
Home » fda 2021 (Page 3)
Category Archives: fda 2021
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Pegcetacoplan
Sequence:
1ICVWQDWGAH RCTXK
Sequence:
1ICVWQDWGAH RCTXK
Sequence Modifications
| Type | Location | Description |
|---|---|---|
| terminal mod. | Lys-15 | C-terminal amide |
| terminal mod. | Lys-15′ | C-terminal amide |
| bridge | Cys-2 – Cys-12 | disulfide bridge, dimer |
| bridge | Lys-15 – Lys-15′ | covalent bridge, dimer |
| bridge | Cys-2′ – Cys-12′ | disulfide bridge, dimer |
| uncommon | Oaa-14 | – |
| uncommon | Oaa-14′ | – |
Pegcetacoplan
ペグセタコプラン;
FDA APPROVED Empaveli, 2021/5/14
Protein Sequence
Sequence Length: 30, 15, 15multichain; modifiedPoly(oxy-1,2-ethanediyl), α-hydro-ω-hydroxy-, 15,15′-diester with N-acetyl-L-isoleucyl-L-cysteinyl-L-valyl-1-methyl-L-tryptophyl-L-glutaminyl-L-α-aspartyl-L-tryptophylglycyl-L-alanyl-L-histidyl-L-arginyl-L-cysteinyl-L-threonyl-2-[2-(2-aminoethoxy)ethoxy]acetyl-N6-carboxy-L-lysinamide cyclic (2→12)-(disulfide)Polymer
Poly(oxy-1,2-ethanediyl), alpha-hydro-omega-hydroxy-, 15,15′-diester with N-acetyl-Lisoleucyl-L-cysteinyl-L-valyl-1-methyl-L-tryptophyl-L-glutaminyl-L-alpha-aspartyl-L-tryptophylglycyl-L-alanyl-L-histidyl-L-arginyl-L-cysteinyl-L-threonyl-2-(2-(2-aminoethoxy)ethoxy)acetyl-N6-carboxy-L-lysinamide cyclic (2�->12)-(disulfide)
O,O’-bis((S2,S12-cyclo(N-acetyl-L-isoleucyl-L-cysteinyl-L-valyl-1-methyl-Ltryptophyl-L-glutaminyl-L-alpha-aspartyl-L-tryptophylglycyl-L-alanyl-L-histidyl-L-arginyl-L-cysteinyl-L-threonyl-2-(2-(2-aminoethoxy)ethoxy)acetyl-L-lysinamide))-N6.15-carbonyl)polyethylene glycol(n = 800-1100)
- APL-2
- WHO 10743
| Formula | C170H248N50O47S4. (C2H4O)n3872.40 g·mol−1 |
|---|---|
| EfficacyDisease | Complement inhibitorParoxysmal nocturnal hemoglobinuria |
| CAS | 2019171-69-6 |
| Comment | Treatment of paroxysmal nocturnal hemoglobinuria (PNH), complement-mediated nephropathies, and age-related macular degeneration (AMD) |
- OriginatorApellis Pharmaceuticals
- ClassAnti-inflammatories; Anti-ischaemics; Antianaemics; Cyclic peptides; Eye disorder therapies; Polyethylene glycols; Urologics
- Mechanism of ActionComplement C3 inhibitors
- Orphan Drug StatusYes – Paroxysmal nocturnal haemoglobinuria; Autoimmune haemolytic anaemia; Glomerulonephritis
- RegisteredParoxysmal nocturnal haemoglobinuria
- Phase IIIAge-related macular degeneration
- Phase IIAmyotrophic lateral sclerosis; Autoimmune haemolytic anaemia; Glomerulonephritis; IgA nephropathy; Lupus nephritis; Membranous glomerulonephritis
- Phase I/IIWet age-related macular degeneration
- DiscontinuedIschaemia
- 02 Jun 2021Apellis Pharmaceuticals plans a phase III trial for Glomerulonephritis in the second half of 2021
- 25 May 2021Top-line efficacy and safety results from the phase III PRINCE trial for Paroxysmal nocturnal haemoglobinuria released by Apellis Pharmaceuticals
- 18 May 2021Registered for Paroxysmal nocturnal haemoglobinuria in USA (SC) – First global approval
Pegcetacoplan, sold under the brand name Empaveli, is a medication used to treat paroxysmal nocturnal hemoglobinuria (PNH).[1][2]
The most common side effects include injection-site reactions, infections, diarrhea, abdominal pain, respiratory tract infection, viral infection, and fatigue.[2]
Paroxysmal nocturnal hemoglobinuria is characterized by red blood cell destruction, anemia (red blood cells unable to carry enough oxygen to tissues), blood clots, and impaired bone marrow function (not making enough blood cells).[1]
Pegcetacoplan is the first treatment for paroxysmal nocturnal hemoglobinuria that binds to complement protein C3.[1] Pegcetacoplan was approved for medical use in the United States in May 2021.[1][3]
Pegcetacoplan is a complement inhibitor indicated in the treatment of paroxysmal nocturnal hemoglobinuria (PNH).5,7 Prior to its FDA approval, patients with PNH were typically treated with the C5 inhibiting monoclonal antibody eculizumab.5 Patients given eculizumab experienced less hemolysis caused by the membrane attack complex, but were still somewhat susceptible to hemolysis caused by C3b opsonization.5,6 Pegcetacoplan was developed out of a need for an inhibitor of complement mediated hemolysis further upstream of C5.5,6 Pegcetacoplan is a pegylated C3 inhibitor that can disrupt the processes leading to both forms of hemolysis that threaten patients with PNH.5
Pegcetacoplan was granted FDA approval on 14 May 2021.7
Medical uses
Pegcetacoplan is indicated to treat adults with paroxysmal nocturnal hemoglobinuria (PNH).[1][2]
EMPAVELI contains pegcetacoplan, a complement inhibitor. Pegcetacoplan is a symmetrical molecule comprised of two identical pentadecapeptides covalently bound to the ends of a linear 40-kiloDalton (kDa) PEG molecule. The peptide portions of pegcetacoplan contain 1-methyl-L-tryptophan (Trp(Me)) in position 4 and amino(ethoxyethoxy)acetic acid (AEEA) in position 14.
The molecular weight of pegcetacoplan is approximately 43.5 kDa. The molecular formula is C1970H3848N50O947S4. The structure of pegcetacoplan is shown below.
 |
EMPAVELI injection is a sterile, clear, colorless to slightly yellowish aqueous solution for subcutaneous use and is supplied in a 20-mL single-dose vial. Each 1 mL of solution contains 54 mg of pegcetacoplan, 41 mg of sorbitol, 0.384 mg of glacial acetic acid, 0.490 mg of sodium acetate trihydrate, and Water for Injection USP. EMPAVELI may also contain sodium hydroxide and/or additional glacial acetic acid for adjustment to a target pH of 5.0.
FDA approves new treatment for adults with serious rare blood disease..
FDA has approved Empaveli (pegcetacoplan) injection to treat adults with paroxysmal nocturnal hemoglobinuria (PNH), a rare, life-threatening blood disease. Empaveli is the first PNH treatment that binds to compliment protein C3.
PNH is characterized by red blood cell destruction, anemia (red blood cells unable to carry enough oxygen to tissues), blood clots, and impaired bone marrow function (not making enough blood cells). The disease affects 1-1.5 people per million. Individuals are typically diagnosed around ages 35 to 40. PNH can be serious, with median survival of 10 years after diagnosis. However, some patients live for decades with only minor symptoms.
PNH is caused by gene mutations that affect red blood cells. Red blood cells in people with these mutations are defective and can be destroyed by the immune system, which causes anemia.
The effectiveness of Empaveli was evaluated in a study enrolling 80 patients with PNH and anemia who had been taking eculizumab, a treatment previously approved for PNH. Patients first completed a four-week period during which they received Empaveli 1,080 mg twice weekly in addition to eculizumab at their previous dose. After the first four weeks, patients were randomly assigned to receive either Empaveli or their current dose of eculizumab for 16 weeks.
After 16 weeks, the severity of anemia was compared in the two treatment groups on the basis of hemoglobin concentration (a laboratory measure of anemia). In both treatment groups, the average hemoglobin was 8.7 g/dL at baseline, indicating severe anemia. (Normal hemoglobin values in adult men are 14 g/dL or above; normal values in adult women are 12 g/dL or above.) During the 16 weeks of treatment, patients in the Empaveli group had an average increase in their hemoglobin of 2.4 g/dL. Meanwhile, patients in the eculizumab group had an average decrease in their hemoglobin of 1.5 g/dL.
Empaveli is available only through a restricted program under a risk evaluation and mitigation strategy. Meningococcal (a type of bacteria) infections can occur in patients taking Empaveli and can become life-threatening or fatal if not treated early. Empaveli may also predispose individuals to serious infections, especially infections caused by encapsulated bacteria. Patients should be monitored for infusion-related reactions. Empaveli can interfere with certain laboratory tests. The most common side effects are injection site reactions, infections, diarrhea, abdominal pain, respiratory tract infection, viral infection, and fatigue.
Empaveli received priority review, fast track and orphan drug designations for this indication.
FDA granted the approval of Empaveli to Apellis Pharmaceuticals.
Adverse effects
Meningococcal (a type of bacteria) infections can occur in people taking pegcetacoplan and can become life-threatening or fatal if not treated early.[1] Pegcetacoplan may also predispose individuals to serious infections, especially infections caused by encapsulated bacteria.[1]
History
The effectiveness of pegcetacoplan was evaluated in a study enrolling 80 participants with paroxysmal nocturnal hemoglobinuria and anemia who had been taking eculizumab, a treatment previously approved for paroxysmal nocturnal hemoglobinuria.[1]
References
- ^ Jump up to:a b c d e f g h i “FDA approves new treatment for adults with serious rare blood disease”. U.S. Food and Drug Administration (FDA). 14 May 2021. Retrieved 14 May 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d https://pi.apellis.com/files/PI_Empaveli.pdf
- ^ “Apellis Announces U.S. Food and Drug Administration (FDA) Approval of Empaveli (pegcetacoplan) for Adults with Paroxysmal Nocturnal Hemoglobinuria (PNH)” (Press release). Apellis Pharmaceuticals. 14 May 2021. Retrieved 14 May 2021 – via GlobeNewswire.
External links
- “Pegcetacoplan”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03500549 for “Study to Evaluate the Efficacy and Safety of APL-2 in Patients With Paroxysmal Nocturnal Hemoglobinuria (PNH)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Empaveli |
| Other names | APL-2 |
| License data | US DailyMed: Pegcetacoplan |
| Routes of administration | Subcutaneous infusion |
| Drug class | Complement inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 2019171-69-6 |
| UNII | TO3JYR3BOU |
| KEGG | D11613 |
| ChEMBL | ChEMBL4298211 |
| Chemical and physical data | |
| Formula | C170H248N50O47S4 |
| Molar mass | 3872.40 g·mol−1 |
/////////Pegcetacoplan, ペグセタコプラン , FDA 2021, APPROVALS 2021, APL-2, WHO 10743, Apellis Pharmaceuticals, Empaveli, priority review, fast track, orphan drug
https://www.sec.gov/Archives/edgar/data/1492422/000156459020007350/apls-10k_20191231.htm
NEW DRUG APPROVALS
ONE TIME
$10.00
Sotorasib

![4-((S)-4-Acryloyl-2-methylpiperazin-1-yl)-6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=137278711&t=l)
Sotorasib
6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-propan-2-ylpyridin-3-yl)-4-[(2S)-2-methyl-4-prop-2-enoylpiperazin-1-yl]pyrido[2,3-d]pyrimidin-2-one
AMG 510
AMG-510
AMG510
| Formula | C30H30F2N6O3 |
|---|---|
| CAS | 2296729-00-3 |
| Mol weight | 560.5944 |
FDA APPROVED, 2021/5/28 Lumakras
Antineoplastic, Non-small cell lung cancer (KRAS G12C-mutated)
ソトラシブ (JAN);
Sotorasib
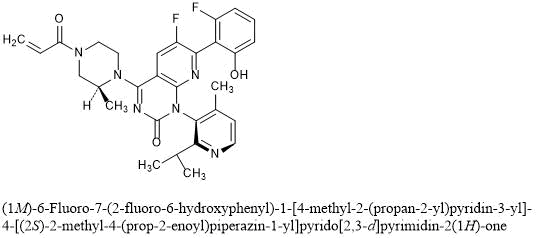
(1M)-6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one
C30H30F2N6O3 : 560.59
[2296729-00-3]
Sotorasib is an inhibitor of the RAS GTPase family. The molecular formula is C30H30F2N6O3, and the molecular weight is 560.6 g/mol. The chemical name of sotorasib is 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2enoyl) piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one. The chemical structure of sotorasib is shown below:
 |
Sotorasib has pKa values of 8.06 and 4.56. The solubility of sotorasib in the aqueous media decreases over the range pH 1.2 to 6.8 from 1.3 mg/mL to 0.03 mg/mL.
LUMAKRAS is supplied as film-coated tablets for oral use containing 120 mg of sotorasib. Inactive ingredients in the tablet core are microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, and magnesium stearate. The film coating material consists of polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and iron oxide yellow.
FDA grants accelerated approval to sotorasib for KRAS G12C mutated NSCLC
On May 28, 2021, the Food and Drug Administration granted accelerated approval to sotorasib (Lumakras™, Amgen, Inc.), a RAS GTPase family inhibitor, for adult patients with KRAS G12C ‑mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA ‑approved test, who have received at least one prior systemic therapy.
FDA also approved the QIAGEN therascreen® KRAS RGQ PCR kit (tissue) and the Guardant360® CDx (plasma) as companion diagnostics for Lumakras. If no mutation is detected in a plasma specimen, the tumor tissue should be tested.
Approval was based on CodeBreaK 100, a multicenter, single-arm, open label clinical trial (NCT03600883) which included patients with locally advanced or metastatic NSCLC with KRAS G12C mutations. Efficacy was evaluated in 124 patients whose disease had progressed on or after at least one prior systemic therapy. Patients received sotorasib 960 mg orally daily until disease progression or unacceptable toxicity.
The main efficacy outcome measures were objective response rate (ORR) according to RECIST 1.1, as evaluated by blinded independent central review and response duration. The ORR was 36% (95% CI: 28%, 45%) with a median response duration of 10 months (range 1.3+, 11.1).
The most common adverse reactions (≥ 20%) were diarrhea, musculoskeletal pain, nausea, fatigue, hepatotoxicity, and cough. The most common laboratory abnormalities (≥ 25%) were decreased lymphocytes, decreased hemoglobin, increased aspartate aminotransferase, increased alanine aminotransferase, decreased calcium, increased alkaline phosphatase, increased urine protein, and decreased sodium.
The recommended sotorasib dose is 960 mg orally once daily with or without food.
The approved 960 mg dose is based on available clinical data, as well as pharmacokinetic and pharmacodynamic modeling that support the approved dose. As part of the evaluation for this accelerated approval, FDA is requiring a postmarketing trial to investigate whether a lower dose will have a similar clinical effect.
View full prescribing information for Lumakras.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), Health Canada, and the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA). The application reviews are ongoing at the other regulatory agencies.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, the Assessment Aid, and the Product Quality Assessment Aid (PQAA), voluntary submissions from the applicant to facilitate the FDA’s assessment. The FDA approved this application approximately 10 weeks ahead of the FDA goal date.
This application was granted priority review, fast-track, breakthrough therapy and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Sotorasib, sold under the brand name Lumakras is an anti-cancer medication used to treat non-small-cell lung cancer (NSCLC).[1][2] It targets a specific mutation, G12C, in the protein KRAS which is responsible for various forms of cancer.[3][4]
The most common side effects include diarrhea, musculoskeletal pain, nausea, fatigue, liver damage and cough.[1][2]
Sotorasib is an inhibitor of the RAS GTPase family.[1]
Sotorasib is the first approved targeted therapy for tumors with any KRAS mutation, which accounts for approximately 25% of mutations in non-small cell lung cancers.[2] KRAS G12C mutations represent about 13% of mutations in non-small cell lung cancers.[2] Sotorasib was approved for medical use in the United States in May 2021.[2][5]
Sotorasib is an experimental KRAS inhibitor being investigated for the treatment of KRAS G12C mutant non small cell lung cancer, colorectal cancer, and appendix cancer.
Sotorasib, also known as AMG-510, is an acrylamide derived KRAS inhibitor developed by Amgen.1,3 It is indicated in the treatment of adult patients with KRAS G12C mutant non small cell lung cancer.6 This mutation makes up >50% of all KRAS mutations.2 Mutant KRAS discovered in 1982 but was not considered a druggable target until the mid-2010s.5 It is the first experimental KRAS inhibitor.1
The drug MRTX849 is also currently being developed and has the same target.1
Sotorasib was granted FDA approval on 28 May 2021.6
Medical uses
Sotorasib is indicated for the treatment of adults with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA-approved test, who have received at least one prior systemic therapy.[1][2]
Clinical development
Sotorasib is being developed by Amgen. Phase I clinical trials were completed in 2020.[6][7][8] In December 2019, it was approved to begin Phase II clinical trials.[9]
Because the G12C KRAS mutation is relatively common in some cancer types, 14% of non-small-cell lung cancer adenocarcinoma patients and 5% of colorectal cancer patients,[10] and sotorasib is the first drug candidate to target this mutation, there have been high expectations for the drug.[10][11][12] The Food and Drug Administration has granted a fast track designation to sotorasib for the treatment of metastatic non-small-cell lung carcinoma with the G12C KRAS mutation.[13]
Chemistry and pharmacology
Sotorasib can exist in either of two atropisomeric forms and one is more active than the other.[10] It selectively forms an irreversible covalent bond to the sulfur atom in the cysteine residue that is present in the mutated form of KRAS, but not in the normal form.[10]
History
Researchers evaluated the efficacy of sotorasib in a study of 124 participants with locally advanced or metastatic KRAS G12C-mutated non-small cell lung cancer with disease progression after receiving an immune checkpoint inhibitor and/or platinum-based chemotherapy.[2] The major outcomes measured were objective response rate (proportion of participants whose tumor is destroyed or reduced) and duration of response.[2] The objective response rate was 36% and 58% of those participants had a duration of response of six months or longer.[2]
The U.S. Food and Drug Administration (FDA) granted the application for sotorasib orphan drug, fast track, priority review, and breakthrough therapy designations.[2] The FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), Health Canada and the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA).[2] The application reviews are ongoing at the other regulatory agencies.[2]
The FDA granted approval of Lumakras to Amgen Inc.[2]
Society and culture
Economics
Sotorasib costs US$17,900 per month.[5]
Names
Sotorasib is the recommended international nonproprietary name (INN).[14]
PAPER
Nature (London, United Kingdom) (2019), 575(7781), 217-223
https://www.nature.com/articles/s41586-019-1694-1
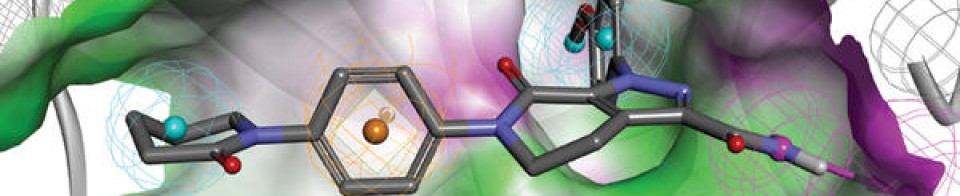
KRAS is the most frequently mutated oncogene in cancer and encodes a key signalling protein in tumours1,2. The KRAS(G12C) mutant has a cysteine residue that has been exploited to design covalent inhibitors that have promising preclinical activity3,4,5. Here we optimized a series of inhibitors, using novel binding interactions to markedly enhance their potency and selectivity. Our efforts have led to the discovery of AMG 510, which is, to our knowledge, the first KRAS(G12C) inhibitor in clinical development. In preclinical analyses, treatment with AMG 510 led to the regression of KRASG12C tumours and improved the anti-tumour efficacy of chemotherapy and targeted agents. In immune-competent mice, treatment with AMG 510 resulted in a pro-inflammatory tumour microenvironment and produced durable cures alone as well as in combination with immune-checkpoint inhibitors. Cured mice rejected the growth of isogenic KRASG12D tumours, which suggests adaptive immunity against shared antigens. Furthermore, in clinical trials, AMG 510 demonstrated anti-tumour activity in the first dosing cohorts and represents a potentially transformative therapy for patients for whom effective treatments are lacking.
Paper
Scientific Reports (2020), 10(1), 11992
PAPER
European journal of medicinal chemistry (2021), 213, 113082.
https://www.sciencedirect.com/science/article/abs/pii/S0223523420310540

KRAS is the most commonly altered oncogene of the RAS family, especially the G12C mutant (KRASG12C), which has been a promising drug target for many cancers. On the basis of the bicyclic pyridopyrimidinone framework of the first-in-class clinical KRASG12C inhibitor AMG510, a scaffold hopping strategy was conducted including a F–OH cyclization approach and a pyridinyl N-atom working approach leading to new tetracyclic and bicyclic analogues. Compound 26a was identified possessing binding potency of 1.87 μM against KRASG12C and cell growth inhibition of 0.79 μM in MIA PaCa-2 pancreatic cancer cells. Treatment of 26a with NCI–H358 cells resulted in down-regulation of KRAS-GTP levels and reduction of phosphorylation of downstream ERK and AKT dose-dependently. Molecular docking suggested that the fluorophenol moiety of 26a occupies a hydrophobic pocket region thus forming hydrogen bonding to Arg68. These results will be useful to guide further structural modification.
PAPER
Journal of Medicinal Chemistry (2020), 63(1), 52-65.
https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01180

KRASG12C has emerged as a promising target in the treatment of solid tumors. Covalent inhibitors targeting the mutant cysteine-12 residue have been shown to disrupt signaling by this long-“undruggable” target; however clinically viable inhibitors have yet to be identified. Here, we report efforts to exploit a cryptic pocket (H95/Y96/Q99) we identified in KRASG12C to identify inhibitors suitable for clinical development. Structure-based design efforts leading to the identification of a novel quinazolinone scaffold are described, along with optimization efforts that overcame a configurational stability issue arising from restricted rotation about an axially chiral biaryl bond. Biopharmaceutical optimization of the resulting leads culminated in the identification of AMG 510, a highly potent, selective, and well-tolerated KRASG12C inhibitor currently in phase I clinical trials (NCT03600883).
AMG 510 [(R)-38]. (1R)-6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1-piperazinyl]-pyrido[2,3-d]pyrimidin-2(1H)-one
………… concentrated in vacuo. Chromatographic purification of the residue (silica gel; 0–100% 3:1 EtOAc–EtOH/heptane) followed by chiral supercritical fluid chromatography (Chiralpak IC, 30 mm × 250 mm, 5 μm, 55% MeOH/CO2, 120 mL/min, 102 bar) provided (1R)-6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1-piperazinyl]pyrido[2,3-d]pyrimidin-2(1H)-one (AMG 510; (R)-38; 2.25 g, 43% yield) as the first-eluting peak. 1H NMR (600 MHz, DMSO-d6) δ ppm 10.20 (s, 1H), 8.39 (d, J = 4.9 Hz, 1H), 8.30 (d, J = 8.9 Hz, 0.5H), 8.27 (d, J = 8.7 Hz, 0.5H), 7.27 (q, J = 8.4 Hz, 1H), 7.18 (d, J = 4.9 Hz, 1H), 6.87 (dd, J = 16.2, 10.8 Hz, 0.5H), 6.84 (dd, J = 16.2, 10.7 Hz, 0.5H), 6.74 (d, J = 8.4 Hz, 1H), 6.68 (t, J = 8.4 Hz, 1H), 6.21 (d, J = 16.2 Hz, 0.5H), 6.20 (d, J = 16.2 Hz, 0.5H), 5.76 (d, J = 10.8 Hz, 0.5H), 5.76 (d, J = 10.7 Hz, 0.5H), 4.91 (m, 1H), 4.41 (d, J = 12.2 Hz, 0.5H), 4.33 (d, J = 12.2 Hz, 1H), 4.28 (d, J = 12.2 Hz, 0.5H), 4.14 (d, J = 12.2 Hz, 0.5H), 4.02 (d, J = 13.6 Hz, 0.5H), 3.69 (m, 1H), 3.65 (d, J = 13.6 Hz, 0.5H), 3.52 (t, J = 12.2 Hz, 0.5H), 3.27 (d, J = 12.2 Hz, 0.5H), 3.15 (t, J = 12.2 Hz, 0.5H), 2.72 (m, 1H), 1.90 (s, 3H), 1.35 (d, J = 6.7 Hz, 3H), 1.08 (d, J = 6.7 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H).
19F NMR (376 MHz, DMSO-d6) δ −115.6 (d, J = 5.2 Hz, 1 F), −128.6 (br s, 1 F).
13C NMR (151 MHz, DMSO-d6) δ ppm 165.0 (1C), 163.4 (1C), 162.5 (1C), 160.1 (1C), 156.8 (1C), 153.7 (1C), 151.9 (1C), 149.5 (1C), 148.3 (1C), 145.2 (1C), 144.3 (1C), 131.6 (1C), 130.8 (1C), 127.9 (0.5C), 127.9 (0.5C), 127.8 (0.5C), 127.7 (0.5C), 123.2 (1C), 122.8 (1C), 111.7 (1C), 109.7 (1C), 105.7 (1C), 105.3 (1C), 51.4 (0.5C), 51.0 (0.5C), 48.9 (0.5C), 45.4 (0.5C), 44.6 (0.5C), 43.7 (0.5C), 43.5 (0.5C), 41.6 (0.5C), 29.8 (1C), 21.9 (1C), 21.7 (1C), 17.0 (1C), 15.5 (0.5C), 14.8 (0.5C).
FTMS (ESI) m/z: [M + H]+ calcd for C30H30F2N6O3 561.24202. Found 561.24150.

d (1R)-6-Fluoro7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1- piperazinyl]-pyrido[2,3-d]pyrimidin-2(1H)-one ((R)-38; AMG 510; 2.25 g, 43% yield) as the first-eluting peak.1 H NMR (600 MHz, DMSO-d6) δ ppm 10.20 (s, 1H), 8.39 (d, J = 4.9 Hz, 1H), 8.30 (d, J = 8.9 Hz, 0.5H), 8.27 (d, J = 8.7 Hz, 0.5H), 7.27 (q, J = 8.4 Hz, 1H), 7.18 (d, J = 4.9 Hz, 1H), 6.87 (dd, J = 16.2, 10.8 Hz, 0.5H), 6.84 (dd, J = 16.2, 10.7 Hz, 0.5H), 6.74 (d, J = 8.4 Hz, 1H), 6.68 (t, J = 8.4 Hz, 1H), 6.21 (d, J = 16.2 Hz, 0.5H), 6.20 (d, J = 16.2 Hz, 0.5H), 5.76 (d, J = 10.8 Hz, 0.5H), 5.76 (d, J = 10.7 Hz, 0.5H), 4.91 (m, 1H), 4.41 (d, J = 12.2 Hz, 0.5H), 4.33 (d, J = 12.2 Hz, 1H), 4.28 (d, J = 12.2 Hz, 0.5H), 4.14 (d, J = 12.2 Hz, 0.5H), 4.02 (d, J = 13.6 Hz, 0.5H), 3.69 (m, 1H), 3.65 (d, J = 13.6 Hz, 0.5H), 3.52 (t, J = 12.2 Hz, 0.5H), 3.27 (d, J = 12.2 Hz, 0.5H), 3.15 (t, J = 12.2 Hz, 0.5H), 2.72 (m, 1H), 1.90 (s, 3H), 1.35 (d, J = 6.7 Hz, 3H), 1.08 (d, J = 6.7 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H).
19F NMR (376 MHz, DMSO-d6) δ –115.6 (d, J = 5.2 Hz, 1 F), –128.6 (br. s., 1 F).
13C NMR (151 MHz, DMSO-d6) δ ppm 165.0 (1C), 163.4 (1C), 162.5 (1C), 160.1 (1C), 156.8 (1C), 153.7 (1C), 151.9 (1C), 149.5 (1C), 148.3 (1C), 145.2 (1C), 144.3 (1C), 131.6 (1C), 130.8 (1C), 127.9 (0.5C), 127.9 (0.5C), 127.8 (0.5C), 127.7 (0.5C), 123.2 (1C), 122.8 (1C), 111.7 (1C), 109.7 (1C), 105.7 (1C), 105.3 (1C), 51.4 (0.5C), 51.0 (0.5C), 48.9 (0.5C), 45.4 (0.5C), 44.6 (0.5C), 43.7 (0.5C), 43.5 (0.5C), 41.6 (0.5C), 29.8 (1C), 21.9 (1C), 21.7 (1C), 17.0 (1C), 15.5 (0.5C), 14.8 (0.5C).
FTMS (ESI) m/z: [M+H]+ Calcd for C30H30F2N6O3 561.24202; Found 561.24150. Atropisomer configuration (R vs. S) assigned crystallographically.The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01180.
PATENT
WO 2021097212
The present disclosure relates to an improved, efficient, scalable process to prepare intermediate compounds, such as compound of Formula 6A, having the structure,
useful for the synthesis of compounds for the treatment of KRAS G12C mutated cancers.
BACKGROUND
[0003] KRAS gene mutations are common in pancreatic cancer, lung adenocarcinoma, colorectal cancer, gall bladder cancer, thyroid cancer, and bile duct cancer. KRAS mutations are also observed in about 25% of patients with NSCLC, and some studies have indicated that KRAS mutations are a negative prognostic factor in patients with NSCLC. Recently, V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations have been found to confer resistance to epidermal growth factor receptor (EGFR) targeted therapies in colorectal cancer; accordingly, the mutational status of KRAS can provide important information prior to the prescription of TKI therapy. Taken together, there is a need for new medical treatments for patients with pancreatic cancer, lung adenocarcinoma, or colorectal cancer, especially those who have been diagnosed to have such cancers characterized by a KRAS mutation, and including those who have progressed after chemotherapy.
Related Synthetic Processes
[0126] The following intermediate compounds of 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one are representative examples of the disclosure and are not intended to be construed as limiting the scope of the present invention.
[0127] A synthesis of Compound 9 and the relevant intermediates is described in U.S. Serial No.15/984,855, filed May 21, 2018 (U.S. Publication No.2018/0334454, November 22, 2018) which claims priority to and the benefit claims the benefit of U.S. Provisional Application No.62/509,629, filed on May 22, 2017, both of which are incorporated herein by reference in their entireties for all purposes. 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one was prepared using the following process, in which the isomers of the final product were isolated via chiral chromatography.
[0128] Step 1: 2,6-Dichloro-5-fluoronicotinamide (Intermediate S). To a mixture of 2,6-dichloro-5-fluoro-nicotinic acid (4.0 g, 19.1 mmol, AstaTech Inc., Bristol, PA) in dichloromethane (48 mL) was added oxalyl chloride (2M solution in DCM, 11.9 mL, 23.8 mmol), followed by a catalytic amount of DMF (0.05 mL). The reaction was stirred at room temperature overnight and then was concentrated. The residue was dissolved in 1,4-dioxane (48 mL) and cooled to 0 °C. Ammonium hydroxide solution (28.0-30% NH3 basis, 3.6 mL, 28.6 mmol) was added slowly via syringe. The resulting mixture was stirred at 0 °C for 30 min and then was concentrated. The residue was diluted with a 1:1 mixture of EtOAc/Heptane and agitated for 5 min, then was filtered. The filtered solids were discarded, and the remaining mother liquor was partially concentrated to half volume and filtered. The filtered solids were washed with heptane and dried in a reduced-pressure oven (45 °C) overnight to provide 2,6-dichloro-5-fluoronicotinamide. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.23 (d, J = 7.9 Hz, 1 H) 8.09 (br s, 1 H) 7.93 (br s, 1 H). m/z (ESI, +ve ion): 210.9 (M+H)+.
[0129] Step 2: 2,6-Dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. To an ice-cooled slurry of 2,6-dichloro-5-fluoronicotinamide (Intermediate S, 5.0 g, 23.9 mmol) in THF (20 mL) was added oxalyl chloride (2 M solution in DCM, 14.4 mL, 28.8 mmol) slowly via syringe. The resulting mixture was heated at 75 °C for 1 h, then heating was stopped, and the reaction was concentrated to half volume. After cooling to 0 °C, THF (20 mL) was added, followed by a solution of 2-isopropyl-4-methylpyridin-3-amine (Intermediate R, 3.59 g, 23.92 mmol) in THF (10 mL), dropwise via cannula. The resulting mixture was stirred at 0 °C for 1 h and then was quenched with a 1:1 mixture of brine and saturated aqueous ammonium chloride. The mixture was extracted with EtOAc (3x) and the combined organic layers were dried over anhydrous sodium sulfate and concentrated to provide 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. This material was used without further purification in the following step. m/z (ESI, +ve ion): 385.1(M+H)+.
[0130] Step 3: 7-Chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione. To an ice-cooled solution of 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (9.2 g, 24.0 mmol) in THF (40 mL) was added KHMDS (1 M solution in THF, 50.2 mL, 50.2 mmol) slowly via syringe. The ice bath was removed and the resulting mixture was stirred for 40 min at room temperature. The reaction was quenched with saturated aqueous ammonium chloride and extracted with EtOAc (3x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% 3:1 EtOAc-EtOH/heptane) to provide 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione.1H NMR (400 MHz, DMSO-d6) δ ppm 12.27 (br s, 1H), 8.48-8.55 (m, 2 H), 7.29 (d, J = 4.8 Hz, 1 H), 2.87 (quin, J = 6.6 Hz, 1 H), 1.99-2.06 (m, 3 H), 1.09 (d, J = 6.6 Hz, 3 H), 1.01 (d, J = 6.6 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -126.90 (s, 1 F). m/z (ESI, +ve ion): 349.1 (M+H)+.
[0131] Step 4: 4,7-Dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. To a solution of 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (4.7 g, 13.5 mmol) and DIPEA (3.5 mL, 20.2 mmol) in acetonitrile (20 mL) was added phosphorus oxychloride (1.63 mL, 17.5 mmol), dropwise via syringe. The resulting mixture was heated at 80 °C for 1 h, and then was cooled to room temperature and concentrated to provide 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. This material was used without further purification in the following step. m/z (ESI, +ve ion): 367.1 (M+H)+.
[0132] Step 5: (S)-tert-Butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. To an ice-cooled solution of 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one (13.5 mmol) in acetonitrile (20 mL) was added DIPEA (7.1 mL, 40.3 mmol), followed by (S)-4-N-Boc-2-methyl piperazine (3.23 g, 16.1 mmol, Combi-Blocks, Inc., San Diego, CA, USA). The resulting mixture was warmed to room temperature and stirred for 1 h, then was diluted with cold saturated aqueous sodium bicarbonate solution (200 mL) and EtOAc (300 mL). The mixture was stirred for an additional 5 min, the layers were separated, and the aqueous layer was extracted with more EtOAc (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% EtOAc/heptane) to provide (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. m/z (ESI, +ve ion): 531.2 (M+H)+.
[0133] Step 6: (3S)-tert-Butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. A mixture of (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (4.3 g, 8.1 mmol), potassium trifluoro(2-fluoro-6-hydroxyphenyl)borate (Intermediate Q, 2.9 g, 10.5 mmol), potassium acetate (3.2 g, 32.4 mmol) and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with dichloromethane (661 mg, 0.81 mmol) in 1,4-dioxane (80 mL) was degassed with nitrogen for 1 min. De-oxygenated water (14 mL) was added, and the resulting mixture was heated at 90 °C for 1 h. The reaction was allowed to cool to room temperature, quenched with half-saturated aqueous sodium bicarbonate, and extracted with EtOAc (2x) and DCM (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-60% 3:1 EtOAc-EtOH/heptane) to provide (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate.1H NMR (400 MHz, DMSO-d6) δ ppm 10.19 (br s, 1 H), 8.38 (d, J = 5.0 Hz, 1 H), 8.26 (dd, J = 12.5, 9.2 Hz, 1 H), 7.23-7.28 (m, 1 H), 7.18 (d, J = 5.0 Hz, 1 H), 6.72 (d, J = 8.0 Hz, 1 H), 6.68 (t, J = 8.9 Hz, 1 H), 4.77-4.98 (m, 1 H), 4.24 (br t, J = 14.2 Hz, 1 H), 3.93-4.08 (m, 1 H), 3.84 (br d, J=12.9 Hz, 1 H), 3.52-3.75 (m, 1 H), 3.07-3.28 (m, 1 H), 2.62-2.74 (m, 1 H), 1.86-1.93 (m, 3 H), 1.43-1.48 (m, 9 H), 1.35 (dd, J = 10.8, 6.8 Hz, 3 H), 1.26-1.32 (m, 1 H), 1.07 (dd, J = 6.6, 1.7 Hz, 3 H), 0.93 (dd, J = 6.6, 2.1 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -115.65 (s, 1 F), -128.62 (s, 1 F). m/z (ESI, +ve ion): 607.3 (M+H)+.
[0134] Step 7: 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one. Trifluoroacetic acid (25 mL, 324 mmol) was added to a solution of (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (6.3 g, 10.4 mmol) in DCM (30 mL). The resulting mixture was stirred at room temperature for 1 h and then was concentrated. The residue was dissolved in DCM (30 mL), cooled to 0 °C, and sequentially treated with DIPEA (7.3 mL, 41.7 mmol) and a solution of acryloyl chloride (0.849 mL, 10.4 mmol) in DCM (3 mL; added dropwise via syringe). The reaction was stirred at 0 °C for 10 min, then was quenched with half-saturated aqueous sodium bicarbonate and extracted with DCM (2x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-100% 3:1 EtOAc-EtOH/heptane) to provide 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one.1H NMR (400 MHz, DMSO-d6) δ ppm 10.20 (s, 1 H), 8.39 (d, J = 4.8 Hz, 1 H), 8.24-8.34 (m, 1 H), 7.23-7.32 (m, 1 H), 7.19 (d, J = 5.0 Hz, 1 H), 6.87 (td, J = 16.3, 11.0 Hz, 1 H), 6.74 (d, J = 8.6 Hz, 1 H), 6.69 (t, J = 8.6 Hz, 1 H), 6.21 (br d, J = 16.2 Hz, 1 H), 5.74-5.80 (m, 1 H), 4.91 (br s, 1 H), 4.23-4.45 (m, 2 H), 3.97-4.21 (m, 1 H), 3.44-3.79 (m, 2 H), 3.11-3.31 (m, 1 H), 2.67-2.77 (m, 1 H), 1.91 (s, 3 H), 1.35 (d, J = 6.8 Hz, 3 H), 1.08 (d, J = 6.6 Hz, 3 H), 0.94 (d, J = 6.8 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ ppm -115.64 (s, 1 F), -128.63 (s, 1 F). m/z (ESI, +ve ion): 561.2 (M+H)+.
[0135] Another synthesis of Compound 9 and the relevant intermediates was described in a U.S. provisional patent application filed November 16, 2018, which is incorporated herein by reference in its entirety for all purposes.
Representative Synthetic Processes
[0136] The present disclosure comprises the following steps wherein the synthesis and utilization of the boroxine intermediate is a novel and inventive step in the manufacture of AMG 510 (Compound 9):
Raw Materials
Step la
[0137] To a solution of 2,6-dichloro-5-fluoro-3-pyridinecarboxylic acid (25kg; 119. lmol) in dichloromethane (167kg) and DMF (592g) was added Oxalyl chloride (18.9kg; 148.9mol) while maintaining an internal temp between 15-20 °C. Additional dichloromethane (33kg) was added as a rinse and the reaction mixture stirred for 2h. The reaction mixture is cooled then quenched with ammonium hydroxide (40.2L; 595.5mol) while maintaining internal temperature 0 ± 10°C. The resulting slurry was stirred for 90min then the product collected by filtration. The filtered solids were washed with DI water (3X 87L) and dried to provide 2,6-dichloro-5-fluoronicotinamide (Compound 1).
Step 1b
[0138] In reactor A, a solution of 2,6-dichloro-5-fluoronicotinamide (Compound 1) (16.27kg; 77.8mol) in dichloromethane (359.5kg) was added oxalyl chloride (11.9kg;
93.8mol) while maintaining temp ≤ 25°C for 75min. The resulting solution was then headed to 40°C ± 3°C and aged for 3h. Using vacuum, the solution was distilled to remove dichloromethane until the solution was below the agitator. Dichloromethane (300 kg) was then added and the mixture cooled to 0 ± 5°C. To a clean, dry reactor (reactor B) was added,2-isopropyl-4-methylpyridin-3-amine (ANILINE Compound 2A) (12.9kg; 85.9mol) followed by dichloromethane (102.6 kg). The ANILINE solution was azeodried via vacuum distillation while maintaining an internal temperature between 20-25 °), replacing with additional dichloromethane until the solution was dry by KF analysis (limit ≤ 0.05%). The solution volume was adjusted to approx. 23L volume with dichloromethane. The dried ANILINE solution was then added to reactor A while maintaining an internal temperature of 0 ± 5°C throughout the addition. The mixture was then heated to 23 °C and aged for 1h. the solution was polish filtered into a clean reactor to afford 2,6-dichloro-5-fluoro-N-((2- isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (Compound 3) as a solution in DCM and used directly in the next step.
Step 2
[0139] A dichloromethane solution of 2,6-dichloro-5-fluoro-N-{[4-methyl-2-(propan-2- yl)pyridin-3-yl]carbamoyl}pyridine-3-carboxamide (UREA (Compound 3)) (15kg contained; 38.9mol) was solvent exchanged into 2-MeTHF using vacuum distillation while maintaining internal temperature of 20-25 °C. The reactor volume was adjusted to 40L and then
additional 2-MeTHF was charged (105.4 kg). Sodium t-butoxide was added (9.4 kg;
97.8mol) while maintaining 5-10 °C. The contents where warmed to 23 °C and stirred for 3h. The contents where then cooled to 0-5C and ammonium chloride added (23.0kg; 430mol) as a solution in 60L of DI water. The mixture was warmed to 20 C and DI water added (15L) and further aged for 30min. Agitation was stopped and the layers separated. The aqueous layer was removed and to the organic layer was added DI water(81.7L). A mixture of conc HCl (1.5kg) and water (9L) was prepared then added to the reactor slowly until pH measured between 4-5. The layers were separated, and the aqueous layer back extracted using 2-MeTHF (42.2kg). The two organic layers combined and washed with a 10% citric acid solution (75kg) followed by a mixture of water (81.7L) and saturated NaCl (19.8 kg). The organic layer was then washed with saturated sodium bicarbonate (75kg) repeating if necessary to achieve a target pH of ≥ 7.0 of the aqueous. The organic layer was washed again with brine (54.7kg) and then dried over magnesium sulfate (5kg). The mixture was filtered to remove magnesium sulfate rinsing the filtered bed with 2-MeTHF (49.2 kg). The combined filtrate and washes where distilled using vacuum to 40L volume. The concentrated solution was heated to 55 °C and heptane (10-12kg) slowly added until cloud point. The solution was cooled to 23 °C over 2h then heptane (27.3 kg) was added over 2h. The product slurry was aged for 3h at 20-25 °C then filtered and washed with a mixture of 2-MeTHF (2.8kg) and heptane (9kg). The product was dried using nitrogen and vacuum to afford solid 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (rac-DIONE (Compound 4)).
Step 3
[0140] To a vessel, an agitated suspension of Compound 4, (1.0 eq.) in 2- methylterahydrofuran (7.0 L/kg) was added (+)-2,3-dibenzoyl-D-tartaric acid (2.0 eq.) under an atmosphere of nitrogen. 2-MeTHF is chiral, but it is used as a racemic mixture. The different enantiomers of 2-MeTHF are incorporated randomly into the co-crystal. The resulting suspension was warmed to 75°C and aged at 75°C until full dissolution was observed (< 30 mins.). The resulting solution was polish filtered at 75°C into a secondary vessel. To the polish filtered solution was charged n-Heptane (2.0 L/kg) at a rate that maintained the internal temperature above 65°C. The solution was then cooled to 60°C, seeded with crystals (0.01 kg/kg) and allowed to age for 30 minutes. The resulting suspension was cooled to 20°C over 4 hours and then sampled for chiral purity analysis by HPLC. To the suspension, n-Heptane (3.0 L/kg) was charged and then aged for 4 hours at 20°C under an atmosphere of nitrogen. The suspension was filtered, and the isolated solids were washed two times with (2:1) n-Heptane:2-methyltetrahydrofuran (3.0 L/kg). The material was dried with nitrogen and vacuum to afford M-Dione:DBTA: Me-THF complex (Compound 4a).
Step 4
[0141] To vessel A, a suspension of disodium hydrogen phosphate (21.1 kg, 2.0 equiv) in DI water (296.8 L, 6.3 L/kg) was agitated until dissolution was observed (≥ 30 min.). To vessel B, a suspension of the M-Dione:DBTA: Me-THF complex (Composition 4a)[46.9 kg (25.9 kg corrected for M-dione, 1.0 equiv.)] in methyl tert-butyl ether (517.8 L, 11.0 L/kg) was agitated for 15 to 30 minutes. The resulting solution from vessel A was added to vessel B, and then the mixture was agitated for more than 3 hours. The agitation was stopped, and the biphasic mixture was left to separate for more than 30 minutes. The lower aqueous phase was removed and then back extracted with methyl tert-butyl ether (77.7 L, 1.7 L/kg). The organic phases were combined in vessel B and dried with magnesium sulfate (24.8 kg, 0.529 kg/kg). The resulting suspension from vessel B was agitated for more than three hours and then filtered into vessel C. To vessel B, a methyl tert-butyl ether (46.9 L, 1.0 L/kg) rinse was charged and then filtered into vessel C. The contents of vessel C were cooled to 10 °C and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until 320-350 kg (6.8-7.5 kg/kg) of methyl tert-butyl ether was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged a second time over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (195.9 L, 4.2 L/kg) was charged a third time over one hour and then sampled for solvent composition by GC analysis. The vessel C suspension continued to agitate for more than one hour. The suspension was filtered, and then washed with a n-Heptane (68.6 L, 1.5 L/kg) rinse from vessel C. The isolated solids were dried at 50°C, and a sample was submitted for stock suitability. Afforded 7-chloro-6-fluoro-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (M-DIONE) Compound 5M.
[0142] The first-generation process highlighted above has been successfully scaled on 200+ kg of rac-dione starting material (Compound 4). In this process, seeding the crystallization with the thermodynamically-stable rac-dione crystal form (which exhibits low solubility) would cause a batch failure. Based on our subsequent studies, we found that increasing the DBTA equivalents and lowering the seed temperature by adjusting heptane
charge schedule improves robustness of the process. The improved process is resistant to the presence of the thermodynamically-stable rac-dione crystal form and promotes successful separation of atropisomers. Subsequent batches will incorporate the improved process for large scale manufacture.
Step 5
Note: All L/kg amounts are relative to M-Dione input; All equiv. amounts are relative to M-Dione input after adjusted by potency.
[0143] M-Dione (Compound 5M, 1.0 equiv.) and Toluene-1 (10.0 L/kg) was charged to Vessel A. The resulting solution was dried by azeotropic distillation under vacuum at 45 °C until 5.0 L/kg of solvents has been removed. The contents of Vessel A were then cooled to 20 °C.
[0144] Vessel C was charged with Toluene-3 (4.5 L/kg), Phosphoryl chloride (1.5 equiv.) and N,N-Diisopropylethylamine-1 (2.0 equiv.) while maintaining the internal temperature below 20 ± 5 °C.
Upon finishing charging, Vessel C was warmed to 30 ± 5 °C. The contents of Vessel A were then transferred to Vessel C over 4 hours while maintaining the internal temperature at 30 ± 5°C. Vessel A was rinsed with Toluene-2 (0.5 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated at 30°C for an additional 3 hours. The contents of Vessel C were cooled to 20 ± 5 °C. A solution of (s)-1-boc-3-methylpiperazine (1.2 equiv.), N,N-Diisopropylethylamine-2 (1.2 equiv.) in isopropyl acetate-1 (1.0 L/kg) was prepared in Vessel D. The solution of Vessel D was charged to vessel C while maintaining a batch temperature of 20 ± 5 °C (Note: Exotherm is observed). Upon the end of transfer, Vessel D was rinsed with additional dichloromethane (1.0 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated for an additional 60 minutes at 20 °C. A solution of sodium bicarbonate [water-1 (15.0 L/kg + Sodium bicarbonate (4.5 equiv.)] was then charged into Vessel C over an hour while maintaining an internal temperature at 20 ± 5 °C throughout the addition. The contents of Vessel C were agitated for at least 12 hours at which point the Pipazoline (Compound 6) product was isolated by filtration in an agitated filter dryer. The cake was washed with water-2 and -3 (5.0 L/kg x 2 times, agitating each wash for 15 minutes) and isopropyl acetate-2 and 3 (5.0 L/kg x 2 times, agitating each wash for 15 min). The cake as dried under nitrogen for 12 hours.
Acetone Re-slurry (Optional):
[0145] Pipazoline (Compound 6) and acetone (10.0 L/kg) were charged to Vessel E. The suspension was heated to 50 °C for 2 hours. Water-4 (10.0 L/kg) was charged into Vessel E over 1 hour. Upon completion of water addition, the mixture was cooled to 20 °C over 1 hour. The contents of Vessel E were filtered to isolate the product, washing the cake with 1:1 acetone/water mixture (5.0 L/kg). The cake was dried under nitrogen for 12 hours.
Step 6
General Note: All equivalents and volumes are reported in reference to Pipazoline input
Note: All L/kg and kg/kg amounts are relative to Pipazoline input
[0146] Reactor A is charged with Pipazoline (Compound 6, 1.0 equiv), degassed 2- MeTHF (9.0 L/kg) and a solution of potassium acetate (2.0 equiv) in degassed water (6.5 L/kg). The resulting mixture is warmed to 75 ± 5 °C and then, charge a slurry of
Pd(dpePhos)Cl2 (0.003 equiv) in 2-MeTHF (0.5 L/kg). Within 2 h of catalyst charge, a solution of freshly prepared Boroxine (Compound 6A, 0.5 equiv) in wet degassed 2-MeTHF (4.0 L/kg, KF > 4.0%) is charged over the course of >1 hour, but < 2 hours, rinsing with an additional portion of wet 2-MeTHF (0.5 L/kg) after addition is complete. After reaction completion ( <0.15 area % Pipazoline remaining, typically <1 h after boroxine addition is complete), 0.2 wt% (0.002 kg/kg) of Biaryl seed is added as a slurry in 0.02 L/kg wet 2- MeTHF, and the resulting seed bed is aged for > 60 min. Heptane (5.0 L/kg) is added over 2 hours at 75 ± 5 °C. The batch is then cooled to 20 ± 5 °C over 2 hours and aged for an additional 2 h. The slurry is then filtered and cake washed with 1 x 5.0L/kg water, 1 x 5.0L/kg 1:1 iPrOH:water followed by 1 x 5.0 L/kg 1:1 iPrOH:heptane (resuspension wash: the cake is resuspended by agitator and allow to set before filtering) . The cake (Biaryl, Compound 7) is then dried under vacuum with a nitrogen sweep.
Note: If the reaction stalls, an additional charge of catalyst and boroxine is required
Step 7 Charcoal Filtration for Pd removal
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Note: All L/kg and kg/kg amounts are relative to crude Biaryl input
[0147] In a clean Vessel A, charge crude Biaryl (1 equiv) and charge DCM (10 L/kg). Agitate content for > 60 minutes at 22 ± 5 °C, observing dissolution. Pass crude Biaryl from Vessel A, through a bag filter and carbon filters at a flux ≤ 3 L2/min/m and collect filtrate in clean Vessel B. Charge DCM rinse (1 L/kg) to Vessel A, and through carbon filters to collect in vessel B.
[0148] From filtrate in Vessel B, pull a solution sample for IPC Pd content. Sample is concentrated to solid and analyzed by ICP-MS. IPC: Pd ≤ 25 ppm with respect to Biaryl. a. If Pd content is greater than 25 ppm with respect to Biaryl on first or second IPC sample, pass solution through carbon filter a second time at ≤ 3 L2/min/m2, rinsing with 1 L/kg DCM; sample filtrate for IPC.
b. If Pd content remains greater than 25 ppm after third IPC, install and condition fresh carbon discs. Pass Biaryl filtrate through refreshed carbon filter, washing with 1 L/kg DCM. Sample for IPC.
[0149] Distill and refill to appropriate concentration. Prepare for distillation of recovered filtrate by concentrating to ≤ 4 L/kg DCM, and recharge to reach 5.25 ± 0.25 L/kg DCM prior to moving into Step 7 Boc-deprotection reaction.
Step 7
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Note: All L/kg and kg/kg amounts are relative to Biaryl input
[0150] To Reactor A was added: tert-butyl (3S)-4-{6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl}-3-methylpiperazine-1-carboxylate (Biaryl) (1.0 equiv), dichloromethane (5.0 L/kg), and the TFA (15.0 equiv, 1.9 L/kg) is charged slowly to maintain the internal temperature at 20 ± 5 °C. The reaction was stirred for 4 h at 20 ± 5 °C.
[0151] To Reactor B was added: potassium carbonate (18.0 equiv), water (20.0 L/kg), and NMP (1.0) to form a homogenous solution. While agitating at the maximum acceptable rate for the equipment, the reaction mixture in A was transferred into the potassium carbonate solution in B over 30 minutes (~ 0.24 L/kg/min rate). The mixture was stirred at 20 ± 5 °C for an additional 12 h.
[0152] The resulting slurry was filtered and rinsed with water (2 x 10 L/kg). The wet cake was dried for 24 h to give 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-4-[(2S)-2-methylpiperazin- 1-yl]-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Des- Boc, Compound 8).
Step 8
Note: All L/kg and kg/kg amounts are relative to Des-Boc input
[0153] Des-Boc (Compound 8, 1.0 equiv) and NMP (4.2 L/kg) are charged to Vessel A under nitrogen, charge the TFA (1.0 equiv.) slowly to maintain the Tr <25 °C. The mixture is aged at 25 °C until full dissolution is observed (about 0.5 hour). The solution is then polish filtered through a 0.45 micron filter into Vessel B, washing with a NMP (0.8 L/kg). The filtrate and wash are combined, and then cooled to 0 °C. To the resulting solution, Acryloyl Chloride (1.3 equiv.) is added while maintaining temperature < 10 C. The reaction mixture is then aged at 5 ±5°C until completed by IPC (ca.1.5 hrs).
Preparation of Aqueous Disodium Phosphate Quench:
[0154] Disodium Phosphate (3.0 equiv) and Water (15.0 L/kg) are charged to Vessel C. The mixture is aged at 25 °C until full dissolution is observed. The solution is warmed to 45 ±5°C. A seed slurry of AMG 510 (0.005 equiv.) in Water (0.4 L/kg) is prepared and added to Vessel C while maintaining temperature at 45 ±5°C.
[0155] The reaction mixture in Vessel B is transferred to Vessel C (quench solution) while maintaining temperature at 45 ±5°C (ca.1 hrs). Vessel B is washed with a portion of NMP (0.5 L/kg). The product slurry is aged for 2 hrs at 45 ±5°C, cooled to 20 °C over 3 hrs, aged at 20 °C for a minimum of 12 hrs, filtered and washed with Water (2 x 10.0 L/kg). The product is dried using nitrogen and vacuum to afford Crude AMG 510 (Compound 9A).
Step 9
General Note: All equivalents and volumes are reported in reference to crude AMG 510 input
Note: All L/kg and kg/kg amounts are relative to Crude AMG 510 input
[0156] Reactor A was charged with 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4- methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1- yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Crude AMG 510) (1.0 equiv), ethanol (7.5 L/kg), and water (1.9 L/kg). The mixture heated to 75 °C and polish filtered into a clean Reactor B. The solution was cool to 45 °C and seeded with authentic milled AMG 510 seed (0.015 േ 0.005
1 Seed performs best when reduced in particle size via milling or with other type of mechanical grinding if mill is not available (mortar/ pestle). Actual seed utilized will be based on seed availability. 1.0- 2.0% is seed is target amount.
kg/kg); the resulting slurry was aged for 30 min. Water (15.0 L/kg) was added over 5h while maintaining an internal temperature > 40 °C; the mixture was aged for an additional 2h.
[0157] The mixture was cooled to 20 °C over 3 hours and aged for 8h, after which the solid was collected by filtration and washed using a mixture of ethanol (2.5 L/kg) and water (5.0 L/kg). The solid was dried using vacuum and nitrogen to obtain 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (AMG 510, Compound 9).
Compound 6A Boroxine Synthesis:
Lithiation/borylation
[0158] Reactor A was charged with THF (6 vol), a secondary amine base, Diisopropylamine (1.4 equiv), and a catalyst, such as triethylamine hydrochloride (0.01 equiv.). The resulting solution was cooled to -70 °C and a first base, n-BuLi (2.5 M in hexane, 1.5 equiv) was slowly added. After addition is complete, a solution of 3-fluoroanisole (1.0 equiv) in THF (6 vol) was added slowly and kept at -70 °C for 5 min. Concurrently or subsequently, a reagent, B(EtO)3 (2.0 equiv), was added slowly and kept at -70 °C for 10 min. The reaction mixture was quenched with an acid, 2N HCl. The quenched reaction mixture was extracted with MTBE (3 x 4 vol). The combined organic phases were concentrated to 1.5-3 total volumes. Heptane (7-9 vol) was added drop-wise and the mixture was cooled to 0-10 °C and stirred for 3 h. The mixture was filtrated and rinsed with heptane (1.5 vol). The solid was dried under nitrogen at < 30 °C to afford (2-fluoro-6-methoxyphenyl)boronic acid.
Demethylation:
Note: All L/kg and kg/kg amounts are relative to (2-fluoro-6-methoxyphenyl)boronic acid input
[0159] To a reactor, charge dichloromethane (solvent, 4.0 L/kg) and an acid, BBr3 (1.2 equiv), and cool to -20 °C. To this solution, a suspension of (2-fluoro-6-methoxyphenyl)boronic acid (1.0 equiv) in dichloromethane (4.0 L/kg) was added into the BBr3/DCM mixture while keeping temperature -15 to -25 °C. The reaction was allowed to proceed for approximately 2 hours while monitored by HPLC [≤1% (2-fluoro-6-methoxyphenyl)boronic acid] before reverse quenching into water (3.0 L/kg). The precipitated solid was then isolated by filtration and slurried with water (3.0 L/kg) on the filter prior to deliquoring. The filtrates were adjusted to pH 4-6 by the addition of sodium bicarbonate. The bottom organic phase was separated and the resulting aqueous layer was washed with dichloromethane (solvent, 5.0 Vol) and adjusted to pH = 1 by addition of concentrated hydrochloric acid. The resulting solids were isolated by filtration, washing the cake with water (2 x 5.0 L/kg)
Purification via Reslurry (required)
[0160] The combined crude solids were charged into a reactor and slurried with 5% EtOH/water (5.0 L/kg) at 20 °C for >1 h. The purified product was then isolated by filtration and rinsed with water (2 x 3 L/kg) before drying on the filter at < 30 °C to with nitrogen/vacuum to afford 2,2′,2”-(1,3,5,2,4,6-trioxatriborinane-2,4,6-triyl)tris(3-fluorophenol) (Boroxine, Compound 6A).
PATENT
WO 2020102730
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020102730
PATENT
US 20180334454
References
- ^ Jump up to:a b c d e “Lumakras- sotorasib tablet, coated”. DailyMed. Retrieved 6 June 2021.
- ^ Jump up to:a b c d e f g h i j k l m n “FDA Approves First Targeted Therapy for Lung Cancer Mutation Previously Considered Resistant to Drug Therapy”. U.S. Food and Drug Administration (FDA). 28 May 2021. Retrieved 28 May 2021. This article incorporates text from this source, which is in the public domain.
- ^ “KRAS mutant-targeting AMG 510”. NCI Drug Dictionary. National Cancer Institute. 2 February 2011. Retrieved 16 November2019.
- ^ Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. (November 2019). “The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity”. Nature. 575 (7781): 217–23. Bibcode:2019Natur.575..217C. doi:10.1038/s41586-019-1694-1. PMID 31666701.
- ^ Jump up to:a b “FDA approves Amgen drug for lung cancer with specific mutation”. CNBC. 28 May 2021. Retrieved 28 May 2021.
- ^ Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. (2020). “KRASG12C inhibition with sotorasib in advanced solid tumors”. N Engl J Med. doi:10.1056/NEJMoa1917239. PMC 7571518.
- ^ Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation ” at ClinicalTrials.gov
- ^ “The Discovery Of Amgen’s Novel Investigational KRAS(G12C) Inhibitor AMG 510 Published In Nature” (Press release). Amgen. 30 October 2019. Retrieved 16 November 2019.
- ^ Irving M (24 December 2019). “Drug targeting common cancer cause enters phase 2 clinical trials”. New Atlas. Retrieved 24 December 2019.
- ^ Jump up to:a b c d Halford B (3 April 2019). “Amgen unveils its KRas inhibitor in human clinical trials: AMG 510 shuts down a mutant version of the cancer target via covalent interaction”. Chemical & Engineering News. 97 (4). Retrieved 16 November 2019.
- ^ Al Idrus A (9 September 2019). “Amgen’s KRAS drug continues to deliver but faces ‘curse’ of high expectations”. fiercebiotech.com. Retrieved 16 November 2019.
- ^ Kaiser J (30 October 2019). “Two new drugs finally hit ‘undruggable’ cancer target, providing hope for treatments”. Science Magazine. AAAS. Retrieved 16 November 2019.
- ^ Astor L (9 September 2019). “FDA Grants AMG 510 Fast Track Designation for KRAS G12C+ NSCLC”. targetedonc.com. Retrieved 16 November 2019.
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 85” (PDF). WHO Drug Information. 35 (1).
Further reading
- Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. (September 2020). “KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors”. N Engl J Med. 383 (13): 1207–17. doi:10.1056/NEJMoa1917239. PMC 7571518. PMID 32955176.
- Lanman BA, Allen JR, Allen JG, Amegadzie AK, Ashton KS, Booker SK, et al. (January 2020). “Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors”. J Med Chem. 63 (1): 52–65. doi:10.1021/acs.jmedchem.9b01180. PMID 31820981.
External links
- “Sotorasib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation (CodeBreaK 100)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Lumakras |
| Other names | AMG 510 |
| License data | US DailyMed: Sotorasib |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2252403-56-6 |
| PubChem CID | 137278711 |
| DrugBank | DB15569 |
| ChemSpider | 72380148 |
| UNII | 2B2VM6UC8G |
| KEGG | D12055 |
| Chemical and physical data | |
| Formula | C30H30F2N6O3 |
| Molar mass | 560.606 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////Sotorasib, ソトラシブ , FDA 2021, APPROVALS 2021, Lumakras, CANCER, ANTINEOPLASTIC, AMG 510, AMG-510, AMG510, AMGEN, priority review, fast-track, breakthrough therapy, orphan drug
CC1CN(CCN1C2=NC(=O)N(C3=NC(=C(C=C32)F)C4=C(C=CC=C4F)O)C5=C(C=CN=C5C(C)C)C)C(=O)C=C
Sotorasib
6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-propan-2-ylpyridin-3-yl)-4-[(2S)-2-methyl-4-prop-2-enoylpiperazin-1-yl]pyrido[2,3-d]pyrimidin-2-one
AMG 510
AMG-510
AMG510
| Formula | C30H30F2N6O3 |
|---|---|
| CAS | 2296729-00-3 |
| Mol weight | 560.5944 |
FDA APPROVED, 2021/5/28 Lumakras
Antineoplastic, Non-small cell lung cancer (KRAS G12C-mutated)
ソトラシブ (JAN);
Sotorasib
(1M)-6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one
C30H30F2N6O3 : 560.59
[2296729-00-3]
Sotorasib is an inhibitor of the RAS GTPase family. The molecular formula is C30H30F2N6O3, and the molecular weight is 560.6 g/mol. The chemical name of sotorasib is 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2enoyl) piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one. The chemical structure of sotorasib is shown below:
|
Sotorasib has pKa values of 8.06 and 4.56. The solubility of sotorasib in the aqueous media decreases over the range pH 1.2 to 6.8 from 1.3 mg/mL to 0.03 mg/mL.
LUMAKRAS is supplied as film-coated tablets for oral use containing 120 mg of sotorasib. Inactive ingredients in the tablet core are microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, and magnesium stearate. The film coating material consists of polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and iron oxide yellow.
FDA grants accelerated approval to sotorasib for KRAS G12C mutated NSCLC
On May 28, 2021, the Food and Drug Administration granted accelerated approval to sotorasib (Lumakras™, Amgen, Inc.), a RAS GTPase family inhibitor, for adult patients with KRAS G12C ‑mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA ‑approved test, who have received at least one prior systemic therapy.
FDA also approved the QIAGEN therascreen® KRAS RGQ PCR kit (tissue) and the Guardant360® CDx (plasma) as companion diagnostics for Lumakras. If no mutation is detected in a plasma specimen, the tumor tissue should be tested.
Approval was based on CodeBreaK 100, a multicenter, single-arm, open label clinical trial (NCT03600883) which included patients with locally advanced or metastatic NSCLC with KRAS G12C mutations. Efficacy was evaluated in 124 patients whose disease had progressed on or after at least one prior systemic therapy. Patients received sotorasib 960 mg orally daily until disease progression or unacceptable toxicity.
The main efficacy outcome measures were objective response rate (ORR) according to RECIST 1.1, as evaluated by blinded independent central review and response duration. The ORR was 36% (95% CI: 28%, 45%) with a median response duration of 10 months (range 1.3+, 11.1).
The most common adverse reactions (≥ 20%) were diarrhea, musculoskeletal pain, nausea, fatigue, hepatotoxicity, and cough. The most common laboratory abnormalities (≥ 25%) were decreased lymphocytes, decreased hemoglobin, increased aspartate aminotransferase, increased alanine aminotransferase, decreased calcium, increased alkaline phosphatase, increased urine protein, and decreased sodium.
The recommended sotorasib dose is 960 mg orally once daily with or without food.
The approved 960 mg dose is based on available clinical data, as well as pharmacokinetic and pharmacodynamic modeling that support the approved dose. As part of the evaluation for this accelerated approval, FDA is requiring a postmarketing trial to investigate whether a lower dose will have a similar clinical effect.
View full prescribing information for Lumakras.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), Health Canada, and the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA). The application reviews are ongoing at the other regulatory agencies.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, the Assessment Aid, and the Product Quality Assessment Aid (PQAA), voluntary submissions from the applicant to facilitate the FDA’s assessment. The FDA approved this application approximately 10 weeks ahead of the FDA goal date.
This application was granted priority review, fast-track, breakthrough therapy and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Sotorasib, sold under the brand name Lumakras is an anti-cancer medication used to treat non-small-cell lung cancer (NSCLC).[1][2] It targets a specific mutation, G12C, in the protein KRAS which is responsible for various forms of cancer.[3][4]
The most common side effects include diarrhea, musculoskeletal pain, nausea, fatigue, liver damage and cough.[1][2]
Sotorasib is an inhibitor of the RAS GTPase family.[1]
Sotorasib is the first approved targeted therapy for tumors with any KRAS mutation, which accounts for approximately 25% of mutations in non-small cell lung cancers.[2] KRAS G12C mutations represent about 13% of mutations in non-small cell lung cancers.[2] Sotorasib was approved for medical use in the United States in May 2021.[2][5]
Sotorasib is an experimental KRAS inhibitor being investigated for the treatment of KRAS G12C mutant non small cell lung cancer, colorectal cancer, and appendix cancer.
Sotorasib, also known as AMG-510, is an acrylamide derived KRAS inhibitor developed by Amgen.1,3 It is indicated in the treatment of adult patients with KRAS G12C mutant non small cell lung cancer.6 This mutation makes up >50% of all KRAS mutations.2 Mutant KRAS discovered in 1982 but was not considered a druggable target until the mid-2010s.5 It is the first experimental KRAS inhibitor.1
The drug MRTX849 is also currently being developed and has the same target.1
Sotorasib was granted FDA approval on 28 May 2021.6
Medical uses
Sotorasib is indicated for the treatment of adults with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA-approved test, who have received at least one prior systemic therapy.[1][2]
Clinical development
Sotorasib is being developed by Amgen. Phase I clinical trials were completed in 2020.[6][7][8] In December 2019, it was approved to begin Phase II clinical trials.[9]
Because the G12C KRAS mutation is relatively common in some cancer types, 14% of non-small-cell lung cancer adenocarcinoma patients and 5% of colorectal cancer patients,[10] and sotorasib is the first drug candidate to target this mutation, there have been high expectations for the drug.[10][11][12] The Food and Drug Administration has granted a fast track designation to sotorasib for the treatment of metastatic non-small-cell lung carcinoma with the G12C KRAS mutation.[13]
Chemistry and pharmacology
Sotorasib can exist in either of two atropisomeric forms and one is more active than the other.[10] It selectively forms an irreversible covalent bond to the sulfur atom in the cysteine residue that is present in the mutated form of KRAS, but not in the normal form.[10]
History
Researchers evaluated the efficacy of sotorasib in a study of 124 participants with locally advanced or metastatic KRAS G12C-mutated non-small cell lung cancer with disease progression after receiving an immune checkpoint inhibitor and/or platinum-based chemotherapy.[2] The major outcomes measured were objective response rate (proportion of participants whose tumor is destroyed or reduced) and duration of response.[2] The objective response rate was 36% and 58% of those participants had a duration of response of six months or longer.[2]
The U.S. Food and Drug Administration (FDA) granted the application for sotorasib orphan drug, fast track, priority review, and breakthrough therapy designations.[2] The FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), Health Canada and the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA).[2] The application reviews are ongoing at the other regulatory agencies.[2]
The FDA granted approval of Lumakras to Amgen Inc.[2]
Society and culture
Economics
Sotorasib costs US$17,900 per month.[5]
Names
Sotorasib is the recommended international nonproprietary name (INN).[14]
PAPER
Nature (London, United Kingdom) (2019), 575(7781), 217-223
https://www.nature.com/articles/s41586-019-1694-1
KRAS is the most frequently mutated oncogene in cancer and encodes a key signalling protein in tumours1,2. The KRAS(G12C) mutant has a cysteine residue that has been exploited to design covalent inhibitors that have promising preclinical activity3,4,5. Here we optimized a series of inhibitors, using novel binding interactions to markedly enhance their potency and selectivity. Our efforts have led to the discovery of AMG 510, which is, to our knowledge, the first KRAS(G12C) inhibitor in clinical development. In preclinical analyses, treatment with AMG 510 led to the regression of KRASG12C tumours and improved the anti-tumour efficacy of chemotherapy and targeted agents. In immune-competent mice, treatment with AMG 510 resulted in a pro-inflammatory tumour microenvironment and produced durable cures alone as well as in combination with immune-checkpoint inhibitors. Cured mice rejected the growth of isogenic KRASG12D tumours, which suggests adaptive immunity against shared antigens. Furthermore, in clinical trials, AMG 510 demonstrated anti-tumour activity in the first dosing cohorts and represents a potentially transformative therapy for patients for whom effective treatments are lacking.
Paper
Scientific Reports (2020), 10(1), 11992
PAPER
European journal of medicinal chemistry (2021), 213, 113082.
https://www.sciencedirect.com/science/article/abs/pii/S0223523420310540
KRAS is the most commonly altered oncogene of the RAS family, especially the G12C mutant (KRASG12C), which has been a promising drug target for many cancers. On the basis of the bicyclic pyridopyrimidinone framework of the first-in-class clinical KRASG12C inhibitor AMG510, a scaffold hopping strategy was conducted including a F–OH cyclization approach and a pyridinyl N-atom working approach leading to new tetracyclic and bicyclic analogues. Compound 26a was identified possessing binding potency of 1.87 μM against KRASG12C and cell growth inhibition of 0.79 μM in MIA PaCa-2 pancreatic cancer cells. Treatment of 26a with NCI–H358 cells resulted in down-regulation of KRAS-GTP levels and reduction of phosphorylation of downstream ERK and AKT dose-dependently. Molecular docking suggested that the fluorophenol moiety of 26a occupies a hydrophobic pocket region thus forming hydrogen bonding to Arg68. These results will be useful to guide further structural modification.
PAPER
Journal of Medicinal Chemistry (2020), 63(1), 52-65.
https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01180
KRASG12C has emerged as a promising target in the treatment of solid tumors. Covalent inhibitors targeting the mutant cysteine-12 residue have been shown to disrupt signaling by this long-“undruggable” target; however clinically viable inhibitors have yet to be identified. Here, we report efforts to exploit a cryptic pocket (H95/Y96/Q99) we identified in KRASG12C to identify inhibitors suitable for clinical development. Structure-based design efforts leading to the identification of a novel quinazolinone scaffold are described, along with optimization efforts that overcame a configurational stability issue arising from restricted rotation about an axially chiral biaryl bond. Biopharmaceutical optimization of the resulting leads culminated in the identification of AMG 510, a highly potent, selective, and well-tolerated KRASG12C inhibitor currently in phase I clinical trials (NCT03600883).
AMG 510 [(R)-38]. (1R)-6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1-piperazinyl]-pyrido[2,3-d]pyrimidin-2(1H)-one
………… concentrated in vacuo. Chromatographic purification of the residue (silica gel; 0–100% 3:1 EtOAc–EtOH/heptane) followed by chiral supercritical fluid chromatography (Chiralpak IC, 30 mm × 250 mm, 5 μm, 55% MeOH/CO2, 120 mL/min, 102 bar) provided (1R)-6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1-piperazinyl]pyrido[2,3-d]pyrimidin-2(1H)-one (AMG 510; (R)-38; 2.25 g, 43% yield) as the first-eluting peak. 1H NMR (600 MHz, DMSO-d6) δ ppm 10.20 (s, 1H), 8.39 (d, J = 4.9 Hz, 1H), 8.30 (d, J = 8.9 Hz, 0.5H), 8.27 (d, J = 8.7 Hz, 0.5H), 7.27 (q, J = 8.4 Hz, 1H), 7.18 (d, J = 4.9 Hz, 1H), 6.87 (dd, J = 16.2, 10.8 Hz, 0.5H), 6.84 (dd, J = 16.2, 10.7 Hz, 0.5H), 6.74 (d, J = 8.4 Hz, 1H), 6.68 (t, J = 8.4 Hz, 1H), 6.21 (d, J = 16.2 Hz, 0.5H), 6.20 (d, J = 16.2 Hz, 0.5H), 5.76 (d, J = 10.8 Hz, 0.5H), 5.76 (d, J = 10.7 Hz, 0.5H), 4.91 (m, 1H), 4.41 (d, J = 12.2 Hz, 0.5H), 4.33 (d, J = 12.2 Hz, 1H), 4.28 (d, J = 12.2 Hz, 0.5H), 4.14 (d, J = 12.2 Hz, 0.5H), 4.02 (d, J = 13.6 Hz, 0.5H), 3.69 (m, 1H), 3.65 (d, J = 13.6 Hz, 0.5H), 3.52 (t, J = 12.2 Hz, 0.5H), 3.27 (d, J = 12.2 Hz, 0.5H), 3.15 (t, J = 12.2 Hz, 0.5H), 2.72 (m, 1H), 1.90 (s, 3H), 1.35 (d, J = 6.7 Hz, 3H), 1.08 (d, J = 6.7 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H).
19F NMR (376 MHz, DMSO-d6) δ −115.6 (d, J = 5.2 Hz, 1 F), −128.6 (br s, 1 F).
13C NMR (151 MHz, DMSO-d6) δ ppm 165.0 (1C), 163.4 (1C), 162.5 (1C), 160.1 (1C), 156.8 (1C), 153.7 (1C), 151.9 (1C), 149.5 (1C), 148.3 (1C), 145.2 (1C), 144.3 (1C), 131.6 (1C), 130.8 (1C), 127.9 (0.5C), 127.9 (0.5C), 127.8 (0.5C), 127.7 (0.5C), 123.2 (1C), 122.8 (1C), 111.7 (1C), 109.7 (1C), 105.7 (1C), 105.3 (1C), 51.4 (0.5C), 51.0 (0.5C), 48.9 (0.5C), 45.4 (0.5C), 44.6 (0.5C), 43.7 (0.5C), 43.5 (0.5C), 41.6 (0.5C), 29.8 (1C), 21.9 (1C), 21.7 (1C), 17.0 (1C), 15.5 (0.5C), 14.8 (0.5C).
FTMS (ESI) m/z: [M + H]+ calcd for C30H30F2N6O3 561.24202. Found 561.24150.
d (1R)-6-Fluoro7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1- piperazinyl]-pyrido[2,3-d]pyrimidin-2(1H)-one ((R)-38; AMG 510; 2.25 g, 43% yield) as the first-eluting peak.1 H NMR (600 MHz, DMSO-d6) δ ppm 10.20 (s, 1H), 8.39 (d, J = 4.9 Hz, 1H), 8.30 (d, J = 8.9 Hz, 0.5H), 8.27 (d, J = 8.7 Hz, 0.5H), 7.27 (q, J = 8.4 Hz, 1H), 7.18 (d, J = 4.9 Hz, 1H), 6.87 (dd, J = 16.2, 10.8 Hz, 0.5H), 6.84 (dd, J = 16.2, 10.7 Hz, 0.5H), 6.74 (d, J = 8.4 Hz, 1H), 6.68 (t, J = 8.4 Hz, 1H), 6.21 (d, J = 16.2 Hz, 0.5H), 6.20 (d, J = 16.2 Hz, 0.5H), 5.76 (d, J = 10.8 Hz, 0.5H), 5.76 (d, J = 10.7 Hz, 0.5H), 4.91 (m, 1H), 4.41 (d, J = 12.2 Hz, 0.5H), 4.33 (d, J = 12.2 Hz, 1H), 4.28 (d, J = 12.2 Hz, 0.5H), 4.14 (d, J = 12.2 Hz, 0.5H), 4.02 (d, J = 13.6 Hz, 0.5H), 3.69 (m, 1H), 3.65 (d, J = 13.6 Hz, 0.5H), 3.52 (t, J = 12.2 Hz, 0.5H), 3.27 (d, J = 12.2 Hz, 0.5H), 3.15 (t, J = 12.2 Hz, 0.5H), 2.72 (m, 1H), 1.90 (s, 3H), 1.35 (d, J = 6.7 Hz, 3H), 1.08 (d, J = 6.7 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H).
19F NMR (376 MHz, DMSO-d6) δ –115.6 (d, J = 5.2 Hz, 1 F), –128.6 (br. s., 1 F).
13C NMR (151 MHz, DMSO-d6) δ ppm 165.0 (1C), 163.4 (1C), 162.5 (1C), 160.1 (1C), 156.8 (1C), 153.7 (1C), 151.9 (1C), 149.5 (1C), 148.3 (1C), 145.2 (1C), 144.3 (1C), 131.6 (1C), 130.8 (1C), 127.9 (0.5C), 127.9 (0.5C), 127.8 (0.5C), 127.7 (0.5C), 123.2 (1C), 122.8 (1C), 111.7 (1C), 109.7 (1C), 105.7 (1C), 105.3 (1C), 51.4 (0.5C), 51.0 (0.5C), 48.9 (0.5C), 45.4 (0.5C), 44.6 (0.5C), 43.7 (0.5C), 43.5 (0.5C), 41.6 (0.5C), 29.8 (1C), 21.9 (1C), 21.7 (1C), 17.0 (1C), 15.5 (0.5C), 14.8 (0.5C).
FTMS (ESI) m/z: [M+H]+ Calcd for C30H30F2N6O3 561.24202; Found 561.24150. Atropisomer configuration (R vs. S) assigned crystallographically.The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01180.
PATENT
WO 2021097212
The present disclosure relates to an improved, efficient, scalable process to prepare intermediate compounds, such as compound of Formula 6A, having the structure,
useful for the synthesis of compounds for the treatment of KRAS G12C mutated cancers.
BACKGROUND
[0003] KRAS gene mutations are common in pancreatic cancer, lung adenocarcinoma, colorectal cancer, gall bladder cancer, thyroid cancer, and bile duct cancer. KRAS mutations are also observed in about 25% of patients with NSCLC, and some studies have indicated that KRAS mutations are a negative prognostic factor in patients with NSCLC. Recently, V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations have been found to confer resistance to epidermal growth factor receptor (EGFR) targeted therapies in colorectal cancer; accordingly, the mutational status of KRAS can provide important information prior to the prescription of TKI therapy. Taken together, there is a need for new medical treatments for patients with pancreatic cancer, lung adenocarcinoma, or colorectal cancer, especially those who have been diagnosed to have such cancers characterized by a KRAS mutation, and including those who have progressed after chemotherapy.
Related Synthetic Processes
[0126] The following intermediate compounds of 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one are representative examples of the disclosure and are not intended to be construed as limiting the scope of the present invention.
[0127] A synthesis of Compound 9 and the relevant intermediates is described in U.S. Serial No.15/984,855, filed May 21, 2018 (U.S. Publication No.2018/0334454, November 22, 2018) which claims priority to and the benefit claims the benefit of U.S. Provisional Application No.62/509,629, filed on May 22, 2017, both of which are incorporated herein by reference in their entireties for all purposes. 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one was prepared using the following process, in which the isomers of the final product were isolated via chiral chromatography.
[0128] Step 1: 2,6-Dichloro-5-fluoronicotinamide (Intermediate S). To a mixture of 2,6-dichloro-5-fluoro-nicotinic acid (4.0 g, 19.1 mmol, AstaTech Inc., Bristol, PA) in dichloromethane (48 mL) was added oxalyl chloride (2M solution in DCM, 11.9 mL, 23.8 mmol), followed by a catalytic amount of DMF (0.05 mL). The reaction was stirred at room temperature overnight and then was concentrated. The residue was dissolved in 1,4-dioxane (48 mL) and cooled to 0 °C. Ammonium hydroxide solution (28.0-30% NH3 basis, 3.6 mL, 28.6 mmol) was added slowly via syringe. The resulting mixture was stirred at 0 °C for 30 min and then was concentrated. The residue was diluted with a 1:1 mixture of EtOAc/Heptane and agitated for 5 min, then was filtered. The filtered solids were discarded, and the remaining mother liquor was partially concentrated to half volume and filtered. The filtered solids were washed with heptane and dried in a reduced-pressure oven (45 °C) overnight to provide 2,6-dichloro-5-fluoronicotinamide. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.23 (d, J = 7.9 Hz, 1 H) 8.09 (br s, 1 H) 7.93 (br s, 1 H). m/z (ESI, +ve ion): 210.9 (M+H)+.
[0129] Step 2: 2,6-Dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. To an ice-cooled slurry of 2,6-dichloro-5-fluoronicotinamide (Intermediate S, 5.0 g, 23.9 mmol) in THF (20 mL) was added oxalyl chloride (2 M solution in DCM, 14.4 mL, 28.8 mmol) slowly via syringe. The resulting mixture was heated at 75 °C for 1 h, then heating was stopped, and the reaction was concentrated to half volume. After cooling to 0 °C, THF (20 mL) was added, followed by a solution of 2-isopropyl-4-methylpyridin-3-amine (Intermediate R, 3.59 g, 23.92 mmol) in THF (10 mL), dropwise via cannula. The resulting mixture was stirred at 0 °C for 1 h and then was quenched with a 1:1 mixture of brine and saturated aqueous ammonium chloride. The mixture was extracted with EtOAc (3x) and the combined organic layers were dried over anhydrous sodium sulfate and concentrated to provide 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. This material was used without further purification in the following step. m/z (ESI, +ve ion): 385.1(M+H)+.
[0130] Step 3: 7-Chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione. To an ice-cooled solution of 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (9.2 g, 24.0 mmol) in THF (40 mL) was added KHMDS (1 M solution in THF, 50.2 mL, 50.2 mmol) slowly via syringe. The ice bath was removed and the resulting mixture was stirred for 40 min at room temperature. The reaction was quenched with saturated aqueous ammonium chloride and extracted with EtOAc (3x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% 3:1 EtOAc-EtOH/heptane) to provide 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione.1H NMR (400 MHz, DMSO-d6) δ ppm 12.27 (br s, 1H), 8.48-8.55 (m, 2 H), 7.29 (d, J = 4.8 Hz, 1 H), 2.87 (quin, J = 6.6 Hz, 1 H), 1.99-2.06 (m, 3 H), 1.09 (d, J = 6.6 Hz, 3 H), 1.01 (d, J = 6.6 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -126.90 (s, 1 F). m/z (ESI, +ve ion): 349.1 (M+H)+.
[0131] Step 4: 4,7-Dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. To a solution of 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (4.7 g, 13.5 mmol) and DIPEA (3.5 mL, 20.2 mmol) in acetonitrile (20 mL) was added phosphorus oxychloride (1.63 mL, 17.5 mmol), dropwise via syringe. The resulting mixture was heated at 80 °C for 1 h, and then was cooled to room temperature and concentrated to provide 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. This material was used without further purification in the following step. m/z (ESI, +ve ion): 367.1 (M+H)+.
[0132] Step 5: (S)-tert-Butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. To an ice-cooled solution of 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one (13.5 mmol) in acetonitrile (20 mL) was added DIPEA (7.1 mL, 40.3 mmol), followed by (S)-4-N-Boc-2-methyl piperazine (3.23 g, 16.1 mmol, Combi-Blocks, Inc., San Diego, CA, USA). The resulting mixture was warmed to room temperature and stirred for 1 h, then was diluted with cold saturated aqueous sodium bicarbonate solution (200 mL) and EtOAc (300 mL). The mixture was stirred for an additional 5 min, the layers were separated, and the aqueous layer was extracted with more EtOAc (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% EtOAc/heptane) to provide (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. m/z (ESI, +ve ion): 531.2 (M+H)+.
[0133] Step 6: (3S)-tert-Butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. A mixture of (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (4.3 g, 8.1 mmol), potassium trifluoro(2-fluoro-6-hydroxyphenyl)borate (Intermediate Q, 2.9 g, 10.5 mmol), potassium acetate (3.2 g, 32.4 mmol) and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with dichloromethane (661 mg, 0.81 mmol) in 1,4-dioxane (80 mL) was degassed with nitrogen for 1 min. De-oxygenated water (14 mL) was added, and the resulting mixture was heated at 90 °C for 1 h. The reaction was allowed to cool to room temperature, quenched with half-saturated aqueous sodium bicarbonate, and extracted with EtOAc (2x) and DCM (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-60% 3:1 EtOAc-EtOH/heptane) to provide (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate.1H NMR (400 MHz, DMSO-d6) δ ppm 10.19 (br s, 1 H), 8.38 (d, J = 5.0 Hz, 1 H), 8.26 (dd, J = 12.5, 9.2 Hz, 1 H), 7.23-7.28 (m, 1 H), 7.18 (d, J = 5.0 Hz, 1 H), 6.72 (d, J = 8.0 Hz, 1 H), 6.68 (t, J = 8.9 Hz, 1 H), 4.77-4.98 (m, 1 H), 4.24 (br t, J = 14.2 Hz, 1 H), 3.93-4.08 (m, 1 H), 3.84 (br d, J=12.9 Hz, 1 H), 3.52-3.75 (m, 1 H), 3.07-3.28 (m, 1 H), 2.62-2.74 (m, 1 H), 1.86-1.93 (m, 3 H), 1.43-1.48 (m, 9 H), 1.35 (dd, J = 10.8, 6.8 Hz, 3 H), 1.26-1.32 (m, 1 H), 1.07 (dd, J = 6.6, 1.7 Hz, 3 H), 0.93 (dd, J = 6.6, 2.1 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -115.65 (s, 1 F), -128.62 (s, 1 F). m/z (ESI, +ve ion): 607.3 (M+H)+.
[0134] Step 7: 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one. Trifluoroacetic acid (25 mL, 324 mmol) was added to a solution of (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (6.3 g, 10.4 mmol) in DCM (30 mL). The resulting mixture was stirred at room temperature for 1 h and then was concentrated. The residue was dissolved in DCM (30 mL), cooled to 0 °C, and sequentially treated with DIPEA (7.3 mL, 41.7 mmol) and a solution of acryloyl chloride (0.849 mL, 10.4 mmol) in DCM (3 mL; added dropwise via syringe). The reaction was stirred at 0 °C for 10 min, then was quenched with half-saturated aqueous sodium bicarbonate and extracted with DCM (2x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-100% 3:1 EtOAc-EtOH/heptane) to provide 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one.1H NMR (400 MHz, DMSO-d6) δ ppm 10.20 (s, 1 H), 8.39 (d, J = 4.8 Hz, 1 H), 8.24-8.34 (m, 1 H), 7.23-7.32 (m, 1 H), 7.19 (d, J = 5.0 Hz, 1 H), 6.87 (td, J = 16.3, 11.0 Hz, 1 H), 6.74 (d, J = 8.6 Hz, 1 H), 6.69 (t, J = 8.6 Hz, 1 H), 6.21 (br d, J = 16.2 Hz, 1 H), 5.74-5.80 (m, 1 H), 4.91 (br s, 1 H), 4.23-4.45 (m, 2 H), 3.97-4.21 (m, 1 H), 3.44-3.79 (m, 2 H), 3.11-3.31 (m, 1 H), 2.67-2.77 (m, 1 H), 1.91 (s, 3 H), 1.35 (d, J = 6.8 Hz, 3 H), 1.08 (d, J = 6.6 Hz, 3 H), 0.94 (d, J = 6.8 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ ppm -115.64 (s, 1 F), -128.63 (s, 1 F). m/z (ESI, +ve ion): 561.2 (M+H)+.
[0135] Another synthesis of Compound 9 and the relevant intermediates was described in a U.S. provisional patent application filed November 16, 2018, which is incorporated herein by reference in its entirety for all purposes.
Representative Synthetic Processes
[0136] The present disclosure comprises the following steps wherein the synthesis and utilization of the boroxine intermediate is a novel and inventive step in the manufacture of AMG 510 (Compound 9):
Raw Materials
Step la
[0137] To a solution of 2,6-dichloro-5-fluoro-3-pyridinecarboxylic acid (25kg; 119. lmol) in dichloromethane (167kg) and DMF (592g) was added Oxalyl chloride (18.9kg; 148.9mol) while maintaining an internal temp between 15-20 °C. Additional dichloromethane (33kg) was added as a rinse and the reaction mixture stirred for 2h. The reaction mixture is cooled then quenched with ammonium hydroxide (40.2L; 595.5mol) while maintaining internal temperature 0 ± 10°C. The resulting slurry was stirred for 90min then the product collected by filtration. The filtered solids were washed with DI water (3X 87L) and dried to provide 2,6-dichloro-5-fluoronicotinamide (Compound 1).
Step 1b
[0138] In reactor A, a solution of 2,6-dichloro-5-fluoronicotinamide (Compound 1) (16.27kg; 77.8mol) in dichloromethane (359.5kg) was added oxalyl chloride (11.9kg;
93.8mol) while maintaining temp ≤ 25°C for 75min. The resulting solution was then headed to 40°C ± 3°C and aged for 3h. Using vacuum, the solution was distilled to remove dichloromethane until the solution was below the agitator. Dichloromethane (300 kg) was then added and the mixture cooled to 0 ± 5°C. To a clean, dry reactor (reactor B) was added,2-isopropyl-4-methylpyridin-3-amine (ANILINE Compound 2A) (12.9kg; 85.9mol) followed by dichloromethane (102.6 kg). The ANILINE solution was azeodried via vacuum distillation while maintaining an internal temperature between 20-25 °), replacing with additional dichloromethane until the solution was dry by KF analysis (limit ≤ 0.05%). The solution volume was adjusted to approx. 23L volume with dichloromethane. The dried ANILINE solution was then added to reactor A while maintaining an internal temperature of 0 ± 5°C throughout the addition. The mixture was then heated to 23 °C and aged for 1h. the solution was polish filtered into a clean reactor to afford 2,6-dichloro-5-fluoro-N-((2- isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (Compound 3) as a solution in DCM and used directly in the next step.
Step 2
[0139] A dichloromethane solution of 2,6-dichloro-5-fluoro-N-{[4-methyl-2-(propan-2- yl)pyridin-3-yl]carbamoyl}pyridine-3-carboxamide (UREA (Compound 3)) (15kg contained; 38.9mol) was solvent exchanged into 2-MeTHF using vacuum distillation while maintaining internal temperature of 20-25 °C. The reactor volume was adjusted to 40L and then
additional 2-MeTHF was charged (105.4 kg). Sodium t-butoxide was added (9.4 kg;
97.8mol) while maintaining 5-10 °C. The contents where warmed to 23 °C and stirred for 3h. The contents where then cooled to 0-5C and ammonium chloride added (23.0kg; 430mol) as a solution in 60L of DI water. The mixture was warmed to 20 C and DI water added (15L) and further aged for 30min. Agitation was stopped and the layers separated. The aqueous layer was removed and to the organic layer was added DI water(81.7L). A mixture of conc HCl (1.5kg) and water (9L) was prepared then added to the reactor slowly until pH measured between 4-5. The layers were separated, and the aqueous layer back extracted using 2-MeTHF (42.2kg). The two organic layers combined and washed with a 10% citric acid solution (75kg) followed by a mixture of water (81.7L) and saturated NaCl (19.8 kg). The organic layer was then washed with saturated sodium bicarbonate (75kg) repeating if necessary to achieve a target pH of ≥ 7.0 of the aqueous. The organic layer was washed again with brine (54.7kg) and then dried over magnesium sulfate (5kg). The mixture was filtered to remove magnesium sulfate rinsing the filtered bed with 2-MeTHF (49.2 kg). The combined filtrate and washes where distilled using vacuum to 40L volume. The concentrated solution was heated to 55 °C and heptane (10-12kg) slowly added until cloud point. The solution was cooled to 23 °C over 2h then heptane (27.3 kg) was added over 2h. The product slurry was aged for 3h at 20-25 °C then filtered and washed with a mixture of 2-MeTHF (2.8kg) and heptane (9kg). The product was dried using nitrogen and vacuum to afford solid 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (rac-DIONE (Compound 4)).
Step 3
[0140] To a vessel, an agitated suspension of Compound 4, (1.0 eq.) in 2- methylterahydrofuran (7.0 L/kg) was added (+)-2,3-dibenzoyl-D-tartaric acid (2.0 eq.) under an atmosphere of nitrogen. 2-MeTHF is chiral, but it is used as a racemic mixture. The different enantiomers of 2-MeTHF are incorporated randomly into the co-crystal. The resulting suspension was warmed to 75°C and aged at 75°C until full dissolution was observed (< 30 mins.). The resulting solution was polish filtered at 75°C into a secondary vessel. To the polish filtered solution was charged n-Heptane (2.0 L/kg) at a rate that maintained the internal temperature above 65°C. The solution was then cooled to 60°C, seeded with crystals (0.01 kg/kg) and allowed to age for 30 minutes. The resulting suspension was cooled to 20°C over 4 hours and then sampled for chiral purity analysis by HPLC. To the suspension, n-Heptane (3.0 L/kg) was charged and then aged for 4 hours at 20°C under an atmosphere of nitrogen. The suspension was filtered, and the isolated solids were washed two times with (2:1) n-Heptane:2-methyltetrahydrofuran (3.0 L/kg). The material was dried with nitrogen and vacuum to afford M-Dione:DBTA: Me-THF complex (Compound 4a).
Step 4
[0141] To vessel A, a suspension of disodium hydrogen phosphate (21.1 kg, 2.0 equiv) in DI water (296.8 L, 6.3 L/kg) was agitated until dissolution was observed (≥ 30 min.). To vessel B, a suspension of the M-Dione:DBTA: Me-THF complex (Composition 4a)[46.9 kg (25.9 kg corrected for M-dione, 1.0 equiv.)] in methyl tert-butyl ether (517.8 L, 11.0 L/kg) was agitated for 15 to 30 minutes. The resulting solution from vessel A was added to vessel B, and then the mixture was agitated for more than 3 hours. The agitation was stopped, and the biphasic mixture was left to separate for more than 30 minutes. The lower aqueous phase was removed and then back extracted with methyl tert-butyl ether (77.7 L, 1.7 L/kg). The organic phases were combined in vessel B and dried with magnesium sulfate (24.8 kg, 0.529 kg/kg). The resulting suspension from vessel B was agitated for more than three hours and then filtered into vessel C. To vessel B, a methyl tert-butyl ether (46.9 L, 1.0 L/kg) rinse was charged and then filtered into vessel C. The contents of vessel C were cooled to 10 °C and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until 320-350 kg (6.8-7.5 kg/kg) of methyl tert-butyl ether was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged a second time over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (195.9 L, 4.2 L/kg) was charged a third time over one hour and then sampled for solvent composition by GC analysis. The vessel C suspension continued to agitate for more than one hour. The suspension was filtered, and then washed with a n-Heptane (68.6 L, 1.5 L/kg) rinse from vessel C. The isolated solids were dried at 50°C, and a sample was submitted for stock suitability. Afforded 7-chloro-6-fluoro-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (M-DIONE) Compound 5M.
[0142] The first-generation process highlighted above has been successfully scaled on 200+ kg of rac-dione starting material (Compound 4). In this process, seeding the crystallization with the thermodynamically-stable rac-dione crystal form (which exhibits low solubility) would cause a batch failure. Based on our subsequent studies, we found that increasing the DBTA equivalents and lowering the seed temperature by adjusting heptane
charge schedule improves robustness of the process. The improved process is resistant to the presence of the thermodynamically-stable rac-dione crystal form and promotes successful separation of atropisomers. Subsequent batches will incorporate the improved process for large scale manufacture.
Step 5
Note: All L/kg amounts are relative to M-Dione input; All equiv. amounts are relative to M-Dione input after adjusted by potency.
[0143] M-Dione (Compound 5M, 1.0 equiv.) and Toluene-1 (10.0 L/kg) was charged to Vessel A. The resulting solution was dried by azeotropic distillation under vacuum at 45 °C until 5.0 L/kg of solvents has been removed. The contents of Vessel A were then cooled to 20 °C.
[0144] Vessel C was charged with Toluene-3 (4.5 L/kg), Phosphoryl chloride (1.5 equiv.) and N,N-Diisopropylethylamine-1 (2.0 equiv.) while maintaining the internal temperature below 20 ± 5 °C.
Upon finishing charging, Vessel C was warmed to 30 ± 5 °C. The contents of Vessel A were then transferred to Vessel C over 4 hours while maintaining the internal temperature at 30 ± 5°C. Vessel A was rinsed with Toluene-2 (0.5 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated at 30°C for an additional 3 hours. The contents of Vessel C were cooled to 20 ± 5 °C. A solution of (s)-1-boc-3-methylpiperazine (1.2 equiv.), N,N-Diisopropylethylamine-2 (1.2 equiv.) in isopropyl acetate-1 (1.0 L/kg) was prepared in Vessel D. The solution of Vessel D was charged to vessel C while maintaining a batch temperature of 20 ± 5 °C (Note: Exotherm is observed). Upon the end of transfer, Vessel D was rinsed with additional dichloromethane (1.0 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated for an additional 60 minutes at 20 °C. A solution of sodium bicarbonate [water-1 (15.0 L/kg + Sodium bicarbonate (4.5 equiv.)] was then charged into Vessel C over an hour while maintaining an internal temperature at 20 ± 5 °C throughout the addition. The contents of Vessel C were agitated for at least 12 hours at which point the Pipazoline (Compound 6) product was isolated by filtration in an agitated filter dryer. The cake was washed with water-2 and -3 (5.0 L/kg x 2 times, agitating each wash for 15 minutes) and isopropyl acetate-2 and 3 (5.0 L/kg x 2 times, agitating each wash for 15 min). The cake as dried under nitrogen for 12 hours.
Acetone Re-slurry (Optional):
[0145] Pipazoline (Compound 6) and acetone (10.0 L/kg) were charged to Vessel E. The suspension was heated to 50 °C for 2 hours. Water-4 (10.0 L/kg) was charged into Vessel E over 1 hour. Upon completion of water addition, the mixture was cooled to 20 °C over 1 hour. The contents of Vessel E were filtered to isolate the product, washing the cake with 1:1 acetone/water mixture (5.0 L/kg). The cake was dried under nitrogen for 12 hours.
Step 6
General Note: All equivalents and volumes are reported in reference to Pipazoline input
Note: All L/kg and kg/kg amounts are relative to Pipazoline input
[0146] Reactor A is charged with Pipazoline (Compound 6, 1.0 equiv), degassed 2- MeTHF (9.0 L/kg) and a solution of potassium acetate (2.0 equiv) in degassed water (6.5 L/kg). The resulting mixture is warmed to 75 ± 5 °C and then, charge a slurry of
Pd(dpePhos)Cl2 (0.003 equiv) in 2-MeTHF (0.5 L/kg). Within 2 h of catalyst charge, a solution of freshly prepared Boroxine (Compound 6A, 0.5 equiv) in wet degassed 2-MeTHF (4.0 L/kg, KF > 4.0%) is charged over the course of >1 hour, but < 2 hours, rinsing with an additional portion of wet 2-MeTHF (0.5 L/kg) after addition is complete. After reaction completion ( <0.15 area % Pipazoline remaining, typically <1 h after boroxine addition is complete), 0.2 wt% (0.002 kg/kg) of Biaryl seed is added as a slurry in 0.02 L/kg wet 2- MeTHF, and the resulting seed bed is aged for > 60 min. Heptane (5.0 L/kg) is added over 2 hours at 75 ± 5 °C. The batch is then cooled to 20 ± 5 °C over 2 hours and aged for an additional 2 h. The slurry is then filtered and cake washed with 1 x 5.0L/kg water, 1 x 5.0L/kg 1:1 iPrOH:water followed by 1 x 5.0 L/kg 1:1 iPrOH:heptane (resuspension wash: the cake is resuspended by agitator and allow to set before filtering) . The cake (Biaryl, Compound 7) is then dried under vacuum with a nitrogen sweep.
Note: If the reaction stalls, an additional charge of catalyst and boroxine is required
Step 7 Charcoal Filtration for Pd removal
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Note: All L/kg and kg/kg amounts are relative to crude Biaryl input
[0147] In a clean Vessel A, charge crude Biaryl (1 equiv) and charge DCM (10 L/kg). Agitate content for > 60 minutes at 22 ± 5 °C, observing dissolution. Pass crude Biaryl from Vessel A, through a bag filter and carbon filters at a flux ≤ 3 L2/min/m and collect filtrate in clean Vessel B. Charge DCM rinse (1 L/kg) to Vessel A, and through carbon filters to collect in vessel B.
[0148] From filtrate in Vessel B, pull a solution sample for IPC Pd content. Sample is concentrated to solid and analyzed by ICP-MS. IPC: Pd ≤ 25 ppm with respect to Biaryl. a. If Pd content is greater than 25 ppm with respect to Biaryl on first or second IPC sample, pass solution through carbon filter a second time at ≤ 3 L2/min/m2, rinsing with 1 L/kg DCM; sample filtrate for IPC.
b. If Pd content remains greater than 25 ppm after third IPC, install and condition fresh carbon discs. Pass Biaryl filtrate through refreshed carbon filter, washing with 1 L/kg DCM. Sample for IPC.
[0149] Distill and refill to appropriate concentration. Prepare for distillation of recovered filtrate by concentrating to ≤ 4 L/kg DCM, and recharge to reach 5.25 ± 0.25 L/kg DCM prior to moving into Step 7 Boc-deprotection reaction.
Step 7
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Note: All L/kg and kg/kg amounts are relative to Biaryl input
[0150] To Reactor A was added: tert-butyl (3S)-4-{6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl}-3-methylpiperazine-1-carboxylate (Biaryl) (1.0 equiv), dichloromethane (5.0 L/kg), and the TFA (15.0 equiv, 1.9 L/kg) is charged slowly to maintain the internal temperature at 20 ± 5 °C. The reaction was stirred for 4 h at 20 ± 5 °C.
[0151] To Reactor B was added: potassium carbonate (18.0 equiv), water (20.0 L/kg), and NMP (1.0) to form a homogenous solution. While agitating at the maximum acceptable rate for the equipment, the reaction mixture in A was transferred into the potassium carbonate solution in B over 30 minutes (~ 0.24 L/kg/min rate). The mixture was stirred at 20 ± 5 °C for an additional 12 h.
[0152] The resulting slurry was filtered and rinsed with water (2 x 10 L/kg). The wet cake was dried for 24 h to give 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-4-[(2S)-2-methylpiperazin- 1-yl]-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Des- Boc, Compound 8).
Step 8
Note: All L/kg and kg/kg amounts are relative to Des-Boc input
[0153] Des-Boc (Compound 8, 1.0 equiv) and NMP (4.2 L/kg) are charged to Vessel A under nitrogen, charge the TFA (1.0 equiv.) slowly to maintain the Tr <25 °C. The mixture is aged at 25 °C until full dissolution is observed (about 0.5 hour). The solution is then polish filtered through a 0.45 micron filter into Vessel B, washing with a NMP (0.8 L/kg). The filtrate and wash are combined, and then cooled to 0 °C. To the resulting solution, Acryloyl Chloride (1.3 equiv.) is added while maintaining temperature < 10 C. The reaction mixture is then aged at 5 ±5°C until completed by IPC (ca.1.5 hrs).
Preparation of Aqueous Disodium Phosphate Quench:
[0154] Disodium Phosphate (3.0 equiv) and Water (15.0 L/kg) are charged to Vessel C. The mixture is aged at 25 °C until full dissolution is observed. The solution is warmed to 45 ±5°C. A seed slurry of AMG 510 (0.005 equiv.) in Water (0.4 L/kg) is prepared and added to Vessel C while maintaining temperature at 45 ±5°C.
[0155] The reaction mixture in Vessel B is transferred to Vessel C (quench solution) while maintaining temperature at 45 ±5°C (ca.1 hrs). Vessel B is washed with a portion of NMP (0.5 L/kg). The product slurry is aged for 2 hrs at 45 ±5°C, cooled to 20 °C over 3 hrs, aged at 20 °C for a minimum of 12 hrs, filtered and washed with Water (2 x 10.0 L/kg). The product is dried using nitrogen and vacuum to afford Crude AMG 510 (Compound 9A).
Step 9
General Note: All equivalents and volumes are reported in reference to crude AMG 510 input
Note: All L/kg and kg/kg amounts are relative to Crude AMG 510 input
[0156] Reactor A was charged with 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4- methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1- yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Crude AMG 510) (1.0 equiv), ethanol (7.5 L/kg), and water (1.9 L/kg). The mixture heated to 75 °C and polish filtered into a clean Reactor B. The solution was cool to 45 °C and seeded with authentic milled AMG 510 seed (0.015 േ 0.005
1 Seed performs best when reduced in particle size via milling or with other type of mechanical grinding if mill is not available (mortar/ pestle). Actual seed utilized will be based on seed availability. 1.0- 2.0% is seed is target amount.
kg/kg); the resulting slurry was aged for 30 min. Water (15.0 L/kg) was added over 5h while maintaining an internal temperature > 40 °C; the mixture was aged for an additional 2h.
[0157] The mixture was cooled to 20 °C over 3 hours and aged for 8h, after which the solid was collected by filtration and washed using a mixture of ethanol (2.5 L/kg) and water (5.0 L/kg). The solid was dried using vacuum and nitrogen to obtain 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (AMG 510, Compound 9).
Compound 6A Boroxine Synthesis:
Lithiation/borylation
[0158] Reactor A was charged with THF (6 vol), a secondary amine base, Diisopropylamine (1.4 equiv), and a catalyst, such as triethylamine hydrochloride (0.01 equiv.). The resulting solution was cooled to -70 °C and a first base, n-BuLi (2.5 M in hexane, 1.5 equiv) was slowly added. After addition is complete, a solution of 3-fluoroanisole (1.0 equiv) in THF (6 vol) was added slowly and kept at -70 °C for 5 min. Concurrently or subsequently, a reagent, B(EtO)3 (2.0 equiv), was added slowly and kept at -70 °C for 10 min. The reaction mixture was quenched with an acid, 2N HCl. The quenched reaction mixture was extracted with MTBE (3 x 4 vol). The combined organic phases were concentrated to 1.5-3 total volumes. Heptane (7-9 vol) was added drop-wise and the mixture was cooled to 0-10 °C and stirred for 3 h. The mixture was filtrated and rinsed with heptane (1.5 vol). The solid was dried under nitrogen at < 30 °C to afford (2-fluoro-6-methoxyphenyl)boronic acid.
Demethylation:
Note: All L/kg and kg/kg amounts are relative to (2-fluoro-6-methoxyphenyl)boronic acid input
[0159] To a reactor, charge dichloromethane (solvent, 4.0 L/kg) and an acid, BBr3 (1.2 equiv), and cool to -20 °C. To this solution, a suspension of (2-fluoro-6-methoxyphenyl)boronic acid (1.0 equiv) in dichloromethane (4.0 L/kg) was added into the BBr3/DCM mixture while keeping temperature -15 to -25 °C. The reaction was allowed to proceed for approximately 2 hours while monitored by HPLC [≤1% (2-fluoro-6-methoxyphenyl)boronic acid] before reverse quenching into water (3.0 L/kg). The precipitated solid was then isolated by filtration and slurried with water (3.0 L/kg) on the filter prior to deliquoring. The filtrates were adjusted to pH 4-6 by the addition of sodium bicarbonate. The bottom organic phase was separated and the resulting aqueous layer was washed with dichloromethane (solvent, 5.0 Vol) and adjusted to pH = 1 by addition of concentrated hydrochloric acid. The resulting solids were isolated by filtration, washing the cake with water (2 x 5.0 L/kg)
Purification via Reslurry (required)
[0160] The combined crude solids were charged into a reactor and slurried with 5% EtOH/water (5.0 L/kg) at 20 °C for >1 h. The purified product was then isolated by filtration and rinsed with water (2 x 3 L/kg) before drying on the filter at < 30 °C to with nitrogen/vacuum to afford 2,2′,2”-(1,3,5,2,4,6-trioxatriborinane-2,4,6-triyl)tris(3-fluorophenol) (Boroxine, Compound 6A).
PATENT
WO 2020102730
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020102730
PATENT
US 20180334454
References
- ^ Jump up to:a b c d e “Lumakras- sotorasib tablet, coated”. DailyMed. Retrieved 6 June 2021.
- ^ Jump up to:a b c d e f g h i j k l m n “FDA Approves First Targeted Therapy for Lung Cancer Mutation Previously Considered Resistant to Drug Therapy”. U.S. Food and Drug Administration (FDA). 28 May 2021. Retrieved 28 May 2021. This article incorporates text from this source, which is in the public domain.
- ^ “KRAS mutant-targeting AMG 510”. NCI Drug Dictionary. National Cancer Institute. 2 February 2011. Retrieved 16 November2019.
- ^ Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. (November 2019). “The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity”. Nature. 575 (7781): 217–23. Bibcode:2019Natur.575..217C. doi:10.1038/s41586-019-1694-1. PMID 31666701.
- ^ Jump up to:a b “FDA approves Amgen drug for lung cancer with specific mutation”. CNBC. 28 May 2021. Retrieved 28 May 2021.
- ^ Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. (2020). “KRASG12C inhibition with sotorasib in advanced solid tumors”. N Engl J Med. doi:10.1056/NEJMoa1917239. PMC 7571518.
- ^ Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation ” at ClinicalTrials.gov
- ^ “The Discovery Of Amgen’s Novel Investigational KRAS(G12C) Inhibitor AMG 510 Published In Nature” (Press release). Amgen. 30 October 2019. Retrieved 16 November 2019.
- ^ Irving M (24 December 2019). “Drug targeting common cancer cause enters phase 2 clinical trials”. New Atlas. Retrieved 24 December 2019.
- ^ Jump up to:a b c d Halford B (3 April 2019). “Amgen unveils its KRas inhibitor in human clinical trials: AMG 510 shuts down a mutant version of the cancer target via covalent interaction”. Chemical & Engineering News. 97 (4). Retrieved 16 November 2019.
- ^ Al Idrus A (9 September 2019). “Amgen’s KRAS drug continues to deliver but faces ‘curse’ of high expectations”. fiercebiotech.com. Retrieved 16 November 2019.
- ^ Kaiser J (30 October 2019). “Two new drugs finally hit ‘undruggable’ cancer target, providing hope for treatments”. Science Magazine. AAAS. Retrieved 16 November 2019.
- ^ Astor L (9 September 2019). “FDA Grants AMG 510 Fast Track Designation for KRAS G12C+ NSCLC”. targetedonc.com. Retrieved 16 November 2019.
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 85” (PDF). WHO Drug Information. 35 (1).
Further reading
- Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. (September 2020). “KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors”. N Engl J Med. 383 (13): 1207–17. doi:10.1056/NEJMoa1917239. PMC 7571518. PMID 32955176.
- Lanman BA, Allen JR, Allen JG, Amegadzie AK, Ashton KS, Booker SK, et al. (January 2020). “Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors”. J Med Chem. 63 (1): 52–65. doi:10.1021/acs.jmedchem.9b01180. PMID 31820981.
External links
- “Sotorasib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation (CodeBreaK 100)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Lumakras |
| Other names | AMG 510 |
| License data | US DailyMed: Sotorasib |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2252403-56-6 |
| PubChem CID | 137278711 |
| DrugBank | DB15569 |
| ChemSpider | 72380148 |
| UNII | 2B2VM6UC8G |
| KEGG | D12055 |
| Chemical and physical data | |
| Formula | C30H30F2N6O3 |
| Molar mass | 560.606 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////Sotorasib, ソトラシブ , FDA 2021, APPROVALS 2021, Lumakras, CANCER, ANTINEOPLASTIC, AMG 510, AMG-510, AMG510, AMGEN, priority review, fast-track, breakthrough therapy, orphan drug
CC1CN(CCN1C2=NC(=O)N(C3=NC(=C(C=C32)F)C4=C(C=CC=C4F)O)C5=C(C=CN=C5C(C)C)C)C(=O)C=C
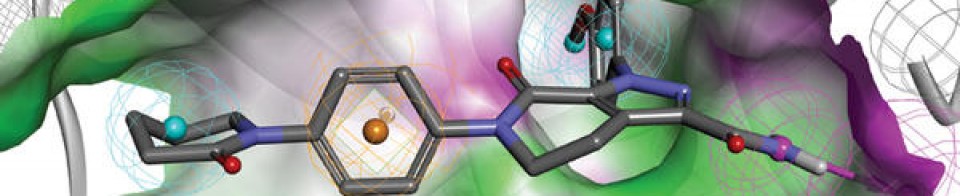
NEW DRUG APPROVALS
ONE TIME
$10.00
Infigratinib phosphate


Infigratinib phosphate
FDA APPR Truseltiq 2021/5/28
インフィグラチニブリン酸塩;
3-(2,6-dichloro-3,5-dimethoxyphenyl)-1-[6-[4-(4-ethylpiperazin-1-yl)anilino]pyrimidin-4-yl]-1-methylurea;phosphoric acid
- BGJ 398
- BGJ-398
- BGJ398
- NVP-BGJ398
- WHO 10032
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Infigratinib acetate | 03D0789NYP | 1310746-17-8 | XHCQHOGMMJKLRU-UHFFFAOYSA-N |
| Infigratinib hydrochloride | WY8VD4RV77 | 1310746-15-6 | VBAIJSJSFCXDJB-UHFFFAOYSA-N |
| Infigratinib mesylate | E223Z0KWCC | 1310746-12-3 | BXJJFNXYWJLBOS-UHFFFAOYSA-N |
| Infigratinib phosphate | 58BH47BV6S | 1310746-10-1 | GUQNHCGYHLSITB-UHFFFAOYSA-N |
International/Other BrandsTruseltiq (BridgeBio Pharma, Inc.)
| Formula | C26H31Cl2N7O3. H3PO4 |
|---|---|
| CAS | 1310746-10-1FREE form 872511-34-7 |
| Mol weight | 658.4706 |
- Originator Novartis
- Developer Array BioPharma; Novartis; Novartis Oncology; QED Therapeutics
- Class Aniline compounds; Antineoplastics; Chlorobenzenes; Methylurea compounds; Phenyl ethers; Piperazines; Pyrimidines; Small molecules
- Mechanism of Action Type 1 fibroblast growth factor receptor antagonists; Type 3 fibroblast growth factor receptor antagonists; Type 4 fibroblast growth factor receptor antagonists; Type-2 fibroblast growth factor receptor antagonists
- Orphan Drug Status Yes – Cholangiocarcinoma
- RegisteredCholangiocarcinoma
- Phase IIIBladder cancer; Urogenital cancer
- Phase IIAchondroplasia; Head and neck cancer
- Phase IBreast cancer
- Phase 0Glioblastoma
- DiscontinuedHaematological malignancies; Malignant melanoma; Solid tumours
- 31 May 2021Clinical development is ongoing in Bladder cancer (QED Therapeutics pipeline, May 2020)
- 28 May 2021Registered for Cholangiocarcinoma (Second-line therapy or greater, Metastatic disease, Inoperable/Unresectable, Late-stage disease) in USA (PO) – First global approval (under Project Orbis using RTOR program)
- 28 May 2021Efficacy and safety data from a phase II trial in Cholangiocarcinoma released by QED Therapeutics
Infigratinib, sold under the brand name Truseltiq, is an anti-cancer medication used to treat cholangiocarcinoma (bile duct cancer).[1][2]
Infigratinib is a receptor tyrosine kinase inhibitor (and more specifically an inhibitor of the fibroblast growth factor receptors FGFR1, FGFR2, FGFR3).[3][1][2] It was designated an orphan drug by the U.S. Food and Drug Administration (FDA) in 2019,[4] and it was approved for medical use in the United States in May 2021.[2]
Infigratinib is a pan-fibroblast growth factor receptor (FGFR) kinase inhibitor. By inhibiting the FGFR pathway, which is often aberrated in cancers such as cholangiocarcinoma, infigratinib suppresses tumour growth.1 Cholangiocarcinoma is the most common primary malignancy affecting the biliary tract and the second most common primary hepatic malignancy.2 Infitratinib is a pan-FGFR inhibitor, as it is an ATP-competitive inhibitor of all four FGFR receptor subtypes.1
On May 28, 2021, the FDA granted accelerated approval to infigratinib – under the market name Truseltiq – for the treatment of previously treated, unresectable locally advanced or metastatic cholangiocarcinoma in adults with a fibroblast growth factor receptor 2 (FGFR2) fusion or another rearrangement as detected by an FDA-approved test.5 This approval follows pemigatinib, another FGFR inhibitor approved by the FDA for the same therapeutic indication.
Infigratinib is indicated for the treatment of previously treated, unresectable locally advanced or metastatic cholangiocarcinoma in adults with a fibroblast growth factor receptor 2 (FGFR2) fusion or another rearrangement as detected by an FDA-approved test.4
Medical uses
Infigratinib is indicated for the treatment of adults with previously treated, unresectable locally advanced or metastatic cholangiocarcinoma (bile duct cancer) with a fibroblast growth factor receptor 2 (FGFR2) fusion or other rearrangement as detected by an FDA-approved test.[1]
PAPER
Journal of Medicinal Chemistry (2011), 54(20), 7066-7083.
https://pubs.acs.org/doi/10.1021/jm2006222

A novel series of N-aryl-N′-pyrimidin-4-yl ureas has been optimized to afford potent and selective inhibitors of the fibroblast growth factor receptor tyrosine kinases 1, 2, and 3 by rationally designing the substitution pattern of the aryl ring. On the basis of its in vitro profile, compound 1h (NVP-BGJ398) was selected for in vivo evaluation and showed significant antitumor activity in RT112 bladder cancer xenografts models overexpressing wild-type FGFR3. These results support the potential therapeutic use of 1h as a new anticancer agent.

PATENT
US 9067896
PATENT
WO 2020243442
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020243442
In 2018, it was estimated that 150,350 new patients would be diagnosed with urinary system cancer: 81,190 urinary bladder; 65,340 kidney and renal pelvis; and, 3,820 ureter and other urinary organs. Excluding non-urothelial kidney cancers, approximately 5 to 10% of all urothelial carcinomas are upper tract urothelial carcinomas (UTUC). The incidence of UTUC is 2 to 3 times greater in men than women (Siegel et al, 2018; Roupret et al, 2015).
[0003] In contrast to invasive urinary bladder cancer (UCB), UTUC has a more aggressive clinical course. At the time of diagnosis, 60% of patients with UTUC have invasive cancer compared to 15% to 25% of patients with UCB (Roupret et al, 2015; Margulis et al., 2009). Thirty-six percent have regional disease and 9% distant disease (Raman et al., 2010). A large retrospective review of 1363 patients with UTUC who underwent radical nephroureterectomy (RNU) at 12 centers demonstrated that 28% of the total population had recurrence after RNU (Margulis et al, 2009).
[0004] To reduce the morbidity and mortality in patients with UTUC, neoadjuvant or adjuvant treatment is needed. The POUT study, a large randomized trial in UTUC supports the use of standard-of-care adjuvant cisplatin-based chemotherapy (Birtle et al., 2020). Because many patients with UTUC will have one remaining kidney following RNU and frequently have other significant co-morbid conditions, cisplatin-based therapy is not well tolerated (NCCN Guidelines Version 3, 2018). Renal function before and after RNU greatly limits the number of patients with UTUC who are eligible for platinum-based neoadjuvant or adjuvant therapy. Therefore, targeted therapies are needed for treating UTUC (Lane et al., 2010).
[0005] Despite demonstrated survival benefit for neoadjuvant treatment of invasive UCB, many patients with invasive UCB are unlikely to receive (neo)adjuvant cisplatin-based chemotherapy, due in part to cisplatin ineligibility (Porter et al., 2011). In addition, residual disease following neoadjuvant therapy is associated with a poor prognosis (Grossman et al, 2003). Therefore,
there remains an unmet need for a substantial proportion of patients with invasive UCB who are ineligible or refuse to receive cisplatin-based adjuvant chemotherapy or who have residual disease following neoadjuvant therapy.
Infigratinib, as depicted in formula (I), is a selective and ATP-competitive pan-fibroblast growth factor receptor (FGFR) kinase inhibitor, also known as 3-(2,6-dichloro-3,5-dimethoxyphenyl)- 1 – { 6- [4-(4-ethyl- 1 -piperazin- 1 -yljphenylamino] -pyrimidinyl-4-yl } – 1 -methylurea. Infigratinib selectively inhibits the kinase activity of FGFR1, FGFR2, FGFR3, and
FGFR4.
PATENT
WO 2011071821
https://patents.google.com/patent/WO2011071821A1/en3-(2,6-Dichloro-3,5-dimethoxy-phenyl)-l-{6-[4-(4-ethyl-piperazin-l-yl)- phenylamino]-pyrimidin-4-yl}-l -methyl-urea (described in USSN 11/570983, filed June 23, 2005, and incorporated by reference in its entirety herein) has the structure of Formula I:

The compound of Formula I is a protein kinase inhibitor and is useful in the treatment of proliferative diseases mediated by protein kinases. In particular, the compound of Formula I inhibits FGFR1, FGFR2, FGFR3, FGFR4, KDR, HER1, HER2, Bcr-Abl, Tie2, and Ret kinases. It is therefore useful in the treatment of cancers including AML, melanocyte neoplasia, breast cancer, colon cancer, lung cancer (especially small-cell lung cancer), cancer of the prostate or Kaposi’s sarcoma.[0003] It is well known that the crystalline form of the active pharmaceutical ingredient (API) of a particular drug is often an important determinant of the drug’s ease of preparation, hygroscopicity, stability, solubility, storage stability, ease of formulation, rate of dissolution in gastrointestinal fluids and in vivo bioavailability. Crystalline forms occur where the same composition of matter crystallizes in a different lattice arrangement resulting in different thermodynamic properties and stabilities specific to the particular crystalline form.Crystalline forms may also include different hydrates or solvates of the same compound. In deciding which form is preferable, the numerous properties of the forms are compared and the preferred form chosen based on the many physical property variables. It is entirely possible that one form can be preferable in some circumstances where certain aspects such as ease of preparation, stability, etc. are deemed to be critical. In other situations, a different form may be preferred for greater dissolution rate and/or superior bioavailability. It is not yet possible to predict whether a particular compound or salt of a compound will form polymorphs, whether any such polymorphs will be suitable for commercial use in a therapeutic composition, or which polymorphs will display such desirable properties.Example 2: Manufacture of the Free Base of the Compound of Formula I

IA. N- [4-(4-ethyl-piperazin- 1 -yl)-phenyl] -N’ -methyl-pyrimidine-4,6-diamine[0077] A mixture of 4-(4-ethylpiperazin-l-yl)-aniline (1 g, 4.88 mmol), (6-chloro- pyrimidin-4-yl)-methyl-amine (1.81 g, 12.68 mmol, 1.3 eq.), and 4N HC1 in dioxane (15 ml) is heated in a sealed tube to 150°C for 5h. The reaction mixture is concentrated, diluted with DCM and a saturated aqueous solution of sodium bicarbonate. The aqueous layer is separated and extracted with DCM. The organic phase is washed with brine, dried (sodium sulfate), filtered and concentrated. Purification of the residue by silica gel column chromatography (DCM/MeOH, 93:7) followed by trituration in diethyl ether affords the title compound as a white solid: ESI-MS: 313.2 [MH]+; tR= 1.10 min (gradient J); TLC: Rf = 0.21 (DCM/MeOH, 93:7).B. 4-(4-Ethylpiperazin- 1 -yl)-aniline[0078] A suspension of l-ethyl-4-(4-nitro-phenyl)-piperazine (6.2 g, 26.35 mmol) and Raney Nickel (2 g) in MeOH (120 mL) is stirred for 7 h at RT, under a hydrogen atmosphere. The reaction mixture is filtered through a pad of celite and concentrated to afford 5.3 g of the title compound as a violet solid: ESI-MS: 206.1 [MH]+; TLC: Rf = 0.15 (DCM/MeOH + 1 % NH3aq, 9:l).C. 1 -Ethyl-4-(4-nitro-phenyl)-piperazine[0079] A mixture of l-bromo-4-nitrobenzene (6 g, 29.7 mmol) and 1-ethylpiperazine (7.6 ml, 59.4 mmol, 2 eq.) is heated to 80°C for 15h. After cooling to RT, the reaction mixture is diluted with water and DCM/MeOH, 9:1. The aqueous layer is separated and extracted with DCM/MeOH, 9:1. The organic phase is washed with brine, dried (sodium sulfate), filtered and concentrated. Purification of the residue by silica gel column chromatography(DCM MeOH + 1 % NH3aq, 9:1) affords 6.2 g of the title compound as a yellow solid: ESI- MS: 236.0 [MH]+; tR= 2.35 min (purity: 100%, gradient J); TLC: Rf = 0.50 (DCM/MeOH + 1 % NH3aq, 9:1).D. (6-chloro-pyrimidin-4-yl)-methyl-amine[0080] This material was prepared by a modified procedure published in the literature (J. Appl. Chem. 1955, 5, 358): To a suspension of commercially available 4,6- dichloropyrimidine (20 g, 131.6 mmol, 1.0 eq.) in isopropanol (60 ml) is added 33% methylamine in ethanol (40.1 ml, 328.9 mmol, 2.5 eq.) at such a rate that the internal temperature does not rise above 50°C. After completion of the addition the reaction mixture was stirred for lh at room temperature. Then, water (50 ml) is added and the suspension formed is chilled in an ice bath to 5°C. The precipitated product is filtered off, washed with cold isopropanol/water 2:1 (45 ml) and water. The collected material is vacuum dried over night at 45°C to afford the title compound as colorless powder: tR = 3.57 min (purity: >99%, gradient A), ESI-MS: 144.3 / 146.2 [MH]+.E. (3-(2,6-Dichloro-3,5-dimethoxy-phenyl)-l-{6-[4-(4-ethyl-piperazin-l-yl)-phenylamino]- pyrimidin-4-yl} – 1 -methyl-urea)[0081] The title compound was prepared by adding 2,6-dichloro-3,5-dimethoxyphenyl- isocyanate (1.25 eq.) to a solution of N-[4-(4-ethyl-piperazin-l-yl)-phenyl]-N’-methyl- pyrimidine-4,6-diamine (2.39 g, 7.7 mmol, 1 eq.) in toluene and stirring the reaction mixture for 1.5h at reflux. Purification of the crude product by silica gel column chromatography (DCM MeOH + 1 % NH3aq, 95:5) affords the title compound as a white solid: ESI-MS: 560.0 / 561.9 [MHf; tR= 3.54 min (purity: 100%, gradient J); TLC: Rf = 0.28 (DCM/MeOH + 1 % NH3aq, 95:5). Analysis: C26H3iN703Cl2, calc. C 55.72% H 5.57% N 17.49% O 8.56% CI 12.65%; found C 55.96% H 5.84% N 17.17% O 8.46% CI 12.57%. The title compound was characterized by XRPD, thermal and other methods as described below. Example 3: Manufacture of the Monophosphoric Acid Salt Form A of the Compound of Formula I.[0082] To a round bottom flask was added 3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-{6-[4- (4-ethylpiperazin-l-yl)phenylamino]-pyrimidine-4-yl}-l -methyl-urea (134 g, 240 mmol) and IPA (2000 ml). The suspension was stirred and heated to 50°C and a solution of phosphoric acid (73.5 g, 750 mmol) in water (2000 ml) added to it portions. The mixture was stirred at 60°C for 30 min. and filtered through a polypropylene pad. The pad was washed with warm IP A/water (1:1, 200 ml) and the filtrates were combined. To this clear solution, IPA (6000 ml) was added and the mixture was stirred under reflux for 20 min, cooled slowly to room temperature (25° C), and stirred for 24 hours. The white salt product was collected by filtration, washed with IPA (2 χ 500 ml) and dried in the oven at 60° C under reduced pressure for two days to provide the phosphate salt (form A) 110 g. Yield 70%. Purity >98% by HPLC. Analysis: C26H34 707C12P, calc. C 47.42% H 5.20% N 14.89% O 17.01% CI 10.77% P 4.70%; found C 47.40% H 5.11% N 14.71% O 17.18% CI 10.73% P 4.87%. The title compound was characterized by XRPD, thermal and other methods as described below.
References
- ^ Jump up to:a b c d “Infigratinib prescribing information” (PDF). U.S. Food and Drug Administration. May 2021.
- ^ Jump up to:a b c “BridgeBio Pharma’s Affiliate QED Therapeutics and Partner Helsinn Group Announce FDA Approval of Truseltiq (infigratinib) for Patients with Cholangiocarcinoma” (Press release). BridgeBio Pharma. 28 May 2021. Retrieved 28 May 2021 – via GlobeNewswire.
- ^ Botrus G, Raman P, Oliver T, Bekaii-Saab T (April 2021). “Infigratinib (BGJ398): an investigational agent for the treatment of FGFR-altered intrahepatic cholangiocarcinoma”. Expert Opinion on Investigational Drugs. 30 (4): 309–316. doi:10.1080/13543784.2021.1864320. PMID 33307867.
- ^ “Infigratinib Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 11 September 2019. Retrieved 30 May 2021.
External links
- “Infigratinib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02150967 for “A Phase II, Single Arm Study of BGJ398 in Patients With Advanced Cholangiocarcinoma” at ClinicalTrials.gov
| Efficacy | Antineoplastic, Angiogenesis inhibitor |
|---|---|
| Disease | Cholangiocarcinoma (FGFR2 fusion or other rearrangement) |
| Clinical data | |
|---|---|
| Trade names | Truseltiq |
| Other names | BGJ-398 |
| License data | US DailyMed: Infigratinib |
| Routes of administration | By mouth |
| Drug class | Tyrosine kinase inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| CAS Number | 872511-34-7 |
| PubChem CID | 53235510 |
| DrugBank | DB11886 |
| ChemSpider | 26333103 |
| UNII | A4055ME1VK |
| KEGG | D11589 |
| CompTox Dashboard (EPA) | DTXSID70236238 |
| Chemical and physical data | |
| Formula | C26H31Cl2N7O3 |
| Molar mass | 560.48 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESCCN1CCN(CC1)C2=CC=C(C=C2)NC3=CC(=NC=N3)N(C)C(=O)NC4=C(C(=CC(=C4Cl)OC)OC)Cl | |
| hideInChIInChI=1S/C26H31Cl2N7O3/c1-5-34-10-12-35(13-11-34)18-8-6-17(7-9-18)31-21-15-22(30-16-29-21)33(2)26(36)32-25-23(27)19(37-3)14-20(38-4)24(25)28/h6-9,14-16H,5,10-13H2,1-4H3,(H,32,36)(H,29,30,31)Key:QADPYRIHXKWUSV-UHFFFAOYSA-N |
////////Infigratinib phosphate, FDA 2021 APPROVALS 2021, Truseltiq, インフィグラチニブリン酸塩 , Orphan Drug, Cholangiocarcinoma, BGJ 398, BGJ-398, BGJ398, NVP-BGJ398, WHO 10032

NEW DRUG APPROVALS
one time
$10.00
Ibrexafungerp citrate, Brexafemme
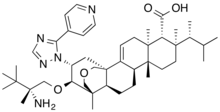
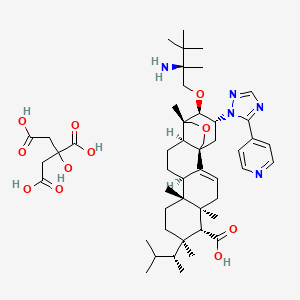

Ibrexafungerp citrate
| アイブレキサフンジェルプクエン酸塩; |
| Formula | C44H67N5O4. C6H8O7 |
|---|---|
| cas | Citrate1965291-08-0 free 1207753-03-4 |
| Mol weight | 922.1574 |
Brexafemme, fda approved 2021, 2021/6/1
Antifungal, Cell wall biosynthesis inhibitor, Treatment of invasive fungal infections due to Candida spp. or Aspergillus spp., vulvovaginal candidiasis
SCY-078 citrate, MK-3118; SCY-078,
- WHO 10597
(1R,5S,6R,7R,10R,11R,14R,15S,20R,21R)-21-[(2R)-2-amino-2,3,3-trimethylbutoxy]-5,7,10,15-tetramethyl-7-[(2R)-3-methylbutan-2-yl]-20-(5-pyridin-4-yl-1,2,4-triazol-1-yl)-17-oxapentacyclo[13.3.3.01,14.02,11.05,10]henicos-2-ene-6-carboxylic acid;2-hydroxypropane-1,2,3-tricarboxylic acid
- Originator Merck & Co; SCYNEXIS
- Class Antifungals; Glycosides; Triterpenes
- Mechanism of ActionBeta-1,3-D glucan synthetase inhibitors
- Orphan Drug StatusYes – Invasive bronchopulmonary aspergillosis; Candidiasis
- RegisteredVulvovaginal candidiasis
- Phase IIICandidiasis
- Phase IIInvasive bronchopulmonary aspergillosis
- Phase IUnspecified
- PreclinicalPneumocystis pneumonia
- 01 Jun 2021Registered for Vulvovaginal candidiasis (In adolescents, In children, In the elderly, In adults) in USA (PO)
- 01 May 2021Ibrexafungerp – SCYNEXIS receives Qualified Infectious Disease Product status for Vulvovaginal candidiasis (Recurrent, Prevention) in USA
- 30 Apr 2021Efficacy data from phase III VANISH-303 and VANISH-306 trials in Vulvovaginal Candidiasis presented at the 2021 American College of Obstetricians and Gynecologists Annual Meeting (ACOG-2021)
Ibrexafungerp, sold under the brand name Brexafemme, is an antifungal medication used to treat vulvovaginal candidiasis (VVC) (vaginal yeast infection).[1] It is taken by mouth.[1]
Ibrexafungerp is a triterpenoid antifungal.[1]
Ibrexafungerp was approved for medical use in the United States in June 2021.[1][2] It is the first approved drug in a novel antifungal class.[2]
Medical uses
Ibrexafungerp is indicated for the treatment of adult and postmenarchal pediatric females with vulvovaginal candidiasis (VVC).[1][2]
Syn
https://www.sciencedirect.com/science/article/abs/pii/S0960894X20307721

Abstract
We previously reported medicinal chemistry efforts that identified MK-5204, an orally efficacious β-1,3-glucan synthesis inhibitor derived from the natural product enfumafungin. Further extensive optimization of the C2 triazole substituent identified 4-pyridyl as the preferred replacement for the carboxamide of MK-5204, leading to improvements in antifungal activity in the presence of serum, and increased oral exposure. Reoptimizing the aminoether at C3 in the presence of this newly discovered C2 substituent, confirmed that the (R) t-butyl, methyl aminoether of MK-5204 provided the best balance of these two key parameters, culminating in the discovery of ibrexafungerp, which is currently in phase III clinical trials. Ibrexafungerp displayed significantly improved oral efficacy in murine infection models, making it a superior candidate for clinical development as an oral treatment for Candida and Aspergillus infections.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
SYN
Bioorg. Med. Chem. Lett. 2021, 32, 127661.
PATENT
WO 2010019204
https://patents.google.com/patent/WO2010019204A1/en
SYN
https://doi.org/10.1021/acs.jmedchem.3c00501
Ibrexafungerp (Brexafemme). Ibrexafungerp (1), formerly SCY-078 or MK-3118 and developed by Scynexis Inc., is a first-in-class triterpenoid antifungal that inhibits the biosynthesis of β-(1,3)
D-glucan in the fungal cell wall. This mechanism of action provides an opportunity for the treatment
of fungal infections that are azole- or echinocandrin-resistant strains. In June 2021, ibrexafungerp received its first approval by the United States Food and Drug Administration (USFDA) for
the treatment of vulvovaginal candidiasis in adult and postmenarchal pediatric females.
24,25 Ibrexafungerp is a semisynthetic derivative of the natural product enfumafungin that
incorporates a pyridine triazole moiety on the core phenanthropyran ring system as well as a pendant 2-amino-2,3,3trimethyl-butyl ether. The drug demonstrates potent, broad spectrum activity against Candida sp. and is orally bioavailable.
As shown in Scheme 1, the synthesis of ibrexafungerp started with the natural product enfumafungin (1.1). The lactol of enfumafungin was reduced using triethylsilane and trifluoroacetic acid to give pyran 1.2. Treatmentwith H2SO4 in methanol resulted in cleavage of the glucose moiety to generate 1.3 in 87%
yield over 2 steps. Carboxylic acid 1.3 was converted to the corresponding benzyl ester upon treatment with benzyl bromide to give compound 1.4in an89%yield. Reaction of 1.4 with (R)
N-sulfonyl aziridine 1.5 (prepared as shown in Scheme 2) in the presence of potassium t-pentylate and the cation complexing agent 18-crown-6 provided ether 1.6 in 78% yield. Metal reduction with sodiumin liquid ammoniaconcurrently removed the N-sulfonyl benzyl groups to generate compound 1.7, which
was converted to hydrazine intermediate 1.8 with anhydrous hydrazine and BF32·OEt 28-30 in 1,2-dichloroethane (DCE). Cyclocondensation of 1.8 with acyl amidine derivative 1.9 upon heating in acetic acid then provided ibrexafungerp (1) in 66% yield.
Thepreparationof(R)-N-sulfonylaziridine1.5 isdescribedin Scheme 2. Condensation of 3,3-dimethylbutan-2-one (1.10)with (R)-p-toluenesulfinamide (1.11) gave an 84% yield of compound 1.12, which cyclized upon treatment with trimethylsulfoxonium chloride and n-butyllithium to give chiral toluenesulfinyl aziridine 1.13 in 64% yield. Oxidation of 1.13 with meta-chloroperoxybenzoic acid afforded the tosyl-pro
tected (R)-alpha-disubstituted aziridine 1.5..
(24) Lee, A. Ibrexafungerp: First approval. Drugs 2021, 81, 1445−
1450.
(25) Jallow, S.; Govender, N. P. Ibrexafungerp: A first-in-class oral
triterpenoid glucan synthase inhibitor. J. Fungi 2021, 7, 163.
(26) Lamoth, F.; Alexander, B. D. Antifungal activities of SCY-078
(MK-3118) and standard antifungal agents against clinical non
aspergillus mold isolates. Antimicrob. Agents Chemother. 2015, 59,
4308−4311
(27) Scorneaux, B.; Angulo, D.; Borroto-Esoda, K.; Ghannoum, M.;
Peel, M.; Wring, S. SCY-078 is fungicidal against Candida species in
time-kill studies. Antimicrob. Agents Chemother. 2017, 61, e01961-16.
(28) Apgar, J. M.; Wilkening, R. R.; Parker, D. L.; Meng, D.;
Wildonger, K. J.; Sperbeck, D.; Greenlee, M. L.; Balkovec, J. M.;
Flattery, A. M.; Abruzzo, G. K.; Galgoci, A. M.; Giacobbe, R. A.; Gill, C.
J.; Hsu, M.-J.; Liberator, P.; Misura, A. S.; Motyl, M.; Nielsen Kahn, J.;
Powles, M.; Racine, F.; Dragovic, J.; Fan, W.; Kirwan, R.; Lee, S.; Liu,
H.; Mamai, A.; Nelson, K.; Peel, M. Ibrexafungerp: an orally active β
1,3-glucan synthesis inhibitor. Bioorg. Med. Chem. Lett. 2021, 32,
127661.
(29) Greenlee, M. L.; Wilkening, R.; Apgar, J.; Sperbeck, D.;
Wildonger, K. J.; Meng, D.; Parker, D. L.; Pacofsky, G. J.; Heasley, B.
H.; Mamai, A.; Nelson, K. Antifungal Agents. WO 2010019204, 2010.
(30) Greenlee, M. L.; Wilkening, R.; Apgar, J.; Wildonger, K. J.; Meng,
D.; Parker, D. L. Antifungal Agents. WO 2010019203A1, 2010.
(31) Imran, M.; Khan, S. A.; Alshammari, M. K.; Alqahtani, A. M.;
Alanazi, T. A.; Kamal, M.; Jawaid, T.; Ghoneim, M. M.; Alshehri, S.;
Shakeel, F. Discovery, development, inventions and patent review of
fexinidazole: The first all-oral therapy for human African trypanoso
miasis. Pharmaceuticals 2022, 15, 128.


SYN
European Journal of Medicinal Chemistry 245 (2023) 114898
The gram-scale synthesis of this drug is demonstrated in Scheme 3 [50]. Starting with triterpene glycoside enfumafungin 14, a reduction of the bridging hemiacetal with triethylsilane provided the intermediate 15, followed by hydrolysis, etherification and benzyl protection, gave compound 16 in 76% yield over 2 steps. Subsequent ring-opening alkylation reaction of 16 with tosyl protected aziridine 17 gave com pound 18, which then underwent Borch reduction to provide the in termediate 19. Treatment of 19 with biaryl 20 in the presence of boron trifluoride diethyl etherate gave rise to the substitution product ibrexafungerp. In this synthetic method, the pyridine-triazolium biaryl and chiral benzene sulfonamide were elegantly introduced into the triterpene enfumafungin through ring-opening and substitution reactions to give the triterpene derivative. These elegant and practical synthetic
methods could be employed as the versatile tools for the synthesis of other drug molecules.
[50] J.M. Apgar, R.R. Wilkening, D.L. Parker, J.D. Meng, K.J. Wildonger, D. Sperbeck,
M.L. Greenlee, J.M. Balkovec, A.M. Flattery, G.K. Abruzzo, A.M. Galgoci, R.
A. Giacobbe, C.J. Gill, M.J. Hsu, P. Liberator, A.S. Misura, M. Motyl, J.N. Kahn,
M. Powles, F. Racine, J. Dragovic, W. Fan, R. Kirwan, S. Lee, H. Liu, A. Mamai,
K. Nelson, M. Peel, Ibrexafungerp: an orally active β-1, 3-glucan synthesis
inhibitor, Bioorg, Med. Chem. Lett. 32 (2021), 127661.

.
References
- ^ Jump up to:a b c d e f g https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214900s000lbl.pdf
- ^ Jump up to:a b c “Scynexis Announces FDA Approval of Brexafemme (ibrexafungerp tablets) as the First and Only Oral Non-Azole Treatment for Vaginal Yeast Infections”. Scynexis, Inc. (Press release). 2 June 2021. Retrieved 2 June 2021.
Further reading
- Azie N, Angulo D, Dehn B, Sobel JD (September 2020). “Oral Ibrexafungerp: an investigational agent for the treatment of vulvovaginal candidiasis”. Expert Opin Investig Drugs. 29 (9): 893–900. doi:10.1080/13543784.2020.1791820. PMID 32746636.
- Davis MR, Donnelley MA, Thompson GR (July 2020). “Ibrexafungerp: A novel oral glucan synthase inhibitor”. Med Mycol. 58 (5): 579–592. doi:10.1093/mmy/myz083. PMID 31342066.
- Petraitis V, Petraitiene R, Katragkou A, Maung BB, Naing E, Kavaliauskas P, et al. (May 2020). “Combination Therapy with Ibrexafungerp (Formerly SCY-078), a First-in-Class Triterpenoid Inhibitor of (1→3)-β-d-Glucan Synthesis, and Isavuconazole for Treatment of Experimental Invasive Pulmonary Aspergillosis”. Antimicrob Agents Chemother. 64 (6). doi:10.1128/AAC.02429-19. PMC 7269506. PMID 32179521.
External links
- “Ibrexafungerp”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03734991 for “Efficacy and Safety of Oral Ibrexafungerp (SCY-078) vs. Placebo in Subjects With Acute Vulvovaginal Candidiasis (VANISH 303)” at ClinicalTrials.gov
- Clinical trial number NCT03987620 for “Efficacy and Safety of Oral Ibrexafungerp (SCY-078) vs. Placebo in Subjects With Acute Vulvovaginal Candidiasis (Vanish 306)” at ClinicalTrials.gov
Ibrexafungerp, also known as SCY-078 or MK-3118, is a novel enfumafungin derivative oral triterpene antifungal approved for the treatment of vulvovaginal candidiasis (VVC), also known as a vaginal yeast infection.1,9 It was developed out of a need to treat fungal infections that may have become resistant to echinocandins or azole antifungals.1 Ibrexafungerp is orally bioavailable compared to the echinocandins caspofungin, micafungin, and anidulafungin; which can only be administered parenterally.1,2 Similar to echinocandins, ibrexafungerp targets the fungal β-1,3-glucan synthase, which is not present in humans, limiting the chance of renal or hepatic toxicity.6,9
Ibrexafungerp was granted FDA approval on 1 June 2021.9
β-1,3-glucan synthase is composed of a catalytic subunit, FKS1 or FKS2, and a GTP-binding regulatory subunit, Rho1.5,6 This synthase is involved in the synthesis of β-1,3-glucan, a fungal cell wall component.6
Ibrexafungerp acts similarly to the echinocandin antifungals, by inhibiting the synthesis of β-1,3-glucan synthase.1,9 While echinocandins bind to the FKS1 domain of β-1,3-glucan synthase, enfumafungin and its derivatives bind at an alternate site which allows them to maintain their activity against fungal infections that are resistant to echinocandins.3,4
Ibrexafungerp has been shown in animal studies to distribute well to vaginal tissue, making it a favourable treatment for vulvovaginal candidiasis.4
- Wring SA, Randolph R, Park S, Abruzzo G, Chen Q, Flattery A, Garrett G, Peel M, Outcalt R, Powell K, Trucksis M, Angulo D, Borroto-Esoda K: Preclinical Pharmacokinetics and Pharmacodynamic Target of SCY-078, a First-in-Class Orally Active Antifungal Glucan Synthesis Inhibitor, in Murine Models of Disseminated Candidiasis. Antimicrob Agents Chemother. 2017 Mar 24;61(4). pii: AAC.02068-16. doi: 10.1128/AAC.02068-16. Print 2017 Apr. [Article]
- Hector RF, Bierer DE: New beta-glucan inhibitors as antifungal drugs. Expert Opin Ther Pat. 2011 Oct;21(10):1597-610. doi: 10.1517/13543776.2011.603899. Epub 2011 Jul 25. [Article]
- Kuhnert E, Li Y, Lan N, Yue Q, Chen L, Cox RJ, An Z, Yokoyama K, Bills GF: Enfumafungin synthase represents a novel lineage of fungal triterpene cyclases. Environ Microbiol. 2018 Sep;20(9):3325-3342. doi: 10.1111/1462-2920.14333. Epub 2018 Sep 13. [Article]
- Larkin EL, Long L, Isham N, Borroto-Esoda K, Barat S, Angulo D, Wring S, Ghannoum M: A Novel 1,3-Beta-d-Glucan Inhibitor, Ibrexafungerp (Formerly SCY-078), Shows Potent Activity in the Lower pH Environment of Vulvovaginitis. Antimicrob Agents Chemother. 2019 Apr 25;63(5). pii: AAC.02611-18. doi: 10.1128/AAC.02611-18. Print 2019 May. [Article]
- Ha YS, Covert SF, Momany M: FsFKS1, the 1,3-beta-glucan synthase from the caspofungin-resistant fungus Fusarium solani. Eukaryot Cell. 2006 Jul;5(7):1036-42. doi: 10.1128/EC.00030-06. [Article]
- Perlin DS: Mechanisms of echinocandin antifungal drug resistance. Ann N Y Acad Sci. 2015 Sep;1354:1-11. doi: 10.1111/nyas.12831. Epub 2015 Jul 17. [Article]
- Wring S, Murphy G, Atiee G, Corr C, Hyman M, Willett M, Angulo D: Clinical Pharmacokinetics and Drug-Drug Interaction Potential for Coadministered SCY-078, an Oral Fungicidal Glucan Synthase Inhibitor, and Tacrolimus. Clin Pharmacol Drug Dev. 2019 Jan;8(1):60-69. doi: 10.1002/cpdd.588. Epub 2018 Jun 27. [Article]
- Ghannoum M, Arendrup MC, Chaturvedi VP, Lockhart SR, McCormick TS, Chaturvedi S, Berkow EL, Juneja D, Tarai B, Azie N, Angulo D, Walsh TJ: Ibrexafungerp: A Novel Oral Triterpenoid Antifungal in Development for the Treatment of Candida auris Infections. Antibiotics (Basel). 2020 Aug 25;9(9). pii: antibiotics9090539. doi: 10.3390/antibiotics9090539. [Article]
- FDA Approved Drug Products: Brexafemme (Ibrexafungerp) Oral Tablet [Link]
| Clinical data | |
|---|---|
| Pronunciation | /aɪˌbrɛksəˈfʌndʒɜːrp/ eye-BREKS-ə-FUN-jurp |
| Trade names | Brexafemme |
| Other names | SCY-078 |
| License data | US DailyMed: Ibrexafungerp |
| Pregnancy category | Contraindicated[1] |
| Routes of administration | oral, intravenous |
| Drug class | Antifungal |
| ATC code | J02AX07 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Pharmacokinetic data | |
| Protein binding | >99%[1] |
| Metabolism | Hydroxylation (CYP3A4) then conjugation (glucuronidation, sulfation)[1] |
| Elimination half-life | 20 hours[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1207753-03-4as citrate: 1965291-08-0 |
| PubChem CID | 46871657as citrate: 137552087 |
| DrugBank | DB12471as citrate: DBSALT003185 |
| UNII | A92JFM5XNUas citrate: M4NU2SDX3E |
| KEGG | D11544as citrate: D11545 |
| ChEMBL | ChEMBL4297513as citrate: ChEMBL4298168 |
| CompTox Dashboard (EPA) | DTXSID901336871 |
| Chemical and physical data | |
| Formula | C44H67N5O4 |
| Molar mass | 730.051 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////Ibrexafungerp citrate, Brexafemme, アイブレキサフンジェルプクエン酸塩 , SCY-078 citrate, UNII-M4NU2SDX3E, M4NU2SDX3E, MK-3118; SCY-078, Orphan Drug, Merck, SCYNEXIS, WHO 10597, ANTI FUNGAL
CC(C)C(C)C1(CCC2(C3CCC4C5(COCC4(C3=CCC2(C1C(=O)O)C)CC(C5OCC(C)(C(C)(C)C)N)N6C(=NC=N6)C7=CC=NC=C7)C)C)C.C(C(=O)O)C(CC(=O)O)(C(=O)O)O
NEW DRUG APPROVALS
ONE TIME
$10.00
Idecabtagene vicleucel
Idecabtagene vicleucel
CAS 2306267-75-2
STN: BLA 125736
An autologous T lymphocyte-enriched cell transduced ex vivo with an anti-BCMA CAR lentiviral vector encoding a chimeric antigen receptor CAR, comprising a CD8 hinge and TM domain, 4-1BB costimulatory domain and CD3ζ signaling domain, targeting human B cell maturation antigen for cancer immunotherapy (Celgene Corp., NJ)
- Bb2121
| Name | Idecabtagene vicleucel (USAN); Abecma (TN) |
|---|---|
| Product | ABECMA (Celgene Corporation) |
| CAS | 2306267-75-2 |
| Efficacy | Antineoplastic, Anti-BCMA CAR-T cell |
| Disease | Multiple myeloma [DS:H00010] |
| Comment | Cellular therapy product |
USFDA 2021/4/21 APPROVED
Dendritic cells (DCs) are antigen-presenting cells (APCs) that process antigens and display them to other cells of the immune system. Specifically, dendritic cells are capable of capturing and presenting antigens on their surfaces to activate T cells such as cytotoxic T cells (CTLs). Further, activated dendritic cells are capable of recruiting additional immune cells such as macrophages, eosinophils, natural killer cells, and T cells such as natural killer T cells.
Despite major advances in cancer treatment, cancer remains one of the leading causes of death globally. Hurdles in designing effective therapies include cancer immune evasion, in which cancer cells escape destructive immunity, as well as the toxicity of many conventional cancer treatments such as radiation therapy and chemotherapy, which significantly impacts a patient’s ability to tolerate the therapy and/or impacts the efficacy of the treatment.
Given the important role of dendritic cells in immunity, derailed dendritic cell functions have been implicated in diseases such as cancer and autoimmune diseases. For example, cancer cells may evade immune detection and destruction by crippling dendritic cell functionality through prevention of dendritic cell recruitment and activation. In addition, dendritic cells have been found in the brain during central nervous system inflammation and may be involved in the pathogenesis of autoimmune diseases in the brain.
One mechanism by which cancers evade immune detection and destruction is by crippling dendritic cell functionality through prevention of dendritic cell (DC) recruitment and activation. Accordingly, there remains a need for cancer therapies that can effectively derail tumor evasion and enhance anti-tumor immunity as mediated, for example, by dendritic cells.
NEW DRUG APPROVALS
ONE TIME
$10.00
DESCRIPTION
ABECMA is a BCMA-directed genetically modified autologous T cell immunotherapy product consisting of a patient’s own T cells that are harvested and genetically modified ex vivo through transduction with an anti-BCMA02 chimeric antigen receptor (CAR) lentiviral vector (LVV). Autologous T cells transduced with the anti-BCMA02 CAR LVV express the anti-BCMA CAR on the T cell surface. The CAR is comprised of a murine extracellular single-chain variable fragment (scFv) specific for recognizing B cell maturation antigen (BCMA) followed by a human CD8α hinge and transmembrane domain fused to the T cell cytoplasmic signaling domains of CD137 (4-1BB) and CD3ζ chain, in tandem. Binding of ABECMA to BCMA-expressing target cells leads to signaling initiated by CD3ζ and 4-1BB domains, and subsequent CAR-positive T cell activation. Antigen-specific activation of ABECMA results in CAR-positive T cell proliferation, cytokine secretion, and subsequent cytolytic killing of BCMA-expressing cells.
ABECMA is prepared from the patient’s peripheral blood mononuclear cells (PBMCs), which are obtained via a standard leukapheresis procedure. The mononuclear cells are enriched for T cells, through activation with anti-CD3 and anti-CD28 antibodies in the presence of IL-2, which are then transduced with the replication-incompetent lentiviral vector containing the anti-BCMA CAR transgene. The transduced T cells are expanded in cell culture, washed, formulated into a suspension, and cryopreserved. The product must pass a sterility test before release for shipping as a frozen suspension in one or more patient-specific infusion bag(s). The product is thawed prior to infusion back into the patient [see DOSAGE AND ADMINISTRATION and HOW SUPPLIED/Storage And Handling].
The ABECMA formulation contains 50% Plasma-Lyte A and 50% CryoStor® CS10, resulting in a final DMSO concentration of 5%.
FDA approves idecabtagene vicleucel for multiple myeloma
On March 26, 2021, the Food and Drug Administration approved idecabtagene vicleucel (Abecma, Bristol Myers Squibb) for the treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody. This is the first FDA-approved cell-based gene therapy for multiple myeloma.
Idecabtagene vicleucel is a B-cell maturation antigen (BCMA)-directed genetically modified autologous chimeric antigen receptor (CAR) T-cell therapy. Each dose is customized using a patient’s own T-cells, which are collected and genetically modified, and infused back into the patient.
Safety and efficacy were evaluated in a multicenter study of 127 patients with relapsed and refractory multiple myeloma who received at least three prior lines of antimyeloma therapies; 88% had received four or more prior lines of therapies. Efficacy was evaluated in 100 patients who received idecabtagene vicleucel in the dose range of 300 to 460 x 106 CAR-positive T cells. Efficacy was established based on overall response rate (ORR), complete response (CR) rate, and duration of response (DOR), as evaluated by an Independent Response committee using the International Myeloma Working Group Uniform Response Criteria for Multiple Myeloma.
The ORR was 72% (95% CI: 62%, 81%) and CR rate was 28% (95% CI 19%, 38%). An estimated 65% of patients who achieved CR remained in CR for at least 12 months.
The idecabtagene vicleucel label carries a boxed warning for cytokine release syndrome (CRS), neurologic toxicities, hemophagocytic lymphohistiocytosis/ macrophage activation syndrome, and prolonged cytopenias. The most common side effects of idecabtagene vicleucel include CRS, infections, fatigue, musculoskeletal pain, and hypogammaglobulinemia.
Idecabtagene vicleucel is approved with a risk evaluation and mitigation strategy requiring that healthcare facilities that dispense the therapy must be specially certified to recognize and manage CRS and nervous system toxicities. To evaluate long-term safety, the FDA is requiring the manufacturer to conduct a post-marketing observational study involving patients treated with idecabtagene vicleucel.
The recommended dose range for idecabtagene vicleucel is 300 to 460 × 106 CAR-positive T cells. View full prescribing information for Abecma.
This application was granted breakthrough therapy designation and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
FDA D.I.S.C.O. Burst Edition: FDA approval of ABECMA (idecabtagene vicleucel) the first FDA approved cell-based gene therapy for the treatment of adult patients with relapsed or refractory multiple myeloma
Welcome back to the D.I.S.C.O., FDA’s Drug Information Soundcast in Clinical Oncology, Burst Edition, brought to you by FDA’s Division of Drug Information in partnership with FDA’s Oncology Center of Excellence. Today we have another quick update on a recent FDA cancer therapeutic approval.
On March 26, 2021, the FDA approved idecabtagene vicleucel (brand name Abecma) for the treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody. This is the first FDA-approved cell-based gene therapy for multiple myeloma.
Idecabtagene vicleucel is a B-cell maturation antigen-directed genetically modified autologous chimeric antigen receptor T-cell therapy. Each dose is customized using a patient’s own T-cells, which are collected and genetically modified, and infused back into the patient.
Safety and efficacy were evaluated in a multicenter study of 127 patients with relapsed and refractory multiple myeloma who received at least three prior lines of antimyeloma therapies, 88% of whom had received four or more prior lines of therapies. Efficacy was evaluated in 100 patients who received idecabtagene vicleucel and was established based on overall response rate, complete response rate, and duration of response, as evaluated by an Independent Response committee using the International Myeloma Working Group Uniform Response Criteria for Multiple Myeloma.
The overall response rate was 72% and complete response rate was 28%. An estimated 65% of patients who achieved complete response remained in complete response for at least 12 months.
The idecabtagene vicleucel label carries a boxed warning for cytokine release syndrome, neurologic toxicities, hemophagocytic lymphohistiocytosis/ macrophage activation syndrome, and prolonged cytopenias. Idecabtagene vicleucel is approved with a risk evaluation and mitigation strategy requiring that healthcare facilities dispensing the therapy must be specially certified to recognize and manage cytokine release syndrome and nervous system toxicities. To evaluate long-term safety, the FDA is requiring the manufacturer to conduct a post-marketing observational study involving patients treated with idecabtagene vicleucel.
Full prescribing information for this approval can be found on the web at www.fda.gov, with key word search “Approved Cellular and Gene Therapy Products”.
Health care professionals should report serious adverse events to FDA’s MedWatch Reporting System at www.fda.gov/medwatch.
Follow the Division of Drug Information on Twitter @FDA_Drug_InfoExternal Link Disclaimer and the Oncology Center of Excellence @FDAOncologyExternal Link Disclaimer. Send your feedback via email to FDAOncology@fda.hhs.gov. Thanks for tuning in today to the DISCO Burst Edition.
PAT
WO 2019148089
In various aspects, the present invention relates to XCR1 binding agents having at least one targeting moiety that specifically binds to XCR1. In various embodiments, these XCR1 binding agents bind to, but do not functionally modulate ( e.g . partially or fully neutralize) XCR1. Therefore, in various embodiments, the present XCR1 binding agents have use in, for instance, directly or indirectly recruiting a XCR1-expressing cell to a site of interest while still allowing the XCR1-expressing cell to signal via XCR1 (i.e. the binding of the XCR1 binding agent does not reduce or eliminate XCR1 signaling at the site of interest). In various embodiments, the XCR-1 binding agent functionally modulates XCR1. In an embodiment, the targeting moiety is a single domain antibody (e.g. VHH, HUMABODY, scFv, on antibody). In various embodiments, the XCR1 binding agent further comprises a signaling agent, e.g., without limitation, an interferon, an interleukin, and a tumor necrosis factor, that may be modified to attenuate activity. In various embodiments, the XCR1 binding agent comprises additional targeting moieties that bind to other targets (e.g. antigens, receptor) of interest. In an embodiment, the other targets (e.g. antigens, receptor) of interest are present on tumor cells. In another embodiment, the other targets (e.g. antigens, receptor) of interest are present on immune cells. In some embodiments, the present XCR1 binding agent may directly or indirectly recruit an immune cell (e.g. a dendritic cell) to a site of action (such as, by way of non-limiting example, the tumor microenvironment). In some embodiments, the present XCR1 binding agent facilitates the presentation of antigens (e.g., tumor antigens) by dendritic cells.
In various embodiments, the present XCR binding agent or targeting moiety of the present chimeric proteins comprises the heavy chain of SEQ ID NO: 223 and/or the light chain of SEQ ID NO: 224, or a variant thereof (e.g. an amino acid sequence having at least about 90%, or at least about 93%, at least about 95%, at least about 97%, at least about 98%, at least about 99%, identity with SEQ ID NO: 223 and/or SEQ ID NO: 224).
In various embodiments, the present XCR binding agent or targeting moiety of the present chimeric proteins comprises a heavy chain CDR 1 of SHNLH (SEQ ID NO: 225), heavy chain CDR 2 of AIYPGNGNTAYNQKFKG (SEQ ID NO: 226), and heavy chain CDR 3 of WGSVVGDWYFDV (SEQ ID NO: 227) and/or a light chain CDR 1 of RSSLGLVHRNGNTYLH (SEQ ID NO: 228), light chain CDR 2 of KVSHRFS (SEQ ID NO: 229), and light chain CDR 3 of SQSTFIVPWT (SEQ ID NO: 230), or a variant thereof (e.g. with four or fewer amino acid substitutions, or with three or fewer amino acid substitutions, or with two or fewer amino acid substitutions, or with one amino acid substitution).
In various embodiments, the present XCR binding agent or targeting moiety of the present chimeric proteins comprises a heavy chain CDR 1 of SHNLH (SEQ ID NO: 225), heavy chain CDR 2 of AIYPGNGNTAYNQKFKG (SEQ ID NO: 226), and heavy chain CDR 3 of WGSVVGDWYFDV (SEQ ID NO: 227).
Illustrative Disease Modifying Therapies
EXAMPLES
Example 1. Identification and Characterization of Human XCR1 Ab AFNs
As used in this Example and associated figures,“AFN” is a chimera of the anti-Xcr1 5G7 antibody and human IFNa2 with an R149A mutation.
AFNs were made based on the 5G7 anti-hXcr1 Ab using the intact (full) Ab or a scFv format.
The 5G7 heavy chain is:
QAYLQQSGAELVRPGASVKMSCKASGYTFTSHNLHWVKQTPRQGLQWIGAIYPGNGNTAYNQKFKGKATLTVD
KSSSTAYMQLSSLTSDDSAVYFCARWGSVVGDWYFDVWGTGTTVTVSSASTKGPSVFPLAPCSRSTSESTAAL
GCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSWTVPSSNFGTQTYTCNVDHKPSNTKVDKTVE
RKCCVECPPCPAPPAAAPSVFLFPPKPKDTLMISRTPEVTCVWDVSHEDPEVQFNWYVDGVEVHNAKTKPREE
QFNSTFRVVSVLTWHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLV
KGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLS
LSPGK (SEQ ID NO: 223)
The 5G7 light chain is:
DWMTQTPLSLPVTLGNQASIFCRSSLGLVHRNGNTYLHWYLQKPGQSPKLLIYKVSHRFSGVPDRFSGSGSGT DFTLKISRVEAEDLGVYFCSQSTHVPWTFGGGTKLEIKRTVAAPSVFIFPPSDEQLKSGTASWCLLNNFYPREAK VQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC (SEQ ID NO: 224)
5G7 Heavy chain CDR 1 is SHNLH (SEQ ID NO: 225), Heavy chain CDR 2 is AIYPGNGNTAYNQKFKG (SEQ ID NO: 226), Heavy chain CDR 3 is WGSVVGDWYFDV (SEQ ID NO: 227). 5G7 Light chain CDR 1 is RSSLGLVHRNGNTYLH (SEQ ID NO: 228), Light chain CDR 2 is KVSHRFS (SEQ ID NO: 229), and Light chain CDR 3 is SQSTHVPWT (SEQ ID NO: 230).
The sequence of hulFNa2(R149A) is:
CDLPQTHSLGSRRTLMLLAQMRKISLFSCLKDRHDFGFPQEEFGNQFQKAETIPVLHEMIQQIFNLFSTKDSSAA WDETLLDKFYTELYQQLNDLEACVIQGVGVTETPLMKEDSILAVRKYFQRITLYLKEKKYSPCAWEVVRAEIMASF SLSTNLQESLRSKE (SEQ ID NO: 231).
In case of the intact Ab AFN, the 5G7 Ab heavy chain was fused to h I FN a2_R149A (human IFNal with a R149A mutation) via a flexible (GGS)2oG-linker and co-expressed with the 5G7 Ab light chain (sequences shown below). 5G7 scFv-AFN was constructed by linking the Ab VL and VH domains via a (GGGS)4 linker and followed by a (GGS)2o-linker and the sequence encoding hlFNa2_R149A. Recombinant proteins, cloned in the pcDNA3.4 expression-vector, were produced in ExpiCHO cells (Thermo Fisher Scientific) and purified on HisPUR spin plates (Thermo Fisher Scientific) according to the manufacturer’s instructions.
To test binding of the AFNs, parental HL1 16 and HL1 16 cells stably expressing hXcrl (HL116-hXcr1) were incubated with a serial dilution AFN for two hours at 4°C. Binding was detected using THE™ HIS antibody-FITC (GenScript) and measured on a MACSQuant X instrument (Miltenyi Biotec) and analysed using the FlowLogic software (Miltenyi Biotec). Data in Figures 1A and 1 B clearly show that both 5G7 Ab-AFN and 5G7 scFv bind specifically to hXcrl expressing cells.
Biological activity was measured on parental HL1 16 cells (an IFN responsive cell-line stably transfected with a p6-16 luciferase reporter) and the derived HL116-hXcr1 cells. Cells were seeded overnight and stimulated for 6 hours with a serial dilution 5G7 AFNs. Luciferase activity was measured on an EnSight Multimode Plate Reader (Perkin Elmer). Data in Figures 2A and 2B clearly illustrate that 5G7 AFNs, in the intact Ab format or as scFv, are clearly more active on cells expressing hXcrl compared to parental cells, illustrating that it is possible to restore signaling of an IFNa2 mutant by specific targeting to hXcrl .
Example 2. Identification and Characterization of Mouse Xcr1 Ab AFNs
As used in this Example and associated figures,“AFN” is a chimera of the anti-Xcr1 MAARX10 antibody and human IFNa2 with Q124R mutation.
Similar to the anti-human Xcr1 Ab, AFNs based on the MARX10 anti-mouse Xcr1 Ab were made, as intact Ab or as scFv. In case of the intact Ab AFN, the MARX10 Ab heavy chain was fused to hlFNa2_Q124R (human IFNa2 with Q124R mutation) via a flexible (GGS)2oG-linker and co-expressed with the MARX10 Ab light chain. scFv-AFN was constructed by linking the Ab VL and VH domains, in VH-VL (scFv(1 )) or VL-VH (scFv(2)) orientation, via a (GGGS)4 linker and followed by a (GGS)2o-linker and h I FN a2_Q 124R.
Selectivity of AFNs (produced and purified as described above for the human Xcr1 Ab AFNs) was tested by comparing binding at 2.5 pg/ml to MOCK or mouse Xcr1 transfected Hek293T cells. Binding was detected using THE™ HIS antibody-FITC (GenScript) and measured on a MACSQuant X instrument (Miltenyi Biotec) and analysed using the FlowLogic software (Miltenyi Biotec). Data in Figure 3 clearly show that all three specifically bind to mXcrl expressing cells.
REF
New England Journal of Medicine (2021), 384(8), 705-716
https://www.rxlist.com/abecma-drug.htm#indications
///////////Idecabtagene vicleucel, breakthrough therapy designation, orphan drug designation, FDA 2021, APPROVALS 2021, Bb2121, Bb , ABECMA
Manufacturer: Celgene Corporation, a Bristol-Myers Squibb Company
Indications:
- Treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody.
Product Information
- Package Insert – ABECMA
- Demographic Subgroup Information – idecabtagene vicleucel [ABECMA]
Refer to Section 1.1 of the Clinical Review Memo for information about participation in the clinical trials and any analysis of demographic subgroup outcomes that is notable.
Supporting Documents
Loncastuximab tesirine


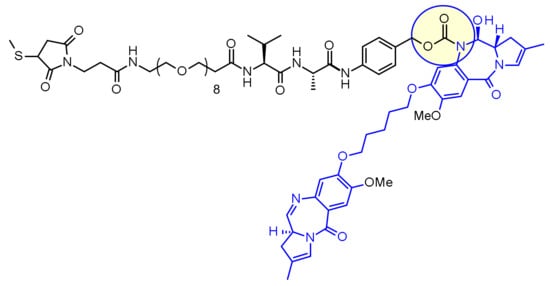
Loncastuximab tesirine
ZYNLONTA FDA APPROVED 2021/4/23
| Formula | C6544H10048N1718O2064S52 |
|---|---|
| Exact mass | 147387.9585 |
| CAS | 1879918-31-6 |
| Efficacy | Antineoplasitc, Anti-CD19 antibody |
| Disease | Diffuse large B-cell lymphoma not otherwise specified [DS:H02434] |
| Comment | Antibody-drug conjugate Treatment of hematological cancers |
ロンカスツキシマブテシリン; ADCT-402, ADCX 19
Immunoglobulin G1, anti-(human CD19 antigen) (human-Mus musculus monoclonal RB4v1.2 γ1-chain), disulfide with human-Mus musculus monoclonal RB4v1.2 κ-chain, dimer, bis(thioether) with N-[31-(3-mercapt-2,5-dioxo-1-pyrrolidinyl)-1,29-dioxo-4,7,10,13,16,19,22,25-octaoxa-28-azahentriacont-1-yl]-L-valyl-N-[4-[[[[(11S,11aS)-8-[[5-[[(11aS)-5,11a-dihydro-7-methoxy-2-methyl-5-oxo-1H-pyrrolo[2,1-c][1,4]benzodiazepin-8-yl]oxy]pentyl]oxy]-11,11a-dihydro-11-hydroxy-7-methoxy-2-methyl-5-oxo-1H-pyrrolo[2,1-c][1,4]benzodiazepin-10(5H)-yl]carbonyl]oxy]methyl]phenyl]-L-alaninamide
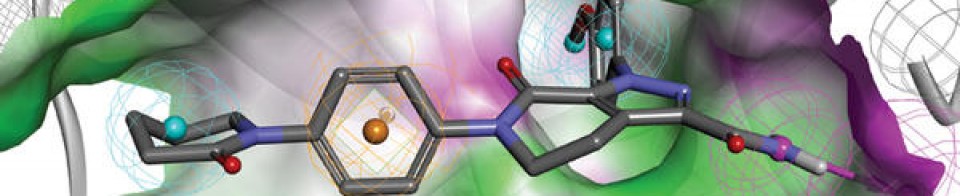
NEW DRUG APPROVALS
ONETIME
$10.00
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized |
| Target | CD19 |
| Clinical data | |
| Trade names | Zynlonta |
| Other names | ADCT-402, loncastuximab tesirine-lpyl |
| License data | US DailyMed: Loncastuximab_tesirine |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| CAS Number | 1879918-31-6 |
| DrugBank | DB16222 |
| ChemSpider | none |
| UNII | 7K5O7P6QIU |
| KEGG | D11338 |
| Chemical and physical data | |
| Formula | C6544H10048N1718O2064S52 |
| Molar mass | 147481.45 g·mol−1 |
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Zynlonta | Injection, powder, lyophilized, for solution | 5 mg/1mL | Intravenous | ADC Therapeutics America, Inc. | 2021-04-30 | Not applicable |  |
Loncastuximab tesirine-lpyl is a CD19-directed antibody and alkylating agent conjugate, consisting of a humanized IgG1 kappa monoclonal antibody conjugated to SG3199, a pyrrolobenzodiazepine (PBD) dimer cytotoxic alkylating agent, through a protease-cleavable valine–alanine linker. SG3199 attached to the linker is designated as SG3249, also known as tesirine.
|
Loncastuximab tesirine-lpyl has an approximate molecular weight of 151 kDa. An average of 2.3 molecules of SG3249 are attached to each antibody molecule. Loncastuximab tesirine-lpyl is produced by chemical conjugation of the antibody and small molecule components. The antibody is produced by mammalian (Chinese hamster ovary) cells, and the small molecule components are produced by chemical synthesis.
ZYNLONTA (loncastuximab tesirine-lpyl) for injection is supplied as a sterile, white to off-white, preservative-free, lyophilized powder, which has a cake-like appearance, for intravenous infusion after reconstitution and dilution. Each single-dose vial delivers 10 mg of loncastuximab tesirine-lpyl, L-histidine (2.8 mg), L-histidine monohydrochloride (4.6 mg), polysorbate 20 (0.4 mg), and sucrose (119.8 mg). After reconstitution with 2.2 mL Sterile Water for Injection, USP, the final concentration is 5 mg/mL with a pH of approximately 6.0.
Loncastuximab tesirine , sold under the brand name Zynlonta, is used for the treatment of large B-cell lymphoma. It is an antibody-drug conjugate (ADC) composed of a humanized antibody targeting the protein CD19, which is expressed in a wide range of B cell hematological tumors.[2] The experimental drug, developed by ADC Therapeutics is being tested in clinical trials for the treatment of B-cell non-Hodgkin lymphoma (NHL) and B-cell acute lymphoblastic leukemia (ALL).
On April 23, 2021, the Food and Drug Administration granted accelerated approval to loncastuximab tesirine-lpyl (Zynlonta, ADC Therapeutics SA), a CD19-directed antibody and alkylating agent conjugate, for adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified, DLBCL arising from low grade lymphoma, and high-grade B-cell lymphoma.
Approval was based on LOTIS-2 (NCT03589469), an open-label, single-arm trial in 145 adult patients with relapsed or refractory DLBCL or high-grade B-cell lymphoma after at least two prior systemic regimens. Patients received loncastuximab tesirine-lpyl 0.15 mg/kg every 3 weeks for 2 cycles, then 0.075 mg/kg every 3 weeks for subsequent cycles. Patients received treatment until progressive disease or unacceptable toxicity.
The main efficacy outcome measure was overall response rate (ORR), as assessed by an independent review committee using Lugano 2014 criteria. The ORR was 48.3% (95% CI: 39.9, 56.7) with a complete response rate of 24.1% (95% CI: 17.4, 31.9). After a median follow-up of 7.3 months, median response duration was 10.3 months (95% CI: 6.9, NE). Of the 70 patients who achieved objective responses, 36% were censored for response duration prior to 3 months.
Most common (≥20%) adverse reactions in patients receiving loncastuximab tesirine-lpyl, including laboratory abnormalities, are thrombocytopenia, increased gamma-glutamyltransferase, neutropenia, anemia, hyperglycemia, transaminase elevation, fatigue, hypoalbuminemia, rash, edema, nausea, and musculoskeletal pain.
The prescribing information provides warnings and precautions for adverse reactions including edema and effusions, myelosuppression, infections, and cutaneous reactions.
The recommended loncastuximab tesirine-lpyl dosage is 0.15 mg/kg every 3 weeks for 2 cycles, then 0.075 mg/kg every 3 weeks for subsequent cycles, by intravenous infusion over 30 minutes on day 1 of each cycle (every 3 weeks). Patients should be premedicated with dexamethasone 4 mg orally or intravenously twice daily for 3 days beginning the day before loncastuximab tesirine-lpyl.
Technology
The humanized monoclonal antibody is stochastically conjugated via a valine-alanine cleavable, maleimide linker to a cytotoxic (anticancer) pyrrolobenzodiazepine (PBD) dimer. The antibody binds to CD19, a protein which is highly expressed on the surface of B-cell hematological tumors[3] including certain forms of lymphomas and leukemias. After binding to the tumor cells the antibody is internalized, the cytotoxic drug PBD is released and the cancer cells are killed. PBD dimers are generated out of PBD monomers, a class of natural products produced by various actinomycetes. PBD dimers work by crosslinking specific sites of the DNA, blocking the cancer cells’ division that cause the cells to die. As a class of DNA-crosslinking agents they are significantly more potent than systemic chemotherapeutic drugs.[4]
Clinical trials
Two phase I trials are evaluating the drug in patients with relapsed or refractory B-cell non-Hodgkin’s lymphoma and relapsed or refractory B-cell acute lymphoblastic leukemia.[5] At the 14th International Conference on Malignant Lymphoma interim results from a Phase I, open-label, dose-escalating study designed to evaluate the treatment of loncastuximab tesirine in relapsed or refractory non-Hodgkin’s lymphoma were presented.[6] Among the patients enrolled at the time of the data cutoff the overall response rate was 61% in the total patient population (42% complete response and 19% partial response) and in patients with relapsing or refractory diffuse large B-cell lymphoma (DLBCL) the overall response rate was 57% (43% complete response and 14% partial response).[7][8]
Orphan drug designation
Loncastuximab tesirine was granted Orphan Drug Designation by the U.S. Food and Drug Administration (FDA) for the treatment of diffuse large B-cell lymphoma and mantle cell lymphoma.[9]
References
- ^ https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761196s000lbl.pdf
- ^ WHO Drug Information: International Nonproprietary Names for Pharmaceutical Substances
- ^ Wang K, Wei G, Liu D (November 2012). “CD19: a biomarker for B cell development, lymphoma diagnosis and therapy”. Experimental Hematology & Oncology. 1 (1): 36. doi:10.1186/2162-3619-1-36. PMC 3520838. PMID 23210908.
- ^ “Pyrrolobenzodiazepine”. ADC Review.
- ^ Clinical trial number NCT02669017 for “ADCT-402 in B-NHL” at ClinicalTrials.gov
- ^ Kahl B, Hamadani M, Caimi PF, Reid EG, Havenith K, He S, Feingold JM, O’Connor O (June 2017). “First clinical results of ADCT‐402, a novel pyrrolobenzodiazepine-based antibody drug conjugate (ADC), in relapsed/refractory B‐cell linage NHL” (PDF). Hematol Oncol. 35 (S2): 49–51. doi:10.1002/hon.2437_33.
- ^ “First clinical results of ADCT-402”. ADC Review.
- ^ Bainbridge K. “Grandfather fighting deadly cancer reveals scans of tumors after testing new drug”. Mirror.
- ^ “ADCT-402 Orphan Drug Designation” (PDF). ADC Therapeutics press release.
External links
- “Loncastuximab tesirine”. Drug Information Portal. U.S. National Library of Medicine.
/////////Loncastuximab tesirine, FDA 2021, APPROVALS 2021, ZYNLONTA, ロンカスツキシマブテシリン, ORPHAN DRUG, ADCT-402, priority review, ADCX 19
ZyCoV-D

ZyCoV-D
CAS 2541524-47-2
DNA vaccine construct encoding a spike protein antigen of SARS-CoV-2 virus (Zydus-Cadila)
UPDATE. APPROVED IN INDIA AUG 2021
http://ctri.nic.in/Clinicaltrials/showallp.php?mid1=51254&EncHid=&userName=ZyCoV-D
bioRxiv (2021), 1-26.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7423510/
| ZyCoV-D | (CTRI/2020/07/026352, 2020, CTRI/2020/07/026352, 2020; Myupchar, 2020) | ZYDUS CADILA |
ZyCoV-D is a genetically engineered DNA plasmid based vaccine encoding for the membrane proteins of the virus. The clinical trials to study the immunogenicity, and safety of the vaccine, will administer three doses at an interval of 28 days in 1048 individuals.
Phase 1/2: CTRI/2020/07/026352
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | DNA |
| Clinical data | |
| Routes of administration |
Intradermal |
| ATC code | None |
| Identifiers | |
| DrugBank | DB15892 |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| SARS-CoV-2 (virus)COVID-19 (disease) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 portal |
ZyCoV-D is a DNA plasmid based COVID-19 vaccine being developed by Cadila Healthcare with support from the Biotechnology Industry Research Assistance Council.
The ZYCOV-D vaccine candidate was developed by Cadila Healthcare Ltd. based in India1. The vaccine was developed using a DNA vaccine platform with a non-replicating and non-integrating plasmid carrying the gene of interest3. Once the plasmid DNA is introduced into host cells and the viral protein is translated, it elicits a strong immune response, stimulating the humoral and cellular components of the immune system3. The DNA vaccine platform offers minimal biosafety requirements, more improved vaccine stability, and lower cold chain requirements3. Phase I clinical trials of this vaccine candidate were completed in July 2020, with the company reporting successful dosing and tolerance1,2. As of August, 2020 the candidate is in Phase II clinical trials1.
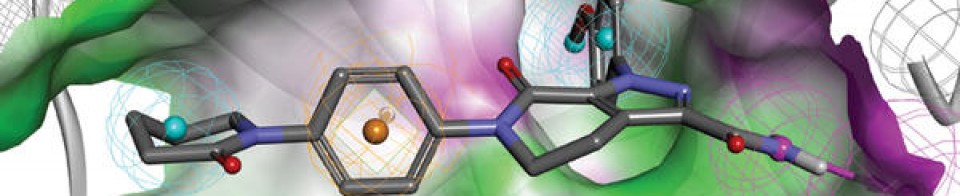
NEW DRUG APPROVALS
ONE TIME
$10.00
Clinical research
Phase I and II trials
In February 2020, Cadila Healthcare decided to develop a DNA plasmid based COVID-19 vaccine at their Vaccine Technology Centre (VTC) in Ahmedabad.[1] The vaccine candidate was able to pass the pre-clinical trials on animal models successfully. A report of the study was made available via bioRxiv.[2] Thereafter, human trials for Phase I and II were approved by the regulator.[3]
The Phase II trials of the vaccine candidate were conducted in over 1,000 volunteers as part of the adaptive Phase I/II multi-centric, dose escalation, randomised, double-blind placebo controlled method.[4][5]
Phase III trials
In November 2020, the company announced it would test the vaccine candidate on 30,000 patients in Phase III trials.[6] The vaccine would be given out in three doses at five sites across four cities of India.[7] In January 2021, the Drugs Controller General of India (DCGI) granted permission to conduct the Phase III clinical trials for 28,216 Indian participants.[8][9]
In April 2021, the company reported that they expected to have initial data for the Phase III trials by May 2021.[10]
Production
On 23 April 2021, production of the ZyCoV-D vaccine was started, with a yearly capacity of 240 million doses. It is expected to get emergency use authorization in May or June.[11]
References
- ^ “Zydus Cadila launches a fast tracked programme to develop vaccine for the novel coronavirus, 2019-nCoV (COVID-19)”(PDF). http://www.zyduscadila.com. Cadila Healthcare.
- ^ Dey A, Rajanathan C, Chandra H, Pericherla HP, Kumar S, Choonia HS, et al. (26 January 2021). “Immunogenic Potential of DNA Vaccine candidate, ZyCoV-D against SARS-CoV-2 in Animal Models”. bioRxiv: 2021.01.26.428240. doi:10.1101/2021.01.26.428240. S2CID 231777527.
- ^ “A prospective, randomized, adaptive, phase I/II clinical study to evaluate the safety and immunogenicity of Novel Corona Virus −2019-nCov vaccine candidate of M/s Cadila Healthcare Limited by intradermal route in healthy subjects”. ctri.nic.in. Clinical Trials Registry India. 15 December 2020. CTRI/2020/07/026352. Archived from the original on 22 November 2020.
- ^ “Zydus Cadila’s ZyCov-D vaccine found to be ‘safe and immunogenic'”. @businessline. The Hindu. 24 December 2020.
- ^ Rawat K, Kumari P, Saha L (February 2021). “COVID-19 vaccine: A recent update in pipeline vaccines, their design and development strategies”. European Journal of Pharmacology. 892: 173751. doi:10.1016/j.ejphar.2020.173751. PMC 7685956. PMID 33245898.
- ^ Thacker T (7 November 2020). “Zydus Cadila to test ZyCoV-D on 30,000 patients in Phase-3 trials”. The Economic Times.
- ^ “Covid 19 vaccine in India: Zydus Cadila begins enrolment for Phase 3 trial of ZyCoV-D in 4 cities”. The Financial Express. 22 January 2021.
- ^ “DBT-BIRAC supported indigenously developed DNA Vaccine Candidate by Zydus Cadila, approved for Phase III clinical trials”. pib.gov.in. Press Information Bureau. 3 January 2021.
- ^ “Novel Corona Virus-2019-nCov vaccine by intradermal route in healthy subjects”. ctri.nic.in. Clinical Trials Registry – India. Retrieved 10 April 2021.
- ^ Das, Sohini (22 April 2021). “Cadila Healthcare testing two-shot regimen for ZyCoV-D, data likely by May”. Business Standard India.
- ^ Writer, Staff (24 April 2021). “Cadila Healthcare starts production of Covid vaccine candidate”. mint. Retrieved 27 April 2021.
Zydus Cadila Covid vaccine close to getting approved in India, says MD Sharvil Patel
In an exclusive interview with India Today TV, Managing Director of Zydus Cadila Dr Sharvil Patel said the company’s Covid vaccine candidate ZyCoV-D against the Covid-19 infection is very close to getting approved in India. They are likely to apply for emergency use authorisation this month.
Ahmedabad-based pharmaceutical company Zydus Cadila is likely to submit the application for emergency use authorisation of its Covid-19 vaccine candidate ‘ZyCoV-D’ in India this month. The company is confident that the vaccine will be approved in May itself. The company plants to produce one crore doses of its ‘painless’ Covid-19 vaccine per month.
If approved, ZyCoV-D will be the fourth vaccine to be used in India’s Covid-19 vaccination drive. Made in India, the company plans to ramp up the vaccine’s production to 3-4 crore doses per month and is already in talks with two other manufacturing companies for the same
Although the vaccine should ideally be stored between 2 and 8 degrees Celsius, it remains stable even at room temperature conditions at 25 degrees Celsius. It is easy to administer, the developers said, and will be administered via intradermal injection.
If approved for emergency use, ZyCoV-D could help India fill the vacuum of vaccine doses currently being experienced in the country’s immunisation drive.
Earlier in April, Zydus Cadila announced that its drug Virafin had received restricted emergency use approval from the Drug Controller General of India for the treatment of mild cases of Covid-19.
In an exclusive interview with India Today TV, Sharvil Patel sheds details on all aspects of the Covid-19 vaccine ZyCoV-D.
When asked the status of Covid vaccine candidate ZyCoV-D and when exactly Zydus Cadila would apply for emergency use authorisation in India, Dr Sharvil Patel said the vaccine was getting very close to getting approved in the country.
“I am very happy to say that India’s first indigenously developed DNA vaccine candidate against Covid, which is our ZyCoV-D, is getting very close to approval,” he said.
“We have almost completed all our recruitment for the clinical trials. We have, by far, recruited the largest number of patients for a Covid vaccine trial in India. The number of volunteers who have been vaccinated as a part of the trial is 28,000,” Sharvil Patel said.
Sharvil Patel also said that his company has also included children in the 12-17 age group for the vaccine trials.
He said, “The recruitment holds very important milestones in terms of cohorts because not only have we included the elderly and those with co-morbidities, but also children in the age group of 12 to 17 years.”
Sharvil Patel said as soon as the efficacy data is obtained, Sydus Cadila will file for emergency use authorisation. As soon as the approval is granted, Zydus Cadila will start production of Covid-19 vaccines from July, he said.
“We hope to see our efficacy data in the middle of May. As soon as we see strong efficacy which correlates to the vaccine’s strong immunogenicity in Phase 2, we will file for emergency use authorization. We hope to produce a good quantity of the vaccine from July onwards to make sure it is available to the people. That is the need of the hour right now,” Sharvil Patel said.
He said by May the company will be in a position to talk to the regulators about the restricted use of the Covid-19 vaccine. “The regulatory process is a rolling one. I believe the regulators look at the data in a short period of time,” Sharvil Patel said.
“We have submitted a lot of data already so that it will aid the regulators once we provide them with the efficacy results. We are, hence, expecting to get the approval in May itself,” Sharvil Patel said.
///////////ZyCoV-D, COVID 19, CORONA VIRUS, VACCINE, INDIA 2021, APPROVALS 2021, SARS-CoV-2
Dostarlimab
(Heavy chain)
EVQLLESGGG LVQPGGSLRL SCAASGFTFS SYDMSWVRQA PGKGLEWVST ISGGGSYTYY
QDSVKGRFTI SRDNSKNTLY LQMNSLRAED TAVYYCASPY YAMDYWGQGT TVTVSSASTK
GPSVFPLAPC SRSTSESTAA LGCLVKDYFP EPVTVSWNSG ALTSGVHTFP AVLQSSGLYS
LSSVVTVPSS SLGTKTYTCN VDHKPSNTKV DKRVESKYGP PCPPCPAPEF LGGPSVFLFP
PKPKDTLMIS RTPEVTCVVV DVSQEDPEVQ FNWYVDGVEV HNAKTKPREE QFNSTYRVVS
VLTVLHQDWL NGKEYKCKVS NKGLPSSIEK TISKAKGQPR EPQVYTLPPS QEEMTKNQVS
LTCLVKGFYP SDIAVEWESN GQPENNYKTT PPVLDSDGSF FLYSRLTVDK SRWQEGNVFS
CSVMHEALHN HYTQKSLSLS LGK
(Light chain)
DIQLTQSPSF LSAYVGDRVT ITCKASQDVG TAVAWYQQKP GKAPKLLIYW ASTLHTGVPS
RFSGSGSGTE FTLTISSLQP EDFATYYCQH YSSYPWTFGQ GTKLEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(Disulfide bridge: H22-H96, H130-L214, H143-H199, H222-H’222, H225-H’225, H257-H317, H363-H421, H’22-H’96, H’130-L’214, H’143-H’199, H’257-H’317, H’363-H’421, L23-L88, L134-L194, L’23-L’88, L’194-L’134)
>Heavy Chain EVQLLESGGGLVQPGGSLRLSCAASGFTFSSYDMSWVRQAPGKGLEWVSTISGGGSYTYY QDSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCASPYYAMDYWGQGTTVTVSSASTK GPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYS LSSVVTVPSSSLGTKTYTCNVDHKPSNTKVDKRVESKYGPPCPPCPAPEFLGGPSVFLFP PKPKDTLMISRTPEVTCVVVDVSQEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTYRVVS VLTVLHQDWLNGKEYKCKVSNKGLPSSIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVS LTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSRLTVDKSRWQEGNVFS CSVMHEALHNHYTQKSLSLSLGK
>Light Chain DIQLTQSPSFLSAYVGDRVTITCKASQDVGTAVAWYQQKPGKAPKLLIYWASTLHTGVPS RFSGSGSGTEFTLTISSLQPEDFATYYCQHYSSYPWTFGQGTKLEIKRTVAAPSVFIFPP SDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLT LSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
References:
- Statement on a Nonproprietary Name Adopted by the USAN Council: Dostarlimab [Link]
Dostarlimab
Immunoglobulin G4, anti-(programmed cell death protein 1 (PDCD1)) (humanized clone ABT1 γ4-chain), disulfide with humanized clone ABT1 κ-chain, dimer
Protein Sequence
Sequence Length: 1314, 443, 443, 214, 214multichain; modified (modifications unspecified)
- GSK-4057190
- GSK4057190
- TSR 042
- TSR-042
- WBP-285
- ANB 011
| Formula | C6420H9832N1680O2014S44 |
|---|---|
| CAS | 2022215-59-2 |
| Mol weight | 144183.6677 |
Jemperli FDA 2021/4/22 AND EMA 2021/4/21
NEW DRUG APPROVALS
ONE TIME
$10.00
Dostarlimab, sold under the brand name Jemperli, is a monoclonal antibody medication used for the treatment of endometrial cancer.[1][2][3][4]
The most common adverse reactions (≥20%) were fatigue/asthenia, nausea, diarrhea, anemia, and constipation.[1][2] The most common grade 3 or 4 adverse reactions (≥2%) were anemia and transaminases increased.[1][2]
Dostarlimab is a programmed death receptor-1 (PD-1)–blocking antibody.[1][2]
Dostarlimab was approved for medical use in the United States in April 2021.[1][2][5]
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Jemperli | Injection | 50 mg/1mL | Intravenous | GlaxoSmithKline LLC | 2021-04-22 | Not applicable | |
Medical uses
Dostarlimab is indicated for the treatment of adults with mismatch repair deficient (dMMR) recurrent or advanced endometrial cancer, as determined by an FDA-approved test, that has progressed on or following prior treatment with a platinum-containing regimen.[1][2]
On April 22, 2021, the Food and Drug Administration granted accelerated approval to dostarlimab-gxly (Jemperli, GlaxoSmithKline LLC) for adult patients with mismatch repair deficient (dMMR) recurrent or advanced endometrial cancer, as determined by an FDA-approved test, that has progressed on or following a prior platinum-containing regimen.
Efficacy was evaluated based on cohort (A1) in GARNET Trial (NCT02715284), a multicenter, multicohort, open-label trial in patients with advanced solid tumors. The efficacy population consisted of 71 patients with dMMR recurrent or advanced endometrial cancer who progressed on or after a platinum-containing regimen. Patients received dostarlimab-gxly, 500 mg intravenously, every 3 weeks for 4 doses followed by 1,000 mg intravenously every 6 weeks.
The main efficacy endpoints were overall response rate (ORR) and duration of response (DOR), as assessed by blinded independent central review (BICR) according to RECIST 1.1. Confirmed ORR was 42.3% (95% CI: 30.6%, 54.6%). The complete response rate was 12.7% and partial response rate was 29.6%. Median DOR was not reached, with 93.3% of patients having durations ≥6 months (range: 2.6 to 22.4 months, ongoing at last assessment).
Serious adverse reactions occurred in 34% of patients receiving dostarlimab-gxly. Serious adverse reactions in >2% of patients included sepsis , acute kidney injury , urinary tract infection , abdominal pain , and pyrexia . The most common adverse reactions (≥20%) were fatigue/asthenia, nausea, diarrhea, anemia, and constipation. The most common grade 3 or 4 adverse reactions (≥2%) were anemia and transaminases increased. Immune-mediated adverse reactions can occur including pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis.
The recommended dostarlimab-gxly dose and schedule (doses 1 through 4) is 500 mg every 3 weeks. Subsequent dosing, beginning 3 weeks after dose 4, is 1,000 mg every 6 weeks until disease progression or unacceptable toxicity. Dostarlimab-gxly should be administered as an intravenous infusion over 30 minutes.
View full prescribing information for Jemperli.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
FDA also approved the VENTANA MMR RxDx Panel as a companion diagnostic device for selecting endometrial cancer patients for treatment with dostarlimab-gxly.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
This application was granted priority review, and breakthrough therapy designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Side effects
Serious adverse reactions in >2% of patients included sepsis, acute kidney injury, urinary tract infection, abdominal pain, and pyrexia.[1][2]
Immune-mediated adverse reactions can occur including pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis.[1][2]
History
Like several other available and experimental monoclonal antibodies, it is a PD-1 inhibitor. As of 2020, it is undergoing Phase I/II and Phase III clinical trials.[6][7][8] The manufacturer, Tesaro, announced prelimary successful results from the Phase I/II GARNET study.[6][9][10]
In 2020, the GARNET study announced that Dostarlimab was demonstrating potential to treat a subset of women with recurrent or advanced endometrial cancer.[11]
April 2021, Dostarlimab is approved for the treatment of recurrent or advanced endometrial cancer with deficient mismatch repair (dMMR), which are genetic anomalies abnormalities that disrupt DNA repair.[12]
On April 22, 2021, the Food and Drug Administration granted accelerated approval to dostarlimab-gxly (Jemperli, GlaxoSmithKline LLC).[1] Efficacy was evaluated based on cohort (A1) in GARNET Trial (NCT02715284), a multicenter, multicohort, open-label trial in patients with advanced solid tumors.[1]
Society and culture
Legal status
On 25 February 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a conditional marketing authorization for the medicinal product Jemperli, intended for the treatment of certain types of recurrent or advanced endometrial cancer.[13] The applicant for this medicinal product is GlaxoSmithKline (Ireland) Limited.[13]
References[
- ^ Jump up to:a b c d e f g h i j k “FDA grants accelerated approval to dostarlimab-gxly for dMMR endometri”. U.S. Food and Drug Administration(FDA) (Press release). 22 April 2021. Retrieved 22 April 2021. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e f g h i “Jemperli- dostarlimab injection”. DailyMed. Retrieved 28 April 2021.
- ^ Statement On A Nonproprietary Name Adopted By The USAN Council – Dostarlimab, American Medical Association.
- ^ World Health Organization (2018). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 119” (PDF). WHO Drug Information. 32 (2).
- ^ “FDA grants accelerated approval for GSK’s Jemperli (dostarlimab-gxly) for women with recurrent or advanced dMMR endometrial cancer” (Press release). GlaxoSmithKline. 22 April 2021. Retrieved 22 April 2021 – via PR Newswire.
- ^ Jump up to:a b Clinical trial number NCT02715284 for “A Phase 1 Dose Escalation and Cohort Expansion Study of TSR-042, an Anti-PD-1 Monoclonal Antibody, in Patients With Advanced Solid Tumors (GARNET)” at ClinicalTrials.gov
- ^ Clinical trial number NCT03981796 for “A Study of Dostarlimab (TSR-042) Plus Carboplatin-paclitaxel Versus Placebo Plus Carboplatin-paclitaxel in Patients With Recurrent or Primary Advanced Endometrial Cancer (RUBY)” at ClinicalTrials.gov
- ^ Clinical trial number NCT03602859 for “A Phase 3 Comparison of Platinum-Based Therapy With TSR-042 and Niraparib Versus Standard of Care Platinum-Based Therapy as First-Line Treatment of Stage III or IV Nonmucinous Epithelial Ovarian Cancer (FIRST)” at ClinicalTrials.gov
- ^ “Data from GARNET study indicates robust activity of dostarlimab in patients with advanced or recurrent endometrial cancer”. Tesaro (Press release). Retrieved 1 January 2020.
- ^ Scalea B (28 May 2019). “Dostarlimab Effective in Endometrial Cancer Regardless of MSI Status”. Targeted Oncology. Retrieved 1 January 2020.
- ^ “GSK Presents New Data from the GARNET Study Demonstrating Potential of Dostarlimab to Treat a Subset of Women with Recurrent or Advanced Endometrial Cancer – Drugs.com MedNews”. Drugs.com. Retrieved 29 April 2020.
- ^ “FDA Approves New Immunotherapy for Endometrial Cancer”. Medscape. Retrieved 23 April 2021.
- ^ Jump up to:a b “Jemperli: Pending EC decision”. European Medicines Agency (EMA) (Press release). 25 February 2021. Retrieved 22 April 2021.
External links
- “Dostarlimab”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02715284 for “Study of TSR-042, an Anti-programmed Cell Death-1 Receptor (PD-1) Monoclonal Antibody, in Participants With Advanced Solid Tumors (GARNET)” at ClinicalTrials.gov
- Kaplon H, Muralidharan M, Schneider Z, Reichert JM: Antibodies to watch in 2020. MAbs. 2020 Jan-Dec;12(1):1703531. doi: 10.1080/19420862.2019.1703531. [Article]
- Temrikar ZH, Suryawanshi S, Meibohm B: Pharmacokinetics and Clinical Pharmacology of Monoclonal Antibodies in Pediatric Patients. Paediatr Drugs. 2020 Apr;22(2):199-216. doi: 10.1007/s40272-020-00382-7. [Article]
- Green AK, Feinberg J, Makker V: A Review of Immune Checkpoint Blockade Therapy in Endometrial Cancer. Am Soc Clin Oncol Educ Book. 2020 Mar;40:1-7. doi: 10.1200/EDBK_280503. [Article]
- Deshpande M, Romanski PA, Rosenwaks Z, Gerhardt J: Gynecological Cancers Caused by Deficient Mismatch Repair and Microsatellite Instability. Cancers (Basel). 2020 Nov 10;12(11). pii: cancers12113319. doi: 10.3390/cancers12113319. [Article]
- FDA Approved Drug Products: Jemperli (dostarlimab-gxly) for intravenous injection [Link]
- FDA News Release: FDA grants accelerated approval to dostarlimab-gxly for dMMR endometrial cancer [Link]
- Statement on a Nonproprietary Name Adopted by the USAN Council: Dostarlimab [Link]
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized |
| Target | PCDP1 |
| Clinical data | |
| Trade names | Jemperli |
| Other names | TSR-042, WBP-285, dostarlimab-gxly |
| License data | US DailyMed: Dostarlimab |
| Routes of administration | Intravenous |
| Drug class | Antineoplastic |
| ATC code | L01XC40 (WHO) |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 2022215-59-2 |
| PubChem SID | 384585344 |
| DrugBank | DB15627 |
| UNII | P0GVQ9A4S5 |
| KEGG | D11366 |
| Chemical and physical data | |
| Formula | C6420H9832N1690O2014S44 |
| Molar mass | 144325.73 g·mol−1 |
/////////Dostarlimab, PEPTIDE, ANTINEOPLASTIC, CANCER, ドスタルリマブ , GSK 4057190, GSK4057190, TSR 042, TSR-042, WBP-285, FDA 2021, EU 2021
DEXMETHYLPHENIDATE
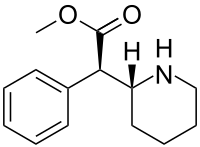
DEXMETHYLPHENIDATE
SynonymsDexmethylphenidate HCl, UNII1678OK0E08, CAS Number19262-68-1, WeightAverage: 269.77
Chemical FormulaC14H20ClNO2
methyl (2R)-2-phenyl-2-[(2R)-piperidin-2-yl]acetate hydrochloride

| CAS Number | 40431-64-9 as HCl: 19262-68-1 |
|---|---|
| PubChem CID | 154101as HCl: 154100 |
| IUPHAR/BPS | 7554 |
| DrugBank | DB06701 as HCl: DBSALT001458 |
| ChemSpider | 135807 as HCl: 135806 |
| UNII | M32RH9MFGPas HCl: 1678OK0E08 |
Trade Name:Focalin® / Attenade®MOA:Norepinephrine-dopamine reuptake inhibitorIndication:Attention deficit hyperactivity disorder (ADHD)Status:ApprovedCompany:Novartis (Originator) , CelgeneSales:$365 Million (Y2015); 
$492 Million (Y2014);
$594 Million (Y2013);
$554 Million (Y2012);
$550 Million (Y2011);ATC Code:N06BA11
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2005-05-26 | New dosage form | Focalin XR | Attention deficit hyperactivity disorder (ADHD) | Capsule, Extended release | 5 mg/10 mg/15 mg/20 mg/25 mg/30 mg/35 mg/40 mg | Novartis | |
| 2001-11-13 | Marketing approval | Focalin | Attention deficit hyperactivity disorder (ADHD) | Tablet | 2.5 mg/5 mg/10 mg | Novartis |
Dexmethylphenidate hydrochloride was approved by the U.S. Food and Drug Administration (FDA) on Nov 13, 2001. It was developed and marketed as Focalin® by Novartis in the US.
Dexmethylphenidate hydrochloride is a norepinephrine-dopamine reuptake inhibitor (NDRI). It is indicated for the treatment of attention deficit hyperactivity disorder (ADHD).
Focalin® is available as tablet for oral use, containing 2.5 mg, 5 mg or 10 mg of Dexmethylphenidate hydrochloride. The recommended dose is 10 mg twice daily, at least 4 hours apart.
NEW DRUG APPROVALS
ONE TIME
$10.00
NDA 212994, AZSTARYS
FDA APPROVE 2021

Drug Product Name Serdexmethylphenidate and Dexmethylphenidate (SDX/d-MPH)
Dosage Form capsule Strength 26.1/5.2 mg SDX/d-MPH 39.2/7.8 mg SDX/d-MPH 52.3/10.4 mg SDX/d-MPH
Route of Administration oral
Rx/OTC Dispensed Rx
Maximum Daily Dose 52.3 mg serdexmethylphenidate /10.4 mg dmethylphenidate as free base or 56 mg serdexmethylphenidate Chlorid
Dexmethylphenidate, sold under the brand name Focalin among others, is a medication used to treat attention deficit hyperactivity disorder (ADHD) in those over the age of five years.[3] If no benefit is seen after four weeks it is reasonable to discontinue its use.[3] It is taken by mouth.[3] The immediate release formulation lasts up to five hours while the extended release formulation lasts up to twelve hours.[4]
Common side effects include abdominal pain, loss of appetite, and fever.[3] Serious side effects may include abuse, psychosis, sudden cardiac death, mania, anaphylaxis, seizures, and dangerously prolonged erection.[3] Safety during pregnancy and breastfeeding is unclear.[5] Dexmethylphenidate is a central nervous system (CNS) stimulant.[6][3] How it works in ADHD is unclear.[3] It is the more active enantiomer of methylphenidate.[3]
Dexmethylphenidate was approved for medical use in the United States in 2001.[1] It is available as a generic medication.[3] In 2018, it was the 156th most commonly prescribed medication in the United States, with more than 3 million prescriptions.[7][8] It is also available in Switzerland.[9]
Reference:1. US6528530B2.
2. J. Org. Chem. 1998, 63, 9628-9629.Route 2
Reference:1. J. Am. Chem. Soc. 1999, 121,6509-6510.Route 3
Reference:1. Org. Process Res. Dev. 2010, 14, 1473–1475.Route 4
Reference:1. J. Med. Chem. 1998, 41,591-601.Route 5
Reference:1. Org. Lett. 1999, 1, 175-178.
2. Organic Syntheses 1985, 63, 206-212.
Four isomers of methylphenidate are possible, since the molecule has two chiral centers. One pair of threo isomers and one pair of erythro are distinguished, from which primarily d-threo-methylphenidate exhibits the pharmacologically desired effects.[102][124] The erythro diastereomers are pressor amines, a property not shared with the threo diastereomers. When the drug was first introduced it was sold as a 4:1 mixture of erythro:threo diastereomers, but it was later reformulated to contain only the threo diastereomers. “TMP” refers to a threo product that does not contain any erythro diastereomers, i.e. (±)-threo-methylphenidate. Since the threo isomers are energetically favored, it is easy to epimerize out any of the undesired erythro isomers. The drug that contains only dextrorotatory methylphenidate is sometimes called d-TMP, although this name is only rarely used and it is much more commonly referred to as dexmethylphenidate, d-MPH, or d-threo-methylphenidate. A review on the synthesis of enantiomerically pure (2R,2′R)-(+)-threo-methylphenidate hydrochloride has been published.[125]Methylphenidate synthesis

Method 1: Methylphenidate preparation elucidated by Axten et al. (1998)[126] via Bamford-Stevens reaction.

Method 2: Classic methylphenidate synthesis[127]

Method 3: Another synthesis route of methylphenidate which applies Darzens reaction to obtain aldehyde as an intermediate. This route is significant for its selectivity.SYNhttps://onlinelibrary.wiley.com/doi/abs/10.1002/jhet.2705SUN
1.9 Synthesis of (R, R), (R, S), (S, S) and (S, R) methyl 2-piperidin-2-yl-phenylacetate hydrochloride (1a, 1b, 1c and 1d)
Compound 8a, 8b, 8c or 8d (400 mg, 1.3 mmol) was dissolved into methanol solution (15 mL), and then thionyl chloride (1 mL) was added drop-wise. The reaction mixture was stirred for 12 hours and concentrated in vacuum; a white solid was precipitated and filtered to afford the final product. (1a. 0.28 g, 82% yield; 1b. 0.30 g, 84% yield; 1c. 0.31 g, 85% yield; 1d. 0.30 g, 84% yield). The characterization data of the four final products had been reported [2] by us in 2016.
SYN
https://patents.google.com/patent/US20040180928A1/en
- Dexmethylphenidate, also known as d-threo-methylphenidate, (R,R)-methylphenidate or (R,R)-α-phenyl-2-piperidineacetic acid methyl ester, having the formula:
- [0029]
is CNS (central nervous system) stimulant that is chemically and pharmacologically similar to the amphetamines. Dexmethylphenidate’s CNS actions is milder than those of the amphetamines and have more noticeable effects on mental activities than on motor activities. - [0030]
It has been reported by Sporzny (1961) that among racemic mixtures of threo and erythro diastereomers of methylphenidate, only threo-isomer displays stimulant properties. Dexmethylphenidate hydrochloride (i.e. the d-threo enantiomer of methylphenidate hydrochloride) has been reported to be 5 to 38 times more active than the corresponding (S,S)-methylphenidate hydrochloride (Prashad 2000). - [0031]
A commercially available drug is sold under the name Focalin™ (Novartis) and it consists of dexmethylphenidate in the form of the hydrochloride salt. This product is orally administered and clinically used in the treatment of narcolepsy and as adjunctive treatment in children with attention deficit disorder (ADD) and attention-deficit hyperactivity disorder (ADHD). - [0032]
A synthesis of dexmethylphenidate hydrochloride was firstly described in U.S. Pat. No. 2,838,519 and include resolution of erythro-α-phenyl-2-piperidineacetamide to obtain enantiopure (2R,2′S)-α-phenyl-2-piperidineacetamide, which was subjected to epimerization, hydrolysis, and esterification as shown in Scheme 1: - [0033]
Related example of preparation of dexmethylphenidate from erythro-α-phenyl-2-piperidineacetamide was described in U.S. Pat. No. 5,936,091. - [0034]
Preparation of dexmethylphenidate through optical resolution of threo-α-phenyl-2-piperidineacetamide was described in U.S. Pat. No. 5,965,734, as shown in Scheme 2: - [0035]
Synthetic methods for the preparation of racemic mixture of threo- and erythro-α-phenyl-2-piperidineacetamides as raw materials for the preparation of dexmethylphenidate were described by Panizzon (1944) and Patric (1982) and in U.S. Pat. Nos. 2,507,631, 2,838,519, 2,957,880 and 5,936,091, and in WO 01/27070. These methods include using sodium amide as base in the nucleophilic substitution of chlorine in 2-chloropyridine with phenylacetonitrile followed by hydrolysis of the formed nitrile and reduction of a pyridine ring to a piperidine one by hydrogenation on PtO 2 catalyst, as shown in Scheme 3: - [0036]
Alternatively, 2-bromopyridine was used instead of 2-chloropyridine by Deutsch (1996). - [0037]
In some other methods threo-methylphenidate was used as the raw material for the preparation of dexmethylphenidate. Threo-methylphenidate may be prepared by a several routes, inter alia by the following two processes: - [0038]
i) by esterification of threo-ritalinic acid which may be prepared from erythro-enriched and threo-α-phenyl-2-piperidineacetamides as shown in Scheme 4: - [0039]
ii) by cyclization of easily available 1-(phenylglyoxylyl)piperidine arenesulfonylhydrazone to (R*,R*)-enriched 7-phenyl-1-azabicyclo[4.2.0]octan-8-one and further converting the β-lactam to threo-methylphenidate hydrochloride, as described by Axten (1998), Corey (1965) and Earle (1969) and in WO 99/36403 and shown in Scheme 5: - [0040]
The resolution of threo-methylphenidate to afford dexmethylphenidate was first reported by Patric (1987) which used (R)-(−)-binaphthyl-2,2′-diyl hydrogen phosphate as the resolving agent. Several new resolutions of threo-methylphenidate have been reported recently by Prashad (1999) and in U.S. Pat. Nos. 6,100,401, 6,121,453, 6,162,919 and 6,242,464 as described in Scheme 6: - [0041]
wherein the chiral acid is one of the following: (R)-(−)-binaphthyl-2,2′-diyl hydrogen phosphate, (−)-menthoxyacetic acid, ditoluoyl-D-tartaric acid or dibenzoyl-D-tartaric acid. - [0042]
Resolution of threo-methylphenidate may be also achieved by enzymatic hydrolysis methods as proposed by Prashad (1998) and in WO 98/25902. Such resolution is described in Scheme 7: - [0043]
Resolution of threo-ritalinic acid hydrochloride with (S)-1-phenylethylamine give complex salt (R,R)-enriched threo-ritalinic acid.HCl.(S)-1-phenylethylamine with 77% ee optical purity of ritalinic acid (U.S. Ser. No. 2002/0019535), Scheme 8:
- [0119]
- [0120]
Gaseous hydrogen chloride was passed through a boiling solution of (R,R)-N-Boc-ritalinic acid (95.4 g, 299 mmol) in methanol (1.5 L). The mixture was stirred for 12 hours under reflux conditions and concentrated to the volume of 250 mL. Toluene (750 mL) was added to the stirred residue, then methanol lo was removed from boiling suspension under normal pressure. The obtained mixture was stirred overnight at 0-5° C. The precipitated solids were filtered off, washed on the filter with toluene (3×50 mL) and dried under reduced pressure to give 78.4 g (97.2% yield) of dexmethylphenidate hydrochloride as white crystals with mp 222-224° C. and [α]D 25 87.0° (c=1, MeOH).
- [0117]
- [0118]
A mixture of crystalline salt of (R,R)-N-Boc-ritalinic acid and (S)-1-phenylamine with [α]D 20 −28.6° (c=1, MeOH) (133.0 g, 302 mmol), ethyl acetate (1.3 L) and solution of citric acid (164.0 g, 845 mmol) in water (1.3 L) was stirred at 15-25° C. for 1.5 hours. The organic layer was separated, washed lo with brine (20 mL), dried over sodium sulfate, filtered and evaporated under reduced pressure to give 95.4 g (99%) of (R,R)-N-B
- [0115]
- [0116]
(S)-1-Phenylethylamine (113.8 g, 0.94 mol, 0.6 eq) was added dropwise to a stirred solution of N-Boc-threo-ritalinic acid (500 g, 1.57 mol, 1 eq) in ethyl acetate (5 L) for 1 hour at 20-40° C. The mixture was stirred for 1 hour at 40° C. and overnight at 5° C. The precipitated solids were filtered off, washed on the lo filter with cold ethyl acetate (2×500 mL) and dried under reduced pressure to give 380 g of white crystals with [α]D 20−23.3° (c=1, MeOH). The salt was twice recrystallized from aqueous methanol. The precipitated crystals were filtered off, washed on the filter with cold aqueous methanol and dried under reduced pressure to a constant weight to give 265 g (33.5% yield) of salt of (R,R)-N-Boc-ritalinic acid and (S)
- [0113]
- [0114]
A mixture of solution of N-Boc-threo-ritalinic acid sodium salt (1700 g, 4.98 mmol), citric acid (1150 g, 5.98 mmol) and water (5 mL) was stirred at 15-25° C. for 0.5 hour and extracted with ethyl acetate (3×4 L). Combined organic extracts were washed with brine (2×3 L), dried over sodium sulfate, filtered and evaporated under reduced pressure to constant weight to give 1560 g (98.1% yield) of N-Boc-threo-ritalinic acid with mp 133-134° C. (EtOAc/hexane) and 99.8% purity by HPLC.
Medical uses
Dexmethylphenidate is used as a treatment for ADHD, usually along with psychological, educational, behavioral or other forms of treatment. It is proposed that stimulants help ameliorate the symptoms of ADHD by making it easier for the user to concentrate, avoid distraction, and control behavior. Placebo-controlled trials have shown that once-daily dexmethylphenidate XR was effective and generally well tolerated.[6]
Improvements in ADHD symptoms in children were significantly greater for dexmethylphenidate XR versus placebo.[6] It also showed greater efficacy than osmotic controlled-release oral delivery system (OROS) methylphenidate over the first half of the laboratory classroom day but assessments late in the day favoured OROS methylphenidate.[6]
CLIP
An Improved and Efficient Process for the Production of Highly Pure Dexmethylphenidate Hydrochloride
Long-Xuan Xing, Cheng-Wu Shen, Yuan-Yuan Sun, Lei Huang, Yong-Yong Zheng,* Jian-Qi Li*
https://onlinelibrary.wiley.com/doi/abs/10.1002/jhet.2705
The present work describes an efficient and commercially viable process for the synthesis of dexmethylphenidate hydrochloride (1), a mild nervous system stimulant. The overall yield is 23% with ~99.9% purity (including seven chemical steps). Formation and control of possible impurities are also described in this report.

(R)-methyl 2-phenyl-2-((R)-piperidin-2-yl)acetate hydrochloride (1). ………… afford 1 as a white solid (107.6 g, 87.3% yield) with 99.50% purity and 99.70% ee. The crude product (107.6 g, 0.4 mol) was further purified by recrystallization from pure water (100 mL) to obtain the qualified product 1 (98.3 g, 91.4% yield) with 99.92 purity and 99.98% ee.
[α] 25 D +85.6 (MeOH, c 1) (lit [4b]. [α] 25 D +84 (MeOH, c 1));
Mp 222-223 C (lit [4b]. Mp 222– 224°C); MS m/z 234 [M + H]+ .
1 H NMR (400Hz, DMSO-d6) δ 1 H NMR (400 MHz, DMSO-d6) δ 9.64 (br, 1H), 8.97 (br, 1H), 7.41-7.26 (m, 5H), 4.18-4.16 (d, J = 9.2Hz, 1H), 3.77-3.75 (m, 1H), 3.66 (s, 3H), 3.25 (m, 1H), 2.94 (m, 1H), 1.67-1.64 (m, 3H), 1.41-1.25 (m, 3H).
13C NMR (100.6 MHz, DMSO-d6) δ 171.3, 134.2, 129.1, 128.6, 128.2, 56.8, 53.3, 52.6, 44.5, 25.7, 21.5, 21.4.
1H-NMR, and 13C-NMR of compound 1………………………………….. 10-11


DEPT,

COSY, NOESY, GHMBC, and HMQC of compound 1……………… 12-14

COSY

NOESY

GHMBC

HMQC
Contraindications
This section is transcluded from Methylphenidate. (edit | history)
Methylphenidate is contraindicated for individuals using monoamine oxidase inhibitors (e.g., phenelzine, and tranylcypromine), or individuals with agitation, tics, glaucoma, or a hypersensitivity to any ingredients contained in methylphenidate pharmaceuticals.[10]
The US Food and Drug Administration (FDA) gives methylphenidate a pregnancy category of C, and women are advised to only use the drug if the benefits outweigh the potential risks.[11] Not enough human studies have been conducted to conclusively demonstrate an effect of methylphenidate on fetal development.[12] In 2018, a review concluded that it has not been teratogenic in rats and rabbits, and that it “is not a major human teratogen”.[13]
Adverse effects
Part of this section is transcluded from Methylphenidate. (edit | history)
Products containing dexmethylphenidate have a side effect profile comparable to those containing methylphenidate.[14]

Addiction experts in psychiatry, chemistry, pharmacology, forensic science, epidemiology, and the police and legal services engaged in delphic analysis regarding 20 popular recreational drugs. Methylphenidate was ranked 13th in dependence, 12th in physical harm, and 18th in social harm.[15]
The most common adverse effects include appetite loss, dry mouth, anxiety/nervousness, nausea, and insomnia. Gastrointestinal adverse effects may include abdominal pain and weight loss. Nervous system adverse effects may include akathisia (agitation/restlessness), irritability, dyskinesia (tics), lethargy (drowsiness/fatigue), and dizziness. Cardiac adverse effects may include palpitations, changes in blood pressure and heart rate (typically mild), and tachycardia (rapid heart rate).[16] Smokers with ADHD who take methylphenidate may increase their nicotine dependence, and smoke more often than before they began using methylphenidate, with increased nicotine cravings and an average increase of 1.3 cigarettes per day.[17] Ophthalmologic adverse effects may include blurred vision and dry eyes, with less frequent reports of diplopia and mydriasis.[18]
There is some evidence of mild reductions in height with prolonged treatment in children.[19] This has been estimated at 1 centimetre (0.4 in) or less per year during the first three years with a total decrease of 3 centimetres (1.2 in) over 10 years.[20][21]
Hypersensitivity (including skin rash, urticaria, and fever) is sometimes reported when using transdermal methylphenidate. The Daytrana patch has a much higher rate of skin reactions than oral methylphenidate.[22]
Methylphenidate can worsen psychosis in people who are psychotic, and in very rare cases it has been associated with the emergence of new psychotic symptoms.[23] It should be used with extreme caution in people with bipolar disorder due to the potential induction of mania or hypomania.[24] There have been very rare reports of suicidal ideation, but some authors claim that evidence does not support a link.[19] Logorrhea is occasionally reported. Libido disorders, disorientation, and hallucinations are very rarely reported. Priapism is a very rare adverse event that can be potentially serious.[25]
USFDA-commissioned studies from 2011 indicate that in children, young adults, and adults there is no association between serious adverse cardiovascular events (sudden death, heart attack, and stroke) and the medical use of methylphenidate or other ADHD stimulants.[26]
Because some adverse effects may only emerge during chronic use of methylphenidate, a constant watch for adverse effects is recommended.[27]
A 2018 Cochrane review found that methylphenidate might be associated with serious side effects such as heart problems, psychosis, and death; the certainty of the evidence was stated as very low and the actual risk might be higher.[28]
Overdose
The symptoms of a moderate acute overdose on methylphenidate primarily arise from central nervous system overstimulation; these symptoms include: vomiting, nausea, agitation, tremors, hyperreflexia, muscle twitching, euphoria, confusion, hallucinations, delirium, hyperthermia, sweating, flushing, headache, tachycardia, heart palpitations, cardiac arrhythmias, hypertension, mydriasis, and dryness of mucous membranes.[29][30] A severe overdose may involve symptoms such as hyperpyrexia, sympathomimetic toxidrome, convulsions, paranoia, stereotypy (a repetitive movement disorder), rapid muscle breakdown, coma, and circulatory collapse.[29][30][31] A methylphenidate overdose is rarely fatal with appropriate care.[31] Following injection of methylphenidate tablets into an artery, severe toxic reactions involving abscess formation and necrosis have been reported.[32]
Treatment of a methylphenidate overdose typically involves the administration of benzodiazepines, with antipsychotics, α-adrenoceptor agonists and propofol serving as second-line therapies.[31]
Addiction and dependence[edit]
| ΔFosB accumulation from excessive drug use Top: this depicts the initial effects of high dose exposure to an addictive drug on gene expression in the nucleus accumbens for various Fos family proteins (i.e., c-Fos, FosB, ΔFosB, Fra1, and Fra2). Bottom: this illustrates the progressive increase in ΔFosB expression in the nucleus accumbens following repeated twice daily drug binges, where these phosphorylated (35–37 kilodalton) ΔFosB isoforms persist in the D1-type medium spiny neurons of the nucleus accumbens for up to 2 months.[33][34] |
Methylphenidate is a stimulant with an addiction liability and dependence liability similar to amphetamine. It has moderate liability among addictive drugs;[35][36] accordingly, addiction and psychological dependence are possible and likely when methylphenidate is used at high doses as a recreational drug.[36][37] When used above the medical dose range, stimulants are associated with the development of stimulant psychosis.[38] As with all addictive drugs, the overexpression of ΔFosB in D1-type medium spiny neurons in the nucleus accumbens is implicated in methylphenidate addiction.[37][39]
Methylphenidate has shown some benefits as a replacement therapy for individuals who are addicted to and dependent upon methamphetamine.[40] Methylphenidate and amphetamine have been investigated as a chemical replacement for the treatment of cocaine addiction[41][42][43][44] in the same way that methadone is used as a replacement drug for physical dependence upon heroin. Its effectiveness in treatment of cocaine or psychostimulant addiction, or psychological dependence has not been proven and further research is needed.[45]
Biomolecular mechanisms
Further information: Addiction § Biomolecular mechanisms
Methylphenidate has the potential to induce euphoria due to its pharmacodynamic effect (i.e., dopamine reuptake inhibition) in the brain’s reward system.[39] At therapeutic doses, ADHD stimulants do not sufficiently activate the reward system, or the reward pathway in particular, to the extent necessary to cause persistent increases in ΔFosB gene expression in the D1-type medium spiny neurons of the nucleus accumbens;[36][39][46] consequently, when taken as directed in doses that are commonly prescribed for the treatment of ADHD, methylphenidate use lacks the capacity to cause an addiction.[36][39][46] However, when methylphenidate is used at sufficiently high recreational doses through a bioavailable route of administration (e.g., insufflation or intravenous administration), particularly for use of the drug as a euphoriant, ΔFosB accumulates in the nucleus accumbens.[36][39] Hence, like any other addictive drug, regular recreational use of methylphenidate at high doses eventually gives rise to ΔFosB overexpression in D1-type neurons which subsequently triggers a series of gene transcription-mediated signaling cascades that induce an addiction.[39][46][47]
Overdose
This section is transcluded from Methylphenidate. (edit | history)
The symptoms of a moderate acute overdose on methylphenidate primarily arise from central nervous system overstimulation; these symptoms include: vomiting, nausea, agitation, tremors, hyperreflexia, muscle twitching, euphoria, confusion, hallucinations, delirium, hyperthermia, sweating, flushing, headache, tachycardia, heart palpitations, cardiac arrhythmias, hypertension, mydriasis, and dryness of mucous membranes.[29][30] A severe overdose may involve symptoms such as hyperpyrexia, sympathomimetic toxidrome, convulsions, paranoia, stereotypy (a repetitive movement disorder), rapid muscle breakdown, coma, and circulatory collapse.[29][30][31] A methylphenidate overdose is rarely fatal with appropriate care.[31] Following injection of methylphenidate tablets into an artery, severe toxic reactions involving abscess formation and necrosis have been reported.[32]
Treatment of a methylphenidate overdose typically involves the administration of benzodiazepines, with antipsychotics, α-adrenoceptor agonists and propofol serving as second-line therapies.[31]
Interactions
This section is transcluded from Methylphenidate. (edit | history)
Methylphenidate may inhibit the metabolism of vitamin K anticoagulants, certain anticonvulsants, and some antidepressants (tricyclic antidepressants, and selective serotonin reuptake inhibitors). Concomitant administration may require dose adjustments, possibly assisted by monitoring of plasma drug concentrations.[48] There are several case reports of methylphenidate inducing serotonin syndrome with concomitant administration of antidepressants.[49][50][51][52]
When methylphenidate is coingested with ethanol, a metabolite called ethylphenidate is formed via hepatic transesterification,[53][54] not unlike the hepatic formation of cocaethylene from cocaine and ethanol. The reduced potency of ethylphenidate and its minor formation means it does not contribute to the pharmacological profile at therapeutic doses and even in overdose cases ethylphenidate concentrations remain negligible.[55][54]
Coingestion of alcohol (ethanol) also increases the blood plasma levels of d-methylphenidate by up to 40%.[56]
Liver toxicity from methylphenidate is extremely rare, but limited evidence suggests that intake of β-adrenergic agonists with methylphenidate may increase the risk of liver toxicity.[57]
Mode of activity
Methylphenidate is a catecholamine reuptake inhibitor that indirectly increases catecholaminergic neurotransmission by inhibiting the dopamine transporter (DAT) and norepinephrine transporter (NET),[58] which are responsible for clearing catecholamines from the synapse, particularly in the striatum and meso-limbic system.[59] Moreover, it is thought to “increase the release of these monoamines into the extraneuronal space.”[2]
Although four stereoisomers of methylphenidate (MPH) are possible, only the threo diastereoisomers are used in modern practice. There is a high eudysmic ratio between the SS and RR enantiomers of MPH. Dexmethylphenidate (d-threo-methylphenidate) is a preparation of the RR enantiomer of methylphenidate.[60][61] In theory, D-TMP (d-threo-methylphenidate) can be anticipated to be twice the strength of the racemic product.[58][62]
| Compd[63] | DAT (Ki) | DA (IC50) | NET (Ki) | NE (IC50) |
|---|---|---|---|---|
| D-TMP | 161 | 23 | 206 | 39 |
| L-TMP | 2250 | 1600 | >10K | 980 |
| DL-TMP | 121 | 20 | 788 | 51 |
Pharmacology
Main article: Methylphenidate § Pharmacology
Dexmethylphenidate has a 4–6 hour duration of effect (a long-acting formulation, Focalin XR, which spans 12 hours is also available and has been shown to be as effective as DL (dextro-, levo-)-TMP (threo-methylphenidate) XR (extended release) (Concerta, Ritalin LA), with flexible dosing and good tolerability.[64][65]) It has also been demonstrated to reduce ADHD symptoms in both children[66] and adults.[67] d-MPH has a similar side-effect profile to MPH[14] and can be administered without regard to food intake.[68]
References
- ^ Jump up to:a b “Focalin- dexmethylphenidate hydrochloride tablet”. DailyMed. 24 June 2020. Retrieved 15 November 2020.
- ^ Jump up to:a b “Focalin XR- dexmethylphenidate hydrochloride capsule, extended release”. DailyMed. 27 June 2020. Retrieved 15 November 2020.
- ^ Jump up to:a b c d e f g h i “Dexmethylphenidate Hydrochloride Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 15 April 2019.
- ^ Mosby’s Drug Reference for Health Professions – E-Book. Elsevier Health Sciences. 2013. p. 455. ISBN 9780323187602.
- ^ “Dexmethylphenidate Use During Pregnancy”. Drugs.com. Retrieved 15 April 2019.
- ^ Jump up to:a b c d Moen MD, Keam SJ (December 2009). “Dexmethylphenidate extended release: a review of its use in the treatment of attention-deficit hyperactivity disorder”. CNS Drugs. 23(12): 1057–83. doi:10.2165/11201140-000000000-00000. PMID 19958043. S2CID 24975170.
- ^ “The Top 300 of 2021”. ClinCalc. Retrieved 18 February 2021.
- ^ “Dexmethylphenidate Hydrochloride – Drug Usage Statistics”. ClinCalc. Retrieved 18 February 2021.
- ^ “Focalin XR”. Drugs.com. Retrieved 15 April 2019.
- ^ “DAYTRANA” (PDF). United States Food and Drug Administration. Noven Pharmaceuticals, Inc. October 2013. Archived (PDF) from the original on 14 July 2014. Retrieved 13 June 2014.
- ^ “Methylphenidate: Use During Pregnancy and Breastfeeding”. Drugs.com. Archived from the original on 2 January 2018.
- ^ Humphreys C, Garcia-Bournissen F, Ito S, Koren G (2007). “Exposure to attention deficit hyperactivity disorder medications during pregnancy”. Canadian Family Physician. 53 (7): 1153–5. PMC 1949295. PMID 17872810.
- ^ Ornoy, Asher (6 February 2018). “Pharmacological Treatment of Attention Deficit Hyperactivity Disorder During Pregnancy and Lactation”. Pharmaceutical Research. 35 (3): 46. doi:10.1007/s11095-017-2323-z. PMID 29411149. S2CID 3663423.
- ^ Jump up to:a b Keating GM, Figgitt DP (2002). “Dexmethylphenidate”. Drugs. 62 (13): 1899–904, discussion 1905–8. doi:10.2165/00003495-200262130-00009. PMID 12215063.
- ^ Nutt D, King LA, Saulsbury W, Blakemore C (March 2007). “Development of a rational scale to assess the harm of drugs of potential misuse”. Lancet. 369 (9566): 1047–53. doi:10.1016/S0140-6736(07)60464-4. PMID 17382831. S2CID 5903121.
- ^ “Ritalin LA® (methylphenidate hydrochloride) extended-release capsules” (PDF). Novartis. Archived from the original (PDF)on 20 July 2011.
- ^ Bron TI, Bijlenga D, Kasander MV, Spuijbroek AT, Beekman AT, Kooij JJ (2013). “Long-term relationship between methylphenidate and tobacco consumption and nicotine craving in adults with ADHD in a prospective cohort study”. European Neuropsychopharmacology. 23 (6): 542–554. doi:10.1016/j.euroneuro.2012.06.004. PMID 22809706. S2CID 23148548.
- ^ Jaanus SD (1992). “Ocular side effects of selected systemic drugs”. Optometry Clinics. 2 (4): 73–96. PMID 1363080.
- ^ Jump up to:a b Cortese S, Holtmann M, Banaschewski T, Buitelaar J, Coghill D, Danckaerts M, et al. (March 2013). “Practitioner review: current best practice in the management of adverse events during treatment with ADHD medications in children and adolescents”. Journal of Child Psychology and Psychiatry, and Allied Disciplines. 54 (3): 227–46. doi:10.1111/jcpp.12036. PMID 23294014.
- ^ Poulton A (August 2005). “Growth on stimulant medication; clarifying the confusion: a review”. Archives of Disease in Childhood. 90 (8): 801–6. doi:10.1136/adc.2004.056952. PMC 1720538. PMID 16040876.
- ^ Hinshaw SP, Arnold LE (January 2015). “ADHD, Multimodal Treatment, and Longitudinal Outcome: Evidence, Paradox, and Challenge”. Wiley Interdisciplinary Reviews. Cognitive Science. 6(1): 39–52. doi:10.1002/wcs.1324. PMC 4280855. PMID 25558298.
- ^ Findling RL, Dinh S (March 2014). “Transdermal therapy for attention-deficit hyperactivity disorder with the methylphenidate patch (MTS)”. CNS Drugs. 28 (3): 217–28. doi:10.1007/s40263-014-0141-y. PMC 3933749. PMID 24532028.
- ^ Kraemer M, Uekermann J, Wiltfang J, Kis B (July 2010). “Methylphenidate-induced psychosis in adult attention-deficit/hyperactivity disorder: report of 3 new cases and review of the literature”. Clinical Neuropharmacology. 33 (4): 204–6. doi:10.1097/WNF.0b013e3181e29174. PMID 20571380. S2CID 34956456.
- ^ Wingo AP, Ghaemi SN (2008). “Frequency of stimulant treatment and of stimulant-associated mania/hypomania in bipolar disorder patients”. Psychopharmacology Bulletin. 41 (4): 37–47. PMID 19015628.
- ^ “Methylphenidate ADHD Medications: Drug Safety Communication – Risk of Long-lasting Erections”. U.S. Food and Drug Administration. 17 December 2013. Archived from the original on 17 December 2013. Retrieved 17 December 2013.
- ^ “FDA Drug Safety Communication: Safety Review Update of Medications used to treat Attention-Deficit/Hyperactivity Disorder (ADHD) in children and young adults”. United States Food and Drug Administration. 20 December 2011. Archived from the original on 30 October 2013. Retrieved 4 November 2013.
• Cooper WO, Habel LA, Sox CM, Chan KA, Arbogast PG, Cheetham TC, Murray KT, Quinn VP, Stein CM, Callahan ST, Fireman BH, Fish FA, Kirshner HS, O’Duffy A, Connell FA, Ray WA (November 2011). “ADHD drugs and serious cardiovascular events in children and young adults”. N. Engl. J. Med. 365 (20): 1896–1904. doi:10.1056/NEJMoa1110212. PMC 4943074. PMID 22043968.
• “FDA Drug Safety Communication: Safety Review Update of Medications used to treat Attention-Deficit/Hyperactivity Disorder (ADHD) in adults”. United States Food and Drug Administration. 15 December 2011. Archived from the original on 30 October 2013. Retrieved 4 November 2013.
• Habel LA, Cooper WO, Sox CM, Chan KA, Fireman BH, Arbogast PG, Cheetham TC, Quinn VP, Dublin S, Boudreau DM, Andrade SE, Pawloski PA, Raebel MA, Smith DH, Achacoso N, Uratsu C, Go AS, Sidney S, Nguyen-Huynh MN, Ray WA, Selby JV (December 2011). “ADHD medications and risk of serious cardiovascular events in young and middle-aged adults”. JAMA. 306 (24): 2673–2683. doi:10.1001/jama.2011.1830. PMC 3350308. PMID 22161946. - ^ Gordon N (1999). “Attention deficit hyperactivity disorder: possible causes and treatment”. International Journal of Clinical Practice. 53(7): 524–8. PMID 10692738.
- ^ Storebø OJ, Pedersen N, Ramstad E, Kielsholm ML, Nielsen SS, Krogh HB, et al. (May 2018). “Methylphenidate for attention deficit hyperactivity disorder (ADHD) in children and adolescents – assessment of adverse events in non-randomised studies”. The Cochrane Database of Systematic Reviews. 5: CD012069. doi:10.1002/14651858.CD012069.pub2. PMC 6494554. PMID 29744873.
- ^ Jump up to:a b c d Noven Pharmaceuticals, Inc. (17 April 2015). “Daytrana Prescribing Information” (PDF). United States Food and Drug Administration. pp. 1–33. Archived (PDF) from the original on 23 June 2015. Retrieved 23 June 2015.
- ^ Jump up to:a b c d Heedes G, Ailakis J. “Methylphenidate hydrochloride (PIM 344)”. INCHEM. International Programme on Chemical Safety. Archived from the original on 23 June 2015. Retrieved 23 June2015.
- ^ Jump up to:a b c d e f Spiller HA, Hays HL, Aleguas A (June 2013). “Overdose of drugs for attention-deficit hyperactivity disorder: clinical presentation, mechanisms of toxicity, and management”. CNS Drugs. 27 (7): 531–543. doi:10.1007/s40263-013-0084-8. PMID 23757186. S2CID 40931380.
The management of amphetamine, dextroamphetamine, and methylphenidate overdose is largely supportive, with a focus on interruption of the sympathomimetic syndrome with judicious use of benzodiazepines. In cases where agitation, delirium, and movement disorders are unresponsive to benzodiazepines, second-line therapies include antipsychotics such as ziprasidone or haloperidol, central alpha-adrenoreceptor agonists such as dexmedetomidine, or propofol. … However, fatalities are rare with appropriate care
- ^ Jump up to:a b Bruggisser M, Bodmer M, Liechti ME (2011). “Severe toxicity due to injected but not oral or nasal abuse of methylphenidate tablets”. Swiss Med Wkly. 141: w13267. doi:10.4414/smw.2011.13267. PMID 21984207.
- ^ Nestler EJ, Barrot M, Self DW (September 2001). “DeltaFosB: a sustained molecular switch for addiction”. Proceedings of the National Academy of Sciences of the United States of America. 98(20): 11042–6. Bibcode:2001PNAS…9811042N. doi:10.1073/pnas.191352698. PMC 58680. PMID 11572966.
Although the ΔFosB signal is relatively long-lived, it is not permanent. ΔFosB degrades gradually and can no longer be detected in brain after 1–2 months of drug withdrawal … Indeed, ΔFosB is the longest-lived adaptation known to occur in adult brain, not only in response to drugs of abuse, but to any other perturbation (that does not involve lesions) as well.
- ^ Nestler EJ (December 2012). “Transcriptional mechanisms of drug addiction”. Clinical Psychopharmacology and Neuroscience. 10 (3): 136–43. doi:10.9758/cpn.2012.10.3.136. PMC 3569166. PMID 23430970.
The 35–37 kD ΔFosB isoforms accumulate with chronic drug exposure due to their extraordinarily long half-lives. … As a result of its stability, the ΔFosB protein persists in neurons for at least several weeks after cessation of drug exposure. … ΔFosB overexpression in nucleus accumbens induces NFκB
- ^ Morton WA, Stockton GG (2000). “Methylphenidate Abuse and Psychiatric Side Effects”. Prim Care Companion J Clin Psychiatry. 2 (5): 159–164. doi:10.4088/PCC.v02n0502. PMC 181133. PMID 15014637.
- ^ Jump up to:a b c d e Malenka RC, Nestler EJ, Hyman SE (2009). “Chapter 15: Reinforcement and Addictive Disorders”. In Sydor A, Brown RY (eds.). Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York: McGraw-Hill Medical. p. 368. ISBN 9780071481274.
Cocaine, [amphetamine], and methamphetamine are the major psychostimulants of abuse. The related drug methylphenidate is also abused, although it is far less potent. These drugs elicit similar initial subjective effects ; differences generally reflect the route of administration and other pharmacokinetic factors. Such agents also have important therapeutic uses; cocaine, for example, is used as a local anesthetic (Chapter 2), and amphetamines and methylphenidate are used in low doses to treat attention deficit hyperactivity disorder and in higher doses to treat narcolepsy (Chapter 12). Despite their clinical uses, these drugs are strongly reinforcing, and their long-term use at high doses is linked with potential addiction, especially when they are rapidly administered or when high-potency forms are given.
- ^ Jump up to:a b Steiner H, Van Waes V (January 2013). “Addiction-related gene regulation: risks of exposure to cognitive enhancers vs. other psychostimulants”. Prog. Neurobiol. 100: 60–80. doi:10.1016/j.pneurobio.2012.10.001. PMC 3525776. PMID 23085425.
- ^ Auger RR, Goodman SH, Silber MH, Krahn LE, Pankratz VS, Slocumb NL (2005). “Risks of high-dose stimulants in the treatment of disorders of excessive somnolence: a case-control study”. Sleep. 28 (6): 667–72. doi:10.1093/sleep/28.6.667. PMID 16477952.
- ^ Jump up to:a b c d e f Kim Y, Teylan MA, Baron M, Sands A, Nairn AC, Greengard P (2009). “Methylphenidate-induced dendritic spine formation and DeltaFosB expression in nucleus accumbens”. Proc. Natl. Acad. Sci. U.S.A. 106 (8): 2915–20. Bibcode:2009PNAS..106.2915K. doi:10.1073/pnas.0813179106. PMC 2650365. PMID 19202072.
Despite decades of clinical use of methylphenidate for ADHD, concerns have been raised that long-term treatment of children with this medication may result in subsequent drug abuse and addiction. However, meta analysis of available data suggests that treatment of ADHD with stimulant drugs may have a significant protective effect, reducing the risk for addictive substance use (36, 37). Studies with juvenile rats have also indicated that repeated exposure to methylphenidate does not necessarily lead to enhanced drug-seeking behavior in adulthood (38). However, the recent increase of methylphenidate use as a cognitive enhancer by the general public has again raised concerns because of its potential for abuse and addiction (3, 6–10). Thus, although oral administration of clinical doses of methylphenidate is not associated with euphoria or with abuse problems, nontherapeutic use of high doses or i.v. administration may lead to addiction (39, 40).
- ^ Elkashef A, Vocci F, Hanson G, White J, Wickes W, Tiihonen J (2008). “Pharmacotherapy of methamphetamine addiction: an update”. Substance Abuse. 29 (3): 31–49. doi:10.1080/08897070802218554. PMC 2597382. PMID 19042205.
- ^ Grabowski J, Roache JD, Schmitz JM, Rhoades H, Creson D, Korszun A (December 1997). “Replacement medication for cocaine dependence: methylphenidate”. Journal of Clinical Psychopharmacology. 17 (6): 485–8. doi:10.1097/00004714-199712000-00008. PMID 9408812.
- ^ Gorelick DA, Gardner EL, Xi ZX (2004). “Agents in development for the management of cocaine abuse”. Drugs. 64 (14): 1547–73. doi:10.2165/00003495-200464140-00004. PMID 15233592. S2CID 5421657. Archived from the original on 1 July 2019. Retrieved 1 July 2019.
- ^ Karila L, Gorelick D, Weinstein A, Noble F, Benyamina A, Coscas S, et al. (May 2008). “New treatments for cocaine dependence: a focused review”. The International Journal of Neuropsychopharmacology. 11 (3): 425–38. doi:10.1017/S1461145707008097. PMID 17927843.
- ^ “NIDA InfoFacts: Understanding Drug Abuse and Addiction”(PDF). 2008. Archived from the original (PDF) on 15 December 2010.
- ^ Shearer J (May 2008). “The principles of agonist pharmacotherapy for psychostimulant dependence”. Drug and Alcohol Review. 27 (3): 301–8. doi:10.1080/09595230801927372. PMID 18368612.
- ^ Jump up to:a b c Nestler EJ (December 2013). “Cellular basis of memory for addiction”. Dialogues in Clinical Neuroscience. 15 (4): 431–43. doi:10.31887/DCNS.2013.15.4/enestler. PMC 3898681. PMID 24459410.
Despite the importance of numerous psychosocial factors, at its core, drug addiction involves a biological process: the ability of repeated exposure to a drug of abuse to induce changes in a vulnerable brain that drive the compulsive seeking and taking of drugs, and loss of control over drug use, that define a state of addiction. … A large body of literature has demonstrated that such ΔFosB induction in D1-type NAc neurons increases an animal’s sensitivity to drug as well as natural rewards and promotes drug self-administration, presumably through a process of positive reinforcement … Another ΔFosB target is cFos: as ΔFosB accumulates with repeated drug exposure it represses c-Fos and contributes to the molecular switch whereby ΔFosB is selectively induced in the chronic drug-treated state.41. … Moreover, there is increasing evidence that, despite a range of genetic risks for addiction across the population, exposure to sufficiently high doses of a drug for long periods of time can transform someone who has relatively lower genetic loading into an addict.4
- ^ Ruffle JK (November 2014). “Molecular neurobiology of addiction: what’s all the (Δ)FosB about?”. The American Journal of Drug and Alcohol Abuse. 40 (6): 428–37. doi:10.3109/00952990.2014.933840. PMID 25083822. S2CID 19157711.
The strong correlation between chronic drug exposure and ΔFosB provides novel opportunities for targeted therapies in addiction (118), and suggests methods to analyze their efficacy (119). Over the past two decades, research has progressed from identifying ΔFosB induction to investigating its subsequent action (38). It is likely that ΔFosB research will now progress into a new era – the use of ΔFosB as a biomarker. …
Conclusions
ΔFosB is an essential transcription factor implicated in the molecular and behavioral pathways of addiction following repeated drug exposure. The formation of ΔFosB in multiple brain regions, and the molecular pathway leading to the formation of AP-1 complexes is well understood. The establishment of a functional purpose for ΔFosB has allowed further determination as to some of the key aspects of its molecular cascades, involving effectors such as GluR2 (87,88), Cdk5 (93) and NFkB (100). Moreover, many of these molecular changes identified are now directly linked to the structural, physiological and behavioral changes observed following chronic drug exposure (60,95,97,102). New frontiers of research investigating the molecular roles of ΔFosB have been opened by epigenetic studies, and recent advances have illustrated the role of ΔFosB acting on DNA and histones, truly as a molecular switch(34). As a consequence of our improved understanding of ΔFosB in addiction, it is possible to evaluate the addictive potential of current medications (119), as well as use it as a biomarker for assessing the efficacy of therapeutic interventions (121,122,124). Some of these proposed interventions have limitations (125) or are in their infancy (75). However, it is hoped that some of these preliminary findings may lead to innovative treatments, which are much needed in addiction.
• Biliński P, Wojtyła A, Kapka-Skrzypczak L, Chwedorowicz R, Cyranka M, Studziński T (2012). “Epigenetic regulation in drug addiction”. Annals of Agricultural and Environmental Medicine. 19(3): 491–6. PMID 23020045.For these reasons, ΔFosB is considered a primary and causative transcription factor in creating new neural connections in the reward centre, prefrontal cortex, and other regions of the limbic system. This is reflected in the increased, stable and long-lasting level of sensitivity to cocaine and other drugs, and tendency to relapse even after long periods of abstinence. These newly constructed networks function very efficiently via new pathways as soon as drugs of abuse are further taken … In this way, the induction of CDK5 gene expression occurs together with suppression of the G9A gene coding for dimethyltransferase acting on the histone H3. A feedback mechanism can be observed in the regulation of these 2 crucial factors that determine the adaptive epigenetic response to cocaine. This depends on ΔFosB inhibiting G9a gene expression, i.e. H3K9me2 synthesis which in turn inhibits transcription factors for ΔFosB. For this reason, the observed hyper-expression of G9a, which ensures high levels of the dimethylated form of histone H3, eliminates the neuronal structural and plasticity effects caused by cocaine by means of this feedback which blocks ΔFosB transcription
• Robison AJ, Nestler EJ (October 2011). “Transcriptional and epigenetic mechanisms of addiction”. Nature Reviews. Neuroscience. 12 (11): 623–37. doi:10.1038/nrn3111. PMC 3272277. PMID 21989194.ΔFosB has been linked directly to several addiction-related behaviors … Importantly, genetic or viral overexpression of ΔJunD, a dominant negative mutant of JunD which antagonizes ΔFosB- and other AP-1-mediated transcriptional activity, in the NAc or OFC blocks these key effects of drug exposure14,22–24. This indicates that ΔFosB is both necessary and sufficient for many of the changes wrought in the brain by chronic drug exposure. ΔFosB is also induced in D1-type NAc MSNs by chronic consumption of several natural rewards, including sucrose, high fat food, sex, wheel running, where it promotes that consumption14,26–30. This implicates ΔFosB in the regulation of natural rewards under normal conditions and perhaps during pathological addictive-like states.
- ^ “Concerta product monograph” (PDF). Janssen Pharmaceuticals. Archived (PDF) from the original on 28 January 2017. Retrieved 4 December 2016.
- ^ Ishii M, Tatsuzawa Y, Yoshino A, Nomura S (April 2008). “Serotonin syndrome induced by augmentation of SSRI with methylphenidate”. Psychiatry and Clinical Neurosciences. 62 (2): 246. doi:10.1111/j.1440-1819.2008.01767.x. PMID 18412855. S2CID 5659107.
- ^ Türkoğlu S (2015). “Serotonin syndrome with sertraline and methylphenidate in an adolescent”. Clinical Neuropharmacology. 38(2): 65–6. doi:10.1097/WNF.0000000000000075. PMID 25768857.
- ^ Park YM, Jung YK (May 2010). “Manic switch and serotonin syndrome induced by augmentation of paroxetine with methylphenidate in a patient with major depression”. Progress in Neuro-Psychopharmacology & Biological Psychiatry. 34 (4): 719–20. doi:10.1016/j.pnpbp.2010.03.016. PMID 20298736. S2CID 31984813.
- ^ Bodner RA, Lynch T, Lewis L, Kahn D (February 1995). “Serotonin syndrome”. Neurology. 45 (2): 219–23. doi:10.1212/wnl.45.2.219. PMID 7854515. S2CID 35190429.
- ^ Patrick KS, González MA, Straughn AB, Markowitz JS (2005). “New methylphenidate formulations for the treatment of attention-deficit/hyperactivity disorder”. Expert Opinion on Drug Delivery. 2(1): 121–43. doi:10.1517/17425247.2.1.121. PMID 16296740. S2CID 25026467.
- ^ Jump up to:a b Markowitz JS, DeVane CL, Boulton DW, Nahas Z, Risch SC, Diamond F, Patrick KS (2000). “Ethylphenidate formation in human subjects after the administration of a single dose of methylphenidate and ethanol”. Drug Metabolism and Disposition. 28 (6): 620–4. PMID 10820132.
- ^ Markowitz JS, Logan BK, Diamond F, Patrick KS (1999). “Detection of the novel metabolite ethylphenidate after methylphenidate overdose with alcohol coingestion”. Journal of Clinical Psychopharmacology. 19 (4): 362–6. doi:10.1097/00004714-199908000-00013. PMID 10440465.
- ^ Patrick KS, Straughn AB, Minhinnett RR, Yeatts SD, Herrin AE, DeVane CL, Malcolm R, Janis GC, Markowitz JS (March 2007). “Influence of ethanol and gender on methylphenidate pharmacokinetics and pharmacodynamics”. Clinical Pharmacology and Therapeutics. 81 (3): 346–53. doi:10.1038/sj.clpt.6100082. PMC 3188424. PMID 17339864.
- ^ Roberts SM, DeMott RP, James RC (1997). “Adrenergic modulation of hepatotoxicity”. Drug Metab. Rev. 29 (1–2): 329–53. doi:10.3109/03602539709037587. PMID 9187524.
- ^ Jump up to:a b Markowitz JS, Patrick KS (June 2008). “Differential pharmacokinetics and pharmacodynamics of methylphenidate enantiomers: does chirality matter?”. Journal of Clinical Psychopharmacology. 28 (3 Suppl 2): S54-61. doi:10.1097/JCP.0b013e3181733560. PMID 18480678.
- ^ Schweri MM, Skolnick P, Rafferty MF, Rice KC, Janowsky AJ, Paul SM (October 1985). “[3H]Threo-(+/-)-methylphenidate binding to 3,4-dihydroxyphenylethylamine uptake sites in corpus striatum: correlation with the stimulant properties of ritalinic acid esters”. Journal of Neurochemistry. 45 (4): 1062–70. doi:10.1111/j.1471-4159.1985.tb05524.x. PMID 4031878. S2CID 28720285.
- ^ Ding YS, Fowler JS, Volkow ND, Dewey SL, Wang GJ, Logan J, et al. (May 1997). “Chiral drugs: comparison of the pharmacokinetics of [11C]d-threo and L-threo-methylphenidate in the human and baboon brain”. Psychopharmacology. 131 (1): 71–8. doi:10.1007/s002130050267. PMID 9181638. S2CID 26046917.
- ^ Ding YS, Gatley SJ, Thanos PK, Shea C, Garza V, Xu Y, et al. (September 2004). “Brain kinetics of methylphenidate (Ritalin) enantiomers after oral administration”. Synapse. 53 (3): 168–75. CiteSeerX 10.1.1.514.7833. doi:10.1002/syn.20046. PMID 15236349. S2CID 11664668.
- ^ Davids E, Zhang K, Tarazi FI, Baldessarini RJ (February 2002). “Stereoselective effects of methylphenidate on motor hyperactivity in juvenile rats induced by neonatal 6-hydroxydopamine lesioning”. Psychopharmacology. 160 (1): 92–8. doi:10.1007/s00213-001-0962-5. PMID 11862378. S2CID 8037050.
- ^ Williard RL, Middaugh LD, Zhu HJ, Patrick KS (February 2007). “Methylphenidate and its ethanol transesterification metabolite ethylphenidate: brain disposition, monoamine transporters and motor activity”. Behavioural Pharmacology. 18 (1): 39–51. doi:10.1097/FBP.0b013e3280143226. PMID 17218796. S2CID 20232871.
- ^ McGough JJ, Pataki CS, Suddath R (July 2005). “Dexmethylphenidate extended-release capsules for attention deficit hyperactivity disorder”. Expert Review of Neurotherapeutics. 5 (4): 437–41. doi:10.1586/14737175.5.4.437. PMID 16026226. S2CID 6561452.
- ^ Silva R, Tilker HA, Cecil JT, Kowalik S, Khetani V, Faleck H, Patin J (2004). “Open-label study of dexmethylphenidate hydrochloride in children and adolescents with attention deficit hyperactivity disorder”. Journal of Child and Adolescent Psychopharmacology. 14(4): 555–63. doi:10.1089/cap.2004.14.555. PMID 15662147.
- ^ Arnold LE, Lindsay RL, Conners CK, Wigal SB, Levine AJ, Johnson DE, et al. (Winter 2004). “A double-blind, placebo-controlled withdrawal trial of dexmethylphenidate hydrochloride in children with attention deficit hyperactivity disorder”. Journal of Child and Adolescent Psychopharmacology. 14 (4): 542–54. doi:10.1089/cap.2004.14.542. PMID 15662146.
- ^ Spencer TJ, Adler LA, McGough JJ, Muniz R, Jiang H, Pestreich L (June 2007). “Efficacy and safety of dexmethylphenidate extended-release capsules in adults with attention-deficit/hyperactivity disorder”. Biological Psychiatry. 61 (12): 1380–7. doi:10.1016/j.biopsych.2006.07.032. PMID 17137560. S2CID 45976373.
- ^ Teo SK, Scheffler MR, Wu A, Stirling DI, Thomas SD, Stypinski D, Khetani VD (February 2004). “A single-dose, two-way crossover, bioequivalence study of dexmethylphenidate HCl with and without food in healthy subjects”. Journal of Clinical Pharmacology. 44 (2): 173–8. doi:10.1177/0091270003261899. PMID 14747426. S2CID 20694072.
External links
- “Dexmethylphenidate”. Drug Information Portal. U.S. National Library of Medicine.
- “Dexmethylphenidate hydrochloride”. Drug Information Portal. U.S. National Library of Medicine.
///////////DEXMETHYLPHENIDATE, FDA 2021, APPROVALS 2021
Cl.[H][C@@](C(=O)OC)(C1=CC=CC=C1)[C@@]1([H])CCCCN1
NEW DRUG APPROVALS
ONE TIME
$10.00
Serdexmethylphenidate

Serdexmethylphenidate
- Molecular FormulaC25H30ClN3O8
- Average mass535.974 Da
CAS 1996626-29-9 base
| 1996626-30-2 chloride |
L-Serine, N-[[1-[[[[(2R)-2-[(1R)-2-methoxy-2-oxo-1-phenylethyl]-1-piperidinyl]carbonyl]oxy]methyl]-3-pyridiniumyl]carbonyl]-, chloride (1:1)
N-[(1-{[({(2R)-2-[(1R)-2-Methoxy-2-oxo-1-phenylethyl]-1-piperidinyl}carbonyl)oxy]methyl}-3-pyridiniumyl)carbonyl]-L-serine chloride
Azstarys, FDA APPROVED, 3/2/2021, Products on NDA 212994, Type 1 – New Molecular Entity and Type 4 – New Combination
Serdexmethylphenidate Chloride (SDX), SDX or KP145


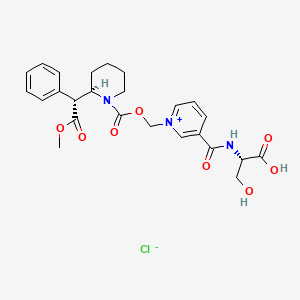
| Molecular Formula | C25H30ClN3O8 |
|---|---|
| Synonyms | UNII-FN54BT298YKP415 ClSerdexmethylphenidate chlorideFN54BT298YSerdexmethylphenidate chloride (USAN) |
| Molecular Weight | 536 g/mol |
CAS 1996626-30-2 chloride
(2S)-3-hydroxy-2-[[1-[[(2R)-2-[(1R)-2-methoxy-2-oxo-1-phenylethyl]piperidine-1-carbonyl]oxymethyl]pyridin-1-ium-3-carbonyl]amino]propanoic acid;chloride
Serdexmethylphenidate is a derivative of dexmethylphenidate created by pharmaceutical company KemPharm. The compound is under investigation for the treatment of ADHD in children, adolescents, and adults as of 2020.[2] The drug was approved for medical use by the FDA in March, 2021. Serdexmethylphenidate is a prodrug which has a delayed onset of action and a prolonged duration of effects compared to dexmethylphenidate, its parent compound.
SCHEME

WO2021173533
US20200237742
WO2019241019
US20190381017
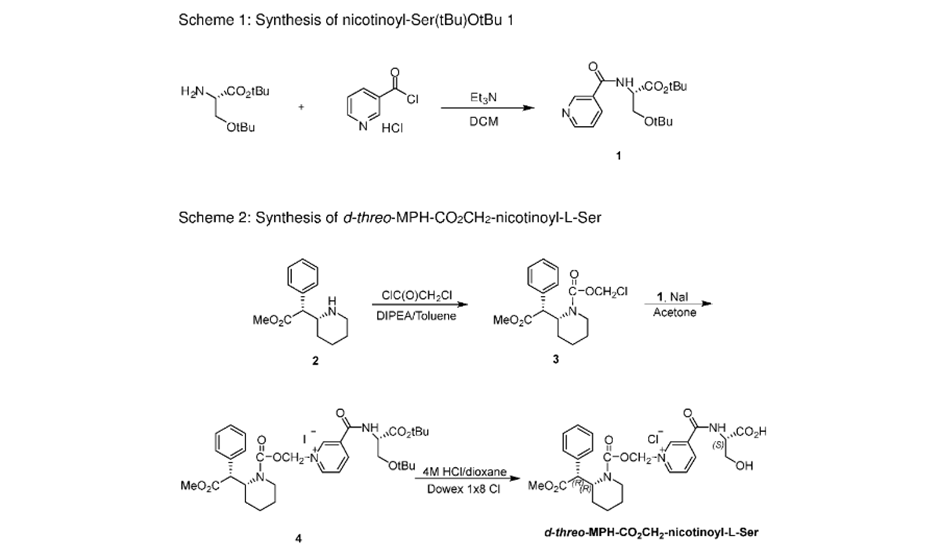
NEW DRUG APPROVALS
ONE TIME
$10.00
Formulations
Serdexmethylphenidate/dexmethylphenidate (Azstarys), a co-formulation of serdexmethylphenidate and dexmethylphenidate, was approved by the Food and Drug Administration (FDA) in March 2021, for the treatment of ADHD in those above six years of age. Co-formulation of serdexmethylphenidate with dexmethylphenidate allows for a more rapid onset of action while still retaining up to 13 hours of therapeutic efficacy.[3][4]
Due to serdexmethylphenidate’s delayed onset and prolonged duration of effects, several dosage forms containing serdexmethylphenidate have been investigated for use as long-acting psychostimulants in the treatment of ADHD. Under the developmental codename KP484, serdexmethylphenidate has been investigated as a “super-extended duration” psychostimulant, with therapeutic efficacy lasting up to 16 hours following oral administration. In 2011, MonoSol Rx entered into a partnership with KenPharm to develop oral films containing KP415.[5]
Abuse potential
The abuse potential of serdexmethylphenidate is theorized to be lower than other psychostimulants because serdexmethylphenidate is an inactive prodrug of dexmethylphenidate, and must undergo enzymatic metabolism prior to exerting any stimulant effects.[6] Common routes of administration used during the abuse of psychostimulants such as insufflation and intravenous injection have little impact on the pharmacokinetics and metabolism of serdexmethylphenidate and do not result in a faster onset of action.[7]
SYN

SYN
US 20200237742
Title(EN) Serdexmethylphenidate Conjugates, Compositions And Methods Of Use Thereof
Abstract
(EN)
The present technology is directed to one or more compositions comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof. The present technology also relates to one or more compositions and oral formulations comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof. The present technology also relates to one or more methods of using compositions comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof. The present technology additionally relates to one or more pharmaceutical kits containing a composition comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof.
| Synthetic Process for Making Serdexmethylphenidate |
| In one embodiment, the protected serdexmethylphenidate intermediate can be prepared as shown in Scheme 4. |
| In an alternate embodiment, the protected serdexmethylphenidate intermediate can be prepared according to Scheme 5. |
PATENT
US 20190381017
Title(EN) Compositions Comprising Serdexmethylphenidate Conjugates And Methods Of Use Thereof
Abstract
(EN)
The present technology is directed to one or more compositions comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof. The present technology also relates to one or more compositions and oral formulations comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof. The present technology also relates to one or more methods of using compositions comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof. The present technology additionally relates to one or more pharmaceutical kits containing a composition comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof.
PATENT
WO 2019241019
PAT
WO 2018107131
WO 2018107132
References
- ^ “Azstarys Prescribing Information” (PDF). United States Food and Drug Administration. Retrieved 18 March 2021.
- ^ “KemPharm’s KP415 and Serdexmethylphenidate (SDX) Prodrug to be Featured in Multiple Sessions at the AACAP 2020 Virtual Meeting”. http://www.globenewswire.com.
- ^ Mickle T. “Prodrugs for ADHD Treatments: Opportunities & Potential to Fill Unmet Medical Needs” (PDF). Retrieved 15 November 2020.
- ^ Eric Bastings, MD (2 March 2021). “NDA 212994 Approval” (PDF). United States Food and Drug Administration. Retrieved 6 March 2021.
- ^ Van Arnum P (1 March 2012). “Meeting Solubility Challenges”. Pharmaceutical Technology. 2012 (2): S6–S8. Retrieved 15 November 2020.
- ^ Mickle T. “Prodrugs for ADHD Treatments: Opportunities & Potential to Fill Unmet Medical Needs” (PDF). Retrieved 15 November 2020.
- ^ Braeckman R (1 October 2018). “Human Abuse Potential of Intravenous Serdexmethylphenidate (SDX), A Novel Prodrug of D-Methylphenidate, in Recreational Stimulant Abusers”. Journal of the American Academy of Child & Adolescent Psychiatry. 57 (10): 176. doi:10.1016/j.jaac.2018.09.141. Retrieved 15 November 2020.
External links
- “Serdexmethylphenidate”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Other names | KP484 |
| License data | US DailyMed: Serdexmethylphenidate |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only |
| Pharmacokinetic data | |
| Bioavailability | 3%[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1996626-30-2 |
| PubChem CID | 134823897 |
| ChemSpider | 81368035 |
| UNII | FN54BT298Y |
| KEGG | D11401 |
| ChEMBL | ChEMBL4298139 |
| Chemical and physical data | |
| Formula | C25H30ClN3O8 |
| Molar mass | 535.98 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
//////////Serdexmethylphenidate, Azstarys, FDA 2021 APPROVALS 2021, SDX, KP 145,
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
NEW DRUG APPROVALS
ONW TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}