1T 28
CAS 1374743-00-6
2′-{[5-(4-Methyl-1-piperazinyl)-2-pyridinyl]amino}-7′,8′-dihydro-6’H-spiro[cyclohexane-1,9′-pyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyrimidin]-6′-one
G1T28, SHR 6390
Spiro[cyclohexane-1,9′(6’H)-pyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyrimidin]-6′-one, 7′,8′-dihydro-2′-[[5-(4-methyl-1-piperazinyl)-2-pyridinyl]amino]-
- 7′,8′-Dihydro-2′-[[5-(4-methyl-1-piperazinyl)-2-pyridinyl]amino]spiro[cyclohexane-1,9′(6’H)-pyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyrimidin]-6′-one
- 2′-[[5-(4-Methylpiperazin-1-yl)pyridin-2-yl]amino}-7′,8′-dihydro-6’H-spiro[cyclohexane-1,9′-pyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyrimidin]-6′-one
UNII:U6072DO9XG
Reduction of Chemotherapy-Induced Myelosuppression
Trilaciclib dihydrochloride
1977495-97-8
In phase II clinical development as a chemoprotectant at G1 Therapeutics for first- or second-line treatment in patients with metastatic triple negative breast cancer, in combination with gemcitabine and carboplatin
![]()
PATENT, WO 2014144326, Compound 89 (also referred to as Compound T)
| WO2014144847A3 | |
| Inventors | Norman E. Sharpless, Jay Copeland Strum, John Emerson Bisi, Patrick Joseph Roberts, Francis Xavier Tavares |
| Applicant | G1 Therapeutics, Inc. |
| Norman Sharpless | |
|---|---|
 |
|
| Born | Norman Edward Sharpless September 20, 1966 Greensboro, North Carolina |
| Nationality | American |
| Other names | Ned Sharpless |
| Occupation | Director, Lineberger Comprehensive Cancer Center Founder, G1 Therapeutics ($GTHX) |
| Notable work | Wellcome Distinguished Professor, American Society of Clinical Investigation Member, Association of American Cancer Institute board of directors, |
NCI Director Dr. Norman E. Sharpless

NCI Director Dr. Norman E. Sharpless, Credit: National Institutes of Health
Norman E. “Ned” Sharpless, M.D., was officially sworn in as the 15th director of the National Cancer Institute (NCI) on October 17, 2017. Prior to his appointment, Dr. Sharpless served as the director of the University of North Carolina (UNC) Lineberger Comprehensive Cancer Center, a position he held since January 2014.
Dr. Sharpless was a Morehead Scholar at UNC–Chapel Hill and received his undergraduate degree in mathematics. He went on to pursue his medical degree from the UNC School of Medicine, graduating with honors and distinction in 1993. He then completed his internal medicine residency at the Massachusetts General Hospital and a hematology/oncology fellowship at Dana-Farber/Partners Cancer Care, both of Harvard Medical School in Boston.
After 2 years on the faculty at Harvard Medical School, he joined the faculty of the UNC School of Medicine in the Departments of Medicine and Genetics in 2002. He became the Wellcome Professor of Cancer Research at UNC in 2012.
Dr. Sharpless is a member of the Association of American Physicians as well as the American Society for Clinical Investigation (ASCI), the nation’s oldest honor society for physician–scientists, and served on the ASCI council from 2011 to 2014. Dr. Sharpless was an associate editor of Aging Cell and deputy editor of the Journal of Clinical Investigation. He has authored more than 150 original scientific papers, reviews, and book chapters, and is an inventor on 10 patents. He cofounded two clinical-stage biotechnology companies: G1 Therapeutics and HealthSpan Diagnostics.
In addition to serving as director of NCI, Dr. Sharpless continues his research in understanding the biology of the aging process that promotes the conversion of normal self-renewing cells into dysfunctional cancer cells. Dr. Sharpless has made seminal contributions to the understanding of the relationship between aging and cancer, and in the preclinical development of novel therapeutics for melanoma, lung cancer, and breast cancer.
| Record ID | Title | Status | Phase |
|---|---|---|---|
| NCT03041311 | Carboplatin, Etoposide, and Atezolizumab With or Without Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Extensive Stage Small Cell Lung Cancer (SCLC) | Recruiting | 2 |
| NCT02978716 | Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Combination With Gemcitabineand Carboplatin in Metastatic Triple Negative Breast Cancer (mTNBC) | Recruiting | 2 |
| NCT02514447 | Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Patients With Previously Treated Extensive Stage SCLC Receiving Topotecan Chemotherapy | Recruiting | 2 |
| NCT02499770 | Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Combination With Etoposide and Carboplatin in Extensive Stage Small Cell Lung Cancer (SCLC) | Active, not recruiting | 2 |
Synthesis
WO 2016040858


Trilaciclib (G1T28)
Trilaciclib is a potential first-in-class short-acting CDK4/6 inhibitor in development to preserve hematopoietic stem cells and enhance immune system function during chemotherapy. Trilaciclib is administered intravenously prior to chemotherapy and has the potential to significantly improve treatment outcomes.
G1 is currently evaluating trilaciclib in four Phase 2 clinical trials: three studies in patients with small-cell lung cancer (SCLC), and one study in patients with triple-negative breast cancer (TNBC). Preliminary data from the SCLC trials were presented at the American Society of Clinical Oncology 2017 Annual Meeting and at the 2016 World Conference on Lung Cancer.
Data from a Phase 1 trial in healthy volunteers were presented at the American Society of Clinical Oncology 2015 Annual Meeting and published in Science Translational Medicine. Trilacicilib has been extensively studied in animals; these preclinical data have been presented at several scientific meetings and published in Molecular Cancer Therapeutics, Science Translational Medicine, and Cancer Discovery.
Trilaciclib is a small molecule, competitive inhibitor of cyclin dependent kinases 4 and 6 (CDK4/6), with potential antineoplastic and chemoprotective activities. Upon intravenous administration, trilaciclib binds to and inhibits the activity of CDK4/6, thereby blocking the phosphorylation of the retinoblastoma protein (Rb) in early G1. This prevents G1/S phase transition, causes cell cycle arrest in the G1 phase, induces apoptosis, and inhibits the proliferation of CDK4/6-overexpressing tumor cells. In patients with CDK4/6-independent tumor cells, G1T28 may protect against multi-lineage chemotherapy-induced myelosuppression (CIM) by transiently and reversibly inducing G1 cell cycle arrest in hematopoietic stem and progenitor cells (HSPCs) and preventing transition to the S phase. This protects all hematopoietic lineages, including red blood cells, platelets, neutrophils and lymphocytes, from the DNA-damaging effects of certain chemotherapeutics and preserves the function of the bone marrow and the immune system. CDKs are serine/threonine kinases involved in the regulation of the cell cycle and may be overexpressed in certain cancer cell types. HSPCs are dependent upon CDK4/6 for proliferation.
Trilaciclib (G1T28) is a CDK4/6 inhibitor in phase II clinical development as a chemoprotectant at G1 Therapeutics for first- or second-line treatment in patients with metastatic triple negative breast cancer, in combination with gemcitabine and carboplatin. Also, phase II trials are ongoing in newly diagnosed, treatment-naive small-cell lung cancer patients, in combination with carboplatin, etoposide, and atezolizumab and phase I trials in previously treated small-cell lung cancer patients, in combination with topotecan.
U.S. Patent Nos. 8,822,683; 8,598,197; 8,598,186, 8,691,830, 8,829,102, 8,822,683, 9, 102,682, 9,499,564, 9,481,591, and 9,260,442, filed by Tavares and Strum and assigned to Gl Therapeutics describe a class of N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amine cyclin dependent kinase inhibitors including those of the formula with variables as defined therein):

U.S. Patent Nos. 9,464,092, 9,487,530, and 9,527,857 which are also assigned to Gl Therapeutics describe the use of the above pyrimidine-based agents in the treatment of cancer.
These patents provide a general synthesis of the compounds that is based on a coupling reaction of a fused chloropyrimidine with a heteroaryl amine to form the central disubstituted amine. Such coupling reactions are sometimes referred to as Buchwald coupling (see WO Ί56 paragraph 127; reference WO 2010/020675). The lactam of the fused chloropyrimidine, for example, a 2-chloro-spirocyclo-pyrrolo[2,3-d]pyrimidine-one such as Intermediate K as shown below can be prepared by dehydration of the corresponding carboxylic acid. The reported process to prepare intermediate IK requires seven steps.

(Intermediate IK; page 60, paragraph 215 of WO Ί56)
WO 2013/148748 (U.S. S.N. 61/617,657) entitled “Lactam Kinase Inhibitors” filed by Tavares, and also assigned to Gl Therapeutics likewise describes the synthesis of N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines via the coupling reaction of a fused chloropyrimidine with a heteroaryl amine to form the central disubstituted amine.
WO 2013/163239 (U.S. S.N. 61/638,491) “Synthesis of Lactams” describes a method for the synthesis of this class of compounds with the variation that in the lactam preparation step, a carboxylic acid can be cyclized with a protected amine in the presence of a strong acid and a dehydrating agent, which can be together in one moiety as a strong acid anhydride. The purported improvement is that cyclization can occur without losing the protecting group on the amine before cyclization. The typical leaving group is “tBOC” (t-butoxycarbonyl). The application teaches (page 2 of WO 2013/163239) that the strong acid is, for example, trifluoroacetic acid anhydride, tribromoacetic acid anhydride, trichloroacetic acid anhydride or mixed anhydrides. An additional step may be necessary to take off the N-protecting group. The dehydrating agent can be a carbodiimide-based compound such as DCC (Ν,Ν-dicyclohexylcarbodiimide), EDC (l-ethyl-3-(3-dimethylaminopropyl)carbodiimide, or DIC (Ν,Ν-diisopropylcarbodiimide). DCC and DIC are in the same class of reagents-carbodiimides. DIC is sometimes considered better because it is a liquid at room temperature, which facilitates reactions.
WO 2015/061407 filed by Tavares and licensed to Gl Therapeutics also describes the synthesis of these compounds via the coupling of a fused chloropyrimidine with a heteroaryl amine to form the central disubstituted amine. WO ‘407 focuses on the lactam production step and in particular describes that the fused lactams of these compounds can be prepared by treating the carboxylic acid with an acid and a dehydrating agent in a manner that a leaving group on the amine is not removed during the amide-forming ring closing step.
Other publications that describe compounds of this general class include the following. WO 2014/144326 filed by Strum et al. and assigned to Gl Therapeutics describes compounds and methods for protection of normal cells during chemotherapy using pyrimidine based CDK4/6 inhibitors. WO 2014/144596 filed by Strum et al. and assigned to Gl Therapeutics describes compounds and methods for protection of hematopoietic stem and progenitor cells against ionizing radiation using pyrimidine based CDK4/6 inhibitors. WO 2014/144847 filed by Strum et al. and assigned to Gl Therapeutics describes HSPC-sparing treatments of abnormal cellular proliferation using pyrimidine based CDK4/6 inhibitors. WO2014/144740 filed by Strum et al. and assigned to Gl Therapeutics describes highly active anti -neoplastic and anti-proliferative pyrimidine based CDK 4/6 inhibitors. WO 2015/161285 filed by Strum et al. and assigned to Gl Therapeutics describes tricyclic pyrimidine based CDK inhibitors for use in radioprotection. WO 2015/161287 filed by Strum et al. and assigned to Gl Therapeutics describes analogous tricyclic pyrimidine based CDK inhibitors for the protection of cells during chemotherapy. WO 2015/161283 filed by Strum et al. and assigned to Gl Therapeutics describes analogous tricyclic pyrimidine based CDK inhibitors for use in HSPC-sparing treatments of RB-positive abnormal cellular proliferation. WO 2015/161288 filed by Strum et al. and assigned to Gl Therapeutics describes analogous tricyclic pyrimidine based CDK inhibitors for use as anti -neoplastic and anti-proliferative agents. WO 2016/040858 filed by Strum et al. and assigned to Gl Therapeutics describes the use of combinations of pyrimidine based CDK4/6 inhibitors with other anti-neoplastic agents. WO 2016/040848 filed by Strum et al. and assigned to Gl Therapeutics describes compounds and methods for treating certain Rb-negative cancers with CDK4/6 inhibitors and topoisomerase inhibitors.
Other biologically active fused spirolactams and their syntheses are described, for example, in the following publications. Griffith, D. A., et al. (2013). “Spirolactam-Based Acetyl-CoA Carboxylase Inhibitors: Toward Improved Metabolic Stability of a Chromanone Lead Structure.” Journal of Medicinal Chemistry 56(17): 7110-7119, describes metabolically stable spirolactams wherein the lactam resides on the fused ring for the inhibition of acetyl-CoA carboxylase. WO 2013/169574 filed by Bell et al. describes aliphatic spirolactams as CGRP receptor antagonists wherein the lactam resides on the spiro ring. WO 2007/061677 filed by Bell et al. describes aryl spirolactams as CGRP receptor antagonists wherein the lactam resides on the spiro ring. WO 2008/073251 filed by Bell et al. describes constrained spirolactam compounds wherein the lactam resides on the spiro ring as CGRP receptor antagonists. WO 2006/031606 filed by Bell et al. describes carboxamide spirolactam compounds wherein the spirolactam resides on the spiro ring as CGRP receptor antagonists. WO 2006/031610, WO 2006/031491, and WO 2006/029153 filed by Bell et al. describe anilide spirolactam compounds wherein the spirolactam resides on the spiro ring; WO 2008/109464 filed by Bhunai et al. describes spirolactam compounds wherein the lactam resides on the spiro ring which is optionally further fused.
Given the therapeutic activity of selected N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines, it would be useful to have additional methods for their preparation. It would also be useful to have new intermediates that can be used to prepare this class of compounds.
PATENT
WO 2014144596
PATENT
Compound 89 (also referred to as Compound T)
| WO2014144847A3 | |
| Inventors | Norman E. Sharpless, Jay Copeland Strum, John Emerson Bisi, Patrick Joseph Roberts, Francis Xavier Tavares |
| Applicant | G1 Therapeutics, Inc. |
EXAMPLES
Intermediates B, E, K, L, 1A, IF and 1CA were synthesized according to US 8,598,186 entitled CDK Inhibitors to Tavares, F.X. and Strum, J.C..
The patents WO 2013/148748 entitled Lactam Kinase Inhibitors to Tavares, F.X., WO 2013/163239 entitled Synthesis of Lactams to Tavares, F.X., and US 8,598,186 entitled CDK Inhibitors to Tavares, F.X. and Strum, J.C. are incorporated by reference herein in their entirety. Example 1
Synthesis of tert-butyl N- [2- [(5-bromo-2-chloro-pyrimidin-4yl)amino] ethyl] carbamate, Compound 1

To a solution of 5-bromo-2,4-dichloropyrimidine (3.2 g, 0.0135 mol) in ethanol (80 mL) was added Hunig’s base (3.0 mL) followed by the addition of a solution of N-(tert- butoxycarbonyl)-l,2-diaminoethane (2.5 g, 0.0156 mole) in ethanol (20 mL). The contents were stirred overnight for 20 hrs. The solvent was evaporated under vacuum. Ethyl acetate (200 mL) and water (100 mL) were added and the layers separated. The organic layer was dried with magnesium sulfate and then concentrated under vacuum. Column chromatography on silica gel using hexane/ethyl acetate (0- 60%) afforded tert-butyl N-[2-[(5-bromo-2-chloro-pyrimidin-4- yl)amino]ethyl]carbamate. 1HNMR (d6-DMSO) δ ppm 8.21 (s, 1H), 7.62 (brs, 1H), 7.27 (brs, 1H), 3.39 (m, 2H), 3.12 (m, 2H), 1.34 (s, 9H). LCMS (ESI) 351 (M + H).
Example 2
Synthesis of tert-butyl N-[2-[[2-chloro-5-(3,3-diethoxyprop-l-ynyl)pyrimidin-4- yl] amino] ethyl] carbamate, Compound 2

To tert-butyl N-[2-[(5-bromo-2-chloro-pyrimidin-4-yl)amino]ethyl]carbamate (1.265 g, 6 mmol) in THF (10 mL) was added the acetal (0.778 mL, 5.43 mmol), Pd(dppf)CH2Cl2 (148 g), and triethylamine (0.757 mL, 5.43 mmol). The contents were degassed and then purged with nitrogen. To this was then added Cul (29 mg). The reaction mixture was heated at reflux for 48 hrs. After cooling, the contents were filtered over CELITE™ and concentrated. Column chromatography of the resulting residue using hexane/ethyl acetate (0- 30%) afforded tert-butyl N- [2- [ [2-chloro-5 -(3 ,3 -diethoxyprop- 1 -ynyl)pyrimidin-4-yl]amino] ethyl] carbamate. 1HNMR (d6-DMSO) δ ppm 8.18 (s, 1H), 7.63 (brs, 1H), 7.40 (brs, 1H), 5.55 (s, 1H), 3.70 (m, 2H), 3.60 (m, 2H), 3.42 (m, 2H), 3.15 (m, 2H), 1.19 – 1.16 (m, 15H). LCMS (ESI) 399 (M + H).
Example 3
Synthesis of tert-butyl N-[2-[2-chloro-6-(diethoxymethyl)pyrrolo[2,3-d]pyrimidin-7- yl] ethyl] carbamate, Compound 3

To a solution of the coupled product (2.1 g, 0.00526 mole) in THF (30 mL) was added TBAF solid (7.0 g). The contents were heated to and maintained at 65 degrees for 2 hrs. Concentration followed by column chromatography using ethyl acetate/hexane (0-50%) afforded tert-butyl N-[2-[2-chloro-6-(diethoxymethyl)pyrrolo[2,3-d]pyrimidin-7-yl]ethyl]carbamate as a pale brown liquid (1.1 g). 1FiNMR (d6-DMSO) δ ppm 8.88 (s, 1H), 6.95 (brs, 1H), 6.69 (s, 1H), 5.79 (s, 1H), 4.29 (m, 2H), 3.59 (m, 4H), 3.34 (m, 1H), 3.18 (m, 1H), 1.19 (m, 9H), 1.17 (m, 6H). LCMS (ESI) 399 (M + H).
Example 4
Synthesis of tert-buty\ N-[2-(2-chloro-6-formyl-pyrrolo [2,3-d] pyrimidin-7- yl)ethyl] carbamate, Compound 4

To the acetal (900 mg) from the preceeding step was added AcOH (8.0 mL) and water
(1.0 mL). The reaction was stirred at room temperature for 16 hrs. Cone, and column chromatography over silica gel using ethyl acetate/hexanes (0- 60%) afforded tert-butyl N-[2-(2- chloro-6-formyl-pyrrolo[2,3-d]pyrimidin-7-yl)ethyl]carbamate as a foam (0.510 g). 1HNMR (d6-DMSO) δ ppm 9.98 (s, 1H), 9.18 (s, 1H), 7.66 (s, 1H), 6.80 (brs, 1H), 4.52 (m, 2H), 4.36 (m, 2H), 1.14 (s, 9H). LCMS (ESI) 325 (M + H).
Example 5
Synthesis of 7- [2-(teri-butoxycarbonylamino)ethyl] -2-chloro-pyrrolo [2,3-d] pyrimidine-6- carboxylic acid, Compound 5

To the aldehyde (0.940 g) from the preceeding step in DMF (4 mL) was added oxone (1.95 g, 1.1 eq). The contents were stirred at room temp for 7 hrs. Silica gel column chromatography using hexane/ethyl acetate (0- 100%) afforded l-\2-(tert- butoxycarbonylamino)ethyl]-2-chloro-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid (0.545 g). 1HNMR (d6-DMSO) δ ppm 9.11 (s, 1H), 7.39 (s, 1H), 4.38 (m, 2H), 4.15 (m, 2H), 1.48 (m, 9H). LCMS (ESI) 341(M + H).
Example 6
Synthesis of methyl 7-[2-(teri-butoxycarbonylamino)ethyl]-2-chloro-pyrrolo[2,3- d]pyrimidine-6-carboxylate, Compound 6

To a solution of 2-chloro-7-propyl-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid (0.545 g, 0.00156 mole) from the preceeding step in toluene (3.5 mL) and MeOH (1 mL) was added TMS- diazomethane (1.2 mL). After stirring overnight at room temperature, the excess of TMS- diazomethane was quenched with acetic acid (3 mL) and the reaction was concentrated under vacuum. The residue was purified by silica gel column chromatography with hexane/ethyl acetate (0- 70%) to afford methyl 7-[2-(tert-butoxycarbonylamino)ethyl]-2-chloro-pyrrolo[2,3- d]pyrimidine-6-carboxylate as an off white solid (0.52 g). 1HNMR (d6-DMSO) δ ppm 9.10 (s, 1H), 7.45 (s, 1H), 6.81 (brs, 1H) 4.60 (m, 2H), 3.91 (s, 3H), 3.29 (m, 2H), 1.18 (m, 9H) LCMS (ESI) 355 (M + H).
Example 7
Synthesis of Chloro tricyclic amide, Compound 7

To methyl 7- [2-(tert-butoxycarbonylamino)ethyl] -2-chloro-pyrrolo [2,3 -d]pyrimidine-6- carboxylate (0.50 g, 0.0014 mole) from the preceeding step in dichloromethane (2.0 mL) was added TFA (0.830 mL). The contents were stirred at room temperature for 1 hr. Concentration under vacuum afforded the crude amino ester which was suspended in toluene (5 mL) and Hunig’s base (0.5 mL). The contents were heated at reflux for 2 hrs. Concentration followed by silica gel column chromatography using hexane/ethyl acetate (0- 50%) afforded the desired chloro tricyclic amide (0.260 g). 1HNMR (d6-DMSO) δ ppm 9.08 (s, 1H), 8.48 (brs, 1H), 7.21 (s, 1H) 4.33 (m, 2H), 3.64 (m, 2H). LCMS (ESI) 223 (M + H).
Example 8
Synthesis of chloro-N-methyltricyclic amide, Compound 8

To a solution of the chloro tricycliclactam, Compound 7, (185 mg, 0.00083 mole) in DMF (2.0 mL) was added sodium hydride (55% dispersion in oil, 52 mg). After stirring for 15 mins, methyl iodide (62 μί, 1.2 eq). The contents were stirred at room temperature for 30 mins. After the addition of methanol (5 mL), sat NaHCOs was added followed by the addition of ethyl acetate. Separation of the organic layer followed by drying with magnesium sulfate and concentration under vacuum afforded the N-methylated amide in quantitative yield. 1FiNMR (d6-DMSO) δ ppm 9.05 (s, 1H), 7.17 (s, 1H) 4.38 (m, 2H), 3.80 (m, 2H), 3.05 (s, 3H). LCMS (ESI) 237 (M + H). Example 9
Synthesis of l-methyl-4-(6-nitro-3-pyridyl)piperazine, Compound 9

To 5-bromo-2-nitropyridine (4.93 g, 24.3 mmole) in DMF (20 mL) was added N- methylpiperazine (2.96 g, 1.1 eq) followed by the addition of DIPEA (4.65 mL, 26.7 mmole). The contents were heated at 90 degrees for 24 hrs. After addition of ethyl acetate (200 mL), water (100 mL) was added and the layers separated. Drying followed by concentration afforded the crude product which was purified by silica gel column chromatography using (0-10%) DCM/Methanol. 1HNMR (d6-DMSO) δ ppm 8.26 (s, 1H), 8.15 (1H, d, J = 9.3 Hz), 7.49 (1H, d, J = 9.4 Hz), 3.50 (m, 4H), 2.49 (m, 4H), 2.22 (s, 3H).
Example 10
Synthesis of 5-(4-methylpiperazin-l-yl)pyridin-2-amine, Compound 10

To l-methyl-4-(6-nitro-3-pyridyl)piperazine (3.4 g) in ethyl acetate (100 mL) and ethanol (100 mL) was added 10%> Pd/C (400 mg) and then the reaction was stirred under hydrogen (10 psi) overnight. After filtration through CELITE™, the solvents were evaporated and the crude product was purified by silica gel column chromatography using DCM/ 7N ammonia in MeOH (0- 5%) to afford 5-(4-methylpiperazin-l-yl)pyridin-2-amine (2.2 g). 1HNMR (d6-DMSO) δ ppm 7.56 (1H, d, J = 3 Hz), 7.13 (1H, m), 6.36 (1H, d, J = 8.8 Hz), 5.33 (brs, 2H), 2.88 (m, 4H), 2.47 (m, 4H), 2.16 (s, 3H).
Example 11
Synthesis of tert-butyl 4-(6-amino-3-pyridyl)piperazine-l-carboxylate, Compound 11

This compound was prepared as described in WO 2010/020675 Al .
Synthesis of Compound 89 (also referred to as Compound T)

Compound 89 was synthesized in a similar manner to that described for compound 78 and was converted to an HCl salt. 1HNMR (600 MHz, DMSO-d6) δ ppm 1.47 (br. s., 6 H) 1.72 (br. s., 2 H) 1.92 (br. s., 2 H) 2.77 (br. s., 3 H) 3.18 (br. s., 2 H) 3.46 (br. s., 2 H) 3.63 (br. s., 2 H) 3.66 (d, J=6.15 Hz, 2 H) 3.80 (br. s., 2 H) 7.25 (s, 1 H) 7.63 (br. s., 2 H) 7.94 (br. s., 1 H) 8.10 (br. s., 1 H) 8.39 (br. s., 1 H) 9.08 (br. s., 1 H) 11.59 (br. s., 1 H). LCMS (ESI) 447 (M + H)
PATENT
WO 2014144740
PATENT
Preparation of Active Compounds
Syntheses
The disclosed compounds can be made by the following general schemes:

Scheme 1
In Scheme 1, Ref-1 is WO 2010/020675 Al; Ref-2 is White, J. D.; et al. J. Org. Chem. 1995, 60, 3600; and Ref-3 Presser, A. and Hufher, A. Monatshefte fir Chemie 2004, 135, 1015.

Scheme 2
In Scheme 2, Ref-1 is WO 2010/020675 Al; Ref-4 is WO 2005/040166 Al; and Ref-5 is Schoenauer, K and Zbiral, E. Tetrahedron Letters 1983, 24, 573.

92


93 
3) Pd/C/H2 ![]()
Scheme 6

Scheme 7
NHfOH

Scheme 8
In Scheme 8, Ref-1 is WO 2010/020675 Al; Ref-2 is WO 2005/040166 Al; and Ref-3 is Schoenauer, K and Zbiral, E. Tetrahedron Letters 1983, 24, 573.
Alternatively, the lactam can be generated by reacting the carboxylic acid with a protected amine in the presence of a strong acid and a dehydrating agent, which can be together in one moiety as a strong acid anhydride. Examples of strong acid anhydrides include, but are not limited to, trifluoroacetic acid anhydride, tribromoacetic acid anhydride, trichloroacetic acid anhydride, or mixed anhydrides. The dehydrating agent can be a carbodiimide based compound such as but not limited to DCC (Ν,Ν-dicyclohexylcarbodiimide), EDC (l-ethyl-3-(3-
dimethylaminopropyl)carbodiimide or DIC (Ν,Ν-diisopropylcarbodiimide). An additional step may be necessary to take off the N-protecting group and the methodologies are known to those skilled in the art.
Alternatively, the halogen moiety bonded to the pyrimidine ring can be substituted with any leaving group that can be displaced by a primary amine, for example to create an intermediate for a final product such as Br, I, F, SMe, SO2Me, SOalkyl, SO2alkyl. See, for Exmaple PCT /US2013/037878 to Tavares.
Other amine intermediates and final amine compounds can be synthesized by those skilled in the art. It will be appreciated that the chemistry can employ reagents that comprise reactive functionalities that can be protected and de-protected and will be known to those skilled in the art at the time of the invention. See for example, Greene, T.W. and Wuts, P.G.M., Greene’s Protective Groups in Organic Synthesis, 4th edition, John Wiley and Sons.

Scheme 9
CDK4/6 Inhibitors of the present invention can be synthesized according to the generalized Scheme 9. Specific synthesis and characterization of the Substituted 2-aminopyrmidines can be found in, for instance, WO2012/061156.
Compounds T, Q, GG, and U were prepared as above and were characterized by mass spectrometry and NMR as shown below:
Compound T
1H NMR (600 MHz, DMSO- d6) ppm 1.47 (br. s., 6 H) 1.72 (br. s., 2 H) 1.92 (br. s., 2 H) 2.77 (br. s., 3 H) 3.18 (br. s., 2 H) 3.46 (br. s., 2 H) 3.63 (br. s., 2 H) 3.66 (d, J=6.15 Hz, 2 H) 3.80 (br. s., 2 H) 7.25 (s, 1 H) 7.63 (br. s., 2 H) 7.94 (br. s., 1 H) 8.10 (br. s., 1 H) 8.39 (br. s., 1 H) 9.08 (br. s., 1 H) 11.59 (br. s., 1 H). LCMS ESI (M + H) 447.
PATENT
Synthesis of N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines. The application appears to be particularly focused on methods for the preparation of trilaciclib and an analog of it. Trilaciclib is the company’s lead CDK4/6 inhibitor presently in phase II trials against small-cell lung cancer and triple negative breast cancer. Interestingly, the company is working on a second CDK4/6 inhibitor, G1T38 , which is in a phase II trial against breast cancer.
GENERAL METHODS
The structure of starting materials, intermediates, and final products was confirmed by standard analytical techniques, including NMR spectroscopy and mass spectrometry. Unless otherwise noted, reagents and solvents were used as received from commercial suppliers. Proton nuclear magnetic resonance spectra were obtained on a Bruker AVANCE 500 at 500 MHz in DMSO-dis. HPLC analyses were performed on a Waters HPLC using the below HPLC method.
HPLC Method
Column: Atlantis T3 (150 χ 4.6, 3 μιη)
Column Temperature: 40°C
Flow Rate: 1 mL/min
Detection: UV @ 275 nm
Analysis Time: 36 min
Mobile Phase A: Water (with 0.1% Trifluoroacetic Acid)
Mobile Phase B : Acetonitrile (with 0.1% Trifluoroacetic Acid)
Sample preparation: dissolve PC sample, wet or dry solid (~1 mg of active compound) in acetonitrile/water (1/1) to achieve complete dissolution.
HPLC Method Gradient

Example 1. General Routes of Synthesis

Scheme 1-1 : Starting from an appropriately substituted halo pyrimidine, compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a substituted spirolactam. In Step 2 the appropriately substituted spirolactam is protected with a group selected from R2. In Step 3 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 3, Step 4, Step 5, or Step 6. Oxidation prior to Step 3 results in undesired byproducts. In Step 4 the hydroxyl group of the fused spirolactam is converted to a leaving group.
In Step 5 the leaving group is dehydrated to afford a compound of Formula IV. In Step 6 the compound of Formula IV is optionally deprotected.

Scheme 1-2: Starting from an appropriately substituted halo pyrimidine compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a substituted spirolactam. In Step 2 the appropriately substituted spirolactam is protected with a group selected from R2. In Step 3 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam of Formula IV. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 3 or Step 4. Oxidation prior to Step 3 results in undesired byproducts. In Step 4 the compound of Formula IV is optionally deprotected.

Scheme 1-3 : Starting from an appropriately substituted alkyl glycinate, compounds of the present invention can be prepared. In Step 1 the appropriately substituted alkyl glycinate is subjected to cyclohexanone and TMSCN in the presence of base to afford a cyano species. In Step 2 the appropriately substituted cyanospecies is reduced and subsequently cyclized to afford a compound of Formula I.
Scheme 1-4

Scheme 1-4: Starting from an appropriately substituted l-(aminomethyl)cyclohexan-l-amine, compounds of the present invention can be prepared. In Step 1 the appropriately substituted l-(aminomethyl)cyclohexan-l -amine is reductively aminated with an aldehyde. In Step 2 the appropriately substituted cyclohexane amine is optionally deprotected (i.e.: the group selected from R2 if not H is optionally replaced by H). In Step 3 the cyclohexane amine is cyclized to afford a compound of Formula I. In Step 4 the compound of Formula I is optionally protected.
1-5

Conversion

Scheme 1-5: Starting from an appropriately substituted halo pyrimidine, compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a
substituted spirolactam. In Step 2 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 2, Step 3, Step 4, or Step 5. Oxidation prior to Step 2 results in undesired byproducts. In Step 3 the hydroxyl group of the fused spirolactam is converted to a leaving group. In Step 4 the leaving group is dehydrated to afford a compound of Formula IV. In Step 5 the compound of Formula IV is optionally deprotected.
S

Scheme 1-6: Starting from an appropriately substituted halo pyrimidine compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a substituted spirolactam. In Step 2 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam of Formula IV. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 2 or Step 3. Oxidation prior to Step 2 results in undesired byproducts. In Step 3 the compound of Formula IV is optionally deprotected.

Scheme 1-7: Starting from compound of Formula IV a CDK4/6 inhibitor can be prepared. In Step 1 a heteroaryl amine is subjected to a base and a compound of Formula IV is added slowly under chilled conditions to afford a nucleophilic substitution reaction. The compound of Formula IV can previously be prepared as described in the schemes herein.
Example 2. Representative Routes of Synthesis
Scheme 2-1

quant, yield 2 steps
isolated

70% yield 2 steps 75% yield 95% yield
isolated isolated isolated
Scheme 2-1 : An ester route is one embodiment, of the present invention. Ideally, the best synthesis scheme would afford crystalline intermediates to provide material of consistent purity without column chromatography, and high yielding steps while using safe and cost effective reagents when possible.
The first step in the ester route is a SNAr nucleophilic substitution of CI group in commercially available ester 3 using spirolactam 4. Due to low reactivity of 4, a reaction temperature of 85-95 °C was required. Because of the temperature requirements, DIPEA and dimethylacetamide were selected as the base and solvent, respectively. The reaction follows second-order kinetics and usually stalls after -85% conversion. Therefore, the reaction was typically stopped after 60 hours by first cooling it to room temperature at which point solid formation was observed. The mixture was then partitioned between MTBE and water and product was filtered with excellent purity with -53% yield of the desired product 5. The obtained
compound 5 was protected with a Boc group using Boc anhydride and DMAP as the catalyst and dichloromethane as the solvent. The intermediate 6 was obtained in a quantitative yield. Due to the semi-solid nature of compound 6, the material was taken to the next step without further purification. The Dieckmann condensation was initially performed with strong bases such as LiHMDS and tBuOK. A similar result to the aldehyde route (Scheme 2-2) was obtained: a partial deprotection of Boc group was observed that required column chromatography. However, the best results were obtained when DBU was used as base and THF as solvent. The reaction outcome was complete, clean conversion of 6 to 7. Moreover, the product crystallized from the reaction mixture upon seeding, and a quantitative yield was obtained for the two steps.
The hydroxyl group of 7 was removed via a two-step procedure. First, compound 7 was converted completely into triflate 8 using triflic anhydride and triethylamine in dichloromethane. The reaction was found to proceed well at 0°C. Due to the potential instability of the triflate intermediate, it was not isolated. It was immediately taken to the next step and reduced with triethylsilane and palladium tetrakis to afford the product 9 after ethyl acetate crystallization in -70% yield. The Boc group of 9 was removed using trifluoroacetic acid in dichloromethane to afford 10. Intermediate 10 was converted into the final sulfone 11 using Oxone™ in acetonitrile/water solvent system.
The obtained sulfone 11 was use-tested in the coupling step and was found to perform well. In conclusion, the route to sulfone 11 was developed which eliminated the use of column chromatography with good to excellent yields on all steps.
Scheme 2-2

Molecular Weight: 421 
Scheme 2-2: The first step of Scheme 2-2 consistently afforded product 13 contaminated with one major impurity found in substantial amount. Thorough evaluation of the reaction impurity profile by LC-MS and 2D MR was performed, which showed the impurity was structurally the condensation of two aldehyde 12 molecules and one molecule of lactam 4. Therefore, column chromatography was required to purify compound 13, which consistently resulted in a modest 30% yield. A solvent screen revealed that sec-butanol, amyl alcohol, dioxane, and tert-butanol can all be used in the reaction but a similar conversion was observed in each case. However, tert-butanol provided the cleanest reaction profile, so it was selected as a solvent for the reaction. Assessing the impact of varying the stoichiometric ratio of 4 and 12 on the reaction outcome was also investigated. The reaction was performed with 4 equivalents of amine 4 in an attempt to disrupt the 2: 1 aldehyde/amine composition of the impurity. The result was only a marginal increase in product 13 formation. The temperature impact on the reaction outcome was evaluated next. The coupling of aldehyde 12 and 4 was investigated at two different temperatures: 50 °C and 40 °C with 1 : 1 ratio of aldehyde/amine. Reactions were checked at 2 and 4 hours and then every 12 hours. The reaction progress was slow at 50°C and was accompanied by growth of other impurities. The reaction at 40°C was much cleaner; however the conversion was lower in the same time period. The mode of addition of the reagents was investigated as well at 80°C with a slow addition (over 6 hours) of either aldehyde 12 or amine 4 to the reaction mixture. The product distribution did not change and an about 1 to 1 ratio was observed between product and impurity when amine 4 was added slowly to the reaction mixture containing aldehyde 12 and
DIPEA at reflux. The product distribution did change when aldehyde 12 was added slowly to the mixture of amine 4 and DIPEA. However, the major product of the reaction was the undesired impurity. Other organic bases were tried as well as different ratios of DIPEA. No product was observed when potassium carbonate was used as a base. The results of the experiments are presented in Table 1 below.
Table 1

Compound 13 was successfully formed in three cases: triethylamine, 2,6-lutidine and DIPEA, with the DIPEA result being the best. The use of Boc protected spirolactam 4 had no effect on the impurity formation as well. Its utilization was speculated to be beneficial in performing the coupling step together with the following step, preparation of compound 14.
The major impurity formed during Step 1 of Scheme 2-2 is:

Chemical Formula:€2)Η;Μ(¾ 6( 2ί>2
Molecular Weight: 527.4903
The second step (Boc protection of the free lactam) proceeded well using DMAP as a catalyst in dichloromethane at room temperature. The product 14 is a thick oil, and, therefore, cannot be purified by crystallization. The Boc protected intermediate 14 was cyclized successfully into the desired pentacyclic structure 10 upon treatment with a strong base such as LiHMDS or tBuOK. Surprisingly, the Boc group was partially removed during the reaction. The level of deprotection was independent from the internal reaction temperature and was positively correlated with excess of base used. Therefore the mixture of the desired product 10 and 10-Boc compound was treated with acid to completely deprotect Boc group. The conversion of methyl sulfide into the final sulfone 11 was carried out with Oxone™. Initially a mixture of methanol and water was used for the reaction. As the result, a partial displacement of sulfone by methoxy group was detected. The methanol was replaced with acetonitrile and the sulfone displacement was eliminated.
In summary, the ester route (Scheme 2-1) is preferred because:
1. Formation of the impurity during the first step of Scheme 2-2 was unavoidable and resulted in yields of < 35%.
2. Column purification was required to isolate intermediate 14.
3. The aldehyde starting material was not commercially available and required two synthetic steps from the corresponding ester.

Scheme 2-3 : Starting with cyclohexanone, compounds of the present invention can be prepared. In Step 1 the methyl glycinate is subjected to cyclohexanone and TMSCN in the presence of tri ethyl amine in DCM to afford 15. In Step 2 15 hydrogenated with hydrogen gas in the presence of catalytic platinum oxide and subsequently undergoes an intramolecular cyclization to afford compound 16 which is used in the schemes above.

Scheme 2-4: Starting with compound 17, compounds of the present invention can be prepared. In Step 1 compound 17 is subjected to ethyl 2-oxoacetate in the presence platinum on carbon and hydrogen gas to afford compound 18. In Step 2 compound 18 is Boc-deprotected with hydrochloric acid. In Step 3 compound 18 is cyclized to afford compound 16 which is used in the schemes above.
Scheme 2-5

11 19
Scheme 2-5: Starting from compound 11 the CDK 4/6 inhibitor 19 can be prepared. In Step 1 5-(4-methylpiperazin-l-yl)pyridin-2-amine is subjected to LiHMDS and compound 11 is added slowly under chilled conditions to afford a nucleophilic substitution reaction and compound 19. Compound 11 can be prepared as described in the schemes herein.

Scheme 2-6: Starting from compound 11 the CDK 4/6 inhibitor 20 can be prepared. In Step 1 5-(4-isopropylpiperazin-l-yl)pyridin-2-amine is subjected to LiHMDS and compound 11 is added slowly under chilled conditions to afford a nucleophilic substitution reaction and compound 20. Compound 11 can be prepared as described in the schemes herein.
Preparation of Compound 5:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet, and reflux condenser was charged with ethyl 4-chloro-2-(methylthio)pyrimidine-5-carboxylate 3 (49.2 g, 0.21 mol, 1.00 equiv.), spirolactam 4 (39.2 g, 0.23 mol, 1.10 equiv.), DIPEA (54.7 g, 0.42 mol, 2.00 equiv.), and DMAc (147.6 mL, 3 vol). The batch was heated to 90-95 °C, and after 60 h, IPC confirmed -14% (AUC) of ethyl 4-chloro-2-(methylthio)pyrimidine-5-carboxylate remained. The batch was cooled to RT, and precipitate formation was observed. The suspension was diluted with MTBE (100 mL, 2 vol) and water (442 mL, 9 vol) and stirred for 2 h at RT. The product was isolated by vacuum filtration and washed with MTBE (49 mL, 1 vol). The solid cake was conditioned for 1 h and dried under vacuum at 40 °C for 16 h to afford compound 5 [41.0 g, 53% yield] as an off-white solid with a purity of >99% AUC. ¾ MR (CDCh): δ 8.76 (d, J = 2.0 Hz, 1H), 6.51-6.29 (br, 1H), 4.33 (q, J = 7.0 Hz, 2H), 3.78 (s, 2H), 3.58 (s, 2H), 2.92 (s, 2H), 2.53 (s, 3H), 1.63-1.37 (m, 12H). LCMS (ESI, m/z = 365.3 [M+H]).
Preparation of Compound 6:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 5 [41.0 g, 0.11 mol, 1.00 equiv.], Boc-anhydride (36.8 g, 0.17 mol, 1.50 equiv.), DMAP (1.37 g, 0.01 mol, 0.10 equiv.), and dichloromethane (287 mL, 7 vol). The batch was stirred for 3 h at RT. IPC confirmed no starting material remained (AUC). The batch was concentrated into a residue under reduced pressure and taken to the next step (a quantitative yield is assumed for this step). An aliquot (200 mg) was purified by column chromatography (heptanes/ethyl acetate 0 to 100%) to afford compound 6. 1H MR (CDCh): δ 8.64 (s, 1H), 4.31 (q, J = 7.0 Hz, 2H), 4.07 (s, 2H), 3.83 (S, 2H), 3.15 (m, 2H), 2.56 (s, 3H), 172 (m, 3H), 1.59 (m, 15H), 1.42 (t, J= 7.0 Hz, 3H). LCMS (ESI, m/z = 465.2 [M+H]).
Preparation of Compound 7:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with compound 6 [residue from a previous step, quantitative yield assumed, 52.2 g, 0.11 mol, 1.00 equiv.], and THF (261 mL, 5 vol). The batch was cooled to 0°C and 1,8-diazabicyclo[5.4.0]un-dec-7-ene (17.1 g, 0.11 mmol, 1.00 equiv.) was added keeping the internal temperature in 0-10°C range. After the addition was complete, the cooling bath was removed and the reaction mixture was allowed to warm up to RT and after 2 h, IPC confirmed no starting material remained. The batch was seeded with the product (1.0 g) and was cooled to 0°C. The slurry was stirred at 0°C for 2 h. The product was isolated by vacuum filtration and washed with cold (0°C) THF (50 mL, 1 vol). The solid cake was conditioned for 1 h and dried under vacuum at 40°C for 16 h to afford 7 [47 g, quantitative yield] as a light orange solid with a purity of >99% AUC. The color of the product changed into yellow once the batch was exposed to air for an extended period of time (~ 1 day). Material was isolated with substantial amount DBU, according to proton NMR. However, it did not interfere with the next step. 1H MR (CDCh): δ 8.71 (s, 1H), 4.03 (s, 2H), 2.57 (s, 3H), 1.85 (m, 10H), 1.51 (s, 9H). LCMS (ESI, m/z = 419.2 [M+H]).
Preparation of Compound 8:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 7 [40.8 g, 0.10 mol, 1.00 equiv.], triethylamine (31.5 g, 0.31 mol, 3.20 equiv.), and dichloromethane (408 mL, 10 vol). The batch was purged with N2 for 15 min and was cooled to 0°C. Triflic anhydride (44.0 g, 0.16 mol, 1.60 equiv.) was added keeping the
internal temperature in 0-10°C range. The batch was stirred at 0°C and after 3 h, IPC confirmed -7.0% (AUC) of 7 remained. [It was speculated that the product was hydrolyzing back into starting material during the analysis.] Once the reaction was deemed complete, the batch was transferred to a 1 L, separatory funnel and was washed with 50% saturated sodium bicarbonate (200 mL, 5 vol). [It was prepared by mixing saturated sodium bicarbonate (100 mL) with water (100 mL)).] The aqueous layer was separated and was extracted with DCM (2×40 mL, 1 vol). The organic layers were combined and concentrated into a residue under reduced pressure and taken to the next step. LCMS (ESI, m/z = 551.6 [M+H]).
Preparation of Compound 9:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with compound 8 [residue from a previous step, quantitative yield assumed, 53.7 g, 0.10 mol, 1.00 equiv.], and THF (110 mL, 2 vol). The solvent was removed under vacuum distillation and the procedure was repeated two times. The flask was charged with triethylsilane (22.7 g, 0.20 mol, 2.00 equiv.), and DMF (268 mL, 5 vol). The batch was degassed by five cycles of evacuation, followed by backfilling with nitrogen. The flask was charged with tetrakis(triphenylphosphine)palladium(0) (11.3 g, 0.01 mol, 0.1 equiv.). The batch was heated to 45-50°C, and after 14 h, IPC confirmed no starting material remained. The batch was transferred to a 500 mL, separatory funnel while still warm. The reaction was partitioned between water (5 vol) and ethyl acetate (5 vol). The aqueous layer was extracted with ethyl acetate (3 x3 vol). The organic layers were combined and concentrated down to 2 volumes. The precipitate was filtered and washed with ethyl acetate (2x 1 vol). The solid cake was conditioned for 1 h and dried under vacuum at 40°C for 16 h to afford 9 [27.5 g, 70% yield] as a yellow solid with a purity of -98% AUC. Proton NMR showed some triphenylphosphine oxide present. ¾ NMR (DMSO-i¾):5 9.01 (s, 1H), 7.40 (s, 1H), 4.30 (s, 2H), 2.58 (m, 2H), 2.58 (s, 3H), 1.81 (m, 5H), 1.51 (s, 9H). LCMS (ESI, m/z = 403.4 [M+H]).
Preparation of Compound 10 from the Scheme 2-1 route:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged 9 (12.8 g, 31.8 mmol, 1.00 equiv.) and dichloromethane (64 mL, 5 vol). Trifluoroacetic acid (18.2 g, 159 mmol, 5.00 equiv.) was added over 20 min and the solution was stirred for 2 h at RT. IPC confirmed reaction was complete. The batch was transferred to a 500 mL, separatory funnel and washed with saturated sodium bicarbonate (200 mL). The aqueous layer was extracted with dichlorom ethane (3 x3 vol). The organic layers were combined and concentrated down to 1 volume. The precipitate was filtered and conditioned for 1 h and dried under vacuum at 40 °C for 16 h to afford 9 [6.72 g, 70% yield] as an off-white solid with a purity of 99.1% AUC. ¾ NMR (DMSO-dis): δ 8.95 (s, 1H), 8.32 (s, 1H), 7.15 (s, 1H), 3.68 (d, J = 1.0 Hz, 2H), 2.86 (m, 2H), 2.57 (s, 3H), 1.92 (m, 2H), 1.73 (m, 3H), 1.39 (m, 3H). LCMS, ESI, m/z = 303.2 [M+H]).
Preparation of Compound 10 from Scheme 2-2 route:
A 50 mL, three-neck flask equipped with a magnetic stirring bar, thermocouple, N2 inlet was charged 14 (680 mg, 1.62 mmol, 1.00 equiv.) and THF (6.8 mL, 10 vol). A I M solution of potassium tert-butoxide (3.2 mL, 3.24 mmol, 2.00 equiv.) in THF was added over 10 min and the solution was stirred for 2 h at RT. IPC confirmed reaction was complete. The batch was acidified with 4 N hydrogen chloride solution in dioxane (2.4 mL, 9.72 mmol, 6.00 equiv.) and stirred for additional 1 h. The batch was transferred to a 500 mL, separatory funnel and washed with saturated sodium bicarbonate (100 mL). The aqueous layer was extracted with ethyl acetate (3 x20 vol). The organic layers were combined and concentrated down to 3volumes and product precipitated. The precipitate was filtered and conditioned for 1 h and dried under vacuum at 40 °C for 16 h to afford 9 [489 mg, quantitative yield] as an off-white solid.
Preparation of Compound 11 :
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 10 (9.00 g, 29.8 mmol, 1.00 equiv.), and acetonitrile (180 mL, 20 vol). A solution of Oxone™ (45.9 g, 0.15 mol, 5.00 equiv.) in water (180 mL, 20 vol) was added to the batch over 20 min. The batch was stirred for 2 h and IPC confirmed the reaction was complete. The batch was concentrated down to ½ of the original volume and was extracted with dichloromethane DCM (4x 10 vol). The organic layers were combined; polish filtered and concentrated down to -10 vol of DCM. The product was slowly crystallized out by addition of heptanes (-30 vol). The mixture was cooled to 0°C and the product was filtered and dried under vacuum at 40 °C for 16 h to afford 11 [9.45 g, 95% yield] as an off-white solid with a purity of >99% AUC. ¾ NMR (CDCb): 5 9.24 (s, 1H), 7.78 (s, 1H), 7.46 (s, 1H), 3.89 (d, J= 2.0 Hz, 2H), 3.43 (s, 3H), 2.98 (m, 2H), 2.10 (m, 2H), 1.86 (m, 3H), 1.50 (m, 3H). LCMS (ESI, m/z = 335.2 [M+H]).
Preparation of Compound 13:
A 250 mL, single-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet, and reflux condenser was charged with 4-chloro-2-(methylthio)pyrimidine-5-carbaldehyde (2.00 g, 10.6 mmol, 1.00 equiv.), spirolactam 4 (1.96 g, 11.7 mmol, 1.10 equiv.), DIPEA (2.74 g, 21.2 mmol, 2.00 equiv.), and fert-butanol (20 mL, 10 vol). The batch was heated to 80-85 °C, and after 24 h, IPC confirmed no aldehyde 12 remained. The batch was cool to RT and concentrated into a residue, which was loaded on silica gel column. The product was eluted with ethyl acetate/heptanes (0% to 100%). The product containing fractions were pulled out and concentrated to afford 13 [0.98 g, 29% yield] as an off-white solid.
Preparation of Compound 14:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 13 [0.98 g, 3.00 mmol, 1.00 equiv.], Boc-anhydride (4.90 g, 21.5 mmol, 7.00 equiv.), DMAP (36 mg, 0.30 mmol, 0.10 equiv.), and dichloromethane (7 mL, 7 vol). The batch was stirred for 3 h at RT. IPC confirmed no starting material remained. The batch was cool to RT and concentrated into a residue, which was loaded on silica gel column. The product was eluted with ethyl acetate/heptanes (0% to 100%). The product containing fractions were pulled out and concentrated to afford 14 [0.98 g, 29% yield] as an off-white solid.
Preparation of Compound 15:
To a suspension of methyl glycinate (500 g, 3.98 mol, 1 eq) in DCM (10 L) was added
TEA dropwise at rt under nitrogen atmosphere, followed by the addition of cyclohexanone (781 g, 7.96 mol, 2 eq) dropwise over 15 min. To the resulting mixture was added TMSCN (591 g, 5.97 mol, 1.5 eq) dropwise over 1 hour while maintaining the internal reaction temperature below 35
°C. After stirred at rt for 2 hrs, the suspension became a clear solution. The progress of the reaction was monitored by H- MR.
When the methyl glycinate was consumed completely as indicated by H-NMR analysis, the reaction was quenched by water (5 L). The layers were separated. The aqueous layer was extracted with DCM (1 L). The combined organic phase was washed with water (5 L X 2) and
dried over Na2S04 (1.5 Kg). After filtration and concentration, 1.24 Kg of crude 15 was obtained as oil.
The crude 15 was dissolved in IPA (4 L). The solution was treated with HC1/IPA solution (4.4 mol/L, 1.1L) at RT. A large amount of solid was precipitated during the addition. The resulting suspension was stirred for 2 hrs. The solid product was collected by vacuum filtration and rinsed with MTBE (800 mL). 819 g of pure 15 was obtained as a white solid. The yield was 88.4%. ¾- MR (300 MHz, CD3OD) 4.20 (s, 2H), 3.88 (s, 3H), 2.30-2.40 (d, J = 12 Hz, 2H), 1.95-2.02 (d, J = 12 Hz, 2H), 1.55-1.85 (m, 5H), 1.20-1.40 (m, 1H).
Preparation of Compound 16:
To a solution of 15 (10 g, 43 mmol) in MeOH (100 mL) was added methanolic hydrochloride solution (2 .44 mol/L, 35.3 mL, 2 eq) and Pt02 (0.5 g, 5 wt %). The reaction suspension was stirred with hydrogen bubble at 40 °C for 6 hours. H- MR analysis showed consumption of 15. To the reaction mixture was added K2CO3 (15 g, 108 mmol, 2.5 eq) and the mixture was stirred for 3 hrs. The suspension was filtered and the filtrate was concentrated to dryness. The residual oil was diluted with DCM (100 mL) and resulting suspension was stirred for 3 hrs. After filtration, the filtrate was concentrated to provide crude 16 (6.6 g) as an oil. The crude 16 was diluted with EtOAc/hexane (1 : 1, 18 mL) at rt for 2 hrs. After filtration, 16 (4 g) was isolated. The obtained 16 was dissolved in DCM (16.7 mL) and hexane (100 mL) was added dropwise to precipitate the product. After further stirred for 1 h, 2.8 g of the pure 16 was isolated as a white solid. The yield was 39%. HPLC purity was 98.3%; MS (ESI): 169.2 (MH+); 1 H-NMR (300 MHz, D2O) 3.23 (s, 3H), 3.07 (s, 3H), 1.37-1.49 (m, 10H).
Preparation of compound 19:
5-(4-methylpiperazin-l-yl)pyridin-2-amine (803.1 g; 3.0 equivalents based on sulfone 11) was charged to a 22 L flask. The flask was blanketed with N2 and anhydrous THF added (12.4 kg). The resulting black-purple solution was cooled in an ice bath to < 5°C. 1M LiHMDS (4.7 L; 1.2 equivalents based on sulfone 11) was added via an addition funnel in three equal additions to keep the temperature below 10°C. Upon the completion of the addition, the reaction mixture was warmed to 16°C. The sulfone 11 (455.1 g; 1.00 equivalents) was added in five additions. Reaction proceeded until HPLC analysis of an IPC sample indicated less than 3% of sulfone 11 remained.
To quench the reaction, the contents of the 22L flask were transferred to a 100 L flask containing water. After stirring for 30 minutes at <30°C, the crude product was collected by filtration, washed with water and dried to afford 19 (387 g, 99.1% purity, 63.7% yield).
Preparation of compound 20:
5-(4-isopropylpiperazin-l-yl)pyridin-2-amine (1976.2 g; 3.0 equivalents based on sulfone 11) was charged to a 50 L flask. The flask was blanketed with N2 and anhydrous THF added (10.7 kg). The resulting black-purple solution was cooled in an ice bath to < 5°C. 1M LiHMDS (9.6 kg; 3.6 equivalents based on sulfone) was added via an addition funnel at a rate to keep the temperature below 10°C. Upon the completion of the addition, the reaction mixture was warmed to 16°C over 120 minutes by removing the ice bath. The sulfone (1000 g; 1.00 mol) was added in five additions. The reaction proceeded until HPLC analysis of an IPC sample indicated less than 1% of sulfone 11 remained. After completion of the reaction, ammonium chloride was added to the reaction mixture. The mixture stirred at < 32°C for at least 30 minutes and the solids collected by filtration to afford 20 (900 g, 99.1% purity, 64.2% yield).
Alternate Route to Spirolactam via cyclohexanone:
Scheme 2-7

26
In one embodiment the spirolactam is made via the synthetic scheme above. By reducing the nitrile group before addition of the glycinate group the reaction sequence proceeds in higher yield. The chemistry used in Step 1 is described in the literature (J. Org. Chem. 2005, 70,8027-8034), and was performed on a kilogram scale. The chemistry to convert Compound 24 into the
spirolactam was also demonstrated on kilogram scale. The Boc protection of Compound 23, is carried out at -70°C in order to limit formation of the di-Boc protected product. Experimental details of a 200 g pilot run are described below.
Step 1

200 g of cyclohexanone 21 was converted to 22 using Ti(Oi-Pr)4 /TMSCN/NH3. After work-up, 213 g of 22 was obtained. The H- MR was clean. The yield was 84%. The titanium salts were removed by vacuum filtration. In one embodiment, the titanium salts are removed by centrifugation or Celite filtration.
Step 2

190 g of 22 was mixed with LAH (2 eq) in MTBE for 30 minutes at 45°C. After work-up, 148 g of crude 23 was obtained.
Step 3

136 g of the crude 23 from step 2 was converted to 24 with 0.9 eq of B0C2O at -70°C. The reaction was completed and worked up. After concentration, 188 g of 24 was obtained. The yield was 86%. The H-NMR and C-NMR spectra confirmed that the compound was pure.
Step 4

188 g of 24 was subjected to methyl 2-bromoacetate and K2CO3 in acetonitrile to afford 25. 247 g of crude 25 was obtained.
Step 5

247 g of 25 was subjected to TFA in DCE heated to reflux to afford 26. After work-up, 112 g of 6 as TFA salt was obtained. H- MR was clean.
Step 6

26 27
Compound 26 was stirred in EtOH in the presence at room temperature overnight to afford 27. In one embodiment DCM is used as the solvent instead of EtOH.
Example 3. Purge of residual palladium from Step 5 Scheme 2-1:
Since palladium was used in Step 5 of Scheme 2-1, the levels of residual Pd present in the subsequent synthetic steps was determined. Table 2 below and Figure 3 show the palladium levels in the isolated solids.
Table 2

The material after Step 5 was isolated containing 1.47% (14700 ppm) of residual palladium. This data represents the highest level of palladium in the worst case scenario. The workup conditions of the latter steps purged nearly all of the palladium and the final product, 19 bis HC1 salt, contained 14 ppm of Pd, which is below the standard 20 ppm guidline. The Pd levels will likely be even lower once the catal st loading is optimized in Step 5.

19
The process developed in this route was a significant improvement over the one used for the first generation synthesis. Overall, the scheme consists of seven steps with five isolations, all by crystallization. No silica column chromatography is employed in the synthesis, which makes the process highly scalable. The process workup conditions can successfully purge the 1.47% of residual palladium after step 5 of Scheme 2-1.
///////////////TRILACICLIB, G1T28, G1T 28, SHR 6390, PHASE 2, G1 Therapeutics, Inc.
CN1CCN(CC1)C2=CN=C(C=C2)NC3=NC=C4C=C5C(=O)NCC6(N5C4=N3)CCCCC6
Ponesimod

Ponesimod
Phase III
MW 460.97, C23 H25 Cl N2 O4 S
A sphingosine-1-phosphate receptor 1 (S1P1) agonist potentially for the treatment of multiple sclerosis.
(Z,Z)-5-[3-chloro-4-(2R)-2,3-dihydroxy-propoxy)-benzylidene]-2-propylimino-3-o-tolylthiazolidin-4-one
- (2Z,5Z)-5-[[3-Chloro-4-[(2R)-2,3-dihydroxypropoxy]phenyl]methylene]-3-(2-methylphenyl)-2-(propylimino)-4-thiazolidinone
- 5-[3-Chloro-4-[((2R)-2,3-dihydroxypropyl)oxy]benz-(Z)-ylidene]-2-((Z)-propylimino)-3-(o-tolyl)thiazolidin-4-one
- ACT 128800
ACT-128800; RG-3477; R-3477
CAS No. 854107-55-4
update 18/3/21 FDA APPROVEDAS PONVORY
SYNTHESIS
Ponesimod
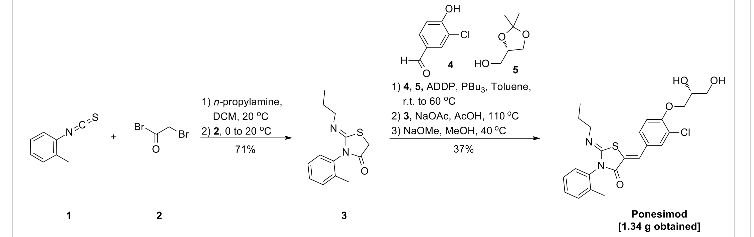

NMR CDCL3 FROM NET
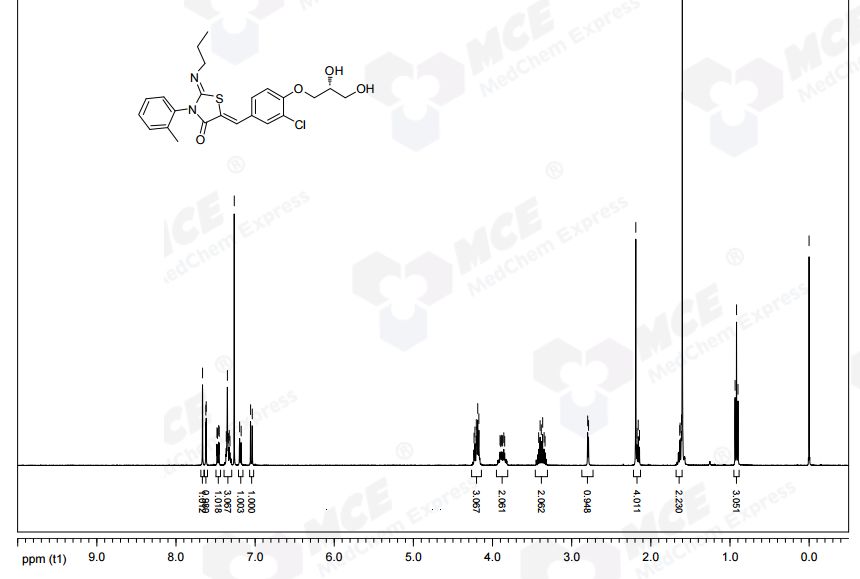
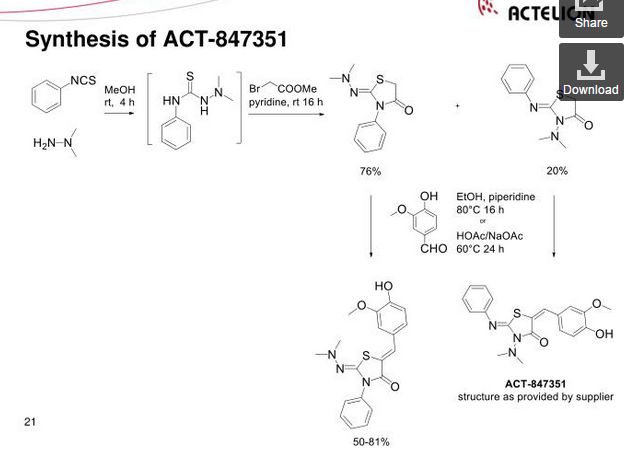
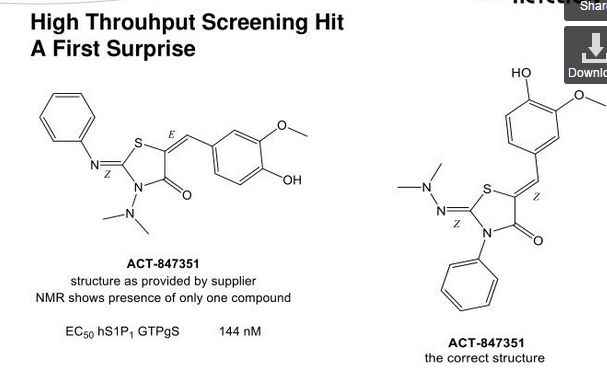
Ponesimod (INN, codenamed ACT-128800) is an experimental drug for the treatment of multiple sclerosis (MS) and psoriasis. It is being developed by Actelion.
The first oral treatment for relapsing multiple sclerosis, the nonselective sphingosine-1-phosphate receptor (S1PR) modulator fingolimod, led to identification of a pivotal role of sphingosine-1-phosphate and one of its five known receptors, S1P1R, in regulation of lymphocyte trafficking in multiple sclerosis. Modulation of S1P3R, initially thought to cause some of fingolimod’s side effects, prompted the search for novel compounds with high selectivity for S1P1R. Ponesimod is an orally active, selective S1P1R modulator that causes dose-dependent sequestration of lymphocytes in lymphoid organs. In contrast to the long half-life/slow elimination of fingolimod, ponesimod is eliminated within 1 week of discontinuation and its pharmacological effects are rapidly reversible. Clinical data in multiple sclerosis have shown a dose-dependent therapeutic effect of ponesimod and defined 20 mg as a daily dose with desired efficacy, and acceptable safety and tolerability. Phase II clinical data have also shown therapeutic efficacy of ponesimod in psoriasis. These findings have increased our understanding of psoriasis pathogenesis and suggest clinical utility of S1P1R modulation for treatment of various immune-mediated disorders. A gradual dose titration regimen was found to minimize the cardiac effects associated with initiation of ponesimod treatment. Selectivity for S1P1R, rapid onset and reversibility of pharmacological effects, and an optimized titration regimen differentiate ponesimod from fingolimod, and may lead to better safety and tolerability. Ponesimod is currently in phase III clinical development to assess efficacy and safety in relapsing multiple sclerosis. A phase II study is also ongoing to investigate the potential utility of ponesimod in chronic graft versus host disease.http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4707431/
Biology and pharmacology of sphingosine-1-phosphate receptor 1
The past decades have witnessed major advances in the treatment of autoimmune and chronic inflammatory diseases. A plethora of novel therapies targeting specific molecules involved in the inflammatory or immune system activation cascades have become available. These have significantly increased our understanding of disease pathogenesis and improved the management of immune-mediated disorders. However, most of the targeted therapies are biological drugs which need to be injected, are eliminated slowly (e.g. over several weeks) and can lose efficacy or tolerability due to their potential immunogenicity. In an attempt to overcome these hurdles, pharmaceutical research has made considerable efforts to develop novel oral targeted therapies for autoimmune and chronic inflammatory diseases.
Sphingosine-1-phosphate receptor 1 (S1P1R) is one of five known G protein-coupled receptors with nanomolar affinity for the lysophospholipid sphingosine-1-phosphate (S1P), which is generated through physiologic metabolism of the cell membrane constituent sphingomyelin by all cells [Brinkmann, 2007]. S1P receptors, including S1P1R, are widely expressed in many tissues [Chun et al. 2010]. S1P1R expression on lymphocytes controls their egress from thymus and secondary lymphoid organs [Cyster and Schwab, 2012]. Lymphocyte egress requires a gradient of S1P concentration, which is established by a high S1P concentration in blood and lymph compared with a low concentration in the interstitial fluid of lymphoid organs [Grigorova et al. 2009].
Synthetic S1P1 receptor modulators disrupt the interaction of the physiologic S1P ligand with S1P1R by promoting initial activation followed by sustained internalization and desensitization of S1P1R [Hla and Brinkmann, 2011; Pinschewer et al. 2011]. Experiments conducted in animal models of transplant rejection, multiple sclerosis, lupus erythematosus, arthritis and inflammatory bowel disease with the first-generation, nonselective S1P receptor modulator, fingolimod, have demonstrated the potential efficacy of this mode of action across several immune-mediated chronic inflammatory conditions [Brinkmann, 2007]. Fingolimod is a structural analog of sphingosine that is phosphorylated in the body by a sphingosine kinase to generate the bioactive form of the drug, fingolimod phosphate, which binds to multiple S1P receptors [Brinkmann, 2007]. Clinical trials in multiple sclerosis (MS) have confirmed the efficacy of fingolimod in relapsing MS, but not in primary progressive disease, and led to the approval of the first oral medication for the treatment of relapsing forms of MS in 2010 [Kappos et al. 2010].
The mechanism of action of fingolimod has increased our understanding of MS pathogenesis. T and B cells, but not natural killer (NK) cells, express functional S1P1R and are affected by fingolimod [Cyster and Schwab, 2012]. Furthermore, S1P1R is differentially expressed and regulated in functionally distinct subsets of lymphocytes and fingolimod has been shown to predominantly affect naïve T cells and central memory T cells (TCM) while sparing effector memory T cells (TEM), and terminally differentiated effector T cells (TE) in patients with relapsing MS [Mehling et al. 2008, 2011]. This has raised the possibility that, at least in MS, retention of TCM cells, which include pro-inflammatory T helper 17 (Th17) cells, by fingolimod may prevent their accumulation in the cerebrospinal fluid (CSF) and subsequent differentiation to TE cells in the central nervous system (CNS) [Hla and Brinkmann, 2011]. The effects of S1P1R modulation on B cells are less well defined. Recent data from patients with relapsing MS have shown predominant reduction of memory B cells and recently activated memory B cells (CD38int-high) in peripheral blood after treatment with fingolimod [Claes et al. 2014; Nakamura et al. 2014]. As memory B cells are implicated in the pathogenesis of MS and other autoimmune diseases, these observations suggest another potential mechanism underlying the therapeutic effects of S1P1R modulators.
Astrocytes, microglia, oligodendrocytes and neurons express various S1P receptors including S1P1R, S1P3R and S1P5R. Fingolimod has been shown to penetrate the CNS tissues and in vitro studies have shown activation of astrocytes and oligodendrocytes by fingolimod [Foster et al. 2007]. Conditional deletion of S1P1R on neural cells in mice reduced the severity of experimental autoimmune encephalomyelitis (EAE) and reductions in the clinical scores were paralleled by decreased demyelination, axonal loss and astrogliosis [Choi et al. 2011]. Unfortunately, there was no beneficial effect in a recently completed, large study of fingolimod in patients with primary progressive MS [Lublin et al. 2015], suggesting that the direct effect on CNS cells alone may not be sufficient. Taken together, these data suggest the possibility of a direct beneficial effect of S1P1R modulation in the brain of patients with relapsing MS [Dev et al. 2008]; however, its contribution to efficacy relative to the immunological effects remains unclear.
Initial studies in rodents suggested that modulation of S1P3R on cardiac myocytes by fingolimod was associated with a reduction of heart rate (HR) by activation of G-protein-coupled inwardly rectifying potassium channels (GIRK) that regulate pacemaker frequency, and the shape and duration of action potentials [Koyrakh et al. 2005; Camm et al. 2014]. Modulation of S1P2R and S1P3R on myofibroblasts by fingolimod was also shown to stimulate extracellular matrix synthesis [Sobel et al. 2013]. Modulation of these receptors on vascular smooth muscle cells appeared to be associated with vasoconstriction, leading to the slight increase in blood pressure observed with fingolimod treatment [Salomone et al. 2003; Watterson et al. 2005; Hu et al. 2006; Lorenz et al. 2007; Kappos et al. 2010]. These observations raised the possibility that some side effects associated with fingolimod treatment could be avoided by more selective S1P1R modulators, thus triggering the search for novel compounds.
Currently, there are several selective S1P1R modulators in clinical development [Gonzalez-Cabrera et al.2014; Subei and Cohen, 2015]. Here we review data and the development status of ponesimod, a selective S1P1R modulator developed by Actelion Pharmaceuticals Ltd.http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4707431/
Ponesimod, a selective, rapidly reversible, orally active, sphingosine-1-phosphate receptor modulator
Ponesimod (ACT-128800 (Z,Z)-5-[3-chloro-4-(2R)-2,3-dihydroxy-propoxy)-benzylidene]-2-propylimino-3-o-tolylthiazolidin-4-one) is a selective, rapidly reversible, orally active, S1P1R modulator. Ponesimod emerged from the discovery of a novel class of S1P1R agonists based on the 2-imino-thiazolidin-4-one scaffold (Figure 1) [Bolli et al. 2010]. Ponesimod activates S1P1R with high potency [half maximal effective concentration (EC50) of 5.7 nM] and selectivity. Relative to the potency of S1P, the potency of ponesimod is 4.4 higher for S1P1R and 150-fold lower for S1P3R, resulting in an approximately 650-fold higher S1P1R selectivity compared with the natural ligand.

Chemical structure of ponesimod, C23H25N2O4CIS (molecular weight 460.98).http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4707431/
Clinical trials
In a 2009–2011 Phase II clinical trial including 464 MS patients, ponesimod treatment resulted in fewer new active brain lesions thanplacebo, measured during the course of 24 weeks.[3][4]
In a 2010–2012 Phase II clinical trial including 326 patients with psoriasis, 46 or 48% of patients (depending on dosage) had a reduction of at least 75% Psoriasis Area and Severity Index (PASI) score compared to placebo in 16 weeks.[3][5]
SEE https://clinicaltrials.gov/ct2/show/NCT02425644
Adverse effects
Common adverse effects in studies were temporary bradycardia (slow heartbeat), usually at the beginning of the treatment,dyspnoea (breathing difficulties), and increased liver enzymes (without symptoms). No significant increase of infections was observed under ponesimod therapy.[3] QT prolongation is detectable but was considered to be too low to be of clinical importance in a study.[6]
Mechanism of action
Like fingolimod, which is already approved for the treatment of MS, ponesimod blocks the sphingosine-1-phosphate receptor. This mechanism prevents lymphocytes (a type of white blood cells) from leaving lymph nodes.[3] Ponesimod is selective for subtype 1 of this receptor, S1P1.[7]
PAPER
Bolli, Martin H.; Journal of Medicinal Chemistry 2010, V53(10), P4198-4211 CAPLUS
2-Imino-thiazolidin-4-one Derivatives as Potent, Orally Active S1P1Receptor Agonists
Drug Discovery Chemistry, Actelion Pharmaceuticals Ltd., Gewerbestrasse 16, CH-4123 Allschwil, Switzerland
J. Med. Chem., 2010, 53 (10), pp 4198–4211
DOI: 10.1021/jm100181s
Publication Date (Web): May 06, 2010
Copyright © 2010 American Chemical Society
*To whom correspondence should be addressed. Phone: + 41 61 565 65 70. Fax: + 41 61 565 65 00. E-mail:martin.bolli@actelion.com.

Sphingosine-1-phosphate (S1P) is a widespread lysophospholipid which displays a wealth of biological effects. Extracellular S1P conveys its activity through five specific G-protein coupled receptors numbered S1P1 through S1P5. Agonists of the S1P1 receptor block the egress of T-lymphocytes from thymus and lymphoid organs and hold promise for the oral treatment of autoimmune disorders. Here, we report on the discovery and detailed structure−activity relationships of a novel class of S1P1 receptor agonists based on the 2-imino-thiazolidin-4-one scaffold. Compound 8bo (ACT-128800) emerged from this series and is a potent, selective, and orally active S1P1 receptor agonist selected for clinical development. In the rat, maximal reduction of circulating lymphocytes was reached at a dose of 3 mg/kg. The duration of lymphocyte sequestration was dose dependent. At a dose of 100 mg/kg, the effect on lymphocyte counts was fully reversible within less than 36 h. Pharmacokinetic investigation of8bo in beagle dogs suggests that the compound is suitable for once daily dosing in humans.
(Z,Z)-5-[3-Chloro-4-((2R)-2,3-dihydroxy-propoxy)-benzylidene]-2-propylimino-3-o-tolyl-thiazolidin-4-one (8bo)
…………..DELETED…………… column chromatography on silica gel eluting with heptane:ethyl acetate 1:4 to give the title compound (1.34 g, 37%) as a pale-yellow foam.
1H NMR (CDCl3): δ 0.94 (t, J = 7.3 Hz, 3 H), 1.58−1.70 (m, 2 H), 2.21 (s, 3 H), 3.32−3.48 (m, 2 H), 3.82−3.95 (m, 3 H), 4.12−4.27 (m, 4 H), 7.07 (d, J = 8.8 Hz, 1 H), 7.21 (d, J = 7.0 Hz, 1 H), 7.31−7.39 (m, 3 H), 7.49 (dd, J = 8.5, 2.0 Hz, 1 H), 7.64 (d, J= 2.0 Hz, 1 H), 7.69 (s, 1 H).
13C NMR (CDCl3): δ 11.83, 17.68, 23.74, 55.42, 63.46, 69.85, 70.78, 133.48, 120.75, 123.71, 127.05, 128.25, 128.60, 129.43, 130.06, 131.13, 131.50, 134.42, 136.19, 146.98, 154.75, 166.12. LC-MS (ES+): tR 0.96 min. m/z: 461 (M + H).
HPLC (ChiralPak AD-H, 4.6 mm × 250 mm, 0.8 mL/min, 70% hexane in ethanol): tR 11.8 min. Anal. (C23H25N2O4SCl): C, H, N, O, S, Cl.
PATENT
WO 2014027330
https://www.google.com/patents/WO2014027330A1?cl=3Den
The present invention relates inter alia to a new process for the preparation of (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one (hereinafter also referred to as the “COMPOUND” or “compound (2)”), especially in crystalline form C which form is described in WO 2010/046835. The preparation of COMPOUND and its activity as immunosuppressive agent is described in WO 2005/054215. Furthermore, WO 2008/062376 describes a new process for the preparation of (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one which can be used as an intermediate in the preparation of COMPOUND.
Example 1 a) below describes such a process of preparing (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one according to WO 2008/062376. According to WO 2008/062376 the obtained (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one can then be transformed into COMPOUND by using standard methods for the alkylation of phenols. Such an alkylation is described in Example 1 b) below. Unfortunately, this process leads to the impurity (2Z,5Z)-5-(3-chloro-4-((1 ,3-dihydroxypropan-2-yl)oxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one which is present in about 2% w/w in the crude product (see Table 1 ) and up to 6 recrystallisations are necessary in order to get this impurity below 0.4% w/w (see Tables 1 and 2) which is the specified limit based on its toxicological qualification.


the obtained (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde (1 ) with 2-[(Z)-propylimino]-3-o-tolyl-thiazolidin-4-one to form (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one (2):

.
The reaction of (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde (1 ) with 2-[(Z)-propylimino]-3-o-tolyl-thiazolidin-4-one can be performed under conditions which are typical for a Knoevenagel condensation. Such conditions are described in the literature for example in Jones, G., Knoevenagel Condensation in Organic Reaction, Wiley: New York, 1967, Vol. 15, p 204; or Prout, F. S., Abdel-Latif, A. A., Kamal, M. R., J. Chem. Eng. Data, 2012, 57, 1881-1886.
2-[(Z)-Propylimino]-3-o-tolyl-thiazolidin-4-one can be prepared as described in WO 2008/062376, preferably without the isolation and/or purification of intermediates such as the thiourea intermediate that occurs after reacting o-tolyl-iso-thiocyanate with n-propylamine. Preferably 2-[(Z)-propylimino]-3-o-tolyl-thiazolidin-4-one obtained according to WO 2008/062376 is also not isolated and/or purified before performing the Knoevenagel condensation, i.e. before reacting 2-[(Z)-propylimino]-3-o-tolyl-thiazolidin-4-one with (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde (1 ), i.e. in a preferred embodiment compound (2) is prepared in a one-pot procedure analogous to that described in WO 2008/062376.
Example 1 : (2Z,5Z)-5-(3-Chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one

a) Preparation of (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one:

Acetic acid solution: To acetic acid (149.2 mL) are added sodium acetate (1 1 .1 1 g, 2.00 eq.) and 3-chloro-4-hydroxybenzaldehyde (10.60 g, 1.00 eq.) at 20 °C. The mixture is stirred at 20 °C until complete dissolution (2 to 3 h).
n-Propylamine (4.04 g, 1.00 eq.) is added to a solution of o-tolyl-iso-thiocyanate (10 g, 1.00 eq.) in dichloromethane (100 mL) at 20 °C. The resulting pale yellow solution is agitated for 40 min at 20 °C before IPC (conversion specification≥ 99.0 %). The reaction is cooled to -2 °C. Bromoacetyl bromide (13.53 g, 1.00 eq.) is added and the resulting solution is stirred for 15 min at -2 °C. Pyridine (10.92 g, 2.05 eq.) is then added slowly at -2 °C. The intensive yellow reaction mixture is stirred for 15 min at -2 °C before IPC (conversion specification≥ 93.0 %). 70 mL of dichloromethane are distilled off under atmospheric pressure and jacket temperature of 60 °C. The temperature is adjusted to 42 °C and the acetic acid solution is added to the reaction mixture. The resulting solution is heated to 58 °C and stirred at this temperature for 15 h before IPC (conversion specification≥ 95 %). 25 mL of solvents are distilled off under vacuum 900 – 500 mbars and jacket temperature of 80 °C. The temperature is adjusted to 60 °C and water (80.1 mL) is added to the reaction mixture over 1 h. The resulting yellow suspension is stirred at 60 °C for 30 min. The suspension is cooled to 20 °C over 1 h and stirred at this temperature for 30 min.
The product is filtered and washed with a mixture of acetic acid (30 mL) and water (16 mL) and with water (50 mL) at 20 °C. The product is dried under vacuum at 50 °C for 40 h to afford a pale yellow solid; yield 25.93 g (78 %).
b) Preparation of crude (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one:
To a suspension of (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one (10.00 g, 1.00 eq.) in ethanol (47.2 mL) is added (R)-3-chloro-1 ,2-
propanediol (3.37 g, 1.18 eq.) at 20 °C. Potassium tert-butoxide (3.39 g, 1.13 eq.) is added in portions at 20 °C. The resulting fine suspension is stirred at 20 °C for 25 min before being heated to reflux (88 °C). The reaction mixture is stirred at this temperature for 24 h before IPC (conversion specification≥ 96.0 %). After cooling down to 60 °C, acetonitrile (28.6 mL) and water (74.9 mL) are added. The resulting clear solution is cooled from 60 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.010 g, 0.001 eq.; crystalline form C can be prepared as described in WO 2010/046835) are added at 50 °C. The suspension is heated from 0 °C to 50 °C, cooled to 0 °C over 6 h and stirred at this temperature for 12 h.
The product is filtered and washed with a mixture of acetonitrile (23.4 mL) and water (23.4 mL) at 0 °C. The product is dried under vacuum at 45 °C for 24 h to afford a pale yellow solid; yield 1 1.91 g (84 %).
c) Purification of (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one:
Recrystallisation I: The crude (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one (10 g) is dissolved in acetonitrile (30 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 12.8 mL).
Recrystallisation II: The wet product is dissolved in acetonitrile (27.0 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 1 1.3 mL).
Recrystallisation III: The wet product is dissolved in acetonitrile (24.3 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4- one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 10.1 mL).
Recrystallisation IV: The wet product is dissolved in acetonitrile (21.9 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 9.1 mL).
Recrystallisation V: The wet product is dissolved in acetonitrile (19.7 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 8.2 mL).
Recrystallisation VI: The wet product is dissolved in acetonitrile (23.9 mL) at 70 °C. Water (20 mL) is added at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h.
During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2- (propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed twice with a mixture of acetonitrile (4.5 mL) and water (4.5 mL) at -10 °C.
The product is dried under vacuum at 45 °C for 24 h to afford a pale yellow solid; yield: 7.0 g (70 %).
Example 2: (R)-3-Chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde

Potassium tert-butoxide (1 18 g, 1.20 eq.) is added to n-propanol (963 mL) followed by 3-chloro-4-hydroxybenzaldehyde (137 g, 1.00 eq.). To the mixture is added (R)-3-chloro-1 ,2-propanediol (126 g, 1.30 eq.). The suspension is heated to 90 °C and stirred at this temperature for 17 h. Solvent (500 mL) is distilled off at 120 °C external temperature and reduced pressure. Water is added (1.1 L) and solvent (500 mL) is removed by distillation. The turbid solution is cooled to 20 °C. After stirring for one hour a white suspension is obtained. Water (500 mL) is added and the suspension is cooled to 10 °C. The suspension is filtered and the resulting filter cake is washed with water (500 mL). The product is dried at 50 °C and reduced pressure to yield 149 g of a white solid (73%), which is (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde in crystalline form A.
Example 3: (R)-3-Chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde
Potassium tert-butoxide (8.60 g, 1.20 eq.) is added to n-propanol (70 mL) below 15 °C, the temperature is allowed to rise. After the addition the temperature is corrected again to below 15 °C before addition of 3-chloro-4-hydroxybenzaldehyde (10 g, 1 .00 eq.). The suspension is heated to 40 °C and stirred for 30 min. (R)-3-Chloro-1 ,2-propanediol (9.18 g, 1.30 eq.) is added at 40 °C. The resulting suspension is heated to 60 °C and stirred at this temperature for 15 h then heated to 94 °C till meeting the IPC-specification (specification conversion≥ 90.0 %). The mixture is cooled to 30 °C and n-propanol is partially distilled off (-50 mL are distilled off) under reduced pressure and a maximum temperature of 50 °C, the jacket temperature is not allowed to raise above 60 °C.
Water (81 mL) is added and a second distillation is performed under the same conditions (24 mL are distilled off). The mixture is heated till homogeneous (maximum 54 °C) and then cooled to 24 °C. At 24 °C the mixture is seeded with crystalline (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde of form A (0.013 g, 0.00085 eq.). How to obtain the crystalline seeds is described in Examples 2 and 5. The reaction mixture is cooled to 0 °C over 7.5 h.
The product is filtered and washed with water (2 x 35 mL) and once with methyl tert-butyl ether (20 mL) at 5 °C. The product is dried under vacuum at 40 °C for 20 h to afford an off-white solid; yield: 10.6 g (72 %), which is (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde in crystalline form A.
Example 4: (2Z,5Z)-5-(3-Chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)- 3-(o-tolyl)thiazolidin-4-one
a) Preparation of crude (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one:
n-Propylamine (5.23 g, 1.32 eq.) is added to a solution of o-tolyl-iso-thiocyanate (10 g, 1.00 eq.) in dichloromethane (100 mL) at 20 °C. The resulting pale yellow solution is agitated for 15 min at 20 °C before IPC (conversion specification≥ 99.0 %). The reaction is cooled to -2 °C. Bromoacetyl bromide (14.88 g, 1.10 eq.) is added and the resulting solution is stirred for 15 min at -2 °C. Pyridine (10.92 g, 2.05 eq.) is then added slowly at -2 °C. The intensive yellow reaction mixture is stirred for 15 min at -2 °C before IPC (conversion specification≥ 93.0 %). Dichloromethane is partially distilled off (66 mL are distilled off) under atmospheric pressure and jacket temperature of 60 °C. Ethanol (1 1 1.4 mL), sodium acetate (12.75 g, 2.30 eq.) and (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde from Example 3 (14.38 g, 0.93 eq.) are added. The remaining dichloromethane and a part of ethanol are distilled off (49.50 mL are distilled off) under atmospheric pressure and jacket temperature up to 85 °C. The reaction mixture (orange suspension) is stirred for 3 – 5 h under reflux (78 °C) before IPC (conversion specification≥ 97.0 %).
Water (88.83 mL) is added and the temperature adjusted to 40 °C before seeding with micronized (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one in crystalline form C (0.075 g, 0.0024 eq.). The reaction mixture is cooled to 0 °C over 5 h, heated up to 40 °C, cooled to 0 °C over 6 h and stirred at this temperature for 2 h.
The product is filtered and washed with a 1 :1 ethanohwater mixture (2 x 48 mL) at 0 °C. The product is dried under vacuum at 45 °C for 10 h to afford a pale yellow solid; yield: 24.71 g (86 %).
b) Purification of (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one:
The crude (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one (10 g) is dissolved in ethanol (40 mL) at 70 °C. The temperature is adjusted at 50 °C for seeding with micronised (2Z,5Z)-5-(3-chloro-4-((R)-2,3- dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one in crystalline form C (0.016 g, 0.0016 eq.). The reaction mixture is cooled from 50 °C to 0 °C over 4 h, heated up to 50 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h.
The product is filtered and washed with ethanol at 0 °C (2 x 12.8 mL). The product is dried under vacuum at 45 °C for 10 h to afford a pale yellow solid; yield: 9.2 g (92 %).
Example 5: Preparation of crystalline seeds of (R)-3-chloro-4-(2,3-dihydroxypropoxy)- benzaldehyde
10 mg of (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde of at least 99.5% purity by 1 H-NMR assay is dissolved in a 4 mL vial by adding 1 mL of pure ethanol (puriss p. a.). The solvent is allowed to evaporate through a small hole in the cap (approx. 2 mm of diameter) of the vial until complete dryness. The white solid residue is crystalline (R)-3-chloro-4-(2,3- dihydroxypropoxy)-benzaldehyde in crystalline form A. Alternatively, methanol or methylisobutylketone (both in puriss p. a. quality) is used. This procedure is repeated until sufficient seeds are made available.

PATENT
WO 2005054215
SEE https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2005054215
| WO2005054215A1 | Nov 16, 2004 | Jun 16, 2005 | Actelion Pharmaceuticals Ltd | 5-(benz- (z) -ylidene) -thiazolidin-4-one derivatives as immunosuppressant agents |
| WO2008062376A2 | Nov 22, 2007 | May 29, 2008 | Actelion Pharmaceuticals Ltd | New process for the preparation of 2-imino-thiazolidin-4-one derivatives |
| WO2010046835A1 | Oct 19, 2009 | Apr 29, 2010 | Actelion Pharmaceuticals Ltd | Crystalline forms of (r) -5- [3-chloro-4- ( 2, 3-dihydroxy-propoxy) -benz [z] ylidene] -2- ( [z] -propylimino) -3-0-tolyl-thiazolidin-4-one |
| Reference | ||
|---|---|---|
| 1 | * | BOLLI, M.H. ET AL.: “2-Imino-thiazolidin-4-one Derivatives as Potent, Orally Active S1P1 Receptor Agonists“, JOURNAL OF MEDICINAL CHEMISTRY, vol. 53, no. 10, 2010, pages 4198-4211, XP55090073, ISSN: 0022-2623, DOI: 10.1021/jm100181s |
References
- “Multiple-dose tolerability, pharmacokinetics, and pharmacodynamics of ponesimod, an S1P1 receptor modulator: Favorable impact of dose up-titration”. The Journal of Clinical Pharmacology 54: 179–88. Feb 2014. doi:10.1002/jcph.244. PMID 24408162.
- “Mass balance, pharmacokinetics and metabolism of the selective S1P1 receptor modulator ponesimod in humans”. Xenobiotica 45: 139–49. Feb 2015. doi:10.3109/00498254.2014.955832. PMID 25188442.
- H. Spreitzer (29 September 2014). “Neue Wirkstoffe – Ponesimod”. Österreichische Apothekerzeitung (in German) (20/2014): 42.
- “Oral ponesimod in relapsing-remitting multiple sclerosis: a randomised phase II trial”. Journal of Neurology, Neurosurgery 85: 1198–208. Nov 2014. doi:10.1136/jnnp-2013-307282. PMC 4215282. PMID 24659797.
- “Oral ponesimod in patients with chronic plaque psoriasis: a randomised, double-blind, placebo-controlled phase 2 trial”. The Lancet 384: 2036–45. Dec 2014. doi:10.1016/S0140-6736(14)60803-5. PMID 25127208.
- “Effect of Ponesimod, a selective S1P1 Receptor Modulator, on the QT Interval in Healthy Subjects”. Basic 116: 429–37. May 2015.doi:10.1111/bcpt.12336. PMID 25287214.
- “Ponesimod”. Actelion. Retrieved 31 October 2014.

ABOUT PONESIMOD
Ponesimod is a potent orally active, selective sphingosine-1-phosphate receptor 1 (S1P1) immunomodulator.
Ponesimod prevents lymphocytes from leaving lymph nodes, thereby reducing circulating blood lymphocyte counts and preventing infiltration of lymphocytes into target tissues. The lymphocyte count reduction is rapid, dose-dependent, sustained upon continued dosing, and quickly reversible upon discontinuation. Initial data suggest that ponesimod does not cause lymphotoxicity by destroying/depleting lymphocytes or interfering with their cellular function. Other blood cells e.g. cells of the innate immune system are largely unaffected. Ponesimod is therefore considered a promising new oral agent for the treatment of a variety of autoimmune disorders.
CURRENT STATUS
OPTIMUM (Oral Ponesimod versus Teriflunomide In relapsing MUltiple sclerosis) is a Phase III multi-center, randomized, double-blind, parallel-group, active-controlled superiority study to compare the efficacy and safety of ponesimod to teriflunomide in patients with relapsing multiple sclerosis (RMS). The study aims to determine whether ponesimod is more efficacious than teriflunomide in reducing relapses. The study is expected to enroll approximately 1’100 patients, randomized in 2 groups in a 1:1 ratio to receive ponesimod 20 mg/day or teriflunomide 14 mg/day, and is expected to last a little over 3 years. An additional study to further characterize the utility and differentiation of ponesimod in multiple sclerosis is being discussed with Health Authorities.
Ponesimod is also evaluated in a Phase II open-label, single-arm, intra-subject dose-escalation study to investigate the biological activity, safety, tolerability, and pharmacokinetics of ponesimod in patients suffering from moderate or severe chronic graft versus host disease (GvHD)inadequately responding to first- or second-line therapy. The study will also investigate the clinical response to ponesimod treatment in these patients. Approximately 30 patients will be enrolled to receive ponesimod in escalating doses of 5, 10, and 20 mg/day over the course of 24 weeks. The study is being conducted at approximately 10 sites in the US and is expected to last approximately 18 months.
AVAILABLE CLINICAL DATA
The decision to move into Phase III development was based on the Phase IIb dose-finding study with ponesimod in patients with relapsing-remitting multiple sclerosis. A total of 464 patients were randomized into this study and the efficacy, safety and tolerability of three ponesimod doses (10, 20, and 40 mg/day) versus placebo, administered once daily for 24 weeks.
The primary endpoint of this study was defined as the cumulative number of new gadolinium-enhancing lesions on T1-weighted magnetic resonance imaging (MRI) scans at weeks 12, 16, 20, and 24 after study drug initiation. A key secondary endpoint of this study was the annualized relapse rate over 24 weeks of treatment. Patients who completed 24 weeks of treatment were offered the opportunity to enter into an extension study. This ongoing trial is investigating the long-term safety, tolerability, and efficacy of 10 and 20 mg/day of ponesimod in patients with relapsing-remitting multiple sclerosis, in a double-blind fashion. The study continues to provide extensive safety and efficacy information for ponesimod in this indication, with some patients treated for more than 6 years.
The safety database from all studies with ponesimod now comprises more than 1,300 patients and healthy volunteers.
MILESTONES
2015 – Phase III program in multiple sclerosis initiated
2011 – Phase IIb dose-finding study in multiple sclerosis successfully completed
2006 – Entry-into-man
2004 – Preclinical development initiated
KEY SCIENTIFIC LITERATURE
Olsson T et al. J Neurol Neurosurg Psychiatr. 2014 Nov;85(11):1198-208. doi: 10.1136/jnnp-2013-307282. Epub 2014 Mar 21
Freedman M.S, et al. Multiple Sclerosis Journal, 2012; 18 (4 suppl): 420 (P923).
Fernández Ó, et al. Multiple Sclerosis Journal, 2012; 18 (4 suppl): 417 (P919).
Piali L, Froidevaux S, Hess P, et al. J Pharmacol Exp Ther 337(2):547-56, 2011
Bolli MH, Abele S, Binkert C, et al. J Med Chem. 53(10):4198-211, 2010
Kappos L et al. N Engl J Med. 362(5):387-401, 2010
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(2Z,5Z)-5-{3-Chloro-4-[(2R)-2,3-dihydroxypropoxy]benzylidene}-3-(2-methylphenyl)-2-(propylimino)-1,3-thiazolidin-4-one
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | 2 main metabolites |
| Biological half-life | 31–34 hrs[1] |
| Excretion | Feces (57–80%, 26% unchanged), urine (10–18%)[2] |
| Identifiers | |
| CAS Number | 854107-55-4 |
| ATC code | none |
| PubChem | CID 11363176 |
| ChemSpider | 9538103 |
| ChEMBL | CHEMBL1096146 |
| Synonyms | ACT-128800 |
| Chemical data | |
| Formula | C23H25ClN2O4S |
| Molar mass | 460.974 g/mol |
////Ponesimod, Phase III , A sphingosine-1-phosphate receptor 1, S1P1 agonist, multiple sclerosis. ACT-128800; RG-3477; R-3477, autoimmune disease, lymphocyte migration, multiple sclerosis, psoriasis, transplantation
CCC/N=C\1/N(C(=O)/C(=C/C2=CC(=C(C=C2)OC[C@@H](CO)O)Cl)/S1)C3=CC=CC=C3C
Finerenone, BAY 94-8862
Finerenone
Finerenone; UNII-DE2O63YV8R; BAY 94-8862; DE2O63YV8R; 1050477-31-0
update FDA approved, 7/9/2021, Kerendia, To reduce the risk of kidney and heart complications in chronic kidney disease associated with type 2 diabetes
| C21H22N4O3 | |
| MW | 378.42438 g/mol |
|---|
(4s)-4-(4-cyano-2-methoxyphenyl)-5-ethoxy-2,8-dimethyl-1,4-dihydro-1-6-naphthyridine-3-carbox-amide
Bayer Corp
![]()
Mineralocorticoid receptor antagonist
phase III in January 2016, for treating diabetic kidney disease and chronic heart failure in patients with worsening chronic cardiac insufficiency
Used as mineralocorticoid receptor antagonist for treating heart failure and diabetic nephropathy.
SYNTHESIS
Finerenone (INN, USAN) (developmental code name BAY-94-8862) is a non-steroidal antimineralocorticoid that is in phase IIIclinical trials for the treatment of chronic heart failure as of October 2015. It has less relative affinity to other steroid hormone receptors than currently available antimineralocorticoids such as eplerenone and spironolactone, which should result in fewer adverse effects like gynaecomastia, impotence, and low sex drive.[1][2]
Pharmacology
Finerenone blocks mineralocorticoid receptors, which makes it a potassium-sparing diuretic.
This table compares inhibitory (blocking) concentrations (IC50, unit: nM) of three antimineralocorticoids. Mineralocorticoid receptor inhibition is responsible for the desired action of the drugs, whereas inhibition of the other receptors potentially leads to side effects. Lower values mean stronger inhibition.[1]
| Spironolactone | Eplerenone | Finerenone | |
|---|---|---|---|
| Mineralocorticoid receptor | 24 | 990 | 18 |
| Glucocorticoid receptor | 2400 | 22,000 | >10,000 |
| Androgen receptor | 77 | 21,200 | >10,000 |
| Progesterone receptor | 740 | 31,200 | >10,000 |
The above-listed drugs have insignificant affinity for the estrogen receptor.
Chemistry
Unlike currently marketed antimineralocorticoids, finerenone is not a steroid but a dihydropyridine derivative.
Research
The drug is also being investigated in early trials for the treatment of diabetic nephropathy.[3]
PAPER
Discovery of BAY 94-8862: A Nonsteroidal Antagonist of the Mineralocorticoid Receptor for the Treatment of Cardiorenal Diseases
Article first published online: 12 JUL 2012
DOI: 10.1002/cmdc.201200081

ChemMedChem
Volume 7, Issue 8, pages 1385–1403, August 2012
Abstract
Aldosterone is a hormone that exerts manifold deleterious effects on the kidneys, blood vessels, and heart which can lead to pathophysiological consequences. Inhibition of the mineralocorticoid receptor (MR) is a proven therapeutic concept for the management of associated diseases. Use of the currently marketed MR antagonists spironolactone and eplerenone is restricted, however, due to a lack of selectivity in spironolactone and the lower potency and efficacy of eplerenone. Several pharmaceutical companies have implemented programs to identify drugs that overcome the known liabilities of steroidal MR antagonists. Herein we disclose an extended SAR exploration starting from cyano-1,4-dihydropyridines that were identified by high-throughput screening. Our efforts led to the identification of a dihydronaphthyridine, BAY 94-8862, which is a potent, selective, and orally available nonsteroidal MR antagonist currently under investigation in a clinical phase II trial.
PATENT
WO2008104306,
http://www.google.co.in/patents/WO2008104306A2?cl=en
Lars Baerfacker, BELOW

Peter Kolkhof, BELOW

Karl-Heinz Schlemmer, Rolf Grosser, Adam Nitsche,Martina Klein, Klaus Muenter, Barbara Albrecht-Kuepper, Elke Hartmann,
EXAMPLES
Example 1
4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2-methyl-l, 4-dihydro-l, 6-naphthyridine-3-carboxamide

100 mg (ca. 0:24 mmol) of the compound from Example 23A are initially charged in 3 ml DMF. Is 2.94 mg Then (0.024 mmol) of 4-N, N-dimethylaminopyridine and 340 ul of ammonia (28 wt .-% – solution in water, 2:41 mmol) and 3 h at 100 0 C temperature. After cooling, the crude product is purified directly by preparative HPLC (eluent: acetonitrile / water with 0.1% formic acid, gradient 20:80 → 95: 5). There are 32 mg (37% d. Th.) The title connection receive.
LC-MS (Method 3): R, = 1:57 min; MS (EIPOS): m / z = 365 [M + H] +
1 H-NMR (300 MHz, DMSOd6): δ = 1:07 (t, 3H), 2.13 (s, 3H), 3.83 (s, 3H), 4:04 (m, 2H), 5:36 (s, IH), 6:42 (d, IH), 6.66 (br. s, 2H), 7.18 (d, IH), 7.29 (dd, IH), 7:38 (d, IH), 7.67 (d, IH), 8.80 (s, IH).
Example 2
4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,7-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carboxamide

640 mg (1.69 mmol) of the compound from Example 27A are initially charged in 30 ml of ethyl acetate, 342 mg (2.11 mmol) l, r-carbonyldiimidazole and then stirred overnight at room temperature. A TLC check (silica gel; mobile phase: cyclohexane / ethyl acetate 1: 1 or dichloromethane / methanol 9: 1) shows complete conversion. The volatile components are removed on a rotary evaporator and the residue taken up in 20 ml DMF. Subsequently, 2.36 ml of ammonia (28 wt .-% – solution in water, 16.87 mmol) was added and the reaction mixture for 8 hours at 50 0 C temperature. The solvent is distilled off under reduced pressure and the residue purified by preparative HPLC (eluent: acetonitrile / water with 0.1% formic acid, gradient 20:80 -> 95: 5). This gives 368 mg (58% d. Th.) Of the title compound.
LC-MS (method 7): R t = 1.91 min; MS (EIPOS): m / z = 379 [M + H] +
1 H-NMR (300 MHz, DMSO-d 6): δ = 1:05 (t, 3H), 2.13 (s, 3H), 2.19 (s, 3H), 3.84 (s, 3H), 4:02 (q, 2H) , 5:32 (s, IH), 6.25 (s, IH), 6.62 (br. s, 2H), 7.16 (d, IH), 7.28 (dd, IH), 7:37 (d, IH), 8.71 (s, IH ).
Example 3
e ‘f 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,7-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carbox- amide [(-) – enantiomer and (+) – enantiomer \

The racemate of Example 2 can be separated on a preparative scale by chiral HPLC into its enantiomers [column: Chiralpak IA, 250 mm x 20 mm; Eluent: methyl tert-butyl ether / methanol 85: 15 (v / v); Flow: 15 ml / min; Temperature: 30 0 C; UV detection: 220 Dm].
(-) – Enantiomer:
HPLC: R, = 5.28 min, ee> 98% [column: Chiralpak IA, 250 mm x 4.6 mm; Eluent: methyl tert-butyl ether / methanol 80:20 (v / v); Flow: 1 ml / min; Temperature: 25 0 C; UV detection: 220 nm];
specific optical rotation (chloroform, nm 589, 19.8 ° C, c = 0.50500 g / 100 ml): -239.3 °.
A single crystal X-ray structural analysis revealed a ^ -configuration at C * for this enantiomer – atom.
(+) – Enantiomer:
HPLC: R = 4:50 min, ee> 99% [column: Chiralpak IA, 250 mm x 4.6 mm; Eluent: methyl tert-butyl ether / methanol 80:20 (v / v); Flow: 1 ml / min; Temperature: 25 ° C; UV detection: 220 nm];
specific optical rotation (chloroform, nm 589, 20 0 C, c = 0.51000 g / 100 ml): + 222.7 °.
Example 4
4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carboxamide

1:46 g (3.84 mmol) of the compound from Example 3oA are introduced into 50 ml of ethyl acetate, 777 mg (4.79 mmol) l, r-carbonyldiimidazole and then stirred overnight at room temperature. A TLC check (silica gel; eluent: ethyl acetate) shows complete conversion. The volatile components are removed on a rotary evaporator and the residue taken up in 20 ml DMF.Then 10.74 ml of ammonia (28 wt% solution in water, 76.8 mmol) was added and the reaction mixture heated for 30 minutes at 100 0 C. The solvent is distilled off under reduced pressure and the residue purified by preparative HPLC (eluent: acetonitrile / water with 0.1% formic acid, gradient 20:80 -> 95: 5). After concentrating the product fractions, the residue in 40 ml of dichloromethane / methanol (1: 1 v / v) and treated with 100 ml of ethyl acetate. The solvent is concentrated to a volume of about 20 ml, whereupon the product crystallized. The precipitate is filtered off and washed with a little diethyl ether.After drying at 40 0 C in a vacuum oven obtained 1:40 g (96%. Th.) The title connection.
LC-MS (Method 3): R, = 1.64 min; MS (EIPOS): m / z = 379 [M + H] +
1 H-NMR (300 MHz, DMSOd6): δ = 1:05 (t, 3H), 2.12 (s, 3H), 2.18 (s, 3H), 3.82 (s, 3H), 3.99-4.07 (m, 2H) , 5:37 (s, IH), 6.60-6.84 (m, 2H), 7.14 (d, IH), 7.28 (dd, IH), 7:37 (d, IH), 7:55 (s, IH), 7.69 (s, IH ).
Example 5
e “M- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carbox- amide [(-) – enantiomer and (+ ) enantiomer]

The racemate of Example 4 can be separated on a preparative scale by chiral HPLC into its enantiomers [column: 680 mm x 40 mm; Silica gel phase based on the chiral selector poly (N-methacryloyl-D-leucine dicyclopropylmethylamide; eluent: ethyl acetate; temperature: 24 ° C; flow: 80 ml / min; UV detection: 260 nm].
(-) – Enantiomer:
HPLC: R = 2:48 min, ee = 99.6% [column: 250 mm x 4.6 mm; Silica gel phase based on the chiral selector poly (N-methacryloyl-D-leucine dicyclopropylmethylamide; eluent: ethyl acetate; temperature: 24 ° C; flow: 2 ml / min; UV detection: 260 nm];
specific optical rotation (chloroform, nm 589, 19.7 ° C, c = 0.38600 g / 100 ml): -148.8 °.
A single crystal X-ray structure analysis showed this enantiomer S configuration at C * – atom.
(+) – Enantiomer:
HPLC: R = 4:04 min, ee = 99.3% [column: 250 mm x 4.6 mm; Silica gel phase based on the chiral selector poly (N-methacryloyl-D-leucine dicyclopropylmethylamide; eluent: ethyl acetate; temperature: 24 ° C; flow: 2 ml / min; UV detection: 260 nm];
specific optical rotation (chloroform, nm 589, 19.8 ° C, c = 0.36300 g / 100 ml): + 153.0 °.
PATENT
The present invention relates to a novel and improved process for preparing 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-1, 4-dihydro- 1, 6-naphthyridine-3-carbox- amide of formula (I)

as well as the preparation and use of crystalline modification I of (4S) – 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-1, 4-dihydro- 1, 6-naphthyridine-3- carbox-amide of formula (I).
The compound of formula (I) acts as a non-steroidal mineralocorticoid receptor antagonist and can be used as agents for the prophylaxis and / or treatment of cardiovascular and renal diseases such as heart failure and diabetic nephropathy.
The compound of formula (I) and their preparation process are described in WO 2008/104306 and ChemMedChem 2012 7, described in 1385, in both publications a detailed discussion of research synthesis is disclosed. A disadvantage of the synthesis described there is the fact that this synthesis is not suitable for another large-scale process, since many steps in very high dilution, at very high reagent surpluses and thus run on a relatively low overall yield. Furthermore, many chromatographic cleanings are necessary, which are usually very expensive and require a high consumption of solvents, are costly and which should therefore be avoided if possible.Some stages can not be realized due to safety and procedural difficulties.
There is therefore a need for an industrially viable synthesis, reproducible in high overall yield, low production costs and high purity provides the compound of formula (I) and complies with all regulatory requirements in order to supply the clinical trials on drug and for subsequent regulatory submission to be used.
With the present invention a very efficient synthesis has been found, which allows to meet the above requirements.
In the publication ChemMedChem 2012 7, in which the research synthesis of the compound of formula (I) disclosed in 1385, the compound of formula (I), starting from vanillin prepared in 10 steps with an overall yield of 3.76% of theory , The compound of formula (I) was obtained by evaporation of the chromatography fractions as an amorphous solid, a defined process Kristalhsations- the stage for polymorphism-setting has not been described.
The following Scheme 1 shows the known process for preparing the compound of formula (I).

(II) (HI) (IV)

(V) (VI)



(XIII) (I)
Scheme 1: synthesis research of the compound of formula (I)
There are used 3 chromatographic purifications, and a chiral chromatography step to separate the enantiomers of the racemate of formula (XIII). The steps run partially in very high dilution and using very large amounts of reagent.
Thus, in particular the sequence of the preparation of the nitrile aldehyde intermediate (VI), which occupies a central role in the synthesis of atom not economically acceptable.
Furthermore, not to apply this process to an industrial scale, since [=> (IV) (III)] and excesses of acrylic acid tert-butyl ester are used for a very expensive reagents such as trifluoromethanesulfonic anhydride. When upscaling the Heck reaction (IV) => (V) formed in the boiler, a plastic similar residue resulting from the polymerization of acrylic acid tert.butyl ester used in excess. This is not acceptable in the technical implementation, there is a risk that there may be a Rührerbruch and it would lead to strong to remove residues in the agitators.
The subsequent cleavage of the double bond with sodium and the highly toxic osmium tetroxide is to be avoided since there is a delay of reaction and thereby caused to a strongly exothermic and connected with that comes a runaway under the test conditions described.
Scheme 2 illustrates the new process of the invention that the compound of formula (I) in 9 levels in 27.7% d. Th. Total yield without a chromatographic
Purification of intermediates supplies.




Scheme 2: According to the Invention for preparing the compound of formula (I).
Examples
example 1
Methyl 4-bromo-2-methoxybenzoate (XV)
3.06 kg (22.12 mol) potassium carbonate are placed in 1 acetone 3.6 and heated to reflux. To this suspension is metered in 1.2 kg of 4-bromo-2-hydroxybenzoic acid (5.53 mol) suspended in 7.8 1 of acetone and rinsed with 0.6 1 acetone. The mixture is heated for one hour under reflux (vigorous evolution of gas!). is boiled for 2.65 kg (21.01 mol) Dimethylsufat over 4 hours then metered. 2.5 hours then is stirred under reflux. The solvent is distilled off to a large extent (up to the stirrability) and returns to 12 1 toluene, then the remaining acetone is distilled off at 110 ° C. There are about 3 1 distillate distilled, these are supplemented by the addition of a further 3 1 toluene to approach. Allow to cool to 20 ° C and are 10.8 1 water were added and agitated vigorously. The organic phase is separated and the aqueous phase extracted again with 6.1 1 of toluene. The combined organic phases are washed with 3 1 of saturated sodium chloride solution, and the toluene phase is concentrated to about 4 first A quantitative analysis by evaporating a subset results converted a yield 1.306 kg (96.4% of theory). The solution is used directly in the next stage.
HPLC method A: RT about 11.9 min.
MS (EIPOS): m / z = 245 [M + H] +
H NMR (400 MHz, CD 2 C1 2 ): δ = 3.84 (s, 3H), 3.90 (s, 3H), 7:12 to 7:20 (m, 2H), 7.62 (d, 1H).
example 2
4-bromo-2-methoxybenzaldehyde (XVI)
It puts 1.936 kg (6.22 mol) 65% Red- Al solution in toluene with 1.25 1 of toluene at -5 ° C before. To this solution was dosed 0.66 kg (6.59 mol) of 1-methylpiperazine and rinsed with 150 ml of toluene, the temperature keeps you here from -7 to -5 ° C.. It is allowed for 30 minutes at 0 ° C. for. This solution is then dosed to a solution of 1.261 kg (5.147 mol) of methyl 4-bromo-2-methoxybenzoate (XV), dissolved in 4 1 of toluene, the temperature is maintained at – 8-0 ° C. Rinse twice with 0.7 1 of toluene and stirred for 1.5 hours at 0 ° C to. For working up, dosed to a 0 ° C cold aqueous sulfuric acid (12.5 1 water + 1.4 kg of conc. Sulfuric acid). The temperature should rise to a maximum of 10 ° C (slow dosage). The pH is, if necessary, by addition of further sulfuric acid to a pH of the first The organic phase is separated and extracted the aqueous phase with 7.6 1 of toluene. The combined organic phases are washed with 5.1 1 of water and then substantially concentrated and the residue taken up with 10 1 DMF. The mixture is concentrated again to about 5 1 volume. A quantitative analysis by evaporating a subset results converted a yield 1.041 kg (94.1% of theory). The solution is used directly in the next stage.
HPLC method A: RT approximately 12.1 min.
MS (EIPOS): m / z = 162 [M + H] +
X H-NMR (CDCl, 400MHz): δ = 3.93 (3H, s), 7.17 (2H, m), 7.68 (1H, d), 10:40 (1H, s)
example 3
4-formyl-3-methoxybenzonitrile (VI)
719 g (3.34 mol) of 4-bromo-2-methoxybenzaldehyde (XVI) as a solution in 4.5 1 of DMF with 313 g (0.74 mol) of potassium hexacyanoferrate (K4 [Fe (CN) 6]) and 354 g submitted (3.34 mol) of sodium carbonate and a further 1.2 1 of DMF and 3.8 g (0.017 mol) of palladium acetate. It is stirred for 3 hours at 120 ° C. Allow to cool to 20 ° C and are 5.7 1 water to approach. It is extracted with 17 1 ethyl acetate, and the aqueous phase is washed again with 17 1 of ethyl acetate to. The organic phases are combined and substantially concentrated with 5 1 of isopropanol was added and concentrated to about 2 1st The mixture is heated to boiling and dripping 2 1 of water.Allow to cool to 50 ° C and are again added 2 1 water. It is cooled to 3 ° C and stirred for one hour at this temperature. The product is filtered and washed with water (2 times 1.2 1) washed. It is dried at 40 ° C under vacuum.
Yield: 469 g (87% of theory.) Of a beige solid.
HPLC method A: RT about 8.3 min.
MS (EIPOS): m / z = 162 [M + H] +
1H-NMR (300 MHz, DMSO-d6): δ = 3.98 (s, 3H), 7:53 (d, 1H), 7.80 (s, 1H), 7.81 (d, 1H), 10:37 (s, 1H).
example 4
2-cyanoethyl 4- (4-cyano-2-methoxyphenyl) -2,8-dimethyl-5-oxo-l, 4,5,6-tett ^
din-3-carboxylate (X)
option A
1.035 kg (6.422 mol) of 4-formyl-3-methoxybenzonitrile (VI), 1.246 kg (8.028 mol) of 2-Cyanefhyl 3-oxobutanoate, 54.6 g (0.642 mol) of piperidine and 38.5 g (0.642 mol) of glacial acetic acid are heated under reflux on a water in 10 1 dichloromethane 6.5 hours. Allow to cool to room temperature and the organic phase was washed 2 times with 5 1 water. Subsequently, the dichloromethane phase is concentrated under atmospheric pressure and the still stirrable residue with 15.47 kg of 2-butanol and 0.717 kg (5.78 mol) of 4-amino-5-methylpyridone added. The residual dichloromethane is distilled off until an internal temperature of 98 ° C is reached. Then, 20 hours, heated under reflux. It is cooled to 0 ° C, can be 4 hours at this temperature is stirred and filtered off the product. It is dried at 40 ° C under vacuum to the carrier gas.
Yield: 2.049 kg (87.6% of theory based on 4-amino-5-methylpyridone, since this component is used in deficiency) of a slightly yellowish colored solid.
HPLC method A: RT about 9.7 min.
MS (EIPOS): m / z = 405 [M + H] +
Ή-NMR (300 MHz, DMSO-d 6 ): δ = 2:03 (s, 3H), 2:35 (s, 3H), 2.80 (m, 2H), 3.74 (s, 3H), 4:04 (m, 1H), 4.11 (m, 1H), 5.20 (s, 1H), 6.95 (s, 1H), 7.23 (dd, 1H), 7:28 to 7:33 (m, 2H), 8.18 (s, 1H), 10.76 (s, 1H) ,
variant B
1.344 kg (8.34 mol) of 4-formyl-3-methoxy-benzonitrile (VI), 71 g (0.834 mol) piperidine and 50.1 g (0.834 mol) of glacial acetic acid are introduced into 6 1 of isopropanol at 30 ° C within 3 hours, a solution of 1.747 kg (11.26 mol) of 2-cyanoethyl 3-oxobutanoate metered in 670 ml of isopropanol. Stirring an hour after at 30 ° C. It is cooled to 0-3 ° C and stirred at 0.5 hours. the product is filtered off and washed 2 times with 450 ml of cold isopropanol to. For yield determination is under vacuum at 50 ° C. (2.413 kg, 97% of theory..); but it is usually due to the high yield continued to work directly with the isopropanol-moist product. For this, the product is taken up with 29 1 of isopropanol and 1.277 kg (7.92
mol) of 4-amino-5-methylpyridone added, followed by 24 internal temperature under about 1.4 bar overpressure in the closed vessel is heated at 100 ° C h. It is cooled by a ramp within 5 h at 0 ° C. stirred for 3 hours at 0 ° C. It is filtered off and washed with 2.1 1 of cold isopropanol. It is dried under vacuum at 60 ° C.
Yield: 2.819 kg (88% of theory based on 4-amino-5-methylpyridone, since this component is used in deficiency) of a slightly yellowish colored solid.
HPLC method A: RT about 9.7 min.
MS (EIPOS): m / z = 405 [M + H] +
Ή-NMR (300 MHz, DMSO-d 6 ): δ = 2:03 (s, 3H), 2:35 (s, 3H), 2.80 (m, 2H), 3.74 (s, 3H), 4:04 (m, 1H), 4.11 (m, 1H), 5.20 (s, 1H), 6.95 (s, 1H), 7.23 (dd, 1H), 7:28 to 7:33 (m, 2H), 8.18 (s, 1H), 10.76 (s, 1H) ,
example 5
2- cyanoethyl-4- (4-cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carboxylate (XI)
2.142 kg (5.3 mol) of 2-cyanoethyl 4- (4-cyano-2-methoxyphenyl) -2,8-dimefhyl-5-oxo-l, 4,5,6-tetrahydro-l, 6-naphthyridin-3 carboxylate (X) and 4.70 kg (29 mol) of triethyl orthoacetate are dissolved in 12.15 1 of dimethylacetamide and 157.5 grams of concentrated sulfuric acid was added. The mixture is heated for 1.5 hours at 115 ° C and then cooled to 50 ° C. At 50 ° C are added dropwise to 30 minutes 12.15 1 water. After complete addition the Titelbelbindung (XI) is treated with 10 g seeded and further added dropwise to 12.15 1 of water over 30 minutes at 50 ° C. It is cooled to 0 ° C (ramp, 2 hours) and stirred for 2 hours at 0 ° C to. The product is filtered, washed 2 times each with 7.7 1 of water and dried in vacuo at 50 ° C.
Yield: 2114.2 g (92.2% of theory) of a slightly yellowish colored solid.
HPLC Method B: RT 10,2 min.
MS (EIPOS): m / z = 433 [M + H] +
X H-NMR (300 MHz, DMSO-d 6 ): δ = 1.11 (t, 3H), 2.16 (s, 3H), 2:42 (s, 3H), 2.78 (m, 2H), 3.77 (s, 3H) , 4:01 to 4:13 (m, 4H), 5:37 (s, 1H), 7.25 (d, 1H), 7:28 to 7:33 (m, 2H), 7.60 (s, 1H), 8:35 (s, 1H).
Alternatively, the reaction in NMP (l-methyl-2-pyrrolidone) may be carried out
2- cyanoethyl-4- (4-cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carboxylate (XI)
2.142 kg (5.3 mol) of 2-cyanoethyl 4- (4-cyano-2-methoxyphenyl) -2,8-dimethyl-5-oxo-l, 4,5,6-tetrahydro-l, 6-naphthyridin-3 carboxylate (X) and 2.35 kg (14.5 mol) of triethyl orthoacetate are in 3.21 kg NMP (l-methyl-2-pyrrolidone) and dissolved 157.5 g of concentrated sulfuric acid was added. The mixture is heated for 1.5 hours at 115 ° C and then cooled to 50 ° C. At 50 ° C are added dropwise to 30 minutes 2.2 1 water. After complete addition the Titelbelbindung (XI) is treated with 10 g seeded and dropped further 4.4 1 of water over 30 minutes at 50 ° C. It is cooled to 0 ° C (ramp, 2 hours) and stirred for 2 hours at 0 ° C to. The product is filtered off, washed 2 times each with 4 1 of water and dried under vacuum at 50 ° C.
Yield: 2180.7 g (95.1% of theory) of a slightly yellowish colored solid.
HPLC Method B: RT 10,2 min.
example 6
4- (4-cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-1, 4-dihydro- 1, 6-naphthyridine-3-carboxylic acid IXM
2.00 kg (4.624 mol) of 2-cyanoethyl 4- (4-cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carboxylate (XI ) are dissolved in a mixture of 12 1 THF and 6 1 of water and cooled to 0 ° C. To this solution, a sodium hydroxide solution is added in drops within 15 minutes at 0 ° C (prepared from 0.82 kg 45% aqueous. NaOH (9.248 mol) and 4.23 1 of water and stirred for 1.5 hours at 0 ° C to . The mixture is extracted 2 times with each 4.8 1 methyl tert-butyl and once with 4.8 1 of ethyl acetate. The aqueous solution is at 0 ° C with dilute hydrochloric acid (prepared from 0.371 kg 37% HCl and 1.51 1 water ) adjusted to pH 7. the mixture is allowed to warm to 20 ° C and adding an aqueous solution of 2.05 kg of ammonium chloride in 5.54 1 water. the mixture is stirred 1 hour at 20 ° C, the product filtered and 2 times with each each 1.5 1 water and washed once with 4 1 acetonitrile. It is dried at 40 ° C under vacuum to the carrier gas.
Yield: 1736.9 g (99% of theory..) Of an almost colorless powder (very slight yellow tinge).
HPLC Method C: RT: about 6.8 min.
MS (EIPOS): m / z = 380 [M + H]
X H-NMR (300 MHz, DMSO-d 6 ): δ = 1.14 (t, 3H), 2.14 (s, 3H), 2:37 (s, 3H), 3.73 (s, 3H), 4:04 (m, 2H) , 5:33 (s, 1H), 7.26 (m, 2H), 7:32 (s, 1H), 7:57 (s, 1H), 8.16 (s, 1H), 11:43 (br. s, 1H).
Alternative workup with toluene for extraction:
4- (4-cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carboxylic-isäure (XII)
2.00 kg (4.624 mol) of 2-cyanoethyl 4- (4-cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carboxylate (XI ) are dissolved in a mixture of 12 1 THF and 6 1 of water and cooled to 0 ° C. To this solution, a sodium hydroxide solution is added in drops within 15 minutes at 0 ° C (prepared from 0.82 kg 45% aqueous. NaOH (9.248 mol) and 4.23 1 of water and stirred for 1.5 hours at 0 ° C to . Add 5 L of toluene and 381.3 g Natiumacetat added and stirred vigorously. Allow to settle the phases and the organic phase is separated. the aqueous phase is adjusted with 10% hydrochloric acid to pH 6.9 (at about pH 9.5 is inoculated with 10 g of the title compound of). After completion of the precipitation of the product for one hour at 0 ° C is stirred and then filtered and washed twice with 4 1 of water and twice with 153 ml of toluene. the mixture is dried at 40 ° C under vacuum to carrier gas (nitrogen, 200 mbar. yield:.. 1719.5 g (98% of theory) of an almost colorless powder (very slight yellow tinge).
HPLC Method C: RT: about 6.8 min).
example 7
4- (4-cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-1, 4-dihydro- 1, 6-naphthyridine-3-carboxamide
1.60 kg (4.22 mol) of 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carboxylic-isäure ( XII) and 958 g (5.91 mol) of 1,1-carbodiimidazole be presented in 8 1 of THF and at 20 ° C 51 g (0.417 mol) of DMAP was added. Stirring for one hour at 20 ° C (gas evolution!) And then heated 2.5 hours 50 ° C. are added to this solution 2.973 kg (18.42 mol) of hexamethyldisilazane and boil for 22 hours under reflux. Man admits further 1.8 1 THF and cooled to 5 ° C. A mixture is prepared from 1.17 1 of THF and 835 g of water is metered in over 3 hours, so that the temperature is between 5 and 20 ° C remains. Then boiled for one hour under reflux, then cooled via a ramp (3 hours) at 0 ° C. and stirred for one hour at this temperature. The product is filtered off and washed 2 times with 2.4 1 THF and twice with 3.2 1 water. It is dried under vacuum at 70 ° C under a carrier gas.
Yield: 1.501 kg (. 94% of theory) of an almost colorless powder (very slight yellow tinge).
HPLC Method B: RT about 6.7 min.
MS (EIPOS): m / z = 379 [M + H]
Ή-NMR (300 MHz, DMSO-d 6 ): δ = 1:05 (t, 3H), 2.12 (s, 3H), 2.18 (s, 3H), 3.82 (s, 3H), 3.99-4.07 (m, 2H ), 5:37 (s, 1H), 6.60-6.84 (m, 2H), 7.14 (d, 1H), 7.28 (dd, 1H), 7:37 (d, 1H), 7:55 (s, 1H), 7.69 (s, 1H).
example 8
(4S) – 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy
carbox-amide (I) as a solution in acetonitrile / Methariol 40:60
Enantiomeric separation on a SMB unit
As a feed solution a solution corresponding to a concentration is used consisting of 50 g racemic 4- (4-cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridin-3 -carbox-amide (XIII) dissolved in 1 liter of a mixture of methanol / acetonitrile 60:40.
There is a SMB unit on a stationary phase: 20 chromatographed μιη Chiralpak AS-V. The pressure is 30 bar, as the eluent a mixture of methanol / acetonitrile 60:40 is used.
9.00 kg of 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carbox-amide (XII) are dissolved in 180 1 a mixture dissolved consisting of methanol / acetonitrile 60:40 and chromatographed by SMB. After concentrating the product-containing fractions, 69.68 liters of a 6.2% solution (corresponding to 4.32 kg (4S) – 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl- 1, 4-dihydro- 1, 6-naphthyridine-3-carbox-amide (I) as a solution in acetonitrile / methanol 40:60).
Yield: 4.32 kg (48% of theory.) Dissolved in 69.68 liters of acetonitrile / methanol 40:60 as a colorless fraction.
Enantiomeric purity:> 98.5% ee (HPLC, method D)
A sample is concentrated in vacuum to give: MS (EIPOS): m / z = 379 [M + H] +
Ή-NMR (300 MHz, DMSO-d 6 ): δ = 1:05 (t, 3H), 2.12 (s, 3H), 2.18 (s, 3H), 3.82 (s, 3H), 3.99-4.07 (m, 2H ), 5:37 (s, 1H), 6.60-6.84 (m, 2H), 7.14 (d, 1H), 7.28 (dd, 1H), 7:37 (d, 1H), 7:55 (s, 1H), 7.69 (s, 1H).
example 9
(4S) – 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carbox-amide (I)
Crystallization and Polymorphism setting
64.52 liters of a 6.2% solution of Example 8 in a mixture Acetonitiril / methanol 40:60 (equal 4.00 kg of compound 1) (1.2 .mu.m) via a filter cartridge and then concentrated at 250 mbar applicable so that the solution is still stirrable. It added 48 1 of ethanol denatured with toluene and distilled again at 250 mbar to stirrability from (Umdestillation on ethanol). They gave an additional 48 1 of ethanol denatured with toluene and then distilled at atmospheric pressure to a total volume of about 14 1 from (jacket temperature 98 ° C). The mixture was cooled via a ramp (4 hours) to 0 ° C, stirred for 2 hours at 0 ° C and filtered by the product from. It was washed twice with 4 1 of cold ethanol and then dried in vacuo at 50 ° C.
Yield: 3.64 kg (91% of theory.) Of a colorless, crystalline powder
Enantiomeric purity: “99% ee (HPLC method D); Retention times / RRT: (4S) – 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carbox-amide (1) ca. 11 min. RRT: 1.00; (4R) – 4- (4-Cyano-2-methoxyphenyl) -5-ethoxy-2,8-dimethyl-l, 4-dihydro-l, 6-naphthyridine-3-carbox-amide (I) is about 9 min ,RRT: 0.82
Purity:> 99.8% (HPLC method B) RT: about 6.7 min.
Content: 99.9% (against an external standard)
specific rotation (chloroform, 589 nm, 19.7 ° C, c = 0.38600 g / 100 ml): – 148.8 °.
MS (EIPOS): m / z = 379 [M + H] +
Ή-NMR (300 MHz, DMSO-d 6 ): δ = 1:05 (t, 3H), 2.12 (s, 3H), 2.18 (s, 3H), 3.82 (s, 3H), 3.99-4.07 (m, 2H ), 5:37 (s, 1H), 6.60-6.84 (m, 2H), 7.14 (d, 1H), 7.28 (dd, 1H), 7:37 (d, 1H), 7:55 (s, 1H), 7.69 (s, 1H).
Melting point: 252 ° C (compound of formula (I) in crystalline form of modification I)
Physico-chemical characterization of compound of formula (I) in crystalline form of modification I
Compound of formula (I) melts in crystalline form of modification I at 252 ° C, ΔΗ = 95 -113 Jg 1 (heating rate 20 K min 1 , Figure 1).
A depression of the melting point was observed as a function of the heating rate.
The melting point decreases at a lower heating rate (eg 2 K min “1 ) because decomposition occurs. There were no other phase transitions. A mass loss of about 0.1% was observed up to a temperature of 175 ° C.


References
- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel Herbst 2015 (German)
- Pitt, B; Anker, S. D.; Böhm, M; Gheorghiade, M; Køber, L; Krum, H; Maggioni, A. P.; Ponikowski, P; Voors, A. A.; Zannad, F; Nowack, C; Kim, S. Y.; Pieper, A; Kimmeskamp-Kirschbaum, N; Filippatos, G (2015). “Rationale and design of MinerAlocorticoid Receptor antagonist Tolerability Study-Heart Failure (ARTS-HF): A randomized study of finerenone vs. Eplerenone in patients who have worsening chronic heart failure with diabetes and/or chronic kidney disease”. European Journal of Heart Failure 17 (2): 224–32.doi:10.1002/ejhf.218. PMID 25678098.
- Bakris, G. L.; Agarwal, R; Chan, J. C.; Cooper, M. E.; Gansevoort, R. T.; Haller, H; Remuzzi, G; Rossing, P; Schmieder, R. E.; Nowack, C; Kolkhof, P; Joseph, A; Pieper, A; Kimmeskamp-Kirschbaum, N; Ruilope, L. M.; Mineralocorticoid Receptor Antagonist Tolerability Study–Diabetic Nephropathy (ARTS-DN) Study Group (2015). “Effect of Finerenone on Albuminuria in Patients with Diabetic Nephropathy: A Randomized Clinical Trial”. JAMA 314 (9): 884–94. doi:10.1001/jama.2015.10081. PMID 26325557.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(4S)-4-(4-Cyano-2-methoxyphenyl)-5-ethoxy-2,8-dimethyl-1,4-dihydro-1,6-naphthyridine-3-carboxamide
|
|
| Clinical data | |
| Legal status |
|
| Routes of administration |
Oral |
| Identifiers | |
| CAS Number | 1050477-31-0 |
| ATC code | None |
| PubChem | CID 60150535 |
| ChemSpider | 28669387 |
| UNII | DE2O63YV8R |
| KEGG | D10633 |
| ChEMBL | CHEMBL2181927 |
| Synonyms | BAY 94-8862 |
| Chemical data | |
| Formula | C21H22N4O3 |
| Molar mass | 378.42 g/mol |
SEE………http://apisynthesisint.blogspot.in/2016/02/finerenone-bay-94-8862.html
////Finerenone , BAYER, PHASE 3, BAY 94-8862
CCOC1=NC=C(C2=C1C(C(=C(N2)C)C(=O)N)C3=C(C=C(C=C3)C#N)OC)C
Fexinidazole Hoe-239

Fexinidazole, Hoe-239
1-Methyl-2-{[4-(methylsulfanyl)phenoxy]methyl}-5-nitro-1H-imidazole
| cas59729-37-2 |
| Molecular formula | C12H13N3O3S |
| Molar mass | 279.31 g mol−1 |
Sanofi (Originator)
University of Dundee
Drugs for Neglected Diseases Initiative
Winkelmann, E.; Raether, W.
Chemotherapeutically active nitro compounds. 4,5-nitroimidazoles. Part III
Arzneim-Forsch Drug Res 1978, 28(5): 739
US 4042705, DE 2531303,
UPDATE 7/16/2021 FDA APPROVES
To treat human African trypanosomiasis caused by the parasite Trypanosoma brucei gambiense
600 MG TABLET ORAL, DRUGS FOR NEGLECTED DISEASES INITIATIVE
US FDA approves fexinidazole as the first all-oral treatment for sleeping sickness
POSTED ON JULY 19
The US Food and Drug Administration (FDA) has approved fexinidazole as the first all-oral treatment for both stages of the Trypanosoma brucei gambiense form of sleeping sickness (Human African trypanosomiasis) in patients 6 years of age and older and weighing at least 20 kg.
Fexinidazole was developed as part of an innovative partnership between the non-profit research and development organization Drugs for Neglected Diseases initiative (DNDi), which conducted the pivotal clinical trials for this treatment, in partnership with the National Sleeping Sickness Programs of the Democratic Republic of Congo (DRC) and Central African Republic (CAR), and Sanofi.
Sleeping sickness is a parasitic disease transmitted by the bite of an infected tse-tse fly. It affects mostly populations living in remote rural areas of sub-Saharan Africa, where about 65 million people are at risk of infection. Left untreated, sleeping sickness is almost always fatal. Through Sanofi’s collaboration the number of sleeping sickness cases reported to the WHO has been reduced by ~97% between 2001 and 2020. DNDi, Sanofi and partners are deeply committed to ensuring access to fexinidazole in all sleeping sickness-endemic countries.
Current treatment options for the disease are effective, but burdensome for patients and health workers due to the need for infusion or injection, requiring hospitalization, especially challenging for people living in remote areas.
“Having a simple, all-oral treatment for sleeping sickness is a dream come true for frontline clinicians,” said Dr Bernard Pécoul, DNDi Executive Director. “We are proud of this latest milestone in our long-term partnership with Sanofi, developed in close collaboration with researchers in countries hard-hit by sleeping sickness.”
Fexinidazole is indicated as a 10-day once-a-day treatment for Trypanosoma brucei gambiense sleeping sickness, the most common form of the disease found in West and Central Africa. Fexinidazole is the first all-oral treatment that works both for the first stage of the disease, as well as the second stage of the disease in which the parasites have crossed the blood-brain barrier, causing patients to suffer from neuropsychiatric symptoms.
“This FDA approval is a key milestone in Sanofi’s long-term commitment to fight sleeping sickness, started 20 years ago alongside the WHO through an ambitious partnership to combat Neglected Tropical Diseases” said Luc Kuykens, Senior Vice President, Sanofi Global Health unit. “Following the positive scientific opinion granted by the European Medicines Agency end 2018, the FDA approval is an important step to revitalize efforts to support the sustainable elimination of the disease”.
As a result of FDA approval, a Tropical Disease Priority Review Voucher (PRV) has been awarded to DNDi. The FDA Tropical Disease PRV Program was established in 2007 to incentivize development of new treatments for neglected tropical diseases, including sleeping sickness. Any benefits from the PRV will be shared between Sanofi and DNDi, which will enable continued investments in innovating for and ensuring access to new health tools for sleeping sickness and other neglected diseases. Sanofi commits to continue to provide the drug free-of-charge to the World Health Organization for distribution to affected countries, as part of a long-term collaboration with WHO.
About Sleeping sickness
Sleeping sickness, or human African trypanosomiasis (HAT), is usually fatal without treatment. Transmitted by the bite of an infected tse-tse fly, following a period with nonspecific symptoms, it evolves to cause neuropsychiatric symptoms, including abnormal behaviour, and a debilitating disruption of sleep patterns that have given this neglected disease its name. About 65 million people in sub-Saharan Africa are at moderate to very high risk of infection.
About DNDi
The Drugs for Neglected Diseases initiative (DNDi) is a collaborative, patient needs-driven, not-for-profit research and development (R&D) organization that develops safe, effective, and affordable treatments for sleeping sickness, leishmaniasis, Chagas disease, filarial infections, mycetoma, paediatric HIV, hepatitis C, and covid-19. Since its inception in 2003, DNDi has delivered eight new treatments, including nifurtimox-eflornithine combination therapy (NECT) for late-stage sleeping sickness, and fexinidazole, the first all-oral drug for sleeping sickness.
Fexinidazole is an antiparasitic agent.[1] It has activity against Trypanosoma cruzi, Tritrichomonas foetus, Trichomonas vaginalis,Entamoeba histolytica,[1] Trypanosoma brucei,[2] and Leishmania donovani.[3] The biologically relevant active metabolites in vivo are the sulfoxide and sulfone [3][4]
Fexinidazole was discovered by the German pharmaceutical company Hoechst AG, but its development as a pharmaceutical was halted in the 1980s.[5] Fexinidazole is now being studied through a collaboration between Sanofi and the Drugs for Neglected Diseases Initiative for the treatment of Chagas disease and human African trypanosomiasis (sleeping sickness).[6][7] Fexinidazole is the first drug candidate for the treatment of advanced-stage sleeping sickness in thirty years.[8]
Fexinidazole is currently in phase II/III clinical development at Drugs for Neglected Diseases Initiative for the oral treatment of African trypanosomiasis (sleeping sickness). In May 2009, Sanofi (formerly known as sanofi-aventis) licensed the drug candidate to Drugs for Neglected Diseases Initiative for the development, manufacturing and distribution as a treatment of human African trypanosomiasis. Once approved, the companies plan to make the drug available on a nonprofit basis.
Fexinidazole was originally developed by a German pharmaceutical company called Hoechst, now part of Sanofi; however, its development was abandoned in the 1980s when the company gave up its tropical disease programs. Fexinidazole is one of a class of drugs known as azoles, like fluconazole, that work against fungi and may work against cancer.
-
Onset of trypanosomiasis is caused by Trypanosoma protozoa and it is said that every year 200,000 to 300,000 of new patients of African sleeping sickness fall sick. At present the number of patients of African sleeping sickness cannot be confirmed due to the low reliability of the investigative data. According to the WHO, at least 150,000 people died of African sleeping sickness in 1996 and it is said that its aftereffect remains in not less than 100,000 people. Beyond that, enormous is the damage to domestic animals caused by a disease called as nagana, and several hundred thousands of cattle which are to be protein sources for people die every year. Further, in the area of about 10,000,000 km2of savanna equal to the United States of America, cattle-breeding is impossible due to Trypanosoma. Thus, African sleeping sickness remarkably damages the health and the economical development of African people, and this is the reason why the WHO adopts the trypanosomiasis as one of the infectious diseases that should be controlled.
-
African sleeping sickness is a protozoal infectious disease by Trypanosoma transmitted through tsetse flies and the protozoa appear in the blood stream in about 10 days after infection. In the initial period of infection the protozoa multiply in the blood stream and give fever, physical weakness, headache, a pain of muscles and joints and a feeling of itching to proceed. On entering the chromic period, the central nerve is affected to show symptoms such as mental confusion and systemic convulsion, and finally the patients lapse into lethargy and are led to death.
-
The trypanosomiasis of domestic animals has Trypanosoma brucei brucei, Trypanosoma evansi, Trypanosoma congolense and Trypanosoma vivax as pathogens and is a communicable disease which affects domestic animals such as horses, cattle, pigs and dogs and, in addition, mice, guinea pigs, rabbits and the like. Particularly, the loss of cattle and horses is greatest and almost fetal, and they are led to anemia, edema, weakening and the like and fall dead in one month after infection.
-
In treating trypanosomiasis, pentamidine, melarsoprol, eflornithine and the like are used and there was a feeling in the 1960s that its eradication might be possible. However, these drugs are old and are gradually losing their efficacy. Particularly, the resistance to melarsoprol of an arsenic agent causes a big problem and the situation is so dire that patients with no efficacy only await death and the development of novel antitrypanosoma agents are strongly desired.
-
Trypanosoma mainly lives in the blood stream of the human body. This bloodstream energy metabolism depends on the glycolytic pathway localized in the organelle characteristic of the protozoa which is called as glycosome and the so-called oxidative phosphorylation does not function. However, in order to efficiently drive this glycolytic pathway, the produced NADH has to be reoxidized, and the glycerol-3-phosphate oxidation system of mitochondria plays an important role in this reoxidation. The terminal oxidase of this oxidation system functions as a quinol oxidase having a reduced ubiquinone as an electron donor and has properties greatly different from those of cytochrome oxidase of an aerobic respiration system which the host has. Particularly, a remarkable point is that the terminal oxidase of the oxidation system is non-sensitive to the cyanide which quickly inhibits the cytochrome oxidase of the host. Then, many researchers centered around Western countries have tried to develop drugs targeting this cyanide resistant oxidase but effective drugs having a selective toxicity have not been obtained.
-
Under these circumstances the present inventors et al. found that isoprenoid based physiologically active substances of ascochlorin, ascofuranone and derivatives thereof, particularly ascofuranone specifically inhibits the glycerol-3-phosphate oxidation system of trypanosome at a very low concentration of the order of nM and filed a patent application (Japanese Patent Publication A No. : H09-165332). They also clarified that acofuranone exhibits a very strong multiplication inhibition effect in the copresence of glycerin (Molecular and Biochemical Parasitology, 81: 127-136, 1996).
In consideration of practical use of ascofuranone, it was found essential to discover agents which replace glycerin and exhibit an effect of the combined use in a small amount, and by using an alkaloid compound having an indole skeleton existing in a plant of the family Simaroubaceae together with ascofuranone, the prolongation of life and recovery effect in African seeping sickness was found and a patent application was filed (Japanese Patent Application No.: 2003-24643, Japanese Patent Publication A No.: 2004-23601).
Method for the preparation of fexinidazole, useful for the treatment of parasitic diseases, visceral leishmaniasis, chagas disease and human African trypanosomiasis. Family members of the product patent, WO2005037759, are expected to expire from October 2024. This to be the first application from Drugs for Neglected Diseases Initiative (DNDi) on this API. DNDi in collaboration with Sanofi, the Swiss Tropical & Public Health Institute and the University of Dundee, is developing fexinidazole, an antiparasitic agent, for treating human African trypanosomiasis (HAT) and visceral Leishmaniasis (VL). By June 2013, phase I clinical studies had been completed and at that time, DNDi was planning to initiate a phase II proof-of-concept study in VL patients in early 2013.

fexinidazole[inn], 59729-37-2, 1-Methyl-2-((4-(methylthio)phenoxy)methyl)-5-nitro-1H-imidazole, Fexinidazol, Fexinidazolum

Chemotherapeutically active nitro compounds. 4,5-Nitroimidazoles. Part III
Synthesis
By condensation of 4 – (methylmercapto) phenol (II) with 1-mehtyl-2-chloromethyl-5-nitroimidazole (I) by means of K2CO3 in DMF (1,2) Description:. Crystals, mp 116 C. References: 1) Raether, W., Winkelman, E.; Chemotherapeutically active nitro compounds 4,5-Nitroimidazoles Part III Arzneim-Forsch 1978, 28 (5):. 739 2) Winkelmann,… E., Raether, W. (Hoechst AG); DE 2531303.
Winkelman, E.; Raether, W.;… Chemotherapeutically active nitro compounds 4,5-Nitroimidazoles Part III Arzneim-Forsch 1978, 28, 5, 739
Arzneim-Forsch1978, 28, (5): 739
………………..
http://www.google.com/patents/EP1681280A1?cl=en
…………..
US 4042705
http://www.google.co.in/patents/US4042705
…………
new patent june 2014
WO-2014079497
Process for preparing fexinidazole – comprising the reaction of 1-methyl-2-hydroxymethyl-5-nitro-imidazole with methanesulfonyl chloride, followed by reaction with 4-methylmercapto-phenol, and further manipulative steps.
1-Methyl-2-hydroxymethyl-5-nitro-imidazole is (I) and 1-methyl-2-(4-methylmercapto-phenyloxymethyl)-5-nitro-imidazole (fexinidazole) is (II) (claim 1, page 12).
The synthesis of (II) via intermediate (I) is described (example 1, pages 6-8).
A process for preparing fexinidazole comprising the reaction of 1-methyl-2-hydroxymethyl-5-nitro-imidazole with methanesulfonyl chloride in the presence of a suspension of powdered alkaline carbonate (eg potassium carbonate) in an anhydrous organic solvent (eg acetone), followed by reaction with 4-methylmercapto-phenol, removal of hydrochloride salt, and isolation and purification is claimed. Also claimed is their use for treating parasitic diseases, visceral leishmaniasis, chagas disease, and human African trypanosomiasis. Fexinidazole is known to be an antiparasitic agent.
|
2-1-1983
|
The activity of fexinidazole (HOE 239) against experimental infections with Trypanosoma cruzi, trichomonads and Entamoeba histolytica.
|
Annals of tropical medicine and parasitology
|
|
|
1-1-1983
|
The use of the 2 substituted 5-nitroimidazole, Fexinidazole (Hoe 239) in the treatment of chronic T. brucei infections in mice.
|
Zeitschrift für Parasitenkunde (Berlin, Germany)
|
|
5-1-2011
|
1-Aryl-4-nitro-1H-imidazoles, a new promising series for the treatment of human African trypanosomiasis.
|
European journal of medicinal chemistry
|
|
|
2-1-2011
|
Compounds containing 2-substituted imidazole ring for treatment against human African trypanosomiasis.
|
Bioorganic & medicinal chemistry letters
|
|
|
1-1-2011
|
Trypanocidal activity of nitroaromatic prodrugs: current treatments and future perspectives.
|
Current topics in medicinal chemistry
|
|
|
12-1-2010
|
Potential new drugs for human African trypanosomiasis: some progress at last.
|
Current opinion in infectious diseases
|
|
|
7-1-2010
|
Cross-resistance to nitro drugs and implications for treatment of human African trypanosomiasis.
|
Antimicrobial agents and chemotherapy
|
|
|
1-1-2010
|
Fexinidazole–a new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness.
|
PLoS neglected tropical diseases
|
|
|
1-1-1999
|
[Use of megazol for the treatment of trypanosomiasis].
|
Médecine tropicale : revue du Corps de santé colonial
|
|
|
11-1-1998
|
A method to assess invasion and intracellular replication of Trypanosoma cruzi based on differential uracil incorporation.
|
Journal of immunological methods
|
|
|
10-1-1996
|
Topical chemotherapy for experimental murine African CNS-trypanosomiasis: the successful use of the arsenical, melarsoprol, combined with the 5-nitroimidazoles, fexinidazole or MK-436.
|
Tropical medicine & international health : TM & IH
|
|
|
6-1-1991
|
Chemotherapy of CNS-trypanosomiasis: the combined use of the arsenicals and nitro-compounds.
|
|
11-15-2013
|
Targeting the human parasite Leishmania donovani: discovery of a new promising anti-infectious pharmacophore in 3-nitroimidazo[1,2-a]pyridine series.
|
Bioorganic & medicinal chemistry
|
|
|
10-1-2013
|
The R enantiomer of the antitubercular drug PA-824 as a potential oral treatment for visceral Leishmaniasis.
|
Antimicrobial agents and chemotherapy
|
|
|
2-1-2013
|
Assessing the essentiality of Leishmania donovani nitroreductase and its role in nitro drug activation.
|
Antimicrobial agents and chemotherapy
|
|
|
9-1-2012
|
Genotoxicity profile of fexinidazole–a drug candidate in clinical development for human African trypanomiasis (sleeping sickness).
|
Mutagenesis
|
|
|
7-15-2012
|
Discovery of nitroheterocycles active against African trypanosomes. In vitro screening and preliminary SAR studies.
|
Bioorganic & medicinal chemistry letters
|
|
|
2-1-2012
|
The anti-trypanosome drug fexinidazole shows potential for treating visceral leishmaniasis.
|
Science translational medicine
|
|
|
1-1-2012
|
Fexinidazole: a potential new drug candidate for Chagas disease.
|
PLoS neglected tropical diseases
|
|
|
1-1-2012
|
Management of trypanosomiasis and leishmaniasis.
|
British medical bulletin
|
|
|
12-1-2011
|
Antitrypanosomal activity of fexinidazole, a new oral nitroimidazole drug candidate for treatment of sleeping sickness.
|
Antimicrobial agents and chemotherapy
|
|
|
6-1-2011
|
Development of novel drugs for human African trypanosomiasis.
|
Future microbiology
|
| US3682951 * | 2 Nov 1970 | 8 Aug 1972 | Searle & Co | 1-{8 {62 -(1-adamantyloxy)halophenethyl{9 {0 imidazoles and congeners |
| US3714179 * | 8 Sep 1970 | 30 Jan 1973 | Searle & Co | 1-alkyl-2-furfurylthioimidazoles and congeners |
| US3796704 * | 16 Aug 1971 | 12 Mar 1974 | Bayer Ag | Phenyl-imidazolylalkanyl derivatives |
| US3828065 * | 11 Dec 1972 | 6 Aug 1974 | Searle & Co | 2-methyl-5-nitro-1-(2-phenylthioethyl)imidazoles |
| US3842097 * | 22 Jan 1973 | 15 Oct 1974 | Searle & Co | 2-(phenoxyalkylthio)imidazoles and congeners |
| US3910925 * | 24 May 1974 | 7 Oct 1975 | Searle & Co | {8 2-(2-Methyl-5-nitro-1-imidazolyl)ethyl{9 benzo(b)pyridyloxy ethers |
| US3922277 * | 14 Nov 1974 | 25 Nov 1975 | Hoechst Ag | (1-Alkyl-5-nitro-imidazolyl-2-alkyl)-pyridyl compounds |
| DE2124103A1 * | 14 May 1971 | 25 Nov 1971 | Title not available |
References
- Raether, W; Seidenath, H (1983). “The activity of fexinidazole (HOE 239) against experimental infections with Trypanosoma cruzi, trichomonads and Entamoeba histolytica”. Annals of Tropical Medicine and Parasitology 77 (1): 13–26. PMID 6411009.
- Jennings, FW; Urquhart, GM (1983). “The use of the 2 substituted 5-nitroimidazole, Fexinidazole (Hoe 239) in the treatment of chronic T. brucei infections in mice”. Zeitschrift für Parasitenkunde 69 (5): 577–581. doi:10.1007/bf00926669. PMID 6636983.
- Wyllie, S; Patterson, S; Stojanovski, FRC; Norval, S; Kime, R; Read, RD; Fairlamb, AH (2012). “The anti-trypanosome drug fexinidazole shows potential for treating visceral leishmaniasis”. Science Translational Medicine 4 (119): 119re1.doi:10.1126/scitranslmed.3003326. PMC 3457684. PMID 22301556.
- Sokolova, AY; Wyllie, S; Patterson, S; Oza, SL; Read, RD; Fairlamb, AH (2010). “Cross-resistance to nitro drugs and implications for treatment of human African trypanosomiasis”. Antimicrobial Agents and Chemotherapy 54 (7): 2893–900. doi:10.1128/AAC.00332-10.PMID 20439607.
- “Jump-Start on Slow Trek to Treatment for a Disease”. New York Times. January 8, 2008.
- “Fexinidazole Progresses into Clinical Development”. DNDi Newsletter. November 2009.
- “Sanofi-aventis and DNDi enter into a Collaboration Agreement on a New Drug for Sleeping Sickness, Fexinidazole”. DNDi. May 18, 2009.
- Torreele, E; Bourdin Trunz, B; Tweats, D; Kaiser, M; Brun, R; Mazué, G; Bray, MA; Pécoul, B (2010). “Fexinidazole–a new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness”. In Boelaert, Marleen. PLOS Neglected Tropical Diseases 4 (12): e923. doi:10.1371/journal.pntd.0000923. PMC 3006138. PMID 21200426.

