
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 423)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
MAA, EU, Boehringer Ingelheim and Eli Lilly and Company announce acceptance of EMA application for investigational Type 2 Diabetes treatment empagliflozin

Empagliflozin
26 March 2013
Boehringer Ingelheim and Eli Lilly and Company today announced the European Medicines Agency (EMA) has accepted for review a marketing authorisation application (MAA) for the investigational sodium glucose cotransporter-2 (SGLT2) inhibitor, empagliflozin, for the treatment of Type 2 Diabetes (T2D) in adults. The acceptance of the MAA marks the beginning of the review process in the European Union for this potential oral diabetes treatment.
A New Drug Application (NDA) for empagliflozin was recently submitted to the Food and Drug Administration (FDA) in the United States for the treatment of Type 2 Diabetes mellitus (T2D) in adults.
Empagliflozin is part of a class of drugs being investigated for the reduction of blood glucose levels in adults with T2D. In clinical trials to date, SGLT2 inhibitors have been shown to reduce blood glucose by removing excess glucose independently of beta cell function and insulin resistance.
* Empagliflozin is an investigational compound. Its safety and efficacy have not yet been fully established
About Diabetes
An estimated 371 million people worldwide have Type 1 and Type 2 Diabetes. Type 2 Diabetes is the most common type, accounting for an estimated 90% of all diabetes cases.Diabetes is a chronic condition that occurs when the body either does not properly produce, or use, the hormone insulin.
Empagliflozin is drug which is being investigated in clinical trials for the oral treatment of type 2 diabetes by Boehringer Ingelheim and Eli Lilly and Company.[1][2] It is an inhibitor of the sodium glucose co-transporter-2 (SGLT-2), which is found almost exclusively in the proximal tubules of nephronic components in the kidneys. SGLT-2 accounts for about 90 percent of glucose reabsorption into the blood. Blocking SGLT-2 causes blood glucose to be eliminated through the urine via the urethra.[3][4]
SGLT-2 inhibitors such as empagliflozin reduce blood glucose by blocking glucose reabsorption in the kidney and thereby excreting glucose (i.e., blood sugar) via the urine.[5]
- Grempler R, Thomas L, Eckhardt M, Himmelsbach F, Sauer A, Sharp DE, Bakker RA, Mark M, Klein T, Eickelmann P (January 2012). “Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors”. Diabetes Obes Metab 14 (1): 83–90. doi:10.1111/j.1463-1326.2011.01517.x. PMID 21985634.
- “Empagliflozin”. clinicaltrials.gov. U.S. National Institutes of Health. Retrieved 22 September 2012.
- Nair S, Wilding JP (January 2010). “Sodium glucose cotransporter 2 inhibitors as a new treatment for diabetes mellitus”. J. Clin. Endocrinol. Metab. 95 (1): 34–42. doi:10.1210/jc.2009-0473. PMID 19892839.
- Bays H (March 2009). “From victim to ally: the kidney as an emerging target for the treatment of diabetes mellitus”. Curr Med Res Opin 25 (3): 671–81. doi:10.1185/03007990802710422. PMID 19232040.
- Abdul-Ghani MA, DeFronzo RA (September 2008). “Inhibition of renal glucose reabsorption: a novel strategy for achieving glucose control in type 2 diabetes mellitus”. Endocr Pract 14 (6): 782–90. PMID 18996802.
Kyowa Hakko Kirin, has received approval for NOURIAST tablets 20 mg (istradefylline), a novel antiparkinsonian agent, has been approved for manufacturing and marketing in Japan

Istradefylline (KW-6002) is a selective antagonist at the A2A receptor. It has been found to be useful in the treatment of Parkinson’s disease.[1] Istradefylline reduces dyskinesia resulting from long-term treatment with classical antiparkinson drugs such as levodopa. Istradefylline is an analog of caffeine.
References
Peter A. LeWitt, MD, M. Guttman, James W. Tetrud, MD, Paul J. Tuite, MD, Akihisa Mori, PhD, Philip Chaikin, PharmD, MD, Neil M. Sussman, MD (2008). “Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces off time in Parkinson’s disease: A double-blind, randomized, multicenter clinical trial (6002-US-005)”. Annals of Neurology 63 (3): 295–302. doi:10.1002/ana.21315. PMID 18306243.
TUE 26 MAR 2013
Japan, Kyorin, Pentasa, mesalazine suppositories launched
PENTASA (mesalamine) for oral administration is a controlled-release formulation of mesalamine, an amino-salicylate anti-inflammatory agent for gastrointestinal use. Chemically, mesalamine is 5-amino-2-hydroxybenzoic acid. It has a molecular weight of 153.14.
The structural formula is:

Cellular Biomedicine Group Marks the Launch of China Clinical Trial for TC-DC Therapy for Hepatocellular Carcinoma

March 21, 2013
Cellular Biomedicine Group announced that in the first week of March 2013, the company launched a clinical trial for TC-DC (Tumor Stem Cell Specific Dendritic Cell) therapy for hepatocellular carcinoma (HCC), the most common type of liver cancer. The clinical trial, which is already in progress, is the result of collaboration between CBMG, California Stem Cell (CSC) and Shanghai’s PLA 85 Hospital. It is the first immune cell clinical trial of its kind in China.
CBMG’s joint venture with CSC grants CBMG an exclusive license from CSC to develop and market CSC’s cancer (TC-DC) technology in Greater China. CBMG receives support from CSC’s California-based team of scientists and medical professionals, including CSC’s Dr. Hans Keirstead .
PLA 85 Hospital is a large general teaching hospital with 12 departments, located in Shanghai and has been granted Class A Hospital status at the Tertiary Level (the highest class). The hospital has over 600 beds with more than 200 professors and associate professors, including many well-known experts who are known for their pioneering work in the diagnosis and treatment of tumors. The principal Investigator of the trial and director of the Liver Disease Center of PLA 85 Hospital, ProfessorChengwei Chen , commented, “When I heard of the success this treatment had in clinical trials for other cancers in the U.S., I was very excited at the prospect of the hope it could bring to the millions of patients in China suffering from HCC. I am happy to lead this endeavor to help as many people as we can.” Said Dr. Steve Liu , Chairman of CBMG, “The launch of this trial is a major milestone for all of the physicians, scientists and other professionals at CBMG, CSC and PLA 85 Hospital who have contributed to this work.”
Multinational Contract Research Organization (CRO) CMIC-GCP has been contracted to manage the trial design and minimize delays.
Hepatocellular Carcinoma
Forty-five percent of the world’s HCC patients are in China, with over 300,000 new patients diagnosed every year. Currently the therapies commonly offered to most patients are surgery and local chemotherapy, with a 2-year recurrence rate of 51% and median survival time of 13 months.
CBMG’s research studies the effects of TC-DC (Tumor Stem Cell Specific Dendritic Cell) therapy. Dr. William Cao , President of CBMG said, “In simplified terms, TC-DC therapy takes a sample of the patient’s own dendritic, or immune cells and a sample of the patient’s tumor stem cells and places them together in the lab. The dendritic cell will learn the characteristics of the tumor stem cells, and is reintroduced to the patient’s body, where it can “train” the immune system to fight and destroy the tumor stem cells, which are the root cause of tumor recurrence and metastasis.”
About Cellular Biomedicine Group
Cellular Biomedicine Group, Inc. develops proprietary cell therapies for the treatment of certain degenerative diseases and cancers. Our developmental stem cell, progenitor cell, and immune cell projects are the result of research and development by scientists and doctors from China and the United States. Our flagship GMP facility, consisting of eight independent cell production lines, is designed, certified and managed according to U.S. standards. To learn more about CBMG, please visit:www.cellbiomedgroup.com
Daiichi Sankyo Receives Approval in Japan for Manufacture and Marketing of PRALIA ®
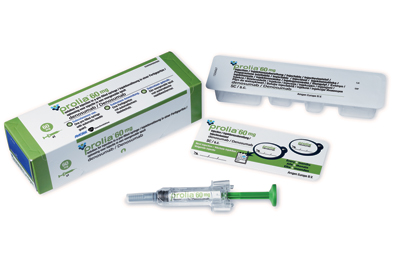
Daiichi Sankyo Receives Approval 25 mar 2013, in Japan for Manufacture and Marketing of PRALIA ® for the osteoporosis treatment PRALIA ®subcutaneous injection 60mg syringe (INN: Denosumab; genetic recombination) for the treatment of osteoporosis.
Denosumab[1] is a fully human monoclonal antibody for the treatment of osteoporosis, treatment-induced bone loss, bone metastases, rheumatoid arthritis, multiple myeloma, and giant cell tumor of bone.[2][3] It was developed by the biotechnology company Amgen.[4]
Denosumab is designed to inhibit RANKL (RANK ligand), a protein that acts as the primary signal for bone removal. In many bone loss conditions, RANKL overwhelms the body’s natural defenses against bone destruction.
In June 2010, denosumab was approved by the U.S. Food and Drug Administration (FDA) for use in postmenopausal women with risk of osteoporosis under the trade name Prolia,[5] and in November 2010, as Xgeva, for the prevention of skeleton-related events in patients with bone metastases from solid tumors.[6] Denosumab is the first RANKL inhibitor to be approved by the FDA.[7] In the summer of 2011 clinical trials were investigating denosumab in giant cell tumors, multiple myeloma with bone metastases, and hypercalcemia of malignancy, and further investigating its dosing and safety.[8]
- Pageau, Steven C. (2009). “Denosumab”. MAbs 1 (3): 210–215. doi:10.4161/mabs.1.3.8592. PMC 2726593.PMID 20065634.
- McClung, Michael R.; Lewiecki, E. Michael; Cohen, Stanley B.; Bolognese, Michael A.; Woodson, Grattan C.; Moffett, Alfred H.; Peacock, Munro; Miller, Paul D. et al. (2006). “Denosumab in Postmenopausal Women with Low Bone Mineral Density”. New England Journal of Medicine 354 (8): 821–31. doi:10.1056/NEJMoa044459. PMID 16495394.
- Ellis, G. K.; Bone, H. G.; Chlebowski, R.; Paul, D.; Spadafora, S.; Smith, J.; Fan, M.; Jun, S. (2008). “Randomized Trial of Denosumab in Patients Receiving Adjuvant Aromatase Inhibitors for Nonmetastatic Breast Cancer”. Journal of Clinical Oncology 26 (30): 4875–82. doi:10.1200/JCO.2008.16.3832.PMID 18725648.
- “Prolia (denosumab)”. Products. Amgen. Retrieved 6 May 2012.
- Matthew Perrone (June 2, 2010). “FDA clears Amgen’s bone-strengthening drug Prolia”. BioScience Technology.
- “Amgen’s Denosumab Cleared by FDA for Second Indication”. 19 Nov 2010.
- “FDA Approves Denosumab for Osteoporosis”. 2 June 2010.
- Russell S. Crawford, BPharm; Morgane C. Diven, PharmD; Laura Yarbro, PharmD (2011). “Denosumab: A Review of Its Pharmacology and Clinical Implications”. Contemporary Oncology 3 (1).
MHLW, sNDA, JAPAN, Daiichi Sankyo has received approval for Anticancer Agent irinotecan hydrochloride hydrate

Irinotecan (Camptosar, Pfizer; Campto, Yakult Honsha) is a drug used for the treatment of cancer.
Irinotecan prevents DNA from unwinding by inhibition of topoisomerase 1. In chemical terms, it is a semisynthetic analogue of the natural alkaloid camptothecin.
Its main use is in colon cancer, in particular, in combination with other chemotherapy agents. This includes the regimen FOLFIRI, which consists of infusional 5-fluorouracil,leucovorin, and irinotecan.
Irinotecan received accelerated approval by the U.S. Food and Drug Administration (FDA) in 1996[1] and full approval in 1998.[2] During development, it was known as CPT-11.
Japan, Shionogi Receives Marketing and Manufacturing Approval of a Drug for Lipodystrophy,METRELEPTIN for Subcutaneous Injection

Metreleptin, an analog of the human hormone leptin, is a unique potential therapy for certain metabolic disorders in patients with rare forms of inherited or acquired lipodystrophy. Lipodystrophy is a very rare condition characterized by loss of subcutaneous fat.
Metreleptin is being studied as a potential therapy for certain metabolic disorders in patients with inherited or acquired lipodystrophy. Metreleptin is believed to work by reducing fat accumulation in organs, caused by the disease, thereby improving insulin sensitivity. Clinical studies have been conducted by investigators at the National Institutes of Health (NIH) and other academic institutions in the US, Europe, and Japan to determine whether metreleptin can improve glycemic control and hypertriglyceridemia in patients with lipodystrophy.
In April 2012, Amylin completed its Biologics License Application (BLA) for metreleptin to treat diabetes and/or hypertriglyceridemia (high levels of triglycerides in the bloodstream) in patients with rare forms of lipodystrophy and requested Priority Review by the FDA.
If approved, metreleptin would be the first therapy indicated specifically for the treatment of diabetes and/or hypertriglyceridemia in patients with inherited or acquired lipodystrophy, and the first approved therapeutic use of a leptin analog.
About Lipodystrophy
Lipodystrophy is a life-threatening, “ultra orphan” rare disease that is estimated to impact a few thousand people worldwide, often with an early age of onset, for which there is a significant unmet medical need. There are currently no approved drugs that treat the underlying cause of the disease.
Fat tissue is a major endocrine organ producing important metabolic hormones such as leptin. People with lipodystrophy lack the required fat tissue for normal metabolic function. This can be partial, affecting select areas of the body, or generalized, affecting nearly the entire body. A lack of fat tissue can lead to relative deficiency of leptin.
Without adequate leptin function, the metabolic system, which regulates food intake and the storage and break-down of dietary fat and carbohydrates, falls out of balance. As a result, fat accumulates in the blood and organs such as liver and muscle, which can lead to life-threatening complications including insulin-resistant diabetes, hypertriglyceridemia (high levels of triglycerides in the bloodstream), acute pancreatitis, and hepatic steatosis or steatohepatitis, also known as fatty liver disease. There are no approved drugs that address the underlying relative leptin deficiency that is believed to contribute in large part to the metabolic abnormalities that occur in lipodystrophy. Currently available therapies for diabetes and hypertriglyceridemia are often rendered marginally effective due to the severity of the condition.
NOVARTIS TOBI Podhaler receives FDA approval for cystic fibrosis patients with Pseudomonas aeruginosa

Tobramycin
MAR 22.2013, FDA approves TOBI Podhaler to treat a type of bacterial lung infection in cystic fibrosispatients
The U.S. Food and Drug Administration today approved TOBI Podhaler (tobramycininhalation powder) for the management of cystic fibrosis patients with Pseudomonas aeruginosa, a bacterium that causes lung infections.
Cystic fibrosis is a genetic disease that affects about 30,000 pediatric and adult patients in the United States. Cystic fibrosis causes the body to produce thick, sticky mucus that builds up in the lungs and blocks airways. The buildup of mucus makes it easy for bacteria like P. aeruginosa to grow and cause a chronic lung infection that, over time, can severely damage the lungs. Many patients with cystic fibrosis are treated with antibiotics using a nebulizer machine.
Tobramycin is an aminoglycoside antibiotic derived from Streptomyces tenebrarius and used to treat various types of bacteria infections, particularly Gram-negative infections. It is especially effective against species of Pseudomonas.
TOBI Podhaler, a plastic, handheld inhaler device, contains a dry powder formulation of tobramycin, an antibiotic used to treat P. aeruginosa infection. The powder is inhaled twice daily using the Podhaler device for 28 days. Patients should then stop TOBI Podhaler therapy for 28 days before resuming again.
“Today’s approval broadens the available delivery mechanism options for patients withcystic fibrosis who require treatment for P. aeruginosa,” said Edward Cox, M.D., M.P.H, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research. “This product is the first dry powder antibacterial drug delivered with a handheld dry powder inhaler.”
Florbetaben (18F), FDA and EMA accept NDA and MAA for Piramal‘s Alzheimer’s imaging agent

Florbetaben (18F)
PHOTO CREDIT-KEGG
902143-01-5 cas no
(18F-AV-1/ZK; BAY-94-9172; 18F-BAY-94-9172; ZK-6013443)

Mr Ajay Piramal, Chairman, Piramal Healthcare
Imaging with amyloid-β PET can potentially aid the early and accurate diagnosis of Alzheimer’s disease. Florbetaben (¹⁸F) is a promising ¹⁸F-labelled amyloid-β-targeted PET tracer in clinical development. We aimed to assess the sensitivity and specificity of florbetaben (¹⁸F) PET in discriminating between patients with probable Alzheimer’s disease and elderly healthy controls.
METHODS:
We did a multicentre, open-label, non-randomised phase 2 study in 18 centres in Australia, Germany, Switzerland, and the USA. Imaging with florbetaben (¹⁸F) PET was done on patients with probable Alzheimer’s disease (age 55 years or older, mini-mental state examination [MMSE] score=18-26, clinical dementia rating [CDR]=0·5-2·0) and age-matched healthy controls (MMSE ≥ 28, CDR=0). Our primary objective was to establish the diagnostic efficacy of the scans in differentiating between patients with probable disease and age-matched healthy controls on the basis of neocortical tracer uptake pattern 90-110 min post-injection. PET images were assessed visually by three readers masked to the clinical diagnosis and all other clinical findings, and quantitatively by use of pre-established brain volumes of interest to obtain standard uptake value ratios (SUVRs), taking the cerebellar cortex as the reference region. This study is registered with ClinicalTrials.gov, number NCT00750282.
FINDINGS:
81 participants with probable Alzheimer’s disease and 69 healthy controls were assessed. Independent visual assessment of the PET scans showed a sensitivity of 80% (95% CI 71-89) and a specificity of 91% (84-98) for discriminating participants with Alzheimer’s disease from healthy controls. The SUVRs in all neocortical grey-matter regions in participants with Alzheimer’s disease were significantly higher (p < 0·0001) compared with the healthy controls, with the posterior cingulate being the best discriminator. Linear discriminant analysis of regional SUVRs yielded a sensitivity of 85% and a specificity of 91%. Regional SUVRs also correlated well with scores of cognitive impairment such as the MMSE and the word-list memory and word-list recall scores (r -0·27 to -0·33, p ≤ 0·021). APOE ɛ4 was more common in participants with positive PET images compared with those with negative scans (65%vs 22% [p=0·027
MAR 21 2013
Piramal Imaging SA, a division of Piramal Enterprises, today announced that the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have accepted its applications for review of the investigational PET amyloid imaging agent [18F] florbetaben. A New Drug Application (NDA) was submitted to the U.S. Food and Drug Administration (FDA) and a Marketing Authorization Application to the EMA for [18F] florbetabenuse in the visual detection of beta-amyloid in the brains of adultswith cognitive impairment who are being evaluated for Alzheimer’s disease and other causes of cognitive decline.[18F] florbetaben binds to beta-amyloid plaques in the human brain, a hallmark characteristic in Alzheimer’s disease.
Today, Alzheimer’s disease is usually diagnosed after a person with a cognitive impairment undergoes an extensive clinical examination which typically includes family and medical history, physical and neurological examinations, laboratory tests, and imaging procedures such as computed tomography (CT) and magnetic resonance imaging (MRI) scans. Still, a definitive diagnosis of Alzheimer’s disease can only be made after death where an autopsy can reveal the presence of beta-amyloid plaques and neurofibrillary tangles in the brain. However, post-mortem studies looking for accumulations of beta-amyloid in the brain have shown that 10 to 30 percent of diagnoses based on clinical examinations are incorrect. [18F] florbetaben is being studied to determine its potential ability to detect beta-amyloid plaquesin living subjects with cognitive impairment.
FLORBETABEN F18
Diagnostic radiopharmaceutical
1. Benzenamine, 4-[(1E)-2-[4-[2-[2-[2-(fluoro-18F)ethoxy]ethoxy]ethoxy]phenyl]
ethenyl]-N-methyl-
2. 4-{(1E)-2-(4-{2-[2-(2-[18F]fluoroethoxy)ethoxy]ethoxy}phenyl)eth- 1-en-1-yl}-N-methylaniline
C21H26[18F]NO3
358.5
Bayer Healthcare
UNII-TLA7312TOI
CAS REGISTRY NUMBER 902143-01-5
https://www.ama-assn.org/resources/doc/usan/florbetaben-f18.pdf

4-[(E)-2-(4-{2-[2-(2-fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline has been labeled with [F-18]fluoride and is claimed by patent application WO2006066104 and members of the corresponding patent family.

4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
The usefulness of this radiotracer for the detection of Αβ plaques have been reported in the literature (W. Zhang et al., Nuclear Medicine and Biology 32 (2005) 799-809; C. Rowe et al., Lancet Neurology 7 (2008) 1 -7).
The synthesis of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)- vinyl]-N-methylaniline has been described before:
a) W. Zhang et al., Nuclear Medicine and Biology 32 (2005) 799-809.

4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
4 mg precursor 2a (2-[2-(2-{4-[(E)-2-{4-[(tert-butoxycarbonyl)(methyl)amino]- phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl methanesulfonate) in 0.2 mL
DMSO were reacted with [F-18]fluoride/kryptofix/potassium carbonate complex. The intermediate was deprotected with HCI and neutralized with
NaOH. The mixture was extracted with ethyl acetate. The solvent was dried and evaporated, the residue was dissolved in acetonitrile and purified by semi-preparative HPLC. 20% (decay corrected), 1 1 % (not corrected for decay) 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N- methylaniline were obtained in 90 min.
WO2006066104
4 mg precursor 2-[2-(2-{4-[(E)-2-{4-[(tert-butoxycarbonyl)(methyl)amino]- phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl methanesulfonate in 0.2 mL DMSO were reacted with [F-18]fluoride/kryptofix/potassium carbonate complex. The intermediates was deprotected with HCI and neutralized with NaOH. The mixture was extracted with ethyl acetate. The solvent was dried and evaporated, the residue was dissolved in acetonitrile and purified by semi- preparative HPLC. 30% (decay corrected), 17% (not corrected for decay) 4- [(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N- methylaniline were obtained in 90 min. to yield N-Boc protected 4-[(E)-2-(4-{2-[2-(2-[F- 18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline. The unreacted perfluorinated precursor was removed using a fluorous phase cartridge.
Deprotection, final purification and formulation to obtain a product, suitable for injection into human is not disclosed. Furthermore, the usefulness (e.g. regarding unwanted F-19/F-18 exchange) of this approach at a higher radioactivity level is not demonstrated. Finally, this method would demand a two-pot setup (first reaction vessel: fluorination, followed by solid-phase- extraction, and deprotection in the second reaction vessel).
However, the focus of the present invention are compounds and methods for an improved “one-pot process” for the manufacturing of 4-[(E)-2-(4-{2-[2-(2- [F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline.
Very recently, further methods have been described:
d) US201001 13763
The mesylate precursor 2a was reacted with [F-18]fluoride species in a solvent mixture consisting of 100 μΙ_ acetonitrile and 500 μΙ_ tertiary alcohol. After fluorination for 10 min at 100-150 °C, the solvent was evaporated. After deprotection (1 N HCI, 5 min, 100-120 °C), the crude product was purified by HPLC (C18 silica, acetonitrile / 0.1 M ammonium formate).
e) H. Wang et al., Nuclear Medicine and Biology 38 (201 1 ) 121 -127
5 mg precursor 2a (2-[2-(2-{4-[(E)-2-{4-[(tert-butoxycarbonyl)(methyl)amino]- phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl methanesulfonate) in 0.5 ml_
DMSO were reacted with [F-18]fluoride/kryptofix/potassium carbonate complex. The intermediate was deprotected with HCI and neutralized with NaOH. The crude product was diluted with acetonitrile / 0.1 M ammonium dformate (6/4) and purified by semi-preparative HPLC. The product fraction was collected, diluted with water, passed through a C18 cartridge and eluted with ethanol, yielding 17% (not corrected for decay) 4-[(E)-2-(4-{2-[2-(2-[F- 18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline within 50 min. In the paper, the conversion of an unprotected mesylate precursor (is described:
5 mg unprotected mesylate precursor (2-{2-[2-(4-{(E)-2-[4- (methylamino)phenyl]vinyl}phenoxy)ethoxy]-ethoxy}ethyl 4- methanesulfonate) in 0.5 ml_ DMSO were reacted with [F- 18]fluoride/kryptofix/potassium carbonate complex. The crude product was diluted with acetonitrile / 0.1 M ammonium formate (6/4) and purified by semi- preparative HPLC. The product fraction was collected, diluted with water, passed through a C18 cartridge and eluted with ethanol, yielding 23% (not corrected for decay) 4-[(E)-2-(4-{2-[2-(2-[F-
18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline within 30 min. Beside the purification by HPLC, a process based on solid-phase-extraction was investigated, but the purity was inferior to that with HPLC purification. So far, one-pot radiolabelings have been performed using a mesylate precursor. It is know, that for F-18 labeling of stilbenes, mesylates have advantages over corresponding tosylates by providing more clean reactions with less amount of by-products (W. Zhang et al. Journal of Medicinal Chemistry 48 (2005) 5980- 5988), whereas the purification starting from the tosylate precursor was tedious and time consuming resulting in a low yield.
In contrast to this teaching of the prior art, we found advantages of tosylate and further arylsulfonate precursors for 4-[(E)-2-(4-{2-[2-(2-[F- 18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline compared to the corresponding mesylate. Less non-radioactive by-products that eluted close to the retention time of 4-[(E)-2-(4-{2-[2-(2-[F-
18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline were found in the crude products if arylsulfonate precursors were used (Example 2 – Example 6) compared to the crude mixture that was obtained after conversion of the mesylate precursor (Example 1 ).
The favorable by-product profile after radiolabeling of tosylate precursor 2b (Figure 10) compared to the radiolabeling of mesylate precursor 2a (Figure 7) supported an improved cartridge based purification (Example 8, Example 9).
…………………
The term “F-18” means fluorine isotope 18F. The term”F-19″ means fluorine isotope 19F. EXAMPLES
Example 1 Radiolabeling of mesylate precursor 2a

4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
Radiolabeling was performed on a remote controlled synthesis module (Tracerlab FXN). Precursor 2a (2 mg) in 0.5 mL DMSO + 0.5 mL acetonitrile was treated with dried potassium carbonate/kryptofix/[F-18]fluoride complex for 6 min at 100 °C. 1 M HCI (1 mL) + 10 mg ascorbic acid was added and the mixture was heated for 4 min at 100 °C. 2M NaOH (0.5 mL), water (6 mL) and ethanol (1 mL) were added and the crude mixture was trapped on a C18 cartridge. The crude product mixture was eluted with acetonitrile and diluted with 0.1 M ammonium formiat buffer (1 mL) + 5 mg ascorbic acid. A sample of the crude product was taken and analyzed by analytical HPLC (Figure 1 ). After purification by semi- preparative HPLC, the product was diluted with water + 5 mg ascorbic acid, trapped on a C18 cartridge and eluted with 1 mL ethanol.
Yield of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)-vinyl]-N- methylaniline: 21 % (corrected for decay).
Example 2 Synthesis and radiolabeling of tosylate precursor 2b


4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
4-Dimethylaminopyridine (26.7 mg) and triethylamine (225 μΙ_) were added to a solution of 1 .0 g terf-butyl {4-[(E)-2-(4-{2-[2-(2- hydroxyethoxy)ethoxy]ethoxy}phenyl)vinyl]phenyl}methylcarbamate (4) in dichloromethane (12 mL) at 0 °C. A solution of p- toluenesulfonyl chloride (417 mg) in dichloromethane (13.5 mL) was added at 0 °C. The resulting mixture was stirred at room temperature over night. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography (silica, 0- 80% ethyl acetate in hexane). 850 mg 2b were obtained as colorless solid.
1 H NMR (300 MHz, CDCI3) δ ppm 1 .46 (s, 9 H), 2.43 (s, 3 H), 3.27 (s, 3 H), 3.59-3.73 (m, 6 H), 3.80- 3.86 (m, 2 H), 4.05-4.19 (m, 2 H), 6.88-7.05 (m, 4 H), 7.21 (d, J = 8.3 Hz, 2 H), 7.32 (d, J = 8.3 Hz, 2 H), 7.39-7-47 (m, 4 H), 7.80 (d, J = 8.3 Hz, 2 H). MS (ESIpos): m/z = 612 (M+H)+
Radiolabeling was performed on a remote controlled synthesis module (Tracerlab FXN). Precursor 2b (2 mg) in 0.5 mL DMSO + 0.5 mL acetonitrile was treated with dried potassium carbonate/kryptofix/[F-18]fluoride complex for 6 min at 100 °C. 1 M HCI (1 mL) + 10 mg ascorbic acid was added and the mixture was heated for 4 min at 100 °C. 2M NaOH (0.5 mL), water (6 mL) and ethanol (1 mL) were added and the crude mixture was trapped on a C18 cartridge. The crude product mixture was eluted with acetonitrile and diluted with 0.1 M ammonium formiat buffer (1 mL) + 5 mg ascorbic acid. A sample of the crude product was taken and analyzed by analytical HPLC (Figure 2). After purification by semi- preparative HPLC, the product was diluted with water + 5 mg ascorbic acid, trapped on a C18 cartridge and eluted with 1 mL ethanol.
Yield of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)-vinyl]-N- methylaniline: 25% (corrected for decay).
Example 3 Synthesis and radiolabeling of 2c (2-[2-(2-{4-[(E)-2-{4-[(tert- butoxycarbonyl)(methyl)amino]phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl
4-bromobenzenesulfonate)

4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline To a stirred solution of 100 mg (0,219 mmol) tert-butyl-{4-[(E)-2-(4-{2-[2-(2- hydroxyethoxy)ethoxy]ethoxy}phenyl)vinyl]phenyl}methylcarbamate
(WO2006/066104) in 2 mL THF was added a solution of 140 mg (0.548 mmol) 4-brombenzene sulfonylchlorid in 3 mL THF drop by drop. The reaction mixture was cooled to 0°C. 156.8 mg (1 .1 mmol) potassium trimethylsilanolat was added. The milky suspension was stirred at 0°C for 2 hours and at 80°C for another 2 hours. The reaction mixture was poured onto ice-cooled water. The aqueous solution was extracted with dichloromethane several times. The combined organic phases were dried with sodium sulphate and concentrated in vacuum. The crude product was purified using silica gel with ethyl acetate/hexane-gradient as mobile phase. The desired product 2c was obtained with 77 mg (0.1 14 mmol, 52.0 % yield).
1 H NMR (300 MHz, CDCI3) δ ppm 1 .39 (s, 10 H) 3.20 (s, 3 H) 3.50 – 3.57 (m, 2 H) 3.57 – 3.61 (m, 2 H) 3.61 – 3.66 (m, 2 H) 3.72 – 3.80 (m, 2 H) 4.02 – 4.10 (m, 2 H) 4.10 – 4.17 (m, 2 H) 6.79 – 6.85 (m, 2 H) 6.91 (d, J=8.10 Hz, 2 H) 7.10 – 7.17 (m, 2 H) 7.32 – 7.41 (m, 5 H) 7.57 – 7.65 (m, 2 H) 7.67 – 7.74 (m, 2 H)
MS (ESIpos): m/z = 676/678 (M+H)+
Radiolabeling was performed on a remote controlled synthesis module (Tracerlab FXN). Precursor 2c (2 mg) in 0.5 mL DMSO + 0.5 mL acetonitrile was treated with dried potassium carbonate/kryptofix/[F-18]fluoride complex for 6 min at 100 °C. 1 M HCI (1 mL) + 10 mg ascorbic acid was added and the mixture was heated for 4 min at 100 °C. 2M NaOH (0.5 mL), water (6 mL) and ethanol (1 mL) were added and the crude mixture was trapped on a C18 cartridge. The crude product mixture was eluted with acetonitrile and diluted with 0.1 M ammonium formiat buffer (1 mL) + 5 mg ascorbic acid. A sample of the crude product was taken and analyzed by analytical HPLC (Figure 3). After purification by semi- preparative HPLC, the product was diluted with water + 5 mg ascorbic acid, trapped on a C18 cartridge and eluted with 1 mL ethanol.
Yield of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)-vinyl]-N- methylaniline: 43% (corrected for decay). Example 4 Synthesis and radiolabeling of 2d (2-[2-(2-{4-[(E)-2-{4-[(tert- butoxycarbonyl)(methyl)amino]phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl
4-(adamantan-1 -yl)benzenesulfonate)

4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
To a stirred solution of 151 mg (0,330 mmol) tert-butyl-{4-[(E)-2-(4-{2-[2-(2- hydroxyethoxy)ethoxy]ethoxy}phenyl)vinyl]phenyl}methylcarbamate
(WO2006/066104), 4.03 mg (0,033 mmol) DMAP und 36.7 mg (363 mmol) triethylamine in 4 mL dichlormethane was added a solution of 97,4 mg (0,313 mmol) 4-(adamantan-1 -yl)benzene sulfonylchloride in 1 mL dichlormethane at 0°C. The reaction mixture was stirred at 0°C for 1 hour and over night at room temperature. 7.3 mg (0,072 mmol) triethylamin und 19.5 mg (0.062 mmol) 4- (adamantan-l -yl)benzenesulfonyl chloride were added to the reaction mixture. The reaction mixture was stirred at room temperature for 3 days. It was concentrated in vacuum. The crude product was purified using silica gel with ethyl acetate/hexane-gradient as mobile phase. The desired product 2d was obtained with 104 mg (0.142 mmol, 43.4% yield).
1 H NMR (300 MHz, CDCI3) δ ppm 1 .51 (s, 9 H), 1 .62 (s, 1 H), 1 .74 – 1 .91 (m, 6 H), 1 .94 (d, J=3.20 Hz, 6 H), 2.16 (br. s., 3 H), 3.31 (s, 3 H), 3.63 – 3.69 (m, 2 H), 3.69 – 3.73 (m, 2 H), 3.76 (dd, J=5.27, 4.52 Hz, 2 H), 3.89 (d, J=4.90 Hz, 2 H), 4.13 – 4.26 (m, 4 H), 6.95 (d, J=8.85 Hz, 2 H), 7.02 (d, J=8.29 Hz, 2 H), 7.25 (d, J=8.48 Hz, 2 H), 7.40 – 7.52 (m, 4 H), 7.55 (m, J=8.67 Hz, 2 H), 7.89 (m, J=8.67 Hz, 2 H)
MS (ESIpos): m/z = 732 (M+H)+
Radiolabeling was performed on a remote controlled synthesis module (Tracerlab FXN). Precursor 2d (2 mg) in 0.5 mL DMSO + 0.5 mL acetonitrile was treated with dried potassium carbonate/kryptofix/[F-18]fluoride complex for 6 min at 100 °C. 1 M HCI (1 mL) + 10 mg ascorbic acid was added and the mixture was heated for 4 min at 100 °C. 2M NaOH (0.5 mL), water (6 mL) and ethanol (1 mL) were added and the crude mixture was trapped on a C18 cartridge. The crude product mixture was eluted with acetonitrile and diluted with 0.1 M ammonium formiat buffer (1 mL) + 5 mg ascorbic acid. A sample of the crude product was taken and analyzed by analytical HPLC (Figure 4). After purification by semi- preparative HPLC, the product was diluted with water + 5 mg ascorbic acid, trapped on a C18 cartridge and eluted with 1 mL ethanol.
Yield of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)-vinyl]-N- methylaniline: 27% (corrected for decay).
Example 5 Synthesis and radiolabeling of 2e (2-[2-(2-{4-[(E)-2-{4-[(tert- butoxycarbonyl)(methyl)amino]phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl
4-cyanobenzenesulfonate)


4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
To a stirred solution of 151 mg (0.330 mmol) tert-butyl-{4-[(E)-2-(4-{2-[2-(2- hydroxyethoxy)ethoxy]ethoxy}phenyl)vinyl]phenyl}methylcarbamate
(WO2006/066104), 4.03 mg (0.033 mmol) DMAP und 36.7 mg (0.363 mmol) triethylamine in 4 mL dichlormethane was added a solution of 63.2 mg (0.313 mmol) 4-cyanobenzenesulfonyl chloride in 1 mL dichlormethane at 0°C. The reaction mixture was stirred over night and concentrated in vacuum. The crude product was purified using silica gel with ethyl acetate/hexane-gradient as mobile phase. The desired product 2e was obtained with 118 mg (0.190 mmol, 57.6 % yield).
1 H NMR (400 MHz, CDCI3) δ ppm 1 .47 (s, 9 H) 3.28 (s, 3 H) 3.58 – 3.63 (m, 2 H) 3.63 – 3.68 (m, 2 H) 3.70 – 3.75 (m, 2 H) 3.81 – 3.87 (m, 2 H) 4.1 1 – 4.18 (m, 2 H) 4.24 – 4.30 (m, 2 H) 6.91 (d, J=8.59 Hz, 2 H) 6.99 (dt, 2 H) 7.22 (d, J=8.34 Hz, 2 H) 7.39 – 7.50 (m, 4 H) 7.83 (m, J=8.59 Hz, 2 H) 7.98 – 8.1 1 (m, 2 H)
MS (ESIpos): m/z = 623 (M+H)+
Radiolabeling was performed on a remote controlled synthesis module (Tracerlab FXN). Precursor 2e (2 mg) in 0.5 mL DMSO + 0.5 mL acetonitrile was treated with dried potassium carbonate/kryptofix/[F-18]fluoride complex for 6 min at 100 °C. 1 M HCI (1 mL) + 10 mg ascorbic acid was added and the mixture was heated for 4 min at 100 °C. 2M NaOH (0.5 mL), water (6 mL) and ethanol (1 mL) were added and the crude mixture was trapped on a C18 cartridge. The crude product mixture was eluted with acetonitrile and diluted with 0.1 M ammonium formiat buffer (1 mL) + 5 mg ascorbic acid. A sample of the crude product was taken and analyzed by analytical HPLC (Figure 5). After purification by semi- preparative HPLC, the product was diluted with water + 5 mg ascorbic acid, trapped on a C18 cartridge and eluted with 1 mL ethanol.
Yield of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)-vinyl]-N- methylaniline: 31 % (corrected for decay).
Example 6 Synthesis and radiolabeling of 2f (2-[2-(2-{4-[(E)-2-{4-[(tert- butoxycarbonyl)(methyl)amino]phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl
2-nitrobenzenesulfonate)


4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- eth oxy} phe nyl )vi ny I] -N -methyla n i I i ne
To a stirred solution of 200 mg (0.437 mmol) tert-butyl-{4-[(E)-2-(4-{2-[2-(2- hydroxyethoxy)ethoxy]ethoxy}phenyl)vinyl]phenyl}methylcarbamate
(WO2006/066104), 5.34 mg (0.044 mmol) DMAP und 47.5 mg (0.470 mmol) triethylamine in 4 mL dichlormethane was added a solution of 92 mg (0,415 mmol) 2-nitrobenzenesulfonyl chloride in 1 mL dichlormethane at 0°C. The reaction mixture was stirred over night and concentrated in vacuum. The crude product was purified with ethyl acetate/hexane-gradient as mobile phase using silica gel. The desired product 2f was obtained with 77 mg (0.1 19 mmol, 59.5 % yield). 1 H NMR (400 MHz, CDCI3) δ ppm 1 .39 (s, 9 H) 3.20 (s, 3 H) 3.55 – 3.63 (m, 4 H) 3.59 (m, 4 H) 3.69 – 3.74 (m, 2 H) 3.75 – 3.80 (m, 2 H) 4.06 (dd, J=5.68, 3.92 Hz,
2 H) 4.32 – 4.37 (m, 2 H) 6.80 – 6.84 (m, 2 H) 6.84 – 6.98 (dt, 2 H) 7.14 (d, J=8.59 Hz, 2 H) 7.35 (d, J=3.03 Hz, 2 H) 7.37 (d, J=2.78 Hz, 2 H) 7.62 – 7.74 (m,
3 H) 8.06 – 8.1 1 (m, 1 H)
Lomitapide (Juxtapid)

FDA approved on 21 December 2012
Lomitapide is a cholesterol-lowering medication. It reduces blood levels of “bad” cholesterol, such as low-density lipoprotein (LDL) or non-high-density liproprotein (non-HDL), as well as a protein that carries bad cholesterol in the blood. Lomitapide is used together with a low-fat diet and other treatments to lower total cholesterol in people with homozygous familial hypercholesterolemia (an inherited type of high cholesterol).
It is not known whether lomitapide will lower your risk of heart disease.
What is the most important information I should know about lomitapide?
Do not use lomitapide if you are pregnant. Some medicines can interact with lomitapide and should not be used at the same time. Tell your doctor about all medicines you use, and those you start or stop using during your treatment with lomitapide.
Grapefruit and grapefruit juice may interact with lomitapide and lead to unwanted side effects. Do…
View original post 1,032 more words
