
Home » CHINA 2021
Category Archives: CHINA 2021
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
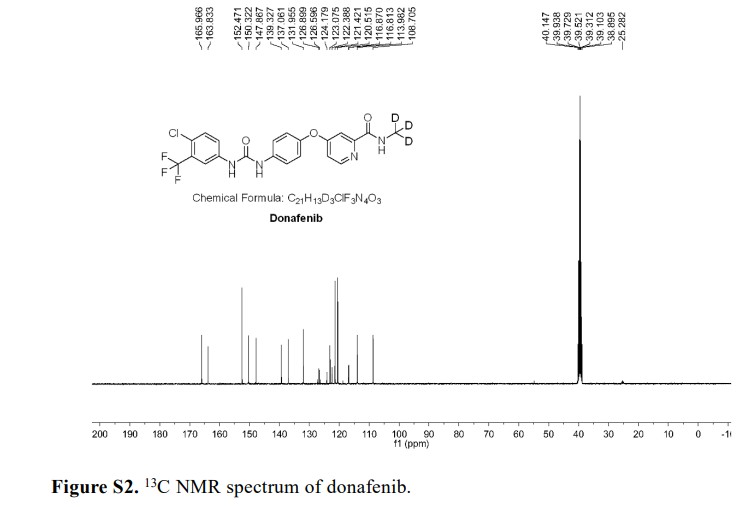
Donafenib
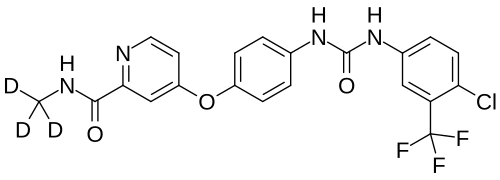
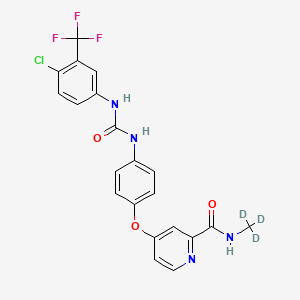
Donafenib
CAS 1130115-44-4, CM-4307, Zepsun, 41XGO0VS1U
4-[4-[[4-chloro-3-(trifluoromethyl)phenyl]carbamoylamino]phenoxy]-N-(trideuteriomethyl)pyridine-2-carboxamide
- 4-(4-(((4-chloro-3-(trifluoromethyl)phenyl)carbamoyl)amino)phenoxy)-N-(2H3)methylpyridine-2-carboxamide
- Sorafenib-d3
- Sorafenib D3
- Donafenib (Sorafenib D3)
- Sorafenib-methyl-d3
- d3-sorafenib
CM-4307 is under investigation in clinical trial NCT03602495 (Donafenib in 131I-Refractory Differentiated Thyroid Cancer).
Donafenib, sold under the brand name Zepsun, is a pharmaceutical drug for the treatment of cancer.
In China, donafenib is approved for the treatment of unresectable hepatocellular carcinoma in patients who have not previously received systemic treatment.[1][2]
Donafenib is a kinase inhibitor that targets Raf kinase and various receptor tyrosine kinases.[3] It is a deuterated derivative of sorafenib with improved pharmacokinetic properties.[4][5]
Donafenib is an orally available multikinase inhibitor that targets Raf kinase and various receptor tyrosine kinases (RTKs), with potential antineoplastic activity. Upon oral administration, donafenib binds to and blocks the activity of Raf kinase, and inhibits Raf-mediated signal transduction pathways. This inhibits cell proliferation in Raf-expressing tumor cells. In addition, this agent may inhibit unidentified RTKs, and thus may further block tumor cell proliferation in susceptible tumor cells. Raf, a serine/threonine protein kinase, plays a key role in the Raf/mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) signaling pathway. Deregulation of this pathway often results in tumor cell proliferation and survival.
SYN
ACS Omega 2021, 6, 5532−5547.
https://pubs.acs.org/doi/10.1021/acsomega.0c05908
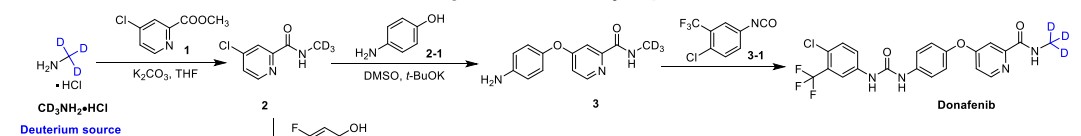
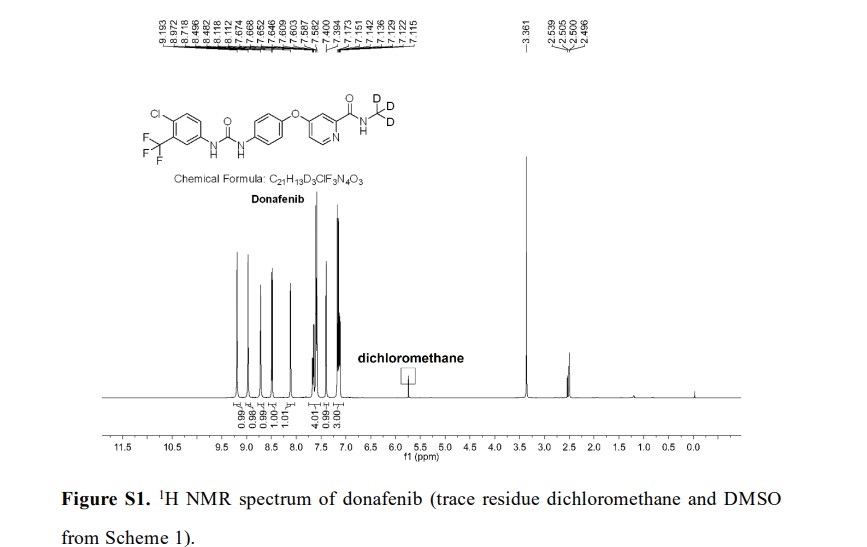
Syn
Donafenib (Zepsun). Donafenib (31), developed by Suzhou Zelgen Biopharmaceuticals, is a deuterated derivative of sorafenib, a multikinase inhibitor for the treatment of advanced hepatocellular carcinoma (HCC). 222 HCC is the most common type of primary liver cancer in adults and the third leading cause of cancer-related deaths worldwide.223,224 Donafenib inhibits Raf kinase and VEGFR tyrosine kinases,
thereby preventing the proliferation of tumor cells. 225 The presence of the deuterated methyl group in donafenib improves metabolic stability with prolonged half-life, lower systemic clearance, and higher systemic exposure.226 Donafenib has been shown to significantly improve the overall survival
of patients with HCC when compared against sorafenib, with favorable safety and tolerability.227 228
In June 2021, donafenib was first approved in China for treating unresectable HCC in patients who have not previously received systemic treatment.
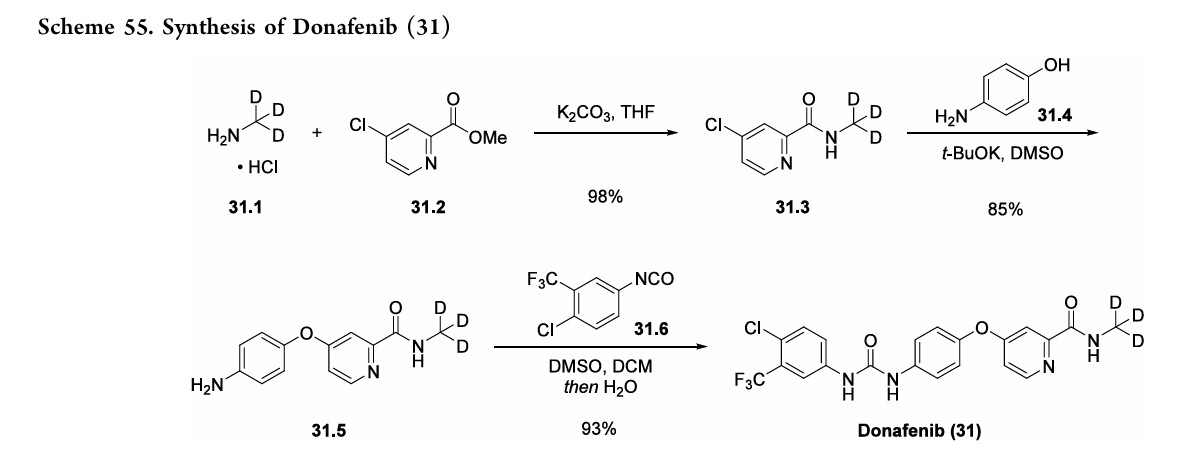
A gram-scale synthesis of donafenib was recently disclosed by Luo and co-workers (Scheme 55).229
The synthetic sequence commenced with amidation of methyl ester 31.2 using methan-d3-amine hydrochloride (31.1) as the deuterium source, affording CD 3-amide 31.3 in high yield (98%). SNAr
displacement with aminophenol 31.4 in DMSO provided diaryl ether 31.5. Finally, reaction of the aniline moiety with isocyanate 31.6 delivered donafenib (31) in 79% yield from 31.3
(222) Mousa, A. B. Sorafenib in the treatment of advanced
hepatocellular carcinoma. Saudi J. Gastroenterol 2008, 14, 40−42.
(223) Forner, A.; Llovet, J. M.; Bruix, J. Hepatocellular carcinoma.
Lancet 2012, 379, 1245−1255.
(224) Vogel, A.; Meyer, T.; Sapisochin, G.; Salem, R.; Saborowski,
A. Hepatocellular carcinoma. Lancet 2022, 400, 1345−1362.
(225) Gong, X.; Qin, S. Study progression of anti-angiogenetic
therapy and its combination with other agents for the treatment of
advanced hepatocellular carcinoma. Hepatobiliary Surg. Nutr. 2018, 7,
466−474.
(226) Zhong, L.; Hou, C.; Zhang, L.; Zhao, J.; Li, F.; Li, W.
Synthesis of deuterium-enriched sorafenib derivatives and evaluation
of their biological activities. Mol. Divers. 2019, 23, 341−350.
(227) Qin, S.; Bi, F.; Gu, S.; Bai, Y.; Chen, Z.; Wang, Z.; Ying, J.; Lu,
Y.; Meng, Z.; Pan, H.; et al. Donafenib versus sorafenib in first-line
treatment of unresectable or metastatic hepatocellular carcinoma: A
randomized, open-label, parallel-controlled phase II-III trial. J. Clin.
Oncol. 2021, 39, 3002−3011.
(228) Keam, S. J.; Duggan, S. Donafenib: First approval. Drugs 2021,
81, 1915−1920.
(229) Li, C.; Zhong, J.; Liu, B.; Yang, T.; Lv, B.; Luo, Y. Study on
typical diarylurea drugs or derivatives in cocrystallizing with strong H
bond acceptor DMSO. ACS Omega 2021, 6, 5532−5547.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- Keam SJ, Duggan S (November 2021). “Donafenib: First Approval”. Drugs. 81 (16): 1915–1920. doi:10.1007/s40265-021-01603-0. PMID 34591285.
- Chen R, Ielasi L, di Carlo A, Tovoli F (February 2023). “Donafenib in hepatocellular carcinoma”. Drugs of Today. 59 (2): 83–90. doi:10.1358/dot.2023.59.2.3507751. hdl:11585/955557. PMID 36811408.
- “Donafenib”. NCI Cancer Dictionary. National Cancer Institute, National Institutes of Health.
- Qin S, Bi F, Gu S, Bai Y, Chen Z, Wang Z, et al. (September 2021). “Donafenib Versus Sorafenib in First-Line Treatment of Unresectable or Metastatic Hepatocellular Carcinoma: A Randomized, Open-Label, Parallel-Controlled Phase II-III Trial”. Journal of Clinical Oncology. 39 (27): 3002–3011. doi:10.1200/JCO.21.00163. PMC 8445562. PMID 34185551.
- Qin S, Bi F, Xu J, Du C, Fan Q, Zhang L, et al. (2020). “P-86 Comparison of the pharmacokinetics of donafenib and sorafenib in patients with advanced hepatocellular carcinoma: An open-label, randomized, parallel-controlled, multicentre phase II/III trial”. Annals of Oncology. 31: S117 – S118. doi:10.1016/j.annonc.2020.04.168.
| Clinical data | |
|---|---|
| Trade names | Zepsun |
| Other names | CM-4307 |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1130115-44-4 |
| PubChem CID | 25191001 |
| DrugBank | DB15414 |
| ChemSpider | 23937167 |
| UNII | 41XGO0VS1U |
| ChEMBL | ChEMBL4297490 |
| CompTox Dashboard (EPA) | DTXSID90648995 |
| Chemical and physical data | |
| Formula | C21H16ClD3F3N4O3 |
| Molar mass | 470.87 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////Donafenib, ZEPSUN, CHINA 2021, APPROVALS 2021, Suzhou Zelgen, 1130115-44-4, CM 4307, 41XGO0VS1U, Sorafenib D3
Olverembatinib

Olverembatinib
1257628-77-5- 3-((1H-pyrazolo[3,4-b]pyridin-5-yl)ethynyl)-4-methyl-N-(4-((4-methylpiperazin-1-yl)methyl)-3-(trifluoromethyl)phenyl)benzamide
- HQP1351
- 4-methyl-N-[4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl]-3-[2-(1H-pyrazolo[3,4-b]pyridin-5-yl)ethynyl]benzamide
- HQP1351 is under investigation in clinical trial NCT03883100 (A Pivotal Study of HQP1351 in Patients of Chronic Myeloid Leukemia in Accelerated Phase With T315I Mutation).
- 4-methyl-N-[4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl]-3-[2-(1H-pyrazolo[3,4-b]pyridin-5-yl)ethynyl]benzamide
- D-824
- GZD824
WeightAverage: 532.571
Monoisotopic: 532.219844002, Chemical FormulaC29H27F3N6O
| Molecular Weight | 724.77 |
|---|---|
| Formula | C31H35F3N6O7S2 |
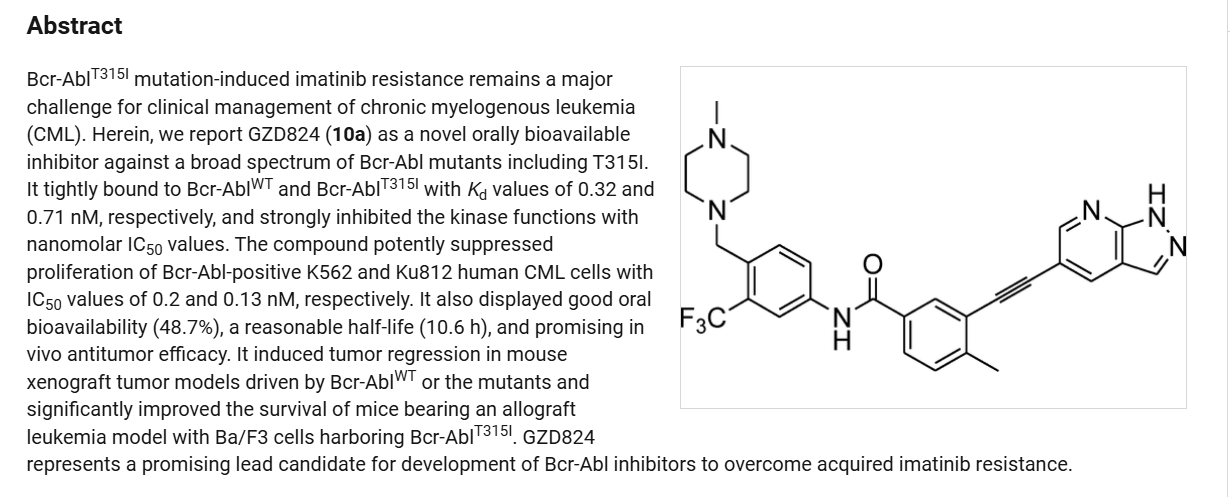
Olverembatinib (GZD824) dimesylate is a potent and orally active pan-Bcr-Abl inhibitor. Olverembatinib dimesylate potently inhibits a broad spectrum of Bcr-Abl mutants. Olverembatinib dimesylate strongly inhibits native Bcr-Abl and Bcr-AblT315I with IC50s of 0.34 nM and 0.68 nM, respectively. Olverembatinib dimesylate has antitumor activity. Olverembatinib (dimesylate) is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
Olverembatinib is a BCR-ABL1 tyrosine kinase inhibitor developed by Ascentage Pharma. In 2021, it was approved in China “for the treatment of adult patients with TKI-resistant chronic-phase CML (CML-CP) or accelerated-phase CML (CML-AP) harbouring the T315I mutation”.[1][2][3]
SYN
Ren, Xiaomei;Pan, Xiaofen;Zhang, Zhang;Wang, Deping;Lu, Xiaoyun;Li, Yupeng;Wen, Donghai;Long, Huoyou;Luo, Jinfeng;Feng, Yubing;Zhuang, Xiaoxi;Zhang, Fengxiang;Liu, Jianqi;Leng, Fang;Lang, Xingfen;Bai, Yang;She, Miaoqin;Tu, Zhengchao;Pan, Jingxuan;Ding, Ke [Journal of Medicinal Chemistry,2013,vol. 56,# 3,p. 879 – 894]
https://pubs.acs.org/doi/10.1021/jm301581y
PATENT
CN 114163434
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN355399053&_cid=P10-MDPKRT-75688-1
| Example |
| The following examples further illustrate but do not limit the present invention. It should be noted that those skilled in the art can make various modifications and improvements without departing from the inventive concept of the present invention, all of which are included in the scope of protection of the present invention. |
| The specific conditions not disclosed in the experimental methods of the following examples can be selected according to conventional methods and conditions, or according to the product instructions. |
| Unless otherwise specified, “room temperature” in the following examples refers to 20°C to 25°C. The term “h” used herein refers to hours. |
| Example 1 |
| Step 1: |
| |
| Under nitrogen, N-methylpyrrolidone (137.6 g) was heated to 30-35°C to obtain the compound of Formula 1 (14.4 g, 1.3 eq) and the compound of Formula 2 (19.14 g, 1 eq). Bis(triphenylphosphate)palladium dichloride (0.46 g, 0.01 eq) and cuprous iodide (0.113 g, 0.01 eq) were added sequentially. Triethylamine (9.45 g, 1.5 eq) was then added under nitrogen. The reaction mixture was heated to 65-75°C and maintained at this temperature for 2 hours. The reaction process was monitored by liquid chromatography-mass spectrometry. The reaction was terminated when the content of the compound of Formula 2 was ≤0.1%. After completion of the reaction, the reaction solution was cooled to 35-45°C and N-acetyl-L-cysteine (1 g, 0.1 eq) was added directly. The reaction was stirred for 4-5 hours. The resulting product was cooled to room temperature, precipitated with water, centrifuged, and washed with pure water to obtain a crude filter cake. The crude filter cake was vacuum-dried and then slurried with a mixture of ethyl acetate and n-heptane (5 mL of the mixed solvent, wherein the volume ratio of ethyl acetate to n-heptane was 1:1) at a rate of 5 mL per gram of crude filter cake. The resulting slurry was vacuum-dried to yield the compound of Formula 3 with a yield of 85.97% and a purity of 98.2%. |
| The NMR data for the compound of Formula 3 are as follows : 1 H NMR (400 MHz, d-DMSO): δ ppm: 8.93 (1H, d, J = 2.0 Hz); 8.63 (1H, d, J = 2.0 Hz); 8.49 (1H, s); 8.11 (1H, d, J = 2.0 Hz); 7.92 (1H, dd, J = 1.6 Hz; J = 8.0 Hz); 7.52 (1H, d, J = 8.0 Hz); 3.88 (3H, s); 2.59 (3H, s); 1.65 (9H, s). |
| Step 2: |
| |
| Under nitrogen, methanol (160 g) and water (50 g) were sequentially added to the compound of formula 3 (20 g, 1.0 eq). The reaction system was stirred at reflux for 18 hours with process control. The resulting product was cooled to room temperature and filtered to obtain a filter cake (no drying required). Recrystallization was performed by adding 10 times the mass of the filter cake in methanol. The resulting mixture was stirred at 60-70°C for 8-10 hours, then cooled to 40-50°C and subjected to a gradient cooling process at a cooling rate of 5°C per 1 to 1.5 hours to slowly form a solid precipitate. The resulting mixture was filtered, the filter cake was washed with methanol, and vacuum dried to obtain the compound of formula 4 in a 91% yield and 99.7% purity. |
| The NMR data for the compound of Formula 4 are as follows : 1 H NMR (400 MHz, d-DMSO): δ ppm: 8.73 (1H, d, J = 2.0 Hz); 8.52 (1H, t, J = 2.0 Hz); 8.21 (1H, d, J = 2.0 Hz); 8.06 (1H, s); 7.86 (1H, dd, J1 = 2.0 Hz; J2 = 8.0 Hz); 7.49 (1H, dd, J1 = 1.6 Hz; J2 = 7.6 Hz); 3.86 (3H, s); 2.56 (3H, s). |
| Step 3: |
| |
| Under nitrogen, THF (448 mL), compound of formula 4 (29.1 g, 1 eq), and compound of formula 5 (24.6 g, 0.9 eq) were added, stirred, and cooled to -65°C to -60°C. At this temperature, potassium tert-butoxide (19 g x 3) was added in batches every 0.5 h. The reaction process was controlled by liquid phase detection. After 2 hours, the reaction temperature was raised to -5 to 0°C. The reaction solution was washed with purified water, stirred for 0.5-1 hour, washed with brine, and separated to obtain an organic phase. N-acetyl-L-cysteine (11.41 g, 0.7 eq) was added to the organic phase, stirred, washed with brine, neutralized, and concentrated under reduced pressure. The resulting filter cake was washed with purified water and made into a slurry. The resulting product was washed again with purified water and dried under vacuum to obtain compound of formula 6 with a yield of 88.2% and a purity of 98.6%. |
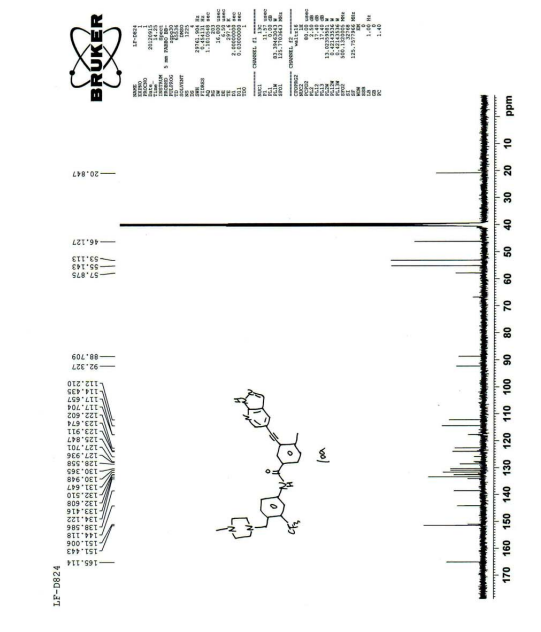
| The NMR data for the compound of formula 6 are as follows : 1 H NMR (400 MHz, d-DMSO): δ ppm: 10.53 (1H, s); 8.75 (d, J = 2.0); 8.53 (d, J = 2.4); 8.24 (1H, s); 8.23 (d, J = 2.4); 8.21 (d, J = 1.6); 8.09 (dd, J1 = 1.6; J2 = 8.4); 7.94 (dd, J1 = 2.0; J2 = 8.0); 7.71 (d, J = 8.8); 7.53 (d, J = 8.0); 3.56 (2H, s); 2.59 (3H, s); 2.34-2.35 (8H, m), 2.16 (3H, s). |
| Its carbon spectrum data are 13 C NMR (100 MHz, d-DMSO): δ ppm: 20.38, 45.65, 52.64, 54.67, 57.41, 88.26, 91.86, 111.76, 113.98, 117.19, 122.14, 123.43, 127.35 (q), 124.30 (q), 128.10, 129.89, 130.49, 131.15, 132.02, 132.13, 132.93, 133.66, 138.15, 143.65, 150.55, 164.64. |
PATENT
CN 101885722
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN84081329&_cid=P10-MDPKML-68458-1
| Example 23 |
| 3-((1H-pyrazolo[3,4-b]pyridine-5-substituted)ethynyl)-4-methyl-N-(4-((4-methylpiperazine-1-substituted)methyl)3-(trifluoromethyl)phenyl)benzamide (D824) |
| (3-((1H-pyrazolo[3,4-b]pyridin-5-yl)ethynyl)-4-methyl-N-(4-((4-methylpiperazin-1-yl)m ethyl)-3-(trifluoromethyl)phenyl)benzamide) |

| The synthesis method is the same as in Example 1. |
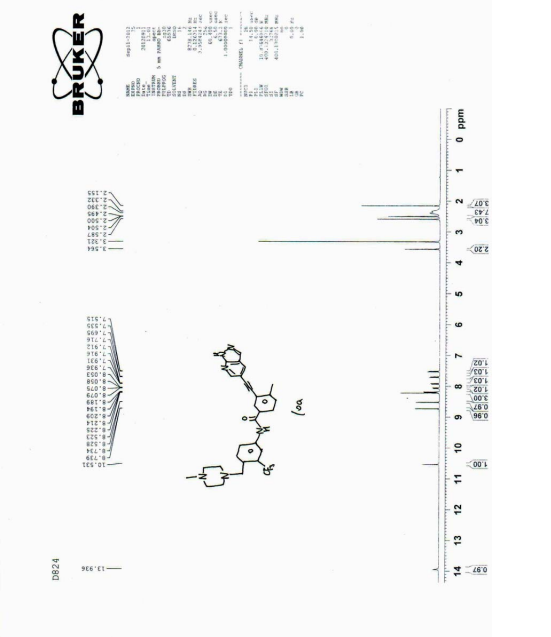
| 1 HNMR (400MHz, d-DMSO), δ13.92 (s, 1H), 10.55 (s, 1H), 8.72 (d, J=2.0Hz, 1H), 8.52 (d, J=2.0Hz, 1H), 8.17 (m, 3H), 8.10 (d, J=8.0Hz, 1H), 7.92 (dd, J=8.0, 2.0Hz, 1H), 7.70 (d, J=8.8Hz, 1H), 7.53 (d, J=8.0Hz, 1H), 3.80 (s, 2H), 3.10 (brs, 8H), 2.71 (s, 3H), 2.57 (s, 3H). |
| MS(ESI), m/z: 533, (M + +H + ). |
SYN
Olverembatinib(24) wasdeveloped by Ascentage Pharma as anorally available, third-generation
tyrosinekinase inhibitor (TKI) for the treatment of chronic myeloid leukemia (CML), acute myeloid leukemia, acute lymphoblastic leukemia (ALL), and solid tumors.167 It received its first approval inChina inNovember 2021 and was approved for use in adults with TKI-resistant CML chronicphaseandCML-acceleratephaseharboringtheT315I “gatekeeper” mutation.168 The current mainstay of CML
treatmentiscenteredaroundTKIs;however,resistancetoTKItherapy, often through BCR-ABL1 kinase domain point mutations, remains a challenge for early generation therapies.169Olverembatinibretainsitsefficacybyfunctioningasan ATP-bindingsiteinhibitorofwild-typeBCR-ABL1kinaseand broadly relatedmutants including T315I, which otherwise confers resistance against all first and second generation TKIs.168
Thesynthesisofolverembatinibhasbeenreportedinseveral patents,170−172 aswell as a journal article173 that details the divergentapproachtorelatedanalogues. Inarecentpatent,170 the synthesis of olverembatinib began with a Sonogashira coupling of commercially available alkyne 24.1 with
bromopyridine24.2toaffordester24.3in98%yield(Scheme43). Cleavage of the N-Boc group was accomplished by refluxingcarbamate24.3inaMeOHandwatermixturetogive pyrazole24.4 in91%yield. AfinalKOtBumediatedamide formation with aniline 24.5 resulted in the isolation of
olverembatinib(24) in88%yield.
(167) Dhillon, S. Olverembatinib: First approval. Drugs 2022, 82,
469−475.
(168) Braun, T. P.; Eide, C. A.; Druker, B. J. Response and resistance
to BCR-ABL1-targeted therapies. Cancer Cell 2020, 37, 530−542.
(169) Shoukier, M.; Kubiak, M.; Cortes, J. Review of new-generation
tyrosine kinase inhibitors for chronic myeloid leukemia. Curr. Oncol.
Rep. 2021, 23, 91.
(170) Wen, J.; Feng, J.; Wu, T.; Cai, M.; Teng, S. Preparation
method of alkynyl containing compound and its intermediate. China
Patent CN 114163434, 2022.
(171) Guo, M.; Wen, J.; Teng, S.; Wu, T.; Feng, J. Preparation of
(trifluoromethylphenyl)(pyrazolo[3,4-b]pyridinylethynyl)benzamide
derivative. China Patent CN 113292556, 2021.
(172) Ding, K.; Wang, D.; Pei, D.; Zhang, Z.; Shen, M.; Luo, K.;
Feng, Y. Heterocyclic alkynylbenzene derivatives as cancer cell line
inhibitors and their preparation, pharmaceutical compositions and use
in the treatment of cancer. China Patent CN 101885722, 2010.
(173) Ren, X.; Pan, X.; Zhang, Z.; Wang, D.; Lu, X.; Li, Y.; Wen, D.;
Long, H.; Luo, J.; Feng, Y.; et al. Identification of GZD824 as an
orally bioavailable inhibitor that targets phosphorylated and non
phosphorylated breakpoint cluster region−abelson (Bcr-Abl) kinase
and overcomes clinically acquired mutation-induced resistance against
imatinib. J. Med. Chem. 2013, 56, 879−894.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Dhillon, Sohita (March 2022). “Olverembatinib: First Approval”. Drugs. 82 (4): 469–475. doi:10.1007/s40265-022-01680-9. PMID 35195876. S2CID 247027755.
- Jiang, Qian; Li, Zongru; Qin, Yazhen; Li, Weiming; Xu, Na; Liu, Bingcheng; Zhang, Yanli; Meng, Li; Zhu, Huanling; Du, Xin; Chen, Suning; Liang, Yang; Hu, Yu; Liu, Xiaoli; Song, Yongping; Men, Lichuang; Chen, Zi; Niu, Qian; Wang, Hengbang; Lu, Ming; Yang, Dajun; Zhai, Yifan; Huang, Xiaojun (18 August 2022). “Olverembatinib (HQP1351), a well-tolerated and effective tyrosine kinase inhibitor for patients with T315I-mutated chronic myeloid leukemia: results of an open-label, multicenter phase 1/2 trial”. Journal of Hematology & Oncology. 15 (1): 113. doi:10.1186/s13045-022-01334-z. PMC 9389804. PMID 35982483.
- Jiang, Qian; Huang, Xiaojun; Chen, Zi; Niu, Qian; Shi, Dayu; Li, Zongru; Hou, Yue; Hu, Yu; Li, Weiming; Liu, Xiaoli; Xu, Na; Song, Yongping; Zhang, Yanli; Meng, Li; Hong, Zhenya; Liu, Bingcheng; Zeng, Shan; Men, Lichuang; Li, Yan; Chen, Suning; Xue, Mengxing; Zhu, Huanling; Li, He; Du, Xin; Lou, Jin; Zhang, Xiaohan; Liang, Yang; Dai, Yujun; Lu, Ming; Wang, Hengbang; Ji, Jiao; Yue, Changai; Yang, Dajun; Zhai, Yifan (5 November 2020). “Novel BCR-ABL1 Tyrosine Kinase Inhibitor (TKI) HQP1351 (Olverembatinib) Is Efficacious and Well Tolerated in Patients with T315I-Mutated Chronic Myeloid Leukemia (CML): Results of Pivotal (Phase II) Trials”. Blood. 136 (Supplement 1): 50–51. doi:10.1182/blood-2020-142142. S2CID 228875477.
| Clinical data | |
|---|---|
| Other names | GZD-824; GZD824 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1257628-77-5 |
| PubChem CID | 51038269 |
| IUPHAR/BPS | 10630 |
| DrugBank | DB16185 |
| ChemSpider | 29395146 |
| UNII | KV1M7Q3CBP |
| ChEMBL | ChEMBL2316582 |
| CompTox Dashboard (EPA) | DTXSID301352011 |
| Chemical and physical data | |
| Formula | C29H27F3N6O |
| Molar mass | 532.571 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
[1]. Ren X, Pan X, Zhang Z, Identification of GZD824 as an orally bioavailable inhibitor that targets phosphorylated and nonphosphorylated breakpoint cluster region-Abelson (Bcr-Abl) kinase and overcomes clinically acquired mutation-induced resistance against imatinib. J Med Chem. 2013 Feb 14;56(3):879-94. [Content Brief]
//////////Olverembatinib, approvals 2021, china 2021, Ascentage Pharma, cancer, HQP1351, HQP 1351, D-824, D 824, KV1M7Q3CBP, GZD824
Chiglitazar
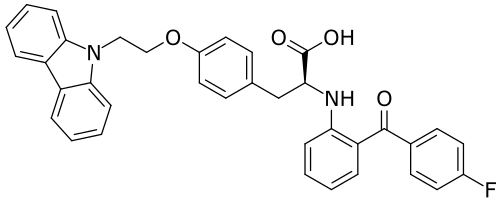
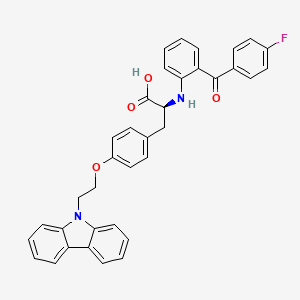
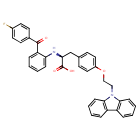
Chiglitazar
CAS 743438-45-1
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Chiglitazar sodium, (S)- | YN12H6OCV6 | 2390374-10-2 | RMVIEXHXRDCWBT-UCRKPPETSA-M |
- CS 038
- Carfloglitazar, (s)-
- E6EJV1J6Y0
- (2S)-3-[4-(2-carbazol-9-ylethoxy)phenyl]-2-[2-(4-fluorobenzoyl)anilino]propanoic acid
- C36H29FN2O4
- 572.6 g/mol
- (2S)-3-[4-(2-carbazol-9-ylethoxy)phenyl]-2-[2-(4-fluorobenzoyl)anilino]propanoic acid
- (2S)-3-(4-(2-CARBAZOL-9-YLETHOXY)PHENYL)-2-(2-(4-FLUOROBENZOYL)ANILINO)PROPANOIC ACID
- (2s)-3-(4-(2-carbazol-9-ylethoxy)phenyl)-2-(2-(4-fluorobenzoyl)anilino)propanoic acid
- Carfloglitazar, (s)-
- L-tyrosine, o-(2-(9h-carbazol-9-yl)ethyl)-n-(2-(4-fluorobenzoyl)phenyl)-
- O-(2-(9h-carbazol-9-yl)ethyl)-n-(2-(4-fluorobenzoyl)phenyl)-l-tyrosine
Chiglitazar was developed by Chipscreen Biosciences and was approved in China for improving glycemic control in adult
patients with type2 diabetes in October2021.
Chiglitazar (trade name Bilessglu) is a drug for the treatment of type 2 diabetes.[1] It is a peroxisome proliferator-activated receptor (PPAR) agonist.
In China, chiglitazar is approved for glycemic control in adult patients with type 2 diabetes when used in combination with diet and exercise.[2]
Chiglitazar is under investigation in clinical trial NCT06125587 (Chiglitazar/metformin in Non-obese Women With PCOS).
SYN
WO 2004048333
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2004048333&_cid=P12-MDMUOB-48741-1
Example 15
Preparation of 2-[(2-(4-fluorobenzoyl)phenyl)amino]-3-[4-(2-carbazolylethoxy)-phenyl]
-propionic acid (compound CS038)
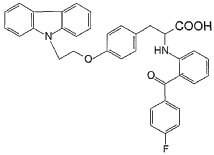
To a solution of 2-[(2-(4-fluorobenzoyl)phenyl)amino]-3-[4-(2-bromoethoxy)-phenyl] -propionic acid methyl ester (0.25 g, 0.49 mmol) and carbazole (0.082 g, 0.49 mmol) in benzene (10 ml) is added tetrabutyl ammonium bromide (0.08 g) and 50% NaOH aqueous solution (0.084 g, 1.08 mmol), then the mixture is heated to reflux for 10 h. After cooled, benzene (30ml) is added, and the mixture is washed with water (3×30 ml). Then the solvent is evaporated under a vacuum. The crude product is purified by silica gel chromatography using CHCl3/MeOH (4:1) as eluent to give the title compound (0.10 g, 36%). HRMS calcd for C36H29FN204: 572.6357. Found: 572.6354. MA calcd for C36H29FN204: C, 75.51%; H, 5.11%; N, 4.89%. Found: C, 75.83%; H, 5.10%; N, 4.90%.
PATENT
US 10640465
https://patentscope.wipo.int/search/en/detail.jsf?docId=US249083802&_cid=P12-MDMUQY-52500-1
The pharmacological activity of the compound is described in Chinese patent application No. CN03126974.5 and U.S. Pat. No. 7,268,157. 2-(2-(4-fluorobenzoyl)phenylamino)-3-(4-(2-(9H-carbazol-9-yl)ethoxy)phenyl)propanoic acid is able to selectively activate PPAR-α, PPAR-γ and PPAR-6, and can be used to treat the diseases associated with metabolic syndrome such as diabetes, hypertension, obesity, insulin resistance, hypertriglyceridemia, hyperglycemia, high cholesterol, arteries atherosclerosis, coronary heart disease, etc. A preparation method of 2-(2-(4-fluorobenzoyl)phenylamino)-3-(4-(2-(9H-carbazol-9-yl)ethoxy)phenyl)propanoic acid is disclosed in Chinese patent application No. CN03126974.5 and U.S. Pat. No. 7,268,157, and the synthetic route thereof is as follows:
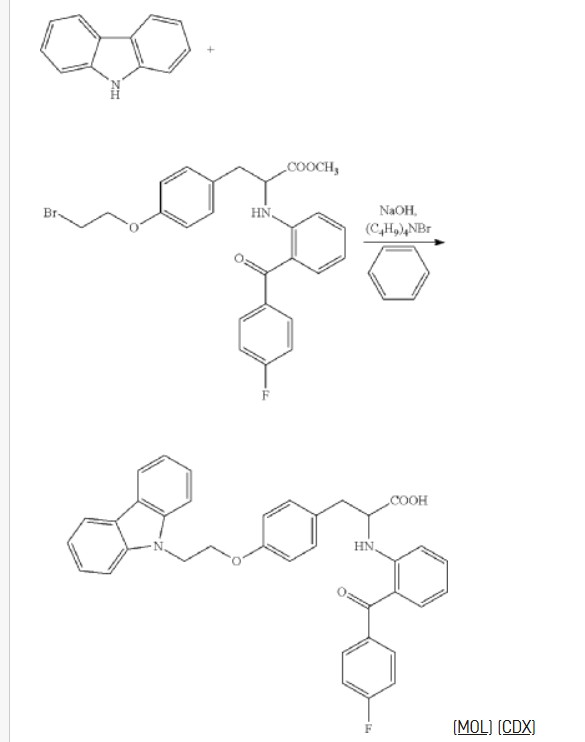
EXAMPLES
Example 1: Preparation of 2-(2-(4-fluorobenzoyl)phenylamino)-3-(4-(2-(9H-carbazol-9-yl)ethoxy)phenyl)propanoic Acid
SYN
J. Med. Chem. 2024, 67, 4376−4418
Chiglitazar (Bilessglu). Chiglitazar (17), a novel nonthiazolidinedione pan-agonist of α, δ, and γ peroxisome proliferator-activated receptors (PPARs), has shown promise for the treatment of type 2 diabetes. 126 Type 2 diabetes impacts over 374 million patients worldwide and continues to
rise in incidence and prevalence globally. 127 Chiglitazar preferentially regulates expression of ANGPTL4 and PDK4 genes, which are involved in glucose and lipid metabolism. 128 Chiglitazar was developed by Chipscreen Biosciences and was approved in China for improving glycemic control in adult
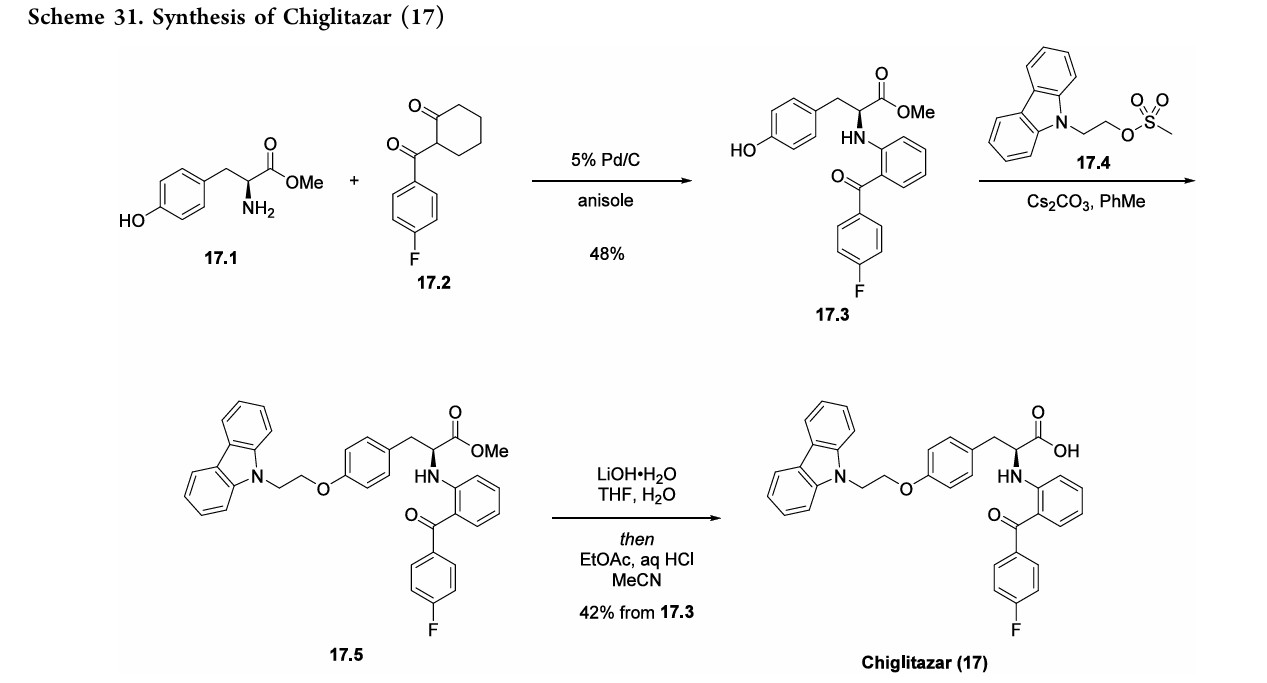
patients with type2 diabetes in October2021.129 Thesynthesisof17beganwithimineformationbetweenL
tyrosine methyl ester (17.1) and 2-(4-fluorobenzoyl) cyclohexanone(17.2)with tandemaromatizationunderPd/C catalysis to generate aniline derivative 17.3 (Scheme31).130,131 Alkylation of the phenol moiety of 17.3 with mesylate17.4furnishedphenyl alkyl etherderivative17.5.132
Hydrolysisof themethylester in17.5withlithiumhydroxide followedbyacidificationwithhydrochloricacidandrecrystal lization fromacetonitrile afforded chiglitazar (17) in 42% overall yield from17.3.Thisprocessdeliveredchiglitazar in 99.4%purityat24gscale.
(126) Ji, L.; Song, W.; Fang, H.; Li, W.; Geng, J.; Wang, Y.; Guo, L.;
Cai, H.; Yang, T.; Li, H.; et al. Efficacy and safety of chiglitazar, a
novel peroxisome proliferator-activated receptor pan-agonist, in
patients with type 2 diabetes: a randomized, double-blind, placebo
controlled, phase 3 trial (CMAP). Sci. Bull. 2021, 66, 1571−1580.
(127) Chatterjee, S.; Khunti, K.; Davies, M. J. Type 2 diabetes.
Lancet 2017, 389, 2239−2251.
(128) Pan, D.-S.; Wang, W.; Liu, N.-S.; Yang, Q.-J.; Zhang, K.; Zhu,
J.-Z.; Shan, S.; Li, Z.-B.; Ning, Z.-Q.; Huang, L.; Lu, X.-P. Chiglitazar
preferentially regulates gene expression via configuration-restricted
binding and phosphorylation inhibition of PPARγ. PPAR Research
2017 2017, 2017, 1−16.
(129) Deeks, E. D. Chiglitazar: First approval. Drugs 2022, 82, 87−
92.
(130) Li, Z.; Lu, X.-P.; Liao, C.; Shi, L.; Liu, Z.; Ma, B. Substituted
arylalcanoic acid derivatives as PPAR pan agonists with potent
antihyperglycemic and antihyperlipidemic activity. WO 2004048333
A1, 2004.
(131) Sutter, M.; Sotto, N.; Raoul, Y.; Métay, E.; Lemaire, M.
Straightforward heterogeneous palladium catalyzed synthesis of aryl
ethers and aryl amines via a solvent free aerobic and non-aerobic
dehydrogenative arylation. Green Chem. 2013, 15, 347−352.
(132) Lu, X.; Li, Z.; Wang, X. Method for preparing phenylalanine
compound. U.S. Patent US 10640465 B2, 2020.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Ji L, Song W, Fang H, Li W, Geng J, Wang Y, et al. (August 2021). “Efficacy and safety of chiglitazar, a novel peroxisome proliferator-activated receptor pan-agonist, in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled, phase 3 trial (CMAP)”. Science Bulletin. 66 (15): 1571–1580. Bibcode:2021SciBu..66.1571J. doi:10.1016/j.scib.2021.03.019. PMID 36654286. S2CID 233650336.
- Deeks ED (January 2022). “Chiglitazar: First Approval”. Drugs. 82 (1): 87–92. doi:10.1007/s40265-021-01648-1. PMID 34846697. S2CID 244716275.
| Clinical data | |
|---|---|
| Trade names | Bilessglu |
| Other names | Carfloglitazar |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 743438-45-1 |
| PubChem CID | 71402018 |
| ChemSpider | 57523239 |
| UNII | E6EJV1J6Y0 |
| ChEMBL | ChEMBL4650349 |
| CompTox Dashboard (EPA) | DTXSID00225352 |
| Chemical and physical data | |
| Formula | C36H29FN2O4 |
| Molar mass | 572.636 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////////Chiglitazar, Chipscreen Biosciences, CHINA 2021, DIABETES, CS 038, Carfloglitazar, (s)-, E6EJV1J6Y0,
Hetrombopag Olamine
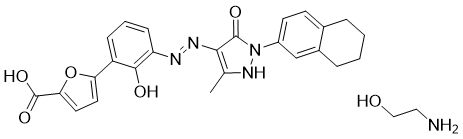
Hetrombopag Olamine, RAFUTROMBOPAG OLAMINE
- Hetrombopag diolamine
- SHR8735 olamine
- Hetrombopag ethanolamine
- SHR-8735 olamine
580.6 g/mol, C29H36N6O7, V45T2I862X
2-aminoethanol;5-[2-hydroxy-3-[[5-methyl-3-oxo-2-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazol-4-yl]diazenyl]phenyl]furan-2-carboxylic acid
CAS 1257792-42-9
1257792-41-8 (free acid) 1257792-41-8 (ethanolamine) 1257792-42-9 (olamine)
Jiangsu Hengrui Pharmaceutical, was approved in China in June 2021 for treatment of adult patients with chronic primary immune thrombocytopenia (ITP) and severe aplastic anemia who have not responded well to other treatments
Hetrombopag Olamine is the orally active ethanolamine salt of hetrombopag, a small-molecule, nonpeptide thrombopoietin receptor (TPO-R or CD110) agonist, with megakaryopoiesis-stimulating activity. Upon oral administration, hetrombopag targets, binds to and stimulates the transmembrane domain of the platelet TPO-R, a member of the hematopoietin receptor superfamily. Activation of TPO-R leads to the proliferation and differentiation of cells in the megakaryocytic lineage and an increase in platelet production. This may prevent or treat chemotherapy-induced thrombocytopenia.
- OriginatorJiangsu Hengrui Medicine Co.
- DeveloperAtridia; Jiangsu Hengrui Medicine Co.
- ClassAntianaemics; Antihaemorrhagics; Aza compounds; Carboxylic acids; Furans; Pyrazolones; Small molecules; Tetrahydronaphthalenes
- Mechanism of ActionThrombopoietin receptor agonists
- Orphan Drug StatusYes – Thrombocytopenia
- MarketedAplastic anaemia; Idiopathic thrombocytopenic purpura
- Phase IIIThrombocytopenia
- No development reportedUnspecified
- 07 Dec 2024Efficacy and adverse events data from a phase-III trial in Aplastic anaemia presented at the 66th American Society of Hematology Annual Meeting and Exposition (ASH-Hem-2024)
- 31 Jul 2024Phase-III clinical trials in Thrombocytopenia in China (PO) (NCT06507436)
- 25 Jul 2024Jiangsu Hengrui Medicine plans a phase III trial in Thrombocytopenia (PO) in July 2024 (NCT06507436)
SYN
CN 113929668
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN349207982&_cid=P21-MDCUSL-44897-1
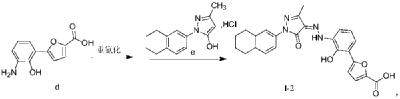
| Example 1. Synthesis of 5-(2-carbonyl-2,3-dihydrobenzoxazol-7-yl)furan-2-carboxylic acid |
| |
| Add purified water to the batching barrel, add 4.0kg of compound a under stirring, then add 10L of hydrochloric acid, stir, pump the material into a 50L reactor, add 10L of purified water to the batching barrel and pump it into the reactor. Turn on stirring, start cooling, the temperature drops to -5~2°C, start adding sodium nitrite aqueous solution (6.4L purified water, 1840g sodium nitrite), keep the temperature in the reactor no higher than 5°C during the process; after adding, continue stirring for 10~20min; add 800g of urea, continue stirring for 10~20min, the obtained diazonium salt solution is ready for use, and the temperature in the whole process is kept no higher than 5°C. |
| 44kg of acetone was pumped into a 200L reactor, and 15.0kg of compound b and 463.5g of copper chloride dihydrate were added in sequence under stirring. The temperature was raised to 30-35°C, and the obtained diazonium salt solution was added. The temperature was maintained at 30-40°C during the period. After the addition was completed, the temperature was maintained at 30-40°C and the reaction was continued with stirring for 1-1.5h. 120.0L of purified water was added, the temperature was raised to 40-45°C, and stirring was continued for a period of time. Filter, wash the filter cake with purified water until the filtrate is neutral, filter again, and collect the filter cake. 80L of purified water was added to the reactor, stirring was started, and the filter cake was added. Sodium hydroxide aqueous solution was added to the reactor to adjust the pH, the pH value was maintained at 8-10 for a period of time, and the filtrate was pumped into the reactor, and the filter was pressed into the material barrel through the filter press. Then 10L of purified water was pumped into the reactor and filtered into the material barrel. The material in the material barrel was pumped into the reactor, and then ethyl acetate was pumped in, stirred, and allowed to stand for 30-40 minutes. The aqueous phase was separated and collected, and the aqueous phase was pumped into the reactor, and the pH was adjusted to 3-4 with hydrochloric acid solution, and the filter cake was washed with purified water until the filtrate was neutral, and then the filter cake was collected. The filter cake was dried to obtain compound c. The yield of this step was 3.59 kg, and the yield was 55%. |
| Example 2: Synthesis of 5-(3-amino-2-hydroxyphenyl)furan-2-carboxylic acid |
| |
| Purified water was pumped into the 50L reactor, stirring was started, 3.53kg of sodium hydroxide was added, and compound c obtained in the previous step was added. Under nitrogen protection, the reaction mixture was heated to reflux in the reactor for reaction. After the reaction, the reaction solution was cooled, the temperature was lowered to 0-10°C, and hydrochloric acid solution was added to adjust the pH value to 5-6. The filter cake was filtered, and the filtrate was washed with purified water until neutral, and then filtered again to collect the filter cake. The filter cake was dried to obtain compound d. The yield in this step was 2.78kg, with a yield of 90%. |
| Example 3. Synthesis of (Z)-5-(2-hydroxy-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazol-4(5H)-ylidene)hydrazino)phenyl)furan-2-carboxylic acid |
| |
| Purified water was added to the batching barrel, and compound d was added in sequence under stirring, and then 6.3L hydrochloric acid was added, and the materials were pumped into a 200L reactor. Purified water was added to the batching barrel again, and then pumped into the reactor. Stirring was started, and the temperature was lowered to -5 to 2°C. Sodium nitrite aqueous solution (sodium nitrite to compound d molar ratio is 1:1) was added, and the internal temperature was kept at no more than 5°C during the process. After the addition was completed, stirring was continued; urea was added, and stirring was continued to obtain a diazonium salt solution for use, and the internal temperature was kept at no more than 5°C during the whole process. |
| Add 36L purified water and 4000g sodium hydroxide to the batching barrel, stir to dissolve, and set aside. Take 26kg of the above sodium hydroxide aqueous solution, add compound e (the molar ratio of compound e to compound d is 0.9:1), stir, and add the resulting solution to the diazonium salt solution, keeping the temperature not exceeding 8°C. Add the above-prepared sodium hydroxide aqueous solution dropwise, adjust the pH to 8-10, and keep the temperature at 5-10°C for 3-4h. Add hydrochloric acid solution dropwise to adjust the pH to 2-3, keep the temperature not exceeding 25°C, filter, wash the filter cake with purified water until the filtrate is neutral, filter again, and collect the filter cake. Pump 48.0kg of tetrahydrofuran aqueous solution (22.5kg tetrahydrofuran, 25.5L purified water) into the reactor, add the above-obtained filter cake, beat, filter, wash the filter cake with tetrahydrofuran aqueous solution, wash the filter cake with purified water, filter again, and collect the filter cake. Dry the filter cake. |
| Ethyl acetate was pumped into the reactor, and the above-obtained materials were added to the reactor for slurrying, and the filter cake was washed with ethyl acetate, and the filter cake was washed until no obvious droplets flowed out of the mirror, and the filter cake was collected and dried to obtain the compound of formula (I-2). The yield in this step was 5.34 kg, and the yield was 97.5%. |
| Example 4. Synthesis of (Z)-5-(2-hydroxy-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazol-4(5H)-ylidene)hydrazino)phenyl)furan-2-carboxylic acid |
| The compound of formula (I-2) was prepared by using a method substantially the same as in Example 3 (except that the equivalent of compound e was adjusted from 0.9 in Example 3 to the current 0.95, other conditions remained unchanged). |
| Comparative Example 1: Synthesis of (Z)-5-(2-hydroxy-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazole-4(5H)-ylidene)hydrazino)phenyl)furan-2-carboxylic acid |
| |
| The compound of formula (I-2) was prepared by using a method substantially the same as in Example 3 (except that the step of adding urea was changed to starch potassium iodide test paper to indicate the reaction endpoint, and other conditions remained unchanged). |
| Test Example 1: Effect of urea on the preparation process of the compound of formula (I-2) |
| HPLC conditions: |
| Chromatographic column: Welch Ultimate |
| Flow rate: 1.0ml/min |
| Injection volume: 10 μl |
| Detector: UV detector |
| Detection wavelength: 251nm |
| Mobile phase: 0.1% trifluoroacetic acid aqueous solution was used as mobile phase A, acetonitrile was used as mobile phase B, and elution was performed at a ratio of 50%/50% of mobile phase A/mobile phase B. |
PATENT
EP 2441457
PATENT
WO 2010142137
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2010142137&_cid=P21-MDCUXF-51461-1
PATENT
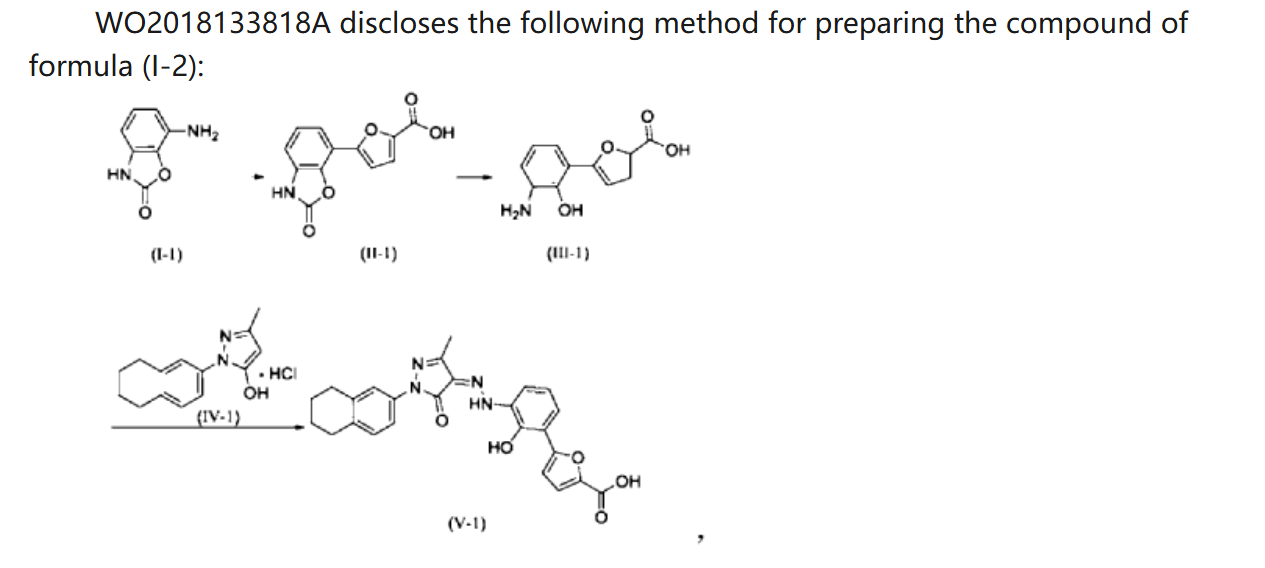
WO 2018133818
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018133818&_cid=P21-MDCUYN-53075-1
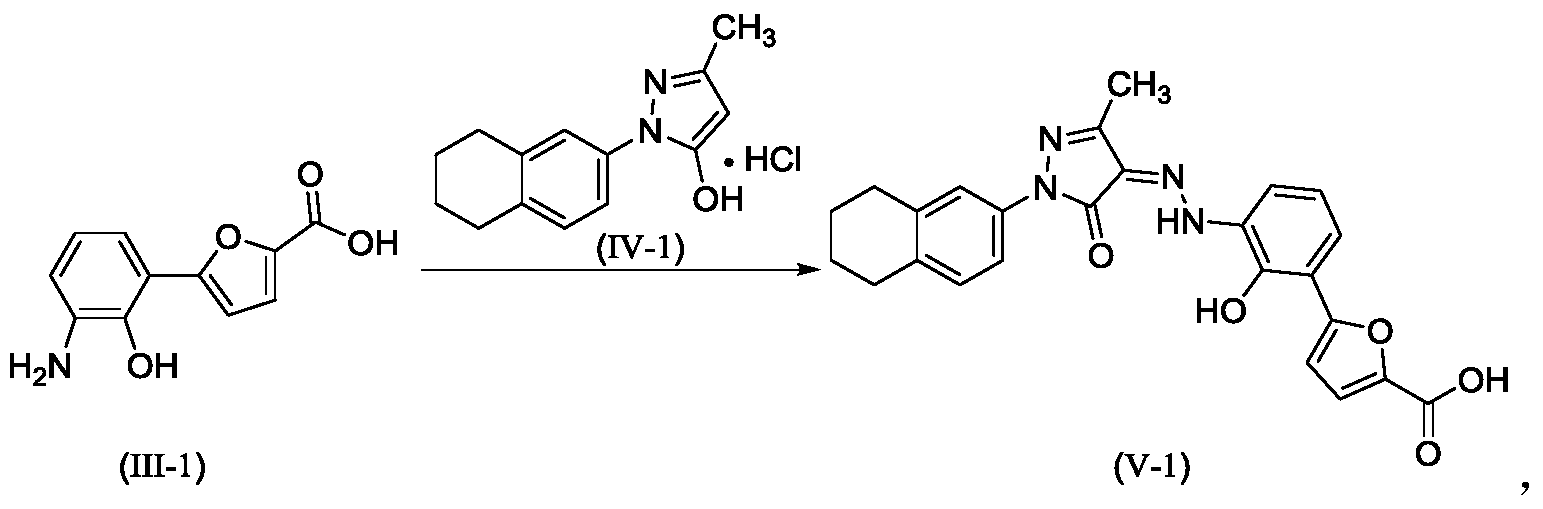
Example 1. Preparation of 3-methyl-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazol-5-ol hydrochloride
[0107]
[0108](5,6,7,8-tetrahydronaphthalene-2-yl)hydrazine hydrochloride (1.3 kg, prepared according to the method in patent application WO2009092276A1) and ethyl acetoacetate (1.17 L) were added to ethyl acetate (5.2 L). The mixture was heated under reflux for 2 hours. The reaction solution was cooled to room temperature, then cooled to 0-5°C, stirred for 1 hour, filtered, and the solid was washed with a small amount of ethyl acetate to obtain a white solid product (1.4 kg, yield 81%).
[0109]
[0110]Example 2. Preparation of (Z)-5-(2-hydroxy-3-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1,5-dihydro-4H-pyrazol-4-ylidene)hydrazino)phenyl)furan-2-carboxylic acid (V-1)
[0111]
[0112]Step 1: Synthesis of intermediate (II-1)
[0113]Purified water (14.80 kg), 7-aminobenzo[d]oxazol-2(3H)-one (2.00 kg, prepared according to the method in patent application WO2005016898A2), and hydrochloric acid (5.33 kg) were added to the reaction kettle, the temperature was raised to 40-45°C, stirred for 10 min, cooled to -3-5°C, and sodium nitrite aqueous solution (sodium nitrite 940 g, water 3.20 kg) was added dropwise, the internal temperature was kept at no more than 5°C, the end point was controlled by starch potassium iodide test paper, and stirring was continued for 15 min;
[0114]Add acetone (28L) to the reactor, then add furoic acid (4.57kg) and cupric chloride dihydrate (232g), stir at 35-40℃ until dissolved, add diazonium salt solution dropwise, keep the internal temperature at 35-40℃, and continue stirring for 1.5h. Add purified water (60L), heat to 35-40℃ and stir for 30min. Filter, wash the filter cake with 45-50℃ purified water. Add the filter cake to purified water (40kg), adjust the pH to 8-9 with 15% sodium hydroxide aqueous solution, filter, adjust the pH of the filtrate to 3-4 with 6mol/L hydrochloric acid, filter, wash the filter cake with purified water, and dry to obtain a solid (1.63kg, yield 50%).
[0115]Step 2: Synthesis of intermediate (III-1)
[0116]The product from the previous step (1.4 kg) and 15% aqueous sodium hydroxide solution (9.7 kg) were heated to reflux under argon protection and reacted for 28 hours. The reaction solution was poured into ice water (5-6 kg), and hydrochloric acid (6N, 3 L) was slowly added to adjust the pH value to 5-6. The temperature was maintained below 20°C. During this period, ethyl acetate was added to eliminate bubbles. The mixture was filtered, washed with purified water, and dried to obtain a solid (1.18 kg, yield 94%).
[0117]Step 3: Synthesis of intermediate (V-1)
[0118]Add the product of the previous step (1.10kg), purified water (27.5kg), and hydrochloric acid (2.92kg) to the reactor in sequence, stir and dissolve, cool to -4 to -1°C, add sodium nitrite aqueous solution (346g sodium nitrite, 5.5kg water), and continue to react for 15min after the addition is completed. Cool to -8 to -5°C. Dissolve sodium hydroxide (1.48kg) in purified water (13.2kg) to obtain a 10% sodium hydroxide aqueous solution. Add 5-methyl-2-(5,6,7,8-tetrahydronaphthalen-2-yl)-2H-pyrazole-3-ol hydrochloride (1.26kg) to the above sodium hydroxide aqueous solution (10kg) to dissolve, and add the resulting solution to the diazonium salt solution at once, keeping the temperature not higher than 10°C. Add the remaining 10% sodium hydroxide aqueous solution, adjust the pH to 8 to 9, naturally heat to 8 to 12°C for reaction, and react for 4h. Add 6N hydrochloric acid, adjust pH=2-3, keep the temperature not more than 20°C, filter, and wash the filter cake with water until pH=6-7. Add the filter cake to 50% tetrahydrofuran aqueous solution (19kg), slurry at room temperature for 2h, filter, wash with 50% tetrahydrofuran aqueous solution, wash with water, and dry. Add ethyl acetate (20kg) to the solid, slurry at 40-45°C for 2h under argon protection, cool to room temperature, filter, wash with ethyl acetate, add the solid to ethyl acetate (20kg), slurry at 40-45°C for 2h under argon protection, cool to room temperature, filter, wash with ethyl acetate, and dry to obtain a solid (2.18kg, yield 95%, purity 99.5%).
[0120]Example 3. Preparation of (Z)-5-(2-hydroxy-3-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1,5-dihydro-4H-pyrazol-4-ylidene)hydrazino)phenyl)furan-2-carboxylic acid ethanolamine salt (1:2)
[0121]
[0122]Preparation of crude product
[0123]The compound of formula (V-1) (1.8 kg) was suspended in a tetrahydrofuran/ethanol (14.5 kg, V/V = 2:1) mixed solvent at room temperature, stirred for 0.5 h, cooled to 10-15 ° C, and a tetrahydrofuran ethanol solution of ethanolamine (479.6 g) (tetrahydrofuran 91 g and ethanol 41 g) was added dropwise. The mixture was naturally heated to room temperature and reacted for 20 h. Filtered, washed with a tetrahydrofuran/ethanol (V/V = 2:1) mixed solvent, washed with ethyl acetate, filtered, and dried to obtain a dark red solid (1.73 kg, yield 76%, purity 99.7%).
[0124]
1H-NMR(500MHz,D 2O+NaOH)δ7.725-7.741(d,1H),7.298-7.316(d,3H),7.183-7.198(d,1H),7.131-7.149(m,2H),6.612-6.643(t,1H),3.574-3.596(t,4H),2.759-2.778(br,4H),2.698-2.721(t,4H),2.428(s,3H),1.772(br,4H).
SYN
J.Med.Chem.2024,67,4376−4418
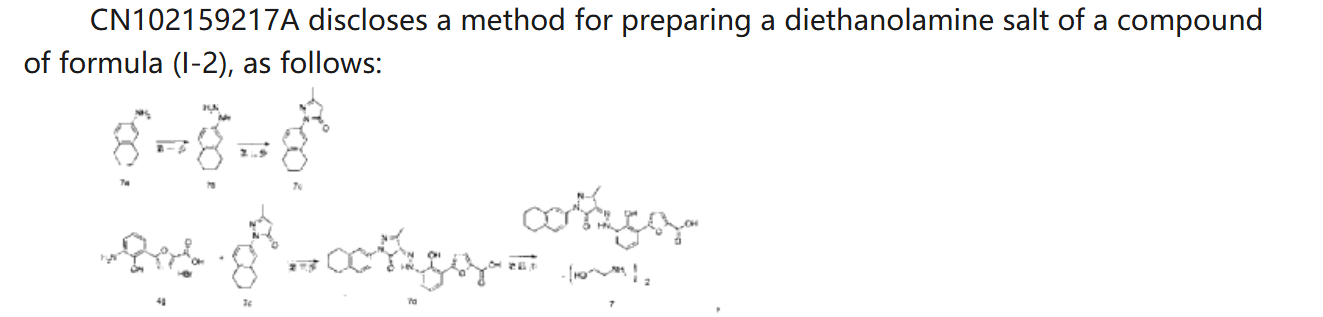
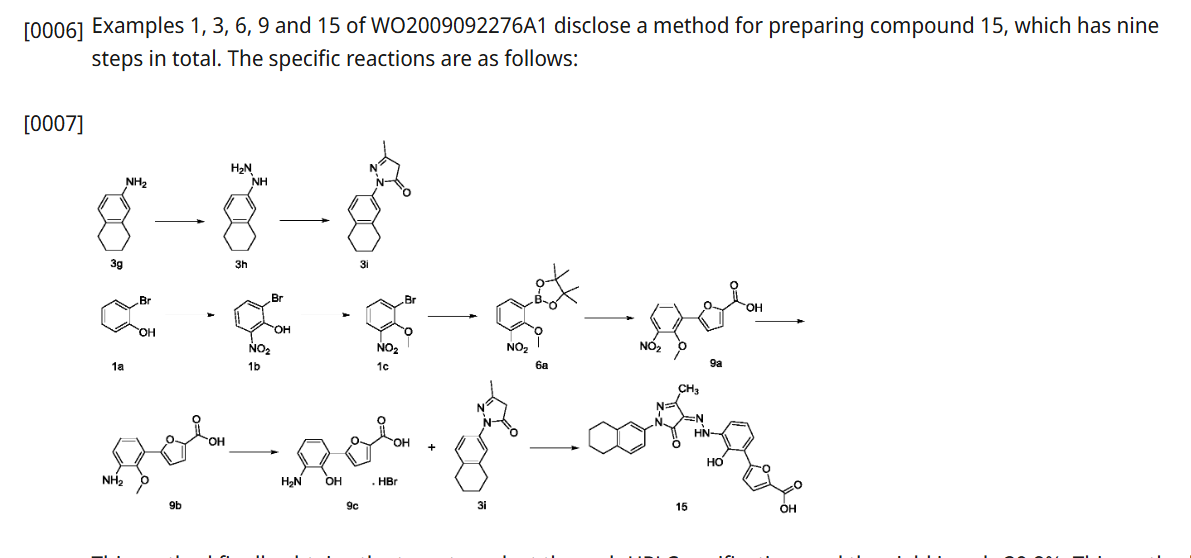
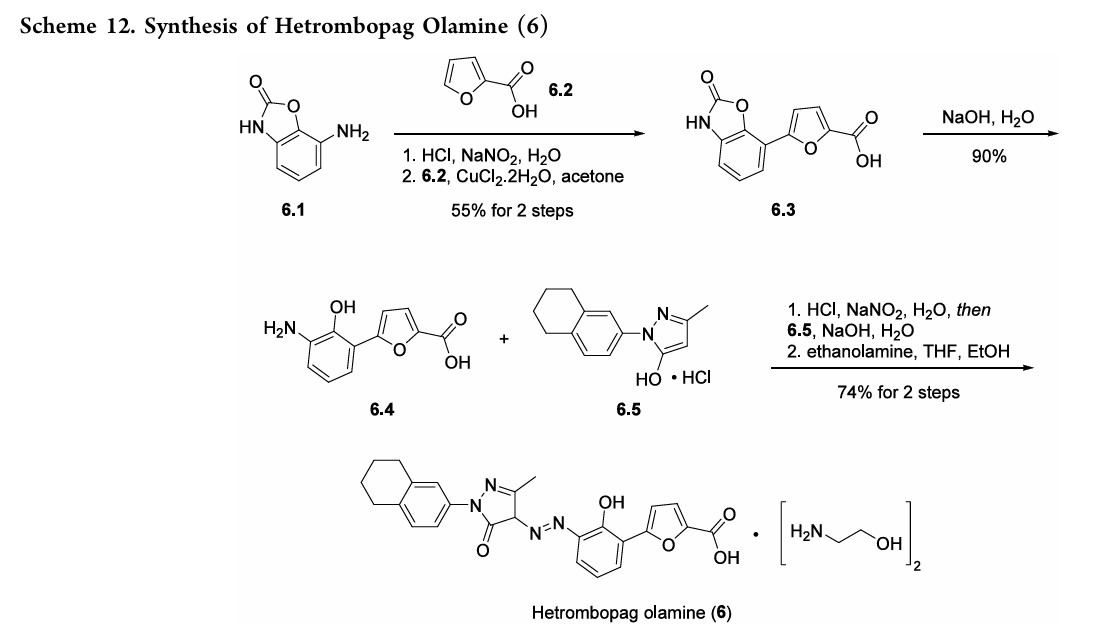
HetrombopagOlamine (Hengqu).
Hetrombopag olamine (6), an oral nonpeptide thrombopoietin receptor
(TpoR)agonistdevelopedby JiangsuHengruiPharmaceutical, was approved in China in June2021 for treatment of adult patients with chronic primary immune thrombocytopenia (ITP) and severe aplastic anemiawhohave not responded well to other treatments.46Hetrombopag, like other TpoR agonists, increases platelet production by binding to the transmembranedomainofTpoRinprogenitorcells, inducing
megakaryocytes.Theeffectisadditivewiththeactionofnative thrombopoietin, whichbinds to the extracellular domainof TpoR.Hetrombopag is structurallyrelatedtoeltrombopag, a previously approvedTpoR, withmodifications to enhance potencyandminimizetoxicity.46−48InaPhaseIIIclinicaltrial, ITPpatients demonstratedadurableplatelet response, with reducedbleedingriskanduseof rescuetherapycomparedto
placebo.49 Akilo-scale, chromatography-freesynthesisofhetrombopag has been reported by researchers at Jiangsu Hengrui Pharmaceutical in the Chinese-language patent literature (Scheme 12).50,51 Commercially available aniline 6.1 was coupledwith furoic acid (6.2) using aMeerwein arylation reaction togive intermediate6.3.This process first involves diazotizationof the anilineusing sodiumnitrite andhydrochloricacid.Ureawasusedtoquenchtheresidualnitrousacid, animprovement thatultimatelygavetheproductwithhigher purity and lower levels of specific impurities; the crude
diazoniumsalt solutionwas carried forwarddirectlywithout furthermanipulation.Furoicacid(6.2)inacetonewastreated withcopper(II)chloridedihydratefollowedbyadditionofthe
diazonium salt solution to affect the arylation. The crude productwaspurifiedbyacid−baseextractionandisolatedby filtrationtoprovide6.3 in55%yield.Basichydrolysisof the
cycliccarbamateunveiledthefreeanilineandphenolmoieties in arene 6.4. Nucleophilic attack of the enolate anion of pyrazolone 6.5 (see Scheme 13) on the diazoniumsalt of aniline6.4 formed the central hydrazonemoiety ina JappKlingemann-like reaction. The crude product was triturated withethylacetatetorapidlyprovidehetrombopagfreebasein
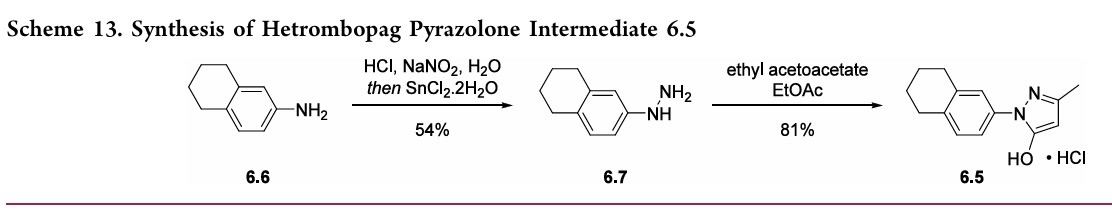
97.5%yield.TreatmentwithethanolamineinTHFandEtOH thengeneratedhetrombopagolamine (6) in76%yieldand 99.7%purity.51 Pyrazolone intermediate6.5was synthesized in two steps
(Scheme 13).52,53 5,6,7,8-Tetrahydronaphthalen-2-yl amine (6.6)was converted to the diazoniumion and reduced in situ to the corresponding hydrazine 6.7 using stannous chloridedihydrate.Condensationof thehydrazinewithethyl acetoacetate in ethyl acetate and in situ cyclization gave pyrazolone6.5.While the synthesis fromaniline6.1 to the activepharmaceutical ingredient(API)6wasreportedonthe
kilo-scale, thesynthesisofpyrazolone6.5wasreportedonlyon gram-scale
(46) Syed, Y. Y. Hetrombopag: First approval. Drugs 2021, 81, 1581−1585.
(47) Xie, C.; Zhao, H.; Bao, X.; Fu, H.; Lou, L. Pharmacological characterization of hetrombopag, a novel orally active human thrombopoietin receptor agonist. J. Cell. Mol. Med. 2018, 22, 5367−5377.
(48) Zheng, L.; Liang, M.-z.; Zeng, X.-l.; Li, C.-z.; Zhang, Y.-f.; Chen, X.-y.; Zhu, X.; Xiang, A.-b. Safety, pharmacokinetics and pharmacodynamics of hetrombopag olamine, a novel TPO-R agonist, in healthy individuals. Basic Clin. Pharmacol. Toxicol. 2017, 121, 414−422.
(49) Mei, H.; Liu, X.; Li, Y.; Zhou, H.; Feng, Y.; Gao, G.; Cheng, P.; Huang, R.; Yang, L.; Hu, J.; Hou, M.; Yao, Y.; Liu, L.; Wang, Y.; Wu, D.; Zhang, L.; Zheng, C.; Shen, X.; Hu, Q.; Liu, J.; Jin, J.; Luo, J.; Zeng, Y.; Gao, S.; Zhang, X.; Zhou, X.; Shi, Q.; Xia, R.; Xie, X.; Jiang, Z.; Gao, L.; Bai, Y.; Li, Y.; Xiong, J.; Li, R.; Zou, J.; Niu, T.; Yang, R.;
Hu, Y. A multicenter, randomized phase III trial of hetrombopag: a novel thrombopoietin receptor agonist for the treatment of immune thrombocytopenia. J. Hematol. Oncol. 2021, 14, 37.
(50) Shi, A.; Diao, A.; Du, Y. Preparation of bicyclic substituted pyrazolone azo derivatives. China Patent CN 113929668, 2022.
(51) Diao, A.; Gao, X.; Bian, L. Method for preparing bicyclo substituted pyrazolone azo derivatives and intermediates. WO 2018133818, 2018.
(52) Tang, P. C.; Lue, H.; Fei, H.; Chen, Y. Preparation of pyrazole derivatives as thrombopoietin receptor agonists. WO 2010142137, 2010.
(53) Tang, P. C.; Lue, H.; Fei, H.; Chen, Y. Salts of bicyclo substituted pyrazolon azo derivatives, preparation method and use
thereof. European Patent EP 2441457, 2014.

.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////Hetrombopag Olamine, CHINA 2021, APPROVALS 2021, Hetrombopag diolamine, SHR 8735 olamine, Hetrombopag ethanolamine, SHR-8735 olamine, V45T2I862X, RAFUTROMBOPAG OLAMINE
Contezolid
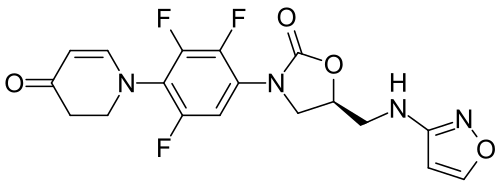
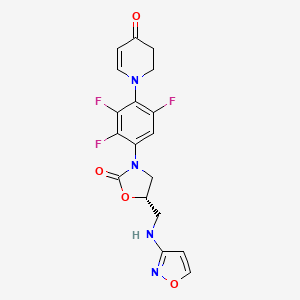

Contezolid
- MRX-I
- 1112968-42-9
- MRX-1
- B669M62ELP
- (5S)-5-[(1,2-oxazol-3-ylamino)methyl]-3-[2,3,5-trifluoro-4-(4-oxo-2,3-dihydropyridin-1-yl)phenyl]-1,3-oxazolidin-2-one
- 4(1H)-Pyridinone, 2,3-dihydro-1-(2,3,6-trifluoro-4-((5S)-5-((3-isoxazolylamino)methyl)-2-oxo-3-oxazolidinyl)phenyl)-
- (5S)-5-[(1,2-oxazol-3-ylamino)methyl]-3-[2,3,5-trifluoro-4-(4-oxo-2,3-dihydropyridin-1-yl)phenyl]-1,3-oxazolidin-2-one
- 4(1H)-Pyridinone, 2,3-dihydro-1-[2,3,6-trifluoro-4-[(5S)-5-[(3-isoxazolylamino)methyl]-2-oxo-3-oxazolidinyl]phenyl]-
- 1-{2,3,6-trifluoro-4-[(5S)-5-{[(1,2-oxazol-3-yl)amino]methyl}-2-oxo-1,3-oxazolidin-3-yl]phenyl}-1,2,3,4-tetrahydropyridin-4-one
- 5-((isoxazol-3-ylamino)methyl)-3-(2,3,5-trifluoro-4-(4-oxo-3,4-dihydropyridin-1(2H)-yl)phenyl)oxazolidin-2-one
- コンテゾリド;
WeightAverage: 408.337
Monoisotopic: 408.104539468
Chemical FormulaC18H15F3N4O4
Shanghai MicuRx Pharmaceutical Co. Ltd
Contezolid was approved for use by the National Medical Products Administration (NMPA) of China in 2021
- OriginatorMicuRx Pharmaceuticals
- ClassAntibacterials; Oxazolidinones; Skin disorder therapies
- Mechanism of ActionProtein synthesis inhibitors
- Phase IIIDiabetic foot; Skin and soft tissue infections
- No development reportedGram-positive infections
- 28 Jan 2025No recent reports of development identified for phase-I development in Gram-positive-infections(In volunteers) in China (IV)
- 28 Jan 2025No recent reports of development identified for phase-I development in Gram-positive-infections(In volunteers) in China (PO)
- 29 Nov 2024Phase-III clinical trials in Skin and soft tissue infections in China (IV), prior to November 2024
Contezolid (trade name Youxitai) is an antibiotic of the oxazolidinone class.[1][2] It is effective against Staphylococcus aureus, methicillin-resistant Staphylococcus aureus (MRSA), Streptococcus pyogenes, Streptococcus agalactiae, and other bacteria.[3]
In 2021, it was approved by the National Medical Products Administration of China for the treatment of complicated skin and soft tissue infections (cSSTI).[3][4]
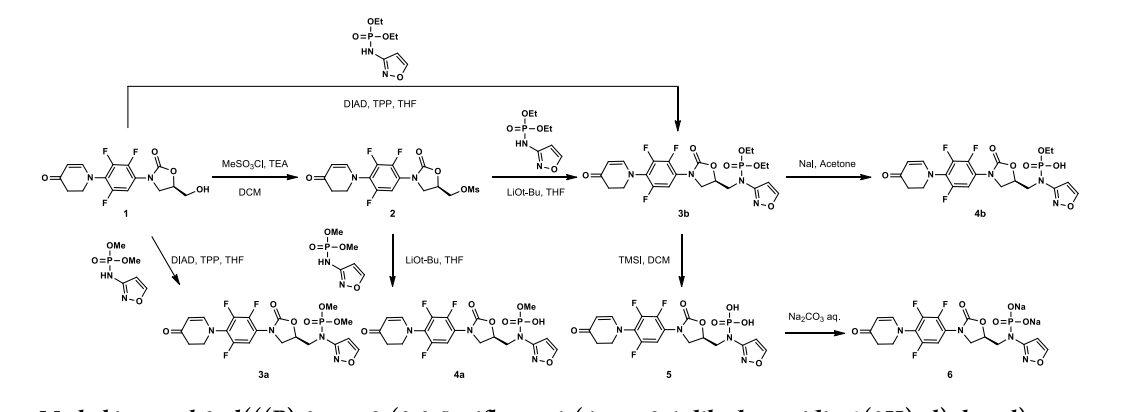
A prodrug of contezolid, contezolid acefosamil, which is formulated for IV administration[5] is in Phase III clinical trials for diabetic foot infection.[6]

Chemical structure of contezolid acefosamil
SYN
https://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.2c00191
Abstract

New oral antibiotic contezolid (CZD) is effective against Gram-positive infections but unsuitable for intravenous (IV) administration due to its modest solubility. To address the medical need for an IV form of CZD, its isoxazol-3-yl phosphoramidate derivatives have been explored, and contezolid acefosamil (CZA, 8), the first representative of a novel O-acyl phosphoramidate prodrug class, has been identified. CZA exhibits high aqueous solubility (>200 mg/mL) and good hydrolytic stability at media pH suitable for IV administration. CZA rapidly converts into the active drug CZD in vivo. In a pharmacokinetic (PK) rat model, the exposure of active drug CZD after IV administration of the prodrug CZA was similar to or higher than that from the IV administration of CZD. The prodrug CZA is bioequivalent to or better than CZD in several preclinical infection models. CZA is likewise active upon its oral administration. To date, CZA has been evaluated in Phase 1 and Phase 2 clinical trials in the USA. It is advancing into further clinical studies including step-down therapy with in-hospital intravenous CZA administration followed by outpatient oral CZD treatment.
SYN
Contezolid (Youxitai). Contezolid (4), also referred to as MRX-I, is an orally administered oxazolidinone
antibacterial agent developed by Shanghai MicuRx Pharmaceutical Co. Ltd. Contezolid was developed to overcome the myelosuppression and monoamine oxidase (MAO) inhibition limitations of the structurally similar linezolid. 32 Contezolid is used to treat complicated skin and soft tissue infections arising
from multidrug-resistant Gram-positive bacterial infections including methicillin-susceptible Staphylococcus aureus (MSSA), methicillin-resistant Staphylococcus aureus (MRSA), Streptococcus pyogenes, Streptococcus agalactiae, and vancomycin-resistant enterococci.3334 Contezolid was approved for use by the National Medical Products Administration (NMPA) of China in 2021.
As with most antibacterial oral therapies, high 35 dosage is required; the drug is given twice daily for 7−14 days.36,37
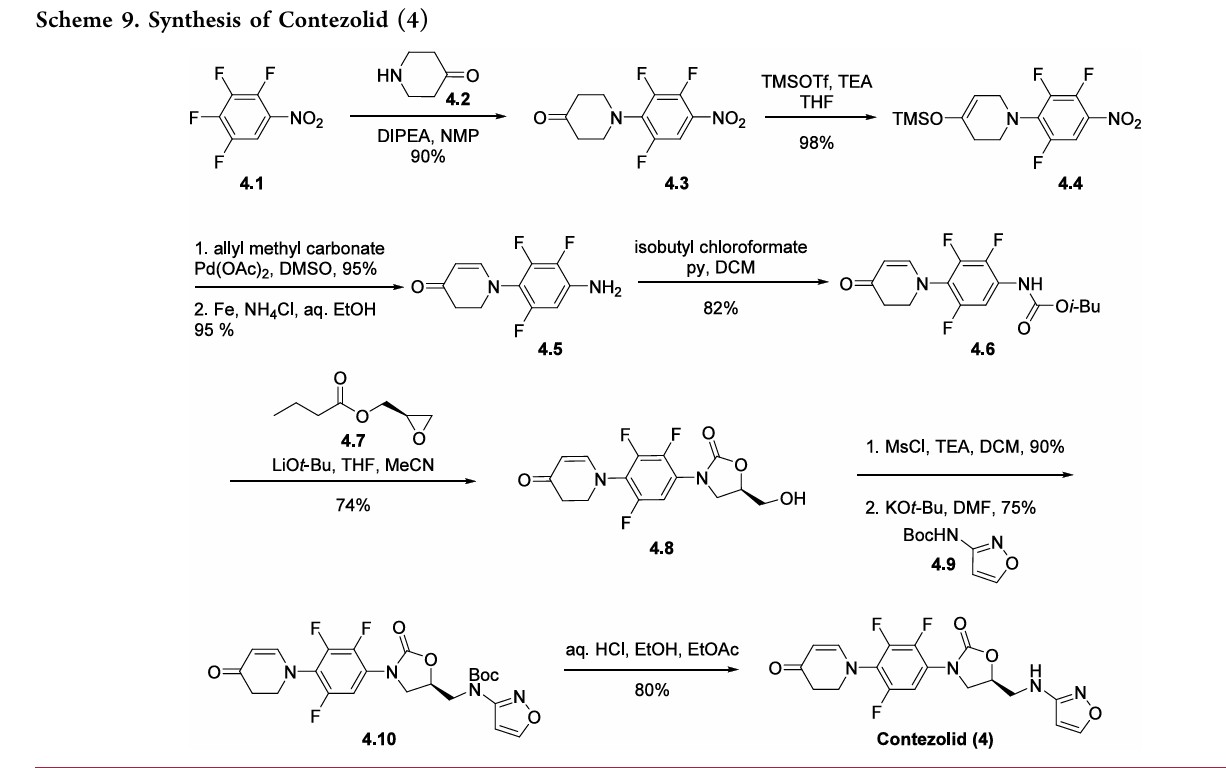
The synthesis of contezolid builds on prior research from other groups.
A sequence developed by Pharmaciawith a facile SN38began Ar reaction between polyfluorinated nitro
benzene 4.1 and piperidine-4-one 4.2 to furnish 4.3 in good yield (Scheme 9). Silyl enol ether formation afforded 4.4, which was subjected to Tsuji’s 39 method to give the α,βunsaturated ketone in excellent yield. Subsequent reduction of the nitro group gave aryl amine 4.5. Treatment of 4.5 with isobutyl chloroformate gave carbamate 4.6, which was treated with optically pure epoxide 4.7 to give xazolidinone 4.8. 38Mesylation of the free alcohol and displacement with N-Bocaminoisoxazole 4.9 afforded the Boc-protected contezolid 4.10. Simple acidic removal of the Boc group provided contezolid 4.
(32) Wang, W.; Voss, K. M.; Liu, J.; Gordeev, M. F. Nonclinical
evaluation of antibacterial oxazolidinones contezolid and contezolid
acefosamil with low serotonergic neurotoxicity. Chem. Res. Toxicol.
2021, 34, 1348−1354.
(33) Hoy, S. M. Contezolid: First approval. Drugs 2021, 81, 1587−
1591.
(34) MicuRx Pharmaceuticals. China NMPA approves MicuRx’s
contezolid for treatment of drug-resistant bacterial infection. http://www.
micurx.com/703.html (accessed 2023-06).
(35) MSD Pharmaceuticals. Usual dosages of commonly prescribed
antibiotics. https://www.msdmanuals.com/en-jp/professional/
multimedia/table/usual-dosages-of-commonly-prescribed-antibioticsa
(accessed 2023-06).
(36) Barbachyn, M. R.; Hutchinson, D. K.; Brickner, S. J.; Cynamon,
M. H.; Kilburn, J. O.; Klemens, S. P.; Glickman, S. E.; Grega, K. C.;
Hendges, S. K.; Toops, D. S.; et al. Identification of a novel
oxazolidinone (U-100480) with potent antimycobacterial activity. J.
Med. Chem. 1996, 39, 680−685.
(37) Im, W. B.; Choi, S. H.; Park, J. Y.; Choi, S. H.; Finn, J.; Yoon, S.
H. Discovery of torezolid as a novel 5-hydroxymethyl-oxazolidinone
antibacterial agent. Eur. J. Med. Chem. 2011, 46, 1027−1039.
(38) Manninen, P. R.; Brickner, S. J. Preparation of N-aryl-5R
hydroxymethyl-2-oxazolidinones from N-aryl carbamates: N-phenyl
(5R)-hydroxymethyl-2-oxazolidinone. Organic Synth 2005, 81, 112.
(39) Tsuji, J.; Minami, I.; Shimizu, I. A novel palladium-catalyzed
preparative method of α,β-unsaturated ketones and aldehydes from
saturated ketones and aldehydes via their silyl enol ethers. Tetrahedron
Lett. 1983, 24, 5635−5638.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Trade names | Youxitai |
| Other names | MRX-I |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1112968-42-9 |
| PubChem CID | 25184541 |
| IUPHAR/BPS | 10795 |
| DrugBank | DB12796 |
| ChemSpider | 34217570 |
| UNII | B669M62ELP |
| KEGG | D11297 |
| ChEMBL | ChEMBL3287379 |
| CompTox Dashboard (EPA) | DTXSID901353186 |
| Chemical and physical data | |
| Formula | C18H15F3N4O4 |
| Molar mass | 408.337 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Gordeev MF, Yuan ZY (June 2014). “New Potent Antibacterial Oxazolidinone (MRX-I) with an Improved Class Safety Profile”. Journal of Medicinal Chemistry. 57 (11): 4487–4497. doi:10.1021/jm401931e. PMID 24694071.
- Zhao X, Huang H, Yuan H, Yuan Z, Zhang Y (May 2022). “A Phase III multicentre, randomized, double-blind trial to evaluate the efficacy and safety of oral contezolid versus linezolid in adults with complicated skin and soft tissue infections”. The Journal of Antimicrobial Chemotherapy. 77 (6): 1762–1769. doi:10.1093/jac/dkac073. PMID 35265985.
- Hoy SM (September 2021). “Contezolid: First Approval”. Drugs. 81 (13): 1587–1591. doi:10.1007/s40265-021-01576-0. PMC 8536612. PMID 34365606.
- Mak E (3 June 2021). “Micurx wins China approval for antibacterial contezolid”. BioWorld.
- Liu J, Wang W, Wang C, Zhang L, Zhang X, Liu S, et al. (July 2022). “Discovery of Antibacterial Contezolid Acefosamil: Innovative O-Acyl Phosphoramidate Prodrug for IV and Oral Therapies”. ACS Medicinal Chemistry Letters. 13 (7): 1030–1035. doi:10.1021/acsmedchemlett.2c00191. PMC 9290071. PMID 35859881.
- “Contezolid acefosamil by MicuRx Pharmaceuticals for Diabetic Foot Infection (DFI): Likelihood of Approval”. GlobalData. 31 May 2023 – via Pharmaceutical Technology.
/////////Contezolid, CHINA 2021, APPROVALS 2021, MRX-I, 1112968-42-9, MRX 1, B669M62ELP, コンテゾリド ,

{kind=link}
{kind=link}
{kind=link}
{kind=link}