


The US Food and Drug Administration has approved AstraZeneca’s Movantik for opioid-induced constipation in adults with chronic non-cancer pain.
Home » FDA 2014 (Page 2)
Category Archives: FDA 2014
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
尼达尼布 ニンテダニブ NINTEDANIB For Idiopathic pulmonary fibrosis

NINTEDANIB, BBIF 1120, Intedanib
Boehringer Ingelheim Corp
As a potential treatment for a range of different solid tumour types
CAS 656247-17-5
CAS 1377321-64-6 (nintedanib bisethanesulfonate)
CAS [656247-18-6] mono ethane sulfonate
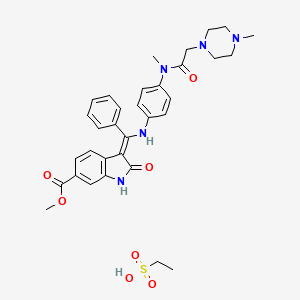
3(Z)-[1-[4-[N-Methyl-N-[2-(4-methylpiperazin-1-yl)acetyl]amino]phenylamino]-1-phenylmethylene]-2-oxo-2,3-dihydro-1H-indole-6-carboxylic acid methyl ester
MW 539.62, MF C31 H33 N5 O4
Launched 2014 USA….Idiopathic pulmonary fibrosis
| chinese, japanese | 尼达尼布 ニンテダニブ |
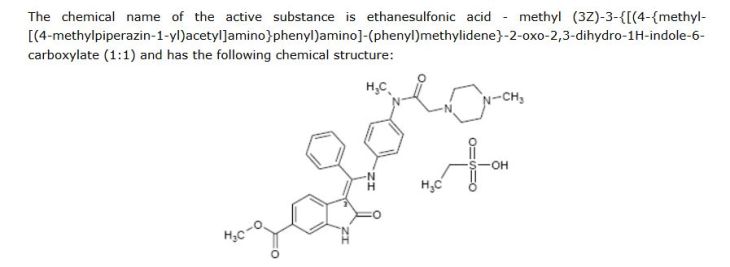
Ethanesulfonic acid – methyl (3Z)-3-{[(4-{methyl[(4-methyl-1-piperazinyl)acetyl]amino}phenyl)amino](phenyl)methylene}-2-oxo-6-indolinecarboxylate (1:1)
Nintedanib esylate
Cas 656247-18-6 [RN]
Methyl (3Z)-3-[({4-[N-methyl-2-(4-methylpiperazin-1-yl)acetamido]phenyl}amino)(phenyl)methylidene]-2-oxo-2,3-dihydro-1H-indole-6-carboxylate ethanesulfonate
Nintedanib esylate [USAN]
(3Z)-2,3-Dihydro-3-[[[4-[methyl[2-(4-methyl-1-piperazinyl)acetyl]amino]phenyl]amino]phenylmethylene]-2-oxo-1H-indole-6-carboxylic acid methyl ester ethanesulfonate
1H-Indole-6-carboxylic acid, 2,3dihydro-3-[[[4-[methyl[(4-methyl-1-piperazinyl)acetyl]amino]phenyl]amino]phenylmethylene]-2-oxo-,methyl ester, (3Z)-, ethanesulfonate (1:1)
Nintedanib esylate, 656247-18-6, UNII-42F62RTZ4G, , NSC753000, NSC-753000, KB-62821
Molecular Formula: C33H39N5O7S Molecular Weight: 649.75706
ニンテダニブエタンスルホン酸塩
Highly crystalline (mp = 305 °C) and exhibits a log P of 3.0 and good aqueous solubility (>20 mg/mL in water)…..J. Med. Chem., 2015, 58 (3), pp 1053–1063
Nintedanib esilate is a bright yellow powder soluble in water. The solubility increases at lower pH and decrease at higher pH due to the non-protonated free base which has a low solubility in water.At room temperature, the active substance exists only in one single crystalline form . The active substance contains no chiral centres. The double bond at C
-3 of the indole moiety allows forE/Zisomerism, but the activesubstance is the Z

Trade Name:Ofev® / Vargatef®
MOA:Tyrosine kinase inhibitor
Indication:Idiopathic pulmonary fibrosis (IPF); Non small cell lung cancer (NSCLC)
In 2011, orphan drug designation was assigned in the U.S. and Japan for the treatment of idiopathic pulmonary fibrosis. In 2013, orphan drug designation was also assigned for the same indication in the E.U. In 2014, a Breakthrough Therapy Designation was assigned to the compound for the treatment of idiopathic pulmonary fibrosis.
Nintedanib (formerly BIBF 1120) is a small molecule tyrosine-kinase inhibitor, targeting vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR) and platelet derived growth factor receptor (PDGFR) being developed by Boehringer Ingelheim as an anti-angiogenesis anti-cancer agent under the trade name Vargatef, and recently approved for treatment of idiopathic pulmonary fibrosis as Ofev.
The use of nintedanib or its salts, particularly its esylate salt is claimed for treating non-small cell lung cancer (NSCLC) in a patient who has received prior treatment with an anti-tumor therapy other than with nintedanib, wherein the patient to be treated is selected for treatment on the basis showing progression of the cancer within a period of 9 months or less after the initiation of said prior treatment. It is also claimed that the compound may be administered in combination with an anti-cancer drug, eg docetaxel. Nintedanib is known to be an antagonist of FGF-1, FGF-2, FGF-3, VEGF-1, VEGF-2, VEGF-3, PDGF-α and PDGF-β receptors.
Use of nintedanib for the treatment of non-small cell lung cancer in a patient who has received prior anti-tumour therapy other than with nintedanib. Boehringer Ingelheim has developed and launched Ofev, an oral capsule formulation of nintedanib, for the treatment of idiopathic pulmonary fibrosis (IPF), hepatic insufficiency and cancer, including metastatic NSCLC, ovarian, prostate and colorectal cancer. In October 2014, the US FDA approved the drug and an NDA was filed in Japan for IPF. Picks up from WO2014049099, claiming pharmaceutical combinations comprising nintedanib and sunitinib.
Nintedanib is an indolinone derivative angiogenesis inhibitor, originated at Boehringer Ingelheim. In 2014, the product candidate was approved and launched in the U.S. for the treatment of idiopathic pulmonary fibrosis, and a positive opinion was received by the EMA for the same indication. Also in 2014, Nintedanib was approved in the E.U. for the oral treatment of locally advanced, metastatic or locally recurrent non-small cell lung cancer (NSCLC) of adenocarcinoma tumour histology after first-line chemotherapy, in combination with docetaxel.The drug candidate is a small-molecule triple kinase inhibitor targeting the angiogenesis kinases (angiokinases) vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR) and platelet-derived growth factor receptor (PDGFR). By allowing the vascularization necessary for the nourishment of tumors, these angiokinases have been implicated in tumor growth, proliferation and metastasis. In previous studies, intedanib potently and selectively inhibited human endothelial cell proliferation and induced apoptosis in human umbilical vein endothelial cells (HUVEC). It showed good oral bioavailability and tolerance, and significant antitumor activity was observed in a number of human tumor xenograft models.
Mechanism of action
Nintedanib is an indolinone-derived drug that inhibits the process of blood vessel formation (angiogenesis). Angiogenesis inhibitors stop the formation and reshaping of blood vessels in and around tumours, which reduces the tumour’s blood supply, starving tumour cells of oxygen and nutrients leading to cell death and tumour shrinkage. Unlike conventional anti-cancer chemotherapy which has a direct cell killing effect on cancer cells, angiogenesis inhibitors starve the tumour cells of oxygen and nutrients which results in tumour cell death. One of the advantages of this method of anti-cancer therapy is that it is more specific than conventional chemotherapy agents, therefore results in fewer and less severe side effects than conventional chemotherapy.
The process of new blood vessel formation (angiogenesis) is essential for the growth and spread of cancers. It is mediated by signaling molecules (growth factors) released from cancer cells in response to low oxygen levels. The growth factors cause the cells of the tumour’s blood vessel to divide and reorganize resulting in the sprouting of new vessels in and around the tumour, improving its blood supply.
Angiogenesis is a process that is essential for the growth and spread of all solid tumours, blocking it prevents the tumour from growing and may result in tumour shrinkage as well as a reduction in the spread of the cancer to other parts of the body. Nintedanib exerts its anti-cancer effect by binding to and blocking the activation of cell receptors involved in blood vessel formation and reshaping (i.e. VEGFR 1-3, FGFR 1-3 AND PDGFRα and β). Inhibition of these receptors in the cells that make up blood vessels (endothelial cells, smooth muscle cells and pericytes) by Nintedanib leads to programmed cell death, destruction of tumor blood vessels and a reduction in blood flow to the tumour. Reduced tumour blood flow inhibits tumor cell proliferation and migration hence slowing the growth and spread of the cancer.[1]
Adverse effects
Preclinical studies have shown that nintedanib binds in a highly selective manner to the ATP binding pocked of its three target receptor families, without binding to similarly shaped ATP domains in other proteins, which reduces the potential for undesirable side effects.[2]
The most common side effects observed with nintedanib were reversible elevation in liver enzymes (10-28% of patients) and gastrointestinal disturbance (up to 50%). Side effects observed with nintedanib were worse with the higher 250 mg dose, for this reason subsequent trials have used the equally clinically effective 200 mg dose.[1][2][3][4][5][6][7][8][9]
Nintedanib inhibits the growth and reshaping of blood vessels which is also an essential process in normal wound healing and tissue repair. Therefore a theoretical side effect of nintedanib is reduced wound healing however, unlike other anti-angiogenic agents, this side effect has not been observed in patients receiving nintedanib.
Studies
Preclinical studies have demonstrated that nintedanib selectively binds to and blocks the VEGF, FGF and PDGF receptors, inhibiting the growth of cells that constitute the walls of blood vessels (endothelial and smooth muscle cells and pericytes) in vitro. Nintedanib reduces the number and density of blood vessels in tumours in vivo, resulting in tumour shrinkage.[1][2] Nintedanib also inhibits the growth of cells that are resistant to existing chemotherapy agents in vitro, which suggests a potential role for the agent in patients with solid tumours that are unresponsive to or relapse following current first line therapy.[10]
Early clinical trials of nintedanib have been carried out in patients with non-small cell lung, colorectal, uterine, endometrial, ovarian and cervical cancer and multiple myeloma.[4][5][7][8][9] These studies reported that the drug is active in patients, safe to administer and is stable in the bloodstream. They identified that the maximum tolerated dose of nintedanib is 20 0 mg when taken once a day.
Clinical studies
In the first human trials, nintedanib halted the growth of tumours in up to 50% of patients with non-small cell lung cancer and 76% of patients with advanced colorectal cancer and other solid tumours.[4][8] A complete response was observed in 1/26 patients with non-small cell lung and 1/7 patients with ovarian cancer treated with nintedanib. A further 2 patients with ovarian cancer had partial responses to nintedanib.[8][9]
Two phase II trials have been carried out assessing the efficacy, dosing and side effects of nintedanib in non-small cell lung and ovarian cancer. These trials found that nintedanib delayed relapse in patients with ovarian cancer by two months[6] and that overall survival of patients with non-small cell lung who received nintedanib was similar to that observed with the FDA approved VEGFR inhibitor sorafenib. These trials also concluded that increasing the dose of the nintedanib has no effect on survival.[3]
SYNTHESIS
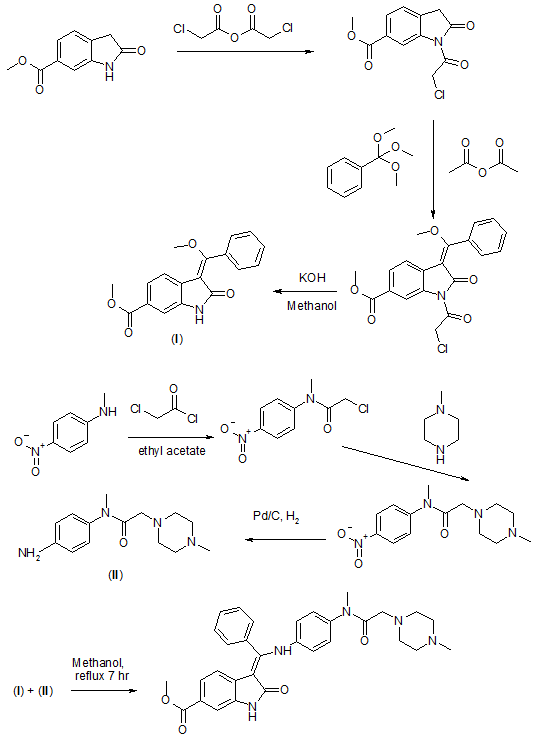
WO2009071523A1
MORE SYNTHESIS
Route 1


Reference:1. WO0127081A1.
2. US6762180B1.
3. J. Med. Chem. 2009, 52, 4466-4480.
Route 2


Reference:1. WO2009071523A1 / US8304541B2.
Route 3


Reference:1. CN104262232A.
Route 4


Reference:1. CN104844499A.
Current clinical trials
Nintedanib is being tested in several phase I to III clinical trials for cancer. Angiogenesis inhibitors such as nintedanib may be effective in a range of solid tumour types including; lung, ovarian, metastatic bowel, liver and brain cancer. Patients are also being recruited for three phase III clinical trials that will evaluate the potential benefit of nintedanib when added to existing 1st line treatments in patients with ovarian.[11] and 2nd line treatment in non-small cell lung cancer [12][13] The phase III trials of nintedanib in lung cancer have been named LUME-Lung 1 and LUME-Lung 2.
Current phase II trials are investigating the effect of nintedanib in patients with metastatic bowel cancer, liver cancer and the brain tumour: glioblastoma multiforme.[14]
Phase III trials are investigating the use of nintedanib in combination with the existing chemotherapy agents permexetred and docetaxel in patients with non-small cell lung cancer,[15] and in combination with carboplatin and paclitaxel as a first line treatment for patients with ovarian cancer.[16]
A phase III clinical trial was underway examining the safety and efficacy of nintedanib on patients with the non-cancerous lung condition idiopathic pulmonary fibrosis.[17] Nintedanib, under the brand name Ofev, was approved by the FDA for treatment of idiopathic pulmonary fibrosis on 15 Oct 2014. [18]
In terms of clinical development, additional phase III clinical trials are ongoing for the treatment of epithelial ovarian cancer, fallopian tube or primary peritoneal cancer, in combination with chemotherapy, and for the treatment of refractory metastatic colorectal cancer. Phase II clinical trials are also ongoing at the company for the treatment of glioblastoma multiforme, previously untreated patients with renal cell cancer, and for the treatment of patients with unresectable malignant pleural mesothelioma. The National Cancer Center of Korea (NCC) is evaluating the compound in phase II studies as second line treatment for small cell lung cancer (SCLC). The Centre Oscar Lambret is also conducting phase II clinical trials for the treatment of breast cancer in combination with docetaxel. Phase II trials are under way at EORTC as second line therapy for patients with either differentiated or medullary thyroid cancer progressing after first line therapy. The compound is also in early clinical development for the treatment of cancer of the peritoneal cavity, hepatocellular carcinoma, acute myeloid leukemia and ovarian cancer. Clinical trials have been completed for the treatment of prostate cancer and for the treatment of colorectal cancer. Boehringer Ingelheim is also conducting phase I/II clinical trials for the treatment of NSCLC and acute myeloid leukemia in addition to low-dose cytarabine. Phase I clinical studies are ongoing at the company for the treatment of epithelial ovary cancer and for the treatment of patients with mild and moderate hepatic impairment. The company had been evaluating the compound in early clinical trials for the treatment of prostate cancer in combination with docetaxel, but recent progress reports for this indication are not available at present.
In 2011, orphan drug designation was assigned in the U.S. and Japan for the treatment of idiopathic pulmonary fibrosis. In 2013, orphan drug designation was also assigned for the same indication in the E.U. In 2014, a Breakthrough Therapy Designation was assigned to the compound for the treatment of idiopathic pulmonary fibrosis.
PAPER
http://pubs.acs.org/doi/full/10.1021/jm501562a
Nintedanib: From Discovery to the Clinic
†Department of Medicinal Chemistry; §Department of Drug Metabolism and Pharmacokinetics; ‡Department of Non-Clinical Drug Safety; ∥Department of Translational Medicine and Clinical Pharmacology; ⊥Department of Respiratory Diseases Research; and #Corporate Division Medicine, TA Oncology, Boehringer Ingelheim Pharma GmbH & Co. KG, 88397 Biberach an der Riss, Germany
○ Clinical Development and Medical Affairs, Respiratory, Boehringer Ingelheim Inc., Ridgefield, Connecticut 06877, United States
∇ Boehringer Ingelheim RCV GmbH & Co. KG, A-1121 Vienna, Austria
J. Med. Chem., 2015, 58 (3), pp 1053–1063
DOI: 10.1021/jm501562a

Nintedanib (BIBF1120) is a potent, oral, small-molecule tyrosine kinase inhibitor, also known as a triple angiokinase inhibitor, inhibiting three major signaling pathways involved in angiogenesis. Nintedanib targets proangiogenic and pro-fibrotic pathways mediated by the VEGFR family, the fibroblast growth factor receptor (FGFR) family, the platelet-derived growth factor receptor (PDGFR) family, as well as Src and Flt-3 kinases. The compound was identified during a lead optimization program for small-molecule inhibitors of angiogenesis and has since undergone extensive clinical investigation for the treatment of various solid tumors, and in patients with the debilitating lung disease idiopathic pulmonary fibrosis (IPF). Recent clinical evidence from phase III studies has shown that nintedanib has significant efficacy in the treatment of NSCLC, ovarian cancer, and IPF. This review article provides a comprehensive summary of the preclinical and clinical research and development of nintedanib from the initial drug discovery process to the latest available clinical trial data.
-
Roth, G. J.; Heckel, A.; Colbatzky, F.; Handschuh, S.; Kley, J.; Lehmann-Lintz, T.; Lotz, R.; Tontsch-Grunt,U.; Walter, R.; Hilberg, F.Design, synthesis, and evaluation of indolinones as triple angiokinase inhibitors and the discovery of a highly specific 6-methoxycarbonyl-substituted indolinone (BIBF 1120) J. Med. Chem.2009, 52, 4466– 4480
-
2.Roth, G. J.; Sieger, P.; Linz, G.; Rall, W.; Hilberg, F.; Bock, T. 3-Z-[1-(4-(N-((4-Methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone monoethanesulphonate and the use thereof as a pharmaceutical composition. WO2004/013099. 2004.
-
3.Merten, J.; Linz, G.; Schnaubelt, J.; Schmid, R.; Rall, W.; Renner, S.; Reichel, C.; Schiffers, R. Process for the manufacture of an indolinone derivative. WO2009/071523. 2009
PAPER
http://pubs.acs.org/doi/abs/10.1021/jm900431g
J. Med. Chem., 2009, 52 (14), pp 4466–4480
DOI: 10.1021/jm900431g

Inhibition of tumor angiogenesis through blockade of the vascular endothelial growth factor (VEGF) signaling pathway is a new treatment modality in oncology. Preclinical findings suggest that blockade of additional pro-angiogenic kinases, such as fibroblast and platelet-derived growth factor receptors (FGFR and PDGFR), may improve the efficacy of pharmacological cancer treatment. Indolinones substituted in position 6 were identified as selective inhibitors of VEGF-, PDGF-, and FGF-receptor kinases. In particular, 6-methoxycarbonyl-substituted indolinones showed a highly favorable selectivity profile. Optimization identified potent inhibitors of VEGF-related endothelial cell proliferation with additional efficacy on pericyctes and smooth muscle cells. In contrast, no direct inhibition of tumor cell proliferation was observed. Compounds 2 (BIBF 1000) and 3 (BIBF 1120) are orally available and display encouraging efficacy in in vivo tumor models while being well tolerated. The triple angiokinase inhibitor 3 is currently in phase III clinical trials for the treatment of nonsmall cell lung cancer.
PATENT
The present invention relates to a beneficial treatment of tumours in patients suffering from NSCLC, and to a clinical marker useful as predictive variable of the responsiveness of tumours in patients suffering from NSCLC. The present invention further relates to a method for selecting patients likely to respond to a given therapy, wherein said method optionally comprises the use of a specific clinical marker. The present invention further relates to a method for delaying disease progression and/or prolonging patient survival of NSCLC patients, wherein said method comprises the use of a specific clinical marker.
The monoethanesulphonate salt form of this compound presents properties which makes this salt form especially suitable for development as medicament. The chemical structure of 3-Z-[l-(4-(N-((4-methyl-piperazin-l-yl)-methylcarbonyl)-N-methyl-amino)-anilino)- 1 -phenyl-methylene] -6-methoxycarbonyl-2-indolinone-monoethanesulphonate (ΓΝΝ name nintedanib esylate) is depicted below as Formula Al .
Formula Al

This compound is thus for example suitable for the treatment of diseases in which angiogenesis or the proliferation of cells is involved. The use of this compound for the treatment of immunologic diseases or pathological conditions involving an
immunologic component is being described in WO 2004/017948, the use for the treatment of, amongst others, oncological diseases, alone or in combination, is being described in WO 2004/096224 and WO 2009/147218, and the use for the treatment of fibrotic diseases is being described in WO 2006/067165.
A method using biomarkers for monitoring the treatment of an individual with the compound 3-Z-[l-(4-(N-((4-methyl-piperazin-l-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-l -phenyl-methylene] -6-methoxycarbonyl-2-indolinone or a pharmaceutically acceptable salt thereof, wherein it is determined if a sample from said individual comprises a biomarker in an amount that is indicative for said treatment, is disclosed in WO 2010/103058.
PATENT
http://www.google.com/patents/US20110201812
The present invention relates to a process for the manufacture of a specific indolinone derivative and a pharmaceutically acceptable salt thereof, namely 3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone and its monoethanesulfonate, to new manufacturing steps and to new intermediates of this process.
The indolinone derivative 3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone and its monoethanesulfonate are known from the following patent applications: WO 01/027081, WO 04/013099, WO 04/017948, WO 04/096224 and WO 06/067165. These patent applications disclose the compound, a process for its manufacture, a specific salt form of this compound and the use of the compound or its salt in a pharmaceutical composition to treat oncological or non-oncological diseases via inhibition of the proliferation of target cells, alone or in combination with further therapeutic agents. The mechanism of action by which the proliferation of the target cells occurs is essentially a mechanism of inhibition of several tyrosine kinase receptors, and especially an inhibition of the vascular endothelial growth factor receptor (VEGFR).




EXAMPLE 1Synthesis of the 6-methoxycarbonyl-2-oxindole in accordance with the process shown in synthesis scheme CSynthesis of benzoic acid, 4-chloro-3-nitro-, methylester
-
- 20 kg of 4-chloro-3-nitro-benzoic acid (99.22 mol) is suspended in 76 L methanol. 5.9 kg thionylchloride (49.62 mol) is added within 15 minutes and refluxed for about 3 hours. After cooling to about 5° C., the product is isolated by centrifugation and drying at 45° C.
- Yield: 19.0 kg (88.8% of theoretical amount)
- Purity (HPLC): 99.8%
Synthesis of propanedioic acid, [4-(methoxycarbonyl)-2-nitrophenyl]-, dimethylester
-
- 12.87 kg of malonic acid, dimethylester (97.41 mol) is added to a hot solution (75° C.) of 10.73 kg sodium-tert.amylate (97.41 mol) in 35 L 1-methyl-2-pyrrolidinone (NMP). A solution of 10 kg benzoic acid, 4-chloro-3-nitro-, methylester (46.38 mol) in 25 L 1-methyl-2-pyrrolidinone is added at 75° C. After stirring for 1.5 hours at about 75° C. and cooling to 20° C., the mixture is acidified with 100 L diluted hydrochloric acid to pH 1. After stirring for 1.5 hours at about 5° C., the product is isolated by centrifugation and drying at 40° C.
- Yield: 13.78 kg (95.4% of theoretical amount)
- Purity (HPLC): 99.9%
- Alternatively, propanedioic acid, [4-(methoxycarbonyl)-2-nitrophenyl]-, dimethylester can be synthesized as follows:
- 33.1 kg of malonic acid, dimethylester (250.6 mol) and 27.0 kg benzoic acid, 4-chloro-3-nitro-, methylester (125.3 mol) are subsequently added to a solution of 45.1 kg sodium-methylate (250.6 mol) in 172 kg 1-methyl-2-pyrrolidinone (NMP) at 20° C. After stirring for 1.5 hours at about 45° C. and cooling to 30° C., the mixture is acidified with 249 L diluted hydrochloric acid. At the same temperature, the mixture is seeded, then cooled to 0° C. and stirred for an additional hour. The resulting crystals are isolated by centrifugation, washed and dryed at 40° C.
- Yield: 37.5 kg (86% of theoretical amount)
- Purity (HPLC): 99.7%
Synthesis of 6-methoxycarbonyl-2-oxindole
A solution of 13 kg propanedioic acid, [4-(methoxycarbonyl)-2-nitrophenyl]-, dimethylester (41.77 mol) in 88 L acetic acid is hydrogenated at 45° C. and under 40-50 psi in the presence of 1.3 kg Pd/C 10%. After standstill of the hydrogenation, the reaction is heated up to 115° C. for 2 hours. The catalyst is filtered off and 180 L water is added at about 50° C. The product is isolated after cooling to 5° C., centrifugation and drying at 50° C.
-
- Yield: 6.96 kg (87.2% of theoretical amount)
- Purity (HPLC): 99.8%
EXAMPLE 2Synthesis of the “chlorimide” (methyl-1-(chloroacetyl)-2-oxoindoline-6-carboxylate)

Method 1
6-methoxycarbonyl-2-oxindole (400 g; 2.071 mol) is suspended in toluene (1200 ml) at room temperature. Chloroacetic anhydride (540 g; 3.095 mol) is added to this suspension. The mixture is heated to reflux for 3 h, then cooled to 80° C. and methyl cyclohexane (600 ml) is added within 30 min. The resulting suspension is further cooled down to room temperature within 60 min. The mother liquor is separated and the solid is washed with ice cold methanol (400 ml). The crystals are dried to afford 515.5 g (93.5%) of the “chlorimide” compound as a white solid. 1H-NMR (500 MHz, DMSO-d6) δ: 8.66 (s, 1H, 6-H); 7.86 (d, J=8.3 Hz, 1H, 8-H); 7.52 (d, J=8.3 Hz, 1H, 9-H); 4.98 (s, 2H, 15-H2); 3.95 (s, 3H, 18-H3); 3.88 (s, 2H, 3-H2). 13C-NMR (126 MHz, DMSO-d6) δ: 174.7 (C-2); 36.0 (C-3); 131.0 (C-4); 140.8 (C-5); 115.7 (C-6); 128.9 (C-7); 126.1 (C-8); 124.6 (C-9); 166.6 (C-10); 165.8 (C-13); 46.1 (C-15); 52.3 (C-18). MS: m/z 268 (M+H)+. Anal. calcd. for C12H10ClNO4: C, 53.85; H, 3.77; Cl, 13.25; N, 5.23. Found: C, 52.18; H, 3.64; Cl, 12.89; N, 5.00.
Method 2
6-Methoxycarbonyl-2-oxindole (10 g; 0.052 mol) is suspended in n-butyl acetate (25 ml) at room temperature. To this suspension a solution of chloroacetic anhydride (12.8 g; 0.037 mol) in n-butyl acetate (25 ml) is added within 3 min. The mixture is heated to reflux for 2 h, then cooled to 85° C. and methyl cyclohexane (20 ml) is added. The resulting suspension is further cooled down to room temperature and stirred for 2 h. The mother liquor is separated and the solid is washed with methanol (400 ml) at ambient temperature. The crystals are dried to afford 12.7 g (91.5%) of the “chlorimide” compound as a slightly yellow solid.
EXAMPLE 3Synthesis of the “chlorenol” (methyl-1-(chloroacetyl)-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate)

Method 1
Methyl-1-(chloroacetyl)-2-oxoindoline-6-carboxylate (12.0 g; 0.045 mol) is suspended in toluene (60 ml) at ambient temperature. Acetic anhydride (16.2 g; 0.157 mol) is added to this suspension. The mixture is heated to not less than 104° C. and trimethyl orthobenzoate (20.0 g; 0.108 mol) is added within 60 min. During the addition period and subsequent stirring at the same temperature for 3 h, volatile parts of the reaction mixture are distilled off. The concentration of the reaction mixture is kept constant by replacement of the distilled part by toluene (40 ml). The mixture is cooled down to 5° C., stirred for 1 h and filtrated. The solid is subsequently washed with toluene (14 ml) and with a mixture of toluene (8 ml) and ethyl acetate (8 ml). After drying, 16.3 g (91.7%) of the “chlorenol” compound are isolated as slightly yellow crystals. 1H-NMR (500 MHz, DMSO-d6) δ: 8.73 (d, J=1.5 Hz, 1H, 6-H); 8.09 (d, J=8.0 Hz, 1H, 9-H); 7.90 (dd, J=8.1; 1.5 Hz, 1H, 8-H); 7.61-7.48 (m, 5H, 21-H, 22-H, 23-H, 24-H, 25-H); 4.85 (s, 2H, 18-H2); 3.89 (s, 3H, 27-H3); 3.78 (s, 3H, 15-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 165.9 (C-2+C16); 103.9 (C-3); 127.4; 128.6; 130.0; 135.4 (C-4+C-5+C-7+C-20); 115.1 (C-6); 126.1 (C-8); 122.5 (C-9); 166.7 (C-10); 173.4 (C-13); 58.4 (C-15); 46.4 (C-18); 128.6 (C-21+C-22+C-24+C-25); 130.5 (C-23); 52.2 (C-27). MS: m/z 386 (M+H)+. Anal. calcd. for C20H16ClNO5: C, 62.27; H, 4.18; Cl, 9.19; N, 3.63. Found: C, 62.21; H, 4.03; Cl, 8.99; N, 3.52.
Method 2
Methyl-1-(chloroacetyl)-2-oxoindoline-6-carboxylate (12.0 g; 0.045 mol) is suspended in xylene (60 ml) at ambient temperature. Acetic anhydride (16.2 g; 0.157 mol) is added to this suspension. The mixture is heated to reflux, trimethyl orthobenzoate (20.0 g; 0.108 mol) is added within 40 min and heating is maintained for 4 h. The mixture is cooled down to 0° C. and the mother liquor is separated. The solid is subsequently washed with xylene (14 ml) and a mixture of xylene (8 ml) and ethyl acetate (8 ml). After drying 14.3 g (81.0%) of the “chlorenol” compound are isolated as yellow crystals.
Method 3
Methyl-1-(chloroacetyl)-2-oxoindoline-6-carboxylate (12.0 g; 0.045 mol) is suspended in toluene (60 ml) at ambient temperature. Acetic anhydride (16.2 g; 0.157 mol) is added to this suspension. The mixture is heated to reflux, trimethyl orthobenzoate (20.0 g; 0.108 mol) is added within 40 min and heating is maintained for 3 h. The mixture is cooled down to 0° C. and the mother liquor is separated. The solid is subsequently washed with toluene (14 ml) and a mixture of toluene (8 ml) and ethyl acetate (8 ml). After drying 15.3 g (87.3%) of the “chlorenol” compound are isolated as fawn crystals.
EXAMPLE 4Synthesis of the “enolindole” (methyl-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate)

Method 1
A solution of potassium hydroxide (0.41 g, 0.006 mol) in methanol (4 ml) is added at 63° C. to a suspension of methyl-1-(chloroacetyl)-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (8.0 g; 0.020 mol) in methanol (32 ml). The mixture is then stirred for 30 min, cooled to 0° C. and stirring is maintained for 2 h. After filtration, the solid is washed with methanol (24 ml) and dried to afford 6.0 g (94.6%) of the “enolindole” compound as yellow crystals. 1H-NMR (500 MHz, CDCl3) δ: 8.08 (s, 1H, 1-H); 7.88 (d, J=7.8 Hz, 1H, 9-H); 7.75 (m, 1H, 8-H); 7.52-7.56 (m, 3H, 18-H, 19-H, 20-H); 7.40-7.45 (m, 3H, 6-H, 17-H, 21-H); 3.92 (s, 3H, 23-H3); 3.74 (s, 3H, 13-H3). 13C-NMR (126 MHz, CDCl3) δ: 168.8 (C-2); 107.4 (C-3); 130.8 (C-4); 138.2 (C-5); 109.4 (C-6); 128.2 and 128.3 (C-7, C-16); 123.5 (C-8); 123.1 (C-9); 170.1 (C-11); 57.6 (C-13); 167.2 (C-14); 128.7 and 128.9 (C-17, C-18, C-20, C-21); 130.5 (C-19); 52.1 (C-23). MS (m/z): 310 (M+H)+. Anal. calcd. for C18H15NO4: C, 69.89; H, 4.89; N, 4.53. Found: C, 69.34; H, 4.92; N, 4.56.
Method 2
A suspension of methyl-1-(chloroacetyl)-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (7.0 g; 0.018 mol) in methanol (28 ml) is heated to reflux. Within 3 min, a solution of sodium methoxide in methanol (0.24 g, 30 (w/w), 0.001 mol) is added to this suspension. The mixture is then stirred for 30 min, cooled to 5° C. and stirring is maintained for 2 h. After filtration, the solid is washed with methanol (9 ml) and dried to afford 5.4 g (89.7%) of the “enolindole” compound as yellow crystals.
Method 3
A suspension of methyl-1-(chloroacetyl)-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (8.0 g; 0.021 mol) in methanol (32 ml) is heated to reflux. A solution of sodium methoxide in methanol (0.74 g, 30% (w/w), 0.004 mol), further diluted with methanol (4 ml), is added dropwise to this suspension. The mixture is then stirred for 90 min, cooled to 0° C. and stirring is maintained for 2 h. After filtration, the solid is washed with methanol (24 ml) and dried to afford 5.9 g (91.2%) of the “enolindole” compound as yellow crystals.
EXAMPLE 5Synthesis of the “chloroacetyl” (N-(4-nitroanilino)-N-methyl-2-chloro-acetamide)

Method 1
A suspension of N-methyl-4-nitroaniline (140 g; 0.920 mol) in ethyl acetate (400 ml) is heated to 70° C. Within 90 min, chloro acetylchloride (114 g; 1.009 mol) is added to this suspension. The resulting solution is then refluxed for 1 h, cooled to 60° C. and methyl cyclohexane (245 ml) is added. The suspension is further cooled down to 0° C. and stirred for 1 h. The reaction mixture is filtrated, washed with methyl cyclohexane (285 ml) and the precipitate is dried to afford 210.4 g (92.7%) of the “chloroacetyl” compound as white crystals. 1H-NMR (500 MHz, DMSO-d6) δ: 8.29 (d, J=8.5 Hz, 2H, 1-H+3-H); 7.69 (d, J=8.5 Hz, 2H, 4-H+6-H); 4.35 (s, 2H, 9-H2); 3.33 (s, 3H, 12-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 124.6 (C-1+C-3); 145.6 (C-2); 127.4 (C-4+C-6); 148.6 (C-5); 165.6 (C-8); 42.7 (C-9); 37.2 (C-12). MS (m/z): 229 (M+H)+. Anal. calcd. for C9H9ClN2O3: C, 47.28; H, 3.97; N, 12.25. Found: C, 47.26; H, 3.99; Cl, 15.73; N, 12.29.
Method 2
A suspension of N-methyl-4-nitroaniline (20.0 g; 0.131 mol) in ethyl acetate (20 ml) is heated to 60° C. Within 15 min, a solution of chloro acetic anhydride (26.0 g; 0.151 mol) in ethyl acetate (60 ml) is added to this suspension. The resulting solution is then refluxed for 1 h, cooled to 75° C. ° C. and methyl cyclohexane (80 ml) is added. After seeding at 60° C., the suspension is further cooled down to 0° C. and stirred for 1 h. The reaction mixture is filtrated, washed with methyl cyclohexane (40 ml) and the precipitate is dried to afford 25.9 g (83.3%) of the “chloroacetyl” compound as grey crystals.
EXAMPLE 6Synthesis of the “nitroaniline” (N-(4-nitrophenyl)-N-methyl-2-(4-methylpiperazin-1-yl)acetamide) and of the “aniline” (N-(4-aminophenyl)-N-methyl-2-(4-methylpiperazin-1-yl)acetamide)

Method 1
A suspension of N-(4-nitroanilino)-N-methyl-2-chloro-acetamide (20.0 g; 0.087 mol) in toluene (110 ml) is heated to 40° C. Within 30 min, 1-methylpiperazine (21.9 g; 0.216 mol) is added dropwise. After purging of the dropping funnel with toluene (5 ml) the reaction mixture is stirred for 2 h at 55° C., cooled to ambient temperature and washed with water (15 ml). The organic layer is diluted with isopropanol (100 ml) and Pd/C (10%; 1.0 g) is added. After subsequent hydrogenation (H2, 4 bar) at 20° C. the catalyst is removed. Approximately ⅘ of the volume of the resulting solution is evaporated at 50° C. The remaining residue is dissolved in ethyl acetate (20 ml) and toluene (147 ml) heated to 80° C., then cooled to 55° C. and seeded. The reaction mixture is further cooled to 0° C. and stirred for 3 h at the same temperature. After filtration, the solid is washed with ice cold toluene (40 ml) and dried to afford 20.2 g (88.0%) of the “aniline” compound as white crystals. 1H-NMR (500 MHz, DMSO-d6) δ: 6.90 (d, J=8.5 Hz, 2H, 4-H+6-H); 6.65 (d, J=8.5 Hz, 2H, 1-H+3-H); 5.22 (2H, 19-H2); 3.04 (s, 3H, 9-H3); 2.79 (s, 2H, 11-H2); 2.32 (m, 4H, 13-H2+17-H2); 2.23 (m, 4H, 14-H2+16-H2); 2.10 (s, 3H, 18-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 114.0 (C-1+C-3); 148.0 (C-2); 127.6 (C-4+C-6); 131.5 (C-5); 168.9 (C-8); 36.9 (C-9); 58.5 (C-11); 52.4 (C-13+C-17); 54.6 (C-14+C-16); 45.7 (C-18). MS (m/z): 263 (M+H)+. Anal. calcd. for C14H22N4O: C, 64.09; H, 8.45; N, 21.36. Found: C, 64.05; H, 8.43; N, 21.39.
Method 2
A suspension of N-(4-nitroanilino)-N-methyl-2-chloro-acetamide (14.5 g; 0.063 mol) in ethyl acetate (65 ml) is heated to 40° C. Within 30 min, 1-methylpiperazine (15.8 g; 0.156 mol) is added dropwise. After purging of the dropping funnel with ethyl acetate (7 ml) the reaction mixture is stirred at 50° C. for 90 min, cooled to ambient temperature and washed with water (7 ml). The organic layer is diluted with isopropanol (75 ml) and dried over sodium sulphate. After separation of the solid, Pd/C (10%; 2.0 g) is added and the solution is hydrogenated (H2, 5 bar) at ambient temperature without cooling. Subsequently the catalyst is removed by filtration and the solvent is evaporated at 60° C. The remaining residue is dissolved in ethyl acetate (250 ml) and recrystallized. After filtration and drying 10.4 g (60.4%) of the “aniline” compound are isolated as white crystals.
EXAMPLE 7Synthesis of the “anilino” (3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone)

Method 1
A suspension of methyl-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (10.0 g; 0.032 mol) and N-(4-aminophenyl)-N-methyl-2-(4-methylpiperazin-1-yl)acetamide (8.6 g; 0.032 mol) in a mixture of methanol (72 ml) and N,N-dimethylformamide (18 ml) is heated to reflux. After 7 h of refluxing the suspension is cooled down to 0° C. and stirring is maintained for additional 2 h. The solid is filtered, washed with methanol (40 ml) and dried to afford 15.4 g (88.1%) of the “anilino” compound as yellow crystals. 1H-NMR (500 MHz, DMSO-d6) δ: 11.00 (s, 1H, 23-H); 12.23 (s, 19-H); 7.61 (t; J=7.1 Hz, 1H, 33-H); 7.57 (t, J=7.5 Hz, 2H, 32-H+34-H); 7.50 (d, J=7.7 Hz, 2H, 31-H+35-H); 7.43 (d, J=1.6 Hz, 1H, 29-H); 7.20 (dd, J=8.3; 1.6 Hz, 1H, 27-H); 7.13 (d, J=8.3 Hz, 2H, 14-H+18-H); 6.89 (d, 8.3 Hz, 2H, 15-H+17-H); 5.84 (d, J=8.3 Hz, 1H, 26-H); 3.77 (s, 3H, 40-H3); 3.06 (m, 3H, 12-H3); 2.70 (m, 2 H, 8-H2); 2.19 (m, 8H, 2-H2, 3-H2, 5-H2, 6-H2); 2.11 (s, 3H, 7-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 54.5 (C-2+C-6); 52.2 (C-3+C-5); 45.6 (C-7); 59.1 (C-8); 168.5 (C-9); 36.6 (C-12); 140.1 (C-13); 127.6 (C-14+C-18); 123.8 (C-17+C-15); 137.0 (C-16); 158.3 (C-20); 97.5 (C-21); 170.1 (C-22); 136.2 (C-24); 128.9 (C-25); 117.2 (C-26); 121.4 (C-27); 124.0 (C-28); 109.4 (C-29); 131.9 (C-30); 128.4 (C-31+C-35); 129.4 (C-32+C-34); 130.4 (C-33); 166.3 (C-37); 51.7 (C-40). MS (m/z): 540 (M+H)+. Anal. calcd. for C31H33N5O4: C, 69.00; H, 6.16; N, 12.98. Found: C, 68.05; H, 6.21; N, 12.81.
Method 2
A suspension of methyl-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (20.0 g; 0.064 mol) and N-(4-aminophenyl)-N-methyl-2-(4-methylpiperazin-1-yl)acetamide (17.1 g; 0.065 mol) in methanol (180 ml) is heated to reflux for 7.5 h. The resulting suspension is cooled down to 10° C. within 1 h and stirring is maintained for 1 h. After filtration, the solid is washed with ice cold methanol (80 ml) and dried to afford 31.0 g (89.0%) of the “anilino” compound as yellow crystals.
EXAMPLE 8Synthesis of the 3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone, monoethanesulfonate

A suspension of 3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone (30.0 g; 0.055 mol) in methanol (200 ml) and water (2.4 ml) is heated to 60° C. Aqueous ethanesulfonic acid (70% (w/w); 8.75 g; 0.056 mol) is added to the reaction mixture. The resulting solution is cooled to 50° C., seeded and then diluted with isopropanol (200 ml). The mixture is further cooled to 0° C. and stirred for 2 h at this temperature. The precipitate is isolated, washed with isopropanol (120 ml) and dried to furnish 35.1 g (97.3%) of the monoethanesulfonate salt of the compound as yellow crystals. 1H-NMR (400 MHz, DMSO-d6) δ: 12.26 (s, 11-H); 10.79 (s, 1H, 1-H); 9.44 (s, 1H, 24-H); 7.64 (m, 1H, 32-H); 7.59 (m, 2H, 31-H+33-H); 7.52 (m, 2H, 30-H+34-H); 7.45 (d, J=1.6 Hz, 1H, 7-H); 7.20 (dd, J=8.2; 1.6 Hz, 1H, 5-H); 7.16 (m, 2H, 14-H+16-H); 6.90 (m, 2H, 13-H+17-H); 5.85 (d, J=8.2 Hz, 1H, 4-H); 3.78 (s, 3H, 37-H3); 3.45-2.80 (broad m, 4H, 23-H2+25-H2); 3.08 (s, 3H, 28-H3); 2.88 (s, 2H, 20-H2); 2.85-2.30 (broad m, 4H, 22-H2+26-H2); 2.75 (s, 3H, 27-H3); 2.44 (q, J=7.4 Hz, 2H, 39-H2); 1.09 (t, J=7.4 Hz, 3H, 38-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 9.8 (C-38); 36.6 (C-28); 42.3 (C-27); 45.1 (C-39); 51.7 (C-37); 48.9 (C-22+C-26); 52.6 (C-23+C-25); 57.5 (C-20); 97.7 (C-3); 109.5 (C-7); 117.3 (C-4); 121.4 (C-5); 123.8 (C-13+C-17); 124.1 (C-6); 127.7 (C-14+C-16); 128.4 (C-30+C-34); 128.8 (C-9); 129.5 (C-31+C-33); 130.5 (C-32); 132.0 (C-29); 168.5 (C-9); 136.3 (C-8); 137.3 (C-12); 139.5 (C-15); 158.1 (C-10); 166.3 (C-35); 168.0 (C-19); 170.1 (C-2). MS (m/z): 540 (M(base)+H)+. Anal. calcd. for C33H39N5O7S: C, 60.17; H, 6.12; N, 10.63; S, 4.87. Found: C, 60.40; H, 6.15; N, 10.70; S, 4.84.
CLIPS

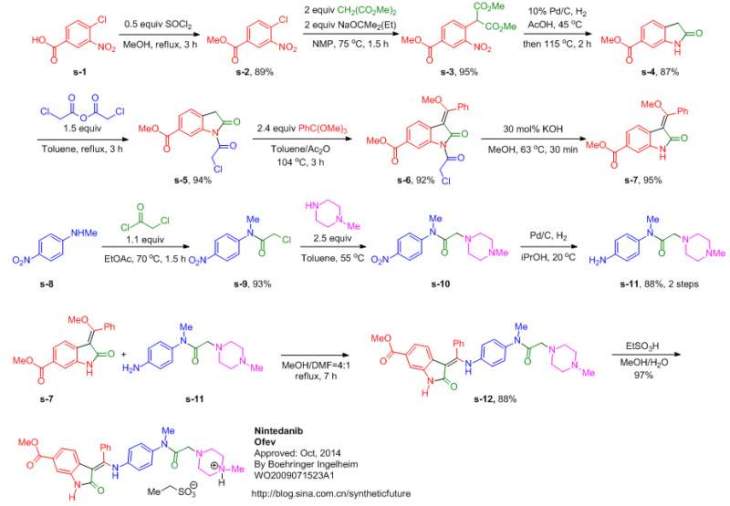
After a classical malonic ester addition to arene 3, the resulting nitro benzene (4) is hydrogenated under acidic conditions, furnishing the 6-methoxycarbonyl-substituted oxindole 5 via decarboxylative cyclization. Condensation of 5 with trimethyl orthobenzoate in acetic anhydride leads to compound 6, one of the two key building blocks of the synthesis. The concomitant N-acetylation of the oxindole activates the scaffold for the condensation reaction.
The aniline side chain (9) can be prepared by a one-pot bromo-acetylation/amination of the para-nitro-phenylamine (7) using bromoacetyl bromide and N-methylpiperazine and a subsequent hydrogenation furnishing 9 as the second key building block. Condensation of both building blocks in an addition–elimination sequence and subsequent acetyl removal with piperidine furnishes 2 as free base (pKa = 7.9), which subsequently is converted into its monoethanesulfonate salt (1). Compound 1 is highly crystalline (mp = 305 °C) and exhibits a log P of 3.0 and good aqueous solubility (>20 mg/mL in water).
CLIPS
see
http://www.yaopha.com/2014/07/09/synthesis-of-vargatef-nintedanib-boehringer-ingelheim-idiopathic-pulmonary-fibrosis-drug/

CLICK ON PIC
Updates………..
“J.Med.Chem” 2009 Vol. 52, page 4466-4480 and the “Chinese Journal of Pharmaceuticals” 2012, Vol. 43, No. 9, page 726-729 reported a further intermediate A and B synthesis, and optimized from the reaction conditions, the reaction sequence, the feed ratio and catalyst selection, etc., so that the above-described synthetic routes can be simplified and reasonable.
PATENT
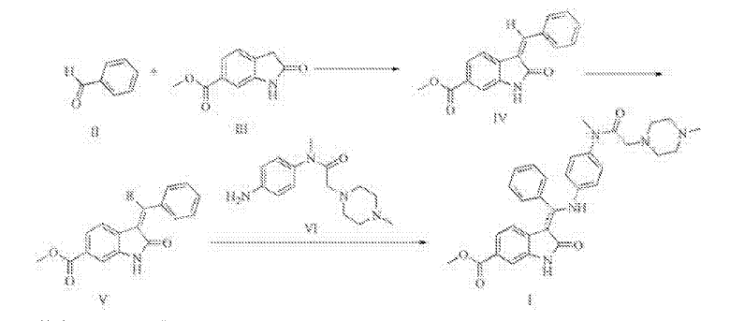
MACHINE TRANSLATED FROM CHINESE
Synthesis of Trinidad Neeb (I),
A 500ml reaction flask was charged 30g of compound V, 22.5g compound of the VI, ethanol 300ml, sodium bicarbonate and 15g, the reaction was heated to reflux for 2 hours, the reaction mixture was added to 600ml of water, there are large amount of solid precipitated, was filtered, the cake washed with 100ml washed once with methanol, a yellow solid 41.9g refined Trinidad Neeb (I). Yield 92.7%.
4 bandit R (400MHz, dmso) δ11 · 97 (s, 1H), 8.38 (s, 1H), 7.97 (dd, J = 11.9, 5.0Hz, 2H), 7.67 (d, J = 8.1Hz, 1H), 7.16 (ddd, J = 26.9, 22.1, 7.0Hz, 5H), 6.85 (d, J = 8.6Hz, 2H), 6.63 (d, J = 8.7Hz, 2H), 3.90 (s, 3H), 2.99 (s, 3H), 2.69 (s, 2H), 2.51-2.24 (m, 8H), 2.20 (s, 3H) MS:. m / z540 (m + 1) + 2 Example: Preparation of compound IV 250ml reaction flask was added 28.7g of 2- oxindole-6-carboxylate, 130ml ethanol, stirred open, then added 30.3ml (31.8g) benzaldehyde, 2.97 mL piperidine was heated to 70 ° C-80 after ° C for 2 hours, allowed to cool to 20 ° C- 30 ° C, the precipitate was filtered, the filter cake was washed with absolute ethanol, 50 ° C 5 hours and dried in vacuo give a yellow solid 38.7g (IV of), yield: 92.4% Preparation of compound V square in 500ml reaction flask was added 30g compound IV, dichloromethane 360ml, cooled with ice water to 0-5 ° C, 71/92 bromine 3.lml (9.7g), drop finished warmed to 20- 30 ° C, 3 hours after the reaction, the reaction solution was washed once with 150ml dichloromethane layer was concentrated oil was done by adding 200ml ethanol crystallization, filtration, 60 ° C and dried under vacuum 36.lg white solid (V ), yield: 93 · 8%.
After Trinidad Technip (I) are synthesized in the reaction flask was added 500ml of 30g compound V, 33.0g compound of the VI, ethanol 300ml, sodium bicarbonate, 15g, was heated to reflux for 2 hours, the reaction mixture was added to 600ml water, there are large amount of solid precipitated, was filtered, the filter cake washed once with 100ml methanol obtained 42.3g of yellow solid was purified by Technip Trinidad (I). Yield 93.6%.
ΧΗNMR (400MHz, dmso) δ11.94 (s, 1Η), 8.36 (s, 1H), 7.96 (dd, J = 11.9, 5.0Hz, 2H), 7.67 (d, J = 8.1Hz, 1H) , 7.16 (ddd, J = 26.9, 22.1, 7.0Hz, 5H), 6.85 (d, J = 8.6Hz, 2H), 6.61 (d, J = 8.7Hz, 2H), 3.90 (s, 3H), 2.99 ( s, 3H), 2.65 (s, 2H), 2.50-2.30 (m, 8H), 2.20 (s, 3H) MS:. m / z540 (m + 1) + square
PATENT
(I) 2.30g, yield 85.3%. Melting point 241 ~ 243 ℃, Mass spectrum (the EI): m / Z 540 (the M + the H), 1 the H NMR (of DMSO D . 6 ): 2.27 (S, 3H), 2.43 (m, 8H), 2.78 (S, 2H) , 3.15 (s, 3H), 3.82 (s, 3H), 5.97 (d, J = 8.3Hz, 1H), 6.77 (d, J = 8.7Hz, 1H), 6.96 (d, J = 8.6Hz, 2H) , 7.32-7.62 (m, 8H), 8.15 (s, 1H), 12.15 (s, 1H).
CLIPS
http://pubs.rsc.org/en/content/articlelanding/2015/ay/c5ay01207d#!divAbstract


Nintedanib
|
|
| Systematic (IUPAC) name | |
|---|---|
| Methyl (3Z)-3-{[(4-{methyl[(4-methylpiperazin-1-yl)acetyl]amino}phenyl)amino](phenyl)methylidene}-2-oxo-2,3-dihydro-1H-indole-6-carboxylate | |
| Clinical data | |
| Trade names | Vargatef, Ofev |
| AHFS/Drugs.com | Consumer Drug Information |
| Pregnancy cat. |
|
| Legal status | |
| Routes | Oral and intravenous |
| Identifiers | |
| CAS number | 656247-17-5 |
| ATC code | None |
| Chemical data | |
| Formula | C31H33N5O4 |
| Mol. mass | 539.6248 g/mol |
References
- Hilberg, F.; G. J. Roth, M. Krssak, S. Kautschitsch, W. Sommergruber, U. Tontsch-Grunt, P. Garin-Chesa, G. Bader, A. Zoephel, J. Quant, A. Heckel, W. J. Rettig (2008). “BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy”. Cancer Res 68 (12): 4774–82. doi:10.1158/0008-5472.CAN-07-6307. ISSN 1538-7445. PMID 18559524.
- Hilberg, F.; U. Tontsch-Grunt, F. Colbatzky, A. Heckel, R. Lotz, J.C.A. van Meel, G.J. Roth (2004). “BIBF1120 a novel, small molecule triple angiokinase inhibitor: profiling as a clinical candidate for cancer therapy”. European Journal of Cancer Supplements 2 (50).
- Reck, M.; R. Kaiser; C. Eschbach; M. Stefanic; J. Love; U. Gatzemeier; P. Stopfer; J. von Pawel (2011). “A phase II double-blind study to investigate efficacy and safety of two doses of the triple angiokinase inhibitor BIBF 1120 in patients with relapsed advanced non-small-cell lung cancer”. Ann Oncol. ISSN 1569-8041.
- Okamoto, I.; H. Kaneda, T. Satoh, W. Okamoto, M. Miyazaki, R. Morinaga, S. Ueda, M. Terashima, A. Tsuya, A. Sarashina, K. Konishi, T. Arao, K. Nishio, R. Kaiser, K. Nakagawa (2010). “Phase I safety, pharmacokinetic, and biomarker study of BIBF 1120, an oral triple tyrosine kinase inhibitor in patients with advanced solid tumors”. Mol Cancer Ther 9 (10): 2825–33. doi:10.1158/1535-7163.MCT-10-0379. ISSN 1538-8514. PMID 20688946.
- Mross, K.; M. Stefanic, D. Gmehling, A. Frost, F. Baas, C. Unger, R. Strecker, J. Henning, B. Gaschler-Markefski, P. Stopfer, L. de Rossi, R. Kaiser (2010). “Phase I study of the angiogenesis inhibitor BIBF 1120 in patients with advanced solid tumors”. Clin Cancer Res 16 (1): 311–9. doi:10.1158/1078-0432.CCR-09-0694. ISSN 1078-0432. PMID 20028771.
- Ledermann, J.A. (2009). “A randomised phase II placebo-controlled trial using maintenance therapy to evaluate the vascular targeting agent BIBF 1120 following treatment of relapsed ovarian cancer (OC)”. J Clin Oncol 27 (15s): (suppl; abstr 5501).
- Kropff, M.; J. Kienast; G. Bisping; W. E. Berdel; B. Gaschler-Markefski; P. Stopfer; M. Stefanic; G. Munzert (2009). “An open-label dose-escalation study of BIBF 1120 in patients with relapsed or refractory multiple myeloma”. Anticancer Res 29 (10): 4233–8. ISSN 1791-7530. PMID 19846979.
- Ellis, P. M.; R. Kaiser; Y. Zhao; P. Stopfer; S. Gyorffy; N. Hanna (2010). “Phase I open-label study of continuous treatment with BIBF 1120, a triple angiokinase inhibitor, and pemetrexed in pretreated non-small cell lung cancer patients”. Clin Cancer Res 16 (10): 2881–9. doi:10.1158/1078-0432.CCR-09-2944. ISSN 1078-0432. PMID 20460487.
- du Bois, A.; J. Huober; P. Stopfer; J. Pfisterer; P. Wimberger; S. Loibl; V. L. Reichardt; P. Harter (2010). “A phase I open-label dose-escalation study of oral BIBF 1120 combined with standard paclitaxel and carboplatin in patients with advanced gynecological malignancies”. Ann Oncol 21 (2): 370–5. doi:10.1093/annonc/mdp506. ISSN 1569-8041. PMID 19889612.
- Xiang, Q. F.; F. Wang; X. D. Su; Y. J. Liang; L. S. Zheng; Y. J. Mi; W. Q. Chen; L. W. Fu (2011). “Effect of BIBF 1120 on reversal of ABCB1-mediated multidrug resistance”. Cell Oncol (Dordr) 34 (1): 33–44. doi:10.1007/s13402-010-0003-7. ISSN 2211-3436.
- “Boehringer Ingelheim – AGO-OVAR 12 / LUME-Ovar 1 Trial Information”. 2011.
- “Boehringer Ingelheim – LUME-Lung 2 Trial Information”. 2011.
- “Boehringer Ingelheim – LUME-Lung 1 Trial Information”. 2011.
- http://clinicaltrials.gov/ct2/results?term=++%09+BIBF+1120&phase=1
- http://clinicaltrials.gov/ct2/show/NCT00805194 Phase III LUME-Lung 1: BIBF 1120 Plus Docetaxel as Compared to Placebo Plus Docetaxel in 2nd Line Non Small Cell Lung Cancer
- http://clinicaltrials.gov/ct2/show/NCT01015118 Phase III BIBF 1120 or Placebo in Combination With Paclitaxel and Carboplatin in First Line Treatment of Ovarian Cancer
- http://clinicaltrials.gov/ct2/show/NCT01335477 Safety and Efficacy of BIBF 1120 at High Dose in Idiopathic Pulmonary Fibrosis Patients II
- “FDA approves Ofev to treat idiopathic pulmonary fibrosis”. 2014.
-
F. Hilberg et al. Cancer Res. 2008, 68, 4774
2. M. Reck et al. Ann. Oncol. 2011, 22, 1374
3. M. Reck et al. J. Clin. Oncol. 2013 (suppl.), Abst LBA8011
4. N. H. Hanna et al. J. Clin. Oncol. 2013, 2013 (suppl.), Abst 8034
5. J.A. Ledermann et al. J. Clin Oncol. 2011, 29, 3798
6. Glioblastoma: A. Muhac et al. J. Neurooncol. 2013, 111, 205
7. O. Bouche et al. Anticancer Res. 2011, 31, 2271
8. T. Eisen et al. J. Clin. Oncol. 2013 (suppl.), Abst. 4506
MORE…………….
Reference:
[6]. Japan PMDA.
[7]. Drug@FDA, NDA205832 Pharmacology Review(s).
[8]. Med. Chem. 2015, 58, 1053-1063.
[9]. Drug@EMA, EMEA/H/C/002569 Vargatef: EPAR-Assessment Report.
[10]. Drug Des. Devel. Ther. 2015, 9, 6407-6419.
[11]. Cancer Res. 2008, 68, 4774-4782.
[12]. J. Med. Chem. 2009, 52, 4466-4480.
Merten, J.; et. al. Process for the manufacture of an indolinone derivative. US20110201812A1
2. Roth, G. J.; et. al. 3-z-[1-(4-(n-((4-methyl-piperazin-1-yl)-methylcarbonyl)-n-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone-monoethanesulphonate and the use thereof as a pharmaceutical composition. WO2004013099A1
3. Roth, G. J.; et. al. Design, Synthesis, and Evaluation of Indolinones as Triple Angiokinase Inhibitors and the Discovery of a Highly Specific 6-Methoxycarbonyl-Substituted Indolinone (BIBF 1120). J Med Chem, 2009, 52(14), 4466-4480. -

ニンテダニブエタンスルホン酸塩
Nintedanib Ethanesulfonate
C31H33N5O4.C2H6O3S : 649.76
[656247-18-6]US7119093 * Jul 21, 2003 Oct 10, 2006 Boehringer Ingelheim Pharma Gmbh & Co. Kg 3-Z-[1-(4-(N-((4-Methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone-monoethanesulphonate and the use thereof as a pharmaceutical composition ///////////////
FDA Approves Blincyto (blinatumomab) for Precursor B-Cell Acute Lymphoblastic Leukemia
 Blinatumomab linking a T cell to a malignant B cell.
Blinatumomab linking a T cell to a malignant B cell.
FDA Approves Blincyto (blinatumomab) for Precursor B-Cell Acute Lymphoblastic Leukemia
December 3, 2014 — The U.S. Food and Drug Administration today
approved Blincyto (blinatumomab) to treat patients with Philadelphia
chromosome-negative precursor B-cell acute lymphoblastic leukemia
(B-cell ALL), an uncommon form of ALL.

Blinatumomab (AMG103) is a drug that has anti-cancer properties. It belongs to a new class of constructed monoclonal antibodies,bi-specific T-cell engagers (BiTEs), that exert action selectively and direct the human immune system to act against tumor cells. Blinatumomab specifically targets the CD19 antigen present on B cells.[1]
The drug was developed by a German-American company Micromet, Inc. in cooperation with Lonza; Micromet was later purchases by Amgen, which has furthered the drug’s clinical trials. In July 2014, the FDA granted breakthrough therapy status to blinatumomab for the treatment of acute lymphoblastic leukemia (ALL).[2] In October 2014, Amgen’s Biologics License Application for blinatumomab was granted priority review designation by the FDA, thus establishing a deadline of May 19, 2015 for completion of the FDA review process.[3]
Structure and mechanism of action
Blinatumomab linking a T cell to a malignant B cell.
Blinatumomab enables a patient’s T cells to recognize malignant B cells. A molecule of blinatumomab combines two binding sites: a CD3site for T cells and a CD19 site for the target B cells. CD3 is part of the T cell receptor. The drug works by linking these two cell types andactivating the T cell to exert cytotoxic activity on the target cell.[4] CD3 and CD19 are expressed in both pediatric and adult patients, making blinatumomab a potential therapeutic option for both pediatric and adult populations.[5]
Therapeutic use
Clinical trials
In a phase 1 clinical study with blinatumomab, patients with non-Hodgkin’s lymphoma showed tumor regression, and in some cases complete remission.[6] There are ongoing phase 1 and phase 2 clinical trials of blinatumomab in patients with acute lymphoblastic leukemia (ALL).[7] One phase II trial for ALL reported good results in 2010 and another is starting.[8]
Adverse effects
Common side effects observed in Phase 2 trials are listed below; they were temporary and typically occurred during the first treatment cycle:[5]
- Flu-like symptoms (i.e. fever, headache, and fatigue)
- Tremor
- Weight increase
- Hypokalemia
- Decrease of blood immunoglobulin
CNS effects were also observed during clinical trials and were treated via a lower dose of blinatumomab, administration of dexamethasone, or treatment discontinuation. Because the side effects were reversible, early monitoring for the CNS symptoms listed below is important:[5]
- Seizure
- Encephalopathy
- Tremor
- Apraxia
- Speech disorders
- Disorientation
Less common side effects include cytokine release syndrome and immunogenicity.[5]
References
- Statement on a Nonproprietary Name adopted by the USAN Council: Blinatumomab
- Amgen Receives FDA Breakthrough Therapy Designation For Investigational BiTE® Antibody Blinatumomab In Acute Lymphoblastic Leukemia
- Amgen’s BiTE® Immunotherapy Blinatumomab Receives FDA Priority Review Designation In Acute Lymphoblastic Leukemia
- Mølhøj, M; Crommer, S; Brischwein, K; Rau, D; Sriskandarajah, M; Hoffmann, P; Kufer, P; Hofmeister, R; Baeuerle, PA (March 2007). “CD19-/CD3-bispecific antibody of the BiTE class is far superior to tandem diabody with respect to redirected tumor cell lysis”. Mol Immunol 44 (8): 1935–43. doi:10.1016/j.molimm.2006.09.032. PMID 17083975.
- Background Information for the Pediatric Subcommittee of the Oncologic Drugs Advisory Committee Meeting 04 December 2012
- Bargou, R; et al. (2008). “Tumor regression in cancer patients by very low doses of a T cell-engaging antibody”. Science 321 (5891): 974–977. doi:10.1126/science.1158545.PMID 18703743.
- ClinicalTrials.gov NCT00560794 Phase II Study of the BiTE Blinatumomab (MT103) in Patients With Minimal Residual Disease of B-precursor Acute ALL
- “Micromet initiates MT103 phase 2 trial in adult ALL patients”. 20 Sep 2010.
External links
| Monoclonal antibody | |
|---|---|
| Type | Bi-specific T-cell engager |
| Source | Mouse |
| Target | CD19, CD3 |
| Clinical data | |
| Legal status |
?
|
| Identifiers | |
| CAS number | 853426-35-4 |
| ATC code | None |
| UNII | 4FR53SIF3A |
| Chemical data | |
| Formula | C2367H3577N649O772S19 |
| Mol. mass | 54.1 kDa |
The U.S. Food and Drug Administration approved Ofev (nintedanib) for the treatment of idiopathic pulmonary fibrosis (IPF).
Nintedanib
FDA approves Ofev to treat idiopathic pulmonary fibrosis
10/15/2014
The U.S. Food and Drug Administration today approved Ofev (nintedanib) for the treatment of idiopathic pulmonary fibrosis (IPF).
read at
see synthesis
https://newdrugapprovals.org/2014/05/21/in-battle-of-ipf-drugs-bis-nintedanib-impresses/

FDA approves Esbriet (pirfenidone ピルフェニドン 吡非尼酮) to treat idiopathic pulmonary fibrosis

The U.S. Food and Drug Administration today approved Esbriet (pirfenidone)
ピルフェニドン 吡非尼酮
for the treatment of idiopathic pulmonary fibrosis (IPF).
read at
SYNTHESIS
Click for synthesis



//////////
FDA Approves Vitekta (elvitegravir) for HIV-1 Infection
FDA Approves Vitekta (elvitegravir) for HIV-1 Infection
September 24, 2014 — The U.S. Food and Drug Administration (FDA) has approved Vitekta (elvitegravir), an integrase strand transfer inhibitor for the combination treatment of human immunodeficiency virus type 1 (HIV-1) infection in treatment-experienced adults.

Elvitegravir
697761-98-1 CAS

Elvitegravir (EVG, formerly GS-9137) is a drug used for the treatment of HIV infection. It acts as an integrase inhibitor. It was developed[1] by the pharmaceutical company Gilead Sciences, which licensed EVG from Japan Tobacco in March 2008.[2][3][4] The drug gained approval by U.S. Food and Drug Administration on August 27, 2012 for use in adult patients starting HIV treatment for the first time as part of the fixed dose combination known as Stribild.[5]
According to the results of the phase II clinical trial, patients taking once-daily elvitegravir boosted by ritonavir had greater reductions in viral load after 24 weeks compared to individuals randomized to receive a ritonavir-boosted protease inhibitor.[6]
。

Human immunodeficiency virus type 1 (HIV-1) is the causative agent of acquired immunodeficiency disease syndrome (AIDS). After over 26 years of efforts, there is still not a therapeutic cure or an effective vaccine against HIV/AIDS. The clinical management of HIV-1 infected people largely relies on antiretroviral therapy (ART). Although highly active antiretroviral therapy (HAART) has provided an effective way to treat AIDS patients, the huge burden of ART in developing countries, together with the increasing incidence of drug resistant viruses among treated people, calls for continuous efforts for the development of anti-HIV-1 drugs. Currently, four classes of over 30 licensed antiretrovirals (ARVs) and combination regimens of these ARVs are in use clinically including: reverse transcriptase inhibitors (RTIs) (e.g. nucleoside reverse transcriptase inhibitors, NRTIs; and non-nucleoside reverse transcriptase inhibitors, NNRTIs), protease inhibitors (PIs), integrase inhibitors and entry inhibitors (e.g. fusion inhibitors and CCR5 antagonists).
- Gilead Press Release Phase III Clinical Trial of Elvitegravir July 22, 2008
- Gilead Press Release Gilead and Japan Tobacco Sign Licensing Agreement for Novel HIV Integrase Inhibitor March 22, 2008
- Shimura K, Kodama E, Sakagami Y, et al. (2007). “Broad Anti-Retroviral Activity and Resistance Profile of a Novel Human Immunodeficiency Virus Integrase Inhibitor, Elvitegravir (JTK-303/GS-9137)”. J Virol 82 (2): 764. doi:10.1128/JVI.01534-07. PMC 2224569. PMID 17977962.
- Stellbrink HJ (2007). “Antiviral drugs in the treatment of AIDS: what is in the pipeline ?”. Eur. J. Med. Res. 12 (9): 483–95. PMID 17933730.
- Sax, P. E.; Dejesus, E.; Mills, A.; Zolopa, A.; Cohen, C.; Wohl, D.; Gallant, J. E.; Liu, H. C.; Zhong, L.; Yale, K.; White, K.; Kearney, B. P.; Szwarcberg, J.; Quirk, E.; Cheng, A. K.; Gs-Us-236-0102 Study, T. (2012). “Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus co-formulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: A randomised, double-blind, phase 3 trial, analysis of results after 48 weeks”.The Lancet 379 (9835): 2439–2448. doi:10.1016/S0140-6736(12)60917-9. PMID 22748591. edit
- Thaczuk, Derek and Carter, Michael. ICAAC: Best response to elvitegravir seen when used with T-20 and other active agents Aidsmap.com. 19 Sept. 2007.

The life cycle of HIV-1. 1. HIV-1 gp120 binds to CD4 and co-receptor CCR5/CXCR4 on target cell; 2. HIV-1 gp41 mediates fusion with target cell; 3. Nucleocapsid containing viral genome and enzymes enters cells; 4. Viral genome and enzymes are released; 5. Viral reverse transcriptase catalyzes reverse transcription of ssRNA, forming RNA-DNA hybrids; 6. RNA template is degraded by ribonuclease H followed by the synthesis of HIV dsDNA; 7. Viral dsDNA is transported into the nucleus and integrated into the host chromosomal DNA by the viral integrase enzyme; 8. Transcription of proviral DNA into genomic ssRNA and mRNAs formation after processing; 9. Viral RNA is exported to cytoplasm; 10. Synthesis of viral precursor proteins under the catalysis of host-cell ribosomes; 11. Viral protease cleaves the precursors into viral proteins; 12. HIV ssRNA and proteins assemble under host cell membrane, into which gp120 and gp41 are inserted; 13. Membrane of host-cell buds out, forming the viral envelope; 14. Matured viral particle is released
Elvitegravir, also known as GS 9137 or JTK 303, is an investigational new drug and a novel oral integrase inhibitor that is being evaluated for the treatment of HIV-1 infection. After HIVs genetic material is deposited inside a cell, its RNA must be converted (reverse transcribed) into DNA. A viral enzyme called integrase then helps to hide HIVs DNA inside the cell’s DNA. Once this happens, the cell can begin producing genetic material for new viruses. Integrase inhibitors, such as elvitegravir, are designed to block the activity of the integrase enzyme and to prevent HIV DNA from entering healthy cell DNA. Elvitegravir has the chemical name: 6-(3-chloro-2-fluorobenzyl)-1-[(S)-1 -hydroxy -methyl-2- methylpropyl]-7-methoxy-4-oxo-1, 4-dihydroquinoline-3-carboxylic acid and has the following structural formula:

WO 2000040561 , WO 2000040563 and WO 2001098275 disclose 4-oxo-1 , 4-dihydro-3- quinoline which is useful as antiviral agents. WO2004046115 provides certain 4- oxoquinoline compounds that are useful as HIV Integrase inhibitors.
US 7176220 patent discloses elvitegravir, solvate, stereoisomer, tautomer, pharmaceutically acceptable salt thereof or pharmaceutical composition containing them and their method of treatment. The chemistry involved in the above said patent is depicted below in the Scheme A. Scheme-A
Toluene, DIPEA
SOCl2 ,COCl (S)-(+)-Valinol
Toluene
,4-Difluoro-5-iodo- benzoic acid


THF
dichlorobis(triphenylphosphine)
palladium argon stream,

Elvitegravir Form ] Elvitegravir (residue) US 7635704 patent discloses certain specific crystalline forms of elvitegravir. The specific crystalline forms are reported to have superior physical and chemical stability compared to other physical forms of the compound. Further, process for the preparation of elvitegravir also disclosed and is depicted below in the Scheme B. The given processes involve the isolation of the intermediates at almost all the stages.
Scheme B
2,
–

Zn THF,
CK Br THF CU “ZnBr dιchlorobis(trιphenylphos
phine)palladium

Elvitegravir WO 2007102499 discloses a compound which is useful as an intermediate for the synthesis of an anti-HIV agent having an integrase-inhibiting activity; a process for production of the compound; and a process for production of an anti-HIV agent using the intermediate.
WO 2009036161 also discloses synthetic processes and synthetic intermediates that can be used to prepare 4-oxoquinolone compounds having useful integrase inhibiting properties.
The said processes are tedious in making and the purity of the final compound is affected because of the number of steps, their isolation, purification etc., thus, there is a need for new synthetic methods for producing elvitegravir which process is cost effective, easy to practice, increase the yield and purity of the final compound, or that eliminate the use of toxic or costly reagents.
US Patent No 7176220 discloses Elvitegravir, solvate, stereoisomer, tautomer, pharmaceutically acceptable salt thereof or pharmaceutical composition containing them and ■ their method of treatment. US Patent No 7635704 discloses Elvitegravir Form II, Form III and processes for their preparation. The process for the preparation of Form Il disclosed in the said patent is mainly by three methods – a) dissolution of Elvitegravir followed by seeding with Form II, b) recrystallisation of Elvitegravir, and c) anti-solvent method.
The process for the preparation of Form III in the said patent is mainly by three methods – a) dissolution of Form Il in isobutyl acetate by heating followed by cooling the reaction mass, b) dissolution of Form Il in isobutyl acetate by heating followed by seeding with Form III, and c) dissolving Form Il in 2-propanol followed by seeding with Form III.
Amorphous materials are becoming more prevalent in the pharmaceutical industry. In order to overcome the solubility and potential bioavailability issues, amorphous solid forms are becoming front-runners. Of special importance is the distinction between amorphous and crystalline forms, as they have differing implications on drug substance stability, as well as drug product stability and efficacy.
An estimated 50% of all drug molecules used in medicinal therapy are administered as salts. A drug substance often has certain suboptimal physicochemical or biopharmaceutical properties that can be overcome by pairing a basic or acidic drug molecule with a counter- ion to create a salt version of the drug. The process is a simple way to modify the properties of a drug with ionizable functional groups to overcome undesirable features of the parent drug. Salt forms of drugs have a large effect on the drugs’ quality, safety, and performance. The properties of salt-forming species significantly affect the pharmaceutical properties of a drug and can greatly benefit chemists and formulators in various facets of drug discovery and development.



chemical synthesis from a carboxylic acid 1 starts after conversion to the acid chloride iodide NIS 2 , and with three condensation 4 . 4 and the amino alcohol 5 addition-elimination reaction occurs 6 , 6 off under alkaline conditions with TBS protected hydroxy get the ring 7 , 7 and zinc reagent 8 Negishi coupling occurs to get 9 , the last 9 hydrolysis and methoxylated
Elvitegravir dimer impurity, WO2011004389A2
Isolation of 1-[(2S)-1-({3-carboxy-6-(3-chloro-2-fluorobenzyl)-1 -[(2S)-I- hydroxy-3-methylbutan-2-yl]-4-oxo-1 , 4-dihydroquinolin-7-yl}oxy)-3- methylbutan-2-yl 6-(3-chloro-2-fluorobenzyl)-7-methoxy-4-oxo-1 , 4-dihydroquinoline-3-carboxylic acid (elvitegravir dimer impurity, 13)
After isolation of the elvitegravir from the mixture of ethyl acetate-hexane, solvent from the filtrate was removed under reduced pressure. The resultant residue purified by column chromatography using a mixture of ethyl acetate-hexane (gradient, 20-80% EtOAc in hexane) as an eluent. Upon concentration of the required fractions, a thick solid was obtained which was further purified on slurry washing with ethyl acetate to get pure elvitegravir dimer impurity (13). The 1H-NMR, 13C-NMR and mass spectral data complies with proposed structure.

1H-NMR (DMSO-Cf6, 300 MHz, ppm) – δ 0.79 (m, d=6.3 Hz, 6H, 20 & 2O’)\ 1.18 & 1.20 (d, J=6.3 Hz & J=6.2 Hz, 6H, 21 & 21′)1, 2.42-2.49 (m, 2H, 19 & 19′), 3.81-3.89 (m, 3H, T & 17’Ha), 3.94-4.01 (m, 1 H, 17’Hb), 4.01 (s, 3H, 23), 4.11 (s, 2H, 7), 4.83-4.85 (m, 3H, 17 & 18′), 5.22 (t, J=4.7 Hz, 1H, OH), 5.41-5.44 (m, 1 H, 18), 6.73-6.78 (t, J=7.1 Hz, 1 H, 11)1‘ 2, 6.92-6.98 (t, J=8.0 Hz, 1H, 3′) 1‘2, 7.12-7.22 (m, 2H, 1 & 3), 7.34-7.39 (m, 1H, 2′),
7.45-7.48 (m, 1 H, 2), 7.49, 7.56 (s, 2H, 15 & 15′), 7.99, 8.02 (s, 2H, 9 & 9′), 8.89, 9.01 (s, 2H, 13 & 13′), 15.30, 15.33 (s, 2H, COOH’ & COOH”).
13C-NMR (DMSO-Cf6, 75 MHz, ppm)- δ 18.87, 19.03 (2OC, 20’C), 19.11 , 19.24 (21 C, 21 ‘C), 27.94 (7’C), 28.40 (7C), 28.91 , 30.08 (19C, 19’C), 56.80(23C), 60.11 (171C), 63.59 (18C), 66.52 (18’C), 68.53 (17C), 97.86, 98.97 (15, 15′), 107.43, 108.16 (12C, 12’C),
118.77, 119.38 (1OC, 10’C), 119.57 (d, J=17.6 Hz, 41C), 119.61 (d, J=17.9 Hz, 4C),
124.88 (d, J=4.3 Hz, 31C), 125.18 (d, J=4.2 Hz, 3C), 126.59, 126.96 (9C1 9’C), 127.14 (8’C), 127.62 (d, J=15.9 Hz, 61C), 127.73 (8C), 127.99 (d, J=15.2 Hz, 6C), 128.66 (2’C),
128.84 (11C), 128.84 (2C), 130.03 (d, J=3.4 Hz, 1C), 142.14, 142.44 (14C, 14’C), 144.37, 145.56 (13C, 131C), 155.24 (d, J=245.1 Hz, 5’C)1 155.61 (d, J=245.1 Hz, 5C),
160.17 (16’C), 162.04 (16C), 166.00, 166.14 (22C, 22’C), 176.17, 176.22 (11C, 111C).
DIP MS: m/z (%)- 863 [M+H]+, 885 [M+Na]+.

MAKE IN INDIA
http://makeinindia.com/sector/pharmaceuticals/

FDA Approves Spiriva Respimat (tiotropium) for the Maintenance Treatment of COPD

Ridgefield, Conn., September 25, 2014 – Boehringer Ingelheim Pharmaceuticals, Inc. announced today that the U.S. Food and Drug Administration (FDA) approved Spiriva Respimat (tiotropium bromide) inhalation spray for the long-term, once-daily maintenance treatment of bronchospasm associated with chronic obstructive pulmonary disease (COPD), including chronic bronchitis and emphysema and to reduce exacerbations in COPD patients. Boehringer Ingelheim anticipates Spiriva Respimat to be available in January 2015.

Spiriva Respimat provides a pre-measured amount of medicine in a slow-moving mist that helps patients inhale the medicine. Spiriva Respimat was developed to actively deliver medication in a way that does not depend of how fast air is breathed in from the inhaler.
READ AT



MAKE IN INDIA
FDA Approves Tybost (cobicistat) for use in the treatment of HIV-1 Infection

Cobicistat, GS-9350
1004316-88-4
| C 40 H 53 N 7 O 5 S 2 |
N-[1(R)-Benzyl-4(R)-[2(S)-[3-(2-isopropylthiazol-4-ylmethyl)-3-methyl]ureido]-4-(4-morpholinyl)butyramido]-5-phenylpentyl]carbamic acid thiazol-5-ylmethyl ester
(1,3-thiazol-5-yl) methyl (5S, 8R, 11R) -8,11-dibenzyl-2-methyl-5-[2 – (morpholin-4-yl) ethyl] -1 – [2 – (propan-2-yl) -1,3-thiazol-4-yl] -3,6-dioxo-2 ,4,7,12-tetraazatridecan-13-oate
cytochrome P450 3A4 (CYP3A4) inhibitor
FDA Approves Tybost (cobicistat) for use in the treatment of HIV-1 Infection
September 24, 2014 — The U.S. Food and Drug Administration (FDA) has approved Tybost (cobicistat), a CYP3A inhibitor used in combination with atazanavir or darunavir for the treatment of human immunodeficiency virus type 1 (HIV-1) infection
Cobicistat is a pharmacokinetic enhancer that works by inhibiting the enzyme (CYP3A) that metabolizes atazanavir and darunavir. It increases the systemic exposure of these drugs and prolongs their effect. Cobicistat is also one of the ingredients in the combination HIV drug Stribild, which was approved by the FDA in August, 2012.
Tybost comes in 150 mg tablets and is administered once daily in combination with the protease inhibitors atazanavir (Reyataz), or darunavir (Prezista).
Because Tybost inhibits CYP3A, other medications metabolized by CYP3A may result in increased plasma concentrations and potentially severe side effects, which may be life-threatening or even fatal. Extra care should be exercised by healthcare professionals to ensure than other medications are reviewed and their concentrations monitored, especially when initiating new medicines or changing doses.
The approval of Tybost was based on the following clinical trials:
•The data to support the use of atazanavir and Tybost were from a phase 2 and 3 trial in treatment-naïve adults comparing atazanavir/cobicistat 300/150 mg and atazanavir/ritonavir 300/100 mg once daily each in combination with Truvada. The atazanavir/cobicistat based regimen was non-inferior to the atazanavir/ritonavir based regimen.
•The data to support the use of cobicistat with darunavir is from a multiple dose trial in healthy subjects comparing the relative bioavailability of darunavir/cobicistat 800/150 mg to darunavir/ritonavir 800/100 mg.
The most common adverse drug reactions observed with Tybost (in combination with atazanavir) in clinical trials were jaundice, ocular icterus, and nausea.
Tybost is a product of Gilead Sciences, Foster City, CA.

Cobicistat (formerly GS-9350) is a licensed drug for use in the treatment of infection with the human immunodeficiency virus (HIV).
Like ritonavir (Norvir), cobicistat is of interest not for its anti-HIV properties, but rather its ability to inhibit liver enzymes that metabolize other medications used to treat HIV, notablyelvitegravir, an HIV integrase inhibitor currently under investigation itself. By combining cobicistat with elvitegravir, higher concentrations of elvitgravir are achieved in the body with lower dosing, theoretically enhancing elvitgravir’s viral suppression while diminishing its adverse side-effects. In contrast with ritonavir, the only currently approved booster, cobicistat has no anti-HIV activity of its own.[1]
Cobicistat, a cytochrome P450 CYP3A4 inhibitor, was approved in the E.U. in 2013 as a pharmacokinetic enhancer of the HIV-1 protease inhibitors atazanavir and darunavir in adults. First launch took place in 2014 in United Kingdom. In 2012, Gilead filed a New Drug Application in the U.S. for the same indication. In April 2013, the FDA issued a Complete Response Letter from the FDA. In 2014 the FDA accepted Gilead’s resubmission.
Cobicistat is a component of the four-drug, fixed-dose combination HIV treatmentelvitegravir/cobicistat/emtricitabine/tenofovir (known as the “Quad Pill” or Stribild).[1][2] The Quad Pill/Stribild was approved by the FDA in August 2012 for use in the United States and is owned by Gilead Sciences.
Cobicistat is a potent inhibitor of cytochrome P450 3A enzymes, including the importantCYP3A4 subtype. It also inhibits intestinal transport proteins, increasing the overall absorption of several HIV medications, including atazanavir, darunavir and tenofovir alafenamide fumarate.[3]
The drug candidate acts as a pharmaco-enhancer to boost exposure of HIV protease inhibitors. In 2011, cobicistat was licensed to Japan Tobacco by Gilead for development and commercialization in Japan as a stand-alone product for the treatment of HIV infection. In 2012, orphan drug designation was assigned in Japan for the pharmacokinetic enhancement of anti-HIV agent.
Oxidative metabolism by cytochrome P450 enzymes is one of the primary mechanisms of drug metabolism.. It can be difficult to maintain therapeutically effective blood plasma levels of drugs which are rapidly metabolized by cytochrome P450 enzymes. Accordingly, the blood plasma levels of drugs which are susceptible to cytochrome P450 enzyme degradation can be maintained or enhanced by co-administration of cytochrome P450 inhibitors, thereby improving the pharmacokinetics of the drug.
While certain drugs are known to inhibit cytochrome P450 enzymes, more and/or improved inhibitors for cytochrome P450 monooxygenase are desirable. Particularly, it would be desirable to have cytochrome P450 monooxygenase inhibitors which do not have appreciable biological activity other than cytochrome P450 inhibition. Such inhibitors can be useful for minimizing undesirable biological activity, e.g., side effects. In addition, it would be desirable to have P450 monooxygenase inhibitors that lack significant or have a reduced level of protease inhibitor activity. Such inhibitors could be useful for enhancing the effectiveness of antiretroviral drugs, while minimizing the possibility of eliciting viral resistance, especially against protease inhibitors.
…………………………….
Cobicistat (GS-9350): A potent and selective inhibitor of human CYP3A as a novel pharmacoenhancer
ACS Med Chem Lett 2010, 1(5): 209
http://pubs.acs.org/doi/abs/10.1021/ml1000257
http://pubs.acs.org/doi/suppl/10.1021/ml1000257/suppl_file/ml1000257_si_001.pdf

Cobicistat (3, GS-9350) is a newly discovered, potent, and selective inhibitor of human cytochrome P450 3A (CYP3A) enzymes. In contrast to ritonavir, 3 is devoid of anti-HIV activity and is thus more suitable for use in boosting anti-HIV drugs without risking selection of potential drug-resistant HIV variants. Compound 3 shows reduced liability for drug interactions and may have potential improvements in tolerability over ritonavir. In addition, 3 has high aqueous solubility and can be readily coformulated with other agents.
1-Benzyl-4-{2-[3-(2-isopropyl-thiazol-4-ylmethyl)-3-methyl-ureido]-4-morpholin-4-yl-butyrylamino}-5-phenyl-pentyl)-carbamic acid thiazol-5-ylmethyl ester (GS-9350)
HPLC (Chiral CelROD-H, Chiral Technologies Inc;heptane/iPrOH = 70/30).
1H NMR (CD3OD)
δ8.98 (1 H, s), 7.82 (1 H, s), 7.25-7.05
(11 H, m), 5.25-5.10 (2 H, m), 4.60-4.50 (2 H, m), 4.21-4.03 (2 H, m), 3.82-3.72 (1
H, m), 3.65-3.65 (4 H, m), 3.35-3.25 (1 H, m), 2.98 (3 H, s), 2.8-2.6 (4 H, m), 2.4-2.2
(6 H, m), 1.95-1.8 (1 H, m), 1.8-1.6 (1 H, m), 1.6-1.4 (4 H, m), 1.42-1.32 (6 H, m).
MS (ESI) m/z: 776.2 (M+H)+.
HRMS calc. for C40H53N7O5S2: 775.355, found: 775.353.
…………………………………
http://www.google.com/patents/CN103694196A?cl=en
CN 103694196
oxidative metabolism by cytochrome P450 enzymes is one of the main mechanisms of drug metabolism, generally by administration of cytochrome P450 inhibitors to maintain or increase the degradation of cytochrome P450 enzymes are sensitive to the drug plasma levels, in order to improve the pharmacokinetics of drugs dynamics, can be used to enhance the effectiveness of anti-retroviral drugs. For example W02008010921 discloses compounds of formula I as a cytochrome P450 monooxygenase specific compounds (Cobicistat):

W02008010921 discloses the synthesis of compounds of formula I with a variety of, as one of the methods of the following routes
Shows:

The reagents used in the method is expensive, and more difficult to remove by-products, long reaction time, high cost, is not conducive to industrial
Production.
W02010115000 on these routes has been improved:

The first step in the route used for the ring-opening reaction reagent trimethylsilyl iodide, trimethylsilyl iodide expensive. W02010115000 reports this step and the subsequent ring-opening reaction of morpholine substitution reaction yield of two steps is not high, only 71%, so that only iodotrimethylsilane a high cost of raw material is not suitable for industrial production.



Preparation of compounds of formula I
Example [0126] Implementation
[0127] I1-a (20g) was dissolved in dichloromethane, was added 50% K0H (5.5g) solution, control the internal temperature does not exceed 25 ° C, TLC analysis ΙΙ-a disappears. Was cooled to O ~ 10 ° C, was added (2R, 5R) -5 – amino-1 ,6 – diphenyl-2 – hexyl-carbamic acid 5 – methyl-thiazole ester hydrochloride (14.8g), stirred for I ~ 2 h, 1 – hydroxybenzotriazole triazole (5.5g), stirred for I h, 1 – ethyl – (3 – dimethylaminopropyl) carbodiimide hydrochloride (15g), and incubated for 5 ~ 10 hours, TLC analysis of the starting material disappeared, the reaction was completed. The reaction was quenched with aqueous acetic acid, methylene chloride layer was separated, washed with saturated aqueous NaHCO3, washed with water, dried and concentrated. By HPLC purity of 99.1%. Adding ethanol, the ethanol was evaporated to give the product compound of part I of a solution in ethanol. Molar yield 88%, LC-MS: M +1 = 777.1 [0128] All publications mentioned in the present invention are incorporated by reference as if each reference was individually incorporated by reference, as cited in the present application. It should also be understood that, after reading the foregoing teachings of the present invention, those skilled in the art that various modifications of the present invention or modifications, and these equivalents falling as defined by the appended claims scope of claims of the present application.
…………………………
US 2014088304
http://www.google.com/patents/US20140088304
International Patent Application Publication Number WO 2008/010921 and International Patent Application Publication Number WO 2008/103949 disclose certain compounds that are reported to be useful to modify the pharmacokinetics of a co-administered drug, e.g. by inhibiting cytochrome P450 monooxygenase. One specific compound identified therein is a compound of the following formula I:

There is currently a need for improved synthetic methods and intermediates that can be used to prepare the compound of formula I and its salts
Schemes 1-4 below.




Preparation of a Compound of Formula IV

Scheme V.
Example 14Preparation of Compound I

To the solution of L-thiazole morpholine ethyl ester oxalate salt XIVa (35.6 kg) in water (66.0 kg) was charged dichloromethane (264 kg), followed by a slow addition of 15 wt % KHCO3 solution (184.8 kg). The resulting mixture was agitated for about 1 hour. The layers were separated and the organic layer was washed with water (132 kg). The organic layer was concentrated under vacuum to dryness. Water (26.5 kg) was charged and the content temperature was adjusted to about 10° C., followed by slow addition of 45% KOH solution (9.8 kg) while maintaining the content temperature at less than or equal to 20° C. The mixture was agitated at less than or equal to 20° C. until the reaction was judged complete by HPLC. The reaction mixture was concentrated under vacuum to dryness and co-evaporated five times with dichloromethane (132 kg each time) under reduced pressure to dryness. Co-evaporation with dichloromethane (132 kg) was continued until the water content was <4% by Karl Fischer titration. Additional dichloromethane (264 kg) was charged and the content temperature was adjusted to −18° C. to −20° C., followed by addition of monocarbamate.HCl salt IXa (26.4 kg). The resulting mixture was agitated at −18° C. to −20° C. for about 1 hour. HOBt (11.4 kg) was charged and the reaction mixture was again agitated at −18° C. to −20° C. for about 1 hour. A pre-cooled solution (−20° C.) of EDC.HCl (21.4 kg) in dichloromethane (396 kg) was added to the reaction mixture while the content temperature was maintained at less than or equal to −20° C. The reaction mixture was agitated at −18° C. to −20° C. until the reaction was judged complete. The content temperature was adjusted to about 3° C. and the reaction mixture quenched with a 10 wt % aqueous citric acid solution (290 kg). The layers were separated and the organic layer was washed once with 15 wt % potassium bicarbonate solution (467 kg) and water (132 kg). The organic layer was concentrated under reduced pressure and then co-evaporated with absolute ethanol.
The product I was isolated as the stock solution in ethanol (35.0 kg product, 76.1% yield).
1H NMR (dDMSO) δ□ 9.05 (s, 1H), 7.85 (s, 1H), 7.52 (d, 1H), 7.25-7.02 (m, 12H), 6.60 (d, 1H), 5.16 (s, 2H), 4.45 (s, 2H), 4.12-4.05 (m, 1H), 3.97-3.85 (m, 1H), 3.68-3.59 (m, 1H), 3.57-3.45 (m, 4H), 3.22 (septets, 1H), 2.88 (s, 3H), 2.70-2.55 (m, 4H), 2.35-2.10 (m, 6H), 1.75 (m, 1H), 1.62 (m, 1H), 1.50-1.30 (m, 4H), 1.32 (d, 6H).
13C NMR (CD3OD) δ 180.54, 174., 160.1, 157.7, 156.9, 153.8, 143.8, 140.1, 140.0, 136.0, 130.53, 130.49, 129.4, 127.4, 127.3, 115.5, 67.7, 58.8, 56.9, 55.9, 54.9, 53.9, 51.6, 49.8, 42.7, 42.0, 35.4, 34.5, 32.4, 32.1, 29.1, 23.7.
Example 13Preparation of L-Thiazole Morpholine Ethyl Ester Oxalate Salt XIVa

To a solution of (L)-thiazole amino lactone XII (33.4 kg) in dichloromethane (89.5 kg) was charged dichloromethane (150 kg) and absolute ethanol (33.4 kg). The content temperature was then adjusted to about 10° C., followed by slow addition of TMSI (78.8 kg) while the content temperature was maintained at less than or equal to 22° C. and agitated until the reaction was judged complete. The content temperature was adjusted to about 10° C., followed by a slow addition of morpholine (49.1 kg) while the content temperature was maintained at less than or equal to 22° C. Once complete, the reaction mixture was filtered to remove morpholine.HI salt and the filter cake was rinsed with two portions of dichloromethane (33.4 kg). The filtrate was washed twice with water (100 kg). The organic layer was concentrated under vacuum to dryness. Acetone (100 kg) was then charged to the concentrate and the solution was concentrated under reduced pressure to dryness. Acetone (233.8 kg) was charged to the concentrate, followed by a slow addition of the solution of oxalic acid (10 kg) in acetone (100 kg). The resulting slurry was refluxed for about 1 hour before cooling down to about 3° C. for isolation. The product XIVa was filtered and rinsed with acetone (66.8 kg) and dried under vacuum at 40° C. to afford a white to off-white solid (40 kg, 71% yield). 1H NMR (CDCl3) δ □7.00 (s, 1H), 6.35 (broad s, 1H), 4.60-4.40 (m, 3H), 4.19 (quartets, 2H), 4.00-3.90 (m, 4H), 3.35-3.10 (m, 7H), 3.00 (s, 3H), 2.40-2.30 (m, 1H), 2.15-2.05 (m, 1H), 1.38 (d, 6H), 1.25 (triplets, 3H).
……………………………………..
W02008010921
http://www.google.co.in/patents/WO2008010921A2?cl=en
Preparation of Example A
Scheme 1

Example A Compound 2
To a solution of Compound 1 (ritonavir) (1.8 g, 2.5 mmol) in 1,2- dichloroethane (15 mL) was added l,l’-thiocarbonyldiimidazole (890 mg, 5.0 mmol). The mixture was heated at 75 SC for 6 hours and cooled to 25 SC. Evaporation under reduced pressure gave a white solid. Purification by flash column chromatography (stationary phase: silica gel; eluent: EtOAc) gave Compound 2 (1.6 g). m/z: 831.1 (M+H)+. Example A
To the refluxing solution of tributyltin hydride (0.78 mL, 2.9 mmol) in toluene (130 mL) was added a solution of Compound 2 (1.6 g, 1.9 mmol) and 2,2′- azobisisobutyronitrile (31 mg, 0.19 mmol) in toluene (30 mL) over 30 minutes. The mixture was heated at 1152C for 6 hours and cooled to 25 BC. Toluene was removed under reduced pressure. Purification by flash column chromatography (stationary phase: silica gel; eluent: hexane/EtOAc = 1/10) gave Example A (560 mg). m/z: 705.2 (M+H)+. 1H-NMR (CDCl3) δ 8.79 (1 H, s), 7.82 (1 H, s), 7.26-7.05 (10 H, m), 6.98 (1 H, s), 6.28 (1 H, m), 6.03 (1 H, m), 5.27 (1 H7 m), 5.23 (2 H, s), 4.45-4.22 (2 H, m), 4.17 (1 H, m), 3.98 (1 H, m), 3.75 (1 H, m), 3.25 (1 H7 m), 2.91 (3 H, s), 2.67 (4 H, m), 2.36 (1 H, m), 1.6-1.2 (10 H, m), 0.85 (6 H, m).
| EP1183026A2 * | 25 May 2000 | 6 Mar 2002 | Abbott Laboratories | Improved pharmaceutical formulations |
| US20060199851 * | 2 Mar 2006 | 7 Sep 2006 | Kempf Dale J | Novel compounds that are useful for improving pharmacokinetics |
| Thiazol-5-ylmethyl N-[1-benzyl-4-[[2-[[(2-isopropylthiazol-4-yl)methyl-methyl-carbamoyl]amino]-4-morpholino-butanoyl]amino]-5-phenyl-pentyl]carbamate | |
| Clinical data | |
|---|---|
| Legal status |
fda approved sept 2014
|
| Identifiers | |
| CAS number | 1004316-88-4 |
| ATC code | V03AX03 |
| PubChem | CID 25151504 |
| ChemSpider | 25084912 |
| UNII | LW2E03M5PG |
| Chemical data | |
| Formula | C40H53N7O5S2 |
| Mol. mass | 776.023 g/mol |
| US7939553 * | Jul 6, 2007 | May 10, 2011 | Gilead Sciences, Inc. | co-administered drug (as HIV protease inhibiting compound, an HIV (non)nucleoside/nucleotide inhibitor of reverse transcriptase, capsid polymerization inhibitor, interferon, ribavirin analog) by inhibiting cytochrome P450 monooxygenase; ureido- or amido-amine derivatives; side effect reduction |
- Highleyman, L.
Elvitegravir “Quad” Single-tablet Regimen Shows Continued HIV Suppression at 48 Weeks
- R Elion, J Gathe, B Rashbaum, and others. The Single-Tablet Regimen of Elvitegravir/Cobicistat/Emtricitabine/Tenofovir Disoproxil Fumarate (EVG/COBI/FTC/TDF; Quad) Maintains a High Rate of Virologic Suppression, and Cobicistat (COBI) is an Effective Pharmacoenhancer Through 48 Weeks. 50th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC 2010). Boston, September 12–15, 2010.
- Lepist, E. -I.; Phan, T. K.; Roy, A.; Tong, L.; MacLennan, K.; Murray, B.; Ray, A. S. (2012). “Cobicistat Boosts the Intestinal Absorption of Transport Substrates, Including HIV Protease Inhibitors and GS-7340, in Vitro”. Antimicrobial Agents and Chemotherapy 56 (10): 5409–5413. doi:10.1128/AAC.01089-12. PMC 3457391. PMID 22850510.
-
Patent No all US
Expiry 5814639 Sep 29, 2015 5814639*PED Mar 29, 2016 5914331 Jul 2, 2017 5914331*PED Jan 2, 2018 5922695 Jul 25, 2017 5922695*PED Jan 25, 2018 5935946 Jul 25, 2017 5935946*PED Jan 25, 2018 5977089 Jul 25, 2017 5977089*PED Jan 25, 2018 6043230 Jul 25, 2017 6043230*PED Jan 25, 2018 6642245 Nov 4, 2020 6642245*PED May 4, 2021 6703396 Mar 9, 2021 6703396*PED Sep 9, 2021 7176220 Nov 20, 2023 7635704 Oct 26, 2026 8148374 Sep 3, 2029
FDA Approves Trulicity (dulaglutide) for Type 2 Diabetes
FDA Approves Trulicity (dulaglutide) for Type 2 Diabetes

DULAGLUTIDE
PRONUNCIATION doo” la gloo’ tide
THERAPEUTIC CLAIM Treatment of type II diabetes
CHEMICAL NAMES
1. 7-37-Glucagon-like peptide I [8-glycine,22-glutamic acid,36-glycine] (synthetic
human) fusion protein with peptide (synthetic 16-amino acid linker) fusion protein with immunoglobulin G4 (synthetic human Fc fragment), dimer
2. [Gly8,Glu22,Gly36]human glucagon-like peptide 1-(7-37)-peptidyltetraglycyl-Lseryltetraglycyl-L-seryltetraglycyl-L-seryl-L-alanyldes-Lys229-[Pro10,Ala16,Ala17]human immunoglobulin heavy constant γ4 chain H-CH2-CH3 fragment, (55-55′:58-58′)-bisdisulfide dimer
- Dulaglutide
- LY 2189265
- LY-2189265
- LY2189265
- UNII-WTT295HSY5
| GLP-1 immunoglobulin G (IgG4) Fc fusion protein with extended activity; a hypoglycemic agent. |
-
7-37-Glucagon-like peptide I (8-glycine,22-glutamic acid,36-glycine) (synthetic human) fusion proteinwith peptide (synthetic 16-amino acid linker) fusion protein with immunoglobulin G4 (synthetic human Fc fragment), dimer
sept 18 2014
The US Food and Drug Administration (FDA) has approved dulaglutide (Trulicity, Eli Lilly & Co), as a once-weekly injection for the treatment of type 2 diabetes.
A member of the glucagon-like peptide-1 receptor agonist class, dulaglutide joins liraglutide (Victoza, Novo Nordisk), exenatide (Byetta, AstraZeneca/Bristol-Myers Squibb), and albiglutide (Tanzeum, GlaxoSmithKline), on the US market.
Once-weekly dulaglutide was approved based on 6 clinical trials involving a total of 3342 patients who received the drug. It was studied as a stand-alone therapy and in combination withmetformin, sulfonylurea, thiazolidinedione, and prandial insulin.
In one trial the once-weekly dulaglutide was non-inferior to daily liraglutide and in another it topped the oral dipeptidyl peptidase-4 (DPP-4) inhibitor sitagliptin (Januvia, Merck).
The most common side effects observed in patients treated with dulaglutide were nausea, diarrhea, vomiting, abdominal pain, and decreased appetite.
Dulaglutide should not be used to treat people with type 1 diabetes, diabetic ketoacidosis, or severe abdominal or intestinal problems, or as first-line therapy for patients who cannot be managed with diet and exercise.
As with others in its class, dulaglutide’s label will include a boxed warning that thyroid C-cell tumors have been observed in rodents but the risk in humans is unknown. The drug should not be used in patients with a personal or family history of medullary thyroid carcinoma (MTC) or multiple endocrine neoplasia type 2.
The FDA is requiring Lilly to conduct the following postmarketing studies for dulaglutide:
• A clinical trial to evaluate dosing, efficacy, and safety in children
• A study to assess potential effects on sexual maturation, reproduction, and central nervous system development and function in immature rats
• An MTC case registry of at least 15 years duration to identify any increase in MTC incidence with the drug
• A clinical trial comparing dulaglutide with insulin glargine on glycemic control in patients with type 2 diabetes and moderate or severe renal impairment
• A cardiovascular outcomes trial to evaluate the drug’s cardiovascular risk profile in patients with high baseline risk for cardiovascular disease.
The FDA approval also comes with a Risk Evaluation and Mitigation Strategy, including a communication plan to inform healthcare professionals about the serious risks associated with the drug.
STRUCTURAL FORMULA
Monomer
HGEGTFTSDV SSYLEEQAAK EFIAWLVKGG GGGGGSGGGG SGGGGSAESK 50
YGPPCPPCPA PEAAGGPSVF LFPPKPKDTL MISRTPEVTC VVVDVSQEDP 100
EVQFNWYVDG VEVHNAKTKP REEQFNSTYR VVSVLTVLHQ DWLNGKEYKC 150
KVSNKGLPSS IEKTISKAKG QPREPQVYTL PPSQEEMTKN QVSLTCLVKG 200
FYPSDIAVEW ESNGQPENNY KTTPPVLDSD GSFFLYSRLT VDKSRWQEGN 250
VFSCSVMHEA LHNHYTQKSL SLSLG 275
Disulfide bridges location
55-55′ 58-58′ 90-150 90′-150′ 196-254 196′-254′
MOLECULAR FORMULA C2646H4044N704O836S18
MOLECULAR WEIGHT 59.67 kDa
MANUFACTURER Eli Lilly and Company
CODE DESIGNATION LY2189265
CAS REGISTRY NUMBER 923950-08-7
http://www.ama-assn.org/resources/doc/usan/dulaglutide.pdf
LY2189265 (dulaglutide), a glucagon-like peptide-1 analog, is a biologic entity being studied as a once-weekly treatment for type 2 diabetes.
Dulaglatuide works by stimulating cells to release insulin only when blood sugar levels are high.
Gwen Krivi, Ph.D., vice president, product development, Lilly Diabetes, said of the drug, “We believe dulaglutide, if approved, can bring significant benefits to people with type 2 diabetes.”
In fact, it might help to control both diabetics’ blood sugar and their high blood pressure.
Eli Lilly CEO John Lechleiter believes the drug has the potential to be a blockbuster. Lilly could be ready to seek approval by 2013.
For more information on dulaglutide clinical studies, click here.
PRESS RELEASES
Data Preseted at 49th EASD Annual Meeting Show Treatment with Lilly’s Investigational Dulaglutide Resulted in Improved Patient-Reported Health Outcomes – September 26, 2013
Lilly Announces Positive Results of Phase III Trials of Dulaglutide in Type 2 Diabetes – April 16, 2013
Lilly Diabetes Announces Positive Results of Phase III Trials of Dulaglutide in Type 2 Diabetes – October 22, 2012
FDA approves AstraZeneca’s constipation drug Movantik
Movantik (naloxegol), an oral once-a-day treatment licensed from Nektar Therapeutics, belongs to a class of drugs called peripherally-acting mu-opioid receptor antagonists, which are used to decrease the constipating effects of opioids. The drug’s safety and effectiveness were established in two trials of 1,352 participants who had taken opioids for at least four weeks for non-cancer related pain and had opioid-induced constipation.
Read more at: http://www.pharmatimes.com/Article/14-09-16/FDA_approves_AstraZeneca_s_constipation_drug_Movantik.aspx#ixzz3DdGiFse8
FDA approves Keytruda for advanced melanoma, First PD-1 blocking drug to receive agency approval

September 4, 2014
FDA Release
The U.S. Food and Drug Administration today granted accelerated approval to Keytruda (pembrolizumab) for treatment of patients with advanced or unresectable melanoma who are no longer responding to other drugs.
Melanoma, which accounts for approximately 5 percent of all new cancers in the United States, occurs when cancer cells form in skin cells that make the pigment responsible for color in the skin. According to the National Cancer Institute, an estimated 76,100 Americans will be diagnosed with melanoma and 9,710 will die from the disease this year.
Keytruda is the first approved drug that blocks a cellular pathway known as PD-1, which restricts the body’s immune system from attacking melanoma cells. Keytruda is intended for use following treatment with ipilimumab, a type of immunotherapy. For melanoma patients whose tumors express a gene mutation called BRAF V600, Keytruda is intended for use after treatment with ipilimumab and a BRAF inhibitor, a therapy that blocks activity of BRAF gene mutations.
“Keytruda is the sixth new melanoma treatment approved since 2011, a result of promising advances in melanoma research,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Many of these treatments have different mechanisms of action and bring new options to patients with melanoma.”
The five prior FDA approvals for melanoma include: ipilimumab (2011), peginterferon alfa-2b (2011), vemurafenib (2011), dabrafenib (2013), and trametinib (2013).
The FDA granted Keytruda breakthrough therapy designation because the sponsor demonstrated through preliminary clinical evidence that the drug may offer a substantial improvement over available therapies. It also received priority review and orphan product designation. Priority review is granted to drugs that have the potential, at the time the application was submitted, to be a significant improvement in safety or effectiveness in the treatment of a serious condition. Orphan product designation is given to drugs intended to treat rare diseases.
The FDA action was taken under the agency’s accelerated approval program, which allows approval of a drug to treat a serious or life-threatening disease based on clinical data showing the drug has an effect on a surrogate endpoint reasonably likely to predict clinical benefit to patients. This program provides earlier patient access to promising new drugs while the company conducts confirmatory clinical trials. An improvement in survival or disease-related symptoms has not yet been established.
Keytruda’s efficacy was established in 173 clinical trial participants with advanced melanoma whose disease progressed after prior treatment. All participants were treated with Keytruda, either at the recommended dose of 2 milligrams per kilogram (mg/kg) or at a higher dose of 10 mg/kg. In the half of the participants who received Keytruda at the recommended dose of 2 mg/kg, approximately 24 percent had their tumors shrink. This effect lasted at least 1.4 to 8.5 months and continued beyond this period in most patients. A similar percentage of patients had their tumor shrink at the 10 mg/kg dose.
Keytruda’s safety was established in the trial population of 411 participants with advanced melanoma. The most common side effects of Keytruda were fatigue, cough, nausea, itchy skin (pruritus), rash, decreased appetite, constipation, joint pain (arthralgia) and diarrhea. Keytruda also has the potential for severe immune-mediated side effects. In the 411 participants with advanced melanoma, severe immune-mediated side effects involving healthy organs, including the lung, colon, hormone-producing glands and liver, occurred uncommonly.
Keytruda is marketed by Merck & Co., based in Whitehouse Station, New Jersey.


Pembrolizumab, Lambrolizumab, MK-3475
STRUCTURAL FORMULA
Heavy chain
QVQLVQSGVE VKKPGASVKV SCKASGYTFT NYYMYWVRQA PGQGLEWMGG 50
INPSNGGTNF NEKFKNRVTL TTDSSTTTAY MELKSLQFDD TAVYYCARRD 100
YRFDMGFDYW GQGTTVTVSS ASTKGPSVFP LAPCSRSTSE STAALGCLVK 150
DYFPEPVTVS WNSGALTSGV HTFPAVLQSS GLYSLSSVVT VPSSSLGTKT 200
YTCNVDHKPS NTKVDKRVES KYGPPCPPCP APEFLGGPSV FLFPPKPKDT 250
LMISRTPEVT CVVVDVSQED PEVQFNWYVD GVEVHNAKTK PREEQFNSTY 300
RVVSVLTVLH QDWLNGKEYK CKVSNKGLPS SIEKTISKAK GQPREPQVYT 350
LPPSQEEMTK NQVSLTCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS 400
DGSFFLYSRL TVDKSRWQEG NVFSCSVMHE ALHNHYTQKS LSLSLGK 447
Light chain
EIVLTQSPAT LSLSPGERAT LSCRASKGVS TSGYSYLHWY QQKPGQAPRL 50′
LIYLASYLES GVPARFSGSG SGTDFTLTIS SLEPEDFAVY YCQHSRDLPL 100′
TFGGGTKVEI KRTVAAPSVF IFPPSDEQLK SGTASVVCLL NNFYPREAKV 150′
QWKVDNALQS GNSQESVTEQ DSKDSTYSLS STLTLSKADY EKHKVYACEV 200′
THQGLSSPVT KSFNRGEC 218′
Disulfide bridges
22-96 22”-96” 23′-92′ 23”’-92”’ 134-218′ 134”-218”’ 138′-198′ 138”’-198”’
147-203 147”-203” 226-226” 229-229” 261-321 261”-321” 367-425 367”-425”
Glycosylation sites (N)
Asn-297 Asn-297”
lambrolizumab, or MK-3475
| C6504H10004N1716O2036S46 (peptide) | |
| MOL. MASS | 146.3 kDa (peptide) |
Pembrolizumab, Lambrolizumab (also known as MK-3475) is a drug in development by Merck that targets the PD-1 receptor. The drug is intended for use in treating metastatic melanoma.
http://www.ama-assn.org/resources/doc/usan/lambrolizumab.pdf structureof lambrolizumab, or MK-3475
https://download.ama-assn.org/resources/doc/usan/x-pub/pembrolizumab.pdf
Statement on a Nonproprietary Name Adopted by the USAN Council. November 27, 2013.
see above link for change in name
may 2, 2013,
An experimental drug from Merck that unleashes the body’s immune system significantly shrank tumors in 38 percent of patients with advanced melanoma, putting the company squarely in the race to bring to market one of what many experts view as the most promising class of drugs in years.
The drugs are attracting attention here at the annual meeting of the American Society of Clinical Oncology, even though they are still in the early stage of testing. Data from drugs developed by Bristol-Myers Squibb and by Roche had already been released.
The drugs work by disabling a brake that prevents the immune system from attacking cancer cells. The brake is a protein on immune system cells called programmed death 1 receptor, or PD-1.
Merck’s study, which was presented here Sunday and also published in the New England Journal of Medicine, involved 135 patients. While tumors shrank in 38 percent of the patients over all, the rate was 52 percent for patients who got the highest dose of the drug, which is called lambrolizumab, or MK-3475.
But that is what is disclosed tonight, as to pembrolizumab, or MK-3475. Wow. With over $44 billion in 2013 worldwide revenue, that disclosure implies (to seasoned SEC lawyers) that spending on this one drug (or, biologic, to be more technical about it — but remember 40 years ago, Merck had no protein chain biologics research & development programs in its pipe — only chemical drug compounds). . . is material, to that number. Normally that would, in turn, mean that the spending is approaching 5 per cent of revenue. So — Merck may be spending $2.2 billion over the next 12 rolling months, on MK-3475. That’s one BIGhairy science bet, given that Whitehouse Station likely already had over $2 billion invested in the program, at year end 2013.


About Pembrolizumab
Pembrolizumab (MK-3475) is an investigational selective, humanized monoclonal anti-PD-1 antibody designed to block the interaction of PD-1 on T-cells with its ligands, PD-L1 and PD-L2, to reactivate anti-tumor immunity. Pembrolizumab exerts dual ligand blockade of PD-1 pathway.
Today, pembrolizumab is being evaluated across more than 30 types of cancers, as monotherapy and in combination. It is anticipated that by the end of 2014, the pembrolizumab development program will grow to more than 24 clinical trials across 30 different tumor types, enrolling an estimated 6,000 patients at nearly 300 clinical trial sites worldwide, including new Phase 3 studies in head and neck and other cancers. For information about Merck’s oncology clinical studies, please click here.
The Biologics License Application (BLA) for pembrolizumab is under priority review with the U.S. Food and Drug Administration (FDA) for the proposed indication for the treatment of patients with advanced melanoma previously-treated with ipilimumab; the PDUFA date is October 28, 2014. Pembrolizumab has been granted FDA’s Breakthrough Therapy designation for advanced melanoma. If approved by the FDA, pembrolizumab has the potential to be the first PD-1 immune checkpoint modulator approved in this class. The company plans to file a Marketing Authorization Application in Europe for pembrolizumab for advanced melanoma in 2014.
About Head and Neck Cancer
Head and neck cancers are a related group of cancers that involve the oral cavity, pharynx and larynx. Most head and neck cancers are squamous cell carcinomas that begin in the flat, squamous cells that make up the thin surface layer (epithelium) of the head and neck (called the). The leading risk factors for head and neck cancer include tobacco and alcohol use. Infection with certain types of HPV, also called human papillomaviruses, is a risk factor for some types of head and neck cancer, specifically cancer of the oropharynx, which is the middle part of the throat including the soft palate, the base of the tongue, and the tonsils. Each year there are approximately 400,000 cases of cancer of the oral cavity and pharynx, with 160,000 cancers of the larynx, resulting in approximately 300,000 deaths.

About Merck Oncology: A Focus on Immuno-Oncology
At Merck Oncology, our goal is to translate breakthrough science into biomedical innovations to help people with cancer worldwide. Harnessing immune mechanisms to fight cancer is the priority focus of our oncology research and development program. The Company is advancing a pipeline of immunotherapy candidates and combination regimens. Cancer is one of the world’s most urgent unmet medical needs. Helping to empower people to fight cancer is our passion. For information about Merck’s commitment to Oncology visit the Oncology Information Center at http://www.mercknewsroom.com/oncology-infocenter.
About Merck
Today’s Merck is a global healthcare leader working to help the world be well. Merck is known as MSD outside the United States and Canada. Through our prescription medicines, vaccines, biologic therapies, and consumer care and animal health products, we work with customers and operate in more than 140 countries to deliver innovative health solutions. We also demonstrate our commitment to increasing access to healthcare through far-reaching policies, programs and partnerships. For more information, visit http://www.merck.com and connect with us on Twitter, Facebook and YouTube.
Hamid, O; Robert, C; Daud, A; Hodi, F. S.; Hwu, W. J.; Kefford, R; Wolchok, J. D.; Hersey, P; Joseph, R. W.; Weber, J. S.; Dronca, R; Gangadhar, T. C.; Patnaik, A; Zarour, H; Joshua, A. M.; Gergich, K; Elassaiss-Schaap, J; Algazi, A; Mateus, C; Boasberg, P; Tumeh, P. C.; Chmielowski, B; Ebbinghaus, S. W.; Li, X. N.; Kang, S. P.; Ribas, A (2013). “Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma”. New England Journal of Medicine 369 (2): 134–44. doi:10.1056/NEJMoa1305133. PMID 23724846

key words
FDA, approved, Keytruda, advanced melanoma, PD-1 blocking drug, pembrolizumab, Lambrolizumab, MK-3475, Monoclonal antibody




{kind=link}