PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Donald Pinto (left) and Michael Orwat (right) work on developing new products for BMS.
Credit: Bristol-Myers Squibb
Ruth R. Wexler, executive director of cardiovascular diseases chemistry at Bristol-Myers Squibb, who led the group that designed and synthesized Eliquis (apixaban) to reduce the risk of stroke in patients with an abnormal heart rhythm called atrial fibrillation, recalls hearing about the drug’s success in late-stage clinical trials for the first time.
“I was at the European Society of Cardiology meeting when the results of ARISTOTLE, our large Phase 3 trial, were announced,” she says. “I was sitting in the audience, and it was just amazing to see the data released for the first time. It blew my mind that the data was that spectacular.”
In the trial, which compared apixaban with the workhorse anticoagulant Coumadin (warfarin), apixaban reduced the risk of stroke in patients with atrial fibrillation by 21%, major bleeding by 31%, and mortality by 11%. Unlike Coumadin, apixaban doesn’t require regular monitoring of the blood.
To reduce the risk of recurring DVT and PE after initial therapy.
Atrial fibrillation
Apixaban is recommended by the National Institute for Health and Clinical Excellence for the prevention of stroke and systemic embolism in people with non-valvular atrial fibrillation and at least one of the following risk factors: prior stroke or transient ischemic attack, age 75 years or older, diabetes mellitus, or symptomatic heart failure.[6]
Apixaban and other newer anticoagulants (dabigatran and rivaroxaban) appear equally effective as warfarin in preventing non-hemorrhagic stroke in people with atrial fibrillation and are associated with lower risk of intracranial bleeding.[7]
Mechanism of action
Apixaban is a highly selective, orally bioavailable, and reversible direct inhibitor of free and clot-bound factor Xa. Factor Xa catalyzes the conversion of prothrombin to thrombin, the final enzyme in the coagulation cascade that is responsible for fibrin clot formation.[10] Apixaban has no direct effect on platelet aggregation, but by inhibiting factor Xa, it indirectly decreases clot formation induced by thrombin.[5]
FDA approval
A new drug application (NDA) for the approval of apixaban was submitted to the FDA by Bristol-Myers Squibb and Pfizer jointly after conclusion of the ARISTOTLE clinical trial in 2011.[11]
Apixaban was approved for the prevention of stroke in people with atrial fibrillation on December 28, 2012.[12] On March 14, 2014, it was approved for the additional use of preventing deep vein thrombosis and pulmonary embolism in people that had recently undergone knee or hip replacement.[13] On August 21, 2014, the FDA approved apixaban for the treatment of recurring deep vein thrombosis and pulmonary embolism.[2]
During development it was known as BMS-562247-01.
Thursday, August 21, 2014 – Bristol-Myers Squibb Company (NYSE: BMY) and Pfizer Inc. (NYSE: PFE) today announced the U.S. Food and Drug Administration (FDA) has approved a Supplemental New Drug Application (sNDA) for Eliquis for the treatment of DVT and PE, and for the reduction in the risk of recurrent DVT and PE following initial therapy. Combined, DVT and PE are known as VTE. It is estimated that every year, approximately 900,000 Americans are affected by DVT and PE.
CAS NO. 503612-47-3, 1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide H-NMR spectral analysis
C-NMR spectral analysis
CAS NO. 503612-47-3, 1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide C-NMR spectral analysis http://www.google.com/patents/WO2012168364A1?cl=en
l-(4-Methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l -yl)phenyl]-4, 5,6,7- tetrahydro- lH-pyrazolo[3,4-c]pyridine-3-carboxyamide of formula (I), also known come apixaban, is a powerful inhibitor of coagulation factor Xa disclosed in US 6,967,208. Said compound is used in the prevention and treatment of thromboembolic disorders.
(I)
US 7, 153,960 discloses a process for the preparation of apixaban wherein the key step is the formation of intermediate (A) by 1 ,3 dipolar cycloaddition reaction between the compounds of formula (B) and (C) and its subsequent conversion to the compound of formula (D) by treatment with an acid. The compound of formula (D), after simple manipulations of functional groups, is converted to apixaban
B C A D
Said patent discloses the preparation of the compounds of formula (B) and (C). While the synthesis of the hydrazone of formula (B) has been known for some time, the preparation of the key intermediate of formula (C) is complex and uses reagents which are expensive and potentially hazardous, such as phosphorus pentachloride (PC15), and drastic reaction conditions.
US 7, 153,960, for example, discloses as preferred the preparation of an enamine intermediate of formula (C) wherein the amine residue NRbRc is a morpholine. The conditions used for the success of the reaction actually involve the use of morpholine as solvent at high temperatures, such as reflux temperature (about 130- 135°C).
The complexity of the known processes for the preparation of the intermediate of formula C, the expense and danger of the reagents and the drastic reaction conditions used make said processes difficult to apply and scale up industrially, especially for the purpose of preparing the intermediates of formula A and D and apixaban.
Example 6. Synthesis of compound of formula (I): l-(4- Methoxyphenyl)-6-[4-(2-oxo-piperidinyl)phenyl]-7-oxo-4,5,6,7-tetrahydro- l//-pyrazolo[3,4-c]pyridine-3-carboxyamide: Apixaban (I)
The compound of formula II, prepared as in Example 5 (17.50 g, 35.82 mmol), is suspended in 100 ml of 33% NH3 and 200 ml of MeOH in a 1L 4-necked flask equipped with coolant, thermometer and magnetic stirrer, in nitrogen atmosphere, and heated to 45°. MeOH (250 ml) is added until completely dissolved, and the solution is left under stirring for 2h. Another addition of 33% NH3 (50 ml) is performed, and the progress of the reaction is monitored by TLC (AcOEt/MeOH 9: 1) and HPLC. After 18h the solvent is evaporated under low pressure, and the solid residue obtained is suspended in 200 ml of H2O and left under stirring for 2h. The white solid is filtered through a Buchner funnel, and washed with H2O (50 ml). The product of formula (I) is stove-dried at 50°C to a constant weight (12.60 g, yield 76%). The HPLC purity of the product exceeds 99%
Apixaban compound of formula- 1 of the present invention is analyzed by HPLC using the following conditions:
Apparatus: A liquid chromatographic system is to be equipped with variable wavelength UV- detector; Column: Zorbax Bonus RP, 250 x 4.6 mm, 5μιη or equivalent; Flow rate: 1.2 ml/min; wavelength: 270 nm; column temperature: 40°C; Injection volume; 5 uL; Run time: 35 minutes; Needle wash: diluent; Diluent: Acetonitrile: water (90: 10 v/v); Elution: Gradient; Mobile phase-A: Buffer; Mobile phase-B: acetonitrile:water (90:10 v/v); Buffer: Weigh accurately about 1.36 g of potassium dihydrogen ortho phosphate in 1000 10 ml of milli-Q water and adjust pH 6.0 with dil KOH solution, then filter through 0.22 μιη nylon membrane filter paper. The following impurities have been observed during the preparation of Apixaban.
Example-1: Preparation of 3-chloro-l-(4-iodophenyI)-5,6-dihydropyridin-2(lH)-one (Formula-6)
Lithium carbonate (4.08 gm) followed by lithium chloride (2.28 gm) were added to a mixture of 3,3-dichloro-l-(4-iodophenyl)piperidin-2-one compound of formula-5 (30 gm) and dimethylformamide (60 ml) at 25-30°C and stirred for 5 min at the same temperature. Heated the reaction mixture to 110-115°C and stirred for 4 hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water was added to the reaction mixture at 25-30°C and stirred for 1 hr at the same temperature. Filtered the precipitated solid and then dried to get the title compound. Yield: 25.0 gm; MR: 120-130°C.
Example-2: Preparation of 3-chIoro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one (Formula-6)
Lithium carbonate (2.99 gm) followed by sodium chloride (2.76 gm) were added to a mixture of 3,3-dichloro-l-(4-iodophenyl)piperidin-2-one compound of formula-5 (50 gm) and dimethylformamide (150 ml) at 30-35°C and stirred for 10 min at the same temperature. Heated the reaction mixture to 110-115°C and stirred for 6 hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water was added to the reaction mixture at 25-30°C and stirred for 1 hr at the same temperature. Filtered the precipitated solid and then dried to get the title compound.
Yield: 42.0 gm; M.R: 120-130°C.
Example-3: Preparation of l-(4-iodophenyl)-3-morpholino-5,6-dihydropyridin-2(lH)-one (Formula-7)
Morpholine (5.09 gm) was added to a mixture of 3-chloro-l-(4-iodophenyl)-5,6-dihydro pyridin-2(lH)-one compound of formula-6 (5 gm) and toluene (5 ml) at 25-30°C and stirred for 5 min at the same temperature. Heated the reaction mixture to 115-120°C and stirred for 3 hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water was added to the reaction mixture at 25-30°C and stirred for 15 hrs at the same temperature. Filtered the precipitated solid and then dried to get the title compound. Yield: 3.8 gm.
Example-4: Preparation of l-(4-iodophenyl)-3-morpholino-5,6-dihydropyridin-2(lH)-one (Formula-7)
Morpholine (28.73 gm) was added to a mixture of 3-chloro-l-(4-iodophenyl)-5,6- dihydropyridin-2(lH)-one compound of formula-6 (50 gm) and toluene (50 ml) at 30-35°C. Heated the reaction mixture to 115-120°C and stirred for 8 hrs at 115-120°C. After completion of the reaction, cooled the reaction mixture to 25-30°C. Methyl tert-butyl ether (100 ml) followed by water were slowly added to the reaction mixture at 25-30°C. Cooled the reaction mixture to 5- 10°C and stirred for 2 hours at 5-10°C. Filtered the precipitated solid and then dried to get the title compound. Yield: 45 gm.
Example-5: Preparation of ethyl 6-(4-iodophenyl)-l-(4-methoxyphenyI)-7-oxo-4,5,6,7-tetra hydro-lH-pyrazoIo[3,4-c]pyridine-3-carboxyIate (FormuIa-13)
A mixture of 3-chloro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one compound of formula-6 (79.2 gm), (Z)-ethyl 2-chloro-2-(2-(4-methoxyphenyl)hydrazono)acetate compound of formula-9 (65 gm) and toluene (450 ml) was heated to 90-100°C and stirred for 5 min at the same temperature. Triethyl amine (72 gm) was slowly added to the reaction mixture at 95-100°C and stirred for 2½ hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water (110 ml) was added to the reaction mixture at 25-30°C and stirred for 8 hrs at the same temperature. Filtered the solid, washed with water and then dried to get the title compound.
Yield: 78.5 gm.
Example-6: Preparation of 5-bromo-N-(4-iodophenyl)pentanamide (Formula-3)
A mixture of 5-bromopentanoic acid (54 g), thionyl chloride (41 g), dimethylformamide (2 ml) and toluene (100 ml) was heated to 40-45°C and stirred for 2 hours at the same temperature. Distilled off the reaction mixture to remove the un-reacted thionyl chloride under reduced pressure at a temperature below 40°C. Toluene (50 ml) was added to the reaction mixture and stirred for 15 minutes. The reaction mixture was cooled to 25-30°C under nitrogen atmosphere and it slowly added to a pre-cooled mixture of 4-iodoaniline compound of formula-2 (50 g) and toluene (350 ml) at 0-5°C. Triethyl amine (29 g) was added to it at 0-5°C. The above reaction mixture containing acid chloride was slowly added to the reaction mixture containing 4- iodoaniline under nitrogen atmosphere and stirred for 2 hours at 0-5°C. Water (250 ml) was added to the reaction mixture and stirred for 2 hours at 0-5°C. Filtered the precipitated solid and then dried to get title compound. Yield: 83 gm; MR: 135-140°C; HPLC purity: 99%.
Example-7: Preparation of 3-chloro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one (Formula-6)
Step-a) Preparation of l-(4-iodophenyl)piperidin-2-one (Formula-4)
Sodium tert-butoxide (18.86 g) was added to a mixture of 5-bromo-N-(4- iodophenyl)pentanamide compound of formula-3 (50 g) and toluene (250 ml) at 0-5°C and stirred for 2 hours at 0-5°C. Water (100 ml) followed by aqueous hydrochloric acid solution (50 ml) were added to the reaction mixture and stirred for 10 minutes at 5-10°C. Both the organic and aqueous layers were separated; the organic layer was washed with water. Distilled off the solvent from the organic layer under reduced pressure at a temperature below 60°C to get title compound as a solid.
Step-b) Preparation of 3,3-dichIoro-l-(4-iodophenyI)piperidin-2-one (Formula-5)
The compound obtained in step-a) was dissolved in dichloromethane (100 ml) and slowly added to a mixture of phosphorous pentachloride (95 g) and dichloromethane (150 ml) at 25- 30°C. The reaction mixture was heated to 35-40°C and stirred for 4 hours at the same temperature. Cooled the reaction mixture to 5-10°C. Chilled water (150 ml) was added to the reaction mixture and stirred for 1.5 hours at 10-15°C. Both the organic and aqueous layers were separated; the organic layer was washed with water followed by 10% aqueous sodium carbonate solution. Distilled off the solvent completely from the organic layer to get title compound as a solid.
Step-c) Preparation of 3-chloro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one (Formula- 6)
To the obtained compound in step-b), dimethylformamide (100 ml), followed by lithium carbonate (2.2 g) and sodium chloride (2.0 g) were added at 25-30°C. The reaction mixture was heated to 115-120°C and stirred for 6 hours at the same temperature. Cooled the reaction mixture to 30-35°C, water (350 ml) was added to it and stirred for 2 hours at 25-30°C. Filtered the precipitated solid and washed with water. Methanol (360 ml) was added to the obtained solid and the reaction mixture was heated to 65-70°C. Stirred the reaction mixture for 20 minutes at the same temperature. Carbon (3.0 g) was added to the reaction mixture and stirred for 20 minutes at 65-70°C. Filtered the reaction mixture through hyflow bed and washed with methanol. Distilled off the solvent from the filtrate under reduced pressure and methanol (300 ml) was added to the residue and stirred for 20 minutes at 25-30°C. Cooled the reaction mixture to -5 to 0°C and stirred for 60 minutes at the same temperature. Filtered the precipitated solid, washed with methanol and then dried to get title compound.
Yield: 25 gm; MR: 115- 120°C: HPLC purity: 98%.
Example-8: Preparation of 3-morpholino-l-(4-(2-oxopiperidin-l-yl)phenyl)-5,6-dihydro pyridin-2(lH)-one (Formula-8)
A mixture of l-(4-iodophenyl)-3-mo holino-5,6-dihydropyridin-2(lH)-one compound of formula-7 (50 g), piperidin-2-one (32.25 g) and o-xylene (75 ml) was stirred for 10 minutes at 25-30°C. Potassium carbonate (27.0 g), followed by copper iodide (7.43 g) were added to the reaction mixture. The reaction mixture was heated to 140-145°C under azeotropic distillation condition and stirred for 6 hours at the same temperature. Cooled the reaction mixture to 35- 40°C, water (175 ml) was slowly added to the reaction mixture at 35-40°C. Cooled the reaction mixture to 10-15°C and ammonia (125 ml) was added to the reaction mixture at 10-15°C. The temperature of the reaction mixture was raised to 25-30°C and stirred for 2 hours at the same temperature. Filtered the precipitated solid, washed with water and then dried to get title compound.
Yield: 35 gm; MR: 195-200°C; HPLC purity: 95%.
Example-9: Preparation of (Z)-ethyl 2-chloro-2-(2-(4-nlethoxyphenyl)hydrazono)acetate (FormuIa-9)
A mixture of 4-methoxyaniline compound of formula- 12 (50 g) and water (150 ml) was cooled to 5-10°C. Hydrochloric acid (100 ml), followed by a solution of sodium nitrite (30.81 g) in water (50 ml) were slowly added to the reaction mixture at 5-10°C and stirred for 2 hours at 5- 10°C to provide diazotized compound. Ethyl acetate (250 ml) was added to the reaction mixture. Ethyl 2-chloro acetoacetate (76.84 g) was slowly added to a mixture of sodium acetate (76.6 g), ethyl acetate (250 ml) and water (150 ml) at 25-30°C and the reaction mixture was stirred for 2 hours at 25-30°C. The reaction mixture was slowly added to the reaction mixture containing diazotized compound at a temperature below 10°C. The temperature of the reaction mixture was raised to 25-30°C and stirred for 16 hours at the same temperature. Both the organic and aqueous layers were separated and the organic layer was washed with 10% aqueous sodium bicarbonate solution followed by 10% aqueous sodium chloride solution. Distilled off the solvent completely from the organic layer under reduced pressure and then co-distilled with toluene. Toluene was added to the obtained compound and stirred for 15 minutes at 25-30°C. Silica-gel was added to the reaction mixture and stirred for 30 minutes at 25-30°C. Filtered the reaction mixture and the solvent from the filtrate was distilled off completely under reduced pressure. Cyclohexane (400 ml) was added to the obtained compound and the reaction mixture was stirred for 60 minutes at 25-30°C. Filtered the precipitated solid, washed with cyclohexane and then dried to get title compound. Yield: 60 gm; MR: 95-100°C; HPLC purity: 99%.
ExampIe-10: Preparation of ethyl l-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-l-yl) phenyl)-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxylate (Formula-11)
A mixture of 3-morpholino-l-(4-(2-oxopiperidin-l-yl)phenyl)-5,6-dihydropyridin-2(lH)- one compound of formula-8 (30 g), sodium carbonate (26.83 g) and acetone (150 ml) was heated to 45-50°C. (Z)-ethyl 2-chloro-2-(2-(4-methoxyphenyl)hydrazono)acetate compound of formula- 9 (32.5 g) was added to the reaction mixture at 45-50°C and stirred for 3 hours at the same temperature. Cooled the reaction mixture to 25-30°C and aqueous hydrochloric acid (50 ml) in 50 ml of water was added to it at 25-30°C. Stirred the reaction mixture for 2 hours at 25-30°C. Water was slowly added to the reaction mixture and stirred for 45 minutes at 25-30°C. Filtered the obtained solid and washed with water. The obtained solid was recrystallized from toluene (150 ml) to get the title compound. Yield: 35 gm; MR: 155-160°C; HPLC purity: 97%.
Example- 11: Preparation of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]- 4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
A mixture of ethyl l-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-l-yl)phenyl)- 4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxylate compound of formula-11 (50 g), formamide (150 ml), sodium methoxide (30 ml) and isopropanol (300 ml) was heated to 65-70°C and stirred for 2 hours at 65-70°C. Cooled the reaction mixture to 0-5°C and stirred for 30 minutes at 0-5°C. Filtered the precipitated solid and washed with isopropanol. Methanol (150 ml) was added to the obtained solid, the reaction mixture was heated to 65-70°C and stirred for 15 minutes at 65-70°C. Cooled the reaction mixture to 0-5°C and stirred for 30 minutes at 0-5°C. Filtered the precipitated solid, washed with methanol and then dried to get title compound. Yield: 35 g. MR: 230-235°C; HPLC purity: 98%.
The PXRD of the crystalline solid obtained from the above example is matches with the PXRD of crystalline form-M of the present invention.
Example-12: Purification of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]- 4,5,6, 7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
1 -(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin- 1 -yl)phenyl]-4,5,6,7-tetrahydro- 1 H- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (100 g) was dissolved in a mixture of dichloromethane (1200 ml) and methanol (200 ml) at 25-30°C. 10% aqueous sodium carbonate solution (200 ml) was added to the reaction mixture and stirred for 15 minutes at 25- 30°C. Both the organic and aqueous layers were separated, methanol (100 ml) was added to the organic layer and again 200 ml of 10% aqueous sodium carbonate solution was added to the reaction mixture. The reaction mixture was stirred for 15 minutes at 25-30°C and separated the organic and aqueous layers. To the organic layer methanol (100 ml) followed by water (200 ml) were added. Both the organic and aqueous layers were separated. The solvent from organic layer was distilled under reduced pressure at a temperature below 40°C. 3000 ml of a mixture of dichloromethane and methanol (in the ratio of 3:7) was added to the crude compound and the reaction mixture was heated to reflux temperature and stirred for 10 minutes. Carbon (10 g) was added to the reaction mixture and stirred for 15 minutes at the reflux temperature. Filtered the reaction mixture through hyflow bed, washed with a mixture of dichloromethane and methanol. The filtrate was cooled to 0-5°C and stirred for 2 hours at 0-5°C. Filtered the precipitated solid and washed with a mixture of dichloromethane and methanol. Isopropanol (1000 ml) was added to the reaction mixture. Heated the reaction mixture to 80-85°C and stirred for 15 minutes. Cooled the reaction mixture to 25-30°C and stirred for 2 hours at 35-30°C. Filtered the precipitated solid, washed with isopropanol and then dried to get title compound.
Yield: 80 gm; MR: 235-240°C.
The PXRD pattern of crystalline solid obtained from the above example is matches with PXRD of crystalline form-M of the present invention.
Example-13: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH- pyrazolo[3,4-c] pyridine-3-carboxamide compound of formula-1 (6.25 gm) was added to isopropanol (400 ml) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Cooled the reaction mixture to 0-5°C and stirred for 60 min the same temperature. Filtered the solid, washed with isopropanol and then dried to get the title compound. Yield: 4.5 gm; Water content: 0.30% w/w. HPLC purity: 99.8%; Acid impurity: 0.02%; Amino acid impurity: Not detected; Chloro impurity: 0.01%; Methyl ester impurity: 0.05%; Ethyl ester impurity: 0.01%; Dehydro impurity: 0.07%.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively.
Example-14: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yI)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
1 -(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin- 1 -yl)phenyl]-4,5,6,7-tetrahydro- 1 H- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (6.25 gm) was added to 50% aqueous isopropanol (60 ml) at 25-30°C. Heated the reaction mixture to 50-60°C and stirred for 4 hrs at the same temperature. Cooled the reaction mixture to 25-30°C and stirred for 60 min at the same temperature. Filtered the solid and then dried to get the title compound.
Yield: 4.1 gm; Water content: 0.35% w/w.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively.
Example-15: Preparation of crystalline form-S of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (34 gm) was added to a mixture of dichloromethane and methanol at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Filtered the reaction mixture and washed with a mixture of dichloromethane and methanol. Cooled the filtrate to 0-5°C and stirred for 60 min at the same temperature. Filtered the precipitated solid and then dried to get the title compound. Yield: 24.0 gm; M.R: 235-245°C; Water content: 7.38% w/w.
The PXRD and DSC of the obtained compound are illustrated in figure-3 and figure-4 respectively.
Example-16: Preparation of crystalline form-N of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3- carboxamide(Formula-l)
A mixture of dichloromethane and ethyl acetate (625 ml, in 3:7 ratio) was added to l-(4- methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c] pyridine-3-carboxamide compound of formula- 1 (6.25 gm) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Cooled the reaction mixture to 0-5°C and stirred for 60 min at the same temperature. Filtered the solid and then dried to get title compound. Yield: 3.9 g; Water content: 5.21% w/w.
The PXRD and DSC of the obtained compound are illustrated in figure-5 and figure-6 respectively.
Example-17: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
1 -(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin- 1 -yl)phenyl]-4,5,6,7-tetrahydro- 1 H- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (34 gm) was added to a mixture of dichloromethane and methanol (1020 ml, in 3:7 ratio) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Filtered the reaction mixture and washed with a mixture of dichloromethane and methanol. Cooled the filtrate to 0- 5°C and stirred for 60 min at the same temperature. Filtered the precipitated solid and added to isopropanol (510 ml). Heated the reaction mixture to reflux temperature and stirred for 15 Minutes at the same temperature. The reaction mixture was cooled to 0-5°C and stirred for 60 minutes at the same temperature. Filtered the solid and then dried to get crystalline form-M of compound of formula-1. Yield: 23 g; Water content: 0.30%w/w.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively.
Example-18: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (34 gm) was added to a mixture of dichloromethane and methanol (1020 ml, in 3:7 ratio) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Filtered the reaction mixture and washed with a mixture of dichloromethane and methanol. Cooled the filtrate to 0- 5°C and stirred for 60 min at the same temperature. Filtered the precipitated solid and added to aq.isopropanol (340 ml). Heated the reaction mixture to 50-60°C and stirred for 15 minutes at the same temperature. The reaction mixture was cooled to 25-35°C and stirred for 60 minutes at the same temperature. Filtered the solid and then dried to get crystalline form-M of compound of formula-1.
Yield: 23 g; Water content: 0.35%w/w.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively
Gómez-Outes, A; Terleira-Fernández, AI; Calvo-Rojas, G; Suárez-Gea, ML; Vargas-Castrillón, E (2013). “Dabigatran, Rivaroxaban, or Apixaban versus Warfarin in Patients with Nonvalvular Atrial Fibrillation: A Systematic Review and Meta-Analysis of Subgroups.”. Thrombosis2013: 640723. PMID24455237.
Enriquez A, Lip GY, Baranchuk A (2015). “Anticoagulation reversal in the era of the non-vitamin K oral anticoagulants”. Europace. doi:10.1093/europace/euv030. PMID25816811.
Frost C, Wang J, Nepal S; et al. (February 2013). “Apixaban, an oral, direct factor Xa inhibitor: single dose safety, pharmacokinetics, pharmacodynamics and food effect in healthy subjects”. Br J Clin Pharmacol75 (2): 476–87. doi:10.1111/j.1365-2125.2012.04369.x. PMID22759198.
The U.S. Food and Drug Administration today approved Cerdelga (eliglustat) for the long-term treatment of adult patients with the Type 1 form of Gaucher disease, a rare genetic disorder.
Gaucher disease occurs in people who do not produce enough of an enzyme called glucocerebrosidase. The enzyme deficiency causes fatty materials to collect in the spleen, liver and bone marrow. The major signs of Gaucher disease include liver and spleen enlargement, low red blood cell counts (anemia), low blood platelet counts and bone problems.
Cerdelga is a hard gelatin capsule containing eliglustat that is taken orally. In patients with Gaucher disease Type 1, the drug slows down the production of the fatty materials by inhibiting the metabolic process that forms them. Type 1 Gaucher disease is estimated to affect about 6,000 people in the United States.
“Today’s approval offers another important treatment option for patients with Type 1 Gaucher disease,” said Amy G. Egan, M.D., M.P.H., deputy director of the Office of Drug Evaluation III in FDA’s Center for Drug Evaluation and Research. “In addition, Cerdelga received orphan drug designation from the FDA, reflecting the agency’s focus and commitment to the development of treatments for rare diseases.”
The safety and effectiveness of Cerdelga were evaluated in two clinical trials with 199 participants with Type 1 Gaucher disease.
In one randomized, double-blind, placebo-controlled, multicenter clinical trial the safety and effectiveness of Cerdelga were evaluated in 40 participants with Type 1 Gaucher’s disease who had not previously received enzyme replacement therapy. Subjects received the drug at a starting dose of 42 mg two times a day, with most receiving a dose of 84 mg two times a day after four weeks. Study participants continued the drug for nine months.
Compared to placebo, treatment with Cerdelga resulted in a greater reduction in spleen volume from baseline to the end of the study (by the 39th week), the trial’s primary endpoint. Cerdelga also resulted in greater improvement in liver volume, blood platelet count, and red blood cell (hemoglobin) level, compared to placebo.
The other trial sought to determine the safety and effectiveness of Cerdelga compared to enzyme replacement therapy in 159 participants with Type 1 Gaucher disease previously treated and stabilized on enzyme replacement therapy. Subjects in the trial received either the enzyme replacement therapy drug imiglucerase or Cerdelga. The trial demonstrated that treatment with Cerdelga resulted in similar stabilization of hemoglobin level, platelet count and spleen and liver volume as imiglucerase.
The most commonly observed side effects in the Cerdelga clinical trials were fatigue, headache, nausea, diarrhea, back pain, pain in extremities, and upper abdominal pain.
Cerdelga is manufactured by Cambridge, Massachusetts-based Genzyme.
The U.S. Food and Drug Administration today approved a new use for Avastin (bevacizumab) to treat patients with persistent, recurrent or late-stage (metastatic) cervical cancer.
Cervical cancer grows in the tissues of the lower part of the uterus known as the cervix. It commonly occurs when human papillomaviruses (HPV), a virus that spreads through sexual contact, cause cells to become cancerous. Although there are two licensed vaccines available to prevent many types of HPV that can cause cervical cancer, the National Cancer Institute estimates that 12,360 American women will be diagnosed with cervical cancer and 4,020 will die from the disease in 2014.
Avastin works by interfering with the blood vessels that fuel the development of cancerous cells. The new indication for cervical cancer is approved for use in combination with chemotherapy drugs paclitaxel and cisplatin or in combination with paclitaxel and topotecan.
“Avastin is the first drug approved for patients with late-stage cervical cancer since the 2006 approval of topotecan with cisplatin,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “It is also the first biologic agent approved for patients with late-stage cervical cancer and was approved in less than four months under the FDA’s priority review program, demonstrating the agency’s commitment to making promising therapies available to patients faster.”
The FDA reviewed Avastin for treatment of patients with cervical cancer under its priority review program because the drug demonstrated the potential to be a significant improvement in safety or effectiveness over available therapy in the treatment of a serious condition. Priority review provides an expedited review of a drug’s application.
The safety and effectiveness of Avastin for treatment of patients with cervical cancer was evaluated in a clinical study involving 452 participants with persistent, recurrent, or late-stage disease. Participants were randomly assigned to receive paclitaxel and cisplatin with or without Avastin or paclitaxel and topotecan with or without Avastin. Results showed an increase in overall survival to 16.8 months in participants who received chemotherapy in combination with Avastin as compared to 12.9 months for those receiving chemotherapy alone.
The most common side effects associated with use of Avastin in patients with cervical cancer include fatigue, decreased appetite, high blood pressure (hypertension), increased glucose in the blood (hyperglycemia), decreased magnesium in the blood (hypomagnesemia), urinary tract infection, headache and decreased weight. Perforations of the gastrointestinal tract and abnormal openings between the gastrointestinal tract and vagina (enterovaginal fistula) also were observed in Avastin-treated patients.
Avastin is marketed by South San Francisco, California-based Genentech, a member of the Roche Group.
Country
Patent Number
Approved
Expires (estimated)
Canada
2286330
2008-06-10
2018-04-03
Canada
2145985
2003-09-16
2012-10-28
Property
Value
Source
melting point
61 °C (FAB fragment), 71 °C (whole mAb)
Vermeer, A.W.P. & Norde, W., Biophys. J. 78:394-404 (2000)
Protein chemical formula
C6538H10034N1716O2033S44
Protein average weight
149 kDa
A recombinant humanized monoclonal IgG1 antibody that binds to and inhibits the biologic activity of human vascular endothelial growth factor (VEGF). Bevacizumab contains human framework regions and the complementarity-determining regions of a murine antibody that binds to VEGF. Bevacizumab is produced in a Chinese Hamster Ovary mammalian cell expression system in a nutrient medium containing the antibiotic gentamicin and has a molecular weight of approximately 149 kilodaltons.
The FDA has approved a new type of sleep drug. This new drug is an orexin receptor antagonist and is the first approved drug of this type. Orexins are chemicals that are involved in regulating the sleep-wake cycle and play a role in keeping people awake. Learn more here:http://go.usa.gov/EcEz
The U.S. Food and Drug Administration today approved Belsomra (suvorexant) tablets for use as needed to treat difficulty in falling and staying asleep (insomnia).
Belsomra is an orexin receptor antagonist and is the first approved drug of this type. Orexins are chemicals that are involved in regulating the sleep-wake cycle and play a role in keeping people awake. Belsomra alters the signaling (action) of orexin in the brain.
Insomnia is a common condition in which a person has trouble falling or staying asleep. It can range from mild to severe, depending on how often it occurs and for how long. Insomnia can cause daytime sleepiness and lack of energy. It also can make a person feel anxious, depressed, or irritable. People with insomnia may have trouble with attentiveness, learning, and memory.
“To assist health care professionals and patients in finding the best dose to treat each individual patient’s sleeplessness, the FDA has approved Belsomra in four different strengths – 5, 10, 15, and 20 milligrams,” said Ellis Unger, M.D., director of the Office of Drug Evaluation I in the FDA’s Center for Drug Evaluation and Research. “Using the lowest effective dose can reduce the risk of side effects, such as next-morning drowsiness.”
Belsomra should be taken no more than once per night, within 30 minutes of going to bed, with at least seven hours remaining before the planned time of waking. The total dose should not exceed 20 mg once daily.
The most commonly reported adverse reaction reported by clinical trial participants taking Belsomra was drowsiness. Medications that treat insomnia can cause next-day drowsiness and impair driving and other activities that require alertness. People can be impaired even when they feel fully awake.
The FDA asked the drug manufacturer, Merck, Sharpe & Dohme Corp., to study next-day driving performance in people who had taken Belsomra. The testing showed impaired driving performance in both male and female participants when the 20 mg strength was taken. Patients using the 20 mg strength should be cautioned against next-day driving or activities requiring full mental alertness. Patients taking lower doses should also be made aware of the potential for next-day driving impairment, because there is individual variation in sensitivity to the drug.
The effectiveness of Belsomra was studied in three clinical trials involving more than 500 participants. In the studies, patients taking the drug fell asleep faster and spent less time awake during the remainder of the night compared to people taking an inactive pill (placebo). Belsomra was not compared to other drugs approved to treat insomnia, so it is not known if there are differences in safety or effectiveness between Belsomra and other insomnia medications.
Like other sleep medicines, there is a risk from Belsomra of sleep-driving and other complex behaviors while not being fully awake, such as preparing and eating food, making phone calls, or having sex. Chances of such activity increase if a person has consumed alcohol or taken other medicines that make them sleepy. Patients or their families should call the prescribing health care professional if this type of activity occurs.
Belsomra will be dispensed with an FDA-approved patient Medication Guide that provides instructions for its use and important safety information. Belsomra is a controlled substance (Schedule-IV) because it can be abused or lead to dependence.
Belsomra is made by Merck, Sharpe & Dohme Corp. of Whitehouse Station, N.J.
A panel of experts at the US Food and Drug Administration has recommended Merck & Co’s insomnia drug suvorexant when given in lower dosages but rejected the higher dose that the company was seeking.———read more at
Suvorexant (MK-4305) is a dual orexin receptor antagonist in development by Merck & Co.[1][2][3] Suvorexant works by turning off wakefulness rather than by inducing sleep.[4] It is not currently approved for commercial use, but it has completed three Phase III trials.[5]The recent FDA review showed that the drug is associated with increased somnolence the next day and users of higher doses had an increased rate of suicidal ideation. [6] It is one of two such compounds currently in development, the other being GlaxoSmithKline‘s SB-649,868.
Ref:Org.Process Res.Dev-2011-15-367.
PAPER
Mangion IK, * Sherry BD, Yin J, Fleitz FJ. Merck & Co., Rahway, USA
Enantioselective Synthesis of a Dual Orexin Receptor Antagonist.Org. Lett. 2012; 14: 3458-3461
OREXINS A AND B ARE EXCITATORY NEUROPEPTIDES THAT STIMULATE WAKEFULNESS. SUVOREXANT IS A DUAL OREXIN RECEPTOR ANTAGONIST THAT IS IN PHASE III CLINICAL TRIALS FOR THE TREATMENT OF INSOMNIA. THE KEY STEP IN THE ASYMMETRIC SYNTHESIS DEPICTED IS A TANDEM ENZYMATIC TRANSAMINATION–ANNULATION SEQUENCE (F → G → H).
A previous synthesis of suvorexant (N. A. Strotman et al. J. Am. Chem. Soc. 2011, 133, 8362) involved an asymmetric Ru-catalyzed reductive amination in the construction of the diazepane ring. The present route benefits from the circumvention of transition-metal catalysis and dichloromethane as solvent.
To a solution of 22.3 g (78 mmol) of the hydrochloride salt of F-1, 15.9 g (78 mmol) A-2, 12.8 g (94 mmol) 1-hydroxy-7-azabenzotriazole, and 43.1 mL (392 mmol) N-methylmorpholine in 300 mL of DMF was added 22.5 g (118 mmol) EDC and the reaction was stirred overnight at room temperature. The reaction was partitioned between EtOAc and saturated aqueous NaHCO3, washed with water, brine, dried over MgSO4, and concentrated by rotary evaporation. The residue was purified by column chromatography on silica gel (EtOAc/hexanes) to provide G-1 as a colorless gum. Data for G-1: LC/MS: rt=2.22 min; m/z (M+H)=434.2 found; 434.2 required.
A round bottom flask containing a solution of 29.6 g (68.3 mmol) G-1 in 300 mL EtOAc and 200 ml MeOH was evacuated under reduced pressure and purged three times with an atmosphere of N2. To the flask was then added 2.4 g of 20% Pd(OH)2on carbon. The flask was again evacuated under reduced pressure and purged three times with an atmosphere of N2, and then three times with H2. The reaction was stirred under an atmosphere of H2 for three days, then filtered through a pad of celite, rinsing with EtOAc followed by MeOH. The filtrate was concentrated to provide G-2 as a white foam. Data for G-2: LC/MS: rt=0.96 & 1.13 min (see two conformers under these conditions); m/z (M+H)=300.0 found; 300.2 required.
To 21.0 g (70.1 mmol) G-2 in 250 mL DMF was added 29.3 mL (210 mmol) triethylamine and 13.2 g (70.1 mmol) D-1 and the mixture was heated in an oil bath at 75° C. for 2 h. After cooling to room temperature, the reaction was diluted with EtOAc, washed with saturated aqueous NaHCO3, water, brine and dried over MgSO4. Following concentration by rotary evaporation, the residue was purified by flash column chromatography (hexanes/EtOAc) to provide a gum. The gum was stirred in a mixture of 150 ml EtOAc and 300 ml hexanes overnight. Filtration provided G-3 as a white solid. Data for G-3: LC/MS: rt=2.29 min; m/z (M+H)=451.1 found; 451.2 required; HRMS (APCI) m/z (M+H) 451.1631 found; 451.1644 required.
(2) Org.Process Res.Dev.2011,15,367 – 375 reported a synthetic route is as follows:
the two lines above has the following disadvantages: starting materials using highly toxic compound methyl vinyl ketone, methyl vinyl ketone to the eyes, skin, mucous membranes and upper respiratory tract irritation strong, easy to operate when used; and finally to preparation suvorexant, the need to chiral separation, is not conducive to industrial production, but low yield.
(3) W02012148553 and J.Am.Chem.Soc.2011,133,8362 – Scheme 8371 report as follows:
The route disadvantages: starting materials using highly toxic compound methyl vinyl ketone, methyl vinyl ketone to the eyes, skin, mucous membranes and upper respiratory tract irritation strong, easy to operate when used; also use a heavy metal catalyst, high cost, and environmentally unfriendly.
(4) Org.Lett, synthetic route Vol.14, N0.13,2012,3458-3461 reported as follows:
The disadvantage of this route: starting materials using highly toxic compound methyl vinyl ketone, methyl vinyl ketone pairs of eyes, skin, mucous membranes and upper respiratory tract irritation strong.; Additional use of biocatalysis, high cost.
(5) Angew.Chem.1nt.Ed.2011,50,11511 – 11515 reported synthetic route is as follows:
The methyl-2- (benzylamino) ethyl ester (20mmol), (R) _3_ ((tert-butoxycarbonyl) amino) butyric acid (21mmol), 1- hydroxybenzotriazole (25mmol), Sodium hydride (24mmol) added to the flask, anhydrous acetone 50ml, was added with stirring 1 (Shu ^ (25 dirty 01), 301:! 411. The reaction was added 10% citric acid solution, extracted with ethyl acetate, 5% Na2CO3 The organic layer was washed with a solution, and saturated brine, MgSO4 dried, filtered and evaporated to dryness, the product obtained from ethyl acetate and petroleum ether (1: 2, volume ratio) was recrystallized to obtain (yield 97%, mp: 107 ° C, [a] 26D =
21.97 (103.76mg / 20ml, MeOH)).
Example 4: [0074] (R) -4- benzyl-7-methyl-1,4-diaza Synthesis heptane-2,5-dione
The 3g (8.2mmol) (R) – methyl _2_ (N- benzyl _3_ ((tert-butoxycarbonyl) amino) butanamide yl) acetate were added to the flask, and dissolved in ethyl acetate was added IOml added 30ml45% of acetate hydrochloride gas, 25 ° C reaction 4h.Evaporated to dryness, and saturated NaHC03 solution, methylene chloride and ethanol (2: 1, volume ratio) was extracted, MgSO4 organic layer was dried and evaporated to dryness to give a pale yellow oil.It was dissolved in 30ml MeOH and dried added 0.487g (9.02mmol) NaOMe, under nitrogen, 10 ° C reaction 4h.Quenched with saturated NH4Cl solution was added 5 ^ Na2CO3 solution, methylene chloride and ethanol (2: 1, volume ratio) was extracted organic layers were combined, MgSO4 dried, rotary evaporated to give a white solid (yield 98.93%, mp: 122_123 ° C , [a] 26D = 33.49 (112.87mg / 20ml, MeOH)).IH NMR (600MHz, DMS0_d6) δ ppm7.77-7.76 (bd, 1H), 7.33-7.25 (m, 5H), 4.59-4.53 (m, 2H), 4.10-4.02 (m, 2H), 3.65-3.62 ( m, 1H), 2.93-2.90 (m, 1H),
2.76-2.72 (m, 1H), 1.14-1.13 (d, 3H); (FIG. 2) MS (ESI) m / z233.10 ([M + H] +) ..
The 3g (8.2mmol) (R) – methyl _2_ (N_ _ _3 benzyl ((tert-butoxycarbonyl) amino) butanamide yl) acetate were added to the flask, dissolved in dichloromethane was added IOml adding 30ml methylene chloride solution containing 10% of CF3COOH of, 25 ° C reaction 4h.Evaporated to dryness and saturated NaHCO3 solution, methylene chloride and ethanol (2: 1, volume ratio) was extracted, MgSO4 organic layer was dried and evaporated to dryness to give a yellow oil.This was dissolved in 50ml of dry toluene, was added 0.156g (6.5mmol) of sodium hydride, 110 ° C reaction 4h.After cooling to room temperature, quenched with saturated NH4Cl solution, 5% Na2CO3 solution is added, methylene chloride and ethanol (2: 1, volume ratio) was extracted organic layers were combined, MgSO4 dried, rotary evaporated to give a white solid 1.83g (yield 90.34 %, mp: 122-123 ° C, [a J26D = 33.49 (112.87mg / 20ml, MeOH)).
The 3g (8.2mmol) (R) – methyl _2_ (N_ _ _3 benzyl ((tert-butoxycarbonyl) amino) butanamide yl) acetate were added to the flask, methanol was added IOml dissolved, 30ml36% methanol solution of hydrochloric acid gas, 25 ° C reaction 4h.Evaporated to dryness, and saturated NaHC03 solution, methylene chloride and ethanol (2: 1, volume ratio) was extracted, MgSO4 organic layer was dried and evaporated to dryness to give a yellow oil.This was dissolved in 50ml of dry toluene, was added 1.7g (12.3mmol) of potassium carbonate, 110 ° C reaction 8h.After cooling to room temperature, quenched with saturated NH4Cl solution, 5% Na2CO3 solution is added, methylene chloride and ethanol (2: 1, volume ratio) was extracted organic layers were combined, MgSO4 dried, rotary evaporated to give a white solid (yield 95.78%, mp: 122_123 ° C, [a] 26D = 33.49 (112.87mg / 20ml, MeOH)).
The 3g (8.2mmol) (R) – methyl _2_ (N_ _ _3 benzyl ((tert-butoxycarbonyl) amino) butanamide yl) acetate were added to the flask, methanol was added IOml dissolved, 30ml of 36% methanol containing hydrochloric acid gas solution, 25 ° C reaction 4h.Evaporated to dryness and saturated NaHCO3 solution, methylene chloride and ethanol (2: 1, volume ratio) was extracted, MgSO4 organic layer was dried and evaporated to dryness to give a yellow oil.Which was dissolved in 30ml of ethyl acetate and dried, was added 0.88g (16.4mmOl) sodium alkoxide, 10 ° C the reaction 6h.Quenched with saturated NH4Cl solution was added 5 ^ Na2CO3 solution, methylene chloride and ethanol (2: 1, volume ratio) was extracted organic layers were combined, MgSO4 dried, rotary evaporated to give a white solid (yield 93%, mp: 122_123 ° C , [a] 26D = 33.49 (112.87mg / 20ml, Me0H)) ο Example 8:
The 3g (8.2mmol) (R) – methyl _2_ (N- benzyl _3_ ((tert-butoxycarbonyl) amino) butanamide yl) acetate were added to the flask, methanol was added IOml dissolved, 30ml hydrochloric acid gas containing 36% methanol solution, 25 ° C reaction 4h.Evaporated to dryness, and saturated NaHC03 solution, methylene chloride and ethanol (2: 1, by volume) to extract, MgS04 organic layer was dried and evaporated to dryness to give a yellow oil.Which was dissolved in 30ml of dry methanol was added 2.07g (20.5mmol) of triethylamine, 60 ° C the reaction 8h.After cooling to room temperature, quenched with saturated NH4Cl solution, 5% Na2CO3 solution is added, methylene chloride and ethanol (2: 1, volume ratio) was extracted organic layers were combined, MgSO4 dried, rotary evaporated to give a white solid (yield 92.68%, mp: 122_123 ° C, [a] 26D = 33.49 (112.87mg / 20ml, MeOH)).
The 3g (8.2mmol) (R) – methyl _2_ (N_ _ _3 benzyl ((tert-butoxycarbonyl) amino) butanamide yl) acetate were added to the flask, methanol was added IOml dissolved, 30ml hydrochloric acid gas containing 36% methanol solution, 25 ° C reaction 4h.Evaporated to dryness, and saturated NaHC03 solution, methylene chloride and ethanol (2: 1, by volume) to extract, MgSO4 organic layer was dried and evaporated to dryness to give a yellow oil.Which was dissolved in 30ml of dry acetonitrile was added 1.38g (12.3mmol) of potassium t-butoxide, 30 ° C the reaction 8h.Quenched with saturated NH4Cl solution was added 5 ^ Na2CO3 solution, methylene chloride and ethanol (2: 1, volume ratio) was extracted organic layers were combined, MgSO4 dried, rotary evaporated to give a white solid (yield 89.86%, mp: 122_123 ° C , [a] 26D = 33.49 (112.87mg / 20ml, MeOH)).
Example 10:
(R) -1- benzyl-5-methyl-1,4-Synthesis diazepan the
A 1.4g (R) -4- benzyl-7-methyl-diaza heptane _2,5_ _1,4_ dione (6mmol) was dissolved in 60ml dry THF, was added portionwise under ice- 1.35g LiAlH4 (36mmol), 25 ° C was stirred for 4h.Cooled to -10 ° C, was added 1.5mlH2O quenched and then 1.5mll5% NaOH, 4.5ml H20, part MgSO4, stirring lh, filtration, spin dried to give 1.2g oil (yield 97.56%, [a] 29D = -5.87 (200.86mg / 20ml, CHCl 3)).ee> 99%, Chrom Techchiral-AGP150 * 4mm Mobile phase: Ammonium dihydrogen sulfate (IM): acetonitrile = 99: 1, column temperature: 30 ° C, flow rate: 0.5ml / Hiin0 IH NMR (600MHz, DMS0_d6) δ ppm7.32-7.20 (m, 5Η), 3.57 (s, 2Η), 3.48 (bs, 1Η), 2.99-2.95 (m, 1Η), 2.86-2.82 (m, 1Η), 2.72-2.68 (m, 1Η ), 2.65-2.61 (m, 1Η), 2.58-2.49 (m, 3Η), 1.75-1.70 (m, 1Η), 1.46-1.41 (m, 1Η), 1.01-1.00 (d, 3Η); (Figure 3 .) MS (ESI) m / z205.10 ([M + H] +) [0095] Example 11:
(R) -1- benzyl-5-methyl-1,4-Synthesis diazepan the
A 1.4g (R) -4- benzyl-7-methyl-diaza heptane _2,5_ _1,4_ dione (6mmol) was dissolved in 60mlTHF TEMPERATURE dropwise 2 equivalents of borane ( 12mm0l), reflux 8h.Cooled to _10 ° C, quenched by addition of methanol, adjusted pH = 3, stirred for 2h, sodium carbonate adjusted to pH = 10, extracted with methylene chloride three times, the combined organic layer, MgSO4 drying, rotary evaporation.(Yield 95.32%, [a] 29D = -5.87 (200.86mg / 20ml, CHCl 3)).[0098] Example 12:
(R) -1- benzyl-5-methyl-1,4-Synthesis diazepan the
The (R) -4- benzyl-7-methyl-1,4-diaza heptane-2,5-dione (5mmol) was dissolved in 15ml dry THF, was added under ice-cooling to a solution of Ig sodium boron (27mmol) in 15ml dry THF hydride was added dropwise a solution of iodine in 20ml THF 12mmol dried under nitrogen, at reflux for 6h.Cooled to (TC, quenched 5ml3N HCl was added, followed by addition of 8ml3NNaOH, liquid separation, the aqueous layer was extracted three times with ether, the combined organic layer was washed with saturated brine, MgSO4 drying, filtration, spin dry (yield 90.34%, [a ] 29D = -5.87 (200.86mg / 20ml, CHC13)).
Example 13:
(R) – (4_-Benzyl-7-methyl-1,4-diazepan-1-yl) (5-methyl _2_ (2H-1,2,3_ three
Synthesis of 2-yl) phenyl) methyl ketone
The 3g (R) -1- benzyl-5-methyl-1,4-diazepane (14.7mmol), 3.66g5_ methyl -2- (2Η-1, 2,3- triazol-2-yl) benzoic acid (18.03mmol) was dissolved in DMF, 2.43gHOBt (18.55mmol), 6ml TEA (42.75mmol), 3.45g EDC (17.99mmol), warmed to 50 ° C, the reaction 2h.Was added a saturated NaHCO3 solution and EA, the aqueous layer was washed three times with EA, the combined organic layers.The organic layer was washed with citric acid solution, the product salified fully into the aqueous phase, the aqueous phase was washed with EA after the addition of sodium carbonate to adjust the pH> 9, EA and washed three times, the organic layers combined, washed with water and saturated brine, MgSO4 dried, rotary dried, PE and EA (4: 1) and recrystallized (yield 98.36%, mp: 108-109 ° C, [α] 31D = -58.37 (202.5mg / 20ml, MeOH)).IH NMR (600MHz, DMS0_d6) δ ppm8.00-7.76 (m, 3H), 7.37-7.17 (m, 7H), 4.40-4.09 (m, 1H), 3.63-3.48 (m, 2H), 3.44-3.02 ( m, 3H), 2.82-2.75 (m, 1H), 2.63-2.47 (m, 1H), 2.63-2.14 (m, 5H), 2.02-1.63 (m, 2H), 1.17-0.99 (m, 3H); (Figure 4) MS (ESI) m / z390.30 ([M + H] +) [0105] Example 14:
The 3g (R) -1- benzyl-5-methyl-1,4-diazepane (14.7mmol), 2.98g5_ methyl -2- (2H-1, 2,3- triazol-2-yl) benzoic acid (14.7mmol) was dissolved in methylene chloride, was added 18.55mmolHOAt, 6ml TEA (42.75mmol), 2.86g CDI (17.64mmol), 30 ° C reaction 4h.Was added a saturated NaHC03 solution and EA, the aqueous layer was washed three times with EA, the combined organic layers.The organic layer was washed with citric acid solution, the product salified fully into the aqueous phase, the aqueous phase was washed with EA after the addition of sodium carbonate to adjust the pH> 9, EA and washed three times, the combined organic layer was washed with saturated brine paint, MgS04 drying, spin dry, PE and EA (4: 1) and recrystallized (yield 96.45%, mp = 108-109 ° C, [a J31D = -58.37 (202.5mg / 20ml, MeOH)).
Example 15:
(R) – (4_-Benzyl-7-methyl-1,4-diazepan-1-yl) (5-methyl _2_ (2H-1,2,3_ triazol – 2- yl) phenyl) -methanone [0110] The 3g (R) -1- benzyl-5-methyl-1,4-diazepane (14.7mmol), 3.28g5_ methyl – 2- (2Η-1,2,3- triazol-2-yl) benzoic acid (16.17mmol) was dissolved in acetone was added 2.43gHOBt (18.55mmol), 6ml TEA (42.75mmol), 3.33gDCC (16.17mmol) After the addition of sodium carbonate, 3 (TC reaction 4h. Saturated NaHCO3 solution was added and EA, the aqueous layer was washed three times with EA, the combined organic layers. The organic layer was washed with citric acid solution, the product salified fully into the aqueous phase, the aqueous phase was washed with EA adjust pH> 9, EA and washed three times, the organic layers combined, washed with water and saturated brine, MgSO4 dried, rotary dried, PE and EA (4: 1) and recrystallized (yield 92.43%, m.ρ .: 108-109 .. , [a J31D = -58.37 (202.5mg / 20ml, MeOH)).
A 2.08g (R) – (4_ _1,4_ Benzyl-7-methyl-diazepan-1-yl) (5_-methyl -2- (2Η-1, 2, 3- triazol-2-yl) phenyl) methyl ketone (7.2mmol) was dissolved in 20ml THF, 10% of the PdC12,50 ° C through the H2 reaction 2h.Filtration, rotary evaporation to give the product (yield 93.24%, [a] 26D = -14.36 (199.12mg / 20ml, MeOH)).
A 2.08g (R) – (4_ _1,4_ Benzyl-7-methyl-diazepan-1-yl) (5_-methyl -2- (2H-1, 2, 3- triazol-2-yl) phenyl) methanone (7.2mmol) was dissolved in 20ml of methanol was added 10% Pd / C, was added ammonium formate (21.6_ο1), the reaction was refluxed for 6h.Filtration, rotary evaporation to give the product (yield 92.68%, [a] 26D = -14.36 (199.12mg / 20ml, MeOH)).
Example 19
Synthesis Suvorexant of
To 0.9g (R) – (7- methyl-1,4-diazepan-1-yl) (methyl 5_ _2_ (2H-1,2,3_ triazol-2 yl) phenyl) methanone (3.0lmmol) of IOml DMF was added 0.57g2, 5- dichlorobenzene and oxazole (3.03mmol), 0.91g TEA (9mmol), heated to 75 ° C, the reaction 2h.Cooled to room temperature, EA dispersion, washed with a saturated NaHCO3 solution, saturated brine, MgSO4 dried, rotary evaporated to give a white solid (yield 93.02%, mp: 128-129 ° C, [a] 3C1.9D = -11.7 (199.99 mg / 20ml, MeOH)).IH NMR (600MHz, DMS0_d6) δ ρρm8.05-7.88 (m, 2Η), 7.82-7.78 (m, 1Η), 7.42-7.25 (m, 2Η), ζ, 06-7.00 (m, IH), 4.29- 4.06 (m, 1Η), 4.01-3.72 (m, 2Η), 3.66-3.49 (m, 2Η), 2.10 (s, 3Η), 2.06-2.01 (m, IH), 1.50 (m, 1Η), 1.78- 1.50 (m, 1Η), 1.14-1.13 (d, 3Η); (FIG. 6) MS (ESI) m / z451.20 ([Μ + Η] +).
The compound of the formula I is disclosed as an antagonist of orexin receptors in US Patent 7,951,797, US Patent Application Publication US 2008/0132490, PCT PatentPublication WO 2008/069997, Cox et al, J. Med. Chem. 2010, 53, 5320-5332, Strotman et al, JACS, 2011, 133(21), 8362-8371, and Baxter et al, Org. Process Res. & Dev., 201 1, 15(2) 367- 375.
This compound is disclosed as having activity in antagonizing the human orexin-1 (OX1) receptor with a Ki of 0.55 nM and in antagonizing the human orexin-2 (0X2) receptor with a Ki of 0.35 nM. The processes disclosed in US Patent 7,951,797, US Patent Application Publication US 2008/0132490, PCT Patent Publication WO 2008/069997, Cox et al, J. Med. Chem. 2010, 53, 5320-5332, Strotman et al, JACS, 201 1, 133(21), 8362-8371, and Baxter et al, Org. Process Res. & Dev., 2011, 15(2) 367-375 are lengthy, suffer from low yields, necessitate multiple protecting groups, rely on chiral chromatography to prepare a single isomer and require microwave technology to prepare the acid intermediate. Relative to the processes disclosed in US Patent 7,951,797, US Patent Application Publication US 2008/0132490, PCT Patent Publication WO 2008/069997, Cox et al, J. Med. Chem. 2010, 53, 5320-5332, Strotman et al, JACS, 2011, 133(21), 8362-8371, and Baxter et al, Org. Process Res. & Dev., 201 1, 15(2) 367- 375, the present invention may provide improved processes for the efficient, scalable, chromatography-free and cost-effective preparation of the formula I, to give higher isolated yield of the subject compound.
EXAMPLE 1
2. DMF
5-Chloro-l,3-benzoxazole-2-thiol (9a)
2-Amino-4-chlorophenol (2.50 kg, 17.4 mol) was charged to a vessel and suspended in water (52 L) and methanol (10.4 L). High dilution was required to prevent slow and difficult filtration of the product. The mixture was stirred, cooled to 0 °C, then thiophosgene (2.00 kg, 17.4 mol) was added to the suspension ensuring that the internal temperature remained at 5 °C throughout the addition. Water (8 L) and methanol (2 L) were added to aid stirring and the slurry was warmed to 13 °C for 1 h, followed by aging at 20 °C for a further 1 h. The slurry was then filtered and the solid washed with water (5 L). The batch was repeated and combined to dry in a vacuum oven (T = 40 °C) for 15 h to give 9-a (5.81 kg, 31.3 mol). The data corresponds to the commercially available material. XH NMR (400 MHz, d6-DMSO): δ 7.51 (d, 1 H, J = 9.2 Hz), 7.307.26 (m, 2 H). 13C NMR (100.6 MHz, d6-DMSO): δ 181.2, 147.4, 133.1, 129.7, 123.9, 1 11.6, 110.8. HRMS (ESI): m/z [M+ + H] calcd for C7H4CINOS: 185.9780; found: 185.9785.
Thiol 9a (10.5 kg, 54.6 mol) was added to a vessel and suspended in DCM (141 kg). Oxalyl chloride (10.4 kg, 82.3 mol) was added (slightly endothermic) followed by DMF (40.0 kg, 547 mol) over 1.25 h, such that the batch temperature was≤ 25 °C. The batch was aged at 20 °C for approximately 30 min, HPLC analysis showed reaction to be complete. The batch was cooled to 10 °C then triethylamine (16.64 kg, 164.4 mol) was added via a sub-surface sample line at such a rate as to maintain a batch temperature of≤ 10 °C. A sub-surface addition protocol was required to prevent build up of triethylamine hydrochloride solid on the walls of the vessel. The batch was cooled to 0 °C, then a solution of N-Boc-ethylenediamine (10.5 kg, 61.2 mol) in DCM (10 kg) was added such that the batch temperature was≤ 10 °C. The reaction was warmed to 20 °C and stirred for 2.5 h, HPLC analysis showed the reaction to be complete. Water (63.6 kg) was charged to the batch and the mixture stirred for 5 min. The layers were separated and the aqueous phase re-extracted with DCM (42.2 kg). The organic solutions were then combined and approximately half of the total DCM volume was distilled from the batch under vacuum whilst maintaining a temperature of≤ 40 °C. MeCN (83.3 kg) was then added and the remaining DCM removed by distillation (0.5 mol % DCM left by XH NMR wrt MeCN). MVK (4.61 kg, 65.8 mol) was added to the batch followed by DBU (4.17 kg, 27.4 mol) such that the temperature was≤ 20 °C. The batch was aged for 10 h at 20 °C then analyzed by HPLC. The reaction was then diluted with water (42.4 kg) and aged for a further 30 min. The mixture was filtered and the slurry washed with MeCN (33.3 kg). The solid was washed with MeCN (-10 L) then dried in a vacuum oven (T = 60 °C) for 22 h. MVK adduct 10 (15.5 kg) was isolated as an off-white solid, mp 145-148 °C. ¾ NMR (400 MHz, CDC13): δ 7.24 (d, 1 H, J = 2.3 Hz), 7.09 (d, 1 H, J = 8.5 Hz), 6.91 (dd, 1 H, J = 8.5, 2.3 Hz), 5.06 (s, 1 H, br), 3.73 (t, 2 H, J = 6.7 Hz), 3.63 (t, 2 H, J = 6.1 Hz), 3.37 (d, 2 H, br), 2.89 (t, 2 H, J = 6.7 Hz), 2.14 (s, 3H), 1.33 (s, 9 H). 13C NMR (100.6 MHz, CDC13): 8 206.7, 163.0, 156.0, 147.4, 144.6, 129.2, 120.3, 116.6, 109.2, 79.4, 49.3, 44.3, 41.9, 39.1, 30.2, 28.3. HRMS (ESI): m/z [M+ + H] calcd for
382.1534; found: 382.1544.
EXAMPLE 2
□ HMDS, THF/hexane (3.6:1.0), -25 to -15 °C; NBS
5-Chlorobenzoxazole (3-2)
To a 250 mL 3-neck round bottom flask equipped with a distillation head, glass stopper, septum, thermocouple and magnetic stir bar was charged 2-amino-4-chlorophenol (20.00 g, 0.139 mol). The solid was dissolved in THF (60 mL) and p-TsOH (0.265 g, 1.39 mmol) was added. The brown solution was warmed to 60 °C over 10 min and aged for 90 min. HPLC assay of the reaction mixture showed 1 LCAP unreacted starting material. The temperature was increased from 60 °C to 74 °C, and at 63 °C solvent distillation began. A total of 58 mL was collected during the first distillation. The mixture was diluted with THF (60 mL) and a total of 67 mL of solvent was removed between 71 and 84 °C. The mixture was again diluted with THF (60 mL) and 61 mL of solvent was removed between 74 and 1 14 °C. The dark brown solution was cooled to room temperature. The final mass of the solution was 27.96 g. Analysis of the crude stream by XH NMR showed 0.1 wt% MeOH present in the sample. XH NMR (500 MHz, CDC13): δ = 8.10 (s, 1H), 7.76 (d, J= 1.5 Hz, 1H), 7.50 (d, J= 8.7 Hz, 1H), 7.36 ppm (dd, J= 8.7, 1.7 Hz, 1H).
A 500 mL 3-neck round bottom flask equipped with a septum, thermocouple, 125 mL addition funnel, inert gas inlet and magnetic stir bar was purged with nitrogen for 10 min. Hexamethyldisilazane (42 mL, 0.20 mol) and THF (78 mL) were charged against positive nitrogen pressure. The addition funnel was charged with a hexane solution of n-butyllithium (78.0 mL, 195 mmol). The amine solution was cooled to -52 °C and n-butyllithium was added over 84 min, resulting in a temperature increase to 12.5 °C over the course of the addition. The resulting lithium hexamethyldisilazide solution was removed from the cooling bath and aged for 30 minutes. To a 500 mL 3 -neck round bottom flask equipped with a septum, thermocouple, inert gas inlet and magnetic stir bar was charged 5-chlorobenzoxazole (20.00 g, 130 mmol). The gray solid was dissolved in THF (100 mL) and the resulting colorless solution was cooled to -25 °C. The freshly prepared lithium hexamethyldisilazide solution was added via cannula over 80 minutes. The temperature of the anion solution was maintained between -25 and -15 °C during the addition. The resulting dark brown solution was aged for 90 minutes between -25 and -15 °C. To a 1000 mL 3-neck round bottom flask equipped with a Claisen adapter, septum,
thermocouple, inert gas inlet, stir rod bearing, and blade was charged THF (100 mL) and N- bromosuccinimide (34.8 g, 195 mmol). The resulting slurry was cooled to -20 °C and the anion solution was added via cannula over 150 minutes. During the addition the anion solution and reaction mixture were maintained between -25 and -15 °C. The resulting brown slurry was removed from the cooling bath and aged for 50 minutes while warming to room temperature. To the resulting bromide slurry was added a solution of ethanolamine (12.6 mL, 208 mmol) in MeCN (38 mL) via syringe pump over 5 hours. During the addition the reaction temperature was maintained between 20 and 27 °C. The resulting brown slurry was aged at room temperature overnight. The reaction mixture was cooled in an ice water bath and the septum replaced with a 50 mL addition funnel charged with concentrated HC1 (32 mL, 390 mmol). The acid solution was added over 10 min, during which time the addition the temperature increased from 10 to 20 °C. The reaction mixture was removed from the ice water bath and aged for 5 min. A 20% (w/w) solution of K2HPO4 in water (170 mL) was added and the resulting biphasic mixture was transferred to a seperatory funnel. The flask was washed with THF (3x, 10 mL) and the washings were added. The aqueous phase was cut; the organic phase was washed with 20% (w/w) K2HPO4 in water (200 mL), separated and analyzed. The crude reaction stream had a total mass of 396.47 g. By quantitative HPLC assayed 25.81 g of 3-3 in the organic phase. XH NMR (500 MHz, DMSO-i¾): δ = 8.17 (t, J= 5.6 Hz, 1H), 7.34 (d, J= 8.4 Hz, 1H), 7.25 (d, J= 1.8 Hz, 1H), 6.97 (dd, J= 8.4, 1.8 Hz, 1H), 4.81 (t, J= 5.4 Hz, 1H), 3.56 (q, J= 5.7 Hz, 2H), 3.35 pm (q, J= 5.8 Hz, 2H).
To a 1000 mL 3-neck round bottom flask equipped with a septum, thermocouple, inert gas inlet and magnetic stir bar was charged 3-3 (25.2 g, 119 mmol). To this flask was added 126 mL DMF, 12.2 mL methyl vinyl ketone (148 mmol) and 0.119 mL 10M NaOH (1.19 mmol). The reaction was then aged for 6 hours, at which time conversion was judged to be complete by HPLC. The solution was diluted with 252 mL iPAc and cooled to 0 °C, then 23.1 mL Et3 (166 mmol) followed by dropwise addition of 12.0 mL methanesulfonyl chloride (154 mmol) over 45 minutes, maintaining internal temperature less than 10 °C. After a further 30 minutes, conversion was judged to be complete by HPLC. The solution was washed with 3x 63 mL 5 w/w% aqueous aHC03 solution, then 66 mL water. After cutting the aqueous layer, the organics were reduced to approximately two volumes or 50 mL iPAc. The organics were then agitated by an overhead stirrer during slow addition of 151 mL n-Heptane over 4 hours. Over this time a crystalline white precipitate developed, and was allowed to stir overnight. At this time there was a thick slurry, which was filtered and washed with 2x 50 mL 90: 10 n- HeptaneTPAc, and after drying with a nitrogen stream over the filter pad, 3-4 was obtained as a white crystalline solid (34.6 g., 96 mmol). ‘H NMR (500 MHz, CDC13): δ = 7.29 (s, 1H), 7.16 (d, J= 8.2 Hz, 1H), 6.97 (d, J= 7.8 Hz, 1H), 4.46 (s, 2H), 3.92 (s, 2H), 3.81 (t, J= 5.9 Hz, 2H), 2.98-2.92 (m, 5H), 2.16 (s, 3H).
EXAMPLE 3
5-Chloro-2-((R)-5-methyl-[l,4]diazepan-l-yl)-benzooxazole hydrochloride (R-11) To a 1000 mL 3 -necked flask was charged isopropylamine hydrochloride (25.8 g., 270 mmol) and 525 mL 0.1 M aqueous triethanolamine solution. To this was added 750 mg pyridoxal 5′-phosphate hydrate (PLP) and 3.0 g of the transaminase polypeptide having the amino acid sequence SEQ ID NO: l and the suspension was stirred until all components dissolved. The transaminase polypeptide having the amino acid sequence SEQ ID NO: 1 was obtained as disclosed in US Patent Publication US 2010/0285541 for the identical sequence “SEQ ID NO: 1 10” therein. The solution was heated to 40 °C and the pH of the solution was adjusted to pH 8.5 with an aqueous 4M solution of isopropylamine. Mesylate 3-4 was added as a 225 mL DMSO solution via syringe over 6 hours, and the resulting mixture stirred for a further 5 hours. At this time, the solution was poured into a 3L separatory funnel and extracted with 1.5 L of 1 : 1 iPAc:IPA. The aqueous layer was cut then extracted again with 750 mL 4: 1 iPAc:IPA. The organics were combined, then washed with 750 mL brine. Then the organics were concentrated with IPA flushing to establish a 45 mL solution in IPA which was then treated with 4.6M HC1 in IPA (9.94 mL, 45.7 mmol) via dropwise addition. The resulting solution was stirred vigorously while 52 mL IP Ac was added slowly over 5 hours, creating a slurry of HQ salt 6. The slurry was then slowly cooled to 0 °C and allowed to stir overnight. At this time the slurry was filtered and dried with a nitrogen stream over the filter pad, providing R-11 as a white crystalline solid (7.80 g., 25.8 mmol). ¾ NMR (500 MHz, CD3OD): δ = 7.13-7.10 (m, 2H), 6.97 (dd, J= 8.2, 1.8 Hz, 1H), 3.99-3.79 (m, 3H), 3.67-3.57 (m, 3H), 3.39-3.33 (m, 1H), 2.24 (s,
1H), 2.12-2.07 (m, 1H), 1.42 (d, J= 6.7 Hz, 3H).
EXAMPLE 4
19 5
5-Methyl-2-[l,2,3]triazol-2-yl-benzoic acid (5) The iodide 19 (6.04 kg, 23.0 mol), THF (45 L) and DMF (9.0 L) were charged to a vessel. Copper iodide (218 g, 1.15 mol) and potassium carbonate (7.94 kg, 57.4 mol) were added and the mixture heated to an internal temperature of 40 °C. 1,2,3-Triazole (3.16 kg, 46.0 mol) was added as a solution in THF (6.0 L) over half an hour (no exotherm) and heating continued to 65 °C (again no exotherm observed) and the reaction monitored by HPLC. Once complete N,N-dimethylethylenediamine (244 mL, 2.30 mol) was added and mixture cooled to RT. Aqueous 3.6 M HC1 (36 L) was added (exotherm) and the mixture extracted twice with ethyl acetate (2 x 30 L). The combined organics were washed with LiCl solution (2 x 20 L). The acid solution assayed for 3.79 kg of 5 (81%) and 4.64 kg of 5 and 20 combined (99%). A solution of acids 5 and 20 (approx. 4.64 kg, 22.9 mol) in THF and EtOAc (approx. 1 10 L) was concentrated to low volume. THF (90 L) was added and the solvent composition checked by XH NMR to ensure most ethyl acetate had been removed. Sodium tert-butoxide (2.42 kg, 25.2 mol) was added slowly as a solid over 1-2 h (slight exotherm), allowing the sodium salt to form and stirred overnight at RT. The liquors showed a 45:55 ratio of product: starting material and the solid was collected by filtration, washed with THF (2 x 20 L) and dried in a vacuum oven (T = 40 °C) for 15 h to afford 4.22 kg of crude sodium salt. The crude sodium salt (4.22 kg, 14.9 mol) was charged to a 50 L vessel and 3.6 M HC1 (21.2 L) was added with cooling. The slurry was then stirred at room temperature for 16 h and the off-white solid isolated by filtration. The cake was washed with water (11 L) and iP Ac/Heptane (2 x 5L), then dried in a vacuum oven (T = 35 °C) for 15 h to give 3.10 kg of crude acid 5 (97.9 LCAP, 92 wt%, corrected weight 2.85 kg, 61% yield from 19). The acid 5 (2.85 kg corrected, 14.0 mol) was charged to a 50 L vessel and EtOAc (28 L) and dilute 0.22 M HC1 (14 L) were added and the mixture stirred until two clear phases resulted. The aqueous layer was removed and the organic layer filtered to remove any particulate matter. The ethyl acetate was reduced to about 8 L and then heptane (15.6 L) was added over 1 h and the liquors sampled to check for appropriate losses. The solid was isolated by filtration, washed with heptane:ethyl acetate (3 : 1 , 4 L) and dried on the filter under nitrogen to give 2.81 kg of acid 5. m.p. 167.5 °C. XH NMR (400 MHz, d6-DMSO): δ 12.09 (br s, 1H), 8.04 (s, 1H), 7.62 (d, 1H, J = 8.4 Hz), 7.58 (d, 1H, J = 1.2 Hz), 7.49 (dd, 1H, J = 8.4, 1.2 Hz), 2.41 (s, 3H). 13C NMR (100.6 MHz, d6-DMSO): δ 168.0, 139.2, 136.4, 135.8, 132.5, 130.3, 128.7, 124.8, 20.9. HRMS (ESI): m/z [M+ + H] calcd for C10H9N3O2: 204.0773; found: 204.0781. EXAMPLE 5
A round bottom flask was charged 6.86 g of 5-methyl-2-[l,2,3]triazol-2-yl- benzoic acid (5) along with 7.0 vol or 70 mis of dry iPAc (KF < 200 ppm) forming a slurry. To this was charged 0.73 g of DMF then the system was purged thoroughly with nitrogen and temperature was set at 20°C-25°C. 5.04 g of oxalyl chloride was added while maintaining 20°C- 25°C and controlling off-gassing since it is extremely vigorous. With the feed of oxalyl chloride the previous slurry dissolved. The batch was aged for 1 hr, sampled for acid chloride formation (< 1 LCAP) and allowed to proceed to amidation. In a separate vessel a solution of potassium carbonate was prepared in 5.0 vol or 50 mL water (note: exotherm). The solution was cooled to 0 °C. When acid chloride (above) was prepared, added 2.5 vol or 25 mL iPAc to the aqueous solution with overhead stirring, then added 10.0 g. amine hydrochloride salt (R-ll) to solution, and stirred for 15 minutes. Then using a cannula, the acid chloride solution was transferred over from separate vessel over the course of 1 hour, maintaining less than 5°C internal temperature. The vessel was flushed with 2.5 vol or 25 mL iPAc and sampled to determine completion. The slurry was heated to 40 °C. Upon reaching 40 °C, 1.5 vol or 15 mL Acetonitrile was and agitated for 5 minutes, and all material went into solution (98% AY observed). Agitation was stopped. After phase separation, the aqueous layer was cut, the organics were stirred with DARCO (10 wt% 6 basis) at 40°C for 3 hours, then filtered hot and taken through to
crystallization. Additional product was recovered from the carbon with an iPAc flush.
The batch was concentrated in iPAc and flushed to 7.5 vol (L/Kg of 1) and heated to 80-85C until complete dissolution. The solution was cooled to 65 °C linearly over 2 hrs, and the agitation speed was adjusted to high. At 65 °C, the solution was charged with 0.3 wt% seed in n-Heptane and aged for 1 hour. After the age and confirmation of the seed bed, the batch was cooled to 45 °C over 2.5 hrs. At this time a solvent switch was conducted at constant volume to a ratio of 90: 10 n-Heptane: iP Ac. The material was filtered hot at 45 °C, the cake was washed with 3 vol (L/Kg of 1) of 90: 10 n-Heptane :iP Ac twice, followed by 3 vol (L/Kg of 1) of n- Heptane twice. The cake was dried at 70 °C under vacuum to give 14.4 g. 1 (31.8 mmol,) as a crystalline white powder.
A reaction vessel was charged with 213.4 g of triazole acid (5) along with 7.4 vol or 2236 mis of dry iPAc (KF < 200 ppm) forming a slurry. To this charge was added 21.93 g of DMF then the system was purged thoroughly with nitrogen and temperature was maintained at 20- 25C. Charged 152.3 g of oxalyl chloride while maintaining 20-25C and control of off-gassing since it is extremely vigorous. With the feed of oxalyl chloride the previous slurry all dissolved. The batch was aged for 1 hr. The reaction was sampled for Acid Chloride formation (< 1 LCAP) and proceeded to distillation. Distillation was conducted down to 11 18 ml or constant volume distillation using 7.4 vol of fresh iPAc under vacuum maintaining less than 30°C.
In a separate vessel prepared a solution of 302.2 g of amine hydrochloride salt (R-ll) in 15.3 vol or 4624 mis of dry iPAc (KF < 200 ppm) to form a slurry. Then transferred the acid chloride solution using a cannula over from a separate vessel followed by flushing the vessel with 6.9 vol or 2085 mis of iPAc. With the amine and acid chloride in the same vessel began addition of 404.8 g of triethylamine. This charge was made over 1 to 4 hrs at a temperature between 20-40C with a desired control of the temperature between 20-30C. Once feed of the TEA was complete, the batch was aged for lhr and then sampled to determine completion.
Once the batch was complete, charged 7.4 vol of water or 2236 mis and then heated the solution to 40C. Once at 40C, the mixture was aged 5 minutes then agitation was stopped. The phases separated but there was an appreciable rag layer so it was allowed to settle and the rag was cut along with the aqueous layer. The aqueous rag was filtered then the aqueous layer was back extracted with 3.5 vol or 1058 ml of iPAc and all iPAc layers were combined.
The batch was recycled in iPAc (~60 g per kg of iPAc) via a Cuno filter (1 bundle per 39 Kg Amine HC1 Salt) for several hours at 40°C. The batch was drummed off through a sparkler filter and additional material was recovered from the carbon with an iPAc flush.
The batch was concentrated in iPAc and flushed to 7.5 vol (L/Kg of product) and heated to 80-85°C until complete dissolution. The mixture was cooled to 65°C linearly over 2 hrs, and agitation speed was adjusted to high from this point forward. At 65°C, the mixture was charged with 0.3 wt% of [(R)-4-(5-chloro-benzooxazol-2-yl)-7-methyl-[l,4]diazepan-l-yl]-(5- methyl-2-[l,2,3]triazol-2-yl-phenyl)-methanone seed in n-Heptane and aged for 1-3 hour. After the age and confirmation of the seed bed, the batch was cooled to 45°C over 2.5 hrs. A solvent switch was conducted at constant volume to a ratio of 90: 10 n-Heptane :iP Ac.
The batch was wet milled to a uniform particle size and filter hot at 45C. The cake was washed with 3 vol (L/Kg of product) of 90: 10 n-Heptane :iP Ac twice, followed by 3 vol (L/Kg of product) of n-heptane twice. The cake was dried at 70°C under vacuum.
Suvorexant (MK-4305) is a potent dual Orexin antagonist under development for the treatment of sleep disorders at Merck. The key transformation is an asymmetric Ru-catalyzed transfer hydrogenation (using a modified Noyori RuCl(p-cymene)(DPEN) complex) of an in-situ generated cyclic imine resulting in the formation of the desired chiral diazepane in 97% yield and 94.5% ee. Mechanistic studies have revealed that CO2 (derived from the formic acid) has pronounced effect on reaction outcome. Studies have determined that the efficiency of the Ru-catalyst, the composition of the resulting amine (via carbamate formation), and the reaction kinetics are mediated by the amount of CO2 generated during the reaction. The efficiency of the reductive-amination can be enhanced by either purging the CO2 or by trapping the newly formed nucleophilic secondary amine.
A new synthetic route to drug candidate 1, a potent and selective dual orexin antagonist for the treatment of sleep disorders, has been developed. The key acyclic precursor 10 was prepared in a one-step process in 75% isolated yield from commercially available starting materials using novel chemistry to synthesize 2-substituted benzoxazoles. A reductive amination was followed by a classical resolution to afford chiral diazepane (R)-11. Finally, coupling of (R)-11 with acid 5 furnished the desired drug candidate 1.
The amine DBT salt 16 (5.67 kg, 9.09 mol) was charged to a vessel and inerted. DCM (28 L) was added, followed by 4 N sodium hydroxide solution (prepared from 10 N NaOH [22.4 L] and water [36 L]). The slurry was then stirred at ambient temperature for 1 h until a solution was obtained. The layers were separated, and the aqueous phase was treated with sodium chloride solution (10.1 kg in 20 L water). DCM (5 L) was then added and the biphasic mixture stirred for 10 min before separating the layers. The combined organic layers were then concentrated under reduced pressure to a 10 L volume. The solution of the free amine was used directly in the next reaction
The triazole acid 5 (13.25 kg, 65.2 mol), DCM (88 L), and DMF (1.35 L, 17.4 mol) were charged to a vessel, and the resulting suspension was cooled to 0 °C. Oxalyl chloride (8.28 kg, 65.2 mol) was added portionwise, keeping the internal temperature between 5 and 10 °C (the anhydride formed above 10 °C), and then the reaction was aged for 30 min at this temperature. HPLC analysis showed acid 5 remained; an additional charge of oxalyl chloride (160 g, 1.26 mol) was made, and the solution stirred at 5 °C for 30 min. A solution of the amine (R)-11 (16.5 kg, 62.1 mol) and triethylamine (13.19 kg, 130.0 mol) in DCM (∼8 L) was added to the acid chloride over 30 min, keeping the internal temperature less than 15 °C. The resulting slurry was aged for 30 min and then quenched by the addition of water (167 L) over 10 min, keeping the internal temperature <15 °C. The lower organic layer was removed and then concentrated under atmospheric pressure to a volume of 100 L. Assay at this stage showed 27.3 kg 1, 98%. The solution was solvent switched to MeCN (∼560 L, 20 mL/g) by distillation under reduced pressure at <50 °C. The MeCN solution was treated with Ecosorb C-941 (2.8 kg) slurried in MeCN (10 L). The resulting slurry was aged for 30 min and then filtered through a Solka Flok pad and a 0.1 um cartridge filter, washing with MeCN (2 × 30 L). The MeCN filtrate was concentrated under reduced pressure at <50 °C to a final volume of ∼112 L. The slurry was cooled to 25 °C and water (280 L) added over 40 min. The resulting slurry was aged at 20 °C for 1 h and then filtered, washing the cake with 5:1 water/MeCN (60 L) followed by water (40 L). The solid was dried in the vacuum oven with nitrogen purge overnight at 50 °C. The final target 1 was isolated as a white solid, 26.72 kg, 95%, 98.5% ee, 99.6 LCAP, mp 153.1 °C.
The 1H NMR data for this compound was extremely complicated due to its existence as four rotamers. These rotamers did not coalesce during high-temperature experiments.(4)
[α]25D −11.8 (c 1.0, MeOH) for a sample of 97.8% ee. HRMS (ESI): m/z [M+ + H] calcd for C23H23ClN6O2: 451.1649; found: 451.1640.
A highly regioselective halogenation of 2-substituted-1,2,3-triazoles was developed via sp2 C–H activation. This method is compatible with halogen atoms, as well as electron-donating and electron-withdrawing groups. Meanwhile, the strategy is also efficient for the synthesis of a key intermediate of Suvorexant.
PAPER
2.1. Synthesis of (R)-methyl 2-(N-benzyl-3-((tert-butoxycarbonyl)amino)butanamido)acetate (3)
To a solution of methyl 2-(benzylamino)acetate (compound 10, 50.14 g,0.28 mol),(R)-3-((tert-butoxycarbonyl)amino)butanoic acid (50.75 g,0.25 mol),1-hydroxy-1H-benzotriazole (41.88 g, 0.31 mol),and dry triethylamine (37.95 g,0.38 mol) in 320 mL of DMF was added EDC hydrochloride (57.51 g,0.30 mol),and the reaction was stirred for 5 h at room temperature. The reaction was partitioned between EtOAc and 10% aqueous citric acid,the layers were separated and the organic was washed with 5% aqueous Na2CO3,then with brine,dried over MgSO4 and concentrated by rotary evaporation. The residue was recrystallized from a mixture solvent (PE:EtOAc = 2:1) to provide compound 3 as a white solid, 83.01 g in 91% yield. Mp: 107 ℃,[α]D 25 22.0 (c0.52,MeOH). 1H NMR (600 MHz,DMSO-d6): δ 7.38-7.23 (m,5H),6.73-6.72 (d,1H, J = 6 Hz),4.75-4.43 (m,2H),4.31-3.95 (m,2H),3.89-3.87 (t,1H, J = 12 Hz),3.64-3.62 (d,3H,J = 12 Hz),2.64-2.50 (m,1H),2.37- 2.23 (m,1H),1.38-1.37 (d,9H,J = 6 Hz),1.08-1.06 (m,3H); MS (ESI) m/z: 365.20 [M+H]+. HR-MS(ESI): m/z [M+H] calcd. for C19H28N2O5: 365.2071; found: 365.2066.
2.2. Synthesis of (R)-4-benzyl-7-methyl-1,4-diazepane-2,5-dione (4)
A solution of compound 3 (15.93 g,43.74 mmol) in 10 mL EtOAc was added 150 mL 45% HCl/EtOAc and the reaction was stirred for 4 h. The solvents were removed by rotary evaporation,and the residue was basified with saturated aqueous NaHCO3,and extracted with CH2Cl2. The organic extracts were concentrated. The residue was dissolved in 150 mL of dehydrated MeOH, treated with CH3ONa (2.84 g,52.49 mmol),and stirred at room temperature overnight (N2 protected,slightly exothermic). The reaction was cooled to room temperature and quenched with aqueous NH4Cl. Most of the solvent was removed and the reaction was then dumped into a separatory funnel containing 5% aqueous Na2CO3 and extracted with CH2Cl2 three times. The organic layers were combined,dried over MgSO4,and concentrated to provide compound 4 as a white solid 9.50 g in 94% yield. Analytical HPLC analysis carried out on Chiralpak AD column (4.6 mm × 250 mm) with 60% EtOH in hexanes (containing 0.1% diethylamine as a modifier),flow rate of 1 mL/min,indicated that intermediate (R)-4 was of >99% ee. Mp: 122-123 ℃. [α]D2533.5 (c 0.56,MeOH). 1H NMR (600 MHz,DMSO-d6): δ 7.77-7.76 (bd,1H,J = 6 Hz),7.33-7.25 (m,5H),4.59-4.53 (m,2H),4.10- 4.02 (m,2H),3.65-3.62 (m,1H),2.93-2.90 (m,1H),2.76-2.72 (m,1H), 1.14-1.13 (d,3H,J = 6 Hz); 13C NMR (150 MHz,DMSO-d6): δ 171.1, 168.4,138.1,128.9,128.0,127.7,53.1,50.6,46.5,40.5,23.3. MS (ESI) m/z: 233.10 [M+H]+. HR-MS(ESI): m/z [M+H] calcd. for C13H16N2O2: 233.1285; found: 233.1289.
2.3. Synthesis of (R)-1-benzyl-5-methyl-1,4-diazepane (6)
A solution of compound 4 (1.40 g,6.0 mmol) in 60 mL THF at 0 ℃ was treated with LiAlH4 (1.36 g,36.0 mmol) in batches. The reaction was slowly warmed to room temperature and stirred for another 4 h. The reaction was then cooled to -10 ℃ and was carefully quenched with 1.5 mL water,then NaOH (1.5 mL,15%) followed by an additional 4.5 mL of water. A portion of MgSO4was added and the mixture was stirred for 1 h before filtered. The filtrate was concentrated to provide light yellow oil 1.10 g in 88% yield. [α]D25 -5.9 (c 1.00,CHCl3),ee >99%,Analytical analysis was performed on Chrom Tech chiral-AGP column (150 mm × 4 mm) with 99% 1 mol/L ammonium dihydrogen phosphate and 1% acetonitrile,at flow rate of 0.5 mL/min with column temperature of 40 ℃. 1H NMR (600 MHz,DMSO-d6): δ 7.32-7.20 (m,5H),3.57 (s, 2H),3.48 (bs,1H),2.99-2.95 (m,1H),2.86-2.82 (m,1H),2.72-2.68 (m,1H),2.65-2.61 (m,1H),2.58-2.49 (m,3H),1.75-1.70 (m,1H), 1.46-1.41 (m,1H),1.01-1.00 (d,3H,J = 6 Hz); 13C NMR (150 MHz, DMSO-d6): δ 140.1,128.9,128.5,127.1,62.5,58.8,52.7,52.6,47.0, 37.5,23.9. MS (ESI) m/z: 205.10 [M+H]+. HR-MS(ESI): m/z [M+H] calcd. for C13H20N2: 205.1699; found: 205.1692.
2.4. Synthesis of (R)-(4-benzyl-7-methyl-1,4-diazepan-1-yl)(5-methyl-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone (7)
To a solution of compound 6 (2.40 g,11.76 mmol),compound 5 (2.86 g,14.11 mmol),1-hydroxy-1H-benzotriazole (1.90 g, 14.11 mmol),and dry triethylamine (3.56 g,35.28 mmol) in 18 mL of dry DMF was added EDC hydrochloride (2.70 g, 14.11 mmol),and the reaction was stirred 2 h at room temperature. The reaction was partitioned between EtOAc and saturated aqueous NaHCO3,the layers were separated and the organic was added to aqueous citric acid stirring for 1 h. Water was added and the mixture was partitioned. Combined the water layers and added saturated aqueous Na2CO3 to regulate pH > 9,then extracted with three portions of EtOAc. The organic layers were combined,dried over MgSO4 and concentrated by rotary evaporation to provide compound 7 as a white power 4.30 g in 93% yield. Mp: 108-109 ℃, [α]D25-58.4 (c 1.01,MeOH). 1HNMR(600 MHz,DMSO-d6): δ 8.00- 7.76 (m,3H),7.37-7.17 (m,7H),4.40-4.09 (m,1H),3.63-3.48 (m, 2H),3.44-3.02 (m,3H),2.82-2.75 (m,1H),2.63-2.47 (m,1H), 2.63-2.14 (m,5H),2.02- 1.63 (m,2H),1.17-0.99 (m,3H); MS (ESI) m/z: 390.30 [M+H]+. HR-MS(ESI): m/z [M+H] calcd. for C23H27N5O: 390.2288; found: 390.2281.
2.5. Synthesis of (R)-(7-methyl-1,4-diazepan-1-yl)(5-methyl-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone (9)
Compound 7 (5.86 g,15.05 mmol) was dissolved in 58 mL MeOH. After a portion of 10% Pd/C was added,the reaction was stirred for 4 h under H2 atmosphere at room temperature. The reaction was filtered through a pad of celite and the filtrate was concentrated to provide compound 9 as a white solid 4.01 g in 89% yield. Mp: 119-121 ℃,[a]D 26 -14.4 (c 1.00,MeOH)). 1H NMR (600 MHz,DMSO-d6): δ 8.24-8.02 (m,2H),7.88-7.29 (m,3H), 4.42-2.50 (m,7H),2.41 (s,3H),2.24-1.98 (m,2H),1.17-0.99 (m, 3H); 13C NMR (150 MHz,DMSO-d6): δ 168.6,138.3,136.9,134.1, 131.1,129.2,128.3,122.5,52.6,49.1,44.4,43.1,37.8,20.8,20.6. MS (ESI)m/z: 300.20 [M+H]+. HR-MS(ESI): m/z [M+H] calcd. for C16H21N5O: 300.1819; found: 300.1812.
2.6. Synthesis of suvorexant
To compound 8 (0.56 g,3 mmol) in 10 mL dry DMF was added TEA (0.91 g,9 mmol) and compound 9 (0.89 g,3 mmol),the mixture was stirred at 75 ℃ for 2 h. After cooling to room temperature,the reaction was diluted with EtOAc,washed with saturated aqueous NaHCO3,water,brine and dried over MgSO4. The residue was recrystallized from i-PrOH/EtOAc to provide a white solid 1.20 g in 90% yield. Mp: 149-150 ℃,[α]D25 -11.6 (c 1.00,MeOH). Analytical HPLC analysis carried out on a Chiralpak AD column (4.6 mm × 250 mm) with 60% EtOH in hexanes (containing 0.1% diethylamine as a modifier) at a flow rate of 1 mL/min,indicated that intermediate (R)-4 was of >99% ee. Mp: 153 ℃,[α]D25 -11.7 (c 1.00,MeOH) [ OPRD REF ],
Baxter, C. A.; Cleator, E.; Brands, K. M. J.; Edwards, J. S.; Reamer, R. A.; Sheen, F. J.; Stewart, G. W.; Strotman, N. A.; Wallace, D. J. (2011). “The First Large-Scale Synthesis of MK-4305: A Dual Orexin Receptor Antagonist for the Treatment of Sleep Disorder”.Organic Process Research & Development15 (2): 367–375. doi:10.1021/op1002853.
“Suvorexant: A Dual Orexin Receptor Antagonist for the Treatment of Sleep Onset and Sleep Maintenance Insomnia.”. Ann Pharmacother49: 477–483. Feb 9, 2015.doi:10.1177/1060028015570467. PMID25667197.
Label: BELSOMRA- Suvorexant Tablet, Film Coated”Label: BELSOMRA- Suvorexant Tablet, Film Coated.” DailyMed. Merck Sharp & Dohme Corp. & the U.S. National Library of Medicine, 01 Aug. 2014. Web. 29 Oct. 2014.
Jacobson, LH; Callander, GE; Hoyer, D (Nov 2014). “Suvorexant for the treatment of insomnia.”. Expert review of clinical pharmacology7 (6): 711–30.doi:10.1586/17512433.2014.966813. PMID25318834.
“Belsomra”. drugs.com. Retrieved 20 February 2015.
“U.S. Food and Drug Administration.” Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. U.S. Food and Drug Administration, 27 Oct. 2014. Web. 30 Oct. 2014.
Suvorexant synthesis There are several ways, the following is a scaled-up process (OPRD, 2011, 15, 367). A compound with sulfur phosgene in ring closure to give 2,2 thiol group with oxalyl chloride to chlorine after conversion to give the intermediate 4 with a primary amine 3 attack, followed by Michael addition occurred with 5 6.6 mesylate de Boc protected After the reductive amination get 7, this is the racemic product. 7 8 after two crystallization with tartaric acid split to give 9 (> 97% ee).Triazole carboxylic acid 10 with 11 to give 12, 12 coupled after conversion to the acid chloride under basic conditions with pH 9 condensation Suvorexant.
The U.S. Food and Drug Administration today approved Cologuard, the first stool-based colorectal screening test that detects the presence of red blood cells and DNA mutations that may indicate the presence of certain kinds of abnormal growths that may be cancers such as colon cancer or precursors to cancer.
Colorectal cancer primarily affects people age 50 and older, and among cancers that affect both men and women, it is the third most common cancer and the second leading cause of cancer-related death in the United States, according to the Centers for Disease Control and Prevention (CDC). Colorectal cancer screening is effective at reducing illness and death related to colon cancer. The CDC estimates that if everyone age 50 or older had regular screening tests as recommended, at least 60 percent of colorectal cancer deaths could be avoided.
Colorectal cancer occurs in the colon (large intestine) or rectum (the passageway that connects the colon to the anus). Most colorectal cancers start as abnormal raised or flat tissue growths on the wall of the large intestine or rectum (polyps). Some very large polyps are called advanced adenomas and are more likely than smaller polyps to progress to cancer.
Using a stool sample, Cologuard detects hemoglobin, a protein molecule that is a component of blood. Cologuard also detects certain mutations associated with colorectal cancer in the DNA of cells shed by advanced adenomas as stool moves through the large intestine and rectum. Patients with positive test results are advised to undergo a diagnostic colonoscopy.
“This approval offers patients and physicians another option to screen for colorectal cancer,” said Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health at the FDA’s Center for Devices and Radiological Health. “Fecal blood testing is a well-established screening tool and the clinical data showed that the test detected more cancers than a commonly used fecal occult test.”
Today’s approval of the Cologuard does not change current practice guidelines for colorectal cancer screening. Stool DNA testing (also called “fecal DNA testing”) is not currently recommended as a method to screen for colorectal cancer by the United States Preventive Services Task Force (USPSTF). Among other guidelines, the USPSTF recommends adults age 50 to 75, at average risk for colon cancer, be screened using fecal occult blood testing, sigmoidoscopy, or colonoscopy.
The safety and effectiveness of Cologuard was established in a clinical trial that screened 10,023 subjects. The trial compared the performance of Cologuard to the fecal immunochemical test (FIT), a commonly used non-invasive screening test that detects blood in the stool. Cologuard accurately detected cancers and advanced adenomas more often than the FIT test. Cologuard detected 92 percent of colorectal cancers and 42 percent of advanced adenomas in the study population, while the FIT screening test detected 74 percent of cancers and 24 percent of advanced adenomas. Cologuard was less accurate than FIT at correctly identifying subjects negative for colorectal cancer or advanced adenomas. Cologuard correctly gave a negative screening result for 87 percent of the study subjects, while FIT provided accurate negative screening results for 95 percent of the study population.
Today the Centers for Medicare & Medicaid Services (CMS) issued a proposed national coverage determination for Cologuard. Cologuard is the first product reviewed through a joint FDA-CMS pilot program known as parallel review where the agencies concurrently review medical devices to help reduce the time between the FDA’s approval of a device and Medicare coverage. This voluntary pilot program is open to certain premarket approval applications for devices with new technologies and to medical devices that fall within the scope of a Part A or Part B Medicare benefit category and have not been subject to a national coverage determination.
“Parallel review allows the last part of the FDA process to run at the same time as the CMS process, cutting as many as six months from the time from study initiation to coverage,” said Nancy Stade, CDRH’s deputy director for policy. “The pilot program is ongoing, but we will apply what we have learned to improve the efficiency of the medical device approval pathway for devices that address an important public health need.”
“This is the first time in history that FDA has approved a technology and CMS has proposed national coverage on the same day,” said Patrick Conway, chief medical officer and deputy administrator for innovation and quality for CMS. “This parallel review represents unprecedented collaboration between the two agencies and industry and most importantly will provide timely access for Medicare beneficiaries to an innovative screening test to help in the early detection of colorectal cancer.”
CMS proposes to cover the Cologuard test once every three years for Medicare beneficiaries who meet all of the following criteria:
age 50 to 85 years,
asymptomatic (no signs or symptoms of colorectal disease including but not limited to lower gastrointestinal pain, blood in stool, positive guaiac fecal occult blood test or fecal immunochemical test), and
average risk of developing colorectal cancer (no personal history of adenomatous polyps, of colorectal cancer, or inflammatory bowel disease, including Crohn’s Disease and ulcerative colitis; no family history of colorectal cancers or an adenomatous polyp, familial adenomatous polyposis, or hereditary nonpolyposis colorectal cancer).
Cologuard is manufactured by Exact Sciences in Madison, Wisconsin.
The U.S. Food and Drug Administration today approved Orbactiv (oritavancin), a new antibacterial drug to treat adults with skin infections.
Orbactiv is approved to treat patients with acute bacterial skin and skin structure infections (ABSSSI) caused by certain susceptible bacteria, includingStaphylococcus aureus (including methicillin-susceptible and methicillin-resistant strains), various Streptococcus species and Enterococcus faecalis. Orbactiv is administered intravenously.
Orbactiv is the third new antibacterial drug approved by the FDA this year to treat ABSSSI. The agency approved Dalvance (dalbavancin) in May 2014 and Sivextro (tedizolid) in June 2014.
“The approval of several new antibacterial drugs this year demonstrates that we are making progress in increasing the availability of treatment options for patients and physicians,” said Edward Cox, M.D., M.P.H, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research. “However, more work is needed in this area, and the FDA remains a committed partner to help promote the development of antibacterial drugs.”
Orbactiv is also the third new drug designated as a Qualified Infectious Disease Product (QIDP) to receive FDA approval. Under the Generating Antibiotic Incentives Now (GAIN) title of the FDA Safety and Innovation Act, Orbactiv was granted QIDP designation because it is an antibacterial or antifungal human drug intended to treat a serious or life-threatening infection.
As part of its QIDP designation, Orbactiv was given priority review, which provides an expedited review of the drug’s application. Orbactiv’s QIDP designation also qualifies it for an additional five years of marketing exclusivity to be added to certain exclusivity periods already provided by the Food, Drug, and Cosmetic Act.
Orbactiv’s safety and efficacy were evaluated in two clinical trials with a total of 1,987 adults with ABSSSI. Participants were randomly assigned to receive Orbactiv or vancomycin. Results showed Orbactiv was as effective as vancomycin for the treatment of ABSSSI.
The most common side effects identified in the clinical trials were headache, nausea, vomiting, the formation of skin and soft tissue abscesses on arms and legs and diarrhea. Orbactiv’s label also includes a warning regarding interference with coagulation tests and interaction with warfarin, a drug used to prevent blood clots.
Orbactiv is marketed by The Medicines Company, based in Parsippany, N.J.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.
Jul 2, 2013 – Inhibits two key steps of cell wall synthesis: – Transglycosylation. – Transpeptidation. • Disrupts bacterial membrane integrity. Differentiated from …
FDA Accepts Filing of NDA for IV Antibiotic Oritavancin with Priority Review
PARSIPPANY, NJ — (Marketwired) — 02/19/14 — The Medicines Company (NASDAQ: MDCO) today announced that the U.S. Food and Drug Administration (FDA) has accepted the filing of a new drug application (NDA) for oritavancin, an investigational intravenous antibiotic, with priority review. The Medicines Company is seeking approval of oritavancin for the treatment of acute bacterial skin and skin structure infections (ABSSSI) caused by susceptible gram-positive bacteria, including methicillin-resistant Staphylococcus aureus (MRSA), administered as a single dose.
In December 2013, the FDA designated oritavancin as a Qualified Infectious Disease Product (QIDP). The QIDP designation provides oritavancin priority review, and an additional five years of exclusivity upon approval of the product for the treatment of ABSSSI. Priority review means the FDA’s goal is to take action on the application within six months, compared to 10 months under standard review. The FDA action date (PDUFA date) for oritavancin is August 6, 2014. Oritavancin (INN, also known as LY333328) is a novel semi-synthetic glycopeptide antibiotic being developed for the treatment of serious Gram-positive infections. Originally discovered and developed by Eli Lilly, oritavancin was acquired by InterMune in 2001 and then by Targanta Therapeuticsin late 2005.[1]
In Dec 2008 the FDA declined to approve it, and an EU application was withdrawn.
In 2009 the development rights were acquired by The Medicine Co. who are running clinical trials for a possible new FDA application in 2013.[2]
Oritavancin is an investigational intravenous antibiotic for which The Medicines Company is seeking approval in the treatment of ABSSSI caused by susceptible gram-positive bacteria, including MRSA. In clinical trials, the most frequently reported adverse events associated with oritavancin were nausea, headache, vomiting and diarrhea. Hypersensitivity reactions have been reported with the use of antibacterial agents including oritavancin.
Oritavancin shares certain properties with other members of the glycopeptide class of antibiotics, which includes vancomycin, the current standard of care for serious Gram-positive infections in the United States and Europe.[4] Data presented at the 47th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC) in September 2007 demonstrated that oritavancin possesses potent and rapid bactericidal activity in vitro against a broad spectrum of both resistant and susceptible Gram positive bacteria, including Staphylococcus aureus, methicillin-resistant Staphylococcus aureus, Enterococci, and Streptococci.[5] Two posters presented at the meeting also demonstrated that oritavancin was more active than either metronidazole or vancomycin against strains of Clostridium difficile tested.[6]
Anthrax : Research presented at the American Society for Microbiology (ASM) 107th Annual General Meeting in May 2007, suggested oritavancin’s potential utility as a therapy for exposure to Bacillus anthracis, the gram-positive bacterium that causes anthrax, having demonstrated efficacy in a mouse model both pre- and post-exposure to the bacterium[7]
oritavancin
The 4′-chlorobiphenylmethyl group disrupts the cell membrane of gram positive bacteria.[8] It also acts by inhibition of transglycosylation and inhibition of transpeptidation.[9]
Results have been presented (in 2003) but possibly not yet published from two pivotal Phase 3 clinical trials testing the efficacy of daily intravenous oritavancin for the treatment of complicated skin and skin-structure infections (cSSSI) caused by Gram-positive bacteria. The primary endpoints of both studies were successfully met, with oritavancin achieving efficacy with fewer days of therapy than the comparator agents (vancomycin followed by cephalexin). In addition, oritavancin showed a significantly improved safety profile with a 19.2 percent relative reduction in the overall incidence of adverse events versus vancomycin/cephalexin (p<0.001) in the second and larger pivotal trial.[10]
A Phase 2 clinical study was planned to run until May 2008 entitled “Single or Infrequent Doses for the Treatment of Complicated Skin and Skin Structure Infections (SIMPLIFI),” evaluating the efficacy and safety of either a single dose of oritavancin or an infrequent dose of oritavancin compared to the previously studied dosing regimen of 200 mg oritavancin given once daily for 3 to 7 days.[11] Results published May 2011.[12]
Regulatory submissions
USA
On February 11, 2008, Targanta submitted a New Drug Application (NDA) to the USFDA seeking approval of oritavancin;[13] in April 2008, the FDA accepted the NDA submission for standard review.[14] On 9 Dec 2008 the FDA said insufficient data for approval of oritavancin had been provided and they requested a further phase 3 clinical study to include more patients with MRSA.[15]
The Medicines Company’s purpose is to save lives, alleviate suffering, and contribute to the economics of healthcare by focusing on 3,000 leading acute/intensive care hospitals worldwide. Its vision is to be a leading provider of solutions in three areas: acute cardiovascular care, surgery and perioperative care, and serious infectious disease care. The company operates in the Americas, Europe and the Middle East, and Asia Pacific regions with global centers today in Parsippany, NJ, USA and Zurich, Switzerland.
“We look forward to working with the FDA during the review process, and sharing the knowledge we have gained in our studies of oritavancin,” said Matthew Wikler, MD, Vice President and Medical Director, Infectious Disease Care for The Medicines Company. “We believe that upon approval, oritavancin, administered as a single dose for the treatment of ABSSSI, will offer new options for both physicians and their patients for the treatment of these infections.”
The oritavancin NDA is based on data from two Phase 3 clinical trials, SOLO I and SOLO II, which were conducted under a Special Protocol Assessment (SPA) agreement with the FDA. These Phase 3 trials evaluated the efficacy and safety of a single 1200mg dose of oritavancin compared to 7 to 10 days of twice-daily vancomycin in adults with ABSSSI, including infections caused by MRSA. The combined SOLO studies were conducted in 1,959 patients (modified intent-to -treat population, or mITT), with 405 of the patients suffering from an ABSSSI with a documented MRSA infection.
Oritavancin inhibits cell wall synthesis by complexing with the terminal D-Ala-D-Ala of a nascent peptidoglycan chain and also to the pentaglycine bridge, thus inhibiting transglyco- sylation and transpeptidation. Unlike other glycopeptides, oritavancin is able to bind to depsipeptides including D-Ala-D-Lac, which fa- cilitates its inhibition of cell wall synthesis even in organisms exhibiting VanA-type resistance. Oritavancin forms homodimers prior to binding to D-Ala-D-Ala or D-Ala-D-Lac, which increases its binding affinity for the target site.The p-chloro-phenylbenzyl side chain of oritavancin interacts with the cell membrane, exerting two beneficial effects. This binding acts to main- tain the antibacterial in a prime position for peptidoglycan interactions and it also imparts oritavancin with the ability to disrupt the bac- terial membrane potential and thus increase membrane permeability.[22,23] Oritavancin has been shown to dissipate membrane potential in both stationary and exponential phase growing bacteria, which is rare and may carry clinical implications in terms of its activity against slowly growing organisms and biofilms. The dual mechanism of action could also theoretically increase effectiveness and reduce the risk of resist- ance selection. In addition to the aforemen- tioned mechanisms, it has also been hypothesized that oritavancin inhibits RNA synthesis.
vancomycin, desmethylvancomycin, eremomycin, teicoplanin (complex of five compounds), dalbavancin, oritavancin, telavancin, and A82846B (LY264826) having structures A, B, C, D, E, F, G and H:
R = B-2-Acetylamido-glucopyraπosyl- Attorney Docket No 33746-704 602
Dalbavancin, oritavancin and telavancin are semisynthetic lipoglycopeptides that demonstrate promise for the treatment of patients with infections caused by multi-drug-resistant Gram-positive pathogens. Each of these agents contains a heptapeptide core, common to all glycopeptides, which enables them to inhibit transglycosylation and transpeptidation (cell wall synthesis). Modifications to the heptapeptide core result in different in vitro activities for the three semisynthetic lipoglycopeptides. All three lipoglycopeptides contain lipophilic side chains, which prolong their half-life, help to anchor the agents to the cell membrane and increase their activity against Gram-positive cocci. In addition to inhibiting cell wall synthesis, telavancin and oritavancin are also able to disrupt bacterial membrane integrity and increase membrane permeability; oritavancin also inhibits RNA synthesis. Enterococci exhibiting the VanA phenotype (resistance to both vancomycin and teicoplanin) are resistant to both dalbavancin and telavancin, whileoritavancin retains activity. Dalbavancin, oritavancin and telavancin exhibit activity against VanB vancomycin-resistant enterococci.
All three lipoglycopeptides demonstrate potent in vitro activity against Staphylococcus aureus and Staphylococcus epidermidis regardless of their susceptibility to meticillin, as well as Streptococcus spp. Both dalbavancin and telavancin are active against vancomycin-intermediate S. aureus (VISA), but display poor activity versus vancomycin-resistant S. aureus (VRSA). Oritavancin is active against both VISA and VRSA. Telavancin displays greater activity against Clostridium spp. than dalbavancin, oritavancin or vancomycin. The half-life of dalbavancin ranges from 147 to 258 hours, which allows for once-weekly dosing, the half-life of oritavancin of 393 hours may allow for one dose per treatment course, while telavancin requires daily administration. Dalbavancin and telavancin exhibit concentration-dependent activity and AUC/MIC (area under the concentration-time curve to minimum inhibitory concentration ratio) is the pharmacodynamic parameter that best describes their activities.
Oritavancin’s activity is also considered concentration-dependent in vitro, while in vivo its activity has been described by both concentration and time-dependent models; however, AUC/MIC is the pharmacodynamic parameter that best describes its activity. Clinical trials involving patients with complicated skin and skin structure infections (cSSSIs) have demonstrated that all three agents are as efficacious as comparators. The most common adverse effects reported with dalbavancin use included nausea, diarrhoea and constipation, while injection site reactions, fever and diarrhoea were commonly observed withoritavancin therapy. Patients administered telavancin frequently reported nausea, taste disturbance and insomnia. To date, no drug-drug interactions have been identified for dalbavancin, oritavancin or telavancin. All three of these agents are promising alternatives for the treatment of cSSSIs in cases where more economical options such as vancomycin have been ineffective, in cases of reduced vancomycin susceptibility or resistance, or where vancomycin use has been associated with adverse events.
Oritavancin diphosphate (oritavancin) is a semi-synthetic lipoglycopeptide derivative of a naturally occurring glycopeptide. Its structure confers potent antibacterial activity against gram-positive bacteria, including vancomycin-resistant enterococci (VRE), methicillin- and vancomycin-resistant staphylococci, and penicillin-resistant streptococci. The rapidity of its bactericidal activity against exponentially-growing S. aureus (≧3-log reduction within 15 minutes to 2 hours against MSSA, MRSA, and VRSA) is one of the features that distinguishes it from the prototypic glycopeptide vancomycin (McKay et al., J Antimicrob Chemother. 63(6):1191-9 (2009), Epub 2009 Apr. 15).
Oritavancin inhibits the synthesis of peptidoglycan, the major structural component of the bacterial cell wall by a mechanism that is shared with glycopeptides, such as vancomycin (Allen et al., Antimicrob Agents Chemother 41(1):66-71 (1997); Cegelski et al., J Mol Biol 357:1253-1262 (2006); Arhin et al., Poster C1-1471: Mechanisms of action of oritavancin in Staphylococcus aureus [poster]. 47th Intersci Conf Antimicro Agents Chemo, Sep. 17-20, 2007; Chicago, Ill.). Oritavancin, like vancomycin, binds to the Acyl-D-Alanyl-D-Alanine terminus of the peptidoglycan precursor, lipid-bound N-acetyl-glucosamine-N-acetyl-muramic acid-pentapeptide (Reynolds, Eur J Clin Microbiol Infect Dis 8(11):943-950 (1989); Nicas and Allen, Resistance and mechanism of action.
In: Nagarajan R, editor. Glycopeptide antibiotics. New York: Marcel Dekker 195-215 (1994); Allen et al., Antimicrob Agents Chemother 40(10):2356-2362 (1996); Allen and Nicas, FEMS Microbiology Reviews 26:511-532 (2003); Kim et al., Biochemistry 45:5235-5250 (2006)). However, oritavancin inhibits cell wall biosynthesis even when the substrate is the altered peptidoglycan precursor that is present in VRE and vancomycin-resistant S. aureus (VRSA). Thus, the spectrum of oritavancin antibacterial activity extends beyond that of vancomycin to include glycopeptide-resistant enterococci and staphylococci (Ward et al., Expert Opin Investig Drugs 15:417-429 (2006); Scheinfeld, J Drugs Dermatol 6:97-103 (2007)). Oritavancin may inhibit resistant bacteria by interacting directly with bacterial proteins in the transglycosylation step of cell wall biosynthesis (Goldman and Gange, Curr Med Chem 7(8):801-820 (2000); Halliday et al., Biochem Pharmacol 71(7):957-967 (2006); Wang et al., Poster C1-1474: Probing the mechanism of inhibition of bacterial peptidoglycan glycotransferases by glycopeptide analogs. 47th Intersci Conf Antimicro Agents Chemo, Sep. 17-20, 2007). Oritavancin also collapses transmembrane potential in gram positive bacteria, leading to rapid killing (McKay et al., Poster C1-682: Oritavancin disrupts transmembrane potential and membrane integrity concomitantly with cell killing in Staphylococcus aureus and vancomycin-resistant Enterococci. 46th Intersci Conf Antimicro Agents Chemo, San Francisco, Calif., Sep. 27-30, 2006). These multiple effects contribute to the rapid bactericidal activity of oritavancin.
Vancomycin(U.S. Patent 3,067,099); A82846A, A82846B, and A82846C (U.S. Patent 5,312,738, European Patent Publication 256,071 A1); PA-42867 factors A, C, and D (U.S. Patent4,946,941 and European Patent Publication 231,111 A2); A83850 (U.S. Patent No. 5,187,082); avoparcm (U.S. Patent 3,338,786 and U.S. Patent 4,322,343); actmoidin, also known as K288 (J. Antibiotics Series A 14:141 (1961); helevecardin (Chem. Abstracts 110:17188 (1989) and Japanese Patent Application 86/157,397); galacardin (Chem. Abstracts 110:17188 (1989) and Japanese Patent Application 89/221,320); and M47767 (European Patent Publication 339,982).
Oritavancin is in clinical development against serious gram-positive infections, where administration of the drug is via intravenous infusion using several dosages administered over a series of days. The development of alternative dosing regimens for the drug could expand treatment options available to physicians. The present invention is directed to novel dosing regimens.
Means for the preparation of the glycopeptide antibiotics, including oritavancin and analogs thereof, may be found, for example, in U.S. Pat. No. 5,840,684,
ORITAVANCIN DIPHOSPHATE
SYNTHESIS
LY-333328 was synthesized by reductocondensation of the glycopeptide antibiotic A82846B (I) with 4′-chlorobiphenyl-4-carboxaldehyde (II) by means of sodium cyanoborohydride in refluxing methanol.
LY-333328 was synthesized by reductocondensation of the glycopeptide antibiotic A82846B (I) with 4′-chlorobiphenyl-4-carboxaldehyde (II) by means of sodium cyanoborohydride in refluxing methanol.
condenser, nitrogen inlet and overhead mechanical stirring apparatus. The flask was charged with pulverized A82846B acetate salt (20.0 g, 1.21 × 10-3 mol) and methanol (1000 mL) under a nitrogen atmosphere. 4′-chlorobiphenylcarboxaldehyde (2.88 g, 1.33 × 10-2 mol, 1.1 eq.) was added to this stirred mixture, followed by methanol (500 mL). Finally, sodium cyanoborohydride (0.84 g, 1.33 × 10-2 mol, 1.1 eq.) was added followed by methanol (500 mL). The resulting mixture was heated to reflux (about 65°C).
After 1 hour at reflux, the reaction mixture attained homogeneity. After 25 hours ac reflux, the heat source was removed and the clear reaction mixture was measured with a pH meter (6.97 at 58.0°C). 1 N NaOH (22.8 mL) was added
dropwise to adjust the pH to 9.0 (at 54.7°C). The flask was equipped with a distillation head and the mixture was concentrated under partial vacuum to a weight of 322.3 grams while maintaining the pot temperature between 40-45°C.
The distillation head was replaced with an addition funnel containing 500 mL of isopropanol (IPA). The IPA was added dropwise to the room temperature solution over 1 hour. After approximately 1/3 of the IPA was added, a granular precipitate formed. The remaining IPA was added at a faster rate after precipitation had commenced. The flask was weighed and found to hold 714.4 grams of the IPA/methanol slurry.
The flask was re-equipped with a still-head and
distilled under partial vacuum to remove the remaining methanol. The resulting slurry (377.8 g) was allowed to chill in the freezer overnight. The crude product was filtered through a polypropylene pad and rinsed twice with 25 mL of cold IPA. After pulling dry on the funnel for 5 minutes, the material was placed in the vacuum oven to dry at 40°C. A light pink solid (22.87 g (theory = 22.43 g) ) was recovered. HPLC analysis versus a standard indicated 68.0% weight percent of Compound 229 (4- [4-chlorophenyl] benzyl-A82846B] in the crude solid, which translated into a
corrected crude yield of 69.3%.
The products of the reaction were analyzed by reverse-phase HPLC utilizing a Zorbax SB-C18 column with ultraviolet light (UV; 230 nm) detection. A 20 minute gradient solvent system consisting of 95% aqueous buffer/5% CH3CN at time=0 minutes to 40% aqueous buffer/60% CH3CN at time=20 minutes was used, where the aqueous buffer was TEAP (5 ml CH3CN, 3 ml phosphoric acid in 1000 ml water).
http://www.farm.ucl.ac.be/Full-texts-FARM/Domenech-2009-1.pdf “Interactions of oritavancin, a new lipoglycopeptide derived from vancomycin, with phospholipid bilayers: Effect on membrane permeability and nanoscale lipid membrane organization” 2009
Scheinfeld, N (2007). “A comparison of available and investigational antibiotics for complicated skin infections and treatment-resistant Staphylococcus aureus and enterococcus“.J Drugs Dermatol.6 (4): 97–103. PMID17373167.
2007 ICAAC Posters: E-1612 “In Vitro Activity Profile of Oritavancin against a Broad Spectrum of Aerobic and Anaerobic Bacterial Pathogens”/E -1613 “In Vitro Activity Profile of Oritavancin (ORI) Against Organisms Demonstrating Key Resistance Profiles to Other Antimicrobial Agents”/E-1614 “In vitro Time Kill Studies of Oritavancin against Drug-resistant Isolates ofStaphylococcus aureus and Enterococci”/E-1615 “Anti-Enterococcal Activity Profile of Oritavancin, a Potent Lipoglycopeptide under Development for Use Against Gram-Positive Infections”/E-1616 “Anti-Streptococcal Activity Profile of Oritavancin, a Potent Lipoglycopeptide under Development for Use Against Gram-Positive Infections”/E-1617 “In Vitro Activity Profile of Oritavancin (ORI) Against Resistant Staphylococcal Populations From a Recent Surveillance Initiative”/E-1620 “Pharmacokinetic Concentrations of Oritavancin Kill Stationary-Phase and Biofilm Staphylococcus aureusIn Vitro.” / Targanta Press Release September 19, 2007
ICAAC 2007 Posters: “In Vitro Susceptibility of Genotypically Distinct Clostridium difficileStrains to Oritavancin” and “Activity of Metronidazole, Vancomycin and Oritavancin Against Epidemic Clostridium difficile Spores” / Targanta Press Release September 19, 2007
ASM 2007 Poster: “Efficacy of Oritavancin in a Murine Model of Bacillus anthracis Spore Inhalation Anthrax” / Targanta Press Release May 24, 2007
ICAAC 2003 Late-breaker poster: “Phase III Trial Comparing 3-7 days of Oritavancin vs. 10-14 days of Vancomycin/Cephalexin in the Treatment of Patients with Complicated Skin and Skin Structure Infections (cSSSI)” / InterMune Press Release September 15, 2003
Human glucosidase, prepro-α-[199-arginine,223-histidine] [1]
Alglucosidase alfa
C4435H6739N1175O1279S32
105270.8020
August 1, 2014
The U.S. Food and Drug Administration today announced the approval of Lumizyme (alglucosidase alfa) for treatment of patients with infantile-onset Pompe disease, including patients who are less than 8 years of age. In addition, the Risk Evaluation and Mitigation Strategy (REMS) known as the Lumizyme ACE (Alglucosidase Alfa Control and Education) Program is being eliminated.
Pompe disease is a rare genetic disorder and occurs in an estimated 1 in every 40,000 to 300,000 births. Its primary symptom is heart and skeletal muscle weakness, progressing to respiratory weakness and death from respiratory failure.
The disease causes gene mutations to prevent the body from making enough of the functional form of an enzyme called acid alpha-glucosidase (GAA). This enzyme is necessary for proper muscle functioning. GAA is used by the heart and muscle cells to convert a form of sugar called glycogen into energy. Without the enzyme action, glycogen builds up in the cells and, ultimately, weakens the heart and muscles. Lumizyme is believed to work by replacing the deficient GAA, thereby reducing the accumulated glycogen in heart and skeletal muscle cells.
Lumizyme, a lysosomal glycogen-specific enzyme, was approved by the FDA in 2010 with a REMS to restrict its use to treatment of patients with late (non-infantile) onset Pompe disease who are 8 years of age and older. The REMS was required to mitigate the potential risk of rapid disease progression in the infantile-onset Pompe disease patients and patients with late onset disease less than 8 years of age, and to communicate the risks of anaphylaxis, severe allergic reactions and severe skin and systemic immune mediated reactions to prescribers and patients.
At the time of Lumizyme’s approval, there were insufficient data to support the safety and efficacy of Lumizyme in the infantile-onset Pompe population, so Lumizyme was approved for use only in late onset Pompe disease patients who are at least 8 years of age. Pompe patients with infantile-onset disease and patients younger than 8 years of age continued treatment with Myozyme, which was already approved. Myozyme and Lumizyme, both manufactured by Genzyme Corporation, are produced from the same cell line at different production scales.
This approval provides access to Lumizyme for all Pompe disease patients, regardless of their age.
The FDA reviewed newly available information and determined that Lumizyme and Myozyme are chemically and biochemically comparable. Consequently, the safety and effectiveness of Lumizyme and Myozyme are expected to be comparable. In addition, a single-center clinical study of 18 infantile-onset Pompe disease patients, aged 0.2 to 5.8 months at the time of first infusion, provides further support that infantile-onset patients treated with Lumizyme will have a similar improvement in ventilator-free survival as those treated with Myozyme.
Because data were submitted supporting approval of Lumizyme for all Pompe patients, a REMS restricting its use to a specific age group is no longer necessary. While the risk of anaphylaxis, severe allergic reactions, and severe cutaneous and immune mediated reactions for Lumizyme still exist, these risks are comparable to Myozyme and are communicated in labeling through the Warnings and Precautions, and a Boxed Warning.
“REMS continue to be vital tools for the agency to employ as we work with companies to address the serious risks associated with drugs and monitor their appropriate and safe use in various health care settings,” said Janet Woodcock, M.D., director of the FDA’s Center for Drug Evaluation and Research. “The agency remains committed to exercising a flexible and responsible regulatory approach that ensures REMS programs are being effectively and efficiently used and not resulting in an unnecessary burden on health care professionals and patients.”
Health care professionals and patients should also be aware:
The Warnings and Precautions section of the Lumizyme product label and the Clinical Studies section of the Lumizyme label have been updated to include the safety information of the drug in infantile-onset Pompe disease patients. This includes information from the currently approved Myozyme label and information from a new, uncontrolled study in which patients with infantile onset disease were treated with Lumizyme.
Lumizyme is approved with a Boxed Warning because of the risk of anaphylaxis, severe allergic reactions, immune-mediated reactions and cardiorespiratory failure.
Health care professionals should continue to refer to the drug prescribing information for the latest recommendations on prescribing Lumizyme and report adverse events to the FDA’s MedWatch program (http://www.fda.gov/Safety/MedWatch/default.htm).
Distribution of Lumizyme will no longer be restricted. Health care professionals, healthcare facilities, and patients will no longer be required to enroll in the Lumizyme REMS program (Lumizyme ACE Program) to be able to prescribe, dispense, or receive Lumizyme.
The most commonly reported side effects for Lumizyme were infusion-related reactions and included severe allergic reactions, hives, diarrhea, vomiting, shortness of breath, itchy skin, skin rash, neck pain, partial hearing loss, flushing, pain in extremities, and chest discomfort.
Myozyme and Lumizyme are marketed by Cambridge, Massachusetts-based Genzyme.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.
Orphan drug pharmaceutical company, Genzyme, markets alglucosidase alfa as “Myozyme”. In 2006, the U.S. Food and Drug Administration (FDA) approved Myozyme as a suitable ERT treatment for children.[2] Some health plans have refused to subsidize Myozyme for adult patients because it lacks approval for treatment in adults, as well as its high cost (US$300,000/yr for life).[4]
On August 1, 2014 the U.S. Food and Drug Administration announced the approval of Lumizyme (alglucosidase alfa) for treatment of patients with infantile-onset Pompe disease, including patients who are less than 8 years of age. In addition, the Risk Evaluation and Mitigation Strategy (REMS) known as the Lumizyme ACE (Alglucosidase Alfa Control and Education) Program is being eliminated. [5]
Side effects
Common observed adverse reactions to alglucosidase alfa treatment are pneumonia, respiratory complications, infections and fever. More serious reactions reported includeheart and lung failure and allergic shock. Myozyme boxes carry warnings regarding the possibility of life-threatening allergic response.[2]
References
^ Jump up to:abcAmerican Medical Association (USAN). “Alglucosidase alfa” (Microsoft Word). STATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL. Retrieved 18 December 2007.
Jump up^Kishnani PS, Corzo D, Nicolino M et al. (2007). “Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease”. Neurology68 (2): 99–109.doi:10.1212/01.wnl.0000251268.41188.04. PMID17151339.
MYOZYME (alglucosidase alfa), a lysosomal glycogen-specific enzyme, consists of the human enzyme acid α-glucosidase (GAA), encoded by the most predominant of nine observed haplotypes of this gene. MYOZYME is produced by recombinant DNA technology in a Chinese hamster ovary cell line. The MYOZYME manufacturing process differs from that for LUMIZYME®, resulting in differences in some product attributes. Alglucosidase alfa degrades glycogen by catalyzing the hydrolysis of α-1,4- and α-1,6- glycosidic linkages of lysosomal glycogen.
Alglucosidase alfa is a glycoprotein with a calculated mass of 99,377 daltons for the polypeptide chain, and a total mass of approximately 110 kilo Daltons, including carbohydrates. Alglucosidase alfa has a specific activity of 3 to 5 U/mg (one unit is defined as that amount of activity that results in the hydrolysis of 1 μmole of synthetic substrate per minute under the specified assay conditions). MYOZYME is intended for intravenous infusion. It is supplied as a sterile, nonpyrogenic, white to off-white, lyophilized cake or powder for reconstitution with 10.3 mL
Sterile Water for Injection, USP. Each 50 mg vial contains 52.5 mg alglucosidase alfa, 210 mg mannitol, 0.5 mg polysorbate 80, 9.9 mg sodium phosphate dibasic heptahydrate, 31.2 mg sodium phosphate monobasic monohydrate. Following reconstitution as directed, each vial contains 10.5 mL reconstituted solution and a total extractable volume of 10 mL at 5.0 mg/mL alglucosidase alfa. MYOZYME does not contain preservatives; each vial is for single use only.
The U.S. Food and Drug Administration today approved Jardiance (empagliflozin) tablets as an addition to diet and exercise to improve glycemic control in adults with type 2 diabetes.
Type 2 diabetes affects approximately 26 million people and accounts for more than 90 percent of diabetes cases diagnosed in the United States. Over time, high blood sugar levels can increase the risk for serious complications, including heart disease, blindness, and nerve and kidney damage.
“Jardiance provides an additional treatment option for the care of patients with type 2 diabetes,” said Curtis J. Rosebraugh, M.D., M.P.H., director of the Office of Drug Evaluation II in the FDA’s Center for Drug Evaluation and Research. “It can be used alone or added to existing treatment regimens to control blood sugar levels in the overall management of diabetes.”
Jardiance is a sodium glucose co-transporter 2 (SGLT2) inhibitor. It works by blocking the reabsorption of glucose (blood sugar) by the kidney, increasing glucose excretion, and lowering blood glucose levels in diabetics who have elevated blood glucose levels. The drug’s safety and effectiveness were evaluated in seven clinical trials with 4,480 patients with type 2 diabetes receiving Jardiance. The pivotal trials showed that Jardiance improved hemoglobin A1c levels (a measure of blood sugar control) compared to placebo.
Jardiance has been studied as a stand-alone therapy and in combination with other type 2 diabetes therapies including metformin, sulfonylureas, pioglitazone, and insulin. Jardiance should not be used: to treat people with type 1 diabetes; in those who have increased ketones in their blood or urine (diabetic ketoacidosis); and in those with severe renal impairment, end stage renal disease, or in patients on dialysis.
The FDA is requiring four postmarketing studies for Jardiance:
Completion of an ongoing cardiovascular outcomes trial.
A pediatric pharmacokinetic/pharmacodynamic study.
A pediatric safety and efficacy study. As part of the safety and efficacy study, the effect on bone health and development will be evaluated.
A nonclinical (animal) juvenile toxicity study with a particular focus on renal development, bone development, and growth.
Jardiance can cause dehydration, leading to a drop in blood pressure (hypotension) that can result in dizziness and/or fainting and a decline in renal function. The elderly, patients with impaired renal function, and patients on diuretics to treat other conditions appeared to be more susceptible to this risk.
The most common side effects of Jardiance are urinary tract infections and female genital infections.
Jardiance is distributed by Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, Connecticut.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.
Olodaterol is a potent agonist of the human β2-adrenoceptor with a high β1/β2 selectivity. Its crystalline hydrochloride salt is suitable for inhalation and is currently undergoing clinical trials in man for the treatment of asthma. Olodaterol has a duration of action that exceeds 24 hours in two preclinical animal models of bronchoprotection and it has a better safety margin compared with formoterol.
Olodaterol hydrochloride [USAN]
Bi 1744 cl
Bi-1744-cl
Olodaterol hydrochloride
Olodaterol hydrochloride [usan]
UNII-65R445W3V9
Boehringer Ingelheim has launched a new chronic obstructive pulmonary disease drug, Striverdi in the UK and Ireland.
Striverdi (olodaterol) is the second molecule to be licenced for delivery via the company’s Respimat Soft Mist inhaler, following the COPD blockbuster Spiriva (tiotropium). The drug was approved in Europe in November based on results from a Phase III programme that included more than 3,000 patients with moderate to very severe disease.http://www.pharmatimes.com/Article/14-07-01/BI_launches_COPD_drug_Striverdi_in_UK_and_Ireland.aspx
Olodaterol hydrochloride is a drug candidate originated by Boehringer Ingelheim. The product, delivered once-daily by the Respimat Soft Mist Inhaler, was first launched in Denmark and the Netherlands in March 2014 for the use as maintenance treatment of chronic obstructive pulmonary disease (COPD), including chronic bronchitis and/or emphysema. In 2013, approval was obtained in Russia and Canada for the same indication, and in the U.S, the product was recommended for approval. Phase III clinical trials for the treatment of COPD are ongoing in Japan.
As of December 2013, olodaterol is not approved for the treatment of asthma. Olodaterol monotherapy was previously evaluated in four Phase 2 studies in asthma patients. However, currently there are no Phase 3 studies planned for olodaterol monotherapy in patients with asthma.
In late January 2013, Olodaterol CAS# 868049-49-4 was the focus of an FDA committee reviewing data for the drug’s approval as a once-daily maintenance bronchodilator to treat chronic obstructive pulmonary disease (COPD), as well as chronic bronchitis and emphysema. The FDA Pulmonary-Allergy Drugs Advisory Committee recommended that the clinical data from the Boehringer Ingelheim Phase III studies be included in their NDA.
Also known as the trade name Striverdi Respimat, Olodaterol is efficacious as a long-acting beta-agonist, which patients self-administer via an easy to use metered dose inhaler. While early statistics from clinical trials of Olodaterol were encouraging, a new set of data was released earlier this week, which only further solidified the effectual and tolerable benefits of this COPD drug.
On September 10, 2013 results from two Phase 3 studies of Olodaterol revealed additional positive results from this formidable COPD treatment. The conclusion from these two 48 week studies, which included over 3,000 patients, showed sizable and significant improvements in the lung function of patients who were dosed with Olodaterol. Patients in the aforementioned studies were administered either a once a day dosage of Olodaterol via the appropriate metered-dose inhaler or “usual care”. The “usual care” included a variety of treatment options, such as inhaled corticosteroids (not Olodaterol), short and long acting anticholinergics, xanthines and beta agonists, which were short acting. The clinical trial participants who were dosed with Olodaterol displayed a rapid onset of action from this drug, oftentimes within the first five minutes after taking this medication. Additionally, patients dispensed the Olodaterol inhaler were successfully able to maintain optimum lung function for longer than a full 24 hour period. The participants who were given Olodaterol experienced such an obvious clinical improvement in their COPD symptoms, and it quickly became apparent that the “usual care” protocol was lacking in efficacy and reliability.
A staggering 24 million patients in the United States suffer from chronic obstructive pulmonary disease, and this patient population is in need of an effectual, safe and tolerable solution. Olodaterol is shaping up to be that much needed solution. Not only have the results from studies of Olodaterol been encouraging, the studies themselves have actually been forward thinking and wellness centered. Boehringer Ingelheim is the first company to included studies to evaluate exercise tolerance in patients with COPD, and compare the data to those patients who were dosed with Olodaterol. By including exercise tolerance as an important benchmark in pertinent data for Olodaterol, Boehringer Ingelheim has created a standard for COPD treatment expectations. The impaired lung function for patients with COPD contributes greatly to their inability to exercise and stay healthy. Patients who find treatments and management techniques to combat the lung hyperinflation that develops during exercise have a distinct advantage to attaining overall good health.
Data has demonstrated that Striverdi, a once-daily long-acting beta2 agonist, significantly improved lung function versus placebo and is comparable to improvements shown with the older LABA formoterol. The NHS price for the drug is £26.35 for a 30-day supply.
Boehringer cited Richard Russell at Wexham Park Hospital as saying that the licensing of Stirverdi will be welcomed by clinicians as it provides another option. He added that the trial results showing improvements in lung function “are particularly impressive considering the study design, which allowed participants to continue their usual treatment regimen. This reflects more closely the real-world patient population”.
Significantly, the company is also developing olodaterol in combination with Spiriva, a long-acting muscarinic antagonist. LAMA/LABA combinations provide the convenience of delivering the two major bronchodilator classes.
Olodaterol is a novel, long-acting beta2-adrenergic agonist (LABA) that exerts its pharmacological effect by binding and activating beta2-adrenergic receptors located primarily in the lungs. Beta2-adrenergic receptors are membrane-bound receptors that are normally activated by endogenous epinephrine whose signalling, via a downstream L-type calcium channel interaction, mediates smooth muscle relaxation and bronchodilation. Activation of the receptor stimulates an associated G protein which then activates adenylate cyclase, catalyzing the formation of cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA). Elevation of these two molecules induces bronchodilation by relaxation of airway smooth muscles. It is by this mechanism that olodaterol is used for the treatment of chronic obstructive pulmonary disease (COPD) and the progressive airflow obstruction that is characteristic of it. Treatment with bronchodilators helps to mitigate associated symptoms such as shortness of breath, cough, and sputum production. Single doses of olodaterol have been shown to improve forced expiratory volume in 1 sec (FEV1) for 24 h in patients with COPD, allowing once daily dosing. A once-a-day treatment with a LABA has several advantages over short-acting bronchodilators and twice-daily LABAs including improved convenience and compliance and improved airflow over a 24-hour period. Despite similarities in symptoms, olodaterol is not indicated for the treatment of acute exacerbations of COPD or for the treatment of asthma.
Adverse effects
Adverse effects generally were rare and mild in clinical studies. Most common, but still affecting no more than 1% of patients, were nasopharyngitis (running nose), dizziness and rash. To judge from the drug’s mechanism of action and from experiences with related drugs, hypertension (high blood pressure), tachycardia (fast heartbeat), hypokalaemia (low blood levels of potassium), shaking, etc., might occur in some patients, but these effects have rarely, if at all, been observed in studies.[1]
Olodaterol is a nearly full β₂-agonist, having 88% intrinsic activity compared to the gold standard isoprenaline. Its half maximal effective concentration (EC50) is 0.1 nM. It has a higher in vitro selectivity for β₂-receptors than the related drugs formoterol and salmeterol: 241-fold versus β₁- and 2299-fold versus β₃-receptors.[2] The high β₂/β₁ selectivity may account for the apparent lack of tachycardia in clinical trials, which is mediated by β₁-receptors on the heart.
Pharmacokinetics
Once bound to a β₂-receptor, an olodaterol molecule stays there for hours – its dissociation half-life is 17.8 hours –, which allows for once-a-day application of the drug[3] like with indacaterol. Other related compounds generally have a shorter duration of action and have to be applied twice daily (e.g. formoterol, salmeterol). Still others (e. g. salbutamol, fenoterol) have to be applied three or four times a day for continuous action, which can also be an advantage for patients who need to apply β₂-agonists only occasionally, for example in an asthma attack.[8]
History
On 29 January 2013 the U.S. Food and Drug Administration (FDA) Pulmonary-Allergy Drugs Advisory Committee (PADAC) recommended that the clinical data included in the new drug application (NDA) for olodaterol provide substantial evidence of safety and efficacy to support the approval of olodaterol as a once-daily maintenance bronchodilator treatment for airflow obstruction in patients with COPD.[9]
On 18 October 2013 approval of olodaterol in the first three European countries – the United Kingdom, Denmark and Iceland – was announced by the manufacturer.[10]
Figure Chemical structures of salmeterol, formoterol, inda- caterol, and emerging once-daily long-acting β2-agonists
Olodaterol hydrochloride was approved for long-term, once-daily maintenance treatment of chronic
obstructive pulmonary disease (COPD) in 2013 in the following countries: Canada, Russia, United
Kingdom, Denmark, and Iceland.142, 143 The drug has been recommended by a federal advisory panel for
approval by the FDA.142, 143 Developed and marketed by Boehringer Ingelheim, olodaterol is a longacting
β2-adrenergic receptor agonist with high selectivity over the β1- and β3-receptors (219- and 1622-fold, respectively).144 Upon binding to and activating the β2-adrenergic receptor in the airway, olodaterol
stimulates adenyl cyclase to synthesize cAMP, leading to the relaxation of smooth muscle cells in the
airway. Administered by inhalation using the Respimat®
Soft Mist inhaler, it delivers significant
bronchodilator effects within five minutes of the first dose and provides sustained improvement in
forced expiratory volume (FEV1) for over 24 hours.143 While several routes have been reported in the
patent and published literature,144-146 the manufacturing route for olodaterol hydrochloride disclosed in
2011 is summarized in Scheme 19 below.147
Commercial 2’,5’-dihydroxyacetophenone (122) was treated with one equivalent of benzyl bromide
and potassium carbonate in methylisobutylketone (MIBK) to give the 5’-monobenzylated product in
76% yield. Subsequent nitration occurred at the 4’-position to provide nitrophenol 123 in 87% yield.
Reduction of the nitro group followed by subjection to chloroacetyl chloride resulted in the construction
of benzoxazine 124 in 82% yield. Next, monobromination through the use of tetrabutylammonium
tribromide occurred at the acetophenone carbon to provide bromoketone 125, and this was followed by
asymmetric reduction of the ketone employing (−)-DIP chloride to afford an intermediate bromohydrin,
which underwent conversion to the corresponding epoxide 126 in situ upon treatment with aqueous
NaOH. This epoxide was efficiently formed in 85% yield and 98.3% enantiomeric excess. Epoxide
126 underwent ring-opening upon subjection to amine 127 to provide amino-alcohol 128 in in 84-90%
yield and 89.5-99.5% enantiomeric purity following salt formation with HCl. Tertiary amine 127 was
itself prepared in three steps by reaction of ketone 129 with methylmagnesium chloride, Ritter reaction
of the tertiary alcohol with acetonitrile, and hydrolysis of the resultant acetamide with ethanolic
potassium hydroxide. Hydrogenative removal of the benzyl ether within 128 followed by
recrystallization with methanolic isopropanol furnished olodaterol hydrochloride (XVI) in 63-70%
yield. Overall, the synthesis of olodaterol hydrochloride required 10 total steps (7 linear) from
commercially available acetophenone 122.
To a solution of 3.6 g 1,1-dimethyl-2-(4-methoxyphenyl)-ethylamine in 100 mL of ethanol at 70 ° C. 7.5 g of (6-benzyloxy-4H-benzo [1,4] oxazin-3-one )-glyoxal added and allowed to stir for 15 minutes. Then within 30 minutes at 10 to 20 ° C. 1 g of sodium borohydride added. It is stirred for one hour, with 10 mL of acetone and stirred for another 30 minutes. The reaction mixture is diluted with 150 mL ethyl acetate, washed with water, dried with sodium sulfate and concentrated. The residue is dissolved in 50 mL of methanol and 100 mL ethyl acetate and acidified with conc. Hydrochloric acid. After addition of 100 mL of diethyl ether, the product precipitates. The crystals are filtered, washed and recrystallized from 50 mL of ethanol. Yield: 7 g (68%; hydrochloride), mp = 232-234 ° C.
b)
6.8 g of the above obtained benzyl compound in 125 mL of methanol with the addition of 1 g of palladium on carbon (5%) was hydrogenated at room temperature and normal pressure. The catalyst is filtered and the filtrate was freed from solvent. Recrystallization of the residue in 50 mL of acetone and a little water, a solid is obtained, which is filtered and washed.
Yield: 5.0 g (89%; hydrochloride), mp = 155-160 ° C.
The (R) – and (S)-enantiomers of Example 3 can be obtained from the racemate, for example, by chiral HPLC (for example, column: Chirobiotic T, 250 x 1.22 mm from the company Astec). As the mobile phase, methanol with 0.05% triethylamine and 0.05% acetic acid. Silica gel with a grain size of 5 microns, to which is covalently bound the glycoprotein teicoplanin can reach as column material used. Retention time (R enantiomer) = 40.1 min, retention time (S-enantiomer) = 45.9 min. The two enantiomers can be obtained by this method in the form of free bases. According to the invention of paramount importance is the R enantiomer of Example 3
Example 1 6-Hydroxy-8-{(1-hydroxy-2-r2-(4-methoxy-phenyl) – 1, 1-dimethyl-ethylamino]-ethyl)-4H-benzor 41oxazin-3-one – Hvdrochlorid
a) l-(5-benzyloxy-2-hydroxy-3-nitro-phenyl)-ethanone
To a solution of 81.5 g (0.34 mol) l-(5-benzyloxy-2-hydroxy-phenyl)-ethanone in 700 ml of acetic acid are added dropwise under cooling with ice bath, 18 mL of fuming nitric acid, the temperature does not exceed 20 ° C. increases. The reaction mixture is stirred for two hours at room temperature, poured onto ice water and filtered. The product is recrystallized from isopropanol, filtered off and washed with isopropanol and diisopropyl ether. Yield: 69.6 g (72%), mass spectroscopy [M + H] + = 288
b) l-(3-Amino-5-benzyloxy-2-hydroxy-phenyl)-ethanone
69.5 g (242 mmol) of l-(5-benzyloxy-2-hydroxy-3-nitro-phenyl)-ethanone are dissolved in 1.4 L of methanol and in the presence of 14 g of rhodium on carbon (10%) as catalyst at 3 bar room temperature and hydrogenated. Then the catalyst is filtered off and the filtrate concentrated. The residue is reacted further without additional purification. Yield: 60.0 g (96%), R f value = 0.45 (silica gel, dichloromethane).
c) 8-acetyl-6-benzyloxy-4H-benzoπ .4] oxazin-3-one
To 60.0 g (233 mmol) of l-(3-Amino-5-benzyloxy-2-hydroxy-phenyl)-ethanone and 70.0 g (506 mmol) of potassium carbonate while cooling with ice bath, 21.0 ml (258 mmol) of chloroacetyl chloride added dropwise. Then stirred overnight at room temperature and then for 6 hours under reflux. The hot reaction mixture is filtered and then concentrated to about 400 mL and treated with ice water. The precipitate is filtered off, dried and purified by chromatography on a short silica gel column (dichloromethane: methanol = 99:1). The product-containing fractions are concentrated, suspended in isopropanol, diisopropyl ether, and extracted with
Diisopropyl ether. Yield: 34.6 g (50%), mass spectroscopy [M + H] + = 298
d) 6-Benzyloxy-8-(2-chloro-acetyl)-4H-benzoFl, 4] oxazin-3-one 13.8 g (46.0 mmol) of 8-benzyloxy-6-Acetyl-4H-benzo [l, 4] oxazin -3-one and 35.3 g (101.5 mmol) of benzyltrimethylammonium dichloriodat are stirred in 250 mL dichloroethane, 84 mL glacial acetic acid and 14 mL water for 5 hours at 65 ° C. After cooling to room temperature, treated with 5% aqueous sodium hydrogen sulfite solution and stirred for 30 minutes. The precipitated solid is filtered off, washed with water and diethyl ether and dried. Yield: 13.2 g (86%), mass spectroscopy [M + H] + = 330/32.
e) 6-Benzyloxy-8-((R-2-chloro-l-hydroxy-ethyl)-4H-benzori ,41-oxazin-3-one The procedure is analogous to a procedure described in the literature (Org. Lett ., 2002, 4, 4373-4376).
To 13:15 g (39.6 mmol) of 6-benzyloxy-8-(2-chloro-acetyl)-4H-benzo [l, 4] oxazin-3-one and 25.5 mg (0:04 mmol) Cρ * RhCl [(S, S) -TsDPEN] (Cp * = pentamethylcyclopentadienyl and TsDPEN = (lS, 2S)-Np-toluenesulfonyl-l ,2-diphenylethylenediamine) in 40 mL of dimethylformamide at -15 ° C and 8 mL of a mixture of formic acid and triethylamine (molar ratio = 5: 2) dropwise. It is allowed for 5 hours at this temperature, stirring, then 25 mg of catalyst and stirred overnight at -15 ° C. The reaction mixture is mixed with ice water and filtered. The filter residue is dissolved in dichloromethane, dried with sodium sulfate and the solvent evaporated. The residue is recrystallized gel (dichloromethane / methanol gradient) and the product in diethyl ether / diisopropyl ether. Yield: 10.08 g (76%), R f value = 00:28 (on silica gel, dichloromethane ethanol = 50:1).
f) 6-Benzyloxy-8-(R-oxiranyl-4H-benzo [“L4] oxazin-3-one 6.10 g (30.1 mmol) of 6-benzyloxy-8-((R)-2-chloro-l-hydroxy- ethyl)-4H-benzo [l, 4] oxazin-3-one are dissolved in 200 mL of dimethylformamide. added to the solution at 0 ° C with 40 mL of a 2 molar sodium hydroxide solution and stirred at this temperature for 4 hours. the reaction mixture is poured onto ice water, stirred for 15 minutes, and then filtered The solid is washed with water and dried to give 8.60 g (96%), mass spectroscopy [M + H] + = 298..
5.25 g (17.7 mmol) of 6-benzyloxy-8-(R)-oxiranyl-4H-benzo [l, 4] oxazin-3-one and 6.30 g (35.1 mmol) of 2 – (4-methoxy-phenyl 1, 1 – dimethyl-ethyl to be with 21 mL
Of isopropanol and stirred at 135 ° C for 30 minutes under microwave irradiation in a sealed reaction vessel. The solvent is distilled off and the residue chromatographed (alumina, ethyl acetate / methanol gradient). The product thus obtained is purified by recrystallization from a mixture further Diethylether/Diisopropylether-. Yield: 5:33 g (63%), mass spectroscopy [M + H] + = 477 h) 6-Hydroxy-8-{(R)-l-hydroxy-2-[2 – (4-methoxy-phenyl)-l, l-dimethyl-ethylamino] – ethyl}-4H-benzo [1, 4, 1 oxazin-3-one hydrochloride
A suspension of 5:33 g (11.2 mmol) of 6-Benyloxy-8-{(R)-l-hydroxy-2-[2 – (4-methoxy-phenyl)-l, l-dimethyl-ethylamino]-ethyl}-4H -benzo [l, 4] oxazin-3-one in 120 mL of methanol with 0.8 g of palladium on carbon (10%), heated to 50 ° C and hydrogenated at 3 bar hydrogen pressure. Then the catalyst is filtered off and the filtrate concentrated. The residue is dissolved in 20 mL of isopropanol, and 2.5 mL of 5 molar hydrochloric acid in isopropanol. The product is precipitated with 200 mL of diethyl ether, filtered off and dried. Yield: 4.50 g (95%, hydrochloride), mass spectroscopy [M + H] + = 387
The discovery of the β2-adrenoceptor agonist (R)-4p designated olodaterol is described. The preclinical profile of the compound suggests a bronchoprotective effect over 24 h in humans.
Van Noord, J. A.; Smeets, J. J.; Drenth, B. M.; Rascher, J.; Pivovarova, A.; Hamilton, A. L.; Cornelissen, P. J. G. (2011). “24-hour Bronchodilation following a single dose of the novel β2-agonist olodaterol in COPD”. Pulmonary Pharmacology & Therapeutics24 (6): 666–672. doi:10.1016/j.pupt.2011.07.006. PMID21839850.edit
The active moiety olodaterol is a selective beta2-adrenergic bronchodilator. The drug substance, olodaterol hydrochloride, is chemically described as 2H-1,4- Benzoxazin-3H(4H)-one, 6-hydroxy-8-[(1R)-1-hydroxy-2-[[2-(4-methoxyphenyl)-1,1-dimethylethyl]-amino]ethyl]-, monohydrochloride. Olodaterol hydrochloride is a white to off-white powder that is sparingly-slightly soluble in water and slightly soluble in ethanol. The molecular weight is 422.9 g/mole (salt): 386.5 g/mole (base), and the molecular formula is C21H26N2O5 x HCl as a hydrochloride. The conversion factor from salt to free base is 1.094.
The structural formula is:
The drug product, STRIVERDI RESPIMAT, is composed of a sterile, aqueous solution of olodaterol hydrochloride filled into a 4.5 mL plastic container crimped into an aluminum cylinder (STRIVERDI RESPIMAT cartridge) for use with the STRIVERDI RESPIMAT inhaler.
Excipients include water for injection, benzalkonium chloride, edetate disodium, and anhydrous citric acid. The STRIVERDI RESPIMAT cartridge is only intended for use with the STRIVERDI RESPIMAT inhaler. The STRIVERDI RESPIMAT inhaler is a hand held, pocket sized oral inhalation device that uses mechanical energy to generate a slow-moving aerosol cloud of medication from a metered volume of the drug solution. The STRIVERDI RESPIMAT inhaler has a yellow-colored cap.
When used with the STRIVERDI RESPIMAT inhaler, each cartridge containing a minimum of 4 grams of a sterile aqueous solution delivers the labeled number of metered actuations after preparation for use. Each dose (1 dose equals 2 actuations) from the STRIVERDI RESPIMAT inhaler delivers 5 mcg olodaterol in 22.1 mcL of solution from the mouthpiece. As with all inhaled drugs, the actual amount of drug delivered to the lung may depend on patient factors, such as the coordination between the actuation of the inhaler and inspiration through the delivery system. The duration of inspiration should be at least as long as the spray duration (1.5 seconds).
Organic chemists from Industry and academics to interact on Spectroscopy techniques for Organic compounds ie NMR, MASS, IR, UV Etc. email me ……….. amcrasto@gmail.com

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











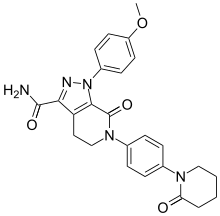



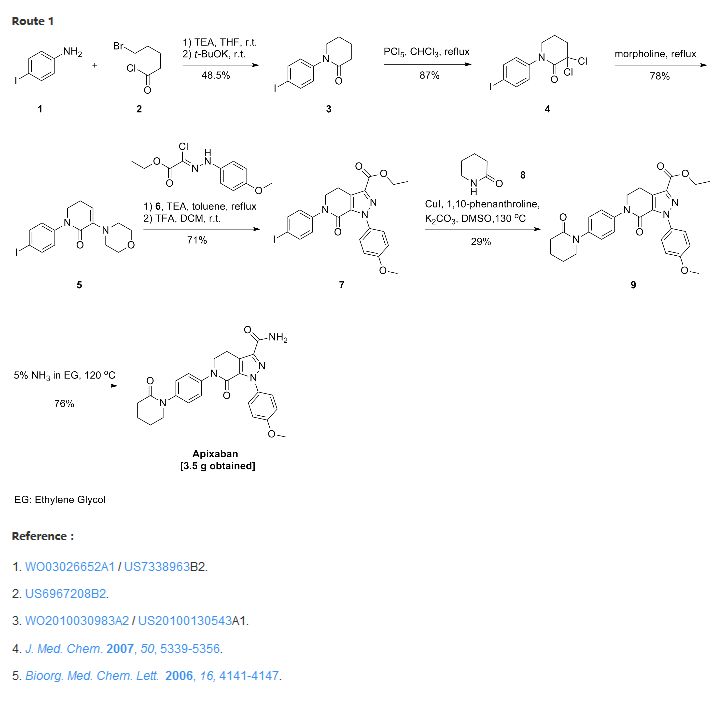



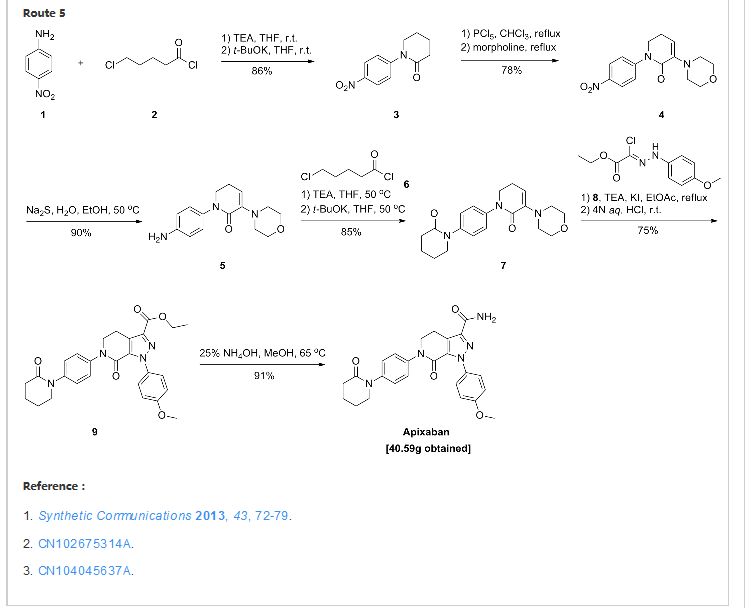
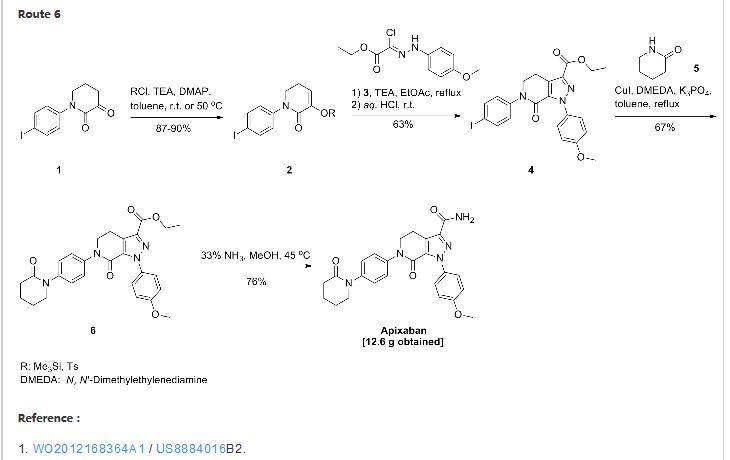



![1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide](https://i0.wp.com/pic8.molbase.net/molpic/ae/b8/543077.png) APIXABAN
APIXABAN





![1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide NMR spectra analysis, Chemical CAS NO. 503612-47-3 NMR spectral analysis, 1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-08-08/000/543/077/503612-47-3-1h.png)
![1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide NMR spectra analysis, Chemical CAS NO. 503612-47-3 NMR spectral analysis, 1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-08-08/000/543/077/503612-47-3-13c.png)






































































 oritavancin
oritavancin oritavancin
oritavancin





 ORITAVANCIN DIPHOSPHATE
ORITAVANCIN DIPHOSPHATE







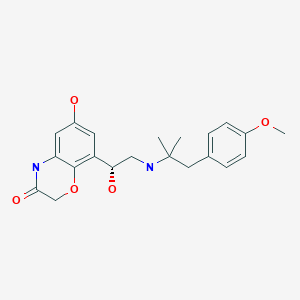
 Olodaterol is a novel, long-acting beta2-adrenergic agonist (LABA) that exerts its pharmacological effect by binding and activating beta2-adrenergic receptors located primarily in the lungs. Beta2-adrenergic receptors are membrane-bound receptors that are normally activated by endogenous epinephrine whose signalling, via a downstream L-type calcium channel interaction, mediates smooth muscle relaxation and bronchodilation. Activation of the receptor stimulates an associated G protein which then activates adenylate cyclase, catalyzing the formation of cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA). Elevation of these two molecules induces bronchodilation by relaxation of airway smooth muscles. It is by this mechanism that olodaterol is used for the treatment of chronic obstructive pulmonary disease (COPD) and the progressive airflow obstruction that is characteristic of it. Treatment with bronchodilators helps to mitigate associated symptoms such as shortness of breath, cough, and sputum production. Single doses of olodaterol have been shown to improve forced expiratory volume in 1 sec (FEV1) for 24 h in patients with COPD, allowing once daily dosing. A once-a-day treatment with a LABA has several advantages over short-acting bronchodilators and twice-daily LABAs including improved convenience and compliance and improved airflow over a 24-hour period. Despite similarities in symptoms, olodaterol is not indicated for the treatment of acute exacerbations of COPD or for the treatment of asthma.
Olodaterol is a novel, long-acting beta2-adrenergic agonist (LABA) that exerts its pharmacological effect by binding and activating beta2-adrenergic receptors located primarily in the lungs. Beta2-adrenergic receptors are membrane-bound receptors that are normally activated by endogenous epinephrine whose signalling, via a downstream L-type calcium channel interaction, mediates smooth muscle relaxation and bronchodilation. Activation of the receptor stimulates an associated G protein which then activates adenylate cyclase, catalyzing the formation of cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA). Elevation of these two molecules induces bronchodilation by relaxation of airway smooth muscles. It is by this mechanism that olodaterol is used for the treatment of chronic obstructive pulmonary disease (COPD) and the progressive airflow obstruction that is characteristic of it. Treatment with bronchodilators helps to mitigate associated symptoms such as shortness of breath, cough, and sputum production. Single doses of olodaterol have been shown to improve forced expiratory volume in 1 sec (FEV1) for 24 h in patients with COPD, allowing once daily dosing. A once-a-day treatment with a LABA has several advantages over short-acting bronchodilators and twice-daily LABAs including improved convenience and compliance and improved airflow over a 24-hour period. Despite similarities in symptoms, olodaterol is not indicated for the treatment of acute exacerbations of COPD or for the treatment of asthma.









 amcrasto@gmail.com
amcrasto@gmail.com

{kind=link}