








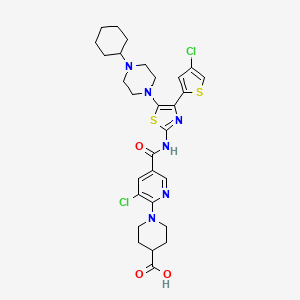
| Patent | Submitted | Granted |
|---|---|---|
| Compound useful for the treatment of degenerative and inflammatory diseases [US8088764] | 2010-12-30 | 2012-01-03 |
| NOVEL COMPOUNDS USEFUL FOR THE TREATMENT OF DEGENERATIVE AND INFLAMMATORY DISEASES [US2011190260] | 2011-08-04 |
Home » 0rphan drug status (Page 16)
Category Archives: 0rphan drug status
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
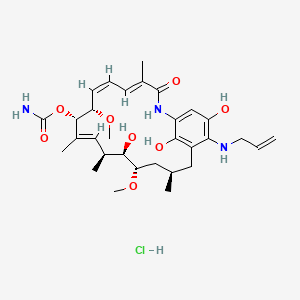
Umbralisib, TGR-1202, a Phosphoinositide-3 kinase delta inhibitor, Rhizen Pharmaceuticals S.A./TG Therapeutics
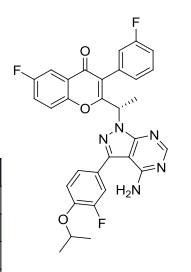

TGR 1202, TGR-1202-101, RP 5264, Umbralisib
AK173784;
(S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
(S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-3-(3-fluorophenyl)-4H-chromen-4-one,
2-[(1S)-1-[4-amino-3-(3-fluoro-4-propan-2-yloxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]ethyl]-6-fluoro-3-(3-fluorophenyl)chromen-4-one
CAS TOSYLATE 1532533-72-4 Umbralisib tosylate
CAS 1532533-67-7, 1514919-95-9
| Molecular Formula: | C31H24F3N5O3 |
|---|---|
| Molecular Weight: | 571.54917 g/mol |
RP-5307
TGR-1202
TGR-1202 PTSA
FU8XW5V3FS (UNII code)
RP-5264 (free base)
A PI3K inhibitor potentially for treatment of chronic lymphocytic leukemia, leukemia,lymphoma,B-cell
TGR‐1202, a next generation PI3K-δ delta inhibitor. TGR-1202 (RP-5264) is a highly specific, orally available, PI3K delta inhibitor, targeting the delta isoform with nanomolar potency and several fold selectivity over the alpha, beta, and gamma isoforms of PI3K.
TG Therapeutics, under license from Rhizen Pharmaceuticals, is developing TGR-1202 (structure shown; formerly RP-5264), a lead from a program of PI3K delta inhibitors, for the potential oral treatment of hematological cancers including Hodgkin lymphoma, non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), B-cell lymphoma and mantle cell lymphoma (MCL)
Incozen Therapeutics Pvt Ltd
TG Therapeutics
TGR-1202 potential to perform as the best PI3K inhibitor in its class and the possible superiority of TG-1101 over Rituxan®.
| Rhizen Pharmaceuticals S.A. | |
| Description | Phosphoinositide 3-kinase (PI3K) delta inhibitor |
Leukemia, chronic lymphocytic PHASE 3, TG Therapeutics
Orphan Drug
Umbralisib is a novel phosphatidylinositol 3-kinase delta (PI3Kdelta) inhibitor under development at TG Therapeutics in phase III clinical trials, in combination with ublituximab, for the treatment of chronic lymphocytic leukemia (CLL) and for the treatment of diffuse large B-cell lymphoma (DLBCL). The company refers to the combination regimen of ublituximab and TGR-1202 as TG-1303. The drug is also in phase II clinical development for the oral treatment of hematologic malignancies, as a single agent or in combination therapy. Phase I clinical trials are ongoing in patients with select relapsed or refractory solid tumors, such as adenocarcinoma of the pancreas, adenocarcinoma of the colon, rectum, gastric and GE junction cancer, and GI Stromal Tumor (GIST).
In 2016, orphan drug designation was assigned to the compound in the U.S. for the treatment of CLL. In 2017, additional orphan drug designation was granted in the U.S. for the treatment of CLL and DLBCL, in combination with ublituximab.
Originated by Rhizen Pharmaceuticals, the product was jointly developed by Rhizen Pharmaceuticals and TG Therapeutics since 2012. In 2014, exclusive global development and commercialization rights (excluding India) were licensed to TG Therapeutics.
CLINICAL TRIALS……….https://clinicaltrials.gov/search/intervention=TGR-1202
B-cell lymphoma; Chronic lymphocytic leukemia; Hematological neoplasm; Hodgkins disease; Mantle cell lymphoma; Non-Hodgkin lymphoma
Phosphoinositide-3 kinase delta inhibitor
SYNTHESIS


Rhizen Pharmaceuticals Announces Out-licensing Agreement for TGR-1202, a Novel Next Generation PI3K-delta Inhibitor
Rhizen to receive upfront payment of $8.0 million — Rhizen to retain global manufacturing and supply rights — Rhizen to retain development and commercialization for India
Rhizen to retain development and commercialization for India
| Source: Rhizen Pharmaceuticals SA
La Chaux-de-Fonds, Switzerland, Sept. 23, 2014 (GLOBE NEWSWIRE) — Rhizen Pharmaceuticals S.A. today announced an out-licensing agreement for TGR-1202, a novel next generation PI3K-delta inhibitor. TG Therapeutics exercised its option for early conversion to a licensing agreement from a 50:50 joint venture partnership.
In exchange for this licensing agreement, TG Therapeutics will pay Rhizen an upfront payment of $8.0 million ($4.0 million in cash and $4.0 million in TG Therapeutics common stock). In addition to the upfront payment, Rhizen will be eligible to receive regulatory filing, approval and sales based milestones in the aggregate of approximately $240 million, and tiered royalties based on net sales.
Swaroop Vakkalanka, Ph.D. and President of Rhizen stated, “We are extremely happy and take pride in discovering a novel, next generation, once-daily PI3K-delta inhibitor under active development led by TG Therapeutics. We are encouraged by the progress of TRG-1202 to date, and the speed at which TG Therapeutics is developing the asset in various hematological malignancies. We look forward to the day this novel drug reaches cancer patients in need of new and safe therapies.”
About Rhizen Pharmaceuticals S.A.:
Rhizen Pharmaceuticals is an innovative, clinical-stage biopharmaceutical company focused on the discovery and development of novel therapeutics for the treatment of cancer, immune and metabolic disorders. Since its establishment in 2008, Rhizen has created a diverse pipeline of proprietary drug candidates targeting several cancers and immune associated cellular pathways. Rhizen is headquartered in La-Chaux-de-Fonds, Switzerland. For additional information, please visit Rhizen’s website, www.rhizen.com.

TGR-1202.with Idelalisib and IPI-145 (left to right) for comparison.
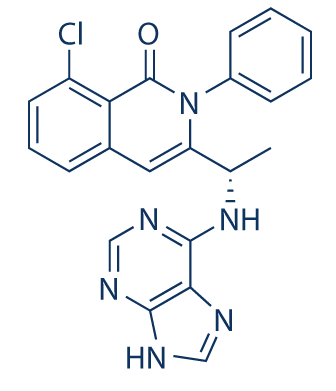
IPI 145
PATENTS
WO 2011055215
http://www.google.com/patents/WO2011055215A2?cl=en

PATENT
WO 2015181728
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015181728
TGR-1202, chemically known as (S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-3-(3-fluorophenyl)-4H-chromen-4-one, has the following chemical structure:

Example 1: Preparation of the PTSA Salt of TGR-1202 (Form A)

7100 g of TGR-1202 was charged in a reactor containing 56.8 litres of acetone and stirred at ambient temperature. 4680 g of p-toluene sulphonic acid was added and the reaction mixture was heated at a temperature of 60-65° C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 142 litres of diethyl ether was then added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass. The solid mass was re-suspended in diethyl ether, stirred for 6 hours, and then filtered to yield a solid mass which was subsequently dissolved in 56.8 litres of acetone, filtered through a HiFlow bed, and concentrated under reduced pressure. The resulting residue mass was stirred with water overnight, then filtered and vacuum dried to yield 6600 g of the PTSA salt of TGR-1202. HPLC: 99.21% and chiral purity of 99.64:0.36 (S:R).
Example 2: Preparation of the PTSA Salt of TGR-1202 (Form B)

1000 g of TGR-1202 was charged in a reactor containing 8 litres of acetone and stirred at ambient temperature. 666 g of p-toluene sulphonic acid was then added and the reaction mixture was heated at a temperature of 60-65 °C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 20 litres of diethyl ether was added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass which was then vacuum dried to yield 1150 g of the PTSA salt of TGR-1202. HPLC: 99.33% and chiral purity: 99.61:0.39 (S:R).
Table 1 lists the XRPD pattern peaks and relative peak intensities for the products of Examples 1 and 2.
TABLE 1

The tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 2 exhibited a Cmax about 2.5 fold and an area under the curve (AUC) about 1.9 fold greater than that of the tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 1. The results are provided in Table 8 below.
TABLE 8

PATENT
WO 2014071125
http://www.google.com/patents/WO2014071125A1?cl=en
formula (A) that is a ΡΒΚδ selective inhibitor,

(A)
Synthesis of Compound of Formula A
Unless otherwise stated, purification implies column chromatography using silica gel as the stationary phase and a mixture of petroleum ether (boiling at 60-80°C) and ethyl acetate or dichloromethane and methanol of suitable polarity as the mobile phases. The term “RT” refers to ambient temperature (25-28°C).
Intermediate 1 : 2-( l-bromoethyl)-6-fluoro-3-f3-fluorophenyl)-4H-chromen-4-one
Step-1 [l-(5-Fluoro-2-hydroxyphenyl)-2-(3-fluorophenyl)ethanone]: 3- Fluorophenylacetic acid (7.33 g, 47.56 mmoles) was dissolved in 25 ml dichloromethane. To this mixture, oxalylchloride (7.54 g, 59.46 mmoles) and DMF (3 drops) were added at 0°C and stirred for 30 min. The solvent was evaporated and dissolved in 25 ml dichloromethane. To this mixture, 4-fluoroanisole (5.00 g, 39.64 mmoles) was added and cooled to 0°C. At 0°C A1C13 (7.95 g, 59.46 mmoles) was added and the reaction mixture was warmed to RT and stirred for 12 hours. The reaction mixture was quenched by the addition of 2N HC1, extracted with ethyl acetate, dried over sodium sulphate and concentrated. The crude product was purified by column chromatography with ethyl acetate :petroleum ether to afford the title compound as colorless solid (4.5 g, 45% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 11.34 (s, 1H), 7.75 (dd, J=9.4, 3.1 Hz, 1H), 7.42 (m, 2H), 7.12 (m, 3H), 7.05 (dd, J=9.0, 4.5 Hz, 1H), 4.47 (s, 2H).
Step-2 [2-Ethyl-6-fiuoro-3-(3-fluorophenyl)-4H-chromen-4-one]: l-(5-Fluoro-2- hydroxyphenyl)-2-(3-fluorophenyl)ethanone obtained from Step-1 (3.00 g, 12.08 mmoles) was placed in a round bottom flask and to this triethylamine (25 ml) and propionic anhydride (4.92 g, 37.82 mmoles) were added, and the mixture was refluxed for 24 hours. After cooling to RT, the reaction mixture was acidified by the addition of IN HC1 solution, extracted with ethyl acetate, washed with sodium bicarbonate solution, dried with sodium sulphate and concentrated. The crude product was purified by column chromatography with ethyl acetate :petroleum ether to afford the title compound as off-yellow solid (1.80 g, 52% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 7.80 (m, 1H), 7.76 (m, 2H), 7.51 (dd, J=8.0, 6.4 Hz), 7.22 (m, 1H), 7.18 (m, 2H), 2.56 (q, J=7.6 Hz, 2H), 1.20 (t, J=7.6 Hz, 3H).
Step-3: To a solution of 2-Ethyl-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one obtained from Step-2 (1.80 g, 6.28 mmoles) in carbon tetrachloride (20 ml), N- bromosuccinimide (1.11 g, 6.28 mmoles) was added and heated to 80°C. Azobisisobutyronitrile (10 mg) was added to the reaction mixture at 80°C. After 12 hours, the reaction mixture was cooled to RT, diluted with dichloromethane and washed with water. The organic layer was dried over sodium sulphate and concentrated under reduced pressure to afford the crude title compound as yellow solid (1.25 g, 55% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 7.91 (dd, J=9.2, 4.3 Hz, 1H), 7.81 (dt, j=8.2, 2.8 Hz, 1H), 7.74 (dd, J=8.3, 3.1 Hz, 1H), 7.57 (m, 1H), 7.32 (dt, J=8.5, 2.4 Hz, 1H), 7.19 (m, 2H), 5.00 (q, J=6.8 Hz, 1H), 1.97 (d, J=6.8 Hz, 3H).
Intermediate 2: 6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

To a solution of Intermediate 1 (15.0 g, 40.84 mmol) in DMSO (150 ml), n-butanol (7.5 ml) was added and heated to 120°C for 3 hours. The reaction mixture was cooled to RT, quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (7.90 g, 64%). 1H-NMR (δ ppm, CDC13, 400 MHz): 7.85 (dd, J = 8.1, 3 Hz, 1H), 7.54 (dd, J = 9.2, 4.2 Hz, 1H), 7.47-7.37 (m, 2H), 7.15-6.98 (m, 3H), 4.74 (quintet, J= 6.8 Hz, 1H), 2.23 (d, J = 7.4 Hz, 1H), 1.54 (d, J = 6.6 Hz, 3H).
Intermediate 3 : 2-acetyl-6-fluoro-3-( 3-fluorophenyl)-4H-chromen-4-one

DMSO (5.60 ml, 79.14 mmol) was added to dichloromethane (40 ml), and cooled to – 78°C, followed by oxalyl chloride (3.40 ml, 39.57 mmol). After 10 min., intermediate 2 (6.00 g, 19.78 mmol) in dichloromethane (54 ml) was added dropwise and stirred for 20 min.
Triethylamine (12 ml) was added and stirred for 1 hour. The reaction mixture was quenched with water and extracted with dichloromethane. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (4.2 g, 71%) which was used as such in the next step.
Intermediate 4: fS)-6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

To intermediate 3 (2.00 g, 6.66 mmol), R-Alpine borane (0.5 M in THF, 20 ml) was added and heated to 60°C for 20 hours. The reaction mixture quenched with 2N HC1, and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (1.51 g, 75%).
Enantiomeric excess: 94.2%, enriched in the fast eluting isomer (retention time: 8.78 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 5: fR)-l-f6-fluoro-3-f3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl 4- chlorobenzoate

To a solution of intermediate 4 (1.45 g, 4.78 mmol) in THF (15 ml), 4-chlorobenzoic acid (0.748 g, 4.78 mmol) and triphenylphosphine (1.88 g, 7.17 mmol) were added and heated to 45°C followed by diisopropylazodicarboxylate (1.4 ml, 7.17 mmol). After 1 hour, the reaction mixture was concentrated and the residue was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (1.81 g, 86%) which was used without purification in the next step. Intermediate 6: fR)-6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

Method A
Intermediate 5 (1.75 g, 3.96 mmol) in methanol (17 ml) was cooled to 10°C, potassium carbonate (0.273 g, 1.98 mmol) was added and stirred for 30 min. The reaction mixture was concentrated, acidified with 2N HCl solution, extracted with ethyl acetate, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (1.05 g, 87% yield). Enantiomeric excess: 93.6%>, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Method B
Step-1 [(R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one]: To l-(5-fluoro-2-hydroxyphenyl)-2-(3-fluorophenyl)ethanone (11.00 g, 44.31 mmol) in dichloromethane, HATU (33.7 g, 88.63 mmol) and R-(+)2-benzyloxypropionic acid (9.58 g, 53.17 mmol) were added and stirred for 10 min. Triethylamine (66.7 ml, 0.47 mol) was added dropwise and stirred at RT for 24 hours. The reaction mixture was quenched with water, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate:
petroleum ether to afford the title compound as a yellow solid (10.5 g, 60%> yield). 1H-NMR (δ ppm, CDCls, 400 MHz): 7.85 (dd, J = 8.1,3 Hz, 1H), 7.58 (dd, J = 9.1, 4.1 Hz, 1H), 7.47-7.39 (m, 1H), 7.39-7.34 (m, 1H), 7.28-7.20 (m, 3H), 7.20-7.14 (m, 2H), 7.16-7.07 (m, 1H), 6.99-6.89 (m, 2H), 4.50-4.31 (m, 3H), 1.56 (d, J = 6.4 Hz, 3H).
Step-2: (R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one obtained in Step-1 (10.5 g, 26.69 mmol) in dichloromethane (110 ml) was cooled to 0°C, aluminium chloride (5.35 g, 40.03 mmol) was added portionwise and stirred at RT for 6 hours. The reaction mixture was quenched with 2N HCl solution, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford intermediate 6 a yellow solid (6.1 g, 76% yield). Enantiomeric excess: 97.7%, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 7: 4-bromo-2-fluoro-l-isopropoxybenzene

To a solution of 4-bromo-3-fluorophenol (10 g, 52.35 mmol) in THF (100ml), isopropyl alcohol (4.8 ml, 62.62 mmol) and triphenylphosphine (20.6 g, 78.52 mmol) were added and heated to 45°C followed by diisopropylazodicarboxylate (15.4 ml, 78.52 mmol). The mixture was refluxed for 1 hour, concentrated and the residue was purified by column
chromatography with ethyl acetate: petroleum ether to afford the title compound as a colorless liquid (13.1 g, 99% yield), which was used without purification in the next step.
Intermediate 8: 2-f3-fluoro-4-isopropoxyphenyl)-4,4,5.,5-tetramethyl-l,3i2-dioxaborolane

Potassium acetate (10.52 g, 107.2 mmol) and bis(pinacolato)diboron (15 g, 58.96 mmol) were added to a solution of intermediate 7 (10.52 g, 107.2 mmol) in dioxane (125 ml), and the solution was degassed for 30 min. [l, -Bis(diphenylphosphino)ferrocene]dichloro palladium(II) CH2CI2 (4.4 g, 5.36 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12 hours, the reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow oil (13.9g, 99%) which was used without purification in the next step.
Intermediate 9: 3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-dlpyrimidin-4-amine

To a solution of 3-iodo-lH-pyrazolo[3,4-d]pyrimidin-4-amine (11.0 g, 42.14 mmol) in DMF (110 ml), ethanol (55 ml) and water (55 ml), intermediate 8 (23.4 g, 84.28 mmol) and sodium carbonate (13.3 g, 126.42 mmol) were added and degassed for 30 min.
Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12 hours, the reaction mixture was filtered through celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
(RS)- 2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
To a solution of intermediate 9 (0.080 g, 0.293 mmol) in DMF (2 ml), potassium carbonate (0.081 g, 0.587 mmol) was added and stirred at RT for 10 min. To this mixture intermediate 1 (0.215 g, 0.587 mmol) was added and stirred for 12 hours. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as a pale yellow solid (0.045 g). MP: 175-177°C. 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 8.20 (s, 1H), 7.85 (dd, J = 81, 3.0 Hz, 1H), 7.48-7.33 (m, 5H), 7.14 (t, J= 8.3 Hz, 1H), 7.02 (m, 2H), 6.90 (m, 1H), 6.10 (q, J = 7.1 Hz, 1H), 5.42 (s, 2H), 4.64 (quintet, J = 6.0 Hz, 1H), 1.99 (d, J = 7.1 Hz, 3H), 1.42 (d, J= 6.1 Hz, 6H).
fS)-2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (“S-isomer”)
To a solution of intermediate 9 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 6 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.15 ml, 0.749 mmol) was added heated to 45°C. After 2 hours, the reaction mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 % yield). MP: 139-142°C. Mass: 571.7 (M+). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64 min.). fR)-2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-ehromen-4-one
To a solution of intermediate 8 (0.284 g, 0.989 mmol) in THF (5.0 ml), intermediate 4 (0.250 g, 0.824 mmol) and tris(4-methoxy)phenylphosphine (0.435 g, 1.23 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.25 ml, 1.23 mmol) was added stirred at RT. After 12 hours, the reaction mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate :
petroleum ether to afford the title compound as an off-white solid (0.105 g, 22 % yield). MP: 145-148°C. Mass: 571.7 (M+). Enantiomeric excess: 95.4% as determined by HPLC on a chiralpak AD-H column, enriched in the late eluting isomer (retention time = 14.83 min.).
PATENT
WO 2014006572
http://www.google.com/patents/WO2014006572A1?cl=en
 B1 IS DESIRED
B1 IS DESIRED
(S)-2- (l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-6- fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (compound-B l)
Intermediate 11
[119] Intermediate 11: 4-bromo-2-fluoro-l-isopropoxybenzene:To a solution of 4-bromo-2- fluorophenol (lOg, 52.35 mmol) in THF (100ml), isopropyl alcohol (4.8ml, 62.62 mmol) and triphenylphosphine (20.6g, 78.52 mmol) were added and heated to 45 C followed by diisopropylazodicarboxylate (15.4ml, 78 52 mmol). The mixture was refluxed for lh, concentrated and the residue was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a colourless liquid (13. lg, 99%) which was used without purification in the next step. Intermediate 12
[120] Intermediate 12: 2-(3-fluoro-4-isopropoxyphenyl)-4,4,5,5-tetramethyl- 1,3,2- dioxaborolane: Potassium acetate (10.52 g, 107.2 mmol) and bis(pinacolato)diboron (15g, 58.96 mmol) were added to a solution of intermediate 11 (10.52 g, 107.2 mmol) in dioxane (125 ml), and the solution was degassed for 30 min. [1,1 ‘- Bis(diphenylphosphino)ferrocene]dichloro palladium(II).CH2Cl2 (4.4g, 5.36 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h the reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow oil (13.9g, 99%) which was used without purification in the next step.
Intermediate 13
[121] Intermediate 13: 3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-4- amine: To a solution of 3-iodo-lH-pyrazolo[3,4-d]pyrimidin-4-amine (11.0 g, 42.14 mmol) in DMF 110 ml), ethanol (55 ml) and water (55 ml), intermediate 12 (23.4 g, 84.28 mmol) and sodium carbonate (13.3 g, 126.42 mmol) were added and degassed for 30 min. Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h, the reaction mixture was filtered though celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
Example Bl
(S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
[127] To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate ( 0.15 ml, 0.749 mmol) was added heated to 45°C. After 2h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 %). MP: 139- 142°C. Mass : 571.7 (M H-NMR (δ ppm, CDC13, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J = 8.2,3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J = 8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J = 8.4 Hz, 1H), 6.11 (q, J = 7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J = 6.1 Hz, 1H), 2.00 (d, J = 7.1Hz, 3H), 1.42 (d, J = 6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64min.).
PATENT
US 2014/0011819 describe the synthesis of TGR-1202 (Example B l)
http://www.google.co.in/patents/US20140011819
Example B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
-
To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.15 ml, 0.749 mmol) was added heated to 45° C. After 2 h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate:petroleum ether to afford the title compound as an off-white solid (0.049 g, 20%). MP: 139-142° C. Mass: 571.7 (M+).1H-NMR (δ ppm, CDCl3, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J=8.2, 3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J=8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J=8.4 Hz, 1H), 6.11 (q, J=7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J=6.1 Hz, 1H), 2.00 (d, J=7.1 Hz, 3H), 1.42 (d, J=6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=10.64 min)
4-Methylbenzenesulfonate Salt of Compound B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one 4-methylbenzenesulfonate
-

-
(S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one 4-methylbenzenesulfonate: To (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (22.7 g, 39.69 mmol) in isopropanol (600 ml), p-toluenesulphonic acid (8.30 g, 43.66 mmol) was added and refluxed for 1 h. The reaction mixture was concentrated, co-distilled with petroleum ether and dried. To the residue water (300 ml) was added and stirred for 30 min. The solid was filtered, washed with petroleum ether and dried under vacuum to afford the title compound as off-white solid (28.2 g, 95%). MP: 138-141° C. 1H-NMR (δ ppm, CDCl3, 400 MHz): 8.11 (s, 1H), 7.85 (dd, J=8.0, 3.0 Hz, 1H), 7.80 (d, J=8.2 Hz, 2H), 7.51 (dd, J=9.3, 4.3 Hz, 1H), 7.45 (dd, J=7.5, 3.1 Hz, 1H), 7.42-7.31 (m, 3H), 7.29 (m, 2H), 7.22 (d, J=8.0 Hz, 2H), 7.16 (t, J=8.3 Hz, 1H), 7.08 (dt, J=8.5, 2.5 Hz, 1H), 6.97 (br s, 1H), 6.88 (br s, 1H), 6.11 (q, J=7.2 Hz, 1H), 4.67 (quintet, J=6.0 Hz, 1H), 2.36 (s, 3H), 2.03 (d, J=7.1 Hz, 3H), 1.43 (d, J=6.0 Hz, 6H). Mass: 572.4 (M++1-PTSA). Enantiomeric excess: 93.4% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=12.35 min.)
Sulphate Salt of Compound B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one sulfate
-

-
(S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one sulphate: To (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (15.0 g, 26.24 mmol) in isopropanol (600 ml) was cooled to 0° C. To this Sulphuric acid (2.83 g, 28.86 mmol) was added and stirred at room temperature for 24 h. The reaction mass was filtered and washed with petroleum ether and dried under vacuum. To the solid, water (150 ml) was added and stirred for 30 min. The solid was filtered, washed with petroleum ether and dried under vacuum to afford the title compound as off-white solid (13.5 g, 76%). MP: 125-127° C. 1H-NMR (δ ppm, CDCl3, 400 MHz): 8.11 (s, 1H), 7.85 (dd, J=8.0, 3.0 Hz, 1H), 7.51 (dd, J=9.2, 4.2 Hz, 1H), 7.45-7.31 (m, 3H), 7.29 (m, 1H), 7.15 (t, J=8.3 Hz, 1H), 7.08 (dt, J=8.5, 2.4 Hz, 1H), 6.96 (br s, 1H), 6.88 (br s, 1H), 6.09 (q, J=7.1 Hz, 1H), 4.676 (quintet, J=6.1 Hz, 1H), 2.01 (d, J=7.1 Hz, 3H), 1.42 (d, J=6.1 Hz, 6H). Mass: 572.2 (M++1-H2SO4). Enantiomeric excess: 89.6% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=12.08 min.)
-
Various other acid addition salts of compound B1 were prepared as provided in Table 1.
-
TABLE 1 Melting Point Acid Method of preparation (° C.) Hydro- Compound B1 (1 eq.) dissolved in THF, 130-132 chloric excess HCl/Et2O was added, the clear acid solution obtained was evaporated completely. The residue obtained was washed with water. p- Compound B1 (1 eq.) dissolved in 138-141° C. Toluene- isopropyl alcohol (IPA), refluxed for sulfonic 30 min., acid (1.1 eq.) in IPA was added, acid the clear solution obtained was evaporated completely. The residue obtained was washed with water. Benzene- Compound B1 (1 eq.) dissolved in IPA, 170-172 sulphonic refluxed for 30 min., acid(1.1 eq.) in IPA acid was added, the clear solution not obtained, the residue was evaporated completely and was washed with water. Maleic Compound B1 (1 eq.) dissolved in IPA, 107-109 acid refluxed for 30 min., acid (1.1 eq.) in IPA was added, the clear solution not obtained, the residue was evaporated completely and was washed with water. Camphor Compound B1 (1 eq.) dissolved in IPA, 120-121 sulfonic refluxed for 30 min., acid (1.1 eq.) in IPA acid was added, the clear solution not obtained, the residue was evaporated completely and was washed with water. Sulphuric Compound B1 (1 eq.) dissolved in IPA, 125-127 acid refluxed for 30 min., acid(1.1 eq.) in IPA was added, the clear solution obtained was evaporated completely. The residue obtained was washed with water.
REFERENCES
WO 2014/006572 and U.S. Patent Publication No. 2014/0011819,
http://www.tgtherapeutics.com/O’ConnorTGR202Single%20AgentEHA&Lugano2015.pdf
-
TGR-1202: Phase I/II started 09/28/2015
Week in Review, Clinical StatusRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) … -
Ublituximab: Phase I/II started 09/28/2015
Week in Review, Clinical StatusLFB S.A., Les Ulis, France TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: Ublituximab (TGTX-1101, TG-1101, LFB-R603) Business: Cancer Molecular target: CD20 Description: Glycoengineered mAb against CD20 … -
The Daily Extra, Company NewsTG Therapeutics Inc. (NASDAQ:TGTX) rose $2.65 (23%) to $14.37 after the company said it received an SPA from FDA for the Phase III UNITY-CLL trial of ublituximab (TG-1101) in combination with TGR-1202 to treat chronic …
-
Targets & Mechanisms: The battle for IRAK 04/23/2015
Nimbus, Aurigene and TG Therapeutics are chasing IRAK4 inhibitors for cancerBC Innovations, Targets & MechanismsNow that Nimbus has put IRAK4 on the map for B cell lymphoma, several companies are closing in with their own inhibitors, and they’re all on track for IND-enabling studies this year. -
TGR-1202: Additional Phase I/II data 01/26/2015
Week in Review, Clinical ResultsRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) … -
Ublituximab: Additional Phase I/II data 01/26/2015
Week in Review, Clinical ResultsLFB S.A., Les Ulis, France TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Ildong Pharmaceutical Co. Ltd. (KSE:000230), Seoul, South Korea Product: Ublituximab (TGTX-1101, TG-1101, LFB-R603) Business: Cancer … -
TGR-1202: Phase I started 12/15/2014
Week in Review, Clinical StatusRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) … -
Rhizen, TG Therapeutics deal 12/08/2014
Week in Review, DealsRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Business: Cancer TG Therapeutics exercised an option under a 2012 deal to license exclusive, worldwide …
| Patent | Submitted | Granted |
|---|---|---|
| NOVEL SELECTIVE PI3K DELTA INHIBITORS [US2014011819] | 2013-07-02 | 2014-01-09 |
| Treatment Of Cancers Using PI3 Kinase Isoform Modulators [US2014377258] | 2014-05-30 | 2014-12-25 |
////////Umbralisib
CC(C)OC1=C(C=C(C=C1)C2=NN(C3=C2C(=NC=N3)N)C(C)C4=C(C(=O)C5=C(O4)C=CC(=C5)F)C6=CC(=CC=C6)F)F
IPI 504, Retaspamycin, Retaspimycin

IPI 504, Retaspamycin, Retaspimycin
CAS 857402-63-2
Cas 857402-23-4 ( Retaspimycin); 857402-63-2 ( Retaspimycin HCl).
MF C31H45N3O8 BASE
MW: 587.32067 BASE
Infinity Pharmaceuticals Inc, INNOVATOR
[(3R,5S,6R,7S,8E,10S,11S,12Z,14E)-6,20,22-trihydroxy-5,11-dimethoxy-3,7,9,15-tetramethyl-16-oxo-21-(prop-2-enylamino)-17-azabicyclo[16.3.1]docosa-1(22),8,12,14,18,20-hexaen-10-yl] carbamate;hydrochloride
17-Allylamino-17-demethoxygeldanamycin Hydroquinone Hydrochloride
| Retaspimycin hydrochloride; 8,21-didehydro-17-demethoxy-18,21-dideoxo-18,21-dihydroxy-17-(2-propenylamino)-geldanamycin monohydrochloride | |
| Application: | A novel, water-soluble, potent inhibitor of heat-shock protein 90 (Hsp90) |
| Molecular Weight: | 624.17 ……….HCl salt |
| Molecular Formula: | C31H46ClN3O8……….HCl salt |
Introduction
IPI-504 is a novel, water-soluble, potent inhibitor of heat-shock protein 90 (Hsp90).
Orphan drug designation was assigned to the compound by the FDA for the treatment of gastrointestinal stromal cancer (GIST).

Retaspimycin Hydrochloride is the hydrochloride salt of a small-molecule inhibitor of heat shock protein 90 (HSP90) with antiproliferative and antineoplastic activities. Retaspimycinbinds to and inhibits the cytosolic chaperone functions of HSP90, which maintains the stability and functional shape of many oncogenic signaling proteins and may be overexpressed or overactive in tumor cells. Retaspimycin-mediated inhibition of HSP90 promotes the proteasomal degradation of oncogenic signaling proteins in susceptible tumor cell populations, which may result in the induction of apoptosis.
Phase I study of Retaspimycin: A phase 1 study of IPI-504 (retaspimycin hydrochloride) administered intravenously twice weekly for 2 weeks at 22.5, 45, 90, 150, 225, 300 or 400 mg/m(2) followed by 10 days off-treatment was conducted to determine the safety and maximum tolerated dose (MTD) of IPI-504 in patients with relapsed or relapsed/refractory multiple myeloma (MM). Anti-tumor activity and pharmacokinetics were also evaluated. Eighteen patients (mean age 60.5 years; median 9 prior therapies) were enrolled. No dose-limiting toxicities (DLTs) were reported for IPI-504 doses up to 400 mg/m(2).
The most common treatment-related adverse event was grade 1 infusion site pain (four patients). All other treatment-related events were assessed as grade 1 or 2 in severity. The area under the curve (AUC) increased with increasing dose, and the mean half-life was approximately 2-4 h for IPI-504 and its metabolites. Four patients had stable disease, demonstrating modest single-agent activity in relapsed or relapsed/refractory MM. (source: Leuk Lymphoma. 2011 Dec;52(12):2308-15.)

Figure Hsp90 protein partners and clients destabilized by Hsp90 inhibition (Jackson et al., 2004).
In a different approach, Infinity Pharmaceuticals has developed IPI504 (retaspimycin or 17-AAG hydroquinone, Figure 4) (Adams et al., 2005; Sydor et al., 2006), a new GA analogue, in which the quinone moiety was replaced by a dihydroquinone one. Indeed, the preclinical data suggested that the hepatotoxicity of 17-AAG was attributable to the ansamycin benzoquinone moiety, prone to nucleophilic attack.
Furthermore, it was recently reported that the hydroquinone form binds Hsp90 with more efficiency than the corresponding quinone form (Maroney et al., 2006). In biological conditions, the hydroquinone form can interconvert with GA, depending on redox equilibrium existing in cell. It has been recently proposed, that NQ01 (NAD(P)H: quinone oxidoreductase) can produce the active hydroquinone from the quinone form of IPI504 (Chiosis, 2006).
However, Infinity Pharmaceuticals showed that if the overexpression of NQ01 increased the level of hydroquinone and cell sensitivity to IPI504, it has no significant effect on its growth inhibitory activity. These results suggest that NQ01 is not a determinant of IPI504 activity in vivo (Douglas et al., 2009).

Figure 4: GA, 17-AAG, 17-DMAG and IPI504.
PATENT
http://www.google.com/patents/EP2321645A1?cl=en
Geldanamycin (IUPAC name ([18S-(4E,5Z,8R*.9R*.10E,12R*.13S*,14R*,l6S*)]- 9- [(aminocarbonyl)oxy]- 13- hydroxy- 8,14,19- trimetoxy- 4,10,12,16- tetramethyl- 2- azabicyclo[16.3.1.]docosa- 4,6.10,18,21- pentan- 3.20,22trion) is a benzoquinone ansamycin antibiotic which may be produced by the bacterium Streptorayces hygroscopicus. Geldanamycin binds specifically to HSP90 (Heat Shock Protein 90) and alters its function.
While Hsp90 generally stabilizes folding of proteins and, in particular in tumor cells, folding of overexpressed/mutant proteins such as v-Src. Bcr-Abl and p53. the Hsp90 inhibitor Geldanamycin induces degradation of such proteins.
The respectiv e formula of geldanamycin is given herein below:

E\en though geldanamycin is a potent antitumor agent, the use of geldanamycin also shows some negathe side-effects (e.g. hepatotoxicity) which led to the dev elopment of geldanamycin analogues/derivatives, in particular analogues/deriv atives containing a derivatisation at the 17 position. Without being bound by theory , modification at the 17 position of geldanamycin may lead to decreases hepatotoxicity.
Accordingly geldanamycin analogues/derivatives which are modified at the 17 position, such as 17-AAG (17-N-Allylamino-17-demethoxygeldanamycin), are preferred in context of the present invention. Also preferred herein are geldanamycin derivatives to be used in accordance with the present invention which are water-soluble or which can be dissoh ed in water completely (at least 90 %. more preferably 95 %. 96 %. 97 %, 98 % and most preferably 99 %). 17-AAG ([QS.5S,6RJS$EΛ0R,l \SΛ2E,14E)-2\- (allylamino)-6-hydroxy-5.11-diraethoxy-
3.7.9,15-tetramethyl-16.20.22-trioxo-17-azabicyclo[16.3.1]docosa-8,12.14,18,21-pentaen-10- yl] carbamate) is. as mentioned above a preferred derivative of geldanamycin. 17- AAG is commercially available under the trade name “Tanespimycin“ (also known as KOS-953) for example from Kosan Biosciences Incorporated (Acquired by Bristol-Myers Squibb Company). Tanespimycin is presently studied in phase II clinical trials for multiple myeloma and breast cancer and is usually administered intravenously.
The respective formula of 17- AAG is given herein below:

Preferred geldanamycin-derh ative (HSP90 inhibitor) to be used in context of the present invention are IPI-504 (also known as retaspiimcin or Mcdi-561 : lnfinin Pharmaceuticals (Medlmmunc/ Astra Zeneca)). Clinical trials on the use of IPI-504 (which is usually administered intravenously) in the treatment of non-small cell lung cancer (NSCLC) and breast cancer are performed. Also alvespimycin hy drochloride (Kosan Biosciences Incorporated Acquired By : Bristol-Myers Squibb Company) is a highly potent, water-soluble and orally acti\e derivative of geldanamycin preferably used in context of the present invention.

IPI-504
PATENT
WO 2005063714
http://www.google.co.ug/patents/WO2005063714A1?cl=en
Example 24
Preparation of Air-stable Hydroquinone Derivatives of the Geldanamycin Family of Molecules
,

17-Allylamino-17-Demethoxygeldanamycin (10.0 g, 17.1 mmol) in ethyl acetate
(200 mL) was stirred vigorously with a freshly prepared solution of 10% aqueous sodium hydrosulfite (200 mL) for 2 h at ambient temperature. The color changed from dark purple to bright yellow, indicating a complete reaction. The layers were separated and the organic phase was dried with magnesium sulfate (15 g). The drying agent was rinsed with ethyl acetate (50 mL). The combined filtrate was acidified with 1.5 M hydrogen chloride in ethyl acetate (12 mL) to pH 2 over 20 min. The resulting slurry was stirred for 1.5 h at ambient temperature. The solids were isolated by filtration, rinsed with ethyl acetate (50 mL) and dried at 40 °C, 1 mm Hg, for 16 h to afford 9.9 g (91%) of off-white solid. Crude hydroquinone hydrochloride (2.5 g) was added to a stirred solution of 5% 0.01 N aq. hydrochloric acid in methanol (5 mL). The resulting solution was clarified by filtration then diluted with acetone (70 mL). Solids appeared after 2-3 min. The resulting slurry was stirred for 3 h at ambient temperature, then for 1 h at 0-5 °C. The solids were isolated by filtration, rinsed with acetone (15 mL) and dried
PAPER
J. Med. Chem., 2006, 49 (15), pp 4606–4615
DOI: 10.1021/jm0603116

17-Allylamino-17-demethoxygeldanamycin (17-AAG)1 is a semisynthetic inhibitor of the 90 kDa heat shock protein (Hsp90) currently in clinical trials for the treatment of cancer. However, 17-AAG faces challenging formulation issues due to its poor solubility. Here we report the synthesis and evaluation of a highly soluble hydroquinone hydrochloride derivative of 17-AAG, 1a (IPI-504), and several of the physiological metabolites. These compounds show comparable binding affinity to human Hsp90 and its endoplasmic reticulum (ER) homologue, the 94 kDa glucose regulated protein (Grp94). Furthermore, the compounds inhibit the growth of the human cancer cell lines SKBR3 and SKOV3, which overexpress Hsp90 client protein Her2, and cause down-regulation of Her2 as well as induction of Hsp70 consistent with Hsp90 inhibition. There is a clear correlation between the measured binding affinity of the compounds and their cellular activities. Upon the basis of its potent activity against Hsp90 and a significant improvement in solubility, 1a is currently under evaluation in Phase I clinical trials for cancer.
17-Allylamino-17-demethoxygeldanamycin Hydroquinone Hydrochloride Ia
17-AAG hydroquinone hydrochloride (1a) as an off-white solid (11 g, 18 mmol, 80% yield). HPLC purity: 99.6%;
IR (neat): 3175, 2972, 1728, 1651, 1581, 1546, 1456, 1392, 1316, 1224, 1099, 1036 cm-1;
1H NMR (CDCl3:d6-DMSO, 6:1, 400 MHz):
δ 10.20 (1H, br), 9.62 (2H, br), 8.53 (1H, s), 8.47 (1H, s), 7.74 (1H, s), 6.72 (1H, d, J= 11.6 Hz), 6.28 (1H, dd, J = 11.6, 11.2 Hz), 5.73 (1H, dddd, J = 17.2, 10.0, 3.2, 2.4 Hz), 5.53 (1H, d, J = 10.4 Hz), 5.49 (1H, dd, J = 10.8, 10.0 Hz), 5.32 (2H, s), 5.04 (1H, d, J = 4.8 Hz), 5.02 (1H, d, J = 16.0 Hz), 4.81 (1H, s), 4.07 (1H, d, J = 9.6 Hz), 3.67 (2H, d, J = 6.4 Hz), 3.31 (1H, d,J = 8.8 Hz), 3.07 (3H, s), 3.07−3.04 (1H, m), 2.99 (3H, s), 2.64 (1H, m), 2.52−2.49 (1H, m), 1.76 (3H, s), 1.61−1.39 (3H, m), 0.78 (3H, d, J = 6.4 Hz), 0.64 (3H, d, J = 7.2 Hz);
13C NMR (CDCl3:d6-DMSO, 6:1, 100 MHz): δ 167.3, 155.8, 143.3, 136.3, 135.0, 134.2, 132.9, 132.1, 128.8, 127.6, 125.9, 125.3, 123.7, 123.0, 115.1, 104.5, 80.9, 80.7, 80.1, 72.5, 56.2, 56.2, 52.4, 34.6, 33.2, 31.1, 27.2, 21.6, 12.1, 12.1, 11.7;
HRMS calculated for C31H45N3O8 (M+ + H): 588.3285, Found 588.3273.
POSTER
Synthesis and biological evaluation of IPI-504, an aqueous soluble analog of 17-AAG and potent inhibitor of Hsp90
MEDI 210 |
| IPI-504 is the hydroquinone hydrochloride salt of 17-allylamino-17-demethoxy-geldanamycin (17-AAG), an Hsp90 inhibitor that is currently in clinical trials for the treatment of cancer.
IPI-504 demonstrates high aqueous solubility (>200 mg/mL). Interestingly, in vitro and in vivo IPI-504 interconverts with 17-AAG and exists in a pH and enzyme-mediated redox equilibrium. This occurs due to oxidation of the hydroquinone (IPI-504) to the quinone (17-AAG) at physiological pH and the reduction of 17-AAG by quinone reductases such as NQO1 to IPI-504. Here we report the design and synthesis of the stabilized hydroquinone IPI-504 and its inhibitory effect against Hsp90 and Grp94. Although IPI-504 was originally designed to be a soluble prodrug of 17-AAG, the hydroquinone is more potent than the quinone in the biochemical Hsp90 binding assay. Various hydroquinone analogs have been prepared to investigate the structure activity relationship of hydroquinone binding to Hsp90. Hydroquinone and quinone forms of 17-AAG metabolites show comparable binding affinities for Hsp90 and in cancer cell lines, hydroquinone analogs elicit specific responses consistent with Hsp90 inhibition. The desirable pharmacological properties as well as in vitro and in vivo activity of our lead compound, IPI-504, has led to the initiation of Phase I clinical trials in multiple myeloma. |
| http://oasys2.confex.com/acs/231nm/techprogram/P945016.HTM |
|
Geldanamycin and Beyond: Progress Toward the Development of HSP90 Inhibitors
8:30 AM-12:05 PM, Wednesday, 29 March 2006 Georgia World Congress Center — Georgia Ballroom 1, OralDivision of Medicinal ChemistryThe 231st ACS National Meeting, Atlanta, GA, March 26-30, 2006 |
References
Synthesis and biological evaluation of IPI-504, an aqueous soluble analog of 17-AAG and potent inhibitor of Hsp90
231st Am Chem Soc (ACS) Natl Meet (March 26-30, Atlanta) 2006, Abst MEDI 210
Design, synthesis, and biological evaluation of hydroquinone derivatives of 17-amino-17-demethoxygeldanamycin as potent, water-soluble inhibitors of Hsp90
J Med Chem 2006, 49(15): 4606
http://www.biotechduediligence.com/retaspamycin-hcl-ipi-504.html
///////////////////Hsp90, IPI-504, infinity pharma, Retaspamycin, Retaspimycin
Defibrotide


Defibrotide sodium is an oligonucleotide mixture with profibrinolytic properties. The chemical name of defibrotide sodium is polydeoxyribonucleotide, sodium salt. Defibrotide sodium is a polydisperse mixture of predominantly single-stranded (ss) polydeoxyribonucleotide sodium salts derived from porcine intestinal tissue having a mean weighted molecular weight of 13-20 kDa, and a potency of 27-39 and 28-38 biological units per mg as determined by two separate assays measuring the release of a product formed by contact between defibrotide sodium, plasmin and a plasmin substrate. The primary structure of defibrotide sodium is shown below.
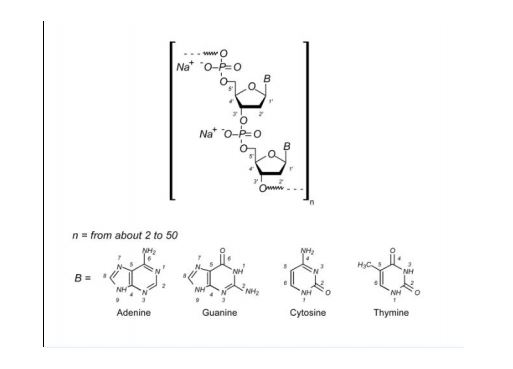
DEFITELIO (defibrotide sodium) injection is a clear, light yellow to brown, sterile, preservative-free solution in a single-patient-use vial for intravenous use. Each milliliter of the injection contains 80 mg of defibrotide sodium and 10 mg of Sodium Citrate, USP, in Water for Injection, USP. Hydrochloric Acid, NF, and/or Sodium Hydroxide, NF, may have been used to adjust pH to 6.8-7.8.
Defibrotide is the sodium salt of a mixture of single-stranded oligodeoxyribonucleotides derived from porcine mucosal DNA. It has been shown to have antithrombotic, anti-inflammatory and anti-ischemic properties (but without associated significant systemic anticoagulant effects). It is marketed under the brand names Dasovas (FM), Noravid, and Prociclide in a variety of countries, but is currently not approved in the USA. The manufacturer is Gentium.
Defibrotide is used to treat or prevent a failure of normal blood flow (occlusive venous disease, OVD) in the liver of patients who have had bone marrow transplants or received certain drugs such as oral estrogens, mercaptopurine, and many others.
In 2012, an IND was filed in Japan seeking approval of the compound for the treatment of veno-occlusive disease.
Approved 3/30/3016 US FDA, defibrotide sodium, (NDA) 208114

To treat adults and children who develop hepatic veno-occlusive disease with additional kidney or lung abnormalities after they receive a stem cell transplant from blood or bone marrow called hematopoietic stem cell transplantation
Polydeoxyribonucleotides from bovine lung or other mamalian organs with molecular weight between 15,000 and 30,000 Da
CAS 83712-60-1
Defibrotide is a polydisperse mixture of oligonucleotides produced by random, chemical cleavage (depolymerisation) of porcine DNA. It is predominantly single stranded, of varying base sequence, lengths and conformations; unfolded, folded or combined. The mean oligonucleotide length is 50 bases with a mean molecular weight of 17 ± 4 kDa. No individually defined component is at more than femtomolar concentration. The only meaningful scientific information that can be obtained about the biochemical nature of defibrotide (aside from determination of percentage of each nucleobase) is a measurement of its average length and its average percentage double stranded character. Therefore, it can be established that this active substance is of highly heterogenic nature.

Defibrotide (Defitelio, Gentium)[1] is a deoxyribonucleic acid derivative (single-stranded) derived from cow lung or porcine mucosa. It is an anticoagulant with a multiple mode of action (see below).
It has been used with antithrombin III.[2]
Jazz Pharmaceuticals plc announced that the FDA has accepted for filing with Priority Review its recently submitted New Drug Application (NDA) for defibrotide. AS ON OCT 2015
Defibrotide is an investigational agent proposed for the treatment of patients with hepatic veno-occlusive disease (VOD), also known as sinusoidal obstruction syndrome (SOS), with evidence of multi-organ dysfunction (MOD) following hematopoietic stem-cell transplantation (HSCT).
Priority Review status is designated for drugs that may offer major advances in treatment or provide a treatment where no adequate therapy exists. Based on timelines established by the Prescription Drug User Fee Act (PDUFA), FDA review of the NDA is expected to be completed by March 31, 2016.
“The FDA’s acceptance for filing and Priority Review status of the NDA for defibrotide is an important milestone for Jazz and reflects our commitment to bringing meaningful medicines to patients who have significant unmet needs,” said Karen Smith, M.D., Ph.D., Global Head of Research and Development and Chief Medical Officer of Jazz Pharmaceuticals. “We look forward to continuing to work closely with the FDA to obtain approval for defibrotide for patients with hepatic VOD with evidence of MOD in the U.S. as quickly as possible, as there are no other approved therapies for treating this rare, often fatal complication of HSCT.”
The NDA includes safety and efficacy data from three clinical studies of defibrotide for the treatment of hepatic VOD with MOD following HSCT, as well as a retrospective review of registry data from the Center for International Blood and Marrow Transplant Research. The safety database includes over 900 patients exposed to defibrotide in the clinical development program for the treatment of hepatic VOD.
The compound was originally developed under a collaboration between Sanofi and Gentium. In December 2001, Gentium entered into a license and supply agreement with Sigma-Tau Pharmaceuticals, pursuant to which the latter gained exclusive rights to distribute, market and sell the product for the treatment of VOD in the U.S. This agreement was expanded in 2005 to include all of North America, Central America and South America.
Defibrotide was granted orphan drug designations from the FDA in July 1985, May 2003 and January 2007 for the treatment of thrombotic thrombocytopenic purpura (TTP), for the treatment of VOD and for the prevention of VOD, respectively. Orphan drug was also received in the E.U. for the prevention and treatment of hepatic veno-occlusive disease (VOD) in 2004 and for the prevention of graft versus host disease (GvHD) in 2013.
Pharmacokinetics
Defibrotide is available as an oral, intravenous, and intramuscular formulation. Its oral bioavailability is in the range of 58-70% of theparenteral forms. T1/2 alpha is in the range of minutes while T1/2 beta is in the range of hours in studies with oral radiolabelleddefibrotide. These data suggest that defibrotide, in spite of its macromolecular nature, is absorbed well after oral administration. Due to the drug’s short half-life, it is necessary to give the daily dose divided in 2 to 4 doses (see below).
In 2014, Jazz Pharmaceuticals (parent of Gentium) acquired the rights of the product in U.S. and in the Americas
Mode of action
The drug appears to prevent the formation of blood clots and to help dissolve blood clots by increasing levels of prostaglandin I2, E2, and prostacyclin, altering platelet activity, increasing tissue plasminogen activator (tPA-)function, and decreasing activity of tissue plasminogen activator inhibitor. Prostaglandin I2 relaxes the smooth muscle of blood vessels and prevents platelets from adhering to each other. Prostaglandin E2 at certain concentrations also inhibits platelet aggregation. Moreover, the drug provides additional beneficial anti-inflammatory and antiischemic activities as recent studies have shown. It is yet unclear, if the latter effects can be utilized clinically (e.g., treatment of ischemic stroke).
Unlike heparin and warfarin, defibrotide appears to have a relatively mild anticoagulant activity, which may be beneficial in the treatment of patients at high risk of bleeding complications. Nevertheless, patients with known bleeding disorders (e.g., hemophilia A) or recent abnormal bleedings should be treated cautiously and under close medical supervision.
The drug was marketed under the brand names Dasovas (FM), Noravid, and Prociclide in a variety of countries. It is currently not approved in the USA. The manufacturer is Gentium.
Defibrotide also received fast track designation from the FDA for the treatment of severe VOD in recipients of stem cell transplants. In 2011, the compound was licensed to Medison Pharma by Gentium in Israel and Palestine. The license covers the management of named-patient sales program and local registration, authorization, marketing, reimbursement and medical affairs for the treatment of peripheral vascular disease.
Usual indications
Defibrotide is used to treat or prevent a failure of normal blood flow (Veno-occlusive disease, VOD) in the liver of patients having had bone marrow transplants or received certain drugs such as oral estrogens, mercaptopurine, and many others. Without intensive treatment, VOD is often a fatal condition, leading to multiorgan failure. It has repeatedly been reported that defibrotide was able to resolve the condition completely and was well tolerated.
Other indications are: peripheral obliterative arterial disease, thrombophlebitis, and Raynaud’s phenomenon. In very high doses, defibrotide is useful as treatment of acute myocardial infarction. The drug may also be used for the pre- and postoperative prophylaxis of deep venous thrombosis and can replace the heparin use during hemodialytic treatments.
It has been investigated for use in treatment of chronic venous insufficiency.[3]
Potential indications in the future
Other recent preclinical studies have demonstrated that defibrotide used in conjunction with Granulocyte Colony-Stimulating Factor (rhG-CSF) significantly increases the number of Peripheral Blood Progenitor Cells (Stem cells). The benefit of this increase in stem cells may be crucial for a variety of clinical indications, including graft engineering procedures and gene therapy programs. This would expand the clinical usefulness of defibrotide to a complete distinct area.
Very recently (since early 2006) combination therapy trials (phase I/II) with defibrotide plus melphalan, prednisone, and thalidomide in patients with multiple myeloma have been conducted. The addition of defibrotide is expected to decrease the myelosuppressive toxicity of melphalan. However, is too early for any definitive results at that stage.
Cautions and contraindications
- The efficacy of the drug has been reported to be poorer in patients with diabetes mellitus.
- Pregnancy: The drug should not be used during pregnancy, because adequate and well controlled human studies do not exist.
- Lactation: No human data is available. In order to avoid damage to the newborn, the nursing mother should discontinue either the drug or breastfeeding, taking into account the importance of treatment to the mother.
- Known Bleeding Disorders or Bleeding Tendencies having occurred recently: Defibrotide should be used cautiously. Before initiation of treatment, the usual coagulation values should be obtained as baseline and regularly controlled under treatment. The patient should be observed regularly regarding local or systemic bleeding events.
Side-effects
Increased bleeding and bruising tendency, irritation at the injection site, nausea, vomiting, heartburn, low blood pressure. Serious allergic reactions have not been observed so far.
Drug interactions
Use of heparin with defibrotide may increase the aPTT, reflecting reduced ability of the body to form a clot. Nothing is known about the concomitant application of other anticoagulants than heparin and dextran containing plasma-expanders, but it can be anticipated that the risk of serious bleeding will be increased considerably.
PATENT
WO 2001078761
G-CSF (CAS registry number 143011-2-7/Merck Index, 1996, page 4558) is a haematopoietic growth factor which is indispensable in the proliferation and differentiation of the progenitor cells of granulocytes; it is a 18-22 kDa glycoprotein normally produced in response to specific stimulation by a variety of cells, including monocytes, fibroblasts and endothelial cells. The term defibrotide (CAS registry number 83712-60-1) normally identifies a polydeoxyribonucleotide obtained by extraction (US 3,770,720 and US 3,899,481) from animal and/or vegetable tissue; this polydeoxyribonucleotide is normally used in the form of a salt of an alkali metal, generally sodium. Defibrotide is used principally for its anti- thrombotic activity (US 3,829,567) although it may be used in different applications, such as, for example, the treatment of acute renal insufficiency (US 4,694,134) and the treatment of acute myocardial ischaemia (US 4,693,995). United States patents US 4,985,552 and US 5,223,609, finally, describe a process for the production of defibrotide which enables a product to be obtained which has constant and well defined physico-chemical characteristics and is also free from any undesired side-effects
References
- “Jazz Pharma Acquiring Gentium for $1B”. Gen. Eng. Biotechnol. News (paper) 34 (2). January 15, 2014. p. 10.
- Haussmann U, Fischer J, Eber S, Scherer F, Seger R, Gungor T (June 2006). “Hepatic veno-occlusive disease in pediatric stem cell transplantation: impact of pre-emptive antithrombin III replacement and combined antithrombin III/defibrotide therapy”. Haematologica 91 (6): 795–800. PMID 16769582.
- Coccheri S, Andreozzi GM, D’Addato M, Gensini GF (June 2004). “Effects of defibrotide in patients with chronic deep insufficiency. The PROVEDIS study”. Int Angiol 23 (2): 100–7.PMID 15507885.
External links
- Palmer KJ, Goa KL. Defibrotide: a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in vascular disorders. Drugs 1993;45:259-94.
- http://www.globalrx.com/medinfo/Defibrotide.htm
- Fisher J, Holland TK, Pescador R, Porta R, Ferro L (January 1996). “Study on pharmacokinetics of radioactive labelled defibrotide after oral or intravenous administration in rats”. Thromb. Res. 81 (1): 55–63. doi:10.1016/0049-3848(95)00213-8. PMID 8747520.
- http://www.gentium.it/Defibrotide.aspx (information provided by manufacturer)
- “Melphalan: profile and news”. Archived from the original on 2007-09-28. (on cytostatic combination therapy)
- Beşişik SK, Oztürk GB, Calişkan Y, Sargin D (March 2005). “Complete resolution of transplantation-associated thrombotic microangiopathy and hepatic veno-occlusive disease by defibrotide and plasma exchange”. Turk J Gastroenterol 16 (1): 34–7. PMID 16252186.
| WO2003101468A1 * | Jun 2, 2003 | Dec 11, 2003 | Guenther Eissner | Method for the protection of endothelial and epithelial cells during chemotherapy |
| US4985552 | Jul 5, 1989 | Jan 15, 1991 | Crinos Industria Farmacobiologica S.P.A. | Process for obtaining chemically defined and reproducible polydeoxyribonucleotides |
| US5223609 | May 26, 1992 | Jun 29, 1993 | Crinos Industria Farmacobiologica S.P.A. | Process for obtaining chemically defined and reproducible polydeoxyribonucleotides |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1999026639A1 * | 24 Nov 1998 | 3 Jun 1999 | Allegheny University Of The He | Methods for mobilizing hematopoietic facilitating cells and hematopoietic stem cells into the peripheral blood |
| EP0317766A1 * | 20 Oct 1988 | 31 May 1989 | Crinos Industria Farmacobiologica S.p.A. | A method for preventing blood coaguli from being formed in the extra-body circuit of dialysis apparatus and composition useful thereof |
| EP0416678A1 * | 10 Aug 1990 | 13 Mar 1991 | Crinos Industria Farmacobiologica S.p.A. | Topical compositions containing Defibrotide |
| US5199942 * | 26 Sep 1991 | 6 Apr 1993 | Immunex Corporation | Method for improving autologous transplantation |
| US5977083 * | 5 Jun 1995 | 2 Nov 1999 | Burcoglu; Arsinur | Method for using polynucleotides, oligonucleotides and derivatives thereof to treat various disease states |
| Reference | ||
|---|---|---|
| 1 | * | CARLO-STELLA, C. (1) ET AL: “Defibrotide significantly enhances peripheral blood progenitor cell mobilization induced by recombinant human granulocyte colony – stimulating factor ( rhG – CSF.” BLOOD, ( NOVEMBER 16, 2000 ) VOL. 96, NO. 11 PART 1, PP. 553A. PRINT. MEETING INFO.: 42ND ANNUAL MEETING OF THE AMERICAN SOCIETY OF HEMATOLOGY SAN FRANCISCO, CALIFORNIA, USA DECEMBER 01-05, 2000 AMERICAN SOCIETY OF HEMATOLOGY. , XP002176349 |
| 2 | * | GURSOY A: “PREPARATION, CHARACTERIZATION AND ANTI-INFLAMMATORY EFFECT OF DEFIBROTIDE LIPOSOMES” PHARMAZIE,DD,VEB VERLAG VOLK UND GESUNDHEIT. BERLIN, vol. 48, no. 7, 1 July 1993 (1993-07-01), pages 549-550, XP000372658 ISSN: 0031-7144 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2005017160A2 * | 12 Aug 2004 | 24 Feb 2005 | Childrens Hosp Medical Center | Mobilization of hematopoietic cells |
| WO2009115465A1 * | 13 Mar 2009 | 24 Sep 2009 | Gentium Spa | Synthetic phosphodiester oligonucleotides and therapeutical uses thereof |
| EP2103689A1 * | 19 Mar 2008 | 23 Sep 2009 | Gentium S.p.A. | Synthetic phosphodiester oligonucleotides and therapeutical uses thereof |
| US7417026 | 12 Aug 2004 | 26 Aug 2008 | Children’s Hospital Medical Center | Mobilization of hematopoietic cells |
| US7915384 | 5 Jan 2009 | 29 Mar 2011 | Children’s Hospital Medical Center | Chimeric peptides for the regulation of GTPases |
| US8242246 | 28 Feb 2011 | 14 Aug 2012 | Children’s Hospital Medical Center | Chimeric peptides for the regulation of GTPases |
| US8674075 | 13 Aug 2012 | 18 Mar 2014 | Children’s Medical Center Corporation | Chimeric peptides for the regulation of GTPases |
| US8980862 | 12 Nov 2010 | 17 Mar 2015 | Gentium S.P.A. | Defibrotide for use in prophylaxis and/or treatment of Graft versus Host Disease (GVHD) |
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
oral, i.m., i.v. |
| Pharmacokinetic data | |
| Bioavailability | 58 – 70% orally (i.v. and i.m. = 100%) |
| Biological half-life | t1/2-alpha = minutes; t1/2-beta = a few hours |
| Identifiers | |
| CAS Registry Number | 83712-60-1 |
| ATC code | B01AX01 |
| DrugBank | DB04932 |
| UNII | 438HCF2X0M |
| KEGG | D07423 |
///////////Approved, 3/30/3016, US FDA, defibrotide sodium, NDA 208114, FDA 2016
Updates……….
FDA approves first treatment for rare disease in patients who receive stem cell transplant from blood or bone marrow
For Immediate Release
March 30, 2016
Release
The U.S. Food and Drug Administration today approved Defitelio (defibrotide sodium) to treat adults and children who develop hepatic veno-occlusive disease (VOD) with additional kidney or lung abnormalities after they receive a stem cell transplant from blood or bone marrow called hematopoietic stem cell transplantation (HSCT). This is the first FDA-approved therapy for treatment of severe hepatic VOD, a rare and life-threatening liver condition.
HSCT is a procedure performed in some patients to treat certain blood or bone marrow cancers. Immediately before an HSCT procedure, a patient receives chemotherapy. Hepatic VOD can occur in patients who receive chemotherapy and HSCT. Hepatic VOD is a condition in which some of the veins in the liver become blocked, causing swelling and a decrease in blood flow inside the liver, which may lead to liver damage. In the most severe form of hepatic VOD, the patient may also develop failure of the kidneys and lungs. Fewer than 2 percent of patients develop severe hepatic VOD after HSCT, but as many as 80 percent of patients who develop severe hepatic VOD do not survive.
“The approval of Defitelio fills a significant need in the transplantation community to treat this rare but frequently fatal complication in patients who receive chemotherapy and HSCT,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research.
The efficacy of Defitelio was investigated in 528 patients treated in three studies: two prospective clinical trials and an expanded access study. The patients enrolled in all three studies had a diagnosis of hepatic VOD with liver or kidney abnormalities after HSCT. The studies measured the percentage of patients who were still alive 100 days after HSCT (overall survival). In the three studies, 38 to 45 percent of patients treated with Defitelio were alive 100 days after HSCT. Based on published reports and analyses of patient-level data, the expected survival rates 100 days after HSCT would be 21 to 31 percent for patients with severe hepatic VOD who received only supportive care or interventions other than Defitelio.
The most common side effects of Defitelio include abnormally low blood pressure (hypotension), diarrhea, vomiting, nausea and nosebleeds (epistaxis). Serious potential side effects of Defitelio that were identified include bleeding (hemorrhage) and allergic reactions. Defitelio should not be used in patients who are having bleeding complications or who are taking blood thinners or other medicines that reduce the body’s ability to form clots.
The FDA granted the Defitelio application priority review status, which facilitates and expedites the development and review of certain drugs in light of their potential to benefit patients with serious or life-threatening conditions. Defitelio also received orphan drug designation, which provides incentives such as tax credits, user fee waivers and eligibility for exclusivity to assist and encourage the development of drugs for rare diseases.
Defitelio is marketed by Jazz Pharmaceuticals based in Palo Alto, California
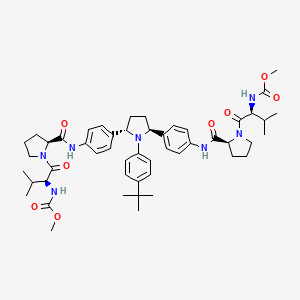
Ombitasvir

Ombitasvir; ABT-267; ABT 267; UNII-2302768XJ8; 1258226-87-7;
| C50H67N7O8 | |
| Molecular Weight: | 894.10908 g/mol |
|---|
Anti-Viral Compounds [US2010317568]
Dimethyl (2S,2′S)-1,1′-((2S,2′S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-Butylphenyl)pyrrolidine-2,5-diyl)bis(4,1-phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-2,1-diyl)dicarbamate
methyl N-[(2S)-1-[(2S)-2-[[4-[(2S,5S)-1-(4-tert-butylphenyl)-5-[4-[[(2S)-1-[(2S)-2-(methoxycarbonylamino)-3-methylbutanoyl]pyrrolidine-2-carbonyl]amino]phenyl]pyrrolidin-2-yl]phenyl]carbamoyl]pyrrolidin-1-yl]-3-methyl-1-oxobutan-2-yl]carbamate
1258226-87-7 [RN]
2:9 hydrate cas= 1456607-70-7…… is the drug substance
ABT-267
Abbvie Inc. innovator
ombitasvir is Dimethyl ([(2S,5S)-1-(4-tert-butylphenyl) pyrrolidine-2,5diyl]bis{benzene-4,1-diylcarbamoyl(2S)pyrrolidine-2,1-diyl[(2S)-3-methyl-1-oxobutane-1,2diyl]})biscarbamate hydrate. The molecular formula is C50H67N7O8•4.5H2O (hydrate) and the molecular weight for the drug substance is 975.20 (hydrate).

Ombitasvir is an antiviral drug for the treatment of hepatitis C virus (HCV) infection. In the United States, it is approved by theFood and Drug Administration for use in combination with paritaprevir, ritonavir and dasabuvir in the product Viekira Pak for the treatment of HCV genotype 1,[1][2] and with paritaprevir and ritonavir in the product Technivie for the treatment of HCV genotype 4.[3][4]
Ombitasvir is in phase II clinical development at AbbVie for the treatment of chronic hepatitis C infection in combination with ABT-450/ritonavir and, in combination with peginterferon alpha-2a/ribavirin (pegIFN/RBV) in treatment naïve Hepatitis C virus (HCV) genotype 1 infected patients.
Ombitasvir is part of a fixed-dose formulation with ABT-450/ritonavir that is approved in the U.S. and the E.U.
Ombitasvir acts by inhibiting the HCV protein NS5A.[5]
In 2013, breakthrough therapy designation was assigned in the U.S. for the treatment of genotype 1 hepatitis C in combination with ABT-450, ritonavir and ABT-333, with and without ribavirin.

DeGoey, DA, Discovery of ABT-267, a Pan-genotypic Inhibitor of HCV NS5A, J. Med. Chem., 2014, 57 (5), pp 2047-2057
http://pubs.acs.org/doi/full/10.1021/jm401398x
http://pubs.acs.org/doi/suppl/10.1021/jm401398x/suppl_file/jm401398x_si_001.pdf

We describe here N-phenylpyrrolidine-based inhibitors of HCV NS5A with excellent potency, metabolic stability, and pharmacokinetics. Compounds with 2S,5S stereochemistry at the pyrrolidine ring provided improved genotype 1 (GT1) potency compared to the 2R,5Ranalogues. Furthermore, the attachment of substituents at the 4-position of the central N-phenyl group resulted in compounds with improved potency. Substitution with tert-butyl, as in compound 38 (ABT-267), provided compounds with low-picomolar EC50 values and superior pharmacokinetics. It was discovered that compound 38 was a pan-genotypic HCV inhibitor, with an EC50 range of 1.7–19.3 pM against GT1a, -1b, -2a, -2b, -3a, -4a, and -5a and 366 pM against GT6a. Compound 38 decreased HCV RNA up to 3.10 log10 IU/mL during 3-day monotherapy in treatment-naive HCV GT1-infected subjects and is currently in phase 3 clinical trials in combination with an NS3 protease inhibitor with ritonavir (r) (ABT-450/r) and an NS5B non-nucleoside polymerase inhibitor (ABT-333), with and without ribavirin.
Dimethyl (2S,2′S)-1,1′-((2S,2′S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-Butylphenyl)pyrrolidine-2,5-diyl)bis(4,1-phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-2,1-diyl)dicarbamate (38)…desired
and
Dimethyl (2S,2′S)-1,1′-((2S,2′S)-2,2′-(4,4′-((2R,5R)-1-(4-tert-Butylphenyl)pyrrolidine-2,5-diyl)bis(4,1-phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-2,1-diyl)dicarbamate (39)…….undesired
…………….. The resulting mixture was stirred at room temperature for 16 h. The mixture was partitioned between ethyl acetate and water, and the organic layer was washed with saturated aqueous NaHCO3, brine (2×) and dried with Na2SO4. The drying agent was filtered off and the solution was concentrated in vacuo to give a crude product that was purified by column chromatography on silica gel, eluting with a solvent gradient of 2–8% methanol in dichloromethane to give a 1:1 mixture of trans-pyrrolidine isomers (290 mg, 96%). The mixture was separated on a Chiralpak AD-H column, eluting with a mixture of 1 part (2:1 isopropanol/ethanol) and 2 parts hexanes (0.1% TFA).
Compound 38 was the first of two stereoisomers to elute (101 mg, 99% ee by chiral HPLC). 1H NMR (400 MHz, DMSO-d6) δ 0.88 (d, J = 6.61 Hz, 6H), 0.93 (d, J = 6.72 Hz, 6H), 1.11 (s, 9H), 1.63 (d, J = 5.42 Hz, 2H), 1.80–2.04 (m, 8H), 2.09–2.19 (m, 2H), 2.44–2.47 (m, 2H), 3.52 (s, 6H), 3.59–3.66 (m, 2H), 3.77–3.84 (m, 2H), 4.02 (t, J = 8.40 Hz, 2H), 4.42 (dd, J = 7.86, 4.83 Hz, 2H), 5.14 (d, J = 6.18 Hz, 2H), 6.17 (d, J = 8.67 Hz, 2H), 6.94 (d, J = 8.78 Hz, 2H), 7.13 (d, J = 8.46 Hz, 4H), 7.31 (d, J= 8.35 Hz, 2H), 7.50 (d, J = 8.35 Hz, 4H), 9.98 (s, 2H).
MS (ESI) m/z 894.9 (M + H)+.
Compound39 was the second of two stereoisomers to elute. 1H NMR (400 MHz, DMSO-d6) δ 0.87 (d, J = 6.51 Hz, 6H), 0.92 (d, J = 6.72 Hz, 6H), 1.11 (s, 9H), 1.63 (d, J = 5.53 Hz, 2H), 1.82–2.04 (m, 8H), 2.09–2.18 (m, 2H), 2.41–2.47 (m, 2H), 3.52 (s, 6H), 3.58–3.67 (m, 2H), 3.75–3.84 (m, 2H), 4.02 (t, J = 7.26 Hz, 2H), 4.43 (dd, J = 7.92, 4.88 Hz, 2H), 5.14 (d, J = 6.18 Hz, 2H), 6.17 (d, J = 8.78 Hz, 2H), 6.94 (d, J = 8.67 Hz, 2H), 7.12 (d, J = 8.46 Hz, 4H), 7.31 (d, J = 8.35 Hz, 2H), 7.49 (d, J = 8.46 Hz, 4H), 9.98 (s, 2H). MS (ESI) m/z 895.0 (M + H)+.
………..
PATENT
WO 2011156578
dimethyl (2S,2,S)-l,l ‘-((2S,2’S)-2,2′-(4,4’-((2S,5S)-l-(4-fert-butylphenyl)pyrrolidine- 2,5-diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3- methyl- l-oxobutane-2,l-diyl)dicarbamate

hereinafter Compound IA),..http://www.google.com/patents/WO2011156578A1?cl=en
……………………………..
PATENT
US 20100317568
https://www.google.co.in/patents/US20100317568
Example 34
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate and
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-ter/’-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate

Example 34A l-(4-fer?-butylphenyl)-2,5-bis(4-nitrophenyl)pyrrolidine The product from Example 1C (3.67 g, 7.51 mmol) and 4-tert-butylaniline (11.86 ml, 75 mmol) in DMF (40 ml) was stirred under nitrogen at 50 °C for 4 h. The resulting mixture was diluted into ethyl acetate, treated with IM HCl, stirred for 10 minutes and filtered to remove solids. The filtrate organic layer was washed twice with brine, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (5% to 30%) to give a solid. The solid was triturated in a minimal volume of 1 :9 ethyl acetate/hexane to give a light yellow solid as a mixture of trans and cis isomers (1.21 g, 36%).
Example 34B 4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)dianiline and 4,4′-((2R,5R)-1-(4-fert- butylphenyl)pyrrolidine-2,5-diyl)dianiline To a solution of the product from Example 34A (1.1 g, 2.47 mmol) in ethanol (20 ml) and
THF (20 ml) was added PtC>2 (0.22 g, 0.97 mmol) in a 50 ml pressure bottle and stirred under 30 psi hydrogen at room temperature for 1 h. The mixture was filtered through a nylon membrane and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (20% to 60%). The title compound eluted as the first of 2 stereoisomers (trans isomer, 0.51 g, 54%).
Example 34C
(2S,2’S)-tert-Butyl 2,2′-(4,4′-((2S,5S)-1-(4-fer/’-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)dipyrrolidine- 1 -carboxylate and (2S,2’S)-tert-Butyl 2,2′- (4,4′-((2R,5R)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)dipyrrolidine-1-carboxylate To a mixture of the product from Example 34B (250 mg, 0.648 mmol), (S)-1-(tert- butoxycarbonyl)pyrrolidine-2-carboxylic acid (307 mg, 1.427 mmol) and HATU (542 mg, 1.427 mmol) in DMSO (10 ml) was added Hunig’s base (0.453 ml, 2.59 mmol). The reaction mixture was stirred at room temperature for 1 h. The mixture was partitioned with ethyl acetate and water. The organic layer was washed with brine, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (10% to 50%) to give the title compound (500 mg, 99%).
Example 34D
(2S,2’S)-N,N’-(4,4′-((2S,5S)-1-(4-ter/’-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))dipyrrolidine-2-carboxamide and (2S,2’S)-N,N’-(4,4′-((2R,5R)-1-(4-tert- butylphenyl)pyrrolidine-2,5-diyl)bis(4,l-phenylene))dipyrrolidine-2-carboxamide To the product from Example 34C (498 mg, 0.638 mmol) in dichloromethane (4 ml) was added TFA (6 ml). The reaction mixture was stirred at room temperature for 1 h and concentrated in vacuo. The residue was partitioned between 3: 1 CHCl3dsopropyl alcohol and saturated aq. NaHCO3. The aqueous layer was extracted by 3: 1 CHCl3:isopropyl alcohol again. The combined organic layers were dried over
filtered and concentrated to give the title compound (345 mg, 93%).
Example 34E Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-fert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate and
Dimethyl (2S,2’S)-1, r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-fert-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate
The product from Example 34D (29.0 mg, 0.050 mmol), (S)-2-(methoxycarbonylamino)-3- methylbutanoic acid (19.27 mg, 0.110 mmol), EDAC (21.09 mg, 0.110 mmol), HOBT (16.85 mg,
0.110 mmol) and N-methylmorpholine (0.027 ml, 0.250 mmol) were combined in DMF (2 ml). The reaction mixture was stirred at room temperature for 3 h. The mixture was partitioned with ethyl acetate and water. The organic layer was washed with brine twice, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (50% to 80%) to give a solid. The solid was triturated with ethyl acetate/hexane to give the title compound (13 mg, 29%). 1H NMR (400 MHz, DMSO-D6) δ ppm 0.85 – 0.95 (m, 12 H) 1.11 (s, 9 H) 1.59 – 1.65 (m, 2 H) 1.79 – 2.04 (m, 8 H) 2.10 – 2.18 (m, 2 H) 2.41-2.46 (m, 2H) 3.52 (s, 6 H)
3.57 – 3.67 (m, 2 H) 3.76 – 3.86 (m, 2 H) 4.00 (t, J=7.56 Hz, 2 H) 4.39 – 4.46 (m, 2 H) 5.15 (d, J=7.00
Hz, 2 H) 6.17 (d, J=7.70 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=7.37 Hz, 4 H) 7.30 (d, J=8.20
Hz, 2 H) 7.50 (d, J=8.24 Hz, 4 H) 9.98 (s, 2 H); (ESI+) m/z 895 (M+H)+. The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Example 35
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate
 ………………desired
………………desiredThe product from Example 34E was purified by chiral chromatography on a Chiralpak AD-H semi-prep column eluting with a 2:1 mixture of hexane:(2: l isopropyl alcohol: EtOH). The title compound was the first of the 2 diastereomers to elute. 1H NMR (400 MHz, DMSO-D6) δ ppm 0.88 (d, J=6.61 Hz, 6 H) 0.93 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.42 Hz, 2 H) 1.80 – 2.04 (m, 8 H) 2.09 – 2.19 (m, 2 H) 2.44 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.59 – 3.66 (m, 2 H) 3.77 – 3.84 (m, 2 H) 4.02 (t, J=8.40 Hz, 2 H) 4.42 (dd, J=7.86, 4.83 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.67 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.50 (d, J=8.35 Hz, 4 H) 9.98 (s, 2 H). The title compound showed an EC50 value of less than about 0.1 nM in HCV Ib- Conl replicon assays in the presence of 5% FBS.
Example 36 Dimethyl (2S,2’S)-1, r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-fert-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate
 …….undesired
…….undesiredThe product from Example 34E was purified by chiral chromatography on a Chiralpak AD-H semi-prep column eluting with a 2:1 mixture of hexane:(2: l isopropyl alcohol: EtOH). The title compound was the second of 2 diastereomers to elute. 1H NMR (400 MHz, DMSO-D6) δ ppm 0.87
(d, J=6.51 Hz, 6 H) 0.92 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.53 Hz, 2 H) 1.82 – 2.04 (m, 8
H) 2.09-2.18 (m, 2 H) 2.41 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.58 – 3.67 (m, 2 H) 3.75 – 3.84 (m, 2 H) 4.02
(t, J=7.26 Hz, 2 H) 4.43 (dd, J=7.92, 4.88 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.78 Hz, 2 H) 6.94 (d, J=8.67 Hz, 2 H) 7.12 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.49 (d, J=8.46 Hz, 4 H)
9.98 (s, 2 H). The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Example 37 Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-fert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate
 ……………desired
……………desiredExample 37A (S)-2,5-dioxopyrrolidin-1-yl 2-(methoxycarbonylamino)-3-methylbutanoate To a mixture of (S)-2-(methoxycarbonylamino)-3-methylbutanoic acid (19.66 g, 112 mmol) and N-hydroxysuccinimide (13.29g, 116 mmol) was added ethyl acetate (250 ml), and the mixture was cooled to 0-5 °C. Diisopropylcarbodiimide (13.88 g, 110 mmol) was added and the reaction mixture was stirred at 0-5 °C for about 1 hour. The reaction mixture was warmed to room temperature. The solids (diisopropylurea by-product) were filtered and rinsed with ethyl acetate. The filtrate was concentrated in vacuo to an oil. Isopropyl alcohol (200 ml) was added to the oil and the mixture was heated to about 50 °C to obtain a homogeneous solution. Upon cooling, crystalline solids formed. The solids were filtered and washed with isopropyl alcohol (3 x 20 ml) and dried to give the title compound as a white solid (23.2 g, 77% yield).
Example 37B
(S)- 1 -((S)-2-(methoxycarbonylamino)-3-methylbutanoyl)pyrrolidine-2-carboxylic acid To a mixture of L-proline (4.44g, 38.6 mmol), water (20 ml), acetonitrile (20 ml) and DIEA (9.5 g, 73.5 mmol) was added a solution of the product from Example 37A (1Og, 36.7 mmol) in acetonitrile (20 inL) over 10 minutes. The reaction mixture was stirred overnight at room temperature. The solution was concentrated under vacuum to remove the acetonitrile. To the resulting clear water solution was added 6N HCl (9 ml) until pH ~ 2 .The solution was transferred to a separatory funnel and 25% NaCl (10 ml) was added and the mixture was extracted with ethyl acetate (75 ml), and then again with ethyl acetate (6 x 20 ml), and the combined extracts were washed with 25% NaCl (2 x 10ml). The solvent was evaporated to give a thick oil. Heptane was added and the solvent was evaporated to give a foam, which was dried under high vacuum. Diethyl ether was added and the solvent was evaporated to give a foam, which was dried under high vacuum to give the title compound (10.67g) as a white solid.
The compound of Example 37B can also be prepreared according to the following procedure: To a flask was charged L- valine (35 g, 299 mmol), IN sodium hydroxide solution (526 ml,
526 mmol) and sodium carbonate (17.42 g, 164 mmol). The mixture was stirred for 15 min to dissolve solids and then cooled to 15 °C. Methyl chloroformate (29.6 g, 314 mmol) was added slowly to the reaction mixture. The mixture was then stirred at rt for 30 min. The mixture was cooled to 15 °C and pH adjusted to -5.0 with concentrated HCl solution. 100 inL of 2-methytetrahydrofuran (2- MeTHF) was added and the adjustment of pH continued until the pH reached ~ 2.0. 150 mL of 2- MeTHF was added and the mixture was stirred for 15 min. Layers were separated and the aqueous layer extracted with 100 mL of 2-MeTHF. The combined organic layer was dried over anhyd Na2SC^ and filtered, and Na2SC^ cake was washed with 50 mL of 2-MeTHF. The product solution was concentrated to ~ 100 mL, chased with 120 mL of IPAc twice. 250 mL of heptanes was charged slowly and then the volume of the mixture was concentrated to 300 mL. The mixture was heated to 45 °C and 160 mL of heptanes charged. The mixture was cooled to rt in 2h, stirred for 30 min, filtered and washed with 2-MeTHF/heptanes mixture (1:7, 80 inL). The wetcake was dried at 55 °C for 24 h to give 47.1 g of Moc-L- VaI-OH product as a white solid (90%).
Moc-L- VaI-OH (15O g, 856 mmol), HOBt hydrate (138 g, 899 mmol) and DMF (1500 ml) were charged to a flask. The mixture was stirred for 15 min to give a clear solution. EDC hydrochloride (172 g, 899 mmol) was charged and mixed for 20 min. The mixture was cooled to 13
°C and (L)-proline benzyl ester hydrochloride (207 g, 856 mmol) charged. Triethylamine (109 g,
1079 mmol) was then charged in 30 min. The resulting suspension was mixed at rt for 1.5 h. The reaction mixture was cooled to 15 °C and 1500 mL of 6.7% NaHCO3 charged in 1.5 h, followed by the addition of 1200 mL of water over 60 min. The mixture was stirred at rt for 30 min, filtered and washed with water/DMF mixture (1 :2, 250 mL) and then with water (1500 mL). The wetcake was dried at 55 °C for 24 h to give 282 g of product as a white solid (90%).
The resulting solids (40 g) and 5% Pd/ Alumina were charged to a Parr reactor followed by THF (160 mL). The reactor was sealed and purged with nitrogen (6 x 20 psig) followed by a hydrogen purge (6 x 30 psig). The reactor was pressurized to 30 psig with hydrogen and agitated at room temperature for approximately 15 hours. The resulting slurry was filtered through a GF/F filter and concentrated to approximately 135 g solution. Heptane was added (120 mL), and the solution was stirred until solids formed. After an addition 2 – 3 hours additional heptane was added drop-wise (240 mL), the slurry was stirred for approximately 1 hour, then filtered. The solids were dried to afford the title compound.
Example 37C
(lR,4R)-1,4-bis(4-nitrophenyl)butane-1,4-diyl dimethanesulfonate
The product from Example 32 (5.01 g, 13.39 mmol) was combined with 2- methyltetrahydrofuran (70 mL) and cooled to -5 °C, and N,N-diisopropylethylamine (6.81 g, 52.7 mmol) was added over 30 seconds. Separately, a solution of methanesulfonic anhydride (6.01 g, 34.5 mmol) in 2-methyltetrahydrofuran (30 mL) was prepared and added to the diol slurry over 3 min., maintaining the internal temperature between -15 °C and -25 °C. After mixing for 5 min at -15 °C, the cooling bath was removed and the reaction was allowed to warm slowly to 23 °C and mixed for 30 minutes. After reaction completion, the crude slurry was carried immediately into the next step.
Example 37D
(2S,5S)-1-(4-tert-butylphenyl)-2,5-bis(4-nitrophenyl)pyrrolidine
To the crude product solution from Example 37C (7.35 g, 13.39 mmol) was added 4-tert- butylaniline (13.4 g, 90 mmol) at 23 °C over 1 minute. The reaction was heated to 65 °C for 2 h. After completion, the reaction mixture was cooled to 23 °C and diluted with 2-methyltetrahydrofuran (100 mL) and 1 M HCl (150 mL). After partitioning the phases, the organic phase was treated with 1 M HCl (140 mL), 2-methyltetrahydrofuran (50 mL), and 25 wt% aq. NaCl (100 mL), and the phases were partitioned. The organic phase was washed with 25 wt% aq. NaCl (50 mL), dried over MgSO/t, filtered, and concentrated in vacuo to approximately 20 mL. Heptane (30 mL) and additional 2- methyltetrahydrofuran were added in order to induce crystallization. The slurry was concentrated further, and additional heptane (40 mL) was slowly added and the slurry was filtered, washing with 2- methyltetrahydrofuran:heptane (1:4, 20 mL). The solids were suspended in MeOH (46 mL) for 3 h, filtered, and the wet solid was washed with additional MeOH (18 mL). The solid was dried at 45 °C in a vacuum oven for 16 h to provide the title compound (3.08 g, 51% 2-step yield).
Example 37E
4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)dianiline
To a 160 ml Parr stirrer hydrogenation vessel was added the product from Example 37D (2 g, 4.49 mmol), followed by 60 ml of THF, and Raney Nickel Grace 2800 (1 g, 50 wt% (dry basis)) under a stream of nitrogen. The reactor was assembled and purged with nitrogen (8 x 20 psig) followed by purging with hydrogen (8 x 30 psig). The reactor was then pressurized to 30 psig with hydrogen and agitation (700 rpm) began and continued for a total of 16 h at room temperature. The slurry was filtered by vacuum filtration using a GF/F Whatman glass fiber filter. Evaporation of the filtrate to afford a slurry followed by the addition heptane and filtration gave the crude title compound, which was dried and used directly in the next step.
Example 37F dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4, l- phenylene)bis(azanediyl)bis(oxomethylene))bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diy 1) die arb amate To a solution of the product from Example 37E (1.64 g, 4.25 mmol) in DMF (20 ml), the product from Example 37B (2.89 g, 10.63 mmol), and HATU (4.04 g, 10.63 mmol) in DMF (15OmL) was added triethylamine (1.07 g, 10.63 mmol), and the solution was stirred at room temperature for 90 min. To the reaction mixture was poured 20 mL of water, and the white precipitate obtained was filtered, and the solid was washed with water (3×5 mL). The solid was blow dried for Ih. The crude material was loaded on a silica gel column and eluted with a gradient starting with ethyl acetate/ heptane (3/7), and ending with pure ethyl acetate. The desired fractions were combined and solvent distilled off to give a very light yellow solid, which was dried at 45 °C in a vacuum oven with nitrogen purge for 15 h to give the title compound (2.3 g, 61% yield). 1H NMR (400 MHz, DMSO- D6) δ ppm 0.88 (d, J=6.61 Hz, 6 H) 0.93 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.42 Hz, 2 H) 1.80 – 2.04 (m, 8 H) 2.09 – 2.19 (m, 2 H) 2.44 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.59 – 3.66 (m, 2 H) 3.77 – 3.84 (m, 2 H) 4.02 (t, J=8.40 Hz, 2 H) 4.42 (dd, J=7.86, 4.83 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.67 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.50 (d, J=8.35 Hz, 4 H) 9.98 (s, 2 H).
Alternately, the product from example 37E (11.7 g, 85 wt%, 25.8 mmol) and the product from example 37B (15.45 g, 56.7 mmol) are suspended in EtOAc (117 mL), diisopropylethylamine (18.67 g, 144 mmol) is added and the solution is cooled to 0 °C. In a separate flask, 1-propanephosphonic acid cyclic anhydride (T3P®) (46.0 g, 50 wt% in EtOAc, 72.2 mmol) was dissolved in EtOAc (58.5 mL), and charged to an addition funnel. The T3P solution is added to the reaction mixture drop-wise over 3-4 h and stirred until the reaction is complete. The reaction is warmed to room temperature,and washed with IM HCl/7.5 wt% NaCl (100 mL), then washed with 5% NaHCO3 (100 mL), then washed with 5% NaCl solution (100 mL). The solution was concentrated to approximately 60 mL, EtOH (300 mL) was added, and the solution was concentrated to 84 g solution.
A portion of the EtOH solution of product (29 g) was heated to 40 °C, and added 134 g 40 w% EtOH in H2O. A slurry of seeds in 58 wt/wt% EtOH/H2O was added, allowed to stir at 40 °C for several hours, then cooled to 0 °C. The slurry is then filtered, and washed with 58wt/wt% EtOH/H2O. The product is dried at 40 – 60 °C under vacuum, and then rehydrated by placing a tray of water in the vacuum oven to give the title compound. The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
……………..
PATENT
http://www.google.com/patents/EP2337781A2?cl=en
Example 34
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate and
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-ter/’-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate

Example 34A l-(4-fer?-butylphenyl)-2,5-bis(4-nitrophenyl)pyrrolidine The product from Example 1C (3.67 g, 7.51 mmol) and 4-tert-butylaniline (11.86 ml, 75 mmol) in DMF (40 ml) was stirred under nitrogen at 50 °C for 4 h. The resulting mixture was diluted into ethyl acetate, treated with IM HCl, stirred for 10 minutes and filtered to remove solids. The filtrate organic layer was washed twice with brine, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (5% to 30%) to give a solid. The solid was triturated in a minimal volume of 1 :9 ethyl acetate/hexane to give a light yellow solid as a mixture of trans and cis isomers (1.21 g, 36%).
Example 34B 4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)dianiline and 4,4′-((2R,5R)-1-(4-fert- butylphenyl)pyrrolidine-2,5-diyl)dianiline To a solution of the product from Example 34A (1.1 g, 2.47 mmol) in ethanol (20 ml) and
THF (20 ml) was added PtC>2 (0.22 g, 0.97 mmol) in a 50 ml pressure bottle and stirred under 30 psi hydrogen at room temperature for 1 h. The mixture was filtered through a nylon membrane and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (20% to 60%). The title compound eluted as the first of 2 stereoisomers (trans isomer, 0.51 g, 54%).
Example 34C
(2S,2’S)-tert-Butyl 2,2′-(4,4′-((2S,5S)-1-(4-fer/’-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)dipyrrolidine- 1 -carboxylate and (2S,2’S)-tert-Butyl 2,2′- (4,4′-((2R,5R)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)dipyrrolidine-1-carboxylate To a mixture of the product from Example 34B (250 mg, 0.648 mmol), (S)-1-(tert- butoxycarbonyl)pyrrolidine-2-carboxylic acid (307 mg, 1.427 mmol) and HATU (542 mg, 1.427 mmol) in DMSO (10 ml) was added Hunig’s base (0.453 ml, 2.59 mmol). The reaction mixture was stirred at room temperature for 1 h. The mixture was partitioned with ethyl acetate and water. The organic layer was washed with brine, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (10% to 50%) to give the title compound (500 mg, 99%).
Example 34D
(2S,2’S)-N,N’-(4,4′-((2S,5S)-1-(4-ter/’-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))dipyrrolidine-2-carboxamide and (2S,2’S)-N,N’-(4,4′-((2R,5R)-1-(4-tert- butylphenyl)pyrrolidine-2,5-diyl)bis(4,l-phenylene))dipyrrolidine-2-carboxamide To the product from Example 34C (498 mg, 0.638 mmol) in dichloromethane (4 ml) was added TFA (6 ml). The reaction mixture was stirred at room temperature for 1 h and concentrated in vacuo. The residue was partitioned between 3: 1 CHCl3dsopropyl alcohol and saturated aq. NaHCO3. The aqueous layer was extracted by 3: 1 CHCl3:isopropyl alcohol again. The combined organic layers were dried over
filtered and concentrated to give the title compound (345 mg, 93%).
Example 34E Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-fert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate and
Dimethyl (2S,2’S)-1, r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-fert-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate
The product from Example 34D (29.0 mg, 0.050 mmol), (S)-2-(methoxycarbonylamino)-3- methylbutanoic acid (19.27 mg, 0.110 mmol), EDAC (21.09 mg, 0.110 mmol), HOBT (16.85 mg,
0.110 mmol) and N-methylmorpholine (0.027 ml, 0.250 mmol) were combined in DMF (2 ml). The reaction mixture was stirred at room temperature for 3 h. The mixture was partitioned with ethyl acetate and water. The organic layer was washed with brine twice, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (50% to 80%) to give a solid. The solid was triturated with ethyl acetate/hexane to give the title compound (13 mg, 29%). 1H NMR (400 MHz, DMSO-D6) δ ppm 0.85 – 0.95 (m, 12 H) 1.11 (s, 9 H) 1.59 – 1.65 (m, 2 H) 1.79 – 2.04 (m, 8 H) 2.10 – 2.18 (m, 2 H) 2.41-2.46 (m, 2H) 3.52 (s, 6 H)
3.57 – 3.67 (m, 2 H) 3.76 – 3.86 (m, 2 H) 4.00 (t, J=7.56 Hz, 2 H) 4.39 – 4.46 (m, 2 H) 5.15 (d, J=7.00
Hz, 2 H) 6.17 (d, J=7.70 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=7.37 Hz, 4 H) 7.30 (d, J=8.20
Hz, 2 H) 7.50 (d, J=8.24 Hz, 4 H) 9.98 (s, 2 H); (ESI+) m/z 895 (M+H)+. The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Example 35
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate
 ………….desired
………….desiredThe product from Example 34E was purified by chiral chromatography on a Chiralpak AD-H semi-prep column eluting with a 2:1 mixture of hexane:(2: l isopropyl alcohol: EtOH). The title compound was the first of the 2 diastereomers to elute. 1H NMR (400 MHz, DMSO-D6) δ ppm 0.88 (d, J=6.61 Hz, 6 H) 0.93 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.42 Hz, 2 H) 1.80 – 2.04 (m, 8 H) 2.09 – 2.19 (m, 2 H) 2.44 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.59 – 3.66 (m, 2 H) 3.77 – 3.84 (m, 2 H) 4.02 (t, J=8.40 Hz, 2 H) 4.42 (dd, J=7.86, 4.83 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.67 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.50 (d, J=8.35 Hz, 4 H) 9.98 (s, 2 H). The title compound showed an EC50 value of less than about 0.1 nM in HCV Ib- Conl replicon assays in the presence of 5% FBS.
Example 36 Dimethyl (2S,2’S)-1, r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-fert-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate
 ……….undesired
……….undesiredThe product from Example 34E was purified by chiral chromatography on a Chiralpak AD-H semi-prep column eluting with a 2:1 mixture of hexane:(2: l isopropyl alcohol: EtOH). The title compound was the second of 2 diastereomers to elute. 1H NMR (400 MHz, DMSO-D6) δ ppm 0.87
(d, J=6.51 Hz, 6 H) 0.92 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.53 Hz, 2 H) 1.82 – 2.04 (m, 8
H) 2.09-2.18 (m, 2 H) 2.41 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.58 – 3.67 (m, 2 H) 3.75 – 3.84 (m, 2 H) 4.02
(t, J=7.26 Hz, 2 H) 4.43 (dd, J=7.92, 4.88 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.78 Hz, 2 H) 6.94 (d, J=8.67 Hz, 2 H) 7.12 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.49 (d, J=8.46 Hz, 4 H)
9.98 (s, 2 H). The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Example 37 Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-fert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate
 ………………desired
………………desiredExample 37A (S)-2,5-dioxopyrrolidin-1-yl 2-(methoxycarbonylamino)-3-methylbutanoate To a mixture of (S)-2-(methoxycarbonylamino)-3-methylbutanoic acid (19.66 g, 112 mmol) and N-hydroxysuccinimide (13.29g, 116 mmol) was added ethyl acetate (250 ml), and the mixture was cooled to 0-5 °C. Diisopropylcarbodiimide (13.88 g, 110 mmol) was added and the reaction mixture was stirred at 0-5 °C for about 1 hour. The reaction mixture was warmed to room temperature. The solids (diisopropylurea by-product) were filtered and rinsed with ethyl acetate. The filtrate was concentrated in vacuo to an oil. Isopropyl alcohol (200 ml) was added to the oil and the mixture was heated to about 50 °C to obtain a homogeneous solution. Upon cooling, crystalline solids formed. The solids were filtered and washed with isopropyl alcohol (3 x 20 ml) and dried to give the title compound as a white solid (23.2 g, 77% yield).
Example 37B
(S)- 1 -((S)-2-(methoxycarbonylamino)-3-methylbutanoyl)pyrrolidine-2-carboxylic acid To a mixture of L-proline (4.44g, 38.6 mmol), water (20 ml), acetonitrile (20 ml) and DIEA (9.5 g, 73.5 mmol) was added a solution of the product from Example 37A (1Og, 36.7 mmol) in acetonitrile (20 inL) over 10 minutes. The reaction mixture was stirred overnight at room temperature. The solution was concentrated under vacuum to remove the acetonitrile. To the resulting clear water solution was added 6N HCl (9 ml) until pH ~ 2 .The solution was transferred to a separatory funnel and 25% NaCl (10 ml) was added and the mixture was extracted with ethyl acetate (75 ml), and then again with ethyl acetate (6 x 20 ml), and the combined extracts were washed with 25% NaCl (2 x 10ml). The solvent was evaporated to give a thick oil. Heptane was added and the solvent was evaporated to give a foam, which was dried under high vacuum. Diethyl ether was added and the solvent was evaporated to give a foam, which was dried under high vacuum to give the title compound (10.67g) as a white solid.
The compound of Example 37B can also be prepreared according to the following procedure: To a flask was charged L- valine (35 g, 299 mmol), IN sodium hydroxide solution (526 ml,
526 mmol) and sodium carbonate (17.42 g, 164 mmol). The mixture was stirred for 15 min to dissolve solids and then cooled to 15 °C. Methyl chloroformate (29.6 g, 314 mmol) was added slowly to the reaction mixture. The mixture was then stirred at rt for 30 min. The mixture was cooled to 15 °C and pH adjusted to -5.0 with concentrated HCl solution. 100 inL of 2-methytetrahydrofuran (2- MeTHF) was added and the adjustment of pH continued until the pH reached ~ 2.0. 150 mL of 2- MeTHF was added and the mixture was stirred for 15 min. Layers were separated and the aqueous layer extracted with 100 mL of 2-MeTHF. The combined organic layer was dried over anhyd Na2SC^ and filtered, and Na2SC^ cake was washed with 50 mL of 2-MeTHF. The product solution was concentrated to ~ 100 mL, chased with 120 mL of IPAc twice. 250 mL of heptanes was charged slowly and then the volume of the mixture was concentrated to 300 mL. The mixture was heated to 45 °C and 160 mL of heptanes charged. The mixture was cooled to rt in 2h, stirred for 30 min, filtered and washed with 2-MeTHF/heptanes mixture (1:7, 80 inL). The wetcake was dried at 55 °C for 24 h to give 47.1 g of Moc-L- VaI-OH product as a white solid (90%).
Moc-L- VaI-OH (15O g, 856 mmol), HOBt hydrate (138 g, 899 mmol) and DMF (1500 ml) were charged to a flask. The mixture was stirred for 15 min to give a clear solution. EDC hydrochloride (172 g, 899 mmol) was charged and mixed for 20 min. The mixture was cooled to 13
°C and (L)-proline benzyl ester hydrochloride (207 g, 856 mmol) charged. Triethylamine (109 g,
1079 mmol) was then charged in 30 min. The resulting suspension was mixed at rt for 1.5 h. The reaction mixture was cooled to 15 °C and 1500 mL of 6.7% NaHCO3 charged in 1.5 h, followed by the addition of 1200 mL of water over 60 min. The mixture was stirred at rt for 30 min, filtered and washed with water/DMF mixture (1 :2, 250 mL) and then with water (1500 mL). The wetcake was dried at 55 °C for 24 h to give 282 g of product as a white solid (90%).
The resulting solids (40 g) and 5% Pd/ Alumina were charged to a Parr reactor followed by THF (160 mL). The reactor was sealed and purged with nitrogen (6 x 20 psig) followed by a hydrogen purge (6 x 30 psig). The reactor was pressurized to 30 psig with hydrogen and agitated at room temperature for approximately 15 hours. The resulting slurry was filtered through a GF/F filter and concentrated to approximately 135 g solution. Heptane was added (120 mL), and the solution was stirred until solids formed. After an addition 2 – 3 hours additional heptane was added drop-wise (240 mL), the slurry was stirred for approximately 1 hour, then filtered. The solids were dried to afford the title compound.
Example 37C
(lR,4R)-1,4-bis(4-nitrophenyl)butane-1,4-diyl dimethanesulfonate
The product from Example 32 (5.01 g, 13.39 mmol) was combined with 2- methyltetrahydrofuran (70 mL) and cooled to -5 °C, and N,N-diisopropylethylamine (6.81 g, 52.7 mmol) was added over 30 seconds. Separately, a solution of methanesulfonic anhydride (6.01 g, 34.5 mmol) in 2-methyltetrahydrofuran (30 mL) was prepared and added to the diol slurry over 3 min., maintaining the internal temperature between -15 °C and -25 °C. After mixing for 5 min at -15 °C, the cooling bath was removed and the reaction was allowed to warm slowly to 23 °C and mixed for 30 minutes. After reaction completion, the crude slurry was carried immediately into the next step.
Example 37D
(2S,5S)-1-(4-tert-butylphenyl)-2,5-bis(4-nitrophenyl)pyrrolidine
To the crude product solution from Example 37C (7.35 g, 13.39 mmol) was added 4-tert- butylaniline (13.4 g, 90 mmol) at 23 °C over 1 minute. The reaction was heated to 65 °C for 2 h. After completion, the reaction mixture was cooled to 23 °C and diluted with 2-methyltetrahydrofuran (100 mL) and 1 M HCl (150 mL). After partitioning the phases, the organic phase was treated with 1 M HCl (140 mL), 2-methyltetrahydrofuran (50 mL), and 25 wt% aq. NaCl (100 mL), and the phases were partitioned. The organic phase was washed with 25 wt% aq. NaCl (50 mL), dried over MgSO/t, filtered, and concentrated in vacuo to approximately 20 mL. Heptane (30 mL) and additional 2- methyltetrahydrofuran were added in order to induce crystallization. The slurry was concentrated further, and additional heptane (40 mL) was slowly added and the slurry was filtered, washing with 2- methyltetrahydrofuran:heptane (1:4, 20 mL). The solids were suspended in MeOH (46 mL) for 3 h, filtered, and the wet solid was washed with additional MeOH (18 mL). The solid was dried at 45 °C in a vacuum oven for 16 h to provide the title compound (3.08 g, 51% 2-step yield).
Example 37E
4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)dianiline
To a 160 ml Parr stirrer hydrogenation vessel was added the product from Example 37D (2 g, 4.49 mmol), followed by 60 ml of THF, and Raney Nickel Grace 2800 (1 g, 50 wt% (dry basis)) under a stream of nitrogen. The reactor was assembled and purged with nitrogen (8 x 20 psig) followed by purging with hydrogen (8 x 30 psig). The reactor was then pressurized to 30 psig with hydrogen and agitation (700 rpm) began and continued for a total of 16 h at room temperature. The slurry was filtered by vacuum filtration using a GF/F Whatman glass fiber filter. Evaporation of the filtrate to afford a slurry followed by the addition heptane and filtration gave the crude title compound, which was dried and used directly in the next step.
Example 37F dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4, l- phenylene)bis(azanediyl)bis(oxomethylene))bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diy 1) die arb amate To a solution of the product from Example 37E (1.64 g, 4.25 mmol) in DMF (20 ml), the product from Example 37B (2.89 g, 10.63 mmol), and HATU (4.04 g, 10.63 mmol) in DMF (15OmL) was added triethylamine (1.07 g, 10.63 mmol), and the solution was stirred at room temperature for 90 min. To the reaction mixture was poured 20 mL of water, and the white precipitate obtained was filtered, and the solid was washed with water (3×5 mL). The solid was blow dried for Ih. The crude material was loaded on a silica gel column and eluted with a gradient starting with ethyl acetate/ heptane (3/7), and ending with pure ethyl acetate. The desired fractions were combined and solvent distilled off to give a very light yellow solid, which was dried at 45 °C in a vacuum oven with nitrogen purge for 15 h to give the title compound (2.3 g, 61% yield). 1H NMR (400 MHz, DMSO- D6) δ ppm 0.88 (d, J=6.61 Hz, 6 H) 0.93 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.42 Hz, 2 H) 1.80 – 2.04 (m, 8 H) 2.09 – 2.19 (m, 2 H) 2.44 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.59 – 3.66 (m, 2 H) 3.77 – 3.84 (m, 2 H) 4.02 (t, J=8.40 Hz, 2 H) 4.42 (dd, J=7.86, 4.83 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.67 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.50 (d, J=8.35 Hz, 4 H) 9.98 (s, 2 H).
Alternately, the product from example 37E (11.7 g, 85 wt%, 25.8 mmol) and the product from example 37B (15.45 g, 56.7 mmol) are suspended in EtOAc (117 mL), diisopropylethylamine (18.67 g, 144 mmol) is added and the solution is cooled to 0 °C. In a separate flask, 1-propanephosphonic acid cyclic anhydride (T3P®) (46.0 g, 50 wt% in EtOAc, 72.2 mmol) was dissolved in EtOAc (58.5 mL), and charged to an addition funnel. The T3P solution is added to the reaction mixture drop-wise over 3-4 h and stirred until the reaction is complete. The reaction is warmed to room temperature,and washed with IM HCl/7.5 wt% NaCl (100 mL), then washed with 5% NaHCO3 (100 mL), then washed with 5% NaCl solution (100 mL). The solution was concentrated to approximately 60 mL, EtOH (300 mL) was added, and the solution was concentrated to 84 g solution.
A portion of the EtOH solution of product (29 g) was heated to 40 °C, and added 134 g 40 w% EtOH in H2O. A slurry of seeds in 58 wt/wt% EtOH/H2O was added, allowed to stir at 40 °C for several hours, then cooled to 0 °C. The slurry is then filtered, and washed with 58wt/wt% EtOH/H2O. The product is dried at 40 – 60 °C under vacuum, and then rehydrated by placing a tray of water in the vacuum oven to give the title compound. The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Intermediates
Example 32
( 1 R,4R)- 1 ,4-bis(4-mtrophenyl)butane- 1 ,4-diol
To (S)-(-)-α,α-diphenyl-2-pyrrohdinemethanol (2 71 g, 10 70 mmol) was added THF (80 mL) at 23 °C The very thin suspension was treated with t11methyl borate (1 44 g, 13 86 mmol) over 30 seconds, and the resulting solution was mixed at 23 °C for 1 h The solution was cooled to 16-19 °C, and N,N-diethylanilme borane (21 45 g, 132 mmol) was added dropwise via syringe over 3-5 mm (caution vigorous H2 evolution), while the internal temperature was maintained at 16-19 °C After 15 mm, the H2 evolution had ceased To a separate vessel was added the product from Example IA (22 04 g, 95 wt%, 63 8 mmol), followed by THF (80 mL), to form an orange slurry After cooling the slurry to 11 °C, the borane solution was transferred via cannula into the dione slurry over 3-5 min During this period, the internal temperature of the slurry rose to 16 °C After the addition was complete, the reaction was maintained at 20-27 °C for an additional 2 5 h After reaction completion, the mixture was cooled to 5 °C and methanol (16 7 g, 521 mmol) was added dropwise over 5-10 mm, maintaining an internal temperature <20 °C (note vigorous H2 evolution) After the exotherm had ceased (ca 10 mm), the temperature was adjusted to 23 °C, and the reaction was mixed until complete dissolution of the solids had occurred Ethyl acetate (300 mL) and 1 M HCl (120 mL) were added, and the phases were partitioned The organic phase was then washed successively with 1 M HCl (2 x 120 mL), H2O (65 mL), and 10% aq NaCl (65 mL) The orgamcs were dried over MgSO4, filtered, and concentrated in vacuo Crystallization of the product occurred during the concentration The slurry was warmed to 50 °C, and heptane (250 inL) was added over 15 min. The slurry was then allowed to mix at 23 °C for 30 min and filtered. The wet cake was washed with 3: 1 heptane:ethyl acetate (75 mL), and the orange, crystalline solids were dried at 45 °C for 24 h to provide the title compound (15.35 g, 99.3% ee, 61% yield), which was contaminated with 11% of the meso isomer (vs. dl isomer).
References
- “VIEKIRA PAK™ (ombitasvir, paritaprevir and ritonavir tablets; dasabuvir tablets), for Oral Use. Full Prescribing Information”(PDF). AbbVie Inc., North Chicago, IL 60064. Retrieved 30 July 2015.
- “FDA approves Viekira Pak to treat hepatitis C”. Food and Drug Administration. December 19, 2014.
- “TECHNIVIE™ (ombitasvir, paritaprevir and ritonavir) Tablets, for Oral Use. Full Prescribing Information” (PDF). AbbVie Inc., North Chicago, IL 60064. Retrieved 28 July 2015.
- “FDA approves Technivie for treatment of chronic hepatitis C genotype 4”. Food and Drug Administration. July 24, 2015.
- Jordan J. Feld, Kris V. Kowdley, Eoin Coakley, Samuel Sigal, David R. Nelson, Darrell Crawford, Ola Weiland, Humberto Aguilar, Junyuan Xiong, Tami Pilot-Matias, Barbara DaSilva-Tillmann, Lois Larsen, Thomas Podsadecki, and Barry Bernstein (2014). “Treatment of HCV with ABT-450/r–Ombitasvir and Dasabuvir with Ribavirin”. N Engl J Med 370: 1594–1603.doi:10.1056/NEJMoa1315722.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
Dimethyl ({(2S,5S)-1-[4-(2-methyl-2-propanyl)phenyl]-2,5-pyrrolidinediyl}bis{4,1-phenylenecarbamoyl(2S)-2,1-pyrrolidinediyl[(2S)-3-methyl-1-oxo-1,2-butanediyl]})biscarbamate
|
|
| Clinical data | |
| Trade names | Viekira Pak (with ombitasvir, paritaprevir, ritonavir and dasabuvir), Technivie (with ombitasvir, paritaprevir, and ritonavir) |
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | not determined |
| Protein binding | ~99.9% |
| Metabolism | amide hydrolysis followed by oxidation |
| Onset of action | ~4 to 5 hours |
| Biological half-life | 21 to 25 hours |
| Excretion | mostly with feces (90.2%) |
| Identifiers | |
| CAS Registry Number | 1258226-87-7 |
| PubChem | CID: 54767916 |
| ChemSpider | 31136214 |
| ChEBI | CHEBI:85183 |
| Synonyms | ABT-267 |
| Chemical data | |
| Formula | C50H67N7O8 |
| Molecular mass | 894.11 g/mol |
rx list
VIEKIRA PAK is ombitasvir, paritaprevir, ritonavir fixed dose combination tablets copackaged with dasabuvir tablets.
Ombitasvir, paritaprevir, ritonavir fixed dose combination tablet includes ahepatitis C virus NS5A inhibitor (ombitasvir), a hepatitis C virus NS3/4Aprotease inhibitor (paritaprevir), and a CYP3A inhibitor (ritonavir) that inhibits CYP3A mediated metabolism of paritaprevir, thereby providing increased plasma concentration of paritaprevir. Dasabuvir is a hepatitis C virus nonnucleoside NS5B palm polymerase inhibitor, which is supplied as separate tablets in the copackage. Both tablets are for oral administration.
Ombitasvir
The chemical name of ombitasvir is Dimethyl ([(2S,5S)-1-(4-tert-butylphenyl) pyrrolidine-2,5diyl]bis{benzene-4,1-diylcarbamoyl(2S)pyrrolidine-2,1-diyl[(2S)-3-methyl-1-oxobutane-1,2diyl]})biscarbamate hydrate. The molecular formula is C50H67N7O8•4.5H2O (hydrate) and the molecular weight for the drug substance is 975.20 (hydrate). The drug substance is white to light yellow to light pink powder, and is practically insoluble in aqueous buffers but is soluble in ethanol. Ombitasvir has the following molecular structure:
![]()
Paritaprevir
The chemical name of paritaprevir is (2R,6S,12Z,13aS,14aR,16aS)-N-(cyclopropylsulfonyl)-6{[(5-methylpyrazin-2-yl)carbonyl]amino}-5,16-dioxo-2-(phenanthridin-6-yloxy)1,2,3,6,7,8,9,10,11,13a,14,15,16,16a-tetradecahydrocyclopropa[e]pyrrolo[1,2-a][1,4] diazacyclopentadecine-14a(5H)-carboxamide dihydrate. The molecular formula is C40H43N7O7S•2H2O (dihydrate) and the molecular weight for the drug substance is 801.91 (dihydrate). The drug substance is white to off-white powder with very low water solubility. Paritaprevir has the following molecular structure:
 |
Ritonavir
The chemical name of ritonavir is [5S-(5R*,8R*,10R*,11R*)]10-Hydroxy-2-methyl-5-(1methyethyl)-1-[2-(1-methylethyl)-4-thiazolyl]-3,6-dioxo-8,11-bis(phenylmethyl)-2,4,7,12tetraazatridecan-13-oic acid,5-thiazolylmethyl ester. The molecular formula is C37H48N6O5S2 and the molecular weight for the drug substance is 720.95. The drug substance is white to off white to light tan powder practically insoluble in water and freely soluble in methanol and ethanol. Ritonavir has the following molecular structure:
![]()
Ombitasvir, Paritaprevir, Ritonavir Fixed-Dose Combination Tablets
Ombitasvir, paritaprevir, and ritonavir film-coated tablets are co-formulated immediate release tablets. The tablet contains copovidone, K value 28,vitamin E polyethylene glycol succinate, propylene glycol monolaurate Type I, sorbitan monolaurate, colloidal silicon dioxide/colloidal anhydrous silica, sodium stearyl fumarate, polyvinyl alcohol, polyethylene glycol 3350/macrogol 3350, talc, titanium dioxide, and iron oxide red. The strength for the tablet is 12.5 mg ombitasvir, 75 mg paritaprevir, 50 mg ritonavir.
Dasabuvir
The chemical name of dasabuvir is Sodium 3-(3-tert-butyl-4-methoxy-5-{6[(methylsulfonyl)amino]naphthalene-2-yl}phenyl)-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-ide hydrate (1:1:1). The molecular formula is C26H26N3O5S•Na•H2O (salt, hydrate) and the molecular weight of the drug substance is 533.57 (salt, hydrate). The drug substance is white to pale yellow to pink powder, slightly soluble in water and very slightly soluble in methanol and isopropyl alcohol. Dasabuvir has the following molecular structure:
 |
Dasabuvir is formulated as a 250 mg film-coated, immediate release tablet containing microcrystalline cellulose (D50-100 um), microcrystalline cellulose (D50-50 um), lactose monohydrate, copovidone, croscarmellose sodium, colloidal silicon dioxide/anhydrous colloidal silica, magnesium stearate, polyvinyl alcohol, titanium dioxide, polyethylene glycol 3350/macrogol 3350, talc, and iron oxide yellow, iron oxide red and iron oxide black. Each tablet contains 270.3 mg dasabuvir sodium monohydrate equivalent to 250 mg dasabuvir.
//////////fda 2014, Ombitasvir, orphan drug, Abbvie Inc.
Tocagen’s Double Action Glioblastoma Treatment Receives FDA Orphan Drug Designation

Toca 511 and Toca FC, developed by Tocagen, is a combination treatment currently being investigated in phase I/II trials for recurrent high grade glioma including the notoriously difficult to treat glioblastoma multiforme. Toca 511 (vocimagene amiretrorepvec) is a nonlytic retroviral replicating vector (RRV) that encodes the transgene cytosine deaminase (CD). This enzyme is used to catalyze the conversion of Toca FC, a novel oral extended-release prodrug 5-fluorocytosine (5-FC) to the active 5-fluorouracil (5-FU). Intravenous or intracranial injection of Toca 511 takes place during initial treatment and 3-7 weeks later the patient starts cyclic administration of Toca FC.1,2,3 The phase I/II trials in humans have shown similar results of patients exceeding the average life expectancy of high grade gliomas.4
Clinical stage immuno-oncology company, Tocagen, Inc., announced the US Food and Drug Administration has granted its primary immuno-oncology candidate orphan drug designation as a promising and much-needed treatment of glioblastoma, the most common form of primary brain cancer. Every year, over 10,000 people are diagnosed with glioblastoma in the United States. The new designation brings the company’s Toca 511 & Toca FC closer to helping patients suffering with this type of tumor. Tocagen is preparing to proceed with a pivotal clinical trials later this year.
ANNA TAN
Glioblastoma is known to be extremely aggressive, with newly diagnosed patients expecting a mere five-year survival rate of less than 5 percent, along with a high likelihood of tumor recurrence despite completion of standard treatment. Once the tumor recurs, the average survival is only 8 months.

Toca 511 is a retroviral replicating vector (RRV) that selectively delivers a gene for the enzyme cytosine deaminase into the tumor. Patients then take oral cycles of Toca FC, a novel formulation of an antifungal drug, which is converted within infected cancer cells into the FDA-approved anticancer drug, 5-fluorouracil (5 FU). Toca 511 & Toca FC work by programming cancer cells to convert the prodrug 5-FC into the anticancer drug 5-FU, effectively causing tumor cell death and stimulating the immune system through a combination of mechanisms.

“There’s an extraordinary need for new treatment options for patients with this devastating disease,” said Harry Gruber, M.D., chief executive officer of Tocagen. “We believe FDA’s granting of both orphan drug and Fast Track designations to Toca 511 & Toca FC will enable us to more efficiently advance our program, which we hope will ultimately offer physicians and patients a new option in the fight against brain cancer.”
ICT-107 is a dendritic cell-based immunotherapy targeting multiple tumor-associated antigens on glioblastoma stem cells. The trial will be a randomized, double-blind, placebo-controlled, and will aim to enroll around 400 HLA-A2 positive patients. The study will be conducted across 120 sites in the US, Canada, and the European Union.
Mechanism of action
Retroviruses, once inside the target cell, use reverse transcriptase to produce DNA from the RNA present in the virus. Toca 511 is based on the gamma retrovirus, murine leukemia (MLV).5 The virus has many innate properties that are suitable for targeted cancer treatment. One of the most important properties is the reproduction mechanism that occurs without cytolysis of the host cell. In non-lytic reproduction, the infected cell continuously forms small buds that are pinched off containing the virus to allow rapid infection. Another property is the requirement for cell division. Infection is limited to mitotically active cells. These two properties present an ideal candidate vector for modification. The lack of cytolysis in the host cell prevents an immune response and the necessity for the cell to be dividing allows localization to cancerous tumors. As an oncolytic agent, the mechanism uses the rapid mitotic activity of the cancerous tumor cells to spread the therapeutic gene in an effective and controlled manner.5 In Toca 511, the insertion of the CD transgene into the active tumor catalyzes the treatment. The expression of CD by the tumor allows intratumoral conversion of 5-FC to 5-FU.6 This allows the cytotoxic 5-FU to be maintained within the tumor cell. A second mechanism of action is proposed based upon recent data. Post-treatment, a systemic anticancer immune response is present that selectively acts against the cancerous cells.4,7
Design
The design of the Toca 511 RRV is based upon the vector design by Logg et al.5 Multiple changes facilitated selection of a clinically efficacious RRV. The original ecotropic envelope was changed to an amphotropic sequence. In the IRES-CD cassette, multiple small repeats were removed to allow for decreased instability during homologous recombination. A restriction site Psi I was placed at the 3′ of IRES for the insertion of the CD transgene. The resulting vector consists of the following, 5′ to 3′: CMV-R-U5, PBS, 5′ SS, gag, pol (with a 3′ SS), 4070A env, IRES, Psi I, yCD2, Not I, PPT, and the U3-R-U5.8
Clinical trials
Toca 511 and Toca FC combination therapy is currently being investigated for recurrent and progressive Grade III or IV glioma.1,2,3 The initial clinical study is the first to use a RRV to facilitate gene transfer into gliomas. In a recent presentation by Tocagen, researchers expressed the safety and efficacy of the therapy in the first two trials. Minimal treatment toxicity was reported. The landmark six and twelve month survival rates were higher than previously published data in both studies.4 Following positive results with the initial two trials, investigation into the intravenous efficacy is currently being determined.7
Preclinical investigations
Two important discoveries that led to the creation of Toca 511/FC treatment are the optimization of yeast CD and modifications to the vector backbone for genomic replication stability. The optimization of the yeast CD involved the modification of the codon sequence at three amino acids to a known preferred human codon sequence. This did not change the amino acid sequence. This resulted in stability at 37°C compared to the previous 26°C. The vector backbone modification at the env-3′ untranslated boundary created a vector with higher fidelity than the wild type.8 In studies of mice with implanted gliomas, Toca 511 and Toca FC therapy resulted in an unprecedented survival rate.6,8 Furthermore, when the mice were re-implanted with the same glioma post-treatment, memory T lymphocytes remained active and the growth was inhibited.6 The combination of these findings led to the clinical candidate that is currently undergoing trials.
References
1. Tocagen Inc. A Phase 1 Ascending Dose Trial of the Safety and Tolerability of Toca 511 in Patients With Recurrent High Grade Glioma. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000- [cited 2014 June 12]. Available from: http://clinicaltrials.gov/show/NCT01156584 NLM Identifier: NCT01156584.
2. Tocagen Inc. A P1 Ascending Dose Trial of Safety and Tolerability of Toca 511, a Retroviral Replicating Vector, Administered to Subjects at the Time of Resection for Recurrent High Grade Glioma & Followed by Treatment With Toca FC, Extended-Release 5-FC. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000- [cited 2014 June 12]. Available from: http://clinicaltrials.gov/show/NCT01470794 NLM Identifier: NCT01470794.
3. Tocagen Inc. A P1 Ascending Dose Trial of the Safety and Tolerability of Toca 511, a Retroviral Replicating Vector, Administered Intravenously Prior to, and Intracranially at the Time of, Subsequent Resection for Recurrent HGG & Followed by Treatment With Extended-Release 5-FC. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000- [cited 2014 June 12]. Available from: http://clinicaltrials.gov/show/NCT01985256 NLM Identifier: NCT01985256.
4. Interim Clinical Data for Tocagen’s Toca 511 & Toca FC in Patients with High Grade Glioma Presented at American Association of Neurological Surgeons Annual Meeting. Tocagen Inc., 10 April 2014. Web. 10 June 2014. .
5. Logg, C. R.; Robbins, M. J. Retroviral Replicating Vectors in Cancer. Methods in Enzymology 2012, 507, 199-228.
6. Ostertag, D.; Amundson, K. K.; Espinoza, F. L.; Martin, B. Brain tumor eradication and prolonged survival from intratumoral conversion of 5-fluorocytosine to 5-flurouracil using a nonlytic retroviral replicating vector. Neuro-Oncology 2012, 14(2), 145-159.
7. Tocagen Doses First Patient Intravenously in Clinical Trial of
Selective Cancer Therapy, Toca 511 & Toca FC. Tocagen Inc., 11 March 2014. Web. 10 June 2014. http://www.tocagen.com/press/tocagen-doses-first-patient-intravenously-in-clinical-trial-of-selective-cancer-therapy-toca-511-toca-fc/
8. Perez, O. D.; Logg, C. R.; Hiraoka, K.; Diago, O. Design and Selection of Toca 511 for Clinical Use: Modified Retroviral Replicating Vector With Improved Stability and Gene Expression. Molecular Therapy 2012, 20(9), 1689-1698.
Anna Tan, RN

Anna Tan, R.N. – Managing Editor | BioNews Services
Anna Tan, R.N.
///
Vandetanib
Vandetanib; 443913-73-3; Zactima; ZD6474; Caprelsa; ZD 6474; ch 331, azd 6474
cas 338992-00-0 free form
338992-48-6 HCl
338992-53-3 monotrifluoroacetate
N-(4-Bromo-2-fluorophenyl)-6-methoxy-7-(1-methylpiperidin-4-ylmethoxy)quinazolin-4-amine
Vandetanib (INN, trade name Caprelsa) is an anti-cancer drug that is used for the treatment of certain tumours of the thyroid gland. It acts as a kinase inhibitor of a number of cell receptors, mainly the vascular endothelial growth factor receptor (VEGFR), theepidermal growth factor receptor (EGFR), and the RET-tyrosine kinase.[1][2] The drug was developed by AstraZeneca.
Orphan drug designation has been assigned in the E.U. for the treatment of medullary thyroid carcinoma. In 2005, orphan drug designation was also assigned in the U.S. for several indications, including treatment of patients with follicular thyroid carcinoma, medullary thyroid carcinoma, anaplastic thyroid carcinoma, and locally advanced and metastatic papillary thyroid carcinoma. In 2013, orphan drug designation has been assigned in Japan as well for the treatment of thyroid cancer.
Approvals and indications
Vandetanib was the first drug to be approved by FDA (April 2011) for treatment of late-stage (metastatic) medullary thyroid cancer in adult patients who are ineligible for surgery.[3] Vandetanib was first initially marketed without a trade name,[4] and is being marketed under the trade name Caprelsa since August 2011.[5]
Vandetanib is an orally active vascular endothelial growth factor receptor-2 (VEGFR-2/KDR) tyrosine kinase inhibitor, originally developed by AstraZeneca, which was filed for approval in the U.S. and the E.U. for the treatment of non-small cell lung cancer (NSCLC) in combination with chemotherapy, in patients previously treated with one prior anticancer therapy.
However, in late 2009 the company withdrew both the U.S and the EU applications. In 2010, AstraZeneca discontinued development of this compound for the treatment of NSCLC. In 2011, the FDA approved vandetanib for the treatment of medullary thyroid cancer. Also in 2011, a positive opinion was assigned to the regulatory application filed in the E.U. for this indication and in Japan was filed for approval.
Final EMA approval was granted in February 2012 and first E.U. launch took place in the U.K. in 2012.
2011 年 4 月 6 by the FDA-approved surgical resection can not be used for locally advanced or metastatic medullary thyroid cancer (medullary thyroid cancer, MTC) of the drug. Vandetanib is vascular endothelial growth factor receptors (vascular endothelial growth factor receptor, VEGFR) and epidermal growth factor receptor (epidermal growth factor receptor, EGFR) antagonists, tyrosine kinase inhibitors (tyrosine kinase inhibitor). Produced by AstraZeneca.
The synthetic route is as follows:
………………
………………………..
……….
Design and structure-activity relationship of a new class of potent VEGF receptor tyrosine kinase inhibitors
J Med Chem 1999, 42(26): 5369
http://pubs.acs.org/doi/abs/10.1021/jm990345w
………………………
Radiosynthesis of [(11)C]Vandetanib and [(11)C]chloro-Vandetanib as new potential PET agents for imaging of VEGFR in cancer
Bioorg Med Chem Lett 2011, 21(11): 3222
Novel 4-anilinoquinazolines with C-7 basic side chains: Design and structure activity relationship of a series of potent, orally active, VEGF receptor tyrosine kinase inhibitors
J Med Chem 2002, 45(6): 1300
A novel approach to quinazolin-4(3H)-one via quinazoline oxidation: An improved synthesis of 4-anilinoquinazolines
Tetrahedron 2010, 66(4): 962
………………………………
CN 104098544
http://www.google.com/patents/CN104098544A?cl=en
Vandetanib is a synthetic Anilinoquinazoline, advanced medullary thyroid cancer can not be used for the treatment of surgical treatment (medullary thyroid cancer), chemical name: 4- (4-bromo-2- fluoroanilino) _6_ methoxy -7 – [(l- methylpiperidin-4-yl) methoxy] quinazoline, having the following structural formula I:

[0004] The present method of synthesizing the compound are as follows:
[0005] US Patent US7173038 AstraZeneca announced the following methods:
[0006] Method One:
[0007]

Method two:

A structure in which the synthesis of compounds of formula as follows:

the process is cumbersome, long synthetic route, therefore a need to provide a new synthetic way to overcome these problems.
An aspect provides a compound having the structure of formula II:

Another aspect provides a process for preparing a compound of the structural formula II, a compound of formula III with a compound of formula IV in the presence of a base to give a compound of the structural formula II,

where Μ for methylphenylsulfonyl, methylsulfonyl.
Example: 4- (4-bromo-2-fluoroanilino) -6_ methoxy-7 – [(1-formyl-4-yl) methoxy] quinazoline preparation
[0026] in 50mL two-neck flask was added 4- (4-bromo-2-fluoroanilino) -6-methoxy-7-hydroxy-quinazoline (3. 64g, 0 · Olmol), 1- formyl- 4-p methylsulfonyloxy- methylpiperazine steep (3. 56g, 0 · 012mol) and potassium carbonate (4. 14g, 0.03mol), yellow turbid solution was stirred and heated to 100 ° C, TLC detection to feed completion of the reaction. Down to room temperature, the reaction mixture was slowly poured into l〇〇mL water, stirred, filtered, then the filter cake was washed with 50mL water, 15mL of ethyl acetate and then slurried, filtered and dried to give a pale green solid 4- (4- bromo-2-fluoroanilino) -6-methoxy -7 – [(l- carboxylic acid piperidin-4-yl) methoxy] quinazoline 3. 9g, 80% yield.
[0027] ^ NMR (400Mz, DMS0): δ = 1 1〇-1 29 (m, 2H), δ = 1 40-1 43 (m, 2H), δ = 2 15 (s,….. 1H), δ = 2. 64-2. 73 (m, 1H), δ = 3. 06-3. 12 (m, 1H), δ = 3. 71-3. 74 (d, 1H), δ = 3. 95 (s, 3H), δ = 4 • 03-4. 05 (d, 2H), δ = 4. 20-4. 23 (d, 1H), δ = 7. 20 (s, 1H), δ = 7. 46-7. 48 (m, 1H), δ = 7. 51-7 • 53 (m, 1H), δ = 7. 65-7. 67 (d, 1H), δ = 7. 80 (s, 1H), δ = 8. 01 (s, 1H), δ = 8. 35 (s, 1H), δ = 9. 54 (s, 1H).
[0028] Example 2: Preparation of 4- (4-bromo-2-fluoroanilino) -6-methoxy-7 – [(1-methyl-piperidin-4-yl) methoxy] quinazoline preparation
[0029] 4- (4-bromo-2-fluoroanilino) in 100mL three-necked flask, 6-methoxy-7 – [(1-formyl-4-yl) methoxy] quinoline oxazoline (0 · 98g, 2. Ommol), zinc (0 · 6g, 4. 4mmol) and tetrahydrofuran (20mL), stirred pale yellow turbid liquid. At room temperature was added portionwise sodium borohydride (0. 15g, 4. OmmoL), little change in the temperature. Heating
……………………………….
CN 104211649
http://www.google.com/patents/CN104211649A?cl=en
Pharmacokinetics
Vandetanib is well absorbed from the gut, reaches peak blood plasma concentrations 4 to 10 hours after application, and has a half-life of 120 hours days on average, per Phase I pharmacokinetic studies. It has to be taken for about three months to achieve a steady-state concentration. In the blood, it is almost completely (90–96%) bound to plasma proteins such as albumin. It is metabolised to N-desmethylvandetanib via CYP3A4 and to vandetanib-N-oxide via FMO1 and 3. Both of these are active metabolites. Vandetanib is excreted via the faeces (44%) and the urine (25%) in form of the unchanged drug and the metabolites.[2][9][10]

Metabolites of vandetanib (top left): N-desmethylvandetanib (bottom left, via CYP3A4), vandetanib-N-oxide (bottom right, via FMO1 andFMO3), both pharmacologically active, and a minor amount of aglucuronide.[10]
Clinical trials
Non-small cell lung cancer
The drug underwent clinical trials as a potential targeted treatment for non-small-cell lung cancer. There have been some promising results from a phase III trial withdocetaxel.[11] There have also been ambivalent results when used with pemetrexed.[12] Another trial with docetaxel was recruiting in July 2009.[13]
AstraZeneca withdrew EU regulatory submissions for vandetanib (under the proposed trade name Zactima) in October 2009 after trials showed no benefit when the drug was administered alongside chemotherapy.[14]
References
- “Definition of vandetanib”. NCI Drug Dictionary. National Cancer Institute.
- “Vandetanib Monograph”. Drugs.com. Retrieved 29 August 2012.
- “FDA approves new treatment for rare form of thyroid cancer”. Retrieved 7 April 2011.
- “FDA approves orphan drug vandetanib for advanced medullary thyroid cancer” (Press release). AstraZeneca. Retrieved 2011-08-17.
- “AstraZeneca announces trade name CAPRELSA® for vandetanib” (Press release). AstraZeneca. Retrieved 2011-08-17.
- Khurana V, Minocha M, Pal D, Mitra AK (March 2014). “Role of OATP-1B1 and/or OATP-1B3 in hepatic disposition of tyrosine kinase inhibitors.”. Drug Metabol Drug Interact.0 (0): 1–11. doi:10.1515/dmdi-2013-0062. PMID 24643910.
- Haberfeld, H, ed. (2012). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag.
- Khurana V, Minocha M, Pal D, Mitra AK (May 2014). “Inhibition of OATP-1B1 and OATP-1B3 by tyrosine kinase inhibitors.”. Drug Metabol Drug Interact. 0 (0): 1–11.doi:10.1515/dmdi-2014-0014. PMID 24807167.
- Martin, P.; Oliver, S.; Kennedy, S. J.; Partridge, E.; Hutchison, M.; Clarke, D.; Giles, P. (2012). “Pharmacokinetics of Vandetanib: Three Phase I Studies in Healthy Subjects”.Clinical Therapeutics 34 (1): 221–237. doi:10.1016/j.clinthera.2011.11.011.PMID 22206795.
- “Clinical Pharmacology Review: Vandetanib” (PDF). US Food and Drug Administration, Center for Drug Evaluation and Research. 20 August 2010. Retrieved29 August 2012.
- “Vandetanib Shows Clinical Benefit When Combined With Docetaxel For Lung Cancer”. ScienceDaily. 3 June 2009.
- “IASLC: Vandetanib Fails to Improve NSCLC Outcomes with Pemetrexed”. Medpage today. 5 Aug 2009.
- Clinical trial number NCT00687297 for “Study of Vandetanib Combined With Chemotherapy to Treat Advanced Non-small Cell Lung Cancer” at ClinicalTrials.gov
- “Zactima”. European Medicines Agency.
External links
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-(4-bromo-2-fluorophenyl)-6-methoxy-7-[(1-methylpiperidin-4-yl)methoxy]quinazolin-4-amine
|
|
| Clinical data | |
| Trade names | Caprelsa |
| AHFS/Drugs.com | Consumer Drug Information |
| MedlinePlus | a611037 |
| Licence data | US FDA:link |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Protein binding | 90–96% |
| Metabolism | CYP3A4, FMO1, FMO3 |
| Biological half-life | 120 hours (mean) |
| Excretion | 44% faeces, 25% urine |
| Identifiers | |
| CAS Registry Number | 443913-73-3 |
| ATC code | L01XE12 |
| PubChem | CID: 3081361 |
| IUPHAR/BPS | 5717 |
| DrugBank | DB08764 |
| ChemSpider | 2338979 |
| UNII | YO460OQ37K |
| ChEBI | CHEBI:49960 |
| ChEMBL | CHEMBL24828 |
| Synonyms | ZD6474 |
| Chemical data | |
| Formula | C22H24BrFN4O2 |
| Molecular mass | 475.354 g/mol |
//////
Vintafolide

Vintafolide, EC-145 , MK-8109
mw 1917.041, cas 742092-03-1, mf C86 H109 N21 O26 S2
(2S)-2-[(4-{[(2-amino-4-oxo-3H-pteridin-6-yl)methyl]amino}phenyl)formamido]-4-{[(1S)-1-{[(1S)-4-carbamimidamido-1-{[(1S)-2-carboxy-1-{[(1S)-2-carboxy-1-{[(1R)-1-carboxy-2-({2-[({[(1R,9R,10S,11R,12R,19R)-12-ethyl-4-[(13S,15R,17S)-17-ethyl-17-hydroxy-13-(methoxycarbonyl)-1,11-diazatetracyclo[13.3.1.04,12.05,10]nonadeca-4(12),5,7,9-tetraen-13-yl]-10,11-dihydroxy-5-methoxy-8-methyl-8,16-diazapentacyclo[10.6.1.01,9.02,7.016,19]nonadeca-2,4,6,13-tetraen-10-yl]formohydrazido}carbonyl)oxy]ethyl}disulfanyl)ethyl]carbamoyl}ethyl]carbamoyl}ethyl]carbamoyl}butyl]carbamoyl}-2-carboxyethyl]carbamoyl}butanoic acid
Vincaleukoblastin-23-oic acid, O4-deacetyl-, 2-[(2-mercaptoethoxy)carbonyl]hydrazide, disulfide with N-[4-[[(2-amino-1,4-dihydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-L-γ-glutamyl-L-α-aspartyl-L-arginyl-L-α-aspartyl-L-α-aspartyl-L-cysteine
Endocyte innovator
Vintafolide is an investigational targeted cancer therapeutic currently under development by Endocyte and Merck & Co.[1] It is a small molecule drug conjugate consisting of a small molecule targeting the folate receptor, which is overexpressed on certain cancers, such as ovarian cancer, and a potent chemotherapy drug, vinblastine.[2] It is being developed with a companion imaging agent, etarfolatide, that identifies patients that express the folate receptor and thus would likely respond to the treatment with vintafolide.[3] A Phase 3 study evaluating vintafolide for the treatment of platinum-resistant ovarian cancer (PROCEED trial) and a Phase 2b study(TARGET trial) in non-small-cell lung carcinoma (NSCLC) are ongoing.[4] Vintafolide is designed to deliver the toxic vinblastine drug selectively to cells expressing the folate receptor using folate targeting.[5]
A Marketing Authorization Application (MAA) filing for vintafolide and etarfolatide for the treatment of patients withfolate receptor-positive platinum-resistant ovarian cancer in combination with doxorubicin, pegylated liposomal doxorubicin (PLD), has been accepted by the European Medicines Agency.[6] The drug received an orphan drug status in Europe in March 2012.[1] Merck & Co. acquired the development and marketing rights to this experimental cancer drug from Endocyte in April 2012.[1] The drug received orphan drug status in Europe in March 2012.[3]Endocyte remains responsible for the development and commercialization of etarfolatide, a non-invasive companion imaging agent used to identify patients expressing the folate receptor that will likely respond to treatment with vintafolide.[4] Vintafolide is designed to deliver the toxic vinblastine drug selectively to cells expressing the folate receptor using folate targeting.[5]
In 2014 Merck and Endocyte stopped a late-stage study of vintafolide in treating ovarian cancer on the recommendation of a data safety monitoring board, saying that the drug failed to improve progression-free survival.[7]
Vintafolide is folate-conjugated with DAVBLH, which is a derivative of the vinca alkaloid vinblastine.Vinblastine is a microtubule-destabilizing agent that binds tubulin and causes M phase-specific cell cycle arrest and apoptosis of mitotically active cells. Vinblastine is an extremely potent chemotherapeutic agent but has significant toxicities including bone marrow suppression, neurotoxicity, gastrointestinal toxicity and vesicant injury.
Endocyte’s desacetylvinblastinehydrazide/folate conjugate (EC-145) is a folate-targeted cytotoxic anticancer drug in early development for the treatment of non-small cell lung cancer (NSCLC) and breast cancer. The compound had been pre-registered in the E.U. by Merck for the treatment of ovarian cancer, but the application was withdrawn due to lack of efficacy.
In 2012, the product was licensed to Merck & Co. by Endocyte for worldwide exclusive development and commercialization. In 2014, however, this license agreement was terminated and Endocyte regained all rights.
Folates can serve as one-carbon donors in reactions that are critical in the de novo biosynthesis of purines and thymidylate, amino acid metabolism and methylation reactions. Folate can enter a cell by two routes: RFC or by membrane-bound FRs. RFC is a bidirectional anion transporter that is the normal entry method for reduced folates in most cells. By contrast, FRs are expressed in a limited distribution in normal tissues but are overexpressed in multiple cancers including ovarian, lung, breast and colorectal cancer. FRs bind folate derivatives with high affinity and mediate their internalization by endocytosis. Given that FRs are not typically expressed on the luminal surface of epithelial cells, making them inaccessible to normal circulation, they are attractive therapeutic targets with limited toxicity. In addition to the therapeutic agent vintafolide, a radiodiagnostic agent (99mTc-etarfolatide [EC20]) has been developed to allow single-photon emission computed tomography (SPECT) imaging to identify FR-expressing tissues (tumors).
In 2012, orphan drug designations were assigned in the E.U. for the treatment of ovarian cancer and to be used with folic acid for the diagnosis of positive folate-receptor status in ovarian cancer. In 2013, orphan drug designation was assigned in the U.S. for the treatment of ovarian cancer.
Vintafolide is a water-soluble derivative of folic acid and the vinca alkaloid DAVLBH. The molecules are connected through a hydrophilic L-peptide spacer and a disulfide linker (Figure 1). The disulfide linker serves as a cleavable bond that is necessary for drug release following receptor mediated endocytosis. The disulfide bond is reduced in the acidic environment of the endosome, leading to efficient release of vinblastine.
Vintafolide.
DAVBLH: Desacetylvinblastine hydrazide

Structure of vintafolide and mechanism of release of the payload in the endosome.

Mechanism of action
Folate is required for cell division, and rapidly dividing cancer cells often express folate receptors in order to capture enough folate to support rapid cell growth. Elevated expression of the folate receptor occurs in many diseases, including other aggressively growing cancers and inflammatory disorders.[8] Vintafolide binds to the folate receptor and is subsequently taken up by the cell through a natural internalization process called endocytosis. Once inside the cell, vintafolide’s linker releases the chemotherapy drug which kills the cell.[3]
……………
Bioorganic & Medicinal Chemistry Letters (2006), 16(19), 5093-5096
http://www.sciencedirect.com/science/article/pii/S0960894X06008079
An efficient synthesis of the folate receptor (FR) targeting conjugate EC145 is described. EC145 is a water soluble derivative of the vitamin folic acid and the potent cytotoxic agent, desacetylvinblastine monohydrazide. Both molecules are connected in regioselective manner via a hydrophilic peptide spacer and a reductively labile disulfide linker.

………approach for the design and regioselective synthesis of a FA-vinca alkaloid conjugate 1 (EC145,BELOW). As indicated in the retrosynthetic scheme, 1 can be assembled by tethering a FA-Spacer unit 2 to the highly potent cytotoxic molecule, desacetylvinblastine monohydrazide 3, via a linker containing a reducible disulfide bond. The latter is important for drug delivery applications since real-time imaging using a fluorescence resonance energy transfer technique has recently demonstrated that reduction-mediated release of the drug cargo from a disulfide linked FA-conjugate efficiently occurs within the endosomes of cancer cells.

-
Scheme 1.
Reagents and conditions: (i) a—Fmoc-Asp(OtBu)-OH, PyBOP, DIPEA, RT, 1 h; b—20% piperidine/DMF, rt, 10 min; (ii) a—Fmoc-Arg(Pbf)-OH, PyBOP, DIPEA, rt, 1 h; b—20% piperidine/DMF, rt, 10 min; (iii) a—Fmoc-Glu-OtBu, PyBOP, DIPEA, rt, 1 h; b—20% piperidine/DMF, rt, 10 min; (iv) N10-TFA-pteroic acid, PyBOP, DIPEA, rt, 1.5 h; (v) TFA/H2O/TIPS/EDT (92.5:2.5:2.5:2.5), rt, 1 h; (vi) aq NH4OH, pH 9.3, rt, 1 h.
- Selected 1H NMR data for 2 (D2O, 300 MHz): δ 8.68 (s, 1H, FA H-7), 7.57 (d, 2H,J = 8.4 Hz, FA H-12 & 16), 6.67 (d, 2H, J = 9 Hz, FA H-13 & 15), 4.40–4.75 (series of m, 5H), 4.35 (m, 2H), 4.16 (m, 1H), 3.02 (m, 2H), 2.55–2.95 (series of m, 8H), 2.42 (m, 2H), 2.00–2.30 (m, 2H), 1.55–1.90 (m, 2H), 1.48 (m, 2H).
- 1H NMR for compound 6 (DMSO-d6, 300 MHz): δ 8.38 (m, 1H), 8.16 (dt, 1H, J = 8 Hz, 1 Hz), 8.02 (dt, 1H, J = 8 Hz, 1 Hz), 7.88 (ddd, 1H, J = 8 Hz, 7 Hz, 1 Hz), 7.7 (m, 2H), 7.63 (ddd, 1H, J = 8 Hz, 7 Hz, 1 Hz,), 7.4–7.2 (br, 1H), 7.2 (m, 1H), 4.72 (t, 2H,J = 6 Hz), 3.36 (t, 2H, J = 6 Hz).
- Selected 1H NMR data for
EC145 (D2O, 300 MHz): δ 8.67 (s, 1H, FA H-7), 7.50 (br s, 1H, VLB H-11′), 7.30–7.40 (br s, 1H, VLB H-14′), 7.35 (d, 2H, J = 7.8 Hz, FA H-12 & 16), 7.25 (m, 1H, VLB H-13′), 7.05 (br s, 1H, VLB H-12′), 6.51 (d, 2H, J = 8.7 Hz, FA H-13 & 15), 6.4 (s, 2H, VLB H-14 & 17), 5.65 (m, 1H, VLB H-7), 5.5 (m, 1H, VLB H-6), 4.15 (m,1H, VLB H-8′), 3.82 (s, 3H, VLB C18′ –CO2CH3), 3.69 (s, 3H, VLB C16 –OCH3), 2.8 (s, 3H, VLB N–CH3), 1.35 (br s, 1H, VLB H-3′), 1.15 (m, 1H, VLB H-2′), 0.9 (t, 3H, J = 7 Hz, VLB H-21′), 0.55 (t, 3H, J = 6.9 Hz, VLB H-21).
 CLICK ON IMAGE FOR CLEAR VIEW
CLICK ON IMAGE FOR CLEAR VIEW
…………
WO 2004069159
http://www.google.com/patents/WO2004069159A2?cl=en
EXAMPLE 16b

The compounds of Examples 16a and 16b were prepared from the peptidyl fragment Pte-Glu-Asp-Arg-Asp-Asp-Cys-OH , prepared according to the general procedure described in Scheme 12. The Michael addition of this peptidyl fragment to the maleimido derivative of seco-CBI-bis-indole resulted in the folate conjugates Example 16a. The peptidyl fragment also reacted with either the thiosulfonate or pyridyldithio-activated vinblastine to form Example 16b. The maleimido derivative of seco-CBI-bis-indole, and the thiosulfonate and pyridyldithio- activated vinblastine intermediates were prepared using the procedures described herein for other examples.
……………..
https://www.google.com/patents/WO2012142281A1?cl=en
Folate-targeted drugs have been developed and are being tested in clinical trials as cancer therapeutics. EC145, also known as vintafolide, comprises a highly potent vinca alkaloid cytotoxic compound, desacetylvinblastine hydrazide (DAVLBH), conjugated to folate. The EC 145 molecule targets the folate receptor found at high levels on the surface of epithelial tumors, including non-small cell lung carcinomas (NSCLC), ovarian, endometrial and renal cancers, and others, including fallopian tube and primary peritoneal carcinomas. It is believed that EC 145 binds to tumors that express the folate receptor delivering the vinca moiety directly to cancer cells while avoiding normal tissue. Thus, upon binding, EC 145 enters the cancer cell via endocytosis, releases DAVLBH and causes cell death or inhibits cell function. EC 145 has the following formula

EC145
and has been accorded the Chemical Abstracts Registry Number 742092-03-1. As used herein, according to the context, the term EC 145 means the compound, or a pharmaceutically acceptable salt thereof; and the compound may be present in a solid, solution or suspension in an ionized form, including a protonated form. EC145 is disclosed in U.S. Patent No. 7,601,332; and particular uses and an aqueous liquid pH 7.4, phosphate-buffered formulation for intravenous administration are disclosed in WO 2011/014821. As described in WO 2011/014821, it is necessary to store the aqueous liquid formulation in the frozen state to ensure its stability. To avoid this necessity, a formulation is needed which has adequate stability at ambient temperature.
As one aspect of the invention described herein, there is provided a pharmaceutical composition of EC145 which is a lyophilized solid which has adequate stability for storage at ambient temperature and which is capable of redissolving in an aqueous diluent prior to administration.
In another aspect of the invention, there is provided a pharmaceutical composition of EC 145 which is an X-ray amorphous solid which has adequate stability for storage at ambient temperature and which is capable of redissolving in an aqueous diluent prior to administration.
| Systematic (IUPAC) name | |
|---|---|
|
N-(4-{[(2-Amino-4-oxo-1,4-dihydropteridin-6-yl)methyl]amino}benzoyl)-L-γ-glutamyl-L-α-aspartyl-L-arginyl-L-α-aspartyl-L-α-aspartyl-L-cysteine disulfide with methyl (5S,7R,9S)-5-ethyl-9-[(3aR,4R,5S,5aR,10bR,13aR)-3a-ethyl-4,5-dihydroxy-8-methoxy-6-methyl-5-({2-[(2-sulfanylethoxy)carbonyl]hydrazinyl}carbonyl)-3a,4,5,5a,6,11,12,13a-octahydro-1H-indolizino[8,1-cd]carbazol-9-yl]-5-hydroxy-1,4,5,6,7,8,9,10-octahydro-2H-3,7-methanoazacycloundecino[5,4-b]indol-9-carboxylate
|
|
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS Registry Number | 742092-03-1 |
| ATC code | L01CA06 |
| ChemSpider | 27444385 |
| Synonyms | EC-145 |
| Chemical data | |
| Formula | C86H109N21O26S2 |
| Molecular mass | 1917 g/mol |
References
- Sridharan, Balaji (Apr 16, 2012). “Endocyte soars on cancer drug deal with Merck”. Reuters.
- Statement on a nonproprietary name adopted by the USAN Council, United States Adopted Names (USAN) Council, 6 April 2012
- Kuo, Phillip H. (February 2013). “Companion Imaging Diagnostics for Targeted Therapies”. Radiology Today 14 (2): 32.
- “Merck, Endocyte in Development Deal”. Drug Development & Discovery magazine. 2012-04-25.
- Dosio, F.; Milla, P.; Cattel, L. (2010). “EC-145, a folate-targeted Vinca alkaloid conjugate for the potential treatment of folate receptor-expressing cancers”. Current opinion in investigational drugs (London, England : 2000) 11 (12): 1424–1433. PMID 21154124.
- “EMA Accepts For Review MAA Filings For Vintafolide And Etarfolatide”. rttnews.com. 2012-11-27.
- Garde, Damian (2014-05-02). “Merck halts study of the billion-dollar cancer drug vintafolide”. Fierce Biotech. Retrieved 21 April 2015.
- “Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay” 338 (2). March 2005. pp. 284–93. doi:10.1016/j.ab.2004.12.026.PMID 15745749.
-
WO2008098970A1 * Feb 13, 2008 Aug 21, 2008 Pf Medicament Anhydrous crystalline vinflunine salts, method of preparation and use thereof as a drug and means of vinflunine purification WO2010150100A1 * Jun 23, 2010 Dec 29, 2010 Entarco Sa The use of spinosyns and spinosyn compositions against diseases caused by protozoans, viral infections and cancer WO2011014821A1 * Jul 30, 2010 Feb 3, 2011 Endocyte, Inc. Folate-targeted diagnostics and treatment US20100247669 * Sep 30, 2010 Cerulean Pharma Inc. Polymer-agent conjugates, particles, compositions, and related methods of use
////////Vintafolide, BMS-753493, DAVBLH, Desacetylvinblastine hydrazide, EC-145 , MK-8109 , phase 2
Pevonedistat

Millennium Pharmaceuticals, Inc. INNOVATOR
Millennium Pharmaceuticals, Inc., a subsidiary of Takeda Pharmaceutical Company Limited,
MLN4924, MLN 4924-003, TAK-924
905579-51-3 BASE
1160295-21-5 HcL
A potent and selective inhibitor of NAE. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. The ubiquitin-proteasome pathway mediates the destruction of unwanted proteins.
(((1S,2S,4R)-4-{4-[(S)-2,3-Dihydro-1H-inden-1-ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl}-2-hydroxycyclopentyl)methyl sulfamate hydrochloride) (pevonedistat), a novel NEDD8-activating enzyme (NAE) inhibitor, has demonstrated in vitro cytotoxic activity against a variety of human malignancies and is currently being developed by Takeda Pharmaceuticals Company Limited as a clinical candidate for the treatment of cancer
In 2011, orphan drug designation was assigned to MLN-4924 for the treatment of MDS and for the treatment of acute myelogenous leukemia.
PHASE 1…….CANCER SOLID TUMOR

………………….
PATENT
http://www.google.com/patents/US20120330013

preparing a compound represented by the following formula 1 by reacting the compound of formula 11 with TFA (step 9):


The retrosynthetic analysis of MLN4924 (1), as the final desired nucleoside, is shown in the following.

MLN 4924 (1) can be synthesized by condensing cyclic sulfate 3 as the glycosyl donor with a purine base. The glycosyl donor 3 can be produced from diol 4, which in turn can be obtained from cyclopentanone 5 via a stereoselective reduction and a regioselective cleavage of the isopropylidene moiety. The cyclopentanone 5 can be synthesized from cyclopentenone 6 by stereoselective reduction. The intermediate cyclopentenone 6 can be easily derived from D-ribose according to our previously published procedure (Jeong, L. S. et al., J. Org. Chem. 2004, 69, 2634-2636).
The synthetic route for the glycosyl donor 3 is shown in the following scheme 1.

Example 1 Preparation of MLN4924 Step 1: Preparation of 6-(tert-butyl-diphenyl-silanyloxymethyl)-2,2-dimethyl-tetrahydro-cyclopenta[1,3]dioxol-4-one (Compound 5)

To a suspension of the compound 6 (20.0 g, 47.1 mmol) in methanol (400 ml) was added 10% palladium on activated carbon (1.0 g), and the mixture was stirred at room temperature overnight under H2 atmosphere. After filtration of the reaction mixture, the solvent was removed and the residue was dissolved in methylene chloride and then filtered through short pad silica gel. Then, the solvent was evaporated to give the compound 5 (20.1 g, 100%) as a colorless syrup.
[α]20 D −28.32 (c 1.49, MeOH); HR-MS (ESI): m/z calcd for C25H32NaO4Si [M+Na]+ 447.1968, Found 447.1956; 1H NMR (400 MHz, CDCl3) δ 7.69 (m, 4H), 7.40 (m, 6H), 4.84 (t, J=4.4 Hz, 1H), 4.22 (dd, J=1.2, 4.8 Hz, 1H), 3.96 (dd, J=8.0, 10.0 Hz, 1H), 3.82 (dd, J=6.8, 10.0 Hz, 1H), 2.37 (m, 1H), 2.30 (ddd, J=1.2, 8.4, and 18.4 Hz, 1H), 2.20 (ddd, J=1.2, 12.0, and 18.4 Hz, 1H), 1.37 (s, 3H), 1.35 (s, 3H), 1.06 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 112.6, 80.5, 77.6, 77.2, 76.9, 63.6, 38.1, 36.9, 27.1, 27.02, 27.01, 25.3, 19.5; Anal. Calcd for C25H32O4Si: C, 70.72; H, 7.60. Found: C, 70.79; H, 7.75.
Step 2: Preparation of 6-(tert-butyl-diphenyl-silanyloxymethyl)-2,2-dimethyl-tetrahydro-cyclopenta[1,3]dioxol-4-ol (Compound 7)

To a suspension of the compound 5 (20.1 g, 47.1 mmol) in methanol (500 ml) were added sodium borohydride (2.17 g, 57.4 mmol) and cerium (III) chloride heptahydrate (21.3 g, 57.2 mmol) at 0° C., and the mixture was stirred at room temperature for 30 min. After the solvent was removed, the residue was partitioned between ethyl acetate and water. The organic layer was then washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=5/1) to give the compound 7 (20.86 g, 98%) as a colorless syrup.
[α]20 D +34.55 (c 0.55, MeOH); HR-MS (ESI): m/z calcd for C25H34NaO4Si [M+Na]+: 449.2124; Found: 449.2110; 1H NMR (400 MHz, CDCl3) δ 7.69 (m, 4H), 7.39 (m, 6H), 4.62 (t, J=5.6 Hz, 1H), 4.44 (t, J=5.6 Hz, 1H), 3.89 (dd, J=6.0, 7.6 Hz, 1H), 3.84 (m, 1H), 3.68 (dd, J=6.4, 10.0 Hz, 1H), 1.91 (m, 2H), 1.26 (m, 1H), 1.42 (s, 3H), 1.33 (s, 3H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.9, 135.8, 134.2, 134.1, 129.8, 129.7, 127.8, 127.7, 110.6, 79.4, 78.9, 77.6, 77.2, 76.9, 72.5, 62.9, 41.6, 33.4, 27.0, 25.9, 27.0, 25.9, 24.4, 19.5; Anal. Calcd for C25H34O4Si: C, 70.38; H, 8.03. Found: C, 70.41; H, 8.08.
Step 3: Preparation of 3-tert-butoxy-4-(tert-butyl-diphenyl-silanyloxymethyl)-cyclopentane-1,2-diol (Compound 4)

To a solution of the compound 7 (20.86 g, 47.12 mmol) in methylene chloride was added trimethylaluminum (2.0 M in toluene, 132.1 ml) at 0° C., and the mixture was stirred at room temperature for 2 days. The mixture was cooled to 0° C., slowly quenched with an aqueous saturated ammonium chloride solution, filtered, and evaporated. The residue was partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=2/1) to give the compound 4 (13.42 g, 62%) as a colorless syrup.
[α]20 D +3.30 (c 0.55, MeOH); HR-MS (ESI): m/z calcd for C26H38NaO4Si [M+Na]+: 465.2437; Found: 465.2423; 1H NMR (400 MHz, CDCl3) δ 7.70 (m, 4H), 7.41 (m, 6H), 4.05 (dd, J=4.4, 7.2 Hz, 1H), 3.93 (m, 1H), 3.72 (m, 2H), 3.59 (dd, J=3.6, 12.0 Hz, 2H), 2.70 (d, J=20.8 Hz, 1H), 2.10 (m, 2H), 1.60 (m, 1H), 1.20 (s, 9H), 1.06 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.9, 133.5, 130.0, 129.9, 127.9, 127.9, 77.6, 77.2, 76.9, 74.9, 73.8, 72.7, 72.1, 63.3, 42.1, 34.0, 28.5, 27.0, 19.4; Anal. Calcd for C26H38O4Si: C, 70.55; H, 8.65. Found: C, 70.61; H, 8.70.
Step 4: Preparation of (4-tert-butoxy-2,2-dioxo-tetrahydro-2-yl-6-cyclopenta[1,3,2]-dioxathiol-5-ylmethoxy)-tert-butyl-diphenyl-silane (Compound 3)

To a solution of the compound 4 (13.42 g, 30.3 mmol) in methylene chloride were added triethyl amine (14.5 ml, 101.0 mmol) and thionyl chloride (3.7 ml, 47.4 mmol) at 0° C., and the reaction mixture was stirred at 0° C. for 10 minutes. The reaction mixture was partitioned between methylene chloride and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=6/1) to give the cyclic sulfite (14.37 g, 97%) as a white foam.
[α]20 D +20.00 (c 0.05, MeOH); HR-MS (ESI): m/z calcd for C26H36NaO5SSi [M+Na]+: 511.1950; Found: 511.1929; 1H NMR (400 MHz, CDCl3) δ 7.64 (m, 4H), 7.40 (m, 6H), 5.23 (m, 1H), 5.04 (dd, J=4.4, 6.0 Hz, 1H), 4.01 (t, J=4.8 Hz, 1H), 3.68 (dd, J=3.6, 10.4 Hz, 1H), 3.56 (dd, J=8.0, 10.4 Hz, 1H), 2.07 (m, 2H), 1.96 (m, 1H), 1.14 (s, 9H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.8, 135.7, 133.9, 133.8, 129.9, 129.9, 127.9, 127.8, 85.7, 83.2, 77.6, 77.2, 76.9, 75.0, 71.1, 62.7, 44.7, 31.4, 28.5, 27.1, 19.4; Anal. Calcd for C26H36O5SSi: C, 63.90; H, 7.42; S, 6.56. Found: C, 63.94; H, 7.45; S, 6.61.
To a solution of the cyclic sulfite obtained above (14.37 g, 29.4 mmol) in the mixture of carbon tetrachloride, acetonitrile and water (1:1:1.5, 210 ml) were added sodium metaperiodate (18.56 g, 56.4 mmol) and ruthenium chloride (1.72 g, 8.25 mmol), and the reaction mixture was stirred at room temperature for 10 minutes. The reaction mixture was partitioned between methylene chloride and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=4/1) to give the compound 3 (13.36 g, 90%) as a white solid.
mp 101-104° C.; [α]20 D −80.00 (c 0.05, MeOH); HR-MS (ESI): m/z calcd for C26H36NaO6SSi [M+Na]+: 527.1900; Found: 527.1881; 1H NMR (400 MHz, CDCl3) δ 7.64 (m, 4H), 7.41 (m, 6H), 5.13 (m, 1H), 4.83 (dd, J=4.4, 6.8 Hz, 1H), 4.13 (t, J=4.0 Hz, 1H), 3.92 (dd, J=6.4, 10.4 Hz, 1H), 3.69 (dd, J=5.2, 10.4 Hz, 1H), 2.11 (m, 2H), 2.02 (m, 1H), 1.15 (s, 9H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.7, 135.0, 133.8, 133.7, 130.0, 128.0, 127.9, 83.5, 82.2, 77.6, 77.2, 76.9, 75.4, 70.4, 70.4, 62.2, 43.9, 31.3, 28.2, 27.1, 26.8, 19.4; Anal. Calcd for C26H36O6SSi: C, 61.87; H, 7.19; S, 6.35. Found: C, 61.91; H, 7.14; S, 6.30.
Step 5: Preparation of 2-tert-butoxy-3-(tert-butyl-diphenyl-silanyloxymethyl)-5-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentanol (Compound 8)

A suspension of N6-indanyl-7-deazaadenine (8.80 g, 35.2 mmol), sodium hydride (1.38 g, 45.7 mmol) and 18-crown-6 (9.11 g, 45.7 mmol) in THF (200 ml) was stirred at 80° C. To the reaction mixture was added a solution for the compound 3 (13.36 g, 26.5 mmol) in THF (150 ml), and the stirring was continued at 80° C. overnight. The reaction mixture was cooled down to 0° C., and conc. HCl was added slowly until pH reaches 1-2. Then the reaction mixture was further stirred at 80° C. for 2 hours. After neutralized with saturated aqueous NaHCO3 solution, the reaction mixture was partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=2/1) to give the compound 8 (11.62 g, 65%) as a white foam.
UV (CH2Cl2) λmax 272.5 nm; [α]20 D −8.89 (c 0.45, MeOH); HR-MS (ESI): m/z calcd for C41H51N4O3Si [M+H]+: 675.3730; Found: 675.3717; 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 7.70 (m, 4H), 7.41 (m, 6H), 6.92 (d, J=3.6 Hz, 1H), 6.29 (d, J=3.2 Hz, 1H), 5.91 (dd, J=7.6, 14.8 Hz, 1H), 5.14 (br d, J=6.8 Hz, 1H), 4.77 (m, 1H), 4.36 (t, J=6.0 Hz, 1H), 4.22 (dd, J=5.2, 10.8 Hz, 1H), 3.84 (dd, J=5.6, 10.4 Hz, 1H), 3.73 (dd, J=8.4, 10.4 Hz, 1H), 3.37 (d, J=5.6 Hz, 1H), 3.06 (m, 1H), 2.95 (m, 1H), 2.75 (m, 1H), 2.75 (m, 1H), 2.58 (m, 1H), 2.38 (m, 1H), 2.15 (m, 1H), 1.98 (m, 1H), 1.65 (s, 1H), 1.55 (s, 1H), 1.16 (s, 9H), 1.07 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 156.4, 151.8, 150.3, 144.1, 143.8, 135.9, 134.0, 129.9, 128.2, 127.9, 127.9, 127.0, 125.1, 124.4, 123.3, 103.8, 97.4, 77.8, 77.6, 77.2, 76.9, 74.9, 72.4, 63.5, 62.1, 56.3, 43.9, 34.9, 30.5, 30.5, 28.5, 27.2, 19.5; Anal. Calcd for C41H50N4O3Si: C, 72.96; H, 7.47; N, 8.30. Found: C, 73.01; H, 7.45; N, 8.36.
Step 6: Preparation of {7-[3-tert-butoxy-4-(tert-butyl-diphenyl-silanyloxymethyl)-cyclopentyl]-7H-pyrrolo[2,3-d]pyrimidin-4-yl}-indan-1-yl-amine (Compound 9)

To a solution of the compound 8 (11.62 g, 17.2 mmol) in methylene chloride (300 ml) were added N,N-dimethylaminopyridine (5.64 g, 51.6 mmol) and phenyl chlorothionocarbonate (4.3 ml, 34.4 mmol), and the reaction mixture was stirred at room temperature overnight. After the solvent was removed, the residue was purified by silica gel column chromatography (hexane/ethyl acetate=6/1) to give the thiocarbonate (13.82 g, 99%) as a white foam.
UV (MeOH) λmax 271.50 nm; [α]20 D +10.00 (c 0.15, MeOH); HR-MS (ESI): m/z calcd for C48H55N4O4SSi [M+H]+: 811.3713; Found: 811.3687; 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 1H), 7.61 (dd, J=1.6, 7.6 Hz, 4H), 7.34 (m, 5H), 7.26 (m, 4H), 7.18 (m, 6H), 6.86 (s, 1H), 6.25 (d, J=3.2 Hz, 1H), 6.00 (dd, J=3.2, 8.4 Hz, 1H), 5.83 (d, J=6.8 Hz, 1H), 5.19 (m, 1H), 5.07 (br s, 1H), 4.48 (t, J=3.6 Hz, 1H), 3.82 (dd, J=7.2, 10.4 Hz, 1H), 3.52 (dd, J=7.2, 10.0 Hz, 1H), 2.99 (m, 1H), 2.88 (m, 2H), 2.69 (m, 2H), 2.18 (dd, J=11.2, 13.6 Hz, 1H), 1.94 (m, 2H), 1.12 (s, 9H), 0.98 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 194.9, 153.5, 152.1, 143.9, 135.9, 135.8, 134.1, 129.9, 129.6, 128.3, 127.9, 127.0, 126.7, 125.1, 124.6, 123.2, 122.0, 87.9, 77.6, 77.2, 76.9, 74.6, 70.4, 63.5, 57.3, 42.8, 35.0, 30.7, 30.5, 29.9, 28.7, 27.1, 19.4; Anal. Calcd for C48H54N4O4SSi: C, 71.08; H, 6.71; N, 6.91; S, 3.95. Found: C, 71.14; H, 6.75; N, 6.95; S, 4.01.
To a solution of the thiocarbonate obtained above (13.82 g, 17.0 mmol) in toluene (200 ml) were added tri-n-butyltinhydride (9.4 ml, 34.1 mmol) and 2,2′-azo-bis-isobutyronitrile (4.32 g, 26.3 mmol), and the reaction mixture was stirred at 110° C. for 1 hour. After the mixture was cooled down, the solvent was removed. The resulting residue was purified by silica gel column chromatography (hexane/ethyl acetate=3/1) to give the compound 9 (9.21 g, 82%) as a white foam.
UV (MeOH) λmax 272.50 nm; [α]20 D −10.00 (c 0.20, MeOH); HR-MS (ESI): m/z calcd for C41H51N4O2Si [M+H]+: 659.3781; Found: 659.3757; 1H NMR (400 MHz, CDCl3) δ 8.41 (s, 1H), 7.69 (m, 4H), 7.41 (m, 6H), 7.29 (m, 2H), 7.23 (m, 2H), 6.92 (d, J=3.6 Hz, 1H), 6.31 (d, J=3.6 Hz, 1H), 5.90 (dd, J=7.2, 14.8 Hz, 1H), 5.38 (m, 1H), 5.15 (br s, 1H), 4.33 (dd, J=5.2, 8.4 Hz, 1H), 3.88 (dd, J=6.4, 10.0 Hz, 1H), 3.68 (dd, J=7.2, 10.4 Hz, 1H), 3.05 (m, 1H), 2.96 (dd, J=7.6, 15.6 Hz, 1H), 2.76 (m, 1H), 2.45 (d, J=5.2 Hz, 1H), 2.29 (m, 2H), 2.06 (m, 1H), 1.95 (m, 2H), 1.55 (s, 1H), 1.13 (s, 9H), 1.06 (s, 9H);13C NMR (100 MHz, CDCl3) δ 156.3, 151.9, 144.1, 143.9, 135.9, 135.8, 134.3, 129.8, 128.2, 127.8, 127.0, 125.1, 124.6, 121.8, 77.6, 77.2, 76.7, 73.5, 72.2, 63.6, 56.4, 52.8, 46.8, 42.8, 34.9, 34.5, 30.5, 28.6, 27.2, 28.7, 19.4; Anal. Calcd for C41H50N4O2Si: C, 74.73; H, 7.65; N, 8.30. Found: C, 74.79; H, 7.61; N, 8.25.
Step 7: Preparation of 2-tert-butoxy-4-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentanol (Compound 10)

To a solution of the compound 9 (9.21 g, 13.97 mmol) in the mixture of THF and pyridine (1:1, 160 ml) was added dropwise pyridine hydrofluoride (18.42 ml, 190.0 mmol) at 0° C., and the reaction mixture was stirred at room temperature for 1 hour. The mixture was neutralized with saturated aqueous NaHCO3 solution and partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. Then, the residue was purified by silica gel column chromatography (hexane/ethyl acetate=1/3) to give the compound 10 (5.63 g, 99%) as a white foam.
UV (MeOH) λmax 273.00 nm; [α]20 D −6.36 (c 1.10, MeOH); HR-MS (ESI): m/z calcd for C25H33N4O2 [M+H]+: 421.2604; Found: 421.2599; 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1H), 7.30 (d, J=7.6 Hz, 1H), 7.22 (d, J=7.2 Hz, 2H), 7.15 (t, J=6.8 Hz, 1H), 6.88 (d, J=3.2 Hz, 1H), 6.23 (d, J=3.6 Hz, 1H), 5.83 (dd, J=7.2, 15.2 Hz, 1H), 5.28 (m, 1H), 5.06 (m, 1H), 4.47 (dd, J=5.6, 10.4 Hz, 1H), 3.78 (m, 1H), 3.70 (m, 1H), 3.24 (t, J=5.2 Hz, 1H), 2.98 (m, 1H), 2.87 (m, 1H), 2.68 (m, 1H), 2.46 (m, 1H), 2.37 (m, 2H), 1.93 (m, 2H), 1.18 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 156.2, 151.8, 147.9, 143.9, 143.9, 128.3, 126.9, 125.1, 124.5, 121.9, 97.7, 77.6, 77.2, 76.9, 75.5, 74.9, 63.4, 56.4, 53.8, 44.2, 42.2, 34.9, 33.2, 30.5, 28.6; Anal. Calcd for C25H32N4O2: C, 71.40; H, 7.67; N, 13.32. Found: C, 71.46; H, 7.60; N, 13.35.
Step 8: Preparation of sulfamic acid 2-tert-butoxy-4-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentylmethyl ester (Compound 11)

Preparation of 2.0 M solution of chlorosulfonamide in acetonitrile: Formic acid (14.15 ml, 166.0 mmol) was added dropwise to chlorosulfonyl isocyanate (32.0 ml, 162.5 mmol) under nitrogen atmosphere at 0° C. When the addition was completed, the mixture was solidified. To the mixture was added acetonitrile (61.3 ml), and the resulting solution was left to stand under nitrogen source at room temperature overnight.
To a solution of the compound 10 (5.63 g, 13.83 mmol) and triethyl amine (9.7 ml, 0.74 mmol) in acetonitrile (278 ml) was added 2.0 M solution of chlorosulfonamide in acetonitrile (13.83 ml, 27.76 mmol) at 0° C., and the reaction mixture was stirred at room temperature for 45 minutes. Additional 2.0 M chlorosulfonamide solution in acetonitrile (13.83 ml, 27.76 mmol) was added and the mixture was stirred at room temperature for 15 minutes. The reaction was quenched with methanol, and the solvent was removed. The residue was purified by silica gel column chromatography (methylene chloride/methanol=20/1) to give the compound 11 (6.37 g, 92%) as a white foam.
UV (MeOH) λmax 273.00 nm; [α]20 D −18.00 (c 0.50, MeOH); HR-MS (ESI): m/z calcd for C25H34N5O4S [M+H]+: 500.2332; Found: 500.2331; 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 7.36 (d, J=7.2 Hz, 1H), 7.29 (d, J=7.2 Hz, 1H), 7.22 (m, 2H), 6.95 (d, J=3.6 Hz, 1H), 6.31 (d, J=3.2 Hz, 1H), 5.89 (d, J=6.4 Hz, 1H), 5.10 (s, 2H), 4.41 (m, 2H), 4.26 (m, 1H), 3.05 (m, 1H), 2.94 (m, 1H), 2.76 (m, 2H), 2.27 (m, 3H), 2.06 (m, 1H), 1.97 (m, 1H), 1.76 (br s, 1H); 13C NMR (100 MHz, CDCl3) δ 156.4, 151.9, 149.9, 143.9, 143.8, 128.3, 126.9, 125.1, 124.5, 121.9, 121.9, 103.5, 97.9, 77.4, 77.2, 76.9, 74.3, 71.9, 71.3, 56.4, 53.1, 49.0, 42.3, 34.9, 34.3, 30.5, 28.6; Anal. Calcd for C25H33N5O4S: C, 60.10; H, 6.66; N, 14.02; S, 6.42. Found: C, 60.15; H, 6.71; N, 13.98; S, 6.39.
Step 9: Preparation of sulfamic acid 2-hydroxy-4-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentylmethyl ester (Compound 1)

A solution of the compound 11 (6.37 g, 12.72 mmol) in 70% trifluoroacetic acid (149.24 ml) was stirred at room temperature for 2 hours. The solvent was removed and the residue was purified by silica gel column chromatography (hexane/ethylene acetate=1/2) to give the compound 1 (5.08 g, 90%) as a white foam. BASE
UV (MeOH) λmax 279.50 nm; [α]20 D −6.41 (c 2.34, MeOH);
HR-MS (ESI): m/z calcd for C21H26N5O4S [M+H]+: 444.1705; Found: 444.1706;
1H NMR (400 MHz, CD3OD) δ 8.17 (d, J=1.6 Hz, 1H), 7.25 (m, 2H), 7.18 (m, 2H), 6.64 (d, J=3.6 Hz, 1H), 5.86 (t, J=7.6 Hz, 1H), 5.46 (m, 1H), 4.49 (d, J=2.8 Hz, 1H), 3.07 (m, 1H), 2.92 (m, 1H), 2.80 (m, 1H), 2.64 (m, 1H), 2.35 (m, 1H), 2.25 (m, 2H), 2.03 (m, 2H);
13C NMR (100 MHz, CD3OD) δ 152.1, 145.3, 144.6, 128.8, 127.6, 125.7, 125.2, 122.6, 100.5, 73.1, 70.9, 56.9, 54.0, 44.8, 43.6, 34.9, 34.6, 31.1;
Anal. Calcd for C21H25N5O4S: C, 56.87; H, 5.68; N, 15.79; S, 7.23. Found: C, 56.91; H, 5.73; N, 15.82; S, 7.26.
…………………….
http://www.google.com/patents/WO2010132110A1?cl=en
((lS,2S,4R)-4-{4-[(lS)-2,3-dihydro-lH-inden-l-ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl }-2-hydroxycyclopentyl)methyl sulfamate (//) is described in Intl. App. Pub. No. WO 07/092213, U.S. App. Pub. No. 2007/0191293, and U.S. App. Pub. No. 2009/0036678. The potassium salt of ((lS,2S,4R)-4-{4-[( 1 S)-2,3-dihydro- 1 H-inden- 1 -ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl } -2-hydroxycyclopentyl)methyl sulfamate is disclosed in Intl. App. Pub. No. WO 07/092213 and U.S. App. Pub. No. 2007/0191293.

(H)
((lS,2S,4R)-4-{4-[(lS)-2,3-dihydro-lH- inden-l-ylamino]-7H-pyπOlo[2,3-d]pyrimidin-7-yl}-2-hydroxycyclopentyl)methyl sulfamate (/):

Step 3: Synthesis of ((lS,2S.4R)-4-(4-r(lS)-2,3-dihydro-lH-inden-l-ylaminol-7H-pyrrolor2.3-dlpyrimidin-7-yl}-2-hvdroxycvclopentyl)methyl sulfamate hydrochloride Form 1
[0158] A reactor was charged with ((lS,2S,4R)-4-{4-[(lS)-2,3-dihydro-lH-inden-l-ylarnino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl }-2-hydroxycyclopentyl)methyl sulfamate (13.4 Kg, 30.2 mol) and 200-proof ethanol (106.2 Kg). The mixture was heated to reflux to afford a clear solution. The mixture was cooled to 50 ± 5 0C and passed through a cartridge filter. 200 proof ethanol (8.9 Kg) was used to rinse the filter. 1.27M hydrogen chloride in ethanol (10.2 Kg) was added via a cartridge filter at a rate to maintain a temperature of 50 ± 5 0C. The mixture was then seeded with Form 1 (67 g). Further 1.27M HCl (10.2 Kg) was added via a cartridge filter at a rate to maintain a temperature of 50 ± 5 0C. The mixture was then stirred at 50 ± 5 0C for about 3 hours. The mixture was then cooled to 20 ± 5 0C over about 3 hours and then stirred for about 2.5 hours. The solid product was then isolated by filtration and washed with 200-proof ethanol (I x 20.4 Kg and 1 x 21.2 Kg). The solids were dried by aspiration on the filter until no supernatant was seen to be collected, and then further dried under reduced pressure at <30 0C to afford the title compound (12.2 Kg) as a white solid determined to be Form 1 by XRPD. IH NMR (300MHz, DMSO, δ): 9.83 (s, IH), 8.34 (s, IH), 7.62 (s, IH), 7.44 (s, 2H), 7.30 (m, 3H), 7.22 (t, IH), 7.07 (s, IH), 5.86 (dd, IH), 5.42 (m, IH), 4.32 (m, IH), 4.21 (dd, IH), 4.02 (dd, IH), 3.04 (m, IH), 2.88 (m, IH), 2.67 (m, 2H), 2.15 (m, 2H), 2.08 (m, 2H), 1.94 (m, IH). XRPD data for Form 1 is shown in FIGURE 1 and Table 1; DSC data is shown in FIGURE 2, and TGA data for Form 1 is shown in FIGURE 3.
…………..
http://www.google.com/patents/WO2007092213A2?cl=en
Example 70: Diastereoisomeric mixture of (lS/2R/4R)-4-{4-[(lS)-2/3-dihydro-lH-inden-l- ylaimnol-ZH-pyrrolop^-dlpyxirnidin-Z-ylJ^-hydroxycyclopentyl s ulf amate and (lRf2S/4S)-4-{4-[(lS)-2,3-dihydro-lH-inden-l-ylaminol-7H-pyrrolo[2,3d]- pyrimidin-7-yl}-2-hydroxycyclopentyl sulfamate (Compounds 1-77 and 1-78)

Step a: Cyclopent-3-en-l-yl methanesulfonate
[0335] 3-Cydopentene-l-ol (0.500 g, 5.94 mmol) was stirred in DCM (95 mL).
Pyridine (2.40 mL), N,N-dimethylaminopyridine (0.10 g, 1.00 mmol) and methanesulfonyl chloride (0.690 mL, 8.92 mmol) were added, and the reaction mixture was stirred at 350C for 4 h. N,N-Dimethylarrιinopyridirιe (0.14 g, 1.2 mmol) and methanesulfonyl chloride (0.69 mL, 8.92 mmol) were added, and the reaction was stirred overnight. TLC indicated complete conversion. The reaction mixture was cooled and concentrated. The residue was purified by silica gel chromatography, eluting with DCM, to afford the title compound as a clear oil (0.660 g, 68%).
Step b: 7-Cyclopent-3-en-l-yl-N-r(lSV2,3-dihydro-lH-inden-l-yn-7H-pyrrolor2,3-rfl- pyrmτidin-4-arnine
[0336] N-[(lS)-2,3-DihydrcHlH-mden-l-yl]-7H-pyrrolo[2/3-d]p3αimidin-4-amine (1.32 g, 5.29 mmol) was azeotroped with toluene and placed under high vacuum for 30 min. N,N-Dimethylformamide (17.7 mL) was added, followed by cesium carbonate (1.99 g, 6.10 mmol). The mixture was stirred at 700C for 10 min. Cyclopent-3-en-l-yl methanesulfonate (0.660 g, 4.07 mmol) in N,N-dimethylformarnide (12.6 mL) was added dropwise. The reaction mixture was heated to 1100C for 1 h. The reaction mixture was cooled, quenched with brine and diluted with H2O. The aqueous layer was extracted with EtOAc (3x), washed with H2O and brine, dried (Na2SO4), filtered, and concentrated. The residue -was purified by via silica gel chromatography, eluting with a gradient of 0 to 5% MeOH in DCM followed by 25 to 50% EtOAc in hexanes, to afford the title compound as a pale brown solid (0.684 g, 53%). LC/MS: R1 = 1.38 min, ES+ 317 (FA standard). Step c: (lR,2S,45)-4-{4-r(lS)-2,3-dihydro-lH-inden-l-ylaininol-7H-pyrrolof2.3- rf1pyrimidin-7-yl}cyclopentane-l,2-diol
[0337] 7-Cyclopent-3-en-l-yl-N-[(lS)-2^-dihyrdo-lH-inden-l-yl]-7H-pyrrolo[2,3- d]pyτimidin-4-amine (0.312 g, 0.986 mmol) was stirred in tert-butyl alcohol (4.9 mL) and H2O (4.9 mL). AD-mix-α (Sigma- Aldrich, 1.4 g) was added, and the suspension was stirred at rt overnight. TLC indicated complete conversion. The reaction was quenched with sodium sulfite (1.48 g, 11.7 mmol), and the mixture was stirred for 5 h. The reaction mixture was diluted with EtOAc and H2O, and the aqueous layer was extracted with EtOAc (2x). The organic layer was dried (Na2SO4), filtered, and concentrated. The residue was purified via silica gel chromatography, eluting with EtOAc, to afford the title compound as a white solid (0.190 g, 55%).
Step d: Diastereoisomeric mixture of (lS,2R,4R)-4-{4-r(15)-23-dihydro-lH-inden-l- ylarninoi^jH-pyrrolofΣ^dlpyrirnidin-y-yll-l-hydroxycyclopentyl sulfamate and (lR,2S,4S)-4-{4-iαSV2,3-dihydro-lH-inden-l-ylarninol-7H-pyrrolor2,3- rf1pyrimidm-7-yl)-2-hydroxycyclopenryl sulfamate (Compounds 1-77 and 1-78)
[0338] (lR,2S,4S)-4-{4-[(lS)-2,3-Dihydro-lH-inden-l-ylarnino]-7H-pyrrolo[2/3- d]pyrimidin-7-ylJcyclopentane-l,2-diol (0.080 g, 0.23 mmol) was azeotroped with toluene and then was dissolved in anhydrous acetonitrile (2.3 mL). Pyridine (0.0369 mL, 0.458 mmol) was added. The reaction mixture was cooled to 00C, and a 2N solution of chlorosulfonamide in acetonitrile (0.144 mL) was added dropwise. The reaction was stirred for 1 h, and then additional 2N chlorosulfonamide in acetonitrile (0.028 mL) was added. After 30 min, additional 2N chlorosulfonamide in acetonitrile (0.0342 mL) was added, and the reaction mixture was stirred for 2 h. The reaction was quenched with methanol, and the mixture was concentrated in vacuo. The residue was purified by preparative thin layer chromatography using DCM:AcCN:MeOH (50:45:5). The relevant band was cut, washed with acetone, filtered, and concentrated to give a mixture of diastereomers as a white solid. (11 mg, 11%). 1H NMR (CDCl3, 400 NMR, δ): 8.36-8.27 (m, IH); 7.38-7.09 (m, 5H); 6.90-6.80 (m, IH); 6.36- 6.20 (m, IH); 5.95-5.76 (m, IH); 5.51-5.22 (m, 2H); 4.83-4.68 (m, IH); 3.87-3.72 (m, IH); 3.12- 2.83 (m, 2H); 2.75-2.53 (m, IH); 2.50-2.14 (m, 2H); 2.08-1.79 (m, 2H) ppm. LC/MS: R, = 1.16 min, ES+ 430 (FA standard).
…………
WO 2012061551
http://www.google.im/patents/WO2012061551A1?cl=en
The compound ((lS,2S,4R)-4-(4-((lS)-2,3-dihydro-lH-inden-l-ylamino)-7H-pyrrolo[2,3-d]- pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl sulfamate:

also known as MLN4924, is an inhibitor of NEDD8-activating enzyme (NAE). Inhibition of NAE has been shown to induce cancer cell death and inhibit the growth of tumors in xenograft models. See, e.g., T.A. Soucy et al., Nature, 2009, 458, 732-737; T.A. Soucy ei al., Clin. Cancer Res., 2009, 15 (12), 3912-3916; and J.E. Brownell et al., Mol. Cell., 2010, 37 (1), 102-111, each of which is hereby incorporated by reference herein in its entirety. MLN4924, pharmaceutical compositions of MLN4924, processes for its synthesis, and polymorphic forms have been described previously. See, e.g., US Patent Appl. Nos. 11/700,614 (Publ. No. 2007/0191293), 12/221,399 (Publ. No. 2009/0036678) and 12/779,331 (Publ. No. 2011/0021544),
……………
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.5b00209, http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00209

A practical synthesis of a novel NEDD8-activating enzyme (NAE) inhibitor pevonedistat (MLN4924) is described. Key steps include an enantioselective synthesis of an amino-diol cyclopentane intermediate containing three chiral centers and a novel, regioselective sulfamoylation using N-(tert-butoxycarbonyl)-N-[(triethylenediammonium)sulfonyl]azanide. The linear process, involving six solid isolations, has been carried out in multiple cGMP productions on 15–30 kg scale to produce pevonedistat in 98% (a/a) chemical purity and 25% overall yield.

((1S,2S,4R)-4-(4-(((S)-2,3-Dihydro-1H-inden-1-yl)amino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl Sulfamate (1)
The reaction yielded 1 (0.285 kg, 58.5%, 93.0% a/a) as an off-white solid.
HPLC retention time of 1 BASE(Method C): 22.6 min;
1H NMR (400 MHz, DMSO) δ 8.19 (s, 1H), 7.77 (d, J = 8.4 Hz, 1H), 7.45 (s, 2H), 7.31–7.26 (m, 2H), 7.22 (t, J = 6.6 Hz, 2H), 7.15 (t, J = 7.2 Hz, 1H), 6.66 (d, J = 3.5 Hz, 1H), 5.92 (q, J = 8.0 Hz, 1H), 5.39 (qd, J = 8.8, 5.7 Hz, 1H), 4.95 (d, J = 3.9 Hz, 1H), 4.42–4.31 (m, 1H), 4.25 (dd, J = 9.7, 7.0 Hz, 1H), 4.07 (dd, J = 9.6, 8.0 Hz, 1H), 3.01 (ddd, J = 15.7, 8.7, 3.0 Hz, 1H), 2.95–2.81 (m, 1H), 2.81–2.65 (m, 1H), 2.58–2.49 (m, 1H), 2.31–1.86 (m, 5H);
13C NMR (100 MHz, DMSO) δ 155.91, 151.18, 149.02, 144.66, 142.98, 127.30, 126.28, 124.49, 124.11, 121.68, 102.83, 98.86, 70.82, 69.37, 54.48, 52.15, 42.58, 42.25, 33.50, 33.26, 29.72;
m/z: 444.4 (M + H)+;
mp: 164–166 °C.
((1S,2S,4R)-4-(4-(((S)-2,3-Dihydro-1H-inden-1-yl)amino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl Sulfamate·Hydrochloride (Pevonedistat)
Pevonedistat (14.0 g, 92.5%, 99.0% a/a) as a white solid.
HPLC retention time of pevonedistat (Method C): 22.6 min;
1H NMR (400 MHz, DMSO) δ 9.70 (s, 1H), 8.39 (s, 1H), 7.63 (s, 1H), 7.45 (s, 2H), 7.41–7.20 (m, 4H), 7.04 (s, 1H), 5.78 (s, 1H), 5.44 (s, 1H), 4.42–4.28 (m, 1H), 4.24 (dd, J = 9.7, 6.9 Hz, 1H), 4.05 (dd, J = 9.6, 8.0 Hz, 1H), 3.18–2.99 (m, 1H), 2.91 (dt, J = 15.6, 7.7 Hz, 1H), 2.81–2.57 (m, 2H), 2.24–1.86 (m, 6H).
13C NMR (100 MHz, DMSO) δ 149.12, 145.71, 143.23, 142.11, 141.30, 128.28, 126.64, 124.97, 124.82, 124.49, 102.57, 101.74, 70.67, 69.22, 57.38, 53.14, 42.52, 42.40, 33.57, 32.56, 29.80;
m/z: 444.4 (M + H)+;
mp: 155–157 °C.
……………..
J. Org. Chem., 2011, 76 (9), pp 3557–3561
DOI: 10.1021/jo2001897

MLN4924 (1), which is in clinical trials as an anticancer agent, was stereoselectively synthesized from d-ribose via a route involving stereoselective reduction, regioselective cleavage of an isopropylidene moiety, and selective displacement of a cyclic sulfate moiety as key steps.
Sulfamic Acid 2-Hydroxy-4-[4-(indan-1-ylamino)pyrrolo[2,3-d]pyrimidin-7-yl]cyclopentylmethyl Ester (1) BASE
purified by silica gel column chromatography (hexane/ethyl acetate = 1/2) to give 1 (5.08 g, 90%) as a white foam:
UV (MeOH) λmax 279.50 nm;
[α]20D −6.41 (c 2.34, MeOH);
HR-MS (ESI) m/z calcd for C21H26N5O4S [M + H]+ 444.1705, found 444.1706;
1H NMR (400 MHz, CD3OD) δ 8.17 (d, J = 1.6 Hz, 1H), 7.25 (m, 2H), 7.18 (m, 2H), 6.64 (d, J = 3.6 Hz, 1H), 5.86 (t, J = 7.6 Hz, 1H), 5.46 (m, 1H), 4.49 (d, J = 2.8 Hz, 1H), 3.07 (m, 1H), 2.92 (m, 1H), 2.80 (m, 1H), 2.64 (m, 1H), 2.35 (m, 1H), 2.25 (m, 2H), 2.03 (m, 2H);
13C NMR (100 MHz, CD3OD) δ 152.1, 145.3, 144.6, 128.8, 127.6, 125.7, 125.2, 122.6, 100.5, 73.1, 70.9, 56.9, 54.0, 44.8, 43.6, 34.9, 34.6, 31.1. Anal. Calcd for C21H25N5O4S: C, 56.87; H, 5.68; N, 15.79; S, 7.23. Found: C, 56.91; H, 5.73; N, 15.82; S, 7.26.



NMR FROM CHEMIETEK
| WO2012061551A1 * | Nov 3, 2011 | May 10, 2012 | Millennium Pharmaceuticals, Inc. | Administration of nedd8-activating enzyme inhibitor |
| WO2013028832A2 * | Aug 23, 2012 | Feb 28, 2013 | Millennium Pharmaceuticals, Inc. | Inhibitors of nedd8-activating enzyme |
| WO2013028832A3 * | Aug 23, 2012 | May 2, 2013 | Millennium Pharmaceuticals, Inc. | Inhibitors of nedd8-activating enzyme |
| US8809356 | Aug 23, 2012 | Aug 19, 2014 | Millennium Pharmaceuticals, Inc. | Inhibitors of NEDD8-activating enzyme |
| Reference | ||
|---|---|---|
| 1 | * | Lee, et. al., Journal of Organic Chemistry (2011), 76(9), 3557-3561. |
1H NMR PREDICT

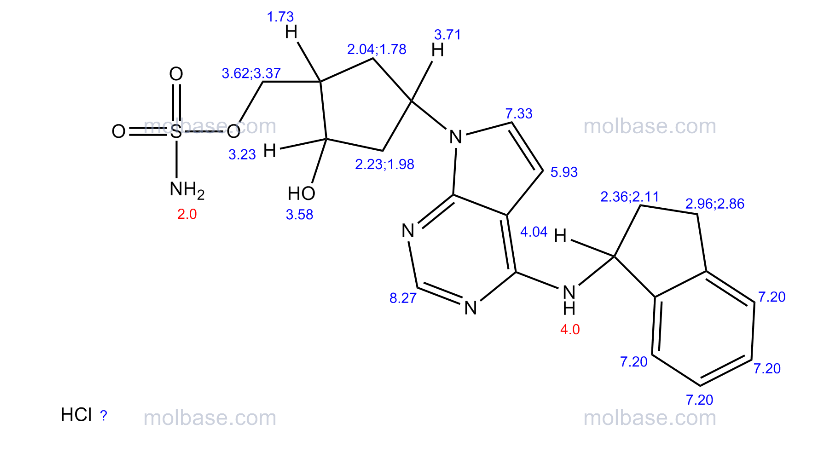
13 C NMR


//////////Pevonedistat, MLN4924, Millennium Pharmaceuticals, TAKEDA, TAK-924 , PHASE 1, orphan drug designation
Avatrombopag

Avatrombopag
AVATROMBOPAG; UNII-3H8GSZ4SQL; AKR-501; E5501; 570406-98-3; AS 1670542
| C29H34Cl2N6O3S2 | |
| Molecular Weight: | 649.65466 g/mol |
|---|
Elemental Analysis: C, 53.61; H, 5.28; Cl, 10.91; N, 12.94; O, 7.39; S, 9.87
1-[3-chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)-1,3-thiazol-2-yl]carbamoyl]pyridin-2-yl]piperidine-4-carboxylic acid,
1-(3-Chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)thiazol-2-yl]carbamoyl]pyridin-2-yl)piperidine-4-carboxylic acid,
1-[3-Chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)thiazol-2-yl]carbamoyl]-2-pyridyl]piperidine-4-carboxylic acid
4-Piperidinecarboxylic acid, 1-[3-chloro-5-[[[4-(4-chloro-2-thienyl)-5-(4-cyclohexyl-1-piperazinyl)-2-thiazolyl]amino]carbonyl]-2-pyridinyl]-
Phase III Clinical Trials
Drugs used in platelet disorders
Idiopathic thrombocytopenic purpura (ITP)
small-molecule thrombopoietin receptor (c-Mpl) agonist that stimulates platelet production
INNOVATOR: YAMANOUCHI PHARMACEUTICAL
DEVELOPER: Eisai

Avatrombopag maleate; UNII-GDW7M2P1IS; E5501 MALEATE; 677007-74-8; YM 477, AKR 501
| C33H38Cl2N6O7S2 | |
| Molecular Weight: | 765.72682 g/mol |
|---|
UNIIGDW7M2P1IS
(Z)-but-2-enedioic acid;1-[3-chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)-1,3-thiazol-2-yl]carbamoyl]pyridin-2-yl]piperidine-4-carboxylic acid
INTRODUCTION
Avatrombopag, also known as AKR-501, YM477, AS 1670542 or E5501, is a novel orally-active thrombopoietin (TPO) receptor agonist. AKR-501 specifically targeted the TPO receptor and stimulated megakaryocytopoiesis throughout the development and maturation of megakaryocytes just as rhTPO did. Daily oral administration of AKR-501 dose-dependently increased the number of human platelets in these mice, with significance achieved at doses of 1 mg/kg and above. The peak unbound plasma concentrations of AKR-501 after administration at 1 mg/kg in NOD/SCID mice were similar to those observed following administration of an active oral dose in human subjects. AKR-501 may be useful in the treatment of patients with thrombocytopenia. (source: Eur J Haematol. 2009 Apr;82(4):247-54).

Avatrombopag is a thrombopoietin receptor (c-Mpl) agonist in phase III clinical evaluation at Eisai for the oral treatment of chronic immune thrombocytopenia (idiopathic thrombocytopenia purpura) and for the treatment of thrombocytopenia associated with liver diseases. Phase II studies are ongoing for the treatment of thrombocytopenia during antiviral therapy (inhibition and maintenance) with Interferon for hepatitis C.
The drug candidate may hold potential in treating thrombocytopenia of diverse etiologies, including idiopathic thrombocytopenic purpura (ITP) and thrombocytopenia of myelodysplastic syndromes (MDS), in combination with or as a substitute for platelet transfusion.
AKR-501, a novel, small-molecule thrombopoietin mimetic being investigated for the treatment of thrombocytopenia. AkaRx is now a wholly-owned subsidiary of Eisai Inc. and Eisai has the exclusive worldwide rights to develop, market and manufacture AKR-501. AKR-501 is an investigational thrombopoietin receptor agonist that, based on preclinical studies, increases platelet production by stimulating megakaryocytic proliferation and differentiation. Eisai is currently conducting Phase II clinical trials of AKR-501 in the United States as a potential treatment for idiopathic thrombocytopenic purpura (ITP) and thrombocytopenia associated with liver diseases (TLD), and has confirmed proof of concept in the clinical studies for ITP. In addition, Eisai will explore the compound’s potential as a treatment for chemotherapy-induced thrombocytopenia (CIT).

E-5501 stimulates the production of thrombopoietin (TPO), a glycoprotein hormone that stimulates the production and differentiation of megakaryocytes, the bone marrow cells that fragment into large numbers of platelets. The drug candidate was originally developed at Yamanouchi, and development responsibilities were passed to AkaRx when it was formed in 2005 as a spin-off following the creation of Astellas Pharma subsequent to the merger of Yamanouchi Pharmaceutical and Fujisawa Healthcare.
In 2007, MGI Pharma was granted a license to E-5501 for the treatment of thrombocytopenia. Eisai eventually gained the rights to the product as results of its acquisition of MGI Pharma. In 2010, Eisai acquired AkaRx. AkaRx is now a wholly-owned subsidiary of Eisai Inc. and Eisai has the exclusive worldwide rights to develop, market and manufacture E-5501. In 2011, orphan drug designation was assigned by the FDA for the treatment of idiopathic thrombocytopenic purpura.
E5501 (or AKR-501 or YM477) is a small molecule agonist c-Mpl, orally available. It is in clinical trials for the treatment of chronic idiopathic thrombocytopenic purpura (ITP). It acts as an agonist of the thrombopoietin receptor active orally, mimicking its biological effect. Thrombocytopenic purpura The is the idiopathic consequence of a low number of platelets (thrombocytopenia) of unknown cause. A very low platelets can even lead to purpura (bruises), or bleeding diathesis.
February 2012: A Phase III, multicenter, randomized, double-blind, controlled against placebo, parallel group, with an open-label extension phase to assess the efficacy and safety of combined oral E5501 to standard treatment for the treatment of thrombocytopenia in adults with chronic immune thrombocytopenia, is underway.
January 2010: Eisai Inc. announced its successful acquisition of the biopharmaceutical company, AkaRx Inc. Following this acquisition, AkaRx became a wholly owned subsidiary of Eisai Inc. Eisai now owns the worldwide exclusive rights to develop , marketing and manufacture AKR-501.
October 2009: Eisai Research Institute of Boston, Inc. (established in 1987) and Eisai Medical Research Inc. (established in 2002) were merged into Eisai Inc. 2005: AkaRx was founded as a spin-out of the merger of Yamanouchi Pharmaceutical Company Ltd. and Fujisawa Pharmaceutical Company Ltd. to form Astellas Pharma Inc. AKR-501 was discovered by Yamanouchi and was licensed to AkaRx as part of the foundation of the company in 2005.
In a Phase I trial in healthy volunteers, 10 mg of AKR-501 for 14 days, increased platelet count by 50%.AKR-501 was well tolerated in both studies, mono- and multi-dose. No adverse effects were reported, even at the highest doses.
……………………
Patent
Compound A is a compound of the present invention has the following chemical structure.

That is, compounds useful as a platelet 增多 agent according to the present invention A, as well as medicaments for the Compound A as an active ingredient, in particular increasing platelets agents and Z or thrombocytopenia treating agent.


………………
PATENT

……………………
JP 2014144916/WO 2013018362
https://www.google.co.in/patents/WO2013018362A1?cl=en
1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)thiazol-2-yl]carbamoyl}pyridin-2-yl)piperidine-4-carboxylic acid as expressed by the following chemical formula (hereinafter referred to as “Compound X”) and pharmaceutically acceptable salts are known to have excellent thrombocytosis effects (patent literature 1, patent literature 2).
[Formula 1]

Patent literature 1 discloses a hydrochloride of compound X as example 16 (hereinafter referred to as “compound X hydrochloride”).
Furthermore, patent literature 2 discloses a maleic acid salt of compound X that has endothermic peaks near 198 degree C and 271 degree C in thermo gravimetric analysis (hereinafter referred to as “maleic acid salt of compound X”). However, patent literature 2 neither discloses nor suggests that the maleic acid salt of compound X exhibits crystal polymorphism.
On the other hand, compounds exhibiting crystal polymorphism demonstrate entirely different effects regardless of being the same compound, because various physical properties including physicochemical properties differ depending on the crystalline form. In pharmaceutical products in particular, if compounds that have different functional effects are expected to have the same effect, a different functional effect than expected will occur, which is thought to induce unexpected circumstances, and therefore there is demand for supply of a drug substance with constant quality. Therefore, when a compound which has crystal polymorphism is used as a medicine, one type of crystal of that compound must always be constantly provided in order to ensure constant quality and constant effects that are required of the medicine.
Under the aforementioned conditions, from the perspective of supplying a drug substance for medicines, there is a need for compound X or crystals of pharmaceutically acceptable salts thereof, which can ensure constant quality and constant effects and which can be stably supplied in mass production such as industrial production or the like, as well as for establishment of a manufacturing method thereof.
International patent publication WO 03/062233 International patent publication WO 2004/029049
The crystals of compound X maleic acid salt disclosed in patent literature 2 (hereinafter referred to as “compound X maleic acid salt A type crystals”) cannot be isolated as compound X maleic acid salt A type crystals when scaled up for mass production using the method disclosed in example 1 of patent literature 2, and therefore must be isolated in a different crystal form. (This other crystal form is referred to as “compound X maleic acid salt B type crystals”). Therefore, the compound X maleic acid salt A type crystals have a possibility that the crystal form will morph depending on the scale of production, and is clearly inappropriate as a drug substance for medicines which require constant quality and constant effects.
Preparation Example 1: Manufacture of Compound X Maleic Acid Salt B Type Crystal
310 mL of a 1 M aqueous solution of sodium hydroxide at room temperature was added to a mixture of 70.0 g of the ethyl ester of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid and 1.2 L of ethanol, the insoluble matter was filtered out, and then washed with 200 mL of ethanol. The reaction solution was stirred for 90 minutes at 60 degree C. After cooling to room temperature, 1.4 L of an aqueous solution containing 24.11 g of maleic acid was added to the solution obtained, and then the precipitate was collected by filtering.
The same operation was repeated and when combined with the previously obtained precipitate, 136.05 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid was obtained.
18.9 g of maleic acid and 2.1 L of 80% ethanol water were added to 88.90 g of the carboxylic acid obtained, and the solution was stirred for one hour at room temperature and for another hour at 100 degree C. After cooling to room temperature and further cooling with ice, the precipitated solid was filtered out to obtain 87.79 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt as a crude product.
6.84 g of maleic acid was added to 231 g of the crude product containing the crude product obtained above and those manufactured in a similar manner, dissolved in 5.5 L of 80% ethanol water, and then the precipitated solid was collected by filtering to obtain 203 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
Example 1: Manufacture of Compound X Maleic Acid Salt C Type Crystals (1)
1.52 L of ethanol, 0.38 L of water, and 15.7 g of maleic acid were added to 78.59 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid, and heated while stirring. After cooling to room temperature and further cooling with ice, the precipitated solid was collected by filtering to obtain 71.60 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt as a crude product.
296 mg of maleic acid was added to 10.0 g of the crude product obtained, dissolved in 60 mL of acetone, 60 mL of DMSO, and 30 mL of water, and then the precipitated solids were collected to obtain 8.41 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
Example 2: Manufacture of Compound X Maleic Acid Salt C Type Crystals (2)
A mixture containing 80.1 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid, 580 mL of DMSO, 580 mL of acetone, 17.2 g of maleic acid, and 290 mL of water was stirred at 69 degree C. The insoluble matter was filtered out, washed with a mixture of 32 mL of DMSO, 32 mL of acetone, and 16 mL of water, and then the filtrate was cooled and the precipitate was collected by filtering. Washing was successively performed using 150 mL of water, 80 mL of acetone, 650 mL of water, and 80 mL of acetone, followed by drying, to obtain 70.66 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
Example 3: Manufacture of Compound X Maleic Acid Salt C Type Crystals (3)
A mixture containing 20 kg of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid, 100 L of DMSO, 100 L of acetone, 4.29 kg of maleic acid, and 50 L of water is stirred at 65 degree C, and then the insoluble matter is filtered out and washed with a mixture of 8 L of DMSO, 8 L of acetone, and 4 L of water, and then the filtrate is cooled, the precipitate is collected by filtering, successively washed using 40 L of acetone, 100 L of water, and 40 L of acetone, and then dried to obtain approximately 20 kg of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
…………………………….

REFERENCES
Garabet, L.; Ghanima, W.; Lee, S.; Mowinckel, M.C.; Liebman, H.; Jonassen, C.M.; Bussel, J.; Sandset, P.M.
Thrombopoietin receptor agonists do no not cause coagulation activation: In patients with immune thrombocytopenia
25th Congr Int Soc Thromb Haemost (ISTH) (June 20-25, Toronto) 2015, Abst PO311-MON
Terrault, N.; Hassanein, T.; Joshi, S.; Lake, J.R.; Sher, L.S.; Vargas, H.E.; McIntosh, J.W.; Tang, S.; Jenkins, T.
Once-daily oral avatrombopag (E5501) prior to elective surgical or diagnostic procedures in patients with chronic liver disease and thrombocytopenia: Results from a phase 2, randomized, double-blind, placebo-controlled study (study 202)
63rd Annu Meet Am Assoc Study Liver Dis (November 9-13, Boston) 2012, Abst
Thiophenyl Triazol-3-one Derivatives As Smooth Muscle relaxers: US6613786 (2003) Priority: US20010336865P, Nov. 2, 2001 (Bristol-Myers Squibb CO, US)
Preparation Of Avatrombopag: 2-Acylaminothiazole derivative or salt thereof: EP1466912 (2004) Priority: JP20020010413, 18 Jan. 2002 (Yamanouchi Pharma Co Ltd, Japan)
Synthesis And Use Of MSE Framework-Type Molecular Sieves: US2009318696 (2009) Priority: US20080214631 20 Jun. 2008 (Exxon Mobil, US).
5,6-Dichloro-Nicotinic Acid Production By Reacting 6-Hydroxy-Nicotinic Acid With Acid Chloride Reacting With Chlorine Products, Then With Acid Chloride And Hydrolysing Products: CH664754 (1988) Priority: CH19850002692, 25 Jun. 1985 (Lonza AG, Switzerland).
David J. Kuter, New Thrombopoietic Growth Factors, Lymphoma and Myeloma Clinical Journal Volume 9, Supplement 3, S347-S356
| WO2003062233A1 | 15 Jan 2003 | 31 Jul 2003 | Yamanouchi Pharma Co Ltd | 2-acylaminothiazole derivative or salt thereof |
| WO2004029049A1 | 29 Sep 2003 | 8 Apr 2004 | Yuuji Awamura | Novel salt of 2-acylaminothiazole derivative |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| EP2764866A1 | 4 Feb 2014 | 13 Aug 2014 | IP Gesellschaft für Management mbH | Inhibitors of nedd8-activating enzyme |
| Patent | Submitted | Granted |
|---|---|---|
| CANCER TREATMENT METHOD [US2011160130] | 2011-06-30 | |
| METHOD FOR STIMULATING PLATELET PRODUCTION [US2011166112] | 2011-07-07 | |
| COMPOSITIONS AND METHODS FOR INCREASING BLOOD PLATELET LEVELS IN HUMANS [US2011224226] | 2011-09-15 | |
| Method of treating viral diseases with combinations of TPO receptor agonist and anti-viral agents [US2012020923] | 2012-01-26 |
| Patent | Submitted | Granted |
|---|---|---|
| 2-Acylaminothiazole derivative or salt thereof [US7638536] | 2005-07-14 | 2009-12-29 |
| Compositions and methods for treating thrombocytopenia [US2007203153] | 2007-08-30 | |
| Novel Combinations [US2009304634] | 2009-12-10 | |
| 2-ACYLAMINOTHIAZOLE DERIVATIVE OR SALT THEREOF [US2010222329] | 2010-09-02 | |
| 2-ACYLAMINOTHIAZOLE DERIVATIVE OR SALT THEREOF [US2010222361] | 2010-09-02 | |
| Compositions and methods for increasing blood platelet levels in humans [US2008039475] | 2008-02-14 | |
| CANCER TREATMENT METHOD [US2009022814] | 2009-01-22 | |
| Compositions and methods for treating thrombocytopenia [US2010041668] | 2010-02-18 | |
| CANCER TREATMENT METHOD [US2010075928] | 2010-03-25 |
///////E 5501, AKR 501, Phase III, eisai, Avatrombopag, y 477, orphan drug, ym 477, AS 1670542, Yamanouchi Pharma Co Ltd, Japan
UPDATE MAY 2018
Avatrombopag
https://newdrugapprovals.org/2015/08/24/avatrombopag/
The U.S. Food and Drug Administration today approved Doptelet (avatrombopag) tablets to treat low blood platelet count (thrombocytopenia) in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure. This is the first drug approved by the FDA for this use.Continue reading.
May 21, 2018
Release
The U.S. Food and Drug Administration today approved Doptelet (avatrombopag) tablets to treat low blood platelet count (thrombocytopenia) in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure. This is the first drug approved by the FDA for this use.
“Patients with chronic liver disease who have low platelet counts and require a procedure are at increased risk of bleeding,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Doptelet was demonstrated to safely increase the platelet count. This drug may decrease or eliminate the need for platelet transfusions, which are associated with risk of infection and other adverse reactions.”
Platelets (thrombocytes) are colorless cells produced in the bone marrow that help form blood clots in the vascular system and prevent bleeding. Thrombocytopenia is a condition in which there is a lower-than-normal number of circulating platelets in the blood. When patients have moderately to severely reduced platelet counts, serious or life-threatening bleeding can occur, especially during invasive procedures. Patients with significant thrombocytopenia typically receive platelet transfusions immediately prior to a procedure to increase the platelet count.
The safety and efficacy of Doptelet was studied in two trials (ADAPT-1 and ADAPT-2) involving 435 patients with chronic liver disease and severe thrombocytopenia who were scheduled to undergo a procedure that would typically require platelet transfusion. The trials investigated two dose levels of Doptelet administered orally over five days as compared to placebo (no treatment). The trial results showed that for both dose levels of Doptelet, a higher proportion of patients had increased platelet counts and did not require platelet transfusion or any rescue therapy on the day of the procedure and up to seven days following the procedure as compared to those treated with placebo.
The most common side effects reported by clinical trial participants who received Doptelet were fever, stomach (abdominal) pain, nausea, headache, fatigue and swelling in the hands or feet (edema). People with chronic liver disease and people with certain blood clotting conditions may have an increased risk of developing blood clots when taking Doptelet.
This product was granted Priority Review, under which the FDA’s goal is to take action on an application within six months where the agency determines that the drug, if approved, would significantly improve the safety or effectiveness of treating, diagnosing or preventing a serious condition.
The FDA granted this approval to AkaRx Inc.
//////////////Doptelet, avatrombopag, fda 2018, akarx, priority review,
Filgotinib

Filgotinib
EU APPROVED 2020/9/24, JYSELECA
JAPAN APPROVED2020/9/25
- C21H23N5O3S
- MW425.504
- Elemental Analysis: C, 59.28; H, 5.45; N, 16.46; O, 11.28; S, 7.54
1206161-97-8
Cyclopropanecarboxamide, N-[5-[4-[(1,1-dioxido-4-thiomorpholinyl)methyl]phenyl][1,2,4]triazolo[1,5-a]pyridin-2-yl]-
G146034
GLPG0634
N-(5-(4-((1,1-dioxidothiomorpholino)methyl)phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl)cyclopropanecarboxamide
Galapagos Nv INNOVATOR
PHASE 3, Crohn’s disease, Rheumatoid arthritis, Ulcerative colitis
Filgotinib is an orally available inhibitor of JAK1/JAK2 and TYK2 in phase III clinical development at Galapagos and Gilead for the treatment of rheumatoid arthritis, moderate or severe Crohn’s disease and ulcerative colitis
IL-6 antagonist; Jak1 tyrosine kinase inhibitor; Tyk2 tyrosine kinase inhibitor; Jak3 tyrosine kinase inhibitor; Jak2 tyrosine kinase inhibitor
Autoimmune disease; Cancer; Colitis; Crohns disease; Inflammatory disease; Neoplasm; Rheumatoid arthritis; Transplant rejection
In 2017, orphan drug designation was assigned to the compound in the U.S. for the treatment of pediatric Crohn’s disease and pediatric ulcerative colitis.
GlaxoSmithKline had been developing filgotinib preclinically for the treatment of rheumatoid arthritis pursuant to a license; however, in 2010, the compound was re-acquired by Galapagos. In 2012, the product was licensed to Abbott for development and marketing. In January 2013, Abbott spun-off its research-based pharmaceutical business into a newly-formed company AbbVie. The license agreement between Galapagos and Abbott was terminated in September 2015, Galapagos regaining all rights to the product. The same year, Galapagos and Gilead entered into a global partnership and Gilead obtained the global rights of codevelopment and commercialization for the treatment of inflammatory diseases
Filgotinib (GLPG0634), by the Belgian biotech company Galápagos NV, is a drug which is currently under investigation for the treatment of rheumatoid arthritis and Crohn’s disease.
Filgotinib (GLPG0634) is an orally-available, selective inhibitor of JAK1 (Janus kinase 1) for the treatment of rheumatoid arthritis and potentially other inflammatory diseases. Filgotinib (GLPG0634) dose-dependently inhibited Th1 and Th2 differentiation and to a lesser extent the differentiation of Th17 cells in vitro. GLPG0634 was well exposed in rodents upon oral dosing, and exposure levels correlated with repression of Mx2 expression in leukocytes. The JAK1 selective inhibitor GLPG0634 (Filgotinib) is a promising novel therapeutic with potential for oral treatment of rheumatoid arthritis and possibly other immune-inflammatory diseases. Filgotinib (GLPG0634) is currently in a Phase 2 study in Crohn’s disease.

Mechanism of action
Filgotinib is a Janus kinase inhibitor with selectivity for subtype JAK1 of this enzyme. It is considered a promising agent as it inhibits JAK1 selectively. Less selective JAK inhibitors (e.g. tofacitinib) are already being marketed. They show long-term efficacy in the treatment of various inflammatory diseases. However, their lack of selectivity leads to dose-limiting side effects.[1] It is thought that inhibition of all JAK isoenzymes is beneficial in rheumatoid arthritis. However, pan-JAK inhibition might also lead to unwanted side effects that might not outweigh its benefits. This is the rationale for the development of newer and more selective inhibitors like filgotinib.
The signal transmission of large numbers of proinflammatory cytokines is dependent on JAK1. Inhibition of JAK2 may also contribute to the efficacy against RA. Nonetheless it is thought that JAK2 inhibition might lead to anemia and thrombopenia by interference witherythropoietin and thrombopoietin and granulocyte-macrophage colony-stimulating factor. Therefore one might prefer to choose a more selective JAK1 inhibitor as a primary therapeutic option. Filgotinib exerts a 30-fold selectivity for JAK1 compared to JAK2.[2] It is however still to be seen to what extent JAK2 inhibition should be avoided.
Novel crystalline forms of filgotinib salts, particularly hydrochloride salt, useful for treating JAK-mediated diseases eg inflammatory diseases, autoimmune diseases, proliferative diseases, allergy and transplant rejection. Galapagos and licensee AbbVie are developing filgotinib, a selective JAK-1 inhibitor, for treating rheumatoid arthritis (RA) and Crohn’s disease (CD). In August 2015, the drug was reported to be in phase 2 clinical development for treating RA and CD. The drug is also being investigated for the treatment of colitis and was discovered as part of the company’s arthritis alliance with GSK; however in August 2010 Galapagos reacquired the full rights. See WO2013189771, claiming use of filgotinib analog for treating inflammatory diseases. Also see WO2010010190 (co-assigned with GSK and Abbott) and WO2010149769 (assigned to Galapagos) claiming filgotinib, generically and specifically, respectively.
Clinical trials and approval
The efficacy of filgotinib is currently studied in a phase2b program (DARWIN trial 1, 2) with involvement of 886 rheumatoid arthritis patients and 180 Crohn’s disease patients.
Phase 1 study
It was shown in phase 1 studies that the pharmacokinetics of filgotinib metabolism is independent of hepatic CYP450 enzymatic degradation. The drug metabolism is however mediated by carboxylesterases. There is no interference reported with the metabolism of methotrexate nor with any of the investigated transport proteins.[3]
Phase 2 study: Proof of concept (2011)
In november 2011 Galápagos released the results of their phase 2 study (identification: NCT01384422, Eudract: 2010-022953-40) in which 36 patients were treated who showed a suboptimal clinical response to methotrexate treatment. Three groups of twelve patients were treated either with 200 mg filgotinib in a single dose, 200 mg divided in two doses or placebo. The primary end-point was the ACR20 score, which monitors improvements in the symptomatology of the patient. After the scheduled 4 weeks of treatment, 83% of the respondents showed an improved ACR20-score. Half of the treated patients showed a complete (or near complete) remission of the disease. There were no reports ofanemia nor changes in lipidemia. The company stated in their press release that filgotinib is the first selective JAK1 inhibitor that shows clinical efficacy. As a result of this study, the company stated that “GLPG0634 shows one of the highest initial response rates ever reported for rheumatoid arthritis treatments”.[4]
DARWIN 1 trial
The DARWIN 1 trial is a 24 week double blind placebo-controlled trial with 599 rheumatoid arthritis patients enrolled. All participants have moderate to severe RA and showed an insufficient response to standard methotrexate treatment. The trial compares three dosages of filgotinib as a once or twice per day regimen. During the trial all participants remain on their methotrexate treatment. According to the company, the results of this trial are expected in July 2015.[5]
DARWIN 2 trial
The DARWIN 2 trial is a double blind placebo-controlled trial with 280 rheumatoid arthritis patients enrolled who show an insufficient response to standard methotrexate treatment. This trial, in contrast to the previous DARWIN 1 trial, methotrexate is discontinued. Therefore, this trial investigates filgotinib as a monotherapy.[6] The recruitment of DARWIN trial 2b ended in november 2014.[7] Preliminary results are expected in the second quarter of 2015 and a full completion of the study is expected in the third quarter of 2015.
DARWIN 3 trial
Patients who complete DARWIN 1 and 2 will be eligible for DARWIN 3.
COSY PREDICT
Time line
- june 2011: results of first phase 2 trial
- november 2014: initiation of DARWIN 1 and 2 trials
- april 2015: expected date of DARWIN 1 trial results
- june 2015: expected date of DARWIN 2 trial results
NMR FROM NET….ABMOLE, DMSOD6
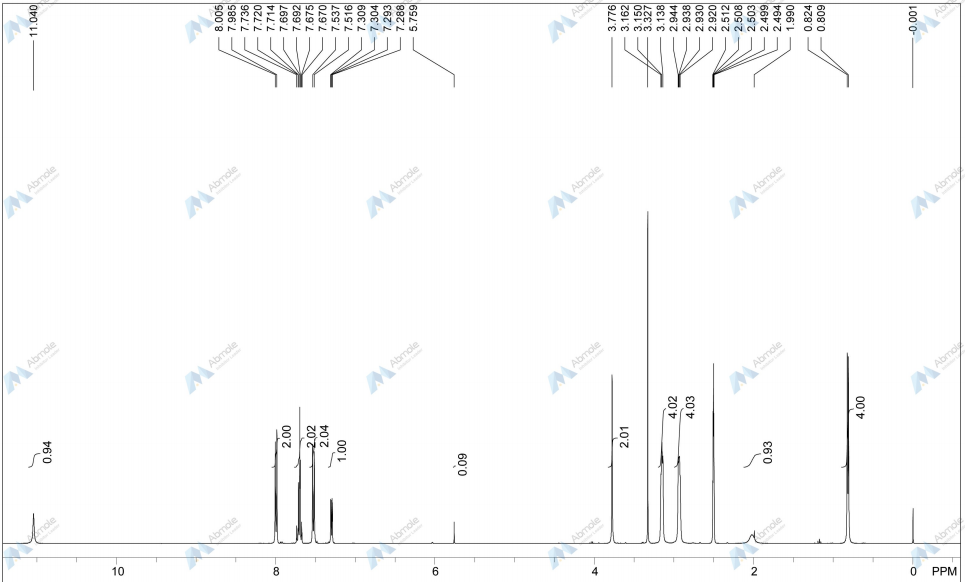
NMR MEDKOO DMSOD6

CHEMIETEK
1H NMR PREDICT


13C NMR PREDICT

……………………
MORE PREDICTS
1H NMR PREDICT
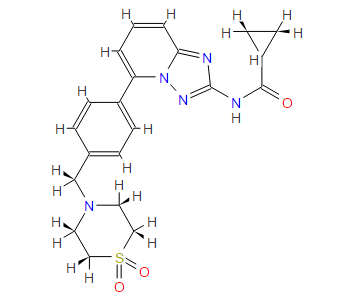
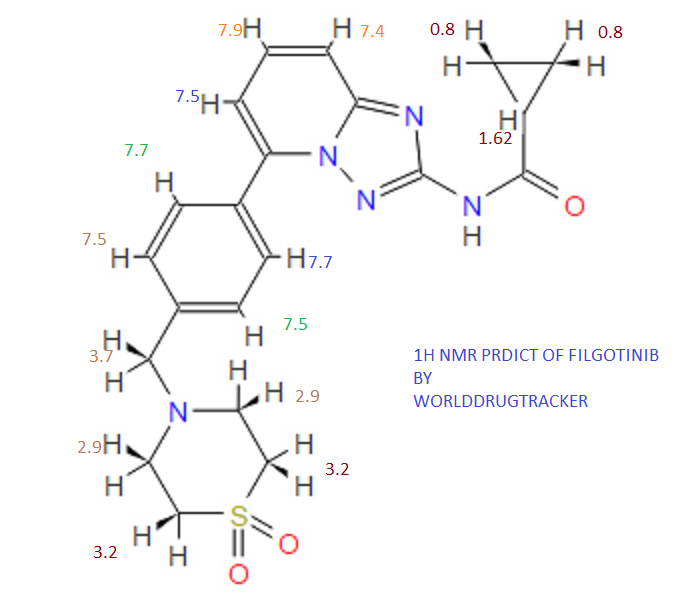
13C NMR PREDICT
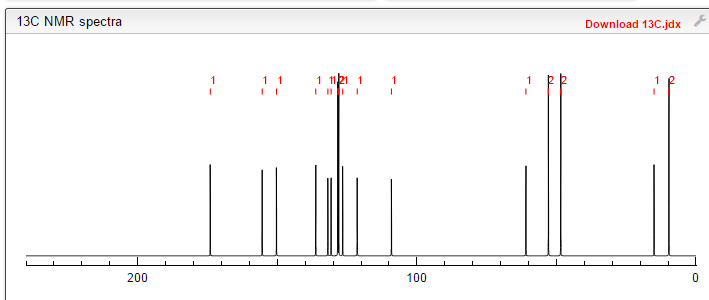
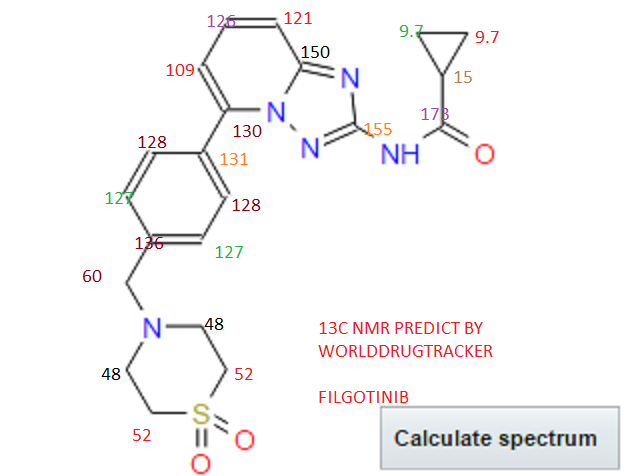
PRODUCT PATENT
http://www.google.com/patents/WO2010149769A1?cl=en
| Applicants: | GALAPAGOS NV [BE/BE]; Generaal De Wittelaan L11/A3 B-2800 Mechelen (BE) (For All Designated States Except US). MENET, Christel Jeanne Marie [FR/BE]; (BE) (For US Only). SMITS, Koen Kurt [BE/BE]; (BE) (For US Only) |
| Inventors: | MENET, Christel Jeanne Marie; (BE). SMITS, Koen Kurt; (BE) |
| International Filing Date: | 25.06.2010 |
ESTIMATED EXP 2030
Condensation of 2-amino-6-bromopyridine (I) with ethoxycarbonyl isothiocyanate (II) in CH2Cl2 gives 1-(6-bromopyridin-2-yl)-3-carboethoxythiourea (III), which upon cyclization with hydroxylamine hydrochloride (IV) in the presence of DIEA in EtOH/MeOH yields 2-amino-5-bromo[1,2,4]triazolo[1,5-a]pyridine (V). N-Acylation of amine (V) with cyclopropanecarbonyl chloride (VI) using Et3N in acetonitrile, and subsequent treatment with methanolic ammonia furnishes the carboxamide (VII) (1-3), which upon Suzuki coupling with 4-(hydroxymethyl)phenylboronic acid (VIII) in the presence of PdCl2(dppf) and K2CO3 in dioxane/H2O at 90 °C, followed by bromination with PBr3 in CHCl3 affords intermediate (IX). Condensation of benzyl bromide derivative (IX) with thiomorpholine-1,1-dioxide (X) using DIEA in CH2Cl2/MeOH yields filgotinib (1,2). Alternatively, condensation of (4-bromomethylphenyl)-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane (XI) with thiomorpholine 1,1-dioxide (X) in the presence of DIEA in CH2Cl2/MeOH gives intermediate (XII), which undergoes Suzuki coupling with aryl bromide (VII) in the presence of PdCl2(dppf) and K2CO3 in dioxane/H2O at 90 °C to afford the target filgotinib
The present invention is based on the discovery that the compound of the invention is able to act as an inhibitor of JAK and that it is useful for the treatment of inflammatory conditions, autoimmune diseases, proliferative diseases, transplantation rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6. In a specific aspect the compound is an inhibitor of JAKl and JAK2. The present invention also provides methods for the production of this compound, a pharmaceutical composition comprising this compound and methods for treating inflammatory conditions, autoimmune diseases, proliferative diseases, transplantation rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6 by administering the compound of the invention.
Accordingly, in a first aspect of the invention, a compound of the invention is provided having a formula (I):

[0017] The compound of the invention is a novel inhibitor of JAK that appears to exhibit a dramatically improved in vivo potency as compared to structurally similar compounds. In a particular embodiment the compound of the invention is an inhibitor of JAKl and JAK2. In particular it appears to exhibit this increase in potency at lower in vivo exposure levels compared to structurally similar compounds. The use of a compound with these improvements is expected to result in a lower dosage requirement (and therefore an improved dosing schedule).
General Synthetic Method Scheme 1

1. RCOCI, Et3N 2. NH3 / MeOH CH3CN, 20 0C 2O 0C

wherein Ar represents phenyl-Ll-heterocycloalkyl, where Ll is a bond, -CH2– or -CO- and the heterocycloalkyl group is optionally substituted.
General
1.1.1 l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2)

(2)
[00117] To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5 0C is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture is then allowed to warm to room temp. (20 0C) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea may be used as such for the next step without any purification. 1H (400 MHz, CDCl3) δ 12.03 (IH, br s, NH), 8.81 (IH, d, J 7.8 Hz, H-3), 8.15 (IH, br s, NH), 7.60 (IH, t, J 8.0 Hz, H-4), 7.32 (IH, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
7.7.2 5-Bromo-[l, 2, 4]triazolo[l, 5-a]pyridin-2-ylamine (3)

[00118] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH
(1 :1, 900 mL) is added N,N-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20 0C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3 h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition Of H2O (250 mL) and filtration. The combined solids are washed successively with H2O (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et2O (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. 1H (400 MHz, DMSO-t/β) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (IH, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 :1, M+H+, 100%).
7.7.3 General procedure for mono-acylation to afford intermediate (4):

[00119] To a solution of the 2-amino-triazolopyridine (3) (7.10 g, 33.3 mmol) in dry CH3CN
(150 mL) at 5 0C is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material (3) is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp, (for 1-16 h) to hydro lyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et2O (50 mL). The solids are collected by filtration, washed with H2O (2x50mL), acetone (50 mL) and Et2O (50 mL), then dried in vacuo to give the required bromo intermediate (4).
Method A
Preparation of compounds of the invention via Suzuki coupling (5):
[00120] An appropriate boronic acid (2eq.) is added to a solution of bromo intermediate (4) in
1 ,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140 0C for 30 min (this reaction can also be carried out by traditional heating in an oil bath at 900C for 16h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters
XBridge Prep Cl 8 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using
MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method B

Bl. 4 4-[2-(Cyclopropanecarbonyl-amino)-[ 1 , 2, 4]triazolo[l, 5-a] pyridin-5-yl] -benzoyl chloride

[00121] 2 Drops of DMF are added to a solution of 4-[2-(cyclopropanecarbonyl-amino)- [l,2,4]triazolo[l,5-a]pyridin-5-yl]-benzoic acid (1 eq) obtained by Method A using 4-carboxyphenylboronic acid in DCM under N2 atmosphere. Then oxalyl chloride (2 eq) is added dropwise to this resulting solution (gas release). The mixture is stirred at room temperature for 2 hours. After completion of the reaction by LCMS, the solvent is removed. The crude acid chloride is used without further purification in next step.
B2. Amide formation (General Method)

[00122] An appropriate amine (1.1 eq) and Et3N (5 eq) are dissolved in DCM under N2 atmosphere and cooled at 00C. The acid chloride (Bl, 1 eq) dissolved in DCM is added dropwise to this solution. The reaction is stirred at room temperature for 16 h. After this time, reaction is complete. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers are filtered and evaporated. The final compound is isolated by preparative HPLC. Preparative HPLC: Waters XBridge Prep C18 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method C

Wherein R3a or R3b together with the nitrogen atom to which they are attached, may form a heterocycloalkyl.
Reductive alkylation (general method)
[00123] An appropriate amine (2 eq.), cyclopropanecarboxylic acid (for example cyclopropanecarboxylic acid [5-(4-formyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridine-2-yl]-amide) prepared by method A (1 eq.) and Ti(OPr)4 are mixed and stirred at room temperature for 3 hrs. The mixture is diluted in ethanol and Na(CN)BH3 (leq.) is added. The resulting solution is stirred at room temperature for 16 hrs. The mixture is diluted in water and filtered. The filtrate is washed with ethanol. The combined solvent phases are evaporated under vacuum. The final compound is isolated by preparative HPLC.
Method D 
wherein R1 and R2 together with the Nitrogen atom to which they are attached, may form a heterocycloalkyl.
Reaction ofalkylation

[00124] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (leq) and Et3N (2 eq) (or AgCO3) are dissolved in DCM/MeOH (4:1 v:v) under N2 and an amine (2 eq) is added dropwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSθ4. Organic layers are filtered and evaporated. The final compound is isolated by flash chromatography.
Suzuki coupling

[00125] The obtained boronic acid (2eq.) is added to a solution of cyclopropanecarboxylic acid
(5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide (4) in 1 ,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140 0C for 30 min (This reaction can also be carried out by traditional heating in an oil bath at 900C for 16h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters XBridge Prep C18 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Synthesis of the compound of the invention and comparative examples
Compound l(the compound of the invention)
Step 1:

[00126] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (leq) and DIPEA
(2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) was added portionwise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhyd. MgS O4. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
Step 2: Suzuki coupling

[00127] 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide
(l.leq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 900C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
[00128] Alternatively, after completion of the reaction, a palladium scavenger such as 1,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cooled down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HCl is added, and after stirring at RT, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at RT, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H2O, treated with a palladium scavenger (e.g. SMOPEX 234) at 500C, the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the title compound as a free base.
Alternative route to Compound l(the compound of the invention):
Step 1:

[00129] 4-(Hydroxymethyl)phenylboronic acid (l.leq.) was added to a s o luti o n o f cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 900C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSθ4 and evaporated in vacuo. The resulting mixture was used without further purification.
Step 2:

[00130] To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)- [l,2,4]triazolo[l,5-a]pyridin-2-yl]-amide (1.0 eq) in chloroform was slowly added phosphorus tribromide (1.0 equiv.). The reaction mixture was stirred at room temperature for 20 hours, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer was dried over anhyd. MgSθ4, filtered and concentrated to dryness. The resulting white residue was triturated in dichloromethane/diethyl ether 2:1 to afford the expected product as a white solid.
Step 3:

[00131] Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin- 2-yl]-amide (leq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (1.1 eq) was added dropwise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was dissolved in DCM, washed with water and dried over anhyd. MgSO^ Organic layers were filtered and evaporated. The final compound was isolated by column chromatography using EtOAc to afford the desired product.
PATENT
WO 2010010190
WO 2013173506
WO 2013189771
WO 2015117980
WO 2015117981
POLYMORPH
CN 105061420
https://encrypted.google.com/patents/CN105061420A?cl=en
JAK inhibitor N-(5-(4-(1,1-dioxothiomorpholinyl)methyl)phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl)cyclopropanecarboxamide, and methods for preparing the four crystal forms, wherein the four crystal forms respectively are a crystal form H1, a crystal form H2, a crystal form H3 and a crystal form H4,
POLYMORPH
E CRYSTAL
CN 105111206
D CRYSTAL
CN 105111207
H CRYSYAL
CN 105198876
G
CN 105198877
F CN 105198878
C CN 105198880
POLYMORPH
WO 2016105453
POLYMORPH
Preparation of solid forms of filgotinib free base
WO 2017012773
The present invention relates to cryst. filgotinib free base (I), a method of its prepn. and a pharmaceutical compn. comprising the same. Thus, reacting the compd. II with thiomorpholine dioxide afforded 92% I (purity of 98.6%) which was then converted into its hydrochloride salt. Pharmaceutical compn. comprising compd. I was disclosed.
POLYMORPH
CN 105669669
The present invention provides a crystal form A, B, D, G and M of N-[5-[4-[(1,1-dioxido-4-thiomorpholinyl)methyl]phenyl][1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide hydrochloride.
PAPER
Future Medicinal Chemistry (2015), 7(2), 203-235. | Language: English, Database: CAPLUSA review. The discovery of the JAK-STAT pathway was a landmark in cell biol. The identification of these pathways has changed the landscape of treatment of rheumatoid arthritis and other autoimmune diseases. The two first (unselective) JAK inhibitors have recently been approved by the US FDA for the treatment of myelofibrosis and rheumatoid arthritis and many other JAK inhibitors are currently in clin. development or at the discovery stage. Research groups have demonstrated the different roles of JAK member and the therapeutic potential of targeting them selectively. ………..
https://www.future-science.com/doi/10.4155/fmc.14.149
PAPER
Journal of Pharmaceutical Sciences (Philadelphia, PA, United States) (2018), 107(6), 1624-1632.
PATENT
US2010/331319 A1, ; Page/Page column 13-14
http://www.google.com/patents/US20100331319
Synthetic Preparation of the Compound of the Invention and Comparative Examples
The compound of the invention and the comparative examples can be produced according to the following scheme.

wherein Ar represents phenyl-L1-heterocycloalkyl, where L1 is a bond, —CH2— or —CO— and the heterocycloalkyl group is optionally substituted.
General 1.1.1 1-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2)

To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5° C. is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture is then allowed to warm to room temp. (20° C.) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea may be used as such for the next step without any purification. 1H (400 MHz, CDCl3) δ 12.03 (1H, br s, NH), 8.81 (1H, d, J=7.8 Hz, H-3), 8.15 (1H, br s, NH), 7.60 (1H, t, J=8.0 Hz, H-4), 7.32 (1H, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
1.1.2 5-Bromo-[1,2,4]triazolo[1,5-a]pyridin-2-ylamine (3)

To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1:1, 900 mL) is added N,N-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20° C.) for 1 h. 1-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3 h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition of H2O (250 mL) and filtration. The combined solids are washed successively with H2O (250 mL), EtOH/MeOH (1:1, 250 mL) and Et2O (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. 1H (400 MHz, DMSO-d6) δ 7.43-7.34 (2H, m, 2×aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1:1, M+H+, 100%).
1.1.3 General Procedure for Mono-Acylation to Afford Intermediate (4)

To a solution of the 2-amino-triazolopyridine (3) (7.10 g, 33.3 mmol) in dry CH3CN (150 mL) at 5° C. is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material (3) is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp. (for 1-16 h) to hydrolyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et2O (50 mL). The solids are collected by filtration, washed with H2O (2×50 mL), acetone (50 mL) and Et2O (50 mL), then dried in vacuo to give the required bromo intermediate (4).
Method A Preparation of Compounds of the Invention Via Suzuki Coupling (5):
An appropriate boronic acid (2 eq.) is added to a solution of bromo intermediate (4) in 1,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140° C. for 30 min (this reaction can also be carried out by traditional heating in an oil bath at 90° C. for 16 h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSO4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters XBridge Prep C18 5 μm ODB 19 mm ID×100 mm L (Part No. 186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method B

B1. 4 4-[2-(Cyclopropanecarbonyl-amino)-[1,2,4]triazolo[1,5-a]pyridin-5-yl]-benzoyl chloride

2 Drops of DMF are added to a solution of 4-[2-(cyclopropanecarbonyl-amino)-[1,2,4]triazolo[1,5-a]pyridin-5-yl]-benzoic acid (1 eq) obtained by Method A using 4-carboxyphenylboronic acid in DCM under N2 atmosphere. Then oxalyl chloride (2 eq) is added dropwise to this resulting solution (gas release). The mixture is stirred at room temperature for 2 hours. After completion of the reaction by LCMS, the solvent is removed. The crude acid chloride is used without further purification in next step.
B2. Amide Formation (General Method)

An appropriate amine (1.1 eq) and Et3N (5 eq) are dissolved in DCM under N2 atmosphere and cooled at 0° C. The acid chloride (B1, 1 eq) dissolved in DCM is added dropwise to this solution. The reaction is stirred at room temperature for 16 h. After this time, reaction is complete. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers are filtered and evaporated. The final compound is isolated by preparative HPLC. Preparative HPLC: Waters XBridge Prep C18 5 μm ODB 19 mm ID×100 mm L (Part No. 186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
…
Synthesis of the Compound of the Invention and Comparative Examples Compound 1 (the Compound of the Invention) Step 1:

2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1-dioxide (2 eq) was added portionwise. The resulting solution was stirred at room temperature for 16 h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
STEP 2: Suzuki coupling

4-[4-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-1,1-dioxide (1.1 eq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-yl)-amide in 1,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90° C. for 16 h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSO4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
Alternatively, after completion of the reaction, a palladium scavenger such as 1,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cooled down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HCl is added, and after stirring at RT, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at RT, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H2O, treated with a palladium scavenger (e.g. SMOPEX 234) at 50° C., the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the title compound as a free base.
Alternative Route to Compound 1 (the Compound of the Invention): Step 1:

4-(Hydroxymethyl)phenylboronic acid (1.1 eq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-yl)-amide in 1,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90° C. for 16 h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSO4 and evaporated in vacuo. The resulting mixture was used without further purification.
Step 2:

To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl]-amide (1.0 eq) in chloroform was slowly added phosphorus tribromide (1.0 equiv.). The reaction mixture was stirred at room temperature for 20 hours, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer was dried over anhyd. MgSO4, filtered and concentrated to dryness. The resulting white residue was triturated in dichloromethane/diethyl ether 2:1 to afford the expected product as a white solid.
Step 3:

Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl]-amide (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1-dioxide (1.1 eq) was added dropwise. The resulting solution was stirred at room temperature for 16 h. After this time, the reaction was complete. The solvent was evaporated. The compound was dissolved in DCM, washed with water and dried over anhyd. MgSO4. Organic layers were filtered and evaporated. The final compound was isolated by column chromatography using EtOAc to afford the desired product.
…………………….
PATENT
Novel salts and pharmaceutical compositions thereof for the treatment of inflammatory disorders
Also claims a method for preparing filgotinib hydrochloride trihydrate. The present filing forms a pair with this week’s filing, WO2015117980, claiming a tablet composition comprising filgotinib hydrochloride.
The compound cyclopropanecarboxylic acid {5-[4-(l,l-dioxo-thiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5-a]pyridin-2-yl -amide (Compound 1), which has the chemical structure:

is disclosed in our earlier application WO 2010/149769 (Menet C. J., 2010) as being an inhibitor of JAK and as being useful in the treatment of inflammatory conditions, autoimmune diseases, proliferative diseases, allergy, transplant rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6 or interferons. Hereafter this compound is named Compound 1. The data presented in WO 2010/149769 demonstrate that despite similar in vitro activities, Compound 1 has unexpectedly high in vivo potency compared with structurally similar compounds.
Example 1. Preparation of Compound 1
1.1. Route 1
1.1.1. 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide

[00205] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) are dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) is added portionwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is extracted with EtOAc and water, washed with brine and dried over anhydrous MgSO i. Organic layers are filtered and evaporated. The final compound is isolated without further purification.
1.1.2. Cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide

1.1.2.1. Step i): l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea
[00206] To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5°C is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction
mixture is then allowed to warm to room temp. (20 °C) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3 x 600 niL) and air-dried to afford the desired product. The thiourea may be used as such for the next step without any purification. lH (400 MHz, CDC13) δ 12.03 (1H, br s), 8.81 (1H, d), 8.15 (1H, br s), 7.60 (1H, t), 7.32 (1H, dd), 4.31 (2H, q), 1.35 (3H, t).
1.1.2.2. Step ii): 5-Bromo-[l,2,4]triazolo[l,5-a]pyridin-2-ylamine
[00207] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1 : 1, 900 mL) is added NN-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20 °C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition of H20 (250 mL) and filtration. The combined solids are washed successively with H20 (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et20 (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. lH (400 MHz, DMSO-i¼) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 : 1, M+H+, 100%).
1.1.2.3. Step Hi): Cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide
[00208] To a solution of the 2-amino-triazolopyridine obtained in the previous step (7.10 g, 33.3 mmol) in dry MeCN (150 mL) at 5°C is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp, (for 1-16 h) to hydro lyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et20 (50 mL). The solids are collected by filtration, washed with H20 (2x50mL), acetone (50 mL) and Et20 (50 mL), then dried in vacuo to give the desired compound.
1.1.3. Compound 1

[00209] 4-[4-(4,4,5,5-Tetramethyl-[l ,3,2]dioxaborolan-2-yl)-benzyl] hiomoφholine , l -dioxide (l . l eq.) is added to a solution of cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4: 1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) are added to the solution. The resulting mixture is then heated in an oil bath at 90°C for 16h under N2. Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhydrous MgS04 and evaporated in vacuo.
[00210] The final compound is obtained after purification by flash chromatography.
[00211] Alternatively, after completion of the reaction, a palladium scavenger such as 1 ,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cool down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HC1 is added, and after stirring at room temperature, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at room temperature, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H20, treated with a palladium scavenger (e.g. SMOPEX 234) at 50°C, the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the desired compound as a free base.
1.2. Route 2
1.2.1. Step 1: cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[l,2, 4]triazolo[l, 5- a] pyridin-2-yl] -amide

[00212] 4-(Hydroxymethyl)phenylboronic acid (l . l eq.) is added to a solution of cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water
(4:1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) are added to the solution. The resulting mixture is then heated in an oil bath at 90°C for 16h under N2. Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhydrous MgS04 and evaporated in vacuo. The resulting mixture is used without further purification.
1.2.2. Step 2: Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5- a Jpyridin-2-ylJ -amide

[00213] To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin-2-yl] -amide (1.0 eq) in chloroform is slowly added phosphorus tribromide (1.0 eq.). The reaction mixture is stirred at room temperature for 20 h, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer is dried over anhydrous MgSO i, filtered and concentrated to dryness. The resulting white residue is triturated in dichloromethane/diethyl ether 2:1 to afford the desired product.
1.2.3. Step 3:

[00214] Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin-2-yl]-amide (l eq) and DIPEA (2 eq) are dissolved in DCM/MeOH (5: 1 v:v) under N2 and thiomorpho line 1,1-dioxide (1.1 eq) is added dropwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is dissolved in DCM, washed with water and dried over anhydrous MgSO i. Organic layers are filtered and evaporated. The final compound is isolated by column chromatography using EtOAc to afford the desired product.
…………………
PATENT
http://www.google.co.in/patents/WO2013189771A1?cl=en
Example 1. Synthesis of the compounds
1.1. Route 1
1.1.1. Synthesis of 5-Bromo-[l,2,4]triazolo[l,5-a]pyridin-2-ylamine (Intermediate 3)

led to 5 °C was added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture was then allowed to warm to room temp. (20 °C) and stirred for 16 h. Evaporation in vacuo gave a solid which was collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea was used as such in the next step without any purification.
[00157] lH (400 MHz, CDC13) δ 12.03 (IH, br s, NH), 8.81 (IH, d, J 7.8 Hz, H-3), 8.15 (IH, br s, NH), 7.60 (IH, t, J 8.0 Hz, H-4), 7.32 (IH, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
1.1.1.2. 5-Bromo-f 1,2, 4]triazolo[ 1 ,5-a] pyridin-2-ylamine (3)
[00158] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1 : 1, 900 mL) was added NN-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture was stirred at room temp. (20 °C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) was then added and the mixture slowly heated to reflux (Note: bleach scrubber was required to quench H2S evolved). After 3 h at reflux, the mixture was allowed to cool and filtered to collect the precipitated solid. Further product was collected by evaporation in vacuo of the filtrate, addition of H20 (250 mL) and filtration. The combined solids were washed successively with H20 (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et20 (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound was used as such in the next step without any purification.
[00159] lH (400 MHz, DMSO-i¼) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 : 1, M+H+, 100%).
1.1.2. Synthesis of 4-[ 4-(4, 4, 5, 5-Tetramethyl-f 1, 3,2] ‘ dioxaborolan-2-yl) -benzyl] ‘- thiomor holine- 1, 1 -dioxide (Intermediate 4)

[00160] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) was added portion wise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhydrous MgSO i. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
1.1.3. Synthesis of 5-[4-(l, l-Dioxothiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5- a ridin-2-ylamine (Formula I)

[00161] 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide (l .leq.) was added to a solution of 5-bromo-[l,2,4]triazolo[l,5-a]pyrid in-2-ylamine (4: 1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90°C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhydrous MgSC>4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
[00162] lH (400 MHz, CDC13) δ 7.94-7.92 (d, 2H), 7.52-7.48 (m, 3H), 7.37-7.34 (m, 1H), 7.02-7.00 (m, 1H), 6.00 (d, 2H), 3.76 (d, 2H), 3.15-3.13 (m, 4H), 2.93-2.91 (m, 4H).
[00163] m/z 358.2 (M+H+, 100%). 1.2. Route 2
1.2.1. Cyclopropanecarboxylic acid {5-[4-(l, l-dioxo-thiomorpholin-4-ylmethyl)-phenylJ- [l,2,4]triazolo[l,5-a]pyridin-2-yl}-amide (Formula II)
[00164] The compound according to Formula II may be synthesized according to the procedure described in WO 2010/149769.
1.2.2. Synthesis of 5-[4-(l, l-Dioxothiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5- aJpyridin-2-ylamine (Formula I)
[00165] The compound according to Formula I can also be produced by hydrolysis of the compound accor ing to Formula II:

[00166] Hydrochloric acid 30% aq (12.06 kg; 3.9 rel. volumes) was added to a slurry of the compound according to Formula II (3.45 kg; 1.0 equiv.) in demineralized water (10.0 kg; 3.0 rel. volumes). Subsequently, a line rinse was performed with demineralized water (3.4 kg; 1.0 rel. volumes). The reaction mixture was heated to 80±5°C for 14.5 h. After completion of the reaction (conversion > 99%>), the reaction mixture was cooled to 20±5°C. The reaction mixture was diluted with demineralized water (6.8 kg; 2.0 rel. volumes) and sodium hydroxide 33%> aq (9.52 kg; 3.7 rel volumes) was dosed at such a rate that the temperature of the reactor contents remained below 35°C. An additional amount of sodium hydroxide 33%> aq (2.55 kg; 1.0 rel. volumes) was needed to get the pH > 10. The product was filtered off, washed twice with demineralized water (1.5 rel. volumes) and dried under vacuum for 1 h, thus yielding the crude compound according to Formula I.
[00167] The crude compound according to Formula I (5.70 kg) was re-slurried in demineralized water (23.0 kg; 8.5 rel. volumes). Hydrochloric acid 30%> aq (1.65 kg; 0.7 rel. volumes) and demineralized water (4.3 kg; 1.6 rel. volumes) were added and the reaction mixture was stirred at 20±5°C for 45 min. As the compound according to Formula I was not dissolved completely, the reaction mixture was stirred at 45±5°C for 1 h. The reaction mixture was filtered and the residue was washed with demineralized water (2.0 kg 0.75 rel. volumes). Sodium hydroxide 33%> aq (1.12 kg; 0.6 rel volumes) was added to the filtrate. An additional amount of sodium hydroxide 33%> aq (1.01 kg) was needed to get the pH > 10. The resulting reaction mixture was stirred at 20±5°C for about 3 h. The product was filtered off, washed twice with demineralized water (4.1 kg; 1.5 rel. volumes), and twice with methyl tert-butyl ether (MTBE; 3.0 kg; 1.5 rel. volumes) and dried under vacuum for 15.5 h on the filter. The product was further dried in a vacuum oven at 40±5°C for 202 h, thus affording the desired compound according to Formula I.
Update
Scheme 1: General S nthesis of Compounds of Formula I or A

Formula A
Scheme 7.

(16) (17) (18)
(18a): R3a=R3b=R2a=R (18b): R3a=R3b=D; R2a 18c): R3a=R3b=H; R2a

References
- Namour, Florence; Diderichsen, Paul Matthias; Cox, Eugène; Vayssière, Béatrice; Van der Aa, Annegret; Tasset, Chantal; Van’t Klooster, Gerben (2015-02-14). “Pharmacokinetics and Pharmacokinetic/Pharmacodynamic Modeling of Filgotinib (GLPG0634), a Selective JAK1 Inhibitor, in Support of Phase IIB Dose Selection”. Clin Pharmacokinet. Epub ahead of print.doi:10.1007/s40262-015-0240-z.
- Van Rompaey, L; Galien, R; Van der Aar, E; Clement-Lacroix, P; Van der Aar, E; Nelles, L; Smets, B; Lepescheux, L; Cristophe, T; Conrath, K; Vandeghinste, N; Vayssiere, B; De Vos, S; Fletcher, S; Brys, R; Van’t Klooster, G; Feyen, J; Menet, C (2013-10-01). “Preclinical characterization of GLPG0634, a selective inhibitor of JAK1 for the treatment of inflammatory diseases”. J Immunol. 191(7). doi:10.4049/jimmunol.1201348.
- http://acrabstracts.org/abstracts/phase-1-and-phase-2-data-confirm-that-glpg0634-a-selective-jak1-inhibitor-has-a-low-potential-for-drug-drug-interactions/
- “Galapagos’ GLPG0634 shows excellent efficacy and safety in rheumatoid arthritis Phase II study” (PDF) (Press release). Retrieved 2015-02-26.
- “Galapagos reports that the last patient in DARWIN 1 has completed 12 weeks of treatment” (PDF) (Press release). Retrieved 2015-02-26.
- “Galapagos completes recruitment for Darwin 1 study with GLPG0634 (filgotinib) in RA”. EuroInvestor. Retrieved 2015-02-26.
- NASDAQ OMX Corporate Solutions. “Galapagos completes recruitment for Darwin 2 monotherapy study with GLPG0634 (filgotinib) in RA”. Yahoo Finance. Retrieved 2015-02-26.
| US8551980 | Nov 17, 2010 | Oct 8, 2013 | Bayer Intellectual Property Gmbh | Substituted triazolopyridines |
| US8796457 | Jun 25, 2010 | Aug 5, 2014 | Galapagos Nv | Compound useful for the treatment of degenerative and inflammatory diseases |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-[5-[4-[(1,1-dioxo-1,4-thiazinan-4-yl)methyl]phenyl]-[1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Biological half-life | 6 hours[1] |
| Identifiers | |
| CAS Registry Number | 1206161-97-8 |
| ATC code | L01XE18 |
| IUPHAR/BPS | 7913 |
| ChemSpider | 28189566 |
| UNII | 3XVL385Q0M |
| ChEMBL | CHEMBL3301607 |
| Chemical data | |
| Formula | C21H23N5O3S |
| Molecular mass | 425.50402 g/mol |
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


Join me on twitter

 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL
/////////Galapagos, GLPG0634, Filgotinib, PHASE 2, orphan drug designation, PHASE 3, Crohn’s disease, Rheumatoid arthritis, Ulceraticolitis
ve SMILES code: O=C(C1CC1)NC2=NN3C(C4=CC=C(CN5CCS(CC5)(=O)=O)C=C4)=CC=CC3=N2
