
Home » cancer (Page 12)
Category Archives: cancer
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Resveratrol remains effective against cancer after the body converts it
A chemical found in red wine remains effective at fighting cancer even after the body’s metabolism has converted it into other compounds. This is an important finding in a new paper published in the journal Science Translational Medicine by Cancer Research UK-funded researchers at the University of Leicester’s Department of Cancer Studies and Molecular Medicine.
The paper reveals that resveratrol – a compound extracted from the skins of red grapes – is not rendered ineffective once it is metabolised by the body.
This is an important development, as resveratrol is metabolised very quickly – and it had previously been thought that levels of the extracted chemical drop too quickly to make it usable in clinical trials.
The new research shows that the chemical can still be taken into cells after it has been metabolised into resveratrol sulfates.
Enzymes within cells are then able to break it down into resveratrol…
View original post 504 more words
FDA Approves Zykadia, Ceritinib, LDK378 for ALK-Positive NSCLC



Acting 4 months ahead of schedule, the FDA has granted an accelerated approval to ceritinib (Zykadia; LDK378) as a treatment for patients with ALK-positive metastatic non-small cell lung cancer (NSCLC) following treatment with crizotinib (Xalkori), based on a single-arm clinical trial demonstrating a durable improvement in overall response rates (ORR).
The approval for the second-generation ALK inhibitor was supported by results from an analysis of 163 patients treated with single-agent ceritinib at 750 mg daily following progression on crizotinib. In these select patients, the ORR was 54.6% with a 7.4-month median duration of response, according to data submitted to the FDA by Novartis, the company developing the drug. Based on these findings, the FDA granted ceritinib a Breakthrough Therapy designation, Priority Review, and orphan product designation.
“Today’s approval illustrates how a greater understanding of the underlying molecular pathways of a disease can lead to the development of specific therapies aimed at these pathways,” Richard Pazdur, MD, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a statement. “It also demonstrates the FDA’s commitment to working cooperatively with companies to expedite a drug’s development, review and approval, reflecting the promise of the breakthrough therapy designation program.”
In the study that was the basis for the approval, the primary endpoint was ORR by RECIST criteria with a secondary outcome measure of duration of response. Treatment with ceritinib resulted in an ORR of 54.6% by investigator assessment with a median duration of response of 7.4 months. By blinded independent central review, the ORR was 43.6% and the duration of response was 7.1 months.
Earlier this year, results from a dose escalation study that examined ceritinib in 130 patients who were untreated or refractory to crizotinib were published in the New England Journal of Medicine. In this analysis for patients who received doses of at least 400 mg (n = 114), the ORR was 58%. Patients who had progressed on crizotinib (n = 80) experienced an ORR of 56% and those who were crizotinib-naïve (n =34) had an ORR of 62%.
The median progression-free survival was 7.0 months and the median duration of response was 8.2 months (95% CI; 6.9-11.4). Additionally, responses were seen in patients with untreated metastatic brain lesions who progressed on prior therapy with crizotinib, the authors of the study noted.
The most frequent adverse events were nausea (82%), diarrhea (75%), vomiting (65%), fatigue (47%) and increased alanine aminotransferase levels (35%). These adverse events were generally mild and resolved when treatment stopped or the dose was reduced.
The most common grade 3 or 4 drug-related adverse events were increased alanine aminotransferase levels (21%), increased aspartate aminotransferase levels (11%), diarrhea (7%) and increased lipase levels (7%), all of which were reversible upon treatment discontinuation.
“Zykadia represents an important treatment option for ALK-positive NSCLC patients who relapse after starting initial therapy with crizotinib,” Alice Shaw, MD, PhD, of the Massachusetts General Hospital (MGH) Cancer Center, lead author of the report, said in a statement. “This approval will affect the way we manage and monitor patients with this type of lung cancer, as we will now be able to offer them the opportunity for continued treatment response with a new ALK inhibitor.”
Two phase III studies are enrolling patients to further explore the efficacy and safety of ceritinib in patients with ALK-positive NSCLC. These studies will likely act as confirmation for the accelerated approval. In the first, ceritinib will be compared with chemotherapy in untreated patients with ALK-rearranged NSCLC (NCT01828099). The second will compare ceritinib to chemotherapy in ALK-positive patients with NSCLC following progression on chemotherapy and crizotinib (NCT01828112).
“The approval of Zykadia less than three and a half years after the first patient entered our clinical trial exemplifies what is possible with a highly focused approach to drug development and strong collaboration,” Alessandro Riva, MD, president of Novartis Oncology ad interim and global head of Oncology Development and Medical Affairs, said in a statement. “The dedication of clinical investigators, patients, the FDA and others has enabled us to bring this medicine to patients in need as swiftly as possible.”
![]()

Nitration of 2-chloro-4-fluoro-1-methylbenzene with KNO3 in the presence of H2SO4 gives 1-chloro-5-fluoro-2-methyl-4-nitrobenzene , which upon condensation with isopropyl alcohol in the presence of Cs2CO3 in 2-PrOH at 60 °C yields 5-isopropoxy-2-methyl-4-nitrobenzene .
Suzuki coupling of chloride with 4-pyridineboronic acid in the presence of Pd2dba3, K3PO4 and SPhos in dioxane/water at 150 °C (microwave irradiation) provides 4-(5-isopropoxy-2-methyl-4-nitrophenyl)pyridine , which is then subjected to global reduction using H2 over PtO2 in the presence of TFA in AcOH to afford 2-isopropoxy-5-methyl-4-piperidin-4-ylaniline .
N-Protection of piperidine with Boc2O in the presence of Et3N in CH2Cl2 furnishes the corresponding carbamate (VIII), which upon Buchwald-Hartwig cross coupling with 2,5-dichloropyrimidine derivative (prepared by condensation of 2-(isopropylsulfonyl)aniline and 2,4,5-trichloropyrimidine in the presence of NaH in DMSO/DMF) in the presence of Pd(OAc)2, Xantphos and Cs2CO3 in THF affords Boc-protected ceritinib . Finally, removal of Boc-group in compound using TFA in CH2Cl2 furnishes the target compound ceritinib
/////////////////

Anaplastic lymphoma kinase (ALK), a member of the insulin receptor superfamily of receptor tyrosine kinases, has been implicated in oncogenesis in hematopoietic and non- hematopoietic tumors. The aberrant expression of full-length ALK receptor proteins has been reported in neuroblastomas and glioblastomas; and ALK fusion proteins have occurred in anaplastic large cell lymphoma. The study of ALK fusion proteins has also raised the possibility of new therapeutic treatments for patients with ALK-positive malignancies. (Pulford et al., Cell. MoI. Life Sci. 61:2939-2953 (2004)).
Focal Adhesion Kinase (FAK) is a key enzyme in the integrin-mediated outside-in signal cascade (D. Schlaepfer et al., Prog Biophys MoI Biol 1999, 71, 43578). The trigger in the signal transduction cascade is the autophosphorylation of Y397. Phosphorylated Y397 is a SH2 docking site for Src family tyrosine kinases; the bound c-Src kinase phosphorylates other tyrosine residues in FAK. Among them, phsophorylated Y925 becomes a binding site for the SH2 site of Grb2 small adaptor protein. This direct binding of Grb2 to FAK is one of the key steps for the activation of down stream targets such as the Ras-ERK2/MAP kinase cascade.
Zeta-chain-associated protein kinase 70 (ZAP-70), a member of the protein tyrosine kinase family, is of potential prognostic importance in chronic lymphocytic leukemia (CLL). ZAP-70, known to be of importance in T and NK cell signaling but absent in normal peripheral B cells, is expressed in the majority of the poorer prognosis unmutated CLL and absent in most cases with mutated IgVH genes. ZAP-70 is also expressed in a minority of other B cell tumors. (Orchard et al., Leuk. Lymphoma 46:1689-98 (2005)). [0006] Insulin- like growth factor (IGF-I) signaling is highly implicated in cancer, with the IGF-I receptor (IGF-IR) as the predominating factor. IGR-IR is important for tumor transformation and survival of malignant cells, but is only partially involved in normal cell growth. Targeting of IGF-IR has been suggested to be a promising option for cancer therapy. (Larsson et al., Br. J. Cancer 92:2097-2101 (2005)).
Because of the emerging disease-related roles of ALK, FAK, ZAP-70 and IGF-IR, there is a continuing need for compounds which may be useful for treating and preventing a disease which responds to inhibition of ALK, FAK, ZAP-70 and/or IGF-IR
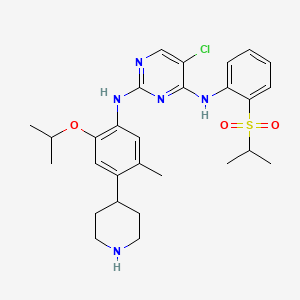
The compound 5-Chloro-N2-(2-isopropoxy-5-methyl-4-piperidin-4-yl-phenyl)-N4-[2- (propane-2-sulfonyl)-phenyl]-pyrimidine-2, 4-diamine, in the form of a free base, of formula

(I)
is an anaplastic lymphoma kinase (ALK) inhibitor, a member of the insulin receptor super family of receptor tyrosine kinases. Compound I was originally described in WO 2008/073687 Al as Example 7, compound 66. WO 2008/073687 Al , however, provides no information about crystalline forms of 5-
Chloro-N2-(2-isopropoxy-5-methyl-4-piperidin-4-yl-phenyl)-N4-[2-(propane-2-sulfonyl)- phenyl]-pyriniidine-2, 4-diamine or its corresponding salts. Crystalline forms of 5-Chloro-N2- (2-isopropoxy-5-methyl-4-piperidin-4-yl-phenyl)-N4-[2-(propane-2-sulfonyl)-phenyl]- pyrimidine-2, 4-diamine have been discovered, which are useful in treating diseases which respond to inhibition of anaplastic lymphoma kinase activity, focal adhesion kinase (FAK), zeta- chain-associated protein kinase 70 (ZAP-70) insulin-like growth factor (IGF-1 or a
combination thereof. The crystalline forms exhibit new physical properties that may be exploited in order to obtain new pharmacological properties, and that may be utilized in the drug product development of 5-Chloro-N2-(2-isopropoxy-5-methyl-4-piperidin-4-yl-phenyl)-N4-[2- (propane-2-sulfonyl)-phenyl]-pyrimidine-2, 4-diamine.
…………………….
WO 2008073687
http://www.google.com/patents/WO2008073687A2?cl=en
Example 7
5-Chloro-N2-(2-isopropoxy-5-methyl-4-piperidin-4-yl-phenyl)-N4-r2-(propane-2-sulfonyl)- phenvH-pyrimidine-2,4-diamine (66)

1 : 4-(5-Isopropoxv-2-methvl-4-nitro-phenyl)-pvridine

[0111] 4-Pyridineboronic acid (147 mg, 1.20 mmol, 1.1 equiv.) is dissolved in a 2:1 v/v mixture of dioxane and H2O (15 mL) and N2 is bubbled through for 5 minutes. Tris(dibenzylidene acetone)dipalladium (0) (100 mg, 0.109 mmol, 0.1 equiv.), 2- dicyclohexylphosphine-2′-6′-dimethoxy biphenyl (112 mg, 0.272 mmol, 0.25 equiv.), 1-chloro- 5-isopropoxy-2-methyl-4-nitro-benzene (Intermediate 4, 250 mg, 1.09 mmol, 1.0 equiv.) and K3PO4 (462 mg, 2.18 mmol, 2.0 equiv.) are added under a N2 blanket. The reaction vessel is sealed and heated with microwave irradiation to 150 0C for 20 min. After cooling to room temperature, the reaction is diluted with ethyl acetate and washed with 1 N aqueous NaOH (2x), the organic layer is then dried over Na2SO4 and filtered. After concentration, the crude product is purified by silica gel chromatography (gradient from hexanes to 30% ethyl acetate in hexanes) to give 4-(5-Isopropoxy-2-methyl-4-nitro-phenyl)-pyridine as a brown solid: ESMS m/z 273.1 (M + H+).
Steps 2 and 3 : 4-(4-Amino-5-isopropoxy-2-methyl-phenyl)-piperidine-l-carboxylic acid tert- butyl ester

[0112] 4-(5-Isopropoxy-2-methyl-4-nitro-phenyl)-pyridine from the previous step(438 mg, 1.61 mmol) dissolved in acetic acid (30 mL) is treated with TFA (0.24 mL, 3.22 mmol) and PtO2 (176 mg, 40% w/w). The reaction mixture is vigorously stirred under 1 atm. H2 for 36 hours. The reaction mixture is filtered and the filtrate is concentrated under vacuum. The resulting residue is diluted with ethyl acetate and washed with 1 N aqueous NaOH (2x), the organic layer is then dried over Na2SO4 and filtered. After concentration, the crude product (391 mg) is dissolved in anhydrous CH2Cl2 (30 mL). TEA is added (0.44 mL, 3.15, 2 equiv.) followed by Boc2O (344 mg, 1.57 equiv, 1 equiv.). The reaction is stirred at room temperature for 30 min. The reaction is concentrated under vacuum. The resulting residue is purified by silica gel chromatography (gradient from hexanes to 30% ethyl acetate in hexanes) to give 4-(4-amino-5- isopropoxy-2-methyl-phenyl)-piperidine-l-carboxylic acid tert-butyl ester as a sticky foam: ESMS m/z 293.1 (M-?Bu+H)+.
Steps 4 and 5
[0113] 4-(4-Amino-5-isopropoxy-2-methyl-phenyl)-piperidine-l-carboxylic acid tert-butyl ester (170 mg, 0.488 mmol) from the previous step, (2,5-Dichloro-pyrimidin-4-yl)-[2-(propane- 2-sulfonyl)-phenyl]-amine (Intermediate 2, 169 mg, 0.488 mmol, 1 equiv.), xantphos (28 mg, 0.049 mmol, 0.1 equiv.), palladium acetate (5.5 mg, 0.024 mmol, 0.05 equiv.), and Cs2CO3 (477 mg, 1.46 mmol, 3 equiv.) are dissolved in anhydrous THF (6 mL). N2 is bubbled through the reaction mixture for 5 minutes and then the reaction vessel is sealed and heated with microwave irradiation to 150 0C for 20 min. The reaction is filtered and the filtrate concentrated under vacuum. After concentration, the crude product is purified by silica gel chromatography (gradient from hexanes to 30% ethyl acetate in hexanes) to give 4-(4-{5-chloro-4-[2-(propane-2- sulfonyl)-phenylamino]-pyrimidin-2-ylamino}-5-isopropoxy-2-methyl-phenyl)-piperidine-l- carboxylic acid tert-butyl ester as a yellow film: ESMS m/z 658.3 (M + H+). This product (105 mg, 0.160 mmol) is dissolved in CH2Cl2 (3 mL) and treated with TFA (3 mL). After 45 min., the reaction is concentrated under vacuum. 1 N HCl in Et2O (5 mL x 2) is added causing the product HCl salt to precipitate. The solvent is removed by decantation. The resulting 5- Chloro-N2-(2-isopropoxy-5-methyl-4-piperidin-4-yl-phenyl)-N4-[2-(propane-2-sulfonyl)- phenyl]-pyrimidine-2,4-diamine (66) is dried under high vacuum, generating an off-white powder: 1H NMR (400 MHz, DMSO-J6+ trace D2O) δ 8.32 (s, IH), 8.27 (d, IH), 7.88 (d, IH), 7.67 (dd, IH), 7.45 (dd, IH), 7.42 (s, IH), 6.79 (s, IH), 4.56-4.48 (m, IH), 3.49-3.32 (m, 3H), 3.10-2.91 (m, 3H), 2.09 (s, 3H), 1.89-1.77 (m, 4H), 1.22 (d, 6H), 1.13 (d, 6H); ESMS m/z 558.1 (M + H+).
 66
66
……………………..
WO 2012082972
http://www.google.com/patents/WO2012082972A1
EXAMPLE 1
Preparation of Form A of 5-chloro-N-(2-isopropoxy-5-methyl-4-(piperidin-4-ylphenyl)-N-2- (isopropylsulfonyl phenyl)-2^-diamine
5-chloro-N-(2-isopropoxy-5-methyl-4-(piperidin-4-ylphenyl)-N-2-(isopropylsulfonyl)phenyl)- 2,4-diamine di-hydrochloride salt
The compound 2-isopropoxy-5-methyl-4-(piperdin-4-yl) aniline dihydrochloride (33.00 g, 85.25 mmol) and 2,5-dichloro-N-(2-(isopropyl sulfonyl )phenyl)pyrimidin-4-amine (32.53 g) was added to a 3 -necked 500-mL round-bottomed flask equipped with mechanical stirring, thermocouple, reflux condenser and N2 inlet-outlet. A solvent, 2-propanol (255.0 g, 325 mL), was added and the mixture to heated to reflux at 82±2 °C and stirred for at least 14 hours. The mixture was cooled to 22±3 °C over 1 hour and stirred at 22±3 °C for 3 hours. The resulting solids were filtered and rinsed with 3 x 40 g (3 x 51 mL) of 2-propanol. The solids were dried at 50±5 °C/10 mbar for 16 hours to yield 44.63 g of 5-chloro-N-(2-isopropoxy-5-methyl-4- (piperidin-4-ylphenyl)-N-2-(isopropylsulfonyl)phenyl)-2,4-diamine di-hydrochloride salt. Chemical Purity (as determined by HPLC): 97.3%. Corrected yield: 71.6%. LOD = 11.60%. The dihydrochloride salt was recrystallized using acetone:water (10:l,v/v). Chemical Purity (as determined by HPLC): 98.8%.
…………….
J Med Chem 2013, 56(14): 5675
http://pubs.acs.org/doi/abs/10.1021/jm400402q

Synthesis of 5-Chloro-N2-(2-isopropoxy-5-methyl-4-piperidin-4-ylphenyl)-N4-[2-(propane-2-sulfonyl)phenyl]pyrimidine-2,4-diamine 15b
|
10-21-2011
|
COMPOUNDS AND COMPOSITIONS AS PROTEIN KINASE INHIBITORS
|
|
|
10-21-2011
|
COMPOUNDS AND COMPOSITIONS AS PROTEIN KINASE INHIBITORS
|
|
|
10-19-2011
|
COMPOUNDS AND COMPOSITIONS AS PROTEIN KINASE INHIBITORS
|
|
|
8-5-2011
|
COMPOUNDS AND COMPOSITIONS AS PROTEIN KINASE INHIBITORS
|
Medical Mushrooms – The Future of Cancer Treatment?
Cancer rates are on the rise worldwide, which means that in coming generations more and more people will have their lives turned inside out with a diagnosis, and with having to turn their attention to battling this new plague. The psychological effects of having your world turned on its so quickly can be devastating, and often put people in a depressed, anxious and negative emotional state.
With so many types of cancers affecting people these days, there is no such thing as a single cure for cancer, because each type is different and will respond to different remedies. Finding the miracle cure often requires an intense search, deviation from standard doctor’s recommendations, a huge investment of time and money, and tremendous amount of hope, belief and faith. Not everything works for every cancer, but, some things consistently aid in the struggle with all cancers, like the right diet…
View original post 1,125 more words
Binimetinib in phase 3 for for the treatment of metastatic or unresectable cutaneous melanoma with NRAS mutations and in combination with LGX-818 in adult patients with BRAF V600


Binimetinib
Array BioPharma Inc;PHASE 3 Cancer, ovary (serous)
Novartis PHASE 3 Melanoma
AGARRY-162
ARRY-438162
MEK-162
MEK-1 protein kinase inhibitor; MEK-2 protein kinase inhibitor
Liver injury; Melanoma; Noonan syndrome; Ovary tumor; Solid tumor
Growth factor-mediated proliferative signals are transmitted from the extracellular environment to the nucleus through several pathways, including the RAS/RAF/ MEK pathway. The RAS/RAF/MEK kinase signal transduction pathway is activated through initial extracellular binding and stimulation of tyrosine receptor kinases (RTKs) by their respective cognate ligands. Upon autophosphorylation of specific tyrosine residues in the cytosolic domain of RTKs, the Grb2-Sos complex translocates to the plasma membrane, and converts the inactive RAS’GDP to active RAS’GTP. The interaction between the Grb2 docking protein and the activated kinases or the phosphorylated receptor associated proteins is mediated by the Src Homology (SH2) domain of the signaling protein that recognizes specific phosphotyrosine sequences. RAS undergoes a conformational change upon guanosine 5 ‘-triphosphate (GTP) binding and causes the recruitment of RAF- 1 to the cytoplasmic membrane where it is phosphorylated by several kinases and simultaneous disphosphorylated at key residues by protein phosphatase-2B. Activated RAF phosphorylates the mitogen- activated protein kinase kinase (MEK) on two serine residues in the activation loop, which results in the activation of this protein kinase. MEK then phosphorylates and activates extracellular signal-regulated kinase (ERK), allowing its translocation to the nucleus where it phosphorylates transcriptional factors permitting the expression of a variety of genes.
The RAS/RAF/MEK signal transduction pathway is deregulated, often through mutations that result in ectopic protein activation, in roughly 1/3 of human cancers. This deregulation in turn results in a wide array of cellular changes that are integral to the etiology and maintenance of a cancerous phenotype including, but not limited to, the promotion of proliferation and evasion of apoptosis (Dhillon et al., Oncogene, 2007, 26: 3279-3290).
Accordingly, the development of small molecule inhibitors of key members of the RAS/ RAF/ MEK signal transduction pathway has been the subject of intense effort within the pharmaceutical industry and oncology community.
MEK is a major protein in the RAS/ RAF/ MEK pathway, which signals toward cell proliferation and survival, and frequently activated in tumors that have mutations in the RAS or RAF oncogenes or in growth receptor tyrosine kinases. MEK is a key player in the RAS/RAF/MEK pathway as it is downstream of RAS and RAF. Despite being only rarely mutated in cancer (Murugan et al., Cell Cycle, 2009, 8: 2122-2124; Sasaki et al., J. Thorac. Oncol., 2010, 5: 597-600), inhibitors of the MEK1 and MEK2 proteins have also been targeted for small molecule inhibition owing to their central position within the RAS/ RAF/ MEK signal transduction pathway signaling cascade (Fremin and Meloche, J. Hematol.
Oncol., 2010, 3:8). Recently a potent MEK inhibitor failed to demonstrate efficacy in clinical trials in patients with advanced non-small cell lung cancer (Haura et al., Clin. Cancer Res., 2010, 16: 2450-2457). The reason for failure in this trial is not clear.
6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxyethyoxy)-amide (hereinafter, “Compound A”) is a benzimidazole compound that is a known potent and selective inhibitor of the MEK1 and MEK2 proteins, and useful in the treatment of hyperproliferative diseases, particularly cancer, in mammals. For example, in a recently published Phase I study of 28 patients suffering from unresectable, locally advanced or metastatic biliary cancer and who had received < 1 prior systemic therapy, oral Compound A treatment (60 mg twice daily) resulted in 1 complete regression, 1 partial regression and 11 stable disease diagnoses after at least 6 weeks of treatment (Finn et al., J. Clin. Oncol. 30, 2012 (Supplement 4, 2012 Gastrointestinal Cancers Symposium, Abstract No. 220). Compound A has also been demonstrated to be effective in the treatment of patients with either BRAFV600 or NRAS-mutant melanoma (Ascierto et al., J. Clin. Oncol. 30, 2012 (Supplement, 2012 ASCO Annual Meeting, Abstract No. 8511).
The compound, as well as a process for its preparation, is disclosed in PCT Pub. No. WO 03/077914
MEK-162, a potent, orally active MEK1/2 inhibitor, is in phase III clinical trials at Array BioPharma and licensee Novartis for the treatment of metastatic or unresectable cutaneous melanoma with NRAS mutations and in combination with LGX-818 in adult patients with BRAF V600. Phase III studies are also under way at Array BioPharma for the treatment of low grade serous carcinomas of the ovary, fallopian tube or primary peritoneum following at least one prior platinum-based chemotherapy regimen and no more than three lines of prior chemotherapy regimens. Novartis and Array BioPharma are also conducting phase II clinical studies for the treatment of locally advanced and unresectable or metastatic malignant cutaneous melanoma, harboring BRAFV600E mutations; in BRAF mutated melanoma in combination with AMG-479 and for the treatment of Noonan’s syndrome, and in non-small cell lung cancer harboring KRAS or EGFR mutation and in combination with erlotinib. MEK-162 is being evaluated in phase I/II as first line treatment of advanced biliary tract carcinoma and for the treatment of adult patients with mutant or wild-type RAS metastatic colorectal cancer. The product is in early clinical trials at Array Biopharma for the treatment of biliary cancer.
According to Array, MEK-162 may also provide broad therapeutic benefits in the treatment of chronic degenerative diseases. However, a phase II trial for the treatment of stable rheumatoid arthritis (RA) did not meet its primary endpoint. Based on these data, the company focused development of MEK-162 solely in oncology.
In 2010, MEK-162 was licensed to Novartis by Array BioPharma for worldwide development. In 2013, orphan drug designation was assigned in Japan for the treatment of malignant melanoma with NRAS or BRAF V600 mutation.
WO-2014063024 DEALS WITH Preparation, crystalline forms, and formulations comprising binimetinib. Binimetinib is a MEK-1/2 inhibitor originally claimed in WO03077914, which Array and Novartis are developing for the treatment of cancer, including melanoma, low-grade serous ovarian cancer, and other solid tumors, as well as Noonan syndrome hypertrophic cardiomyopathy and hepatic impairment. See also WO2014018725 for the most recent filing on the agent
//////////////////////////
WO 03/077914
http://www.google.com/patents/WO2003077914A1?cl=en
Schemes 1-4.
Scheme 1


Scheme la

Scheme 2

Scheme 3

17 18
Scheme 4

25
Scheme 5


General synthetic methods which may be referred to for preparing some of the compounds of the present invention are provided in PCT published application number WO 00/42022 (published July 20, 2000). The foregoing patent application is incorporated herein by reference in its entirety.
similar ie chloro instead of fluoro
Example 52

6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid (2-hydroxy-ethoxy)-amide (lOcc) Step A: 3-Chloro-2,4-difluoro-5-nitro-benzoic acid 2a
3-Chloro-2,4-difluoro-benzoic acid la (3.00 g, 15.6 mmol) is added to a stirred solution of concentrated H2SO4 (16 mL) and fuming nitric acid (0.85 mL, 20.3 mmol). After 3 hours a precipitate forms. The yellow slurry is poured onto ice water (100 mL). The aqueous mixture is extracted with diethyl ether (3x). The organic extracts are dried (Na2SO4) and concentrated under reduced pressure to give 3.50 g (95%) of clean desired product as a pale yellow solid.
Step B: 4-Amino-3-chloro-2-fluoro-5-nitro-benzoic acid 3a
Ammonium hydroxide solution (6.88 g, -30% in water, 58.9 mmol) is added to a solution of 3-chloro-2,4-difluoro-5-nitro-benzoic acid 2a (3.5 g, 14.7 mmol) in water (16 mL) at 0 °C with stirring. Upon completion of the ammonium hydroxide addition the reaction mixture is warmed to room temperature. After 5 hours the reaction mixture is cooled to 0 °C and concentrated HCl is carefully added until the pH of the reaction mixture is near zero. The solid is collected by filtration and washed with water and diethyl ether. The solids are transferred to a round bottom flask as a solution in MeOH and EtOAc and concentrated under reduced pressure to give 2.96 g of a yellow solid. The filtrate is partitioned between diethyl ether and water and the organic layer is washed with brine. The combined organic extracts are dried (Na2SO ) and concentrated under reduced pressure to give 0.65 g of product. Recovered a total of 3.61 g (104%) of pure desired product, that is carried forward without further purification.
Step C: 4~Amino-3-chloro-2-fluoro-5-nitro-benzoic acid methyl ester 4a
To a stirred solution of 4-amino-3-chloro-2-fluoro-5-nitro-benzoic acid 3a (3.61 g, 15.4 mmol) in THF (30 mL) and MeOH (10 mL), TMS diazomethane (9.23 mL, 2.0 M solution in hexanes, 18.5 mmol) is added. After completion of reaction, the reaction mixture is concentrated via rotary evaporation with acetic acid in the trap. The recovered oily solid is triturated with diethyl ether to provide 1.51 g of a yellow solid. The filtrate is concentrated and triturated with diethyl ether to give an additional 0.69 g of yellow solid. A total of 2.20 g (57%) of pure desired product is recovered.
Step D: 4-Amino-3-chloro-5-nitro-2-phenylamino-benzoic acid methyl ester 5c
4-Amino-3-chloro-2-fluoro-5-nitro-benzoic acid methyl ester 4a (2.20 g, 8.84 mmol) is suspended in MeOH (9.4 mL) and aniline (3.22 mL, 35.4 mmol) is added. The reaction mixture is heated to reflux with stirring under a nitrogen atmosphere. After 19 hours, the reaction is complete. Distilled water (3.22 mL) is added to the reaction mixture and refluxing is continued for one hour. The reaction mixture is cooled to 0 °C in an ice bath for 20 minutes. The reaction mixture is filtered and washed with 3:10 distilled water/MeOH (65 mL total) and then with MeOH. The solid is dissolved with CH2C12 and concentrated under reduced pressure to give 2.40 g (84%) of pure desired product. MS APCI (-) m/z 320.3 (M-l) detected.
Step E: 4, 5-Diamino-3-chloro-2-phenylamino-benzoic acid methyl ester 6b
4-Amino-3-chloro-5-nitro-2-phenylamino-benzoic acid methyl ester 5c (0.50 g, 1.55 mmol) is dissolved into 2:1 EtOH/MeOH (15.5 mL). Saturated aqueous NH4C1 (15 mL), Zn powder (1.02 g, 15.6 mmol), and THF (10 mL) are added. After stirring for 20 hours, the reaction mixture is diluted with CH C12/THF and water. The organic layer is washed with water (3x). The combined organic extracts are dried (Na2SO4) and concentrated under reduced pressure. The solids are triturated with ether to give 0.32 g (70%) clean desired product. Step F: 7-Chloro-6-phenylamino-3H-benzoimidazole-5-carboxylic acid methyl ester 7c
4,5-Diamino-3-chloro-2-phenylamino-benzoic acid methyl ester 6b (0.32 g, 1.09 mmol) and formamidine acetate (72 mg, 1.64 mmol) in EtOH (36 mL) are heated, with stirring, to 80 °C. After 44 hours, the reaction mixture is cooled to room temperature and diluted with EtOAc and washed with water (3x), saturated NaHCO3, and brine. The combined organic extracts are dried (Na2SO4) and concentrated under reduced pressure to give 0.33 g (99%) clean desired product as a solid. MS APCI (+) m/z 302.3 (M+l) detected.
Step G: 6-(4-Bromo-phenylamino)-7-chloro-3H-benzoimidazole-5-carboxylic acid methyl ester 8g
7-Chloro-6-phenylamino-3H-benzoimidazole-5-carboxylic acid methyl ester 7c (0.327 g, 1.08 mmol) is dissolved into DMF (16 mL) and NBS (0.193 g, 1.08 mmol) is added. After one hour, the reaction mixture is quenched by the addition of saturated aqueous NaHSO3. The reaction mixture is then partitioned between EtOAc/THF and water. The organic layer is washed with water and brine. The combined organic extracts are dried (Na2SO ) and concentrated under reduced pressure. The recovered solid is triturated with ether to give 0.225 g (54%) pure desired product. MS ESI (+) m/z 382, 384 (M+, Br pattern) detected.
Step H: 6-(4-Bromo-2-chloro-phenylamino)- 7 -chloro-3H-benzoimidazole-5 -carboxylic acid methyl ester lOdd 6-(4-Bromo-phenylamino)-7-chloro-3H-benzoimidazole-5-carboxylic acid methyl ester 8g (0.225 g, 0.591 mmol) is dissolved in DMF (2 mL) and NCS (79 mg, 0.591 mmol) is added. After the NCS is in solution concentrated HCl (0.005 mL, 0.059 mmol) is added. After 2 hours, sodium bicarbonate, water and NaHSO3 are added to the reaction mixture. Solids are filtered and washed with water and ether to give 0.141 g (57%) of clean desired product as a tan solid. MS APCI (-) m/z 414, 416 (M-, Br pattern) detected.
Step I: 6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid methyl ester lOee
6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3H-benzoimidazole-5-carboxylic acid methyl ester lOdd (0.141 g, 0.34 mmol), potassium carbonate (0.141 g, 1.02 mmol), and iodomethane (0.063 mL, 1.02 mmol) are dissolved in dimethylformamide (3 mL). After 20 hours, the reaction mixture is diluted with EtOAc and washed with water (3x), potassium carbonate, and brine. The organic layer is dried (Na2SO4) and concentrated to a brown oil. The N3 and Nl alkylated regioisomers are separated by flash chromatography (EtOAc). The recovery of the N3 alkylated regioisomer is 20.4 mg (28%). MS ESI (+) m/z 428, 430 (M+, Br pattern) detected.
Step J: 6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid 10 ff
6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid methyl ester lOee (21 mg, 0.048 mmol) is dissolved into 2:1 THF/water (1.2 mL) and NaOH (0.190 mL, 1.0 M aqueous solution, 0.190 mmol) is added. After stirring for 4 hours the reaction is diluted with water and acidified to pH 2 by addition of 1.0 M HCl. The mixture is then extracted with 3:1 EtOAc/THF (3x), dried (Na2SO ) and concentrated to give quantitative yield of desired prodcut as a white solid. MS APCI (+) m/z 414, 416 (M+, Br pattern) detected.
Step K: 6-(4-Bromo-2’chloro-phenylamino)- 7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid (2-vinyloxy-ethoxy) -amide lOgg
6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid lOff (32 mg, 0.077 mmol), O-(2-vinyloxy-ethyl)-hydroxylamine (0.010 mL, 0.092 mmol), HOBt (13 mg, 0.093 mmol), triethylamine (0.011 mL, 0.077 mmol), and EDCI (19 mg, 0.10 mmol) are dissolved into dimethylformamide (1.0 mL) and allowed to stir under a nitrogen atmosphere at room temperature for 24 hours. The reaction mixture is diluted with EtOAc, washed with water (3x), 10% potassium carbonate (2x), saturated ammonium chloride, brine, dried (Na2SO4), and concentrated under reduced pressure to give 39 mg of 85% pure material. MS APCI (-) m/z 497, 501 (M-, Br pattern) detected.
Step L: 6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid (2-hydroxy-ethoxy)-amide lOcc
Hydrochloric acid (0.78 mL, 1.0 M aqueous solution, 0.78 mmol) is added to a suspension of 6-(4-bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H- benzoimidazole-5-carboxylic acid lOgg (2-vinyloxy-ethoxy)-amide (39 mg, 0.078 mmol) in MeOH (1 mL). After one hour, the reaction mixture is neutralized to pH 7 and concentrated under reduced pressure. The solids are dissolved in EtOAc, washed with brine, dried (Na SO4), and concentrated under reduced pressure. Flash chromatography (20:1 CH2Cl2/MeOH) provides 9 mg (23%) of pure product: MS APCI (+) m/z 473, 475 (M+, Br pattern) detected; 1H NMR (400 MHz, CDC13) δ 8.30 (s, IH), 8.08 (s, IH), 7.57
(d, IH), 7.15 (dd, IH), 6.21 (d, IH), 3.97 (s, 3H) 3.86 (m, 2H), 3.57 (m, 2H).
actual is below
Example 18
The following compounds are prepared by methods similar to those described in
Example 10 by using methyl ester 8d and the appropriate alkylating agent (Step A) and
the appropriate hydroxylamine (Step C):

/////////////////////
COMPD A
Example 1. Preparation of 6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-

Compound 1 Compound 3
In an inertized (N2) reaction vessel at internal temperature 20°C and under exclusion of humidity and air, Compound 1 (1.0 eq.) and Compound 2 (1.2 eq.) are reacted in the presence of cesium carbonate (2.4 eq.), tris(dibenzylidenaceton) dipalladium(O) (0.035 eq.) and Xantphos (0.07 eq.) in a mixture of toluene and 1 ,4-dioxane at internal temperature of 99°C. After 8 hours, the mixture is cooled to internal temperature of 60°C.
Subsequently, dimethylformamide (DMF), filter aid (CEFOK) and activated charcoal (EKNS) are added, and the mixture is stirred and cooled to internal temperature of 35 °C. The solids are filtered off and washed with a mixture of dimethylformamide and toluene. To the filtrate, which contains the product Compound 3, is introduced at internal temperature of
25 °C hydrogen chloride gas (CLC) whereupon the HQ salt of Compound 3 crystallizes. The palladium residue mainly remains in solution. After warming to 60 °C and cooling to 0°C, the solids are filtered using a centrifuge and are washed with a mixture of toluene and dimethylformamide.
The damp Compound 3 HC1 salt is charged to a reactor (equipped with pH probe) together with dimethylformamide and is heated to 60°C. By adding a 4 wt% of aqueous tripotassium phosphate solution, the pH is adjusted to a pH range of 6.8-7.6 (with a target of pH 7.2) while Compound 3 crystallizes as free base. After cooling to 22°C and stirring, the solids are filtered using a centrifuge and are washed with drinking water. The moist solids are dried at 50 °C under vacuum to give dry, crude Compound 3.
In order to remove residual palladium, dry, crude Compound 3 is dissolved in dimethylformamide at internal temperature of 60°C and stirred together with Smopex-234 (commercially available from Johnson Matthey) and activated charcoal for 90 minutes. The solids are filtered off at internal temperature of 60°C and are washed with
dimethylformamide. To the filtrate are added drinking water and Compound 3 seed crystals. More drinking water is added while Compound 3 crystallizes. After cooling to internal temperature of 20 °C, the solids are filtered using a centrifuge and are washed with a mixture of deionized water and dimethylformamide and with deionized water. The moist solids are dried at 50°C under vacuum, providing 6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid methyl ester (Compound 3).
Example 2. Preparation of 6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid-(2-tert-butoxyethoxy)-amide
A. “One-pot” Synthesis

Compound 3 Intermediate 1
t-Bu-O. /\ ^ H2
(Compound 4)

Compound 5
In an inertized reaction vessel at internal temperature 20-25 °C under nitrogen, 6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid methyl ester (Compound 3, 1.0 eq.) is added to a mixture of DMF and THF. To this slurry, a solution of potassium trimethylsilanolate (1.05 eq.) in THF is added to the mixture at internal temperature of 25 °C over a period of about 40 minutes, and the resulting mixture is stirred for about 1 hour, providing a potassium salt solution of Intermediate 1. A THF/methanol mixture is then sequentially distilled off from the mixture at 85-120°C during about 2 hours.
The potassium salt solution is then added to a suspension of CDI (1.25 eq.) and imidazole hydrochloride (1.40 eq.) in THF at internal temperature of 25 °C over a period of about 1 hour. The resulting mixture is then stirred for approximately 1 hour at 50°C, and the following imidazolide intermediate

The imidazolide intermediate is not further isolated.
Subsequently, 1.2 eq. of 0-(2-tert-butoxyethyl)hydroxylamine (Compound 4, CAS No. 1023742-13-3, available from suppliers such as Huhu Technology, Inc.®) is added over a period of about 30 minutes at 50°C and stirred for 1.5 hours. Demineralized water is then added at 50°C, producing a precipitate. After cooling to 20°C and stirring for about 3-16 hours, the slurry is filtered off, washed with THF/ demineralized water (1 :2) in 2 portions and with demineralized water in three portions, and dried at 50°C / <70 mbar for about 17 hours, providing 6-(4-bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid-(2-tert-butoxyethoxy)-amide (Compound 5) as monohydrate.
B. A synthesis method with isolation of the intermediate of step a) from the reaction mixture of step a) prior to the reaction of step b)
Alternatively, 6-(4-bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5 -carboxylic acid-(2-tert-butoxyethoxy)-amide (Compound 5) can be made by the synthesis method as shown below. Compound 3, which is a methyl ester, is first converted to a carboxylic acid, which is then isolated by a crystallization to form Compound
6. Compound 6 is then coupled with Compound 4 to form Compound 5 as monohydrate.
The crystallization step in this method removes starting materials such as Compound 1, process impurities, and the dba ligand from the prior catalyst before the coupling reaction with Compound 4, and at the same time maintains the overall yield of the synthesis.


6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-memy acid In an inertized (N2) reaction vessel at internal temperature of 60°C, Compound 3 (1.0 eq.) is dissolved in DMF and stirred with a fiber, which is sold under the trademark
SMOPEX 234, and activated charcoal for the removal of palladium to not more than 100 ppm. The fiber and activated charcoal are removed by filtration at 60°C and washed with DMF.
The filtrate (containing Compound 3) is transferred to a second inertized (N2) reaction vessel and cooled to an internal temperature of 30°C. A thin suspension can form at this point of time. 30% sodium hydroxide (1.1 eq.) and water (for rinsing) are added, and the resulting reaction mixture is vigorously stirred for 3 hours at an internal temperature of 30 °C. The methyl ester is saponified. Conversion is checked by an IPC (HPLC). As soon as the IPC criterion is met, a filter aid, which is sold under the trademark HYFLO, is added. The mixture is stirred for 15 minutes and then filtered at 30°C via a plate filter and polish filter to a third reaction inertized (N2) vessel.
An aqueous HC1 solution 7.5 % is added to the clear filtrate in the third vessel at an internal temperature of 30 °C until a pH value of 8 is reached. Then the solution is seeded at an internal temperature of 30°C with Compound 6, and an aqueous HC1 solution 7.5 % is added under vigorous stirring until a pH value of pH 2.8 is reached. The product gradually crystalizes. The suspension is cooled over 60 min to an internal temperature of 25 °C and
water is added. The suspension is stirred for at least 4 hours at an internal temperature of 25°C.
The resulting solid is collected by centrifugation or filtration. The filter cake is first washed with DMF/water 1 :1 (w/w) and then with water, discharged and dried in a vacuum at 50°C. The water content is controlled by IPC. The crystalline product Compound 6 is discharged as soon as the IPC criterion is met.
6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid- (2-tert-butoxyethoxy) – amide
An inertized (N2) reaction vessel is charged with Compound 6 (1.0 eq.), DMF, and
THF at room temperature. The suspension is heated to 25 °C under stirring with flow of nitrogen. After CDI (1.13 eq.) is added, the suspension can get thinner and slight evolution of gases can be observed. After the suspension finally becomes a solution, it is then monitored by IPC (HPLC).
As soon as the IPC (HPLC) criterion is met, the reaction mixture is heated to 50°C over 20 minutes and imidazole hydrochloride (0.3 eq.) is added, forming a solution of
Intermediate 2.
To the solution of Intermediate 2, Compound 4 (1.3 eq.) is added over 60 minutes at internal temperature of 50°C under stirring at a speed of 300 rpm with flow of nitrogen. As soon as the IPC (HPLC) criterion is met, the mixture is cooled to 20-25 °C over 30 minutes. The mixture is then stored at ambient temperature overnight under nitrogen without stirring. DMF is added to the mixture followed by heating it to 50 °C over 30 minutes. Complete conversion of Intermediate 2 to Compound 5 is confirmed by IPC (HPLC).
Water is added to the mixture at internal temperature of 50 °C over 20 minutes. Then the solution is seeded with Compound 5. After stirring at 50 °C for 60 minutes, more water is added to the suspension at 50 °C over 90 minutes. After vigorous stirring, the suspension is cooled to 20 °C over 2 hours and filtered. The filter cake is washed twice with THF/water (v/v: 1 :2) at 20 °C, and twice with water at 20 °C. Finally, the filter cake is dried at 50 °C under vacuum to provide 6-(4-bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid-(2-tert-butoxyethoxy)-amide (Compound 5) as monohydrate.
Example 3. Preparation of 6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxyethyoxy)-amide (Compound A)

Compound 5 Compound A
6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid-(2-tert-butoxyethoxy)-amide (Compound 5) monohydrate is added in 3 portions to a premixed solution of Acetonitrile and excess Phosphoric acid (85 % aqueous solution) at internal temperature 20-25 °C. After stirring for about 15 minutes, the suspension is heated to internal temperature 50-53 °C. The suspension is maintained at this temperature for 6 hours, cooled to internal temperature 20-25 °C. The mixture is then heated to internal temperature 35-37°C and diluted with Ethanol- Water (3 :1 v/v). EKNS and CEFOK are added, the reaction mixture is stirred approximately 15 minutes and filtered over a funnel coated with CEFOK. The filtrate is cooled to approximately 30°C. 3 N aqueous potassium hydroxide (ΚΟΗ) is added to the cooled filtrate over a period of 90 minutes until a pH- value of about 8.1 is reached. The suspension is heated to internal temperature 60-63 °C, stirred at this temperature for a period of about 2 hours, cooled to 20-23 °C over a period of about 45 minutes, filtered over a funnel, and dried at 50°C pressure <100 mbar over a period of about 17 hours, providing 6-(4-bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxyethyoxy)-amide (Compound A) as a white powder.
Example 4. Preparation of Crystallized 6-(4-bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxyethyoxy)-amide (Compound A) In a dry vessel at room temperature, Compound A is added to a premixed solvent solution of methanol/THF/water (35/35/30 w/w). The suspension is heated to internal temperature 53-55°C, and the resulting solution is hot filtered by deep and membrane filtration (via a paper filter and PTFE membrane) at internal temperature 53-56°C. The clear solution is stirred and cooled to 47-48°C, and the seed crystals suspension (i.e., seed crystals of crystallized Compound A in water, 10% m/m) is added (0.2 to 0.5% of crystallized Compound A expected yield mass). After about 20 minutes, water is slowly added within 25 hours (33.3% within 15 hours and 66.6% within 10 hours with at least 10 minute stirring after addition of water) to obtain a final ratio of methanol THF/water (20/20/60 w/w). After the water is added, the suspension is cooled down to internal temperature 3-5 °C within 10 hours and stirred for 0.5 hours. The white suspension is filtered over a sinter glass nutsche (75 ml, diameter = 6 cm, pore 3) suction filter and washed once with ice cold methanol/THF/water (15/15/70 w/w at 2-4 °C), and two times with ice cold water (2-4 °C). Drying takes place in a vacuum oven dryer at 20°C for 10 hours, and then at 40°C for 10 hours, and then at 60°C for at least 12 hours with pressure < lOmbar, providing crystallized Compound A.
Example 5. Pharmaceutical Composition
Crystallized Compound A is formulated as indicated in Table 1 :
Table 1

* The weight of the drug substance is taken with reference to the dried substance (100%) on the basis of assayed value. The difference in weight is adjusted by the amount of lactose monohydrate.
** The Opadry II is combined with the sterile water to make a 12% w/w Opadry II (85F) film coat suspension, which is then sprayed onto the core tablet.
*** Removed during processing
Upon mixing of the tablet core components, the pharmaceutical composition is converted into a tablet form by direct compression. The formed tablet may be further coated with the tablet coating provided above.
Radiation therapy to treat uterine cancer linked with increased risk of bladder cancer later in life
Radiation therapy used to treat uterine cancer may increase a patient’s risk of developing bladder cancer. That is the conclusion of a recent study published in BJU International. The findings indicate the importance of monitoring patients for potential signs of bladder cancer to ensure early diagnosis and treatment.
In the United States, uterine cancer is the fourth most common cancer in women, with an estimated 49,560 women diagnosed in 2013. In addition to surgery, 38 percent ofpatients undergo pelvic radiation therapy to decrease uterine cancer recurrence. Studies have found that women treated with radiation therapy for uterine cancer, like men who received radiation therapy for prostate cancer, have an increased risk of developing bladder cancer later in life.
To investigate the issue, Guan Wu, MD, PhD, of the University of Rochester Medical Center, and his colleagues analyzed the records of 56,681 patients diagnosed with uterine cancer as their…
View original post 176 more words
An easier, safer, and more accurate treatment for pancreatic cancer

(a) A single axial slice of the pancreas from the pre-treatment CT scans is overlaid with computed contours of light fluence levels around the fiber location. This was simulated using blood content information for tissue absorption from contrast CT. (b) A volume rendering of the blood vessels around the pancreas overlaid with the light dose map in the fiber location, in the same patient. Please see supplementary material at stacks.iop.org/PMB/59/1911/mmedia. Credit: NCCC
Using CT scans with contrast enhancement, Dartmouth researchers measured treatment response to pancreatic cancer photodynamic therapy (PDT) according to a paper published in Physics in Medicine and Biology.
The research team at Dartmouth set out to reduce the imaging obstacles for PDT, a minimally invasive and nontoxic treatment for cancer. “This study implies that treatment response can be reliably predicted using contrast CT. This would represent a major breakthrough in PDT for pancreas cancer that allows…
View original post 466 more words
New method to analyse how cancer cells die

A team from The University of Manchester – part of the Manchester Cancer Research Centre – have found a new method to more efficiently manufacture a chemical used to monitor cancer cells.
The technique could lead to clearer and better quality images on PET scans.
The number of cells within tissue is controlled through apoptosis – a process where cells shrink and their components break up, also known as programmed cell death. Cancer is often characterised by a disruption to the normal process of this cell death.
Being able to study this process accurately would allow doctors to more effectively diagnose and monitor cancer and to test and develop new treatments designed to kill cancer cells.
Ideally, cell death would be measured non-invasively to avoid surgery and current methods are focused on using radioactive tracers – molecules that are taken up in regions of tissue where cells are…
View original post 281 more words
Cobimetinib in phase 3 for metastatic melanoma


Cobimetinib
934660-93-2 cas ………….(S )enantiomer desired
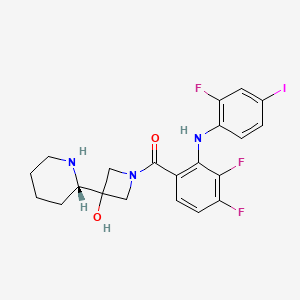
[3,4-Difluoro-2-(2-fluoro-4-iodoanilino)phenyl]{3-hydroxy-3-[(2S)-piperidin-2-yl]azetidin-1-yl} methanone
l-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2S)-piperidin-2- yl]azetidin-3-ol
1-[3,4-Difluoro-2-(2-fluoro-4-iodophenylamino)phenyl]-1-[3-hydroxy-3-[2(S)-piperidinyl]azetidin-1-yl]methanone
 Cobimetinib (racemate)
Cobimetinib (racemate) Cobimetinib (R-enantiomer)
Cobimetinib (R-enantiomer)CAS No: 934660-94-3
cobimetinib fumarate [USAN]
Molecular Formula: C46H46F6I2N6O8
Average mass: 1178.692261 Da
(2E)-2-Butendisäure –{3,4-difluor-2-[(2-fluor-4-iodphenyl)amino]phenyl}{3-hydroxy-3-[(2S)-2-piperidinyl]-1-azetidinyl}methanon (1:2) [German] [ACD/IUPAC Name]
{3,4-Difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}{3-hydroxy-3-[(2S)-2-piperidinyl]-1-azetidinyl}methanone (2E)-2-butenedioate (2:1) [ACD/IUPAC Name]
1369665-02-0 [RN]
Acide (2E)-2-butènedioïque – {3,4-difluoro-2-[(2-fluoro-4-iodophényl)amino]phényl}{3-hydroxy-3-[(2S)-2-pipéridinyl]-1-azétidinyl}méthanone (1:2) [French][ACD/IUPAC Name]
Bis({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}{3-hydroxy-3-[(2S)-piperidin-2-yl]azetidin-1-yl}methanone) (2E)-but-2-enedioate
Methanone, [3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl][3-hydroxy-3-[(2S)-2-piperidinyl]-1-azetidinyl]-, (2E)-2-butenedioate (2:1) (salt
Click to access cobimetinib-fumarate.pdf
Cobimetinib (GDC-0973, XL-518) is a MEK inhibitor being developed by Exelixis and Roche. It is being studied in combination withvemurafenib to treat several cancers, including melanoma.
Cobimetinib is an inhibitor of MEK kinase, which is an enzyme that the mitogen-activated protein kinase (MAPK). This compound was originated by Exelixis and is being developed by Genentech. Currently, Cobimetinb is in phase III trials at Genentech for the treatment of metastatic melanoma and in phase I clinical trials for the treatment of solid tumors. Cobimetinib has received an orphan drug designation in the U.S. for treatment of stage IIb, IIc, III, and IV melanoma with BRAFV600E mutation.
GDC-0973 (XL-518; GDC 0973) is a selective inhibitor of MEK GDC-0973 is also known as mitogen activated protein kinase kinase (MAPKK), is a key component of the RAS / RAF / MEK / ERK pathway, which. is frequently activated in human tumors.
Inappropriate activation of the MEK / ERK pathway promotes cell growth in the absence of exogenous growth factors.
The ERK/MAP kinase cascade is a key mechanism subject to dysregulation in cancer and is constitutively activated or highly upregulated in many tumor types. Mutations associated with upstream pathway components RAS and Raf occur frequently and contribute to the oncogenic phenotype through activation of MEK and then ERK. Inhibitors of MEK have been shown to effectively block upregulated ERK/MAPK signaling in a range of cancer cell lines and have further demonstrated early evidence of efficacy in the clinic for the treatment of cancer. Guided by structural insight, a strategy aimed at the identification of an optimal diphenylamine-based MEK inhibitor with an improved metabolism and safety profile versus PD-0325901 led to the discovery of development candidate 1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2S)-piperidin-2-yl]azetidin-3-ol (XL518, GDC-0973) (1). XL518 exhibits robust in vitro and in vivo potency and efficacy in preclinical models with sustained duration of action and is currently in early stage clinical trials.
Process for the preparation of MEK inhibitors, relates cobimetinib. Genentech and its parent company Roche, under license from Exelixis, are developing cobimetinib, for the treatment of solid tumors, including melanoma, which is in phase 3 trials as of April 2014. The drug was originally disclosed in WO2007044515. For a previous filing on MEK inhibitors, see WO2008124085.
patent
http://www.google.com/patents/WO2007044515A1?cl=en
EXAMPLE 22(a) and 22(b) l-({3,4-difluoro-2-[(2-fluoro-4-iodophenyϊ)amino]phenyl}carbonyI)-3-[(2R)-piperidin-2- yl]azetidin-3-ol

l-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2S)-piperidin-2- yl]azetidin-3-ol
 desired
desired[00334] To a solution of 1 , 1 -dimethylethyl 2-(3 -hydroxy- 1 –
{[(phenylmethyl)oxy]carbonyl}azetidin-3-yl)piperidine-l-carboxylate (368 mg, 0.94 mmol), prepared using procedures similar to those described in Reference 5, in dichloromethane (5 mL) was added DMAP (115 mg, 0.94 mmol) and the resulting solution was cooled to O0C. (i?)-(-)-α-Methoxy-α-trifluoromethylphenylacetyl chloride (105 μL, 0.56 mmol) was added to the solution by syringe and the mixture was allowed to warm to room temperature then stirred an additional 12 hours. The solution was then partitioned with saturated aqueous soldium bicarbonate and the organic phase dried over anhydrous magnesium sulfate then filtered and concentrated to an oily residue.
Silica gel flash chromatography using hexanes:ethyl acetate 3:1 as eluent afforded the less polar 1,1 -dimethyl ethyl (2R)-2-(l- {[(phenylmethyl)oxy]carbonyl}-3-{[(2i?)-3,3,3-trifluoro-2-(methyloxy)-2- phenylpropanoyl]oxy}azetidin-3-yl)piperidine-l-carboxylate (27.5 mg, 5% yield), the more polar 1 , 1 -dimethylethyl (2S)-2-(l -{ [(phenylmethyl)oxy]carbonyl} -3-{ [(2i?)-3,3,3-trifluoro-2- (methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-l-carboxylate (105 mg, 19% yield) and starting material (253 mg, 69% recovery).
[00335] The starting material thus recovered was taken into dichloromethane (3 mL) followed by addition of DMAP (115 mg, 0.94 mmol) and (i?)-(-)-α-methoxy-α- trifluoromethylphenylacetyl chloride (105 μL, 0.56 mmol) and the mixture was allowed to stir at room temperature over 12 hours. Proceeding as before afforded combined 1,1- dimethylethyl (2R)-2-(l-{[(phenylmethyl)oxy]carbonyl}-3-{[(2i?)-3,3,3-trifluoro- 2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-l-carboxylate (46.6 mg, 8% yield), the more polar 1,1 -dimethylethyl (25)-2-(l-{[(phenylmethyl)oxy]carbonyl}-3-{[(2i?)- 3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-l-carboxylate (228 mg, 41% yield) and starting material (100.8 mg, 27% recovery). [00336] The starting material thus recovered was taken into tetrahydrofuran: dichloromethane (1 :1, 2 mL) followed by addition of DMAP (47 mg, 0.39 mmol) and (R)-(-)-α-methoxy-α-trifluoromethylphenylacetyl chloride (80 μL, 0.43 mmol) and the mixture was heated to 60 0C over 12 hours.
Proceeding as before afforded combined less polar 1,1-dimethylethyl (2i?)-2-(l-{[(phenylmethyl)oxy]carbonyl}-3-{[(2i?)-3,3,3- trifluoro-2-(methyloxy)-2-phenylpropanoyl] oxy } azetidin-3 -yl)piperidine- 1 -carboxylate ( 144 mg, 26 % yield). The chiral ester derivatives thus obtained were again subject to silica gel flash chromatography using hexanes:ethyl acetate 3:1 as eluent to give the pure less polar 1,1-dimethylethyl (2i?)-2-(l-{[(phenylmethyl)oxy]carbonyl}-3-{[(2i?)-3,3,3-trifluoro-2-
(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-l-carboxylate (122.8 mg, 22% yield) and the more polar 1,1-dimethylethyl (2£)-2-(l-{[(phenylmethyl)oxy]carbonyl}-3- { [(2R)-3 ,3 ,3 -trifluoro-2-(methyloxy)-2-phenylpropanoyl] oxy } azetidin-3-yl)piperidine- 1 – carboxylate {111.6 mg, 32% yield) both as colorless amorphous residues. [00337] l,l-Dimethylethyl (2i?)-2-(l-{[(phenylmethyl)oxy]carbonyl}-3-{[(2i?)-3,3,3- trifluoro-2-(methyloxy)-2-phenylρropanoyl]oxy}azetidin-3-yl)piperidine-l-carboxylate (122.8 mg, 0.21 mmol) was taken into methanol (4 mL) followed by addition of IM aqueous sodium hydroxide (1 mL) and the resulting solution was stirred for one hour at room temperature.
The solution was then partitioned with ethyl acetate and IN aqueous hydrochloric acid. The organic layer was washed with brine, dried over anhydrous magnesium sulfate then filtered and concentrated. The residue was purified by silica gel flash chromatography using hexanes:ethyl acetate 2:1 to give 1,1-dimethylethyl (2i?)-2-(3- hydroxy- 1 – { [(phenylmethyl)oxy] carbonyl } azetidin-3 -yl)piperidine- 1 -carboxylate (60.8 mg, 81% yield) a colorless amorphous solid. 1,1-dimethylethyl (2ιS)-2-(3-hydroxy-l- {[(phenylmethyl)oxy]carbonyl}azetidin-3-yl)piperidine-l-carboxylate (87.4 mg, 75% yield) was prepared analogously.
[00338] 1 , 1 -Dimethylethyl (2i?)-2-(3-hydroxy- 1 – { [(phenylmethyl)oxy] carbonyl } azetidin- 3 -yl)piperidine-l -carboxylate (60.8 mg, 0.16 mmol) and 10% Pd/C (30 mg) were taken into methanol (2 mL) and the mixture hydrogenated at ambient pressure for one hour. The suspension was then filtered through a celite pad and concentrated then dried in vacuo to a colorless solid. The solid amine was taken into THF (1 mL) followed by addition of DIPEA (42 μL, 0.24 mmol) and 3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]benzoyl fluoride (63 mg, 0.16 mmol), prepared using procedures similar to those described in Reference 1, and the mixture stirred at room temperature for 30 minutes.
The reaction mixture was partitioned with ethyl acetate and 1 N aqueous hydrochloric acid and the organic layer washed with brine, dried over anhydrous magnesium sulfate then filtered and concentrated. Purification of the residue by silica gel flash chromatography using hexanes: ethyl acetate 3:2 as eluent afforded 1,1-dimethylethyl (2i?)-2-[l-({3,4-difluoro-2-[(2-fluoro-4- iodophenyl)amino]phenyl}carbonyl)-3-hydroxyazetidin-3-yl]piperidine-l -carboxylate (74.9 mg, 74% yield) as an amorphous solid. 1,1 -Dimethylethyl (2i?)-2-[l-({3,4-difluoro-2-[(2- fluoro~4-iodophenyl)amino]phenyl}carbonyl)-3-hydroxyazetidin-3-yl]piperidine-l- carboxylate 1R NMR (400 MHz, CDCl3): 8.53 (br s, 0.5H), 8.40 (br s, 0.5H), 7.41-7.38 (dd, IH), 7.34-7.31(dt, IH), 7.17-7.14 (m, IH), 6.86-6.79 (m, IH), 6.63-6.587 (m, IH), 4.24-3.90 (m, 4H), 3.37-3.23 (m, IH), 2.90-2.80 (m, IH), 1.85-1.54 (m, 7H), 1.43 (s, 9H); MS (EI) for C26H29F3IN3O4: 576 (M-C4H9 4).
[00339] 1 , 1 -dimethylethyl (2R)-2-[l -({3,4-difluoro-2-[(2-fluoro-4- iodopheny^aminojphenylJcarbonyO-S-hydroxyazetidin-S-yljpiperidine-l-carboxylate (74.9 mg, 0.12 mmol) was taken into methanol (1 mL) followed by addition of 4 N HCl in dioxane (1 mL) and the solution was stirred at room temperature for one hour. The solution was then concentrated and the residue partitioned with chloroform and saturated aqueous sodium bicarbonate.
The organic layer was washed with brine, dried over anhydrous sodium’ sulfate then filtered and concentrated. Purification of the residue by silica gel flash chromatography using ethyl acetate then concentrated aqueous ammonia in chloroform and methanol (0.1 :10:1) as eluents afforded l-({3,4-difluoro-2-[(2-fluoro-4- iodophenyl)amino]phenyl}carbonyl)-3-[(2i?)-piperidin-2-yl]azetidin-3-ol (57.3 mg) as a colorless amorphous solid.
The free base was taken into methanol (1 mL) then brought to about pH 1 by addition of 4 N HCl in dioxane and the solution concentrated. The residue was triturated with ethyl ether to afford a suspension. The solid was collected by filtration to afford l-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2i?)- piperidin-2-yl]azetidin-3-ol hydrochloride salt (49 mg, 72% yield) as a colorless solid. 1H NMR (400 MHz, CDCl3): 8.43-8.39 (d, IH), 7.41-7.38 (dd, IH), 7.33-7.31(dt, IH), 7.14- 7.10 (m, IH), 6.84-6.80 (m, IH), 6.63-6.57 (m, IH), 4.12-3.99 (m, 4H), 3.10-3.08 (d, IH), 2.72-2.69 (d, IH), 2.64-2.62 (m, IH), 1.61-1.58 (m, 2H), 1.36-1.16 (m, 4H); MS (EI) for C21H2IF3IN3O2: 532 (MH+).

………………….
http://www.google.com/patents/WO2014027056A1?cl=en
[3,4-difluoro-2-[(2-fluoro-4- iodophenyl)amino]phenyl][3-hydroxy-3-[(2S)-2-piperidinyl]-l-azetidinyl]methanone.
GDC-0973 has the chemical structure:

[00156] Compound II may be prepared following the methods described in
US2009/0156576 (the contents of which are hereby incorporated by reference). Compound II has the following CAS Registry Number: 934660-93-2 .
http://www.google.com/patents/US20090156576
Example 22(a) and 22(b) 1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2R)-piperidin-2-yl]azetidin-3-ol

and 1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2S)-piperidin-2-yl]azetidin-3-ol

To a solution of 1,1-dimethylethyl 2-(3-hydroxy-1-{[(phenylmethyl)oxy]carbonyl}azetidin-3-yl)piperidine-1-carboxylate (368 mg, 0.94 mmol), prepared using procedures similar to those described in Reference 5, in dichloromethane (5 mL) was added DMAP (115 mg, 0.94 mmol) and the resulting solution was cooled to 0° C. (R)-(−)-α-Methoxy-α-trifluoromethylphenylacetyl chloride (105 μL, 0.56 mmol) was added to the solution by syringe and the mixture was allowed to warm to room temperature then stirred an additional 12 hours. The solution was then partitioned with saturated aqueous sodium bicarbonate and the organic phase dried over anhydrous magnesium sulfate then filtered and concentrated to an oily residue. Silica gel flash chromatography using hexanes:ethyl acetate 3:1 as eluent afforded the less polar 1,1-dimethylethyl (2R)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (27.5 mg, 5% yield), the more polar 1,1-dimethylethyl (2S)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (105 mg, 19% yield) and starting material (253 mg, 69% recovery).
The starting material thus recovered was taken into dichloromethane (3 mL) followed by addition of DMAP (115 mg, 0.94 mmol) and (R)-(−)-α-methoxy-α-trifluoromethylphenylacetyl chloride (105 μL, 0.56 mmol) and the mixture was allowed to stir at room temperature over 12 hours. Proceeding as before afforded combined 1,1-dimethylethyl (2R)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (46.6 mg, 8% yield), the more polar 1,1-dimethylethyl (2S)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (228 mg, 41% yield) and starting material (100.8 mg, 27% recovery).
The starting material thus recovered was taken into tetrahydrofuran:dichloromethane (1:1, 2 mL) followed by addition of DMAP (47 mg, 0.39 mmol) and (R)-(−)-α-methoxy-α-trifluoromethylphenylacetyl chloride (80 μL, 0.43 mmol) and the mixture was heated to 60° C. over 12 hours. Proceeding as before afforded combined less polar 1,1-dimethylethyl (2R)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (144 mg, 26% yield). The chiral ester derivatives thus obtained were again subject to silica gel flash chromatography using hexanes:ethyl acetate 3:1 as eluent to give the pure less polar 1,1-dimethylethyl (2R)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (122.8 mg, 22% yield) and the more polar 1,1-dimethylethyl (2S)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (177.6 mg, 32% yield) both as colorless amorphous residues.
1,1-Dimethylethyl (2R)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (122.8 mg, 0.21 mmol) was taken into methanol (4 mL) followed by addition of 1M aqueous sodium hydroxide (1 mL) and the resulting solution was stirred for one hour at room temperature. The solution was then partitioned with ethyl acetate and 1N aqueous hydrochloric acid. The organic layer was washed with brine, dried over anhydrous magnesium sulfate then filtered and concentrated. The residue was purified by silica gel flash chromatography using hexanes:ethyl acetate 2:1 to give 1,1-dimethylethyl (2R)-2-(3-hydroxy-1-{[(phenylmethyl)oxy]carbonyl}azetidin-3-yl)piperidine-1-carboxylate (60.8 mg, 81% yield) a colorless amorphous solid. 1,1-dimethylethyl (2S)-2-(3-hydroxy-1-{[(phenylmethyl)oxy]carbonyl}azetidin-3-yl)piperidine-1-carboxylate (87.4 mg, 75% yield) was prepared analogously.
1,1-Dimethylethyl (2R)-2-(3-hydroxy-1-{[(phenylmethyl)oxy]carbonyl}azetidin-3-yl)piperidine-1-carboxylate (60.8 mg, 0.16 mmol) and 10% Pd/C (30 mg) were taken into methanol (2 mL) and the mixture hydrogenated at ambient pressure for one hour. The suspension was then filtered through a celite pad and concentrated then dried in vacuo to a colorless solid. The solid amine was taken into THF (1 mL) followed by addition of DIPEA (42 μL, 0.24 mmol) and 3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]benzoyl fluoride (63 mg, 0.16 mmol), prepared using procedures similar to those described in Reference 1, and the mixture stirred at room temperature for 30 minutes. The reaction mixture was partitioned with ethyl acetate and 1 N aqueous hydrochloric acid and the organic layer washed with brine, dried over anhydrous magnesium sulfate then filtered and concentrated. Purification of the residue by silica gel flash chromatography using hexanes:ethyl acetate 3:2 as eluent afforded 1,1-dimethylethyl (2R)-2-[1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-hydroxyazetidin-3-yl]piperidine-1-carboxylate (74.9 mg, 74% yield) as an amorphous solid. 1,1-Dimethylethyl (2R)-2-[1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-hydroxyazetidin-3-yl]piperidine-1-carboxylate 1H NMR (400 MHz, CDCl3): 8.53 (br s, 0.5H), 8.40 (br s, 0.5H), 7.41-7.38 (dd, 1H), 7.34-7.31 (dt, 1H), 7.17-7.14 (m, 1H), 6.86-6.79 (m, 1H), 6.63-6.587 (m, 1H), 4.24-3.90 (m, 4H), 3.37-3.23 (m, 1H), 2.90-2.80 (m, 1H), 1.85-1.54 (m, 7H), 1.43 (s, 9H); MS (EI) for C26H29F3IN3O4: 576 M-C4H9 +).
1,1-dimethylethyl (2R)-2-[1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-hydroxyazetidin-3-yl]piperidine-1-carboxylate (74.9 mg, 0.12 mmol) was taken into methanol (1 mL) followed by addition of 4 N HCl in dioxane (1 mL) and the solution was stirred at room temperature for one hour. The solution was then concentrated and the residue partitioned with chloroform and saturated aqueous sodium bicarbonate. The organic layer was washed with brine, dried over anhydrous sodium sulfate then filtered and concentrated. Purification of the residue by silica gel flash chromatography using ethyl acetate then concentrated aqueous ammonia in chloroform and methanol (0.1:10:1) as eluents afforded 1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2R)-piperidin-2-yl]azetidin-3-ol (57.3 mg) as a colorless amorphous solid. The free base was taken into methanol (1 mL) then brought to about pH 1 by addition of 4 N HCl in dioxane and the solution concentrated. The residue was triturated with ethyl ether to afford a suspension. The solid was collected by filtration to afford 1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2R)-piperidin-2-yl]azetidin-3-ol hydrochloride salt (49 mg, 72% yield) as a colorless solid. 1H NMR (400 MHz, CDCl3): 8.43-8.39 (d, 1H), 7.41-7.38 (dd, 1H), 7.33-7.31 (dt, 1H), 7.14-7.10 (m, 1H), 6.84-6.80 (m, 1H), 6.63-6.57 (m, 1H), 4.12-3.99 (m, 4H), 3.10-3.08 (d, 1H), 2.72-2.69 (d, 1H), 2.64-2.62 (m, 1H), 1.61-1.58 (m, 2H), 1.36-1.16 (m, 4H); MS (EI) for C21H21F3IN3O2: 532 (MH+).
………………………………..
Novel carboxamide-based allosteric MEK inhibitors: Discovery and optimization efforts toward XL518 (GDC-0973)
ACS Med Chem Lett 2012, 3(5): 416
http://pubs.acs.org/doi/abs/10.1021/ml300049d
http://pubs.acs.org/doi/abs/10.1021/ml300049d
|
8-17-2011
|
Methods of using MEK inhibitors
|
|
|
3-30-2011
|
Azetidines as MEK Inhibitors for the Treatment of Proliferative Diseases
|
|
|
9-29-2010
|
Azetidines as MEK Inhibitors for the Treatment of Proliferative Diseases
|
|
|
3-26-2010
|
Methods of Using PI3K and MEK Modulators
|
FDA Approves Cyramza, ramucirumab (IMC-1121B) for Stomach Cancer
April 21, 2014 — The U.S. Food and Drug Administration today approved Cyramza (ramucirumab) to treat patients with advanced stomach cancer or gastroesophageal junction adenocarcinoma, a form of cancer located in the region where the esophagus joins the stomach.
Stomach cancer forms in the tissues lining the stomach and mostly affects older adults. According to the National Cancer Institute, an estimated 22,220 Americans will be diagnosed with stomach cancer and 10,990 will die from the disease, this year.
Cyramza is an angiogenesis inhibitor that blocks the blood supply to tumors. It is intended for patients whose cancer cannot be surgically removed (unresectable) or has spread (metastatic) after being treated with a fluoropyrimidine- or platinum-containing therapy.
“Although the rates of stomach cancer in the United States have decreased over the past 40 years, patients require new treatment options, particularly when they no longer respond to other therapies,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Cyramza is new treatment option that has demonstrated an ability to extend patients’ lives and slow tumor growth.”
Cyramza’s safety and effectiveness were evaluated in a clinical trial of 355 participants with unresectable or metastatic stomach or gastroesophageal junction cancer. Two-thirds of trial participants received Cyramza while the remaining participants received a placebo. The trial was designed to measure the length of time participants lived before death (overall survival).
Results showed participants treated with Cyramza experienced a median overall survival of 5.2 months compared to 3.8 months in participants receiving placebo. Additionally, participants who took Cyramza experienced a delay in tumor growth (progression-free survival) compared to participants who were given placebo. Results from a second clinical trial that evaluated the efficacy of Cyramza plus paclitaxel (another cancer drug) versus paclitaxel alone also showed an improvement in overall survival.
Common side effects experienced by Cyramza-treated participants during clinical testing include diarrhea and high blood pressure.
The FDA reviewed Cyramza under its priority review program, which provides an expedited review for drugs that have the potential, at the time the application was submitted, to be a significant improvement in safety or effectiveness in the treatment of a serious condition. Cyramza was also granted orphan product designation because it is intended to treat a rare disease or condition.
Cyramza is marketed by Indianapolis-based Eli Lilly.
Source: FDA
old article
Eli Lilly’s third-quarter earnings fell 9 percent compared with last year, when the maker of Cymbalta and Cialis booked a sizeable revenue-sharing payment from a former drug developer partner.
The Indianapolis company beat Wall Street expectations for the quarter and narrowed its earnings forecast for the year.
Lilly also said Wednesday that the U.S. Food and Drug Administration will give its stomach cancer treatment ramucirumab a priority review, which means the drugmaker will learn about its fate inside of eight months rather than a year, which is the norm.
read at
http://www.dddmag.com/news/2013/10/eli-lillys-profit-slides-gets-priority-review
cut paste old article
Eli Lilly and Co. announced that results from the Phase 3 REGARD trial of ramucirumab (IMC-1121B) as a single agent in patients with advanced gastric cancer who have had disease progression after initial chemotherapy were published today in The Lancet. REGARD is the first Phase 3 study with either a single-agent biologic or an anti-angiogenic therapy to show improved overall survival and progression-free survival in advanced gastric cancer patients.
READ ALL AT
Ramucirumab (IMC-1121B)[1] is a fully human monoclonal antibody (IgG1) being developed for the treatment of solid tumors. It is directed against the vascular endothelial growth factor receptor 2 (VEGFR2). By binding to VEGFR2 it works as a receptor antagonist blocking the binding of vascular endothelial growth factor (VEGF) to VEGFR2. VEGFR2 is known to mediate the majority of the downstream effects of VEGF inangiogenesis.
Ramucirumab is being tested in several phase III clinical trials for the treatment of metastatic gastric adenocarcinoma,[2] non-small cell lung cancer,[3] among other types of cancer. On September 26, 2013 Eli Lilly announced that its Phase III study for ramucirumab failed to hit its primary endpoint on progression-free survival among women with metastatic breast cancer.[4][5]
This drug was developed by ImClone Systems Inc. It was isolated from a native phage display library from Dyax.
- Statement On A Nonproprietary Name Adopted By The USAN Council – Ramucirumab, American Medical Association.
- ClinicalTrials.gov NCT01170663 A Study of Paclitaxel With or Without Ramucirumab in Metastatic Gastric Adenocarcinoma (RAINBOW)
- ClinicalTrials.gov NCT01168973 A Study in Second Line Non Small Cell Lung Cancer
- ClinicalTrials.gov NCT00703326 Phase III Study of Docetaxel + Ramucirumab or Placebo in Breast Cancer
- Fierce Biotech. “In another stinging setback, Eli Lilly’s ramucirumab fails PhIII breast cancer study”. Retrieved 27 September 2013.

