
Home » cancer (Page 8)
Category Archives: cancer
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Pevonedistat

Millennium Pharmaceuticals, Inc. INNOVATOR
Millennium Pharmaceuticals, Inc., a subsidiary of Takeda Pharmaceutical Company Limited,
MLN4924, MLN 4924-003, TAK-924
905579-51-3 BASE
1160295-21-5 HcL
A potent and selective inhibitor of NAE. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. The ubiquitin-proteasome pathway mediates the destruction of unwanted proteins.
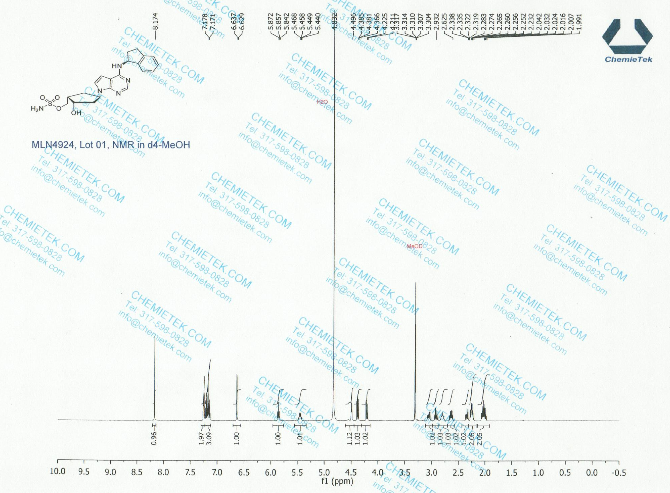
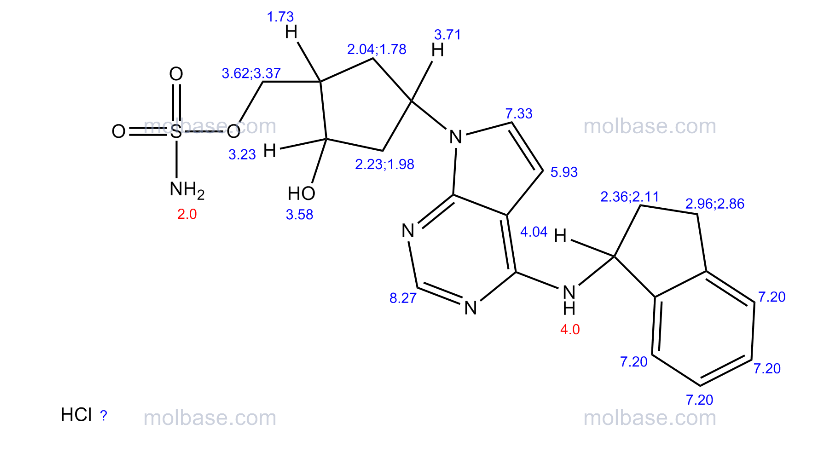
(((1S,2S,4R)-4-{4-[(S)-2,3-Dihydro-1H-inden-1-ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl}-2-hydroxycyclopentyl)methyl sulfamate hydrochloride) (pevonedistat), a novel NEDD8-activating enzyme (NAE) inhibitor, has demonstrated in vitro cytotoxic activity against a variety of human malignancies and is currently being developed by Takeda Pharmaceuticals Company Limited as a clinical candidate for the treatment of cancer
In 2011, orphan drug designation was assigned to MLN-4924 for the treatment of MDS and for the treatment of acute myelogenous leukemia.
PHASE 1…….CANCER SOLID TUMOR

………………….
PATENT
http://www.google.com/patents/US20120330013

preparing a compound represented by the following formula 1 by reacting the compound of formula 11 with TFA (step 9):


The retrosynthetic analysis of MLN4924 (1), as the final desired nucleoside, is shown in the following.

MLN 4924 (1) can be synthesized by condensing cyclic sulfate 3 as the glycosyl donor with a purine base. The glycosyl donor 3 can be produced from diol 4, which in turn can be obtained from cyclopentanone 5 via a stereoselective reduction and a regioselective cleavage of the isopropylidene moiety. The cyclopentanone 5 can be synthesized from cyclopentenone 6 by stereoselective reduction. The intermediate cyclopentenone 6 can be easily derived from D-ribose according to our previously published procedure (Jeong, L. S. et al., J. Org. Chem. 2004, 69, 2634-2636).
The synthetic route for the glycosyl donor 3 is shown in the following scheme 1.

Example 1 Preparation of MLN4924 Step 1: Preparation of 6-(tert-butyl-diphenyl-silanyloxymethyl)-2,2-dimethyl-tetrahydro-cyclopenta[1,3]dioxol-4-one (Compound 5)

To a suspension of the compound 6 (20.0 g, 47.1 mmol) in methanol (400 ml) was added 10% palladium on activated carbon (1.0 g), and the mixture was stirred at room temperature overnight under H2 atmosphere. After filtration of the reaction mixture, the solvent was removed and the residue was dissolved in methylene chloride and then filtered through short pad silica gel. Then, the solvent was evaporated to give the compound 5 (20.1 g, 100%) as a colorless syrup.
[α]20 D −28.32 (c 1.49, MeOH); HR-MS (ESI): m/z calcd for C25H32NaO4Si [M+Na]+ 447.1968, Found 447.1956; 1H NMR (400 MHz, CDCl3) δ 7.69 (m, 4H), 7.40 (m, 6H), 4.84 (t, J=4.4 Hz, 1H), 4.22 (dd, J=1.2, 4.8 Hz, 1H), 3.96 (dd, J=8.0, 10.0 Hz, 1H), 3.82 (dd, J=6.8, 10.0 Hz, 1H), 2.37 (m, 1H), 2.30 (ddd, J=1.2, 8.4, and 18.4 Hz, 1H), 2.20 (ddd, J=1.2, 12.0, and 18.4 Hz, 1H), 1.37 (s, 3H), 1.35 (s, 3H), 1.06 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 112.6, 80.5, 77.6, 77.2, 76.9, 63.6, 38.1, 36.9, 27.1, 27.02, 27.01, 25.3, 19.5; Anal. Calcd for C25H32O4Si: C, 70.72; H, 7.60. Found: C, 70.79; H, 7.75.
Step 2: Preparation of 6-(tert-butyl-diphenyl-silanyloxymethyl)-2,2-dimethyl-tetrahydro-cyclopenta[1,3]dioxol-4-ol (Compound 7)

To a suspension of the compound 5 (20.1 g, 47.1 mmol) in methanol (500 ml) were added sodium borohydride (2.17 g, 57.4 mmol) and cerium (III) chloride heptahydrate (21.3 g, 57.2 mmol) at 0° C., and the mixture was stirred at room temperature for 30 min. After the solvent was removed, the residue was partitioned between ethyl acetate and water. The organic layer was then washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=5/1) to give the compound 7 (20.86 g, 98%) as a colorless syrup.
[α]20 D +34.55 (c 0.55, MeOH); HR-MS (ESI): m/z calcd for C25H34NaO4Si [M+Na]+: 449.2124; Found: 449.2110; 1H NMR (400 MHz, CDCl3) δ 7.69 (m, 4H), 7.39 (m, 6H), 4.62 (t, J=5.6 Hz, 1H), 4.44 (t, J=5.6 Hz, 1H), 3.89 (dd, J=6.0, 7.6 Hz, 1H), 3.84 (m, 1H), 3.68 (dd, J=6.4, 10.0 Hz, 1H), 1.91 (m, 2H), 1.26 (m, 1H), 1.42 (s, 3H), 1.33 (s, 3H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.9, 135.8, 134.2, 134.1, 129.8, 129.7, 127.8, 127.7, 110.6, 79.4, 78.9, 77.6, 77.2, 76.9, 72.5, 62.9, 41.6, 33.4, 27.0, 25.9, 27.0, 25.9, 24.4, 19.5; Anal. Calcd for C25H34O4Si: C, 70.38; H, 8.03. Found: C, 70.41; H, 8.08.
Step 3: Preparation of 3-tert-butoxy-4-(tert-butyl-diphenyl-silanyloxymethyl)-cyclopentane-1,2-diol (Compound 4)

To a solution of the compound 7 (20.86 g, 47.12 mmol) in methylene chloride was added trimethylaluminum (2.0 M in toluene, 132.1 ml) at 0° C., and the mixture was stirred at room temperature for 2 days. The mixture was cooled to 0° C., slowly quenched with an aqueous saturated ammonium chloride solution, filtered, and evaporated. The residue was partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=2/1) to give the compound 4 (13.42 g, 62%) as a colorless syrup.
[α]20 D +3.30 (c 0.55, MeOH); HR-MS (ESI): m/z calcd for C26H38NaO4Si [M+Na]+: 465.2437; Found: 465.2423; 1H NMR (400 MHz, CDCl3) δ 7.70 (m, 4H), 7.41 (m, 6H), 4.05 (dd, J=4.4, 7.2 Hz, 1H), 3.93 (m, 1H), 3.72 (m, 2H), 3.59 (dd, J=3.6, 12.0 Hz, 2H), 2.70 (d, J=20.8 Hz, 1H), 2.10 (m, 2H), 1.60 (m, 1H), 1.20 (s, 9H), 1.06 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.9, 133.5, 130.0, 129.9, 127.9, 127.9, 77.6, 77.2, 76.9, 74.9, 73.8, 72.7, 72.1, 63.3, 42.1, 34.0, 28.5, 27.0, 19.4; Anal. Calcd for C26H38O4Si: C, 70.55; H, 8.65. Found: C, 70.61; H, 8.70.
Step 4: Preparation of (4-tert-butoxy-2,2-dioxo-tetrahydro-2-yl-6-cyclopenta[1,3,2]-dioxathiol-5-ylmethoxy)-tert-butyl-diphenyl-silane (Compound 3)

To a solution of the compound 4 (13.42 g, 30.3 mmol) in methylene chloride were added triethyl amine (14.5 ml, 101.0 mmol) and thionyl chloride (3.7 ml, 47.4 mmol) at 0° C., and the reaction mixture was stirred at 0° C. for 10 minutes. The reaction mixture was partitioned between methylene chloride and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=6/1) to give the cyclic sulfite (14.37 g, 97%) as a white foam.
[α]20 D +20.00 (c 0.05, MeOH); HR-MS (ESI): m/z calcd for C26H36NaO5SSi [M+Na]+: 511.1950; Found: 511.1929; 1H NMR (400 MHz, CDCl3) δ 7.64 (m, 4H), 7.40 (m, 6H), 5.23 (m, 1H), 5.04 (dd, J=4.4, 6.0 Hz, 1H), 4.01 (t, J=4.8 Hz, 1H), 3.68 (dd, J=3.6, 10.4 Hz, 1H), 3.56 (dd, J=8.0, 10.4 Hz, 1H), 2.07 (m, 2H), 1.96 (m, 1H), 1.14 (s, 9H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.8, 135.7, 133.9, 133.8, 129.9, 129.9, 127.9, 127.8, 85.7, 83.2, 77.6, 77.2, 76.9, 75.0, 71.1, 62.7, 44.7, 31.4, 28.5, 27.1, 19.4; Anal. Calcd for C26H36O5SSi: C, 63.90; H, 7.42; S, 6.56. Found: C, 63.94; H, 7.45; S, 6.61.
To a solution of the cyclic sulfite obtained above (14.37 g, 29.4 mmol) in the mixture of carbon tetrachloride, acetonitrile and water (1:1:1.5, 210 ml) were added sodium metaperiodate (18.56 g, 56.4 mmol) and ruthenium chloride (1.72 g, 8.25 mmol), and the reaction mixture was stirred at room temperature for 10 minutes. The reaction mixture was partitioned between methylene chloride and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=4/1) to give the compound 3 (13.36 g, 90%) as a white solid.
mp 101-104° C.; [α]20 D −80.00 (c 0.05, MeOH); HR-MS (ESI): m/z calcd for C26H36NaO6SSi [M+Na]+: 527.1900; Found: 527.1881; 1H NMR (400 MHz, CDCl3) δ 7.64 (m, 4H), 7.41 (m, 6H), 5.13 (m, 1H), 4.83 (dd, J=4.4, 6.8 Hz, 1H), 4.13 (t, J=4.0 Hz, 1H), 3.92 (dd, J=6.4, 10.4 Hz, 1H), 3.69 (dd, J=5.2, 10.4 Hz, 1H), 2.11 (m, 2H), 2.02 (m, 1H), 1.15 (s, 9H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.7, 135.0, 133.8, 133.7, 130.0, 128.0, 127.9, 83.5, 82.2, 77.6, 77.2, 76.9, 75.4, 70.4, 70.4, 62.2, 43.9, 31.3, 28.2, 27.1, 26.8, 19.4; Anal. Calcd for C26H36O6SSi: C, 61.87; H, 7.19; S, 6.35. Found: C, 61.91; H, 7.14; S, 6.30.
Step 5: Preparation of 2-tert-butoxy-3-(tert-butyl-diphenyl-silanyloxymethyl)-5-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentanol (Compound 8)

A suspension of N6-indanyl-7-deazaadenine (8.80 g, 35.2 mmol), sodium hydride (1.38 g, 45.7 mmol) and 18-crown-6 (9.11 g, 45.7 mmol) in THF (200 ml) was stirred at 80° C. To the reaction mixture was added a solution for the compound 3 (13.36 g, 26.5 mmol) in THF (150 ml), and the stirring was continued at 80° C. overnight. The reaction mixture was cooled down to 0° C., and conc. HCl was added slowly until pH reaches 1-2. Then the reaction mixture was further stirred at 80° C. for 2 hours. After neutralized with saturated aqueous NaHCO3 solution, the reaction mixture was partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=2/1) to give the compound 8 (11.62 g, 65%) as a white foam.
UV (CH2Cl2) λmax 272.5 nm; [α]20 D −8.89 (c 0.45, MeOH); HR-MS (ESI): m/z calcd for C41H51N4O3Si [M+H]+: 675.3730; Found: 675.3717; 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 7.70 (m, 4H), 7.41 (m, 6H), 6.92 (d, J=3.6 Hz, 1H), 6.29 (d, J=3.2 Hz, 1H), 5.91 (dd, J=7.6, 14.8 Hz, 1H), 5.14 (br d, J=6.8 Hz, 1H), 4.77 (m, 1H), 4.36 (t, J=6.0 Hz, 1H), 4.22 (dd, J=5.2, 10.8 Hz, 1H), 3.84 (dd, J=5.6, 10.4 Hz, 1H), 3.73 (dd, J=8.4, 10.4 Hz, 1H), 3.37 (d, J=5.6 Hz, 1H), 3.06 (m, 1H), 2.95 (m, 1H), 2.75 (m, 1H), 2.75 (m, 1H), 2.58 (m, 1H), 2.38 (m, 1H), 2.15 (m, 1H), 1.98 (m, 1H), 1.65 (s, 1H), 1.55 (s, 1H), 1.16 (s, 9H), 1.07 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 156.4, 151.8, 150.3, 144.1, 143.8, 135.9, 134.0, 129.9, 128.2, 127.9, 127.9, 127.0, 125.1, 124.4, 123.3, 103.8, 97.4, 77.8, 77.6, 77.2, 76.9, 74.9, 72.4, 63.5, 62.1, 56.3, 43.9, 34.9, 30.5, 30.5, 28.5, 27.2, 19.5; Anal. Calcd for C41H50N4O3Si: C, 72.96; H, 7.47; N, 8.30. Found: C, 73.01; H, 7.45; N, 8.36.
Step 6: Preparation of {7-[3-tert-butoxy-4-(tert-butyl-diphenyl-silanyloxymethyl)-cyclopentyl]-7H-pyrrolo[2,3-d]pyrimidin-4-yl}-indan-1-yl-amine (Compound 9)

To a solution of the compound 8 (11.62 g, 17.2 mmol) in methylene chloride (300 ml) were added N,N-dimethylaminopyridine (5.64 g, 51.6 mmol) and phenyl chlorothionocarbonate (4.3 ml, 34.4 mmol), and the reaction mixture was stirred at room temperature overnight. After the solvent was removed, the residue was purified by silica gel column chromatography (hexane/ethyl acetate=6/1) to give the thiocarbonate (13.82 g, 99%) as a white foam.
UV (MeOH) λmax 271.50 nm; [α]20 D +10.00 (c 0.15, MeOH); HR-MS (ESI): m/z calcd for C48H55N4O4SSi [M+H]+: 811.3713; Found: 811.3687; 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 1H), 7.61 (dd, J=1.6, 7.6 Hz, 4H), 7.34 (m, 5H), 7.26 (m, 4H), 7.18 (m, 6H), 6.86 (s, 1H), 6.25 (d, J=3.2 Hz, 1H), 6.00 (dd, J=3.2, 8.4 Hz, 1H), 5.83 (d, J=6.8 Hz, 1H), 5.19 (m, 1H), 5.07 (br s, 1H), 4.48 (t, J=3.6 Hz, 1H), 3.82 (dd, J=7.2, 10.4 Hz, 1H), 3.52 (dd, J=7.2, 10.0 Hz, 1H), 2.99 (m, 1H), 2.88 (m, 2H), 2.69 (m, 2H), 2.18 (dd, J=11.2, 13.6 Hz, 1H), 1.94 (m, 2H), 1.12 (s, 9H), 0.98 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 194.9, 153.5, 152.1, 143.9, 135.9, 135.8, 134.1, 129.9, 129.6, 128.3, 127.9, 127.0, 126.7, 125.1, 124.6, 123.2, 122.0, 87.9, 77.6, 77.2, 76.9, 74.6, 70.4, 63.5, 57.3, 42.8, 35.0, 30.7, 30.5, 29.9, 28.7, 27.1, 19.4; Anal. Calcd for C48H54N4O4SSi: C, 71.08; H, 6.71; N, 6.91; S, 3.95. Found: C, 71.14; H, 6.75; N, 6.95; S, 4.01.
To a solution of the thiocarbonate obtained above (13.82 g, 17.0 mmol) in toluene (200 ml) were added tri-n-butyltinhydride (9.4 ml, 34.1 mmol) and 2,2′-azo-bis-isobutyronitrile (4.32 g, 26.3 mmol), and the reaction mixture was stirred at 110° C. for 1 hour. After the mixture was cooled down, the solvent was removed. The resulting residue was purified by silica gel column chromatography (hexane/ethyl acetate=3/1) to give the compound 9 (9.21 g, 82%) as a white foam.
UV (MeOH) λmax 272.50 nm; [α]20 D −10.00 (c 0.20, MeOH); HR-MS (ESI): m/z calcd for C41H51N4O2Si [M+H]+: 659.3781; Found: 659.3757; 1H NMR (400 MHz, CDCl3) δ 8.41 (s, 1H), 7.69 (m, 4H), 7.41 (m, 6H), 7.29 (m, 2H), 7.23 (m, 2H), 6.92 (d, J=3.6 Hz, 1H), 6.31 (d, J=3.6 Hz, 1H), 5.90 (dd, J=7.2, 14.8 Hz, 1H), 5.38 (m, 1H), 5.15 (br s, 1H), 4.33 (dd, J=5.2, 8.4 Hz, 1H), 3.88 (dd, J=6.4, 10.0 Hz, 1H), 3.68 (dd, J=7.2, 10.4 Hz, 1H), 3.05 (m, 1H), 2.96 (dd, J=7.6, 15.6 Hz, 1H), 2.76 (m, 1H), 2.45 (d, J=5.2 Hz, 1H), 2.29 (m, 2H), 2.06 (m, 1H), 1.95 (m, 2H), 1.55 (s, 1H), 1.13 (s, 9H), 1.06 (s, 9H);13C NMR (100 MHz, CDCl3) δ 156.3, 151.9, 144.1, 143.9, 135.9, 135.8, 134.3, 129.8, 128.2, 127.8, 127.0, 125.1, 124.6, 121.8, 77.6, 77.2, 76.7, 73.5, 72.2, 63.6, 56.4, 52.8, 46.8, 42.8, 34.9, 34.5, 30.5, 28.6, 27.2, 28.7, 19.4; Anal. Calcd for C41H50N4O2Si: C, 74.73; H, 7.65; N, 8.30. Found: C, 74.79; H, 7.61; N, 8.25.
Step 7: Preparation of 2-tert-butoxy-4-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentanol (Compound 10)

To a solution of the compound 9 (9.21 g, 13.97 mmol) in the mixture of THF and pyridine (1:1, 160 ml) was added dropwise pyridine hydrofluoride (18.42 ml, 190.0 mmol) at 0° C., and the reaction mixture was stirred at room temperature for 1 hour. The mixture was neutralized with saturated aqueous NaHCO3 solution and partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. Then, the residue was purified by silica gel column chromatography (hexane/ethyl acetate=1/3) to give the compound 10 (5.63 g, 99%) as a white foam.
UV (MeOH) λmax 273.00 nm; [α]20 D −6.36 (c 1.10, MeOH); HR-MS (ESI): m/z calcd for C25H33N4O2 [M+H]+: 421.2604; Found: 421.2599; 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1H), 7.30 (d, J=7.6 Hz, 1H), 7.22 (d, J=7.2 Hz, 2H), 7.15 (t, J=6.8 Hz, 1H), 6.88 (d, J=3.2 Hz, 1H), 6.23 (d, J=3.6 Hz, 1H), 5.83 (dd, J=7.2, 15.2 Hz, 1H), 5.28 (m, 1H), 5.06 (m, 1H), 4.47 (dd, J=5.6, 10.4 Hz, 1H), 3.78 (m, 1H), 3.70 (m, 1H), 3.24 (t, J=5.2 Hz, 1H), 2.98 (m, 1H), 2.87 (m, 1H), 2.68 (m, 1H), 2.46 (m, 1H), 2.37 (m, 2H), 1.93 (m, 2H), 1.18 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 156.2, 151.8, 147.9, 143.9, 143.9, 128.3, 126.9, 125.1, 124.5, 121.9, 97.7, 77.6, 77.2, 76.9, 75.5, 74.9, 63.4, 56.4, 53.8, 44.2, 42.2, 34.9, 33.2, 30.5, 28.6; Anal. Calcd for C25H32N4O2: C, 71.40; H, 7.67; N, 13.32. Found: C, 71.46; H, 7.60; N, 13.35.
Step 8: Preparation of sulfamic acid 2-tert-butoxy-4-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentylmethyl ester (Compound 11)

Preparation of 2.0 M solution of chlorosulfonamide in acetonitrile: Formic acid (14.15 ml, 166.0 mmol) was added dropwise to chlorosulfonyl isocyanate (32.0 ml, 162.5 mmol) under nitrogen atmosphere at 0° C. When the addition was completed, the mixture was solidified. To the mixture was added acetonitrile (61.3 ml), and the resulting solution was left to stand under nitrogen source at room temperature overnight.
To a solution of the compound 10 (5.63 g, 13.83 mmol) and triethyl amine (9.7 ml, 0.74 mmol) in acetonitrile (278 ml) was added 2.0 M solution of chlorosulfonamide in acetonitrile (13.83 ml, 27.76 mmol) at 0° C., and the reaction mixture was stirred at room temperature for 45 minutes. Additional 2.0 M chlorosulfonamide solution in acetonitrile (13.83 ml, 27.76 mmol) was added and the mixture was stirred at room temperature for 15 minutes. The reaction was quenched with methanol, and the solvent was removed. The residue was purified by silica gel column chromatography (methylene chloride/methanol=20/1) to give the compound 11 (6.37 g, 92%) as a white foam.
UV (MeOH) λmax 273.00 nm; [α]20 D −18.00 (c 0.50, MeOH); HR-MS (ESI): m/z calcd for C25H34N5O4S [M+H]+: 500.2332; Found: 500.2331; 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 7.36 (d, J=7.2 Hz, 1H), 7.29 (d, J=7.2 Hz, 1H), 7.22 (m, 2H), 6.95 (d, J=3.6 Hz, 1H), 6.31 (d, J=3.2 Hz, 1H), 5.89 (d, J=6.4 Hz, 1H), 5.10 (s, 2H), 4.41 (m, 2H), 4.26 (m, 1H), 3.05 (m, 1H), 2.94 (m, 1H), 2.76 (m, 2H), 2.27 (m, 3H), 2.06 (m, 1H), 1.97 (m, 1H), 1.76 (br s, 1H); 13C NMR (100 MHz, CDCl3) δ 156.4, 151.9, 149.9, 143.9, 143.8, 128.3, 126.9, 125.1, 124.5, 121.9, 121.9, 103.5, 97.9, 77.4, 77.2, 76.9, 74.3, 71.9, 71.3, 56.4, 53.1, 49.0, 42.3, 34.9, 34.3, 30.5, 28.6; Anal. Calcd for C25H33N5O4S: C, 60.10; H, 6.66; N, 14.02; S, 6.42. Found: C, 60.15; H, 6.71; N, 13.98; S, 6.39.
Step 9: Preparation of sulfamic acid 2-hydroxy-4-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentylmethyl ester (Compound 1)

A solution of the compound 11 (6.37 g, 12.72 mmol) in 70% trifluoroacetic acid (149.24 ml) was stirred at room temperature for 2 hours. The solvent was removed and the residue was purified by silica gel column chromatography (hexane/ethylene acetate=1/2) to give the compound 1 (5.08 g, 90%) as a white foam. BASE
UV (MeOH) λmax 279.50 nm; [α]20 D −6.41 (c 2.34, MeOH);
HR-MS (ESI): m/z calcd for C21H26N5O4S [M+H]+: 444.1705; Found: 444.1706;
1H NMR (400 MHz, CD3OD) δ 8.17 (d, J=1.6 Hz, 1H), 7.25 (m, 2H), 7.18 (m, 2H), 6.64 (d, J=3.6 Hz, 1H), 5.86 (t, J=7.6 Hz, 1H), 5.46 (m, 1H), 4.49 (d, J=2.8 Hz, 1H), 3.07 (m, 1H), 2.92 (m, 1H), 2.80 (m, 1H), 2.64 (m, 1H), 2.35 (m, 1H), 2.25 (m, 2H), 2.03 (m, 2H);
13C NMR (100 MHz, CD3OD) δ 152.1, 145.3, 144.6, 128.8, 127.6, 125.7, 125.2, 122.6, 100.5, 73.1, 70.9, 56.9, 54.0, 44.8, 43.6, 34.9, 34.6, 31.1;
Anal. Calcd for C21H25N5O4S: C, 56.87; H, 5.68; N, 15.79; S, 7.23. Found: C, 56.91; H, 5.73; N, 15.82; S, 7.26.
…………………….
http://www.google.com/patents/WO2010132110A1?cl=en
((lS,2S,4R)-4-{4-[(lS)-2,3-dihydro-lH-inden-l-ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl }-2-hydroxycyclopentyl)methyl sulfamate (//) is described in Intl. App. Pub. No. WO 07/092213, U.S. App. Pub. No. 2007/0191293, and U.S. App. Pub. No. 2009/0036678. The potassium salt of ((lS,2S,4R)-4-{4-[( 1 S)-2,3-dihydro- 1 H-inden- 1 -ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl } -2-hydroxycyclopentyl)methyl sulfamate is disclosed in Intl. App. Pub. No. WO 07/092213 and U.S. App. Pub. No. 2007/0191293.

(H)
((lS,2S,4R)-4-{4-[(lS)-2,3-dihydro-lH- inden-l-ylamino]-7H-pyπOlo[2,3-d]pyrimidin-7-yl}-2-hydroxycyclopentyl)methyl sulfamate (/):

Step 3: Synthesis of ((lS,2S.4R)-4-(4-r(lS)-2,3-dihydro-lH-inden-l-ylaminol-7H-pyrrolor2.3-dlpyrimidin-7-yl}-2-hvdroxycvclopentyl)methyl sulfamate hydrochloride Form 1
[0158] A reactor was charged with ((lS,2S,4R)-4-{4-[(lS)-2,3-dihydro-lH-inden-l-ylarnino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl }-2-hydroxycyclopentyl)methyl sulfamate (13.4 Kg, 30.2 mol) and 200-proof ethanol (106.2 Kg). The mixture was heated to reflux to afford a clear solution. The mixture was cooled to 50 ± 5 0C and passed through a cartridge filter. 200 proof ethanol (8.9 Kg) was used to rinse the filter. 1.27M hydrogen chloride in ethanol (10.2 Kg) was added via a cartridge filter at a rate to maintain a temperature of 50 ± 5 0C. The mixture was then seeded with Form 1 (67 g). Further 1.27M HCl (10.2 Kg) was added via a cartridge filter at a rate to maintain a temperature of 50 ± 5 0C. The mixture was then stirred at 50 ± 5 0C for about 3 hours. The mixture was then cooled to 20 ± 5 0C over about 3 hours and then stirred for about 2.5 hours. The solid product was then isolated by filtration and washed with 200-proof ethanol (I x 20.4 Kg and 1 x 21.2 Kg). The solids were dried by aspiration on the filter until no supernatant was seen to be collected, and then further dried under reduced pressure at <30 0C to afford the title compound (12.2 Kg) as a white solid determined to be Form 1 by XRPD. IH NMR (300MHz, DMSO, δ): 9.83 (s, IH), 8.34 (s, IH), 7.62 (s, IH), 7.44 (s, 2H), 7.30 (m, 3H), 7.22 (t, IH), 7.07 (s, IH), 5.86 (dd, IH), 5.42 (m, IH), 4.32 (m, IH), 4.21 (dd, IH), 4.02 (dd, IH), 3.04 (m, IH), 2.88 (m, IH), 2.67 (m, 2H), 2.15 (m, 2H), 2.08 (m, 2H), 1.94 (m, IH). XRPD data for Form 1 is shown in FIGURE 1 and Table 1; DSC data is shown in FIGURE 2, and TGA data for Form 1 is shown in FIGURE 3.
…………..
http://www.google.com/patents/WO2007092213A2?cl=en
Example 70: Diastereoisomeric mixture of (lS/2R/4R)-4-{4-[(lS)-2/3-dihydro-lH-inden-l- ylaimnol-ZH-pyrrolop^-dlpyxirnidin-Z-ylJ^-hydroxycyclopentyl s ulf amate and (lRf2S/4S)-4-{4-[(lS)-2,3-dihydro-lH-inden-l-ylaminol-7H-pyrrolo[2,3d]- pyrimidin-7-yl}-2-hydroxycyclopentyl sulfamate (Compounds 1-77 and 1-78)

Step a: Cyclopent-3-en-l-yl methanesulfonate
[0335] 3-Cydopentene-l-ol (0.500 g, 5.94 mmol) was stirred in DCM (95 mL).
Pyridine (2.40 mL), N,N-dimethylaminopyridine (0.10 g, 1.00 mmol) and methanesulfonyl chloride (0.690 mL, 8.92 mmol) were added, and the reaction mixture was stirred at 350C for 4 h. N,N-Dimethylarrιinopyridirιe (0.14 g, 1.2 mmol) and methanesulfonyl chloride (0.69 mL, 8.92 mmol) were added, and the reaction was stirred overnight. TLC indicated complete conversion. The reaction mixture was cooled and concentrated. The residue was purified by silica gel chromatography, eluting with DCM, to afford the title compound as a clear oil (0.660 g, 68%).
Step b: 7-Cyclopent-3-en-l-yl-N-r(lSV2,3-dihydro-lH-inden-l-yn-7H-pyrrolor2,3-rfl- pyrmτidin-4-arnine
[0336] N-[(lS)-2,3-DihydrcHlH-mden-l-yl]-7H-pyrrolo[2/3-d]p3αimidin-4-amine (1.32 g, 5.29 mmol) was azeotroped with toluene and placed under high vacuum for 30 min. N,N-Dimethylformamide (17.7 mL) was added, followed by cesium carbonate (1.99 g, 6.10 mmol). The mixture was stirred at 700C for 10 min. Cyclopent-3-en-l-yl methanesulfonate (0.660 g, 4.07 mmol) in N,N-dimethylformarnide (12.6 mL) was added dropwise. The reaction mixture was heated to 1100C for 1 h. The reaction mixture was cooled, quenched with brine and diluted with H2O. The aqueous layer was extracted with EtOAc (3x), washed with H2O and brine, dried (Na2SO4), filtered, and concentrated. The residue -was purified by via silica gel chromatography, eluting with a gradient of 0 to 5% MeOH in DCM followed by 25 to 50% EtOAc in hexanes, to afford the title compound as a pale brown solid (0.684 g, 53%). LC/MS: R1 = 1.38 min, ES+ 317 (FA standard). Step c: (lR,2S,45)-4-{4-r(lS)-2,3-dihydro-lH-inden-l-ylaininol-7H-pyrrolof2.3- rf1pyrimidin-7-yl}cyclopentane-l,2-diol
[0337] 7-Cyclopent-3-en-l-yl-N-[(lS)-2^-dihyrdo-lH-inden-l-yl]-7H-pyrrolo[2,3- d]pyτimidin-4-amine (0.312 g, 0.986 mmol) was stirred in tert-butyl alcohol (4.9 mL) and H2O (4.9 mL). AD-mix-α (Sigma- Aldrich, 1.4 g) was added, and the suspension was stirred at rt overnight. TLC indicated complete conversion. The reaction was quenched with sodium sulfite (1.48 g, 11.7 mmol), and the mixture was stirred for 5 h. The reaction mixture was diluted with EtOAc and H2O, and the aqueous layer was extracted with EtOAc (2x). The organic layer was dried (Na2SO4), filtered, and concentrated. The residue was purified via silica gel chromatography, eluting with EtOAc, to afford the title compound as a white solid (0.190 g, 55%).
Step d: Diastereoisomeric mixture of (lS,2R,4R)-4-{4-r(15)-23-dihydro-lH-inden-l- ylarninoi^jH-pyrrolofΣ^dlpyrirnidin-y-yll-l-hydroxycyclopentyl sulfamate and (lR,2S,4S)-4-{4-iαSV2,3-dihydro-lH-inden-l-ylarninol-7H-pyrrolor2,3- rf1pyrimidm-7-yl)-2-hydroxycyclopenryl sulfamate (Compounds 1-77 and 1-78)
[0338] (lR,2S,4S)-4-{4-[(lS)-2,3-Dihydro-lH-inden-l-ylarnino]-7H-pyrrolo[2/3- d]pyrimidin-7-ylJcyclopentane-l,2-diol (0.080 g, 0.23 mmol) was azeotroped with toluene and then was dissolved in anhydrous acetonitrile (2.3 mL). Pyridine (0.0369 mL, 0.458 mmol) was added. The reaction mixture was cooled to 00C, and a 2N solution of chlorosulfonamide in acetonitrile (0.144 mL) was added dropwise. The reaction was stirred for 1 h, and then additional 2N chlorosulfonamide in acetonitrile (0.028 mL) was added. After 30 min, additional 2N chlorosulfonamide in acetonitrile (0.0342 mL) was added, and the reaction mixture was stirred for 2 h. The reaction was quenched with methanol, and the mixture was concentrated in vacuo. The residue was purified by preparative thin layer chromatography using DCM:AcCN:MeOH (50:45:5). The relevant band was cut, washed with acetone, filtered, and concentrated to give a mixture of diastereomers as a white solid. (11 mg, 11%). 1H NMR (CDCl3, 400 NMR, δ): 8.36-8.27 (m, IH); 7.38-7.09 (m, 5H); 6.90-6.80 (m, IH); 6.36- 6.20 (m, IH); 5.95-5.76 (m, IH); 5.51-5.22 (m, 2H); 4.83-4.68 (m, IH); 3.87-3.72 (m, IH); 3.12- 2.83 (m, 2H); 2.75-2.53 (m, IH); 2.50-2.14 (m, 2H); 2.08-1.79 (m, 2H) ppm. LC/MS: R, = 1.16 min, ES+ 430 (FA standard).
…………
WO 2012061551
http://www.google.im/patents/WO2012061551A1?cl=en
The compound ((lS,2S,4R)-4-(4-((lS)-2,3-dihydro-lH-inden-l-ylamino)-7H-pyrrolo[2,3-d]- pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl sulfamate:

also known as MLN4924, is an inhibitor of NEDD8-activating enzyme (NAE). Inhibition of NAE has been shown to induce cancer cell death and inhibit the growth of tumors in xenograft models. See, e.g., T.A. Soucy et al., Nature, 2009, 458, 732-737; T.A. Soucy ei al., Clin. Cancer Res., 2009, 15 (12), 3912-3916; and J.E. Brownell et al., Mol. Cell., 2010, 37 (1), 102-111, each of which is hereby incorporated by reference herein in its entirety. MLN4924, pharmaceutical compositions of MLN4924, processes for its synthesis, and polymorphic forms have been described previously. See, e.g., US Patent Appl. Nos. 11/700,614 (Publ. No. 2007/0191293), 12/221,399 (Publ. No. 2009/0036678) and 12/779,331 (Publ. No. 2011/0021544),
……………

A practical synthesis of a novel NEDD8-activating enzyme (NAE) inhibitor pevonedistat (MLN4924) is described. Key steps include an enantioselective synthesis of an amino-diol cyclopentane intermediate containing three chiral centers and a novel, regioselective sulfamoylation using N-(tert-butoxycarbonyl)-N-[(triethylenediammonium)sulfonyl]azanide. The linear process, involving six solid isolations, has been carried out in multiple cGMP productions on 15–30 kg scale to produce pevonedistat in 98% (a/a) chemical purity and 25% overall yield.

((1S,2S,4R)-4-(4-(((S)-2,3-Dihydro-1H-inden-1-yl)amino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl Sulfamate (1)
((1S,2S,4R)-4-(4-(((S)-2,3-Dihydro-1H-inden-1-yl)amino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl Sulfamate·Hydrochloride (Pevonedistat)

MLN4924 (1), which is in clinical trials as an anticancer agent, was stereoselectively synthesized from d-ribose via a route involving stereoselective reduction, regioselective cleavage of an isopropylidene moiety, and selective displacement of a cyclic sulfate moiety as key steps.
Sulfamic Acid 2-Hydroxy-4-[4-(indan-1-ylamino)pyrrolo[2,3-d]pyrimidin-7-yl]cyclopentylmethyl Ester (1) BASE



| WO2012061551A1 * | Nov 3, 2011 | May 10, 2012 | Millennium Pharmaceuticals, Inc. | Administration of nedd8-activating enzyme inhibitor |
| WO2013028832A2 * | Aug 23, 2012 | Feb 28, 2013 | Millennium Pharmaceuticals, Inc. | Inhibitors of nedd8-activating enzyme |
| WO2013028832A3 * | Aug 23, 2012 | May 2, 2013 | Millennium Pharmaceuticals, Inc. | Inhibitors of nedd8-activating enzyme |
| US8809356 | Aug 23, 2012 | Aug 19, 2014 | Millennium Pharmaceuticals, Inc. | Inhibitors of NEDD8-activating enzyme |
| Reference | ||
|---|---|---|
| 1 | * | Lee, et. al., Journal of Organic Chemistry (2011), 76(9), 3557-3561. |
1H NMR PREDICT

13 C NMR


//////////Pevonedistat, MLN4924, Millennium Pharmaceuticals, TAKEDA, TAK-924 , PHASE 1, orphan drug designation
ASLAN Pharmaceuticals Gains Orphan Designation for Rare Cancer Drug ASLAN001 (varlitinib)

(R)-N4-[3-Chloro-4-(thiazol-2-ylmethoxy)-phenyl]-N6-(4-methyl-4,5-dihydro-oxazol-2-yl)-quinazoline-4,6-diamine

ASLAN001 , Varlitinib
C22H19ClN6O2S
Molecular Weight: 466.94
Elemental Analysis: C, 56.59; H, 4.10; Cl, 7.59; N, 18.00; O, 6.85; S, 6.87
CAS: 845272-21-1 (Varlitinib); 1146629-86-8 (Varlitinib tosylate).
ASLAN001; ASLAN-001; ASLAN 001; AR 00334543; ARRY-334543; ARRY334543; ARRY-543; ARRY543; ARRY 543.
(R)-N4-(3-chloro-4-(thiazol-2-ylmethoxy)phenyl)-N6-(4-methyl-4,5-dihydrooxazol-2-yl)quinazoline-4,6-diamine.
(R)-4-[[3-Chloro-4-[(thiazol-2-yl)methoxy]phenyl]amino]-6-[(4-methyl-4,5-dihydrooxazol-2-yl)amino]quinazoline
4,6-Quinazolinediamine, N4-[3-chloro-4-(2-thiazolylmethoxy)phenyl]-N6-[(4R)-4,5-dihydro-4-methyl-2-oxazolyl]-
ASLAN Pharmaceuticals, a Singapore-based drugmaker, announced The Food and Drug Administration (FDA) gave an orphan drug designation on August 13 to its pan-HER inhibitor ASLAN001 (varlitinib), a drug candidate created to treat a destructive form of bile duct cancer called cholangiocarcinoma that has no known cure. ………http://www.dddmag.com/news/2015/08/aslan-pharmaceuticals-gains-orphan-designation-rare-cancer-drug
Current developer: Array Biopharma Inc,
![]()
Varlitinib, also known as ARRY-543 and ASLAN001, is an orally bioavailable inhibitor of the epidermal growth factor receptor family with potential antineoplastic activity.
Varlitinib (ASLAN-001) is an oncolytic drug in phase II clinical trials at ASLAN Pharmaceuticals for the treatment of gastric cancer and for the treatment of metastatic breast cancer in combination with capecitabine. Clinical development is also ongoing for the treatment of solid tumors in combination with cisplatin/FU and cisplatin/capecitabine. The product had been in phase I/II clinical trials at Array BioPharma for the treatment of patients with advanced pancreatic cancer. Phase II clinical trials had also been ongoing for the treatment of solid tumors. No recent development has been reported for this research
Varlitinib selectively and reversibly binds to both EGFR (ErbB-1) and Her-2/neu (ErbB-2) and prevents their phosphorylation and activation, which may result in inhibition of the associated signal transduction pathways, inhibition of cellular proliferation and cell death. EGFR and Her-2 play important roles in cell proliferation and differentiation and are upregulated in various human tumor cell types. Due to the dual inhibition of both EGFR and Her-2, this agent may be therapeutically more effective than agents that inhibit EGFR or Her-2 alone.
The drug is a dual inhibitor of the ErB-2 and EGFR receptor kinases, both of which have been shown to stimulate aberrant growth, prolong survival and promote differentiation of many tumor types. The compound behaves as a reversible ATP-competitive inhibitor with nanomolar potency both in vitro and in cell-based proliferation assays.
In 2011, the compound was licensed to Aslan Pharmaceuticals by Array BioPharma worldwide for the treatment of solid tumors, initially targeting patients with gastric cancer through a development program conducted in Asia.
In 2015, orphan drug designation was assigned to the compound in the U.S. for the treatment of cholangiocarcinoma.
SEE NMR ………….http://www.medkoo.com/Product-Data/Varlitinib/Varlitinib-QC-KB20121128web.pdf
……………..
https://www.google.co.in/patents/US20050043334
Example 52
(R)-N4-[3-Chloro-4-(thiazol-2-ylmethoxy)-phenyl]-N6-(4-methyl-4,5-dihydro-oxazol-2-yl)-quinazoline-4,6-diamine
Prepared using (R)-2-aminopropan-1-o1. MS APCI (+) m/z 467, 469 (M+1, Cl pattern) detected; 1H NMR (400 mHz, DMSO-D6) δ 9.53 (s, 1H), 8.47 (s, 1H), 8.09 (s, 1H), 7.86 (d, 1H), 7.81 (d, 1H), 7.77 (d, 1H), 7.69 (m, 3H), 7.32 (d, 1H), 7.02 (s, 1H), 5.54 (s, 2H), 4.47 (m, 1H), 3.99 (m, 1H), 3.90 (m, 1H), 1.18 (d, 3H).
Example 53

(S)-N4-[3-Chloro-4-(thiazol-2-ylmethoxy)-phenyl]-N6-(4-methyl-4,5-dihydro-oxazol-2-yl)-quinazoline-4,6-diamine
Prepared using (S)-2-amino-propan-1-o1. MS APCI (+) m/z 467, 469 (M+1, Cl pattern) detected; 1H NMR (400 mHz, DMSO-D6) δ 9.53 (s, 1H), 8.47 (s, 1H), 8.09 (s, 1H), 7.86 (d, 1H), 7.81 (d, 1H), 7.77 (d, 1H), 7.69 (m, 3H), 7.32 (d, 1H), 7.02 (s, 1H), 5.54 (s, 2H), 4.47 (m, 1H), 3.99 (m, 1H), 3.90 (m, 1H), 1.18 (d, 3H).
………………
PATENT
http://www.google.co.in/patents/WO2005016346A1?cl=en
Example 52

R VN4-r3-Chloro-4-(‘thiazol-2-v-metho-xy)-phenyll-N6-(4-methyl-4,5-dihvdro-oxazol- 2-yl)-quinazoUne-4,6-diamine
[00194] Prepared using (R)-2-aminopropan- 1 -ol. MS APCI (+) m/z 467, 469
(M+l, CI pattern) detected; 1H NMR (400 mHz, DMSO-D6) δ 9.53 (s, IH), 8.47 (s, IH), 8.09 (s, IH), 7.86 (d, IH), 7.81 (d, IH), 7.77 (d, IH), 7.69 (m, 3H), 7.32 (d, IH), 7.02 (s, IH), 5.54 (s, 2H), 4.47 (m, IH), 3.99 (m, IH), 3.90 (m, IH), 1.18 (d, 3H). Example 53

(S)-N4-|“3-Chloro-4- thiazol-2-ylmethoxy)-phenyll-N6-(‘4-methyl-4,5-dihvdro-oxazol- 2-yl)-quinazoline-4,6-diamine [00195] Prepared using (S)-2-amino-propan- 1 -ol. MS APCI (+) m z 467, 469
(M+l, CI pattern) detected; 1H NMR (400 mHz, DMSO-D6) δ 9.53 (s, IH), 8.47 (s, IH), 8.09 (s, IH), 7.86 (d, IH), 7.81 (d, IH), 7.77 (d, IH), 7.69 (m, 3H), 7.32 (d, IH), 7.02 (s, IH), 5.54 (s, 2H), 4.47 (m, IH), 3.99 (m, IH), 3.90 (m, IH), 1.18 (d, 3H).
………
CAUTION a very similar molecule but not same
 NOTE……..METHYL NEXT TO OXYGEN ATOM
NOTE……..METHYL NEXT TO OXYGEN ATOM
Design, Synthesis and Bioactivities Evaluation of Novel Quinazoline Analogs Containing Oxazole Units
DOI: 10.1002/cjoc.201400271,…………http://onlinelibrary.wiley.com/doi/10.1002/cjoc.201400271/abstract;jsessionid=04211D7526D97240F44E4355C6C57F50.f01t01
CAUTION …THIS IS NOT SAME
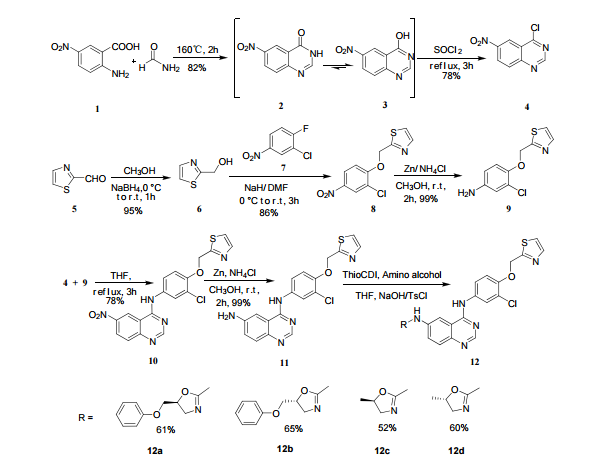
A novel type of quinazoline derivatives, which were designed by the combination of quinazoline as the backbone and oxazole scaffold as the substituent, have been synthesized and their biological activities were evaluated for anti-proliferative activities and EGFR inhibitory potency. Compound 12b demonstrated the most potent inhibitory activity (IC50=0.95 µmol/L for EGFR), which could be optimized as a potential EGFR inhibitor in the further study. The structures of the synthesized quinazoline analogs and all intermediates were comfirmed by 1H and 13C NMR, 2D NMR spectra, IR spectra and MS spectra.
12c: Employing the same method as above, compound 12c was prepared and the amino alcohol was (S)-2-amino-propan-1-ol. Yellow solid, yield 52 %. m.p. 243-244 °C; [α] 20D =﹢22.5 ° (c 1.0, CH3CN); 1 H NMR (DMSO-D6): δ 9.54 (s, 1 H), 8.46 (s, 1 H), 8.06 (s, 2 H), 7.85 (d, 2 H, J=3.3 Hz), 7.79 (d, 2 H, J=3.3 Hz), 7.75 (d, 1 H, J=8.9 Hz), 7.64 (d, 1 H, J=8.3 Hz), 7.30 (d, 1 H, J=9.0 Hz), 5.54 (s, 2 H), 4.76 (m, 1 H), 3.72 (s, 1 H), 3.19 (s, 1 H), 1.34 (d, 3 H, J=6.15 Hz). 13C NMR (DMSO-D6) δ: 165.8, 156.9, 152.0, 148.8, 145.3, 142.6, 134.3, 128.7, 128.0, 123.5, 121.7, 121.3, 121.0, 115.6, 114.6, 72.5, 67.7, 63.0, 29.8, 29.0, 20.0, 13.9. IR (KBr) ν: 3439, 3278, 3101, 2925, 1660, 1631, 1601, 1557, 1500, 1428, 1404, 1384, 1329, 1291, 1257, 1225, 1052 cm-1. Anal. calcd for C22H19N6O2SCl: C 55.59, H 4.10, N 18.00, O 6.85; found C 55.55, H 4.13, N 18.02, O 6.78; MS (ESI) m/z: 467.2 (M+H).
12d: Employing the same method as above, compound 12d was prepared and the amino alcohol was (R)-2-amino-propan-1-ol. Yellow solid, yield 60%. m.p. 242-243 °C; [α] 20D = ﹣22.3 ° (c 1.0, CH3CN); 1 H NMR (DMSO-D6): δ 9.52 (s, 1 H), 8.80 (s, 1 H), 8.52 (dd, 1 H, J=2.7 Hz, J=8.9 Hz), 8.45 (s, 1 H), 8.30 (s, 1 H), 8.07 (s, 1 H), 7.85 (d, 1 H, J=3.2 Hz), 7.79 (d, 1 H, J=3.2 Hz), 7.75 (s, 1 H), 7.63 (d, 1 H, J=8.2 Hz), 7.31 (d, 1 H, J=9.0 Hz), 5.53 (s, 2 H), 4.76 (m, 1 H), 3.81 (s, 1 H), 3.19 (s, 1 H), 1.34 (d, 3 H, J=6.2 Hz). 13C NMR (DMSO-D6) δ: 165.8, 156.9, 152.0, 148.8, 145.3, 142.6, 134.3, 128.7, 128.0, 123.5, 121.7, 121.3, 121.0, 115.6, 114.6, 72.5, 67.7, 63.0, 29.8, 29.0, 20.0, 13.9. IR (KBr) ν: 3439, 3278, 3101, 2925, 1660, 1631, 1601, 1557, 1500, 1428, 1404, 1384, 1329, 1291, 1257, 1225, 1052 cm-1. Anal. calcd for C22H19N6O2SCl: C 55.59, H 4.10, N 18.00, O 6.85; found C 55.55, H 4.13, N 18.02, O 6.78; MS (ESI) m/z: 467.20 (M+H).
The above paper allows you to synthesize the key amino int 11 ………N4-(3-chloro-4-(thiazol-2-ylmethoxy)phenyl)quinazoline-4,6-diamine (11)
this can be applied to varlitinib till int 11
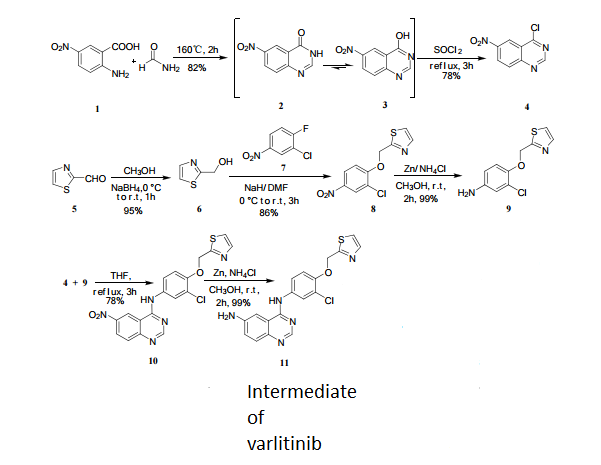
6-Nitro-4-hydroxyquinazoline (3)
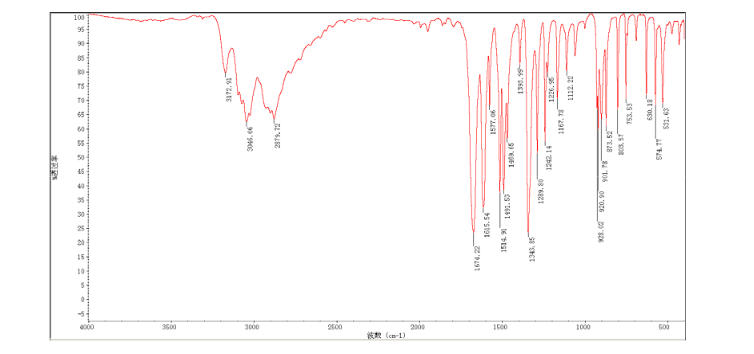
2-amino-5-nitrobenzoic acid (5.46 g, 30 mmol) was added to a 250 mL flask equipped with a reflux condenser. Then 50 mL formamide was added. The mixture was heated with vigorous stirring at 160 °C for 3 h. After cooling the solution was poured in ice-water to give 3 in almost pure form (Yellow solid 4.70 g, yield 82.0%). m.p. 317-318 °C; 1 H NMR (DMSO-d6): δ 12.74 (1 H, s, OH, exchangeable), 8.78 (1 H, d, J=2.4 Hz), 8.53 (1 H, dd, J=2.6 Hz, 9.0 Hz), 8.30 (s, 1 H), 7.84 (1 H, d, J=9.0 Hz); 13C NMR (DMSO-d6) δ: 160.1, 152.9, 148.9, 145.0, 129.1, 128.3, 122.7, 121.9. IR (KBr) ν: 3172, 3046, 2879, 1674, 1615, 1577, 1514, 1491, 1469, 1343, 1289, 1242, 1167, 1112, 928, 920, 901, 803, 753, 630, 574, 531 cm-1. Anal. calcd for C8H5N3O3: C 50.27, H 2.64, N 21.98; found C 50.30, H 2.65, N 21.96; MS (ESI) m/z: 189.97 (M-H).
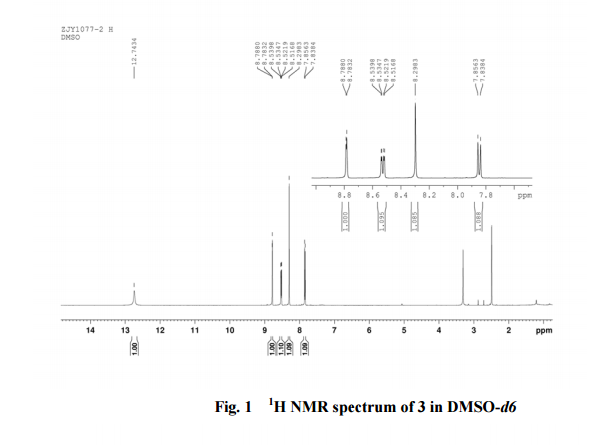
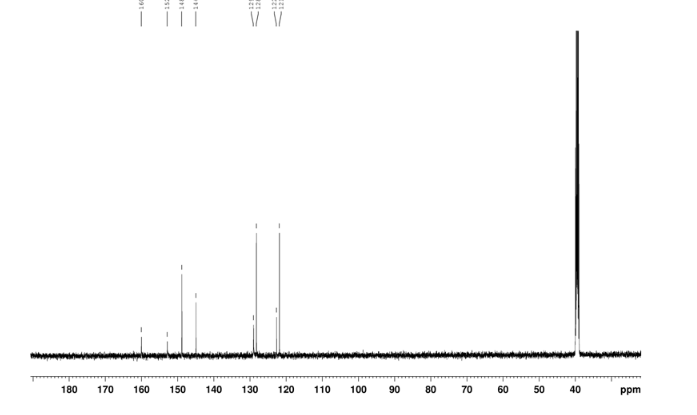 13C NMR OF 3 IN DMSOD6
13C NMR OF 3 IN DMSOD6
IR
4-chloro-6-Nitroquinazoline (4)
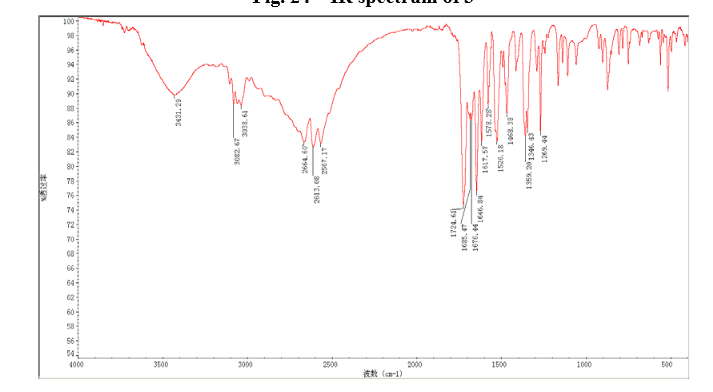
In a 100 mL flask equipped with a reflux condenser, 6-nitroquinazolin-4-one (2.86 g, 15 mmol) and thionyl chloride (SOCl2) 25 mL were added. The mixture was heated under reflux with vigorous stirring for 2 h. After the solution was clear, the reaction mixture was heated for another 2 h. Then, 150 mL of ice MeOH was dropped into it carefully, the mixture was extracted with CH2Cl2. The organic layer was S3 dried under MgSO4, filtered and the solvent removed to give 4-chloro-6-nitroquinazoline (4). Yellow solid 2.45 g, yield 78%. m.p. 134-135 °C; 1 H NMR (DMSO-d6): δ 8.80 (1 H, d, J=3.0 Hz), 8.54(1 H, dd, J=2.7 Hz, 9.0 Hz), 8.35(s, 1 H), 7.87 (1 H, d, J= 9.0 Hz); 13C NMR (DMSO-d6) δ: 160.0, 152.5, 149.1, 145.1, 128.7, 128.4, 122.7, 122.0. IR (KBr) ν: 3431, 3082, 3038, 2664, 2613, 2567, 1724, 1685, 1676, 1646, 1617, 1578, 1526, 1468, 1359, 1346, 1269 cm-1. Anal. calcd for C8H4N3O2Cl: C 45.84, H 1.92, N 20.05, O 15.27; found C 45.81, H 1.97, N 20.02, O 15.21; MS (ESI) m/z: 207.96 (M-H).
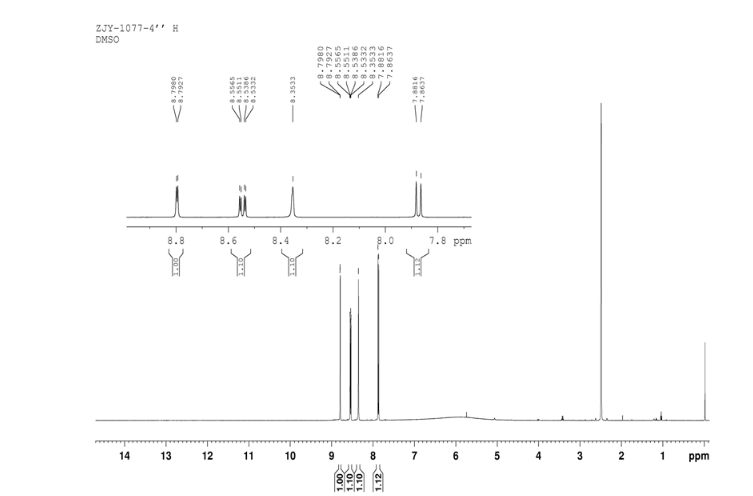 4 nmr dmsod6
4 nmr dmsod6
13C NMR OF4 IN DMSOD6
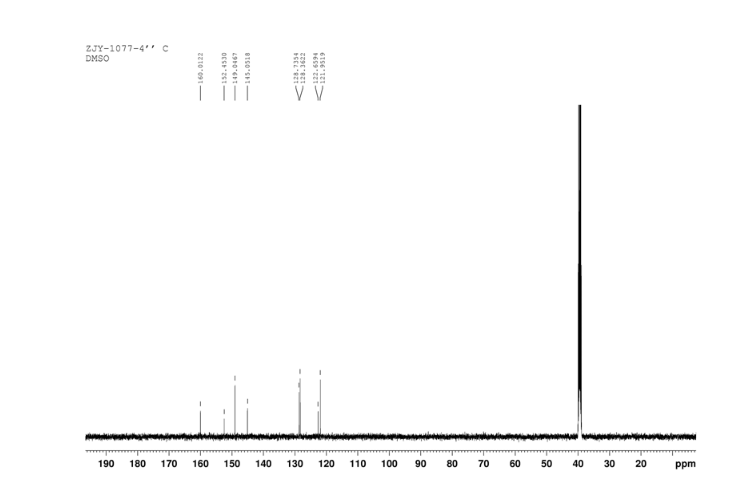
IR
Thiazol-2-yl-methano1 (6)
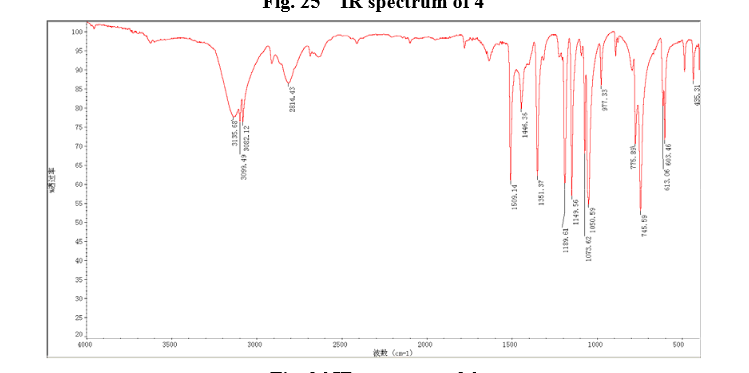
Sodium borohydride (16.0 g, 140 mmol) was added to a stirred solution of thiazole-2-carbaldehyde (24.2 g, 214 mmol) in MeOH (400 mL) at 0 °C . The reaction mixture was warmed to room temperature. After 1 hour, the reaction mixture was quenched by the addition of water and the organics were removed by concentration. The resulting aqueous mixture was extracted with EtOAc. The combined organic extracts were dried under Na2SO4 and concentrated to give thiazol-2-yl-methano1 (23.39 g, 95%). bp:75-76 °C (0.2 mmHg) [lit.[19] bp:70-80 °C (0.2 mmHg)]; m. p. 63-64 °C. 1 H NMR (CDCl3) δ 4.91 (s, 2 H), 5.1(br, l H), 7.28(d, 1 H, J=3.2 Hz), 7.68 (d, 1 H, J=2.9 Hz). IR (KBr) ν: 3135, 3099, 3082, 2814, 1509, 1446, 1351, 1189, 1149, 1073, 1050, 977, 775, 745, 613, 603 cm-1. Anal. calcd for C4H5NOS: C 41.72, H 4.38, N 12.16; found C 41.74, H 4.33, N 12.18; MS (ESI) m/z: 116.11 (M+H).
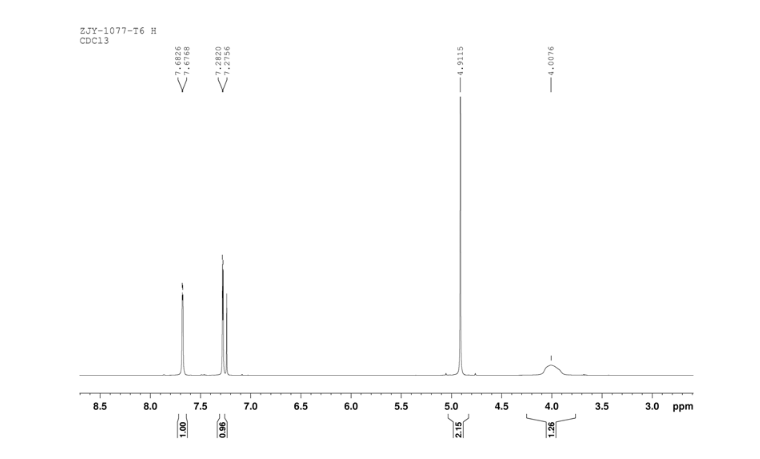 6 in dmsod6 1H NMR
6 in dmsod6 1H NMR
2-((2-Chloro-4-nitrophenoxy)methyl)thiazole (8)
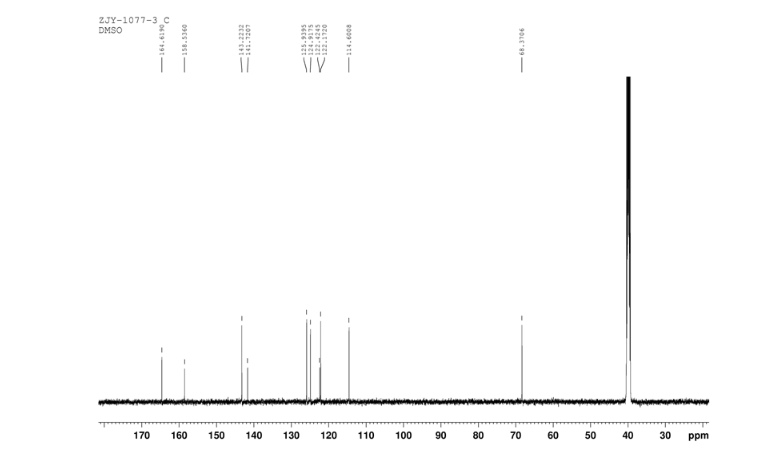
2-(2-chloro-4-nitro-phenoxymethy1)-thiazole was prepared by adding thiazol-2-yl-methanol (5.48 g, 47.65 mmol) to a slurry of sodium hydride (2.42 g of a 60% dispersion in oil, 60.5 mmol) in THF (50 ml) at 0 °C After several minutes, 2-chloro-1-fluoro- 4-nitro-benzene (7.58 g, 43.60 mmol) was added and the reaction mixture warmed to room temperature. The reaction mixture was stirred at room temperature for 3 h, and 60 °C for 16 h. After cooling to room temperature, the reaction mixture was poured into 300 mL water. The resulting precipitate was collected by filtration, washed with water, and dried in vacuo to give 2-(2- chloro-4-nitrophenoxymethy1)-thiazole (11.06 g, 86%) which was used in next step without further purification. m.p. 170-171 °C; 1 H NMR (DMSO-d6): δ 8.35 (1 H, d, J=2.8 Hz), 8.25 (1 H, dd, J=2.8 Hz, 9.15 Hz), 7.87 (1 H, d, J=3.3 Hz), 7.83(1 H, d, J=3.3 Hz), 7.54 (1 H, d, J=9.2 Hz), 5.73(s, 1 H); 13C NMR (DMSO-d6) δ: 164.2, 158.5, 143.2, 141.7, 125.9, 124.9, 122.4, 122.2, 114.6, 68.4; IR (KBr) ν: 3112, 3009, 1587, 1509, 1500, 1354, 1319, 1284, 1255, 1154, 1125, 1054, 1006, 894, 780, 746, 728 cm-1. Anal. calcd for C10H7N2O3SCl: C 44.37, H 2.61, N 10.35, O 17.73; found C 44.31, H 2.67, N 10.29; MS (ESI) m/z: 268.89 (M-H).
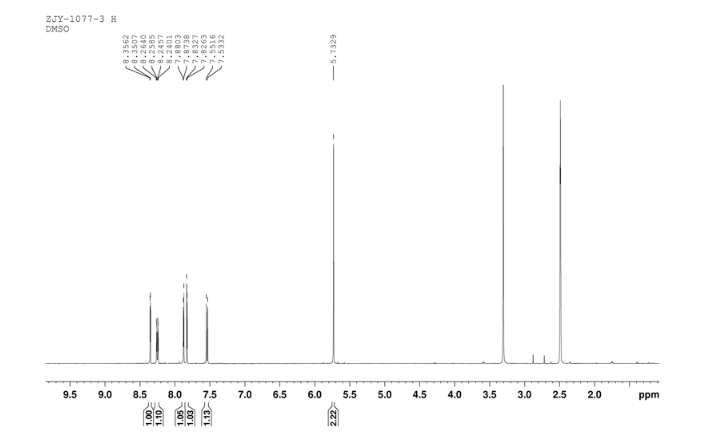 1H NMR 8 DMSOD6
1H NMR 8 DMSOD6
13C NMR OF 8 IN DMSOD6
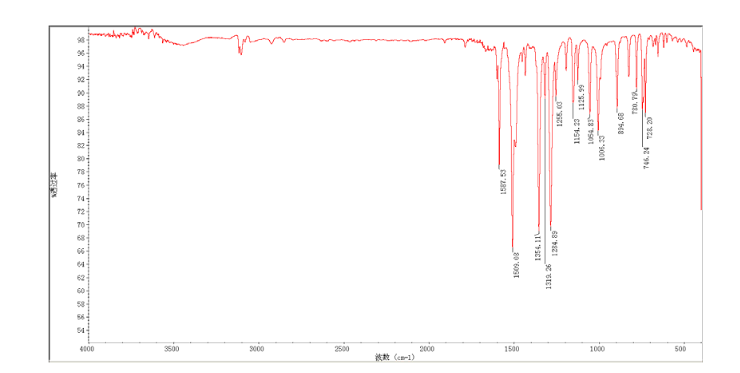
3-Chloro-4-(thiazol-2-ylmethoxy)aniline (9)
In a flask equipped with a reflux condenser, the compound 8 15.00 g (55.6 mmol), reduced zinc powder 14.44 g (222.0 mmo1, 4 eq), saturated ammonia chloride (5 mL) and methanol (100 mL) were mixed. The mixture was stirred at a temperature of 40 °C for 1.5 h. Then the zinc powder was filtered off, the filtrate was concentrated to obtain yellow solid 13.21 g, yield 99%. m.p. 60-61 °C; 1 H NMR (DMSO-d6): δ 7.80 (1 H, d, J=3.3 Hz), 7.75 (1 H, d, J=3.3 Hz), 6.96 (1 H, d, J=8.8 Hz), 6.64(1 H, d, J=2.7 Hz), 6.46 (1 H, dd, J=2.7 Hz, J=8.7 Hz), 5.30 (s, 2 H), 5.04 (s, 2 H, NH2, exchangeable); 13C NMR (DMSO-d6) δ: 166.8, 145.1, 144.1, 142.80, 123.1, 121.5, 117.7, 115.2, 113.6, 69.1. IR (KBr) ν: 3322, 3192, 3112, 1607, 1499, 1457, 1436, 1291, 1274, 1221, 1191, 1144, 1057, 1027, 857, 797, 767, 733, 584 cm-1. Anal. calcd for C10H9N2OSCl: C 49.90, H 3.77, N 11.64, O 6.65; found C 49.95, H 3.76, N 11.66, O 6.60; MS (ESI) m/z: 239.01 (M-H).
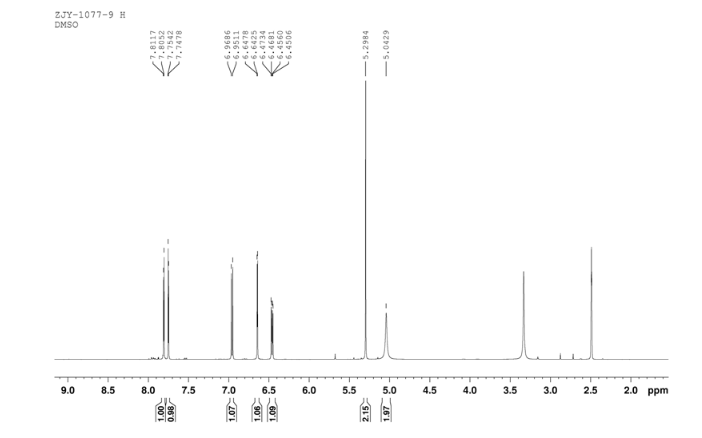 1H NMR DMSOD6 OF 9
1H NMR DMSOD6 OF 9
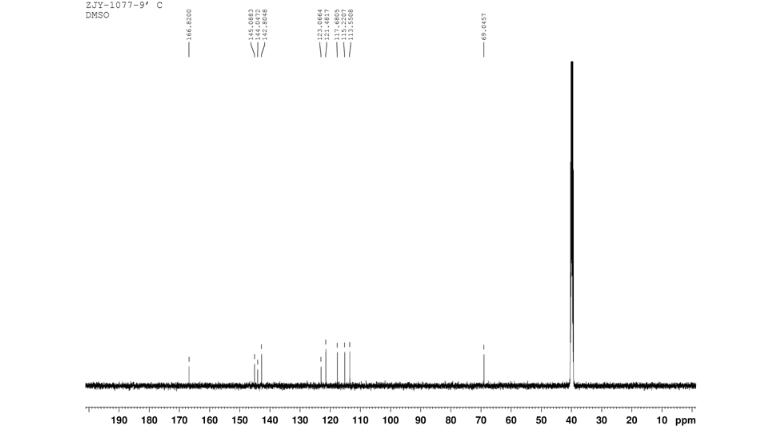 13C NMR OF 9 IN DMSOD6
13C NMR OF 9 IN DMSOD6
N-(3-chloro-4-(thiazol-2-ylmethoxy)phenyl)-6-nitro- quinazolin-4-amine(10)
In a flask equipped with a reflux condenser, 6-nitro-4-chloro- quinazoline 8.0 g (38.3 mmol) and 3-Chloro-4-(thiazol-2-ylmethoxy)aniline 8.9 g (37.0 mmol) were dissolved into 150 mL of THF, and the solution was refluxed for 3 h.Then a lot of yellow solid was deposited. Then it was filtered affording to yellow solid 12.8 g, yield 81%. m.p. 183-184 °C (decompose); 1 H NMR (DMSO-d6): δ 11.97(s, 1 H, exchangeable), 9.84 (s, 1 H), 9.00 (s, 1 H), 8.76 (1 H, d, J=9.1 Hz), 8.12-8.14 (m, 1 H), 7.94 (1 H, d, J=2.3 Hz), 7.87 (1 H, d, J=3.2 Hz), 7.81 (1 H, d, J=3.2 Hz), 7.44 (1 H, d, J=9.0 Hz), 7.69 (1 H, dd, J=2.5 Hz, J=8.9 Hz), 5.61 (s, 2 H); 13C NMR (DMSO-d6) δ: 166.8, 145.1, 144.1, 142.8, 123.1, 121.5, 117.7, 115.2, 113.7, 69.1. IR (KBr) ν: 3442, 3100, 1636, 1618, 1570, 1552, 1523, 1492, 1442, 1400, 1377, 1344, 1301, 1267, 1069, 805 cm-1. Anal. calcd for C18H12N5O3SCl: C 52.24, H 2.92, N 16.92, O 11.60; found C 52.26, H 2.93, N 16.96, O 11.58; MS (ESI) m/z: 412.84 (M-H).
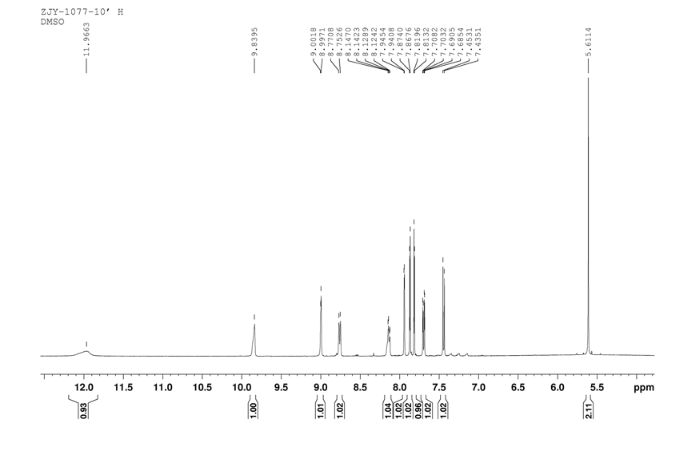 1H NMR DMSOD6 OF 10
1H NMR DMSOD6 OF 10
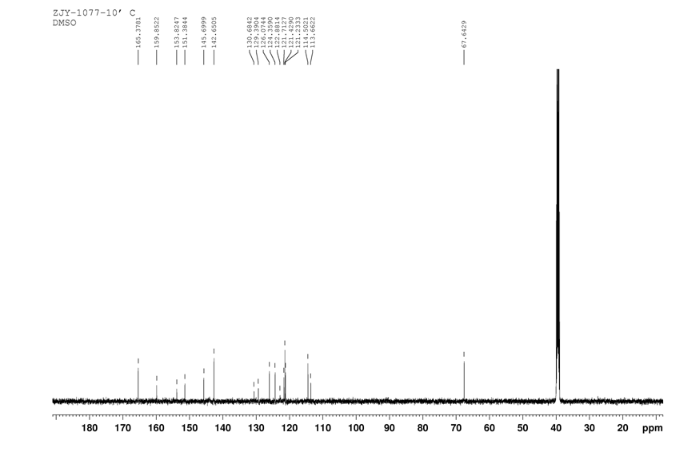 13C NMR OF 10 IN DMSOD6
13C NMR OF 10 IN DMSOD6
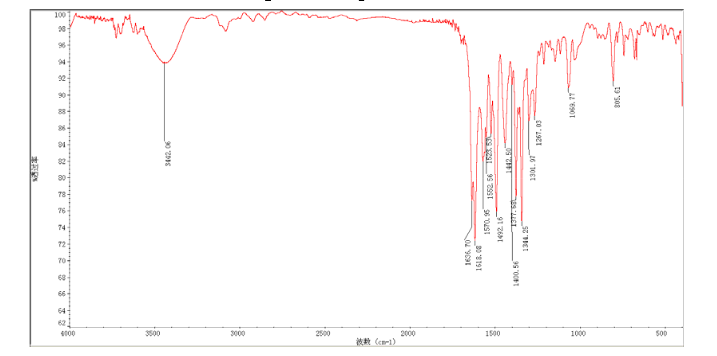
N4-(3-chloro-4-(thiazol-2-ylmethoxy)phenyl)quinazoline-4,6-diamine (11)
In a flask equipped with a reflux condenser, the compound 10 5.00 g (12.1 mmol), reduced zinc powder 3.2 g (48.5 mmo1, 4 eq), saturated ammonia chloride (3 mL) and methanol (60 mL) were mixed. The mixture was stirred at room temperature for 30 min. Then the zinc powder was filtered off, the filtrate was concentrated to obtain yellow solid 4.58 g, yield 98%. m.p. 197-198 °C (decompose); 1 H S4 NMR (DMSO-d6): δ 9.33(s, 1 H, exchangeable), 8.31 (s, 1 H), 8.05 (d, 1 H, J=2.6 Hz), 7.85 (d, 1 H, J=3.3 Hz), 7.79 (1 H, d, J=3.3 Hz), 7.73 (1 H, dd, J=2.5 Hz, J=9.0 Hz), 7.51 (1 H, d, J=8.9 Hz), 7.30 (1 H, d, J=2.4 Hz), 7.29 (1 H, d, J=4.7 Hz), 7.23 (1 H, dd, J=2.3 Hz, J=8.9 Hz), 5.57 (s, 2 H, exchangeable), 5.52 (s, 2 H); 13C NMR (DMSO-d6) δ: 165.9, 155.8, 149.7, 148.5, 147.3, 142.6, 142.5, 134.6, 128.7, 123.6, 123.2, 121.4, 121.3, 121.1, 116.5, 114. 7, 100.9, 67.8. IR (KBr) ν: 3443, 3358, 3211, 3100, 1631, 1596, 1577, 1560, 1530, 1494, 1431, 1383, 1217, 910 cm-1. Anal. calcd for C18H14N5OSCl: C 56.32, H 3.68, N 18.24, O 4.17; found C 56.34, H 3.70, N 18.22, O 4.14; MS (ESI) m/z: 382.66 (M-H).
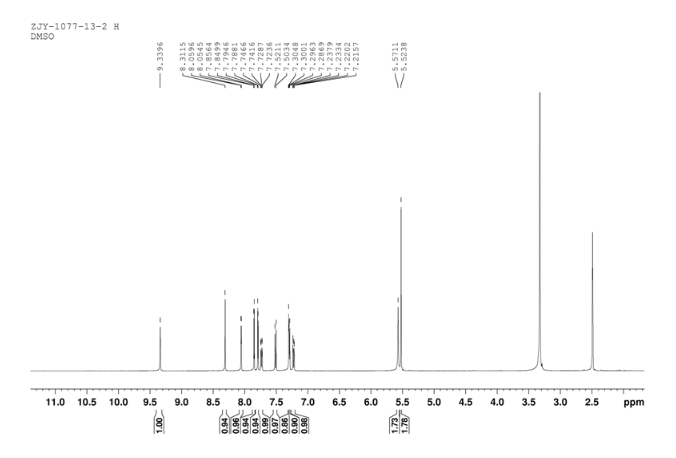 11 1HNMR DMSOD6
11 1HNMR DMSOD6
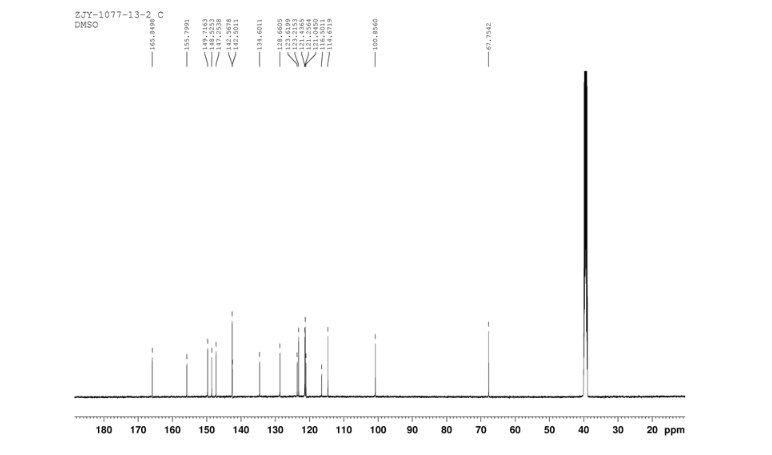 13C NMR OF 11 IN DMSOD6
13C NMR OF 11 IN DMSOD6
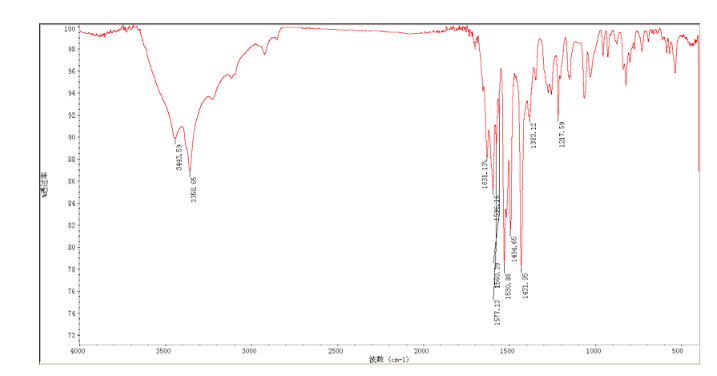
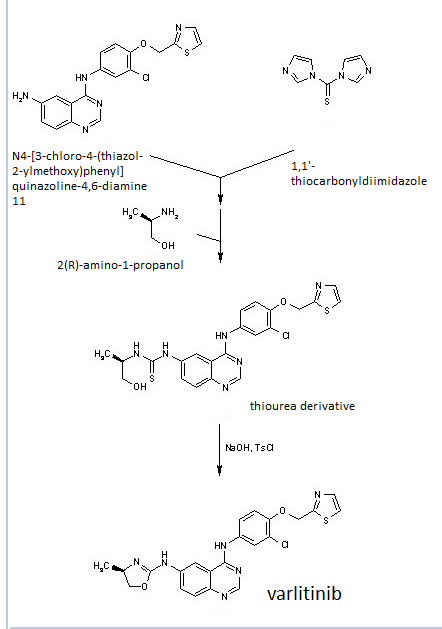
Construction finally as per patent ……….US20050043334
Treatment of N4-[3-chloro-4-(thiazol-2-ylmethoxy)phenyl]quinazoline-4,6-diamine (11) with 1,1′-thiocarbonyldiimidazole , followed by condensation with 2(R)-amino-1-propanol in THF/CH2Cl2 affords thiourea derivative , which finally undergoes cyclization in the presence of TsCl and NaOH in THF/H2O to furnish varlitinib .
-
ASLAN Pharmaceuticals
-
Address: 10 Bukit Pasoh Rd, Singapore 089824Phone:+65 6222 4235


 carl fith
carl fith
Mr Carl Firth, CEO, Aslan Pharmaceuticals, Singapore (left) and Mr Dan Devine, CEO, Patrys, Australia (right)
///////ASLAN001, varlitinib, ASLAN Pharmaceuticals, Orphan Designation, ARRY-534, ARRY-334543 , PHASE 2, ORPHAN DRUG DESIGNATION, array
AXITINIB


Axitinib (AG013736; trade name Inlyta) is a small molecule tyrosine kinase inhibitor developed by Pfizer. It has been shown to significantly inhibit growth of breast cancer in animal (xenograft) models[2] and has shown partial responses in clinical trials with renal cell carcinoma (RCC)[3] and several other tumour types.[4] It was approved by the U.S. Food and Drug Administration after showing a modest increase in progression-free survival,[5] though there have been reports of fatal adverse effects.[6]
Axitinib, a small-molecule indazole derivative chemically known as (E)-N-methyl-2-(3-(2-(pyridin-2-yl)-vinyl)-1H-indazol-6-ylthio)benzamide developed by Pfizer, was approved in January 2012 by the U.S. FDA with the trade name Inlyta. It selectively inhibits vascular endothelial growth factor receptors for the treatment of renal cell carcinoma
On January 27, 2012, axitinib was approved with the trade name INLYTA for treatment of patients in the United States with advanced renal cell carcinoma after failure of one prior systemic therapy.
It has received FDA (27 January 2012), EMA (13 September 2012), MHRA (3 September 2012) and TGA (26 July 2012) approval for use as a treatment for renal cell carcinoma.[11][12][13][14]
A study published in 2015[15] showed that axitinib effectively inhibits a mutated gene (BCR-ABL1[T315I]) that is common in chronic myeloid leukemias and adult acute lymphoblastic leukemias which have become resistant to other tyrosine kinase inhibitors likeimatinib. This is one of the first examples of a new indication for an existing drug being discovered by screening known drugs using a patient’s own cells.

The discovery and development of an efficient synthesis route to axinitib is reported. The first-generation route researched by Pfizer implemented two Pd-catalyzed coupling reactions as key steps. In this work, the development of Heck-type and C–S coupling reactions catalyzed by CuI is briefly described, using an economial and practical protocol. Aspects of this route, such as selecting optimal ligands, solvent, and other conditions, are discussed in detail. The scale-up experiment was carried out to provide more than 300 g of active pharmaceutical ingredients of axitinib in Form XLI with 99.9% purity in 39% yield. In short, we provide a new choice of synthesis route to axitinib, through two copper-catalyzed coupling reactions with good yield.
(E)-N-Methyl-2-(3-(2-(pyridin-2-yl)vinyl)-1H-indazol-6-ylthiol)benzamide (Axitinib) Form XLI (326.4 g in 96% yield with purity 99.91%). Residual Cu content was determined to be 2.2 ppm by atomic absorption spectroscopy: mp 227.7 °C;
1H NMR (300 MHz, DMSO-d6) δ 13.27 (s, 1H), 8.60 (d, J = 4.8 Hz, 1H), 8.29 (d, J = 5.4 Hz, 1H), 8.18 (d, J = 8.5 Hz, 1H), 7.94 (d, J = 16.4 Hz, 1H), 7.81 (t, J = 7.5 Hz, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.63–7.44 (m, 3H), 7.29 (p, J = 7.4, 6.6 Hz, 3H), 7.19 (d, J = 8.5 Hz, 1H), 7.08 (d, J = 7.4 Hz, 1H), 2.78 (d, J = 4.6 Hz, 3H);
13C NMR (75 MHz, DMSO-d6) δ 167.89, 154.86, 149.54, 142.01, 141.86, 136.92, 136.88, 135.67, 132.52, 130.32, 129.99, 129.25, 127.80, 126.15, 125.59, 123.66, 122.68, 122.50, 121.79, 120.29, 114.76, 26.13.
………………………..
Axitinib (Axitinib, AG-013736, CAS: 319460-85-0) is a Pfizer research and development by the United States of new, mainly targeting VEGFR kinase GABA, inhibiting angiogenesis anticancer small molecule drug, trade name Inlyta, for other systems therapy for advanced renal cell carcinoma (Renal Cell Carcinoma, RCC), 2008 has been approved in the domestic clinical, and Pfizer’s cancer drug Sutent another similar imatinib (Sunitinib) , Axitinib also potent and selective multi-targeted tyrosine kinase inhibitor, can inhibit the vascular endothelial growth factor receptor (Vascular EndothelialGrowth Factor Rec India tor, VEGFR), including VEGFl receptor, VECF2 receptors and VECF3 receptor, can inhibit platelet-derived growth factor receptor (Platelet-derived growth factor receptor, PDGFR) and c_KIT. Axitinib is called sunitinib second generation, better than sunitinib adverse reactions.
Axitinib (II) chemical name 6- [2_ (methylcarbamoyl) phenylsulfanyl] -3-E- [2_ (Batch-2-yl) ethenyl] indazole structural formula as follows:

Axitinib (II)
Assi synthesis method for Nepal mainly in the following three ways:
(I) Patent US20060094881 (Agouron Pharmaceuticals), EP2163544 (Pfizer) reported the first synthesis method Axitinib to 3,6-diiodo-indazole as a starting material, first-iodo-6-position is substituted mercapto group, protection of the NH group, then the Heck reaction occurs (pyridine-2-yl) vinyl 3-position, after deprotection Axitinib whole synthesis route is as follows:

Axitinib Scheme I
This method although the synthesis route is shorter, but the catalyst and reagents used relatively expensive and require purified through the column, the total yield is low, is not conducive to industrial production.
[0004] (2) The second method of synthesis Axitinib e.g. W00102369 (Agouron Pharmaceuticals), US6531491 (Agouron Pharmaceuticals) reported in 6-nitro-indazole as a starting material, the 3-position first iodo, followed by the protecting group NH, Suzuki coupling reaction with boronic acid to give 3- styryl styryl-position, a nitro group reduced to an amino group, an amino diazotization reaction was iodo, the 3-position of the styrene-based ozone of the obtained aldehyde, followed by Wittig reaction to give the 3-position (pyridin-2-yl) ethenyl, 6-position is substituted mercapto iodine, alkaline hydrolysis then amidated, and finally deprotection Axitinib, the entire reaction formula as follows:

Axitinib Scheme 2
The method of synthesis route is long, harsh reaction conditions, complex process, the total yield is low, does not apply to industrial production.
[0005] (3) The third method is W02006048745 (Pfizer) discloses to 6-nitro-indazole as a starting material, the 3-position iodo first, followed by the protecting group NH, 3- bits Heck coupling reaction, a nitro group reduced to an amino group, an amino diazotization reaction was iodo, iodo-6-position is substituted mercapto group, and finally deprotected to give Axitinib, the entire reaction is as follows:
This method has an advantage over the first two methods, it is possible to enlarge the production, but the reaction was not complete in the reaction step, will generate new impurities through the column needs to be purified.
SYNTHESIS

aReagents and conditions: (a) I2, K2CO3, DMF; (b) CH2Cl2, CH3SO3H, dihydrofuran; (c) compound B, i-Pr2EtN, Pd(OAc)2, (o-Tol)3P, DMF; (d) iron, EtOH, NH4Cl; (e) AcOH, NaNO2, CH2Cl2, I2/KI; (f) compound C, Pd(dppf)Cl2, Cs2CO3, DMF; (h) 1, p-TsOH, MeOH; 2, NaHCO3; (i) AcOH, MeOH, Pd removal, recrystallization.
http://www.google.com/patents/WO2006048745A1?cl=en
Example 15: Final deprotectioπ step to produce 6-r2-(methylcarbamoyl)phenylsulfanyll-3-E-f2- (pyridine-2-yl)ethenyllindazole

N-1 THP 6-[2-(methylcarbamoyl)phenylsulfanyl]-3-E-[2-(pyridine-2-yl)ethenyl]indazole (355 g) was suspended in 2,485 ml_ of methanol, after which p-toluenesulfonic acid monohydrate (718 g) was added. The mixture was then heated to 65 0C (hard reflux) for 4 hours under argon while the reaction was monitored by HPLC (gluco method). Heating continued until less than 1% of the N-1 THP protected starting material persisted. The heating was then removed and the reaction was cooled to room temperature. The solid was filtered and the wet cake was washed with methanol (2 volumes, 710 mL) then the solids were rinsed with ethyl acetate (2 volumes, 710 mL). The wet cake was transferred to a reactor containing sodium bicarbonate (126.84 g), deionized water (1800 mL), and ethyl acetate (975 mL), which was then stirred for 2 hours at 2O0C. The solids were filtered and washed with 5 volumes of deionized water (1800 mL), then with 2 volumes of ethyl acetate (760 mL), and then dried in a vacuum oven at 400C for 16 hours. The isolated yield for the reaction was 92.5% (274 g). The isolated material was identified as crystalline Form III free base (0.5 ethyl acetate solvate). 1H NMR, 300 MHz, (DMSO-D6), ppm; 13.35 (1 H, s), 8.60 (1 H, d, J=3.8 Hz), 8.39 (1 H, m), 8.23 (1 H, d, J=8.5 Hz), 7.95 (1 H, d, J=16.4 Hz), 7.82 (1 H, ddd, J=7.7, 7.6, 1.8 Hz), 7.67 (1 H, d, J=7.8 Hz), 7.60 (a H, s), 7.57 (1 H, d, J=16.4 Hz), 7.49 (1 H, dd, J=7.1 , 1.6 Hz), 7.35-7.26 (3 H, m), 7.19 (1 H, d, J=8.4 Hz), 7.04 (1 H, d, J=7.8 Hz), 2.77 (3 H, d, J=4.6 Hz). 13C NMR, 75 MHz, (DMSO-D6) ppm: 168.23, 155.18, 149.81 , 142.35, 142.22, 137.31 , 136.00, 132.89, 130.64, 130.36, 129.51 , 128.14, 126.50, 125.93, 124.08, 123.01 , 122.85, 122.12, 120.642, 115.08, 26.45.
Example 21 : Preparation of 6-F2-(methylcarbamovDphenylsulfanyll-3-Z-r2-(pyridine-2- vDethenyllindazole

To a 100 ml_ 3-neck flask containing a solution of 0.95 g of 6-[2- (methylcarbamoyl)phenylsulfanyl]-3-[2-(pyridine-2-yl)ethynyl]indazole was added 2.5 g of phenyliodide diacetate followed by 1.0 mL of H2NNH2 H2O. After the bubbling had settled, more phenyliodide diacetate and H2NNH2 H2O were added in small portions, until LC/MS indicated the disappearance of 6-[2-(methylcarbamoyl)phenylsulfanyl]-3-[2-(pyridine-2-yl)ethynyl]indazole and the formation of 6-[2-(methylcarbamoyl)phenylsuIfanyl]-3-Z-[2-(pyridine-2-yl)ethenyl]indazole. Example 22: Palladium removal and polymorph control of 6-[2-(methylcarbamoyl)phenylsulfanvn- 3-E-r2-(pyridine-2-vDethenyllindazole

4) MeOH, reflux
Polymorph Form IV
5) HOAc/Xylenes
To a 12 L 3-neck flask, equipped with a mechanical stirrer, was added 160.20 g of 6-[2- (methylc’arbamoyl)phenylsulfanyl]-3-E-[2-(pyridine-2-yl)ethenyl]indazole and 1.6 L of DMA and 1.6 L of THF. After stirring for 20 minutes, the mixture became homogeneous. To the clear solution was added 800.99 g of 10% cysteine-silica and the resulting mixture was allowed to stir at room temperature overnight.
The mixture was filtered through a medium sintered glass fritted funnel, and the cake was washed with a solution of 500 mL of DMA and 500 mL of THF. The cake was further washed with 2.0 L of THF and the filtrate was collected into a separate flask. The volatile parts in the latter filtrate were removed in vacuo and the residue was combined with the main filtrate. The combined filtrate was recharged back into the 12 L flask, followed by 800 g of 10% cysteine-silica. The flask was equipped with a mechanical stirrer and stirred over the weekend at room temperature. The mixture was then filtered through a medium sintered glass fritted funnel and the silica was washed with a mixture of solvents of 500 ml. of DMA and 500 ml_ of THF, followed by 3.0 L of THF. The volatile parts in the filtrate were removed in vacuo and the remaining solution was transferred to a 22 L 3-neck flask and treated with 12 L of water (added over a 20 minute period of time), a thick precipitate formed at this stage. After stirring overnight, the mixture was filtered and the cake was washed with 2.0 L of water and sucked dry.
The cake was charged to a 5 L 3-neck flask, followed by 1.6 L of THF and 160 mL of DMF. The flask was equipped with a mechanical stirrer, a reflux condenser and the mixture was heated at reflux for 8 hours. After cooling overnight, the mixture was filtered through sharkskin filter paper and sucked dry. The cake was charged to a 5 L 3-neck flask and 1.6 L of MeOH was added. The flask was equipped with a mechanical stirrer, a water condenser and the contents were heated at reflux for 6 hours. After cooling overnight, the mixture was filtered through sharkskin filter paper and sucked dry.
The cake was dissolved into 1.6 L of HOAc with the assistance of gentle heating in the water bath of a rotary evaporator. The solution was filtered through #3 filter paper and the total volume of the filtrate was reduced to ~500 mL in volume on the rotary evaporator at 60 °C/60 mmHg. At this stage, the bulk of the mixture remained a yellow solution and a small amount of precipitate formed. To the flask was charged 500 mL of xylenes (precipitate formed) and the total volume was reduced to -500 mL in volume on the rotary evaporator at 60°C/60 mmHg. The process was repeated two more times. After cooling, the mixture was filtered, the cake was washed with 500 mL of xylenes and sucked dry. The cake was transferred to a glass dish and further dried at 80°C/27 inch vacuum overnight.
The cake was off-white in color and weighed 108.38g. X-ray powder diffraction analysis indicated that a crystalline form was present, which was characterized as Form IV by a powder X- ray diffraction pattern comprising peaks at the following approximate diffraction angles (20): 8.9, 12.0, 14.6, 15.2, 15.7, 17.8, 19.2, 20.5, 21.6, 23.2, 24.2, 24.8, 26.2, and 27.5.
While the invention has been illustrated by reference to specific and preferred embodiments, those skilled in the art will recognize that variations and modifications may be made through routine experimentation and practice of the invention. Thus, the invention is intended not to be limited by the foregoing description, but to be defined by the appended claims and their equivalents.
………………………..
Chekal, B. P.; Guinness, S. M.; Lillie, B. M.; McLaughlin, R. W.; Palmer, C. W.; Post, R. J.; Sieser, J. E.; Singer, R. A.; Sluggett, G. W.; Vaidyanathan, R.; Withbroe, G. Org. Process Res. Dev. 2014, 18, 266 http://pubs.acs.org/doi/abs/10.1021/op400088k

The manufacturing process of axitinib (1) involves two Pd-catalyzed coupling reactions, a Migita coupling and a Heck reaction. Optimization of both of these pivotal bond-formation steps is discussed as well as the approach to control impurities in axitinib. Essential to the control strategy was the optimization of the Heck reaction to minimize formation of impurities, in addition to the development of an efficient isolation of crude axitinib to purge impurities.
Babu, S.; Dagnino, R., Jr.; Ouellette, M. A.; Shi, B.; Tian, Q.; Zook, S. E. PCT Int. Appl. WO/2006/048745, 2006.
…………………..

………………………………
http://www.google.com/patents/CN103570696A?cl=en

formula:

A Axitinib intermediate (1) production method, based on 6-nitro-indazole as a starting material, in the first catalyst is reacted with 3,4-dihydro -2H- pyran, bits of NH the protecting group tetrahydro -2H- pyran-2-yl, then the three iodide, to give the key intermediate in high yield 3-iodo-6-nitro-1- (tetrahydro -2H- pyrazol pyran-2-yl) -1H- indazole (I), comprising the following synthetic steps:
(1) 6-nitro-indazole dissolved in an aprotic solvent, and 3,4-dihydro -2H- pyran catalyst, 6-nitro-indazole in the catalyst and the 3,4-dihydro -2H – pyran reaction, the protecting group NH-position, was prepared to give 6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, the reaction equation is:

Wherein the 3,4-dihydro -2H- pyran an amount of 3 equivalents wide;
Aprotic solvent is acetonitrile, ethyl acetate, toluene or xylene;
The catalyst is 2,3-dichloro-5,6-dicyano-p-benzoquinone, p-toluenesulfonic acid or methanesulfonic acid;
The reaction temperature is 7 (T90 ° C, the reaction time is 1 to 4 hours;
(2) 6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole dissolved in a polar aprotic solvent, iodine was added and the acid-binding agent, an inorganic base, to afford 3- iodo-6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (I), the reaction equation is:

Wherein the polar aprotic solvent is N, N- dimethylformamide (DMF), N, N- dimethylacetamide, N, N- diethylformamide, N, N- diethyl-acetamide ;
Inorganic base acid binding agent is potassium carbonate, sodium carbonate, potassium hydroxide, sodium hydroxide, potassium bicarbonate, sodium bicarbonate, cesium carbonate, lithium hydroxide;
The reaction temperature is 2 (T40 ° C, the reaction time is 8 to 20 hours.
[0009] A Axitinib intermediate (1) in preparation for the Nepalese Asif application, based on intermediate (1) and 2-vinyl pyridine Heck coupling reaction, followed sequentially nitro reduction and the diazotization reaction of iodine, and finally with a 2-mercapto–N- methylbenzamide was prepared by deprotection docking axitinib, including the following synthetic steps:
(I) Intermediate (1) and be given 2_ vinylpyridine Jie Heck coupling reaction to give (E) _6_ nitro _3- [2_ (P than-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, the reaction equation is:

(2) (E) -6- nitro-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1Η- nitro indazole group reduction reaction, to give (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, The reaction equation is:

(3) (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1Η- indazole diazo of the iodide to give (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole The reaction equation is:

(4) (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1Η- indazole with 2- mercapto-methylbenzamide reaction -N-, to give (E) -N- methyl-2 – {[3- (2- (pyridin-2-yl) ethenyl) -1- (tetrahydro -2H- pyrazol pyran-2-yl) -1H- indazol-6-yl] thio} benzamide, the reaction equation is:

(5) (E) -N- methyl-2- {[3- (2- (pyridin-2-yl) ethenyl) -1- (tetrahydro -2H- pyran-2-yl) -1H- indazol-6-yl] thio} benzamide deprotected Axitinib (II), the reaction equation is:

Example 1
A Assi intermediates for preparing Nigeria, comprising the steps of:
Synthesis of (I) 6- nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole
A 5L reaction flask was added acetonitrile (2L), followed by addition of 6-nitro-indazole (163.1g, 1.0mol), 3, 4- dihydro -2H- pyran (168.2g, 2.0mol), 2,3- dichloro-5,6-dicyano-p-benzoquinone (22.7g, 0.1mol), was heated to 820C under reflux for 2 hours to complete the reaction, cooled to room temperature, rotary evaporated to dryness, added water and dichloromethane 2L 2L, stirring I hour, delamination, the organic phase washed with brine, dried over anhydrous sodium sulfate, filtered, and rotary evaporated to dryness, and then dissolved in acetonitrile and 2L, stirring ice-salt bath chilled to _5 ° C for 2 hours, suction filtered, the filter cake washed with a small amount of cold acetonitrile, recrystallized from ethanol, 60 ° C and dried in vacuo 12 hours to give an off-white solid, 6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, 236.3 g, yield 95.6%, m.p. 110 ~ 120 ° C, 1Η NMR (CDCl3): δ 1.30-1.83 (m, 6Η, Η3, _Η5,), 3.82-3.93 (m, 2Η, Η6 ‘), 5.86 (m , 1Η, Η2 ‘), 8.10-8.12 (m, 2Η, Η3, Η5), 8.31 (m, 1Η; Η4), 8.55 (s, 1Η, Η7);
The reaction equation is as follows:

(2) 3-iodo-6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (I),
5L reaction flask in DMF 700mL, followed by addition of 6-nitro-_1_ (tetrahydro -2H- pyran-2-yl) -1H- indazole (225.0g, 0.91mol, l.0eq) and potassium carbonate ( 251.6g, 1.82mol, 2.0eq), ice-cooled (10 ° C or less), followed by stirring, iodine (415.8g, 1.64mol, 1.8eq) was dissolved in DMF 300mL, was added dropwise to the reaction system, addition time 2 hours , the reaction system was stirred at 25 ° C for 16 hours to complete the reaction, sodium thiosulfate was added (223.0g, 1.41mol, 1.55eq) and 1.50g of potassium carbonate aqueous solution (1.5L), while maintaining the internal temperature 30 ° C Hereinafter, stirred for 30 minutes at room temperature, water was added with stirring 2L, solid precipitated, stirred for 30 minutes at room temperature, suction filtered, the filter cake was washed with water, 60 ° C and dried in vacuo 12 hours to give a pale yellow solid (Ι), 326.5g, yield 96.2%, m.p. 135 ~ 137 ° C / H NMR (DMS0_d6): δ 1.60-1.61 (m, 2H, H4,, H5 ‘), 1.73-1.76 (m, 1H, H5’), 2.01-2.04 (m, 2H, H3 ‘, H4’), 2.35-2.38 (m, 1H, H3 ‘), 3.81-3.87 (m, 2H, H6’), 6.11-6.14 (dd, 1H, H2 ‘), 7.70-7.72 (d , 1H, H4),
8.05-8.07 (dd, 1H, H5), 8.79 (s, 1H, H7).
The reaction equation is as follows:

A Axitinib intermediate (1) in the preparation for the Nepalese Asif applications, including the following synthetic steps:
Synthesis of (I) (E) -6- nitro-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole
A 5L reaction flask was added DMF (2L), followed by addition of the intermediate (1) (312.0g, 0.84mol), 2- vinylpyridine (127.5g, 1.21mol), N, N- diisopropylethylamine ( 205.3g, 1.59mol), tri-o-tolylphosphine (22.3g, 0.073mol) and palladium chloride (4.9g, 0.028mol), nitrogen, and heated to 100 ° C for 12 hours to complete the reaction, cooled to 45 ° C, isopropanol was added 1L, stirring at 45 ° C for 30 minutes, diluted with water and 5L, stirring at room temperature for I h, suction filtered, washed with water, isopropanol was added to the filter cake 1.2L, stirred at 55 ° C for 30 minutes, then stirred at room temperature for 30 minutes, suction filtered, the filter cake washed with cold isopropanol, 50 ° C and dried under vacuum for 12 hours to give (E) -6- nitro-3- [2- (pyridin-2 – yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, 275.3g, 94.0% yield, m.p. 175 ~ 176 ^, ¾ NMR (DMSO-Cl6): δ 1.63-1.64 (m, 2H, H4 ‘, H5’), 1.79-1.81 (m, 1H, H5 ‘), 2.05-2.07 (m, 2H, H3’, H4 ‘), 2.44-2.50 (m, 1H , H3 ‘), 3.86-3.90 (m, 2H, H6’), 6.15-6.18 (dd, 1H, H2 ‘), 7.30-7.33 (dd, 1H, pyridine H5), 7.65-7.69 (d, 1H, J = 16Hz, vinyl H2), 7.72-7.74 (d, 1H, pyridine H4), 7.82-7.86 (m, 1H, pyridine H3), 7.96-8.00 (d, 1H, J = 16Hz, vinyl HI), 8.07 -8.10 (dd, 1H, H4), 8.44-8.46 (d, 1H, H5), 8.63-8.64 (d, 1H, pyridine H6), 8.77-8.78 (d, 1H, H7);
The reaction equation is as follows:

Synthesis of (2) (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2Η-) -1H- indazole
5L reaction flask in ethanol HOOmLdjC 1000mL and ammonium chloride (300.0g, 5.61mol), was dissolved with stirring, followed by addition of (E) -6- nitro-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (255.0g, 0.73mol), was added iron powder (162.6g, 2.91mol), heated to 50 ° C the reaction was stirred for 2 hours to completion of the reaction, was cooled to 22 ° C, tetrahydrofuran 2L, stirred for I hour at room temperature, filtered through Celite, the filter cake washed with tetrahydrofuran and the filtrate was rotary evaporated to dryness, cooled to room temperature, water was added 2L, stirred for I hour at room temperature, pumping filtered, the filter cake washed with petroleum ether, 50 ° C and dried under vacuum for 12 hours to give a pale yellow solid 206.5g, (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1Η- indazole, yield 88.6%, m.p. 162 ~ 164 ° C / H NMR (CDCl3): δ 1.63-1.77 (m, 2H, H4 ‘, H5 ‘), 2.02-2.06 (m, 1H, H5’), 2.17-2.18 (m, 1H, H4 ‘), 2.55-2.60 (m, 1H, H3’) 3.70-3.72 (m, 2H, H3 ‘, H6 ‘), 3.91 (s, 2H, NH2), 4.04-4.07 (m, 1H, H6’), 5.57-5.60 (dd, 1H, H2 ‘), 6.64-6.66 (dd, 1H, H5), 6.74-6.75 (d, 1H, H7), 7.13-7.16 (dd, 1H, pyridine H5), 7.48-7.50 (d, 1H, pyridine H4), 7.49-7.53 (d, 1H, J = 16Hz, vinyl H2), 7.64 -7.68 (m, 1H, pyridine H3), 7.78-7.82 (d, 1H, J = 16Hz, vinyl Hl), 7.82-7.83 (d, 1H, H4), 8.60-8.61 (d, 1H, pyridine H6) ;
The reaction equation is as follows:

Synthesis of (3) (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1Η- indazole
A 5L reaction flask was added 600mL of water and sodium nitrite (70.2g, 1.02mol), stirred and dissolved, and cooled to (TC, (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl ] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (200.0g, 0.62mol) was dissolved in glacial acetic acid 1.3L, dropwise added to the system dropwise over I h, a solution process maintain an internal temperature of 0 ° C, the same temperature for I hour, dropping HCl solution (concentrated hydrochloric acid 112mL, water 200mL) at O ° C, the dropping time of 10 minutes, with the temperature for I h, TLC plate tracking point diazonium salt formation reaction (PE: EA = 1: 1). dropwise 800mL dichloromethane between 0 ° C, the dropping time of 5 minutes, potassium iodide (207.3g, l.25mol) and iodine (79.2g, 0.31mol) was dissolved water 600mL, in (TC dropwise added to the system at the same temperature for 2 hours to complete the reaction. The reaction mixture was poured into the system to 20% sodium thiosulfate solution (2L) and dichloromethane SOOmL and stirred, layered , the aqueous phase was extracted with dichloromethane frozen (2x800mL), dichloromethane phases were combined burning, 3M sodium hydroxide solution was added dropwise 3.5L, adjust the aqueous phase pH = 9 ~ 12, and water was added ammonia 200mL 400mL, stirred for 30 minutes , separated and the aqueous phase was extracted with dichloromethane (2×1.2L), the organic phases were combined, rotary evaporated to dryness, and purified through silica gel to give (E) -6- iodo-3- [2- (pyridin-2-yl ) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, 176.0g, 65.4% yield, m.p. 142 ~ 143 ° C, 1H NMR (DMS0_d6): δ 1.58- 1.61 (m, 2H, H4 ‘, H5,) 1.72-1.78 (m, 1H, H5,), 1.97-2.04 (m, 2H, H3,, H4,), 2.38-2.44 (m, 1H, H3,) , 3.79-3.81 (m, 1H, H6,), 3.88-3.90 (m, 1H, H6,), 5.91-5.94 (dd, 1H, H2,), 7.29-7.31 (m, 1H, pyridine H5), 7.56 -7.60 (d, 1H ,, J = 16Hz, vinyl H2), 7.57-7.59 (m, 1H, pyridine H4), 7.69-7.71 (d, 1H, pyridine H3), 7.80-7.84 (m, 1H, H4 ), 7.89-7.93 (d, 1H, J = 16Hz, vinyl HI), 8.01-8.03 (d, 1H, H5), 8.25 (s, 1H, H7), 8.61-8.62 (d, 1H, pyridine H6) ;
The reaction equation is as follows:

(4) (E) -N- methyl-2 – {[3- (2- (pyridin-2-yl) ethenyl) _1_ (tetrahydro -2H- pyran-2-yl) -1H- indazole 6-ylthio} benzamide]
A 5L reaction flask was added DMF (1750mL) and (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1H- indazole (175.0g, 0.41mol), nitrogen, was added [1, I, – bis (diphenylphosphino) ferrocene] dichloropalladium dichloromethane complex (14.9g, 0.018mmol ), cesium carbonate (198.3g, 0.61mol) and dichloromethane 20mL, was added 2-mercapto -N- methylbenzamide (84.9g, 0.5Imol), heated to 80 ° C for 16 hours to complete the reaction, spin distilled was removed DMF, cooled to room temperature, ethyl acetate was added 3L, water 4L, stirred for 40 minutes, the organic phase was separated, washed with brine, layered, dried over sodium sulfate, filtered, and rotary evaporated to dryness, to give (E) -N- methyl-2 – {[3- (2- (pyridin-2-yl) ethenyl) -1- (tetrahydro -2H- pyran-2-yl) -1H- indazol-6-yl] thio } benzamide, 165.6g, a yield of 86.7%, the melting point of 142 ~ 143 ° C;
The reaction equation is as follows:

(5) Synthesis of axitinib
In a 2L reaction flask was added (E) -N- methyl-2 – {[3- (2- (pyridin-2-yl) ethenyl) _1_ (tetrahydro -2H- pyran-2-yl) -1H – indazol-6-yl] thio} benzamide (150.0g, 0.32mol), p-toluenesulfonic acid monohydrate (303.2g, 1.59mol), methanol (800mL) and water (150mL), nitrogen, heated to 65 ° C for 4 hours, spin evaporated to dryness and ethanol (800mL), 65 ° C was stirred for I hour, the ethanol was removed by rotary evaporation, then repeated three times, TLC spot plate tracking reaction (petroleum ether: ethyl acetate = 1: 1). Completion of the reaction, cooled to room temperature, rotary evaporated to dryness, water was added 500mL, stirred for I h, filtered, and the filter cake was washed with methanol and ice, and then added to the reaction vessel, ethyl acetate was added 450mL, stirred at 65 ° C 30 minutes. cooled to room temperature, suction filtered, the filter cake washed with ethyl acetate and freeze paint, water paint, 50 ° C and dried under vacuum for 12 hours to give a white solid 117.5g, Axitinib (II), yield 95.4%, HPLC purity 98.8 % / H NMR (DMS0_d6): δ 2.78 (d, 3H, CH3), 7.05 (dd, 1H), 7.19 (dd, 1H), 7.36-7.23 (m, 3H), 7.50 (dd, 1H), 7.58 ( d, 1H), 7.61 (s, 1H), 7.66 (d, 1H), 7.85-7.76 (m, 1H), 7.96 (d, 1H, J = 16Hz), 8.21 (d, 1H), 8.39 (q, 1H), 8.61 (d, 1H), 13.35 (s, 1H).
The reaction equation is as follows:

Example 2
A Assi intermediates for preparing Nigeria, comprising the steps of:
Synthesis of (1) 6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole
A 5L reaction flask was added ethyl acetate (2L), followed by addition of 6-nitro-indazole (163.14g, 1.0mol), 3, 4- dihydro -2H- pyran (210.3g, 2.5mol), toluene acid (20.7g, 0.12mol), heated to 78 ° C under reflux for 3 hours to complete the reaction, cooled to room temperature, rotary evaporated to dryness, added water and dichloromethane 2L 2L, stirred for I hour, stratification, the organic phase was washed with brine, dried over anhydrous sodium sulfate, filtered, and rotary evaporated to dryness, and then dissolved in acetonitrile and 2L, stirring ice-salt bath chilled to _5 ° C for 2 hours, suction filtered, the filter cake washed with a small amount of cold acetonitrile, recrystallized from ethanol , 60 ° C and dried in vacuo 12 hours to give an off-white solid 223.3g, 6- nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, yield 90.3%, m.p. 110 ^ 11 TC;
The reaction equation is as follows:

(2) 3-iodo-6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (I),
5L reaction flask in DMF 700mL, followed by addition of 6-nitro-_1_ (tetrahydro -2H- pyran-2-yl) -1H- indazole (200.0g, 0.81mol, l.0eq) and sodium hydroxide (64.7g, 1.62mol, 2.0eq), ice-cooled (10 ° C or less), followed by stirring, iodine (369.6g, 1.46mol, 1.8eq) was dissolved in DMF 300mL, was added dropwise to the reaction system, addition time 2 hours, the reaction system was stirred at 25 ° C for 12 hours to complete the reaction, sodium thiosulfate was added (198.2g, 1.25mol, 1.55eq) and 1.50g of potassium carbonate aqueous solution (1.5L), while maintaining the temperature of 30 ° C or less, and stirred for 30 minutes at room temperature, water was added with stirring 2L, solid precipitated, stirred for 30 minutes at room temperature, suction filtered, the filter cake was washed with water, 60 ° C and dried in vacuo 12 hours to give a pale yellow solid
(1), 294.3g, 97.5% yield, m.p. 136 ~ 137. . .
[0014] The reaction equation is as follows:

A Axitinib intermediate (1) in the preparation for the Nepalese Asif applications, including the following synthetic steps:
Synthesis (1) (E) -6- nitro-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2Η-) -1H- indazole
A 5L reaction flask was added DMF (2L), followed by addition of the intermediate (1) (312.0g, 0.84mol), 2- vinylpyridine (127.5g, 1.21mol), N, N- diisopropylethylamine ( 205.3g, 1.59mol), tri-o-tolylphosphine (22.3g, 0.073mol) and palladium chloride (4.9g, 0.028mol), nitrogen, and heated to 100 ° C for 12 hours to complete the reaction, cooled to 45 ° C, isopropanol was added 1L, stirring at 45 ° C for 30 minutes, diluted with water and 5L, stirring at room temperature for I h, suction filtered, washed with water, isopropanol was added to the filter cake 1.2L, stirred at 55 ° C for 30 minutes, then stirred at room temperature for 30 minutes, suction filtered, the filter cake washed with cold isopropanol, 50 ° C and dried under vacuum for 12 hours to give (E) -6- nitro-3- [2- (pyridin _2 _-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, 275.3g, 94.0% yield, m.p. 175 ~ 176 ^, ¾ NMR (DMSO-Cl6): δ 1.63-1.64 (m, 2H, H4 ‘, H5’), 1.79-1.81 (m, 1H, H5 ‘), 2.05-2.07 (m, 2H, H3’, H4 ‘), 2.44-2.50 (m, 1H , H3 ‘), 3.86-3.90 (m, 2H, H6’), 6.15-6.18 (dd, 1H, H2 ‘), 7.30-7.33 (dd, 1H, pyridine H5), 7.65-7.69 (d, 1H, J = 16Hz, vinyl H2), 7.72-7.74 (d, 1H, pyridine H4), 7.82-7.86 (m, 1H, pyridine H3), 7.96-8.00 (d, 1H, J = 16Hz, vinyl HI), 8.07 -8.10 (dd, 1H, H4), 8.44-8.46 (d, 1H, H5), 8.63-8.64 (d, 1H, pyridine H6), 8.77-8.78 (d, 1H, H7);
The reaction equation is as follows:

Synthesis of (2) (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole
5L reaction flask in ethanol HOOmLdjC 1000mL and ammonium chloride (300.0g, 5.61mol), was dissolved with stirring, followed by addition of (E) -6- nitro-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (255.0g, 0.73mol), was added iron powder (162.6g, 2.91mol), heated to 50 ° C the reaction was stirred for 2 hours to completion of the reaction, was cooled to 22 ° C, tetrahydrofuran 2L, stirred for I hour at room temperature, filtered through Celite, the filter cake washed with tetrahydrofuran and the filtrate was rotary evaporated to dryness, cooled to room temperature, water was added 2L, stirred for I hour at room temperature, pumping filtered, the filter cake washed with petroleum ether, 50 ° C and dried under vacuum for 12 hours to give a pale yellow solid 206.5g, (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1Η- indazole, yield 88.6%, m.p. 162 ~ 164 ° C / H NMR (CDCl3): δ 1.63-1.77 (m, 2H, H4 ‘, H5 ‘), 2.02-2.06 (m, 1H, H5’), 2.17-2.18 (m, 1H, H4 ‘), 2.55-2.60 (m, 1H, H3’) 3.70-3.72 (m, 2H, H3 ‘, H6 ‘), 3.91 (s, 2H, NH2), 4.04-4.07 (m, 1H, H6’), 5.57-5.60 (dd, 1H, H2 ‘), 6.64-6.66 (dd, 1H, H5), 6.74-6.75 (d, 1H, H7), 7.13-7.16 (dd, 1H, pyridine H5), 7.48-7.50 (d, 1H, pyridine H4), 7.49-7.53 (d, 1H, J = 16Hz, vinyl H2), 7.64 -7.68 (m, 1H, pyridine H3), 7.78-7.82 (d, 1H, J = 16Hz, vinyl Hl), 7.82-7.83 (d, 1H, H4), 8.60-8.61 (d, 1H, pyridine H6) ;
The reaction equation is as follows:

Synthesis of (3) (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1Η- indazole
A 5L reaction flask was added 600mL of water and sodium nitrite (70.2g, 1.02mol), stirred and dissolved, and cooled to (TC, (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl ] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (200.0g, 0.62mol) was dissolved in glacial acetic acid 1.3L, dropwise added to the system dropwise over I h, a solution process maintain an internal temperature of 0 ° C, the same temperature for I hour, dropping HCl solution (concentrated hydrochloric acid 112mL, water 200mL) at O ° C, the dropping time of 10 minutes, with the temperature for I h, TLC plate tracking point diazonium salt formation reaction (PE: EA = 1: 1). dropwise 800mL dichloromethane between 0 ° C, the dropping time of 5 minutes, potassium iodide (207.3g, l.25mol) and iodine (79.2g, 0.31mol) was dissolved water 600mL, in (TC dropwise added to the system at the same temperature for 2 hours to complete the reaction. The reaction mixture was poured into the system to 20% sodium thiosulfate solution (2L) and dichloromethane SOOmL and stirred, layered , the aqueous phase was extracted with dichloromethane frozen (2x800mL), dichloromethane phases were combined burning, 3M sodium hydroxide solution was added dropwise 3.5L, adjust the aqueous phase pH = 9 ~ 12, and water was added ammonia 200mL 400mL, stirred for 30 minutes , separated and the aqueous phase was extracted with dichloromethane (2×1.2L), the organic phases were combined, rotary evaporated to dryness, and purified through silica gel to give (E) -6- iodo-3- [2- (pyridin-2-yl ) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, 176.0g, 65.4% yield, m.p. 142 ~ 143 ° C, 1H NMR (DMS0_d6): δ 1.58- 1.61 (m, 2H, H4 ‘, H5,) 1.72-1.78 (m, 1H, H5,), 1.97-2.04 (m, 2H, H3,, H4,), 2.38-2.44 (m, 1H, H3,) , 3.79-3.81 (m, 1H, H6,), 3.88-3.90 (m, 1H, H6,), 5.91-5.94 (dd, 1H, H2,), 7.29-7.31 (m, 1H, pyridine H5), 7.56 -7.60 (d, 1H ,, J = 16Hz, vinyl H2), 7.57-7.59 (m, 1H, pyridine H4), 7.69-7.71 (d, 1H, pyridine H3), 7.80-7.84 (m, 1H, H4 ), 7.89-7.93 (d, 1H, J = 16Hz, vinyl HI), 8.01-8.03 (d, 1H, H5), 8.25 (s, 1H, H7), 8.61-8.62 (d, 1H, pyridine H6) ;
The reaction equation is as follows:

(4) (E) -N- methyl-2 – {[3- (2- (pyridin-2-yl) ethenyl) _1_ (tetrahydro -2H- pyran-2-yl) -1H- indazole 6-ylthio} benzamide]
A 5L reaction flask was added DMF (1750mL) and (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1H- indazole (175.0g, 0.41mol), nitrogen, was added [1, I, – bis (diphenylphosphino) ferrocene] dichloropalladium dichloromethane complex (14.9g, 0.018mmol ), cesium carbonate (198.3g, 0.61mol) and dichloromethane 20mL, was added 2-mercapto -N- methylbenzamide (84.9g, 0.5Imol), heated to 80 ° C for 16 hours to complete the reaction, spin distilled was removed DMF, cooled to room temperature, ethyl acetate was added 3L, water 4L, stirred for 40 minutes, the organic phase was separated, washed with brine, layered, dried over sodium sulfate, filtered, and rotary evaporated to dryness, to give (E) -N- methyl-2 – {[3- (2- (pyridin-2-yl) ethenyl) -1- (tetrahydro -2H- pyran-2-yl) -1H- indazol-6-yl] thio } benzamide, 165.6g, a yield of 86.7%, the melting point of 142 ~ 143 ° C;
The reaction equation is as follows:

(5) Synthesis of axitinib
In a 2L reaction flask was added (E) -N- methyl-2 – {[3- (2- (pyridin-2-yl) ethenyl) _1_ (tetrahydro -2H- pyran-2-yl) -1H – indazol-6-yl] thio} benzamide (150.0g, 0.32mol), p-toluenesulfonic acid monohydrate (303.2g, 1.59mol), methanol (800mL) and water (150mL), nitrogen, heated to 65 ° C for 4 hours, spin evaporated to dryness and ethanol (800mL), 65 ° C was stirred for I hour, the ethanol was removed by rotary evaporation, then repeated three times, TLC spot plate tracking reaction (petroleum ether: ethyl acetate = 1: 1). Completion of the reaction, cooled to room temperature, rotary evaporated to dryness, water was added 500mL, stirred for I h, filtered, and the filter cake was washed with methanol and ice, and then added to the reaction vessel, ethyl acetate was added 450mL, stirred at 65 ° C 30 minutes. cooled to room temperature, suction filtered, the filter cake washed with ethyl acetate and freeze paint, water paint, 50 ° C and dried under vacuum for 12 hours to give a white solid 117.5g, Axitinib (II), yield 95.4%, HPLC purity 98.8 % / H NMR (DMS0_d6): δ 2.78 (d, 3H, CH3), 7.05 (dd, 1H), 7.19 (dd, 1H), 7.36-7.23 (m, 3H), 7.50 (dd, 1H), 7.58 ( d, 1H), 7.61 (s, 1H), 7.66 (d, 1H), 7.85-7.76 (m, 1H), 7.96 (d, 1H, J = 16Hz), 8.21 (d, 1H), 8.39 (q, 1H), 8.61 (d, 1H), 13.35 (s, 1H).
The reaction equation is as follows:

Example 3
A Assi intermediates for preparing Nigeria, comprising the steps of:
Synthesis of (1) 6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole
5L reaction flask in toluene (2L), followed by addition of 6-nitro-indazole (163.lg, 1.0mol), 3,4- dihydro -2H- pyran (193.5g, 2.3mol), methanesulfonic acid (14.4g, 0.15mol), heated to 85 ° C under reflux for 3.5 hours, to complete the reaction, cooled to room temperature, rotary evaporated to dryness, added water and dichloromethane 2L 2L, stirred for I hour, stratification, the organic phase was washed with brine wash, dried over anhydrous sodium sulfate, filtered, and rotary evaporated to dryness, and then dissolved in acetonitrile and 2L, stirring ice-salt bath chilled to _5 ° C for 2 hours, suction filtered, the filter cake washed with a small amount of cold acetonitrile and paint, and recrystallized from ethanol , 60 ° C and dried in vacuo 12 hours to give an off-white solid, 6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, 234.4g, 94.8% yield, m.p. 111 ~ 112.. ;
The reaction equation is as follows:

(2) 3-iodo-6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (I),
5L reaction flask in DMF 700mL, followed by addition of 6-nitro-_1_ (tetrahydro -2H- pyran-2-yl) -1H- indazole (225.0g, 0.91mol, 1.0eq) and potassium hydroxide ( 102.lg, 1.82mol, 2.0eq), ice-cooled below 10 ° C, with stirring, iodine (415.8g, 1.64mol, 1.8eq) was dissolved in DMF 300mL, was added dropwise to the reaction system dropwise over 2 hours, The reaction system was stirred at 30 ° C for 10 hours to complete the reaction, sodium thiosulfate was added (223.0g, 1.41mol, 1.55eq) and 1.50g of potassium carbonate aqueous solution (1.5L), while maintaining the internal temperature below 30 ° C , stirred for 45 minutes at room temperature, water was added with stirring 2L, solid precipitated, stirred for 45 minutes at room temperature, suction filtered, the filter cake was washed with water, 60 ° C and dried in vacuo 12 hours to give a pale yellow solid
(1), 317.2g, 93.4% yield, m.p. 135 ~ 136 ° C.
The reaction equation is as follows:

A Axitinib intermediate (1) in the preparation for the Nepalese Asif applications, including the following synthetic steps:
Synthesis (1) (E) -6- nitro-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole
A 5L reaction flask was added DMF (2L), followed by addition of the intermediate (1) (312.0g, 0.84mol), 2- vinylpyridine (127.5g, 1.21mol), N, N- diisopropylethylamine ( 205.3g, 1.59mol), tri-o-tolylphosphine (22.3g, 0.073mol) and palladium chloride (4.9g, 0.028mol), nitrogen, and heated to 100 ° C for 12 hours to complete the reaction, cooled to 45 ° C, isopropanol was added 1L, stirring at 45 ° C for 30 minutes, diluted with water and 5L, stirring at room temperature for I h, suction filtered, washed with water, isopropanol was added to the filter cake 1.2L, stirred at 55 ° C for 30 minutes, then stirred at room temperature for 30 minutes, suction filtered, the filter cake washed with cold isopropanol, 50 ° C and dried under vacuum for 12 hours to give (E) -6- nitro-3- [2- (pyridin _2 _-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, 275.3g, 94.0% yield, m.p. 175 ~ 176 ^, ¾ NMR (DMSO-Cl6): δ 1.63-1.64 (m, 2H, H4 ‘, H5’), 1.79-1.81 (m, 1H, H5 ‘), 2.05-2.07 (m, 2H, H3’, H4 ‘), 2.44-2.50 (m, 1H , H3 ‘), 3.86-3.90 (m, 2H, H6’), 6.15-6.18 (dd, 1H, H2 ‘), 7.30-7.33 (dd, 1H, pyridine H5), 7.65-7.69 (d, 1H, J = 16Hz, vinyl H2), 7.72-7.74 (d, 1H, pyridine H4), 7.82-7.86 (m, 1H, pyridine H3), 7.96-8.00 (d, 1H, J = 16Hz, vinyl HI), 8.07 -8.10 (dd, 1H, H4), 8.44-8.46 (d, 1H, H5), 8.63-8.64 (d, 1H, pyridine H6), 8.77-8.78 (d, 1H, H7);
The reaction equation is as follows:

Synthesis of (2) (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole
5L reaction flask in ethanol HOOmLdjC 1000mL and ammonium chloride (300.0g, 5.61mol), was dissolved with stirring, followed by addition of (E) -6- nitro-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (255.0g, 0.73mol), was added iron powder (162.6g, 2.91mol), heated to 50 ° C the reaction was stirred for 2 hours to completion of the reaction, was cooled to 22 ° C, tetrahydrofuran 2L, stirred for I hour at room temperature, filtered through Celite, the filter cake washed with tetrahydrofuran and the filtrate was rotary evaporated to dryness, cooled to room temperature, water was added 2L, stirred for I hour at room temperature, pumping filtered, the filter cake washed with petroleum ether, 50 ° C and dried under vacuum for 12 hours to give a pale yellow solid 206.5g, (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1Η- indazole, yield 88.6%, m.p. 162 ~ 164 ° C / H NMR (CDCl3): δ 1.63-1.77 (m, 2H, H4 ‘, H5 ‘), 2.02-2.06 (m, 1H, H5’), 2.17-2.18 (m, 1H, H4 ‘), 2.55-2.60 (m, 1H, H3’) 3.70-3.72 (m, 2H, H3 ‘, H6 ‘), 3.91 (s, 2H, NH2), 4.04-4.07 (m, 1H, H6’), 5.57-5.60 (dd, 1H, H2 ‘), 6.64-6.66 (dd, 1H, H5), 6.74-6.75 (d, 1H, H7), 7.13-7.16 (dd, 1H, pyridine H5), 7.48-7.50 (d, 1H, pyridine H4), 7.49-7.53 (d, 1H, J = 16Hz, vinyl H2), 7.64 -7.68 (m, 1H, pyridine H3), 7.78-7.82 (d, 1H, J = 16Hz, vinyl Hl), 7.82-7.83 (d, 1H, H4), 8.60-8.61 (d, 1H, pyridine H6) ;
The reaction equation is as follows:

Synthesis of (3) (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1Η- indazole
A 5L reaction flask was added 600mL of water and sodium nitrite (70.2g, 1.02mol), stirred and dissolved, and cooled to (TC, (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl ] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (200.0g,
0.62mol) was dissolved in glacial acetic acid 1.3L, dropwise added to the system dropwise over I hour, added dropwise to maintain the internal temperature process 0 ° C, the same temperature for I h, HCl solution was added dropwise at O ° C (112mL of concentrated hydrochloric acid , water 200mL), was added dropwise for 10 minutes, with the temperature for I h, TLC plate tracking point diazonium salt formation reaction (PE: EA = 1: 1). Solution of methylene chloride at 0 ° C and 800mL, dropping time of 5 minutes, potassium iodide (207.3g, l.25mol) and iodine (79.2g, 0.31mol) dissolved in water 600mL, at (TC dropwise added to the system, same temperature for 2 hours to complete the reaction. The reaction system was poured into a mixture of 20% sodium thiosulfate solution (2L) and dichloromethane SOOmL and stirred, layers were separated, the aqueous phase was extracted with dichloromethane frozen (2x800mL ), methylene chloride phases were combined burning, 3M sodium hydroxide solution was added dropwise 3.5L, adjust the aqueous phase pH = 9 ~ 12, and water was added ammonia 200mL 400mL, stirred for 30 minutes, separated and the aqueous phase extracted with dichloromethane ( 2×1.2L), the organic phases were combined, rotary evaporated to dryness, and purified through silica gel to give (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H – pyran-2-yl) -1H- indazole, 176.0g, 65.4% yield, m.p. 142 ~ 143 ° C, 1H NMR (DMS0_d6): δ 1.58-1.61 (m, 2H, H4 ‘, H5,) 1.72-1.78 (m, 1H, H5,), 1.97-2.04 (m, 2H, H3,, H4,), 2.38-2.44 (m, 1H, H3,), 3.79-3.81 (m, 1H, H6,) , 3.88-3.90 (m, 1H, H6,), 5.91-5.94 (dd, 1H, H2,),
7.29-7.31 (m, 1H, pyridine H5), 7.56-7.60 (d, 1H ,, J = 16Hz, vinyl H2), 7.57-7.59 (m, 1H, pyridine H4), 7.69-7.71 (d, 1H, pyridine H3), 7.80-7.84 (m, 1H, H4), 7.89-7.93 (d, 1H, J = 16Hz, vinyl HI), 8.01-8.03 (d, 1H, H5), 8.25 (s, 1H, H7 ), 8.61-8.62 (d, 1H, pyridine H6); reaction equation is as follows:

(4) (E) -N- methyl-2 – {[3- (2- (pyridin-2-yl) ethenyl) _1_ (tetrahydro -2H- pyran-2-yl) -1H- indazole 6-ylthio} benzamide]
A 5L reaction flask was added DMF (1750mL) and (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1H- indazole (175.0g, 0.41mol), nitrogen, was added [1, I, – bis (diphenylphosphino) ferrocene] dichloropalladium dichloromethane complex (14.9g, 0.018mmol ), cesium carbonate (198.3g, 0.61mol) and dichloromethane 20mL, was added 2-mercapto -N- methylbenzamide (84.9g, 0.5Imol), heated to 80 ° C for 16 hours to complete the reaction, spin distilled was removed DMF, cooled to room temperature, ethyl acetate was added 3L, water 4L, stirred for 40 minutes, the organic phase was separated, washed with brine, layered, dried over sodium sulfate, filtered, and rotary evaporated to dryness, to give (E) -N- methyl-2 – {[3- (2- (pyridin-2-yl) ethenyl) -1- (tetrahydro -2H- pyran-2-yl) -1H- indazol-6-yl] thio } benzamide, 165.6g, a yield of 86.7%, the melting point of 142 ~ 143 ° C;
The reaction equation is as follows:
(5) Synthesis of axitinib
In a 2L reaction flask was added (E) -N- methyl-2 – {[3- (2- (pyridin-2-yl) ethenyl) _1_ (tetrahydro -2H- pyran-2-yl) -1H – indazol-6-yl] thio} benzamide (150.0g, 0.32mol), p-toluenesulfonic acid monohydrate (303.2g, 1.59mol), methanol (800mL) and water (150mL), nitrogen, heated to 65 ° C for 4 hours, spin evaporated to dryness and ethanol (800mL), 65 ° C was stirred for I hour, the ethanol was removed by rotary evaporation, then repeated three times, TLC spot plate tracking reaction (petroleum ether: ethyl acetate = 1: 1). Completion of the reaction, cooled to room temperature, rotary evaporated to dryness, water was added 500mL, stirred for I h, filtered, and the filter cake was washed with methanol and ice, and then added to the reaction vessel, ethyl acetate was added 450mL, stirred at 65 ° C 30 minutes. cooled to room temperature, suction filtered, the filter cake washed with ethyl acetate and freeze paint, water paint, 50 ° C and dried under vacuum for 12 hours to give a white solid 117.5g, Axitinib (II),
yield 95.4%, HPLC purity 98.8 % / H NMR (DMS0_d6): δ 2.78 (d, 3H, CH3), 7.05 (dd, 1H), 7.19 (dd, 1H), 7.36-7.23 (m, 3H), 7.50 (dd, 1H), 7.58 ( d, 1H), 7.61 (s, 1H), 7.66 (d, 1H), 7.85-7.76 (m, 1H), 7.96 (d, 1H, J = 16Hz), 8.21 (d, 1H), 8.39 (q, 1H), 8.61 (d, 1H), 13.35 (s, 1H).
The reaction equation is as follows:



SEE NMR……….
http://orgspectroscopyint.blogspot.in/2015/07/axitinib.html
………..
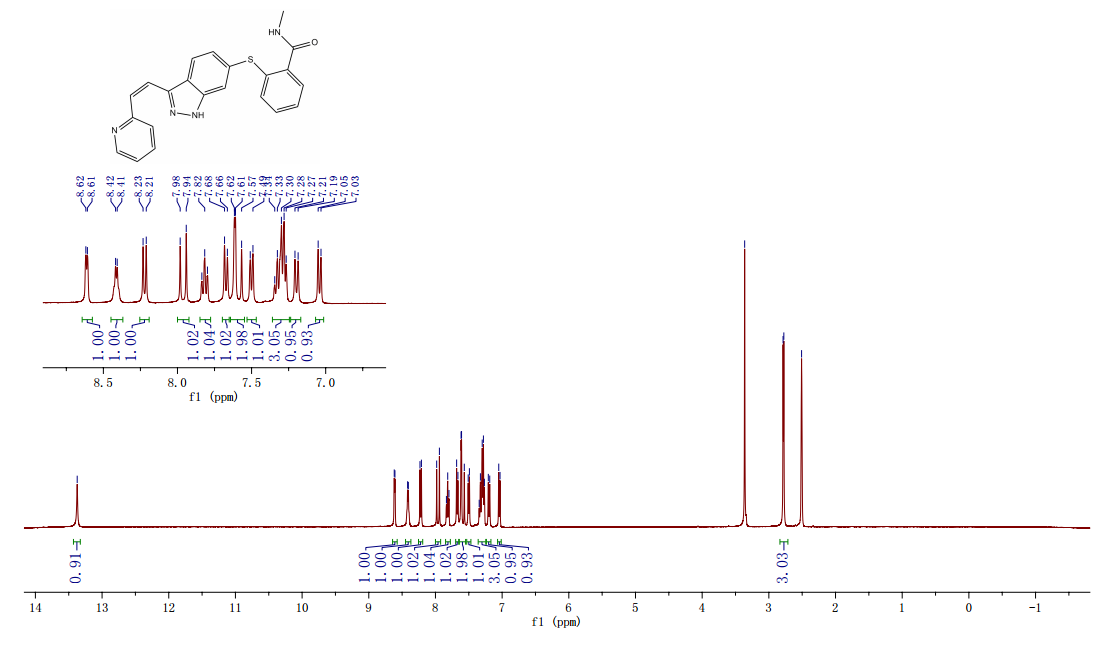

http://dmd.aspetjournals.org/content/suppl/2014/03/07/dmd.113.056531.DC1/Supplemental__Data_Figures_56531.pdf

MASS

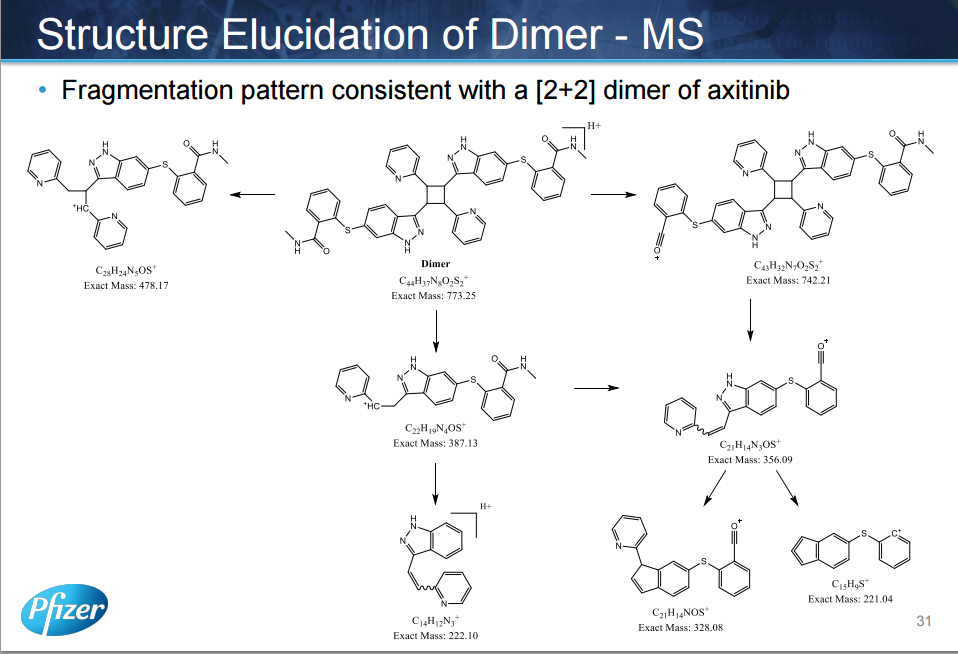

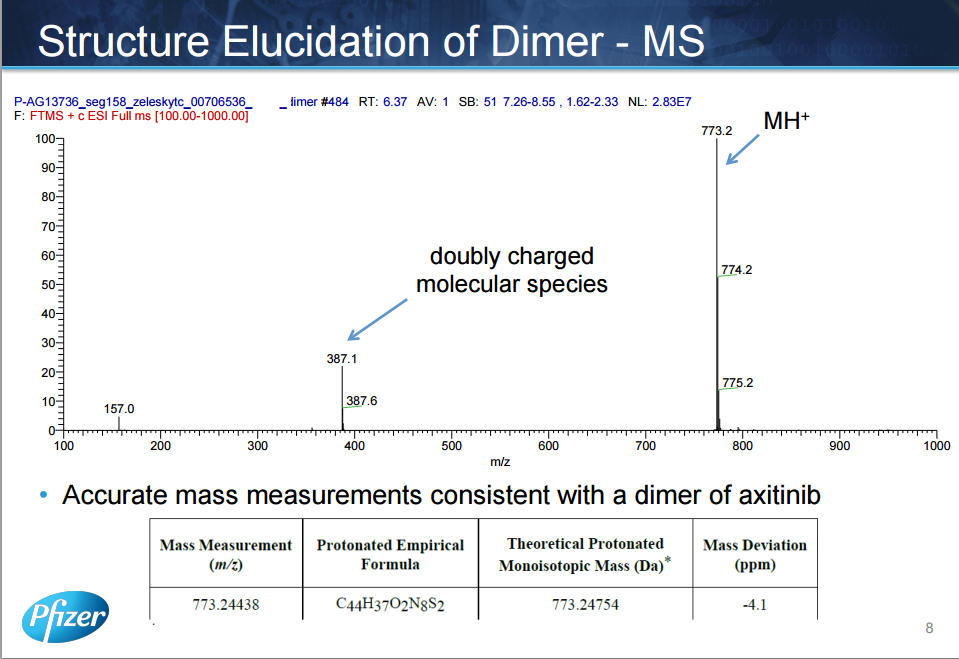

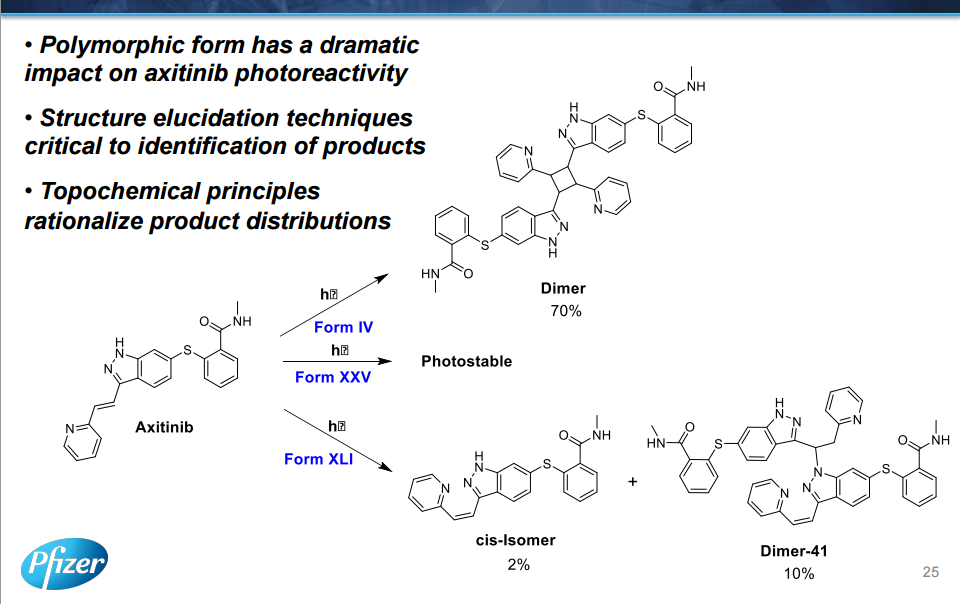
References
- “Inlyta (axitinib) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 25 January 2014.
- Wilmes, LJ; Pallavicini, MG; Fleming, LM; Gibbs, J; Wang, D; Li, KL; Partridge, SC; Henry, RG; Shalinsky, DR; Hu-Lowe, D; Park, JW; McShane, TM; Lu, Y; Brasch, RC; Hylton, NM (April 2007). “AG-013736, a novel inhibitor of VEGF receptor tyrosine kinases, inhibits breast cancer growth and decreases vascular permeability as detected by dynamic contrast-enhanced magnetic resonance imaging”. Magnetic Resonance Imaging 25 (3): 319–27. doi:10.1016/j.mri.2006.09.041. PMID 17371720.
- Rini, B; Rixe, O; Bukowski, R; Michaelson, MD; Wilding, G; Hudes, G; Bolte, O; Steinfeldt, H; Reich, SD; Motzer, R (June 2005). “AG-013736, a multi-target tyrosine kinase receptor inhibitor, demonstrates anti-tumor activity in a Phase 2 study of cytokine-refractory, metastatic renal cell cancer (RCC)”. Journal of Clinical Oncology ASCO Annual Meeting Proceedings 23 (16S): 4509.
- Rugo, HS; Herbst, RS; Liu, G; Park, JW; Kies, MS; Steinfeldt, HM; Pithavala, YK; Reich, SD; Freddo, JL; Wilding, G (August 2005). “Phase I trial of the oral antiangiogenesis agent AG-013736 in patients with advanced solid tumors: pharmacokinetic and clinical results”(PDF). Journal of Clinical Oncology 23 (24): 5474–83. doi:10.1200/JCO.2005.04.192.PMID 16027439.
- “FDA Approves Inlyta for Advanced Renal Cell Carcinoma”. Drugs.com. January 27, 2012.
- John Fauber, Elbert Chu (Oct 27, 2014). “The Slippery Slope: Is a Surrogate Endpoint Evidence of Efficacy?”. Milwaukee Journal Sentinel/MedPage Today.
- Spano, JP; Chodkiewicz, C; Maurel, J; Wong, R; Wasan, H; Barone, C; Létourneau, R; Bajetta, E; Pithavala, Y; Bycott, P; Trask, P; Liau, K; Ricart, AD; Kim, S; Rixe, O (June 2008). “Efficacy of gemcitabine plus axitinib compared with gemcitabine alone in patients with advanced pancreatic cancer: an open-label randomised phase II study”. Lancet 371(9630): 2101–2108. doi:10.1016/S0140-6736(08)60661-3. PMID 18514303.
- “Pfizer pancreatic cancer drug fails, trial halted”. Reuters. January 30, 2009.
- “Pfizer’s Phase III Trial in mRCC Turns Up Positive Results”. 19 Nov 2010.
- “ODAC Unanimously Supports Axitinib for Renal Cell Carcinoma”. 7 Dec 2011.
- “INLYTA (axitinib) tablet, film coated [Pfizer Laboratories Div Pfizer Inc]”. DailyMed. Pfizer Laboratories Div Pfizer Inc. September 2013. Retrieved 25 January 2014.
- “Inlyta : EPAR – Product Information” (PDF). European Medicines Agency. Pfizer Ltd. 17 December 2013. Retrieved 25 January 2014.
- “Inlyta 1 mg 3mg, 5 mg & 7mg film-coated tablets – Summary of Product Characteristics (SPC)”. electronic Medicines Compendium. Pfizer Limited. 5 December 2013. Retrieved25 January 2014.
- “PRODUCT INFORMATION INLYTA (axitinib)” (PDF). TGA eBusiness Services. Pfizer Australia Pty Ltd. 5 July 2013. Retrieved 25 January 2014.
- Tea Pemovska,Eric Johnson,Mika Kontro,Gretchen A. Repasky,Jeffrey Chen,Peter Wells,Ciarán N. Cronin,Michele McTigue,Olli Kallioniemi,Kimmo Porkka,Brion W. Murray & Krister Wennerberg. “Axitinib effectively inhibits BCR-ABL1(T315I) with a distinct binding conformation”. Nature. doi:10.1038/nature14119.
- “FDA Prescribing Information” (PDF). 30 Jan 2012.
- Escudier, B; Gore, M. “Axitinib for the Management of Metastatic Renal Cell Carcinoma” (PDF). Drugs in R&d 11 (2): 113–126. doi:10.2165/11591240-000000000-00000. PMC 3585900. PMID 21679004.
- Zhang Y (Jan 2014). “Screening of kinase inhibitors targeting BRAF for regulating autophagy based on kinase pathways.”. J Mol Med Rep 9 (1): 83–90.doi:10.3892/mmr.2013.1781. PMID 24213221.
- [1] http://www.cancer.gov/cancertopics/druginfo/axitinib[2] http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm289439.htm[3] Kosugi M, Shimizu T, T. Migita, Chemistry Letters , 1978 , pp 13-14.[4] Organic Process Research & Development 2008 , 12, 869? 876.[5] Furstner A. Chem. Commun ., 2008 , 2873? 2875.[6] Thorarensen A. , Synlett , 2010 , 2 pp 219 – 222.
[7] http://en.wikipedia.org/wiki/Heck_reaction – where you can find the reaction mechanism and many other useful information.
[8] Aoyama, T., Synthesis , 2004 , 8 pp 1183-1186.
Honokiol, from magnolia bark, shuts down cancer cells in lab
 |
|
| Magnolia virginiana |
Honokiol, from magnolia bark, shuts down cancer cells in lab
Compound in magnolia may combat head and neck cancers
Honokiol, from magnolia bark, shuts down cancer cells in lab


Magnolias are prized for their large, colorful, fragrant flowers. Does the attractive, showy tree also harbor a potent cancer fighter?
Yes, according to a growing number of studies, including one from VA and the University of Alabama at Birmingham that is now online in the journal Oncotarget.
The study focused on squamous cell head and neck cancers, a scourge among those who use tobacco and alcohol. According to the National Cancer Institute, at least 3 in 4 head and neck cancers are caused by the use of tobacco and alcohol. The cancers have only a 50 percent survival rate, killing some 20,000 Americans each year.
Enter honokiol–chemical formula C18H18O2. As one of the major active compounds in magnolia extract, the phytochemical has been used for centuries in traditional Chinese and Japanese medicine to treat anxiety and other conditions. More recently, scientists have been discovering that the compound, found in magnolia bark, is a wily and versatile adversary of cancer. It seems to exploit many biochemical pathways to shrink tumors of various types, or to keep them from growing in the first place.

The Alabama scientists have now shown how it works against head and neck cancers: It blocks a protein called epidermal growth factor receptor, or EGFR. Prior research has found that almost all head and neck cancer cells display an over-abundance of the protein, and it had been suggested in the literature as a potential target.
The VA-UAB team says, based on its lab studies, that honokiol binds more strongly with EGFR than does the drug gefitinib (sold as Iressa), which is commonly used to treat head and neck cancers.
The researchers tested honokiol on cell lines derived from human cancers of the oral cavity, larynx, tongue, and pharynx. In all cases, the botanical shut down the aberrant cells. The team also tested it against tumors implanted into mice, with similar results.

Senior author Dr. Santosh K. Katiyar and his colleagues wrote, “Conclusively, honokiol appears to be an attractive bioactive small molecule phytochemical for the management of head and neck cancer which can be used either alone or in combination with other available therapeutic drugs.”
Katiyar has published extensively in the past on other natural substances that work against tumors, especially skin cancer. Some of his recent work has focused on compounds in green tea, for example, and grape seed proanthocyanidins.
Purification
There are several methods for purifying and isolating honokiol. In nature, honokiol exists with its structural isomer magnolol, which differs from honokiol only by the position of onehydroxyl group. Because of the very similar properties of magnolol and honokiol, purification has often been limited to a HPLC or electromigration. However, methods developed in 2006 by workers in the lab of Jack L. Arbiser, took advantage of the proximity of the phenolic hydroxyl groups in magnolol, which form a protectable diol, to generate amagnolol acetonide (Figure 1), with a subsequent simple purification via flash chromatography over silica.[4]
Figure 1

Magnolol and Honokiol are normally inseparable. Honokiol is easily separable from the protected magnolol acetonide
Additionally a rapid separation approach was published in the Journal of Chromatography A in 2007. The process uses high-capacity high-speed countercurrent chromatography(high-capacity HSCCC).[5] Through this method honokiol can be separated and purified to above 98% purity with a high yield in under an hour.
Honokiol is a lignan isolated from the bark, seed cones, and leaves of trees belonging to the genus Magnolia. It has been identified as one the chemical compounds in some traditional eastern herbal medicines along with magnolol, 4-O-methylhonokiol, andobovatol.
Traditional medicine

Seed Cone
Extracts from the bark or seed cones of the Magnolia tree have been widely used in traditional medicine in China, Korea, and Japan.[2]
Houpu has traditionally been used in Eastern medicine as analgesic and to treat anxiety and mood disorders.[2][6] However, it has been shown to treat a number of other conditions. In China, magnolia bark is called Houpu and is most commonly taken from the Magnolia obovata and the Magnolia officinalis species.[7] Some Chinese traditional formulas containing Houpu include Banxia Houpu Tang (半夏厚朴丸), Xiao Zhengai Tang, Ping Wei San(平胃散) and Shenmi Tang.[2] Japanese Kampo formulas include, Hange-koboku-to (半夏厚朴湯) and Sai-boku-to (柴朴湯).[2][6]

Seeds
Modern medicine
In the late 1990s, honokiol saw a revival in interest as a potent and highly tolerable antitumorigenic and neurotrophiccompound.
Alternative medicine
Currently there are a large number of supplements containing honokiol on the market, and its use has been widely well received among practitioners of new age, homeopathic, and holistic medicine
|
Stereo image
|
||
|
||
|
||
|
||
|
||
| Mature Magnolia fruit just starting to open, with a few seeds visible |

 |
|
| Names | |
|---|---|
| IUPAC name
2-(4-hydroxy-3-prop-2-enyl-phenyl)- 4-prop-2-enyl-phenol
|
|
| Other names
houpa, hnk
|
|
| Identifiers | |
| 35354-74-6 |
|
| ChEMBL | ChEMBL16901 |
| ChemSpider | 65254 |
| Jmol-3D images | Image |
| KEGG | C10630 |
| PubChem | 72303 |
| Properties | |
| C18H18O2 | |
| Molar mass | 266.334 g/mol |
| Appearance | White solid |
| sparingly (25 °C) | |
| Related compounds | |
|
Related biphenols
|
diethylstilbestrol, dihydroxyeugenol |
|
Related compounds
|
magnolol. 4-O-Methylhonokiol |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
.JPG)
Magnolia seeds and fruit on a tree in northern Argentina

The root and stem bark of Magnolia has been used as a traditional Chinese medicine for the treatment of thrombotic stroke, gastrointestinal complaints, and anxiety. Honokiol (HNK), a substituted biphenyl and an active component isolated and purified from Magnolia, has anti-oxidant, antithrombosis, antibacterial, neurotrophic, xanthine oxidase inhibitory, and anxiolytic effects (Taira et al., Free Radic Res Commun. 1993;19 Suppl l:S71-77; Teng et al. Thromb Res. 1988;50:757-765; Clark et al., J. Pharm. Sci. 1981;70:951-952; Chang et al., Anticancer Res. 1994;14:501-506; Kuribara et al., J. Pharm Pharmacol. 1998;50:819-826; Esumi et al., Bioorg & Medicinal Chem Let 2004, 14: 2621-25).
In the early 1990s, reports of HNK’s anticancer effects were published. In 1994, Hirano et al (Life Sci. 1994;55(13): 1061-9) examined the anti leukemic-cell efficacy of 28 naturally occurring and synthetic flavonoids and 11 naturally occurring ligands on human promyelocytic leukemic cell line HL-60, and cytotoxicity of these compounds was compared with four clinical anti-cancer agents. HNK was identified as one of the most potent compounds in this screen, with an IC50 value less than 100 ng/ml. In 1998, Hibasami et al. demonstrated that HNK induced apoptosis in human lymphoid leukemia Molt 4B cells (Hibasami et al., Int. J. MoI. Med. 1998).
HNK has also been found to induce apoptosis in human squamous cell lung cancer CH27 cells (Yang SE, et al Biochem Pharmacol. 2002;63:1641-1651) and in human colorectal RKO cells (Wang et al World J Gastroenterol. 2004; 10:2205-2208). In 2004, Chen et al. (World J Gastroenterol. 2004; 10: 3459-3463) reported that HNK was effective in an in vivo animal model of human colon cancer by inhibiting tumor growth and prolonging the lifespan of tumor bearing mice.
Honokiol is an inhibitor of angiogenesis and antitumor activity in vivo. HNK can cause apoptosis in tumor cells and inhibit angiogenesis through blocking phosphorylation of vascular endothelial growth factor receptor 2 (VEGFR2), the major mitogenic and chemoattractant endothelial growth factor (Bai et al. (2003) J. Biol. Chem. 278, 35501- 35507). Honokiol also exhibits direct antitumor activity through induction of apoptosis through tumor necrosis factor apoptosis-inducing ligand (TRAIL/ Apo2L) signaling and has been found to be highly effective against angiosarcoma in nude mice in vivo (Bai et al. (2003) J. Biol. Chem. 278, 35501-35507).
Esumi et al. (Biorganic & Medicinal Chemistry Letters (2004) 14: 2621-2625) describe a synthesis method to produce HNK. This report also evaluates the structure activity relationship of O-methylated and/or its hydrogenated analogs of HNK in an in vitro neurotrophic assay. Esumi et al. conclude that the 5-allyl and 4′-hydroxyl groups are essential for the neurotrophic activity of HNK.
PCT Publication No. WO 02/076393 and U.S. Publication No. 2004/0105906 to Emory University describe pharmaceutical compositions and methods of treating conditions such angiogenic-, neoplastic-, and cancer-related conditions and skin conditions by administration of honokiol-type and/or magnolol-type compounds, as shown in Figures 1-4. For example, such compositions comprise at least one compound of formula Al :

AI wherein R1, R2, R3, R4, R5, R1, R2, R3, R4, and R’5 can be independently selected from groups that include, but are not limited to, hydrogen, hydroxyl groups, amides, amines, hydrocarbons, halogenated hydrocarbons, cyclic hydrocarbons, cyclic heterocarbons, halogenated cyclic heterocarbons, benzyl, halogenated benzyl, organo selenium compounds, sulfides, carbonyl, thiol, ether, dinitrogen ring compounds, thiophenes, pyridines, pyrroles, imidazoles, and pyrimidines. Honokiol-type and magnolol-type compounds are shown to inhibit SVR cell proliferation.
In November of 2004, Arbiser et al. reported that honokiol inhibited the growth of multuple myeloma cell lines via induction of Gl growth arrest, followed by apoptosis with IC50 values at 48h of 5 to 10 μg/mL. It was also reported that honokiol inhibited growth of doxorubin (Dox)-resistant (RPMI-Dox40), mephalan resistant (RPMI-LR5) and dexamethasone (Dex)-resistant (MM. IR) cell lines. It was suggested that the mechanism of honokiol triggered cytotoxicity is the honokiol induced increased expressin of Bax and Bad, down-regulated Mc-I protein expression, followed by caspase-8/9/3 cleavage, (Arbiser, J. et al. Poster at the American Society of Hematology Annual Meeting, 2004. Abstract published online November 4, 2004).
In July of 2005, Battle et al. reported that honokiol induces caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia (B-CLL) cells (Blood. July 2005; 106:690- 697). Honokiol induced caspase-dependent cell death in all of the B-CLL cells examined, which were primary tumor cells derived from B-CLL patients, and was more toxic toward B- CLL cells than to normal mononuclear cells. The honokiol-induced apoptosis was characterized by the activation of caspase-3, -8, and -9 and cleavage of poly(adenosine diphosphate-ribose) polymerase (PARP). It was also reported that honokiol enhanced cytotoxicity induced by fludarabine, cladribine, or chlorambucil.
In September 2005, Ishitsuka et al reported that honokiol overcomes conventional drug resistance in human multiple myeloma by induction of caspase-dependent and – independent apoptosis (Blood, 1 September 2005, Vol. 106, No. 5, pp.1794-1800). HNK induced cytotoxicity in human multiple myeloma (MM) cell lines and tumor cells from patients with relapsed refractory MM through induction of apoptosis via both caspase- dependent and -independent pathways. HNK also enhanced MM cell cytotoxicity and apoptosis induced by bortezomib.
It is an object of the present invention to provide new compounds, compositions, methods and uses for the treatment of disorders associated with angiogenesis, cell proliferation, tumor growth, tumorogenesis, and myeloma.

the intermediates for the synthesis of honokiol are 3-allyl-4- hydroxybenzeneboronate 5 and 4-allyl-2-bromophenol 9. The boronate 5 can be prepared from 2-iodophenol 1 by bromination, followed by Suzuki coupling to introduce the allyl group, and boronation under Suzuki conditions. Compound 9 can be prepared from 4- iodophenol 6 by bromination and allylation (Suzuki coupling). The coupling of 5 and 9 under Suzuki conditions can yield honokiol from Suzuki coupling, not other allyl-oriented products from the Heck reaction, as it was shown that Suzuki coupling can succeed in the presence of C=C double bond (see Miyaura, N.; Suzuki, A. (1995), Chem. Rev. 95, 2457- 2483; and Suzuki, A. (1999), J. Organometal. Chem. 576, 147-168, and the references cited therein). Thus, honokiol and derivatives can be synthesized from commercially available starting materials in 6 steps (Scheme 1). Scheme 1

Treatment of honokiol with TMS-diazomethane in methanol results in mono- and di- methylated compounds I-III, and hydrogenation of honokiol with Wilkinson’s catalyst yields di- and tetrahydrohonokiols VI-VIII, as reported by Esumi, T. et al. (2004), Bioorg. Med. Chern. Lett. 14, 2621-2625. The amino and fluoro analogues (IV and V) can be constructed from iodoacetanilide under Suzuki coupling conditions. From 2-iodoacetanilide 10, after bromination, allylation, and boronation, the boronated intermediate 13 can be prepared. The other bromo intermediate 16 can be prepared from 4-iodoacetanilide 14 via bromination and allylation. The coupling of boronate 13 and bromide 16 under Suzuki conditions can afford, after deprotection, the compound IV. Diazotization followed by Schiemann reaction can convert the amino analogue TV to fluoro analogue V (Scheme 2).

The dimethoxy honokiol derivative, III, can also be prepared, for example, by the treatment of honokiol with potassium carbonate, iodomethane. (Scheme 2a). The hydrogenated honokiol analog can alternatively be prepared by the hydrogenation of honokiol with sodium borohydride and nickel(II) chloride to yields tetrahydrohonokiols VI- Vπi. (Scheme 2a). Scheme 2a

The preparation of the vinyl analogue IX is based on combining the Wittig reaction with Suzuki coupling. The intermediate aldehyde 18 can be prepared from 4-iodophenol 17 via the Reimer-Tiemann reaction, while 3-bromo-4-hydroxybenzenealdehyde 23 can be prepared from para-hydroxybenzoic ester 21 via bromination and reduction. The Wittig reaction of these two aldehydes can yield the corresponding vinyl substituted benzenes 19 and 24. Compound 19 can afford the boronate 20, which can be coupled with 24, to yield the compound IX (Scheme 3).

Reagents and conditions: (a) CHCl3, aq. NaOH, 70 °C; (b) Ph3PCH3Br1 n-BuLi, THF; (c) PdC!2(dppf), dppf, KOAc, dioxane, bis(pinacoato)diboron, 80 °C; (d) DIBALH, -70 °C; (e) PdCI2(dppf), dppf, K3PO4, dioxane, reflux.
For the synthesis of honokiol analogues with changed positions of the allyl or hydroxyl groups, the boronate 5, and the bromophenols 4 and 9 can be used as intermediates. Suzuki coupling of one of these intermediates with an appropriate halide or boronate can provide the compounds X-XVII. Compounds X-XII and XTV-XV can be prepared by Suzuki coupling of boronate 5 with an appropriate halide. Halide 25, needed for compound X, can be prepared from 2-bromo-6-iodophenol 2 via allylation, while the intermediate, 5-allyl-2- bromophenol 29 for compound XI, can be furnished from 3-iodophenol 26 via bromination and allylation. The preparation of halide 5-allyl-3-bromophenol 33, an intermediate for the synthesis of compound XIV, requires an organothallium reagent. The thallation of 3- bromophenol 30 followed by treatment with iodide can yield 3-bromo-5-iodophenol 32. After allylation, the allyl-substituted intermediate 33 can be prepared. The synthesis of compound XII can begin with 2-iodoacetanilide 10, via sulfonation, nitration, and reduction to obtain the intermediate 36. Aniline 36, after diazotization, followed by acid and base treatments, will afford 2-amino-3-iodophenol 37. Diazotization, Sandmeyer reaction, and allylation of compound 37 will yield halide 39. By a coupling reaction of these halides (25, 29, 33, and 39) with boronate 5, these compounds (X-XII, and XTV) can be prepared. Compound XV can be synthesized by Suzuki coupling of halide 4 with boronate 5 (Scheme 4).
Scheme 4

Alternatively, compounds X, XV, and XVII can be synthesized by an allylation- Claisen pathway. Biphenol compounds can be reacted first with potassium carbonate and allyl bromide, followed by reaction with BCl3 to yield honokiol-like compounds, for example, X, XV, and XVII. (Scheme 4a). To a cooled solution (O0C with an ice bath) of diallyl starting material (1 eq.) in dry diehloromethane (Concentration of the solution : 0.1 mol.L“1) was added dropwise a solution Of BCl3 (IM in diehloromethane; 1.5 eq. = 0.75 eq for each allyl group). The reaction is then stirred at O0C until disappearance of the starting material on TLC (If after 15 minutes, the reaction is not complete, 1 more equivalent of BCl3 can be added). After hydrolysis with water (about same volume than diehloromethane), the two layers are separated. The organic layer is washed again with water, dried under MgSO4 then evaporated under vacuum. The residue is finally purified by column chromatography to give the di- hydroxy derivative . Scheme 4a

Bromide 9 is also a useful intermediate for coupling with some boronates. For example, Suzuki coupling of bromide 9 with boronate 42, which is prepared from 4-bromo-3- iodophenol 40 via allylation and boronation, can yield the compound XIII. Similarly, the coupling between bromide 9 and boronate 43 can afford the compound XVT. The compound XVII can be prepared from 4-allyl-2-bromophenol 9 via boronation followed by Suzuki coupling with 2-allyl-6-bromophenol 25 (Scheme 5).

The compounds XVIII and XIX can be synthesized from commercially available bisphenol 45 and the dihydroxynaphthalene-disulfuric acid salt 47. Thus, the bisphenol 45, through the Williamson reaction and Claisen rearrangement, can be converted to compound XVπi. Similarly, desulfonation of dihydroxynaphthalene-disulfuric acid salt .47, followed by the Williamson reaction and Claisen rearrangement, can produce the compound XIX (Scheme 6).Scheme 6

Dioxolane compounds can be prepared from magnoliol by reaction of magnoliol with 2,2′-dimethoxypropane and p-toluenesulfonic acid. (Scheme 7). This synthesis also provides a method of separating mixtures of honokiol and magnoliol. Scheme 7

The following examples are offered by way of illustration and not by way of limitation.
……..
http://www.google.com/patents/US20080300298

BEZ 235 (NVP-BEZ235), Dactolisib

BEZ235 (NVP-BEZ235)Dactolisib
4-[2,3-dihydro-3-methyl-2-oxo-8-(3-quinolinyl)-1H-imidazo[4, 5-c]quinolin-1-yl]-α,α-dimethyl-benzeneacetonitrile
2-methyl-2-{4-[3-methyl-2-oxo-8-(quinolin-3-yl)-1H,2H,3H-imidazo[4,5-c]quinolin-1-yl]phenyl}propanenitrile
2-Methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)- phenyl]-propionitrile
Chemical Formula: C30H23N5O
CAS Number: 915019-65-7
Molecular Weight: 469.54
PHASE 2, NOVARTIS
CANCER, BLADDER
NVP-BEZ235 is a dual inhibitor of phosphatidylinositol 3-kinase (P13K)and the downstream mammalian target of rapamycin (mTOR) by binding to the ATP-binding cleft of these enzymes. It specifically blocks the dysfunctional activation of the P13K pathway and induce G(1) arrest. NPV-BEZ235 has been shown to inhibit VEGF induced cell proliferation and survival in vitro and VEGF induced angiogenesis in vivo. It has also been shown to inhibit the growth of human cancer in animal models.
BEZ-235 is an orally active phosphatidylinositol 3-kinase (PI3K) inhibitor in early clinical trials at Novartis for the treatment of advanced breast cancer, renal cell carcinoma, solid tumors and castration-resistant prostate cancer. Phase I clinical trials were also under way at the company for the treatment of glioma, however, no developments in this indication has been reported. Phase II clinical trials are ongoing at Johann Wolfgang Goethe Universität for the treatment of relapsed or refractory acute leukemia.
PI3Ks perform various functions, promoting cell growth, proliferation, differentiation, motility, survival and intracellular trafficking. Mutations leading to increased activity of PI3Ks, including faulty production or action of PI3K antagonists, have been found in many cancers.

……………………………..
WO 2006122806
http://www.google.com/patents/WO2006122806A2?cl=en
![]()
…………………………..
WO 2008064093
2-methyl-2-[4-(3-methyl- 2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile of formula I (compound I),

Example 1
2-Methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)- phenyl]-propionitrile

In a suitable lab glass reactor are placed 45.0 g of starting 2[4-(8-bromo-3-methyl-2-oxo-2,3- dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]2-methyl-propionitrile together with 2.25 g of bistriphenylphosphine’palladium dichloride in 445 ml N,N-dimethylformamide. This mixture is heated to 95 0C and then a solution of 22.2 g of 3-quinoline boronic acid in a mixture of 225 ml DMF, 300 ml H2O and 60 g of KHCO3 is added. This mixture is heated for 2 h at 95 0C. Then 1080 ml H2O are added. The product 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl- 2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]propionitrile precipitates. The mixture is cooled within 1.5 h to 0 – 5 °C. After stirring at that temperature for 2 h the crude product is filtered and washed with 300 ml H2O. This product is dried in vacuo at 60 0C for 18 h, to yield crude product.
40 g of this crude product is dissolved in 200 ml formic acid at 60 0C. 8 g of active charcoal and Smopex 234 are added. The mixture is stirred at 60 0C for 1 h, the charcoal is filtered, the residue washed with 80 ml formic acid and then 175 ml formic acid are distilled off in vacuo. Then 320 ml methanol are added and the mixture is heated at reflux for 3 h. The purified product precipitates from the reaction mixture. The mixture is cooled to 0 – 5 0C within 1 h, then stirred 2 h at that temperature is finally filtered and washed with 80 ml cold methanol. This recrystallisation procedure is repeated again. Finally the twice recrystallised material is dried in vacuo at 60 0C to yield purified 2-Methyl-2-[4-(3-methyl-2-oxo-8-quinolin- 3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]propionitrile.
Example 1a 5-Bromo-2-(2-nitro-vinylamino)-benzoic acid

A suspension of 25 g (16 mmol) of 2-amino-5-bromo-benzoic acid (Fluka, Buchs, Switzerland) in H2O-HCI (37%) (10:1) is stirred for 8 h and then filtered (solution A). 8.17 g (255 mmol) of nitromethane (Fluka, Buchs, Switzerland) are added over 10 min to an ice- bath cooled mixture of 35 g of ice and 15.3 g (382 mmol) of NaOH. After stirring for 1 h at 0 0C and 1 h at rt, the solution is added at 0 0C to 28 g of ice and 42 ml of HCI (37%) (solution B). Solutions A and B are combined and the reaction mixture is stirred for 18 h at rt. The yellow precipitate is filtered off, washed with H2O and dried in vacuo at 400C to give the title compound. ES-MS: 287, 289 (M + H)+, Br pattern; 1H NMR (DMSO-d6): δ 13.7-14.6/br s (1 H), 12.94/d (1 H), 8.07/d (1 H), 8.03/dd (1 H), 7.83/dd (1 H), 7.71/d (1 H), 6.76/d (1 H).
Example 1b 6-Bromo-3-nitro-quinolin-4-ol

29 g (101 mmol) of 5-bromo-2-(2-nitro-vinylamino)-benzoic acid (Example 1a) and 11.9 g (121 mmol) of potassium acetate in 129 ml (152 mmol) of acetic anhydride are stirred for 1.5 h at 120 0C. The precipitate is filtered off and washed with acetic acid until the filtrate is colorless, then is washed with H2O and dried in vacuo to give the title compound. ES-MS: 269, 271 (M + H)+, Br pattern; analytical HPLC: W= 2.70 min (Grad 1).
Example 1c 6-Bromo-4-chloro-3-nitro-quinoline

20 g (74.3 mmol) of 6-bromo-3-nitro-quinolin-4-ol (Example 1b) in 150 ml (1.63 mol) of POCI3 are stirred for 45 min at 120 °C. The mixture is cooled to rt and poured slowly into ice- water. The precipitate is filtered off, washed with ice-cold water, and dissolved in CH2CI2. The organic phase is washed with cold brine, and the aqueous phase is discarded. After drying over MgSO4, the organic solvent is evaporated to dryness to provide the title compound. 1H NMR (CDCI3): J9.20/S (1H), 8.54/d (1H), 8.04/d (1H), 7.96/dd (1H); analytical HPLC: W= 4.32 min (Grad 1).
Example 1d 2-Methyl-2-(4-nitro-phenyl)-propionitrile
O .

To 15 g (92.5 mmol) of (4-nitro-phenyl)-acetonitrile (Fluka, Buchs, Switzerland), 1.64 mg (5.09 mmol) of tetrabutylammonium bromide (Fluka, Buchs, Switzerland) and 43.3 g (305 mmol) of iodomethane in 125 mL of CH2CI2 are added 1O g (250 mmol) of NaOH in 125 ml of water. The reaction mixture is stirred for 20 h at RT. After this time, the organic layer is separated, dried over MgSO4, and evaporated to dryness. The residue is dissolved in diethylether and treated with black charcoal for 30 min, filtered over Celite and evaporated in vacuo to give the title compound as a pale yellow solid. Analytical HPLC: tret= 3.60 minutes (Grad 1).Example 1e (2-(4-Amino-phenyl)-2-methyl-propionitrile

16 g (84.1 mmol) of 2-methyl-2-(4-nitro-phenyl)-propionitrile (Example 1d) and 4.16 g of Raney-Ni are shacked in 160 ml of THF-MeOH (1:1) under 1.1 bar of H2 for 12 h at rt. After completion of the reaction, the catalyst is filtered-off and the filtrate is evaporated to dryness. The residue is purified by flash chromatography on silica gel (hexane-EtOAc 3:1 to 1:2) to provide the title compound as an oil. ES-MS: 161 (M + H)+; analytical HPLC: tret= 2.13 minutes (Grad 1).
Example 1f 2-[4-(6-Bromo-3-nitro-quinolin-4-ylamino)-phenyl]-2-methyl-propionitrile

18 g (62.6 mmol) of 6-bromo-4-chloro-3-nitro-quinoline (Example 1c) and 11 g (68.9 mmol) of (2-(4-amino-phenyl)-2-methyl-propionitrile (Example 1e) are dissolved in 350 ml of acetic acid and stirred for 2 h. After this time, water is added and the yellow precipitate is filtered off and washed with H2O. The solid is dissolved in EtOAc-THF (1 :1), washed with sat. aqueous NaHCO3 and dried over MgSO4. The organic phase is evaporated to dryness to give the title compound as a yellow solid. ES-MS: 411 , 413 (M + H)+, Br pattern; analytical HPLC: tret= 3.69 min (Grad 1).
Example 1q 2-[4-(3-Amino-6-bromo-quinolin-4-ylamino)-phenyl]-2-methyl-propionitrile

24 g (58.4 mmol) of 2-[4-(6-bromo-3-nitro-quinolin-4-ylamino)-phenyl]-2-methyl-propionitrile (Example 1e) is shacked in 300 ml of MeOH-THF (1:1) under 1.1 bar of H2 in the presence of 8.35 g of Raney-Ni for 1 h. After completion of the reaction, the catalyst is filtered off and the filtrate is evaporated to dryness to give the title compound as a yellow foam. ES-MS: 381 , 383 (M + H)+, Br pattern; analytical HPLC: W= 3.21 min (Grad 1).
Example 1h
2-[4-(8-Bromo-2-oxo-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-2-methyl- propionitrile

A solution of 5 g (13.1 mmol) of 2-[4-(3-amino-6-bromo-quinolin-4-ylamino)-phenyl]-2- methyl-propionitrile (Example 1g) and 1.59 g (15.7 mmol) of triethylamine in 120 ml CH2CI2 is added over 40 min to a solution of 2.85 g (14.4 mmol) of trichloromethyl chloroformate (Fluka, Buchs, Switzerland) in 80 ml of CH2CI2 at 00C with an ice-bath. The reaction mixture is stirred for 20 min at this temperature then is quenched with sat. aqueous NaHCO3, stirred for 5 min and extracted with CH2CI2. The organic layer is dried over Na2SO4, filtered and evaporated in vacuo to give crude title compound as a brownish solid. ES-MS: 407, 409 (M + H)+, Br pattern; analytical HPLC: tret= 3.05 min (Grad 1). Example 1i
2-[4-(8-Bromo-3-methyl-2-oxo-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-2- methyl-propionitrile

To a solution of 3.45 g (8.47 mmol) of 2-[4-(8-bromo-2-oxo-2,3-dihydro-imidazo[4,5- c]quinolin-1-yl)-phenyl]-2-methyl-propionitrile (Example 1h), 1.8 g (12.7 mmol) of iodomethane (Fluka, Buchs, Switzerland) and 273 mg (0.847 mmol) of tetrabutylammonium bromide (Fluka, Buchs, Switzerland) in 170 ml of CH2CI2 is added a solution of 508 mg (12.7 mmol) of NaOH (Fluka, Buchs, Switzerland) in 85 ml of H2O. The reaction mixture is stirred for 2 days and 900 mg (6.35 mmol) of iodomethane and 254 mg (6.35 mmol) of NaOH in 5 ml of H2O are added. The reaction mixture is stirred for 1 day at rt . After this time, the reaction is quenched with H2O and extracted with CH2CI2 (2*). The organic layer is washed with brine, dried over Na2SO4, filtered and evaporated in vacuo to give the title compound as a beige solid. ES-MS: 421 , 423 (M + H)+, Br pattern; analytical HPLC: tret= 3.15 min (Grad 1).
Example 2
2-Methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)- phenyl]propionitrile p-toluenesulfonate salt
26.5 g of 2-Methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1- yl)-phenyl]propionitrile are placed together with 55 ml formic acid into a glass reactor. This mixture is heated to 60 0C to get a clear solution. This solution is clearfiltered and washed with 36 ml formic acid. Then formic acid is distilled off until the volume of the residual solution is 55 ml. Then a solution of 11.3 g of p-toluenesulfonic acid in 228 ml acetone is added at 50 0C, followed by further addition of 822 ml acetone within 30 minutes. The salt precipitates from the reaction mixture. The mixture is cooled to 0 0C within 2 h, stirred at that temperature for 3 h, is then filtered and washed with 84 ml acetone. The product‘ is dried at 60 0C in vacuo for 18 h to yield 29.8 g (82.4 %) of the 2-Methyl-2-[4-(3-methyl-2-oxo-8- quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]propionitrile p-toluenesulfonate salt (crystalline form A). The crystalline forms of the present invention are synthesized in accordance with the following examples which are illustrative without limiting the scope of the present invention.
Example 3:
Preparation of form A of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro- imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile
Form A of compound I can be manufactured in the following way: 241 g of free base are dissolved 2.4 I acetic acid at 50 0C. The solution is clearfiltered, washed with 250 ml acetic acid and then at 50 0C 7.2 I of water are added. The free base starts precipitating. The mixture is cooled within 1 h to 25 0C, is then filtered and washed with 10 I H2O. The free base is then dried in vacuo at 50 0C over night to yield 204 g of free base.
References
| WO2005054237A1 | 19 Nov 2004 | 16 Jun 2005 | Hans-Georg Capraro | 1h-imidazoquinoline derivatives as protein kinase inhibitors |
| WO2006122806A2 | 18 May 2006 | 23 Nov 2006 | Novartis Ag | 1,3-dihydro-imidazo [4,5-c] quinolin-2-ones as lipid kinase inhibitors |
| CL11872006A | Title not available |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2009118324A1 * | 24 Mar 2009 | 1 Oct 2009 | Novartis Ag | 5imidazoquinolines and pyrimidine derivatives as potent modulators of vegf-driven angiogenic processes |
| WO2013049300A1 * | 27 Sep 2012 | 4 Apr 2013 | Dana-Farber Cancer Institute, Inc. | Method of treating mucoepidermoid carcinoma |
| WO2013152717A1 | 9 Apr 2013 | 17 Oct 2013 | Shanghai Yunyi Healthcare Management Co., Ltd. | Fused pyrimidine compound, and preparation method, intermediate, composition, and uses thereof |
| EP2474323A2 * | 24 Mar 2009 | 11 Jul 2012 | Novartis AG | Imidazoquinolines and pyrimidine derivatives as potent modulators of vegf-driven angiogenic processes |
| US8476294 | 2 Jun 2010 | 2 Jul 2013 | Novartis Ag | 1H-imidazo[4,5-c]quinolinone derivatives |
ZSTK 474

4-[4-[2-(difluoromethyl)benzimidazol-1-yl]-6-morpholin-4-yl-1,3,5-triazin-2-yl]morpholine
ZSTK474; 475110-96-4; 4,4′-(6-(2-(Difluoromethyl)-1H-benzo[d]imidazol-1-yl)-1,3,5-triazine-2,4-diyl)dimorpholine; ZSTK-474; ZSTK 474; TCMDC-137004;
2-(2-Difluoromethylbenzimidazol-1-yl)-4,6-bis(morpholino)-1,3,5-triazine
2-(2-difluoromethylbenzimidazol-1-yl)-4,6-dimorpholino-1,3,5-triazine
Zenyaku Kogyo (Innovator)
phase2………Treatment of Solid Tumors Therapy
ZSTK474 is a cell permeable and reversible P13K inhibitor with an IC₅₀ at 6nm. It was identified as part of a screening library, selected for its ability to block tumor cell growth. ZSTK474 has shown strong antitumor activities against human cancer xenographs when administered orally to mice without a significant toxic effect.
Phosphatidylinositol 3-kinase (PI3K) has been implicated in a variety of diseases including cancer. A number of PI3K inhibitors have recently been developed for use in cancer therapy. ZSTK474 is a highly promising antitumor agent targeting PI3K. We previously reported that ZSTK474 showed potent inhibition against four class I PI3K isoforms but not against 140 protein kinases.
However, whether ZSTK474 inhibits DNA-dependent protein kinase (DNA-PK), which is structurally similar to PI3K, remains unknown. To investigate the inhibition of DNA-PK, we developed a new DNA-PK assay method using Kinase-Glo. The inhibition activity of ZSTK474 against DNA-PK was determined, and shown to be far weaker compared with that observed against PI3K. The inhibition selectivity of ZSTK474 for PI3K over DNA-PK was significantly higher than other PI3K inhibitors, namely NVP-BEZ235, PI-103 and LY294002.
Other Names: ZSTK-474
Chemical Formula: C19H21F2N7O2
CAS Number: 475110-96-4
Molecular Weight: 417.41
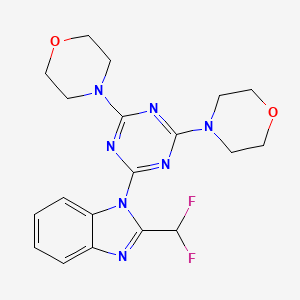
WO 2002088112
http://www.google.co.in/patents/EP1389617A1?cl=en
The condensation of 2,4-dichloro-6-(4-morpholinyl)-1,3,5-triazine

with 2-(difluoromethyl)-1H-benzimidazole by means of K2CO3 in DMF gives

2-chloro-4-[2-(difluoromethyl)-1H-benzimidazol-1-yl]-6-(4-morpholinyl)-1,3,5-triazine ,

which is then condensed with morpholine by means of K2CO3 in DMF to afford the target trisubstituted triazine.
aReagents and conditions: (i) K2CO3, DMF, room temp; (ii) morpholine, DMF or THF, room temp; (iii) NaH or K2CO3, DMF or DMSO, 120 °C.

- 2-(2-difluoromethylbenzimidazol-1-yl)-4,6-dimorpholino-1,3,5-triazine(compound 19)
Melting point: 211-214°C
NMR(CDCl3) δ : 3.79(8H, t, J=4Hz), 3.88(8H, t, J=4Hz), 7.3-7.4(2H, m), 7.56(1H, t, J=53Hz), 7.88(1H, d, J=7Hz), 8.32(1H, d, J=7Hz)
MS m/z: 417(M+
……………………
1H NMR (CDCl3) δ 8.33 (dd, J = 7.3, 1.4 Hz, 1H), 7.89 (dd, J = 7.2, 1.5 Hz, 1H), 7.56 (t, JHF= 53.6 Hz, 1H), 7.46–7.37 (m, 2H), 3.91–3.86 (m, 8H), 3.81–3.76 (m, 8H).
TRIAZINE, PYRIMIDINE AND PYRIDINE ANALOGS AND THEIR USE AS THERAPEUTIC AGENTS AND DIAGNOSTIC PROBES [US2011275762]2011-11-10
| Patent | Submitted | Granted |
|---|---|---|
| Heterocyclic compound and antitumor agent containing the same as active ingredient [US7071189] | 2004-06-17 | 2006-07-04 |
| Treatment of prostate cancer, melanoma or hepatic cancer [US2007244110] | 2007-10-18 | |
| Heterocyclic compound and antitumor agent containing the same as effective ingredient [US7307077] | 2006-11-02 | 2007-12-11 |
| IMMUNOSUPPRESSIVE AGENT AND ANTI-TUMOR AGENT COMPRISING HETEROCYCLIC COMPOUND AS ACTIVE INGREDIENT [US7750001] | 2008-05-15 | 2010-07-06 |
| PYRIMIDINYL AND 1,3,5-TRIAZINYL BENZIMIDAZOLES AND THEIR USE IN CANCER THERAPY [US2011009405] | 2011-01-13 | |
| SUBSTITUTED PYRIMIDINES AND TRIAZINES AND THEIR USE IN CANCER THERAPY [US2011053907] | 2011-03-03 | |
| IMMUNOSUPPRESSIVE AGENT AND ANTI-TUMOR AGENT COMPRISING HETEROCYCLIC COMPOUND AS ACTIVE INGREDIENT [US2010267700] | 2010-10-21 | |
| AMORPHOUS BODY COMPOSED OF HETEROCYCLIC COMPOUND, SOLID DISPERSION AND PHARMACEUTICAL PREPARATION EACH COMPRISING THE SAME, AND PROCESS FOR PRODUCTION OF THE SAME [US8227463] | 2010-09-30 | 2012-07-24 |
| PYRAZOLO[1,5-a]PYRIDINES AND THEIR USE IN CANCER THERAPY [US2010226881] | 2010-09-09 | |
| PYRIMIDINYL AND 1,3,5-TRIAZINYL BENZIMIDAZOLE SULFONAMIDES AND THEIR USE IN CANCER THERAPY [US2010249099] | 2010-09-30 |
…………..
Zenyaku Kogyo






Evofosfamide

Evofosfamide, HAP-302 , TH-302
 |
|
| Names | |
|---|---|
| IUPAC name
(1-Methyl-2-nitro-1H-imidazol-5-yl)methyl N,N’-bis(2-bromoethyl)phosphorodiamidate
|
|
| Other names
TH-302; HAP-302
|
|
| Identifiers | |
| 918633-87-1 |
|
| ChemSpider | 10157061 |
| Jmol-3D images | Image |
| PubChem | 11984561 |
| Properties | |
| C9H16Br2N5O4P | |
| Molar mass | 449.04 g·mol−1 |
| 6 to 7 g/l | |
TH-302 is a nitroimidazole-linked prodrug of a brominated derivative of an isophosphoramide mustard previously used in cancer drugs
evofosfamide (first disclosed in WO2007002931), useful for treating cancer.
Threshold Pharmaceuticals and licensee Merck Serono are codeveloping evofosfamide, the lead in a series of topoisomerase II-inhibiting hypoxia-activated prodrugs and a 2-nitroimidazole-triggered bromo analog of ifosfamide, for treating cancer, primarily soft tissue sarcoma and pancreatic cancer (phase 3 clinical, as of April 2015).
In November 2014, the FDA granted Fast Track designation to the drug for the treatment of previously untreated patients with metastatic or locally advanced unresectable soft tissue sarcoma.
Evofosfamide (INN,[1] USAN;[2] formerly known as TH-302) is an investigational hypoxia-activated prodrug that is in clinical development for cancer treatment. The prodrug is activated only at very low levels of oxygen (hypoxia). Such levels are common in human solid tumors, a phenomenon known as tumor hypoxia.[3]
Evofosfamide is being evaluated in clinical trials for the treatment of multiple tumor types as a monotherapy and in combination with chemotherapeutic agents and other targeted cancer drugs
Discovered at Threshold, TH-302 is a hypoxia-activated prodrug (HAP) designed to exploit low oxygen levels in hypoxic tumor regions. Therapeutics that specifically target resistant hypoxic zones could provide significant additional antitumor activity and clinical benefit over current chemotherapeutic and radiation therapies.
Evofosfamide (TH-302) was developed by Threshold Pharmaceuticals Inc. (Threshold).[4] The company is located in South San Francisco, CA, USA.
In 2012, Threshold signed a global license and co-development agreement for evofosfamide with Merck KGaA, Darmstadt, Germany, which includes an option for Threshold to co-commercialize eofosfamide in the United States. Threshold is responsible for the development of evofosfamide in the soft tissue sarcoma indication in the United States. In all other cancer indications, Threshold and Merck KGaA are developing evofosfamide together.[5] From 2012 to 2013, Merck KGaA paid 110 million US$ for upfront payment and milestone payments to Threshold. Additionally, Merck KGaA covers 70% of all evofosfamide development expenses.[6]
Discovered at Threshold, TH-302 is a hypoxia-activated prodrug (HAP) designed to exploit low oxygen levels in hypoxic tumor regions. Therapeutics that specifically target resistant hypoxic zones could provide significant additional antitumor activity and clinical benefit over current chemotherapeutic and radiation therapies.
History
| Date | Event |
|---|---|
| Jun 2005 | Threshold files evofosfamide (TH-302) patent applications in the U.S.[49] |
| Jun 2006 | Threshold files a evofosfamide (TH-302) patent application in the EU and in Japan[50] |
| Sep 2011 | Threshold starts a Phase 3 trial (TH-CR-406) of evofosfamide in combination with doxorubicin in patients with soft tissue sarcoma |
| Feb 2012 | Threshold signs an agreement with Merck KGaA to co-develop evofosfamide |
| Apr 2012 | A Phase 2b trial (TH-CR-404) of evofosfamide in combination with gemcitabine in patients with pancreatic cancer meets primary endpoint |
SEE
WO2007002931
http://www.google.com/patents/WO2007002931A2?cl=en
Example 8
Synthesis of Compounds 25, 26 [0380] To a solution of 2-bromoethylammmonium bromide (19.4 g) in DCM (90 mL) at – 1O0C was added a solution OfPOCl3 (2.3 mL) in DCM (4 mL) followed by addition of a solution of TEA (14.1 mL) in DCM (25 mL). The reaction mixture was filtered, the filtrate concentrated to ca. 30% of the original volume and filtered. The residue was washed with DCM (3×25 mL) and the combined DCM portions concentrated to yield a solid to which a mixture of THF (6 mL) and water (8 mL) was added. THF was removed in a rotary evaporator, the resulting solution chilled overnight in a fridge. The precipitate obtained was filtered, washed with water (10 mL) and ether (30 mL), and dryed in vacuo to yield 2.1 g of:

Isophosphoramide mustard

can be synthesized employing the method provided in Example 8, substituting 2- bromoethylammmonium bromide with 2-chloroethylammmonium chloride. Synthesis of Isophosphoramide mustard has been described (see for example Wiessler et al., supra).
The phosphoramidate alkylator toxin:

was transformed into compounds 24 and 25, employing the method provided in Example 6 and the appropriate Trigger-OH.
Example 25
Synthesis of l-N-methyl-2-nitroimidazole-5-carboxylis acid

A suspension of the nitro ester (39.2 g, 196.9 rnmol) in IN NaOH (600 mL) and water (200 mL) was stirred at rt for about 20 h to give a clear light brown solution. The pH of the reaction mixture was adjusted to about 1 by addition of cone. HCl and the reaction mixture extracted with EA (5 x 150 mL). The combined ethyl acetate layers were dried over MgS O4 and concentrated to yield l-N-methyl-2-nitroimidazole-5-carboxylis acid (“nitro acid”) as a light brown solid (32.2 g, 95%). Example 26
Synthesis of l-N-methyl-2-nitroimidazole-5-carboxylis acid

A mixture of the nitro acid (30.82 g, 180.23 mmol) and triethylamine (140 niL, 285 mmol) in anhydrous THF (360 mL) was stirred while the reaction mixture was cooled in a dry ice-acetonitrile bath (temperature < -20 0C). Isobutyl chloroformate (37.8 mL, 288 mmol) was added drop wise to this cooled reaction mixture during a period of 10 min and stirred for 1 h followed by the addition of sodium borohydride (36 g, 947 mmol) and dropwise addition of water during a period of 1 h while maintaining a temperature around or less than O0C. The reaction mixture was warmed up to O0C. The solid was filtered off and washed with THF. The combined THF portions were evaporated to yield l-N-methyl-2- nitroimidazole-5-methanol as an orange solid (25 g) which was recrystallized from ethyl acetate.
……………………………………….
WO-2015051921

EXAMPLE 1

1
N-Formylsarcosine ethyl ester 1 (1 ,85 kg) was dissolved in toluene (3,9 kg) and ethyl formate (3,28 kg) and cooled to 10 °C. A 20 wt-% solution of potassium tert-butoxide (1 ,84 kg) in tetrahydrofuran (7,4 kg) was added and stirring was continued for 3h. The reaction mixture was extracted 2x with a solution of sodium chloride in water (10 wt-%) and the combined water extracts were washed lx with toluene.
Aqueous hydrogen chloride (25% wt-%; 5,62 kg) was added to the aqueous solution, followed by ethylene glycol (2,36 kg). The reaction mixture was heated to 55-60 °C for lh before only the organic solvent residues were distilled off under vacuum.
Aqueous Cyanamide (50 wt-%, 2,16 kg) was then added at 20 °C, followed by sodium acetate (3,04 kg). The resulting reaction mixture was heated to 85-90 °C for 2h and cooled to 0-5 °C before a pH of ~ 8-9 was adjusted via addition of aqueous sodium hydroxide (32% wt-%; 4,1 kg). Compound 3 (1,66 kg; 75%) was isolated after filtration and washing with water.
Ή-NMR (400 MHz, d6-DMSO): δ= 1,24 (3H, t, J= 7,1 Hz); 3,53 (3H, s); 4,16 (2H, q, J= 7,0 Hz) ; 6,15 (s, 2 H); 7,28 (s, 1H).
HPLC (Rt = 7,7 min): 97,9% (a/a).
REFERENCES
1
- WHO Drug Information; Recommended INN: List 73
- 2
- Adopted Names of the United States Adopted Names Council
- 3
- Duan J; Jiao, H; Kaizerman, J; Stanton, T; Evans, JW; Lan, L; Lorente, G; Banica, M et al. (2008). “Potent and Highly Selective Hypoxia-Activated Achiral Phosphoramidate Mustards as Anticancer Drugs”. J. Med. Chem. 51 (8): 2412–20. doi:10.1021/jm701028q. PMID 18257544.
- 4
- Website of Threshold Pharmaceuticals Inc.
- 5
- Threshold Pharmaceuticals and Merck KGaA Announce Global Agreement to Co-Develop and Commercialize Phase 3 Hypoxia-Targeted Drug TH-302 – Press release from 3 February 2012
- 6
Threshold Pharmaceuticals Form 8-K from 3 Nov 2014
DHAKA BANGLADESH




 .
.
Steamers and ferries in Sadarghat Port
 Kawran Bazar
Kawran Bazar
 .
.
 Dry fish sellers at the Karwan Dry Fish Market (Bazar), Dhaka, Bangladesh.
Dry fish sellers at the Karwan Dry Fish Market (Bazar), Dhaka, Bangladesh.

Tazemetostat

Tazemetostat
Current developer: Epizyme, Inc., Cambridge, MA 02139.
EPZ-6438 (Tazemetostat)
CAS: 1403254-99-8
HBR 1467052-75-0
タゼメトスタット臭化水素酸塩
Current developer: Epizyme, Inc., Cambridge, MA 02139.
EPZ-6438 (Tazemetostat)
CAS: 1403254-99-8
HBR
Chemical Formula: C34H44N4O4
Exact Mass: 572.33626
USFDA APPROVED 23/1/2020 AS HBR SALT, TAZVERIK, EPIZYME
N-((4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[1,1′-biphenyl]-3-carboxamide
SIMLES: O=C(C1=CC(C2=CC=C(CN3CCOCC3)C=C2)=CC(N(CC)C4CCOCC4)=C1C)NCC5=C(C)C=C(C)NC5=O
(1,1′-Biphenyl)-3-carboxamide, N-((1,2-dihydro-4,6-dimethyl-2-oxo-3-pyridinyl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(4-morpholinylmethyl)-
N-((4,6-Dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-5-(ethyl(oxan-4-yl)amino)-4-methyl-4′-((morpholin-4-yl)methyl)(1,1′-biphenyl)-3-carboxamide
UNII-Q40W93WPE1
Tazemetostat, sold under the brand name Tazverik, is a medication used for the treatment of adults and adolescents aged 16 years and older with metastatic (when cancer cells spread to other parts of the body) or locally advanced (when cancer has grown outside the organ it started in, but has not yet spread to distant parts of the body) epithelioid sarcoma not eligible for complete resection (surgically removing all of a tissue, structure, or organ).[1]
Tazemetostat is a cancer drug that acts as a potent selective EZH2 inhibitor.[2]
Tazemetostat blocks activity of the EZH2 methyltransferase, which may help keep the cancer cells from growing.[1] Most cases of epithelioid sarcoma begin in the soft tissue under the skin of an extremity, though it can start in other areas of the body.[1] Surgical removal is considered the main treatment when the cancer is localized to one area of the body.[1] Chemotherapy or radiation may also be given.[1] However, there is a high likelihood for local and regional spread of the disease even with treatment and approximately 50% of patients have metastatic disease at the time of diagnosis.[1] Metastatic disease is considered life-threatening to the patient.[1]
The most common side effects are pain, fatigue, nausea, decreased appetite, vomiting and constipation.[1] People taking tazemetostat are at increased risk of developing secondary malignancies including: T-cell lymphoblastic lymphoma (a type of blood cancer that affects the lymphatic system usually found in the lymph nodes), myelodysplastic syndrome (a disorder resulting from poorly formed or dysfunctional blood cells) and acute myeloid leukemia (a cancer of the blood and bone marrow).[1]
According to the NCI Drug Dictionary, “tazemetostat is an orally available, small molecule selective and S-adenosyl methionine (SAM) competitive inhibitor of histone methyl transferase EZH2, with potential antineoplastic activity. Upon oral administration, tazemetostat selectively inhibits the activity of both wild-type and mutated forms of EZH2. Inhibition of EZH2 specifically prevents the methylation of histone H3 lysine 27 (H3K27). This decrease in histone methylation alters gene expression patterns associated with cancer pathways and results in decreased tumor cell proliferation in EZH2 mutated cancer cells. EZH2, which belongs to the class of histone methyltransferases (HMTs), is overexpressed or mutated in a variety of cancer cells and plays a key role in tumor cell proliferation.”[3]
History
The U.S. Food and Drug Administration (FDA) approved tazemetostat in January 2020,[1] based on the results of a clinical trial (NCT02601950) enrolling 62 subjects with metastatic or locally advanced epithelioid sarcoma.[1][4] During the clinical trial, subjects received 800 milligrams (mg) of tazemetostat twice a day until the disease progressed or the subject reached an unacceptable level of toxicity.[1][4] Tumor response assessments were performed every eight weeks during the clinical trial.[1] The trial measured how many subjects experienced complete or partial shrinkage (by a certain amount) of their tumors during treatment (overall response rate).[1] The overall response rate was 15%, with 1.6% of subjects having a complete response and 13% having a partial response.[1] Of the nine subjects that had a response, six (67%) subjects had a response lasting six months or longer.[1]
The trial was conducted at 22 sites in France, United Kingdom, Taiwan, Italy, Canada, Belgium, and the United States.[4]
The FDA granted the application for tazemetostat accelerated approval and orphan drug designation.[1] The FDA granted the approval of Tazverik to Epizyme Inc.[1]
PATENT
PRODUCT PAT
US 8410088 EXP 21/1/2034
WO 2012142504
US 9090562 EXP 13/4/32
SEE Proceedings of the National Academy of Sciences of the United States of America (2013), 110(19), 7922-7927, S7922/1-S7922/5….http://www.pnas.org/content/110/19/7922.abstract
http://www.epizyme.com/wp-content/uploads/2014/11/Ribrag-ENA-FINAL.pdf
Tazemetostat, also known as EPZ-6438, is a potent, selective, and orally bioavailable small-molecule inhibitor of EZH2 enzymatic activity. EPZ-6438 induces apoptosis and differentiation specifically in SMARCB1-deleted MRT cells.
Treatment of xenograft-bearing mice with EPZ-6438 leads to dose-dependent regression of MRTs with correlative diminution of intratumoral trimethylation levels of lysine 27 on histone H3, and prevention of tumor regrowth after dosing cessation.
These data demonstrate the dependency of SMARCB1 mutant MRTs on EZH2 enzymatic activity and portend the utility of EZH2-targeted drugs for the treatment of these genetically defined cancers. EPZ-6438 is currently in clinical trials.
Epizyme, Inc., Eisai R&D Management Co.Ltd.
Epizyme is developing tazemetostat, a lead from several small molecule EZH2 inhibitors, for treating cancer (phase 1 clinical, as of April 2015). Japanese licensee Eisai was developing the program for the potential oral treatment of cancers, including non-Hodgkin’s lymphoma; however, in March 2015, Epizyme regained worldwide, ex-Japan, rights to the program.
It appeared that Eisai was planning to investigate the program in Japan .
WO-2015057859 From, Eisai Research Institute; Epizyme Inc, indicates Novel crystalline polymorphic form C of tazemetostat, useful for treating an EZH2-mediated cancer, including non-Hodgkin’s lymphoma and breast cancer.
see WO2013155317, claiming novel hydrobromide salt of tazemetostat.
PREDICT

………………………………….
PATENT
WO 2012142504
http://www.google.com/patents/WO2012142504A1?cl=en

Example 44: Synthesis of N-((4,6-dimethyl-2-oxo-l ,2-dihydropyridin-3- yl)methyl)-5-(ethyl (tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(moφholinomethyl)-[l , – biphenyl]-3-carboxamide

Compound 44
[Step 1 : Synthesis of 5-brom -2-methyl-3-nitrobenzoic acid

To stirred solution of 2-methyl-3-nitrobenzoic acid ( 100 g, 552 mmol) in cone. H2S04 (400 mL), 1 ,3-dibromo-5,5-dimethyl-2,4-imidazolidinedione (88 g, 308 mmol) was added in a portion wise manner at room temperature and the reaction mixture was then stirred at room temperature for 5 h. The reaction mixture was poured onto ice cold water, the precipitated solid was filtered off, washed with water and dried under vacuum to afford the desired compound as a solid ( 140 g, 98%). The isolated compound was taken directly into the next step. Ή NMR (DMSO-4$, 400 MHz) δ 8.31 (s, 1 H), 8.17 (s, 1 H), 2.43 (s, 3H).
Step 2: Synthesis of methyl -bromo-2-methyl-3-nitrobenzoate

To a stirred solution of 5-bromo-2-methyl-3-nitrobenzoic acid (285 g, 1 105 mmol) in DMF (2.8L) at room temperature was added sodium carbonate (468 g, 4415 mmol) followed by addition of methyl iodide (626.6 g, 4415 mmol). The resulting reaction mixture was heated at 60 °C for 8 h. After completion (monitored by TLC), the reaction mixture was filtered (to remove sodium carbonate) and washed with ethyl acetate ( 1 L X 3). The combined filtrate was washed with water (3L X 5) and the aqueous phase was back extracted with ethyl acetate (1L X 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the title compound as a solid (290g, 97% yield). The isolated compound was taken directly into the next step. Ή NMR (CDC13, 400 MHz) δ 8.17 (s, 1H), 7.91 (s, 1H), 3.96 (s, 3H), 2.59 (s, 3H).
Step 3: Synthesis of methyl 3-amino-5-bromo-2-methylbenzoate

To a stirred solution of methyl 5-bromo-2-methyl-3-nitrobenzoate (290 g,
1058 mmol) in ethanol (1 .5L) was added aqueous ammonium chloride (283 g, 5290 mmol dissolved in 1.5L water). The resulting mixture was stirred at 80°C to which iron powder (472 g, 8451 mmol) was added in a portion wise manner. The resulting reaction mixture was heated at 80 °C for 12 h. Upon completion as determined by TLC, the reaction mixture was hot filtered over celite® and the celite bed was washed with methanol (5L) followed by washing with 30% MeOH in DCM (5L). The combined filtrate was concentrated in-vacuo, the residue obtained was diluted with aqueous sodium bicarbonate solution (2L) and extracted with ethyl acetate (5L X 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the title compound as a solid (220 g, 85%). The compound was taken directly into the next step. Ή NMR (CDC13, 400 MHz) δ 7.37 (s, 1 H), 6.92 (s, 1 H), 3.94 (s, 3H), 3.80 (bs, 2H), 2.31 (s, 3H).
Step 4: Synthesis of methyl 5-bromo-2-methyl-3-((tetrahydro-2H-pyran-4-yl) amino) benzoate

To a stirred solution of methyl 3-amino-5-bromo-2-methylbenzoate (15 g, 61 .5 mmol) and dihydro-2H-pyran-4(3)-one (9.2 g, 92 mmol) in dichloroethane (300 mL) was added acetic acid (22 g, 369 mmol) and the reaction mixture stirred at room temperature for 15 minutes, then the reaction mixture was cooled to 0°C and sodium triacetoxyborohydnde (39 g, 184 mmol) was added. The reaction mixture was stirred overnight at room temperature. Upon completion of the reaction as determined by TLC, aqueous sodium bicarbonate solution was added to the reaction mixture until a pH of 7-8 was obtained. The organic phase was separated and the aqueous phase was extracted with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography (100-200 mesh silica gel) eluting with ethyl acetate: hexane to afford the desired compound as a solid ( 14 g, 69%). ‘H NMR (DMSO-<fc, 400 MHz) δ 7.01 (s, 1 H), 6.98 (s, 1 H), 5.00 (d, 1 H, J=7.6 Hz), 3.84-3.87 (m, 2H), 3.79 (s, 31 1), 3.54-3.56 (mf 1 H), 3.43 (L 21 1, J 12 Hz), 2.14 (s. 31 1). 1 . 1 – 1 .84 (m: 211). 1 .47- 1 .55 (m, 2H).
Step 5: Synthesis of methyl 5-bromo-3-(ethyl (tetrahydro-2H-pyran-4-yl) amino)-2- methylbenzoate

To a stirred solution of methyl 5-bromo-2-methyl-3-((tetrahydro-2H-pyran-4-yl) amino) benzoate (14 g, 42.7 mmol) in dichloroethane (150 mL) was added acetaldehyde (3.75 g, 85.2 mmol) and acetic acid ( 15.3 g, 256 mmol). The resulting reaction mixture was stirred at room temperature for 15 minutes. The mixture was cooled to 0 °C and sodium
triacetoxyborohydnde (27 g, 128 mmol) was added. The reaction mixture was stirred at room temperature for 3 hours. Upon completion of the reaction as determined by TLC, aqueous sodium bicarbonate solution was added to the reaction mixture until a pH 7-8 was obtained, the organic phase was separated and the aqueous phase was extracted with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography (100- 200 mesh silica gel) eluting with ethyl acetate: hexane to afford the desired compound as a viscous liquid (14 g, 93%). Ή NMR (DMSO-cfo 400 MHz) δ 7.62 (s, 1 H), 7.52 (s, 1 H), 3.80 (bs, 5H), 3.31 (t, 2H), 2.97-3.05 (m, 2H), 2.87-2.96 (m, 1 H), 2.38 (s, 3H), 1.52-1.61 (m, 2H), 1 .37-1.50 (m, 2H), 0.87 (t, 3H, J=6.8 Hz).
Step 6: Synthesis of 5-bromo-N-((4, 6-dimethyl-2-oxo-l , 2-dihydropyridin-3-yl) methyl)-3 -(ethyl (tetrahydro-2H-pyra -4-yl) amino)-2-methylbenzamide

To a stirred solution of 5-bromo-3-(ethyl (tetrahydro-2H-pyran-4-yl) amino)-2- methylbenzoate (14 g, 39.4 mmol) in ethanol ( 100 mL) was added aqueous NaOH (2.36 g, 59.2 mmol in 25mL water) and the resulting mixture was stirred at 60 °C for 1 h. Upon completion of the reaction as determined by TLC, the solvent was removed under reduced pressure and the residue obtained was acidified with IN HC1 until a pH 7 was obtained and then aqueous citric acid solution was added until a pH 5-6 was obtained. The aqueous layer was extracted with 10% MeOH in DCM (200mL X 3), the combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give the respective acid (14 g, 100%).
The above acid (14 g, 40.9 mmol) was then dissolved in DMSO (70 mL) and 3- (amino methyl)-4, 6-dimethylpyridin-2( l H)-one ( 12.4 g, 81 .9 mmol) was added to it. The reaction mixture was stirred at room temperature for 15 minutes, then PYBOP (31.9 g, 61.4 mmol) was added and stirring was continued for overnight at room temperature. Upon completion of the reaction as determined by TLC, the reaction mixture was poured onto ice- cold water (700 mL), stirred for 30 minutes and the precipitated solid was collected by filtration, washed with water (500 mL) and air dried. The solid obtained was stirred with acetonitrile (75mL X 2), filtered and air dried. The solid obtained was again stirred with 5% MeOH in DCM ( l OOmL), filtered and dried completely under vacuum to afford the title compound as a solid ( 14 g, 74 %). Ή NMR (DMSO- 6, 400 MHz) δ 1 1.47 (s, 1 H), 8.23 (t, 1 H), 7.30 (s, 1 H), 7.08 (s, 1 H), 5.85 (s, 1 H), 4.23 (d, 2H, J=4.4 Hz), 3.81 (d, 2H, J=l 0.4 Hz), 3.20-3.26 (m, 2H), 3.00-3.07 (m, I H), 2.91 -2.96 (m, 2H), 2.18 (s, 3H), 2.14 (s, 3H), 2.10 (s, 3H), 1.58-1.60 (m, 2H), 1.45-1.50 (m, 2H), 0.78 (t, 3H, J=6.8 Hz).
Step 7: Synthesis of N-((4, 6-dimethyl-2-oxo-l , 2-dihydropyridin-3-yl) methyl)-5- (ethyl (tetrahydro-2H-pyran-4-yl) amino)-4-methyl-4′-(morpholinomethyl)-[l , l ‘-biphenyl]-3- carboxamide
 TITLE COMPD
TITLE COMPDTo a stirred solution of 5-bromo-N-((4, 6-dimethyl-2-oxo-l , 2-dihydropyridin-3-yl) methyl)-3-(ethyl (tetrahydro-2H-pyran-4-yl) amino)-2-methylbenzamide (14 g, 29.5 mmol) in dioxane/ water mixture (70 mL/ 14 mL) was added 4-(4-(4, 4, 5, 5-tetramethyl- l , 3, 2- dioxaborolan-2-yl) benzyl) morpholine (13.4 g, 44.2 mmol) followed by addition of Na2C03 (1 1 .2 g, 106.1 mmol). The solution was purged with argon for 15 minutes and then Pd (PPh3)4 (3.40 g, 2.94 mmol) was added and the solution was again purged with argon for a further 10 min. The reaction mixture was heated at 100°C for 4 h. After completion (monitored by TLC), the reaction mixture was diluted with water and extracted with 10% MeOH/DCM.
The combined organic layers were dried over anhydrous sodium sulphate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography (100- 200 mesh silica gel) eluting with methanol: DCM to the title compound as a solid (12 g, 71 %).
Analytical Data: LCMS: 573.35 (M + 1 )+; HPLC: 99.5% (@ 254 nm) (R,;3.999; Method: Column: YMC ODS-A 1 50 mm x 4.6 mm x 5 μ; Mobile Phase: A; 0.05% TFA in water/ B; 0.05% TFA in acetonitrile; Inj. Vol : 10 μΐ, Col. Temp.: 30 °C; Flow rate: 1 .4 mL/min.;
Gradient: 5% B to 95% B in 8 min, Hold for 1 .5 min, 9.51 -12 min 5% B);
Ή NMR (DMSO-i 6, 400 MHz) 5 1 1 .46 (s, I H), 8. 19 (t, 1 H), 7.57 (d, 2H, J=7.2 Hz), 7.36-7.39 (m, 3H), 7.21 (s, I H), 5.85 (s, I H), 4.28 (d, 2H, J=2.8 Hz), 3.82 (d, 2H, J=9.6 Hz), 3.57 (bs, 4H), 3.48 (s, 2H), 3.24 (t, 2H, J=10.8Hz), 3.07-3.09 (m, 2H), 3.01 (m, I H), 2.36 (m, 4H), 2.24 (s, 3H), 2.20 (s, 3H), 2.10 (s, 3H), 1 .64-1 .67 (m, 2H), 1 .51 – 1 .53 (m, 2H), 0.83 (t, 3H, J=6.4 Hz).
TRIHYDROCHLORIDE
Step 8: Synthesis of N-((4,6-dimethyl-2-oxo-l ,2-dihydropyridin-3-yl)methyl)-5- (ethyl (tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[ 1 , 1 ‘-biphenyl]-3- carboxamide trihydrochloride

N-((4, 6-dimethyl-2-oxo-l , 2-dihydropyridin-3-yl) methyl)-5-(ethyl (tetrahydro- 21 l-pyran-4-yl) amino)-4-methyI-4′-(niorpholinornethyl)-[ 1 , 1 ‘-biphenyl]-3-carboxamide ( 12 g, 21.0 mmol) was dissolved in methanolic HC1 (200 mL) and stirred at room temperature for 3 h. After three hours of stirring, the reaction mixture was concentrated under reduced pressure. The solid obtained was stirred with ether ( l OOmL X 2) to afford the desired salt as a solid ( 1 1 g, 77 %).
Analytical Data of the tri-HCl salt: LCMS: 573.40 (M + 1 )+; HPLC: 99.1 % (@ 254 nm) (R,;3.961 ; Method: Column: YMC ODS-A 150 mm x 4.6 mm x 5 μ; Mobile Phase: A; 0.05% TFA in water/ B; 0.05% TFA in acetonitrile; Inj. Vol: 10 pL, Col. Temp.: 30 °C; Flow rate: 1.4 mL/min.; Gradient: 5% B to 95% B in 8 min, Hold for 1.5 min, 9.51 -12 min 5% B);
Ή NMR (D20 400 MHz) δ 7.92 (bs, I H,) 7.80 (s, I H), 7.77 (d, 2H, J=8 Hz), 7.63 (s, I H), 7.61 (s, I H), 6.30 (s, I H), 4.48 (s, 2H), 4.42 (s, 2H), 4.09-4.1 1 (m, 4H), 3.95-3.97 (m, 2H), 3.77 (t, 3H, J=10.4 Hz), 3.44-3.47 (m, 3H), 3.24-3.32 (m, 3H), 2.42 (s, 3H), 2.35 (s, 3H), 2.26 (s, 3H), 2.01 (m, 2H), 1 .76 (m, 2H), 1 .04 (t, 3H, J=6.8 Hz).
…………………………………………
PATENT
WO2013155317
http://www.google.com/patents/WO2013155317A1?cl=en
N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3- yl)methyl)-5-(ethyl (tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’- biphenyl] -3-carboxamide hydrobromide:

N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-5-(ethyl
(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’-biphenyl]-3- carboxamide hydrobromide:

As used herein, “Compound I” refers to N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3- yl)methyl)-5-(ethyl (tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’- biphenyl]-3-carboxamide. The hydrobromide of Compound I can be used to inhibit the histone methyltransferase activity of EZH2, either in a subject or in vitro. The hydrobromide of Compound I can also be used to treat cancer in a subject in need thereof.
Scheme 1

……………………………………..Compound I Compound I – HBr
HPLC
HPLC was conducted on an Agilent 1200 HPLC quaternary pump, low pressure mixing, with an in-line degasser. Analytical method conditions: 8 μΐ^ sample (20 mg of ER-581982-06 diluted with 50 mL of a methanol to provide approximately 0.4 mg/mL solution) was injected onto a Agilent Zorbax Eclipse XDB-C18 (4.6 x 150 mm, 3.5 um), Chromatography conditions: mobile phase A, water with 5mM ammonium formate; mobile phase B, 5 mM ammonium formate in 50/45/5 acetonitrile/methanol/water; flow rate, 1.5 ml/min.; gradient: isocratic at 10% B from 0 to 3 min; linear increase to 70% B from 3 to 7 min; isocratic at 70% B from 7 to 12 min; linear increase to 100% B from 12 to 15 min isocratic at 100% B from 15 to 20 min;
column temperature, 35 °C; detection, UV 230 nm. Approximate retention time of Compound I = 10.7 min.
Synthesis of Polymorph A

5-bromo-2-methyl-3-nitrobenzoic acid stirred solution of 2-methyl-3-nitrobenzoic acid (100 g, 552 mmol) in cone. H2S04 (400 mL), l,3-dibromo-5,5-dimethyl-2,4- imidazolidinedione (88 g, 308 mmol) was added in a portion wise manner at room temperature and the reaction mixture was then stirred at room temperature for 5 h. The reaction mixture was poured onto ice cold water, the precipitated solid was filtered off, washed with water and dried under vacuum to afford the desired compound as a solid (140 g, 98%). The isolated compound was taken directly into the next step. 1H NMR (DMSO-J6, 400 MHz) δ 8.31 (s, 1H), 8.17 (s, 1H), 2.43 (s, 3H).

Methyl 5-bromo-2-methyl-3-nitrobenzoate To a stirred solution of 5-bromo-2- methyl-3-nitrobenzoic acid (285 g, 1105 mmol) in DMF (2.8L) at room temperature was added sodium carbonate (468 g, 4415 mmol) followed by addition of methyl iodide (626.6 g, 4415 mmol). The resulting reaction mixture was heated at 60 °C for 8 h. After completion (monitored by TLC), the reaction mixture was filtered (to remove sodium carbonate) and washed with ethyl acetate (1L X 3). The combined filtrate was washed with water (3L X 5) and the aqueous phase was back extracted with ethyl acetate (1L X 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the title compound as a solid (290g, 97% yield). The isolated compound was taken directly into the next step. 1H NMR (CDC13, 400 MHz) δ 8.17 (s, 1H), 7.91 (s, 1H), 3.96 (s, 3H), 2.59 (s, 3H).

Methyl 3-amino-5-bromo-2-methylbenzoate (1) To a stirred solution of methyl 5- bromo-2-methyl-3-nitrobenzoate (290 g, 1058 mmol) in ethanol (1.5L) was added aqueous ammonium chloride (283 g, 5290 mmol dissolved in 1.5L water). The resulting mixture was stirred at 80°C to which iron powder (472 g, 8451 mmol) was added in a portion wise manner. The resulting reaction mixture was heated at 80 °C for 12 h. Upon completion as determined by TLC, the reaction mixture was hot filtered over celite® and the celite bed was washed with methanol (5L) followed by washing with 30% MeOH in DCM (5L). The combined filtrate was concentrated in- vacuo, the residue obtained was diluted with aqueous sodium bicarbonate solution (2L) and extracted with ethyl acetate (5L X 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the title compound as a solid (220 g, 85%). The compound was taken directly into the next step. 1H
NMR (CDCI3, 400 MHz) δ 7.37 (s, 1H), 6.92 (s, 1H), 3.94 (s, 3H), 3.80 (bs, 2H), 2.31 (s, 3H).

Methyl 5-bromo-2-methyl-3-((tetrahydro-2H-pyran-4-yl) amino) benzoate (2) A reactor was charged with methyl 3-amino-5-bromo-2-methylbenzoate (455.8 g, 1.87 mol), 1,2- Dichloroethane (4.56 L), and acetic acid (535 ml, 9.34 mol). To the mixture were added dihydro-2H-pyran-4(3H)-one (280 g, 2.80 mol) and sodium triacetoxyborohydride (594 g, 2.80 mol) maintaining the internal temperature below 40 °C. The mixture was stirred at 25 °C for 2.5 h and then the reaction was quenched with a solution of sodium hydroxide (448 g, 11.20 mol) in water (5.61 L). After stirring for 20 minutes at ambient temperature, the organic layer was separated and the aqueous layer was extracted with ethyl acetate (3.65 L). The organic layers were combined, washed with brine (1.5 L), and concentrated under vacuum.
The residue was treated with ethyl acetate (1.8 L) and heated to 65-70 °C. The mixture was stirred at 65-70 °C for 15 minutes to give a clear solution and then treated with n-heptane (7.3 L) maintaining the temperature between 60-70 °C. Once the heptane was completely added to the solution, the mixture was held at 65-70 °C for 15 minutes and then allowed to cool to 18- 22 °C over 3 h. The resulting suspension was stirred at 18-22 °C for 4 h, cooled to 0-5 °C over 1 h, and held at 0-5 °C for 2 h. The precipitate was filtered, washed twice with n-heptane (1.4 L), and dried under vacuum to give the title compound (540 g, 88%). The XRPD pattern of this compound is shown in Figure 17.

Methyl 5-bromo-3-(ethyl (tetrahydro-2H-pyran-4-yl) amino)-2-methylbenzoate (3)
To a stirred solution of methyl 5-bromo-2-methyl-3-((tetrahydro-2H-pyran-4-yl) amino) benzoate (14 g, 42.7 mmol) in dichloroethane (150 mL) was added acetaldehyde (3.75 g, 85.2 mmol) and acetic acid (15.3 g, 256 mmol). The resulting reaction mixture was stirred at room temperature for 15 minutes. The mixture was cooled to 0 °C and sodium triacetoxyborohydride (27 g, 128 mmol) was added. The reaction mixture was stirred at room temperature for 3 hours. Upon completion of the reaction as determined by TLC, aqueous sodium bicarbonate solution was added to the reaction mixture until a pH 7-8 was obtained, the organic phase was separated and the aqueous phase was extracted with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography (100-200 mesh silica gel) eluting with ethyl acetate: hexane to afford the desired compound as a viscous liquid (14 g, 93%). 1H NMR DMSO-d6, 400 MHz) δ 7.62 (s, 1H), 7.52 (s, 1H), 3.80 (bs, 5H), 3.31 (t, 2H), 2.97-3.05 (m, 2H), 2.87-2.96 (m, 1H), 2.38 (s, 3H), 1.52-1.61 (m, 2H), 1.37-1.50 (m, 2H), 0.87 (t, 3H, J=6.8 Hz).

Methyl 5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-
[l,l’-biphenyl]-3-carboxylate (4): A mixture of methyl 5-bromo-3-(ethyl(tetrahydro-2H-pyran- 4-yl)amino)-2-methylbenzoate (580 g, 1.63 mol), 4-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan- 2-yl)benzyl)morpholine (592 g, 1.95 mol), 1,4-dioxane (3.86 L), sodium carbonate (618 g, 5.83 mol), and water (771 ml) was degassed by bubbling nitrogen through the mixture at 20 °C for 20 minutes and treated with tetrakis(triphenylphosphine)palladium(0) (14.11 g, 12.21 mmol). The resulting mixture was degassed for an additional 20 minutes and then heated to 87-89 °C for 17 h. After cooling to 20 °C, the mixture was diluted with ethyl acetate (5.80 L) and a solution of (R)-2-Amino-3-mercaptopropionic acid (232 g) in water (2.320 L). After stirring for 1 h at 20 °C, the organic layer was separated and washed again with a solution of (R)-2-Amino-3- mercaptopropionic acid (232 g) in water (2.320 L). The aqueous layers were combined and extracted with ethyl acetate (5.80 L). The organic layers were combined, washed with a solution of sodium hydroxide (93 g) in water (2.32 L), and concentrated under vacuum at 35 °C to give the title compound as an orange oil (1.21 kg, 164% yield).

5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’- biphenyl]-3-carboxylic acid (5): Methyl 5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′- (morpholinomethyl)-[l,l’-biphenyl]-3-carboxylate (69.0 g, 152.5 mmol) (based on the theoretical yield from the previous step) was suspended in ethanol (380 mL) and treated with a solution of sodium hydroxide (24.84 g, 621.0 mmol) in water (207 mL). The mixture was stirred at 40°C for 18 h. After cooling to 0-5 °C, the mixture was neutralized to pH 6.5 with 1 N hydrochloric acid (580 mL) maintaining the temperature below 25 °C. Then, the mixture was extracted twice with a mixture of dichloromethane (690 mL) and methanol (69.0 mL). The organic layers were combined and concentrated under vacuum to give a crude product as a yellow solid (127g).
The crude product was dissolved in 2-methyltetrahydrofuran (656 mL) at 70 °C and then treated with IPA (828 mL). The mixture was allowed to cool to rt over 3-4 h and then stirred overnight at rt. The precipitate was filtered, washed twice with IPA (207 mL), and dried under vacuum to give the title compound as an off white solid (53.54 g, 80%). The XRPD pattern of this compound is shown in Figure 9.

N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-5-(ethyl(tetrahydro-2H- pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’-biphenyl]-3-carboxamide
(Compound I): A mixture of 5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′- (morpholinomethyl)-[l,l’-biphenyl]-3-carboxylic acid (540 g, 1.23 mol) and 3-(aminomethyl)- 4,6-dimethyl-dihydro-pyridin-2(lH)-one hydrochloride (279 g, 1.48 mol) was suspended in DMSO (2.70 L) and treated with triethylamine (223 ml, 1.60 mol). The mixture was stirred at 25 °C for 30 min and treated with EDC-HC1 (354 g, 1.85 mol) and HOBT hydrate (283 g, 1.85 mol). The reaction mixture was stirred at rt for 16 h. After addition of triethylamine (292 ml, 2.09 mol), the mixture was cooled to 15 °C, diluted with water (10.1 L) maintaining the temperature below 30 °C, and stirred at 19-25 °C for 4 h. The resulting precipitate was filtered, washed twice with water (2.70 L), and dried under vacuum to give a crude product (695 g, wt-wt analysis = 78%).
For the further purification of the product, recrystallization was conducted. A crude product (20.00 g, 34.92 mmol) was suspended in a mixture of ethanol (190 ml) and water (10.00 ml) and heated to 75°C until a clear solution was obtained. The solution was allowed to cool to rt overnight. The precipitate was filtered, washed twice with a mixture of ethanol (30.0 ml) and water (30.0 ml), and dried under vacuum at 35 °C to give the title compound as an off white solid (14.0 g, 70% recovery from the crude and 90% yield based on wt-wt assay).

4-((3′-(((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′- (ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[l,l’-biphenyl]-4-yl)methyl)morpholin- 4-ium bromide (Polymorph A): A crude N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3- yl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)am
biphenyl]-3-carboxamide (595 g, 464 g based on wt-wt assay, 810.3 mmol) was suspended in ethanol (3.33 L). After heating to 70 °C, the mixture was treated with 48% aqueous HBr (97 ml, 850.8 mmol) and stirred at 70 °C for 30 min. The resulting orange-red solution was treated with ethyl acetate (3.33 L) maintaining the temperature above 60 °C. The mixture was slowly cooled to rt over 18 h. The mixture was cooled to 0 °C over 1 h and stirred at that temperature for 5.5 h. The resulting precipitate was filtered, washed twice with ethyl acetate (1.39 L), and dried under vacuum to give the title compound as an off white solid (515 g, 97% yield).
Recrystallization of Polymorph A: 4-((3′-(((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3- yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[l,l’-biphenyl]-4- yl)methyl)morpholin-4-ium bromide (0.50 g, 0.77 mmol; 95.6% pure by HPLC) was suspended in ethanol (3.0 mL) and heated to 80 °C until a clear solution was obtained. To the solution was added MTBE (5.0 mL) slowly. The resulting solution was allowed to cool to 18-22 °C over 3 h and stirred at 18-22 °C for 15 h. The precipitate was filtered, washed twice with MTBE (2 mL) and dried under vacuum to give 0.45 g of the title compound (89% recovery, 96.6% pure by HPLC).
Compound I is protonated at the nitrogen of the morpholino substituent, providing a monohydrobromide of Compound I having the following structure:

This particular monohydrobromide can be referred to as “4-((3′-(((4,6-dimethyl-2-oxo- l,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′- methyl-[l, -biphenyl]-4-yl)methyl)morpholin-4-ium bromide.” Figure 11 depicts the X-ray crystal structure of this particular salt form.
…………………………………………………………..
see
WO-2015057859
Eisai Research Institute; Epizyme Inc
Novel crystalline polymorphic form C of tazemetostat, useful for treating an EZH2-mediated cancer, including non-Hodgkin’s lymphoma and breast cancer.
…………………
Synthesis
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| USA | Tazverik | Epizyme, 2020 |
Formulations
- oral tabs. and suspension
References
-
- Knutson, S. K. et al., Proc. Natl. Acad. Sci USA, (2013) 110(19), 7922-7927.
- WO 2012 142504 (Epizyme/Eisai Co; 18.10.2012; appl. 13.4.2012; USA-prior. 13.4.2011).
- WO 2013 155317 (Epizyme/Eisai Co; 17.10.2013; appl. 11.4.2013).
- WO 2015 057859 (Epizyme/Eisai Co; 23.4.2015; appl. 15.10.2014; USA-prior. 16.10.2013).
-
EZH2 inhibitors for treating lymphona:
- WO 2015 195848 (Epizyme; 23.12.2015; appl. 17.6.2015; USA-prior. 17.6.2014).
////////
PAPER
RSC Advances (2015), 5(33), 25967-25978
http://pubs.rsc.org/en/content/articlelanding/2015/ra/c5ra02365c#!divAbstract
DOI: 10.1039/C5RA02365C
The histone lysine methyltransferase EZH2 has been implicated as a key component in cancer aggressiveness, metastasis and poor prognosis. This study discovered a new class of hexahydroisoquinolin derivatives as EZH2 inhibitors. A structure–activity relationship study showed that the steric hindrance was important to the activity for EZH2. A preliminary optimization study led to the discovery of several potent compounds with low nanomolar to sub-nanomolar potency for EZH2. Biological evaluation indicated that SKLB1049 was a highly potent with improved solubility compared to EPZ6438, SAM-competitive, and cell-active EZH2 inhibitor that decreased global H3K27me3 in SU-DHL-6 and Pfeiffer lymphoma cells in a concentration- and time-dependent manner. Further study indicated that SKLB1049 caused cell arrest in G0/G1 phase. These compounds would be useful as chemical tools to further explore the biology of EZH2 and provided us with a start point to develop new EZH2 inhibitors.

|
In vitro protocol: |
Proc Natl Acad Sci U S A. 2013 May 7;110(19):7922-7. |
|
In vivo protocol: |
Proc Natl Acad Sci U S A. 2013 May 7;110(19):7922-7. |
|
References |
1: Knutson SK, Warholic NM, Johnston LD, Klaus CR, Wigle TJ, Iwanowicz D, Littlefield BA, Porter-Scott M, Smith JJ, Moyer MP, Copeland RA, Pollock RM, Kuntz KW, Raimondi A, Keilhack H. Synergistic Anti-Tumor Activity of EZH2 Inhibitors and Glucocorticoid Receptor Agonists in Models of Germinal Center Non-Hodgkin Lymphomas. PLoS One. 2014 Dec 10;9(12):e111840. doi: 10.1371/journal.pone.0111840. eCollection 2014. PubMed PMID: 25493630; PubMed Central PMCID: PMC4262195.
2: Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, Kadowaki T, Uesugi M, Kuznetsov G, Kumar N, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Waters NJ, Smith JJ, Porter-Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Uenaka T, Pollock RM, Kuntz KW, Yokoi A, Keilhack H. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther. 2014 Apr;13(4):842-54. doi: 10.1158/1535-7163.MCT-13-0773. Epub 2014 Feb 21. PubMed PMID: 24563539
3. Inhibitors of human histone methyltransferase EZH2, and methods of use thereof for treating cancer. By Kuntz, Kevin W.; Knutson, Sarah K.; Wigle, Timothy James Nelson . From U.S. Pat. Appl. Publ. (2013), US 20130040906 A1 20130214.
4. Aryl-or heteroaryl-substituted benzamide compounds as anticancer agents and their preparation By Kuntz, Kevin Wayne; Chesworth, Richard; Duncan, Kenneth William; Keilhack, Heike; Warholic, Natalie; Klaus, Christine; Zheng, Wanjun; Seki, Masashi; Shirotori, Syuji; Kawano, Satoshi From PCT Int. Appl. (2012), WO 2012142504 A1 20121018.
5: Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Pollock RM, Kuntz KW, Keilhack H. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013 May 7;110(19):7922-7. doi: 10.1073/pnas.1303800110. Epub 2013 Apr 25. PubMed PMID: 23620515; PubMed Central PMCID: PMC3651445.
| WO2013155317A1 * | Apr 11, 2013 | Oct 17, 2013 | Epizyme, Inc. | Salt form of a human hi stone methyltransf erase ezh2 inhibitor |
| WO2013155464A1 * | Apr 12, 2013 | Oct 17, 2013 | Epizyme, Inc. | Combination therapy for treating cancer |
| WO2014049488A1 * | Sep 16, 2013 | Apr 3, 2014 | Pfizer Inc. | Benzamide and heterobenzamide compounds |
| WO2014062732A1 * | Oct 15, 2013 | Apr 24, 2014 | Epizyme, Inc. | Substituted benzene compounds |
| WO2014062733A2 * | Oct 15, 2013 | Apr 24, 2014 | Epizyme, Inc. | Substituted benzene compounds |
| WO2014172044A1 * | Mar 14, 2014 | Oct 23, 2014 | Epizyme, Inc. | Substituted benzene compounds |
| WO2015004618A1 * | Jul 9, 2014 | Jan 15, 2015 | Glaxosmithkline Intellectual Property (No.2) Limited | Enhancer of zeste homolog 2 inhibitors |
| WO2015010049A1 * | Jul 18, 2014 | Jan 22, 2015 | Epizyme, Inc. | Substituted benzene compounds |
| WO2015010078A2 | Jul 18, 2014 | Jan 22, 2015 | Epizyme, Inc. | Substituted 6,5-fused bicyclic heteroaryl compounds |
| WO2011140325A1 * | May 5, 2011 | Nov 10, 2011 | Glaxosmithkline Llc | Indazoles |
| WO2012142504A1 * | Apr 13, 2012 | Oct 18, 2012 | Eisai Co., Ltd. | Aryl-or heteroaryl-substituted benzene compounds |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014062720A2 * | Oct 15, 2013 | Apr 24, 2014 | Epizyme, Inc. | Methods of treating cancer |
| WO2011140324A1 * | May 5, 2011 | Nov 10, 2011 | Glaxosmithkline Llc | Indoles |
| WO2011140325A1 * | May 5, 2011 | Nov 10, 2011 | Glaxosmithkline Llc | Indazoles |
| WO2012005805A1 * | May 5, 2011 | Jan 12, 2012 | Glaxosmithkline Llc | Azaindazoles |
| US4522811 | Jul 8, 1982 | Jun 11, 1985 | Syntex (U.S.A.) Inc. | Serial injection of muramyldipeptides and liposomes enhances the anti-infective activity of muramyldipeptides |
| US5763263 | Jul 24, 1996 | Jun 9, 1998 | Dehlinger; Peter J. | Method and apparatus for producing position addressable combinatorial libraries |
| US7563589 | May 27, 2005 | Jul 21, 2009 | The University Of North Carolina At Chapel Hill | Including EED, EZH2 and SUZ12 wherein the reconstituted complex has histone methyltransferase (HMTase) activity for lysine 27 of histone H3 (H3-K27); cancer |
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r “FDA approves first treatment option specifically for patients with epithelioid sarcoma, a rare soft tissue cancer”. U.S. Food and Drug Administration (FDA) (Press release). 23 January 2020. Retrieved 23 January 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Lue JK, Amengual JE (October 2018). “Emerging EZH2 Inhibitors and Their Application in Lymphoma”. Curr Hematol Malig Rep. 13 (5): 369–382. doi:10.1007/s11899-018-0466-6. PMID 30112706. S2CID 52010283.
- ^ “Tazemetostat”. NCI Drug Dictionary. National Cancer Institute.
- ^ Jump up to:a b c “Drug Trials Snapshots: Tazverik”. U.S. Food and Drug Administration (FDA). 23 January 2020. Retrieved 22 February 2020. This article incorporates text from this source, which is in the public domain.
External links
- “Tazemetostat”. Drug Information Portal. U.S. National Library of Medicine.
 |
|
| Clinical data | |
|---|---|
| Trade names | Tazverik |
| Other names | EPZ-6438 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620018 |
| License data |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C34H44N4O4 |
| Molar mass | 572.750 g·mol−1 |
| 3D model (JSmol) | |
Epizyme®, Inc.
400 Technology Square, 4th Floor
Cambridge, MA 02139
Phone: (617) 229-5872
Fax: (617) 349-0707
contact@Epizyme.com
![]()



 Jason Rhodes (left) has been appointed to president of Epizyme Inc.,
Jason Rhodes (left) has been appointed to president of Epizyme Inc.,

100 Technology Square

K 912, NC 6300, Epirubicin nano



Cancer Research UK

Irish Cancer Society
Macmillan Cancer Support
NHS Evidence
 Epirubicin – Substance Summary
Epirubicin – Substance Summary
PubChem
MedlinePlus
– See more at: http://www.cancerindex.org/Epirubicin#sthash.BBioCC6K.dpuf
PHASE 1 JAPAN SOLID TUMOURS
DNA/RNA Synthesis Inhibitor
WITH Nano Carrier Co.,Ltdhttp://pdf.irpocket.com/C4571/qnwX/eFou/vG1J.pdf
KOWA COMPANY LTD
CAS FREE FORM. 56420-45-2
Smiles
- O=C2c1c(O)c5c(c(O)c1C(=O)c3cccc(OC)c23)C[C@@](O)(C(=O)CO)C[C@@H]5O[C@@H]4O[C@H]([C@H](O)[C@@H](N)C4)C
- NMR
- http://file.selleckchem.com/downloads/nmr/S122302-Epirubicin-Hydrochloride-NMR-Selleck.pdf
- http://www.medkoo.com/Product-Data/Epirubicin/Epirubicin-QC-TZC20130604web.pdf
-
Epirubicin CAS No.: 56420-45-2 Synonyms: - 4′-Epi-DX;
- Epirubicina;
- WP 697;
- Pidorubicin;
- 4′-epiadriamycin;
- Adriblastin;
- epi-dx;
- Epiadriamycin;
- farmorubicin;
- imi28;
- Farmarubicin;
Formula: C27H29NO11 Exact Mass: 543.17400
NC-6300, an epirubicin-incorporating micelle, extends the antitumor effect and reduces the cardiotoxicity of epirubicin.
Epirubicin is widely used to treat various human tumors. However, it is difficult to achieve a sufficient antitumor effect because of dosage limitation to prevent cardiotoxicity. We hypothesized that epirubicin-incorporating micelle would reduce cardiotoxicity and improve the antitumor effect. NC-6300 comprises epirubicin covalently bound to PEG polyaspartate block copolymer through an acid-labile hydrazone bond. The conjugate forms a micellar structure of 40-80 nm in diameter in an aqueous milieu. NC-6300 (10, 15 mg/kg) and epirubicin (10 mg/kg) were given i.v. three times to mice bearing s.c. or liver xenograft of human hepatocellular carcinoma Hep3B cells. Cardiotoxicity was evaluated by echocardiography in C57BL/6 mice that were given NC-6300 (10 mg/kg) or epirubicin (10 mg/kg) in nine doses over 12 weeks. NC-6300 showed a significantly potent antitumor effect against Hep3B s.c. tumors compared with epirubicin. Moreover, NC-6300 also produced a significantly longer survival rate than epirubicin against the liver orthotopic tumor of Hep3B. With respect to cardiotoxicity, epirubicin-treated mice showed significant deteriorations in fractional shortening and ejection fraction. In contrast, cardiac functions of NC-6300 treated mice were no less well maintained than in control mice. This study warrants a clinical evaluation of NC-6300 in patients with hepatocellular carcinoma or other cancers.

K-912(NC-6300)の概要 K-912(NC-6300)は、世界的に幅広く使用されているアントラサイクリン系の抗が ん剤の一つであるエピルビシンを内包したミセル化ナノ粒子製剤で、その特性により、 エピルビシンの有する心毒性の軽減が期待できます。さらに、pH 応答性システムを採 用することで、腫瘍細胞内でのエピルビシンの放出量を高め、既存のエピルビシンに比 べより強力な抗腫瘍効果が期待できます。
Epirubicin is an anthracycline drug used for chemotherapy. It can be used in combination with other medications to treat breast cancer in patients who have had surgery to remove the tumor. It is marketed by Pfizer under the trade name Ellence in the US andPharmorubicin or Epirubicin Ebewe elsewhere.
Similarly to other anthracyclines, epirubicin acts by intercalating DNA strands. Intercalation results in complex formation which inhibits DNA and RNA synthesis. It also triggers DNA cleavage by topoisomerase II, resulting in mechanisms that lead to cell death. Binding to cell membranes and plasma proteins may be involved in the compound’s cytotoxic effects. Epirubicin also generates free radicalsthat cause cell and DNA damage.
Epirubicin is favoured over doxorubicin, the most popular anthracycline, in some chemotherapy regimens as it appears to cause fewer side-effects. Epirubicin has a different spatial orientation of the hydroxyl group at the 4′ carbon of the sugar – it has the opposite chirality – which may account for its faster elimination and reduced toxicity. Epirubicin is primarily used against breast and ovarian cancer, gastric cancer, lung cancer and lymphomas.
Development history
The first trial of epirubicin in humans was published in 1980.[1] Upjohn applied for approval by the U.S. Food and Drug Administration(FDA) in node-positive breast cancer in 1984, but was turned down because of lack of data.[2] It appears to have been licensed for use in Europe from around this time however.[3] In 1999 Pharmacia (who had by then merged with Upjohn) received FDA approval for the use of epirubicin as a component of adjuvant therapy in node-positive patients.
Patent protection for epirubicin expired in August 2007.
References
- Bonfante, V; Bonadonna, G; Villani, F; Martini, A (1980). “Preliminary clinical experience with 4-epidoxorubicin in advanced human neoplasia”. Recent results in cancer research 74: 192–9. PMID 6934564. PM6934564.
- “On Target”.
- According to the proprietary database iddb.com
External links
- http://www.bccancer.bc.ca/HPI/DrugDatabase/DrugIndexPro/Epirubicin.htm
- http://www.pfizerpro.com/page_not_found?rid=/wyeth_html/home/minisites/oncology/ellence/pi/description.html
1H NMR PREDICT

13C NMR PREDICT

COSY
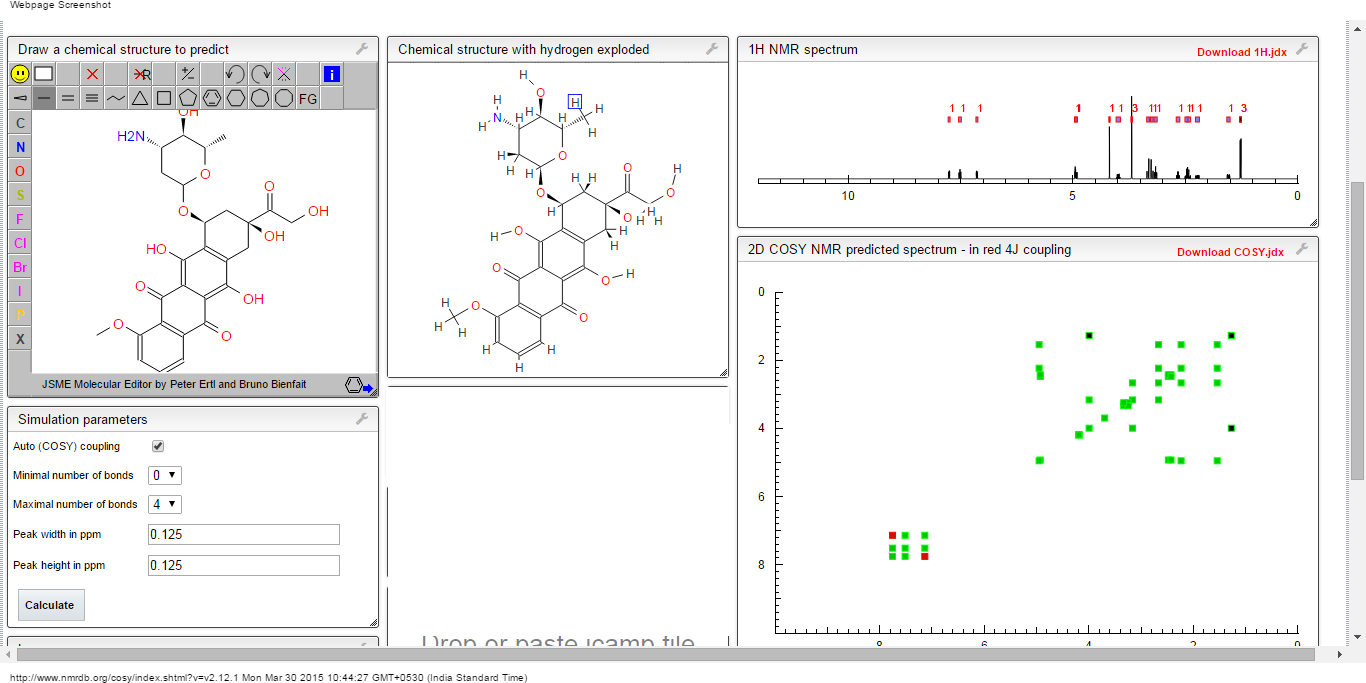
1H NMR

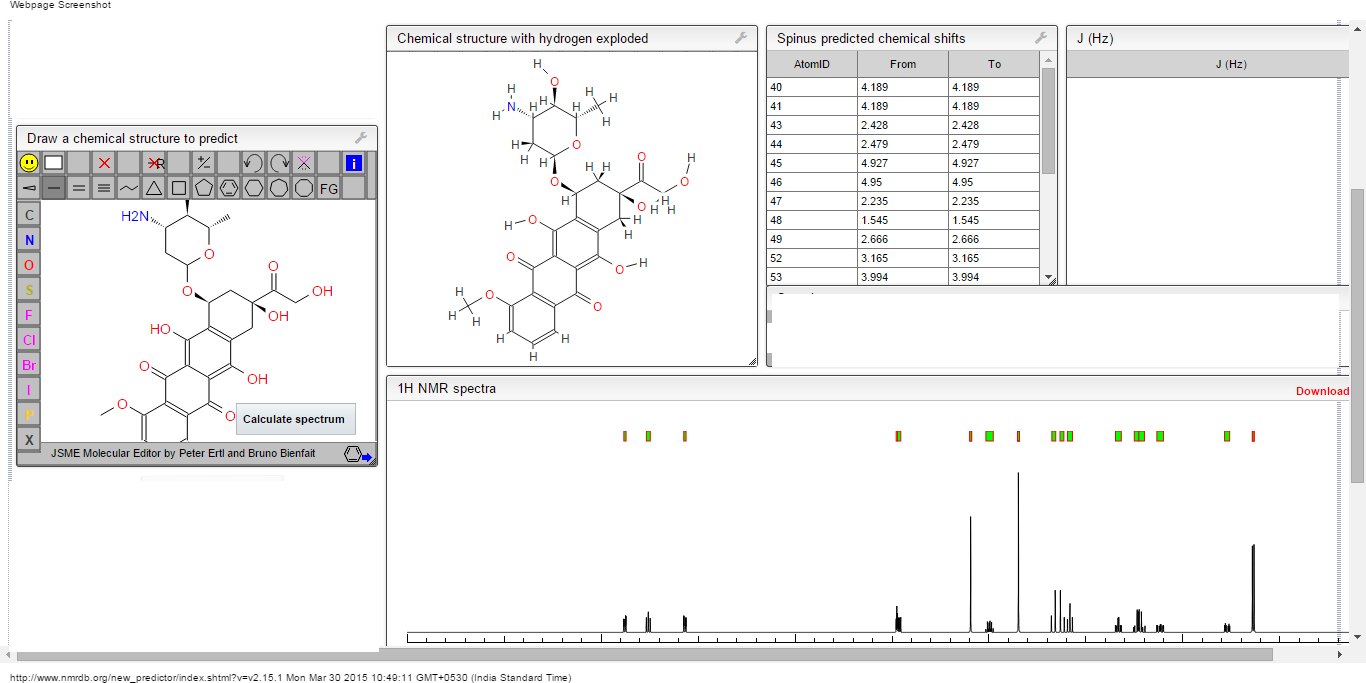
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (8R,10S)-10-((2S,4S,5R,6S)-4-amino-5-hydroxy-6-methyltetrahydro-2H-pyran-2-yl)-6,8,11-trihydroxy-8-(2-hydroxyacetyl)-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione | |
| Clinical data | |
| Trade names | Ellence |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a603003 |
| Intravenous | |
| Pharmacokinetic data | |
| Bioavailability | NA |
| Protein binding | 77% |
| Metabolism | Hepatic glucuronidationand oxidation |
| Excretion | Biliary and renal |
| Identifiers | |
| 56420-45-2 |
|
| L01DB03 | |
| PubChem | CID 41867 |
| DrugBank | DB00445 |
| ChemSpider | 38201 |
| UNII | 3Z8479ZZ5X |
| KEGG | D07901 |
| ChEBI | CHEBI:47898 |
| ChEMBL | CHEMBL417 |
| Chemical data | |
| Formula | C27H29NO11 |
| 543.519 g/mol | |
KOWA COMPANY LTD




P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

Dabrafenib mesylate, GSK 2118436, ダブラフェニブ 达拉菲尼, An antineoplastic agent that inhibits BRAF kinase


DABRAFENIB
ダブラフェニブ
达拉菲尼,
1195765-45-7 BASE
1195768-06-9 cas of mesylate
Benzenesulfonamide, N-[3-[5-(2-amino-4-pyrimidinyl)-2-(1,1-dimethylethyl)-4-thiazolyl]-2-fluorophenyl]-2,6-difluoro-
MW 519.56 BASE
MF C23 H20 F3 N5 O2 S2 BASE
- Dabarefenib
- Dabrafenib
- GSK 2118436
- Tafinlar
- UNII-QGP4HA4G1B
US FDA APPROVAL….Date of Approval: May 29, 2013
update
| Product details | |
|---|---|
| Name |
Tafinlar
|
| Agency product number |
EMEA/H/C/002604
|
| Active substance |
dabrafenib mesilate
|
| International non-proprietary name (INN) or common name |
dabrafenib
|
| Therapeutic area (MeSH) |
Melanoma
|
| Anatomical therapeutic chemical (ATC) code |
L01EC02
|
| Publication details | |
|---|---|
| Marketing-authorisation holder |
Novartis Europharm Limited
|
| Date of issue of marketing authorisation valid throughout the European Union |
26/08/2013
|
An orally bioavailable inhibitor of B-raf (BRAF) protein with potential antineoplastic activity. Dabrafenib selectively binds to and inhibits the activity of B-raf, which may inhibit the proliferation of tumor cells which contain a mutated BRAF gene. B-raf belongs to the the raf/mil family of serine/threonine protein kinases and plays a role in regulating the MAP kinase/ERKs signaling pathway, which may be constitutively activated due to BRAF gene mutations
Dabrafenib (trade name Tafinlar) is a drug for the treatment of cancers associated with a mutated version of the gene BRAF. Dabrafenib acts as an inhibitor of the associated enzyme B-Raf, which plays a role in the regulation of cell growth. Dabrafenib has clinical activity with a manageable safety profile in clinical trials of phase 1 and 2 in patients with BRAF(V600)-mutated metastatic melanoma.[1][2]
The Food and Drug Administration approved dabrafenib as a single agent treatment for patients with BRAF V600E mutation-positive advanced melanoma on May 30, 2013.[3] Clinical trial data demonstrated that resistance to dabrafinib and other BRAF inhibitors occurs within 6 to 7 months.[4] To overcome this resistance, the BRAF inhibitor dabrafenib was combined with the MEK inhibitor trametinib.[4] As a result of this research, on January 8, 2014, the FDA approved the combination of dabrafenib and trametinib for the treatment of patients with BRAF V600E/K-mutant metastatic melanoma.[5]

Inhibitor of BRAF(V600) mutants
| Active Ingredient: | DABRAFENIB MESYLATE |
| Dosage Form;Route: | CAPSULE;ORAL |
| Proprietary Name: | TAFINLAR |
| Applicant: | GLAXOSMITHKLINE |
| Strength: | EQ 75MG BASE |
| NDA Application Number: | N202806 |
| Product Number: | 002 |
| Approval Date: | May 29, 2013 |
| Reference Listed Drug | Yes |
| RX/OTC/DISCN: | RX |
Patent Data
| Appl No | Prod No | Patent No | Patent Expiration |
Drug Substance Claim |
Drug Product Claim |
Patent Use Code |
Delist Requested |
|---|---|---|---|---|---|---|---|
| N202806 | 002 | 7994185 | Jan 20, 2030 | Y | Y | U – 1406 | |
| N202806 | 002 | 8415345 | Jan 20, 2030 | Y | Y | U – 1406 |
Exclusivity Data
| NDA Appl No | Prod No | Exclusivity Code | Exclusivity Expiration |
|---|---|---|---|
| N202806 | 002 | I – 678 | Jan 8, 2017 |
| N202806 | 002 | ODE | Jan 9, 2021 |
| N202806 | 002 | NCE | May 29, 2018 |
| N202806 | 002 | ODE | May 29, 2020 |
PDF……http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/202806s000lbl.pdf
TERMS
I 678, TRAMETINIB, IN COMBINATION WITH DABRAFENIB, FOR THE TREATMENT OF PATIENTS WITH UNRESECTABLE OR METASTATIC MELANOMA WITH BRAF V600E OR V600K MUTATIONS AS DETECTED BY AN FDA-APPROVED TEST
ODE ORPHAN DRUG EXCLUSIVITY
NCE NEW CHEMICAL ENTITY
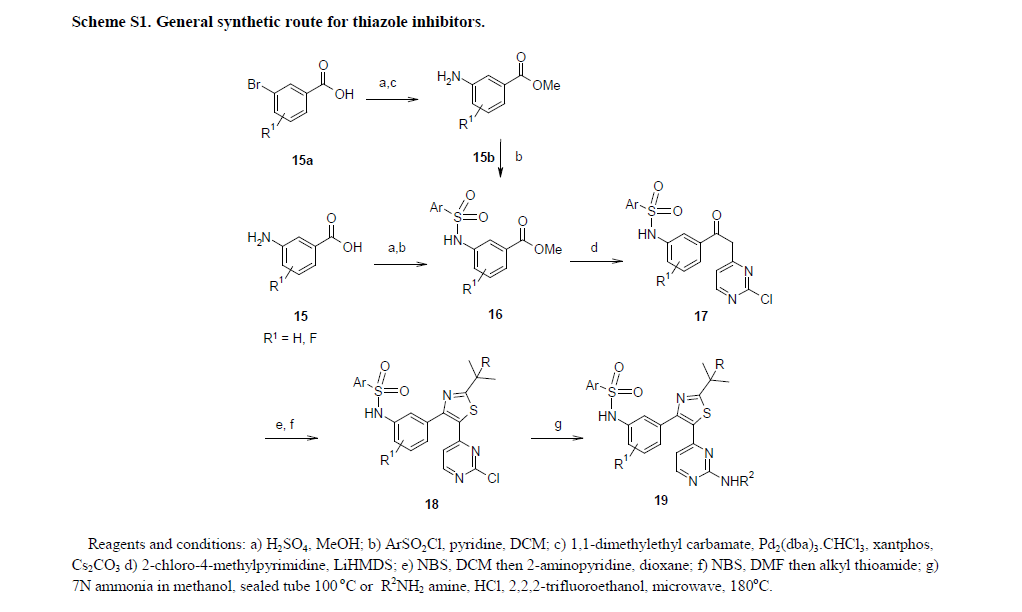

DABRAFENIB SYNTHESIS
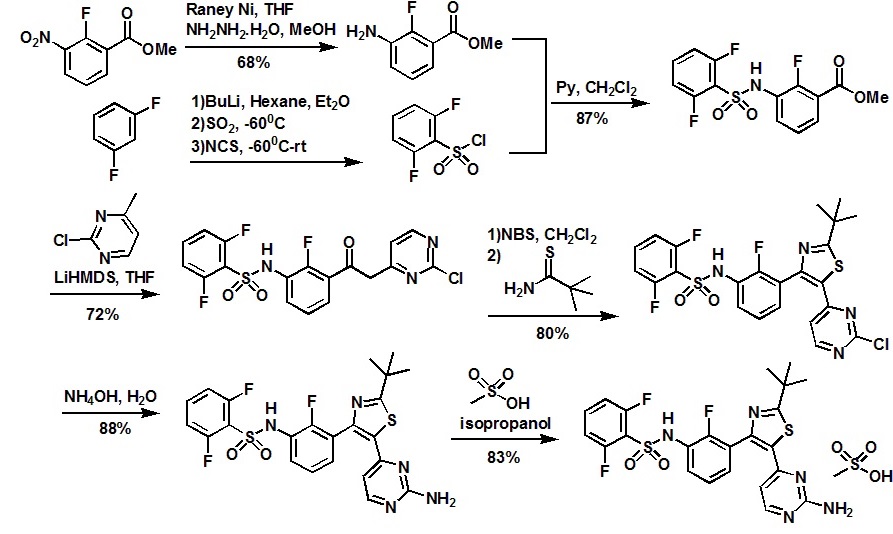
WILL BE UPDATED
—
http://www.google.com/patents/WO2011047238A1?cl=en
Method 1 : Compound B (first crystal form) – A/-{3-[5-(2-Amino-4-pyrimidinyl)-2-(1 ,1 dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide

A suspension of A/-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]- 2-fluorophenyl}-2,6-difluorobenzenesulfonamide (196 mg, 0.364 mmol) and ammonia in methanol 7M (8 ml, 56.0 mmol) was heated in a sealed tube to 90 °C for 24 h. The reaction was diluted with DCM and added silica gel and concentrated. The crude product was chromatographed on silica gel eluting with 100% DCM to 1 :1 [DCM:(9:1 EtOAc:MeOH)]. The clean fractions were concentrated to yield the crude product. The crude product was repurified by reverse phase HPLC (a gradient of acetonitrile:water with 0.1 %TFA in both). The combined clean fractions were concentrated then partitioned between DCM and saturated NaHCO3. The DCM layer was separated and dried over Na2SO4. The title compound, /V-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1 – dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide was obtained (94 mg, 47% yield). 1 H NMR (400 MHz, DMSO-c/6) δ ppm 10.83 (s, 1 H), 7.93 (d, J=5.2 Hz, 1 H), 7.55 – 7.70 (m, 1 H), 7.35 – 7.43 (m, 1 H), 7.31 (t, J=6.3 Hz, 1 H), 7.14 – 7.27 (m, 3 H), 6.70 (s, 2 H), 5.79 (d, J=5.13 Hz, 1 H), 1 .35 (s, 9 H). MS (ESI): 519.9 [M+H]+.
Method 2: Compound B (alternative crystal form) – A/-{3-[5-(2-Amino-4-pyrimidinyl)-2- (1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide 19.6 mg of A/-{3-[5-(2-Amino-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (may be prepared in accordance with example 58a) was combined with 500 L of ethyl acetate in a 2-mL vial at room temperature. The slurry was temperature-cycled between 0-40°C for 48 hrs. The resulting slurry was allowed to cool to room temperature and the solids were collected by vacuum filtration. The solids were analyzed by Raman, PXRD, DSC/TGA analyses, which indicated a crystal form different from the crystal form resulting from Example 58a, above. Method 3: Compound B (alternative crystal form, large batch) – A/-{3-[5-(2-amino-4- pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide

tep A: methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate

Methyl 3-amino-2-fluorobenzoate (50 g, 1 eq) was charged to reactor followed by dichloromethane (250 mL, 5 vol). The contents were stirred and cooled to ~15°C and pyridine (26.2 mL, 1 .1 eq) was added. After addition of the pyridine, the reactor contents were adjusted to ~15°C and the addition of 2,6-diflurorobenzenesulfonyl chloride (39.7 mL, 1 .0 eq) was started via addition funnel. The temperature during addition was kept <25°C. After complete addition, the reactor contents were warmed to 20-25°C and held overnight. Ethyl acetate (150 mL) was added and dichloromethane was removed by distillation. Once distillation was complete, the reaction mixture was then diluted once more with ethyl acetate (5 vol) and concentrated. The reaction mixture was diluted with ethyl acetate (10 vol) and water (4 vol) and the contents heated to 50-55°C with stirring until all solids dissolve. The layers were settled and separated. The organic layer was diluted with water (4 vol) and the contents heated to 50-55° for 20-30 min. The layers were settled and then separated and the ethyl acetate layer was evaporated under reduced pressure to ~3 volumes. Ethyl Acetate (5 vol.) was added and again evaporated under reduced pressure to ~3 volumes.
Cyclohexane (9 vol) was then added to the reactor and the contents were heated to reflux for 30 min then cooled to 0 °C. The solids were filtered and rinsed with cyclohexane (2 x 100 mL). The solids were air dried overnight to obtain methyl 3-{[(2,6- difluorophenyl)sulfonyl]amino}-2-fluorobenzoate (94.1 g, 91 %).
Step B: A/-{3-[(2-chloro-4-pyhmidinyl)acetyl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide

Methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate (490 g, 1 equiv.), prepared generally in accordance with Step A, above, was dissolved in THF (2.45 L, 5 vols) and stirred and cooled to 0-3 °C. 1 M lithium bis(trimethylsilyl)amide in THF (5.25 L, 3.7 equiv.) solution was charged to the reaction mixture followed addition of 2- chloro-4-methylpyrimidine (238 g, 1 .3 equiv.) in THF (2.45 L, 5 vols). The reaction was then stirred for 1 hr. The reaction was quenched with 4.5M HCI (3.92 L, 8 vols). The aqueous layer (bootom layer) was removed and discarded. The organic layer was concentrated under reduced pressure to ~2L. IPAC (isopropyl acetate) (2.45L) was added to the reaction mixture which was then concentrated to ~2L. IPAC (0.5L) and MTBE (2.45 L) was added and stirred overnight under N2. The solids were filtered. The solids and mother filtrate added back together and stirred for several hours. The solids were filtered and washed with MTBE (~5 vol). The solids were placed in vacuum oven at 50 °C overnight. The solids were dried in vacuum oven at 30 °C over weekend to obtain A/-{3-[(2-chloro-4-pyhmidinyl)acetyl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide (479 g, 72%).
Step C: A/-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide

To a reactor vessel was charged /V-{3-[(2-chloro-4-pyrimidinyl)acetyl]-2-fluorophenyl}- 2,6-difluorobenzenesulfonamide (30 g, 1 eq) followed by dichloromethane (300 mL). The reaction slurry was cooled to ~10°C and N-bromosuccinimide (“NBS”) (12.09 g, 1 eq) was added in 3 approximately equal portions, stirring for 10-15 minutes between each addition. After the final addition of NBS, the reaction mixture was warmed to ~20°C and stirred for 45 min . Water (5 vol) was then added to the reaction vessel and the mixture was stirred and then the layers separated. Water (5 vol) was again added to the dichloromethane layer and the mixture was stirred and the layers separated. The dichloromethane layers were concentrated to -120 mL. Ethyl acetate (7 vol) was added to the reaction mixture and concentrated to -120 mL. Dimethylacetamide (270 mL) was then added to the reaction mixture and cooled to ~10°C. 2,2- Dimethylpropanethioamide (1 .3 g, 0.5 eq) in 2 equal portions was added to the reactor contents with stirring for ~5 minutes between additions. The reaction was warmed to 20-25 °C. After 45 min, the vessel contents were heated to 75°C and held for 1 .75 hours . The reaction mixture was then cooled to 5°C and water (270 ml) was slowly charged keeping the temperature below 30°C. Ethyl acetate (4 vol) was then charged and the mixture was stirred and layers separated. Ethyl acetate (7 vol) was again charged to the aqueous layer and the contents were stirred and separated. Ethyl acetate (7 vol) was charged again to the aqueous layer and the contents were stirred and separated. The organic layers were combined and washed with water (4 vol) 4 times and stirred overnight at 20-25°C. The organic layers were then concentrated under heat and vacuum to 120 mL. The vessel contents were then heated to 50°C and heptanes (120 mL) were added slowly. After addition of heptanes, the vessel contents were heated to reflux then cooled to 0°C and held for ~2 hrs. The solids were filtered and rinsed with heptanes (2 x 2 vol). The solid product was then dried under vacuum at 30°C to obtain /V-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonannide (28.8 g, 80%).
Step D: A/-{3-[5-(2-amino-4-pyhmidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonannide
In 1 gal pressure reactor, a mixture of A/-{3-[5-(2-chloro-4-pyrinnidinyl)-2-(1 ,1 – dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (120 g) prepared in accordance with Step C, above, and ammonium hydroxide (28-30%, 2.4 L, 20 vol) was heated in the sealed pressure reactor to 98-103 °C and stirred at this temperature for 2 hours. The reaction was cooled slowly to room temperature (20 °C) and stirred overnight. The solids were filtered and washed with minimum amount of the mother liquor and dried under vacuum. The solids were added to a mixture of EtOAc (15 vol)/ water (2 vol) and heated to complete dissolution at 60-70 °C and the aqueous layer was removed and discarded. The EtOAC layer was charged with water (1 vol) and neutralized with aq. HCI to ~pH 5.4-5.5. and added water (1 vol). The aqueous layer was removed and discarded at 60-70 °C. The organic layer was washed with water (1 vol) at 60-70 °C and the aqueous layer was removed and discarded. The organic layer was filtered at 60 °C and concentrated to 3 volumes. EtOAc (6 vol) was charged into the mixture and heated and stirred at 72 °C for 10 min , then cooled to 20°C and stirred overnight. EtOAc was removed via vacuum distillation to concentrate the reaction mixture to ~3 volumes. The reaction mixture was maintained at ~65-70°C for ~30mins. Product crystals having the same crystal form as those prepared in Example 58b (and preparable by the procedure of Example 58b), above, in heptanes slurry were charged. Heptane (9 vol) was slowly added at 65-70 °C. The slurry was stirred at 65-70 °C for 2- 3 hours and then cooled slowly to 0-5°C. The product was filtered, washed with
EtOAc/heptane (3/1 v/v, 4 vol) and dried at 45°C under vacuum to obtain A/-{3-[5-(2- amino-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide (102.3 g, 88%).
Method 4: Compound B (mesylate salt) – A/-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1 – dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide methanesulfonate

To a solution of /V-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonannide (204 mg, 0.393 mmol) in isopropanol (2 ml_), methanesulfonic acid (0.131 ml_, 0.393 mmol) was added and the solution was allowed to stir at room temperature for 3 hours. A white precipitate formed and the slurry was filtered and rinsed with diethyl ether to give the title product as a white crystalline solid (210 mg, 83% yield). 1 H NMR (400 MHz, DMSO-c/6) δ ppm 10.85 (s, 1 H) 7.92 – 8.05 (m, 1 H) 7.56 – 7.72 (m, 1 H) 6.91 – 7.50 (m, 7 H) 5.83 – 5.98 (m, 1 H) 2.18 – 2.32 (m, 3 H) 1 .36 (s, 9 H). MS (ESI): 520.0 [M+H]+.
Method 5: Compound B (alternative mesylate salt embodiment) – A/-{3-[5-(2-amino-4- pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide methanesulfonate
A/-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}- 2,6-difluorobenzenesulfonamide (as may be prepared according to example 58a) (2.37g, 4.56 mmol) was combined with pre-filtered acetonitrile (5.25 vol, 12.4 ml_). A pre-filtered solution of mesic acid (1 .1 eq., 5.02 mmol, 0.48 g) in H2O (0.75 eq., 1 .78 ml_) was added at 20°C. The temperature of the resulting mixture was raised to 50- 60°C while maintaining a low agitation speed. Once the mixture temperature reached to 50-60°C, a seed slurry of A/-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1 -dimethylethyl)-1 ,3- thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide methanesulfonate (1 .0 %w/w slurried in 0.2 vol of pre-filtered acetonitrile) was added, and the mixture was aged while agitating at a speed fast enough to keep solids from settling at 50-60°C for 2 hr. The mixture was then cooled to 0-5°C at 0.25°C/min and held at 0-5°C for at 6 hr. The mixture was filtered and the wet cake was washed twice with pre-filtered
acetonitrile. The first wash consisted of 14.2 ml (6 vol) pre-filtered acetonitrile and the second wash consisted of 9.5 ml (4 vol) pre-filtered acetonitrile. The wet solid was dried at 50°C under vacuum, yielding 2.39 g (85.1 % yield) of product. Typically, the salts of the present invention are pharmaceutically acceptable salts.
May 29, 2013 — GlaxoSmithKline plc announced today that the U.S. Food and Drug Administration (FDA) has approved Tafinlar (dabrafenib). Tafinlar is indicated as a single-agent oral treatment for unresectable melanoma (melanoma that cannot be removed by surgery) or metastatic melanoma (melanoma which has spread to other parts of the body) in adult patients with BRAF V600E mutation. Tafinlar is not indicated for the treatment of patients with wild-type BRAF melanoma. The mutation must be detected by an FDA-approved test, such as the companion diagnostic assay from bioMérieux S.A., THxID™-BRAF.
Among those with metastatic melanoma, approximately half have a BRAF mutation, which is an abnormal change in a gene that can enable some melanoma tumours to grow and spread
Tafinlar is approved for patients with the BRAF V600E mutation, which accounts for approximately 85 percent of all BRAF V600 mutations in metastatic melanoma.
GSK will be making Tafinlar available for prescription no later than in the early third quarter of 2013.
In 2010, GSK entered a collaboration with bioMérieux to develop a companion diagnostic test to detect BRAF V600 (V600E and V600K) gene mutations found in several cancers, including melanoma. bioMérieux has received FDA pre-market approval of THxID™-BRAF. Currently, it is the only FDA-approved test that detects the V600K mutation.
The primary outcome measure was the estimation of the overall intracranial response rate (OIRR) in each cohort. The OIRR for Cohort A was 18 percent (95% CI: 9.7, 28.2). For Cohort B, the OIRR was also 18 percent (95% CI: 9.9, 30.0). The median duration of response was 4.6 months (95% CI: 2.8, Not Reached) and 4.6 months (95% CI: 1.9, 4.6) in Cohort A and Cohort B, respectively.
Melanoma is the most serious and deadly form of skin cancer. According to statistics from the National Cancer Institute, in 2013 there will be an estimated 9,480 deaths resulting from melanoma in the United States. When melanoma spreads in the body, the disease is called metastatic melanoma.Approximately half of all people with metastatic melanoma have a BRAF mutation, which is an abnormal change in a gene that can enable some melanoma tumours to grow and spread.
One in two patients worldwide with metastatic melanoma is expected to survive for a year after diagnosis, while in the U.S., the five-year survival rate was 16 percent (2003-2009).The median age of a newly diagnosed metastatic melanoma patient is almost a decade younger than other cancers.
Tafinlar (dabrafenib) is now approved for the treatment of adult patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test. Limitation of use: Tafinlar is not recommended for use in patients with wild-type BRAF melanoma.
Tafinlar is not approved or licensed in Europe and may not be approved in other parts of the world for the treatment of patients with BRAF V600 mutation-positive unresectable melanoma or metastatic melanoma.
Dabrafenib mesylate is a kinase inhibitor. The chemical name for dabrafenib mesylate is N-{3-[5-(2-Amino-4-pyrimidinyl)-2-(1,1-dimethylethyl)-1,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzene sulfonamide, methanesulfonate salt. It has the molecular formula C23H20F3N5O2S2•CH4O3S and a molecular weight of 615.68. Dabrafenib mesylate has the following chemical structure:
 |
Dabrafenib mesylate is a white to slightly colored solid with three pKas: 6.6, 2.2, and -1.5. It is very slightly soluble at pH 1 and practically insoluble above pH 4 in aqueous media.
TAFINLAR (dabrafenib) capsules are supplied as 50-mg and 75-mg capsules for oral administration. Each 50-mg capsule contains 59.25 mg dabrafenib mesylate equivalent to 50 mg of dabrafenib free base. Each 75-mg capsule contains 88.88 mg dabrafenib mesylate equivalent to 75 mg of dabrafenib free base.
The inactive ingredients of TAFINLAR are colloidal silicon dioxide, magnesium stearate, and microcrystalline cellulose. Capsule shells contain hypromellose, red iron oxide (E172), and titanium dioxide (E171).
Dabrafenib mesylate
1195768-06-9 cas of mesylate
N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide;methanesulfonic acid
Chemical structure

………………….
WO2015003571
达拉菲尼甲磺酸盐的新晶型及其制备方法
Deficiencies of the prior art W, the main object of the present invention is to provide as follows has a better stability in an aqueous or aqueous systems Dara Feeney 曱

For purposes of this invention, the present invention provides Dallas Phoenix mesylate Form IV (hereinafter referred to as “Form IV”) and its preparation method. The type IV crystal is a hydrate; preferably each 摩尔达拉菲 Nepal mesylate contains about 1.5 moles of water.
Using Cu- Κα radiation, the crystalline form IV of X-ray powder diffraction pattern at diffraction angles 2Θ of 4.7 ± 0.2 °, 9.2 ± 0.2. , 12.8 ± 0.2. , 13.8 ± 0.2. 15.0 0.2 soil. And 16.3 ± 0.2 ° of the characteristic peaks.
Preferably, the crystalline form IV of the X-ray powder diffraction pattern at diffraction angles 2Θ of 4.7 ± 0.2. , 9.2 ± 0.2. , 12.8 ± 0 · 2. , 13 · 8 ± 0 · 2 o , 15.0 ± 0.2. , 16.3 ± 0 · 2. , 18.0 ± 0.2. , 18 · 6 ± 0.2. , 20 · 6 ± 0.2. , 22.9 ± 0.2 °, 23.8 ± 0.2. And 24.3 ± 0.2. Department characteristic peaks
Form IV of FIG differential scanning calorimetry (DSC) show: sample 151~ 105 ° C there is a large endothermic peak (solvent peak), the sample after dehydration melting range of 132 ~ 148 ° C, then at 200 ° C ~ 245 ° C with a heat transfer crystal peak at 249 ° C and finally melted.
The crystalline form IV has the following advantageous properties:
1) left at room temperature for one month and stable, stable for 1 month at room temperature for -97% RH;
2) known Form I in water suspension was stirred for 15 minutes into the free base monohydrate; and Form IV in water suspension was stirred for 15 minutes remain for 曱 salt Form IV, After stirring overnight converted to the free base monohydrate Form IV described more conducive to maintaining the solubility of the sample is larger than the free base state 曱 sulfonates, Form IV has a better stability in water / aqueous system or sex.
3) 0 to 22 hours compared to the elution amount, any detection points of Form IV of elution volume than the known polymorph I of the elution amount. Description Form IV has a better solubility and bioavailability.
4) 0 to 120 minutes elution amount compared to any detection points of Form IV gum Nang elution volume than the known polymorph I of the dissolution of glue Nang. Description Form IV gum Nang has better dissolution.
The Form IV was prepared using any one of the following methods:
1) The Dallas Feeney known mesylate polymorph I was dissolved in a mixed solution of tetrahydrofuran Yue alcohol, volatile crystallization, and then the precipitated crystals were separated and dried to obtain the Form IV;
The Yue alcohol and tetrahydrofuran in a volume ratio of 0.1 to 100: 1, preferably 0.5~50: 1, more preferably 0.5~5: 1;
2) The Dallas Phoenix Yue sulfonates known polymorph I was dissolved in acetone, volatile crystallization, and the precipitated crystals
Separated, dried, to give the Form IV;
3) The Dallas Phoenix 曱 known polymorph I salt is dissolved in isopropanol, after the addition of polyacrylic acid, volatile crystallization, and then the precipitated crystals were separated and dried to obtain the Form IV;
The polyacrylic acid in an amount of polymorph I of the known amount of 0.1% wt~10% wt, preferably
0.5% wt ~ 10% wt, more preferably 2% wt ~ 5% wt; an average molecular weight of the polyacrylic acid is 2000-5000.
Preparation of the above three methods, the known Dara Feeney 曱 sulfonate polymorph I at room temperature in an amount corresponding to its solubility in a solution of 0.1 to 1 times, preferably 0.5 to 1 times, more preferably 0.8 to 1 times;
The crystallization temperature of room temperature ~ 40 ° C, preferably at room temperature; the crystallization time is 1~14 days, preferably for two days; the dry, you can not vacuum or pressure, the pressure is preferably less than 0.09Mpa; temperature of 30 ° C ~ 120 ° C, preferably 4 (TC ~ 80 ° C, more preferably 40 ° C ~ 60 ° C; for 10 to 72 hours, preferably 10~48 hours, more preferably from 10- 24 hours;
4) The Dallas Phoenix Yue sulfonate polymorph Form II or V is placed to give the Form IV;
The placement of room temperature ~ 40 ° C, preferably room temperature; placement time from 15 minutes to 7 days, preferably
One day;
5) The temperature rise Dara Feeney 曱 sulfonate polymorph II to 120 ° C and then spontaneously cooled to room temperature to obtain the crystalline form
IV;
The preparation of Form I of Preparation Example 1 known
Methods Patent Document WO2009 / 137391 or CN200980126781.6 Example 58a and 58d known polymorph I. Preparation Specifically:
The N- {3- [5- (2- chloro-4-pyrimidinyl) -2- (1,1-Yue-yl-ethyl) -1,3-thiazol-4-yl] – 2-fluorophenyl 2,6-difluorophenyl sulfonamide (196 mg, 0.364mmol) and 7M ammonia in methanol (8ml, 56mmol) was added to a 25 ml autoclave, heated to 90 ° C for 24 hours, TLC showed the starting material the reaction was complete, The reaction system was cooled to room temperature, the solvent was concentrated and the residue was dry column chromatography to obtain N- {3- [5- (2- amino-4-pyrimidinyl) -2- (1,1-dimethylethyl ) -1,3-thiazol-4-yl] -2-fluorophenyl} -2,6-difluorobenzenesulfonamide 90 mg, yield: 45%.
The N- {3- [5- (2- amino-4-pyrimidinyl) -2- (1,1-Yue-yl-ethyl) -1,3-thiazol-4-yl] -2-fluorophenyl 2,6-difluorophenyl sulfonamide (204 mg, 0.393mmol) in isopropanol (2 mL) was added 曱 acid (0.131 ml, 0.393mmol) and the solution was stirred at room temperature for 3 hours. A white precipitate formed and the slurry was filtered and washed with diethyl ether to give N- [3- [5- (2- amino-4-pyrimidinyl) -2- (t-butyl) -4-thiazol-yl] -2-fluoro phenyl] -2,6-difluorobenzenesulfonamide 曱 sulphonates crystalline solid (221 mg, 87% yield) as a white.
1HNM (400MHz, DMSO-d6)5 ppm 10.85(s, lH)7.92-8.05(m, 1H), 7.56-7.72(m, 1H), 6.91-7.50(m, 7H), 5.83-5.98(m, 1H) , 2.18-2.32(m, 3H) , 1.36(s, 9H)。
Preparation of crystal form obtained X-ray powder diffraction pattern shown in Figure 10. Report is consistent with the patent document WO2009 / 137391 or CN200980126781.6.
DSC chart is shown in Fig. Show: Known polymorphs I melt away as 247 ° C~250 ° C.
TGA spectrum shown in Figure 12. Show: decomposition temperature of 261 ° C.
Example 1
Take 10.02 mg polymorph IV (Example 7 Preparation) in 5 ml glass vial, add 0.5 ml of water, ultrasonic resulting suspension stirred at room temperature for 15 minutes, after centrifugation without drying, the present invention is to obtain crystalline form II. The yield was 10.00 mg; 99% yield.
X-ray powder diffraction pattern shown in Figure 6.
TGA pattern shown in Figure 7. Show: Form II at 50 ° C before the weight loss of about 4.6% (about 1.5 water), 50 ° C ~ 155 ° C 1.4% weight loss (about 0.5 water), the decomposition temperature of 287 ° C.
………………….
PATENT
http://www.google.com/patents/WO2009137391A2?cl=en
WO 2009137391
Example 58a: Λ/-{3-r5-(2-Amino-4-pyrimidinylV2-(1.1-dimethylethylV1.3-thiazol-4-yll-2- fluorophenyl}-2,6-difluorobenzenesulfonamide

Following a procedure analogous to the procedure described in Example 51, Step B using Λ/-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (196 mg, 0.364 mmol) and ammonia in methanol 7M (8 ml, 56.0 mmol) and heating to 90 0C for 24 h, the title compound, Λ/-{3- [5-(2-amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide was obtained (94 mg, 47% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.83 (s, 1 H), 7.93 (d, J=5.2 Hz, 1 H), 7.55 – 7.70 (m, 1 H), 7.35 –
7.43 (m, 1 H), 7.31 (t, J=6.3 Hz, 1 H), 7.14 – 7.27 (m, 3 H), 6.70 (s, 2 H), 5.79 (d, J=5.13 Hz, 1 H), 1.35 (s, 9 H). MS (ESI): 519.9 [M+H]+.
Example 58b: Λ/-{3-r5-(2-Amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yll-2- fluorophenyl}-2,6-difluorobenzenesulfonamide
19.6 mg of Λ/-{3-[5-(2-Amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (may be prepared in accordance with example 58a) was combined with 500 μl_ of ethyl acetate in a 2-mL vial at room temperature. The slurry was temperature-cycled between 0-400C for 48 hrs. The resulting slurry was allowed to cool to room temperature and the solids were collected by vacuum filtration. The solids were analyzed by Raman, PXRD, DSC/TGA analyses, which indicated a crystal form different from the crystal form resulting from Example 58a, above. Example 58c: Λ/-{3-r5-(2-amino-4-pyrimidinylV2-(1.1-dimethylethylV1.3-thiazol-4-yll-2- fluorophenyl}-2,6-difluorobenzenesulfonamide

Step A: methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate

Methyl 3-amino-2-fluorobenzoate (50 g, 1 eq) was charged to reactor followed by dichloromethane (250 ml_, 5 vol). The contents were stirred and cooled to ~15°C and pyridine (26.2 ml_, 1.1 eq) was added. After addition of the pyridine, the reactor contents were adjusted to ~15°C and the addition of 2,6-diflurorobenzenesulfonyl chloride (39.7 ml_, 1.0 eq) was started via addition funnel. The temperature during addition was kept <25°C. After complete addition, the reactor contents were warmed to 20-250C and held overnight. Ethyl acetate (150 ml.) was added and dichloromethane was removed by distillation. Once distillation was complete, the reaction mixture was then diluted once more with ethyl acetate (5 vol) and concentrated. The reaction mixture was diluted with ethyl acetate (10 vol) and water (4 vol) and the contents heated to 50- 55°C with stirring until all solids dissolve. The layers were settled and separated.
The organic layer was diluted with water (4 vol) and the contents heated to 50-55° for 20-30 min. The layers were settled and then separated and the ethyl acetate layer was evaporated under reduced pressure to ~3 volumes. Ethyl Acetate (5 vol.) was added and again evaporated under reduced pressure to ~3 volumes. Cyclohexane (9 vol) was then added to the reactor and the contents were heated to reflux for 30 min then cooled to 0 0C. The solids were filtered and rinsed with cyclohexane (2 x 100 ml_). The solids were air dried overnight to obtain methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2- fluorobenzoate (94.1 g, 91 %).
Step B: Λ/-{3-[(2-chloro-4-pyrimidinyl)acetyl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide

Methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate (490 g, 1 equiv.), prepared generally in accordance with Step A, above, was dissolved in THF (2.45 L, 5 vols) and stirred and cooled to 0-3 0C. 1 M lithium bis(trimethylsilyl)amide in THF (5.25 L, 3.7 equiv.) solution was charged to the reaction mixture followed addition of 2-chloro-4- methylpyrimidine (238 g, 1.3 equiv.) in THF (2.45 L, 5 vols). The reaction was then stirred for 1 hr. The reaction was quenched with 4.5M HCI (3.92 L, 8 vols). The aqueous layer (bootom layer) was removed and discarded.
The organic layer was concentrated under reduced pressure to ~2L. IPAC (isopropyl acetate) (2.45L) was added to the reaction mixture which was then concentrated to ~2L. IPAC (0.5L) and MTBE (2.45 L) was added and stirred overnight under N2. The solids were filtered. The solids and mother filtrate added back together and stirred for several hours. The solids were filtered and washed with MTBE (~5 vol). The solids were placed in vacuum oven at 50 0C overnight. The solids were dried in vacuum oven at 30 0C over weekend to obtain Λ/-{3-[(2-chloro-4-pyrimidinyl)acetyl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide (479 g, 72%).
Step C: Λ/-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide

To a reactor vessel was charged Λ/-{3-[(2-chloro-4-pyrimidinyl)acetyl]-2-fluorophenyl}- 2,6-difluorobenzenesulfonamide (30 g, 1 eq) followed by dichloromethane (300 ml_). The reaction slurry was cooled to ~10°C and N-bromosuccinimide (“NBS”) (12.09 g, 1 eq) was added in 3 approximately equal portions, stirring for 10-15 minutes between each addition. After the final addition of NBS, the reaction mixture was warmed to ~20°C and stirred for 45 min . Water (5 vol) was then added to the reaction vessel and the mixture was stirred and then the layers separated. Water (5 vol) was again added to the dichloromethane layer and the mixture was stirred and the layers separated.
The dichloromethane layers were concentrated to -120 ml_. Ethyl acetate (7 vol) was added to the reaction mixture and concentrated to -120 ml_. Dimethylacetamide (270 ml.) was then added to the reaction mixture and cooled to -1O0C. 2,2-Dimethylpropanethioamide (1.3 g, 0.5 eq) in 2 equal portions was added to the reactor contents with stirring for -5 minutes between additions. The reaction was warmed to 20-25 0C. After 45 min, the vessel contents were heated to 75°C and held for 1.75 hours . The reaction mixture was then cooled to 5°C and water (270 ml) was slowly charged keeping the temperature below 300C. Ethyl acetate (4 vol) was then charged and the mixture was stirred and layers separated. Ethyl acetate (7 vol) was again charged to the aqueous layer and the contents were stirred and separated.
Ethyl acetate (7 vol) was charged again to the aqueous layer and the contents were stirred and separated. The organic layers were combined and washed with water (4 vol) 4 times and stirred overnight at 20-250C. The organic layers were then concentrated under heat and vacuum to 120 ml_. The vessel contents were then heated to 500C and heptanes (120 ml.) were added slowly. After addition of heptanes, the vessel contents were heated to reflux then cooled to 0°C and held for -2 hrs. The solids were filtered and rinsed with heptanes (2 x 2 vol). The solid product was then dried under vacuum at 300C to obtain Λ/-{3-[5-(2-chloro-4-pyrimidinyl)- 2-(1 , 1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (28.8 g, 80%).
Step D:
Λ/-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide
In 1 gal pressure reactor, a mixture of Λ/-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1- dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (120 g) prepared in accordance with Step C, above, and ammonium hydroxide (28-30%, 2.4 L, 20 vol) was heated in the sealed pressure reactor to 98-103 0C and stirred at this temperature for 2 hours. The reaction was cooled slowly to room temperature (20 0C) and stirred overnight. The solids were filtered and washed with minimum amount of the mother liquor and dried under vacuum. The solids were added to a mixture of EtOAc (15 vol)/ water (2 vol) and heated to complete dissolution at 60-70 0C and the aqueous layer was removed and discarded. The EtOAC layer was charged with water (1 vol) and neutralized with aq. HCI to ~pH 5.4-5.5. and added water (1vol). The aqueous layer was removed and discarded at 60-70 0C.
The organic layer was washed with water (1 vol) at 60-70 0C and the aqueous layer was removed and discarded. The organic layer was filtered at 60 0C and concentrated to 3 volumes. EtOAc (6 vol) was charged into the mixture and heated and stirred at 72 0C for 10 min , then cooled to 2O0C and stirred overnight. EtOAc was removed via vacuum distillation to concentrate the reaction mixture to ~3 volumes.
The reaction mixture was maintained at -65-7O0C for ~30mins. Product crystals having the same crystal form as those prepared in Example 58b (and preparable by the procedure of Example 58b), above, in heptanes slurry were charged. Heptane (9 vol) was slowly added at 65-70 0C. The slurry was stirred at 65-70 0C for 2-3 hours and then cooled slowly to 0-50C. The product was filtered, washed with EtOAc/heptane (3/1 v/v, 4 vol) and dried at 45°C under vacuum to obtain Λ/-{3-[5-(2- amino-4-pyrimidinyl)-2-(1 , 1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide (102.3 g, 88%).
Example 58d:
Λ/-{3-r5-(2-amino-4-pyrimidinvn-2-(1.1-dimethylethylV1.3-thiazol-4-yll-2- fluorophenyl}-2,6-difluorobenzenesulfonamide methanesulfonate
 MESYLATE
MESYLATETo a solution of Λ/-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (204 mg, 0.393 mmol) in isopropanol (2 ml_), methanesulfonic acid (0.131 ml_, 0.393 mmol) was added and the solution was allowed to stir at room temperature for 3 hours. A white precipitate formed and the slurry was filtered and rinsed with diethyl ether to give the title product as a white crystalline solid (210 mg, 83% yield).
1H NMR (400 MHz, DMSO-d6) δ ppm 10.85 (s, 1 H) 7.92 – 8.05 (m, 1 H) 7.56 – 7.72 (m, 1 H) 6.91 – 7.50 (m, 7 H) 5.83 – 5.98 (m, 1 H) 2.18 – 2.32 (m, 3 H) 1.36 (s, 9 H). MS (ESI): 520.0 [M+H]+.WO2009137391
…………………………………………………………………
PAPER
ACS Medicinal Chemistry Letters (2013), 4(3), 358-362.
http://pubs.acs.org/doi/abs/10.1021/ml4000063

…………………………………………………………….
Patent
http://www.google.com/patents/WO2014158467A1?cl=en

Step C : N- {3-[5-(2-chloro-4-pyrimidinyl)-2-(l , 1 -dimethylethyl)-l ,3-thiazol-4-yl]- 2-fluorophenyl}-2,6-difluorobenzenesulfonamide

To a reactor vessel was charged N- {3-[(2-chloro-4-pyrimidinyl)acetyl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (30 g, 1 eq) followed by dichloromethane (300 mL). The reaction slurry was cooled to ~10°C and N-bromosuccinimide (“NBS”) (12.09 g, 1 eq) was added in 3 approximately equal portions, stirring for 10-15 minutes between each addition. After the final addition of NBS, the reaction mixture was warmed to ~20°C and stirred for 45 min . Water (5 vol) was then added to the reaction vessel and the mixture was stirred and then the layers separated. Water (5 vol) was again added to the dichloromethane layer and the mixture was stirred and the layers separated. The dichloromethane layers were concentrated to -120 mL. Ethyl acetate (7 vol) was added to the reaction mixture and concentrated to -120 mL. Dimethylacetamide (270 mL) was then added to the reaction mixture and cooled to ~10°C. 2,2-Dimethylpropanethioamide (1.3 g, 0.5 eq) in 2 equal portions was added to the reactor contents with stirring for ~5 minutes between additions. The reaction was warmed to 20-25 °C. After 45 min, the vessel contents were heated to 75°C and held for 1.75 hours . The reaction mixture was then cooled to 5°C and water (270 ml) was slowly charged keeping the temperature below 30°C. Ethyl acetate (4 vol) was then charged and the mixture was stirred and layers separated. Ethyl acetate (7 vol) was again charged to the aqueous layer and the contents were stirred and separated. Ethyl acetate (7 vol) was charged again to the aqueous layer and the contents were stirred and separated. The organic layers were combined and washed with water (4 vol) 4 times and stirred overnight at 20-25°C. The organic layers were then concentrated under heat and vacuum to 120 mL. The vessel contents were then heated to 50°C and heptanes (120 mL) were added slowly. After addition of heptanes, the vessel contents were heated to reflux then cooled to 0°C and held for ~2 hrs. The solids were filtered and rinsed with heptanes (2 x 2 vol). The solid product was then dried under vacuum at 30°C to obtain N-{3-[5-(2-chloro-4-pyrimidinyl)-2-(l,l-dimethylethyl)-l,3- thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (28.8 g, 80%).
Compound B is disclosed and claimed, along with pharmaceutically acceptable salts thereof, as being useful as an inhibitor of BRaf activity, particularly in the treatment of cancer, in PCT patent application PCT/US09/42682. Compound B is embodied by Examples 58a through 58e of the application. The PCT application was published on 12 November 2009 as publication WO2009/137391, and is hereby incorporated by reference.
Suitably, Compound B may be prepared according to the methods below:
Method 1 : Compound B (first crystal form) – N-{3-[5-(2-Amino-4-pyrimidinyl)-2- (1,1 -dimethylethyl)- 1 ,3-thiazol-4-yl]- -fluorophenyl} -2,6-difluorobenzenesulfonamide

A suspension of N-{3-[5-(2-chloro-4-pyrimidinyl)-2-(l,l-dimethylethyl)-l,3- thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (196 mg, 0.364 mmol) and ammonia in methanol 7M (8 ml, 56.0 mmol) was heated in a sealed tube to 90 °C for 24 h. The reaction was diluted with DCM and added silica gel and concentrated. The crude product was chromatographed on silica gel eluting with 100% DCM to 1 : 1 [DCM: (9: 1 EtOAc:MeOH)]. The clean fractions were concentrated to yield the crude product. The crude product was repurified by reverse phase HPLC (a gradient of acetonitrile: water with 0.1%TFA in both). The combined clean fractions were concentrated then partitioned between DCM and saturated NaHC03. The DCM layer was separated and dried over Na2S04. The title compound, N-{3-[5-(2-amino-4-pyrimidinyl)-2-(l,l-dimethylethyl)- l,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide was obtained (94 mg, 47% yield). 1H NMR (400 MHz, DMSO- 6) δ ppm 10.83 (s, 1 H), 7.93 (d, J=5.2 Hz, 1 H), 7.55 – 7.70 (m, 1 H), 7.35 – 7.43 (m, 1 H), 7.31 (t, J=6.3 Hz, 1 H), 7.14 – 7.27 (m, 3 H), 6.70 (s, 2 H), 5.79 (d, J=5.13 Hz, 1 H), 1.35 (s, 9 H). MS (ESI): 519.9 [M+H]+.
Method 2: Compound B (alternative crystal form) – N-{3-[5-(2-Amino-4- pyrimidinyl)-2-(l,l-dimethylethyl)-l,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide 19.6 mg of N-{3-[5-(2-Amino-4-pyrimidinyl)-2-(l,l- dimethylethyl)- 1 ,3-thiazol-4-yl]-2-fluorophenyl} -2,6-difluorobenzenesulfonamide (may be prepared in accordance with example 58a) was combined with 500 L of ethyl acetate in a 2-mL vial at room temperature. The slurry was temperature-cycled between 0-40°C for 48 hrs. The resulting slurry was allowed to cool to room temperature and the solids were collected by vacuum filtration. The solids were analyzed by Raman, PXRD, DSC/TGA analyses, which indicated a crystal form different from the crystal form resulting from Example 58a, above.
Method 3: Compound B (alternative crystal form, large batch) – N-{3-[5-(2-amino- 4-pyrimidinyl)-2-(l , 1 -dimethylethyl)- 1 ,3-thiazol-4-yl]-2-fluorophenyl} -2,6- difluorobenzenesulfonamide

Step D : N-{3-[5-(2-amino-4-pyrimidinyl)-2-(l,l-dimethylethyl)-l,3-thiazol-4-yl]-
2-fluorophenyl}-2,6-difluorobenzenesulfonamide
In 1 gal pressure reactor, a mixture of N-{3-[5-(2-chloro-4-pyrimidinyl)-2-(l,l- dimethylethyl)- 1 ,3-thiazol-4-yl]-2-fluorophenyl} -2,6-difluorobenzenesulfonamide ( 120 g) prepared in accordance with Step C, above, and ammonium hydroxide (28-30%, 2.4 L, 20 vol) was heated in the sealed pressure reactor to 98-103 °C and stirred at this temperature for 2 hours. The reaction was cooled slowly to room temperature (20 °C) and stirred overnight. The solids were filtered and washed with minimum amount of the mother liquor and dried under vacuum. The solids were added to a mixture of EtOAc (15 vol)/ water (2 vol) and heated to complete dissolution at 60-70 °C and the aqueous layer was removed and discarded. The EtOAC layer was charged with water (1 vol) and neutralized with aq. HC1 to ~pH 5.4-5.5. and added water (lvol). The aqueous layer was removed and discarded at 60-70 °C. The organic layer was washed with water (1 vol) at 60-70 °C and the aqueous layer was removed and discarded. The organic layer was filtered at 60 °C and concentrated to 3 volumes. EtOAc (6 vol) was charged into the mixture and heated and stirred at 72 °C for 10 min , then cooled to 20°C and stirred overnight. EtOAc was removed via vacuum distillation to concentrate the reaction mixture to ~3 volumes. The reaction mixture was maintained at ~65-70°C for ~30mins. Product crystals having the same crystal form as those prepared in Example 58b (and preparable by the procedure of Example 58b), above, in heptanes slurry were charged. Heptane (9 vol) was slowly added at 65-70 °C. The slurry was stirred at 65-70 °C for 2-3 hours and then cooled slowly to 0- 5°C. The product was filtered, washed with EtO Ac/heptane (3/1 v/v, 4 vol) and dried at 45°C under vacuum to obtain N-{3-[5-(2-amino-4-pyrimidinyl)-2-(l,l-dimethylethyl)-l,3- thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (102.3 g, 88%>).
MESYLATE
Method 4: Compound B (mesylate salt) – N-{3-[5-(2-amino-4-pyrimidinyl)-2-(l,l- dimethylethyl)- 1 ,3-thiazol-4-yl]-2-fluorophenyl} -2,6-difluorobenzenesulfonamide methanesulfonate

To a solution of N-{3-[5-(2-amino-4-pyrimidinyl)-2-(l,l-dimethylethyl)-l,3- thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (204 mg, 0.393 mmol) in isopropanol (2 mL), methanesulfonic acid (0.131 mL, 0.393 mmol) was added and the solution was allowed to stir at room temperature for 3 hours. A white precipitate formed and the slurry was filtered and rinsed with diethyl ether to give the title product as a white crystalline solid (210 mg, 83% yield). 1H NMR (400 MHz, DMSO- 6) δ ppm 10.85 (s, 1 H) 7.92 – 8.05 (m, 1 H) 7.56 – 7.72 (m, 1 H) 6.91 – 7.50 (m, 7 H) 5.83 – 5.98 (m, 1 H) 2.18 – 2.32 (m, 3 H) 1.36 (s, 9 H). MS (ESI): 520.0 [M+H]+.
Method 5: Compound B (alternative mesylate salt embodiment) – N-{3-[5-(2- amino-4-pyrimidinyl)-2-(l , 1 -dimethylethyl)-l ,3-thiazol-4-yl]-2-fluorophenyl} -2,6- difluorobenzenesulfonamide methanesulfonate
N- {3-[5-(2-amino-4-pyrimidinyl)-2-(l , 1 -dimethylethyl)- 1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (as may be prepared according to example 58a) (2.37g, 4.56 mmol) was combined with pre-filtered acetonitrile (5.25 vol, 12.4 mL). A pre-filtered solution of mesic acid (1.1 eq., 5.02 mmol, 0.48 g) in H20 (0.75 eq., 1.78 mL) was added at 20°C. The temperature of the resulting mixture was raised to 50-60°C while maintaining a low agitation speed. Once the mixture temperature reached to 50- 60°C, a seed slurry of N-{3-[5-(2-amino-4-pyrimidinyl)-2-(l,l-dimethylethyl)-l,3-thiazol- 4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide methanesulfonate (1.0 %w/w slurried in 0.2 vol of pre-filtered acetonitrile) was added, and the mixture was aged while agitating at a speed fast enough to keep solids from settling at 50-60°C for 2 hr. The mixture was then cooled to 0-5°C at 0.25°C/min and held at 0-5°C for at 6 hr. The mixture was filtered and the wet cake was washed twice with pre-filtered acetonitrile. The first wash consisted of 14.2 ml (6 vol) pre-filtered acetonitrile and the second wash consisted of 9.5 ml (4 vol) pre-filtered acetonitrile. The wet solid was dried at 50°C under vacuum, yielding 2.39 g (85.1% yield) of product
Dara Phoenix (Dabrafenib) by the British GlaxoSmithKline (GSK) has developed Sisu threonine protein kinase (BRAF) inhibitor, as monotherapy ro ー kinds of clothes capsules for carrying BRAF V600E mutation surgical unresectable melanoma or metastatic melanoma treatment of adult patients, Dara Phoenix mesylate in May 2013 was approved by the US Food and Drug Administration (FDA), and is listed on the United States, the trade name Tafinlar (Da Feina). Since the European Medicines Agency (EMA) Committee for Medicinal Products for human use (CHMP) positive evaluation of Tafinlar, making the drug is expected to become after Roche’s Weiluofeini (Vemurafinib) to enter the European market, following a second BRAF inhibitors.
The chemical name Phoenix Dallas: N- [3- [5- (2- amino-4-pyrimidinyl) -2_ (tert-butyl) ~ ~ thiazol-4-yl] _2_ fluorophenyl] – 2,6_-difluorobenzenesulfonamide.

World Patent No. W02009137391, No. W02011047238 and W02012148588 number reported Dallas and Phoenix and its medicinal value synthesis method of the composition. According to the structural characteristics of Dara Phoenix and its analogues, the synthesis of such substances currently have A, B and C are three routes.

A more common route is the synthetic route, by reaction of 3-amino-2-fluorobenzoate (IX) first and 2,6_-difluorobenzene sulfonyl chloride (III) to amidation reaction occurs sulfonamide intermediate ( X); intermediate (X) with 2-chloro-4-methyl pyrimidine (XI) The condensation reaction occurs under the action of a strong base to give the intermediate (XII); intermediate (XII) to give the intermediate bromo
(XIII); intermediate (XIII) with 2,2_ dimethyl thiopropionamide (VI) to give the cyclized intermediate (XIV); and finally, the intermediate (XIV) by ammonolysis to afford the title compound Dallas Phoenix (I).

Different [0009] B is the first route by reaction of 3-amino-2-fluorobenzoate (IX) amino group protection, and thus condensation, cyclization, and bromo; then be obtained by deprotection of the amino group and the sulfonamide Intermediate (XIV); similarly, the intermediate
(XIV) obtained by ammonolysis target compound Dara Phoenix (I).

c route design features that first aminolysis reaction, and then give the desired product by deprotection and amino sulfonamide reaction. Clearly, this design is suitable for the route of these substituted amino ー aminolysis reaction, and for compounds such as Dallas Phoenix having pyrimidinylamino structure is not applicable. The reason is that if there are two aromatic amino groups will make the final sulfonamide ー reaction step to lose selectivity.

Example IV: the reaction flask was added N- [3- (5- formyl-2-t-butyl-ko -4_ thiazolyl) -2_ fluorophenyl] -2,6_ difluoro benzenesulfonamide (VIII) (5.4g, 11.5mmol), N, N- dimethylformamide dimethyl acetal (DMF-DMA) (2.74g, 23mmol) and xylene 50mL, heated to 140 ° C. About every four hours methanol was distilled out of the resulting reaction system, the reaction takes about 24 hours in total, the end of the reaction was detected by TLC. Cool, add hexane 40mL, have produced a yellow solid, filtered, and dried solids obtained after January nitrate melon (1.36,11.5mmol), sodium hydroxide (0.46g, 11.5mmol) and n-Ding enjoy 5OmL, warmed to 120 ° C, The reaction for 12 inches, TLC the reaction was complete. Cooling, with a crystal precipitated crystallized slowly for 3 inches, and filtered. The filter cake starched water, filtered and dried to yield an off-white solid Dara Phoenix (I) 3.58g, yield 60%.
References
- Gibney, G. T.; Zager, J. S. (2013). “Clinical development of dabrafenib in BRAF mutant melanoma and other malignancies”. Expert Opinion on Drug Metabolism & Toxicology 9 (7): 1. doi:10.1517/17425255.2013.794220. PMID 23621583.
- Huang, T.; Karsy, M.; Zhuge, J.; Zhong, M.; Liu, D. (2013). “B-Raf and the inhibitors: From bench to bedside”. Journal of Hematology & Oncology 6: 30. doi:10.1186/1756-8722-6-30. PMC 3646677. PMID 23617957.
- “GSK melanoma drugs add to tally of U.S. drug approvals”. Reuters. May 30, 2013.
- “Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations” 367 (18). New England Journal of Medicine. November 1, 2012. pp. 1694–703. doi:10.1056/NEJMoa1210093. PMC 3549295. PMID 23020132.
“Dabrafenib/Trametinib Combination Approved for Advanced Melanoma”. OncLive. January 9, 2013.
Updates
Dabrafenib prediction
1H NMR PREDICT

![N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide NMR spectra analysis, Chemical CAS NO. 1195765-45-7 NMR spectral analysis, N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2015-01-11/001/566/1566739_1h.png)
13C NM PREDICT
![N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide NMR spectra analysis, Chemical CAS NO. 1195765-45-7 NMR spectral analysis, N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2015-01-11/001/566/1566739_13c.png)
COSY NMR PREDICT

HMBC, HSBC NMR PREDICT
INTERMEDIATES
INT 1
 Methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate;Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoate, 1195768-23-0
Methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate;Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoate, 1195768-23-0
 methyl 3-bromo-2-fluorobenzylate; PC3663; fluorobromobenzoic acid methyl ester;
methyl 3-bromo-2-fluorobenzylate; PC3663; fluorobromobenzoic acid methyl ester; methyl 3-amino-2-fluorobenzoate, CAS No. 1195768-18-3
methyl 3-amino-2-fluorobenzoate, CAS No. 1195768-18-3 Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoate, CAS No. 1195768-19-4
Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoate, CAS No. 1195768-19-4 CAS No. 1042055-86-6 Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoate
CAS No. 1042055-86-6 Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoateSYN 1
![]()
 DABARAFENIB
DABARAFENIB
GLAXOSMITHKLINE LLC; HOOS, Axel; GRESHOCK, Joel Patent: WO2014/66606 A2, 2014 ; Location in patent: Page/Page column 20; 24 ;
SYN 2

![]()
WO2011/47238 A1, ;
SYN 3

![]()
ACS Medicinal Chemistry Letters, , vol. 4, # 3 p. 358 – 362
SYN 4
![]()
ACS Medicinal Chemistry Letters, , vol. 4, # 3 p. 358 – 362
SYN 5
![]()
ACS Medicinal Chemistry Letters, , vol. 4, # 3 p. 358 – 362
SYN 6
WO2011/47238 A1, ;
SYN 7
methyl 3-amino-2-fluorobenzoate, CAS No. 1195768-18-3WO2011/47238
WO2015003571
达拉菲尼甲磺酸盐的新晶型及其制备方法
Brief Description
Figure 1 Form IV of the present invention an X-ray powder diffraction pattern.
Figure 2 is a schematic diagram Form IV of DSC language.
Figure 3 Form IV of the present invention TGA profiles.
Figure 4 is a dynamic water adsorption of Form IV of the invention, FIG.
Figure 5 Form IV of the present invention 1HNMR spectrum.
Figure 6 of the present invention, Form II X-ray powder diffraction pattern.
Figure 7 of the present invention, Form II TGA profiles.
Figure 8 Form III of the present invention an X-ray powder diffraction pattern.
Figure 9 Form V of the present invention, an X-ray powder diffraction pattern.
Figure 58a and in Example 10 in accordance with Patent Document WO2009 / 137391 or in CN200980126781.6
58d method described for the preparation of polymorph I of the known X-ray powder diffraction pattern.
Figure 11 is in accordance with Patent Document WO2009 / 137391 or CN200980126781.6 in the method of Example 58a and 58d described for the preparation of polymorph I of the known DSC pattern.
12 is in accordance with Patent Document WO2009 / 137391 or CN200980126781.6 in the method of Example 58a and 58d described for the preparation of polymorph I of the known TGA profiles.
Figure 13 is a known polymorph I in Comparative Example 1 in various stages XRPD comparison chart with the sample from top to bottom in the order of: Dara Phoenix free base hydrate, a known polymorph I in water was stirred for 15 After minutes to obtain a sample, and a known polymorph I.
Figure 14 Form IV in the present invention Comparative Example 1 each stage XRPD comparison chart with the sample from top to bottom in the order of: Form IV, Form IV in water with stirring for 15 minutes to obtain a sample, Form IV After stirring overnight in water to obtain a sample, as well as the free base of the hydrate Dara Phoenix. Figure 15 is a Comparative Form IV polymorph I of the known elution compared to the situation in Figure 1 (A to Form IV, ■ known Form 1).
Figure 16 is known in the polymorph I of Comparative Example 2 in various stages XRPD comparison chart (figure from top to bottom as follows: Form I is known API by “wet granulation” process of granulation (excluding section 3-step tablet) obtained by the sample, the known polymorphs I and amount of excipients formulated physically mixed formulation obtained sample, lactose monohydrate and microcrystalline cellulose according to Formulation physical sample after mixing, Dara Feeney free base hydrate, as well as known Form 1).
17 is a crystalline form IV according to the present invention in Comparative Example 2 in various stages XRPD comparison chart (from top to bottom as follows: In Form IV according to API “wet granulation” process of granulation (not included in Step 3 tableting) after the sample obtained, Form IV and excipients Formulation amount by physically mixing the obtained sample, the sample lactose monohydrate and microcrystalline cellulose according to Formulation after physical mixing, and Form IV).
 |
|
| Systematic (IUPAC) name | |
|---|---|
| N-{3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide | |
| Clinical data | |
| Trade names | Tafinlar |
|
|
| Legal status | |
| Identifiers | |
| CAS number | 1195765-45-7 |
| ATC code | L01XE23 |
| PubChem | CID 44462760 |
| ChemSpider | 25948204 |
| ChEBI | CHEBI:75045 |
| ChEMBL | CHEMBL2028663 |
| Chemical data | |
| Formula | C23H20F3N5O2S2 |
| Molecular mass | 519.56 g/mol |
