
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 431)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA Grants QIDP and Fast Track Designations for Cubist’s Late-Stage Antibiotic Candidates
- QIDP granted for ceftolozane/tazobactam in HABP/VABP and cUTI
- Fast track status provided for ceftolozane/tazobactam (CXA-201) in cIAI
February 28, 2013
LEXINGTON, Mass., Cubist Pharmaceuticals, Inc. today announced that the U.S. Food and Drug Administration (FDA) has designated the company’s late-stage antibiotic candidate, ceftolozane/tazobactam, as a Qualified Infectious Disease Product (QIDP) for the indications of Hospital-Acquired Bacterial Pneumonia (HABP)/Ventilator-Associated Bacterial Pneumonia (VABP) and Complicated Urinary Tract Infections (cUTI).
Additionally, the company received from the FDA notification that Cubist’s antibiotic candidates, ceftolozane/tazobactam and surotomycin, have been granted Fast Track status in their previously granted QIDP indications, Complicated Intra-Abdominal Infections (cIAI) and Clostridium difficile-Associated Diarrhea (CDAD) respectively.
“We are excited to receive the QIDP and Fast Track designations for ceftolozane/tazobactam and surotomycin, which further reinforce the importance the FDA places on helping to advance critically needed antibiotics,” said Steven Gilman, Ph.D., Executive Vice President of Research and Development and Chief Scientific Officer of Cubist Pharmaceuticals. “In a very short period of time, the GAIN Act has shown its value in helping to incentivize antibiotic development.”
The QIDP designation for ceftolozane/tazobactam will enable Cubist to benefit from certain incentives for the development of new antibiotics, including priority review, eligibility for Fast Track status, and if ceftolozane/tazobactam is ultimately approved by the FDA, a five year extension of Hatch-Waxman exclusivity. These incentives are provided under the Generating Antibiotic Incentives Now Act (GAIN Act), which received strong bipartisan support in Congress and was signed into law by President Obama in July 2012 as part of the FDA Safety and Innovation Act (FDASIA), the fifth authorization of the Prescription Drug User Fee Act.
Ceftolozane/tazobactam is currently being studied in pivotal Phase 3 trials as a potential first-line intravenous therapy for the treatment of cIAI and cUTI caused by Gram-negative pathogens, including those caused by multi-drug resistant Pseudomonas aeruginosa. Cubist expects to initiate a Phase 3 VABP program for ceftolozane/tazobactam by mid-year. Surotomycin, a rapidly bactericidal lipopeptide, is currently in Phase 3 being studied as a potential treatment for patients with a severe and sometimes life-threatening diarrhea caused by CDAD.
About The GAIN Act
The GAIN Act, Title VIII (Sections 801 through 806) of the FDASIA, provides pharmaceutical and biotechnology companies with incentives to develop new antibacterial and antifungal drugs for the treatment of life-threatening infectious diseases caused by drug resistant pathogens. Qualifying pathogens are defined by the GAIN Act to include multi-drug resistant Gram-negative bacteria, including Pseudomonas, Acinetobacter, Klebsiella, and Escherichia coli species; resistant Gram-positive pathogens, including methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus; multi-drug resistant tuberculosis; and Clostridium difficile.
About Gram-negative bacteria
The diseases caused by Gram-negative bacteria include intra-abdominal infections, urinary tract infections, pneumonia, peritonitis, septicemia, neonatal meningitis, and burn and wound infections. In the US in 2003, Gram-negative bacteria were associated with many of the most frequent types of hospital-acquired infections including 71% of urinary tract infections, 65% of pneumonia episodes, 34% of surgical site infections, and 24% of bloodstream infections. Important Gram-negative bacteria include Pseudomonas, Escherichia coli, Klebsiella, and Acinetobacter.
About CDAD
CDAD is a disease caused by an overgrowth of, and toxin production by C. difficile, a Gram-positive bacterium naturally found in the lower gastrointestinal tract. This overgrowth is caused by the use of antibiotics for the treatment of common community and hospital acquired infections. Many antibiotics cure the underlying infection but, as a consequence, disrupt the natural balance of intestinal bacteria which allows C. difficile to overgrow. The overgrown C. difficile bacteria produce enterotoxin and cytotoxin, two proteins that can lead to potentially life-threatening severe diarrhea and sepsis (blood infection). CDAD rates and severity are increasing, due in part to the spread of a new strain with increased virulence and greater resistance to fluoroquinolones, a standard of care treatment. According to an article in the October 2008 issue of the New England Journal of Medicine, during the mid- and late-1990s, the reported incidence of C. difficile infections in acute care hospitals in the United States remained stable at 30 to 40 cases per 100,000. However in 2001, this number rose to almost 50, with subsequent increases to the point that the number of cases that were reported in 2005 (84 per 100,000) was nearly three times the 1996 rate (31 per 100,000).
About Cubist
Cubist Pharmaceuticals, Inc. is a biopharmaceutical company focused on the research, development, and commercialization of pharmaceutical products that address significant unmet medical needs in the acute care environment. Cubist is headquartered in Lexington, Mass. Additional information can be found at Cubist’s web site at www.cubist.com.
ceftolozane
 credit —kegg
credit —kegg
http://www.ama-assn.org/resources/doc/usan/ceftolozane.pdf
89293-68-3 cas no
………………………………………………………………………………………………………………………
tazobactum

cas no89786-04-9
Tazobactam is a compound that inhibits the action of bacterial beta-lactamases. It is combined with the extended spectrum beta-lactam antibiotic piperacillin in the drug Tazocin (also Zosyn, Piprataz)one of the preferred antibiotic treatment for CAP caused by P. aeruginosa. It broadens the spectrum of piperacillin by making it effective against organisms that express beta-lactamase and would normally degrade piperacillin.
Tazobactam sodium is a derivative of the penicillin nucleus and is a penicillanic acid sulfone.
Taiho Pharma seeks Japanese nod to manufacture,market novel anti-tumour agent TAS-102

TRIFLURIDINE

TIPIRACIL
TAS-102 is an anti-cancer drug under development for colorectal cancer.[1]
| Combination of | |
|---|---|
| Trifluridine | cytotoxin |
| Tipiracil | thymidine phosphorylase inhibitor |
Clinical trials
A phase II trial reported in 2011[2] and a phase III trial is due to end in 2014.[1][3]
Mechanism
TAS-102 consists of the cytotoxin trifluridine and the thymidine phosphorylase inhibitor (TPI) tipiracil.[4] Trifluridine is incorporated into DNA during DNA synthesis and inhibits tumor cell growth. Tipiracil protects trifluridine from being broken down when taken orally.
| February 28, 2013, |
|
Taiho Pharmaceutical Co., Ltd. has submitted an application to the Japanese Ministry of Health, Labour and Welfare for approval of the manufacture and marketing of the novel oral nucleoside anti-tumour agent TAS-102 (combination of trifluorothymidine [FTD] and tipiracil hydrochloride [TPI]). Taiho is seeking approval of TAS-102 for the indication of unresectable, advanced, recurrent colorectal cancer. The application for approval is based on the results of a phase II clinical trial (Study 10040030) conducted at 20 facilities throughout Japan. It was a randomized, double-blind comparative study of TAS-102 and a placebo involving 172 patients with unresectable, advanced, recurrent colorectal cancer that was refractory to the standard chemotherapy of at least two or more regimens containing fluoropyrimidine, irinotecan, and oxaliplatin. The results indicated that the group administered TAS-102 had improved overall survival rates (median overall survival: 9.0 months vs. 6.6 months) and a significantly reduced risk of mortality (HR: 0.56, p=0.0011). The most frequently reported adverse drug reaction with a CTCAE grade of 3 or higher was neutropenia. Grade 3 or higher diarrhea, fatigue, nausea, and other adverse reactions were no more than 10 per cent. Taiho Pharmaceutical is currently proceeding with a global phase III clinical trial of TAS-102 in a similar colorectal cancer population (RECOURSE) with the ultimate goal of global registration and commercialization of the agent. Taiho Pharmaceutical believes that TAS-102 will make a significant contribution to cancer patients and will continue its development efforts to broaden its use. TAS-102 is an anti-tumour agent composed of a combination of trifluorothymidine (FTD), a nucleoside that incorporates into DNA and inhibits a variety of genetic functions required for the proliferation of cancer cells, and tipiracil hydrochloride (TPI), an inhibitor of thymidine phosphorylase (which degrades FTD) that maintains an effective blood concentration of FTD. TAS-102 is administered twice daily to achieve a total daily dose of 70mg/m2 for five days followed by two days of rest and then repeated a second time. This is followed by a 14-day rest period to make a 28-day schedule for one course. |
- “New Drug for Colorectal Cancer Shows Promise in Phase II Trial”. 28 Aug 2012.
- “Novel Drug TAS-102 Makes Headway in Refractory Colorectal Cancer”. 4 Oct 2011.
- “Phase II study of TAS-102 for pretreated metastatic colorectal cancer”. 29 Aug 2012.
- “A novel combination antimetabolite, TAS-102, exhibits antitumor activity in FU-resistant human cancer cells through a mechanism involving FTD incorporation in DNA.”. Sept 2004.
|
|
|---|---|
| TRIFLURIDINE | |
| 1-[4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-5- (trifluoromethyl) pyrimidine-2,4-dione |
Trifluridine (also called trifluorothymidine or TFT) is an anti-herpesvirus antiviral drug, used primarily on the eye. It was sold under the trade name, Viroptic, by Glaxo Wellcome, now merged into GlaxoSmithKline. The brand is now owned by Monarch Pharmaceuticals, which is wholly owned by King Pharmaceuticals.
It is a nucleoside analogue, a modified form of deoxyuridine, similar enough to be incorporated into viral DNA replication, but the -CF3 group added to the uracil component blocks base pairing.
It is a component of the experimental anti-cancer drug TAS-102.
TIPIRACIL
| NAME | 5-chloro-6-[(2-iminopyrrolidin-1-yl)methyl]pyrimidine-2,4-(1H,3H)-dione | |||
| CAS | 183204-74-2 | |||
| MOL F | C9H11ClN4O2 | |||
| STR | |
|||
| USE | potentiator of antineoplastics; | |||
Taiho Pharmaceutical, a subsidiary of Otsuka Holdings Co., Ltd., is an R&D-driven specialty pharma focusing on the three fields of oncology, allergies and immunology, and urology.
Hyaluronan initiates chondrogenesis mainly via CD44 in human adipose derived stem cells.

Source
1Kaohsiung Medical University.
Abstract
Cell-matrix adhesion is one of the important interactions that regulate stem cell survival, self-renewal, and differentiation. Our previous report indicated that a microenvironment enriched with hyaluronan (HA) initiated and enhanced chondrogenesis in human adipose derived stem cells (hADSCs). We further hypothesize that HA-induced chondrogenesis in hADSCs is mainly due to the interaction of HA and CD44 (HA-CD44), a cell surface receptor of HA. The HA-CD44 interaction was tested by examining the mRNA expression of hyaluronidase-1 (Hyal-1) and chondrogenic marker genes (SOX-9, collagen type II, and aggrecan) in hADSCs cultured on HA-coated wells. Cartilaginous matrix formation, sulfated glycosaminoglycan (sGAG) and collagen productions by hADSCs affected by HA-CD44 interaction were tested in a 3D fibrin hydrogel. About 99.9% of hADSCs possess CD44. The mRNA expressions of Hyal-1 and chondrogenic marker genes were up-regulated by HA in hADSCs on HA-coated wells. Blocking HA-CD44 interaction by anti-CD44 antibody completely inhibited Hyal-1 expression and reduced chondrogenic marker gene expression, which indicates that HA induced chondrogenesis in hADSCs mainly acts through HA-CD44 interaction. A two-hour pre-incubation and co-culture of cells with HA in hydrogel (HA/fibrin hydrogel) not only assisted in hADSC survival but also enhanced expression of Hyal-1 and chondrogenic marker genes. Higher levels of sGAG and total collagen were also found in HA/fibrin hydrogel group. Immunocytochemistry showed more collagen type II but less collagen type X in HA/fibrin than in fibrin hydrogels. Our results indicate that signaling triggered by HA-CD44 interaction significantly contributes to HA-induced chondrogenesis and may be applied to ADSC-based cartilage regeneration.
Hyaluronan (also called hyaluronic acid or hyaluronate or HA) is an anionic,nonsulfated glycosaminoglycan distributed widely throughout connective,epithelial, and neural tissues. It is unique among glycosaminoglycans in that it is nonsulfated, forms in the plasma membrane instead of the Golgi, and can be very large, with its molecular weight often reaching the millions.[2] One of the chief components of the extracellular matrix, hyaluronan contributes significantly to cell proliferation and migration, and may also be involved in the progression of some malignant tumors.
The average 70 kg (154 lbs) person has roughly 15 grams of hyaluronan in the body, one-third of which is turned over (degraded and synthesized) every day.[3]Hyaluronic acid is also a component of the group A streptococcal extracellularcapsule,[4] and is believed to play a role in virulence.[5][6]
- Hyaluronate Sodium in the ChemIDplus database, consulté le 12 février 2009
- Frasher, J.R.E et al’; Laurent, T. C.; Laurent, U. B. G. (1997).“Hyaluronan: its nature, distribution, functions and turnover”(PDF). Journal of Internal Medicine 242 (1): 27–33.doi:10.1046/j.1365-2796.1997.00170.x. PMID 9260563. Retrieved 2009-06-05.
- Stern R (August 2004). “Hyaluronan catabolism: a new metabolic pathway”. Eur J Cell Biol 83 (7): 317–25.doi:10.1078/0171-9335-00392. PMID 15503855.
- Sugahara, K.; N.B. Schwartz and A. Dorfman (1979).“Biosynthesis of hyaluronic acid by Streptococcus“. Journal of Biological Chemistry 254 (14): 6252–6261. PMID 376529.
- Wessels, M.R.; A.E. Moses, J.B. Goldberg and T.J. DiCesare (1991). “Hyaluronic acid capsule is a virulence factor for mucoid group A streptococci”. PNAS 88 (19): 8317–8321.doi:10.1073/pnas.88.19.8317. PMC 52499.PMID 1656437.
- Schrager, H.M.; J.G. Rheinwald and M.R. Wessels (1996).“Hyaluronic acid capsule and the role of streptococcal entry into keratinocytes in invasive skin infection”. Journal of Clinical Investigation 98 (9): 1954–1958. doi:10.1172/JCI118998.PMC 507637. PMID 8903312.
Hyaluronic Acid


Novartis’ Ilaris canakizumab has become the first biologic drug to be approved in the EU to treat the symptoms of gouty arthritis
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | IL-1β |

Novartis’ Ilaris has become the first biologic drug to be approved in the EU to treat the symptoms of gouty arthritis in another gain for the interleukin-1 beta inhibitor.
march01,2013
First biologic drug approved for condition in Europe
The European Commission (EC) cleared llaris (canakizumab) for the treatment of adult patients who have suffered at least three gouty arthritis attacks in the previous 12 months, but who are unsuitable for treatment with non-steroidal anti-inflammatory drugs (NSAIDs) and colchicine or repeated courses of corticosteroids.
Gouty arthritis – commonly known as gout – is an “excruciating condition”, according to Novartis division head David Epstein, who noted that Ilaris offers new hope to patients who do not currently have treatment options.
Data from two phase III trials of Ilaris in acute gouty arthritis attacks showed that patients treated with the drug experienced significantly greater pain relief compared to the injectable steroid triamcinolone acetonide, while most adverse events were mild to moderate in severity.
The most frequent side effects were infections, and particularly upper respiratory tract infections and nasopharyngitis.
Ilaris was launched in the US and EU in 2009 as a treatment for an auto-inflammatory condition called cryopyrin-associated periodic syndrome (CAPS). The rarity of that condition has meant sales have been relatively small, coming in at $72m last year, albeit a 56 per cent gain over 2011.
Gouty arthritis is a much bigger market for the drug and, along with a juvenile arthritis indication Novartis is pursuing, could push Ilaris towards blockbuster status with sales in excess of $1bn a year.
“Our vision is to realise the potential of Ilaris wherever IL-1 beta plays a key role and available treatment options don’t give patients the help they need,” said Epstein.
EU approval comes after the US FDA knocked back Ilaris for gouty arthritis, saying in 2011 that Novartis needed to provide more data on the drug’s risk-benefit profile, specifically its potential to leave patients vulnerable to infections.
Gout has been a tricky indication for drug developers to crack, with the FDA turning down another CAPS treatment – Regeneron’s IL-1 inhibitor Arcalyst (rilonacept) – in 2012 on the grounds of inadequate safety data and concern about a risk of malignancy.
One success came in 2010 when Savient secured approval for its Krystexxa (pegloticase) drug as a second-line treatment after oral xanthine oxidase inhibitors in patients with severe debilitating chronic tophaceous gout.
However, the drug has failed to make significant inroads because of a high price and tendency to stimulate neutralising antibodies that limit its therapeutic effect, according to Decision Resources.
There is still a great demand for safer and more effective therapies with the phase III pipeline featuring another potential blockbuster in the form of AstraZeneca/Ardea Biosciences URAT1 inhibitor lesinurad.
Canakinumab (INN, trade name Ilaris, previously ACZ885)[1] is a human monoclonal antibody targeted at interleukin-1 beta. It has no cross-reactivity with other members of the interleukin-1 family, including interleukin-1 alpha.[2]
Canakinumab was approved for the treatment of cryopyrin-associated periodic syndromes (CAPS) by the US FDA on June 2009[3] and by the European Medicines Agency in October 2009.[4] CAPS is a spectrum of autoinflammatory syndromes including familial cold autoinflammatory syndrome, Muckle–Wells syndrome, and neonatal-onset multisystem inflammatory disease.
Canakinumab was being developed by Novartis for the treatment of rheumatoid arthritis but this trial has been discontinued.[5] Canakinumab is also in phase I clinical trials as a possible treatment for chronic obstructive pulmonary disease.[6]
References
- Dhimolea, Eugen (2010). “Canakinumab”. MAbs 2 (1): 3–13. doi:10.4161/mabs.2.1.10328. PMC 2828573. PMID 20065636.
- Lachmann, HJ; Kone-Paut I, Kuemmerle-Deschner JB et al. (4 June 2009). “Use of canakinumab in the cryopyrin-associated periodic syndrome”. New Engl J Med 360 (23): 2416–25. doi:10.1056/NEJMoa0810787. PMID 19494217.
- “New biological therapy Ilaris approved in US to treat children and adults with CAPS, a serious life-long auto-inflammatory disease” (Press release). Novartis. 18 June 2009. Retrieved 28 July 2009.
- Wan, Yuet (29 October 2009). “Canakinumab (Ilaris) and rilonacept (Arcalyst) approved in EU for treatment of cryopyrin-associated periodic syndrome”. National electronic Library for Medicines. Retrieved 14 April 2010.
- “clinicaltrials.gov, Identifier NCT00784628: Safety, Tolerability and Efficacy of ACZ885 (Canakinumab) in Patients With Active Rheumatoid Arthritis”. Retrieved 2010-08-21.
- Yasothan U, Kar S (2008). “Therapies for COPD”. Nat Rev Drug Discov 7 (4): 285. doi:10.1038/nrd2533.

Monoclonal Antibody Therapeutic Uses
Otsuka receives FDA approval for ABILIFY MAINTENA to treat schizophrenia
 |
|
|---|---|
| 7-{4-[4-(2,3-Dichlorophenyl)piperazin-1-yl]butoxy}-3,4-dihydroquinolin-2(1H)-one |
aripiprazole
mar 1, 2013
Otsuka Pharmaceutical Co., Ltd. (Otsuka) and H. Lundbeck A/S (Lundbeck) announced the U.S. Food and Drug Administration (FDA) has approved ABILIFY MAINTENA™ (aripiprazole) for extended- release injectable suspension, an intramuscular (IM) depot formulation indicated for the treatment of schizophrenia.
ABILIFY MAINTENA is the first dopamine D2 partial agonist approved as a once- monthly injection. It contributes a new treatment option to address the ongoing need for relapse prevention in patients with schizophrenia – a chronic, debilitating disease.
Efficacy was demonstrated in a 52-week, placebo-controlled, double-blind, randomized-withdrawal, Phase 3 maintenance trial of ABILIFY MAINTENA in patients with schizophrenia. The time to relapse was the primary endpoint. In the trial, ABILIFY MAINTENA>1 In a key secondary endpoint, the percentage of subjects experiencing relapse (i.e., meeting clinical trial criteria for exacerbation of psychotic symptoms/relapse) was also significantly lower with ABILIFY MAINTENA compared to placebo at the end of the study (10% vs. 40%, respectively; p<0.0001). Additional support for efficacy was derived from oral aripiprazole trials.
Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. ABILIFY MAINTENA is not approved for the treatment of patients with dementia-related psychosis. ABILIFY MAINTENA is contraindicated in patients with a known hypersensitivity reaction to aripiprazole. Reactions have ranged from pruritus/urticaria to anaphylaxis (see Important Safety Information below).
ABILIFY MAINTENA will be the first commercialized product from the long-term global alliance between Otsuka and Lundbeck to develop CNS medicines worldwide. The companies expect the product will start becoming available in the U.S. on March 18.
Aripiprazolebrand names: Abilify, Aripiprex) is a partial dopamine agonist of the second generation class of atypical antipsychoticswith additional antidepressant properties that is used in the treatment of schizophrenia,bipolar disorder, and clinical depression. It was approved by the U.S. Food and Drug Administration (FDA) for schizophrenia on November 15, 2002 and the European Medicines Agency on 4 June 2004; for acute manic and mixed episodes associated with bipolar disorder on October 1, 2004; as an adjunct for major depressive disorder on November 20, 2007; and to treat irritability in children with autism on 20 November 2009.[1][2] Aripiprazole was developed by Otsuka in Japan, and in the United States,Otsuka America markets it jointly with Bristol-Myers Squibb.
EU OKs Lundbeck’s Selincro, Nalmefene to cut alcoholic urges

Nalmefene
17-cyclopropylmethyl-4,5α-epoxy-6-methylenemorphinan-3,14-diol
march 1 2013
Lundbeck will be celebrating news that European regulators have issued a green light for Selincro, making it the first therapy approved for the reduction of alcohol consumption in dependent adults.
Selincro (nalmefene) is a unique dual-acting opioid system modulator that acts on the brain’s motivational system, which is dysregulated in patients with alcohol dependence.
The once daily pill has been developed to be taken on days when an alcoholic feels at greater risk of having a drink, in a strategy that aims to reduce – rather than stop – alcohol consumption, which some experts believe is a more realistic goal.
Clinical trials of the drug have shown that it can reduce alcohol consumption by approximately 60% after six months treatment, equating to an average reduction of nearly one bottle of wine per day.
In March last year, data was published from two Phase III trials, ESENSE 1 and ESENSE 2, showing that the mean number of heavy drinking days decreased from 19 to 7 days/month and 20 to 7 days/month, while TAC fell from 85 to 43g/day and from 93 to 30g/day at month six. However, the placebo effect was also strong in the studies.
According to Anders Gersel Pedersen, Executive Vice President and Head of Research & Development at Lundbeck, Selincro “represents the first major innovation in the treatment of alcohol dependence in many years,” and he added that its approval “is exciting news for the many patients with alcohol dependence who otherwise may not seek treatment”.
Alcohol dependence is considered a major public health concern, and yet it is both underdiagnosed and undertreated, highlighting the urgent need for better management of the condition.
In Europe, more than 90% of the 14 million patients with alcohol dependence are not receiving treatment, but research suggests that treating just 40% of these would save 11,700 lives each year.
The Danish firm said it expects to launch Selincro in its first markets in mid-2013, and that it will provide the drug as part of “a new treatment concept that includes continuous psychosocial support focused on the reduction of alcohol consumption and treatment adherence”.
Nalmefene (Revex), originally known as nalmetrene, is an opioid receptor antagonistdeveloped in the early 1970s, and used primarily in the management of alcoholdependence, and also has been investigated for the treatment of other addictions such aspathological gambling and addiction to shopping.
Nalmefene is an opiate derivative similar in both structure and activity to the opiate antagonist naltrexone. Advantages of nalmefene relative to naltrexone include longer half-life, greater oral bioavailability and no observed dose-dependent liver toxicity. As with other drugs of this type, nalmefene can precipitate acute withdrawal symptoms in patients who are dependent on opioid drugs, or more rarely when used post-operatively to counteract the effects of strong opioids used in surgery.
Nalmefene differs from naltrexone by substitution of the ketone group at the 6-position of naltrexone with a methylene group (CH2), which considerably increases binding affinity to the μ-opioid receptor. Nalmefene also has high affinity for the other opioid receptors, and is known as a “universal antagonist” for its ability to block all three.
- US patent 3814768, Jack Fishman et al, “6-METHYLENE-6-DESOXY DIHYDRO MORPHINE AND CODEINE DERIVATIVES AND PHARMACEUTICALLY ACCEPTABLE SALTS”, published 1971-11-26, issued 1974-06-04
- Barbara J. Mason, Fernando R. Salvato, Lauren D. Williams, Eva C. Ritvo, Robert B. Cutler (August 1999). “A Double-blind, Placebo-Controlled Study of Oral Nalmefene for Alcohol Dependence”. Arch Gen Psychiatry 56 (8): 719.
- Clinical Trial Of Nalmefene In The Treatment Of Pathological Gambling
- http://www.fda.gov/cder/foi/label/2000/20459S2lbl.pdf
- “Efficacy of Nalmefene in Patients With Alcohol Dependence (ESENSE1)”. “Lundbeck submits Selincro in EU; Novo Nordisk files Degludec in Japan”. thepharmaletter. 22 December 2011.
- Nalmefene Hydrochloride Drug Information, Professional
Phase 3 Amicus in collaboration with GlaxoSmithKline (GSK) is developing the investigational pharmacological chaperone migalastat HCl for the treatment of Fabry disease
 CAS Number:75172-81-5
CAS Number:75172-81-5-
3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride (1:1), (2R,3S,4R,5S)-
- Molecular Structure:

- Formula:C6H14ClNO4
- Molecular Weight:199.63
- Synonyms:3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride, (2R,3S,4R,5S)- (9CI);3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride, [2R-(2a,3a,4a,5b)]-;Migalastat hydrochloride;Galactostatin hydrochloride;(2S,3R,4S,5S)-2-(hydroxymethyl)piperidine-3,4,5-triol hydrochloride;
- Melting Point:260 °C
- Boiling Point:382.7 °C at 760 mmHg
- Flash Point:185.2 °C
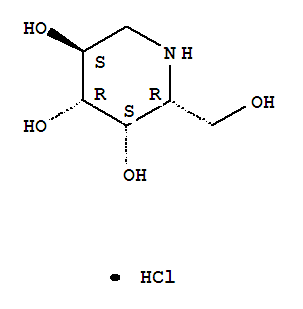
end feb 2013
About Amicus Therapeutics
Amicus Therapeutics is a biopharmaceutical company at the forefront of therapies for rare and orphan diseases. The Company is developing orally-administered, small molecule drugs called pharmacological chaperones, a novel, first-in-class approach to treating a broad range of human genetic diseases. Amicus’ late-stage programs for lysosomal storage disorders include migalastat HCl monotherapy in Phase 3 for Fabry disease; migalastat HCl co-administered with enzyme replacement therapy (ERT) in Phase 2 for Fabry disease; and AT2220 co-administered with ERT in Phase 2 for Pompe disease.
About Migalastat HCl
Amicus in collaboration with GlaxoSmithKline (GSK) is developing the investigational pharmacological chaperone migalastat HCl for the treatment of Fabry disease. Amicus has commercial rights to all Fabry products in the United States and GSK has commercial rights to all of these products in the rest of world.
As a monotherapy, migalastat HCl is designed to bind to and stabilize, or “chaperone” a patient’s own alpha-galactosidase A (alpha-Gal A) enzyme in patients with genetic mutations that are amenable to this chaperone in a cell-based assay. Migalastat HCl monotherapy is in Phase 3 development (Study 011 and Study 012) for Fabry patients with genetic mutations that are amenable to this chaperone monotherapy in a cell-based assay. Study 011 is a placebo-controlled study intended primarily to support U.S. registration, and Study 012 compares migalastat HCl to ERT to primarily support global registration.
For patients currently receiving ERT for Fabry disease, migalastat HCl in combination with ERT may improve ERT outcomes by keeping the infused alpha-Gal A enzyme in its properly folded and active form thereby allowing more active enzyme to reach tissues.2 Migalastat HCl co-administered with ERT is in Phase 2 (Study 013) and migalastat HCl co-formulated with JCR Pharmaceutical Co. Ltd’s proprietary investigational ERT (JR-051, recombinant human alpha-Gal A enzyme) is in preclinical development.
About Fabry Disease
Fabry disease is an inherited lysosomal storage disorder caused by deficiency of an enzyme called alpha-galactosidase A (alpha-Gal A). The role of alpha-Gal A within the body is to break down specific lipids in lysosomes, including globotriaosylceramide (GL-3, also known as Gb3). Lipids that can be degraded by the action of α-Gal are called “substrates” of the enzyme. Reduced or absent levels of alpha-Gal A activity leads to the accumulation of GL-3 in the affected tissues, including the kidneys, heart, central nervous system, and skin. This accumulation of GL-3 is believed to cause the various symptoms of Fabry disease, including pain, kidney failure, and increased risk of heart attack and stroke.
It is currently estimated that Fabry disease affects approximately 5,000 to 10,000 people worldwide. However, several literature reports suggest that Fabry disease may be significantly under diagnosed, and the prevalence of the disease may be much higher.
2. Benjamin, et al., Molecular Therapy: April 2012, Vol. 20, No. 4, pp. 717–726.
http://clinicaltrials.gov/show/NCT01458119
http://www.docstoc.com/docs/129812511/migalastat-hcl
| Chemical Name: | DEOXYGALACTONOJIRIMYCIN, HYDROCHLORIDE |
| Synonyms: | DGJ;Amigal;Unii-cly7m0xd20;GALACTOSTATIN HCL;DGJ, HYDROCHLORIDE;Migalastat hydrochloride;Galactostatin hydrochloride;DEOXYGALACTONOJIRIMYCIN HCL;1-DEOXYGALACTONOJIRIMYCIN HCL;1,5-dideoxy-1,5-imino-d-galactitol |

Lumacaftor, VX-809 an experimental drug for the treatment of Late-Stage cystic fibrosis, being developed by Vertex Pharmaceuticals

3-{6-{[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino}-3-methylpyridin-2-yl}benzoic acid
26,FEB 2013
syn at >>>>>>>https://newdrugapprovals.org/2013/07/28/3274/
Vertex Pharmaceuticals announced Tuesday night the design of two phase III studies for its combination therapy to treat the most common form of cystic fibrosis. The studies will each run for six months, so results could be ready as early as the end of 2013 or during first half of 2014.
The studies announced Tuesday will evaluate the two different doses of an experimental medicine VX-809 in combination with Kalydeco. Each study will enroll 500 cystic fibrosis patients randomized to either the VX-809/Kalydeco arms or a placebo for six months of treatment. The studies’ primary endpoint will be the relative improvement in lung function of VX-809/Kalydeco compared to placebo.
Last fall, Vertex presented data from a phase II study demonstrating that a 600 mg dose of VX-809 and Kalydeco worked synergistically to improve lung function in cystic fibrosis patients with the F508del mutation compared to placebo. This same dose combination will be tested in the phase III study along with a higher 800 mg (actually, 400 mg given twice a day) dose of VX-809 plus Kalydeco.
Vertex also announced new data from this phase II study on Tuesday night showing similar lung function improvements between the 800 mg and 600 mg doses of VX-809. For this reason, the higher dose was included in the phase III studies.
Along with the two phase III studies in adult patients, Vertex will also conduct a six-month study of the combination therapy in pediatric patients ages 6 to 11. This study, along with the data from the adult studies, may be used to expand the combination therapy’s approval into younger patients.
In January, FDA anointed Kalydeco and VX-809 with Breakthrough Therapy Designation as part of the agency’s efforts to accelerate the development and approval of drugs for serious and life-threatening disease. Vertex did not say whether Breakthrough Designation played a specific role in the VX-809/Kalydeco phase III program but the relatively short six-month duration of the studies plus the ability to test the combination in children at the same time does accelerate the development of the combination therapy. If the data from the studies are positive, the drugs could be approved sooner than expected and for more patients.
Lumacaftor (USAN, codenamed VX-809) is an experimental drug for the treatment of cystic fibrosis, being developed by Vertex Pharmaceuticals. The drug is designed to be effective in patients that have the F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR), the defective protein that causes the disease. F508del, meaning that the amino acid phenylalanine in position 508 is missing, is found in about 60% of cystic fibrosis patients.[1]
Interim results from a Phase II clinical trial indicate that patients with the most common form of genetic mutation causing cystic fibrosis homozygous F508del had an 8.5% increase in lung function (FEV1) after 56 days on a combination of lumacaftor and ivacaftor (Kalydeco).[2]
- Merk; Schubert-Zsilavecz. (in German)Pharmazeutische Zeitung 156 (37): 24–27.
- Vertex Pharmaceuticals. May 29,2012.
- syn at >>>>>>>https://newdrugapprovals.org/2013/07/28/3274/
- syn at >>>>>>>https://newdrugapprovals.org/2013/07/28/3274/
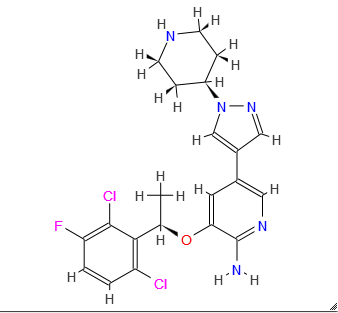
Pfizer Gains China Approval of Kinase-Specific Lung Cancer Drug, Xalkori (crizotinib)

Xalkori, crizotinib,
(PF-02341066)
3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine
Crizotinib; 877399-52-5; Xalkori; PF-2341066; PF-02341066; (R)-crizotinib; 877399-52-5
| Molecular Formula: | C21H22Cl2FN5O |
|---|---|
| Molecular Weight: | 450.336683 g/mol |
Crizotinib an inhibitor of receptor tyrosine kinase for the treatment of non-small cell lung cancer (NSCLC). Verification of the presence of ALK fusion gene is done by Abbott Molecular’s Vysis ALK Break Apart FISH Probe Kit. This verification is used to select for patients suitable for treatment. FDA approved in August 26, 2011.
Crizotinib (1), an anaplastic lymphoma kinase (ALK) receptor tyrosine kinase inhibitor approved by the U.S. Food and Drug Administration in 2011, is efficacious in ALK and ROS positive patients
Feb 25, 2013
Pfizer has been granted China approval for Xalkori (crizotinib), an innovative treatment for patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) that is anaplastic lymphoma kinase (ALK) positive. The ALK-positive variation, which comprises between 3% and 5% of all NSCLC tumors, must be proved by a biomarker test. Pfizer said China’s approval came just eleven months after it submitted a new drug application to the SFDA for Xalkori
Crizotinib (trade name Xalkori,[1] Pfizer), is an anti-cancer drug acting as an ALK (anaplastic lymphoma kinase) and ROS1 (c-ros oncogene 1) inhibitor, approved for treatment of some non-small cell lung carcinoma (NSCLC) in the US and some other countries, and undergoing clinical trials testing its safety and efficacy in anaplastic large cell lymphoma, neuroblastoma, and other advanced solid tumors in both adults and children.[2]
- FDA approves Xalkori with companion diagnostic for a type of late-stage lung cancer. U.S. Food and Drug Administration.http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm269856.htm
- ClinicalTrials.gov NCT00932451 An Investigational Drug, PF-02341066, Is Being Studied In Patients With Advanced Non-Small Cell Lung Cancer With A Specific Gene Profile Involving The Anaplastic Lymphoma Kinase (ALK) Gene
Crizotinib the core structure is a substituted pyridine, the 3 – position of the ether as a chiral center adjacent, so with Mitsunobu reaction to complete, as is a typical Mitsunobu SN2 reaction, the reaction chiral center occurs in reverse, so easy to control, no racemization occurs. Pyridine substituted at position 5 by Suzuki reaction constructed.
Compound 1 The activation of the hydroxyl groups of methanesulfonyl chloride, and then with a 4 – iodopyrazole reaction 2 , 2 to 4 Suzuki reaction conversion can be used, but will generate a large quantity of the reaction product of their coupling, the first 2 converted to a Grignard reagent, and then with a boronic acid ester of 3 reaction 4 .

……………………
http://www.specchemonline.com/articles/view/biocatalyst-breakthroughs#.VTcW9yxabEs
http://www.google.com/patents/WO2014020467A2?cl=en
(R)-3-[l-(2,6-Dichloro-3-fluoro-phenyl)-ethoxy]-5-(l-piperidin-4-yl-lH-py- razol-4-yl)-pyridin-2-ylamine, also known as Crizotinib, is represented by the Formula (I):

Formula (I)
Crizotinib is a potent small-molecule inhibitor of c-Met/HGFR (hepatocyte growth factor receptor) kinase and ALK (anaplastic lymphoma kinase) activity. Enantiomerically pure compound of formula I was first disclosed in US Patent No. 7,858,643. Additionally, the racemate of compound of formula I was disclosed in U.S. patent application 2006/0128724, both of these references discloses similar methods for the synthesis of Compound of Formula I.
Conventionally, the compounds of formula I are prepared by reacting Bis(pinacolato)diboron with protected 5-bromo-3-[l-(2,6-dichloro-3-fluoro-phenyl)-ethoxy]-pyridin-2-ylamine in the presence of Pd catalyst. The obtained product after deprotection is reacted with N- protected 4-(4-bromo-pyrazol-l-yl)-piperidine in the presence of Pd Catalyst. The obtained product is filtered through celite pad and purified by Column Chromatography. The final product of formula I was obtained by deprotection of the purified compound by using HCl/dioxane. US Patent No. 7,858,643 provides enantiomerically pure aminoheteroaryl compounds, particularly aminopyridines and aminopyrazines, having protein tyrosine kinase activity. More particularly, US 7,858,643 describes process for the preparation of 3-[(lR)-l-(2,6- dichloro-3-fluorophenyl)ethoxy]-5-(l-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine. The Scheme is summarized below in Scheme- 1 :


Scheme-1
wherein, “Boc” means tert-butoxycarbonyl; and a) (Boc)2, DMF, Dimethylaminopyridine b) Pd(dppf)Cl2, KOAc, Dichloromethane; c) HC1, Dioxane, Dichloromethane; d) Pd(PPh3)2Cl2, Na2C03, DME/H20; e) 4M HCl/Dioxane, Dichloromethane
A similar process has been disclosed in the U.S. patent application 2006/0128724 for the preparation of Crizotinib. J. Jean Cui et. al. in J. Med. Chem. 2011, 54, 6342-6363, also provides a similar process for the preparation of Crizotinib and its derivatives.
However, above mentioned synthetic process requires stringent operational conditions such as filtration at several steps through celite pad. Also column chromatography is required at various steps which is not only tedious but also results in significant yield loss. Another disadvantage of above process involves extensive use of palladium catalysts, hence metal scavengers are required to remove palladium content from the desired product at various steps which makes this process inefficient for commercial scale.
Yet another disadvantage of above process is the cost of Bis(pinacolato)diboron. This reagent is used in excess in the reaction mixture resulting in considerable cost, especially during large-scale syntheses.
US Patent No. 7,825,137 also discloses a process for the preparation of Crizotinib where Boc protected 4-(4-iodo-pyrazol-l-yl)-piperidine is first reacted with Bis(pinacolato)diboron in the presence of Pd catalyst. The reaction mixture is filtered through a bed of celite and the obtained filtrate is concentrated and purified by silica gel chromatography to give to form tert-butyl-4-[4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l-yl]piperidine-l- carboxylate. To this compound, 5-bromo-3-[l-(2,6-dichloro-3-fluoro-phenyl)-ethoxy]- pyridin-2-ylamine is added in the presence of a Pd catalyst. The reaction mixture is stirred for 16h at 87°C. The reaction mixture is filtered through celite pad and the concentrated filtrate is purified on silica gel column to obtain (4-{6-amino-5-[(R)-l-(2,6-dichloro-3-fluoro- phenyl)-ethoxy]-pyri- din-3-yl}-pyrazol-l-yl)-piperidine-l-carboxylic acid tert-butyl ester of 95% purity. To the solution of resulting compound in dichloromethane 4N HCl/Dioxane is added and thereby getting the reaction suspension is filtered in Buchner funnel lined with filter paper. The obtained solid is dissolved in HPLC water and pH is adjusted to 10 with the addition of Na2C03 Compound is extracted using dichloroform and is purified on a silica gel column by eluting with CH2Cl2 MeOH/NEt3 system to obtain Crizotinib. The scheme is summarized below in scheme 2:

Formula (i) Formula (ii)

Formula (iii) Formula (ii) ula (iv)

Formula (v) Formula (I)
Scheme-2
Preparation of Crizotinib:
To a stirred solution of Tert-butyl 4-(4-{ 6-amino-5-[(li?)-l-(2,6-dichloro-3- fluorophenyl)ethoxy]pyridin-3 -yl } – lH-pyrazol- 1 -yl)piperidine- 1 -carboxylate (material obtained in Example 3) (l.Og, 0.00181 moles) in dichloromethane (-13 ml) at 0°C was added 4.0 M dioxane HQ (6.7 ml, 0.0272 moles). Reaction mixture was stirred at room temperature for 4h. After the completion of reaction monitored by TLC, solid was filtered and washed with dichloromethane (10 ml). The obtained solid was dissolved in water (20 ml); aqueous layer was extracted with dichloromethane (10×2). The pH of aqueous layer was adjusted to 9-10 with Na2C03 and compound was extracted with dichloromethane (10 x 3), combined organic layers were washed with water (20 ml), evaporated under vacuum to get solid product. The solid was stirred with ether (10 ml), filtered off, washed well with ether, dried under vacuum to get Crizotinib.
Yield: 0.45g (55 %)
HPLC Purity: 99.35 %
1HNMR (400 MHz, CDC13) δ: 7.76 (d, J = 1.6 Hz, 1H), 7.56 (s, 1H), 7.49 (s, 1H), 7.30 (dd, J = 9.2 Hz), 7.0 (m, 1H), 6.86 (d, J = 1.6 Hz, 1H), 6.09 ( q, J= 6.8 Hz, 1H), 4.75 (brs, 1H), 4.19 (m, 1H), 3.25 (m, 2H), 2.76 (m, 2H), 2.16 (m, 2H), 1.92 (m, 2H), 1.85 (d, J= 6.8 Hz, 3H), 1.67 (brs, 1H)
…………………………
http://www.sciencedirect.com/science/article/pii/S0040403914000872
Abstract
A novel approach for the synthesis of Crizotinib (1) is described. In addition, new efficient procedures have been developed for the preparation of (S)-1-(2,6-dichloro-3-fluorophenyl)ethanol (2) and tert-butyl 4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)piperidine-1-carboxylate (4), the key intermediates required for the synthesis of Crizotinib.
Graphical abstract

- …………………
- http://www.sciencedirect.com/science/article/pii/S0040403911021745
-
Abstract
4-(4-Iodo-1H-pyrazol-1-yl)piperidine is a key intermediate in the synthesis of Crizotinib. We report a robust three-step synthesis that has successfully delivered multi-kilogram quantities of the key intermediate. The process includes nucleophilic aromatic substitution of 4-chloropyridine with pyrazole, followed by hydrogenation of the pyridine moiety and subsequent iodination of the pyrazole which all required optimization to ensure successful scale-up.
Graphical abstract

……………………

A robust six-step process for the synthesis of crizotinib, a novel c-Met/ALK inhibitor currently in phase III clinical trials, has been developed and used to deliver over 100 kg of API. The process includes a Mitsunobu reaction, a chemoselective reduction of an arylnitro group, and a Suzuki coupling, all of which required optimization to ensure successful scale-up. Conducting the Mitsunobu reaction in toluene and then crystallizing the product from ethanol efficiently purged the reaction byproduct. A chemoselective arylnitro reduction and subsequent bromination reaction afforded the key intermediate 6. A highly selective Suzuki reaction between 6 and pinacol boronate 8, followed by Boc deprotection, completed the synthesis of crizotinib 1.
3-[(1R)-1-(2,6-Dichloro-3-fluorophenyl)ethoxy]-5-[1-(piperidin-4-yl)-1H-pyrazol-4-yl]pyridin-2-amine 1
crizotinib1 (20.7 kg, 80%) as a white solid.
Mp 192 °C;
1H NMR (400 MHz, CDCl3) δ: 7.78 (d, J = 1.8 Hz, 1H), 7.58 (s, 1H), 7.52 (s, 1H), 7.31 (dd, J = 9.0, 4.9 Hz, 1H), 7.06 (m, 1H), 6.89 (d, J = 1.7 Hz, 1H), 6.09 (q, 1H), 4.79 (br s, 2H), 4.21 (m, 1H), 3.26 (m, 2H), 2.78 (m, 2H), 2.17 (m, 2H), 1.90 (m, 2H), 1.87 (d, J = 6.7 Hz, 3H), 1.63 (br s, 1H).
13C NMR (100.6 MHz, CDCl3) δ: 157.5 (d, J = 250.7 Hz), 148.9, 139.8, 137.0, 135.7, 135.6, 129.9, 129.0 (d, J = 3.7 Hz), 122.4, 122.1 (d, J = 19.0 Hz), 119.9, 119.3, 116.7 (d, J = 23.3 Hz), 115.0, 72.4, 59.9, 45.7, 34.0, 18.9.
LC-MS: found m/z 450.0, 451.0, 452.0, 453.0, 454.0, 455.0.
Anal. Calcd for C21H22Cl2FN5O: C, 56.01; H, 4.92; N, 15.55. Found: C, 56.08; H, 4.94; N, 15.80.
Cui, J. J.; Botrous, I.; Shen, H.; Tran-Dube, M. B.; Nambu, M. D.; Kung, P.-P.; Funk, L. A.; Jia, L.; Meng, J. J.; Pairish, M. A.; McTigue, M.; Grodsky, N.; Ryan, K.; Alton, G.; Yamazaki, S.; Zou, H.; Christensen, J. G.; Mroczkowski, B.Abstracts of Papers; 235th ACS National Meeting, New Orleans, LA, United States, April 6–10, 2008.


![3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine NMR spectra analysis, Chemical CAS NO. 877399-52-5 NMR spectral analysis, 3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-29/000/437/336/877399-52-5-1h.png)
![3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine NMR spectra analysis, Chemical CAS NO. 877399-52-5 NMR spectral analysis, 3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-29/000/437/336/877399-52-5-13c.png)
| WO2006021881A2 * | 15 Aug 2005 | 2 Mar 2006 | Pfizer | Pyrazole-substituted aminoheteroaryl compounds as protein kinase inhibitors |
| WO2006021884A2 * | 15 Aug 2005 | 2 Mar 2006 | Pfizer | Enantiomerically pure aminoheteroaryl compounds as protein kinase inhibitors |
| WO2013181251A1 * | 29 May 2013 | 5 Dec 2013 | Ratiopharm Gmbh | Crizotinib hydrochloride salt in crystalline |
| EP2620140A1 * | 26 Jan 2012 | 31 Jul 2013 | ratiopharm GmbH | Crizotinib containing compositions |
-
WO2010048131A1 * Oct 20, 2009 Apr 29, 2010 Vertex Pharmaceuticals Incorporated C-met protein kinase inhibitors WO2011042389A2 * Oct 4, 2010 Apr 14, 2011 Bayer Cropscience Ag Phenylpyri(mi)dinylazoles US7825137 Nov 23, 2006 Nov 2, 2010 Pfizer Inc. Method of treating abnormal cell growth US7858643 Aug 26, 2005 Dec 28, 2010 Agouron Pharmaceuticals, Inc. Crizotinib, a c-Met protein kinase inhibitor anticancer agent; 3-[(R)-1-(2,6-dichloro-3-fluoro-phenyl)-ethoxy]-5-(1-piperidin-4-yl-1H-pyrazol-4-yl)-pyridin-2-ylamine is crizotinib US20060128724 Aug 26, 2005 Jun 15, 2006 Agouron Pharmaceuticals, Inc. Pyrazole-substituted aminoheteroaryl compounds as protein kinase inhibitors 1 J. JEAN CUI J. MED. CHEM. vol. 54, 2011, pages 6342 – 6363 2 ORG. PROCESS RES. DEV. vol. 15, 2011, pages 1018 – 1026 3 * PIETER D. DE KONING ET AL: “Fit-for-Purpose Development of the Enabling Route to Crizotinib (PF-02341066)“, ORGANIC PROCESS RESEARCH & DEVELOPMENT, vol. 15, no. 5, 16 September 2011 (2011-09-16), pages 1018-1026, XP055078841, ISSN: 1083-6160, DOI: 10.1021/op200131n
Pernix Therapeutics Holdings, Inc., a specialty pharmaceutical company, today announced that its subsidiary, Hawthorn Pharmaceuticals, Inc., has received FDA approval of a NDA for Vituz Oral Solution (hydrocodone bitartrate and chlorpheniramine maleate).
Hydrocodone bitartrate is morphinan-6-one, 4,5-epoxy-3-methoxy-17-methyl-, (5α)-, [R-(R*,R*)]-2,3-dihydroxybutanedioate (1:1), hydrate (2:5); also known as 4,5α-Epoxy-3-methoxy-17-methylmorphinan-6-one tartrate (1:1) hydrate (2:5); a fine white crystal or crystalline powder, which is derived from the opium alkaloid, thebaine; and may be represented by the following structural formula:
 |
Hydrocodone Bitartrate
C18H21N03•C4H606•2.5 H20
Molecular weight = 494.5
Chlorpheniramine maleate is 2-pyridinepropanamine, γ-(4-chlorophenyl)-N,N-dimethyl-, (Z)-2-butenedioate (1:1) and has the following chemical structure:
 |
Chlorpheniramine Maleate
C16H19C1N2•C4H404
Molecular weight = 390.86
Feb 28, 2013 – Pernix Therapeutics Holdings, Inc., a specialty pharmaceutical company, today announced that its subsidiary, Hawthorn Pharmaceuticals, Inc., has received U.S. Food and Drug Administration (FDA) approval of a new drug application (NDA) for Vituz Oral Solution (hydrocodone bitartrate and chlorpheniramine maleate). Vituz is indicated for the relief of cough and symptoms associated with upper respiratory allergies or a common cold in adults 18 years of age and older.
Cooper Collins, President and CEO of Pernix, said, “Vituz broadens our cough and cold product line and is our first NDA approved by the FDA, since we closed the acquisition of Hawthorn and Cypress at the end of December 2012. We look forward to the launch of this new treatment option for cough and cold symptoms, which is expected prior to the fall of this year.”
