
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 35)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
SC-52458, FORASARTAN

SC-52458, FORASARTAN
- Molecular FormulaC23H28N8
- Average mass416.522 Da
PHASE 2, PFIZER, HYPERTENSION
145216-43-9[RN]
5-[(3,5-Dibutyl-1H-1,2,4-triazol-1-yl)methyl]-2-[2-(1H-tetrazol-5-yl)phenyl]pyridine
5-[(3,5-dibutyl-1H-1,2,4-triazol-1-yl)methyl]-2-[2-(2H-1,2,3,4-tetrazol-5-yl)phenyl]pyridine
форасартан[Russian][INN], فوراسارتان[Arabic][INN], 福拉沙坦[Chinese][INN]
065F7WPT0B[DBID]
7415[DBID]
UNII-065F7WPT0B[DBID]
SC 52458[DBID]
Type-1 angiotensin II receptor
Forasartan, otherwise known as the compound SC-52458, is a nonpeptide angiotensin II receptor antagonist (ARB, AT1 receptor blocker).[2][3][4][5]
Forasartan, a specific angiotensin II antagonist, is used alone or with other antihypertensive agents to treat hypertension. Forasartan competes with angiotensin II for binding at the AT1 receptor subtype. As angiotensin II is a vasoconstrictor which also stimulates the synthesis and release of aldosterone, blockage of its effects results in a decreases in systemic vascular resistance.
Indications
Forasartan is indicated for the treatment of hypertension[6] and, similar to other ARBs, it protects the kidneys from kidney blood vessel damage caused by increased kidney blood pressure by blocking renin–angiotensin system activation.[7]
Administration
Forasartan is administered in the active oral form [6] which means that it must go through first pass metabolism in the liver. The dose administered ranges between 150 mg-200 mg daily.[6] Increasing to more than 200 mg daily does not offer significantly greater AT1 receptor inhibition.[6] Forasartan is absorbed quickly in the GI, and within an hour it becomes significantly biologically active.[6] Peak plasma concentrations of the drug are reached within one hour.[6]
Contraindications
Negative side effects of Forasartan are similar to other ARBs, and include hypotension and hyperkalemia.[8] There are no drug interactions identified with forasartan.[6]
Bioorganic & Medicinal Chemistry Letters (1994), 4(1), 99-104
PATENT
EP508445
https://worldwide.espacenet.com/patent/search/family/024755845/publication/EP0508445A1?q=EP508445A1
PATENT
WO1992018092
Example 2
2-[5-[(3,5-dibutyl-1H-1,2,4-triazol-1-yI)methyl]- 2-pyridinyl]benzoic acid
Step 1 : Preparation of 2-bromo-5-picoline .
A solution of 1500 mL (14 mol) of 48%
hydrobromic acid was cooled to 10 °C and 300 g (2.8 mol) of 2-amino-5-picoline (Aldrich) was added slowly. The
solution was maintained at or below 0 °C while 450 mL (8.8 mol) of bromime was added dropwise. After the bromine addition was complete, a solution of 500 g (7.3 mol) of sodium nitrite in 1000 mL of water was added slowly over 6 h. The reaction pH was adjusted by the careful addition of 1500 mL (56 mol) of 50% sodium hydroxide at such a rate that the temperature was maintained below 30 °C. The product precipitated from the nearly colorless reaction mixture; filtration gave 450 g (94%) of 2-bromo-5-picoline as a yellow powder: mp 38-40 °C; NMR 7.27 (s, 1H), 7.28 (s, 1H), 7.12 (br s, 1H).
Step 2 : Preparation of N-methyl-N-tertbutylbenzamide.
Under nitrogen, 96.7 g (1.1 mol) of N-methyl-N-tertbutylamine and 111 g (1.1 mol) of triethylamine was dissolved in 1050 mL of anhydrous tetrahydrofuran (THF).
The solution was cooled to 0 °C and treated with 140.6 σ (1.0 mol) of benzoyl chloride. The reaction was allowed to slowly warm to ambient temperature and stir overnight.
Filtration and subsequent concentration in vacuo of the filtrate gave the crude product which was purified by sublimation (65 °, 0.2 torr) to give 184 g (96%) of
colorless N-methyl-N-tertbutybenzamide: mp 80.5-82.0 °C; NMR (CDCI3) δ1.52 (s, 9H), 2.87 (s, 3H), 7.34-7.40 (m, 3H), 7.40-7.46 (m, 2H).
Step 3 : Preparation of 2-(N-methyl-N-tertbutylcarboxamido)phenyIboronic acid.
Under nitrogen, a solution of 50.0 g (262 mmol) of N-methyl-N-tertbutylbenzamide from step 2 and 44 ml (2S2 mmol) of tetramethylethylenediamine (TMEDA) in 3350 mL of anhydrous THF was cooled to -78 °C and slowly treated with 262 mmol of sec-butyllithium in cyclohexane. After 1 h at -78 °C, the reaction was treated with 45 mL (393 mmol) of trimethyl borate and allowed to slowly warm to ambient temperature overnight with stirring. The reaction was concentrated in vacuo; the residue was dissolved in IK sodium hydroxide and extracted with methylene chloride. The pH of the aqueous phase was adjusted to six with dilute hydrochloric acid and extracted with methylene chloride; the organic layer was dried (MgSO4) and concentrated in vacuo to give 55.7 g (90%) of a 80:20 mixture of syn/anti isomers of 2-(N-methyl-N-tertbutylcarboxamido)phenyIboronic acid as a pale yellow glass: NMR (CDCI3) δ 1.30 (s, syn C(CH3)3, 7.3H), 1.54 (s, anti 0(0.3)3, 1.7H), 2.81 (s, anti CH3, 0.6H), 2.94 (s, syn CH3, 2.4H), 7.29-7.46 (m, 3H), 7.95-8.01 (m, 1H).
step 4 : Preparation of N-methyl-N-tertbwtyl-2-(5-methyl-2-pyridinyl)benzamide.
Under nitrogen, 4.44 g (25.8 mmcl) cf 2-bromo-5-picoline from step 1 in 60 mL of toluene was treated with 6.75 g (29 mmol) of 2- (N-methyl-N- tertbutylcarboxamido)phenyIboronic acid from step 3, 1.0 g of tetrakis (triphenylphosphine)palladium zero, 26 mL of ethanol, and 29 mL of 2M sodium carbonate; this mixture was heated to reflux and vigorously stirred for 24 h. The reaction was partitioned between water and ether; the organic layer was separated, dried (MgSθ4), and
concentrated in vacuo. Purification by silica gel
chromatography (Waters Prep-500A) using ethyl
acetate/hexane (1:2) gave 6.51 g (90%) of N-methyl-N- tertbutyl-2-(5-methyl-2-pyridinyl)benzamide as an oil : NMR (CDCI3) δ 1.40 (s, 9H), 2.33 (s, 3H), 2.61 (s, 3H), 7.27- 7.33 (m, 1H), 7.35-7.41 (m, 2H), 7.47-7.51 (m, 2H), 7.60- 7.66 (m, 1H), 8.43 (br s, 1H).
Step 5 : Preparation of sodium 2-(5-methyl-2- pyridinyl)benzoate.
Under nitrogen, 6.5 g (23 mmol) of N-methyl-N- tertbutyl-2-(6-methyl-3-pyridinyl)benzamide from step 4 was treated with 65 mL of anhydrous trifluoroacetic acid (TFA) at reflux for 6 h. The reaction was concentrated in vacuo and the residue dissolved in water. The pH was adjusted to 10 with aqueous sodium hydroxide and lyophilized to give the sodium salt of 2- (5-methyl-2-pyridinyl)benzoic acid as a colorless solid: NMR [CDCI3/CF3CO2H (97:3)] δ 2.62 (s, 3H), 7.42-7.48 (m, 1H), 7.67-7.80 (m, 3H), 8.18-8.24 (m, 1H), 8.28 (dd, J=8 and 2 HZ, 1H), 7.67-7.80 (m, 3H), 8.18-8.24 (m, 1H), 8.28 (dd, J=8 and 2 Hz, 1H), 8.61 (s, 1H) ; MS (FAB) m/e (rel intensity) 214 (20), 196 (100); HRMS.
Calc’d for M+H: 214.0868. Found: 214.0846.
step 6 : Preparation of ethyl 2-(5-methyl-2-pyridinyl)benzoate.
Under nitrogen, the crude sodium salt from step 5 was suspended in 50 mL of chloroform and treated with 9 mL (103 mmol) of oxalyl chloride. The reaction was stirred for 72 h, filtered under nitrogen, and concentrated in vacuo; the residue was dissolved in absolute ethanol.
Concentration in vacuo gave 2.0 g (8 mmol) of ethyl 2-(5-methyl-2-pyridinyl)benzoate as a brown oil: NMR (CDCI3) δ 1.09 (t, J=7 Hz, 3H), 2.36 (s, 3H), 4.15 (q, J=7 Hz, 2H), 7.34 (d, J=8 Hz, 1H), 7.38-7.48 (m, 1H), 7.48-7.58 (m, 3H), 7.80 (d, J=8 Hz, 1H), 8.46 (s, 1H).
Step 7 : Preparation of ethyl 2-(5-bromomethyl-2-pyridinyl)benzoate.
Under nitrogen, the crude ethyl 2-(5-methyl-2-pyridinyl)benzoate from step 6 was treated with 1.7 g (9.5 mmol) of NBS and 160 mg (0.66 mmol) of benzoyl peroxide in 145 mL of anhydrous carbon tetrachloride at reflux for 2.5 h. The reaction was filtered under nitrogen and
concentrated in vacuo to give crude ethyl 2-(5-bromomethyl-2-pyridinyl)benzoate; no purification was attempted.
step 8 : Preparation of ethyl 2-[5-[(3,5-dibutyl-1H- 1 , 2 , 4-triazol-1 -yl )methy] 1 -2-pyridinyl ] benzoate .
Under nitrogen, 630 mg (3.5 mmol) of 3,5-dibutyl-1H-1,2,4-triazole from step 3 of Example 1 was added in small portions to 5.4 mmol of sodium hydride in 8 mL of DMF; stirring was continued until hydrogen evolution had ceased. The anion solution was cooled to 0 °C and treated with a solution of the crude ethyl 2-(5-bromomethyl-2-pyridinyl)benzoate from step 7 in 10 mL of DMF. The reaction was stirred at ambient temperature overnight, quench with 1 mL of absolute ethanol, and concentrated in vacuo; the resulting residue was redisolved in methylene chloride, filtered, and reconcentrated in vacuo to give crude ethyl 2-[5-[(3,5-dibutyl-1H-1,2,4-triazol-1-yl)methyl]-2-pyridinyl]benzoate.
step 9 : Preparation of 2- [5- [ (3, 5-dibutyl-1H-1 , 2, 4 -triazol-1-yl)methyl]-2-pyridinyllbenzoic acid.
A 1.0 g sample of the crude ethyl 2-[5-[(3,5-dibutyl-1H-1,2,4-triazol-1-yl)methyl]-2-pyridinyl]benzoate from step 8 in 10 mL of water was treated with 3 mL of 101 aqueous sodium hydroxide and stirred at ambient temperature overnight. The reaction mixture was washed with 30 mL of ether and the pH adjusted to six with dilute hydrochloric acid. Purification by reverse phase chromatography (Waters Deltaprep-3000) using isocratic acetonitrile/water (28:72) (0.05% TFA) gave 5 mg of 2-[5-[(3,5-dibutyl-1H-1,2,4-triazol-1-yl)methyl]-2-pyridinyl]benzoic acid: NMR (D2O + NaO3S(CH2)3 Si(CH3)3] δ 0.80 (t, J=7 Hz, 3H), 0.86 (t, J=7 Hz, 3H), 1.19-1.33 (m, 4H), 1.54-1.68 (m, 4H), 2.65 (t, J=7 Hz, 2H), 2.82 (t, _ϊ=7 Hz, 2H), 5.43 (s, 2H), 7.45-7.59 (m, 5H), 7.64 (dd, J=8 and 2 Hz, 1H), 8.37-8.45 (m, 1H); MS (FAB) m/e (rel intensity) 393 (80), 375 (30), 212 (40), 182 (100); HRMS. Calc’d for M+Li: 399.2373. Found:
399.2374.
Example 3
5-[2-[5-[(3,5-dibutyl-1H-1,2,4-triazol-1-yl)methyl]- 2-pyridinyl]phenyl]-1H-tetrazole
Step 1 : Preparation of 2-bromo-5-bromomethylpyridine.
A solution of 296.3 g (1.72 mol) of 2-bromo-5-picoline from step 1 of Example 2 in 6000 mL of carbon tetrachloride was treated with 306.5 g (1.72 mol) of N-bromosuccinimide (NBS) and 28.3 g (173 mmol) of
azobisisobutyronitrile (AIBN). The reaction was stirred at reflux under nitrogen for 3 h, filtered, and concentrated in vacuo providing 476 g of crude 2-bromo-5-bromomethylpyridine as a brownish yellow solid (NMR indicates that this material is only 60% monobromomethyl product): NMR (CDCI3 δ 4.42 (s, 2H), 7.48 (d, .J=9 Hz, 1H), 7.60 (dd, J=9 and 3 Hz, 1H), 8.37 (d, J=3 Hz, 1H).
Step 2: Preparation of 2-bromo-5-[(3.5-dibutyl-1H-1,2,4-triazol-1-yl)methyl]pyridine.
Under nitrogen, 3.15 g (17 mmol) of solid 3,5-dibutyl-1H-1,2,4-triazole from step 3 of Example 1 was added in small portions to 33 mmol of sodium hydride in 31 ml of dimethylformamide (DMF); stirring was continued until hydrogen evolution had ceased. The anion solution was cooled to 0 °C and treated with a solution of 7.9 g (19 mmol) of crude 2-bromo-5-bromomethylpyridine from step 1 in 10 ml of dry DMF. The reaction was allowed to warm to ambient temperature and stir overnight. Methanol (10 ml) was added to destroy any unreacted sodium hydride and the
DMF was removed in vacuo. The residue was dissolved in ethyl acetate, washed with water, and dried (MgSO4).
Silica gel chromatography (Waters Prep-500A) using ethyl acetate/hexane (60:40) gave 4.8 g (47%) of 2-bromo-5-[(3,5- dibutyl-1H-1,2,4-triazol-1-yl)methyl]pyridine as an oil: NMR (CDCI3) δ 0.88 (t, J=7 Hz, 1H), 0.92 (t, J=7 Hz, 1H), 1.27-1.44 (m, 4H), 1.59-1.76 (m, 4H), 2.60-2.71 (m, 4H), 5.18 (s, 2H), 7.35 (dd, J=8 and 3 Hz), 7.46 (d, J=8 Hz, 1H), 8.23 (d, .1=3 Hz, 1H).
Step 3: Preparation of 5-[2-[5-[(3,5-dibutyl-1H-1,2,4- triazol-1-yl)methyl]-2-pyridinyl]phenyl]-1H-tetrazole.
Under nitrogen, 1.03 g (2.9 mmol) of 2-bromo-5- [(3,5-dibutyl-1H-1,2,4-triazol-1-yl)methyl]pyridine from step 2 and 2.46 g (5.7 mmol) of 2-(N-triphenyImethyltetrazol-5-yl)phenyIboronic acid from step 5 of Example 1 were treated with 1.0 g (0.86 mmol) of tetrakis (triphenyl-phosphine)palladium zero, 15 mL of toluene, 10 mL of ethanol, and 6.3 mL of 2M aqueous sodium carbonate. The reaction mixture was heated to reflux and vigorously stirred overnight. The product was purified by reverse phase chromatography (Waters Deltaprep-3000) using acetonitrile/water (20-40:80-60) (0.05% TFA). The pure fractions (by analytical HPLC) were combined, the
acetonitrile removed in vacuo, the pH adjusted to four with dilute sodium hydroxide, and the resulting suspension extracted 4 times with ether. The extracts were combined, dried (MgSθ4), and concentrated in vacuo to give 340 mg (28%) of 5-[2-[5-[(3,5-dibutyl-1H-1,2,4-triazol-1-yl)methyl]-2-pyridinyl]phenyl-1H-tetrazole as a colorless solid: mp 139-141 °C; NMR (CD3OD) δ 0.90 (t, J=7 Hz, 3H), 0.93 (t, J=7 Hz, 3H), 1.29-1.44 (m, 4H), 1.58-1.75 (m, 4H), 2.65 (t, J=7 Hz, 2H), 2.81 (t, J=7 Hz, 2H), 5.40 (s, 2H), 7.47 (d, J=8 Hz, 1H), 7.61-7.77 (m, 5H), 8.33 (d, J=2 Hz, 1H); MS (FAB) m/e (rel intensity) 417 (100), 208 (30); HRMS. Calc’d for M+H: 417.2515. Found: 417.2527.
PATENT
WO2001076573
////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Pharmacology
The angiotensin II receptor, type 1
Angiotensin II binds to AT1 receptors, increases contraction of vascular smooth muscle, and stimulates aldosterone resulting in sodium reabsorption and increase in blood volume.[9] Smooth muscle contraction occurs due to increased calcium influx through the L-type calcium channels in smooth muscle cells during the plateau component, increasing the intracellular calcium and membrane potential which sustain depolarization and contraction.[10]
Effects
Forasartan is a competitive and reversible ARB that competes with the angiotensin II binding site on AT1[11] and relaxes vascular smooth muscle,[10] resulting in decreased blood pressure. Forasartan has a high affinity for the AT1 receptor (IC50=2.9 +/- 0.1nM).[12] In dogs, it was found to block the pressor response of Angiotensin II with maximal inhibition, 91%.[10] Forasartan administration selectively inhibits L-type calcium channels in the plateau component of the smooth muscle cells, favoring relaxation of the smooth muscle.[10] Forasartan also decreases heart rate by inhibiting the positive chronotropic effect of high frequency preganglionic stimuli.[13]
Scarce use
Even though experiments have been conducted on rabbits,[6] guinea pigs,[10] dogs [14] and humans,[6][13] forasartan is not a popular drug of choice for hypertension due to its short duration of action; forasartan is less effective than losartan.[6] Research demonstrates that forasartan is also significantly less potent than losartan.[6]
See also
References
- ^ Bräse, Stefan; Banert, Klaus (2010). Organic Azides: Syntheses and Applications. New York: Wiley. p. 38. ISBN 978-0-470-51998-1.
- ^ Knox C, Law V, Jewison T, Liu P, Ly S, Frolkis A, et al. (January 2011). “DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs”. Nucleic Acids Research. DrugBank. 39 (Database issue): D1035-41. doi:10.1093/nar/gkq1126. PMC 3013709. PMID 21059682.
- ^ Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, et al. (January 2008). “DrugBank: a knowledgebase for drugs, drug actions and drug targets”. Nucleic Acids Research. 36 (Database issue): D901-6. doi:10.1093/nar/gkm958. PMC 2238889. PMID 18048412.
- ^ Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, et al. (January 2006). “DrugBank: a comprehensive resource for in silico drug discovery and exploration”. Nucleic Acids Research. 34 (Database issue): D668-72. doi:10.1093/nar/gkj067. PMC 1347430. PMID 16381955.
- ^ Olins GM, Corpus VM, Chen ST, McMahon EG, Palomo MA, McGraw DE, et al. (October 1993). “Pharmacology of SC-52458, an orally active, nonpeptide angiotensin AT1 receptor antagonist”. Journal of Cardiovascular Pharmacology. 22 (4): 617–25. doi:10.1097/00005344-199310000-00016. PMID 7505365. S2CID 93468.
- ^ Jump up to:a b c d e f g h i j k Hagmann M, Nussberger J, Naudin RB, Burns TS, Karim A, Waeber B, Brunner HR (April 1997). “SC-52458, an orally active angiotensin II-receptor antagonist: inhibition of blood pressure response to angiotensin II challenges and pharmacokinetics in normal volunteers”. Journal of Cardiovascular Pharmacology. 29 (4): 444–50. doi:10.1097/00005344-199704000-00003. PMID 9156352.
- ^ Naik P, Murumkar P, Giridhar R, Yadav MR (December 2010). “Angiotensin II receptor type 1 (AT1) selective nonpeptidic antagonists–a perspective”. Bioorganic & Medicinal Chemistry. 18 (24): 8418–56. doi:10.1016/j.bmc.2010.10.043. PMID 21071232.
- ^ Ram CV (August 2008). “Angiotensin receptor blockers: current status and future prospects”. The American Journal of Medicine. 121 (8): 656–63. doi:10.1016/j.amjmed.2008.02.038. PMID 18691475.
- ^ Higuchi S, Ohtsu H, Suzuki H, Shirai H, Frank GD, Eguchi S (April 2007). “Angiotensin II signal transduction through the AT1 receptor: novel insights into mechanisms and pathophysiology”. Clinical Science. 112 (8): 417–28. doi:10.1042/cs20060342. PMID 17346243.
- ^ Jump up to:a b c d e Usune S, Furukawa T (October 1996). “Effects of SC-52458, a new nonpeptide angiotensin II receptor antagonist, on increase in cytoplasmic Ca2+ concentrations and contraction induced by angiotensin II and K(+)-depolarization in guinea-pig taenia coli”. General Pharmacology. 27 (7): 1179–85. doi:10.1016/s0306-3623(96)00058-4. PMID 8981065.
- ^ Olins GM, Chen ST, McMahon EG, Palomo MA, Reitz DB (January 1995). “Elucidation of the insurmountable nature of an angiotensin receptor antagonist, SC-54629”. Molecular Pharmacology. 47 (1): 115–20. PMID 7838120.
- ^ Csajka C, Buclin T, Fattinger K, Brunner HR, Biollaz J (2002). “Population pharmacokinetic-pharmacodynamic modelling of angiotensin receptor blockade in healthy volunteers”. Clinical Pharmacokinetics. 41 (2): 137–52. doi:10.2165/00003088-200241020-00005. PMID 11888333. S2CID 13185772.
- ^ Jump up to:a b Kushiku K, Yamada H, Shibata K, Tokunaga R, Katsuragi T, Furukawa T (January 2001). “Upregulation of immunoreactive angiotensin II release and angiotensinogen mRNA expression by high-frequency preganglionic stimulation at the canine cardiac sympathetic ganglia”. Circulation Research. 88 (1): 110–6. doi:10.1161/01.res.88.1.110. PMID 11139482.
- ^ McMahon EG, Yang PC, Babler MA, Suleymanov OD, Palomo MA, Olins GM, Cook CS (June 1997). “Effects of SC-52458, an angiotensin AT1 receptor antagonist, in the dog”. American Journal of Hypertension. 10 (6): 671–7. doi:10.1016/s0895-7061(96)00500-6. PMID 9194514.
| Clinical data | |
|---|---|
| Other names | SC-52458 |
| Pregnancy category | Not assigned |
| Routes of administration | Oral |
| ATC code | C09CA (WHO) |
| Legal status | |
| Legal status | Development halted, never marketed[1] |
| Pharmacokinetic data | |
| Elimination half-life | 1–2 hours |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 145216-43-9 |
| PubChem CID | 132706 |
| DrugBank | DB01342 |
| ChemSpider | 117146 |
| UNII | 065F7WPT0B |
| KEGG | D04243 |
| ChEBI | CHEBI:141552 |
| ChEMBL | ChEMBL315021 |
| CompTox Dashboard (EPA) | DTXSID70162942 |
| Chemical and physical data | |
| Formula | C23H28N8 |
| Molar mass | 416.533 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////////SC-52458, FORASARTAN, форасартан , فوراسارتان , 福拉沙坦 , PHASE 2, PFIZER, HYPERTENSION
CCCCC1=NN(CC2=CN=C(C=C2)C2=CC=CC=C2C2=NNN=N2)C(CCCC)=N1

NEW DRUG APPROVALS
one time
$10.00
Adagrasib
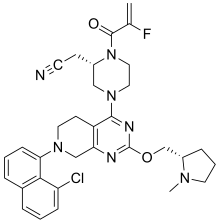
Adagrasib
| Formula | C32H35ClFN7O2 |
|---|---|
| cas | 2326521-71-3 |
| Mol weight | 604.1174 |
| Antineoplastic | |
| Disease | Non-small cell lung cancer |
|---|
| 2022/12/12 |
FDA APPROVED, KRAZATI (Mirati Therapeutics)
- MRTX-849
- MRTX849
- KRAS G12C inhibitor MRTX849
Adagrasib, sold under the brand name Krazati, is an anticancer medication used to treat non-small cell lung cancer.[1][2] Adagrasib is an inhibitor of the RAS GTPase family.[1] It is taken by mouth.[1] It is being developed by Mirati Therapeutics.[1][3]
The most common adverse reactions include diarrhea, nausea, fatigue, vomiting, musculoskeletal pain, hepatotoxicity, renal impairment, dyspnea, edema, decreased appetite, cough, pneumonia, dizziness, constipation, abdominal pain, and QTc interval prolongation.[2] The most common laboratory abnormalities include decreased lymphocytes, increased aspartate aminotransferase, decreased sodium, decreased hemoglobin, increased creatinine, decreased albumin, increased alanine aminotransferase, increased lipase, decreased platelets, decreased magnesium, and decreased potassium.[2]
It was approved for medical use in the United States in December 2022.[1][3]
Synthesis Reference
Fell, Jay B et al. “Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer.” Journal of medicinal chemistry vol. 63,13 (2020): 6679-6693. doi:10.1021/acs.jmedchem.9b02052
Journal of Medicinal Chemistry (2020), 63(13), 6679-6693
PATENT
WO2020101736 https://patents.google.com/patent/WO2020101736A1/en
EXAMPLE 7

2-[(2S)-4-[7-(8-chloro-1-naphthyl)-2-[[(2S)-1-methylpyrrolidin-2-yl]methoxy]-6,8-dihydro-5H- pyrido[3,4-d]pyrimidin-4-yl]-1-(2-fluoroprop-2-enoyl)piperazin-2-yl]acetonitrile
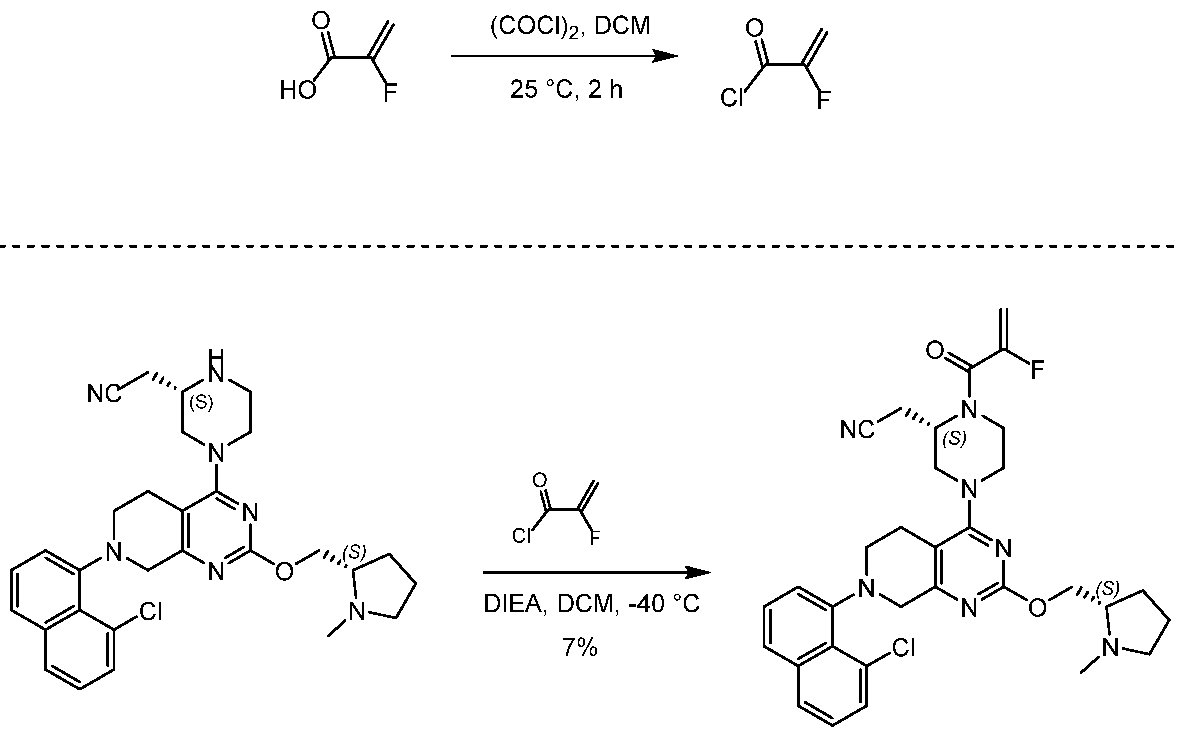
[0432] 2-fluoroprop-2-enoyl chloride. To a solution of 2-fluoroprop-2-enoic acid (400 mg, 4.44 mmol, 1 eq) in DCM (4 mL) was added (COCl)2 (846 mg, 6.66 mmol, 583 µL, 1.5 eq) and DMF (32.5 mg, 444 umol, 34.2 µL, 0.1 eq). The mixture was stirred at 25 °C for 2 hrs. The reaction mixture was concentrated under reduced pressure to remove a part of solvent and give a residue in DCM. Compound 2-fluoroprop-2-enoyl chloride (400 mg, crude) was obtained as a yellow liquid and used into the next step without further purification. [0433] Step A: 2-[(2S)-4-[7-(8-chloro-1-naphthyl)-2-[[(2S)-1- methylpyrrolidin-2-yl]methoxy]- 6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]-1-(2-fluoroprop-2-enoyl)piperazin-2- yl]acetonitrile. To a solution of 2-[(2S)-4-[7-(8-chloro-1-naphthyl)-2-[[(2S)- 1-methylpyrrolidin- 2-yl]methoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-4-yl]piperazin-2-yl]acetonitrile (300 mg, 528 umol, 1 eq, HCl) in DCM (5 mL) was added DIEA (1.73 g, 13.4 mmol, 2.33 mL, 25.4 eq) and 2-fluoroprop-2-enoyl chloride (286 mg, 2.64 mmol, 5 eq) in DCM (5 mL). The mixture was stirred at 0 °C for 1 hour. The reaction mixture was concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (Al2O3, Dichloromethane/Methanol = 10/1 to 10/1). The residue was purified by prep-HPLC (column: Gemini 150 * 25 5u; mobile phase: [water (0.05% ammonia hydroxide v / v) – ACN]; B%: 55% – 85%, 12min). The residue was purified by prep-HPLC (column: Phenomenex Synergi C18 150 * 30mm * 4um; mobile phase: [water (0.225% FA) – ACN]; B%: 20% – 50%, 10.5min). The residue was concentrated under reduced pressure to remove ACN, and then lyophlization. Title compound 2-[(2S)-4-[7-(8-chloro- 1-naphthyl)-2-[[(2S)-1- methylpyrrolidin-2-yl]methoxy]-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin- 4-yl]-1-(2-fluoroprop-2-enoyl)piperazin-2-yl]acetonitrile (EXAMPLE 7, 24.1 mg, 36.7 umol, 7% yield, 99.1% purity, FA) was obtained as a brown solid. [0434] SFC condition: “AD – 3S_3_5_40_3ML Column: Chiralpak AD – 3 100 × 4.6mm I.D., 3um Mobile phase: methanol (0.05% DEA) in CO2 from 5% to 40% Flow rate: 3mL/min Wavelength: 220nm”. [0435] 1H NMR (400 MHz, Acetic) d = 7.82 (d, J = 8.0 Hz, 1H), 7.69 (d, J = 8.0 Hz, 1H), 7.56 (d, J = 7.6 Hz, 1H), 7.49 (t, J = 7.6 Hz, 1H), 7.41 – 7.30 (m, 2H), 5.58 – 5.25 (m, 2H), 5.17 – 4.59 (m, 4H), 4.57 – 4.28 (m, 3H), 4.24 – 3.78 (m, 4H), 3.67 – 3.13 (m, 7H), 3.08 (br d, J = 2.4 Hz, 3H), 2.98 (br d, J = 6.4 Hz, 1H), 2.83 – 2.61 (m, 1H), 2.45 – 2.29 (m, 1H), 2.24 – 2.08 (m, 3H).
PATENT
US20190144444 https://patents.google.com/patent/US20190144444A1/en
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Adagrasib (MRTX849) is an oral, small-molecule KRAS inhibitor developed by Mirati Therapeutics. KRAS mutations are highly common in cancer and account for approximately 85% of all RAS family mutations.5 However, the development of KRAS inhibitors has been challenging due to their high affinity for guanosine triphosphate (GTP) and guanosine diphosphate (GDP), as well as the lack of a clear binding pocket.1 Adagrasib targets KRASG12C, one of the most common KRAS mutations, at the cysteine 12 residue and inhibits KRAS-dependent signalling.2 In a phase I/IB clinical study that included patients with KRASG12C-mutated advanced solid tumors (NCT03785249), adagrasib exhibited anti-tumor activity. The phase II of the same study showed that in patients with KRASG12C-mutated non-small-cell lung cancer (NSCLC), adagrasib was efficient without new safety signals.2,3,6
In February 2022, the FDA accepted a new drug application (NDA) for adagrasib for the treatment of patients with previously treated KRASG12C–positive NSCLC.7 In December 2022, the FDA granted accelerated approval to adagrasib for the treatment of KRASG12C-mutated locally advanced or metastatic NSCLC who have received at least one prior systemic therapy.8,9 Adagrasib joins sotorasib as another KRASG12C inhibitor approved by the FDA.4
Medical uses
Adagrasib is indicated for the treatment of adults with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA approved test, who have received at least one prior systemic therapy.[1][2][4]
History
Approval by the US Food and Drug Administration (FDA) was based on KRYSTAL-1, a multicenter, single-arm, open-label clinical trial (NCT03785249) which included participants with locally advanced or metastatic non-small cell lung cancer with KRAS G12C mutations.[2] Efficacy was evaluated in 112 participants whose disease has progressed on or after platinum-based chemotherapy and an immune checkpoint inhibitor, given either concurrently or sequentially.[2]
The FDA granted the application for adagrasib fast-track, breakthrough therapy, and orphan drug designations.[2]
Research
It is undergoing clinical trials.[5][6][7][8][9][10]
References
- ^ Jump up to:a b c d e f g https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/216340s000lbl.pdf
- ^ Jump up to:a b c d e f g h “FDA grants accelerated approval to adagrasib for KRAS G12C-mutated NSC”. U.S. Food and Drug Administration (FDA). 12 December 2022. Retrieved 14 December 2022.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b “Mirati Therapeutics Announces U.S. FDA Accelerated Approval of Krazati (adagrasib) as a Targeted Treatment Option for Patients with Locally Advanced or Metastatic Non-Small Cell Lung Cancer (NSCLC) with a KRASG12C Mutation” (Press release). Mirati Therapeutics Inc. 12 December 2022. Retrieved 13 December 2022 – via MultiVu.
- ^ https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2022/216340Orig1s000ltr.pdf This article incorporates text from this source, which is in the public domain.
- ^ Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. (January 2020). “The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients”. Cancer Discovery. 10 (1): 54–71. doi:10.1158/2159-8290.CD-19-1167. PMC 6954325. PMID 31658955.
- ^ Fell JB, Fischer JP, Baer BR, Blake JF, Bouhana K, Briere DM, et al. (July 2020). “Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer”. Journal of Medicinal Chemistry. 63 (13): 6679–6693. doi:10.1021/acs.jmedchem.9b02052. PMID 32250617.
- ^ Thein KZ, Biter AB, Hong DS (January 2021). “Therapeutics Targeting Mutant KRAS”. Annual Review of Medicine. 72: 349–364. doi:10.1146/annurev-med-080819-033145. PMID 33138715. S2CID 226242453.
- ^ Christensen JG, Olson P, Briere T, Wiel C, Bergo MO (August 2020). “Targeting Krasg12c -mutant cancer with a mutation-specific inhibitor”. Journal of Internal Medicine. 288 (2): 183–191. doi:10.1111/joim.13057. PMID 32176377.
- ^ Dunnett-Kane V, Nicola P, Blackhall F, Lindsay C (January 2021). “Mechanisms of Resistance to KRASG12C Inhibitors”. Cancers. 13 (1): 151. doi:10.3390/cancers13010151. PMC 7795113. PMID 33466360.
- ^ Jänne PA, Riely GJ, Gadgeel SM, Heist RS, Ou SI, Pacheco JM, et al. (July 2022). “Adagrasib in Non–Small-Cell Lung Cancer Harboring a KRASG12C Mutation”. New England Journal of Medicine. 387 (2): 120–131. doi:10.1056/NEJMoa2204619. PMID 35658005. S2CID 249352736.
External links
- “Adagrasib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03785249 for “Phase 1/2 Study of MRTX849 in Patients With Cancer Having a KRAS G12C Mutation KRYSTAL-1” at ClinicalTrials.gov
///////Adagrasib, KRAZATI, FDA 2022, APPROVALS 2022, MRTX-849, MRTX849, Mirati Therapeutics
[H][C@@]1(COC2=NC3=C(CCN(C3)C3=CC=CC4=C3C(Cl)=CC=C4)C(=N2)N2CCN(C(=O)C(F)=C)[C@@]([H])(CC#N)C2)CCCN1C

NEW DRUG APPROVALS
ONE TIME
$10.00
Nirsevimab
(Heavy chain)
QVQLVQSGAE VKKPGSSVMV SCQASGGLLE DYIINWVRQA PGQGPEWMGG IIPVLGTVHY
GPKFQGRVTI TADESTDTAY MELSSLRSED TAMYYCATET ALVVSETYLP HYFDNWGQGT
LVTVSSASTK GPSVFPLAPS SKSTSGGTAA LGCLVKDYFP EPVTVSWNSG ALTSGVHTFP
AVLQSSGLYS LSSVVTVPSS SLGTQTYICN VNHKPSNTKV DKRVEPKSCD KTHTCPPCPA
PELLGGPSVF LFPPKPKDTL YITREPEVTC VVVDVSHEDP EVKFNWYVDG VEVHNAKTKP
REEQYNSTYR VVSVLTVLHQ DWLNGKEYKC KVSNKALPAP IEKTISKAKG QPREPQVYTL
PPSREEMTKN QVSLTCLVKG FYPSDIAVEW ESNGQPENNY KTTPPVLDSD GSFFLYSKLT
VDKSRWQQGN VFSCSVMHEA LHNHYTQKSL SLSPGK
(Light chain)
DIQMTQSPSS LSAAVGDRVT ITCQASQDIV NYLNWYQQKP GKAPKLLIYV ASNLETGVPS
RFSGSGSGTD FSLTISSLQP EDVATYYCQQ YDNLPLTFGG GTKVEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(Disulfide bridge: H22-H96, H153-H209, H229-L214, H235-H’235, H238-H’238, H270-H330, H376-H434, H’22-H’96, H’153-H’209, H’229-L’214, H’270-H’330, H’376-H’434, L23-L88, L’23-L’88, L134-L194, L’134-L’194)
>Heavy_chain QVQLVQSGAEVKKPGSSVMVSCQASGGLLEDYIINWVRQAPGQGPEWMGGIIPVLGTVHY GPKFQGRVTITADESTDTAYMELSSLRSEDTAMYYCATETALVVSETYLPHYFDNWGQGT LVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFP AVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHTCPPCPA PELLGGPSVFLFPPKPKDTLYITREPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKP REEQYNSTYRVVSVLTVLHQDWLEGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTL PPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLT VDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
>Light_chain DIQMTQSPSSLSAAVGDRVTITCQASQDIVNYLNWYQQKPGKAPKLLIYVASNLETGVPS RFSGSGSGTDFSLTISSLQPEDVATYYCQQYDNLPLTFGGGTKVEIKRTVAAPSVFIFPP SDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLT LSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
Nirsevimab
EMS APPROVED 2022/10/31, Beyfortus, AstraZeneca AB
| Formula | C6494H10060N1708O2050S46 |
|---|---|
| CAS | 1989556-22-0 |
| Mol weight | 146334.5658 |
Monoclonal antibody
Prevention of respiratory syncytial virus infection
- Immunoglobulin g1-kappa, anti-(human respiratory syncytial virus fusion glycoprotein f0 (protein f))human monoclonal antibody.gamma.1 heavy chain (1-456) (human vh (homo sapiens ighv1-69*01(ighd)-ighj4*01 (90.1%)) (8.8.19) (1-126) -homo sapiens ighg1*03
- Immunoglobulin g1, anti-(human respiratory syncytial virus fusion protein)(human monoclonal med18897 .gamma.1-chain), disulfide with monoclonal med18897 .kappa.-chain, dimer
Synthesis Reference
Khan, AA et al. (2020) Dosage regimens for and compositions including anti-rsv antibodies. (U.S. Patent No. 2020/0347120 A1). U.S. Patent and Trademark Office. https://patentimages.storage.googleapis.com/6b/d2/10/a841b66e0c90cf/US20200347120A1.pdf
Nirsevimab, sold under the brand name Beyfortus, is a human recombinant monoclonal antibody with activity against respiratory syncytial virus, or RSV for infants.[2][3] It is under development by AstraZeneca and Sanofi.[2][3] Nirsevimab is designed to bind to the fusion protein on the surface of the RSV virus.[4][5]
The most common side effects reported for nirsevimab are rash, pyrexia (fever) and injection site reactions (such as redness, swelling and pain where the injection is given).[6]
Nirsevimab was approved for medical use in the European Union in November 2022.[1][7]
Nirsevimab (MEDI8897) is a recombinant human immunoglobulin G1 kappa (IgG1ĸ) monoclonal antibody used to prevent respiratory syncytial virus (RSV) lower respiratory tract disease in neonates and infants.6 It binds to the prefusion conformation of the RSV F protein, a glycoprotein involved in the membrane fusion step of the viral entry process, and neutralizes several RSV A and B strains.6,1 Compared to palivizumab, another anti-RSV antibody, nirsevimab shows greater potency at reducing pulmonary viral loads in animal models. In addition, nirsevimab was developed as a single-dose treatment for all infants experiencing their first RSV season, whereas palivizumab requires five monthly doses to cover an RSV season.5 This is due to a modification in the Fc region of nirsevimab that grants it a longer half-time compared to typical monoclonal antibodies.1,6
On November 2022, nirsevimab was approved by the EMA for the prevention of RSV lower respiratory tract disease in newborns and infants during their first RSV season.6
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | F protein of RSV |
| Clinical data | |
| Trade names | Beyfortus |
| Other names | MED-18897, MEDI8897 |
| Routes of administration | Intramuscular |
| ATC code | None |
| Legal status | |
| Legal status | EU: Rx-only [1] |
| Identifiers | |
| CAS Number | 1989556-22-0 |
| PubChem SID | 384585358 |
| DrugBank | DB16258 |
| UNII | VRN8S9CW5V |
| KEGG | D11380 |
| ChEMBL | ChEMBL4297575 |
| Chemical and physical data | |
| Formula | C6494H10060N1708O2050S46 |
| Molar mass | 146336.58 g·mol−1 |
Adverse effects
No major hypersensitivity reactions have been reported, and adverse events of grade 3 or higher were only reported in 8% (77 of 968) of participants in clinical trial NCT02878330.[8][4]
Pharmacology
Mechanism of action
Nirsevimab binds to the prefusion conformation of the RSV fusion protein, i.e. it binds to the site at which the virus would attach to a cell; effectively rendering it useless. It has a modified Fc region, extending the half-life of the drug in order for it to last the whole RSV season.[4]
History
The opinion by the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) is based on data from two randomized, double-blind, placebo-controlled multicenter clinical trials that investigated the efficacy and safety of nirsevimab in healthy preterm (premature) and full-term infants entering their first respiratory syncytial virus (RSV) season.[6] These studies demonstrated that nirsevimab prevents lower respiratory tract infection caused by RSV requiring medical attention (such as bronchiolitis and pneumonia) in term and preterm infants during their first RSV season.[6]
The safety of nirsevimab was also evaluated in a phase II/III, randomized, double‑blind, multicenter trial in infants who were born five or more weeks prematurely (less than 35 weeks gestation) at higher risk for severe RSV disease and infants with chronic lung disease of prematurity (i.e. long-term respiratory problems faced by babies born prematurely) or congenital heart disease.[6] The results of this study showed that nirsevimab had a similar safety profile compared to palivizumab (Synagis).[6]
Society and culture
Legal status
On 15 September 2022, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Beyfortus, intended for the prevention of respiratory syncytial virus (RSV) lower respiratory tract disease in newborns and infants.[9][6] Beyfortus was reviewed under EMA’s accelerated assessment program.[9] The applicant for this medicinal product is AstraZeneca AB.[9] Nirsevimab was approved for medical use in the European Union in November 2022.[1][7]
Research
Nirsevimab is being investigated as an experimental vaccine against respiratory syncytial virus, RSV, in the general infant population.[2][3] The MELODY study is an ongoing, randomized, double-blind, placebo-controlled to evaluate the safety and efficacy of nirsevimab in late preterm and term infants. Initial results have been promising, with nirsevimab reducing LRTI (lower respiratory tract infections) by 74.5% compared to placebo in infants born at term or late preterm.[5][10][11]
Ongoing trials for nirsevimab are:
- “Evaluate the Safety and Efficacy of Nirsevimab in Healthy Preterm and Term Infants in China (CHIMES)”.
- “A Study to Evaluate the Safety and Efficacy of MEDI8897 for the Prevention of Medically Attended Lower Respiratory Tract Infection Due to Respiratory Syncytial Virus in Healthy Late Preterm and Term Infants (MELODY)”.
- “Evaluate the Safety and Tolerability, for Nirsevimab in Immunocompromised Children (MUSIC)”.
References
- ^ Jump up to:a b c “Beyfortus”. Union Register of medicinal products. 3 November 2022. Retrieved 6 November 2022.
- ^ Jump up to:a b c “Nirsevimab demonstrated protection against respiratory syncytial virus disease in healthy infants in Phase 3 trial” (Press release). Sanofi. 26 April 2021. Archived from the original on 27 December 2021. Retrieved 27 December 2021.
- ^ Jump up to:a b c “Nirsevimab MELODY Phase III trial met primary endpoint of reducing RSV lower respiratory tract infections in healthy infants” (Press release). AstraZeneca. 26 April 2021. Archived from the original on 26 December 2021. Retrieved 27 December 2021.
- ^ Jump up to:a b c Griffin MP, Yuan Y, Takas T, Domachowske JB, Madhi SA, Manzoni P, et al. (Nirsevimab Study Group) (July 2020). “Single-Dose Nirsevimab for Prevention of RSV in Preterm Infants”. The New England Journal of Medicine. 383 (5): 415–425. doi:10.1056/NEJMoa1913556. PMID 32726528. S2CID 220876651.
- ^ Jump up to:a b Hammitt LL, Dagan R, Yuan Y, Baca Cots M, Bosheva M, Madhi SA, et al. (March 2022). “Nirsevimab for Prevention of RSV in Healthy Late-Preterm and Term Infants”. The New England Journal of Medicine. 386 (9): 837–846. doi:10.1056/NEJMoa2110275. PMID 35235726. S2CID 247220023.
- ^ Jump up to:a b c d e f “New medicine to protect babies and infants from respiratory syncytial virus (RSV) infection”. European Medicines Agency (EMA) (Press release). 16 September 2022. Archived from the original on 19 September 2022. Retrieved 18 September 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “Beyfortus approved in the EU for the prevention of RSV lower respiratory tract disease in infants”. AstraZeneca (Press release). 4 November 2022. Retrieved 6 November 2022.
- ^ Clinical trial number NCT02878330 at ClinicalTrials.gov
- ^ Jump up to:a b c “Beyfortus: Pending EC decision”. European Medicines Agency (EMA). 15 September 2022. Archived from the original on 19 September 2022. Retrieved 18 September 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Zacks Equity Research (25 March 2022). “Pfizer’s (PFE) RSV Jab Gets Another Breakthrough Therapy Tag”. Nasdaq. Archived from the original on 8 April 2022. Retrieved 8 April 2022.
- ^ “Nirsevimab significantly protected infants against RSV disease in Phase III MELODY trial”. AstraZeneca (Press release). 3 March 2022. Retrieved 6 November 2022.
////////////Nirsevimab, EU 2022, APPROVALS 2022, PEPTIDE, Monoclonal antibody, respiratory syncytial virus infection, ANTIVIRAL, 1989556-22-0, MED-18897, MEDI8897, AstraZeneca AB

NEW DRUG APPROVALS
ONE TIME
$10.00
Olutasidenib

Olutasidenib
- FT-2102
- FT2102
C18H15ClN4O2
354.79
CAS1887014-12-1
Rezlidhia (Forma Therapeutics)
SYN Caravella JA, et al. Structure-Based Design and Identification of FT-2102 (Olutasidenib), a Potent Mutant-Selective IDH1 Inhibitor. J Med Chem. 2020 Feb 27;63(4):1612-1623. doi: 10.1021/acs.jmedchem.9b01423. Epub 2020 Feb 12.
FDA 12/1/2022, To treat adults with relapsed or refractory acute myeloid leukemia with a susceptible isocitrate dehydrogenase-1 (IDH1) mutation, Rezlidhia
Olutasidenib, sold under the brand name Rezlidhia, is an anticancer medication used to treat relapsed or refractory acute myeloid leukemia.[1][2] Olutasidenib is an isocitrate dehydrogenase-1 (IDH1) inhibitor.[1] It is taken by mouth.[1]
Olutasidenib was approved for medical use in the United States in December 2022.[1][2][3][4]
Medical uses
Olutasidenib is indicated for the treatment of adults with relapsed or refractory acute myeloid leukemia with a susceptible isocitrate dehydrogenase-1 (IDH1) mutation as detected by an FDA-approved test.[1][2]
Society and culture
Names
Olutasidenib is the international nonproprietary name.[5]
Olutasidenib is an isocitrate dehydrogenase-1 (IDH1) inhibitor indicated for the treatment of patients with relapsed or refractory acute myeloid leukemia with a susceptible IDH1 mutation as detected by an FDA-approved test.
Olutasidenib (FT-2102) is a selective and potent isocitrate dehydrogenase-1 (IDH1) inhibitor approved by the FDA in December 2022.5,6 It is indicated for the treatment of relapsed or refractory acute myeloid leukemia (AML) in patients with a susceptible IDH1 mutation as determined by an FDA-approved test.5 IDH1 mutations are common in different types of cancer, such as gliomas, AML, intrahepatic cholangiocarcinoma, chondrosarcoma, and myelodysplastic syndromes (MDS), and they lead to an increase in 2-hydroxyglutarate (2-HG), a metabolite that participates in tumerogenesis.1,2 Olutasidenib inhibits the mutated IDH1 specifically, and provides a therapeutic benefit in IDH1-mutated cancers.1,5
Other IDH1 inhibitors, such as ivosidenib, have also been approved for the treatment of relapsed or refractory AML.3,4 Olutasidenib is orally bioavailable and capable of penetrating the blood-brain barrier, and is also being evaluated for the treatment of myelodysplastic syndrome (MDS), as well as solid tumors and gliomas (NCT03684811).4
SYN
https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01423

a Reagents and conditions: (a) DIEA, DMSO, 80−110 °C, 16 h, 67%; (b) (R)-2-methylpropane-2-sulfinamide, CuSO4, 55 °C, DCE, 16 h, 81%; (c) MeMgBr, DCM, −50 to −60 °C, 3 h, 63%; (d) 1 N HCl, dioxane, reflux, 16 h, >98%, 98.4% ee; (e) m-CPBA, CHCl3, reflux, 4 days, 52%; (f) Ac2O, reflux, 3 days, 60%; (g) K2CO3, MeOH, 4 h, 92%; (h) MeI, K2CO3, DMF, 45 min, 67%.
1H NMR (300 MHz,
DMSO-d6) δ 12.07 (s, 1 H), 7.71−7.76 (m, 2 H), 7.51 (dd, J = 8.79,
2.35 Hz, 1 H), 7.31 (d, J = 8.79 Hz, 1 H), 6.97 (d, J = 7.92 Hz, 1 H),
6.93 (d, J = 7.92 Hz, 1 H), 5.95 (d, J = 7.92 Hz, 1 H), 4.62−4.75 (m,
1 H), 3.58 (s, 3 H), 1.50 (d, J = 6.74 Hz, 3 H); 13C NMR (75 MHz,
DMSO-d6) δ 161.0, 155.9, 141.4, 136.6, 135.0, 133.4, 129.8, 126.7,
125.8, 120.1, 119.4, 116.7, 115.1, 104.5, 103.7, 47.4, 34.0, 20.3; LCMS
(method 2) >95% purity; tR 10.18 min; m/z 355, 357 [M + H]+
;
HRMS (ESI) calcd for C18H16ClN4O2 [M + H]+ 355.0962 found
356.0956.
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Trade names | Rezlidhia |
| Other names | FT-2102 |
| License data | US DailyMed: Olutasidenib |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 1887014-12-1 |
| PubChem CID | 118955396 |
| IUPHAR/BPS | 10319 |
| DrugBank | DB16267 |
| ChemSpider | 72380144 |
| UNII | 0T4IMT8S5Z |
| KEGG | D12483 |
| ChEMBL | ChEMBL4297610 |
| PDB ligand | PWV (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C18H15ClN4O2 |
| Molar mass | 354.79 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
References
- ^ Jump up to:a b c d e f https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215814s000lbl.pdf
- ^ Jump up to:a b c d https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2022/215814Orig1s000ltr.pdf This article incorporates text from this source, which is in the public domain.
- ^ “Rigel Announces U.S. FDA Approval of Rezlidhia (olutasidenib) for the Treatment of Adult Patients with Relapsed or Refractory Acute Myeloid Leukemia with a Susceptible IDH1 Mutation”. Rigel Pharmaceuticals, Inc. (Press release). 1 December 2022. Retrieved 2 December 2022.
- ^ “Rigel Announces U.S. FDA Approval of Rezlidhia (olutasidenib) for the Treatment of Adult Patients with Relapsed or Refractory Acute Myeloid Leukemia with a Susceptible IDH1 Mutation” (Press release). Rigel Pharmaceuticals. 1 December 2022. Retrieved 2 December 2022 – via PR Newswire.
- ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 82”. WHO Drug Information. 33 (3). hdl:10665/330879.
Further reading
- Liu X, Gong Y (2019). “Isocitrate dehydrogenase inhibitors in acute myeloid leukemia”. Biomarker Research. 7: 22. doi:10.1186/s40364-019-0173-z. PMC 6806510. PMID 31660152.
- Watts JM, Baer MR, Yang J, Prebet T, Lee S, Schiller GJ, et al. (November 2022). “Olutasidenib alone or with azacitidine in IDH1-mutated acute myeloid leukaemia and myelodysplastic syndrome: phase 1 results of a phase 1/2 trial”. The Lancet Haematology. doi:10.1016/S2352-3026(22)00292-7. PMID 36370742. S2CID 253471380.
External links
- “Olutasidenib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02719574 for “Open-label Study of FT-2102 With or Without Azacitidine or Cytarabine in Patients With AML or MDS With an IDH1 Mutation” at ClinicalTrials.gov
/////////////Olutasidenib, FDA 2022, APPROVALS 2022, Rezlidhia, FT-2102, FT 2102

NEW DRUG APPROVALS
ONE TIME
$10.00
ABY 737
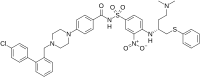
ABT-737
| Molecular Weight | 813.43 |
|---|---|
| Formula | C42H45ClN6O5S2 |
| CAS No. | 852808-04-9 |
ABT-737 is a small molecule drug that inhibits Bcl-2 and Bcl-xL, two members of the Bcl-2 family of evolutionarily-conserved proteins that share Bcl-2 Homology (BH) domains. First developed as a potential cancer chemotherapy,[1] it was subsequently identified as a senolytic (a drug that selectively induces cell death in senescent cells).[2]
The Bcl-2 family is most notable for their regulation of apoptosis, a form of programmed cell death, at the mitochondrion; Bcl-2 and Bcl-xL are anti-apoptotic proteins. Because many cancers have mutations in these genes that allow them to survive, scientists began working to develop drugs that would inhibit this pathway in the 1990s.[1] ABT-737 was one of the earliest of a series of drugs developed by Abbott Laboratories (now Abbvie) to target this pathway, based on their resolution of the 3D structure of Bcl-xL and studies using high-field solution nuclear magnetic resonance (NMR) that revealed how the BH domains of these proteins interacted with their targets.[1]
ABT-737 was superior to previous BCL-2 inhibitors given its higher affinity for Bcl-2, Bcl-xL and Bcl-w. In vitro studies showed that primary cells from patients with B-cell malignancies are sensitive to ABT-737.[3] In animal models, it improved survival, caused tumor regression, and cured a high percentage of mice.[4] In preclinical studies utilizing patient xenografts, ABT-737 showed efficacy for treating lymphoma and other blood cancers.[5]
Unfortunately, ABT-737 is not bioavailable after oral administration, leading to the development of navitoclax (ABT-263) as an orally-available derivative with similar activity on small cell lung cancer (SCLC) cell lines.[1][6] Navitoclax entered clinical trials,[1][6] and showed promise in haematologic cancers, but was stalled when it was found to cause thrombocytopenia (severe loss of platelets), which was discovered to be caused by the platelets’ requirement for Bcl-xL for survival.[1]
Subsequently, it was reported that ABT-737 specifically induces apoptosis in senescent cells in vitro and in mouse models.[2]
ABT-737, a BH3 mimetic, is a potent Bcl-2, Bcl-xL and Bcl-w inhibitor with EC50s of 30.3 nM, 78.7 nM, and 197.8 nM, respectively. ABT-737 induces the disruption of the BCL-2/BAX complex and BAK-dependent but BIM-independent activation of the intrinsic apoptotic pathway. ABT-737 induces autophagy and has the potential for acute myeloid leukemia (AML) research.
PATENT
PATENT
CN113248415
PATENT
US20070015787
Journal of Medicinal Chemistry (2007), 50(4), 641-662
https://pubs.acs.org/doi/10.1021/jm061152t

////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Names | |
|---|---|
| Preferred IUPAC name4-{4-[(4′-Chloro[1,1′-biphenyl]-2-yl)methyl]piperazin-1-yl}-N-(4-{[(2R)-4-(dimethylamino)-1-(phenylsulfanyl)butan-2-yl]amino}-3-nitrobenzene-1-sulfonyl)benzamide | |
| Identifiers | |
| CAS Number | 852808-04-9 |
| 3D model (JSmol) | Interactive image |
| ChEBI | CHEBI:47575 |
| ChemSpider | 9403232 |
| PubChemCID | 11228183 |
| UNII | Z5NFR173NV |
| CompTox Dashboard (EPA) | DTXSID7042641 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C42H45ClN6O5S2 |
| Molar mass | 813.43 g·mol−1 |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). |
References
- ^ Jump up to:a b c d e f Croce, Carlo M; Reed, John C (October 2016). “Finally, An Apoptosis-Targeting Therapeutic for Cancer”. Cancer Research. 76 (20): 5914–5920. doi:10.1158/0008-5472.CAN-16-1248. PMC 5117672. PMID 27694602.
- ^ Jump up to:a b Yosef, Reut; Pilpel, Noam; Tokarsky-Amiel, Ronit; Biran, Anat; Ovadya, Yossi; Cohen, Snir; Vadai, Ezra; Dassa, Liat; Shahar, Elisheva; Condiotti, Reba; Ben-Porath, Ittai; Krizhanovsky, Valery (2016). “Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL”. Nature Communications. 7: 11190. Bibcode:2016NatCo…711190Y. doi:10.1038/ncomms11190. PMC 4823827. PMID 27048913.
- ^ Vogler, Meike, et al. “Bcl-2 inhibitors: small molecules with a big impact on cancer therapy.” Cell Death & Differentiation 16.3 (2008): 360–367.
- ^ Oltersdorf, Tilman; Elmore, Steven W.; Shoemaker, Alexander R.; Armstrong, Robert C.; Augeri, David J.; Belli, Barbara A.; Bruncko, Milan; Deckwerth, Thomas L.; Dinges, Jurgen; Hajduk, Philip J.; Joseph, Mary K.; Kitada, Shinichi; Korsmeyer, Stanley J.; Kunzer, Aaron R.; Letai, Anthony; Li, Chi; Mitten, Michael J.; Nettesheim, David G.; Ng, ShiChung; Nimmer, Paul M.; O’Connor, Jacqueline M.; Oleksijew, Anatol; Petros, Andrew M.; Reed, John C.; Shen, Wang; Tahir, Stephen K.; Thompson, Craig B.; Tomaselli, Kevin J.; Wang, Baole; Wendt, Michael D.; Zhang, Haichao; Fesik, Stephen W.; Rosenberg, Saul H. (2005). “An inhibitor of Bcl-2 family proteins induces regression of solid tumours”. Nature. 435 (7042): 677–81. Bibcode:2005Natur.435..677O. doi:10.1038/nature03579. PMID 15902208. S2CID 4335635.
- ^ Hann CL, Daniel VC, Sugar EA, Dobromilskaya I, Murphy SC, Cope L, Lin X, Hierman JS, Wilburn DL, Watkins DN, Rudin CM (April 2008). “Therapeutic efficacy of ABT-737, a selective inhibitor of BCL-2, in small cell lung cancer”. Cancer Research. 68 (7): 2321–8. doi:10.1158/0008-5472.can-07-5031. PMC 3159963. PMID 18381439.
- ^ Jump up to:a b Hauck P, Chao BH, Litz J, Krystal GW (April 2009). “Alterations in the Noxa/Mcl-1 axis determine sensitivity of small cell lung cancer to the BH3 mimetic ABT-737”. Mol Cancer Ther. 8 (4): 883–92. doi:10.1158/1535-7163.MCT-08-1118. PMID 19372561. Retrieved 9 September 2019.
///////////ABT-737, ABT 737
CN(CC[C@@H](NC1=CC=C(C=C1[N+]([O-])=O)S(NC(C2=CC=C(C=C2)N3CCN(CC3)CC4=CC=CC=C4C5=CC=C(C=C5)Cl)=O)(=O)=O)CSC6=CC=CC=C6)C

NEW DRUG APPROVALS
ONE TIME
$9.00
Tolebrutinib, SAR 442168
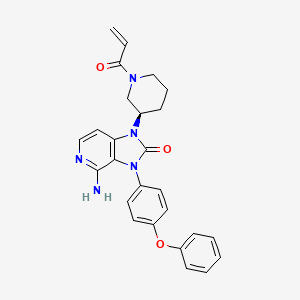
Tolebrutinib
SAR442168
- Treatment of Multiple Sclerosis (MS)
CAS 1971920-73-6
PRN 2246, example 3 [WO2016196840A1]
C26H25N5O3,
| 455.5 |
4-amino-3-(4-phenoxyphenyl)-1-[(3R)-1-prop-2-enoylpiperidin-3-yl]imidazo[4,5-c]pyridin-2-one
4-amino-3-(4-phenoxyphenyl)-1-[(3R)-1-prop-2-enoylpiperidin-3-yl]imidazo[4,5-c]pyridin-2-one
(R)-1-(1-Acryloylpiperidin-3-yl)-4-amino-3-(4-phenoxyphenyl)-1H-imidazo[4,5-c]pyridin-2(3H)-one
4-amino-3-(4-phenoxyphenyl)-1-[(3R)-1-(prop-2-
enoyl)piperidin-3-yl]-1,3-dihydro-2H-imidazo[4,5-
Tolebrutinib (R&D code SAR442168), developed by Principia and later acquired by Sanofi and included in its product line, Tolebrutinib is a BTK inhibitor used to treat cancer, autoimmune diseases such as multiple sclerosis and myasthenia gravis, inflammatory diseases and thromboembolic diseases, etc.,
Tolebrutinib is an orally bioavailable, brain-penetrant, selective, small molecule inhibitor of Bruton’s tyrosine kinase (BTK), with potential immunomodulatory and anti-inflammatory activities. Upon oral administration, tolebrutinib is able to cross the blood-brain barrier and inhibits the activity of BTK both peripherally and in the central nervous system (CNS). This prevents the activation of the B-cell antigen receptor (BCR) signaling pathway, and the resulting immune activation and inflammation. The inhibition of BTK activity also prevents microglial inflammatory signaling in the CNS, and the resulting immune activation, neuroinflammation and neurodegeneration. BTK, a cytoplasmic tyrosine kinase and member of the Tec family of kinases, plays an important role in B lymphocyte development, activation, signaling, proliferation and survival. In addition to B cells, BTK is also expressed in innate immune cells, including macrophages and microglia, and plays an important role in the regulation of microglial inflammatory signaling.
BTK, a member of the Tec family non-receptor tyrosine kinases, is essential for B cell signaling downstream from the B-cell receptor. It is expressed in B cells and other hematopoietic cells such as monocytes, macrophages and mast cells. It functions in various aspects of B cell function that maintain the B cell repertoire (see Gauld S. B. et al., B cell antigen receptor signaling: roles in cell development and disease. Science,
296: 1641 -2. 2002.) B cells pay a role in rheumatoid arthritis (see Perosa F., et ai, CD20-depleting therapy in autoimmune diseases: from basic research to the clinic. / Intern Med. 267:260-77. 2010 and Dorner T, et at. Targeting B cells in immune-mediated
inflammatory disease: a comprehensive review of mechanisms of action and identification of biomarkers. Pharmacol The 125:464-75. 2010 and Honigberg, L., et. ai, The selective BTK inhibitor PCI-32765 blocks B cell and mast cell activation and prevents mouse collagen indiced arthritis. Clin. Immunol. 127 SI :S 111. 2008) and in other autoimmune diseases such as systemic lupus erythematosus and cancers (see Shlomchik M. J., et. ai, The role of B cells in lpr/lpr-induced autoimmunity. /. Exp Med. 180:1295-1306. 1994; Honigberg L. A., The Braton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. 107: 13075-80. 2010; and Mina-Osorio P, et al., Suppression of
glomerulonephritis in lupus-prone NZB x NZW mice by RN486, a selective inhibitor of Bruton’s tyrosine kinase. Arthritis Rheum. 65: 2380-91. 2013).
There is also potential for BTK inhibitors for treating allergic diseases (see Honigberg, L., et. al., The selective BTK inhibitor PCI-32765 blocks B cell and mast cell activation and prevents mouse collagen indiced arthritis. Clin. Immunol. 127 SI :S111. 2008). It was noted that the irreversible inhibitor suppresses passive cutaneous anaphylaxis (PCA) induced by IgE antigen complex in mice. These findings are in agreement with those noted with BTK-mutant mast cells and knockout mice and suggest that BTK inhibitors may be useful for the treatment of asthma, an IgE-dependent allergic disease of the airway.
Accordingly, compounds that inhibit BTK would be useful in treatment for diseases such as autoimmune diseases, inflammatory diseases, and cancer.

PATENT
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016196840
Example 3
Synthesis of (R)-l-(l-acryloylpiperidin-3-yl)-4-amino-3-(4-phenoxyphenyl)-lH- imidazo[4,5-c]pyridin-2(3H)-one
Into a 100-mL round-bottom flask, was placed (R)-4-amino-3-(4-phenoxyphenyl)-l-(piperidin-3-yl)-lH-imidazo[4,5-c]pyridin-2(3H)-one (150 mg, 0.37 mmol, 1.00 equiv), DCM-CH30H (6 mL), TEA (113 mg, 1.12 mmol, 3.00 equiv). This was followed by the addition of prop-2-enoyl chloride (40.1 mg, 0.44 mmol, 1.20 equiv) dropwise with stirring at OoC in 5 min. The resulting solution was stirred for 2 h at 0 °C. The resulting mixture was concentrated under vacuum. The residue was applied onto a silica gel column with dichloromethane/methanol (30: 1). The crude product (100 mg) was purified by Prep-HPLC with the following conditions (Column, XBridge Prep CI 8 OBD
Column,5um, 19*150mm; mobile phase, water with 0.05%TFA and ACN (25.0% ACN up to 45.0% in 8 min). 54.5 mg product of (R)-l-(l -acryloylpiperidin-3-yl)-4-amino-3-(4-phenoxyphenyl)-lH-imidazo[4,5-c]pyridin-2(3H)-one was obtained as a white solid. LC-MS m/z: 465.2 (M+l)
Step 2
Into a 25-mL round-bottom flask was placed tert-butyl (3R)-3-[4-[(E)-[(dimethy]amino)-methylidene]-amino]-2-oxo-3-(4-phenoxyphenyl)-lH,2H,3H-imidazo[4,5-c]pyridin-l -yl]piperidine- l-carboxylate (150 mg, 0.27 mmol, 1.00 equiv), 1,4-dioxane (6 mL), and hydrogen chloride (3 mL). The resulting solution was stirred overnight at 50° C. The reaction mixture was quenched with water. The pH of the solution was adjusted to 9 with sodium bicarbonate. The resulting solution was extracted with dichloromethane:CH3OH=10: 1 and the organic layers were combined. The resulting mixture was washed with sodium chloride and the organic layers were combined, dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was applied onto a silica gel column and eluted with dichloromethane/methanol (30: 1) to give 80 mg (74%) of 4-amino-3-(4-phenoxyphenyl)-l -[(3R)-piperidin-3-yl]-lH,2H,3H-imidazo[4,5-c]pyridin-2-one as a light yellow solid.
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
/////////Tolebrutinib, SAR 442168, PRN 2246, GTPL10625, BTK’168, EX-A4699, BDBM50557487, WHO 11268, Multiple Sclerosis, (MS),

NEW DRUG APPROVALS
ONE TIME
$10.00
Mirvetuximab soravtansine-gynx
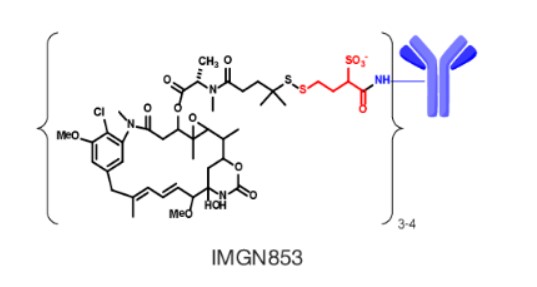
Mirvetuximab soravtansine-gynx
FDA 11/14/2022,To treat patients with recurrent ovarian cancer that is resistant to platinum therapy
| Elahere |
FDA Approves Mirvetuximab Soravtansine-gynx for FRα+ Platinum-resistant Ovarian Cancer
https://www.biochempeg.com/article/315.html
4846-85a8-48171ab38275
FDA Approves Mirvetuximab Soravtansine-gynx for FRα+ Platinum-resistant Ovarian Cancer
November 15, 2022
The FDA has granted accelerated approval to mirvetuximab soravtansine-gynx (Elahere) for the treatment of select patients with folate receptor α–positive, platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer.
The FDA has granted accelerated approval to mirvetuximab soravtansine-gynx (Elahere) for the treatment of adult patients with folate receptor α (Frα)–positive, platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer, who have received 1 to 3 prior systemic treatment regimens.1-3
The regulatory agency also gave the green light to the VENTANA FOLR1 (FOLR-2.1) RxDx Assay for use as a companion diagnostic device to identify patients who are eligible to receive the agent. Testing can be done on fresh or archived tissue. Newly diagnosed patients can be tested at diagnosis to determine whether this agent will be an option for them at the time of progression to platinum resistance.
The decision was supported by findings from the phase 3 SORAYA trial (NCT04296890), in which mirvetuximab soravtansine elicited a confirmed investigator-assessed objective response rate (ORR) of 31.7% (95% CI, 22.9%-41.6%); this included a complete response rate of 4.8% and a partial response rate of 26.9%. Moreover, the median duration of response (DOR) was 6.9 months (95% CI, 5.6-9.7) per investigator assessment.
“The approval of Elahere is significant for patients with FRα-positive platinum-resistant ovarian cancer, which is characterized by limited treatment options and poor outcomes,” Ursula Matulonis, MD, chief of the Division of Gynecologic Oncology at the Dana-Farber Cancer Institute, professor of medicine at the Harvard Medical School, and SORAYA co-principal investigator, stated in a press release. “Elahere impressive anti-tumor activity, durability of response, and overall tolerability observed in SORAYA demonstrate the benefit of this new therapeutic option, and I look forward to treating patients with Elahere.”
The global, single-arm SORAYA trial enrolled a total of 106 patients with platinum-resistant ovarian cancer whose tumors expressed high levels of FRα. Patients were allowed to have received up to 3 prior lines of systemic treatment, and all were required to have received bevacizumab (Avastin).
If patients had corneal disorders, ocular conditions in need of ongoing treatment, peripheral neuropathy that was greater than grade 1 in severity, or noninfectious interstitial lung disease, they were excluded.
Study participants received intravenous mirvetuximab soravtansine at 6 mg/kg once every 3 weeks until progressive disease or unacceptable toxicity. Investigators conducted tumor response assessments every 6 weeks for the first 36 weeks, and every 12 weeks thereafter.
Confirmed investigator-assessed ORR served as the primary end point for the research, and the key secondary end point was DOR by RECIST v1.1 criteria.
In the efficacy-evaluable population (n = 104), the median age was 62 years (range, 35-85). Ninety-six percent of patients were White, 2% were Asian, and 2% did not have their race information reported; 2% of patients were Hispanic or Latino. Regarding ECOG performance status, 57% of patients had a status of 0 and the remaining 43% had a status of 1.
Ten percent of patients received 1 prior line of systemic treatment, 39% received 2 prior lines, and 50% received 3 or more prior lines. All patients previously received bevacizumab, as required, and 47% previously received a PARP inhibitor.
The safety of mirvetuximab soravtansine was evaluated in all 106 patients. The median duration of treatment with the agent was 4.2 months (range, 0.7-13.3).
The all-grade toxicities most commonly experienced with mirvetuximab soravtansine included vision impairment (50%), fatigue (49%), increased aspartate aminotransferase (50%), nausea (40%), increased alanine aminotransferase (39%), keratopathy (37%), abdominal pain (36%), decreased lymphocytes (35%), peripheral neuropathy (33%), diarrhea (31%), decreased albumin (31%), constipation (30%), increased alkaline phosphatase (30%), dry eye (27%), decreased magnesium (27%), decreased leukocytes (26%), decreased neutrophils (26%), and decreased hemoglobin (25%).
Thirty-one percent of patients experienced serious adverse reactions with the agent, which included intestinal obstruction (8%), ascites (4%), infection (3%), and pleural effusion (3%). Toxicities proved to be fatalfor 2% of patients, and these included small intestinal obstruction (1%) and pneumonitis (1%).
Twenty percent of patients required dose reductions due to toxicities. Eleven percent of patients discontinued treatment with mirvetuximab soravtansine because of adverse reactions. Toxicities that resulted in more than 2% of patients discontinuing treatment included intestinal obstruction (2%) and thrombocytopenia (2%). One patient discontinued because of visual impairment.
References
- ImmunoGen announces FDA accelered approval of Elahere (mirvetuximab soravtansine-gynx) for the treatment of platinum-resistant ovarian cancer. News release. ImmunoGen Inc. November 14, 2022. Accessed November 14, 2022. http://bit.ly/3GgrCwL
- FDA grants accelerated approval to mirvetuximab soravtansine-gynx for FRα positive, platinum-resistant epithelial ovarian, fallopian tube, or peritoneal cancer. News release. FDA. November 14, 2022. Accessed November 14, 2022. http://bit.ly/3UP742w
- Elahere (mirvetuximab soravtansine-gynx). Prescribing information; ImmunoGen Inc; 2022. Accessed November 14, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761310s000lbl.pdf

NEW DRUG APPROVALS
ONE TIME
$10.00
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
//////////Mirvetuximab soravtansine-gynx, FDA 2022, APPROVALS 2022, recurrent ovarian cancer,
| Elahere |
TEREVALEFIM

TEREVALEFIM
Molecular Formula
- C9-H8-N2-S
Molecular Weight
- 176.2382
RN: 1070881-42-3
UNII: GG91UXK2M5
- 5-((E)-2-Thiophen-2-yl-vinyl)-lh-pyrazole
- 1H-Pyrazole, 3-((1E)-2-(2-thienyl)ethenyl)-
- ANG-3777
- SNV-003
- OriginatorAngion Biomedica
- ClassAnti-ischaemics; Antifibrotics; Heart failure therapies; Pyrazoles; Small molecules; Thiophenes; Urologics; Vascular disorder therapies
- Mechanism of ActionProto oncogene protein c met stimulants
- Orphan Drug StatusYes – Renal failure
- Phase IIIDelayed graft function
- Phase IIAcute kidney injury; Acute lung injury; Renal failure
- PreclinicalBrain injuries
- No development reportedHeart failure
- DiscontinuedHepatic fibrosis; Myocardial infarction; Stroke
- 02 Aug 2022Vifor Pharma has been acquired by CSL and renamed to CSL Vifor
- 14 Dec 2021Efficacy and adverse events data of a phase II GUARD trial in Acute kidney injury released by the company
- 26 Oct 2021Top-line efficacy and adverse events data from the phase III trial GIFT (Graft Improvement Following Transplant) trial in Delayed graft function released by Angion Biomedica and Vifor Pharma
Terevalefim, an hepatocyte growth factor (HGF) mimetic, selectively activates the c-Met receptor.
PATENT
WO 2004/058721
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2004058721
PATENT
PCT Application No. PCT/US2003/040917, filed December 19, 2003 and published as WO2004/058721 on July 15, 2004, the entirety of which is hereby incorporated by reference, describes certain compounds that act as HGF/SF mimetics . Such compounds include terevalefim:
Terevalefim has been demonstrated to be remarkably useful for treatment of a variety of conditions including, for example, fibrotic liver disease, ischemia-reperfusion injury, cerebral infarction, ischemic heart disease, renal disease, lung fibrosis, damaged and/or ischemic organs, transplants or grafts, stroke, cerebrovascular disease, and renal fibrosis, among others (see, for example, WO 2004/058721, WO 2010/005580, US 2011/0230407, US 7879898, and WO 2009/064422, each of which is hereby incorporated by reference.) Exemplary methods of using terevalefim for, eg, treating delayed graft function after kidney transplantation and acute lung injury, are described in WO 2021/087392 and WO 2021/183774, each of which is hereby incorporated by reference. In particular, Terevalefim is or has been the subject of clinical trials for delayed graft function in recipients of a deceased donor kidney (Clinicaltrials.gov identifier: NCT02474667), acute kidney injury after cardiac surgery involving cardiopulmonary bypass (Clinicaltrials.gov identifier: NCT02771509), and COVID -19 pneumonia (Clinicaltrials.gov identifier: NCT04459676). Without wishing to be bound by any particular theory, it is believed that terevalefim’s HGF mimetic capability imparts a variety of beneficial attributes and activities.
[0035] Terevalefim has a CAS Registry No. of 1070881-42-3 and is also known by at least the following names:
● 3-[(1E)-2-(thiophen-2-yl)ethen-1-yl]-1H-pyrazole; and
● (E)-3-[2-(2-thienyl)vinyl]-1H-pyrazole.
Synthesis of Terevalefim
[0057] In some embodiments, the present disclosure provides methods for preparing compounds useful as HGF/SF mimetics, such as terevalefim. A synthesis of terevalefim is described in detail in Example 7 of WO 2004/058721 (“the ‘721 Synthesis”). The ‘721 Synthesis is depicted in Scheme 1:
The ‘721 Synthesis includes certain features which are not desirable for preparation of terevalefim at scale and/or with consistency and/or with suitable purity for use in humans. For example, the ‘721 Synthesis includes preparation of aldehyde compound 1.2, a viscous oil that is difficult to purify with standard techniques. Additionally, the ‘721 Synthesis uses a diethoxyphosphorylacetaldehyde tosylhydrazone reagent in step 1-2. As such, step 1-2 has poor atom economy and results in multiple byproducts that must be purified away from the final product of terevalefim. Step 1-2 also uses sodium hydride, a highly reactive base that can be difficult to control and often results in byproducts that must be purified away from the final product of terevalefim. Such purification steps can be costly and time-consuming. In some embodiments, the present disclosure encompasses the recognition that one or more features of the ‘721 Synthesis can be improved to increase yield and/or increase reliability and/or increase scale and/or reduce byproducts. In some embodiments, the present disclosure provides such a synthesis, as detailed herein.
[0059] In some embodiments, the present disclosure provides a synthesis of terevalefim as depicted in Scheme 2:
Scheme 2
wherein X and R 1 are defined below and in classes and subclasses as described herein.
[0060] It will be appreciated that compounds described herein, eg, compounds in Scheme 2, may be provided and/or utilized in a salt form. For example, compounds which contain a basic nitrogen atom may form a salt with a suitable acid. Alternatively and/or additionally, compounds which contain an acidic moiety, such as a carboxylic acid group, may form a salt with a suitable base. Suitable counterions are well known in the art, eg, see generally, March ‘s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, MB Smith and J.
March, 5 th Edition, John Wiley & Sons, 2001. All forms of the compounds in Scheme 2 are contemplated by and within the scope of the present disclosure.
Step 2-1 of Scheme 2
[0061] Step 2-1 includes a condensation-elimination reaction between commercially available thiophene-2-carboxaldehyde (1.1) and acetone to provide an α,β-unsaturated ketone compound (2.1).
[0062] In some embodiments, the present disclosure provides a method comprising steps of:
(i) providing compound 1.1:
(ii) contacting compound 1.1 with acetone in the presence of a suitable base,
to compound provide 2.1:
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
///////TEREVALEFIM, ANG-3777, SNV-003, Phase 3, Delayed graft function
C(=C\c1cccs1)/c2cc[nH]n2

NEW DRUG APPROVALS
ONE TIME
$10.00
RELACORILANT
Relacorilant
- Molecular FormulaC27H22F4N6O3S
- Average mass586.561 Da
CAS 1496510-51-0
Phase III
[(4aR)-1-(4-fluorophenyl)-6-(1-methylpyrazol-4-yl)sulfonyl-4,5,7,8-tetrahydropyrazolo[3,4-g]isoquinolin-4a-yl]-[4-(trifluoromethyl)pyridin-2-yl]methanone
релакорилант[Russian][INN]
ريلاكوريلانت[Arabic][INN]
瑞拉可兰[Chinese][INN]
- OriginatorCorcept Therapeutics
- ClassAntineoplastics; Fluorine compounds; Isoquinolines; Ketones; Organic sulfur compounds; Pyrazoles; Pyridines; Small molecules
- Mechanism of ActionGlucocorticoid receptor antagonists
- Orphan Drug StatusYes – Pancreatic cancer; Cushing syndrome
- Phase IIICushing syndrome; Ovarian cancer; Pancreatic cancer
- Phase IIFallopian tube cancer; Peritoneal cancer; Prostate cancer
- Phase I/IISolid tumours
- Phase IAdrenocortical carcinoma
Most Recent Events
- 09 Sep 2022Subgroup analysis efficacy data from a phase-II trial in Ovarian cancer presented at the 47th European Society for Medical Oncology Congress (ESMO-2022)
- 29 Jun 2022Phase-III clinical trials in Ovarian cancer (Combination therapy, Recurrent, Second-line therapy or greater) in USA (PO)
- 06 Jun 2022Corcept Therapeutics announces intentions to submit a NDA for Ovarian cancer
Relacorilant (developmental code name CORT-125134) is an antiglucocorticoid which is under development by Corcept Therapeutics for the treatment of Cushing’s syndrome.[1] It is also under development for the treatment of solid tumors and alcoholism.[1][2] The drug is a nonsteroidal compound and acts as an antagonist of the glucocorticoid receptor.[1] As of December 2017, it is in phase II clinical trials for Cushing’s syndrome and phase I/II clinical studies for solid tumors, while the clinical phase for alcoholism is unknown.[1]
Relacorilant is an orally available antagonist of the glucocorticoid receptor (GR), with potential antineoplastic activity. Upon administration, relacorilant competitively binds to and blocks GRs. This inhibits the activity of GRs, and prevents both the translocation of the ligand-GR complexes to the nucleus and gene expression of GR-associated genes. This decreases the negative effects that result from excess levels of endogenous glucocorticoids, like those seen when tumors overproduce glucocorticoids. In addition, by binding to GRs and preventing their activity, inhibition with CORT125134 also inhibits the proliferation of GR-overexpressing cancer cells. GRs are overexpressed in certain tumor cell types and promote tumor cell proliferation.
SCHEME

CLIP
https://europepmc.org/article/pmc/pmc8175224
Relacorilant (CORT125134)118) is being developed by Corcept Therapeutics, Inc. It is an orally active, high-affinity, selective antagonist of the glucocorticoid receptor that may benefit from the modulation of cortisol activity. In structural optimization, the introduction of a trifluoromethyl group to the 4-position on the pyridyl moiety was found to increase HepG2 tyrosine amino transferase assay potency by a factor of four. Relacorilant is currently being evaluated in a phase II clinical study in patients with Cushing’s syndrome.119)
2-Bromo-4-(trifluoromethyl)pyridine (17) prepared from (E)-4-ethoxy-1,1,1-trifluorobut-3-en-2-one is employed as a key intermediate for the preparation of relacorilant as shown in Scheme 31.120)

Scheme31. Synthesis of relacorilant.118)
118) H. Hunt, T. Johnson, N. Ray and I. Walters (Corcept Therapeutics, Inc.): PCT Int. Appl. WO2013/177559 (2013).
119) H. J. Hunt, J. K. Belanoff, I. Walters, B. Gourdet, J. Thomas, N. Barton, J. Unitt, T. Phillips, D. Swift and E. Eaton: Identification of the Clinical Candidate (R)-(1-(4-Fluorophenyl)-6-((1-methyl-1H-pyrazol-4-yl)sulfonyl)-4,4a,5,6,7,8-hexahydro-1H-pyrazolo[3,4-g]isoquinolin-4a-yl)(4-(trifluoromethyl)pyridin-2-yl)methanone (CORT125134): A Selective Glucocorticoid Receptor (GR) Antagonist. J. Med. Chem. 60, 3405–3421 (2017). [Abstract] [Google Scholar]
120) B. Lehnemann, J. Jung and A. Meudt (Archimica GmbH): PCT Int. Appl. WO 2007/000249 (2007).
PAPER
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.7b00162
The nonselective glucocorticoid receptor (GR) antagonist mifepristone has been approved in the U.S. for the treatment of selected patients with Cushing’s syndrome. While this drug is highly effective, lack of selectivity for GR leads to unwanted side effects in some patients. Optimization of the previously described fused azadecalin series of selective GR antagonists led to the identification of CORT125134, which is currently being evaluated in a phase 2 clinical study in patients with Cushing’s syndrome.

PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2013177559


SYN

////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Cushing’s syndrome (CS) is a metabolic disorder caused by chronic hypercortisolism. CS is associated with cardiovascular, metabolic, skeletal and psychological dysfunctions and can be fatal if left untreated. The first-line treatment for all forms of CS is a surgery. However, medical therapy has to be chosen if surgical resection is not an option or is deemed ineffective. Currently available therapeutics are either not selective and have side effects or are only available as an injection (pasireotide).
References
- ^ Jump up to:a b c d “Relacorilant – Corcept Therapeutics – AdisInsight”.
- ^ Veneris JT, Darcy KM, Mhawech-Fauceglia P, Tian C, Lengyel E, Lastra RR, Pejovic T, Conzen SD, Fleming GF (2017). “High glucocorticoid receptor expression predicts short progression-free survival in ovarian cancer”. Gynecol. Oncol. 146 (1): 153–160. doi:10.1016/j.ygyno.2017.04.012. PMC 5955699. PMID 28456378.
External links
| Clinical data | |
|---|---|
| Other names | CORT-125134 |
| Routes of administration | By mouth |
| Drug class | Antiglucocorticoid |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1496510-51-0 |
| PubChem CID | 73051463 |
| ChemSpider | 57617720 |
| UNII | 2158753C7E |
| KEGG | D11336 |
| Chemical and physical data | |
| Formula | C27H22F4N6O3S |
| Molar mass | 586.57 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
//////////////Relacorilant, Phase III , Orphan Drug, Cushing syndrome, Ovarian cancer, Pancreatic cancer, релакорилант , ريلاكوريلانت , 瑞拉可兰 ,
CN1C=C(C=N1)S(=O)(=O)N2CCC3=CC4=C(CC3(C2)C(=O)C5=NC=CC(=C5)C(F)(F)F)C=NN4C6=CC=C(C=C6)F

NEW DRUG APPROVALS
ONE TIME
$10.00
Tremelimumab
(Light chain)
DIQMTQSPSS LSASVGDRVT ITCRASQSIN SYLDWYQQKP GKAPKLLIYA ASSLQSGVPS
RFSGSGSGTD FTLTISSLQP EDFATYYCQQ YYSTPFTFGP GTKVEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(Heavy chain)
QVQLVESGGG VVQPGRSLRL SCAASGFTFS SYGMHWVRQA PGKGLEWVAV IWYDGSNKYY
ADSVKGRFTI SRDNSKNTLY LQMNSLRAED TAVYYCARDP RGATLYYYYY GMDVWGQGTT
VTVSSASTKG PSVFPLAPCS RSTSESTAAL GCLVKDYFPE PVTVSWNSGA LTSGVHTFPA
VLQSSGLYSL SSVVTVPSSN FGTQTYTCNV DHKPSNTKVD KTVERKCCVE CPPCPAPPVA
GPSVFLFPPK PKDTLMISRT PEVTCVVVDV SHEDPEVQFN WYVDGVEVHN AKTKPREEQF
NSTFRVVSVL TVVHQDWLNG KEYKCKVSNK GLPAPIEKTI SKTKGQPREP QVYTLPPSRE
EMTKNQVSLT CLVKGFYPSD IAVEWESNGQ PENNYKTTPP MLDSDGSFFL YSKLTVDKSR
WQQGNVFSCS VMHEALHNHY TQKSLSLSPG K
(Disulfide bridge: L23-L88, L134-L194, L214-H139, H22-H96, H152-H208, H265-H325, H371-H429, H227-H’227, H228-H’228, H231-H’231, H234-H’234)

Fab fragment of tremelimumab (blue) binding CTLA-4 (green). From PDB entry 5GGV.
Tremelimumab
| Formula | C6500H9974N1726O2026S52 |
|---|---|
| CAS | 745013-59-6 |
| Mol weight | 146380.4722 |
FDA APPROVED2022/10/21, Imjudo
PEPTIDE, CP 675206
| Antineoplastic, Immune checkpoint inhibitor, Anti-CTLA4 antibody | |
| Disease | Hepatocellular carcinoma |
|---|
Tremelimumab (formerly ticilimumab, CP-675,206) is a fully human monoclonal antibody against CTLA-4. It is an immune checkpoint blocker. Previously in development by Pfizer,[1] it is now in investigation by MedImmune, a wholly owned subsidiary of AstraZeneca.[2] It has been undergoing human trials for the treatment of various cancers but has not attained approval for any.
Imjudo (tremelimumab) in combination with Imfinzi approved in the US for patients with unresectable liver cancer
PUBLISHED24 October 2022
24 October 2022 07:00 BST
Approval based on HIMALAYA Phase III trial results which showed single priming dose of Imjudo added to Imfinzi reduced risk of death by 22% vs. sorafenib
AstraZeneca’s Imjudo (tremelimumab) in combination with Imfinzi (durvalumab) has been approved in the US for the treatment of adult patients with unresectable hepatocellular carcinoma (HCC), the most common type of liver cancer. The novel dose and schedule of the combination, which includes a single dose of the anti-CTLA-4 antibody Imjudo 300mg added to the anti-PD-L1 antibody Imfinzi 1500mg followed by Imfinzi every four weeks, is called the STRIDE regimen (Single Tremelimumab Regular Interval Durvalumab).
The approval by the US Food and Drug Administration (FDA) was based on positive results from the HIMALAYA Phase III trial. In this trial, patients treated with the combination of Imjudo and Imfinzi experienced a 22% reduction in the risk of death versus sorafenib (based on a hazard ratio [HR] of 0.78, 95% confidence interval [CI] 0.66-0.92 p=0.0035).1 Results were also published in the New England Journal of Medicine Evidence showing that an estimated 31% of patients treated with the combination were still alive after three years, with 20% of patients treated with sorafenib still alive at the same duration of follow-up.2
Liver cancer is the third-leading cause of cancer death and the sixth most commonly diagnosed cancer worldwide.3,4 It is the fastest rising cause of cancer-related deaths in the US, with approximately 36,000 new diagnoses each year.5,6
Ghassan Abou-Alfa, MD, MBA, Attending Physician at Memorial Sloan Kettering Cancer Center (MSK), and principal investigator in the HIMALAYA Phase III trial, said: “Patients with unresectable liver cancer are in need of well-tolerated treatments that can meaningfully extend overall survival. In addition to this regimen demonstrating a favourable three-year survival rate in the HIMALAYA trial, safety data showed no increase in severe liver toxicity or bleeding risk for the combination, important factors for patients with liver cancer who also have advanced liver disease.”
Dave Fredrickson, Executive Vice President, Oncology Business Unit, AstraZeneca, said: “With this first regulatory approval for Imjudo, patients with unresectable liver cancer in the US now have an approved dual immunotherapy treatment regimen that harnesses the potential of CTLA-4 inhibition in a unique combination with a PD-L1 inhibitor to enhance the immune response against their cancer.”
Andrea Wilson Woods, President & Founder, Blue Faery: The Adrienne Wilson Liver Cancer Foundation, said: “In the past, patients living with liver cancer had few treatment options and faced poor prognoses. With today’s approval, we are grateful and optimistic for new, innovative, therapeutic options. These new treatments can improve long-term survival for those living with unresectable hepatocellular carcinoma, the most common form of liver cancer. We appreciate the patients, their families, and the broader liver cancer community who continue to fight for new treatments and advocate for others.”
The safety profiles of the combination of Imjudo added to Imfinzi and for Imfinzi alone were consistent with the known profiles of each medicine, and no new safety signals were identified.
Regulatory applications for Imjudo in combination with Imfinzi are currently under review in Europe, Japan and several other countries for the treatment of patients with advanced liver cancer based on the HIMALAYA results.
Notes
Liver cancer
About 75% of all primary liver cancers in adults are HCC.3 Between 80-90% of all patients with HCC also have cirrhosis.7 Chronic liver diseases are associated with inflammation that over time can lead to the development of HCC.7
More than half of patients are diagnosed at advanced stages of the disease, often when symptoms first appear.8 A critical unmet need exists for patients with HCC who face limited treatment options.8 The unique immune environment of liver cancer provides clear rationale for investigating medications that harness the power of the immune system to treat HCC.8
HIMALAYA
HIMALAYA was a randomised, open-label, multicentre, global Phase III trial of Imfinzi monotherapy and a regimen comprising a single priming dose of Imjudo 300mg added to Imfinzi 1500mg followed by Imfinzi every four weeks versus sorafenib, a standard-of-care multi-kinase inhibitor.
The trial included a total of 1,324 patients with unresectable, advanced HCC who had not been treated with prior systemic therapy and were not eligible for locoregional therapy (treatment localised to the liver and surrounding tissue).
The trial was conducted in 181 centres across 16 countries, including in the US, Canada, Europe, South America and Asia. The primary endpoint was overall survival (OS) for the combination versus sorafenib and key secondary endpoints included OS for Imfinzi versus sorafenib, objective response rate and progression-free survival (PFS) for the combination and for Imfinzi alone.
Imfinzi
Imfinzi (durvalumab) is a human monoclonal antibody that binds to the PD-L1 protein and blocks the interaction of PD-L1 with the PD-1 and CD80 proteins, countering the tumour’s immune-evading tactics and releasing the inhibition of immune responses.
Imfinzi was recently approved to treat patients with advanced biliary tract cancer in the US based on results from the TOPAZ-1 Phase III trial. It is the only approved immunotherapy in the curative-intent setting of unresectable, Stage III non-small cell lung cancer (NSCLC) in patients whose disease has not progressed after chemoradiotherapy and is the global standard of care in this setting based on the PACIFIC Phase III trial.
Imfinzi is also approved in the US, EU, Japan, China and many other countries around the world for the treatment of extensive-stage small cell lung cancer (ES-SCLC) based on the CASPIAN Phase III trial. In 2021, updated results from the CASPIAN trial showed Imfinzi plus chemotherapy tripled patient survival at three years versus chemotherapy alone.
Imfinzi is also approved for previously treated patients with advanced bladder cancer in several countries.
Since the first approval in May 2017, more than 100,000 patients have been treated with Imfinzi.
As part of a broad development programme, Imfinzi is being tested as a single treatment and in combinations with other anti-cancer treatments for patients with SCLC, NSCLC, bladder cancer, several gastrointestinal (GI) cancers, ovarian cancer, endometrial cancer, and other solid tumours.
Imfinzi combinations have also demonstrated clinical benefit in metastatic NSCLC in the POSEIDON Phase III trial.
Imjudo
Imjudo (tremelimumab) is a human monoclonal antibody that targets the activity of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4). Imjudo blocks the activity of CTLA-4, contributing to T-cell activation, priming the immune response to cancer and fostering cancer cell death.
Beyond HIMALAYA, Imjudo is being tested in combination with Imfinzi across multiple tumour types including locoregional HCC (EMERALD-3), SCLC (ADRIATIC) and bladder cancer (VOLGA and NILE).
Imjudo is also under review by global regulatory authorities in combination with Imfinzi and chemotherapy in 1st-line metastatic NSCLC based on the results of the POSEIDON Phase III trial, which showed the addition of a short course of Imjudo to Imfinzi plus chemotherapy improved both overall and progression-free survival compared to chemotherapy alone.
AstraZeneca in GI cancers
AstraZeneca has a broad development programme for the treatment of GI cancers across several medicines spanning a variety of tumour types and stages of disease. In 2020, GI cancers collectively represented approximately 5.1 million new diagnoses leading to approximately 3.6 million deaths.9
Within this programme, the Company is committed to improving outcomes in gastric, liver, biliary tract, oesophageal, pancreatic, and colorectal cancers.
Imfinzi (durvalumab) is being assessed in combinations in oesophageal and gastric cancers in an extensive development programme spanning early to late-stage disease across settings.
The Company aims to understand the potential of Enhertu (trastuzumab deruxtecan), a HER2-directed antibody drug conjugate, in the two most common GI cancers, colorectal and gastric cancers. Enhertu is jointly developed and commercialised by AstraZeneca and Daiichi Sankyo.
Lynparza (olaparib) is a first-in-class PARP inhibitor with a broad and advanced clinical trial programme across multiple GI tumour types including pancreatic and colorectal cancers. Lynparza is developed and commercialised in collaboration with MSD (Merck & Co., Inc. inside the US and Canada).
AstraZeneca in immuno-oncology (IO)
Immunotherapy is a therapeutic approach designed to stimulate the body’s immune system to attack tumours. The Company’s immuno-oncology (IO) portfolio is anchored in immunotherapies that have been designed to overcome evasion of the anti-tumour immune response. AstraZeneca is invested in using IO approaches that deliver long-term survival for new groups of patients across tumour types.
The Company is pursuing a comprehensive clinical trial programme that includes Imfinzi as a single treatment and in combination with Imjudo (tremelimumab) and other novel antibodies in multiple tumour types, stages of disease, and lines of treatment, and where relevant using the PD-L1 biomarker as a decision-making tool to define the best potential treatment path for a patient.
In addition, the ability to combine the IO portfolio with radiation, chemotherapy, and targeted small molecules from across AstraZeneca’s oncology pipeline, and from research partners, may provide new treatment options across a broad range of tumours.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company’s focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Mechanism of action
Tremelimumab aims to stimulate an immune system attack on tumors. Cytotoxic T lymphocytes (CTLs) can recognize and destroy cancer cells. However, there is also an inhibitory mechanism (immune checkpoint) that interrupts this destruction. Tremelimumab turns off this inhibitory mechanism and allows CTLs to continue to destroy the cancer cells.[3] This is immune checkpoint blockade.
Tremelimumab binds to the protein CTLA-4, which is expressed on the surface of activated T lymphocytes and inhibits the killing of cancer cells. Tremelimumab blocks the binding of the antigen-presenting cell ligands B7.1 and B7.2 to CTLA-4, resulting in inhibition of B7-CTLA-4-mediated downregulation of T-cell activation; subsequently, B7.1 or B7.2 may interact with another T-cell surface receptor protein, CD28, resulting in a B7-CD28-mediated T-cell activation unopposed by B7-CTLA-4-mediated inhibition.
Unlike Ipilimumab (another fully human anti-CTLA-4 monoclonal antibody), which is an IgG1 isotype, tremelimumab is an IgG2 isotype.[4][5]
Clinical trials
Melanoma
Phase 1 and 2 clinical studies in metastatic melanoma showed some responses.[6] However, based on early interim analysis of phase III data, Pfizer designated tremelimumab as a failure and terminated the trial in April 2008.[1][7]
However, within a year, the survival curves showed separation of the treatment and control groups.[8] The conventional Response Evaluation Criteria in Solid Tumors (RECIST) may underrepresent the merits of immunotherapies. Subsequent immunotherapy trials (e.g. ipilimumab) have used the Immune-Related Response Criteria (irRC) instead.
Mesothelioma
Although it was designated in April 2015 as orphan drug status in mesothelioma,[9] tremelimumab failed to improve lifespan in the phase IIb DETERMINE trial, which assessed the drug as a second or third-line treatment for unresectable malignant mesothelioma.[10][11]
Non-small cell lung cancer
In a phase III trial, AstraZeneca paired tremelimumab with a PD-L1 inhibitor, durvalumab, for the first-line treatment of non-small cell lung cancer.[12] The trial was conducted across 17 countries, and in July 2017, AstraZeneca announced that it had failed to meet its primary endpoint of progression-free survival.[13]
References
- ^ Jump up to:a b “Pfizer Announces Discontinuation of Phase III Clinical Trial for Patients with Advanced Melanoma”. Pfizer.com. 1 April 2008. Retrieved 5 December 2015.
- ^ Mechanism of Pathway: CTLA-4 Inhibition[permanent dead link]
- ^ Antoni Ribas (28 June 2012). “Tumor immunotherapy directed at PD-1”. New England Journal of Medicine. 366 (26): 2517–9. doi:10.1056/nejme1205943. PMID 22658126.
- ^ Tomillero A, Moral MA (October 2008). “Gateways to clinical trials”. Methods Find Exp Clin Pharmacol. 30 (8): 643–72. doi:10.1358/mf.2008.30.5.1236622. PMID 19088949.
- ^ Poust J (December 2008). “Targeting metastatic melanoma”. Am J Health Syst Pharm. 65 (24 Suppl 9): S9–S15. doi:10.2146/ajhp080461. PMID 19052265.
- ^ Reuben, JM; et al. (1 Jun 2006). “Biologic and immunomodulatory events after CTLA-4 blockade with tremelimumab in patients with advanced malignant melanoma”. Cancer. 106 (11): 2437–44. doi:10.1002/cncr.21854. PMID 16615096. S2CID 751366.
- ^ A. Ribas, A. Hauschild, R. Kefford, C. J. Punt, J. B. Haanen, M. Marmol, C. Garbe, J. Gomez-Navarro, D. Pavlov and M. Marsha (May 20, 2008). “Phase III, open-label, randomized, comparative study of tremelimumab (CP-675,206) and chemotherapy (temozolomide [TMZ] or dacarbazine [DTIC]) in patients with advanced melanoma”. Journal of Clinical Oncology. 26 (15S): LBA9011. doi:10.1200/jco.2008.26.15_suppl.lba9011.[permanent dead link]
- ^ M.A. Marshall, A. Ribas, B. Huang (May 2010). “Evaluation of baseline serum C-reactive protein (CRP) and benefit from tremelimumab compared to chemotherapy in first-line melanoma”. Journal of Clinical Oncology. 28 (15S): 2609. doi:10.1200/jco.2010.28.15_suppl.2609.[permanent dead link]
- ^ FDA Grants AstraZeneca’s Tremelimumab Orphan Drug Status for Mesothelioma [1]
- ^ “Tremelimumab Fails Mesothelioma Drug Trial”. Archived from the original on 2016-03-06. Retrieved 2016-03-06.
- ^ AZ’ tremelimumab fails in mesothelioma trial
- ^ “AstraZeneca’s immuno-oncology combo fails crucial Mystic trial in lung cancer | FierceBiotech”.
- ^ “AstraZeneca reports initial results from the ongoing MYSTIC trial in Stage IV lung cancer”.
///////////Tremelimumab, Imjudo, APPROVALS 2022, FDA 2022, PEPTIDE, CP 675206, Antineoplastic, Immune checkpoint inhibitor, Anti-CTLA4 antibody

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}