
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 293)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Sun Pharma has bought Ranbaxy for $4 billion to create the world’s fifth-biggest generic drugmaker.
 Dilip sanghvi, sun pharma promoter
Dilip sanghvi, sun pharma promoter
The move will make the company the largest pharma firm in India, while Daiichi Sankyo – majority owner of Ranbaxy – will become the second largest shareholder in Sun Pharma with a 9% stake and the right to nominate one director to Sun Pharma’s Board of Directors. http://www.pharmatimes.com/Article/14-04-07/Sun_buys_Ranbaxy_for_4_billion.aspx
Read more at: http://www.pharmatimes.com/Article/14-04-07/Sun_buys_Ranbaxy_for_4_billion.aspx#ixzz2yGIjkMob



Dilip Shanghvi, Managing Director of Sun Pharma said in a release, “Ranbaxy has a significant presence in the Indian pharma market and in the US where it offers a broad portfolio of ANDAs and first-to-file opportunities. In high-growth emerging markets, it provides a strong platform which is highly complementary to Sun Pharma’s strengths,”
Under the agreement, Ranbaxy shareholders will get 0.8 shares of Sun Pharma for each Ranbaxy share.
Arun Sahwney, managing director and chief executive officer of Ranbaxy said in a statement, “Sun Pharma has a proven track record of creating significant long-term shareholder value and successfully integrating acquisitions into its growing portfolio of assets,”
Who Will Benefit?
Daiichi Sankyo Co. Ltd is the parent company of Ranbaxy as they acquired it from previous promoters and investors. As soon as Ranbaxy was acquired, their plants came under a scanner from US Food and Drug Administration (FDA), which troubled Daiichi as their own reputation was under stake.
Now, they will be the most relived entity as Sun Pharma will manage all such cases pertaining to Ranbaxy. Daiichi will now control 9% of Sun Pharma as a result of the current acquisition.
Insiders are claiming that Daiichi will sell this 9% stake as well and come out of the business all together.
Ranbaxy shareholders have cheered this latest development as their shares have gained since the announcement of this deal.
New ammunition in the fight against type 2 diabetes
Gastric banding can play a vital role in the treatment of type 2 diabetes in people who are overweight and not obese, according to new research.
he Monash University study, led by Emeritus Professor Paul O’Brien and Dr John Wentworth from the Centre for Obesity Research and Education (CORE), has determined that weight loss surgery (gastric banding) for overweight people with diabeteshad a profound impact on the illness.
The research has been released today in the prestigious medical journal The Lancet Diabetes and Endocrinology.
“This is the first randomised controlled trial demonstrating that treatment of type 2 diabetes in overweight people by substantial weight loss is safe and hugely beneficial,” Professor O’Brien said. “As there are no alternative options that can achieve such a result, this study indicates a potentially attractive path for the overweight person with diabetes and for those providing the care.”
The study…
View original post 234 more words
Cuba may have found cure for cancer

Cuban doctors have filed Wednesday in Havana, the result of 14 years of research, a solution of antitumor peptides whose natural analogue is able to offer positive dynamics in cancer treatments
http://youthandeldersja.wordpress.com/2014/03/22/cuba-may-have-found-cure-for-cancer/
Stem Cells from Muscle Can Repair Nerve Damage After Injury, Pitt Researchers Show

In some ground-breaking research, scientists have been able to use stem cells derived from human muscle tissue to repair nerve damage and restore function after injury to sciatic nerves.
And, even after 12 weeks, the regenerated nerve looked and functioned as a normal nerve.
Link to Story:
http://www.upmc.com/media/NewsReleases/2014/Pages/pitt-study-stem-cells-repair-nerve-damage.aspx
Buserelin a luteinizing hormone-releasing hormone (LHRH) agonist
 Buserelin
Buserelin
57982-77-1 cas no
(2S)-N-[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2R)-1-[[(2S)-1-[[(2S)-5-(diaminomethylideneamino)-1-[(2S)-2-(ethylcarbamoyl)pyrrolidin-1-yl]-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-[(2-methylpropan-2-yl)oxy]-1-oxopropan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-3-(1H-imidazol-5-yl)-1-oxopropan-2-yl]-5-oxopyrrolidine-2-carboxamide
6-[O-(1,1-dimethylethyl)-D-serine]-9-(N-ethyl-L-prolinamide)-10-deglycinamideluteinizing hormone-releasing factor (pig)

Buserelin is a luteinizing hormone-releasing hormone (LHRH) agonist, a synthetic hormone which stimulates the pituitary gland’s gonadotrophin-releasing hormone receptor (GnRHR). It is used in prostate cancer treatment.
Buserelin stimulates the pituitary gland’s gonadotrophin-releasing hormone receptor (GnRHR). Buserelin desensitizes the GnRH receptor, reducing the amount of LH and testosterone. However, there is a concomitant surge in LH and testosterone levels with the decrease in androgens, so antiandrogens must administered.
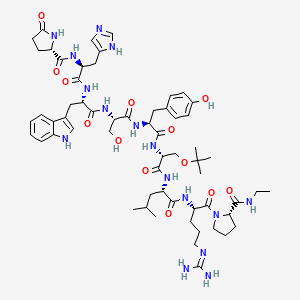
Buserelin is a Gonadotropin-releasing hormone agonist (GnRH agonist). The drug’s effects are dependent on the frequency and time course of administration. GnRH is released in a pulsatile fashion in the postpubertal adult. Initial interaction of any GnRH agonist, such as buserelin, with the GnRH receptor induces release of FSH and LH by gonadotrophes. Long-term exposure to constant levels of buserelin, rather than endogenous pulses, leads to downregulation of the GnRH receptors and subsequent suppression of the pituitary release of LH and FSH.
Like other GnRH agonists, buserelin may be used in the treatment of hormone-responsive cancers such as prostate cancer or breast cancer, estrogen-dependent conditions (such as endometriosis or uterine fibroids), and in assisted reproduction.
It is normally delivered via a nasal spray, but is also available as an injection.
Buserelin acetate is marketed by Sanofi-Aventis under the brand name Suprefact and a generic form of Buserelin is now produced by CinnaGen under the brand name CinnaFact.
Buserelin is also marketed under the brand name Metrelef. Metrelef is approved to treat patients with endometriosis by suppression of ovarian hormone production. In ovulation induction Metrelef is used as a pituitary blockade as an adjunct togonadotrophin administration.

Buserelin, a synthetic gonadotropin-releasing hormone (GRH) agonist, specifically binds to GRH receptor presented at anter iorpituitary and increases or decreases the number of receptors in hypophysis through auto- regulation mechanism (G. Tolis et al., Tumor Growth Inhibition in Patients with Prostatic Carcinoma Treated with Luteinizing Hormone-Feleasing Hormone Agonists, Proc. Natl. Acad. Sci. , 79, pl658, 1982).
<5> The synthetic methods for preparing peptides are divided into two methods, i.e., liquid phase synthesis and solid phase synthesis. The liquid phase peptide synthesis of which all the reagents reacts together under the solution phase by being dissolved in the solution, has been reported to show rapid reaction rate however it has disadvantages such as the difficulty in separating and purification of the products. In a while, solid phase peptide synthesis which have been developed based on the theory of R. B. Merrifield, has been reported to have various advantages comparing with the former method for example, convenient to isolation and purification, the ‘applicability to automation (Bodanszky et al, In Peptide Synthesis, John Wiley & Sons, 1976). Lots of peptide synthetic resins have been developed to synthesize various peptides after the publication of the theory of R. B. Merrifield till now. For example, chloromethyl polystyrene resin had been developed by Merrifield and Wang resin having 4-alkoxybenzyl alcohol had been developed with modifying the former resin to overcome the disadvantages thereof at the early stage. Various resins to improve the disadvantages of conventional resins have been developed after then and the representative resins among those resins are trityl group introduced 2-chlorotrityl resin and rink amide resin which can provide amide group from the carboxyl terminal of peptide under mild cleavage condition, respectively.
<6> At the early stage, the simple structured type-peptides have been synthesized using by the resins however the complex structured type peptides showing various physiological activities have been synthesized mainly. The peptides comprising unnatural amino acids have been synthesized by chemical synthetic method since the peptides could not be prepared by enzymatic synthesis. Among them, the peptides comprising D-amino acid or aza-amino acid have been reported to have potent physiological activities and further to be developed as a medicine (USP Nos. 6,624,290; 6,069,163; 5,965,538; and 4,634,715). However, the novel method for preparing LH-RH such as goserelin or GnRH peptides using by solid phase synthesis has been still need till now since previously known methods, for example, the methods disclosed in USP No. 5,602,231; EP No. 0518655; USP No. 6,879,289; and USP No. 3,914,412, have been reported to have unsolved problems such as a limit to obtain pure product etc.
http://www.google.com/patents/WO2008044890A1?cl=en
Example 4: Preparation of buserelin
<98> Ig of 2-chlroro trityl chloride resin showing 0.9 mM/g of substitution rate was swollen with 10ml of DMF and the reaction mixture mixed with 768 mg of Fmoc-Arg (N02)-0H (1.74 mM) and 271 microliter of DIC (1.74 mM) was added thereto to react together. The resulted resin was treated with 20% piperidine to remove the Fmoc residue and the reaction mixture mixed with 615 mg of Fmoc-Leu-OH (1.74 mM) and 271 microliter of DIC (1.74 mM) was added thereto to react together with a similar way to the above-described method. After washing the resin, the resulted resin was treated with 20% piperidine to remove the Fmoc residue and the reaction mixture mixed with 670 mg of Fmoc-D- SeKtBu)-OH (1.74 mM) and 271 microliter of DIC (1.74 mM) was added thereto again to react together with a similar way to the above-described method. After washing the resin, the resulted resin was treated with 20% piperidine to remove the Fmoc residue and the reaction mixture mixed with 859 mg of Fmoc-Tyr(OBzI)-OH (1.74 mM) and 271 microliter of DIC (1.74 mM) was added thereto again to react together with a similar way to the above-described method. After washing the resin, the resulted resin was treated with 20% piperidine to remove the Fmoc residue and the reaction mixture mixed with 726 mg of Fmoc-Ser(OBzI)-OH (1.74 mM) and 271 microliter of DIC (1.74 mM) was added thereto again to react together with a similar way to the above- described method. After washing the resin, the resulted resin was treated with 20% piperidine to remove the Fmoc residue and the reaction mixture mixed with 742 mg of Fmoc-Trp-0H (1.74 mM) and 271 microliter of DIC (1.74 mM) was added thereto again to react together with a similar way to the above- described method. After washing the resin, the resulted resin was treated with 20% piperidine to remove the Fmoc residue and the reaction mixture mixed with 1.078g of Fmoc-His(Fmoc)-0H (1.74 mM) and 271 microliter of DIC (1.74 mM) was added thereto again to react together with a similar way to the above-described method. After washing the resin, the resulted resin was treated with 20% piperidine to remove the Fmoc residue and the reaction mixture mixed with 244 mg of Pyr-OH (1.74 mM) and 271 microliter of DIC (1.74 mM) was added thereto again to react together with a similar way to the above-described method.
<99> The resin was washed again and 2ml of 1% TFA (Trifluoroacetic acid)/DCM (dichloromethane) per 70mg of peptide resin was added to the resin, eluted to release the peptide from the resin and the elute was collected with 200 microliter of pyridine. The above-described step was repeated five times. The resin was washed with DCM (dichloromethane) and methanol and the elute was collected with the former elute. The elute was concentrated with evaporation and ether was added thereto to obtain the precipitated peptide. The precipitated peptide was performed to coupling reaction with 305 mg of Pro- NH-CH2CH3 (2.4mM) and 303mg of DIC (2.4 niM) in the presence of DCM
(dichloromethane) solvent. The solution was subjected to concentration with evaporator. The resulting concentrate was dissolved in EtOAc, washed with saturated NaHCOs solution, distilled water, 5% citrate solution and dried with anhydrous MgS(V The remaining MgS04 was discarded with filtration and the filtrate was concentrated with evaporation. The benzyl group and Cbz group among the side chain protecting group in the peptide were removed through catalytic hydrogen transfer reaction using by Pd/C and ammonium formate in the presence of methanol. The resulting peptide was purified with reverse phase column chromatography (Shimadzu H-kit, acetonitrile^water= 22:78 → 32:68, 1% increase/min) to isolate pure buserelin (Yield: 40%).
new patent
Solid state method for the preparation of buserelin, an LHRH analog useful for the treatment of sexual dysfunction, ovulation, puberty retardation and cancer. Method is under basic conditions and increases yield and purity. This appears to be the first PCT application from Hybio with this target, however several Chinese national filings have been published. Pan, Ma and Yuan are named on several previous solid phase synthesis PCT applications, most recently WO2013117135.
| US5212288 * | Feb 8, 1991 | May 18, 1993 | Syntex (U.S.A.) Inc. | Temporary minimal protection synthesis of serine-containing polypeptides |
| US5510460 * | May 26, 1995 | Apr 23, 1996 | Zeneca Limited | Peptide process |
| US5602231 * | May 26, 1995 | Feb 11, 1997 | Zeneca Limited | Process for making peptides |
| US6028172 * | Feb 10, 1998 | Feb 22, 2000 | Mallinckrodt Inc. | Reactor and method for solid phase peptide synthesis |
| US6897289 * | May 5, 2000 | May 24, 2005 | Lipotec, S.A. | Peptide synthesis procedure in solid phase |
Relugolix (TAK-385) in phase 2 By Takeda for the treatment of endometriosis and uterine fibroids
Relugolix (TAK-385)
1-[4-[1-(2,6-Difluorobenzyl)-5-(dimethylaminomethyl)-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl]-3-methoxyurea
N-(4-(1-(2,6-difluorobenzyl)-5-((dimethylamino)methyl)-3-(6-methoxy-3-pyridazinyl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl)phenyl)-N’-methoxyurea
CAS NO 737789-87-6
-
C29-H27-F2-N7-O5-S
- 623.6383
Synonyms
-
N-(4-(1-((2,6-Difluorophenyl)methyl)-5-((dimethylamino)methyl)-1,2,3,4-tetrahydro-3-(6-methoxy-3-pyridazinyl)-2,4-dioxothieno(2,3-d)pyrimidin-6-yl)phenyl)-N’-methoxyurea
-
TAK-385
-
UNII-P76B05O5V6
Systematic Name
-
Urea, N-(4-(1-((2,6-difluorophenyl)methyl)-5-((dimethylamino)methyl)-1,2,3,4-tetrahydro-3-(6-methoxy-3-pyridazinyl)-2,4-dioxothieno(2,3-d)pyrimidin-6-yl)phenyl)-N’-methoxy-
TAK-385 is a luteinizing hormone-releasing hormone (LH-RH) receptor antagonist administered orally. By preventing LH-RH from binding with the LH-RH receptor in the anterior pituitary gland and suppressing the secretion of luteinizing hormone (LH) and follicle stimulation hormone (FSH) from the anterior pituitary gland, TAK-385 controls the effect of LH and FSH on the ovary, reduces the level of estrogen in blood, which is known to be associated with the development of endometriosis and uterine fibroids, and is expected to improve the symptoms of these disorders.
- *1 The hormone that controls the secretion of LH and FSH, gonadotropic hormones, secreted from the anterior pituitary gland.
- *2 A hormone that is secreted from the anterior pituitary gland by the action of LH-RH and encourages follicular maturation, ovulation and luteinization by acting on the ovaries.
- *3 A hormone that is secreted from the anterior pituitary gland by the action of LH-RH and encourages follicular maturation by stimulating the ovaries.
TAK-385, an oral antagonist of gonadotropin-releasing hormone (GnRH), was originated by Takeda. It is in phase II clinical trials for the treatment of endometriosis and for the treatment of uterine fibroids (myoma). Phase I clinical trials are also underway for the treatment of prostate cancer.
 Relugolix (TAK-385)
Relugolix (TAK-385)
…………….
http://www.google.co.in/patents/EP1591446A1?cl=en
(Production Method 1)

- (Production method 2)
-


- Production of N-(4-(1-(2,6-difluorobenzyl)-5-((dimethylamino)methyl)-3-(6-methoxy-3-pyridazinyl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl)phenyl)-N’-methoxyurea
-

-
The similar reaction as described in Example 4 by using the compound (100 mg, 0.164 mmol) obtained in Reference Example 54 and methyl iodide (0.010 ml, 0.164 mmol) gave the title compound (17.3 mg, 17 %) as colorless crystals.
1 H-NMR(CDCl3) δ: 2.15 (6H, s), 3.6-3.8 (2H, m), 3.82 (3H, s), 4.18 (3H, s), 5.35 (2H, s), 6.92 (2H, t, J = 8.2 Hz), 7.12 (1H, d, J = 8.8 Hz), 7.2-7.65 (7H, m), 7.69 (1H, s).

……………
Discovery of 1-{4-[1-(2,6-difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (TAK-385) as a potent, orally active, non-peptide antagonist of the human gonadotropin-releasing hormone receptor
J Med Chem 2011, 54(14): 4998. http://pubs.acs.org/doi/full/10.1021/jm200216q
1-{4-[1-(2,6-Difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (16b)
 tak 385
tak 385
Click to access jm200216q_si_001.pdf
…………………….
new patent
Method for the production of TAK-385 or its salt and crystals starting from 6-(4-aminophenyl)-1-(2,6-difluorobenzyl)-5-dimethylaminomethyl-3-(6-methoxypyridazin-3-yl) thieno[2,3-d] pyrimidine-2,4 (1H,3H)-dione or its salt. Takeda Pharmaceutical is developing relugolix (TAK-385), an oral LHRH receptor antagonist analog of sufugolix, for the treatment of endometriosis and uterine fibroids. As of April 2014, the drug is in Phase 2 trails. See WO2010026993 claiming method for improving the oral absorption and stability of tetrahydro-thieno[2,3-d]pyrimidin-6-yl]-phenyl)-N’-methoxy urea derivatives.
references
Discovery of TAK-385, a thieno[2,3-d]pyrimidine-2,4-dione derivative, as a potent and orally bioavailable nonpeptide antagonist of gonadotropin releasing hormone (GnRH) receptor
238th ACS Natl Meet (August 16-20, Washington) 2009, Abst MEDI 386
Discovery of 1-{4-[1-(2,6-difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (TAK-385) as a potent, orally active, non-peptide antagonist of the human gonadotropin-releasing hormone receptor
J Med Chem 2011, 54(14): 4998. http://pubs.acs.org/doi/full/10.1021/jm200216q
Ulipristal acetate for emergency contraception

Ulipristal acetate
17alpha-Acetoxy-11beta-[4-(dimethylamino)phenyl]-19-norpregna-4,9-diene-3,20-dione
(8S,11S,13S,14R,17R)-17-Acetoxy-11-[4-(dimethylamino)phenyl]-19-norpregna-4,9-diene-3,20-dione
REVIEW.http://www.fsrh.org/pdfs/ellaOneNewProductReview1009.pdf
126784-99-4 CAS
HRP-2000
PGL-4001
RTI-3021-012
UPA-UF
VA-2914
CHECK OUT NEW PATENTS BELOW NEW PATENTS IN 2014
WO-2014050105 Amorphous ulipristal acetate, ASKA Pharmaceutical Co Ltd
WO-2014050106 Crystalline polymorphic form of ulipristal acetate
WO-2014050107 Crystalline polymorphic form of ulipristal acetate
Ulipristal acetate (trade name EllaOne in the European Union, Ella in the U.S. for contraception,[1] and Esmya for uterine fibroid) is a selective progesterone receptor modulator (SPRM).

Medical uses
Emergency contraception
For emergency contraception[2] a 30 mg tablet is used within 120 hours (5 days) after an unprotected intercourse or contraceptive failure.[3] It has been shown to prevent about 60% of expected pregnancies,[4] and prevents more pregnancies than emergency contraception with levonorgestrel.[5] Ulipristal acetate is available by prescription for emergency contraception in over 50 countries, with access through pharmacists without a prescription being tested in the United Kingdom.[6][7][8][9] Emergency contraception (EC) is a woman’s second chance for primary prevention of pregnancy.
A reproductive-age woman is a candidate for emergency contraception if she seeks care within 120 hours of unprotected intercourse (UPI), which is the window of pregnancy risk associated with a given act of intercourse based upon the estimated lifespan of sperm in the genital tract (Wilcox et al, 1995). Current hormonal methods of emergency contraception prevent at least half of expected pregnancies if taken within 72 hours of UPI (Von Hertzen et al, 1998).
Levonorgestrel at a total dose of 1.5 mg (taken in a single dose or two 0.75 mg doses 12 hours apart) is the current standard for hormonal emergency contraception and is licensed for use up to 72 hours after UPI. Clinical trials involving levonorgestrel used for emergency contraception more than 72 hours after intercourse do not conclusively establish efficacy rates because of insufficient sample size. Nevertheless, these studies reveal a trend towards markedly higher failure rates when levonorgestrel is taken 48 hours or more after unprotected intercourse (von Hertzen et al, 1998; Von Hertzen et al, 2002).
This trend may be explained by levonorgestrel mode of action for emergency contraception. Levonorgestrel acts by interfering with the LH peak but does not appear to interfere with the ovulatory process when taken close to ovulation, a time when intercourse is most likely to lead to fertilization (Croxatto et al, 2004; Marions et al, 2004; Wilcox et al, 2004). For a woman who presents for emergency contraception more than 72 hours after intercourse, the only currently available method proven to be highly effective is insertion of a copper contraceptive intra-uterine device (IUD). However, IUDs are not widely available in many countries and insertion can only be performed by a trained clinician. Furthermore, many women decline IUD insertion as a method of emergency contraception because the procedure is invasive, is relatively expensive and has a risk of complications including uterine perforation on insertion (Grimes et al, 2004). Additionally, many women seeking emergency contraception are not seeking a long acting contraceptive method.
There is, therefore, a need for a new hormonal emergency contraceptive that can be used and is highly effective up to 120 hours after UPI. Ulipristal acetate (also known as CDB-2914) is a selective progesterone receptor modulator that inhibits or delays ovulation in a dose-dependent fashion (Stratton et al, 2000). In a double-blind non-inferiority trial, ulipristal acetate was shown to be as efficacious as levonorgestrel for preventing pregnancy when used within 72 hours of UPI (Creinin et al, 2006). Moreover, study data suggest improved efficacy in preventing pregnancy from 48 to 72 hours when levonorgestrel efficacy markedly wanes. ulipristal acetate for use in providing post coital contraception in a female subject between about 3 to about 5 days, or between about 72 to about 120 hours, after unprotected intercourse.
A subject of the invention is thus a method for providing post coital contraception in a female subject, comprising providing the subject with a therapeutically effective amount of ulipristal acetate, between about 3 to about 5 days, or between about 72 to about 120 hours, after unprotected intercourse. It is further provided a kit comprising i) a dosage form comprising ulipristal acetate and ii) a printed matter stating that ulipristal acetate may be taken within 120 hours or 5 days after unprotected intercourse Any woman of reproductive age may need post-coital or emergency contraception at some point to avoid an unintended pregnancy. It is meant to be used in situations of unprotected intercourse, such as: when no contraceptive has been used;
when there is a contraceptive failure or incorrect use, including: – condom breakage, slippage, or incorrect use; – non-compliance with dosage regimen for combined oral contraceptive pills; – non-compliance with dosage regimen for progestogen-only pill (minipill); – more than two weeks late for a progestogen-only contraceptive injection (depot- medroxyprogesterone acetate or norethisterone enanthate); – more than seven days late for a combined estrogen-plus-progestogen monthly injection; – dislodgment, delay in placing, or early removal of a contraceptive hormonal skin patch or ring; – dislodgment, breakage, tearing, or early removal of a diaphragm or cervical cap; – failed coitus interruptus (e.g., ejaculation in vagina or on external genitalia); – failure of a spermicide tablet or film to melt before intercourse; – miscalculation of the periodic abstinence method or failure to abstain on fertile day of cycle; – IUD expulsion; or in cases of sexual assault when the woman was not protected by an effective contraceptive method.
Uliprisnil acetate, originally developed at the Research Triangle Institute, is a selective progesterone receptor modulator (SPRM) first launched in the E.U. in 2009 by HRA Pharma as emergency contraception within 120 hours (5 days) of unprotected sexual intercourse or contraceptive failure. The company filed for approval of this indication in the U.S. in 2009 and approval was obtained in 2010. In 2012, the product was approved in the E.U. for the pre-operative treatment of moderate to severe symptoms of uterine fibroids in adult women of reproductive age. First E.U. commercialization took place in Germany in March 2012 followed by the U.K. in April. The compound is being developed in phase II clinical trials at the National Institutes of Health (NIH) for the treatment of uterine fibroids and premenstrual syndrome (PMS). Two formulations of uliprisnil are in early clinical trials at the Population Council for the prevention of pregnancy: a vaginal ring and an intrauterine delivery system (IUS). Watson conducted phase III clinical studies for the treatment of women with anemia associated with uterine leiomyoma, however the development has been discontinued.
Uliprisnil acetate is a well-known steroid that possesses antiprogestational and antiglucocorticoid activity. In preclinical studies, the growth of lead follicles exposed to a midfollicular dose of the compound was delayed in a dose-related fashion, indicating that the compound may have an additional mechanism of action involving progesterone or estrogen antagonism.
In 2007, uliprisnil acetate was licensed to PregLem by HRA Pharma in Europe for the treatment of gynecological disorders excluding contraception. A license for North American was granted to HRA in 2010. In 2010, the compound was licensed to Watson (now Actavis) by HRA Pharma for the commercialization in the U.S. for use as emergency contraception. Also in 2010, Watson (now Actavis) obtained a license to uliprisnil for the treatment of uterine fibroids. In 2011, the product was licensed to Gedeon Richter by HRA Pharma for marketing and distribution in China, Russia and (Commonwealth of Independent States) CIS republics for the treatment of uterine myoma.
Treatment of uterine fibroids
Ulipristal acetate is used for pre-operative treatment of moderate to severe symptoms of uterine fibroids in adult women of reproductive age in a daily dose of a 5 mg tablet.[10] Treatment of uterine fibroids with ulipristal acetate for 13 weeks effectively controlled excessive bleeding due to uterine fibroids and reduced the size of the fibroids.[11][12][13] Two intermittent 3-month treatment courses of ulipristal acetate 10 mg resulted in amenorrhea at the end of the first treatment course in 79.5%, at the end of the second course in 88.5% of subjects. Mean myoma volume reduction observed during the first treatment course (−41.9%) was maintained during the second one (−43.7%).[10
Adverse effects
Common side effects include abdominal pain and temporary menstrual irregularity or disruption. Headache and nausea were observed under long-term administration (12 weeks), but not after a single dose.[3]
Interactions
Ulipristal acetate is metabolized by CYP3A4 in vitro. Ulipristal acetate is likely to interact with substrates of CYP3A4, like rifampicin, phenytoin, St John’s wort, carbamazepine or ritonavir, therefore concomitant use with these agens is not recommended.[10][14] It might also interact with hormonal contraceptives and progestogens such as levonorgestrel and other substrates of the progesterone receptor, as well as with glucocorticoids.[10]
Contraindications
Ulipristal acetate should not be taken by women with severe liver diseases[3] because of its CYP mediated metabolism. It has not been studied in women under the age of 18.[15]
Pregnancy
Unlike levonorgestrel, and like mifepristone, ulipristal acetate is embryotoxic in animal studies.[16] Before taking the drug, a pregnancy must be excluded.[3] The EMA proposed to avoid any allusion to a possible use as an abortifacient in the package insert to avert off-label use.[17] It is unlikely that ulipristal acetate could effectively be used as an abortifacient, since it is used in much lower doses (30 mg) than the roughly equipotent mifepristone (600 mg), and since mifepristone has to be combined with a prostaglandin for the induction of abortion.[18] However, data on embryotoxicity in humans are very limited, and it is not clear what the risk for an abortion or for teratogenicity (birth defects) is. Of the 29 women studied who became pregnant despite taking ulipristal acetate, 16 had induced abortions, six had spontaneous abortions, six continued the pregnancies, and one “was lost to follow-up“.[19]
Lactation
It is not recommended to breast feed within 36 hours of taking the drug since it is not known whether ulipristal acetate or its metabolites are excreted into the breast milk.[3][20]
Pharmacokinetics
In animal studies, the drug was quickly and nearly completely absorbed from the gut. Intake of food delays absorption, but it is not known whether this is clinically relevant.[21] Ulipristal acetate is metabolized in the liver, most likely by CYP3A4, and to a small extent by CYP1A2 and CYP2D6. The two main metabolites have been shown to be pharmacologically active, but less than the original drug. The main excretion route is via the faeces.[22]
Pharmacodynamics
As a SPRM, ulipristal acetate has partial agonistic as well as antagonistic effects on the progesterone receptor. It also binds to the glucocorticoid receptor, but has no relevant affinity to the estrogen, androgen and mineralocorticoid receptors.[23] Phase II clinical trials suggest that the mechanism might consist of blocking or delaying ovulation and of delaying the maturation of the endometrium.[24]
History
Ulipristal acetate was granted marketing authorization by the European Medicines Agency (EMA) in March 2009.[25] The U.S. Food and Drug Administration approved the drug for use in the United States on 13 August 2010,[26] following the FDA advisory committee’s recommendation.[27][28] Watson Pharmaceuticals announced the availability of ulipristal acetate in the United States on 1 December 2010, in retail pharmacies, clinics, and one on-line pharmacy, KwikMed.[29] Amorphous ulipristal acetate. ASKA is developing ulipristal acetate in Japan under license from HRA Pharma for the treatment of uterine fibroids and for emergency contraception. In March 2014, it was in phase II for both indications (in Japan). Also see the co-published WO2014050106 and WO2014050107. Crystalline polymorphic form C of ulipristal acetate.
Also claims its method of preparation. Appears to be the first filing from the assignee on this API, which was developed by HRA Pharma under license from the RTI, indicated in the US as an emergency contraceptive for prevention of pregnancy. In May 2011, ASKA signed an exclusive licensing agreement with HRA Pharma to develop and commercialize the API . In November 2013, ASKA had begun phase II development for emergency contraception and uterine fibroids [1339186] in Japan. Also see concurrently published WO2014050105 and WO2014050107. Crystalline polymorphic form B of ulipristal acetate. Also claims process for the preparation and composition comprising the same. Useful for the treatment of uterine leiomyoma.
Appears to be the first filing from the assignee on this API, see concurrently published WO2014050105 and WO2014050106. The drug was developed by HRA Pharma under license from the RTI, indicated in the US as an emergency contraceptive for prevention of pregnancy. In May 2011, ASKA signed an exclusive licensing agreement with HRA Pharma to develop and commercialize the API In November 2013, ASKA had begun phase II development in Japan for emergency contraception and uterine fibroids Buccal forms or devices are also useful, such as those described in U.S. patent application 20050208129 , herein incorporated by reference. U.S. patent application 20050208129 describes a prolonged release bioadhesive mucosal therapeutic system containing at least one active principle, with an active principle dissolution test of more than 70% over 8 hours and to a method for its preparation.
Said bioadhesive therapeutic system comprises quantities of natural proteins representing at least 50% by weight of active principle and at least 20% by weight of said tablet, between 10% and 20% of a hydrophilic polymer, and compression excipients, and comprising between 4% and 10% of an alkali metal alkylsulphate to reinforce the local availability of active principle and between 0.1 % and 1% of a monohydrate sugar.
Ulipristal acetate, formerly known as CDB-2914, designates within the context of this application 17α-acetoxy-11β-[4-N,N-dimethylamino-phenyl)-19-norpregna-4,9-diene-3,20-dione, represented by formula I:

Ulipristal acetate, and methods for its preparation, are described e.g., in U.S. Pat. Nos. 4,954,490; 5,073,548, and 5,929,262, as well as in international patent applications WO2004/065405 and WO2004/078709. Ulipristal acetate possesses antiprogestational and antiglucocorticoidal activity, and has been proposed for contraception, in particular for emergency contraception, and for the therapy of various hormonal diseases.
(Steroids, 2000,65, 395 ~ 400; US5929262A; CN1298409A; CN101466723A). Reaction is as follows:

Properties of this compound are further described in Blithe et al, Steroids. 2003 68(10-13):1013-7. So far, clinical trials have been conducted using oral capsules of ulipristal acetate (Creinin et al, Obstetrics & Gynecology 2006; 108:1089-1097; Levens et al, Obstet Gynecol. 2008, 111(5):1129-36). In order to increase the properties and clinical benefit of this molecule, there is a need for improved formulations thereof
-
Ulipristal acetate, formerly known as CDB-2914, is 17α-acetoxy-11β-[4-N, N-dimethylamino-phenyl)-19-norpregna- 4, 9-diene-3, 20-dione, represented by formula I:

-
It is a well-known steroid, more specifically a 19-norprogesterone, which possesses antiprogestational and antiglucocorticoidal activity. This compound, and methods for its preparation, are described in U. S. Patent Nos. 4,954, 490,5 , 073,548 , and 5,929, 262 , and international patent applications WO2004/065405 and WO2004/078709 . Properties of this compound are further described in Blithe et al, 2003.
-
Metabolites of CDB-2914, include those described in Attardi et al, 2004 , e.g. monodemethylated CDB-2914 (CDB-3877) ;didemethylated CDB-2914 (CDB-3963) ; 17alpha-hydroxy CDB-2914 (CDB-3236) ; aromatic A-ring derivative of CDB-2914 (CDB-4183).




-
It is now proposed to use ulipristal acetate or a metabolite thereof for treating uterine fibroids, more particularly for reducing or stopping bleeding in a patient afflicted with uterine fibroids, reducing the size of uterine fibroids and/or reducing uterine volume More particularly the inventors have shown in a randomized, placebo-controlled, double blinded, parallel trial, that ulipristal acetate significantly reduces fibroid volume after 3 months, and stops bleeding
-
Ulipristal acetate or a metabolite thereof alleviates symptoms of uterine fibroids, including bleeding, pelvic pain, pressure.
-
Ulipristal acetate or a metabolite thereof is useful for preventing or treating anemia in patients afflicted with uterine fibroids.
-
It is also useful for preventing or treating leiomyosarcomas and for preventing dissemination of uterine fibroids to other organs.





………………
synthesis
http://www.google.com.br/patents/US5929262
CA2216737A1, EP0817793A2, WO1996030390A2, WO1996030390A3
The United States Of America As Represented By The Department Of Health And Human Services 
 EXAMPLE 7 The Preparation of the Compound of Formula (I) (17α-Acetoxy-11β-(4-N,N-dimethylaminophenyl)-19-norpregna-4,9-diene-3,20-dione) From the Compound of Formula (VIII) 340 mL of acetic acid (5.92 mol) were added to a well stirred mixture containing 834 mL of trifluoroacetic anhydride (5.92 mol) in 2,300 mL of methylene chloride under argon. After stirring for 30 minutes at room temperature, 51.3 g of p-toluenesulfonic acid (0.26 mol) were added, and the mixture was chilled to 0 methylene chloride solution containing 128.3 g of the compound of formula (VIII) (0.30 mol) were added, and the reaction mixture was stirred at 0 cautious addition of a 4.5N potassium carbonate solution until the pH was in the range of 7.0-7.5. The reaction mixture was diluted with water and extracted with methylene chloride. The methylene chloride extracts were washed with water and brine, combined, and dried over sodium sulfate.
EXAMPLE 7 The Preparation of the Compound of Formula (I) (17α-Acetoxy-11β-(4-N,N-dimethylaminophenyl)-19-norpregna-4,9-diene-3,20-dione) From the Compound of Formula (VIII) 340 mL of acetic acid (5.92 mol) were added to a well stirred mixture containing 834 mL of trifluoroacetic anhydride (5.92 mol) in 2,300 mL of methylene chloride under argon. After stirring for 30 minutes at room temperature, 51.3 g of p-toluenesulfonic acid (0.26 mol) were added, and the mixture was chilled to 0 methylene chloride solution containing 128.3 g of the compound of formula (VIII) (0.30 mol) were added, and the reaction mixture was stirred at 0 cautious addition of a 4.5N potassium carbonate solution until the pH was in the range of 7.0-7.5. The reaction mixture was diluted with water and extracted with methylene chloride. The methylene chloride extracts were washed with water and brine, combined, and dried over sodium sulfate.
Evaporation of the solvent gave the acetate of formula (I) as a thick syrup. The above syrup was dissolved in 300 mL of isopropyl alcohol and evaporated. The dissolution and evaporation were repeated three times. Finally, the remaining solid, which retained isopropyl alcohol as solvent of recrystallization, was dissolved in ethyl acetate and evaporated to give a stable foam. The foam was quickly dissolved in ether, and this solution was set aside to crystallize. The solid that formed was collected by filtration, washed with ether, and dried in vacuo to yield 105.7 g of the compound of formula (I) as yellow crystals in 75% yield;
m.p. 183-185 1735 and 1714(–C═O), 1664 and 1661 (conjugated –C═O), 1563, 1518, 1441, 1351, 1305, 1252, 1203, 1171; NMR (CDCl.sub.3) δ0.38 (s, 18-CH.sub.3), 2.10 (s, 17-OAc), 2.14 (s, 21-CH.sub.3), 2.92 (s, –N(CH.sub.3).sub.2, 4.44 (d, C-11 H), 5.83 (br. s, C-4 H), 6.71 and 7.07 (d, aromatic H); MS(EI) m/z (relative intensity) 475(M.sup.+, 41), 134(18), 121 (100). Analysis calculated for C.sub.30 H.sub.37 NO.sub.4 : C, 75.76; H, 7.84; N, 2.94. Found. C, 75.80; H 7.96; N, 3.09.
……………..
SYNTHESIS http://www.google.com/patents/WO2004065405A1?cl=en
CA2514169A1, CA2514169C, CN1753905A, CN100354300C, EP1602662A1, EP2348033A2, EP2348033A3
EXAMPLE 1 Preparation of 17α-aceto-d-llβ-(4-N, N-dimetüaminofeniI)-19-norpregna-4 ,9-dien-3,20-dione [VA-2914] Raw Were charged 38.5 g of 3,3 – (l ,2-etanodioxi)-5α-hydroxy-llβ-(4-N, N-dimethylaminophenyl)-17α-acetoxy-19-norpregna-9-en-20-one [carbinol acetate] purified in a flask under nitrogen atmosphere at a temperature between 20 ° C and 22 ° C, and added 385 ml of deionized water and 17.91 g of HKSO. The resulting suspension was stirred until complete dissolution, for about 4 hours. The end of the reaction was determined by thin layer chromatography (TLC). Then added 3.85 g of neutral Al 2 O 3, stirred for 30 minutes, the suspension was filtered and the insolubles were washed with 38.5 ml of deionized water. To the filtrate were added 325 ml of ethyl acetate and the pH was adjusted to a constant value between 7.0 and 7.2 with sodium bicarbonate solution to 7% w / v. The phases were allowed to decant for 15 minutes and, after checking the absence of the final product therein by means of TLC, the phases were separated, discarding the aqueous phase. The resultant organic phase was added 192.5 ml of deionized water, stirred for 10 minutes and the phases were allowed to decant for 15 minutes.
After verifying the absence of aqueous phase final product by TLC, the phases were separated, discarding the aqueous phase. The resulting organic phase was concentrated under vacuum to a residue and obtained approximately 28 g of 17α-acetoxy-llβ-(4-N, N-dimethylaminophenyl)-19-norpregna-4,9-dien-3 ,20-dione [NA -2914] raw. EXAMPLE 2 Isopropanol hemisolvate obtaining 17α-acetoxy-llβ-(4-Ν, Ν-dimethylaminophenyl)-19-norpregna-4 ,9-dien-3 ,20-dione The crude 17α-acetoxy-l lβ-(4-Ν, Ν-dimethylaminophenyl)-19-norpregna-4 ,9-dien-3,20-dione obtained in Example 1 was added 2 x 38.5 ml isopropanol concentrating vacuum to a residue both times. The finally obtained solid was added 77 ml of isopropanol and heated until dissolved. Then allowed to cool to a temperature between 0 ° C and 5 ° C, and the temperature was maintained for 1 h. The resulting suspension was filtered and the cake washed with cold isopropanol.
The yield achieved was 96% molar (5.5% isopropanol content). Isopropanol hemisolvate obtemdo NA-2914 has been characterized by IR spectroscopy, DSC and XRD, as indicated in the description, and has the characteristics indicated therein and shown in Figures 1-3.
…………….
A new and efficient method for the synthesis of Ulipristal acetate
http://www.sciencedirect.com/science/article/pii/S0039128X14000634


In this study, we describe another new and efficient route for preparing Ulipristal acetate. The 1,4-addition compound 5 was greatly improved after the starting material ketone 1 was underwent epoxidation, cyanation, hydroxyl group protection and Grignard addition. The synthetic procedure is only 6 steps and the total yield is about 27.4%, which is much suitable for industrial process.
We have succeeded in finding another convenient and efficient synthetic route for the synthesis of Ulipristal acetate with a good yield.
•The yield of 11β-substituted isomer was greatly improved.
•The 17β-carbonitrile compound was obtained with high purity after the reaction.
•The yield of once Grignard addition dione was greatly improved.
•These synthetic procedures are much suitable for industrial process.
…………….
Volume 78, Issues 12–13, 11 December 2013, Pages 1293–1297
http://www.sciencedirect.com/science/article/pii/S0039128X13002122
We set out to describe a new and efficient route for preparing Ulipristal acetate with a good yield. The selected epoxidization conditions gave out 80% of 5α,10α-epoxide 2a in the two diastereoisomers which greatly improved the yield of 11β-substituted isomer 4a. And phenyl–sulfinyl compound 6 was synthesized from ketone 5 directly treated with phenylsulfenyl chloride in the presence of triethylamine. These synthetic procedures is only 8 steps, less than currently reported in the literature, but more suitable for industrial process.

………..
http://www.google.com/patents/CN103145787A?cl=en
Reaction is as follows:

………..
WO2013063859A1
http://www.google.com/patents/WO2013063859A1?cl=en
Preparation of related reports Uli Division acetate compounds as follows:
1, U.S. Patent US4954490 methods, (see Reaction Scheme 1),
The method is based on the 3 – methoxy -19 – norpregn-1, 3,5 (10), 17 (20) – tetraene as a starting material, in turn by the addition, oxidation, reduction, hydrolysis, addition and elimination, oxidation of 17-hydroxy-19 – norpregn left the -4,9 – 31 women -3, 20 – dione (Compound V2), and then condensed by ethylene glycol, epoxidized-chloroperbenzoic acid, Bonus format, acid hydrolysis, acetylation of 10-step reaction by Uli acetate SECRETARY (Compound 1), and a melting point of 118-121 ° C the product was obtained by recrystallization with methanol ^. Due to the method, the step length, but difficult to obtain a starting material, the complexity of the reaction conditions, the required intermediate product was purified by column chromatography, the total yield is only 0.62%, Gao costs, the instability of the resulting product is not suitable pharmaceutically acceptable. And is not suitable for industrial production.
Reaction Formula I:


2, U.S. Patent No. US5929262 discloses his method Another method for preparing acetic acid Uli Division (see Reaction Scheme II), the reaction of formula II:

The method is based on 3,3 – ethylenedioxy-17) 8 – cyano -19 – norpregn -5 (10)-9 (11) – dien-17 alcohol (compound III) as a starting material, , first with dimethyl chloromethyl silane protected hydroxy, and then at the cryogenic-70Ό obtained by acid hydrolysis with the DBB / LI reagents After the reaction, a condensation reaction with ethylene glycol ketal, epoxy reaction, then the format of the reaction, The acid hydrolysis reaction and the acetylation reaction to obtain the target object and sequentially by treatment with isopropanol, ethyl acetate and crystallized from ether to obtain a yellow product with a melting point of 183-185Ό. The method expensive starting materials prices, harsh reaction conditions, need to be ultra-low temperature and water and oxygen reaction, high cost of low yield (total yield of about 14%), and therefore not suitable for industrial production.
3, World Patent WO2004078709 discloses a method for preparing (see Reaction Scheme III), the method the Πα hydroxy _19_ norpregn _ 4, 9 (10) _ diene-_ _ 3, 17-dione (Compound V2 ), followed by acetylation of 3 – bit carbonyl condensation, epoxy, Bonus format, acid hydrolyzed to give the target. Although the steps are shorter, but a starting material is from Compound VI was prepared by hydrolysis under acidic conditions to obtain a total yield of about 11.8% (starting from the compound VI operator), the actual reaction step is longer, lower yield, higher cost not suitable for industrial production.

In this method, 3,3 – ethylenedioxy -19 – norpregn -5 (10), 9 (11) – dien-17 – one (referred to as 3 – ketal compound II) as a starting material, by the addition of acetylene, benzene sub-sulfonyl chloride, and then by hydrolysis of sodium methoxide, acid hydrolysis, condensation of ethylene glycol, epoxy, Grignard reaction, acid hydrolysis and acetylation reaction of 9-step reaction to obtain a target object, isopropoxy alcohol crystallization with ethanol and water was heated at 70 ° C after 14h excluding solvate crystal. The method uses a greater risk of acetylene and odor of benzene times sulfonyl chloride, especially benzene times, unstable sulfonyl chloride, easy storage, decomposition of impurities involved in the reaction leads to a low yield, and benzene of times sulfonyl chloride of environmental pollution Further crystallization prolonged heating will produce new impurities, the total yield of 13.8% -15.8%, high cost, is not suitable for industrial production.
The existing methods, the methods 1, 2 and 4 are related to the preparation of compound VI, and also the starting materials in Method 3 Hydrolysis of compound VI is obtained. SUMMARY OF THE INVENTION
Technical problems to be solved by the present invention is to overcome these drawbacks,, study design Uli acetate Secretary industrialization production methods.
WO2013063859A1
http://www.google.com/patents/WO2013063859A1?cl=en


………
|
Ulipristal Acetate intermediates(7)
|
![19-Norpregn-9-ene-3,20-dione, 11-[4-(dimethylamino)phenyl]-5,17-dihydroxy-, cyclic 3,20-bis(1,2-ethanediyl acetal), (5α,11β)-](https://i0.wp.com/www.lanospharma.com/images/19-Norpregn-9-ene-3%2C20-dione%2C%2011-%5B4-%28dimethylamino%29phenyl%5D-5%2C17-dihydroxy-%2C%20cyclic%203%2C20-bis%281%2C2-ethanediyl%20acetal%29%2C%20%285%CE%B1%2C11%CE%B2%29-.gif)

![19-Norpregna-4,9-diene-3,20-dione, 17-(acetyloxy)-11-[4-(methylamino)phenyl]-, (11β)-](https://i0.wp.com/www.lanospharma.com/images/19-Norpregna-4%2C9-diene-3%2C20-dione%2C%2017-%28acetyloxy%29-11-%5B4-%28methylamino%29phenyl%5D-%2C%20%2811%CE%B2%29-.gif)




…………
Bioorganic & Medicinal Chemistry
Keywords: Synthesis. New drug molecules. New chemical entities. Medicine. Therapeutic agents. a b s t r a c t …. 1153. 22. Ulipristal acetate (ellaOne®).
…………. FORMULATION http://www.google.com/patents/WO2011091892A1?cl=en 
References
- “FDA approves ella™ tablets for prescription emergency contraception” (Press release). FDA. 13 August 2010. Retrieved 2013-06-12.
- Creinin, MD; Schlaff, W; Archer, DF; Wan, L; Frezieres, R; Thomas, M; Rosenberg, M; Higgins, J (2006). “Progesterone receptor modulator for emergency contraception: a randomized controlled trial”. Obstetrics and gynecology 108 (5): 1089–97. doi:10.1097/01.AOG.0000239440.02284.45. PMC 2853373. PMID 17077229.
- “Summary of Product Characteristics: ellaOne 30 mg tablet”. Retrieved 20 November 2010.
- “European Public Assessment Report for Ellaone. Summary for the public”. EMA. 2009. p. 2. Retrieved 22 November 2009.
- Glasier, A. F.; Cameron, S. T.; Fine, P. M.; Logan, S. J.; Casale, W.; Van Horn, J.; Sogor, L.; Blithe, D. L.; Scherrer, B.; Mathe, H.; Jaspart, A.; Ulmann, A.; Gainer, E. (2010). “Ulipristal acetate versus levonorgestrel for emergency contraception: A randomised non-inferiority trial and meta-analysis”. The Lancet 375 (9714): 555–562. doi:10.1016/S0140-6736(10)60101-8. PMID 20116841.
- Trussell, James; Cleland, Kelly (February 13, 2013). “Dedicated emergency contraceptive pills worldwide”. Princeton: Office of Population Research at Princeton University, Association of Reproductive Health Professionals. Retrieved March 25, 2014.
- ICEC (2014). “EC pill types and countries of availability, by brand”. New York: International Consortium for Emergency Contraception (ICEC). Retrieved March 25, 2014.
- HRA Pharma (March 2013). “Countries where ellaOne was launched”. Paris: HRA Pharma. Retrieved March 25, 2014.
- ECEC (2014). “Emergency contraception availability in Europe”. New York: European Consortium for Emergency Contraception (ECEC). Retrieved March 25, 2014. “Ulipristal acetate Emergency Contraception Pills (UPA ECPs), while available in most European countries since 2010, are not yet available in Albania, Estonia, Macedonia, Malta, Switzerland and Turkey. For now UPA ECPs are sold with a prescription in all countries, although provision without a prescription is currently being tested in the United Kingdom.”
- “Summary of Product Characteristics: Esmya 5mg tablet”. Retrieved 20 Febr 2014.
- Nieman, L. K.; Blocker, W.; Nansel, T.; Mahoney, S.; Reynolds, J.; Blithe, D.; Wesley, R.; Armstrong, A. (2011). “Efficacy and tolerability of CDB-2914 treatment for symptomatic uterine fibroids: A randomized, double-blind, placebo-controlled, phase IIb study”. Fertility and Sterility 95 (2): 767–772.e1–772. doi:10.1016/j.fertnstert.2010.09.059. PMID 21055739.
- Levens, E. D.; Potlog-Nahari, C.; Armstrong, A. Y.; Wesley, R.; Premkumar, A.; Blithe, D. L.; Blocker, W.; Nieman, L. K. (2008). “CDB-2914 for Uterine Leiomyomata Treatment”. Obstetrics & Gynecology 111 (5): 1129–1136. doi:10.1097/AOG.0b013e3181705d0e. PMC 2742990. PMID 18448745.
- Jacques Donnez; Tetyana F. Tatarchuk, Philippe Bouchard, Lucian Puscasiu, Nataliya F. Zakharenko, Tatiana Ivanova, Gyula Ugocsai, Michal Mara, Manju P. Jilla, Elke Bestel, Paul Terrill, Ian Osterloh, and Ernest Loumaye, for the PEARL I Study Group. “Ulipristal Acetate versus Placebo for Fibroid Treatment before Surgery”. New England Journal of Medicine. doi:10.1056/NEJMoa1103182. PMID 22296075.
- CHMP (2009:12, 14)
- CHMP (2009:33, 43)
- CHMP (2009:16)
- CHMP (2009:41)
- RCOG (2004). The Care of Women Requesting Induced Abortion : Evidence-based clinical guideline number 7 (PDF). London: RCOG Press. ISBN 1-904752-06-3. Archived from the original on 27 February 2008.
- CHMP (2009:37)
- CHMP (2009:43)
- CHMP (2009:12, 20)
- CHMP (2009:13–14, 21)
- Attardi, B.; Burgenson, J.; Hild, S.; Reel, J. (2004). “In vitro antiprogestational/antiglucocorticoid activity and progestin and glucocorticoid receptor binding of the putative metabolites and synthetic derivatives of CDB-2914, CDB-4124, and mifepristone”. The Journal of Steroid Biochemistry and Molecular Biology 88 (3): 277–288. doi:10.1016/j.jsbmb.2003.12.004. PMID 15120421.
- CHMP (2009:22–23)
- CHMP (2009). “Assessment Report for Ellaone”. EMA. Retrieved 22 November 2009.
- “FDA grants approval of ella for emergency contraception” (Press release). HRA Pharma. 13 August 2010. Retrieved 2010-08-15.
- Emma Hitt (18 June 2010). “FDA Panel Gives Ulipristal Acetate Unanimous Positive Vote for Emergency Contraception Indication”. Retrieved 2010-06-22.
- Harris, Gardiner (14 August 2010). “F.D.A. Approves 5-Day Emergency Contraceptive”. The New York Times. Retrieved 14 August 2010.
- Watson PR (1 December 2010). “Watson Launches ella(R)(ulipristal acetate)”. Retrieved 12 January 2010.\
| WO2004065405A1 | Jan 21, 2004 | Aug 5, 2004 | Crystal Pharma S A | Method of obtaining 17$g(a)-acetoxy-11$g(b)-(4-n,n-dimethylaminophenyl)-19-norpregna-4,9-diene-3,20-dione |
| WO2004078709A2 | Feb 13, 2004 | Sep 16, 2004 | Hyun K Kim | METHOD FOR PREPARING 17 α-ACETOXY-11β-(4-N,N-DIMETHYLAMINOPHENYL)-19-NORPREGNA-4,9-DIENE-3,20-DIONE, INTERMEDIATES THEREOF, AND METHODS FOR THE PREPARATION OF SUCH INTERMEDIATES |
| US4954490 | Jun 23, 1988 | Sep 4, 1990 | Research Triangle Institute | 11 β-substituted progesterone analogs |
| US5073548 | Apr 3, 1990 | Dec 17, 1991 | Research Triangle Institute | 11 β-substituted progesterone analogs |
| US5929262 | Mar 30, 1995 | Jul 27, 1999 | The United States Of America As Represented By The Department Of Health And Human Services | Method for preparing 17α-acetoxy-11β-(4-N, N-dimethylaminophyl)-19-Norpregna-4,9-diene-3, 20-dione, intermediates useful in the method, and methods for the preparation of such intermediates |
| US20050208129 | Apr 25, 2005 | Sep 22, 2005 | Bioalliance Pharma | Prolonged release bioadhesive therapeutic systems |
| WO2001074840A2 * | Mar 16, 2001 | Oct 11, 2001 | Carmie K Acosta | 17-alpha-substituted-11-beta-substituted-4-aryl and 21-substituted 19-norpregna 21-substituted 19-norpregnadienedione as antiprogestational agents |
| CN101466723A * | May 18, 2007 | Jun 24, 2009 | 吉瑞工厂 | Industrial process for the synthesis of 17a-acetoxy-11ss-[4-(n,n-dimethyl-amino)- phenyl]-19-norpregna-4,9-diene-3,20-dione and new intermediates of the process |
| CN102516345A * | Nov 1, 2011 | Jun 27, 2012 | 上海优拓医药科技有限公司 | Preparation method of ulipristal acetate and key intermediate thereof |
| US5929262 * | Mar 30, 1995 | Jul 27, 1999 | The United States Of America As Represented By The Department Of Health And Human Services | Method for preparing 17α-acetoxy-11β-(4-N, N-dimethylaminophyl)-19-Norpregna-4,9-diene-3, 20-dione, intermediates useful in the method, and methods for the preparation of such intermediates |

Biochips for better cancer therapy

Cancer is the second leading cause of disease-related death in the United States, and may overtake heart disease without aggressive new therapies. One promising area of cancer treatment is photodynamic therapy (PDT), which combines the agents of a photosensitive drug, light, and oxygen to attack cancerous tumors and lesions locally in the targeted region of the body by selective optical illumination.
Research being conducted by Prof. Euisik Yoon’s group aims to dramatically accelerate progress in PDT. And it is being accomplished through a lab-on-a-chip measuring about the size of a quarter. At the heart of this biochip is a 5x5mm testing area that will test the interaction of the drug, light, and oxygen simultaneously, generating results in a fraction of the time of current testing practices.
“In cancer research doctors are always looking for better drugs,” explained Dr. Xia Lou, a postdoctoral fellow in Prof. Yoon’s group. “But there has…
View original post 392 more words
Clinafloxacin from kyorin

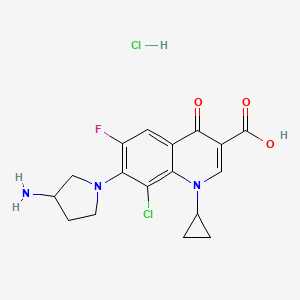
Clinafloxacin
7-(3-Aminopyrrolidin-1-yl)-8-chloro-1-cyclopropyl-6-fluoro-4-oxoquinoline-3-carboxylic acid
7-(3-Amino-1-pyrrolidinyl)-8-chloro-1-cyclopropyl-6-fluoro-l,4-dihydro-4-oxo-3-quinolinecarboxylic acid
(±)-7-(3-Amino-1-pyrrolidinyl)-8-chloro-1-cyclopropyl-6-fluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid
105956-99-8 cas no
Clinafloxacin (INN) is a fluoroquinolone antibiotic. Its use is associated with phototoxicity and hypoglycaemia.[1]
Clinafloxacin is a novel quinolone with wide activity against the plethora of microorganisms encountered in intraabdominal infections.
Clinafloxacin is a chlorofluoroquinolone with excellent bioavailability and activity against gram-positive, gram-negative, and anaerobic pathogens . Typical MICs for α-streptococci are 0.06–0.12 µg/mL . MIC90 values for methicillin-resistant Staphylococcus aureus (MRSA) average 1.0 µg/mL. The MIC90 for enterococci is typically 0.5 µg/mL . Both intravenous and oral formulations have been developed . Several studies have demonstrated the efficacy of clinafloxacin monotherapy for serious infections Clinafloxacin was also active in animal models of endocarditis, including endocarditis due to ciprofloxacin-resistant S. aureus infection .



-
A mixture of 8-chloro-1-cyclopropyl-6,7-difluoro-1,4-di- hydro-4-oxo-3-quinolinecarboxylic acid (0.6 g), anhydrous acetonitrile (6 ml), 3-aminopyrrolidine (0.35 g) and DBU (0.31 g) was refluxed for an hour. Then, 3-aminopyrrolidine (0.2 g) was more added and further refluxed for 2 hours. After cooling, the resulting precipitate was collected by filtration, dissolved in water (9 ml) containing sodium hydroxide (0.12 g) and neutralized with acetic acid. The resulting precipitate was collected by filtration and washed with water and acetonitrile successively to give the title compound (0.52 g) as colorless powder, mp 237-238 °C (decompd.).
-
Analysis (%) for C17H17ClFN3O3·H2O, Calcd. (Found): C, 53.20 (52.97); H, 4.99 (4.62); N, 10.95 (10.83).
- Example 28 7-(3-Amino-1-pyrrolidinyl)-8-chloro-1-cyclopropyl-6-fluoro-l,4-dihydro-4-oxo-3-quinolinecarboxylic acid
Example 29 7-(3-Amino-1-pyrrolidinyl)-8-chloro-1-cyclopropyl-6-fluoro-l,4-dihydro-4-oxo-3-quinolinecarboxylic acid hydrochloride
-
To a suspension of 7-(3-amino-1-pyrrolidinyl)-8-chloro-1-cyclopropyl-6-fluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid (100 mg) in ethanol (2 ml) was added 0.2 ml of ethanol solution of hydrogen chloride (7.0 mmol HC1/ml) and then the mixture was concentrated. The resulting residue was recrystallized from methanol to give the title compound (79 mg) as light yellow prisms, mp 263-265 °C (decompd.).
-
Analysis (%) for C17H17ClFN3O3.HCl, Calcd. (Found): C, 50.76 (50.50); H, 4.51 (4.44); N, 10.45 (10.38).
…………………..
J. Med. Chem., 23, 1358 (1980)

-
structural formula D

may be readily prepared from the known starting material methyl 5-oxo-l-(phenylmethyl)-3-pyrrolidinecarboxylate, A, [J. Org. Chem., 26, 1519 (1961)] by the following reaction sequence.

-
The compound wherein R3 is hydrogen, namely 3-pyrrolidinemethanamine, has been reported in J. Org. Chem., 26, 4955 (1961).


References
- Rubinstein, E. (2001). “History of quinolones and their side effects.”. Chemotherapy. 47 Suppl 3: 3–8; discussion 44–8.doi:10.1159/000057838. PMID 11549783.
| EP0106489A2 * | Sep 6, 1983 | Apr 25, 1984 | Warner-Lambert Company | Antibacterial agents |
| EP0153163A2 * | Feb 15, 1985 | Aug 28, 1985 | Warner-Lambert Company | 7-Substituted-1-cyclopropyl-6,8-difluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acids; 7-substituted-1-cyclopropyl-1,4-dihydro-6-fluoro-4-oxo-1,8-naphthyridine-3-carboxylic acids; their derivatives; and a process for preparing the compounds |
| BE899399A1 * | Title not available | |||
| GB2057440A * | Title not available |
| Examples of | ||
| reported trade | ||
| names for products | ||
| containing the 6- | ||
| 6-Fluoroquinolin- | fluoroquinolin- | |
| 4(1H)-one | 4(1H)-one | Structure |
| amifloxacin |
 |
|
| balofloxacin |
 |
|
| ciprofloxacin | Cipro®, Ciprobay, & Ciproxin |
 |
| clinafloxacin |
 |
|
| danofloxacin | Advocin & Advocid |
 |
| difloxacin | Dicural® & Vetequinon |
 |
| enrofloxacin | Baytril® |
 |
| fleroxacin | Megalone |
 |
| flumequine | Flubactin |
 |
| garenoxacin |
 |
|
| gatifloxacin | Tequin® & Zymar® |
 |
| grepafloxacin | Raxar |
 |
| ibafloxacin |
 |
|
| levofloxacin | Levaquin®, Gatigol, Tavanic, Lebact, Levox, & Cravit |
 |
| lomefloxacin | Maxaquin® |
 |
| marbofloxacin | Marbocyl® & Zenequin |
 |
| moxifloxacin | Avelox® & Vigamox® |
 |
| nadifloxacin | Acuatin, Nadoxia, & Nadixa |
 |
| norfloxacin | Noroxin®, Lexinor, Quinabic, & Janacin |
 |
| ofloxacin | Floxin®, Oxaldin, & Tarivid |
 |
| orbifloxacin | Orbax® & Victas |
 |
| pazufloxacin |
 |
|
| pefloxacin |
 |
|
| pradofloxacin |
 |
|
| prulifloxacin |
 |
|
| rufloxacin | Uroflox |
 |
| sarafloxacin | Floxasol, Saraflox, Sarafin |
 |
| sitafloxacin |
 |
|
| sparfloxacin | Zagam |
 |
| temalioxacin | Omniflox |
 |
| enoxacin | Penetrex & Enroxil |
 |
| gemifloxacin | Factive |
 |
| tosufloxacin |
 |
|
| trovafloxacin | Trovan |
 |
