
Home » APPROVALS 2021
Category Archives: APPROVALS 2021
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
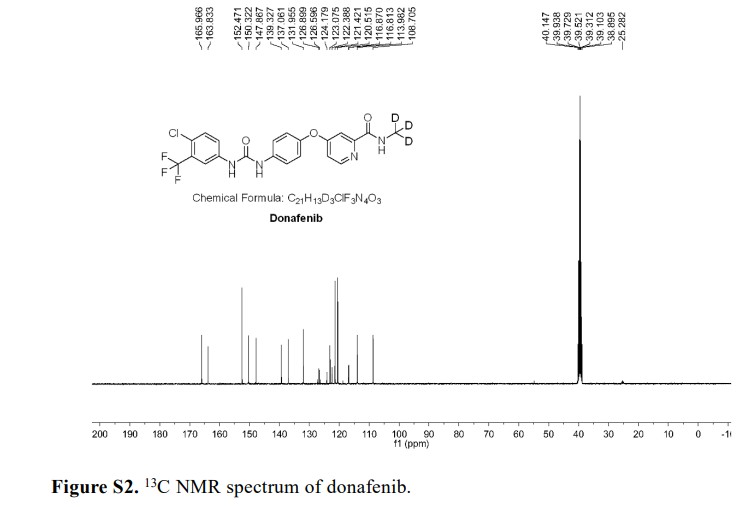
Donafenib
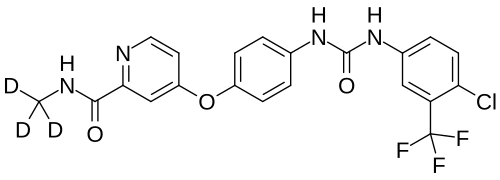
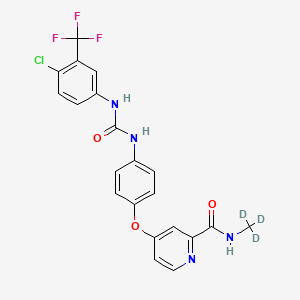
Donafenib
CAS 1130115-44-4, CM-4307, Zepsun, 41XGO0VS1U
4-[4-[[4-chloro-3-(trifluoromethyl)phenyl]carbamoylamino]phenoxy]-N-(trideuteriomethyl)pyridine-2-carboxamide
- 4-(4-(((4-chloro-3-(trifluoromethyl)phenyl)carbamoyl)amino)phenoxy)-N-(2H3)methylpyridine-2-carboxamide
- Sorafenib-d3
- Sorafenib D3
- Donafenib (Sorafenib D3)
- Sorafenib-methyl-d3
- d3-sorafenib
CM-4307 is under investigation in clinical trial NCT03602495 (Donafenib in 131I-Refractory Differentiated Thyroid Cancer).
Donafenib, sold under the brand name Zepsun, is a pharmaceutical drug for the treatment of cancer.
In China, donafenib is approved for the treatment of unresectable hepatocellular carcinoma in patients who have not previously received systemic treatment.[1][2]
Donafenib is a kinase inhibitor that targets Raf kinase and various receptor tyrosine kinases.[3] It is a deuterated derivative of sorafenib with improved pharmacokinetic properties.[4][5]
Donafenib is an orally available multikinase inhibitor that targets Raf kinase and various receptor tyrosine kinases (RTKs), with potential antineoplastic activity. Upon oral administration, donafenib binds to and blocks the activity of Raf kinase, and inhibits Raf-mediated signal transduction pathways. This inhibits cell proliferation in Raf-expressing tumor cells. In addition, this agent may inhibit unidentified RTKs, and thus may further block tumor cell proliferation in susceptible tumor cells. Raf, a serine/threonine protein kinase, plays a key role in the Raf/mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) signaling pathway. Deregulation of this pathway often results in tumor cell proliferation and survival.
SYN
ACS Omega 2021, 6, 5532−5547.
https://pubs.acs.org/doi/10.1021/acsomega.0c05908
Syn
Donafenib (Zepsun). Donafenib (31), developed by Suzhou Zelgen Biopharmaceuticals, is a deuterated derivative of sorafenib, a multikinase inhibitor for the treatment of advanced hepatocellular carcinoma (HCC). 222 HCC is the most common type of primary liver cancer in adults and the third leading cause of cancer-related deaths worldwide.223,224 Donafenib inhibits Raf kinase and VEGFR tyrosine kinases,
thereby preventing the proliferation of tumor cells. 225 The presence of the deuterated methyl group in donafenib improves metabolic stability with prolonged half-life, lower systemic clearance, and higher systemic exposure.226 Donafenib has been shown to significantly improve the overall survival
of patients with HCC when compared against sorafenib, with favorable safety and tolerability.227 228
In June 2021, donafenib was first approved in China for treating unresectable HCC in patients who have not previously received systemic treatment.
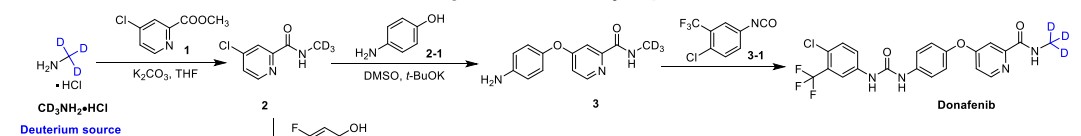
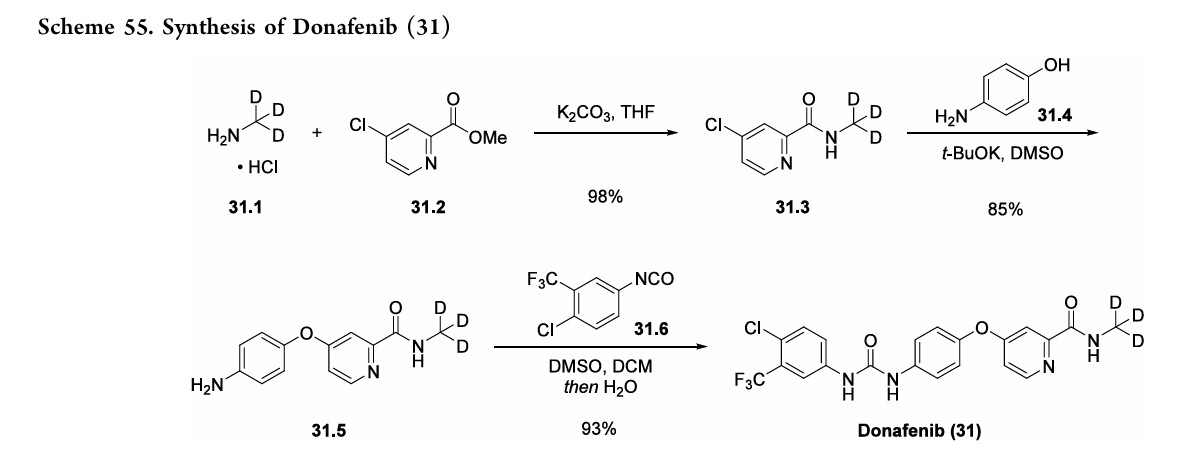
A gram-scale synthesis of donafenib was recently disclosed by Luo and co-workers (Scheme 55).229
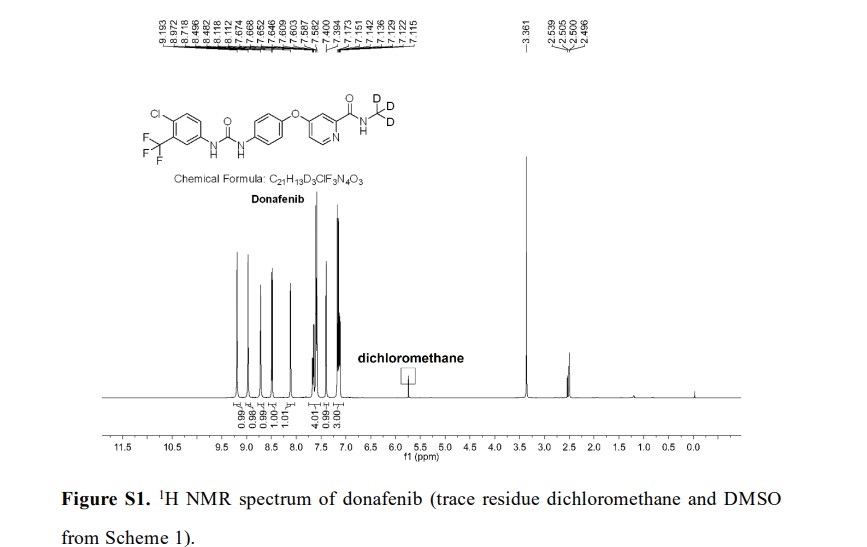
The synthetic sequence commenced with amidation of methyl ester 31.2 using methan-d3-amine hydrochloride (31.1) as the deuterium source, affording CD 3-amide 31.3 in high yield (98%). SNAr
displacement with aminophenol 31.4 in DMSO provided diaryl ether 31.5. Finally, reaction of the aniline moiety with isocyanate 31.6 delivered donafenib (31) in 79% yield from 31.3
(222) Mousa, A. B. Sorafenib in the treatment of advanced
hepatocellular carcinoma. Saudi J. Gastroenterol 2008, 14, 40−42.
(223) Forner, A.; Llovet, J. M.; Bruix, J. Hepatocellular carcinoma.
Lancet 2012, 379, 1245−1255.
(224) Vogel, A.; Meyer, T.; Sapisochin, G.; Salem, R.; Saborowski,
A. Hepatocellular carcinoma. Lancet 2022, 400, 1345−1362.
(225) Gong, X.; Qin, S. Study progression of anti-angiogenetic
therapy and its combination with other agents for the treatment of
advanced hepatocellular carcinoma. Hepatobiliary Surg. Nutr. 2018, 7,
466−474.
(226) Zhong, L.; Hou, C.; Zhang, L.; Zhao, J.; Li, F.; Li, W.
Synthesis of deuterium-enriched sorafenib derivatives and evaluation
of their biological activities. Mol. Divers. 2019, 23, 341−350.
(227) Qin, S.; Bi, F.; Gu, S.; Bai, Y.; Chen, Z.; Wang, Z.; Ying, J.; Lu,
Y.; Meng, Z.; Pan, H.; et al. Donafenib versus sorafenib in first-line
treatment of unresectable or metastatic hepatocellular carcinoma: A
randomized, open-label, parallel-controlled phase II-III trial. J. Clin.
Oncol. 2021, 39, 3002−3011.
(228) Keam, S. J.; Duggan, S. Donafenib: First approval. Drugs 2021,
81, 1915−1920.
(229) Li, C.; Zhong, J.; Liu, B.; Yang, T.; Lv, B.; Luo, Y. Study on
typical diarylurea drugs or derivatives in cocrystallizing with strong H
bond acceptor DMSO. ACS Omega 2021, 6, 5532−5547.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- Keam SJ, Duggan S (November 2021). “Donafenib: First Approval”. Drugs. 81 (16): 1915–1920. doi:10.1007/s40265-021-01603-0. PMID 34591285.
- Chen R, Ielasi L, di Carlo A, Tovoli F (February 2023). “Donafenib in hepatocellular carcinoma”. Drugs of Today. 59 (2): 83–90. doi:10.1358/dot.2023.59.2.3507751. hdl:11585/955557. PMID 36811408.
- “Donafenib”. NCI Cancer Dictionary. National Cancer Institute, National Institutes of Health.
- Qin S, Bi F, Gu S, Bai Y, Chen Z, Wang Z, et al. (September 2021). “Donafenib Versus Sorafenib in First-Line Treatment of Unresectable or Metastatic Hepatocellular Carcinoma: A Randomized, Open-Label, Parallel-Controlled Phase II-III Trial”. Journal of Clinical Oncology. 39 (27): 3002–3011. doi:10.1200/JCO.21.00163. PMC 8445562. PMID 34185551.
- Qin S, Bi F, Xu J, Du C, Fan Q, Zhang L, et al. (2020). “P-86 Comparison of the pharmacokinetics of donafenib and sorafenib in patients with advanced hepatocellular carcinoma: An open-label, randomized, parallel-controlled, multicentre phase II/III trial”. Annals of Oncology. 31: S117 – S118. doi:10.1016/j.annonc.2020.04.168.
| Clinical data | |
|---|---|
| Trade names | Zepsun |
| Other names | CM-4307 |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1130115-44-4 |
| PubChem CID | 25191001 |
| DrugBank | DB15414 |
| ChemSpider | 23937167 |
| UNII | 41XGO0VS1U |
| ChEMBL | ChEMBL4297490 |
| CompTox Dashboard (EPA) | DTXSID90648995 |
| Chemical and physical data | |
| Formula | C21H16ClD3F3N4O3 |
| Molar mass | 470.87 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////Donafenib, ZEPSUN, CHINA 2021, APPROVALS 2021, Suzhou Zelgen, 1130115-44-4, CM 4307, 41XGO0VS1U, Sorafenib D3
Chiglitazar
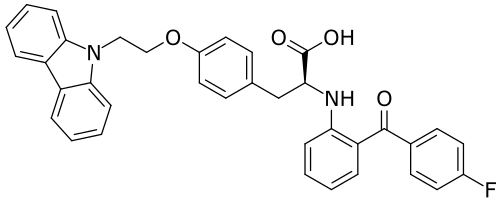
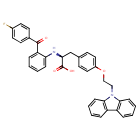
Chiglitazar
CAS 743438-45-1
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Chiglitazar sodium, (S)- | YN12H6OCV6 | 2390374-10-2 | RMVIEXHXRDCWBT-UCRKPPETSA-M |
- CS 038
- Carfloglitazar, (s)-
- E6EJV1J6Y0
- (2S)-3-[4-(2-carbazol-9-ylethoxy)phenyl]-2-[2-(4-fluorobenzoyl)anilino]propanoic acid
- C36H29FN2O4
- 572.6 g/mol
- (2S)-3-[4-(2-carbazol-9-ylethoxy)phenyl]-2-[2-(4-fluorobenzoyl)anilino]propanoic acid
- (2S)-3-(4-(2-CARBAZOL-9-YLETHOXY)PHENYL)-2-(2-(4-FLUOROBENZOYL)ANILINO)PROPANOIC ACID
- (2s)-3-(4-(2-carbazol-9-ylethoxy)phenyl)-2-(2-(4-fluorobenzoyl)anilino)propanoic acid
- Carfloglitazar, (s)-
- L-tyrosine, o-(2-(9h-carbazol-9-yl)ethyl)-n-(2-(4-fluorobenzoyl)phenyl)-
- O-(2-(9h-carbazol-9-yl)ethyl)-n-(2-(4-fluorobenzoyl)phenyl)-l-tyrosine
Chiglitazar was developed by Chipscreen Biosciences and was approved in China for improving glycemic control in adult
patients with type2 diabetes in October2021.
Chiglitazar (trade name Bilessglu) is a drug for the treatment of type 2 diabetes.[1] It is a peroxisome proliferator-activated receptor (PPAR) agonist.
In China, chiglitazar is approved for glycemic control in adult patients with type 2 diabetes when used in combination with diet and exercise.[2]
Chiglitazar is under investigation in clinical trial NCT06125587 (Chiglitazar/metformin in Non-obese Women With PCOS).
SYN
WO 2004048333
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2004048333&_cid=P12-MDMUOB-48741-1
Example 15
Preparation of 2-[(2-(4-fluorobenzoyl)phenyl)amino]-3-[4-(2-carbazolylethoxy)-phenyl]
-propionic acid (compound CS038)
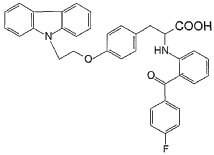
To a solution of 2-[(2-(4-fluorobenzoyl)phenyl)amino]-3-[4-(2-bromoethoxy)-phenyl] -propionic acid methyl ester (0.25 g, 0.49 mmol) and carbazole (0.082 g, 0.49 mmol) in benzene (10 ml) is added tetrabutyl ammonium bromide (0.08 g) and 50% NaOH aqueous solution (0.084 g, 1.08 mmol), then the mixture is heated to reflux for 10 h. After cooled, benzene (30ml) is added, and the mixture is washed with water (3×30 ml). Then the solvent is evaporated under a vacuum. The crude product is purified by silica gel chromatography using CHCl3/MeOH (4:1) as eluent to give the title compound (0.10 g, 36%). HRMS calcd for C36H29FN204: 572.6357. Found: 572.6354. MA calcd for C36H29FN204: C, 75.51%; H, 5.11%; N, 4.89%. Found: C, 75.83%; H, 5.10%; N, 4.90%.
PATENT
US 10640465
https://patentscope.wipo.int/search/en/detail.jsf?docId=US249083802&_cid=P12-MDMUQY-52500-1
The pharmacological activity of the compound is described in Chinese patent application No. CN03126974.5 and U.S. Pat. No. 7,268,157. 2-(2-(4-fluorobenzoyl)phenylamino)-3-(4-(2-(9H-carbazol-9-yl)ethoxy)phenyl)propanoic acid is able to selectively activate PPAR-α, PPAR-γ and PPAR-6, and can be used to treat the diseases associated with metabolic syndrome such as diabetes, hypertension, obesity, insulin resistance, hypertriglyceridemia, hyperglycemia, high cholesterol, arteries atherosclerosis, coronary heart disease, etc. A preparation method of 2-(2-(4-fluorobenzoyl)phenylamino)-3-(4-(2-(9H-carbazol-9-yl)ethoxy)phenyl)propanoic acid is disclosed in Chinese patent application No. CN03126974.5 and U.S. Pat. No. 7,268,157, and the synthetic route thereof is as follows:
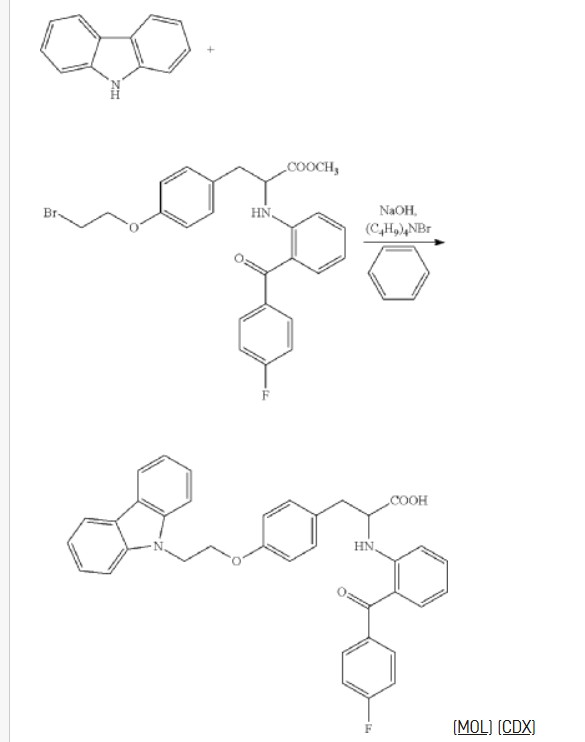
EXAMPLES
Example 1: Preparation of 2-(2-(4-fluorobenzoyl)phenylamino)-3-(4-(2-(9H-carbazol-9-yl)ethoxy)phenyl)propanoic Acid
SYN
J. Med. Chem. 2024, 67, 4376−4418
Chiglitazar (Bilessglu). Chiglitazar (17), a novel nonthiazolidinedione pan-agonist of α, δ, and γ peroxisome proliferator-activated receptors (PPARs), has shown promise for the treatment of type 2 diabetes. 126 Type 2 diabetes impacts over 374 million patients worldwide and continues to
rise in incidence and prevalence globally. 127 Chiglitazar preferentially regulates expression of ANGPTL4 and PDK4 genes, which are involved in glucose and lipid metabolism. 128 Chiglitazar was developed by Chipscreen Biosciences and was approved in China for improving glycemic control in adult
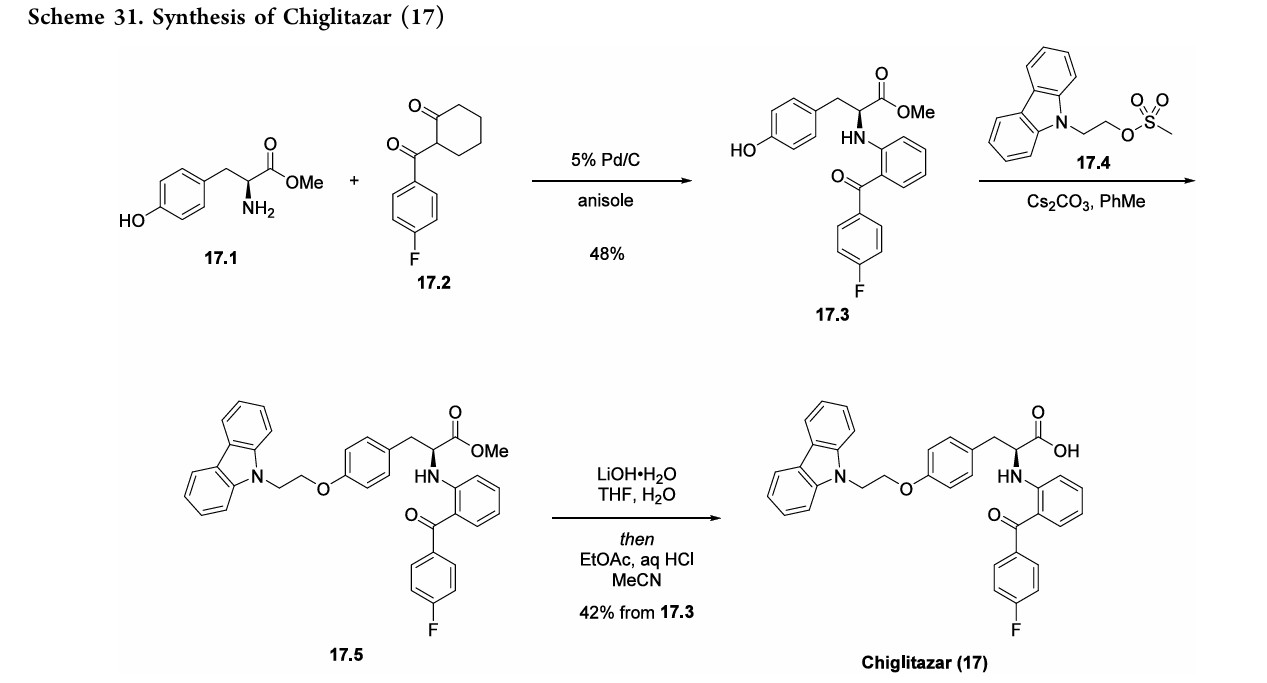
patients with type2 diabetes in October2021.129 Thesynthesisof17beganwithimineformationbetweenL
tyrosine methyl ester (17.1) and 2-(4-fluorobenzoyl) cyclohexanone(17.2)with tandemaromatizationunderPd/C catalysis to generate aniline derivative 17.3 (Scheme31).130,131 Alkylation of the phenol moiety of 17.3 with mesylate17.4furnishedphenyl alkyl etherderivative17.5.132
Hydrolysisof themethylester in17.5withlithiumhydroxide followedbyacidificationwithhydrochloricacidandrecrystal lization fromacetonitrile afforded chiglitazar (17) in 42% overall yield from17.3.Thisprocessdeliveredchiglitazar in 99.4%purityat24gscale.
(126) Ji, L.; Song, W.; Fang, H.; Li, W.; Geng, J.; Wang, Y.; Guo, L.;
Cai, H.; Yang, T.; Li, H.; et al. Efficacy and safety of chiglitazar, a
novel peroxisome proliferator-activated receptor pan-agonist, in
patients with type 2 diabetes: a randomized, double-blind, placebo
controlled, phase 3 trial (CMAP). Sci. Bull. 2021, 66, 1571−1580.
(127) Chatterjee, S.; Khunti, K.; Davies, M. J. Type 2 diabetes.
Lancet 2017, 389, 2239−2251.
(128) Pan, D.-S.; Wang, W.; Liu, N.-S.; Yang, Q.-J.; Zhang, K.; Zhu,
J.-Z.; Shan, S.; Li, Z.-B.; Ning, Z.-Q.; Huang, L.; Lu, X.-P. Chiglitazar
preferentially regulates gene expression via configuration-restricted
binding and phosphorylation inhibition of PPARγ. PPAR Research
2017 2017, 2017, 1−16.
(129) Deeks, E. D. Chiglitazar: First approval. Drugs 2022, 82, 87−
92.
(130) Li, Z.; Lu, X.-P.; Liao, C.; Shi, L.; Liu, Z.; Ma, B. Substituted
arylalcanoic acid derivatives as PPAR pan agonists with potent
antihyperglycemic and antihyperlipidemic activity. WO 2004048333
A1, 2004.
(131) Sutter, M.; Sotto, N.; Raoul, Y.; Métay, E.; Lemaire, M.
Straightforward heterogeneous palladium catalyzed synthesis of aryl
ethers and aryl amines via a solvent free aerobic and non-aerobic
dehydrogenative arylation. Green Chem. 2013, 15, 347−352.
(132) Lu, X.; Li, Z.; Wang, X. Method for preparing phenylalanine
compound. U.S. Patent US 10640465 B2, 2020.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Ji L, Song W, Fang H, Li W, Geng J, Wang Y, et al. (August 2021). “Efficacy and safety of chiglitazar, a novel peroxisome proliferator-activated receptor pan-agonist, in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled, phase 3 trial (CMAP)”. Science Bulletin. 66 (15): 1571–1580. Bibcode:2021SciBu..66.1571J. doi:10.1016/j.scib.2021.03.019. PMID 36654286. S2CID 233650336.
- Deeks ED (January 2022). “Chiglitazar: First Approval”. Drugs. 82 (1): 87–92. doi:10.1007/s40265-021-01648-1. PMID 34846697. S2CID 244716275.
| Clinical data | |
|---|---|
| Trade names | Bilessglu |
| Other names | Carfloglitazar |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 743438-45-1 |
| PubChem CID | 71402018 |
| ChemSpider | 57523239 |
| UNII | E6EJV1J6Y0 |
| ChEMBL | ChEMBL4650349 |
| CompTox Dashboard (EPA) | DTXSID00225352 |
| Chemical and physical data | |
| Formula | C36H29FN2O4 |
| Molar mass | 572.636 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////////Chiglitazar, Chipscreen Biosciences, CHINA 2021, DIABETES, CS 038, Carfloglitazar, (s)-, E6EJV1J6Y0,
Hetrombopag Olamine
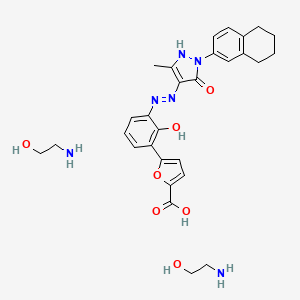
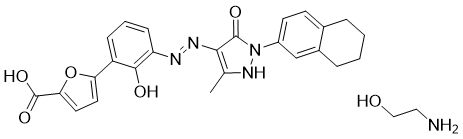
Hetrombopag Olamine, RAFUTROMBOPAG OLAMINE
- Hetrombopag diolamine
- SHR8735 olamine
- Hetrombopag ethanolamine
- SHR-8735 olamine
580.6 g/mol, C29H36N6O7, V45T2I862X
2-aminoethanol;5-[2-hydroxy-3-[[5-methyl-3-oxo-2-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazol-4-yl]diazenyl]phenyl]furan-2-carboxylic acid
CAS 1257792-42-9
1257792-41-8 (free acid) 1257792-41-8 (ethanolamine) 1257792-42-9 (olamine)
Jiangsu Hengrui Pharmaceutical, was approved in China in June 2021 for treatment of adult patients with chronic primary immune thrombocytopenia (ITP) and severe aplastic anemia who have not responded well to other treatments
Hetrombopag Olamine is the orally active ethanolamine salt of hetrombopag, a small-molecule, nonpeptide thrombopoietin receptor (TPO-R or CD110) agonist, with megakaryopoiesis-stimulating activity. Upon oral administration, hetrombopag targets, binds to and stimulates the transmembrane domain of the platelet TPO-R, a member of the hematopoietin receptor superfamily. Activation of TPO-R leads to the proliferation and differentiation of cells in the megakaryocytic lineage and an increase in platelet production. This may prevent or treat chemotherapy-induced thrombocytopenia.
- OriginatorJiangsu Hengrui Medicine Co.
- DeveloperAtridia; Jiangsu Hengrui Medicine Co.
- ClassAntianaemics; Antihaemorrhagics; Aza compounds; Carboxylic acids; Furans; Pyrazolones; Small molecules; Tetrahydronaphthalenes
- Mechanism of ActionThrombopoietin receptor agonists
- Orphan Drug StatusYes – Thrombocytopenia
- MarketedAplastic anaemia; Idiopathic thrombocytopenic purpura
- Phase IIIThrombocytopenia
- No development reportedUnspecified
- 07 Dec 2024Efficacy and adverse events data from a phase-III trial in Aplastic anaemia presented at the 66th American Society of Hematology Annual Meeting and Exposition (ASH-Hem-2024)
- 31 Jul 2024Phase-III clinical trials in Thrombocytopenia in China (PO) (NCT06507436)
- 25 Jul 2024Jiangsu Hengrui Medicine plans a phase III trial in Thrombocytopenia (PO) in July 2024 (NCT06507436)
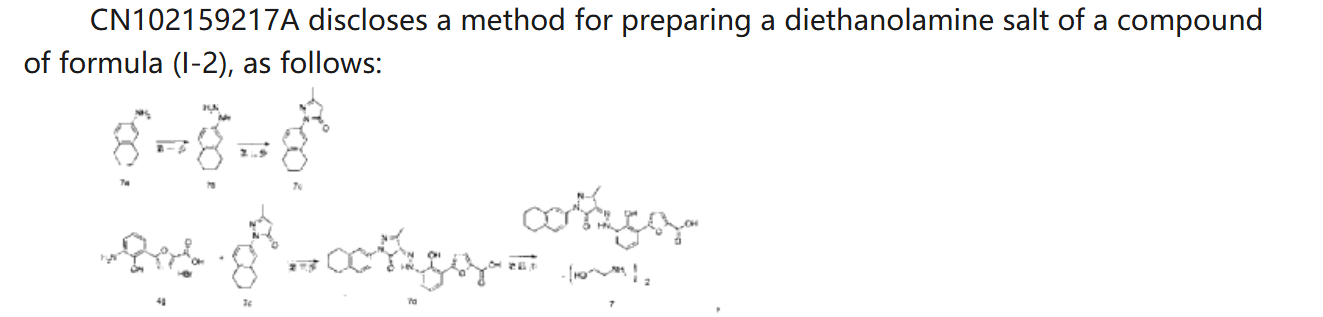
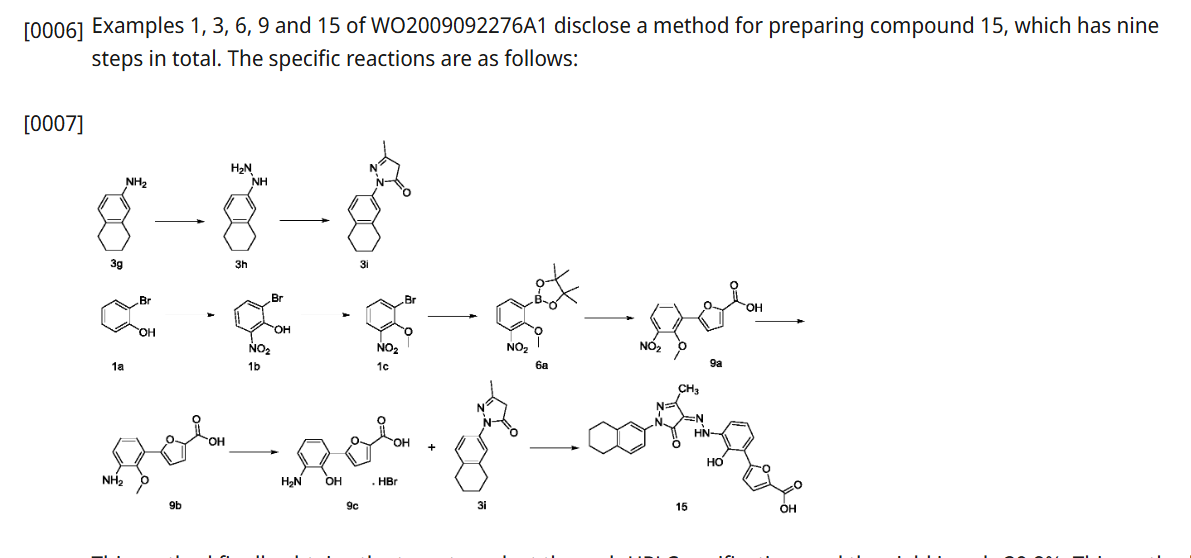
SYN
CN 113929668
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN349207982&_cid=P21-MDCUSL-44897-1
| Example 1. Synthesis of 5-(2-carbonyl-2,3-dihydrobenzoxazol-7-yl)furan-2-carboxylic acid |
| |
| Add purified water to the batching barrel, add 4.0kg of compound a under stirring, then add 10L of hydrochloric acid, stir, pump the material into a 50L reactor, add 10L of purified water to the batching barrel and pump it into the reactor. Turn on stirring, start cooling, the temperature drops to -5~2°C, start adding sodium nitrite aqueous solution (6.4L purified water, 1840g sodium nitrite), keep the temperature in the reactor no higher than 5°C during the process; after adding, continue stirring for 10~20min; add 800g of urea, continue stirring for 10~20min, the obtained diazonium salt solution is ready for use, and the temperature in the whole process is kept no higher than 5°C. |
| 44kg of acetone was pumped into a 200L reactor, and 15.0kg of compound b and 463.5g of copper chloride dihydrate were added in sequence under stirring. The temperature was raised to 30-35°C, and the obtained diazonium salt solution was added. The temperature was maintained at 30-40°C during the period. After the addition was completed, the temperature was maintained at 30-40°C and the reaction was continued with stirring for 1-1.5h. 120.0L of purified water was added, the temperature was raised to 40-45°C, and stirring was continued for a period of time. Filter, wash the filter cake with purified water until the filtrate is neutral, filter again, and collect the filter cake. 80L of purified water was added to the reactor, stirring was started, and the filter cake was added. Sodium hydroxide aqueous solution was added to the reactor to adjust the pH, the pH value was maintained at 8-10 for a period of time, and the filtrate was pumped into the reactor, and the filter was pressed into the material barrel through the filter press. Then 10L of purified water was pumped into the reactor and filtered into the material barrel. The material in the material barrel was pumped into the reactor, and then ethyl acetate was pumped in, stirred, and allowed to stand for 30-40 minutes. The aqueous phase was separated and collected, and the aqueous phase was pumped into the reactor, and the pH was adjusted to 3-4 with hydrochloric acid solution, and the filter cake was washed with purified water until the filtrate was neutral, and then the filter cake was collected. The filter cake was dried to obtain compound c. The yield of this step was 3.59 kg, and the yield was 55%. |
| Example 2: Synthesis of 5-(3-amino-2-hydroxyphenyl)furan-2-carboxylic acid |
| |
| Purified water was pumped into the 50L reactor, stirring was started, 3.53kg of sodium hydroxide was added, and compound c obtained in the previous step was added. Under nitrogen protection, the reaction mixture was heated to reflux in the reactor for reaction. After the reaction, the reaction solution was cooled, the temperature was lowered to 0-10°C, and hydrochloric acid solution was added to adjust the pH value to 5-6. The filter cake was filtered, and the filtrate was washed with purified water until neutral, and then filtered again to collect the filter cake. The filter cake was dried to obtain compound d. The yield in this step was 2.78kg, with a yield of 90%. |
| Example 3. Synthesis of (Z)-5-(2-hydroxy-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazol-4(5H)-ylidene)hydrazino)phenyl)furan-2-carboxylic acid |
| |
| Purified water was added to the batching barrel, and compound d was added in sequence under stirring, and then 6.3L hydrochloric acid was added, and the materials were pumped into a 200L reactor. Purified water was added to the batching barrel again, and then pumped into the reactor. Stirring was started, and the temperature was lowered to -5 to 2°C. Sodium nitrite aqueous solution (sodium nitrite to compound d molar ratio is 1:1) was added, and the internal temperature was kept at no more than 5°C during the process. After the addition was completed, stirring was continued; urea was added, and stirring was continued to obtain a diazonium salt solution for use, and the internal temperature was kept at no more than 5°C during the whole process. |
| Add 36L purified water and 4000g sodium hydroxide to the batching barrel, stir to dissolve, and set aside. Take 26kg of the above sodium hydroxide aqueous solution, add compound e (the molar ratio of compound e to compound d is 0.9:1), stir, and add the resulting solution to the diazonium salt solution, keeping the temperature not exceeding 8°C. Add the above-prepared sodium hydroxide aqueous solution dropwise, adjust the pH to 8-10, and keep the temperature at 5-10°C for 3-4h. Add hydrochloric acid solution dropwise to adjust the pH to 2-3, keep the temperature not exceeding 25°C, filter, wash the filter cake with purified water until the filtrate is neutral, filter again, and collect the filter cake. Pump 48.0kg of tetrahydrofuran aqueous solution (22.5kg tetrahydrofuran, 25.5L purified water) into the reactor, add the above-obtained filter cake, beat, filter, wash the filter cake with tetrahydrofuran aqueous solution, wash the filter cake with purified water, filter again, and collect the filter cake. Dry the filter cake. |
| Ethyl acetate was pumped into the reactor, and the above-obtained materials were added to the reactor for slurrying, and the filter cake was washed with ethyl acetate, and the filter cake was washed until no obvious droplets flowed out of the mirror, and the filter cake was collected and dried to obtain the compound of formula (I-2). The yield in this step was 5.34 kg, and the yield was 97.5%. |
| Example 4. Synthesis of (Z)-5-(2-hydroxy-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazol-4(5H)-ylidene)hydrazino)phenyl)furan-2-carboxylic acid |
| The compound of formula (I-2) was prepared by using a method substantially the same as in Example 3 (except that the equivalent of compound e was adjusted from 0.9 in Example 3 to the current 0.95, other conditions remained unchanged). |
| Comparative Example 1: Synthesis of (Z)-5-(2-hydroxy-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazole-4(5H)-ylidene)hydrazino)phenyl)furan-2-carboxylic acid |
| |
| The compound of formula (I-2) was prepared by using a method substantially the same as in Example 3 (except that the step of adding urea was changed to starch potassium iodide test paper to indicate the reaction endpoint, and other conditions remained unchanged). |
| Test Example 1: Effect of urea on the preparation process of the compound of formula (I-2) |
| HPLC conditions: |
| Chromatographic column: Welch Ultimate |
| Flow rate: 1.0ml/min |
| Injection volume: 10 μl |
| Detector: UV detector |
| Detection wavelength: 251nm |
| Mobile phase: 0.1% trifluoroacetic acid aqueous solution was used as mobile phase A, acetonitrile was used as mobile phase B, and elution was performed at a ratio of 50%/50% of mobile phase A/mobile phase B. |
PATENT
EP 2441457
PATENT
WO 2010142137
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2010142137&_cid=P21-MDCUXF-51461-1
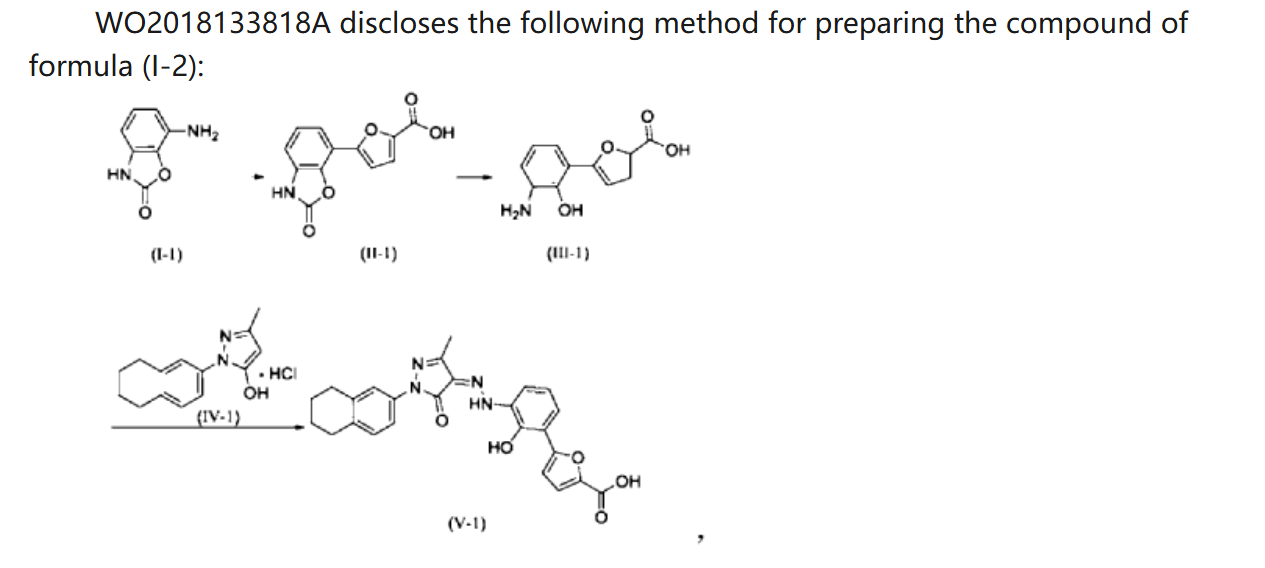
PATENT
WO 2018133818
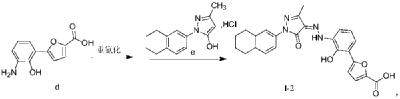
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018133818&_cid=P21-MDCUYN-53075-1
Example 1. Preparation of 3-methyl-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1H-pyrazol-5-ol hydrochloride
[0107]
[0108](5,6,7,8-tetrahydronaphthalene-2-yl)hydrazine hydrochloride (1.3 kg, prepared according to the method in patent application WO2009092276A1) and ethyl acetoacetate (1.17 L) were added to ethyl acetate (5.2 L). The mixture was heated under reflux for 2 hours. The reaction solution was cooled to room temperature, then cooled to 0-5°C, stirred for 1 hour, filtered, and the solid was washed with a small amount of ethyl acetate to obtain a white solid product (1.4 kg, yield 81%).
[0109]
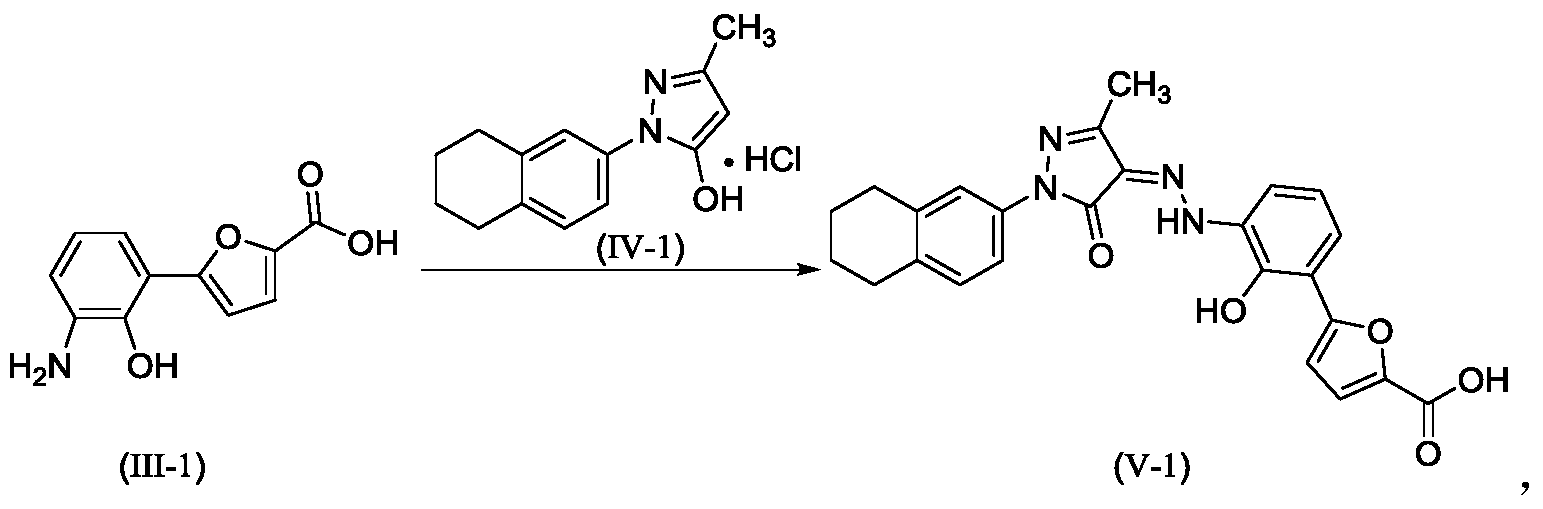
[0110]Example 2. Preparation of (Z)-5-(2-hydroxy-3-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1,5-dihydro-4H-pyrazol-4-ylidene)hydrazino)phenyl)furan-2-carboxylic acid (V-1)
[0111]
[0112]Step 1: Synthesis of intermediate (II-1)
[0113]Purified water (14.80 kg), 7-aminobenzo[d]oxazol-2(3H)-one (2.00 kg, prepared according to the method in patent application WO2005016898A2), and hydrochloric acid (5.33 kg) were added to the reaction kettle, the temperature was raised to 40-45°C, stirred for 10 min, cooled to -3-5°C, and sodium nitrite aqueous solution (sodium nitrite 940 g, water 3.20 kg) was added dropwise, the internal temperature was kept at no more than 5°C, the end point was controlled by starch potassium iodide test paper, and stirring was continued for 15 min;
[0114]Add acetone (28L) to the reactor, then add furoic acid (4.57kg) and cupric chloride dihydrate (232g), stir at 35-40℃ until dissolved, add diazonium salt solution dropwise, keep the internal temperature at 35-40℃, and continue stirring for 1.5h. Add purified water (60L), heat to 35-40℃ and stir for 30min. Filter, wash the filter cake with 45-50℃ purified water. Add the filter cake to purified water (40kg), adjust the pH to 8-9 with 15% sodium hydroxide aqueous solution, filter, adjust the pH of the filtrate to 3-4 with 6mol/L hydrochloric acid, filter, wash the filter cake with purified water, and dry to obtain a solid (1.63kg, yield 50%).
[0115]Step 2: Synthesis of intermediate (III-1)
[0116]The product from the previous step (1.4 kg) and 15% aqueous sodium hydroxide solution (9.7 kg) were heated to reflux under argon protection and reacted for 28 hours. The reaction solution was poured into ice water (5-6 kg), and hydrochloric acid (6N, 3 L) was slowly added to adjust the pH value to 5-6. The temperature was maintained below 20°C. During this period, ethyl acetate was added to eliminate bubbles. The mixture was filtered, washed with purified water, and dried to obtain a solid (1.18 kg, yield 94%).
[0117]Step 3: Synthesis of intermediate (V-1)
[0118]Add the product of the previous step (1.10kg), purified water (27.5kg), and hydrochloric acid (2.92kg) to the reactor in sequence, stir and dissolve, cool to -4 to -1°C, add sodium nitrite aqueous solution (346g sodium nitrite, 5.5kg water), and continue to react for 15min after the addition is completed. Cool to -8 to -5°C. Dissolve sodium hydroxide (1.48kg) in purified water (13.2kg) to obtain a 10% sodium hydroxide aqueous solution. Add 5-methyl-2-(5,6,7,8-tetrahydronaphthalen-2-yl)-2H-pyrazole-3-ol hydrochloride (1.26kg) to the above sodium hydroxide aqueous solution (10kg) to dissolve, and add the resulting solution to the diazonium salt solution at once, keeping the temperature not higher than 10°C. Add the remaining 10% sodium hydroxide aqueous solution, adjust the pH to 8 to 9, naturally heat to 8 to 12°C for reaction, and react for 4h. Add 6N hydrochloric acid, adjust pH=2-3, keep the temperature not more than 20°C, filter, and wash the filter cake with water until pH=6-7. Add the filter cake to 50% tetrahydrofuran aqueous solution (19kg), slurry at room temperature for 2h, filter, wash with 50% tetrahydrofuran aqueous solution, wash with water, and dry. Add ethyl acetate (20kg) to the solid, slurry at 40-45°C for 2h under argon protection, cool to room temperature, filter, wash with ethyl acetate, add the solid to ethyl acetate (20kg), slurry at 40-45°C for 2h under argon protection, cool to room temperature, filter, wash with ethyl acetate, and dry to obtain a solid (2.18kg, yield 95%, purity 99.5%).
[0120]Example 3. Preparation of (Z)-5-(2-hydroxy-3-(2-(3-methyl-5-oxo-1-(5,6,7,8-tetrahydronaphthalen-2-yl)-1,5-dihydro-4H-pyrazol-4-ylidene)hydrazino)phenyl)furan-2-carboxylic acid ethanolamine salt (1:2)
[0121]
[0122]Preparation of crude product
[0123]The compound of formula (V-1) (1.8 kg) was suspended in a tetrahydrofuran/ethanol (14.5 kg, V/V = 2:1) mixed solvent at room temperature, stirred for 0.5 h, cooled to 10-15 ° C, and a tetrahydrofuran ethanol solution of ethanolamine (479.6 g) (tetrahydrofuran 91 g and ethanol 41 g) was added dropwise. The mixture was naturally heated to room temperature and reacted for 20 h. Filtered, washed with a tetrahydrofuran/ethanol (V/V = 2:1) mixed solvent, washed with ethyl acetate, filtered, and dried to obtain a dark red solid (1.73 kg, yield 76%, purity 99.7%).
[0124]
1H-NMR(500MHz,D 2O+NaOH)δ7.725-7.741(d,1H),7.298-7.316(d,3H),7.183-7.198(d,1H),7.131-7.149(m,2H),6.612-6.643(t,1H),3.574-3.596(t,4H),2.759-2.778(br,4H),2.698-2.721(t,4H),2.428(s,3H),1.772(br,4H).
SYN
J.Med.Chem.2024,67,4376−4418
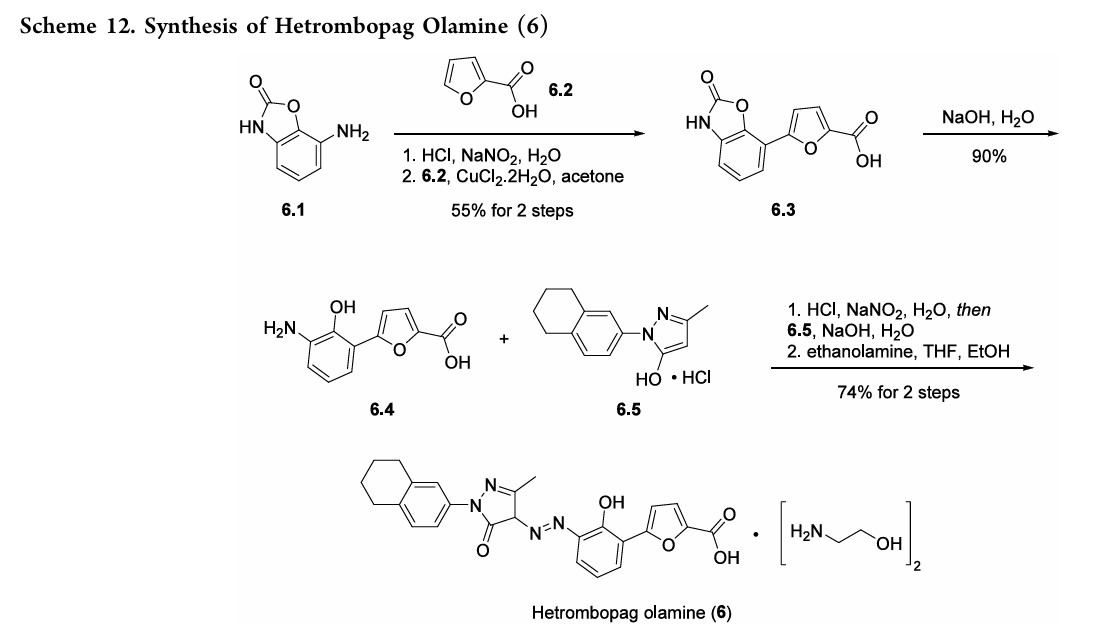
HetrombopagOlamine (Hengqu).
Hetrombopag olamine (6), an oral nonpeptide thrombopoietin receptor
(TpoR)agonistdevelopedby JiangsuHengruiPharmaceutical, was approved in China in June2021 for treatment of adult patients with chronic primary immune thrombocytopenia (ITP) and severe aplastic anemiawhohave not responded well to other treatments.46Hetrombopag, like other TpoR agonists, increases platelet production by binding to the transmembranedomainofTpoRinprogenitorcells, inducing
megakaryocytes.Theeffectisadditivewiththeactionofnative thrombopoietin, whichbinds to the extracellular domainof TpoR.Hetrombopag is structurallyrelatedtoeltrombopag, a previously approvedTpoR, withmodifications to enhance potencyandminimizetoxicity.46−48InaPhaseIIIclinicaltrial, ITPpatients demonstratedadurableplatelet response, with reducedbleedingriskanduseof rescuetherapycomparedto
placebo.49 Akilo-scale, chromatography-freesynthesisofhetrombopag has been reported by researchers at Jiangsu Hengrui Pharmaceutical in the Chinese-language patent literature (Scheme 12).50,51 Commercially available aniline 6.1 was coupledwith furoic acid (6.2) using aMeerwein arylation reaction togive intermediate6.3.This process first involves diazotizationof the anilineusing sodiumnitrite andhydrochloricacid.Ureawasusedtoquenchtheresidualnitrousacid, animprovement thatultimatelygavetheproductwithhigher purity and lower levels of specific impurities; the crude
diazoniumsalt solutionwas carried forwarddirectlywithout furthermanipulation.Furoicacid(6.2)inacetonewastreated withcopper(II)chloridedihydratefollowedbyadditionofthe
diazonium salt solution to affect the arylation. The crude productwaspurifiedbyacid−baseextractionandisolatedby filtrationtoprovide6.3 in55%yield.Basichydrolysisof the
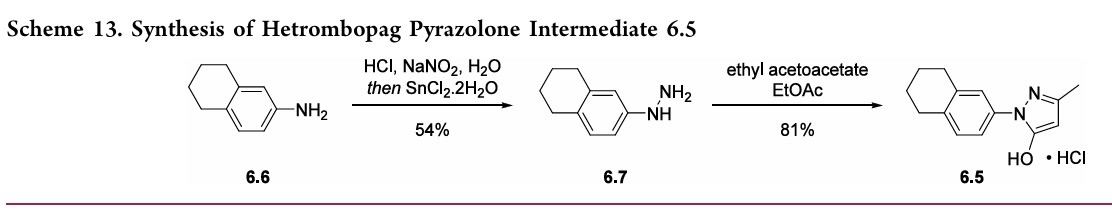
cycliccarbamateunveiledthefreeanilineandphenolmoieties in arene 6.4. Nucleophilic attack of the enolate anion of pyrazolone 6.5 (see Scheme 13) on the diazoniumsalt of aniline6.4 formed the central hydrazonemoiety ina JappKlingemann-like reaction. The crude product was triturated withethylacetatetorapidlyprovidehetrombopagfreebasein
97.5%yield.TreatmentwithethanolamineinTHFandEtOH thengeneratedhetrombopagolamine (6) in76%yieldand 99.7%purity.51 Pyrazolone intermediate6.5was synthesized in two steps
(Scheme 13).52,53 5,6,7,8-Tetrahydronaphthalen-2-yl amine (6.6)was converted to the diazoniumion and reduced in situ to the corresponding hydrazine 6.7 using stannous chloridedihydrate.Condensationof thehydrazinewithethyl acetoacetate in ethyl acetate and in situ cyclization gave pyrazolone6.5.While the synthesis fromaniline6.1 to the activepharmaceutical ingredient(API)6wasreportedonthe
kilo-scale, thesynthesisofpyrazolone6.5wasreportedonlyon gram-scale
(46) Syed, Y. Y. Hetrombopag: First approval. Drugs 2021, 81, 1581−1585.
(47) Xie, C.; Zhao, H.; Bao, X.; Fu, H.; Lou, L. Pharmacological characterization of hetrombopag, a novel orally active human thrombopoietin receptor agonist. J. Cell. Mol. Med. 2018, 22, 5367−5377.
(48) Zheng, L.; Liang, M.-z.; Zeng, X.-l.; Li, C.-z.; Zhang, Y.-f.; Chen, X.-y.; Zhu, X.; Xiang, A.-b. Safety, pharmacokinetics and pharmacodynamics of hetrombopag olamine, a novel TPO-R agonist, in healthy individuals. Basic Clin. Pharmacol. Toxicol. 2017, 121, 414−422.
(49) Mei, H.; Liu, X.; Li, Y.; Zhou, H.; Feng, Y.; Gao, G.; Cheng, P.; Huang, R.; Yang, L.; Hu, J.; Hou, M.; Yao, Y.; Liu, L.; Wang, Y.; Wu, D.; Zhang, L.; Zheng, C.; Shen, X.; Hu, Q.; Liu, J.; Jin, J.; Luo, J.; Zeng, Y.; Gao, S.; Zhang, X.; Zhou, X.; Shi, Q.; Xia, R.; Xie, X.; Jiang, Z.; Gao, L.; Bai, Y.; Li, Y.; Xiong, J.; Li, R.; Zou, J.; Niu, T.; Yang, R.;
Hu, Y. A multicenter, randomized phase III trial of hetrombopag: a novel thrombopoietin receptor agonist for the treatment of immune thrombocytopenia. J. Hematol. Oncol. 2021, 14, 37.
(50) Shi, A.; Diao, A.; Du, Y. Preparation of bicyclic substituted pyrazolone azo derivatives. China Patent CN 113929668, 2022.
(51) Diao, A.; Gao, X.; Bian, L. Method for preparing bicyclo substituted pyrazolone azo derivatives and intermediates. WO 2018133818, 2018.
(52) Tang, P. C.; Lue, H.; Fei, H.; Chen, Y. Preparation of pyrazole derivatives as thrombopoietin receptor agonists. WO 2010142137, 2010.
(53) Tang, P. C.; Lue, H.; Fei, H.; Chen, Y. Salts of bicyclo substituted pyrazolon azo derivatives, preparation method and use
thereof. European Patent EP 2441457, 2014.

.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////Hetrombopag Olamine, CHINA 2021, APPROVALS 2021, Hetrombopag diolamine, SHR 8735 olamine, Hetrombopag ethanolamine, SHR-8735 olamine, V45T2I862X, RAFUTROMBOPAG OLAMINE
Contezolid
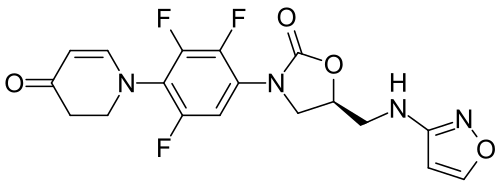
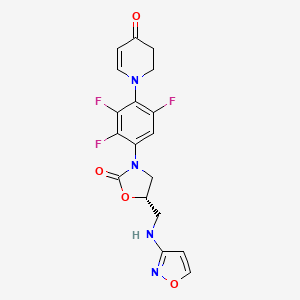

Contezolid
- MRX-I
- 1112968-42-9
- MRX-1
- B669M62ELP
- (5S)-5-[(1,2-oxazol-3-ylamino)methyl]-3-[2,3,5-trifluoro-4-(4-oxo-2,3-dihydropyridin-1-yl)phenyl]-1,3-oxazolidin-2-one
- 4(1H)-Pyridinone, 2,3-dihydro-1-(2,3,6-trifluoro-4-((5S)-5-((3-isoxazolylamino)methyl)-2-oxo-3-oxazolidinyl)phenyl)-
- (5S)-5-[(1,2-oxazol-3-ylamino)methyl]-3-[2,3,5-trifluoro-4-(4-oxo-2,3-dihydropyridin-1-yl)phenyl]-1,3-oxazolidin-2-one
- 4(1H)-Pyridinone, 2,3-dihydro-1-[2,3,6-trifluoro-4-[(5S)-5-[(3-isoxazolylamino)methyl]-2-oxo-3-oxazolidinyl]phenyl]-
- 1-{2,3,6-trifluoro-4-[(5S)-5-{[(1,2-oxazol-3-yl)amino]methyl}-2-oxo-1,3-oxazolidin-3-yl]phenyl}-1,2,3,4-tetrahydropyridin-4-one
- 5-((isoxazol-3-ylamino)methyl)-3-(2,3,5-trifluoro-4-(4-oxo-3,4-dihydropyridin-1(2H)-yl)phenyl)oxazolidin-2-one
- コンテゾリド;
WeightAverage: 408.337
Monoisotopic: 408.104539468
Chemical FormulaC18H15F3N4O4
Shanghai MicuRx Pharmaceutical Co. Ltd
Contezolid was approved for use by the National Medical Products Administration (NMPA) of China in 2021
- OriginatorMicuRx Pharmaceuticals
- ClassAntibacterials; Oxazolidinones; Skin disorder therapies
- Mechanism of ActionProtein synthesis inhibitors
- Phase IIIDiabetic foot; Skin and soft tissue infections
- No development reportedGram-positive infections
- 28 Jan 2025No recent reports of development identified for phase-I development in Gram-positive-infections(In volunteers) in China (IV)
- 28 Jan 2025No recent reports of development identified for phase-I development in Gram-positive-infections(In volunteers) in China (PO)
- 29 Nov 2024Phase-III clinical trials in Skin and soft tissue infections in China (IV), prior to November 2024
Contezolid (trade name Youxitai) is an antibiotic of the oxazolidinone class.[1][2] It is effective against Staphylococcus aureus, methicillin-resistant Staphylococcus aureus (MRSA), Streptococcus pyogenes, Streptococcus agalactiae, and other bacteria.[3]
In 2021, it was approved by the National Medical Products Administration of China for the treatment of complicated skin and soft tissue infections (cSSTI).[3][4]
A prodrug of contezolid, contezolid acefosamil, which is formulated for IV administration[5] is in Phase III clinical trials for diabetic foot infection.[6]

Chemical structure of contezolid acefosamil
SYN
https://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.2c00191
Abstract

New oral antibiotic contezolid (CZD) is effective against Gram-positive infections but unsuitable for intravenous (IV) administration due to its modest solubility. To address the medical need for an IV form of CZD, its isoxazol-3-yl phosphoramidate derivatives have been explored, and contezolid acefosamil (CZA, 8), the first representative of a novel O-acyl phosphoramidate prodrug class, has been identified. CZA exhibits high aqueous solubility (>200 mg/mL) and good hydrolytic stability at media pH suitable for IV administration. CZA rapidly converts into the active drug CZD in vivo. In a pharmacokinetic (PK) rat model, the exposure of active drug CZD after IV administration of the prodrug CZA was similar to or higher than that from the IV administration of CZD. The prodrug CZA is bioequivalent to or better than CZD in several preclinical infection models. CZA is likewise active upon its oral administration. To date, CZA has been evaluated in Phase 1 and Phase 2 clinical trials in the USA. It is advancing into further clinical studies including step-down therapy with in-hospital intravenous CZA administration followed by outpatient oral CZD treatment.
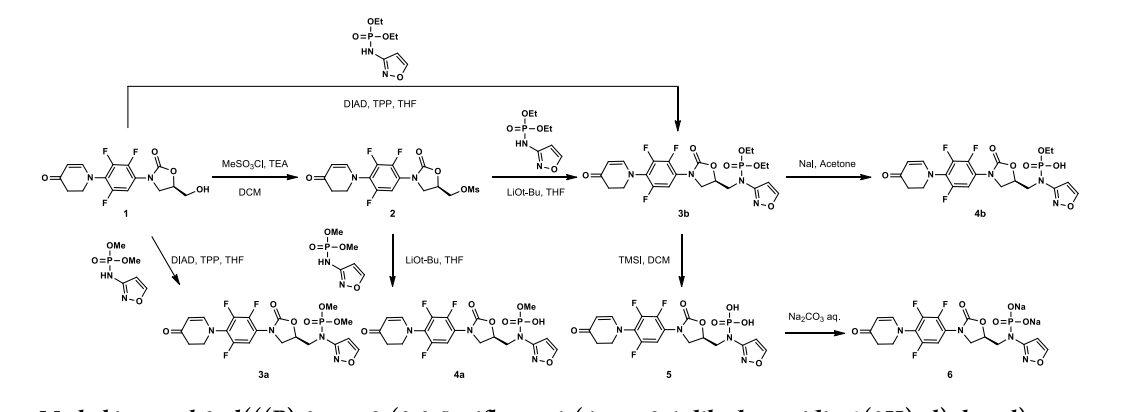
SYN
Contezolid (Youxitai). Contezolid (4), also referred to as MRX-I, is an orally administered oxazolidinone
antibacterial agent developed by Shanghai MicuRx Pharmaceutical Co. Ltd. Contezolid was developed to overcome the myelosuppression and monoamine oxidase (MAO) inhibition limitations of the structurally similar linezolid. 32 Contezolid is used to treat complicated skin and soft tissue infections arising
from multidrug-resistant Gram-positive bacterial infections including methicillin-susceptible Staphylococcus aureus (MSSA), methicillin-resistant Staphylococcus aureus (MRSA), Streptococcus pyogenes, Streptococcus agalactiae, and vancomycin-resistant enterococci.3334 Contezolid was approved for use by the National Medical Products Administration (NMPA) of China in 2021.
As with most antibacterial oral therapies, high 35 dosage is required; the drug is given twice daily for 7−14 days.36,37
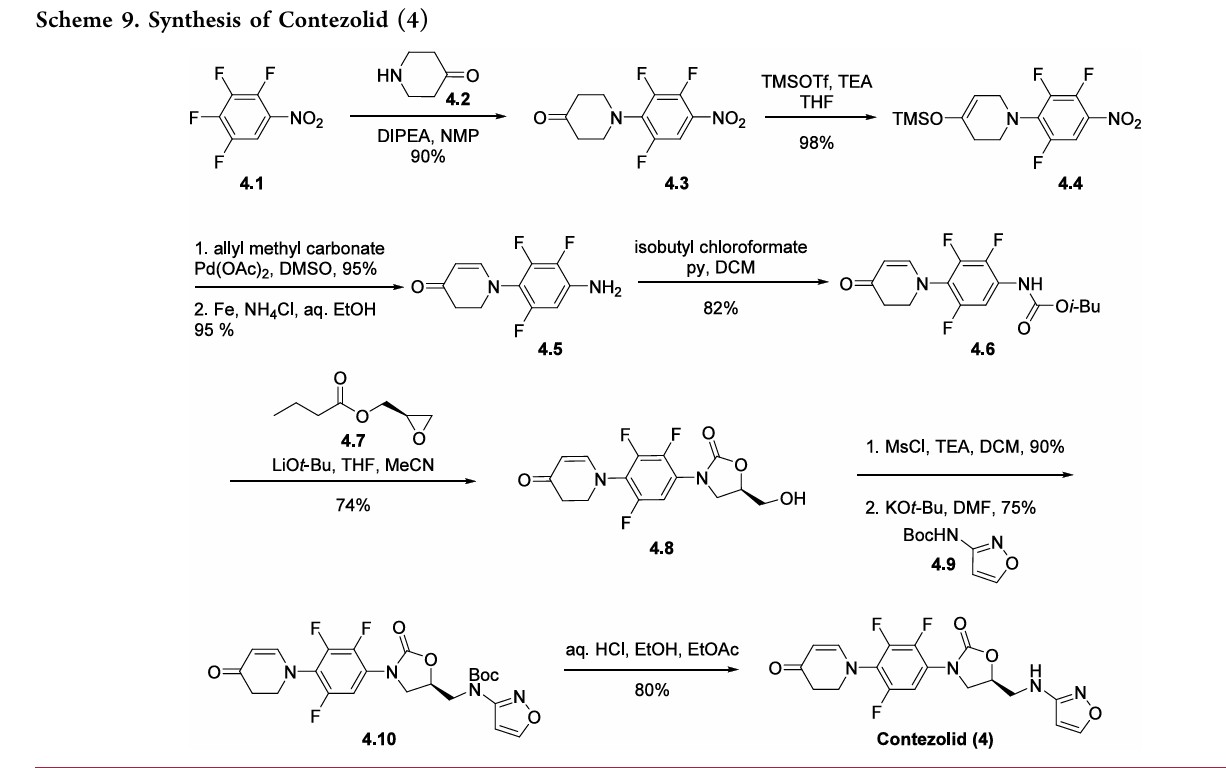
The synthesis of contezolid builds on prior research from other groups.
A sequence developed by Pharmaciawith a facile SN38began Ar reaction between polyfluorinated nitro
benzene 4.1 and piperidine-4-one 4.2 to furnish 4.3 in good yield (Scheme 9). Silyl enol ether formation afforded 4.4, which was subjected to Tsuji’s 39 method to give the α,βunsaturated ketone in excellent yield. Subsequent reduction of the nitro group gave aryl amine 4.5. Treatment of 4.5 with isobutyl chloroformate gave carbamate 4.6, which was treated with optically pure epoxide 4.7 to give xazolidinone 4.8. 38Mesylation of the free alcohol and displacement with N-Bocaminoisoxazole 4.9 afforded the Boc-protected contezolid 4.10. Simple acidic removal of the Boc group provided contezolid 4.
(32) Wang, W.; Voss, K. M.; Liu, J.; Gordeev, M. F. Nonclinical
evaluation of antibacterial oxazolidinones contezolid and contezolid
acefosamil with low serotonergic neurotoxicity. Chem. Res. Toxicol.
2021, 34, 1348−1354.
(33) Hoy, S. M. Contezolid: First approval. Drugs 2021, 81, 1587−
1591.
(34) MicuRx Pharmaceuticals. China NMPA approves MicuRx’s
contezolid for treatment of drug-resistant bacterial infection. http://www.
micurx.com/703.html (accessed 2023-06).
(35) MSD Pharmaceuticals. Usual dosages of commonly prescribed
antibiotics. https://www.msdmanuals.com/en-jp/professional/
multimedia/table/usual-dosages-of-commonly-prescribed-antibioticsa
(accessed 2023-06).
(36) Barbachyn, M. R.; Hutchinson, D. K.; Brickner, S. J.; Cynamon,
M. H.; Kilburn, J. O.; Klemens, S. P.; Glickman, S. E.; Grega, K. C.;
Hendges, S. K.; Toops, D. S.; et al. Identification of a novel
oxazolidinone (U-100480) with potent antimycobacterial activity. J.
Med. Chem. 1996, 39, 680−685.
(37) Im, W. B.; Choi, S. H.; Park, J. Y.; Choi, S. H.; Finn, J.; Yoon, S.
H. Discovery of torezolid as a novel 5-hydroxymethyl-oxazolidinone
antibacterial agent. Eur. J. Med. Chem. 2011, 46, 1027−1039.
(38) Manninen, P. R.; Brickner, S. J. Preparation of N-aryl-5R
hydroxymethyl-2-oxazolidinones from N-aryl carbamates: N-phenyl
(5R)-hydroxymethyl-2-oxazolidinone. Organic Synth 2005, 81, 112.
(39) Tsuji, J.; Minami, I.; Shimizu, I. A novel palladium-catalyzed
preparative method of α,β-unsaturated ketones and aldehydes from
saturated ketones and aldehydes via their silyl enol ethers. Tetrahedron
Lett. 1983, 24, 5635−5638.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Trade names | Youxitai |
| Other names | MRX-I |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1112968-42-9 |
| PubChem CID | 25184541 |
| IUPHAR/BPS | 10795 |
| DrugBank | DB12796 |
| ChemSpider | 34217570 |
| UNII | B669M62ELP |
| KEGG | D11297 |
| ChEMBL | ChEMBL3287379 |
| CompTox Dashboard (EPA) | DTXSID901353186 |
| Chemical and physical data | |
| Formula | C18H15F3N4O4 |
| Molar mass | 408.337 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Gordeev MF, Yuan ZY (June 2014). “New Potent Antibacterial Oxazolidinone (MRX-I) with an Improved Class Safety Profile”. Journal of Medicinal Chemistry. 57 (11): 4487–4497. doi:10.1021/jm401931e. PMID 24694071.
- Zhao X, Huang H, Yuan H, Yuan Z, Zhang Y (May 2022). “A Phase III multicentre, randomized, double-blind trial to evaluate the efficacy and safety of oral contezolid versus linezolid in adults with complicated skin and soft tissue infections”. The Journal of Antimicrobial Chemotherapy. 77 (6): 1762–1769. doi:10.1093/jac/dkac073. PMID 35265985.
- Hoy SM (September 2021). “Contezolid: First Approval”. Drugs. 81 (13): 1587–1591. doi:10.1007/s40265-021-01576-0. PMC 8536612. PMID 34365606.
- Mak E (3 June 2021). “Micurx wins China approval for antibacterial contezolid”. BioWorld.
- Liu J, Wang W, Wang C, Zhang L, Zhang X, Liu S, et al. (July 2022). “Discovery of Antibacterial Contezolid Acefosamil: Innovative O-Acyl Phosphoramidate Prodrug for IV and Oral Therapies”. ACS Medicinal Chemistry Letters. 13 (7): 1030–1035. doi:10.1021/acsmedchemlett.2c00191. PMC 9290071. PMID 35859881.
- “Contezolid acefosamil by MicuRx Pharmaceuticals for Diabetic Foot Infection (DFI): Likelihood of Approval”. GlobalData. 31 May 2023 – via Pharmaceutical Technology.
/////////Contezolid, CHINA 2021, APPROVALS 2021, MRX-I, 1112968-42-9, MRX 1, B669M62ELP, コンテゾリド ,
Difamilast
Difamilast
FDA 2026, APPROVALS 2026, difamilast, Adquey, 2/12/2026, To treat mild to moderate atopic dermatitis
PMDA Moizerto, JAPAN APPROVED 2021/9/27
ジファミラスト
ディファミラスト;
地法米司特
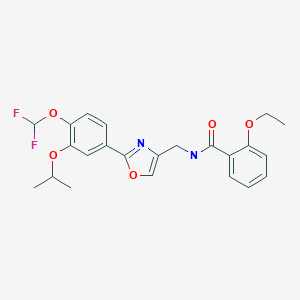
N-({2-[4-(difluoromethoxy)-3-(propan-2-yloxy)phenyl]-1,3- oxazol-4-yl}methyl)-2-ethoxybenzamide
OPA-15406
| Formula |
C23H24F2N2O5
|
|---|---|
| CAS |
937782-05-3
|
| Mol weight |
446.4439
|
MM 36; MM-36-Medimetriks-Pharmaceuticals; Moizerto; OPA-15406
| Efficacy |
Anti-inflammatory, Phosphodiesterase IV inhibitor
|
|---|---|
| Comment |
Treatment of atopic dermatitis
|
OriginatorOtsuka Pharmaceutical Development & Commercialization- DeveloperMedimetriks Pharmaceuticals; Otsuka Pharmaceutical Development & Commercialization
- ClassBenzamides; Nonsteroidal anti-inflammatories; Oxazoles; Skin disorder therapies
- Mechanism of ActionType 4 cyclic nucleotide phosphodiesterase inhibitors
- RegisteredAtopic dermatitis
- 27 Sep 2021Registered for Atopic dermatitis (In adolescents, In children, In adults) in Japan (Topical)
- 11 Nov 2020Otsuka Pharmaceutical completes a phase III trial in Atopic dermatitis (In children, In adolescents, In adults) in Japan (Topical) (NCT03961529)
- 28 Sep 2020Preregistration for Atopic dermatitis in Japan (In children, In adolescents, In adults) (Topical)

Difamilast is under investigation in clinical trial NCT01702181 (A Safety Study to Evaluate the Use and Effectiveness of a Topical Ointment to Treat Adults With Atopic Dermatitis).
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019194211&_cid=P10-MLOK7B-25338-2
Synthesis of Oxazole Compound (Type A Crystal)
Compound (5) (white powder) was prepared in accordance with the method disclosed in Example 352 of PTL 1 (WO2007/058338).
SYN
https://www.chemicalbook.com/article/how-is-difamilast-synthesised.htm
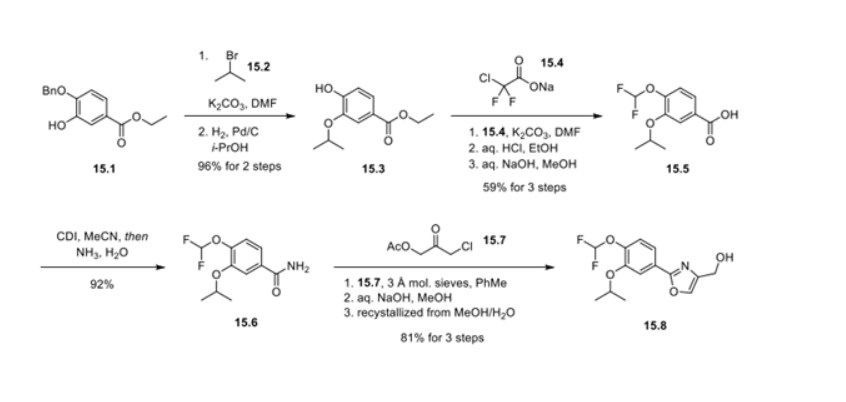
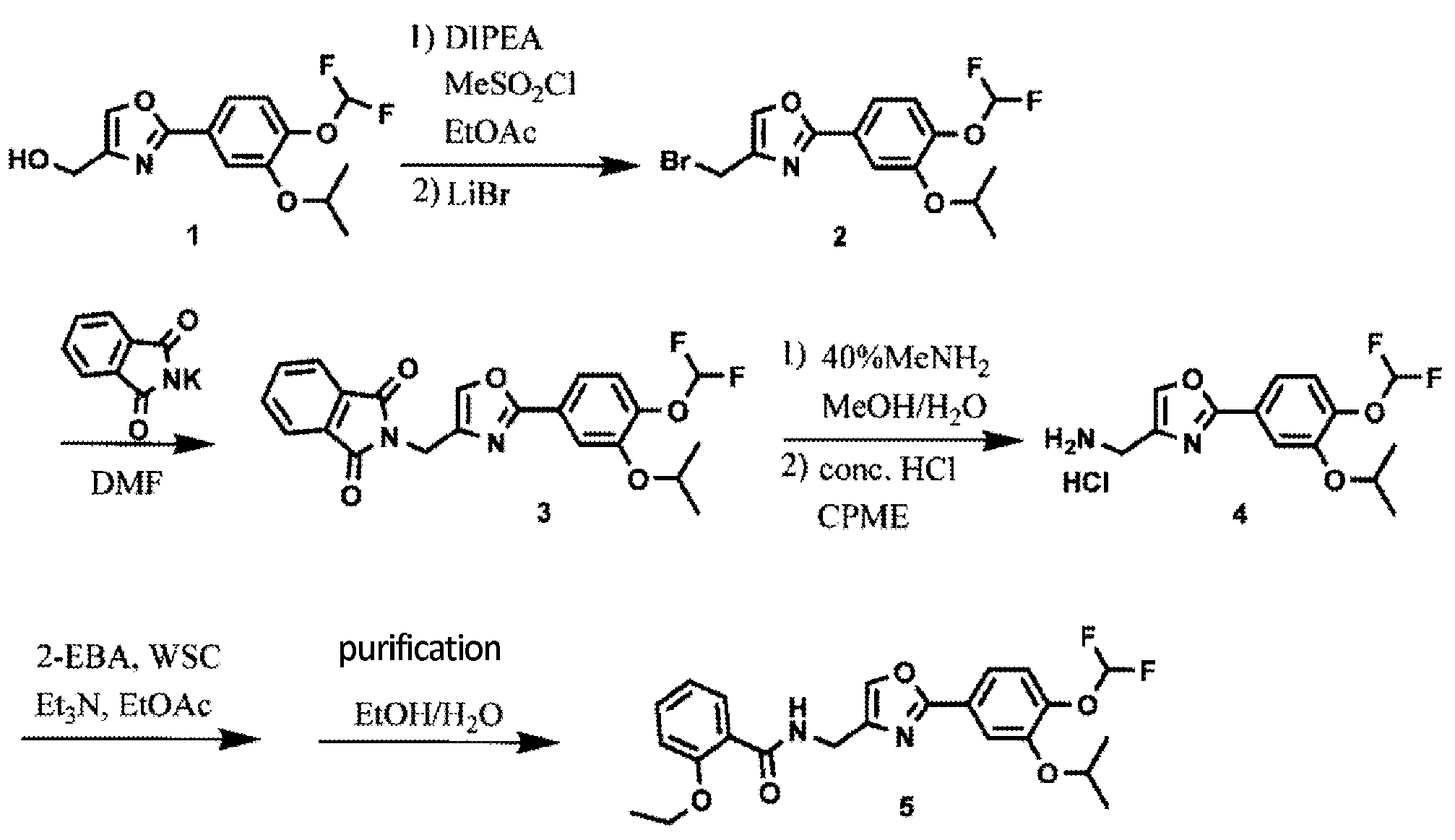
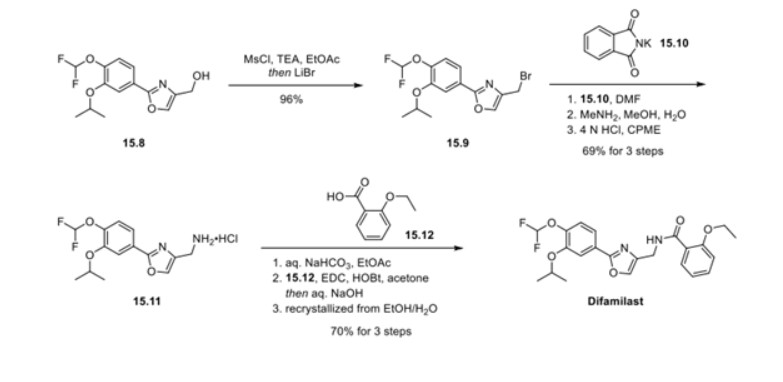
Synthesis of difamilast commenced with the monobenzylated protocatechuic acid ethyl ester 15.1. Phenol 15.1 was first converted into the corresponding isopropyl ether, which was subsequently debenzylated under palladium-catalyzed hydrogenation conditions to generate the phenolic intermediate 15.3. Difluoromethylation of 15.3 was accomplished by introducing sodium chlorodifluoroacetate 15.4 in the presence of potassium carbonate at an elevated temperature. The decarboxylative C− O bond-forming reaction presumably proceeded via a difluorocarbene species. The difluoromethylated product was treated with acid followed by ester hydrolysis under a basic medium to furnish benzoic acid derivative 15.5. Benzoic acid 15.5 was subsequently transformed into benzamide 15.6 via a benzoyl imidazole intermediate. Condensation of benzamide 15.6 with 1-acetoxy-3-chloroacetone 15.7 produced an oxazole derivative, which was subsequently saponified and recrystallized from 50% aqueous MeOH to generate alcohol 15.8.
First, an activation−displacement process transformed alcohol 15.8 into bromide 15.9 via a mesylate intermediate. Alkyl bromide 15.9 was then treated with potassium phthalimide to incorporate the nitrogen center via an SN2-type displacement. Methylamine-mediated phthalimide deprotection and subsequent salt formation produced amine 15.11 as a hydrochloride salt in 69% yield over 3 steps. Finally, hydrochloride salt 15.11 was treated with aqueous sodium bicarbonate to generate a free amine, which was subjected to amide bond formation with 2-ethoxybenzoic acid 15.12 to deliver difamilast after recrystallization from aqueous EtOH.
PATENT
JP 2021059538
https://patentscope.wipo.int/search/en/detail.jsf?docId=JP322244172&_cid=P20-L1WXG6-04592-1
patcit 2 : International Publication No. 2014/034958 (Japanese Publication No. 2015-528433 )
patcit 3 : International Publication No. 2017/115780
Compound (5) (white powder) was prepared by the method described in Example 352 of Patent Document 1 (International Publication No. 2007/088383).
N−({2−[4−(difluoromethoxy)−3−isopropoxyphenyl]oxazol−4−yl}methyl)−2−ethoxybenzamide
: white powder.
1H NMR (400 MHz, CDCl3): δ = 8.56 (br s,
1H, NH), 8.23 (dd, J = 7.6 Hz, 1.6 Hz, 1H, ArH), 7.66 (s, 1H, ArH), 7.63 (d, J = 2.0 Hz, 1H, ArH), 7.58 (dd, J = 8.4 Hz, 2.0 Hz, 1H, ArH), 7.44−7.39 (m, 1H, ArH), 7.21 (d, J = 8.0 Hz, 1H, ArH), 7.08−7.04 (m, 1H, ArH), 6.94 (d, J = 8.0 Hz, 1H, ArH), 6.61 (t, J = 75.2 Hz, 1H, CHF 2), 4.68 (sept, J = 6.0 Hz, 1H, CH), 4.62
(d, J = 6.0 Hz, 2H, CH 2), 4.17 (q, J = 6.93, 2H, CH 2), 1.48 (t, J = 7.2 Hz, 3H,
CH 3), 1.39 (d, J = 5.6 Hz, 6H, 2CH 3).
Using the obtained B-type crystal as a seed crystal, it was examined to further prepare a B-type crystal. Specifically,
B-type crystals were prepared as follows according to the method described in Patent Document 3 (International Publication No. 2017/115780).
aqueous sodium hydroxide solution were added to the organic layer, the temperature was adjusted again to 40 to 50 ° C., the liquid was separated, and the organic layer was concentrated under reduced pressure. 50 mL of ethanol, 20 mL of water, 6 mL of a 25% aqueous sodium hydroxide solution, and 0.6 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. Activated carbon was removed by filtration, washed with 12 mL of ethanol, the filtrate was cooled, and 10 mg of B-type crystals (seed crystals) were added to precipitate crystals. Precipitated crystals were collected by filtration and dried at 60 ° C. to obtain 18.38 g (yield 88.18%) of crystals of compound (5).
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007058338&_cid=P10-MLOK7P-25609-1
Example 352
Using the compound obtained in Example 347 and 2-bromopropane, white powdery N-[2-(4-difluoromethoxy-3-isopropoxyphenyl)oxazol-4-ylmethyl]-2-ethoxybenzamide was obtained following the procedure of Example 348.
1H-NMR (CDCl3) δ: 8.57 (1H, br s), 8.24 (1H, dd, J = 7.5, 1.8 Hz), 7.67 (1H, s), 7.65-7.57 (2H, m), 7.46-7.40 (1H, m), 7.26-7.21 (1H, m), 7.08 (1H, t, J = 7.5 Hz), 6.95 (1H, d, J = 8.4 Hz), 6.63 (1H, t, J = 75 Hz), 4.74-4.62 (3H, m), 4.19 (2H, q, J = 6.9 Hz), 1.49 (3H, t, J = 6.9 Hz), 1.40 (6H, d, J = 6.3 Hz)
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017115780
Compound (3) was produced in accordance with the following reaction scheme.
1H-NMR (CDCl 3) δ: 7.70 (2H,dd,J = 6.4 Hz,2.0 Hz),7.22 (1H,d,J = 9.2 Hz),6.66 (1H,t,J = 74.8 Hz),4.66(1H,sept,J = 6.0 Hz),1.39 (6H,d,J = 6.0 Hz).
Production Example 2: Production 2 of Compound (3)
Compound (3) was produced in accordance with the following reaction scheme.
Compound (7) was produced in accordance with the following reaction scheme.
Compound (11) was produced in accordance with the following reaction scheme.
20.00 g (66.8 mmol) of compound (7) and 17.28 g (134 mmol) of N,N-diisopropylethylamine were added to 300 ml of ethyl acetate, and the mixture was cooled. 11.48 g (100 mmol) of methanesulfonyl chloride was poured in and stirred at 10 to 30°C for 1 hour. 17.41 g (200 mmol) of lithium bromide was added thereto and reacted at 20 to 35°C for 1 hour. 100 ml of water was added to the reaction solution, and the mixture was partitioned, followed by concentration of the organic layer under reduced pressure. 300 ml of ethyl acetate was added to the concentrated residue to dissolve the residue, and the solution was again concentrated under reduced pressure. 200 ml of N,N-dimethylformamide and 17.33 g (93.6 mmol) of potassium phthalimide were added to the concentrated residue and reacted at 75 to 85°C for 1 hour. 200 ml of water was added to the reaction solution to precipitate crystals. The precipitated crystals were collected by filtration and dried at 80°C, thereby obtaining 25.90 g (yield: 90.5%) of compound (9) as a white powder.
15.00 g (35.0 mmol) of compound (9) was mixed with 30 ml of a 40% methylamine aqueous solution, 30 ml of methanol, and 75 ml of water, and reacted under reflux for 30 minutes. 150 ml of cyclopentyl methyl ether (CPME) and 15 ml of a 25% sodium hydroxide aqueous solution were added to the reaction solution, and the temperature was adjusted to 65 to 75°C, followed by partitioning. A mixture of 150 ml of water and 7.50 g of sodium chloride was added to the organic layer, and the temperature was adjusted to 65 to 75°C again, followed by partitioning. 3.75 ml of concentrated hydrochloric acid was added to the organic layer to precipitate crystals. The precipitated crystals were collected by filtration and dried at 60°C, thereby obtaining 11.95 g (yield: quant.) of compound (10) as a white powder.
13.30 g (39.7 mmol) of compound (10) was mixed with 3.83 g (37.8 mmol) of triethylamine and 108 ml of ethyl acetate, and stirred at 20 to 30°C for 1 hour. 9.78 g (58.9 mmol) of 2-ethoxybenzoic acid and 11.28 g (58.8 mmol) of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (WSC) were added to the reaction solution, and reacted at 20 to 30°C for 1 hour. 54 ml of water and 5.4 ml of concentrated hydrochloric acid were added to the reaction solution, and the temperature was adjusted to 40 to 50°C, followed by partitioning. 54 ml of water and 5.4 ml of a 25% sodium hydroxide aqueous solution were added to the organic layer, and the temperature was adjusted to 40 to 50°C again. The mixture was partitioned, and the organic layer was concentrated under reduced pressure. 45 ml of ethanol, 18 ml of water, 5.4 ml of a 25% sodium hydroxide aqueous solution, and 0.54 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. The activated carbon was removed by filtration, and the filtrate was washed with 11 ml of ethanol. The filtrate was cooled, and a seed crystal was added thereto to precipitate crystals. The precipitated crystals were collected by filtration and dried at 35°C, thereby obtaining 12.88 g (72.6%) of compound (11) as a white powder.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019194211
*DIPEA: Diisopropylethylamine, CPME: Cyclopentyl methyl ether,
DMF: N,N-dimethylformamide, 2-EBA: 2-Ethoxybenzoic acid,
WSC: 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
Analysis was conducted to further prepare the type B crystal using the obtained type B crystal as a seed crystal. More specifically, the type B crystal was prepared as follows, in accordance with the method disclosed in PTL 3 (WO2017/115780).
PATENT
WO2014034958A1
WO2007058338A2
WO2007058338A9
WO2007058338A3
US9181205B2
US2015239855A1
USRE46792E
US2020078340A1
US2017216260A1
US2019070151A1
US2009221586A1
US8637559B2
US2014100226A1
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
/////////////Difamilast, JAPAN 2021, APPROVALS 2021, ジファミラスト , MM 36, MM-36-Medimetriks-Pharmaceuticals, Moizerto, OPA-15406, OPA 15406, 地法米司特
O=C(NCC1=COC(C2=CC=C(OC(F)F)C(OC(C)C)=C2)=N1)C3=CC=CC=C3OCC
INTrmediate No.CAS No.DIFAM-001177429-27-5DIFAM-00293652-48-3DIFAM-0031574285-26-9DIFAM-00470-23-5DIFAM-0051574285-28-1DIFAM-0061574285-30-5DIFAM-0071574285-32-7DIFAM-0081574285-36-1DIFAM-0091574285-38-3DIFAM-010DIFAM-0111574285-40-7DIFAM-0121574285-43-0DIFAM-013134-11-2Difamilast937782-05-3
Tixagevimab
| (Heavy chain) QMQLVQSGPE VKKPGTSVKV SCKASGFTFM SSAVQWVRQA RGQRLEWIGW IVIGSGNTNY AQKFQERVTI TRDMSTSTAY MELSSLRSED TAVYYCAAPY CSSISCNDGF DIWGQGTMVT VSSASTKGPS VFPLAPSSKS TSGGTAALGC LVKDYFPEPV TVSWNSGALT SGVHTFPAVL QSSGLYSLSS VVTVPSSSLG TQTYICNVNH KPSNTKVDKR VEPKSCDKTH TCPPCPAPEF EGGPSVFLFP PKPKDTLYIT REPEVTCVVV DVSHEDPEVK FNWYVDGVEV HNAKTKPREE QYNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKALPASIEK TISKAKGQPR EPQVYTLPPS REEMTKNQVS LTCLVKGFYP SDIAVEWESN GQPENNYKTT PPVLDSDGSF FLYSKLTVDK SRWQQGNVFS CSVMHEALHN HYTQKSLSLS PGK (Light chain) EIVLTQSPGT LSLSPGERAT LSCRASQSVS SSYLAWYQQK PGQAPRLLIY GASSRATGIP DRFSGSGSGT DFTLTISRLE PEDFAVYYCQ HYGSSRGWTF GQGTKVEIKR TVAAPSVFIF PPSDEQLKSG TASVVCLLNN FYPREAKVQW KVDNALQSGN SQESVTEQDS KDSTYSLSST LTLSKADYEK HKVYACEVTH QGLSSPVTKS FNRGEC (Disulfide bridge: H22-H96, H101-H106, H150-H206, H216-L216, H232-H’232, H235-H’235, H267-H327, H373-H431, H’22-H’96, H’101-H’106, H’150-H’206, H’226-L’216, H’267-H’327, H’373-H’431, L23-L89, L136-L196, L’23-L’89, L’136-L’196) |
Tixagevimab
FDA 2021, 2021/12/8
ANTI VIRAL, CORONA VIRUS, PEPTIDE
Monoclonal antibody
Treatment and prevention of SARS-CoV-2 infection
| Formula | C6488H10034N1746O2038S50 |
|---|---|
| CAS | 2420564-02-7 |
| Mol weight | 146704.817 |
- 2196
- AZD-8895
- AZD8895
- COV2-2196
- Tixagevimab
- Tixagevimab [INN]
- UNII-F0LZ415Z3B
- WHO 11776
- OriginatorVanderbilt University
- DeveloperAstraZeneca; INSERM; National Institute of Allergy and Infectious Diseases
- ClassAntivirals; Monoclonal antibodies
- Mechanism of ActionVirus internalisation inhibitors
- RegisteredCOVID 2019 infections
- 24 Dec 2021Pharmacodynamics data from a preclinical trial in COVID-2019 infections released by AstraZeneca
- 16 Dec 2021Pharmacodynamics data from a preclinical trial in COVID-2019 infections released by AstraZeneca
- 10 Dec 2021Registered for COVID-2019 infections (In the elderly, Prevention, In adults) in USA (IM) – Emergency Use Authorization
Tixagevimab/cilgavimab is a combination of two human monoclonal antibodies, tixagevimab (AZD8895) and cilgavimab (AZD1061) targeted against the surface spike protein of SARS-CoV-2[4][5] used to prevent COVID-19. It is being developed by British-Swedish multinational pharmaceutical and biotechnology company AstraZeneca.[6][7] It is co-packaged and given as two separate consecutive intramuscular injections (one injection per monoclonal antibody, given in immediate succession).[2]
/////////////////////////////////////////////////////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Development
In 2020, researchers at Vanderbilt University Medical Center discovered particularly potent monoclonal antibodies, isolated from COVID-19 patients infected with a SARS-CoV-2 circulating at that time. Initially designated COV2-2196 and COV2-2130, antibody engineering was used to transfer their SARS-CoV-2 binding specificity to IgG scaffolds that would last longer in the body, and these engineered antibodies were named AZD8895 and AZD1061, respectively (and the combination was called AZD7442).[8]
To evaluate the antibodies’ potential as monoclonal antibody based prophylaxis (prevention), the ‘Provent’ clinical trial enrolled 5,000 high risk but not yet infected individuals and monitored them for 15 months.[9][10] The trial reported that those receiving the cocktail showed a 77% reduction in symptomatic COVID-19 and that there were no severe cases or deaths. AstraZeneca also found that the antibody cocktail “neutralizes recent emergent SARS-CoV-2 viral variants, including the Delta variant“.[7]
In contrast to pre-exposure prophylaxis, the Storm Chaser study of already-exposed people (post-exposure prophylaxis) did not meet its primary endpoint, which was prevention of symptomatic COVID-19 in people already exposed. AZD7442 was administered to 1,000 volunteers who had recently been exposed to COVID.[9]
Regulatory review
In October 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) started a rolling review of tixagevimab/cilgavimab, which is being developed by AstraZeneca AB, for the prevention of COVID-19 in adults.[11]
Also in October 2021, AstraZeneca requested Emergency Use Authorization for tixagevimab/cilgavimab to prevent COVID-19 from the U.S. Food and Drug Administration (FDA).[12][13]
Emergency use authorization
On 14 November 2021, Bahrain granted emergency use authorization.[14]
On 8 December 2021, the U.S. Food and Drug Administration (FDA) granted emergency use authorization of this combination to prevent COVID-19 (before exposure) in people with weakened immunity or who cannot be fully vaccinated due to a history of severe reaction to coronavirus vaccines.[15] The FDA issued an emergency use authorization (EUA) for AstraZeneca’s Evusheld (tixagevimab co-packaged with cilgavimab and administered together) for the pre-exposure prophylaxis (prevention) of COVID-19 in certain people aged 12 years of age and older weighing at least 40 kilograms (88 lb).[2] The product is only authorized for those individuals who are not currently infected with the SARS-CoV-2 virus and who have not recently been exposed to an individual infected with SARS-CoV-2.[2]
References
- ^ “Evusheld- azd7442 kit”. DailyMed. Retrieved 4 January 2022.
- ^ Jump up to:a b c d “Coronavirus (COVID-19) Update: FDA Authorizes New Long-Acting Monoclonal Antibodies for Pre-exposure Prevention of COVID-19 in Certain Individuals”. U.S. Food and Drug Administration (FDA) (Press release). 8 December 2021. Retrieved 9 December 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ O’Shaughnessy, Jacqueline A. (20 December 2021). “Re: Emergency Use Authorization 104” (PDF). Food and Drug Administration. Letter to AstraZeneca Pharmaceuticals LP | Attention: Stacey Cromer Berman, PhD. Archived from the original on 29 December 2021. Retrieved 18 January 2022.
- ^ “IUPHAR/BPS Guide to PHARMACOLOGY”. IUPHAR. 27 December 2021. Retrieved 27 December 2021.
- ^ “IUPHAR/BPS Guide to PHARMACOLOGY”. IUPHAR. 27 December 2021. Retrieved 27 December 2021.
- ^ Ray, Siladitya (21 August 2021). “AstraZeneca’s Covid-19 Antibody Therapy Effective In Preventing Symptoms Among High-Risk Groups, Trial Finds”. Forbes. ISSN 0015-6914. Archived from the original on 21 August 2021. Retrieved 18 January 2022.
- ^ Jump up to:a b Goriainoff, Anthony O. (20 August 2021). “AstraZeneca Says AZD7442 Antibody Phase 3 Trial Met Primary Endpoint in Preventing Covid-19”. MarketWatch. Archived from the original on 21 August 2021. Retrieved 18 January 2022.
- ^ Dong J, Zost SJ, Greaney AJ, Starr TN, Dingens AS, Chen EC, et al. (October 2021). “Genetic and structural basis for SARS-CoV-2 variant neutralization by a two-antibody cocktail”. Nature Microbiology. 6 (10): 1233–1244. doi:10.1038/s41564-021-00972-2. ISSN 2058-5276. PMC 8543371. PMID 34548634.
- ^ Jump up to:a b Haridy, Rich (23 August 2021). “”Game-changing” antibody cocktail prevents COVID-19 in the chronically ill”. New Atlas. Retrieved 23 August 2021.
- ^ “AZD7442 PROVENT Phase III prophylaxis trial met primary endpoint in preventing COVID-19”. AstraZeneca (Press release). 20 August 2021. Retrieved 15 October 2021.
- ^ “EMA starts rolling review of Evusheld (tixagevimab and cilgavimab)”. European Medicines Agency. 14 October 2021. Retrieved 15 October 2021.
- ^ “AZD7442 request for Emergency Use Authorization for COVID-19 prophylaxis filed in US”. AstraZeneca US (Press release). 5 October 2021. Retrieved 15 October 2021.
- ^ “AZD7442 request for Emergency Use Authorization for COVID-19 prophylaxis filed in US”. AstraZeneca (Press release). 5 October 2021. Retrieved 15 October 2021.
- ^ Abd-Alaziz, Moaz; Elhamy, Ahmad (14 November 2021). Macfie, Nick (ed.). “Bahrain authorizes AstraZeneca’s anti-COVID drug for emergency use”. Reuters. Archived from the original on 23 November 2021. Retrieved 18 January 2022.
- ^ Mishra, Manas; Satija, Bhanvi (8 December 2021). Dasgupta, Shounak (ed.). “U.S. FDA authorizes use of AstraZeneca COVID-19 antibody cocktail”. Reuters. Archived from the original on 13 January 2022. Retrieved 18 January 2022.
External links
“Tixagevimab”. Drug Information Portal. U.S. National Library of Medicine.
- “Cilgavimab”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT04625972 for “Phase III Double-blind, Placebo-controlled Study of AZD7442 for Post-exposure Prophylaxis of COVID-19 in Adults (STORM CHASER)” at ClinicalTrials.gov
- Clinical trial number NCT04625725 for “Phase III Double-blind, Placebo-controlled Study of AZD7442 for Pre-exposure Prophylaxis of COVID-19 in Adult. (PROVENT)” at ClinicalTrials.gov
| Tixagevimab (teal, right) and cilgavimab (purple, left) binding the spike protein RBD. From PDB: 7L7E. | |
| Combination of | |
|---|---|
| Tixagevimab | Monoclonal antibody |
| Cilgavimab | Monoclonal antibody |
| Clinical data | |
| Trade names | Evusheld |
| Other names | AZD7442 |
| License data | US DailyMed: Tixagevimab |
| Routes of administration | Intramuscular |
| ATC code | J06BD03 (WHO) |
| Legal status | |
| Legal status | US: ℞-only via emergency use authorization[1][2][3] |
| Identifiers | |
| KEGG | D12262 |
| Clinical data | |
|---|---|
| Drug class | Antiviral |
| ATC code | None |
| Identifiers | |
| CAS Number | 2420564-02-7 |
| DrugBank | DB16394 |
| UNII | F0LZ415Z3B |
| KEGG | D11993 |
| Chemical and physical data | |
| Formula | C6488H10034N1746O2038S50 |
| Molar mass | 146706.82 g·mol−1 |
| Clinical data | |
|---|---|
| Drug class | Antiviral |
| ATC code | None |
| Identifiers | |
| CAS Number | 2420563-99-9 |
| DrugBank | DB16393 |
| UNII | 1KUR4BN70F |
| KEGG | D11994 |
| Chemical and physical data | |
| Formula | C6626H10218N1750O2078S44 |
| Molar mass | 149053.44 g·mol−1 |
/////////////////Tixagevimab, ANTI VIRAL, CORONA VIRUS, PEPTIDE, Monoclonal antibody, SARS-CoV-2 , WHO 11776, 2196, AZD-8895, AZD 8895, COV2-2196, COVID 19
NEW DRUG APPROVALS
ONE TIME
$10.00
Tezepelumab-ekko

(Heavy chain)
QMQLVESGGG VVQPGRSLRL SCAASGFTFR TYGMHWVRQA PGKGLEWVAV IWYDGSNKHY
ADSVKGRFTI TRDNSKNTLN LQMNSLRAED TAVYYCARAP QWELVHEAFD IWGQGTMVTV
SSASTKGPSV FPLAPCSRST SESTAALGCL VKDYFPEPVT VSWNSGALTS GVHTFPAVLQ
SSGLYSLSSV VTVPSSNFGT QTYTCNVDHK PSNTKVDKTV ERKCCVECPP CPAPPVAGPS
VFLFPPKPKD TLMISRTPEV TCVVVDVSHE DPEVQFNWYV DGVEVHNAKT KPREEQFNST
FRVVSVLTVV HQDWLNGKEY KCKVSNKGLP APIEKTISKT KGQPREPQVY TLPPSREEMT
KNQVSLTCLV KGFYPSDIAV EWESNGQPEN NYKTTPPMLD SDGSFFLYSK LTVDKSRWQQ
GNVFSCSVMH EALHNHYTQK SLSLSPGK
(Light chain)
SYVLTQPPSV SVAPGQTARI TCGGNNLGSK SVHWYQQKPG QAPVLVVYDD SDRPSWIPER
FSGSNSGNTA TLTISRGEAG DEADYYCQVW DSSSDHVVFG GGTKLTVLGQ PKAAPSVTLF
PPSSEELQAN KATLVCLISD FYPGAVTVAW KADSSPVKAG VETTTPSKQS NNKYAASSYL
SLTPEQWKSH RSYSCQVTHE GSTVEKTVAP TECS
(Disulfide bridge: H22-H96, H136-L213, H149-H205, H224-H’224, H225-H’225, H228-H’228, H231-H’231, H262-H322, H368-H426, H’22-H’96, H’136-L’213, H’149-H’205, H’262-H’322, H’368-H’426, L22-L87, L136-L195, L’22-L’87, L’136-L’195)
Tezepelumab-ekko
テゼペルマブ (遺伝子組換え)
| Formula | C6400H9844N1732O1992S52 |
|---|---|
| CAS | 1572943-04-4 |
| Mol weight | 144588.4306 |
PEPTIDE
UD FDA APPROVED, 12/17/2021, To treat severe asthma as an add-on maintenance therapy , Tezspire
Monoclonal antibody
Treatment of asthma and atopic dermatitis
Tezepelumab, sold under the brand name Tezspire, is a human monoclonal antibody used for the treatment of asthma.[4][5]
It blocks thymic stromal lymphopoietin (TSLP),[2] an epithelial cytokine that has been suggested to be critical in the initiation and persistence of airway inflammation.[6]
It was approved for medical use in the United States in December 2021.[2][3]
Medical uses
Tezepelumab is indicated for the add-on maintenance treatment of people aged twelve years and older with severe asthma.[2]
Research
In Phase III trials, tezepelumab demonstrated efficacy compared to placebo for patients with severe, uncontrolled asthma.[7][8]
Structural studies by X-ray crystallography showed that Tezepelumab competes against a critical part of the TSLPR binding site on TSLP.[1]
It is being studied for the treatment of chronic obstructive pulmonary disease, chronic rhinosinusitis with nasal polyps, chronic spontaneous urticaria and eosinophilic esophagitis (EoE).[3]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Jump up to:a b Verstraete K, Peelman F, Braun H, Lopez J, Van Rompaey D, Dansercoer A, et al. (April 2017). “Structure and antagonism of the receptor complex mediated by human TSLP in allergy and asthma”. Nature Communications. 8 (1): 14937. Bibcode:2017NatCo…814937V. doi:10.1038/ncomms14937. PMC 5382266. PMID 28368013.
- ^ Jump up to:a b c d https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761224s000lbl.pdf
- ^ Jump up to:a b c “Tezspire (tezepelumab) approved in the US for severe asthma”. AstraZeneca (Press release). 17 December 2021. Retrieved 17 December 2021.
- ^ Marone G, Spadaro G, Braile M, Poto R, Criscuolo G, Pahima H, et al. (November 2019). “Tezepelumab: a novel biological therapy for the treatment of severe uncontrolled asthma”. Expert Opinion on Investigational Drugs. 28 (11): 931–940. doi:10.1080/13543784.2019.1672657. PMID 31549891. S2CID 202746054.
- ^ Matera MG, Rogliani P, Calzetta L, Cazzola M (February 2020). “TSLP Inhibitors for Asthma: Current Status and Future Prospects”. Drugs. 80 (5): 449–458. doi:10.1007/s40265-020-01273-4. PMID 32078149. S2CID 211194472.
- ^ “Tezepelumab granted Breakthrough Therapy Designation by US FDA”. AstraZeneca (Press release). 7 September 2018.
- ^ “Studies found for: Tezepelumab”. ClinicalTrials.Gov. National Library of Medicine, National Institutes of Health, U.S. Department of Health and Human Services.
- ^ Menzies-Gow A, Corren J, Bourdin A, Chupp G, Israel E, Wechsler ME, et al. (May 2021). “Tezepelumab in Adults and Adolescents with Severe, Uncontrolled Asthma”. New England Journal of Medicine. 384 (19): 1800–09. doi:10.1056/NEJMoa2034975. PMID 33979488. S2CID 234484931.
External links
- “Tezepelumab”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02054130 for “Study to Evaluate the Efficacy and Safety of MEDI9929 (AMG 157) in Adult Subjects With Inadequately Controlled, Severe Asthma” at ClinicalTrials.gov
- Clinical trial number NCT03347279 for “Study to Evaluate Tezepelumab in Adults & Adolescents With Severe Uncontrolled Asthma (NAVIGATOR)” at ClinicalTrials.gov
| Structural basis for inhibition of TSLP-signaling by Tezepelumab (PDB 5J13)[1] | |
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | thymic stromal lymphopoietin (TSLP) |
| Clinical data | |
| Trade names | Tezspire |
| Other names | MEDI9929, AMG 157, tezepelumab-ekko |
| License data | US DailyMed: Tezepelumab |
| Routes of administration | Subcutaneous |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [2][3] |
| Identifiers | |
| CAS Number | 1572943-04-4 |
| DrugBank | DB15090 |
| ChemSpider | None |
| UNII | RJ1IW3B4QX |
| KEGG | D11771 |
| Chemical and physical data | |
| Formula | C6400H9844N1732O1992S52 |
| Molar mass | 144590.40 g·mol−1 |
////////////Tezepelumab-ekko, Tezspire, PEPTIDE, APPROVALS 2021, FDA 2021, Monoclonal antibody
, asthma, atopic dermatitis, ANTI INFLAMATORY, テゼペルマブ (遺伝子組換え)

NEW DRUG APPROVALS
ONE TIME
$10.00
Efgartigimod alfa-fcab
DKTHTCPPCP APELLGGPSV FLFPPKPKDT LYITREPEVT CVVVDVSHED PEVKFNWYVD
GVEVHNAKTK PREEQYNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKALPA PIEKTISKAK
GQPREPQVYT LPPSRDELTK NQVSLTCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS
DGSFFLYSKL TVDKSRWQQG NVFSCSVMHE ALKFHYTQKS LSLSPGK
(Disulfide bridge: 6-6′, 9-9′, 41-101, 147-205, 41′-101′, 147′-205′)
Efgartigimod alfa-fcab
| Formula | C2310H3554N602O692S14 |
|---|---|
| CAS | 1821402-21-4 |
| Mol weight | 51279.464 |
US FDA APPROVED 12/17/2021, To treat generalized myasthenia gravis
Press Release, Vyvgart, BLA 761195
| エフガルチギモドアルファ (遺伝子組換え) |
PEPTIDE
Treatment of IgG-driven autoimmune diseases
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-myasthenia-gravis
FDA Approves New Treatment for Myasthenia Gravis
Approval is the First of a New Class of Medication for this Rare, Chronic, Autoimmune, Neuromuscular DiseaseFor Immediate Release:December 17, 2021
The U.S. Food and Drug Administration today approved Vyvgart (efgartigimod) for the treatment of generalized myasthenia gravis (gMG) in adults who test positive for the anti-acetylcholine receptor (AChR) antibody.
Myasthenia gravis is a chronic autoimmune, neuromuscular disease that causes weakness in the skeletal muscles (also called voluntary muscles) that worsens after periods of activity and improves after periods of rest. Myasthenia gravis affects voluntary muscles, especially those that are responsible for controlling the eyes, face, mouth, throat, and limbs. In myasthenia gravis, the immune system produces AChR antibodies that interfere with communication between nerves and muscles, resulting in weakness. Severe attacks of weakness can cause breathing and swallowing problems that can be life-threatening.
“There are significant unmet medical needs for people living with myasthenia gravis, as with many other rare diseases,” said Billy Dunn, M.D., director of the Office of Neuroscience in the FDA’s Center for Drug Evaluation and Research. “Today’s approval is an important step in providing a novel therapy option for patients and underscores the agency’s commitment to help make new treatment options available for people living with rare diseases.”
Vyvgart is the first approval of a new class of medication. It is an antibody fragment that binds to the neonatal Fc receptor (FcRn), preventing FcRn from recycling immunoglobulin G (IgG) back into the blood. The medication causes a reduction in overall levels of IgG, including the abnormal AChR antibodies that are present in myasthenia gravis.
The safety and efficacy of Vyvgart were evaluated in a 26-week clinical study of 167 patients with myasthenia gravis who were randomized to receive either Vyvgart or placebo. The study showed that more patients with myasthenia gravis with antibodies responded to treatment during the first cycle of Vyvgart (68%) compared to those who received placebo (30%) on a measure that assesses the impact of myasthenia gravis on daily function. More patients receiving Vyvgart also demonstrated response on a measure of muscle weakness compared to placebo.
The most common side effects associated with the use of Vyvgart include respiratory tract infections, headache, and urinary tract infections. As Vyvgart causes a reduction in IgG levels, the risk of infections may increase. Hypersensitivity reactions such as eyelid swelling, shortness of breath, and rash have occurred. If a hypersensitivity reaction occurs, discontinue the infusion and institute appropriate therapy. Patients using Vyvgart should monitor for signs and symptoms of infections during treatment. Health care professionals should administer appropriate treatment and consider delaying administration of Vyvgart to patients with an active infection until the infection is resolved.
The FDA granted this application Fast Track and Orphan Drug designations. The FDA granted the approval of Vyvgart to argenx BV.
///////////efgartigimod alfa-fcab, Vyvgart, FDA 2021,APPROVALS 2021, myasthenia gravis, argenx BV, Fast Track, Orphan Drug, PEPTIDE,
| エフガルチギモドアルファ (遺伝子組換え) |

NEW DRUG APPROVALS
one time
$10.00
Regdanvimab

| (Heavy chain) QITLKESGPT LVKPTQTLTL TCSFSGFSLS TSGVGVGWIR QPPGKALEWL ALIDWDDNKY HTTSLKTRLT ISKDTSKNQV VLTMTNMDPV DTATYYCARI PGFLRYRNRY YYYGMDVWGQ GTTVTVSSAS TKGPSVFPLA PSSKSTSGGT AALGCLVKDY FPEPVTVSWN SGALTSGVHT FPAVLQSSGL YSLSSVVTVP SSSLGTQTYI CNVNHKPSNT KVDKRVEPKS CDKTHTCPPC PAPELLGGPS VFLFPPKPKD TLMISRTPEV TCVVVDVSHE DPEVKFNWYV DGVEVHNAKT KPREEQYNST YRVVSVLTVL HQDWLNGKEY KCKVSNKALP APIEKTISKA KGQPREPQVY TLPPSRDELT KNQVSLTCLV KGFYPSDIAV EWESNGQPEN NYKTTPPVLD SDGSFFLYSK LTVDKSRWQQ GNVFSCSVMH EALHNHYTQK SLSLSPGK (Light chain) ELVLTQPPSV SAAPGQKVTI SCSGSSSNIG NNYVSWYQQL PGTAPKLLIY DNNKRPSGIP DRFSGSKSGT SATLGITGLQ TGDEADYYCG TWDSSLSAGV FGGGTELTVL GQPKAAPSVT LFPPSSEELQ ANKATLVCLI SDFYPGAVTV AWKADGSPVK AGVETTKPSK QSNNKYAASS YLSLTPEQWK SHRSYSCQVT HEGSTVEKTV APTECS (Disulfide bridge: H22-H97, H155-H211, H231-L215, H237-H’237, H240-H’240, H272-H332, H378-H436, H’22-H’97, H’155-H’211, H’231-L’215, H’272-H’332, H’378-H’436, L22-L89, L138-L197, L’22-L’89, L’138-L’197) |
>Regdanvimab light chain: ELVLTQPPSVSAAPGQKVTISCSGSSSNIGNNYVSWYQQLPGTAPKLLIYDNNKRPSGIP DRFSGSKSGTSATLGITGLQTGDEADYYCGTWDSSLSAGVFGGGTELTVLGQPKAAPSVT LFPPSSEELQANKATLVCLISDFYPGAVTVAWKADGSPVKAGVETTKPSKQSNNKYAASS YLSLTPEQWKSHRSYSCQVTHEGSTVEKTVAPTECS
>Regdanvimab heavy chain: QITLKESGPTLVKPTQTLTLTCSFSGFSLSTSGVGVGWIRQPPGKALEWLALIDWDDNKY HTTSLKTRLTISKDTSKNQVVLTMTNMDPVDTATYYCARIPGFLRYRNRYYYYGMDVWGQ GTTVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHT FPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHTCPPC PAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKT KPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVY TLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSK LTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
Regdanvimab
レグダンビマブ;
EMA APPROVED, 2021/11/12, Regkirona
Treatment of adults with coronavirus disease 2019 (COVID-19)
MONOCLONAL ANTIBODY, ANTI VIRAL, PEPTIDE
CAS: 2444308-95-4, CT-P59
Regdanvimab, sold under the brand name Regkirona, is a human monoclonal antibody used for the treatment of COVID-19.[1] The antibody is directed against the spike protein of SARS-CoV-2. It is developed by Celltrion.[2][3] The medicine is given by infusion (drip) into a vein.[1][4]
The most common side effects include infusion-related reactions, including allergic reactions and anaphylaxis.[1]
Regdanvimab was approved for medical use in the European Union in November 2021.[1]
Regdanvimab is a monoclonal antibody targeted against the SARS-CoV-2 spike protein used to treat patients with COVID-19 who are at risk of progressing to severe COVID-19.
Regdanvimab (CT-P59) is a recombinant human IgG1 monoclonal antibody directed at the receptor binding domain (RBD) of the SARS-CoV-2 spike protein.4 It blocks the interaction between viral spike proteins and angiotensin-converting enzyme 2 (ACE2) that allows for viral entry into the cell, thereby inhibiting the virus’ ability to replicate. Trials investigating the use of regdanvimab as a therapeutic candidate for the treatment of COVID-19 began in mid-2020.1,3 It received its first full approval in South Korea in September 2021,3 followed by the EU in November 2021.5
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Synthesis Reference
Kim C, Ryu DK, Lee J, Kim YI, Seo JM, Kim YG, Jeong JH, Kim M, Kim JI, Kim P, Bae JS, Shim EY, Lee MS, Kim MS, Noh H, Park GS, Park JS, Son D, An Y, Lee JN, Kwon KS, Lee JY, Lee H, Yang JS, Kim KC, Kim SS, Woo HM, Kim JW, Park MS, Yu KM, Kim SM, Kim EH, Park SJ, Jeong ST, Yu CH, Song Y, Gu SH, Oh H, Koo BS, Hong JJ, Ryu CM, Park WB, Oh MD, Choi YK, Lee SY: A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat Commun. 2021 Jan 12;12(1):288. doi: 10.1038/s41467-020-20602-5.
Celltrion’s Monoclonal Antibody Treatment regdanvimab, Approved by the European Commission for the Treatment of COVID-19
- The European Commission (EC) granted marketing authorisation for Celltrion’s regdanvimab following positive opinion by the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) last week (11/11/2021)
- Celltrion continues to discuss supply agreements with regulatory agencies and contractors in more than 30 countries in Europe, Asia and LATAM to accelerate global access to regdanvimab
- The use of regdanvimab across the Republic of Korea is rapidly increasing to address the ongoing outbreaks
November 14, 2021 08:04 PM Eastern Standard Time
INCHEON, South Korea–(BUSINESS WIRE)–Celltrion Group announced today that the European Commission (EC) has approved Regkirona (regdanvimab, CT-P59), one of the first monoclonal antibody treatments granted marketing authorisation from the European Medicines Agency (EMA). The EC granted marketing authorisation for adults with COVID-19 who do not require supplemental oxygen and who are at increased risk of progressing to severe COVID-19. The decision from the EC follows a positive opinion by the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) on November 11th, 2021.1
“Today’s achievement, coupled with CHMP positive opinion for regdanvimab, underscores our ongoing commitment to addressing the world’s greatest health challenges,” said Dr. HoUng Kim, Ph.D., Head of Medical and Marketing Division at Celltrion Healthcare. “Typically, the recommendations from the CHMP are passed on to the EC for rapid legally binding decisions within a month or two, however, given the unprecedented times, we have received the EC approval within a day. As part of our global efforts to accelerate access, we have been communicating with the governments and contractors in 30 countries in Europe, Asia and LATAM. We will continue working with all key stakeholders to ensure COVID-19 patients around the world have access to safe and effective treatments.”
Monoclonal antibodies are proteins designed to attach to a specific target, in this case the spike protein of SARS-CoV-2, which works to block the path the virus uses to enter human cells. The EC approval is based on the global Phase III clinical trial involving more than 1,315 people to evaluate the efficacy and safety of regdanvimab in 13 countries including the U.S., Spain, and Romania. Data showed regdanvimab significantly reduced the risk of COVID-19 related hospitalisation or death by 72% for patients at high-risk of progressing to severe COVID-19.
Emergency use authorisations are currently in place in Indonesia and Brazil, and the monoclonal antibody treatment is fully approved in the Republic of Korea. In the U.S., regdanvimab has not yet been approved by the Food and Drug Administration (FDA), but the company is in discussion with the FDA to submit applications for an Emergency Use Authorisation (EUA).
As of November 12th, 2021, more than 22,587 people have been treated with regdanvimab in 129 hospitals in the Republic of Korea.
Notes to Editors:
About Celltrion Healthcare
Celltrion Healthcare is committed to delivering innovative and affordable medications to promote patients’ access to advanced therapies. Its products are manufactured at state-of-the-art mammalian cell culture facilities, designed and built to comply with the US FDA cGMP and the EU GMP guidelines. Celltrion Healthcare endeavours to offer high-quality cost-effective solutions through an extensive global network that spans more than 110 different countries. For more information please visit: https://www.celltrionhealthcare.com/en-us.
About regdanvimab (CT-P59)
CT-P59 was identified as a potential treatment for COVID-19 through screening of antibody candidates and selecting those that showed the highest potency in neutralising the SARS-CoV-2 virus. In vitro and in vivo pre- clinical studies showed that CT-P59 strongly binds to SARS-CoV-2 RBD and significantly neutralise the wild type and mutant variants of concern. In in vivo models, CT-P59 effectively reduced the viral load of SARS-CoV-2 and inflammation in lung. Results from the global Phase I and Phase II/III clinical trials of CT-P59 demonstrated a promising safety, tolerability, antiviral effect and efficacy profile in patients with mild-to-moderate symptoms of COVID-19.2 Celltrion also has recently commenced the development of a neutralising antibody cocktail with CT-P59 against new emerging variants of SARS-CoV-2.
Medical uses
In the European Union, regdanvimab is indicated for the treatment of adults with COVID-19 who do not require supplemental oxygen and who are at increased risk of progressing to severe COVID-19.[1]
Society and culture
Names
Regdanvimab is the proposed international nonproprietary name (pINN).[5]
Legal status
In March 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) started a rolling review of data on regdanvimab.[6][7] In October 2021, the EMA started evaluating an application for marketing authorization for the monoclonal antibody regdanvimab (Regkirona) to treat adults with COVID-19 who do not require supplemental oxygen therapy and who are at increased risk of progressing to severe COVID 19.[8] The applicant is Celltrion Healthcare Hungary Kft.[8] The European Medicines Agency (EMA) concluded that regdanvimab can be used for the treatment of confirmed COVID-19 in adults who do not require supplemental oxygen therapy and who are at high risk of progressing to severe COVID-19.[4]
In November 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) recommended granting a marketing authorization in the European Union for regdanvimab (Regkirona) for the treatment of COVID-19.[9][10] The company that applied for authorization of Regkirona is Celltrion Healthcare Hungary Kft.[10] Regdanvimab was approved for medical use in the European Union in November 2021.[1]
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Spike protein of SARS-CoV-2 |
| Clinical data | |
| Trade names | Regkirona |
| Other names | CT-P59 |
| License data | EU EMA: by INN |
| Routes of administration | Intravenous infusion |
| ATC code | None |
| Legal status | |
| Legal status | EU: Rx-only [1] |
| Identifiers | |
| CAS Number | 2444308-95-4 |
| DrugBank | DB16405 |
| UNII | I0BGE6P6I6 |
| KEGG | D12241 |
- Tuccori M, Ferraro S, Convertino I, Cappello E, Valdiserra G, Blandizzi C, Maggi F, Focosi D: Anti-SARS-CoV-2 neutralizing monoclonal antibodies: clinical pipeline. MAbs. 2020 Jan-Dec;12(1):1854149. doi: 10.1080/19420862.2020.1854149. [Article]
- Kim C, Ryu DK, Lee J, Kim YI, Seo JM, Kim YG, Jeong JH, Kim M, Kim JI, Kim P, Bae JS, Shim EY, Lee MS, Kim MS, Noh H, Park GS, Park JS, Son D, An Y, Lee JN, Kwon KS, Lee JY, Lee H, Yang JS, Kim KC, Kim SS, Woo HM, Kim JW, Park MS, Yu KM, Kim SM, Kim EH, Park SJ, Jeong ST, Yu CH, Song Y, Gu SH, Oh H, Koo BS, Hong JJ, Ryu CM, Park WB, Oh MD, Choi YK, Lee SY: A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat Commun. 2021 Jan 12;12(1):288. doi: 10.1038/s41467-020-20602-5. [Article]
- Syed YY: Regdanvimab: First Approval. Drugs. 2021 Nov 1. pii: 10.1007/s40265-021-01626-7. doi: 10.1007/s40265-021-01626-7. [Article]
- EMA Summary of Product Characteristics: Regkirona (regdanvimab) concentrate for solution for intravenous infusion [Link]
- EMA COVID-19 News: EMA recommends authorisation of two monoclonal antibody medicines [Link]
- EMA CHMP Assessment Report: Celltrion use of regdanvimab for the treatment of COVID-19 [Link]
- Protein Data Bank: Crystal Structure of COVID-19 virus spike receptor-binding domain complexed with a neutralizing antibody CT-P59 [Link]
References
- ^ Jump up to:a b c d e f g “Regkirona EPAR”. European Medicines Agency. Retrieved 12 November 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Celltrion Develops Tailored Neutralising Antibody Cocktail Treatment with CT-P59 to Tackle COVID-19 Variant Spread Using Its Antibody Development Platform” (Press release). Celltrion. 11 February 2021. Retrieved 4 March 2021 – via Business Wire.
- ^ “Celltrion Group announces positive top-line efficacy and safety data from global Phase II/III clinical trial of COVID-19 treatment candidate CT-P59” (Press release). Celltrion. 13 January 2021. Retrieved 4 March 2021 – via Business Wire.
- ^ Jump up to:a b “EMA issues advice on use of regdanvimab for treating COVID-19”. European Medicines Agency. 26 March 2021. Retrieved 15 October 2021.
- ^ World Health Organization (2020). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 124 – COVID-19 (special edition)” (PDF). WHO Drug Information. 34 (3): 660–1.
- ^ “EMA starts rolling review of Celltrion antibody regdanvimab for COVID-19” (Press release). European Medicines Agency (EMA). 24 February 2021. Retrieved 4 March 2021.
- ^ “EMA review of regdanvimab for COVID-19 to support national decisions on early use” (Press release). European Medicines Agency (EMA). 2 March 2021. Retrieved 4 March 2021.
- ^ Jump up to:a b “EMA receives application for marketing authorisation Regkirona (regdanvimab) treating patients with COVID-19”. European Medicines Agency. 4 October 2021. Retrieved 15 October 2021.
- ^ “Regkirona: Pending EC decision”. European Medicines Agency. 11 November 2021. Retrieved 11 November 2021.
- ^ Jump up to:a b “COVID-19: EMA recommends authorisation of two monoclonal antibody medicines”. European Medicines Agency (EMA) (Press release). 11 November 2021. Retrieved 11 November 2021.
Further reading
- Kim C, Ryu DK, Lee J, Kim YI, Seo JM, Kim YG, et al. (January 2021). “A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein”. Nature Communications. 12 (1): 288. doi:10.1038/s41467-020-20602-5. PMC 7803729. PMID 33436577.
External links
- “Regdanvimab”. Drug Information Portal. U.S. National Library of Medicine.
///////////Regdanvimab, Regkirona, MONOCLONAL ANTIBODY, ANTI VIRAL, EU 2021, APPROVALS 2021, EMA 2021, COVID 19, CORONAVIRUS, PEPTIDE, レグダンビマブ , CT-P59, CT P59

NEW DRUG APPROVALS
ONE TIME
$10.00
Diroximel fumarate

Diroximel fumarate
ジロキシメルフマル酸エステル;
| Formula |
C11H13NO6
|
|---|---|
| CAS | 1577222-14-0 |
| Mol weight |
255.224
|
2021/11/15 EMA APPROVED, VUMERITY
Treatment of multiple sclerosis
Diroximel fumarate, sold under the brand name Vumerity, is a medication used for the treatment of relapsing forms of multiple sclerosis (MS).[1][3][4]
Diroximel fumarate was approved for medical use in the United States in October 2019,[5] and in the European Union in November 2021.[2]
History
This drug was formulated by Alkermes in collaboration with Biogen.[6]
Society and culture
Legal status
Diroximel fumarate was approved for medical use in the United States in October 2019.[5]
On 16 September 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Vumerity, intended for the treatment of adults with relapsing remitting multiple sclerosis.[7] The applicant for this medicinal product is Biogen Netherlands B.V.[7] It was approved for medical use in the European Union in November 2021.[2]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
PATENT
US 8669281
https://patents.google.com/patent/US8669281B1/en
PATENT
WO 2014152494
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014152494
2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate (14)
2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate 14 was synthesized following general procedure 1 (1.03 g, 35 %).
1H NMR (400 MHz, DMSO): δ 6.81 (2H, dd, J = 15.8 Hz); 4.36 (2H, t, J = 5.3 Hz); 3.84 (2H, t, J = 5.1 Hz); 3.80 (3H, s); 2.73 (4H, s). [M+H]+ = 256.07.
General Procedure 1
To a mixture of monomethyl fumarate (MMF) (1.0 equivalent) and HBTU (1.5 equivalents) in DMF (25 ml per g of MMF) was added Hünigs base (2.0 equivalents). The dark brown solution was stirred for 10 minutes, where turned into a brown suspension, before addition of the alcohol (1.0 – 1.5 equivalents). The reaction was stirred for 18 hours at room temperature. Water was added and the product extracted into ethyl acetate three times. The combined organic layers were washed with water three times, dried with magnesium sulphate, filtered and concentrated in vacuo at 45 ºC to give the crude product. The crude product was purified by silica chromatography and in some cases further purified by trituration with diethyl ether to give the clean desired ester product. All alcohols were either commercially available or made following known literature procedures.
As an alternative to HBTU (N,N,N’,N’-Tetramethyl-O-(1H-benzotriazol-1 -yl)uronium hexafluorophosphate), any one of the following coupling reagents can be used: EDCI/HOBt (N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride/hydroxybenzotriazole hydrate); COMU ((1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate); TBTU (O-(benzotriazol-1 -yl)-N,N,N’,N’-tetramethyluronium tetrafluoroborate); TATU (O-(7-azabenzotriazole-1-yl)-1,1 ,3,3-tetramethyluronium tetrafluoroborate); Oxyma (ethyl (hydroxyimino)cyanoacetate); PyBOP ((benzotriazol-1 -yloxy)tripyrrolidinophosphonium hexafluorophosphate); HOTT (5-(1-oxido-2-pyridyl)-N,N,N’,N’-tetramethylthiuronium hexafluorophosphate); FDPP (pentafluorophenyl diphenylphosphinate); T3P (propylphosphonic anhydride); DMTMM (4-(4,6-dimethoxy-1,3,5-triazin-2-y1)-4-methylmorpholinium tetrafluoroborate); PyOxim ([ethyl
cyano(hydroxyimino)acetato-O2]tri-1-pyrrolidinylphosphonium hexafluorophosphate); TSTU (N,N,N’,N’-tetramethyl-O-(N-succinimidyl)uronium tetrafluoroborate); TDBTU (O-(3,4-dihydro-4-oxo-1,2,3-benzotriazin-3-yl)-N,N,N’,N’-tetramethyluronium tetrafluoroborate); TPTU (O-(2-oxo-1(2H)pyridyl)-N,N,N’,N’-tetramethyluronium tetrafluoroborate); TOTU (O-[(ethoxycarbonyl)cyanomethylenamino]-N,N,N’,N’-tetramethyluronium tetrafluoroborate); IIDQ (isobutyl 1,2-dihydro-2-isobutoxy- 1-quinolinecarboxylate); or PyCIU
(chlorodipyrrolidinocarbenium hexafluorophosphate),
As an alternative to Hünig’s base (diisopropylethylamine), any one of the following amine bases can be used: triethylamine; tributylamine; triphenylamine; pyridine; lutidine (2,6-dimethylpyridine); collidine (2,4,6-trimethylpyridine); imidazole; DMAP (4-(dimethylamino)pyridine); DABCO (1 ,4-diazabicyclo[2.2.2]octane); DBU (1 ,8-
diazabicyclo[5.4.0]undec-7-ene); DBN (1,5-diazabicyclo[4.3.0]non-5-ene); or proton sponge® (N,N,N’,N’-tetramethyl-1 ,8-naphthalenediamine).
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
PATENT
WO 2016124960
PATENT
WO 2017108960
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017108960
Example 3b: Synthesis of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester
Procedure A:
Distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (3 g; 20.96 mmol) and maleic acid anhydride (2.26 g; 23.1 mmol) in toluene (10 mL) were heated to 60°C under stirring for 29 hours. The temperature was raised to 80°C and heated for another 19 hours. Acetyl chloride (0.3 mL; 4.2 mmol) was added and heating (80°C) was continued for 24 hours. The reaction mixture was cooled to RT. The biphasic system was separated, the upper layer was discarded. The lower layer (viscous oil) crystallized. The crystallized compound was suspended in acetone (50 mL) and stirred for 15 minutes before being filtrated off. The product was dried at 50°C for 5 hours and 8 mbar to yield the 1st crop (1.65 g). The mother liquor was evaporated and the obtained oil/solid was suspended in acetone (5 mL) and stirred overnight at RT. The product was filtrated off and dried at 50°C for 5 hours and 8 mbar to yield the 2nd crop (1.41 g). The mother liquor was evaporated and the obtained oil/solid was suspended in a mixture of diethylether/acetone (5 mL/1 mL) and stirred overnight at RT. The product was filtrated off and dried at 8mbar/50°C for 3 hours (3rd crop, 0.37 g).<a name=”
Yield: 3.43 g (68% of theory)
Purity: 1st crop 96.8 area%; 2nd crop 96.0 area-%; 3rd crop 85.4 area-% (HPLC/UV, method A, λ=200nm; tr: 3.8 min.)
1H NMR (400 MHz, DMSO-d6) δ ppm: 2.61 (s, 4 H) 3.66 (t, J=5.47 Hz, 2 H) 4.23 (t, J=5.47 Hz, 2 H) 6.51 – 6.72 (m, 2 H) 6.60 (s, 1 H) 6.63 (s, 1 H) 13.21 (br s, 1 H)
Procedure B:
Reaction performed in a reactor (Mettler Toledo, Optimax):
Distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (20 g; 0.14 mol) and maleic acid anhydride (15 g; 0.15 mol) in toluene (70 mL) were heated to 80°C under stirring (150 rpm) for 29 hours. Acetyl chloride (2 mL; 0.03 mol) was added and heating (80°C) was continued overnight. Stirring speed was raised to 200 rpm) after 15.5 hours (at 80°C) (product precipitated upon raising stirring speed. The reaction mixture was cooled to 20°C within 1 hour, directly after highering stirring speed. The reaction mixture was stirred for 4 hours, before being filtrated off. The filtrated precipitate was washed with toluene (30 mL) and then with heptane (70 mL), the product was dried at 60°C and 18 mbar. The crude product (26.26 g) with -90% purity was suspended in a mixture of acetone (30 mL)/heptane (30 mL) and stirred at RT for 2 days. The product was filtrated off, washed with heptane (30 mL) and dried at 50°C and 7 mbar.
Yield: 24.12 g (72% of theory)
Purity: 97.4 area-% at 200 nm
Procedure C:
a) Ethylene carbonate (8.89 g; 0.1 mol), succinimide (10 g; 0.1 mol) and sodium carbonate (0.53 g, 5 mmol) were heated to 100°C, the temperature was hold overnight. The product was cooled down yielding a brownish solid (13.73 g) which was grinded in a mortar.
b) 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (10 g, 69.9 mmol) from sequence a) and maleic acid anhydride (6.85 g; 69.9 mmol) in toluene (33 mL) were heated to 80°C under stirring for 23 hours. Acetyl chloride (0.5 mL; 7 mmol) was added and heating<a name=”
(80°C) was continued overnight. Heating was stopped and after stirring for another 2 hours the product was filtered off. The product was dried for 2 hours at 60°C and 8 mbar, yielding 15.82 g of crude product.
purity: 63 area-% at 200nm; 80 area-% at 220 nm
Procedure D
a) Ethylene carbonate (44.43 g; 0.5 mol), succinimide (50 g; 0.5 mol) and sodium carbonate (2.67 g; 25 mmol) were heated to 100°C. The reaction mixture was stirred at 100°C for overnight. The mixture was cooled to RT, yielding 72.4 g of the raw product.
40 g of the raw product were suspended in ethylacetate (40 mL) and heated to reflux for 30 minutes. The turbid mixture was cooled to RT and left stirring O/N. The product was filtrated off and dried under vacuum at RT to yield 29.19 g.
b) 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (10 g; 69.9 mmol) from sequence a) and maleic acid anhydride (6.85 g; 69.9 mmol) in toluene (30 mL) were heated to 80°C under stirring. Acetyl chloride (0.5 mL; 7 mmol) was added after 19 hours and heating (80°C) was continued overnight. Heating was stopped and stirring was continued for 2 days. The product was filtrated off and dried at 23 mbar and 60°C.
purity: 82 area% at 200 nm; 91 area-% at 220 nm
Procedure E:
a) Succinimide (500 g; 5.0 mol), ethylene carbonate (444.34 g; 5.0 mol) and sodium carbonate (26.74 g; 0.25 mol) were mixed and slowly heated to 130°C under stirring for 7 hours. The product was distilled via vacuum distillation to yield the product as colourless substance (628.14 g; 87% of theory)
b) The distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (150 g; 1.05 mol) from sequence a) and maleic acid anhydride (102.76 g; 1.05 mol) in toluene (350 mL) were heated to 80°C under stirring for 23 hours. Acetyl chloride (7 mL; 0.01 mol) was added and heating (80°C) was continued. After 6 hours, the reaction mixture was cooled to 20°C within 30 minutes. The product was filtrated off and washed with toluene (200 mL), yielding 221.8 g of a white crystalline product (crude product).
purity: 91 area% at 200 nm; 92 area-% at 220 nm<a name=”
Procedure F:
a) Ethylene carbonate (9.78 g; 0.11 mol), succinimide (10 g; 0.10 mol) and triethylamine (0.7 mL; 5mmol) were heated to 98°C. The reaction mixture was stirred at this temperature overnight. The mixture was cooled to RT, yielding a colourless liquid, which crystallizes upon standing at RT to a colorless solid (14.89 g).
b) The crude 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione from sequence a) (5 g; 35 mmol) and maleic acid anhydride (3.43 g; 35 mmol) in toluene (25 mL) were heated to 80°C under stirring for 24 hours. Acetyl chloride (0.25 mL; 3.5 mmol) was added and heating (80°C) was continued for ~4 hours. The reaction mixture was cooled to RT. The product was filtrated off washed with toluene and dried at 50°C and 8 mbar for 3 hours. Yield: 6.52 g (77%)purity: 93 area% at 200 nm; 94 area-% at 220 nm
Procedure F’
Ethylene carbonate (161.50 g, 1.834 mol) was melted at 50°C in a reactor, succinimide (173.07 g, 1.747 mol) and Et3N (12.2 mL, 87.350 mmol) were added and the reaction mixture was warmed up to 90°C and stirred for 24h. Reaction mixture was cooled to 50°C, 500 mL of acetone was added, followed by addition of maleic anhydride (164.19 g, 1.674 mol) and Et3N (10.15 mL, 72.772 mmol). Reaction mixture was stirred at 50-55°C for 4h, cooled to 0°C and stirred for 20h. Resulting white suspension was filtered off and solid was washed with cold acetone (2×50 mL) and dried for 6h at 50°C and 30 mbar to afford crystalline (Z)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid.
Yield: 274 g (65%)
Purity: 97.23 area % at 200 nm
Procedure F”
(Z)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid (250 g, 1.036 mol) was suspended in acetone (500 mL) in 1-L reactor, acetyl chloride (5.53 mL, 77.736 mmol) was added drop wise at 20-25°C and reaction mixture was warmed up to 50-55°C and stirred for 20h. Reaction mixture was cooled to 0°C and stirred for 3h. Resulting white suspension was filtered off and solid was washed with cold acetone <a name=”(2×50 mL) and dried for 6h at 50°C and 30 mbar to afford crystalline (E)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid (Formula II).
Yield: 231.3 g (92.5%)
Purity: 99.47 area % at 200 nm
Summary:
Procedure B and E, using distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione, showed purities of -90-91 area-% of the crude product, ongoing crystallization of the target compound could improve the purity to -97% also shown in procedure A. Distillation of 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione needs harsh conditions (Ex. 3a; procedure A). Using the crude 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione, produced with Na2CO3 lead to low product purities of 63 area-% (procedure C).
Crystallization of 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (procedure D) lead to product purities comparable to procedure A, B and E with distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione, but crystallization is compounded by a significant product loss of – 25%.
The raw 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione could be used without any disadvantageous impact on product quality by substituting Na2CO3 with triethylamine as shown in procedure F with a purity of 93 area-%.
Procedure G
Two experiments were performed in parallel:
Each with 1 g (7 mmol) 1-(2-hydroxy-ethyl)-pyrrolidine-2,5-dione and 0.75 g (7.7 mmol) maleic acid anhydride in 6 mL acetonitrile in screw capped vials. To one of the reaction mixtures was given 0.1 mL triethylamine. Both mixtures were stirred at RT. Samples were taken and investigated by NMR (in DMSO).
product formation after 1 hour (quantified by NMR):
mixture without triethylamine: 0%
mixture with triethylamine: 55%<a name=”
product formation after 2 hours:
mixture without triethylamine: 0%
mixture with triethylamine: 71 %
Procedure H (isolation of cis intermediate):
1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (5 g; 35 mmol) and maleic acid anhydride (3.43 g; 35 mmol) in toluene (30 mL) were heated to 80°C under stirring for -24 hours. The reaction was cooled to RT, first a biphasic layer was observed, then the product solidified (sticking to glass wall and stirrer). The product was filtrated off after 2.5 hours of stirring, washed with toluene (50 mL) and dried under vacuum. The dried product was milled and suspended again in toluene (60 mL) at RT, after 30 minutes the product was filtrated off and dried under atmospheric conditions to yield 7.24 g of the cis intermediate (86% of theory). The intermediate product was suspended in toluene (30 mL) and heated to 80°C, acetyl chloride (0.25 mL; 3.5 mmol) was added and heating (80°C) was continued for 5 hours. The reaction mixture was cooled to RT and stirred for 2 hours. The product was filtrated off, washed with toluene (30 mL) and dried at 50°C and 8 mbar O/N.
purity: 95.6 area-% at 200nm; (0.2% of Impurity I)
Procedure H (without isolation of cis intermediate):
1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (5 g; 35 mmol) and maleic acid anhydride (3.43 g; 35 mmol) in toluene (30 mL) were heated to 80°C under stirring for 24 hours. Acetyl chloride (0.25 mL; 3.5 mmol) was added and heating (80°C) was continued for ~4 hours. The reaction mixture was cooled to RT. The product was filtrated off washed with toluene (30 mL) and dried at 50°C and 8 mbar for 3 hours.
purity: 93.2 area-% at 200nm; (1.3% of Impurity I)
Procedure I (scale-up without cis isolation)
Maleic acid (959.09 g; 9.8 mol) was added to a reactor under stirring, which was already loaded with toluene (7 L), then 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione<a name=”
(1400 g; 9.8 mol) was added. Then the mixture was heated to 76°C within ~1 h (up to ~50°C the mixture is a suspension with the tendency of conglomeration of solids, very difficult consistency) at 50°C a turbid solution resulted. Stirring was continued at 80°C for 2 days. Acetyl chloride (138 mL; 1.96 mol) was added under enhanced stirring at 80°C. After -5-10 minutes a crystalline precipitate was formed, which transformed into a pasty/syrupy solid, sticking to reactor walls (difficult handling). Heating was continued overnight (reaction completed after 5 hours as IPC showed). Mixture is still an emulsion, seeding was added and the product precipitated. Stirring at 80°C was continued for ~2 hours then the mixture was cooled to RT. The solid was filtrated off and dried at 50°C and 12 mbar overnight to yield 1818.74 g of the product.
purity: 96.34 area-% at 218 nm; (1.5% of Impurity I)
Procedure J:
2L flask (reaction volume ~1 L): Succinimide (460 g; 4.6 mol), ethylene carbonate (450 g; 5.1 mol) and triethylamine (32 mL; 0.23 mol) were heated to 85°C under stirring overnight. Temperature was raised to 95°-97C and heating was continued O/N. The mixture was cooled to 50°C. Acetonitrile (1600 mL) was charged into a 10 L reactor. To the reaction mixture was added acetonitrile (1000 mL) at 50°C and the solution was transferred to the reactor (reactor T ~22°C), triethylamine (35 mL) was added, then maleic acid anhydride (500.81 g; 5.1 mol). The mixture was heated to 55°C for 5.5 hours. A part of the solvent was distilled off (~1200 mL). Then toluene (1200 mL) was added. The mixture was heated to 90°C. The mixture was cooled to 50°C. At 60°C (clear solution), seeding was added ~300 mg, after -3 minutes a suspension resulted. The mixture was further cooled down to 20°C within 10 hours and kept on stirring O/N. The white crystalline product was filtrated off, washed with toluene (1000 mL) and dried at 55°C and 9 mbar for 2 h to yield 908.99 g (81% yield).
905 g of the isolated, crystallized product was suspended in acetonitrile (2.9 L). Acetyl chloride (23 mL) was added and the mixture was heated to 80°C (clear, colorless solution) for 4 hours. Toluene (1000 mL) was added and the mixture was cooled to RT within 2 hours (linear). The mixture was further cooled to 0°C within 60 minutes. The <a name=”product was filtrated off and washed with toluene (1000 mL). The product was dried overnight at 9 mbar and 50°C.
Yield: 742.06 g (66%)
purity: 99.9 area% at 200 nm
Summary:
Isolation of the cis intermediate leads to a significantly lower content of impurities, in particular of Impurity I. Toluene as solvent leads to disadvantageous conditions regarding consistency of the reaction mixture (procedure H). The use of acetonitrile or acetone (procedure I/F) leads to improved reaction conditions and product quality.
Example 3c
Preparation of (E)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid (Formula II) from ethylene carbonate and succinimide (without isolation of
intermediates)
Procedure
Ethylene carbonate (161.50 g, 1.834 mol) was melted at 50°C in an 1-L reactor, succinimide (173.07 g, 1.747 mol) and Et3N (24.4 mL, 0.175 mol) were added and the reaction mixture was warmed up to 90-92°C and stirred for 24h. Distillation column<a name=”
was set up on the reactor and the remaining Et3N was distilled off. Reaction mixture was cooled to 40-45°C, 500 mL of acetone was added, followed by addition of maleic anhydride (184 g, 1.878 mol) and Et3N (10.96 mL, 78.615 mmol). Reaction was stirred at 40°C for 6h (precipitation occurred after 3h), cooled to 20-25°C and acetyl chloride (20.86 mL, 0.293 mol) was added drop wise. Reaction mixture was then warmed up to 50-55°C and stirred for 20h. Orange solution crystallized upon seeding. Reaction mixture was cooled to 0°C and stirred for 3h. Resulting white suspension was filtered off and solid was washed with cold acetone (2×200 mL) and dried for 6h at 50°C and 30 mbar to afford (E)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid. Yield: 352.8 g (83.7%)
Purity: 99.69 area % at 200 nm
Example 4: Synthesis of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester
Procedure A:
The starting material (E)-But-2-enedioic acid mono-[2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl] ester (5 g; 20 mmol) was suspended in dichloromethane (60 mL) and cooled to 0°C, triethylamine (3.16 mL; 22.8 mmol) was added, resulting a clear solution. To this solution methylchloroformate (3.3 mL; 20.7 mmol) was carefully added within 30 minutes via syringe (reaction very exothermic). After 15 min of stirring at 0°C, DMAP (0.25 g; 2.1 mmol) was added into the reaction mixture at 0°C, stirring was continued for 3 hours at 0°C. The reaction mixture was poured into water (200 mL) and additional dichloromethane (100 mL) was added. The organic layer was separated and the aqueous layer was extracted once again with dichloromethane (50 mL). The combined organic layers were washed with brine (50 mL). The solvent was evaporated at 52°C. To the brown oil, which solidified, was added acetone (20 mL) and the mixture was stirred overnight. The product was filtrated off (white solid, part I) (2.73 g) and to the mother <a name=”liquor silica was added, the mixture was evaporated. Acetone (50 mL) was added and silica was filtrated off. The solvent was evaporated and diethylether (30 mL) was added to the solid, the mixture was stirred for ~1 hour. The product was filtrated off (part II) (1.6 g).
Overall yield: 4.33 g (82%)
Purity: part I 100 area-% at 200 nm; part II 97.96 area-% at 200 nm
Procedure A’
(E)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid (Formula II) (200 g, 0.829 mol) was suspended in acetone (2000 mL) in 3-L reactor at 20-25°C and cooled to 0°C. Et3N (150.31 mL, 1.078 mol) was added drop wise at 0-5°C. Into resulting solution, methyl chloroformate (83.27 mL, 1.072 mol) was added drop wise at 0-5°C. Reaction mixture was warmed up to 45°C and stirred for 2h. Upon completion, reaction mixture was cooled to 20-25°C and water (600 mL) was added drop wise with maintaining the temperature at 20-25°C resulting with off white to yellowish solution. pH was adjusted to 7 with 1M HCl. One more volume of water was added and pH corrected if needed. Part of acetone from the reaction mixture (5 volumes or 1000 mL) was distilled off under diminished pressure and reactor walls were washed with 1 more volume of water (200 mL), thus resulting in a solution of acetone/water mixture 1:1 (total 10 volumes). Reaction mixture was gradually cooled to 0°C and stirred for 20h. Resulting white suspension was filtered off and solid was washed with cold water (2×200 mL) and dried for 6h at 50°C and 30 mbar to afford crude 2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate (Formula I).
Yield: 183.7 g (86.8%)
Purity: 100.00 area % at 200 nm
Crude 2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate (170 g) was suspended in acetone (850 mL) at 20-25°C and warmed up to 50°C resulting with colorless solution. Water (850 mL) was added in portions at 50°C and solution was cooled gradually. Crystallization started at 32°C. Reaction mixture was stirred at crystallization temperature for 30 minutes and cooled further to 0°C, stirred at 0°C for 2h and resulting <a name=”white suspension was filtered off and solid was washed with cold water (2×170 mL) and dried for 6h at 50°C and 30 mbar to afford crystalline 2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate.
Yield: 152.5 g (89.7%)
Purity: 100.00 area % at 200 nm
Procedure B:
The starting material (5 g, 20 mmol) was suspended in toluene (25 mL). Acetyl chloride (0.29 mL) and methanol (2.5 mL) were added, the reaction mixture was heated to 55°C and stirred for 3 hours. The reaction mixture was poured into water (100 mL) and extracted with ethylacetate (100 mL). The organic layer was separated and dried over sodium sulfate. The solvent was evaporated (crude product 4.7 g, main impurities dimetylfumarate (13%) and fumaric acid (1%) (HPLC at 200 nm)).
Yield: 4.7 g (88%)
Purity: 82.1 area-% at 200 nm
Procedure C:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester in polymorphic form A; short: (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form A
The starting material (without isolation of (Z)-But-2-enedioic acid mono-[2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl] ester) (30 g; 0.12 mol) was suspended in dichloromethane (DCM, 160 mL) and cooled to 0°C, triethylamine (TEA, 19 mL; 0.14 mol) was added, resulting a clear solution. To this solution methyl chloroformate (19.74 mL; 0.12 mol) was added carefully within 30 minutes via syringe. Stirring was continued for ~2 hours. Water (200 mL) was added to the reaction mixture and stirring was continued for 5-10 minutes. The organic layer was separated and the aqueous layer was washed with another portion of DCM (100 mL). The combined organic layers were dried over sodium sulfate, before being evaporated. To the crude product was added acetone (50 mL) and the mixture was stirred for 3 hours before being filtered off. The product was washed with heptane (50 mL) and dried at 50°C and 21 mbar for 1 h.<a name=”
Yield: 20.52 g (65%)
Purity: 98.7 area-% at 220 nm; (0.3% of Impurity I)
XRPD diffraction peaks: 7.1, 11.6, 13.5, 13.7, 16.3, 16.7, 18.0, 18.4, 21.1, 22.1, 23.1, 23.9, 24.4, 25.5, 27.0, 27.5, 28.0, 28.6, 30.8, 31.2, 31.9, 32.3, 33.7, 34.2, 34.4, 34.9, 35.1, 35.7, 36.0, 36.8, 38.3, 40.1, 40.5, 41.7, 42.4, 43.0, 43.4, 45.0, 45.3, 46.2, 46.4, 47.0, 48.6, 49.4, 49.9, 52.0 + 0.2 degrees two theta.
The Form A according to Procedure C showed a habitus as depicted in Figure 7a
Procedure D:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form A The starting material (10 g; 41.5 mmol) was suspended in toluene (70 mL) at 23°C, triethylamine (TEA; 6.3 mL; 45.6 mmol) was added. Methyl chloroformate (6.58 mL; 41.5 mmol) was slowly added within -30 minutes. After stirring for 2 hours water (40 mL) was added and shortly after acetone (110 mL), stirring was continued for ~2 minutes. The organic layer was separated and washed with brine (15 mL). After drying over sodium sulfate, the solvent was evaporated, yielding a slightly grey solid as crude product (9.42 g). The raw product was suspended in acetone (20 mL) and heptane (20 mL). The mixture was heated to reflux for 15 minutes resulting in a clear solution with just a small amount of solid. The mixture was cooled to RT and stirred overnight (precipitation started at 45°C, cooling: flask left in cooling oil bath ~lh to RT). The resulting product showed polymorphic form A.
Yield: 7.83 g (74%)
Purity: 99.4 area-% at 200 nm
The form A according to Procedure D showed a habitus as depicted in Figure 7b<a name=”
Procedure E:
The starting material (1 g; 4.15 mmol) was suspended in dichloromethane (50 mL) at RT. Methyl chloroformate (0.64 mL; 8.3 mmol) was added and stirring was continued overnight, in process control by HPLC showed no conversion.
Procedure F:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form A
The starting material (7 g; 0.03 mol) and Na2CO3 were suspended in ethylacetate (50 mL). To the suspension was added methyl chloroformate (3.37 mL; 0.04 mol) in one portion. The reaction mixture was heated to 70°C. The temperature was kept for 15.5 h. The reaction mixture was cooled to 20°C and ethyl acetate (70 mL) was added to the white suspension. The solids were filtrated off and the ethyl acetate layer was washed with water (40 mL), dried over Na2S04 and evaporated to yield 6.4 g of the white crystalline crude product.
The crude product was suspended in a mixture of ethylacetate (10 mL) and heptane (10 mL). The suspension was heated to reflux for 30 minutes, then cooled to 23°C and stirred overnight. The product was filtrated off and dried at 8 mbar and 50°C overnight.
Yield: 5.62 g (75%)
Purity: 99.4 area-% at 200 nm
The form A according to Procedure E showed a habitus as depicted in Figure 7c
Procedure G:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester in polymorphic form B; short: (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form B
(A) 9 g of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methylester was heated to 115°C. The melted compound was stirred for -20 minutes and then dropped into a precooled mortar (0°C).<a name=”
Purity of form B: 98.8 area-% at 200nm
XRPD-pattern: (Figure 4)
(B) 3.00 g of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester (form A) was suspended in 150 mL of dibutyl ether. Suspension was heated to 120° C while mixing. Solution was left at 25°C for 2 days. Crystallized material was filtered and dried at 23 °C at 12 mbar.
XRPD-pattern: (Figure 4′)
A measure of the relative volume change of a solid as a response to pressure change is called compressibility. An API should exhibit good compressibility which is dependent on the polymorphic state.
Experimental data:
The compressibility of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester form B and form A was assessed using a die and a flat-faced punch fitted on a TA-XT2 Texture analyser (Stable Micro Systems Ltd., Godalming, UK). 200 mg of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester sample is compressed in a steel mould (with the rate of displacement 0.03 mm/s). Cyclic procedure (similar to tapping) was performed: compressing, then retracting, relaxation for 15 s and then repeated compressive steps (altogether 10 steps). Each step exerts 0.2 MPa pressure on to the sample. Sample density is calculated by dividing the weight by the sample volume for each cycle. Maximum density is reached within 10 steps. Measurements were performed in duplicates for each sample, results are expressed as an average of duplicate measurements.
Results:
<a name=”
Form B of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester exhibits a higher density at compression, indicating superior compressibility compared to form A.
Procedure H:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester in polymorphic form C; short: (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form C
(E)-But-2-enedioic acid mono-[2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl] ester (10 g; 41.5 mmol) was suspended in dichloromethane (DCM; 100 mL) and cooled to 0°C, triethylamine (TEA; 6.3 mL; 45.6 mmol) was added, resulting a clear solution. To the reaction mixture was added methyl chloroformate (6.58 mL; 41.5 mmol) within 30 minutes via a syringe pump. After 15 min of stirring at 0°C, DMAP (0.51 g; 4 mmol) was added into the reaction mixture at 0°C. The resulting solution was stirred at 0°C for 2.5 hours, then the cold suspension was poured into water (70 mL), the reactor was washed with further DCM (20 mL), which was added also to the DCM/water mixture. The organic layer was separated and washed with HCl (32% aq) (5 mL) in water (60 mL), then with water (50 mL) and finally with brine (50 mL). To the obtained deep red to brown solution was added silica (40-63 um) and the mixture was stirred for 5 minutes, before being filtered off to yield a colorless solution, which was evaporated to yield a colorless oil (crude product). The obtained oil was dissolved in a mixture of ethyl acetate/heptane (1/4) (20 mL). The mixture was stirred for 2 days before being filtered off. The product was dried under vacuum.
Yield: 2.87 g (26%)
Purity: 90.9 area-% at 200 nm
XRPD diffraction peaks: 11.2, 11.8, 13.0, 13.6, 13.6, 16.8, 18.1, 19.6, 20.6, 21.2, 21.5, 22.3, 23.2, 23.7, 24.3, 24.4, 25.2, 25.6, 26.5, 27.6, 28.4, 29.1, 30.3, 31.1, 32.0, 33.1, 33.8, 36.1, 36.7, 37.5, 38.4, 38.9, 41.6, 42.5, 43.2, 44.8, 46.5, 48.7, 49.6, 49.9 + 0.2 degrees two theta.<a name=”
Procedure I:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester in polymorphic form D; short: (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form D
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form B (1 g) was suspended in acetonitrile (3 mL). The suspension was stirred for 7 days in a closed screw cap vials followed by slow evaporation of the solvent under ambient conditions within 3 days.
Purity of form D: 96.3 area-% at 200 nm
XRPD diffraction peaks: 6.9, 11.7, 13.6, 13.9, 16.4, 16.9, 18.2, 20.9, 21.3, 22.3, 23.3, 24.0, 24.6, 25.7, 27.5, 27.7, 31.0, 31.3, 32.1, 32.4, 33.9, 35.3, 35.7, 38.4, 41.9, 42.7, 43.1, 43.6, 44.4, 46.5, 48.9 + 0.2 degrees two theta.
Procedure J:
The starting material (obtained via isolation of (Z)-But-2-enedioic acid mono-[2-(2, 5-dioxo-pyrrolidin-1-yl)-ethyl] ester) (400 g; 1.7 mol) and Na2CO3 (264 g; 2.5 mol) were suspended in ethylacetate (2.7 L). To the suspension was added methyl chloroformate (193 mL; 2.5 mol) at 20°C. The reaction mixture was heated to 45°C within 90 minutes (linear heated). The mixture was kept on stirring for 5.5 hours. Ethylacetate (4 L) was added to the white suspension (at 45 °C). The suspension was stirred for 15 minutes before being filtrated off (45 °C suspension). The reactor was rinsed with another portion of ethylacetate (1 L). The filtrated solids were discarded. To the ethylacetate solution was added a mixture of HClaq (32%) (50 mL) and water (1 L) and the mixture was vigorously stirred for 10 minutes (at 35°C). Then the ethylacetate layer was separated (at ~35°C). The ethylacetate layer was transferred back to the reactor and stirred over sodium sulfate for 30 minutes, sodium sulfate was filtrated off and the ethylacetate layer was reduced to 900 mL. The suspension was transferred into a 3 L flask, equipped with a KPG stirrer and reflux condenser. The mixture was heated to reflux (stirring speed 160 rpm), the suspension was stirred until a clear solution was obtained (-30 minutes). Then heptane (550 mL) was added dropwise within 30 minutes<a name=”
under reflux conditions. Then the mixture (still solution) was slowly cooled to RT. The mixture was stirred O/N. The product was filtrated off and the filter cake was rinsed with heptane (500 mL) to yield the crystalline product (362.56 g; 86%).
purity: 99.8 area% at 218 nm (no Impurity I).
Alternative Procedure: Synthesis of (E)-But-2-enedioic acid 2-(2,5-dioxo- pyrrolidin-1-yl) -ethyl ester methyl ester
Procedure A
Monomethylfumarate (20 g, 0.15 mmol) was suspended in dry dichloromethane (400 mL) at RT, 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid hydrochloride (32.42 g, 0.17 mol), N-(2-hydroxyethyl)succinimide (21.57 g, 0.15 mol) and dimethylaminopyridine (0.94 g, 7.7 mmol) were added. The solution was stirred O/N at RT. The formed yellow solution was diluted with dichloromethane (300 mL) and washed twice with water (2×500 mL). The organic layer was dried over sodium sulfate and concentrated under reduced pressure. To the crude product was added methyl tert. butyl ether (850 mL) and the reaction mixture was refluxed for 2.5 hours, cooled to RT, then filtrated and heated to reflux again for ~2 hours. After cooling to RT, the mixture was stored at ~5°C for 4 days. The white precipitate was filtrated off and washed with isopropylacetate (25 mL). The crystalline product was dried at 50°C and 7 mbar.
Yield: 10.8 g (28%)
Procedure B
Monomethylfumarate (1.5 g; 11.5 mmol) was suspended in dry DCM (30 mL) at 0°C. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid hydrochloride (2.47 g; 12.8 mmol), N- (2-hydroxyethyl)succinimide (1.62 g; 11.3 mmol) and DMAP (0.07 g; 0.6 mmol) were added. The solution was stirred overnight at RT. The formed yellow solution was diluted with DCM (50 mL) and washed with water twice (2×35 mL). The organic layer<a name=”
was dried over sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash chromatography (n-heptane:ethyl acetate 1:1->1:2). The final product showed polymorphic form A. The form A according to alternative Procedure B showed a prismatic habitus as depicted in Figure 7d
Yield: 2.3 g (78%)
Purity: 99.5 area-% at 200 nm
Example 5: Kinetic investigations
Monomethyl maleate was prepared in analogy to WO 2014/197860. Samples of 13.2 grams of monomethyl maleate in 50 mL of toluene and 0.1 equivalents of the isomerization catalyst were reacted at 80°C. Samples were taken after the given times and analyzed by HPLC at 200 nm. The absorbance ratio of monomethyl fumarate (3.8 min.) to monomethyl maleate (2.8 min.) was taken as conversion parameter. The results are shown in Figure 1. As it can be seen from Figure 1 the conversion of monomethyl maleate to monomethyl fumarate in the presence of is TMS (trimethylsilylchloride) is advantageously enhanced compared to the one in the presence of AcCl (acetyl chloride).
Example 6: Yield determination
Six samples of 13.2 g (0.1 mol) monomethyl maleate were diluted with toluene (50 mL) and 0.1 eq of the isomerization catalyst (trimethylsilylchloride or acetyl chloride) were added, three samples with trimethylsilylchloride and three samples with acetyl chloride. The resulting reaction mixtures were heated to the temperatures of 45 °C, 51°C and 80°C. After 22 hours the reaction mixtures were cooled to room temperature, the product was filtrated off and dried at 50°C/8-16 mbar overnight. The results are shown in Figure 2 As it can be seen from Figure 2 the isolated yields of the conversion of monomethyl maleate to monomethyl fumarate in the presence of TMS (trimethylsilylchloride) is at any
PATENT
CN 110698442
PATENT
WO 2021053476
PATENT
IN 201921037120
PATENT
WO 2021074842
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021074842
The drug compound having the adopted name “Diroximel fumarate” has chemical name: 2-(2,5-Dioxopyrrolidin-l-yl)ethyl methyl fumarate as below.
Diroximel fumarate is an investigational, novel oral fumarate with a distinct chemical structure and developed by Alkermes pic, for the treatment of relapsing-remitting multiple sclerosis (RRMS) and is currently under review by U.S. Food and Drug Administration. Biogen, under an exclusive license from Alkermes, intends to market Diroximel fumarate under the brand name VUMERITY™.
US 8669281 B1 first disclosed Diroximel fumarate, its preparation, composition and use thereof for treating multiple sclerosis. US 10080733 B2 further discloses the crystalline solid form of Diroximel fumarate having an X-ray powder diffraction pattern comprising 2Q peaks at 11.6, 21.0, 24.3, 27.4, and 27.9 ±0.2 2Q.
WO 2017/108960 A1 also discloses various alternative synthetic approaches to make Diroximel fumarate and crystalline solid forms thereof, designated as Polymorphic forms A to D.
Hence, there remains a need for alternate solid forms of Diroximel fumarate and preparative processes thereof, exhibiting desired bioavailability and stability. Hence, it is desirable to provide a viable solid form of Diroximel fumarate. The known processes for the preparation of Diroximel fumarate are not viable at industrial scale due to the use of expensive reagents and catalyst such as coupling agents disclosed in US 8669281 Bl, with very low yields. Hence, there remains a need for the improved process to make Diroximel fumarate.
In another aspect, the present application provides a process for the preparation of Diroximel fumarate, comprising the step of esterification of monomethyl fumarate with (2,5-dioxopyrrolidin-l-yl)ethanol in the presence of an acid halide.
n another aspect, the present application provides a process for the preparation of Diroximel fumarate, comprising the step of esterification of (E)-4-(2-(2,5-dioxopyrrolidin-l-yl)ethoxy)-4-oxobut-2-enoic acid with methylation agent selected from the group consisting of 2,2-dimethoxypropane, trimethyl orthoformate and dimethyl carbonate.
Diroximel Fumarate
Example-7: Preparation of l-(2-hydroxyethyl)pyrrolidine-2,5-dione
A mixture of succinimide (100 g), ethylene carbonate (70.6 mL) and triethylamine (14 mL) was heated to 90 °C and stirred at the same temperature for 24 hours. The reaction mixture was cooled to 0 °C; methyl /ert-butyl ether (300 mL) was added and the resulting mixture was stirred for 30 minutes at the same temperature. The solid was filtered and dried under vacuum for 5 minutes. The solid was combined with ethyl acetate (100 mL) at 0 °C and stirred at the same temperature for 30 minutes. The solid was filtered and dried in rotatory vacuum dryer at 40 °C for 30 minutes to obtain 142.5 g of the title compound as off-white solid with HPLC purity of 99.6%.
Example-8: Preparation of Diroximel fumarate
To a mixture of (E)-4-methoxy-4-oxobut-2-enoic acid (4.0 g) and dichloromethane (40 mL) at 5 °C, Oxalyl chloride (5.85 g) was added slowly in 10 minutes, then a drop of DMF was added at the same temperature and allowed the reaction mixture to warm up to 27 °C. After complete evolution of the gas, solvent was evaporated from the reaction mixture. To a mixture of l-(2-hydroxyethyl)pyrrolidine-2,5-dione (5.06 g) and dichloromethane (35 mL), diisopropylethylamine (DIPEA) (9.93 g) was added and cooled the reaction mixture to 5 °C. The former mixture of (E)-4-methoxy-4-oxobut-2-enoic acid chloride in dichloromethane was slowly added to this later mixture at 5 °C for 20 minutes and stirred at the same temperature for 1 hour. The reaction mixture was quenched with saturated ammonium chloride solution and the organic layer was separated. Organic layer was washed with 10% citric acid solution and then with brine solution. The solvent from the separated organic layer was evaporated completely at 30 °C and the resultant solid was combined with acetone (15 mL) at 27 °C and stirred for 8 hours at the same temperature. The solid was filtered and the cake was washed with chilled acetone (3 mL) and then with cyclohexane (4 mL). The wet solid was dried at 40 °C under vacuum to obtain 3.3 g of the title compound with HPLC purity of 99.95 %
Example-9: Preparation of Diroximel fumarate
To a mixture of (E)-4-methoxy-4-oxobut-2-enoic acid (100.0 g) and dichloromethane (1000 mL) at 5 °C, Oxalyl chloride (117 g) was added slowly in 15 minutes, then catalytic DMF (1 mL) was added slowly at the same temperature and allowed the reaction mixture to warm up to 27 °C. After complete evolution of the gas, solvent was evaporated from the reaction mixture. To a mixture of l-(2-hydroxyethyl)pyrrolidine-2,5-dione (110 g) and dichloromethane (900 mL), diisopropylethylamine (DIPEA) (139 g) was added and cooled the reaction mixture to -5 °C. The former mixture of (E)-4-methoxy-4-oxobut-2-enoic acid chloride in dichloromethane (100 mL) was slowly added to this later mixture at – 5 °C for 60 minutes and stirred at the same temperature for 1 hour. The reaction mixture was quenched with water and the organic layer was separated. Organic layer was washed with 10% citric acid solution, 10% NaHC03 solution and then with brine solution. The solvent from the separated organic layer was evaporated completely at 30 °C and the resultant solid was combined with acetone (400 mL) at 27 °C. The reaction mixture was heated to 45 °C and stirred at the same temperature for 1 hour. The mixture was cooled to 27 °C and stirred for 8 hours at the same temperature. The solid was filtered and the cake was washed with methanol (200 mL). The wet solid was dried at 45 °C under vacuum to obtain 120 g of the title compound with HPLC purity of 99.97 %
Example-10: Preparation of (E)-4-(2-(2,5-dioxopyrrolidin-l-yl)ethoxy)-4-oxobut-2-enoic acid
A mixture of succinimide (100 g), ethylene carbonate (70.6 mL) and triethylamine (14 mL) was heated to 90 °C and stirred at the same temperature for 24 hours. The reaction mixture was cooled to 50 °C and triethylamine was removed by evaporation under vacuum. The reaction mixture was heated to 90 °C to distill out the traces of triethylamine under vacuum. The reaction mixture was cooled to 40 °C. Acetone (300 mL), maleic anhydride (106.2 g) and triethylamine (6.31 mL) were added. The resulting mixture was stirred at 40 °C for 6 hours. The mixture was cooled to 20 °C and acetyl chloride (12 mL) was added slowly over a period of 30 minutes. The mixture was slowly heated to 50 °C and stirred for 20 hours at the same temperature followed 2 hours at 0 °C. The solid was filtered and washed with cold acetone (2 x 120 mL).The wet solid was dried at 40 °C for 2 hours to obtain 194.2 g of the title compound as white solid with HPLC purity of 99.55%
Example-11: Preparation of Diroximel fumarate
Diroximel Fumarate
To a mixture of (E)-4-(2-(2,5-dioxopyrrolidin-l-yl)ethoxy)-4-oxobut-2-enoic acid (5 g) and 2,2-dimethoxypropane (50 mL) at 29 °C, concentrated hydrochloric acid (1 mL) and water (5 mL) were added and stirred at the same temperature for 17 hours at the same temperature. The pH of the reaction mixture was adjusted to 7 with a saturated aqueous solution of NaHCCh and the solvent was evaporated completely at 40 °C. To the resultant solid, water (50 mL) was added at 29 °C and stirred for 15 minutes. The solid was filtered and dried under vacuum at 29 °C for 5 hours. The resultant solid was combined with methyl /er/-butyl ether (50 mL) at 29 °C and stirred for 20 hours at the same temperature. The solid obtained was filtered and washed with diethyl ether (20 mL). The wet solid was dried under vacuum for 3 hours at 29 °C to obtain 3.3 g of the title compound as white solid with HPLC purity of 98.11%
Example-12: Preparation of Amorphous solid dispersion of Diroximel fumarate with Copovidone
Diroximel fumarate (100 mg) and Copovidone (500 mg) were dissolved in acetone (30 mL) at 30 °C. The clear solution was filtered to make it particle free and the solvent was evaporated in a rotavapor at 45 °C under reduced pressure to obtain the title amorphous solid dispersion. The solid dispersion (100 mg) obtained was combined with Syloid (500 mg) and ground for 20 minutes to obtain the admixture of title compound. XRPD: Amorphous.
Example-13: Crystallization of Diroximel fumarate
Diroximel fumarate (20 g) was dissolved in acetone (80 mL) at 43 °C and methyl tert. butyl ether (30 mL) was added to the clear solution. A suspension of crystalline Diroximel fumarate seed (0.25 g) in methyl tert. butyl ether (10 m) was added at 40 °C and stirred the mixture at the same temperature for 30 minutes. Methyl tert. butyl ether (280 mL) was added slowly for 2 hours at 41 °C. The mixture was cooled to 25 °C in 3
hours and then to 0 °C in 1 hour. The mixture was stirred at 0 °C for 1 hour and the solid was filtered. The wet solid was washed with methyl tert. butyl ether (40 mL) and dried at 42 °C for 6 hours to obtain the title compound.
PXRD: Crystalline; Malvern particle size: Dv (10) 7.776 pm, Dv (50) 31.292 pm & Dv (90) 133.437 pm
Example-14: Crystallization of Diroximel fumarate
Diroximel fumarate (20 g) was dissolved in acetone (80 mL) at 43 °C and DM water (100 mL) was added to the clear solution. A crystalline Diroximel fumarate seed (0.20 g) was added at 42 °C and stirred the mixture at the same temperature for 10 minutes. DM water (100 mL) was added slowly at 41 °C. The mixture was cooled to 28 °C in 1 hour. The mixture was stirred at 28 °C for 2 hour and the solid was filtered to obtain the title compound.
PXRD: Crystalline; Malvern particle size: Dv (10) 8.59 pm, Dv (50) 61.08 pm & Dv (90) 187.07 pm
Example-15: Crystallization of Diroximel fumarate
Diroximel fumarate (20 g) was dissolved in acetone (80 mL) at 45 °C and DM water (400 mL) was added to the clear solution. The mixture was cooled to 30 °C in 1 hour and the solid was filtered to obtain the title compound.
PXRD: Crystalline; Malvern particle size: Dv (10) 7.22 pm, Dv (50) 45.5 pm &
Dv (90) 136.7 pm
Example-16: Crystallization of Diroximel fumarate
Diroximel fumarate (20 g) was dissolved in Isopropyl acetate (360 mL) at 55 °C and cooled to 28 °C. A crystalline Diroximel fumarate seed (0.25 g) was added at 28 °C and cool to 5 °C. The mixture was stirred for 1 hour at the same temperature and the solid was filtered to obtain the title compound.
PXRD: Crystalline; Malvern particle size: Dv (10) 7.3 pm, Dv (50) 43.18 pm & Dv (90) 133.56 pm
PATENT
IN 201941042131
References
- ^ Jump up to:a b “Vumerity- diroximel fumarate capsule”. DailyMed. Retrieved 1 February 2021.
- ^ Jump up to:a b c “Vumerity EPAR”. European Medicines Agency. 14 September 2021. Retrieved 24 November 2021.
- ^ Wang Y, Bhargava P (July 2020). “Diroximel fumarate to treat multiple sclerosis”. Drugs of Today. 56 (7): 431–437. doi:10.1358/dot.2020.56.7.3151521. PMID 32648853. S2CID 220471534.
- ^ Kourakis S, Timpani CA, de Haan JB, Gueven N, Fischer D, Rybalka E (October 2020). “Dimethyl Fumarate and Its Esters: A Drug with Broad Clinical Utility?”. Pharmaceuticals (Basel, Switzerland). 13 (10): 306. doi:10.3390/ph13100306. PMC 7602023. PMID 33066228.
- ^ Jump up to:a b “Drug Approval Package: Vumerity”. U.S. Food and Drug Administration (FDA). 21 April 2020. Retrieved 1 February 2021.
- ^ “Diroximel fumarate”.
- ^ Jump up to:a b “Vumerity: Pending EC decision”. European Medicines Agency. 15 September 2021. Retrieved 17 September 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
External links
- “Diroximel fumarate”. Drug Information Portal. U.S. National Library of Medicine.
 |
|
| Clinical data | |
|---|---|
| Trade names | Vumerity |
| Other names | ALKS-8700 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620002 |
| License data |
|
| Routes of administration |
By mouth |
| ATC code |
|
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C11H13NO6 |
| Molar mass | 255.226 g·mol−1 |
| 3D model (JSmol) | |
/////////Diroximel fumarate, EU 2021, EMA 2021, APPROVALS 2021, VUMERITY, ジロキシメルフマル酸エステル , K0N0Z40J3W, RDC-5108, дироксимела фумарат , ديروكسيميل فومارات , 富马地罗昔美 ,

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}