
Home » ANTIBODIES (Page 5)
Category Archives: ANTIBODIES
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Glenmark Pharmaceuticals Ltd. through its Swiss Subsidiary receives USD 5 Mn. as milestone fee payment from Sanofi

Glenmark Pharmaceuticals Ltd. through its Swiss Subsidiary receives USD 5 Mn. as milestone fee payment from Sanofi
Total Payment received for GBR 500 monoclonal antibody programme from Sanofi is USD 55 Mn
MUMBAI, April 15, 2014: Glenmark Pharmaceuticals Ltd. has informed the Stock Exchange today that the company through its Swiss subsidiary has received USD 5 million as
milestone payment from Sanofi on a collaboration of its VLA2 (alpha2-beta1) integrin monoclonal antibody. GBR 500 is a first-in-class therapeutic monoclonal antibody for chronicautoimmune disorders.
Glenmark has received from Sanofi already USD 50 Mn as an upfront payment in FY2011-12. Hence, the total amount received by Glenmark from Sanofi for its first in class VLA-2monoclonal antibody is USD 55 million
read at
http://www.moneycontrol.com/stocks/stock_market/corp_notices.php?autono=790416
(copy paste on browser)
MD and CEO Mr Glenn Saldanha
old updates
Glenmark GBR 500 enters into Phase II clinical development for ulcerative colitis
17 September 2012
Glenmark Pharmaceuticals, a wholly-owned subsidiary of Glenmark Pharmaceuticals, has commenced the Phase II study of GBR 500 for ulcerative colitis.
GBR 500, an antagonist of the VLA2 (alpha2-beta1) integrin, is a first-in-class therapeutic monoclonal antibody for chronic autoimmune disorders.
The randomised, double-blind, placebo-controlled study will investigate the efficacy and safety of GBR 500 in patients with moderate to severe ulcerative colitis (UC).
Glenmark Pharmaceuticals chief scientific officer Dr Michael Buschle said that UC represents an area of substantial unmet medical need, despite treatment advances in recent years.
“We’re pleased with the continued progress of our partnership with Sanofi and excited about the commencement of this trial,” Buschle said.
The trial, which will be conducted at multiple clinical sites in North America and Europe, is expected to involve approximately 84 patients.
Patients participating in the study will receive multiple doses of either GBR 500 or placebo, administered over a period of several weeks.
Glenmark has completed Phase I of GBR 500 in the US, won licensing rights to all therapeutic indications from Sanofi and is conducting the clinical development programme.
The trial is part of a strategic global collaboration between Glenmark and Sanofi to investigate GBR 500 for the treatment of chronic inflammatory disorders.
MUMBAI, India, May 16, 2011
Glenmark Pharmaceuticals Out-Licenses Novel Monoclonal Antibody, GBR 500, to Sanofi
Combined Upfront and Potential Development, Regulatory and Commercial Milestone Payments Could Total US$613 Mn
MUMBAI, India, May 16, 2011 /PRNewswire-FirstCall/ — Glenmark Pharmaceuticals S.A (GPSA), a wholly owned subsidiary of Glenmark Pharmaceuticals Limited India (GPL), announced today that it has entered into an agreement with Sanofi to grant Sanofi a license for the development and commercialization of GBR 500, a novel monoclonal antibody for the treatment of Crohn’s Disease and other inflammatory conditions. The transaction is expected to close in the coming month subject to customary closing conditions, including the expiration or early termination of the waiting period under the HSR Antitrust Improvements Act.
Under the terms of the agreement, Glenmark will receive an upfront payment of US$ 50 million, of which US$ 25 million will be paid upon closing of the transaction and US$ 25 million, which is contingent upon Sanofi’s positive assessment of certain data to be provided by Glenmark. In addition, Glenmark could receive potential success-based development, regulatory and commercial milestone payments. The total of these payments could reach US$613 Mn. In addition, Glenmark is eligible to receive tiered double-digit royalties on sales of products commercialized under the license.
GBR 500 is an antagonist of the VLA-2 (alpha2-beta1) integrin. It is a first-in-class therapeutic monoclonal antibody and has established proof of concept in animal models across a range of anti-inflammatory conditions. Glenmark has completed Phase I dosing of GBR 500 in the US and the drug has been well tolerated with a good pharmacokinetic profile. Plans are in place to initiate clinical proof of concept studies in Crohn’s Disease. Sanofi has licensed the rights to all therapeutic indications.
“There continues to be a strong medical need for safer and more efficacious products for the treatment of Inflammatory Diseases,” said Elias Zerhouni, M.D., President, Global Research & Development, Sanofi. “GBR500 brings an innovative approach to Sanofi’s Immuno-Inflammation portfolio, which we believe may address a significant gap in treating Inflammatory Diseases which would be of huge benefit to patients”.
Glenn Saldanha MD and CEO of GPL, “This collaboration on a novel first-in-class monoclonal antibody validates Glenmark’s world-class innovative R&D capabilities in the drug discovery arena. We are pleased to have this second licensing collaboration with Sanofi, one of the largest pharmaceutical companies in the world and the first one from Glenmark in the field of novel biologics”.
Amgen Drug Evolocumab Hits Endpoint of Cholesterol Reduction
 Amgen announced that the Phase 3 TESLA (Trial Evaluating PCSK9 Antibody in Subjects with LDL Receptor Abnormalities) trial evaluating evolocumab met its primary endpoint of the percent reduction from baseline at week 12 in low-density lipoprotein cholesterol (LDL-C). The percent reduction in LDL-C, or “bad” cholesterol, was clinically meaningful and statistically significant………….read at
Amgen announced that the Phase 3 TESLA (Trial Evaluating PCSK9 Antibody in Subjects with LDL Receptor Abnormalities) trial evaluating evolocumab met its primary endpoint of the percent reduction from baseline at week 12 in low-density lipoprotein cholesterol (LDL-C). The percent reduction in LDL-C, or “bad” cholesterol, was clinically meaningful and statistically significant………….read at
| Monoclonal antibody | |
|---|---|
| Source | Human |
| Target | PCSK9 |
| Clinical data | |
| Legal status | ? |
| Identifiers | |
| CAS number | 1256937-27-5 |
| ATC code | None |
| Chemical data | |
| Formula | C6242H9648N1668O1996S56 |
| Mol. mass | 141.8 kDa |
Evolocumab[1] is a monoclonal antibody designed for the treatment of hyperlipidemia.[2] Evolocumab is a fully human monoclonal antibody that inhibits proprotein convertase subtilisin/kexin type 9 (PCSK9).

PCSK9 is a protein that targets LDL receptors for degradation and thereby reduces the liver’s ability to remove LDL-C, or “bad” cholesterol, from the blood.
Evolocumab, being developed by Amgen scientists, is designed to bind to PCSK9 and inhibit PCSK9 from binding to LDL receptors on the liver surface. In the absence of PCSK9, there are more LDL receptors on the surface of the liver to remove LDL-C from binding to LDL receptors on the liver surface. In the absence of PCSK9, there are more LDL receptors on the surface of the liver to remove LDL-C from the blood.

On 23 January 2014 Amgen announced that the Phase 3 GAUSS-2 (Goal Achievement After Utilizing an Anti-PCSK9 Antibody in Statin Intolerant Subjects-2) trial evaluating evolocumab in patients with high cholesterol who cannot tolerate statins met its co-primary endpoints: the percent reduction from baseline in low-density lipoprotein cholesterol (LDL-C) at week 12 and the mean percent reduction from baseline in LDL-C at weeks 10 and 12. The mean percent reductions in LDL-C, or “bad” cholesterol, compared to ezetimibe were consistent with results observed in the Phase 2 GAUSS study.[3]
The GAUSS-2 trial evaluated safety, tolerability and efficacy of evolocumab in 307 patients with high cholesterol who could not tolerate effective doses of at least two different statins due to muscle-related side effects. Patients were randomized to one of four treatment groups: subcutaneous evolocumab 140 mg every two weeks and oral placebo daily; subcutaneous evolocumab 420 mg monthly and oral placebo daily; subcutaneous placebo every two weeks and oral ezetimibe 10 mg daily; or subcutaneous placebo monthly and oral ezetimibe 10 mg daily.
Safety was generally balanced across treatment groups. The most common adverse events (> 5 percent in evolocumab combined group) were headache (7.8 percent evolocumab; 8.8 percent ezetimibe), myalgia (7.8 percent evolocumab; 17.6 percent ezetimibe), pain in extremity (6.8 percent evolocumab; 1.0 percent ezetimibe), and muscle spasms (6.3 percent evolocumab; 3.9 percent ezetimibe).

Evolocumab, a PCSK9 inhibitor, was safe and effective at lowering low-density lipoprotein cholesterol (LDL-C) after one year of treatment, according to a study published online Nov. 19 inCirculation and presented simultaneously at the American Heart Association scientific session in Dallas.
The Open-Label Study of Long-term Evaluation Against LDL-C (OSLER) trial took place at 156 study centers around the world that participated in at least one of four phase 2 studies of between October 2011 and June 2012. Evolocumab is a PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibitor made by Amgen.
Investigators led by Michael J. Koren, MD, of the Jacksonville Center for Clinical Research in Florida, randomized 1,104 participants in a 2:1 ratio to receive either evolocumab (420 mg every four weeks) plus standard-of-care therapy (based on guidelines for treatment of hypercholesterolemia) or evolocumab alone, which served as the control. After 12 weeks, lipid results were unblinded and investigators were able to adjust standard-of-care therapy in either group.
The main efficacy objective was to determine the effects of longer-term evolocumab therapy on cholesterol levels and the main safety endpoints included incidence of adverse events, serious adverse events and adverse events resulting in discontinuation of the drug.
Patients who received evolocumab for the first time in the OSLER study had an average LDL-C reduction of 52.3 percent at one year. Patients previously dosed with evolocumab in a prior trial and were in the evolocumab and standard-of-care group in OSLER had an average LDL-C reduction of 52.1 percent at the end of the study compared with 50.4 percent at baseline. Patients who terminated evolocumab when they entered OSLER had their LDL-C levels returned to around their baseline.
Adverse events occurred in 73.1 percent of the standard-of-care group and 81.4 percent of the evolocumab plus standard-of-care group. The researchers determined that 5.6 percent of adverse events were related to evolocumab. Serious adverse events occurred in 6.3 percent of the control group and 7.1 percent in the combination group.
The authors explained that their findings offer more insight into the use of this class of drugs to lower LDL-C in at-risk patients.
“Challenging patients such as those who fail to reach current lipid goals despite maximum doses of highly effective statin agents or those with well-documented statin intolerance are thus logical populations for treatment with PCSK9 inhibitors,” they concluded.

References
- World Health Organization (2012). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 108” (PDF). WHO Drug Information 26 (4).
- Statement On A Nonproprietary Name Adopted By The USAN Council – Evolocumab
- Estel Grace Masangkay, “Amgen Phase III GAUSS-2 Trial of Evolocumab (AMG 145) Meets Co-Primary Endpoints Of LDL Cholesterol Reduction”, Bioresearch Online (January 24 2014)

Romosozumab (AMG 785) shines in phase II for osteoporosis
Romosozumab (AMG 785) is a humanized monoclonal antibody that targets sclerostin for the treatment of osteoporosis.[1]
Romosozumab was originally discovered by Celltech (now owned by UCB).[2] Celltech entered in a partnership with Amgen in 2002 for the product’s development.[3] As of January 2014, Phase 3 clinical trials are recruiting patients.[4]
- “Statement On A Nonproprietary Name Adopted By The USAN Council: Romosozumab”. American Medical Association.
- Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003 Dec 1;22(23):6267-76.
- Celltech group Annual Report and Accounts 2002
- ClinicalTrials.gov: Romosozumab
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized (from mouse) |
| Target | Sclerostin |

Sclerostin / Source: Wikimedia Commons and JMROL
Biloine Young • Thu, November 1st, 2012
Good news for postmenopausal women came from a report given by Michael R. McClung, M.D., at the annual meeting of the American Society for Bone and Mineral Research. McClung and colleagues have found an antibody that targets the Wnt signaling pathway and its osteocyte-regulating molecule sclerostin, which increases bone formation while decreasing bone resorption, according to Nancy Walsh, staff writer for MedPage Today.
Walsh reports that one year of treatment with the antibody romosozumab (formerly AMG 785) led to an 11.3% absolute increase in bone mineral density (BMD) in postmenopausal women with low BMD (body mass index). That compared with BMD increases of only 7% with teriparatide (Forteo), 4% with alendronate (Fosamax), and no change with a placebo. “The discovery of sclerostin as an osteocyte-mediated stimulator of osteoblast function and bone formation opened the door for considering the inhibition of this protein and regulator as a target for osteoporosis treatment,” McClung said.
To further explore the therapeutic potential of this antibody, the researchers conducted a Phase II study that enrolled 419 women whose lumbar spine, total hip, or femoral neck T-scores were between −2 and −3.5. The mean age of the participating women was 67. Researchers randomized participants to receive romosozumab in dosages of 70 mg, 140 mg, or 210 mg each month, 140 or 210 mg every three months, or a placebo.
The total hip increase in BMD with romosozumab was 4.1% at 12 months, which was approximately double that seen with alendronate and teriparatide. The researchers also saw changes in biomarkers of bone metabolism, McClung noted. The pattern seen with romosozumab, McClung said, was increases for serum P1NP, favoring bone formation, and decreases in serum CTX, suggesting a slowing of bone resorption after one week of treatment.
Adverse events were similar in the treatment groups. The most common was back or extremity pain. Serious adverse events occurred in 9.8% of the romosozumab groups and in 14% of the placebo group. The only treatment-related adverse events that occurred in the romosozumab groups were injection site reactions, but these were mild and did not lead to a discontinuation of treatment, said McClung.
Amgen/UCB osteoporosis drug shines in Phase II
Amgen and UCB have been boosted by promising mid-stage data for their investigational osteoporosis drug romosozumab.
A Phase II trial, the results from which have been published in the New England Journal of Medicine,showed that romosozumab demonstrated a significant increase in bone mineral density. Specifically, the trial demonstrated that, compared with placebo, treatment for 12 months with the anti-sclerostin biologic significantly increased BMD at the lumbar spine, total hip and femoral neck.
Amgen and UCB noted that significant increases were also observed in the first BMD assessment at three months and moreover, in exploratory analyses, increases observed at the lumbar spine and hip “were significantly greater than those observed with current treatments”, namely Merck & Co’s Fosamax (alendronate) and Eli Lilly’s Forteo (teriparatide).
Iris Loew-Friedrich, chief medical officer at UCB, noted that romosozumab is designed to stimulate bone formation, “which makes it different from most available treatments that reduce bone resorption”. She added that “we are encouraged by the emerging efficacy and safety profile, and look forward to further investigating its potential in the ongoing global Phase III clinical programme”. Final data from the latter, which will enroll up to 10,000 patients, are expected by the end of 2015.
Sean Harper, Amgen R&D chief, noted that broken bones due to osteoporosis are common “yet the seriousness of this health event remains underappreciated, with only two in ten women receiving follow-up testing or treatment after they have broken a bone”. He added that “with its bone-forming ability, romosozumab may result in new treatment strategies”.
If all goes well in Phase III, many observers believe romosozumab could be a blockbuster.
Links


DR ANTHONY MELVIN CRASTO Ph.D
Farletuzumab

Farletuzumab
Farletuzumab (MORAb-003) is a monoclonal antibody[1] which is being investigated for the treatment of ovarian cancer.[2][3]
This drug was developed by Morphotek, Inc.
It is targeted at FR-alpha which is overexpressed in some cancers such as ovarian cancer.
USAN FARLETUZUMAB
PRONUNCIATION far” le tooz’ oo mab
THERAPEUTIC CLAIM Treatment of cancer
CHEMICAL NAMES
1. Immunoglobulin G1, anti-(human receptor FR-α (folate receptor α)) (human-mouse monoclonal MORAb-003 heavy chain), disulfide with human-mouse monoclonal MORAb-003 κ-chain, dimer
2. Immunoglobulin G1, anti-(human folate receptor alpha (ovarian tumor-associated antigen Mov18)); humanized mouse monoclonal MORAb-003 γ1 heavy chain (222-217′)-disulfide with humanized mouse monoclonal MORAb-003 κ light chain (228-228”:231-231”)-bisdisulfide dimer
MOLECULAR FORMULA C6466H9928N1716O2020S42
MOLECULAR WEIGHT 145.4 kDa
MANUFACTURER Morphotek, Inc.
CODE DESIGNATION MORAb-003
CAS REGISTRY NUMBER 896723-44-7
Farletuzumab, a humanized monoclonal antibody that targets the folate receptor alpha (FRα), could potentially be used in the treatment of patients with relapsed ovarian cancer, according to the results of a recent open-label phase II trial.Armstrong and colleagues investigated the efficacy of farletuzumab as a single agent or in combination with standard chemotherapy in patients with relapsed ovarian cancer following first-line therapy.

Farletuzumab is a humanized IgG1 monoclonal antibody that targets
the human folate receptor FRα, which is overexpressed in most ovarian
epithelial cancers. It is being developed by Morphotek (now part of
Eisai) for the treatment of ovarian cancer, with regulatory submissions
in 2012.
The pivotal Phase III study in ovarian cancer began
in March 2009; Phase II studies in other indications have since begun.
The 900-patient Phase III study is evaluating two doses of
farletuzumab as an add-on to the standard treatment regimen of
carboplatin and a taxane; this study is completed in
September 2012. A 165-patient study in lung adenocarcinoma began in
December 2010. The initial Phase I study in 25 patients with epithelial
ovarian cancers showed farletuzumab to be well tolerated, with evidence
of efficacy in 36% of the patients (Konner et al. 2010).22
Phase II data from a 54-patient study were presented at the 2008 ASCO meeting, with at least some evidence of efficacy seen in 90% of the treated patients.
Farletuzumab represents one of a number of new treatment options
being developed for the treatment of ovarian cancer, with several other
modalities such as kinase inhibition or PARP inhibition also showing
promise. However, the available evidence suggests that farletuzumab
is likely to represent a significant enhancement in the subset of ovarian
cancer patients at which it has been targeted. If it becomes widely
accepted as a component of the platinum-based treatment regimen, then
it can be expected to be a significant commercial success.
- Statement On A Nonproprietary Name Adopted By The Usan Council – Farletuzumab, American Medical Association.
- ClinicalTrials.gov: Phase II Efficacy and Safety Study of MORAb-003 in Platinum-Resistant or Refractory Relapsed Ovarian Cancer
- ClinicalTrials.gov: Phase III Efficacy and Safety of MORAb-003 in Subjects With Platinum-sensitive Ovarian Cancer in First Relapse
…………………
Monoclonal antibody (mAbs) 2013

2013——-29 monoclonal antibody (mAbs) drugs are in Phase III clinical development.
While around 350 therapeutic mAbs are currently in clinical development globally, only 28 had entered active Phase 2/3 or Phase 3 studies as of January 2013, Additionally one mAb mixture was under evaluation in Phase III.
Historically, mAbs that target antigens relevant to cancer have comprised approximately 50% of the mAb clinical pipeline,
but in 2013 the picture has changed: 66% or 19 of the antibodies to watch in 2013 are for non-cancer indications.

The non-cancer mAbs include alirocumab (Regeneron; Sanofi, hypercholesterinemia);
AMG 145 (Amgen, hypercholesterinemia),
epratuzumab (UCB, SLE),
gantenerumab (Roche; Alzheimer’s disease),
gevokizumab (Xoma/Servier, Non-infectious uveitis),
itolizumab (Biocon, Plaque psoriasis), ixekizumab (Eli Lilly and Co., psoriasis),
lebrikizumab (Roche/Genentech, rheumatoid arthritis),
mepolizumab (GSK, Asthma, COPD etc.),
ocrelizumab (Roche/Genentech, multiple sclerosis),
reslizumab (Teva, Eosinophilic asthma), romosozumab (Amgen, Postmenopausal osteoporosis),
sarilumab (Regeneron; Sanofi, rheumatoid arthritis),
secukinumab (Novartis, rheuma, psoriasis),
sirukumab (Janssen R&D LLC, rheumatoid arthritis),
solanezumab (Eli Lilly and Co., Alzheimer’s disease),
tabalumab (Eli Lilly and Co., rheuma, SLE)
and
vedolizumab (Millenium, Ulcerative colitis; Crohn disease).
The mixture of actoxumab and bezlotoxumab (MK-3415A, Merck & Co.) is being evaluated in two Phase 3 studies as a treatment for Clostridium difficile infection.
The ten cancer mAbs are:
elotuzumab (Bristol-Myers Squibb, Abbott, multiple myeloma),
farletuzumab (Morphotek, ovarian cancer),
inotuzumab ozogamicin (Pfizer; UCB, ALL, NHL),
naptumomab estafenatox (Active Biotech, renal cell carcinoma),
necitumumab (ImClone LLC, NSCL),
nivolumab (Bristol-Myers Squibb, NSCL, renal cell carcinoma),
obinutuzumab (Roche/Genetech, Diffuse large B cell lymphoma, CLL, NHL),
onartuzumab (Roche/Genetech, NSCL cancer; gastric cancer),
racotumomab (CIMAB; Laboratorio Elea S.A.C.I.F. y A, NSCL),
and ramucirumab (ImClone LLC, Gastric; liver, breast, colorectal, NSCL cancers).
Antibody
Epratuzumab
Epratuzumab
Epratuzumab is a humanised anti-CD22 monoclonal antibody under investigation (clinical development phase III) for its efficacy in SLE. CD22 is a B cell specific surface protein that is considered to be involved in B cell function.
| Expected indication | Systemic lupus erythematosus |
| R&D stage | Phase 3 ongoing (started in December 2010) |
| Next milestone | Phase 3 results (H1 2014) |
| Quick facts |
|
Epratuzumab is a humanized monoclonal antibody. Potential uses may be found inoncology and in treatment of inflammatory autoimmune disorders, such as lupus (SLE).[1][2] The manufacturers in August 2009 announced success in early trials against SLE.[3]
Epratuzumab binds to the glycoprotein CD22 of mature and malignant B-cells.
- Epratuzumab, a humanized monoclonal antibody targeting CD22: characterization of in vitro properties Clinical Cancer Research Vol. 9, September 1, 2003 free full text
- Dose-Fractionated Radioimmunotherapy in Non-Hodgkin’s Lymphoma Using DOTA-Conjugated, 90Y-Radiolabeled, Humanized Anti-CD22 Monoclonal Antibody, Epratuzumab Clinical Cancer Research Vol. 11, July 15, 2005 free full text
- Reuters: UCB and Immunomedics Announce Positive Results for Epratuzumab Phase IIb Study in Systemic Lupus Erythematosus (SLE)
Epratuzumab is a humanized IgG1 antibody that acts as an antagonist of the CD22 receptor present on B cells. UCB is currently enrolling patients for the 2 Phase III trials, EMBODY-1 and EMBODY-2. The primary objective of both studies is to measure the percent of subjects meeting treatment response criteria at week 48 among those patients with moderate to severe SLE. Epratuzumab is dosed at either 600 mg per week or 1200 mg every other week administered over four 12-week treatment cycles.
The cumulative dose for both treatment arms is 2400 mg for each of the 4-week dosing periods. The estimated primary completion date is January 2014 for both EMBODY-1 and EMBODY-2. –
UCB pipeline. UCB Web site. www.ucb.com/rd/pipeline/new-development/epratuzumab. Published July 10, 2010. Accessed June 18, 2011
Brussels (Belgium), June 13th 2013, 0700 CEST – UCB today announced new data from an open-label extension (SL0008) of the EMBLEM™ phase 2b study evaluating the long-term effects of epratuzumab treatment in adult patients with moderate-to-severe systemic lupus erythematosus (SLE). The primary outcome of the open-label extension was to assess the safety of epratuzumab in patients with SLE.4
Relative to the 12 week, double-blind, placebo-controlled EMBLEM™ study, data from the open-label, long-term extension identified no new safety or tolerability signals.1 In addition, relative to EMBLEM™ baseline values, secondary outcome data indicated that the efficacy of epratuzumab as measured by reduction in disease activity was maintained over two years.2 Secondary outcome data also indicated that relative to EMBLEM™ baseline values, treatment over two years with epratuzumab was associated with decreases in corticosteroid use in patients receiving >7.5 mg/day.1 These data were presented this week at the European League Against Rheumatism 2013 Congress in Madrid, Spain.
Epratuzumab, licensed from Immunomedics Inc. (NASDAQ: IMMU), is an investigational medicine and the first CD-22/B-Cell receptor (BCR) targeted monoclonal antibody to be evaluated in clinical studies for the treatment of SLE. Also known as lupus, SLE is a complex, systemic autoimmune disease that affects many different organ systems, including the skin, joints, lungs, kidneys and blood.3,5
“In EMBLEM™, a dose-ranging, phase 2b study, reduction in disease activity was observed in patients treated with epratuzumab,” said Professor Daniel J Wallace MD, Clinical Professor of Medicine, Cedars-Sinai Medical Center, California, US. “This double-blind study had a relatively short 12-week, placebo-controlled, treatment period and it was important to accumulate long-term data on epratuzumab in the treatment of SLE. The phase 2b extension study adds new two year open-label data on epratuzumab to that already available from the 12-week, randomized, controlled study.”
EMBLEM™ was designed to identify a suitable dosing regimen for epratuzumab.6 A total of 227 patients with moderate-to-severe SLE received either: placebo, epratuzumab cumulative dose of 200 mg (100 mg every other week), 800 mg (400 mg every other week), 2400 mg (600 mg weekly), 2400 mg (1200 mg every other week) or 3600 mg (1800 mg every other week).3,6 In the open-label extension 203 patients from any arm of the EMBELM™ study received 1200 mg epratuzumab at weeks 0 and 2 of 12-week cycles.1,2,7
Data on epratuzumab presented at EULAR 2013
Evaluation of the safety profile of long-term epratuzumab treatment in patients with moderate-to-severe SLE1
Safety variables were primary outcome measures in SL0008 and included duration of exposure, adverse events, infusion reactions and infections.
Exposure to epratuzumab was a median 845 days over a median 10 treatment cycles. Adverse events (AEs) caused discontinuation in 29 (14.3%) patients. The most common serious AEs were SLE flare (3.4%), lupus nephritis (2%) and symptomatic cholelithiasis (1.5%). The most common infections/infestations were urinary tract infection (24.6%) and upper respiratory tract infection (23.2%). There were no opportunistic infections and no patterns of specific serious or severe infections.
Evaluation of long-term efficacy of epratuzumab as measured by reduction in disease activity in patients with moderate-to-severe SLE2
Secondary outcome measures in SL0008 included efficacy as measured by reduction in disease activity, and assessed by: British Isles Lupus Assessment Group (BILAG) improvement, SLE disease activity index (SLEDAI) score, Physician Global Assessment (PGA) score and combined treatment response defined as BILAG improvement without worsening, no SLEDAI worsening and no PGA worsening, relative to EMBLEM™ baseline.
The median BILAG total score was 25.0 at EMBLEM™ baseline and 9.0 at week 108. The score was 14.0 at SL0008 screening. Median SLEDAI score was 12.0 at EMBLEM™ baseline and 4.0 at week 108. The score was 10.0 at SL0008 screening. The median PGA score was 50.0 at EMBLEM™ baseline and 17.5 at week 108 with a score of 31.0 at SL0008 screening.
The proportion of patients achieving the combined treatment response was 32.5% at SL0008 screening (n=203) and 60.3% at week 108 (n=116).
Effect of corticosteroid use of long-term epratuzumab treatment in patients with moderate-to-severe SLE1
Corticosteroid doses were monitored throughout SL0008 and was a secondary outcome measure.
Median corticosteroid dose at EMBLEM™ baseline and SL0008 screening was 10.0 mg/day. At week 116, this was 5 mg/day (n=112). Data indicated that treatment over two years with epratuzumab was associated with decreases in corticosteroid use in patients receiving >7.5 mg/day with a corresponding increase in the proportion of patients receiving lower doses or no longer receiving corticosteroids.
The proportion of patients requiring 7.5-20 mg/day and >20 mg/day decreased (49.8% and 10.8% at baseline and 33.9% and 8.0% respectively, at week 116) and the proportion of patients receiving >0–7.5mg/day or no longer receiving corticosteroids increased (33.5% and 5.9% at baseline and 45.5% and 12.5% respectively, at week 116).
Ramucirumab Trial Shows Improved OS in Gastric Cancer
Eli Lilly and Co. announced that results from the Phase 3 REGARD trial of ramucirumab (IMC-1121B) as a single agent in patients with advanced gastric cancer who have had disease progression after initial chemotherapy were published today in The Lancet. REGARD is the first Phase 3 study with either a single-agent biologic or an anti-angiogenic therapy to show improved overall survival and progression-free survival in advanced gastric cancer patients.
READ ALL AT
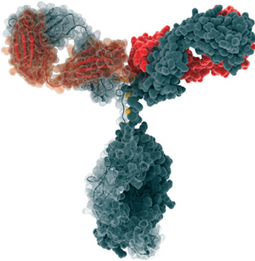
Ramucirumab (IMC-1121B)[1] is a fully human monoclonal antibody (IgG1) being developed for the treatment of solid tumors. It is directed against the vascular endothelial growth factor receptor 2 (VEGFR2). By binding to VEGFR2 it works as a receptor antagonist blocking the binding of vascular endothelial growth factor (VEGF) to VEGFR2. VEGFR2 is known to mediate the majority of the downstream effects of VEGF inangiogenesis.
Ramucirumab is being tested in several phase III clinical trials for the treatment of metastatic gastric adenocarcinoma,[2] non-small cell lung cancer,[3] among other types of cancer. On September 26, 2013 Eli Lilly announced that its Phase III study for ramucirumab failed to hit its primary endpoint on progression-free survival among women with metastatic breast cancer.[4][5]
This drug was developed by ImClone Systems Inc. It was isolated from a native phage display library from Dyax.
- Statement On A Nonproprietary Name Adopted By The USAN Council – Ramucirumab, American Medical Association.
- ClinicalTrials.gov NCT01170663 A Study of Paclitaxel With or Without Ramucirumab in Metastatic Gastric Adenocarcinoma (RAINBOW)
- ClinicalTrials.gov NCT01168973 A Study in Second Line Non Small Cell Lung Cancer
- ClinicalTrials.gov NCT00703326 Phase III Study of Docetaxel + Ramucirumab or Placebo in Breast Cancer
- Fierce Biotech. “In another stinging setback, Eli Lilly’s ramucirumab fails PhIII breast cancer study”. Retrieved 27 September 2013.
Secukinumab
Secukinumab is an anti-IL17A drug being investigated for a number of inflammatory conditions. For plaque psoriasis, Novartis is planning to evaluate a dose of 150 mg subcutaneously compared with placebo.
The primary outcome measure of the planned Phase III trial named ERASURE is to evaluate the efficacy in patients with moderate to severe chronic plaque-type psoriasis. Novartis is also planning to evaluate secukinumab dosed at either 150 or 300 mg versus Enbrel (enterecept) 50 mg in a Phase III trial entitled FIXTURE.
Final data collection for the primary outcome measures in both ERASURE and FIXTURE are anticipated in March 2013.
Secukinumab is a human monoclonal antibody designed for the treatments of uveitis,rheumatoid arthritis, and psoriasis. It targets member A from the cytokine family ofinterleukin 17.[1][2]
Secukinumab was developed by Novartis Pharma AG and has completed Phase II clinical trials for plaque psoriasis in 2011.[3]
CAS registry numbers
- 875356-43-7 (heavy chain)
- 875356-44-8 (light chain)
- ^ “Statement On A Nonproprietary Name Adopted By The USAN Council: Secukinumab”. American Medical Association.
- ^ Hueber, W.; Patel, D. D.; Dryja, T.; Wright, A. M.; Koroleva, I.; Bruin, G.; Antoni, C.; Draelos, Z.; Gold, M. H.; Psoriasis Study, P.; Durez, P. P.; Tak, J. J.; Gomez-Reino, C. S.; Rheumatoid Arthritis Study, R. Y.; Foster, C. M.; Kim, N. S.; Samson, D. S.; Falk, D.; Chu, Q. D.; Callanan, K.; Nguyen, A.; Uveitis Study, F.; Rose, K.; Haider, A.; Di Padova, F. (2010). “Effects of AIN457, a Fully Human Antibody to Interleukin-17A, on Psoriasis, Rheumatoid Arthritis, and Uveitis”. Science Translational Medicine 2 (52): 52ra72.doi:10.1126/scitranslmed.3001107. PMID 20926833. edit
- ^ Papp K.A. et al. ‘Secukinumab efficacy and safety preliminary results from a phase II subcutaneous dose-ranging study in the treatment of moderate-to-severe plaque psoriasis.’ Presented at: 20th Congress of the European Academy of Dermatology and Venereology; 20-24 October, 2011; Lisbon, Portugal.

Certolizumab pegol – FDA gave green light to UCB’s Cimzia to treat psoriatic arthritis

Certolizumab pegol
The US Food and Drug Administration has approved UCB’s Cimzia for the treatment of adults with psoriatic arthritis, the third indication approved by the agency.
The UCB’s biologic drug Cimzia is already on the market for rheumatoid arthritis and Crohn’s disease in both US and Europe. Cimzia, also known as Certolizumab pegol, is a monoclonal antibody directed against tumor necrosis factor alpha. It is a PEGylated Fab’ fragment of a humanized TNF inhibitor monoclonal antibody
read all at http://www.pharmatopics.com/2013/09/fda-gave-green-light-to-ucbs-cimzia-to-treat-psoriatic-arthritis/
Certolizumab pegol (CDP870, tradename Cimzia) is a therapeutic monoclonal antibody to tumor necrosis factor alpha (TNF-α), for the treatment of Crohn’s disease and rheumatoid arthritis, manufactured by UCB.

certolizumab pegol is a monoclonal antibody directed against tumor necrosis factor alpha. More precisely, it is a PEGylated Fab’fragment of a humanized TNF inhibitor monoclonal antibody.
Polyethylene glycol does not cross the placenta, so it should be safe in pregnancy.

Positive results have been demonstrated in two phase III trials (PRECiSE 1 and 2) of certolizumab pegol versus placebo in moderate to severe active Crohn’s disease. In addition, data from both trials suggest it is well tolerated. As yet its efficacy has not been directly compared to other anti-TNF-α agents.
Preliminary results of the RAPID 1 and 2 phase III studies were also reportedly positive.
In 2013, a phase 3 double blind randomized placebo-controlled study found significantly positive result in patient self-reported questionnaires, with rapid improvement of function and pain reduction.
On April 22, 2008, the U.S. Food and Drug Administration (FDA) approved Cimzia for use in the United States for the treatment of Crohn’s disease in people who did not respond sufficiently or adequately to standard therapy.
On June 26, 2009, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMEA) issued a positive opinion recommending that the European Commission grant a marketing authorisation for Cimzia for the treatment of rheumatoid arthritis only – the CHMP refused approval for the treatment of Crohn’s disease. The marketing authorisation was granted to UCB Pharma SA on October 1, 2009.



Daclizumab
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized (from mouse) |
| Target | CD25 |
 DACLIZUMAB,
DACLIZUMAB,

Daclizumab is a humanized monoclonal antibody indicated in the United States for prophylaxis of acute organ rejection in patients receiving renal transplants.
It was marketed as Zenepax, but discontinued by Roche in 2009 due to diminishing market demand for that indication. Biogen Idec is currently conducting phase III trials for daclizumab in MS. A phase III trial started in March 2010 is being conducted to determine efficacy of preventing MS relapse.
Study dosing of daclizumab is 150 mg subcutaneously once every 4 weeks versus interferon beta-1a (Avonex) 30 mg intramuscularly given once weekly for 96 to 144 weeks.
Daclizumab (Zenapax®) (molecular wt = 144 kd.) is a humanized monoclonal antibody (IgG1) produced by recombinant DNA technology. It gained FDA approval in Dec 1997. It is known by several other names including HAT (Humanized Anti-Tac), SMART anti-Tac, anti-CD25, and humanized anti-IL2-receptor. It was developed and patented by Protein Design Laboratories (Mountain View, CA) and it is marketed by Hoffman LaRoche (Nutley, NJ ).
Daclizumab is a composite of human (90%) and murine (10%) antibody sequences. In the model below, the murine portions are shown in red and dark blue; the rest of the molecule (gray color) represents the human sequence
The study is aiming for enrollment of 1500 patients and is expected to be complete in January 2014.
more info
Daclizumab (trade name Zenapax) is a therapeutic humanized monoclonal antibody. It is used to prevent rejection in organ transplantation, especially in kidney transplants. The drug is also under investigation for the treatment of multiple sclerosis.
Daclizumab works by binding to CD25, the alpha subunit of the IL-2 receptor of T cells. The drug is marketed in the US, but not in Europe.
Uses
Prevention of organ transplants
Daclizumab is given in multiple doses, the first 1 hour before the transplant operation and 5 further doses given at two week intervals after the transplant. These saturate the receptors and prevent T cell activation and thus prevent formation of antibodiesagainst the transplant.
Like the similar drug basiliximab, daclizumab reduces the incidence and severity of acute rejection in kidney transplantation without increasing the incidence of opportunistic infections.
Daclizumab usage may also be indicated in place of a calcineurin-inhibitor (ciclosporin or tacrolimus) during the early phase after kidney transplantation, when the kidney is recovering and vulnerable to calcineurin-inhibitor toxicity. This has been shown to be beneficial in non-heart beating donor kidney transplantation.
In the United Kingdom, the National Institute for Health and Clinical Excellence (NICE) has recommended its use be considered for all kidney transplant recipients.[citation needed]
Multiple sclerosis
In 2006 it began a Phase II clinical trial that finished in 2007 as a possible multiple sclerosis (MS) treatment. Participants were nine patients with multiple sclerosis not controlled with interferon. Daclizumab was effective in reducing lesions and improving clinical scores.[1] As of June 2013, the drug is in Phase III trials for this indication.[2]
Autoimmune diseases
Daclizumab has also been used to slow the progression of autoimmune diseases, particularly that of birdshot chorioretinopathy.[3]
Common side effects with a frequency of at least 10% include sleeplessness, tremor, headache, arterial hypertension, dyspnoea, gastrointestinal side effects and oedema. In rare cases, the drug can cause severe anaphylaxis.[4]
Daclizumab must not be administered to lactating women.[4]
History
Daclizumab was developed by PDL Biopharma, building on research at the National Institutes of Health (NIH).[5] Since December 1997, it is marketed by Hoffmann-La Roche in the US.
In April 2008, Hoffmann-La Roche submitted an application to have its marketing authorisation withdrawn in the EU for commercial reasons. The drug faced diminishing market demand, according to the company. There were no safety concerns with its use. As of January 2009, its marketing authorisation has been withdrawn and the product discontinued completely.[6][7]
- Rose JW, Burns JB, Bjorklund J, Klein J, Watt HE, Carlson NG (2007). “Daclizumab phase II trial in relapsing and remitting multiple sclerosis: MRI and clinical results”.Neurology 69 (8): 785–789. doi:10.1212/01.wnl.0000267662.41734.1f.PMID 17709711.
- ClinicalTrials.gov NCT01462318 An Immunogenicity and Pharmacokinetics (PK) Study of DAC HYP Prefilled Syringe in Relapsing Remitting Multiple Sclerosis (RRMS) (OBSERVE)
- Sobrin L, Huang JJ, Christen W, Kafkala C, Choopong P, Foster CS (2008). “Daclizumab for treatment of birdshot chorioretinopathy”. Arch Ophthalmol. 126 (2): 186–191. doi:10.1001/archophthalmol.2007.49. PMID 18268208.
- “EPAR for Zenapax”. European Medicines Agency. 2007.
- Tsurushita, N.; Hinton, P. R.; Kumar, S. (2005). “Design of humanized antibodies: From anti-Tac to Zenapax”. Methods 36 (1): 69–83.doi:10.1016/j.ymeth.2005.01.007. PMID 15848076. edit
- British National Formulary, Edition 57
- EMEA: Withdrawal of the marketing authorisation in the European Union

{kind=link}