
Home » FDA 2018 (Page 6)
Category Archives: FDA 2018
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
GS 9883, Bictegravir an HIV-1 integrase inhibitor
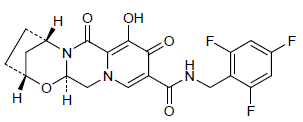

GS 9883, bictegravir
CAS 1611493-60-7
PHASE 3
HIV-1 integrase inhibitor
(2R,5S,13aR)-8-hydroxy-7,9-dioxo-N-[(2,4,6-trifluorophenyl)methyl]-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide
2,5-Methanopyrido(1′,2′:4,5)pyrazino(2,1-b)(1,3)oxazepine-10-carboxamide, 2,3,4,5,7,9,13,13a-octahydro-8-hydroxy-7,9-dioxo-N-((2,4,6-trifluorophenyl)methyl)-, (2R,5S,13aR)-
(2R,5S,13aR)-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluorobenzyl)-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide
(2 ,5S,13aI )-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluoroheoctahydro-2,5-methanopyrido[ 1 ‘,2’:4,5]pyrazino[2, 1 -b][ 1 ,3]oxazepine- 10-carboxamide
MF C21H18F3N3O5,
| MW | 449.37993 g/mol |
|---|
UNII-8GB79LOJ07; 8GB79LOJ07

BICTEGRAVIR
- 16 Nov 2015 Phase-III clinical trials in HIV-1 infections (Combination therapy, Treatment-naive) in USA (PO) (Gilead Pipeline, November 2015)
- 01 Jul 2015 Gilead Sciences completes a phase I trial in HIV-1 infections in USA and New Zealand (NCT02400307)
- 01 Apr 2015 Phase-I clinical trials in HIV-1 infections (In volunteers) in New Zealand (PO) (NCT02400307)
UPDATE Biktarvy (bictegravir/emtricitabine/tenofovir alafenamide); Gilead; For the treatment of HIV-1 infection in adults, Approved February 2018
Human immunodeficiency virus infection and related diseases are a major public health problem worldwide. Human immunodeficiency virus type 1 (HIV-1) encodes three enzymes which are required for viral replication: reverse transcriptase, protease, and integrase. Although drugs targeting reverse transcriptase and protease are in wide use and have shown effectiveness, particularly when employed in combination, toxicity and development of resistant strains have limited their usefulness (Palella, et al. N. Engl. J Med. (1998) 338:853-860; Richman, D. D. Nature (2001) 410:995-1001). Accordingly, there is a need for new agents that inhibit the replication of HIV and that minimize PXR activation when co-administered with other drugs.
Certain polycyclic carbamoylpyridone compounds have been found to have antiviral activity, as disclosed in PCT/US2013/076367. Accordingly, there is a need for synthetic routes for such compounds.
SYNTHESIS
WO 2014100323

PATENTS
xample 42
Preparation of Compound 42
(2 ,5S,13aI )-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluorohe
octahydro-2,5-methanopyrido[ 1 ‘,2’:4,5]pyrazino[2, 1 -b][ 1 ,3]oxazepine- 10-carboxamide

42

Step 1
l-(2,2-dimethoxyethyl)-5-methoxy-6-(methoxycarbonyl)-4-oxo-l ,4-dihydropyridine-3-carboxylic acid (3.15 g, 10 mmol) in acetonitrile (36 mL) and acetic acid (4 mL) was treated with methanesuffhnic acid (0.195 mL, 3 mmol) and placed in a 75 deg C bath. The reaction mixture was stirred for 7 h, cooled and stored at -10 °C for 3 days and reheated to 75 °C for an additional 2 h. This material was cooled and carried on crude to the next step.
Step 2
Crude reaction mixture from step 1 (20 mL, 4.9 mmol) was transferred to a flask containing (lR,3S)-3-aminocyclopentanol (0.809 g, 8 mmol). The mixture was diluted with acetonitrile (16.8 mL), treated with potassium carbonate (0.553 g, 4 mmol) and heated to 85 °C. After 2 h, the reaction mixture was cooled to ambient temperature and stirred overnight. 0.2M HQ (50 mL) was added, and the clear yellow solution was extracted with dichloromethane (2×150 mL). The combined organic layers were dried over sodium sulfate, filtered and concentrated to 1.49 g of a light orange solid. Recrystallization from dichloimethane:hexanes afforded the desired intermediate 42 A: LC S-ESI (m/z): [M+H]+ calculated for Ci5Hi7N206: 321.1 1 ; found: 321.3.
Step 3
Intermediate 42-A (0.225 g, 0.702 mmol) and (2,4,6-trifluorophenyl)methanamine (0.125 g, 0.773 mmol) were suspended in acetonitrile (4 mL) and treated with N,N-diisopropylethylamine (DIPEA) (0.183 mmol, 1.05 mmol). To this suspension was added (dimethyiammo)- V,A/-dimethyi(3H-[l ,2,3]triazolo[4,5-&]pyridm~3-yiox.y)methammimum hexafluorophosphate (HATU, 0.294 g, 0.774 mmol). After 1.5 hours, the crude reaction mixture was taken on to the next step. LfJMS-ESlT (m/z): [M+H calculated for (\ ,l l.,, i \\:0< : 464.14; found: 464.2.
Step 4
To the crude reaction mixture of the previous step was added MgBr2
(0.258 g, 1.40 mmol). The reaction mixture was stirred at 50 °C for 10 minutes, acidified with 10% aqueous HC1, and extract twice with dichloromethane. The combined organic phases were dried over MgS04, filtered, concentrated, and purified by silica gel chromatography (EtOH/dichlormethane) followed by HPLC (ACN H2O with 0.1 % TFA modifier) to afford compound 42: 1H~ M (400 MHz, DMSO-</6) δ 12.43 (s, 1H), 10.34 (t, J = 5.7 Hz, IH), 8.42 (s, 1H), 7.19 (t, J = 8.7 Hz, 2H), 5.43 (dd, ./’ 9.5, 4.1 Hz, I H), 5.08 (s, i l l ). 4.66 (dd, ./ 12.9, 4.0 Hz, IH), 4.59 (s, 1 1 1 ). 4.56 4.45 (m, 2H), 4.01 (dd, J = 12.7, 9.7 Hz, IH), 1.93 (s, 4H), 1.83 (d, J —— 12.0 Hz, I H),
1.56 (dt, J = 12.0, 3.4 Hz, I H). LCMS-ESI+ (m/z): [M+H]+ calculated for { · Ί ί ] ΝΓ :Χ.¾ϋ : 450.13; found: 450.2.
PATENT
WO2015177537
PATENT
WO2015196116
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015196116&redirectedID=true
PATENT
WO2015196137
PATENT
http://www.google.com/patents/US20140221356
Example 42 Preparation of Compound 42 (2R,5S,13aR)-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluorobenzyl)-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide

Step 1
-
1-(2,2-dimethoxyethyl)-5-methoxy-6-(methoxycarbonyl)-4-oxo-1,4-dihydropyridine-3-carboxylic acid (3.15 g, 10 mmol) in acetonitrile (36 mL) and acetic acid (4 mL) was treated with methanesulfonic acid (0.195 mL, 3 mmol) and placed in a 75 deg C. bath. The reaction mixture was stirred for 7 h, cooled and stored at −10° C. for 3 days and reheated to 75° C. for an additional 2 h. This material was cooled and carried on crude to the next step.
Step 2
-
Crude reaction mixture from step 1 (20 mL, 4.9 mmol) was transferred to a flask containing (1R,3S)-3-aminocyclopentanol (0.809 g, 8 mmol). The mixture was diluted with acetonitrile (16.8 mL), treated with potassium carbonate (0.553 g, 4 mmol) and heated to 85° C. After 2 h, the reaction mixture was cooled to ambient temperature and stirred overnight. 0.2M HCl (50 mL) was added, and the clear yellow solution was extracted with dichloromethane (2×150 mL). The combined organic layers were dried over sodium sulfate, filtered and concentrated to 1.49 g of a light orange solid. Recrystallization from dichlormethane:hexanes afforded the desired intermediate 42A: LCMS-ESI+ (m/z): [M+H]+ calculated for C15H17N2O6: 321.11; found: 321.3.
Step 3
-
Intermediate 42-A (0.225 g, 0.702 mmol) and (2,4,6-trifluorophenyl)methanamine (0.125 g, 0.773 mmol) were suspended in acetonitrile (4 mL) and treated with N,N-diisopropylethylamine (DIPEA) (0.183 mmol, 1.05 mmol). To this suspension was added (dimethylamino)-N,N-dimethyl(3H-[1,2,3]triazolo[4,5-b]pyridin-3-yloxy)methaniminium hexafluorophosphate (HATU, 0.294 g, 0.774 mmol). After 1.5 hours, the crude reaction mixture was taken on to the next step. LCMS-ESI+ (m/z): [M+H]+ calculated for C22H21F3N3O5: 464.14; found: 464.2.
Step 4
-
To the crude reaction mixture of the previous step was added MgBr2 (0.258 g, 1.40 mmol). The reaction mixture was stirred at 50° C. for 10 minutes, acidified with 10% aqueous HCl, and extract twice with dichloromethane. The combined organic phases were dried over MgSO4, filtered, concentrated, and purified by silica gel chromatography (EtOH/dichlormethane) followed by HPLC (ACN/H2O with 0.1% TFA modifier) to afford compound 42: 1H-NMR (400 MHz, DMSO-d6) δ 12.43 (s, 1H), 10.34 (t, J=5.7 Hz, 1H), 8.42 (s, 1H), 7.19 (t, J=8.7 Hz, 2H), 5.43 (dd, J=9.5, 4.1 Hz, 1H), 5.08 (s, 1H), 4.66 (dd, J=12.9, 4.0 Hz, 1H), 4.59 (s, 1H), 4.56-4.45 (m, 2H), 4.01 (dd, J=12.7, 9.7 Hz, 1H), 1.93 (s, 4H), 1.83 (d, J=12.0 Hz, 1H), 1.56 (dt, J=12.0, 3.4 Hz, 1H). LCMS-ESI+ (m/z): [M+H]+ calculated for C21H19F3N3O5: 450.13; found: 450.2.
PATENT
General Scheme I:

General Scheme II:



General Scheme II
General Scheme III:


General Scheme III
General Scheme IV:


G-1
General Scheme V:

II
EXAMPLES
In order for this invention to be more fully understood, the following examples are set forth. These examples are for the purpose of illustrating embodiments, and are not to be construed as limiting the scope of this disclosure in any way. The reactants used in the examples below may be obtained either as described herein, or if not described herein, are themselves either commercially available or may be prepared from commercially available materials by methods known in the art.
In one embodiment, a multi-step synthetic method for preparing a compound of Formula I is provided, as set forth below. In certain embodiments, each of the individual steps of the Schemes set forth below is provided. Examples and any combination of two or more successive steps of the below Examples are provided.
A. Acylation and amidation of Meldrum ‘s acid to form C-la:

[0520] In a reaction vessel, Meldrum’s acid (101 g, 1.0 equivalent) and 4-dimethylaminopyridine (1.8 g, 0.2 equivalents) were combined with acetonitrile (300 mL). The resulting solution was treated with methoxyacetic acid (6.2 mL, 1.2 equivalents). Triethylamine (19.4 mL, 2.0 equivalents) was added slowly to the resulting solution, followed by pivaloyl chloride (9.4 mL, 1.1 equivalents). The reaction was then heated to about 45 to about 50 °C and aged until consumption of Meldrum’s acid was deemed complete.
A separate reaction vessel was charged with acetonitrile (50 mL) and J-la (13.4 g, 1.2 equivalents). The resulting solution was treated with trifluoroacetic acid (8.0 mL, 1.5 equivalents), and then this acidic solution was added to the acylation reaction in progress at about 45 to about 50 °C.
The reaction was allowed to age for at least 18 hours at about 45 to about 50 °C, after which time the solvent was removed under reduced pressure. The crude residue was dissolved in ethyl acetate (150 mL), and the organic layer was washed with water. The combined aqueous layers were extracted with ethyl acetate. The combined organic layers were washed with saturated sodium bicarbonate solution, and the combined bicarbonate washes were back extracted with ethyl acetate. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The resulting crude material was purified twice via silica gel chromatography to yield C-la.
lH NMR (400 MHz, CDC13): δ 7.12 (br, 1H), 6.66 (app t, J= 8.1 Hz, 2H), 4.50 (app d, J= 5.7 Hz, 2H), 4.08 (s, 2H), 3.44 (s, 2H), 3.40 (s, 3H). 13C NMR (100 MHz, CDC13): δ 203.96, 164.90, 162.37 (ddd, J= 250.0, 15.7, 15.7 Hz), 161.71 (ddd, J = 250.3, 14.9, 10.9 Hz), 110.05 (ddd, J= 19.7, 19.7, 4.7 Hz), 100.42 (m), 77.58, 59.41, 45.71, 31.17 (t, J= 3.5 Hz). LCMS, Calculated: 275.23, Found: 275.97 (M).
I l l
B. Alkylation of C-la to form E-la:

A solution of C-la (248 mg, 1.0 equivalent) and 2-methyl tetrahydrofuran (1.3 niL) was treated with N,N-dimethylformamide dimethylacetal (0.1 mL, 1.1 equivalent) and stirred at room temperature overnight (~14 hours). The reaction was treated with aminoacetaldehyde dimethyl acetal (0.1 mL, 1.0 equivalents), and was allowed to age for about 2 hours, and then was quenched via the addition of 2 Ν HC1
(1.5 mL).
The reaction was diluted via the addition of ethyl acetate, and phases were separated. The aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The crude residue was purified via silica gel chromatography to yield E-la.
1H NMR (400 MHz, CDC13): δ 10.85 (s, 1H), 9.86 (s, 1H), 8.02 (d, J= 13.1 Hz, 1H), 6.65 (dd, J= 8.7, 7.7 Hz, 2H), 4.53 (d, J= 3.9 Hz, 2H), 4.40 (t, J= 5.1 Hz, 1H), 4.18 (s, 2H), 3.42 (s, 6H), 3.39 (m, 2H), 3.37 (s, 3H). 13C MR (100 MHz, CDC13): δ 193.30, 169.15, 162.10 (ddd, J= 248.9, 15.5, 15.5 Hz), 161.7 (ddd, J =
250.0, 14.9, 1 1.1 Hz), 161.66, 1 11.08 (ddd J= 19.9, 19.9, 4.7 Hz) 103.12, 100.29 (ddd, J= 28.1, 17.7, 2.3 Hz), 76.30, 58.83, 54.98, 53.53, 51.57, 29.89 (t, J= 3.3 Hz). LCMS, Calculated: 390.36, Found: 390.92 (M).
c. Cyclization of E-la to form F-la:

E-1a F-1a
] E-la (0.2 g, 1.0 equivalent), dimethyl oxalate (0.1 g, 2.5 equivalents) and methanol (1.5 mL) were combined and cooled to about 0 to about 5 °C. Sodium methoxide (0.2 mL, 30% solution in methanol, 1.75 equivalents) was introduced to the reaction slowly while keeping the internal temperature of the reaction below about 10 °C throughout the addition. After the addition was completed the reaction was heated to about 40 to about 50 °C for at least 18 hours.
After this time had elapsed, the reaction was diluted with 2 N HC1 (1.5 mL) and ethyl acetate (2 mL). The phases were separated, and the aqueous phase was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over magnesium sulfate, filtered, and solvent was removed under reduced pressure. The resulting crude oil was purified via silica gel chromatography to afford F-la.
lR NMR (400 MHz, CDC13): δ 10.28 (t, J= 5.5 Hz, 1H), 8.38 (s, 1H), 6.66 – 6.53 (m, 2H), 4.58 (d, J= 5.6 Hz, 2H), 4.43 (t, J= 4.7 Hz, 1H), 4.00 (d, J= 4.7 Hz, 2H), 3.92 (s, 3H), 3.88 (s, 3H), 3.32 (s, 6H). 13C NMR (100 MHz, CDC13): δ 173.08, 163.81, 162.17, 162.14 (ddd, J= 249.2, 15.6, 15.6 Hz), 161.72 (ddd, J= 250.5, 15.0, 10.9 Hz), 149.37, 144.64, 134.98, 119.21, 1 10.53 (ddd, J= 19.8, 4.7, 4.7 Hz), 102.70, 100.22 (m), 60.68, 56.75, 55.61, 53.35, 30.64. LCMS, Calculated: 458.39, Found: 459.15 (M+H).
D. Alkylation and cyclization of C-la to form F-la:
1 . DMFDMA

C-1a NaOMe, MeOH, 40 °C F-1a
To a reaction vessel were added C-la (245 mg, 1.0 equivalent) and N,N-dimethylformamide dimethylacetal (0.5 mL, 4.3 equivalent). The reaction mixture was agitated for approximately 30 minutes. The reaction was then treated with 2-methyl tetrahydrofuran (2.0 mL) and aminoacetaldehyde dimethyl acetal (0.1 mL, 1.0 equivalent). The reaction was allowed to age for several hours and then solvent was removed under reduced pressure.
The resulting material was dissolved in methanol and dimethyl oxalate was added (0.3 g, 2.5 equivalents). The reaction mixture was cooled to about 0 to about 5 °C, and then sodium methoxide (0.4 mL, 30% solution in methanol, 1.75 equivalents) was introduced to the reaction slowly. After the addition was completed the reaction was heated to about 40 to about 50 °C.
After this time had elapsed, the reaction was cooled to room temperature and quenched via the addition of 2 Ν HC1 (1.5 mL). The reaction was then diluted with ethyl acetate, and the resulting phases were separated. The aqueous layer was extracted with ethyl acetate. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The crude residue was purified via silica gel chromatography to yield F-la.
lR NMR (400 MHz, CDC13): δ 10.28 (t, J= 5.5 Hz, 1H), 8.38 (s, 1H), 6.66 – 6.53 (m, 2H), 4.58 (d, J= 5.6 Hz, 2H), 4.43 (t, J= 4.7 Hz, 1H), 4.00 (d, J= 4.7 Hz, 2H), 3.92 (s, 3H), 3.88 (s, 3H), 3.32 (s, 6H). 13C NMR (100 MHz, CDC13): δ 173.08, 163.81, 162.17, 162.14 (ddd, J= 249.2, 15.6, 15.6 Hz), 161.72 (ddd, J= 250.5, 15.0, 10.9 Hz), 149.37, 144.64, 134.98, 119.21, 1 10.53 (ddd, J= 19.8, 4.7, 4.7 Hz), 102.70, 100.22 (m), 60.68, 56.75, 55.61, 53.35, 30.64. LCMS, Calculated: 458.39, Found: 459.15 (M+H).
E. Condensation of F-la with N-la to form G-la:

K2C03, MeCN, 75 °C
To a reaction vessel were added F-la (202 mg, 1.0 equivalent) and acetonitrile (1.4 mL). The resulting solution was treated with glacial acetic acid (0.2 mL, 6.0 equivalents) and methane sulfonic acid (0.01 mL, 0.3 equivalents). The reaction was then heated to about 70 to about 75 °C.
After 3 hours, a solid mixture of N-la (0.128g, 1.5 equivalents) and potassium carbonate (0.2 g, 2.7 equivalents) was introduced to the reaction at about 70 to about 75 °C. After the addition was completed, the reaction was allowed to progress for at least about 1 hour.
After this time had elapsed, water (1.4 mL) and dichloromethane (1.4 mL) were introduced to the reaction. The phases were separated, and the aqueous layer was extracted with dichloromethane. The combined organic layers were dried over magnesium sulfate, then were filtered and concentrated under reduced pressure. The resulting crude material was purified via silica gel chromatography to obtain G-la.
lR NMR (400 MHz, CDC13): δ 10.23 (t, J= 5.5 Hz, 1H), 8.39 (s, 1H), 6.60 (t, J= 8.1 Hz, 2H), 5.29 (dd, J= 9.5, 3.7 Hz, 2H), 4.57 (d, J= 5.4 Hz, 3H), 4.33 (dd, J = 12.8, 3.8 Hz, 1H), 4.02 – 3.87 (m, 1H), 3.94 (s, 3H), 2.06 – 1.88 (m, 4H), 1.78 (dd, J = 17.2, 7.5 Hz, 1H), 1.55 – 1.46 (m, 1H). 13C MR (100 MHz, CDC13): δ 174.53, 163.75, 162.33 (dd, J= 249.4, 15.7, 15.7 Hz), 161.86 (ddd, J= 250.4, 14.9, 10.9 Hz), 154.18, 154.15, 142.44, 129.75, 1 18.88, 1 10.58 (ddd, J= 19.8, 4.7, 4.7 Hz), 100.42 (m), 77.64, 74.40, 61.23, 54.79, 51.13, 38.31, 30.73, 29.55, 28.04. LCMS, Calculated: 463.14, Found: 464.15 (M+H).
Γ. Deprotection of G-la to form a compound of Formula la:
G-la (14 g) was suspended in acetonitrile (150 mL) and dichloromethane (150 mL). MgBr2 (12 g) was added. The reaction was heated to 40 to 50 °C for approximately 10 min before being cooled to room temperature. The reaction was poured into 0.5M HC1 (140 mL) and the layers separated. The organic layer was washed with water (70 mL), and the organic layer was then concentrated. The crude product was purified by silica gel chromatography (100% dichloromethane up to 6% ethanol/dichloromethane) to afford la.
REFERENCES
| Patent | Submitted | Granted |
|---|---|---|
| POLYCYCLIC-CARBAMOYLPYRIDONE COMPOUNDS AND THEIR PHARMACEUTICAL USE [US2014221356] | 2013-12-19 | 2014-08-07 |
| US9216996 | Dec 19, 2013 | Dec 22, 2015 | Gilead Sciences, Inc. | Substituted 2,3,4,5,7,9,13,13a-octahydropyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepines and methods for treating viral infections |
see full gravir series at…………..http://medcheminternational.blogspot.in/p/ravir-series.html
//////////
C1CC2CC1N3C(O2)CN4C=C(C(=O)C(=C4C3=O)O)C(=O)NCC5=C(C=C(C=C5F)F)F
OR
c1c(cc(c(c1F)CNC(=O)c2cn3c(c(c2=O)O)C(=O)N4[C@H]5CC[C@H](C5)O[C@@H]4C3)F)F
![]()

BICTEGRAVIR, NEW PATENT, WO 2018005328, CONCERT PHARMA
WO2018005328) DEUTERATED BICTEGRAVIR
CONCERT PHARMACEUTICALS, INC.
TUNG, Roger, D.; (US)

Concert CEO Roger Tung
Novel deuterated forms of bictegravir is claimed. Gilead Sciences is developing the integrase inhibitor bictegravir as an oral tablet for the treatment of HIV-1 infection.
This invention relates to deuterated forms of bictegravir, and pharmaceutically acceptable salts thereof. In one aspect, the invention provides a compound of Formula (I) or a pharmaceutically acceptable salt thereof, wherein each of Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, Y11a, and Y11b is independently hydrogen or deuterium; provided that if each Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, and Y11 is hydrogen, then Y11b is deuterium.

Many current medicines suffer from poor absorption, distribution, metabolism and/or excretion (ADME) properties that prevent their wider use or limit their use in certain indications. Poor ADME properties are also a major reason for the failure of drug candidates in clinical trials. While formulation technologies and prodrug strategies can be employed in some cases to improve certain ADME properties, these approaches often fail to address the underlying ADME problems that exist for many drugs and drug candidates. One such problem is rapid metabolism that causes a number of drugs, which otherwise would be highly effective in treating a disease, to be cleared too rapidly from the body. A possible solution to rapid drug clearance is frequent or high dosing to attain a sufficiently high plasma level of drug. This, however, introduces a number of potential treatment problems such as poor patient compliance with the dosing regimen, side effects that become more acute with higher doses, and increased cost of treatment. A rapidly metabolized drug may also expose patients to undesirable toxic or reactive metabolites.
[3] Another ADME limitation that affects many medicines is the formation of toxic or biologically reactive metabolites. As a result, some patients receiving the drug may experience toxicities, or the safe dosing of such drugs may be limited such that patients receive a suboptimal amount of the active agent. In certain cases, modifying dosing intervals or formulation approaches can help to reduce clinical adverse effects, but often the formation of such undesirable metabolites is intrinsic to the metabolism of the compound.
[4] In some select cases, a metabolic inhibitor will be co-administered with a drug that is cleared too rapidly. Such is the case with the protease inhibitor class of drugs that are used to treat HIV infection. The FDA recommends that these drugs be co-dosed with ritonavir, an inhibitor of cytochrome P450 enzyme 3A4 (CYP3A4), the enzyme typically responsible for their metabolism (see Kempf, D.J. et al., Antimicrobial agents and chemotherapy, 1997, 41(3): 654-60). Ritonavir, however, causes adverse effects and adds to the pill burden for HIV patients who must already take a combination of different drugs. Similarly, the
CYP2D6 inhibitor quinidine has been added to dextromethorphan for the purpose of reducing rapid CYP2D6 metabolism of dextromethorphan in a treatment of pseudobulbar affect. Quinidine, however, has unwanted side effects that greatly limit its use in potential combination therapy (see Wang, L et al., Clinical Pharmacology and Therapeutics, 1994, 56(6 Pt 1): 659-67; and FDA label for quinidine at http://www.accessdata.fda.gov).
[5] In general, combining drugs with cytochrome P450 inhibitors is not a satisfactory strategy for decreasing drug clearance. The inhibition of a CYP enzyme’s activity can affect the metabolism and clearance of other drugs metabolized by that same enzyme. CYP inhibition can cause other drugs to accumulate in the body to toxic levels.
[6] A potentially attractive strategy for improving a drug’s metabolic properties is deuterium modification. In this approach, one attempts to slow the CYP-mediated metabolism of a drug or to reduce the formation of undesirable metabolites by replacing one or more hydrogen atoms with deuterium atoms. Deuterium is a safe, stable, non-radioactive isotope of hydrogen. Compared to hydrogen, deuterium forms stronger bonds with carbon. In select cases, the increased bond strength imparted by deuterium can positively impact the ADME properties of a drug, creating the potential for improved drug efficacy, safety, and/or tolerability. At the same time, because the size and shape of deuterium are essentially identical to those of hydrogen, replacement of hydrogen by deuterium would not be expected to affect the biochemical potency and selectivity of the drug as compared to the original chemical entity that contains only hydrogen.
[7] Over the past 35 years, the effects of deuterium substitution on the rate of metabolism have been reported for a very small percentage of approved drugs (see, e.g., Blake, MI et al, J Pharm Sci, 1975, 64:367-91; Foster, AB, Adv Drug Res 1985, 14:1-40 (“Foster”); Kushner, DJ et al, Can J Physiol Pharmacol 1999, 79-88; Fisher, MB et al, Curr Opin Drug Discov Devel, 2006, 9:101-09 (“Fisher”)). The results have been variable and unpredictable. For some compounds deuteration caused decreased metabolic clearance in vivo. For others, there was no change in metabolism. Still others demonstrated increased metabolic clearance. The variability in deuterium effects has also led experts to question or dismiss deuterium modification as a viable drug design strategy for inhibiting adverse metabolism (see Foster at p.35 and Fisher at p.101).
[8] The effects of deuterium modification on a drug’s metabolic properties are not predictable even when deuterium atoms are incorporated at known sites of metabolism. Only by actually preparing and testing a deuterated drug can one determine if and how the rate of metabolism will differ from that of its non-deuterated counterpart. See, for example, Fukuto et al. (J. Med. Chem.1991, 34, 2871-76). Many drugs have multiple sites where metabolism is possible. The site(s) where deuterium substitution is required and the extent of deuteration necessary to see an effect on metabolism, if any, will be different for each drug.
Exemplary Synthesis
[72] Deuterium-modified analogs of bictegravir can be synthesized by means known in the art of organic chemistry. For instance, using methods described in US Patent No.9,216,996 (Haolun J. et al., assigned to Gilead Sciences, Inc. and incorporated herein by reference), using deuterium-containing reagents provides the desired deuterated analogs.
[73] Such methods can be carried out utilizing corresponding deuterated and optionally, other isotope-containing reagents and/or intermediates to synthesize the compounds delineated herein, or invoking standard synthetic protocols known in the art for introducing isotopic atoms to a chemical structure.
[74] A convenient method for synthesizing compounds of Formula I is depicted in the Schemes below.
 [75] A generic scheme for the synthesis of compounds of Formula I is shown in Scheme 1 above. In a manner analogous to the procedure described in Wang, H. et al. Org. Lett.2015, 17, 564-567, aldol condensation of compound 1 with appropriately deuterated compound 2 affords enamine 3. Enamine 3 is then reacted with primary amine 4 to afford enamine 5, which then undergoes cyclization with dimethyl oxalate followed by ester hydrolysis to provide carboxylic acid 7.
[75] A generic scheme for the synthesis of compounds of Formula I is shown in Scheme 1 above. In a manner analogous to the procedure described in Wang, H. et al. Org. Lett.2015, 17, 564-567, aldol condensation of compound 1 with appropriately deuterated compound 2 affords enamine 3. Enamine 3 is then reacted with primary amine 4 to afford enamine 5, which then undergoes cyclization with dimethyl oxalate followed by ester hydrolysis to provide carboxylic acid 7.
[76] In a manner analogous to the procedure described in US 9,216,996, acetal deprotection of carboxylic acid 7 followed by cyclization with appropriately deuterated aminocyclopentanol 9 provides carboxylic acid intermediate 10. Amide coupling with appropriately deuterated benzylamine 11 followed by deprotection of the methyl ether ultimately affords a compound of Formula I in eight overall steps from compound 1.
[77] Use of appropriately deuterated reagents allows deuterium incorporation at the Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, Y11a, and Y11bpositions of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at any Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, Y11a, and/or Y11b.
[78] Appropriately deuterated intermediates 2a and 2b, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents as exemplified in Scheme 2 below.
S h 2 S th i f C d 2 d 2b

[79] Synthesis of compound 2a (wherein Y3=H) by acetal formation of N,N-dimethylformamide (DMF) with dimethylsulfate has been described in Mesnard, D. et. al. J. Organomet. Chem.1989, 373, 1-10. Replacing DMF with N,N-dimethylformamide-d1 (98-99 atom % D; commercially available from Cambridge Isotope Laboratories) in this reaction would thereby provide compound 2b (wherein Y3=D).
[80] Use of appropriately deuterated reagents allows deuterium incorporation at the Y3 position of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at Y3.
[81] Appropriately deuterated intermediates 4a-4d, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents as exemplified in Scheme 3 below.

[82] As described in Malik, M. S. et. al. Org. Prep. Proc. Int.1991, 26, 764-766, acetaldehyde is converted to alkylhalide 14a via reaction with chlorine gas and subsequent acetal protection with CaCl2 in methanol. As described in CN 103739506, reaction of 14a with aqueous ammonia and then sodium hydroxide provides primary amine 4a (wherein Y9=Y10a=Y10b=H). Replacing acetaldehyde with acetaldehyde-d1, acetaldehyde-2,2,2-d3, or acetaldehyde-d4 (all commercially available from CDN Isotopes with 98-99 atom % D) in the sequence then provides access to compounds 4b (Y9=D, Y10a=Y10b=H), 4c (Y9=H,
Y10a=Y10b=D) and 4d (Y9=Y10a=Y10b=D) respectively (Schemes 3b-d).
[83] Use of appropriately deuterated reagents allows deuterium incorporation at the Y9, Y10a, and Y10b positions of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at any Y9, Y10a, and/or Y10b.
[84] Appropriately deuterated intermediates 9a-9d, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents as exemplified in Scheme 4 below.
 [85] Following the procedures described by Gurjar, M. et. al. Heterocycles, 2009, 77, 909-925, meso-diacetate 16a is prepared in 2 steps from cyclopentadiene. Desymmetrization of 16a is then achieved enzymatically by treatment with Lipase as described in Specklin, S. et. al. Tet. Lett.201455, 6987-6991, providing 17a which is subsequently converted to aminocyclopentanol 9a (wherein Y4a=Y4b=Y5a=Y5b=Y6=Y7a=Y7b=Y8=H) via a 3 step sequence as reported in WO 2015195656.
[85] Following the procedures described by Gurjar, M. et. al. Heterocycles, 2009, 77, 909-925, meso-diacetate 16a is prepared in 2 steps from cyclopentadiene. Desymmetrization of 16a is then achieved enzymatically by treatment with Lipase as described in Specklin, S. et. al. Tet. Lett.201455, 6987-6991, providing 17a which is subsequently converted to aminocyclopentanol 9a (wherein Y4a=Y4b=Y5a=Y5b=Y6=Y7a=Y7b=Y8=H) via a 3 step sequence as reported in WO 2015195656.
[86] As depicted in Scheme 4b, aminocyclopentanol 9b (Y4a=Y4b=Y5a=Y5b=Y6=Y7a=Y7b= Y8=D) is obtained through an analogous synthetic sequence using cyclopentadiene-d6 and performing the penultimate hydrogenation with D2 in place of H2. Cyclopentadiene-d6 is prepared according to the procedure described in Cangoenuel, A. et. al. Inorg. Chem.2013, 52, 11859-11866.
[87] Alternatively, as shown in Scheme 4c, the meso-diol obtained in Scheme 4a is oxidized to the diketone following the procedure reported by Rasmusson, G.H. et. al. Org. Syn.1962, 42, 36-38. Subsequent mono-reduction with sodium borodeuteride and CeCl3 then affords the D1-alcohol in analogy to the method described in WO 2001044254 for the all-protio analog using sodium borohydride. Reduction of the remaining ketone using similar conditions provides the meso-D2-diol in analogy to the method reported in Specklin, S. et. al. Tet. Lett.2014, 55, 6987-6991 for the all protio analog using sodium borohydride. The meso-D2-diol is then converted to 9c (Y4a=Y4b=Y5a=Y5b=Y7a=Y7b=H, Y6=Y8=D) following the same procedures outlined in Scheme 4a.
[88] Likewise, the meso-diol obtained in Scheme 4b may be converted to 9d
(Y4a=Y4b=Y5a=Y5b=Y7a=Y7b=D, Y6=Y8=H) in an analogous manner as depicted in Scheme 4d. The use of deuterated solvents such as D2O or MeOD may be considered to reduce the risk of D to H exchange for ketone containing intermediates.
[89] Use of appropriately deuterated reagents allows deuterium incorporation at the Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, and Y8 positions of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at any Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, and/or Y8.
[90] Appropriately deuterated intermediates 11a-11d, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents exemplified in Scheme 5 below.
Scheme 5. Synthesis of Benzylamines 11a-11d

//////////////////
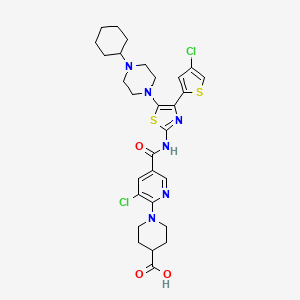
Avatrombopag

| C29H34Cl2N6O3S2 | |
| Molecular Weight: | 649.65466 g/mol |
|---|
Elemental Analysis: C, 53.61; H, 5.28; Cl, 10.91; N, 12.94; O, 7.39; S, 9.87
1-[3-chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)-1,3-thiazol-2-yl]carbamoyl]pyridin-2-yl]piperidine-4-carboxylic acid,
1-(3-Chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)thiazol-2-yl]carbamoyl]pyridin-2-yl)piperidine-4-carboxylic acid,
1-[3-Chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)thiazol-2-yl]carbamoyl]-2-pyridyl]piperidine-4-carboxylic acid
4-Piperidinecarboxylic acid, 1-[3-chloro-5-[[[4-(4-chloro-2-thienyl)-5-(4-cyclohexyl-1-piperazinyl)-2-thiazolyl]amino]carbonyl]-2-pyridinyl]-
Phase III Clinical Trials
Drugs used in platelet disorders
Idiopathic thrombocytopenic purpura (ITP)
small-molecule thrombopoietin receptor (c-Mpl) agonist that stimulates platelet production
INNOVATOR: YAMANOUCHI PHARMACEUTICAL
DEVELOPER: Eisai

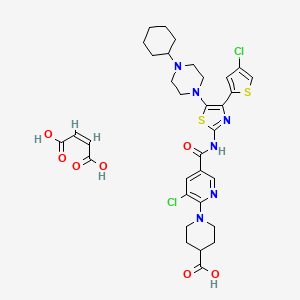
| C33H38Cl2N6O7S2 | |
| Molecular Weight: | 765.72682 g/mol |
|---|
UNIIGDW7M2P1IS
(Z)-but-2-enedioic acid;1-[3-chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)-1,3-thiazol-2-yl]carbamoyl]pyridin-2-yl]piperidine-4-carboxylic acid
INTRODUCTION
Avatrombopag, also known as AKR-501, YM477, AS 1670542 or E5501, is a novel orally-active thrombopoietin (TPO) receptor agonist. AKR-501 specifically targeted the TPO receptor and stimulated megakaryocytopoiesis throughout the development and maturation of megakaryocytes just as rhTPO did. Daily oral administration of AKR-501 dose-dependently increased the number of human platelets in these mice, with significance achieved at doses of 1 mg/kg and above. The peak unbound plasma concentrations of AKR-501 after administration at 1 mg/kg in NOD/SCID mice were similar to those observed following administration of an active oral dose in human subjects. AKR-501 may be useful in the treatment of patients with thrombocytopenia. (source: Eur J Haematol. 2009 Apr;82(4):247-54).

Avatrombopag is a thrombopoietin receptor (c-Mpl) agonist in phase III clinical evaluation at Eisai for the oral treatment of chronic immune thrombocytopenia (idiopathic thrombocytopenia purpura) and for the treatment of thrombocytopenia associated with liver diseases. Phase II studies are ongoing for the treatment of thrombocytopenia during antiviral therapy (inhibition and maintenance) with Interferon for hepatitis C.
The drug candidate may hold potential in treating thrombocytopenia of diverse etiologies, including idiopathic thrombocytopenic purpura (ITP) and thrombocytopenia of myelodysplastic syndromes (MDS), in combination with or as a substitute for platelet transfusion.
AKR-501, a novel, small-molecule thrombopoietin mimetic being investigated for the treatment of thrombocytopenia. AkaRx is now a wholly-owned subsidiary of Eisai Inc. and Eisai has the exclusive worldwide rights to develop, market and manufacture AKR-501. AKR-501 is an investigational thrombopoietin receptor agonist that, based on preclinical studies, increases platelet production by stimulating megakaryocytic proliferation and differentiation. Eisai is currently conducting Phase II clinical trials of AKR-501 in the United States as a potential treatment for idiopathic thrombocytopenic purpura (ITP) and thrombocytopenia associated with liver diseases (TLD), and has confirmed proof of concept in the clinical studies for ITP. In addition, Eisai will explore the compound’s potential as a treatment for chemotherapy-induced thrombocytopenia (CIT).

E-5501 stimulates the production of thrombopoietin (TPO), a glycoprotein hormone that stimulates the production and differentiation of megakaryocytes, the bone marrow cells that fragment into large numbers of platelets. The drug candidate was originally developed at Yamanouchi, and development responsibilities were passed to AkaRx when it was formed in 2005 as a spin-off following the creation of Astellas Pharma subsequent to the merger of Yamanouchi Pharmaceutical and Fujisawa Healthcare.
In 2007, MGI Pharma was granted a license to E-5501 for the treatment of thrombocytopenia. Eisai eventually gained the rights to the product as results of its acquisition of MGI Pharma. In 2010, Eisai acquired AkaRx. AkaRx is now a wholly-owned subsidiary of Eisai Inc. and Eisai has the exclusive worldwide rights to develop, market and manufacture E-5501. In 2011, orphan drug designation was assigned by the FDA for the treatment of idiopathic thrombocytopenic purpura.
E5501 (or AKR-501 or YM477) is a small molecule agonist c-Mpl, orally available. It is in clinical trials for the treatment of chronic idiopathic thrombocytopenic purpura (ITP). It acts as an agonist of the thrombopoietin receptor active orally, mimicking its biological effect. Thrombocytopenic purpura The is the idiopathic consequence of a low number of platelets (thrombocytopenia) of unknown cause. A very low platelets can even lead to purpura (bruises), or bleeding diathesis.
February 2012: A Phase III, multicenter, randomized, double-blind, controlled against placebo, parallel group, with an open-label extension phase to assess the efficacy and safety of combined oral E5501 to standard treatment for the treatment of thrombocytopenia in adults with chronic immune thrombocytopenia, is underway.
January 2010: Eisai Inc. announced its successful acquisition of the biopharmaceutical company, AkaRx Inc. Following this acquisition, AkaRx became a wholly owned subsidiary of Eisai Inc. Eisai now owns the worldwide exclusive rights to develop , marketing and manufacture AKR-501.
October 2009: Eisai Research Institute of Boston, Inc. (established in 1987) and Eisai Medical Research Inc. (established in 2002) were merged into Eisai Inc. 2005: AkaRx was founded as a spin-out of the merger of Yamanouchi Pharmaceutical Company Ltd. and Fujisawa Pharmaceutical Company Ltd. to form Astellas Pharma Inc. AKR-501 was discovered by Yamanouchi and was licensed to AkaRx as part of the foundation of the company in 2005.
In a Phase I trial in healthy volunteers, 10 mg of AKR-501 for 14 days, increased platelet count by 50%.AKR-501 was well tolerated in both studies, mono- and multi-dose. No adverse effects were reported, even at the highest doses.
……………………
Patent
Compound A is a compound of the present invention has the following chemical structure.

That is, compounds useful as a platelet 增多 agent according to the present invention A, as well as medicaments for the Compound A as an active ingredient, in particular increasing platelets agents and Z or thrombocytopenia treating agent.


………………
PATENT

……………………
JP 2014144916/WO 2013018362
https://www.google.co.in/patents/WO2013018362A1?cl=en
1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)thiazol-2-yl]carbamoyl}pyridin-2-yl)piperidine-4-carboxylic acid as expressed by the following chemical formula (hereinafter referred to as “Compound X”) and pharmaceutically acceptable salts are known to have excellent thrombocytosis effects (patent literature 1, patent literature 2).
[Formula 1]

Patent literature 1 discloses a hydrochloride of compound X as example 16 (hereinafter referred to as “compound X hydrochloride”).
Furthermore, patent literature 2 discloses a maleic acid salt of compound X that has endothermic peaks near 198 degree C and 271 degree C in thermo gravimetric analysis (hereinafter referred to as “maleic acid salt of compound X”). However, patent literature 2 neither discloses nor suggests that the maleic acid salt of compound X exhibits crystal polymorphism.
On the other hand, compounds exhibiting crystal polymorphism demonstrate entirely different effects regardless of being the same compound, because various physical properties including physicochemical properties differ depending on the crystalline form. In pharmaceutical products in particular, if compounds that have different functional effects are expected to have the same effect, a different functional effect than expected will occur, which is thought to induce unexpected circumstances, and therefore there is demand for supply of a drug substance with constant quality. Therefore, when a compound which has crystal polymorphism is used as a medicine, one type of crystal of that compound must always be constantly provided in order to ensure constant quality and constant effects that are required of the medicine.
Under the aforementioned conditions, from the perspective of supplying a drug substance for medicines, there is a need for compound X or crystals of pharmaceutically acceptable salts thereof, which can ensure constant quality and constant effects and which can be stably supplied in mass production such as industrial production or the like, as well as for establishment of a manufacturing method thereof.
International patent publication WO 03/062233 International patent publication WO 2004/029049
The crystals of compound X maleic acid salt disclosed in patent literature 2 (hereinafter referred to as “compound X maleic acid salt A type crystals”) cannot be isolated as compound X maleic acid salt A type crystals when scaled up for mass production using the method disclosed in example 1 of patent literature 2, and therefore must be isolated in a different crystal form. (This other crystal form is referred to as “compound X maleic acid salt B type crystals”). Therefore, the compound X maleic acid salt A type crystals have a possibility that the crystal form will morph depending on the scale of production, and is clearly inappropriate as a drug substance for medicines which require constant quality and constant effects.
Preparation Example 1: Manufacture of Compound X Maleic Acid Salt B Type Crystal
310 mL of a 1 M aqueous solution of sodium hydroxide at room temperature was added to a mixture of 70.0 g of the ethyl ester of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid and 1.2 L of ethanol, the insoluble matter was filtered out, and then washed with 200 mL of ethanol. The reaction solution was stirred for 90 minutes at 60 degree C. After cooling to room temperature, 1.4 L of an aqueous solution containing 24.11 g of maleic acid was added to the solution obtained, and then the precipitate was collected by filtering.
The same operation was repeated and when combined with the previously obtained precipitate, 136.05 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid was obtained.
18.9 g of maleic acid and 2.1 L of 80% ethanol water were added to 88.90 g of the carboxylic acid obtained, and the solution was stirred for one hour at room temperature and for another hour at 100 degree C. After cooling to room temperature and further cooling with ice, the precipitated solid was filtered out to obtain 87.79 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt as a crude product.
6.84 g of maleic acid was added to 231 g of the crude product containing the crude product obtained above and those manufactured in a similar manner, dissolved in 5.5 L of 80% ethanol water, and then the precipitated solid was collected by filtering to obtain 203 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
Example 1: Manufacture of Compound X Maleic Acid Salt C Type Crystals (1)
1.52 L of ethanol, 0.38 L of water, and 15.7 g of maleic acid were added to 78.59 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid, and heated while stirring. After cooling to room temperature and further cooling with ice, the precipitated solid was collected by filtering to obtain 71.60 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt as a crude product.
296 mg of maleic acid was added to 10.0 g of the crude product obtained, dissolved in 60 mL of acetone, 60 mL of DMSO, and 30 mL of water, and then the precipitated solids were collected to obtain 8.41 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
Example 2: Manufacture of Compound X Maleic Acid Salt C Type Crystals (2)
A mixture containing 80.1 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid, 580 mL of DMSO, 580 mL of acetone, 17.2 g of maleic acid, and 290 mL of water was stirred at 69 degree C. The insoluble matter was filtered out, washed with a mixture of 32 mL of DMSO, 32 mL of acetone, and 16 mL of water, and then the filtrate was cooled and the precipitate was collected by filtering. Washing was successively performed using 150 mL of water, 80 mL of acetone, 650 mL of water, and 80 mL of acetone, followed by drying, to obtain 70.66 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
Example 3: Manufacture of Compound X Maleic Acid Salt C Type Crystals (3)
A mixture containing 20 kg of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid, 100 L of DMSO, 100 L of acetone, 4.29 kg of maleic acid, and 50 L of water is stirred at 65 degree C, and then the insoluble matter is filtered out and washed with a mixture of 8 L of DMSO, 8 L of acetone, and 4 L of water, and then the filtrate is cooled, the precipitate is collected by filtering, successively washed using 40 L of acetone, 100 L of water, and 40 L of acetone, and then dried to obtain approximately 20 kg of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
…………………………….

REFERENCES
Garabet, L.; Ghanima, W.; Lee, S.; Mowinckel, M.C.; Liebman, H.; Jonassen, C.M.; Bussel, J.; Sandset, P.M.
Thrombopoietin receptor agonists do no not cause coagulation activation: In patients with immune thrombocytopenia
25th Congr Int Soc Thromb Haemost (ISTH) (June 20-25, Toronto) 2015, Abst PO311-MON
Terrault, N.; Hassanein, T.; Joshi, S.; Lake, J.R.; Sher, L.S.; Vargas, H.E.; McIntosh, J.W.; Tang, S.; Jenkins, T.
Once-daily oral avatrombopag (E5501) prior to elective surgical or diagnostic procedures in patients with chronic liver disease and thrombocytopenia: Results from a phase 2, randomized, double-blind, placebo-controlled study (study 202)
63rd Annu Meet Am Assoc Study Liver Dis (November 9-13, Boston) 2012, Abst
Thiophenyl Triazol-3-one Derivatives As Smooth Muscle relaxers: US6613786 (2003) Priority: US20010336865P, Nov. 2, 2001 (Bristol-Myers Squibb CO, US)
Preparation Of Avatrombopag: 2-Acylaminothiazole derivative or salt thereof: EP1466912 (2004) Priority: JP20020010413, 18 Jan. 2002 (Yamanouchi Pharma Co Ltd, Japan)
Synthesis And Use Of MSE Framework-Type Molecular Sieves: US2009318696 (2009) Priority: US20080214631 20 Jun. 2008 (Exxon Mobil, US).
5,6-Dichloro-Nicotinic Acid Production By Reacting 6-Hydroxy-Nicotinic Acid With Acid Chloride Reacting With Chlorine Products, Then With Acid Chloride And Hydrolysing Products: CH664754 (1988) Priority: CH19850002692, 25 Jun. 1985 (Lonza AG, Switzerland).
David J. Kuter, New Thrombopoietic Growth Factors, Lymphoma and Myeloma Clinical Journal Volume 9, Supplement 3, S347-S356
| WO2003062233A1 | 15 Jan 2003 | 31 Jul 2003 | Yamanouchi Pharma Co Ltd | 2-acylaminothiazole derivative or salt thereof |
| WO2004029049A1 | 29 Sep 2003 | 8 Apr 2004 | Yuuji Awamura | Novel salt of 2-acylaminothiazole derivative |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| EP2764866A1 | 4 Feb 2014 | 13 Aug 2014 | IP Gesellschaft für Management mbH | Inhibitors of nedd8-activating enzyme |
| Patent | Submitted | Granted |
|---|---|---|
| CANCER TREATMENT METHOD [US2011160130] | 2011-06-30 | |
| METHOD FOR STIMULATING PLATELET PRODUCTION [US2011166112] | 2011-07-07 | |
| COMPOSITIONS AND METHODS FOR INCREASING BLOOD PLATELET LEVELS IN HUMANS [US2011224226] | 2011-09-15 | |
| Method of treating viral diseases with combinations of TPO receptor agonist and anti-viral agents [US2012020923] | 2012-01-26 |
| Patent | Submitted | Granted |
|---|---|---|
| 2-Acylaminothiazole derivative or salt thereof [US7638536] | 2005-07-14 | 2009-12-29 |
| Compositions and methods for treating thrombocytopenia [US2007203153] | 2007-08-30 | |
| Novel Combinations [US2009304634] | 2009-12-10 | |
| 2-ACYLAMINOTHIAZOLE DERIVATIVE OR SALT THEREOF [US2010222329] | 2010-09-02 | |
| 2-ACYLAMINOTHIAZOLE DERIVATIVE OR SALT THEREOF [US2010222361] | 2010-09-02 | |
| Compositions and methods for increasing blood platelet levels in humans [US2008039475] | 2008-02-14 | |
| CANCER TREATMENT METHOD [US2009022814] | 2009-01-22 | |
| Compositions and methods for treating thrombocytopenia [US2010041668] | 2010-02-18 | |
| CANCER TREATMENT METHOD [US2010075928] | 2010-03-25 |
///////E 5501, AKR 501, Phase III, eisai, Avatrombopag, y 477, orphan drug, ym 477, AS 1670542, Yamanouchi Pharma Co Ltd, Japan
UPDATE MAY 2018
Avatrombopag
https://newdrugapprovals.org/2015/08/24/avatrombopag/
The U.S. Food and Drug Administration today approved Doptelet (avatrombopag) tablets to treat low blood platelet count (thrombocytopenia) in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure. This is the first drug approved by the FDA for this use.Continue reading.
May 21, 2018
Release
The U.S. Food and Drug Administration today approved Doptelet (avatrombopag) tablets to treat low blood platelet count (thrombocytopenia) in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure. This is the first drug approved by the FDA for this use.
“Patients with chronic liver disease who have low platelet counts and require a procedure are at increased risk of bleeding,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Doptelet was demonstrated to safely increase the platelet count. This drug may decrease or eliminate the need for platelet transfusions, which are associated with risk of infection and other adverse reactions.”
Platelets (thrombocytes) are colorless cells produced in the bone marrow that help form blood clots in the vascular system and prevent bleeding. Thrombocytopenia is a condition in which there is a lower-than-normal number of circulating platelets in the blood. When patients have moderately to severely reduced platelet counts, serious or life-threatening bleeding can occur, especially during invasive procedures. Patients with significant thrombocytopenia typically receive platelet transfusions immediately prior to a procedure to increase the platelet count.
The safety and efficacy of Doptelet was studied in two trials (ADAPT-1 and ADAPT-2) involving 435 patients with chronic liver disease and severe thrombocytopenia who were scheduled to undergo a procedure that would typically require platelet transfusion. The trials investigated two dose levels of Doptelet administered orally over five days as compared to placebo (no treatment). The trial results showed that for both dose levels of Doptelet, a higher proportion of patients had increased platelet counts and did not require platelet transfusion or any rescue therapy on the day of the procedure and up to seven days following the procedure as compared to those treated with placebo.
The most common side effects reported by clinical trial participants who received Doptelet were fever, stomach (abdominal) pain, nausea, headache, fatigue and swelling in the hands or feet (edema). People with chronic liver disease and people with certain blood clotting conditions may have an increased risk of developing blood clots when taking Doptelet.
This product was granted Priority Review, under which the FDA’s goal is to take action on an application within six months where the agency determines that the drug, if approved, would significantly improve the safety or effectiveness of treating, diagnosing or preventing a serious condition.
The FDA granted this approval to AkaRx Inc.
//////////////Doptelet, avatrombopag, fda 2018, akarx, priority review,
Lusutrombopag….Oral thrombopoietin (TPO) mimetic
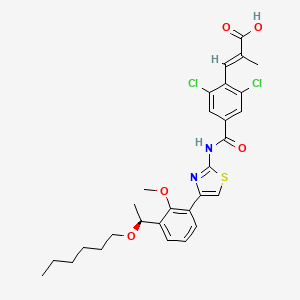
Lusutrombopag
Update…..FDA approved july2018
(E)-3-[2,6-dichloro-4-[[4-[3-[(1S)-1-hexoxyethyl]-2-methoxyphenyl]-1,3-thiazol-2-yl]carbamoyl]phenyl]-2-methylprop-2-enoic acid
(S)-(-)-(E)-3-(2,6-dichloro-4-{4-[3-(1-hexyloxyethyl)-2-methyloxyphenyl]thiazol-2-ylcarbamoyl}phenyl)-2-methylacrylic acid
(2E)-3-{2,6-Dichloro-4-[(4-{3-[(1S)-1-(hexyloxy)ethyl]-2-methoxyphenyl}-1,3-thiazol-2-yl)carbamoyl]phenyl}-2-methylacrylic acid
UNII 6LL5JFU42F, CAS 1110766-97-6,
D10476, MW591.546 , [US2010267783], MF C29H32Cl2N2O5S, S-888711
Shionogi & Co., Ltd., 塩野義製薬株式会社 INNOVATOR
Optically active compound (C-3B) Melting point: 142-145°C…………….EP2184279B1
NMR (DMSO-d6) δ ppm: 12.97 (brs, 1H), 8.29 (s, 2H), 7.90 (dd, 1H, J = 1.8 Hz, 7.5 Hz), 7.72 (s, 1H), 7.35 – 7.40 (m, 2H), 7.26 (t, 1H, J = 7.5 Hz), 4.82 (q, 1H, J = 6.3 Hz), 3.62 (s, 3H), 3.16 – 3.37 (m, 2H), 1.69 (s, 3H), 1.18 – 1.51 (m, 11H), 0.82-0.87 (m, 3H) Optical rotation -4.5 degrees (DMSO, c = 1.001, 25°C)………….EP2184279B1
Optical rotation: -7.0 ± 0.5 degrees (CHCl3, c = 1.040, 21°C), NMR (CDCl3) δ ppm: 0.87 (3H, t, J = 6.8 Hz), 1.2 – 1.4 (6H, m), 1.48 (3H, d, J = 6.4 Hz), 1.52 – 1.64 (2H, m), 1.86 (3H, d, J = 1.4Hz)), 3.35 (2H, t, J = 6.7Hz), 3.55 (3H, s), 4.87 (1H, q, J = 6.3 Hz), 7.25 (1H, t, J = 7.7 Hz), 7.41 (1H, s), 7.49 (1H, dd, J = 7.9 Hz, J = 1.6 Hz), 7.51 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.65 (1H, d, J = 1.4 Hz), 8.33 (2H, s), 13.4 (2H, brs)………EP2184279B1
Thrombopoietin receptor agonist, Oral thrombopoietin (TPO) mimetic
- 24 Mar 2015 Shionogi plans a phase III trial in Thrombocytopenia (in patients with chronic liver disease) in USA (NCT02389621)
- 31 Dec 2014 Preregistration for Thrombocytopenia in Japan (PO)
- 08 Nov 2013 Phase II development is ongoing in the US and the Europe
Process for preparing intermediates of an optically active 1,3-thiazole containing thrombopoietin receptor agonist Also claims crystalline forms of lusutrombopag intermediates and a process for preparing lusutrombopag. Shionogi is developing lusutrombopag, a small-molecule thrombopoietin mimetic, as an oral tablet formulation for treating thrombocytopenia.
In December 2014, an NDA was submitted in Japan. In May 2015, the drug was listed as being in phase III development for thrombocytopenia in the US and Europe.

The lusutrombopag, a low molecular-human thrombopoietin receptor agonist, its chemical formula, “(E) -3- [2,6-Dichloro-4- [4- [3 – [(S) -1-hexyloxyethyl] – 2-methoxyphenyl] -thiazol- 2-ylcarbamoyl] -phenyl] is a -2-methylacrylic acid “. lusutrombopag is represented by the following chemical structural formula.

Eltrombopag is represented by the following chemical structural formula.

Avatrombopag is represented by the following chemical structural formula.
Totrombopag choline is represented by the following chemical structural formula.



Optically active compound (C-3B)Melting point: 142-145°C DESIRED
NMR (DMSO-d6) δ ppm: 12.97 (brs, 1H), 8.29 (s, 2H), 7.90 (dd, 1H, J = 1.8 Hz, 7.5 Hz), 7.72 (s, 1H), 7.35 – 7.40 (m, 2H), 7.26 (t, 1H, J = 7.5 Hz), 4.82 (q, 1H, J = 6.3 Hz), 3.62 (s, 3H), 3.16 – 3.37 (m, 2H), 1.69 (s, 3H), 1.18 – 1.51 (m, 11H), 0.82-0.87 (m, 3H) Optical rotation -4.5 degrees (DMSO, c = 1.001, 25°C)
Example 4: Synthesis of (C-3B)

First step: Synthesis of (S)-1-(3-bromo-2-methyloxyphenyl)ethane-1-ol (17)
Using the same method as that of the first step of Example 3, the compound (17) was obtained from the compound (16) at a yield 77%.
-
-
Optical rotation: -23.5 ± 0.6 degrees (CHCl3, c = 1.050, 21°C)
NMR (CDCl3) θ ppm: 1.49 (3H, d, J = 6.6 Hz), 2.33 (1H, brs), 3.88 (3H, s), 5.19 (1H, q, J = 6.4 Hz), 7.01 (1H, t, J = 7.9 Hz), 7.40 (1H, dd, J = 7.7 Hz, J = 1.1 Hz), 7.46 (1H, dd, J = 8.0 Hz, J = 1.4 Hz)
-
Second step: Synthesis of (S)-1-bromo-3-(1-hexyloxyethyl)-2-methyloxybenzene (18)
-
-
Using the same method as that of the second step of Example 3, the compound (18) was obtained from the compound (17) at a yield of 96%.
Optical rotation: -29.8 ± 0.6 degrees (CHCl3, c = 1.055, 21°C)
NMR (CDCl3) δ ppm: 0.87 (3H, t, J = 6.8 Hz), 1.2 – 1.4 (6H, m), 1.42 (3H, d, J = 6.5 Hz), 1.54 (2H, m), 3.29 (2H, m), 3.85 (3H, s), 4.78 (1H, q, J = 6.4 Hz), 7.02 (1H, t, J = 7.9 Hz), 7.39 (1H, dd, J = 7.8 Hz, J = 1.7 Hz), 7.45 (1H, dd, J = 7.9 Hz, J = 1.7 Hz)
-
Third step and fourth step: Synthesis of (S)-4-(3-(1-hexyloxyethyl)-2-methyloxyphenyl)thiazole-2-amine (20)
-
-
Using the same method as that of the fourth step of Example 3, the compound (19) was obtained from the compound (18), subsequently according to the same method as that of the fourth step, the compound (20) was obtained.
-
Compound (19)
-
-
NMR (CDCl3) δ ppm: 0.87 (3H, t, J = 6.9 Hz), 1.2-1.4 (6H, m), 1.45 (3H, d, J = 6.6 Hz), 1.55 (2H, m), 3.29 (2H, m), 3.78 (3H, s), 4.73 (2H, m), 4.80 (1H, q, J = 6.4 Hz), 7.24 (1H, t, J = 7.8Hz), 7.52 (1H, dd, J = 7.7 Hz, J = 1.8 Hz), 7.65 (1H, dd, J = 7.7 Hz, J = 1.8 Hz)
-
Compound (20)
-
Optical rotation: -4.2 ± 0.4 degrees (DMSO, c = 1.025, 21°C)
NMR (CDCl3) δ ppm: 0.84 (3H, t, J = 7.0 Hz), 1.2 – 1.3 (6H, m), 1.35 (3H, d, J = 6.5 Hz), 1.48 (2H, m), 3.25 (2H, m), 3.61 (3H, s), 4.78 (1H, q, J = 6.4 Hz), 6.99 (2H, brs), 7.05 (1H, s), 7.16 (1H, t, J = 7.7 Hz), 7.27 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.81 (1H, dd, J = 7.6 Hz, J = 1.9 Hz) -
Fifth step: Synthesis of ethyl (S)-(E)-3-(2,6-dichloro-4-(4-(3-(1-hexyloxyethyl)-2-metyloxyphenyl)thiazol-2-ylcarbamoyl)phenyl)-2-methylacrylate (21)
-
-
Using the same method as that of the fifth step of Example 3, the compound (21) was obtained from the compound (20) at a yield of 94%.
Optical rotation: +4.7 ± 0.4 degrees (CHCl3, c = 1.07, 21°C)
NMR (CDCl3 ) δ ppm: 0.87 (3H, t, J = 6.9 Hz), 1.2 – 1.35 (6H, m), 1.38 (3H, t, J = 7.1
Hz), 1.44 (3H, d, J = 6.4 Hz), 1.57 (2H, m), 1.77 (3H, d, J = 1.4 Hz), 3.30 (2H, m), 3.59 (3H, s), 4.31 (2H, q, J = 7.1 Hz), 4.83 (1H, q, J = 6.4 Hz), 7.17 (1H, t, J = 7.7 Hz), 7.42 (1H, d, J = 1.7 Hz), 7.42 (1H, dd, J = 7.7 Hz, J = 1.8 Hz), 7.51 (1H, s), 7.67 (1H, dd, J = 7.6 Hz, J = 1.7 Hz), 7.89 (2H, s), 10.30 (1H, brs)
-
Sixth step: Synthesis of (S)-(E)-3-(2,6-dichloro-4-(4-(3-(1-hexyloxyethyl)-2-metyloxyphenyl)thiazol-2-ylcarbamoyl)phenyl)-2-methylacrylic acid (C-3B)
-
Using the same method as that of the sixth step of Example 3, the compound (C-3B) was obtained from the compound (21) at a yield of 80%.Optical rotation: -7.0 ± 0.5 degrees (CHCl3, c = 1.040, 21°C)
NMR (CDCl3) δ ppm: 0.87 (3H, t, J = 6.8 Hz), 1.2 – 1.4 (6H, m), 1.48 (3H, d, J = 6.4 Hz), 1.52 – 1.64 (2H, m), 1.86 (3H, d, J = 1.4Hz)), 3.35 (2H, t, J = 6.7Hz), 3.55 (3H, s), 4.87 (1H, q, J = 6.3 Hz), 7.25 (1H, t, J = 7.7 Hz), 7.41 (1H, s), 7.49 (1H, dd, J = 7.9 Hz, J = 1.6 Hz), 7.51 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.65 (1H, d, J = 1.4 Hz), 8.33 (2H, s), 13.4 (2H, brs) -
Results of powder X-ray deffraction are shown in Fig. 5.
-
Diffraction angle of main peak: 2θ = 17.8, 21.1, 22.5, 23.3, 24.1, and 24.4 degrees
WO2005014561/EP1655291A1
https://www.google.co.in/patents/EP1655291A1?cl=en
WO2014003155, claiming a composition comprising lusutrombopag, useful for treating thrombocytopenia.
https://www.google.co.in/patents/US20150148385?cl=en
.
Methods respectively for producing optically active compound having agonistic activity on thrombopoietin receptors and intermediate of said compound
(Step 1) Synthesis of compound (VII ‘) under a nitrogen atmosphere, it was dissolved compound 1 (2.00kg) in 1,2-dimethoxyethane (28.0kg). 25% LDA tetrahydrofuran – heptane – ethyl benzene solution (13.20kg) was added dropwise over 1 hour at -55 ℃, and stirred for 30 minutes. It was added dropwise over 40 minutes to 1,2-dimethoxyethane (3.0kg) solution of N- formyl morpholine (3.74kg) at -55 ℃, and stirred for 1 hour. 1,2-dimethoxyethane (3.0kg) solution of 2-phosphono-propanoic acid triethyl (3.74kg) was added dropwise over 45 minutes at 0 ℃, and stirred for 2 hours. 35% aqueous solution of sulfuric acid (15.8kg) was added dropwise over 40 minutes to the reaction solution. Water (16.0kg) was added and extracted. The resulting organic layer was washed with water (8.0kg), and the solvent was evaporated under reduced pressure. Acetonitrile (16.0kg) was added, and the mixture was stirred for 1 hour at 25 ℃, and the mixture was stirred and cooled to 0 ℃ 5 hours and 30 minutes. The precipitated crystals were collected by filtration, and washed with 5 ℃ acetonitrile (3.2kg). The resulting crystals it was dissolved in acetonitrile (16.0kg) at 75 ℃. It was cooled to 60 ℃, and the mixture was stirred for 30 minutes. Over 1 hour and then cooled to 30 ℃, and the mixture was stirred for 45 minutes. Over 40 minutes and then cooled to 5 ℃, and the mixture was stirred for 3 hours.The precipitated crystals were collected by filtration, and washed with 5 ℃ acetonitrile (3.2kg). The resulting crystals it was dissolved in acetonitrile (13.0kg) at 75 ℃. It was cooled to 60 ℃, and the mixture was stirred for 30 minutes. Furthermore, up to 30 ℃ over 1 hour and then cooled and stirred for 70 minutes. Over 30 minutes and then cooled to 5 ℃, and the mixture was stirred for 4 hours. I precipitated crystals were collected by filtration. Washed with 5 ℃ acetonitrile (3.2kg), and dried to give the compound (VII ‘) (1.63kg, 51.2% yield). NMR (CDCl 3 ) delta ppm: 8.07 (s, 2H), 7.47 (s, 1H), 4.32 (Q, 2H, J = 7.0 Hz), 1.79 (s, 3H), 1.38 (t, 3H, J = 7.0 Hz) Results of powder X-ray diffraction and I shown in Figure 1 and Table 3. [Table 3] In the powder X-ray diffraction spectrum, diffraction angle (2θ): 8.1 ± 0.2 °, 16.3 ± 0.2 °, 19.2 ± 0.2 °, 20.0 ± 0. 2 °, the peak was observed at 24.8 ± 0.2 °, and 39.0 ± 0.2 ° degrees.
(Synthesis of Compound (XI ‘))
(Step 2) Synthesis of Compound 4 under a nitrogen atmosphere over Compound 3 (3.00kg) and 1mol / L isopropylmagnesium chloride in tetrahydrofuran (11.40kg) 1 hour at 25 ℃ in The dropped, and stirred for 2 hours. 1mol / L isopropylmagnesium chloride in tetrahydrofuran solution (0.56kg) was added at 25 ℃, and stirred for 2 hours. To the reaction mixture N- methoxymethyl -N- methylacetamide the (1.45kg) was added dropwise over at 25 ℃ 40 minutes, and stirred for 80 minutes. 7% hydrochloric acid (9.7kg) was added to the reaction mixture, and the mixture was extracted with toluene (11.0kg). The resulting organic layer twice with water (each 7.5kg) washed, the solvent was evaporated under reduced pressure to give Compound 4 (2.63kg). NMR (CDCl 3 ) delta ppm: 7.69 (dd, 1H, J = 7.7 Hz, J = 1.5 Hz), 7.55 (dd, 1H, J = 7.7 Hz, J = 1.5 Hz), 7.05 (t, 1H, J = 7.7 Hz), 3.88 (s, 3H), 2.64 (s, 3H) ppm:
(Step 3) Synthesis of Compound 5 Under a nitrogen atmosphere, chloro [(1S Compound 4 (2.63kg), 2S) -N- ( p- toluenesulfonyl) -1,2-diphenyl-ethane diamine] (p- cymene) ruthenium (II) (28.6g), it was added to tetrahydrofuran (1.3kg) and triethylamine (880.0g). Formic acid (570.0g) was added dropwise over 6 hours at 40 ℃, and stirred for 1 hour. In addition 3.5% hydrochloric acid (14.4kg) to the reaction mixture, and the mixture was extracted with toluene (13.0kg).The organic layer was washed with 3.5% hydrochloric acid (14.4kg) and water (7.5kg), the solvent was concentrated under reduced pressure to obtain a toluene solution of Compound 5 (4.44kg).
(Step 4) Synthesis of Compound 6 under a nitrogen atmosphere, it was a potassium hydroxide (6.03kg) was dissolved in water (6.0kg). To the solution, it added tetrabutylammonium bromide (182.0g) and toluene solution of Compound 5 (4.44kg). 1-bromo-hexane (2.79kg) was added dropwise over 1 hour at 60 ℃, and the mixture was stirred for 4 hours. And extracted by adding water (4.4kg) to the reaction solution. The resulting organic layer was filtered through powdered cellulose and extracted with toluene (3.0kg) and water (7.6kg) to the filtrate. The solvent it was evaporated under reduced pressure from the organic layer. Toluene operation of evaporated under reduced pressure and the solvent by the addition of a (7.8kg) was repeated five times to obtain a toluene solution of Compound 6 (10.0kg).
(Step 5) Synthesis of Compound 7 under a nitrogen atmosphere, magnesium powder (301.0g), in tetrahydrofuran (1.3kg), the compound in toluene (6.4kg) and 1mol / L isopropylmagnesium chloride in tetrahydrofuran (432.0g) 6 In addition of the toluene solution (0.50kg) at 30 ℃, and the mixture was stirred for 2 hours. Toluene solution of Compound 6 (9.50kg) was added dropwise over 3 hours at 50 ℃, and stirred for 2 hours. 1-bromo-hexane (746.0g) was added at 50 ℃, and the mixture was stirred for 1 hour. It was added dropwise over 1 hour at 5 ℃ toluene (5.3kg) solution of 2-chloro -N- methoxy -N- methyl-acetamide (1.78kg), and stirred for 1 hour. 3.7% hydrochloric acid (16.7kg) was added to the reaction mixture, and the mixture was extracted. The obtained organic layer was washed with water (15.0kg), and concentrated under reduced pressure to give a toluene solution of Compound 7 (8.25kg).
(Step 6) Synthesis of Compound (II ‘) under a nitrogen atmosphere, thiourea (1.03kg), in ethanol (1.2kg) and 65 ℃ toluene solution of compound 7 (8.25kg) in toluene (6.3kg) over 3 hours was added dropwise and stirred for 2 hours. The reaction solution was extracted by adding 0.7% hydrochloric acid (30.6kg), and washed twice with water (30.0kg). Ethanol in the organic layer (9.5kg), and extracted by addition of heptane (10.0kg) and 3.5% hydrochloric acid (5.9kg). The resulting aqueous layer with 4% hydrochloric acid (1.5kg) and ethanol (3.5kg) merged the aqueous layer was extracted from the organic layer, the ethanol was washed with heptane (10.0kg) (3.1kg) It was added. 8% aqueous sodium hydroxide (6.0kg) was added dropwise over at 5 ℃ 30 minutes, and stirred for 20 minutes. 8% aqueous sodium hydroxide (5.8kg) was added dropwise over a period at 5 ℃ 15 minutes.The precipitated crystals were collected by filtration, washed with 45% aqueous ethanol (10.9kg) and water (15.0kg) (crude crystals of Compound (II ‘)). The resulting crude crystals were dissolved in 50 ℃ in ethanol (8.1kg), over a period of 1 hour and then cooled to 10 ℃, and the mixture was stirred for 30 minutes. Water (10.0kg) over 2 hours was added dropwise and stirred for 30 minutes. The precipitated crystals were collected by filtration, washed with 50% aqueous ethanol (7.5kg) and water (10.0kg) (crystals of the compound after recrystallization from ethanol / water system (II ‘)). The resulting crystals were dissolved at 55 ℃ in toluene (1.6kg) and heptane (1.3kg), over 1 hour and cooled to 20 ℃, and stirred for 30 minutes. Heptane (6.3kg) over a period of 30 minutes was added dropwise and stirred for 15 minutes. The obtained crystals precipitated were collected by filtration, washed with a mixed solvent of toluene (0.3kg) and heptane (2.3kg), and dried to give compound (II ‘) (1.67kg, 44.5% yield) a (crystalline compound after recrystallization from toluene / heptane system (II ‘)).
NMR (CDCl 3 ) delta ppm: 0.84 (3H, t, J = 7.0 Hz), 1.2 – 1.3 (6H, M), 1.35 (3H, D, J = 6.5 Hz), 1.48 (2H, M), 3.25 ( 2H, m), 3.61 (3H, s), 4.78 (1H, q, J = 6.4 Hz), 6.99 (2H, brs), 7.05 (1H, s), 7.16 (1H, t, J = 7.7 Hz), 7.27 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.81 (1H, dd, J = 7.6 Hz, J = 1.9 Hz) it is shown in Figure 2 and Table 4 the results of powder X-ray diffraction. [Table 4] In the powder X-ray diffraction spectrum, diffraction angle (2θ): 12.5 ± 0.2 °, 13.0 ± 0.2 °, 13.6 ± 0.2 °, 16.4 ± 0. 2 °, 23.0 ± 0.2 °, a peak was observed at 24.3 ± 0.2 ° degrees. Above, each of the compounds (II ‘) of the crude crystals, the ethanol / compound after recrystallization from water (II’) crystals and toluene / heptane compound after recrystallization from (II ‘) crystallographic purity of the results of the , Fig. 3, I 4 and 5 as well as Table 5. [Table 5](HPLC was measured by the above method A.) As shown in the results of the above table, as compared to recrystallization from ethanol / water, recrystallized with toluene / heptane system, compounds having a high optical purity it is possible to manufacture a crystal of (II ‘). Next, the above-mentioned compound (II ‘) of the crude crystals, the ethanol / compound after recrystallization from water (II’) crystals and toluene / heptane compound after recrystallization from (II ‘) results of crystals of HPLC of the respectively, Fig. 6, I 7 and 8 and Table 6. [Table 6] (units, .N.D shows the peak area of the (%). is, .HPLC to indicate not detected was measured by the above method B.) As shown in the results of Table, with ethanol / water system Compared to recrystallization, recrystallization from toluene / heptane system is found to be efficiently remove organic impurities A and organic impurities B.
(Step 7) Compound ‘Synthesis of DMSO adduct of (VIII) Under a nitrogen atmosphere, the compound (II ‘) (1.50kg) and compound (VII’) (1.43kg) in ethyl acetate (17.6kg) and triethylamine (1.09kg) were sequentially added, was dissolved.Diphenyl phosphorochloridate the (1.46kg) was added dropwise over 1 hour at 50 ℃, and the mixture was stirred for 3 hours. The reaction mixture was cooled to 25 ℃, after the addition of 2.6% hydrochloric acid (8.1kg), and extracted. The resulting organic layer to 6.3% aqueous solution of sodium hydroxide (3.2kg) and 14% aqueous sodium carbonate (5.2kg) was added and stirred for 20 minutes. Adjusted to pH7.5 with 8.3% hydrochloric acid and extracted. The organic layer it was washed with 4.8% sodium chloride aqueous solution (11.0kg). DMSO and (16.5kg) was added, and the mixture was concentrated under reduced pressure.DMSO and (5.8kg) was added, over a period at 40 ℃ 30 minutes was added dropwise water (0.9kg), and stirred for 1 hour. Over a period of 30 minutes, cooled to 25 ℃, and the mixture was stirred for 30 minutes. Over at 25 ℃ 30 minutes was added dropwise water (1.4kg), and the precipitated crystals were collected by filtration. After washing with 90% DMSO solution (10.0kg) and water (27.0kg), to obtain crystals of DMSO adduct and dried to Compound (VIII ‘) (2.98kg, 95.2% yield).
1H-NMR (CDCl 3 ) delta: 0.87 (t, J = 6.8 Hz, 3H), 1.20-1.34 (M, 6H), 1.37 (t, J = 7.1 Hz, 3H), 1.44 (D, J = 6.5 Hz , 3H), 1.52-1.59 (m, 2H), 1.77 (d, J = 1.3Hz, 3H), 2.62 (s, 6H), 3.28-3.34 (m, 2H), 3.59 (s, 3H), 4.31 ( q, J = 7.1Hz, 2H), 4.83 (q, J = 6.5Hz, 1H), 7.16 (t, J = 7.7Hz, 1H), 7.40-7.43 (m, 2H), 7.51 (s, 1H), 7.68 (dd, J = 7.7, 1.8Hz, 1H), 7.92 (d, J = 1.3Hz, 2H), 10.58 (s, 1H). The results of the powder X-ray diffraction and I are shown in Figure 9 and Table 7. [Table 7]
In the powder X-ray diffraction spectrum, diffraction angle (2θ): 5.2 ° ± 0.2 °, 7.0 ° ± 0.2 °, 8.7 ° ± 0.2 °, 10.5 ° ± 0.2 °, 12.3 ° ± 0.2 °, 14.0 ° ± 0.2 °, 15.8 ° ± 0.2 °, 19.3 ° ± 0.2 °, 22.5 ° peak was observed to ± 0.2 ° and 24.1 ° ± 0.2 °. TG / DTA analysis result it is shown in Figure 10. Then, each result of HPLC of concentrated dry solid and the above DMSO adduct crystals described in the following Reference Examples 1, 11 and 12, 13 and 14, and I are shown in Table 8. [Table 8] (unit, .HPLC showing peak areas of (%) was measured by the above methods C.) As shown in the results of the above Table, when compared with the extract, DMSO adduct of the compound (VIII ‘) The in the crystal, less residual organic impurities D, and it found to be about 56% removal.
(Step 8) under nitrogen atmosphere, DMSO adduct of the compound (VIII ‘) and (2.50kg) it was dissolved in ethanol (15.8kg). 24% sodium hydroxide aqueous solution (1.97kg) was added dropwise over a period at 45 ℃ 30 minutes to the solution and stirred for 3 hours. The reaction mixture was cooled to 25 ℃, water was added (20.0kg) and ethanol (7.8kg). 18% hydrochloric acid (2.61kg) was added dropwise over at 25 ℃ 30 minutes, followed by addition of seed crystals prepared according to the method described in Patent Document 23. After stirring for 3 hours and allowed to stand overnight. Thereafter, the precipitated crystals were collected by filtration, to give after washing with 50% aqueous ethanol solution (14.2kg), and dried to a compound (XI ‘) (1.99kg, 93.9% yield).
NMR (CDCl 3 ) delta ppm: 0.87 (3H, t, J = 6.8 Hz), 1.2 – 1.4 (6H, M), 1.48 (3H, D, J = 6.4 Hz), 1.52 – 1.64 (2H, M), 1.86 (3H, d, J = 1.4Hz), 3.35 (2H, t, J = 6.7Hz), 3.55 (3H, s), 4.87 (1H, q, J = 6.3 Hz), 7.25 (1H, t, J = 7.7 Hz), 7.41 (1H, s), 7.49 (1H, dd, J = 7.9 Hz, J = 1.6 Hz), 7.51 (1H, dd, J = 7.5 Hz, J = 1.8 Hz), 7.65 (1H, d, J = 1.4 Hz), 8.33 (2H, s), 13.4 (2H, brs) I is shown in Figure 15 the results of powder X-ray diffraction.
Patent Document 1: JP-A-10-72492 JP
Patent Document 2: WO 96/40750 pamphlet
Patent Document 3: JP-A-11-1477 JP
Patent Document 4: Japanese Unexamined Patent Publication No. 11-152276
Patent Document 5: International Publication No. 00/35446 pamphlet
Patent Document 6: JP-A-10-287634 JP
Patent Document 7: WO 01/07423 pamphlet
Patent Document 8: International Publication WO 01/53267 pamphlet
Patent Document 9: International Publication No. 02 / 059 099 pamphlet
Patent Document 10: International Publication No. 02/059100 pamphlet
Patent Document 11: International Publication No. 02/059100 pamphlet
Patent Document 12: International Publication No. 02/062775 pamphlet
Patent Document 13: International Publication No. 2003/062233 pamphlet
Patent Document 14: International Publication No. 2004/029049 pamphlet
Patent Document 15: International Publication No. 2005/007651 pamphlet
Patent Document 16: International Publication No. 2005/014561 pamphlet
Patent Document 17: JP 2005-47905 Japanese
patent Document 18: Japanese Patent Publication No. 2006-219480
Patent Document 19: Japanese Patent Publication No. 2006-219481
Patent Document 20: International Publication No. 2007/004038 pamphlet
Patent Document 21: International Publication No. 2007/036709 pamphlet
Patent Document 22: International Publication No. 2007/054783 pamphlet
Patent Document 23: International Publication No. 2009/017098 pamphlet
Non-Patent Document 1: Proceedings of the National Akademyi of Science of the United State of America (…. Proc Natl Acad Sci USA) 1992, Vol. 89, p 5640-5644.
Non-Patent Document 2: Journal of Organic (.. J. Org Chem) Chemistry 1984, Vol. 49, p 3856-3857.
Non-Patent Document 3: (.. J. Org Chem). Journal of Organic Chemistry, 1992, Vol. 57, p 6667-6669
Non-Patent Document 4:. Shinretto (Synlett) 2004 year Vol. 6, p 1092-1094
| 101 | Discovery and biological evaluation of Lusutrombopag (S-888711) as a novel nonpeptide drug candidate for thrombocytopenia Masami Takayama, Hajime Yamada, Hiroshi Takemoto, Takeshi Shiota, Yoshikazu Tanaka, Noriko Yamane, Kouji Takahashi, Naoki Oyabu, Kenji Kuwabara, Itsuki Oshima, Kenzo Koizumi, Hiroshi Yoshida, Ayumu Nogami, Tomomi Yamada, Yutaka Yoshida, Takami Murashi, Shinichiro Hara. |
| 101 – Discovery and biological evaluation of Lusutrombopag (S-888711) as a novel nonpeptide drug candidate for thrombocytopenia
Masami Takayama1, masami.takayama@shionogi.co.jp, Hajime Yamada3, Hiroshi Takemoto2, Takeshi Shiota2, Yoshikazu Tanaka2, Noriko Yamane2, Kouji Takahashi2, Naoki Oyabu3, Kenji Kuwabara3, Itsuki Oshima2, Kenzo Koizumi3, Hiroshi Yoshida3, Ayumu Nogami3, Tomomi Yamada3, Yutaka Yoshida3, Takami Murashi3, Shinichiro Hara2. (1) Department of Strategic Research Planning Offices, Shionogi & CO., LTD, Toyonaka, Osaka 561-0825, Japan, (2) Department of Innovative Drug Discovery Research Laboratories, Shionogi & CO.,LTD, Toyonaka, Osaka 561-0825, Japan, (3) Department of Medicinal Research Laboratories, Shionogi & CO., LTD, Toyonaka, Osaka 561-0825, Japan As a drug candidate of thrombocytopenia, Lusutrombopag (S-888711) is in Phase III clinical trial stage right now. It is been proven that Lusutrombopag (S-888711) is excellent property in safety and efficacy by clinical trials. In this meeting, we will present in detail about the history of drug discovery of Lusutrombopag.Because Lusutrombopag (S-888711) acts specifically to human TPO receptor, we prepared TPOR-Ki/Shi mice expressing a mouse-human chimeric TPOR for evaluating the efficacy. This TPOR-Ki/Shi mice worked very well as an evaluation model of drug efficacy, so we were able to select Lusutrombopag from many candidate compounds. In this meeting, we will present the results of the efficacy in TPOR-Ki/Shi mice of Lusutrombopag and the similar drug (Eltrombopag). |
update………..
FDA approves lusutrombopag for thrombocytopenia in adults with chronic liver disease
https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm615348.htm
synthesis………..https://newdrugapprovals.org/2015/08/20/lusutrombopag-oral-thrombopoietin-tpo-mimetic/
On July 31, 2018, the Food and Drug Administration approved lusutrombopag (Mulpleta, Shionogi Inc.) for thrombocytopenia in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure.
Approval was based on two randomized, double-blind, placebo-controlled trials (L-PLUS 1 and L-PLUS 2, NCT02389621) involving 312 patients with chronic liver disease and severe thrombocytopenia who were undergoing an invasive procedure and had a platelet count less than 50 x 109/L. Patients were randomized 1:1 to receive 3 mg of lusutrombopag or placebo once daily for up to 7 days.
In L-PLUS 1, 78% of patients (38/49) receiving lusutrombopag required no platelet transfusion prior to the primary invasive procedure, compared with 13% (6/48) who received placebo (95% CI for treatment difference: 49%, 79%; p<0.0001). In L-PLUS 2, 65% (70/108) of patients who received lusutrombopag required no platelet transfusion prior to the primary invasive procedure or rescue therapy for bleeding from randomization through 7 days after the procedure, compared with 29% (31/107) receiving placebo (95% CI for treatment difference: 25%, 49%; p<0.0001).
The most common adverse reaction in ≥ 3% of patients was headache.
The recommended lusutrombopag dosage is 3 mg orally once daily with or without food for 7 days.
View full prescribing information for Mulpleta.
FDA granted this application priority review and fast track designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Healthcare professionals should report all serious adverse events suspected to be associated with the use of any medicine and device to FDA’s MedWatch Reporting System or by calling 1-800-FDA-1088.
Follow the Oncology Center of Excellence on Twitter @FDAOncology.
Check out recent approvals at the OCE’s podcast, Drug Information Soundcast in Clinical Oncology.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


Join me on twitter

 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL
//////
phase 3, shionogi, japan, lusutrombopag, S 888711
CCCCCCOC(C)C1=CC=CC(=C1OC)C2=CSC(=N2)NC(=O)C3=CC(=C(C(=C3)Cl)C=C(C)C(=O)O)Cl
