
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 288)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Novel Oxazolidinone Antibacterial Candidate FYL-67 …..(S)-N-((3-(3-Fluoro-4-(4-(pyridin-2-yl)-1H-pyrazol-1-yl)phenyl)-2-oxo-oxazolidin-5-yl)methyl)acetamide

cas no 1416314-55-0
C20 H18 F N5 O3
FYL-67 IS HYDROCHLORIDE
(S)-N-((3-(3-Fluoro-4-(4-(pyridin-2-yl)-1H-pyrazol-1-yl)phenyl)-2-oxo-oxazolidin-5-yl)methyl)acetamide
N-[[(5S)-3-[3-fluoro-4-[4-(2-pyridinyl)-1H-pyrazol-1-yl]phenyl]-2-oxo-5-oxazolidinyl]methyl]-Acetamide,
(S)-N-((3-(3-fluoro-4-(4-(pyridin-2-yl)-1H-pyrazol-1-yl) phenyl)-2-oxooxazolidin-5-yl)methyl)acetamide.
| Inventores | Youfu LUO, 罗有福, Zhenling WANG, 王震玲,Yuquan Wei, 魏于全 |
| Requerente | Si Chuan University, 四川大学 |
The discovery and application of antibiotics is one of the greatest achievements of mankind in the 20th century, the field of medicine, called a revolution of the history of the human fight against illness. Since then, the field of medicine into a bacterial disease caused by greatly reducing the golden age. Today, however, due to the widespread use of antibiotics or even abuse, the growing problem of bacterial resistance, humans are gradually approaching the “post-antibiotic era, the efficacy of antibiotics is gradually reduced. Clinical have been found on many new drug-resistant strains of methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant enterococci (VRE), penicillin-resistant Streptococcus pneumoniae (PRSP) has seriously jeopardize the clinical treatment , the number of varieties of drugs less.
The compounds of the oxazolidinone linezolid was in the United States in 2000, mainly used in clinical acquired pneumonia, soft tissue infections, can also be used for the surgical treatment of infectious diseases, bones, lungs, cerebrospinal fluid permeability pharmacokinetic and tissue concentrations. Domestic and foreign the oxazolidinone drug development is a hot field
WO 2012171479
http://www.google.st/patents/WO2012171479A1?cl=en



The object compound (S N-{[3 – (3 – fluoro-4 – (4 – (2 – pyridyl) pyrazol-yl) phenyl) -2 – oxo-oxazol the embankment -5 – yl] methanone yl}
Weigh 150mg of the compound (26f), was dissolved with 10 ml of anhydrous THF was added under nitrogen protection, an ice water bath 154.1 mg t-BuOLi, ice-water bath after stirring for 5 minutes, 149.9 mg Compound 11, followed by ice-water bath was removed, go reaction at room temperature for 36 hours the reaction was stopped, by adding 10 mL of methylene chloride and 10 ml of water and 22μί acetic acid, stirred for 1 minute, the liquid separation, the aqueous phase was extracted with dichloromethane three times, the organic phases were combined, dried and purified by column chromatography to give the product ( 130 white solid 58 mg of yield of 38.2%.
1H-MR (400 MHz, CDC1 3): δ 8.61 (d, J = 4Hz, IH), 8.52 (d, J = 6.8Hz, 2.4H), 8.22 (s, IH), 7.94 (t, J = 8.8 Hz, IH), 7.77-7.69 (m, 2H), 7.55 (d, J = 8Hz, IH), 7.27-7.26 (m, IH), 7.18-7.15 (m, IH), 6.06 (t, J = 6Hz , IH), 4.86-4.80 (m, IH), 4.11 (t, J = 9.2Hz, IH), 3.86-3.82 (m, IH), 3.78-3.62 (m, 2H), 2.04 (s, 3H 😉 .
13 C-MR (DMSO-e): δ 170.51, 154.47, 152.94, 151.26, 149.94, 139.70, 139.15, 137.43 129.96, 125.61, 125.19, 123.42, 122.19, 120.38, 114.52, 106.68, 72.29, 47.70, 41.84, 22.91.
ESI-MSm / z 418.08 (M + Na +).
………………….
Nanoscale (2013), 5(1), 275-283
Carrier-free nanoassemblies of a novel oxazolidinone compound FYL-67 display antimicrobial activity on methicillin-resistant Staphylococcus aureus
E-mail: luo_youfu@scu.edu.cn, wangzhenling2007@126.com;
Fax: +86-28-85164060 ;
Tel: +86-28-85164063
DOI: 10.1039/C2NR32505E
In this work, a novel oxazolidinone compound FYL-67 was synthesized, and the obtained FYL-67 could form nanoassemblies in aqueous solution by a self-assembly method without using any carrier, organic solvent, or surfactant. The prepared FYL-67 nanoassemblies had a particle size of 264.6 ± 4.3 nm. The FYL-67 nanoassemblies can be lyophilized into a powder form without any cryoprotector or excipient, and the re-dissolved FYL-67 nanoassemblies are stable and homogeneous. The in vitro release profile showed a significant difference between rapid release of free FYL-67 and much slower and sustained release of FYL-67 nanoassemblies. In vitro susceptibility tests were conducted in three strains of methicillin-susceptibleStaphylococcus aureus (MSSA) and three strains of methicillin-resistant Staphylococcus aureus(MRSA), using linezolid as a positive control. FYL-67 nanoassemblies exhibited excellent in vitro activity, with a minimum inhibitory concentration (MIC) value of 0.5 μg mL−1 against MRSA. In the in vitro post-antibiotic effect (PAE) evaluation, FYL-67 nanoassemblies showed a more powerful effect than linezolid. Besides, in vitro cytotoxicity tests indicated that FYL-67 nanoassemblies had a very low cytotoxicity on HEK293 cells and L02 cells. Furthermore, in both MSSA and MRSA systemic infection mouse models, FYL-67 nanoassemblies showed a lower ED50 than linezolid. In a murine model of MRSA systemic infection, FYL-67 nanoassemblies displayed an ED50 of less than 4.0 mg kg−1, which is 2.3-fold better than that oflinezolid. Our findings suggested that the FYL-67 nanoassemblies may be a potential drugcandidate in MRSA therapy.

 |
||
| Fig. 1 Synthetic route of the novel compound FYL-67. (i) 2-(pyridin-2-yl)malonaldehyde, p-TsOH (cat.),ethanol, reflux, 2 h; (ii) Fe, HCl, 95% ethanol, 1 h; (iii) Cbz–Cl, K2CO3, CH2Cl2, 2 h; (iv) (S)-1-acetamido-3-chloropropan-2-yl acetate, LiOt-Bu, THF, r.t.; (v) HCL (g), acetone, ethyl ether | ||
1H-NMR (400 MHz, CDCl3): δ 8.61 (d, J = 4 Hz, 1H), 8.52 (d, J = 6.8 Hz, 2.4H), 8.22 (s, 1H), 7.94 (t, J = 8.8 Hz, 1H), 7.77–7.69 (m, 2H), 7.55 (d, J = 8 Hz, 1H), 7.27–7.26 (m, 1H), 7.18–7.15 (m, 1H), 6.06 (t, J = 6 Hz, 1H), 4.86–4.80 (m, 1H), 4.11 (t, J = 9.2 Hz, 1H), 3.86–3.82 (m, 1H), 3.78–3.62 (m, 2H), 2.04 (s, 3H).
13C-NMR (DMSO-d6): δ 170.51, 154.47, 152.94, 151.26, 149.94, 139.70, 139.15, 137.43, 129.96, 125.61, 125.19, 123.42, 122.19, 120.38, 114.52, 106.68, 72.29, 47.70, 41.84, 22.91.
ESI-MS m/z418.08 (M + Na+).
1H-NMR (400 MHz, DMSO-d6) δ: 9.33 (s, 1H), 8.80 (s, 1H), 8.74 (d, J = 5.6 Hz, 1H), 8.45 (t, J = 7.2 Hz, 1H), 8.38–8.31 (m, 2H), 7.90 (t, J = 8.8 Hz, 1H), 7.81 (dd, J = 2.4 Hz, J = 16.4 Hz, 1H), 7.76 (t,J = 6.0 Hz, 1H); 7.55 (dd, J = 1.6 Hz, J = 8.8 Hz, 1H), 4.83–4.76 (m, 1H), 4.60 (br s, 1H), 4.20 (t, J = 8.8 Hz, 1H), 3.91–3.82 (m, 1H), 3.45 (t, J = 5.2 Hz, 2H), 1.85 (s, 3H);
13C-NMR (DMSO-d6) δ: 170.51, 154.47, 152.94, 151.26, 149.94, 139.70, 139.15, 137.43, 129.96, 125.61, 125.19, 123.42, 122.19, 120.38, 114.52, 106.68, 72.29, 47.70, 41.84, 22.91;
HR-MS(TOF) m/z calcd for C20H18FN5O3 [M + Cl−]: 430.1082, found: 430.1085; for C20H18FN5O3 [M + H+]: 396.1472, found: 396.1472.
PAPER

A concise, environmentally benign, and cost-effective route was developed for the large-scale preparation of 1, a novel oxazolidinone antibacterial candidate. The key intermediate 2-(1-(2-fluoro-4-nitrophenyl)-1H-pyrazol-4-yl)pyridine 7 was prepared with high purity by mild deamination of the regioisomeric mixture 21. The mixture was prepared from a nucleophilic SNAr reaction by selective C–N coupling of the secondary amine functionality of 4-(pyridin-2-yl)-1H-pyrazol-3-amine 14 with 1,2-difluoro-4-nitrobenzene 10 in optimized conditions with the primary amine group remaining intact. The gaseous nitrogen release rate and reaction mixture temperature of the deamination step can be well controlled by altering the feeding manner, thereby providing safety guarantees. The optimized synthetic strategy of 1 with an overall yield of 27.6%, including seven sequential transformations by only five solid–liquid isolations, significantly improved the product separation workup. The strategy bypassed time-consuming and laborious procedures for any intermediate involved as well as for the final API. This study presents a process enabling the rapid delivery of a multikilogram quantity of API with high purity.
\
(S)-N-((3-(3-Fluoro-4-(4-(pyridin-2-yl)-1H-pyrazol-1-yl)phenyl)-2-oxo-oxazolidin-5-yl)methyl)acetamide (1)
-
Brickner, S. J.; Hutchinson, D. K.; Barbachyn, M. R.; Manninen, P. R.; Ulanowicz, D. A.; Garmon, S. A.; Grega, K. C.; Hendges, S. K.; Toops, D. S.; Ford, C. W.; Zurenko, G. E.J. Med. Chem. 1996, 39, 673– 679
-
(a) Gong, C. Y.; Yang, T.; Yang, X. Y.; Liu, Y. Y.; Ang, W.; Tang, J. Y.; Pi, W. Y.; Xiong, L.; Chang, Y.; Ye, W. W.; Wang, Z. L.; Luo, Y. F.; Zhao, X.; Wei, Y. Q. Nanoscale. 2013, 5, 275–283(b) Luo, Y. F.; Wang, Z. L.; Wei, Y. Q.; Geng, F. WO/2012/171479,2012.
WO2008143649A2 * 4 Dez 2007 27 Nov 2008 Das Jagattaran Novel oxazolidinone compounds as antiinfective agents CN1172484A * 29 Jan 1996 4 Fev 1998 法玛西雅厄普约翰美国公司 Hetero-aromatic ring substituted phenyloxazolidinone antimicrobials
Evacetrapib, LY2484595 for Treatment of high cholesterol and preventing cardiac events

Evacetrapib, LY2484595
Evacetrapib is an experimental drug being investigated to raise high-density lipoprotein cholesterol (HDL-C) via inhibition of the cholesteryl ester transfer protein (CETP)
Trans-4-({(5S)-5-[{[3,5-bis(trifluoromethyl)phenyl]methyl}(2-methyl-2H-tetrazol-5- yl)amino]-7,9-dimethyl-2,3,4,5-tetrahydro-1H-benzazepin-1-yl}methyl) cyclohexanecarboxylic acid
trans-4-[[(5S)-5-[[[3 ,5- bis(trifluoromethyl)phenyl]methyl] (2-methyl-2H-tetrazol-5-yl)amino]-2, 3,4,5- tetrahydro-7,9-dimethyl- IH- 1 -benzazepin- 1 -yl]methyl]-cyclohexanecarboxylic acid
trans-4-[5(S)-[N-[3,5-Bis(trifluoromethyl)benzyl]-N-(2-methyl-2H-tetrazol-5-yl)amino]-7,9-dimethyl-2,3,4,5-tetrahydro-1H-1-benzazepin-1-ylmethyl]cyclohexanecarboxylic acid
1186486-62-3 is cas
UNII-51XWV9K850
-
C31-H36-F6-N6-O2
- 638.6534
- lily……….. .innovator
Evacetrapib is a drug under development by Eli Lilly & Company (investigational name LY2484595) that inhibits cholesterylester transfer protein, which transfers and thereby increases high-density lipoprotein and lowers low-density lipoprotein. It is thought that modifying lipoprotein levels modifies the risk of cardiovascular disease.[1]
The first CETP inhibitor, torcetrapib, was unsuccessful because it increased levels of the hormone aldosterone and increased blood pressure,[2] which led to excess cardiac events when it was studied.[2] Evacetrapib does not have the same effect.[1] When studied in a small clinical trial in people with elevated LDL and low HDL, significant improvements were noted in their lipid profile.[3]
LY-2484595 is in phase III clinical trials at Lilly for the treatment of high-risk vascular disease and in phase II for the treatment of dyslipidemia.
Evacetrapib is one of two CETP inhibitors currently being evaluated (the other being anacetrapib).[1] Two other CETP inhibitors (torcetrapib and dalcetrapib) were discontinued during trials due to increased deaths and little identifiable cardiovascular benefit (despite substantial increases in HDL). Some hypothesize that CETP inhibitors may still be useful in the treatment of dyslipidemia, though significant caution is warranted.[2]

……………………………..
http://www.google.com/patents/WO2006002342A1?cl=en
Intermediate Preparation Scheme 1


Preparation Scheme 2

Intermediate Preparation Scheme 3




Scheme 7

Scheme 8

Scheme 11


…………………
http://www.google.com/patents/WO2011002696A1?cl=en
trans-4-[[(5S)-5-[[[3 ,5- bis(trifluoromethyl)phenyl]methyl] (2-methyl-2H-tetrazol-5-yl)amino]-2, 3,4,5- tetrahydro-7,9-dimethyl- IH- 1 -benzazepin- 1 -yl]methyl]-cyclohexanecarboxylic acid, (identified according to its Chemical Abstracts Index Name (referred to herein as BCCA) having the structure of Formula I illustrated below, and pharmaceutically acceptable salts of this compound.

I
The compound, BCCA, can be a free acid (referred to herein as BCCA free acid), or a pharmaceutically acceptable salt thereof, as a solvate (referred herein as BCCA’solvate) and a hydrate (referred to herein as BCCA ‘hydrate). The solvate molecules include water (as the hydrate), methanol, ethanol, formic acid, acetic acid, and isopropanol.
Scheme 1
(MeO) SO


Scheme 2

Scheme 3 : Alternate method for preparing BCCA

Preparation 11 Preparation 12

Preparation 13 Preparation 14 Preparation 15

Preparation 16

Preparation 17
Example 16
Scheme 4

………….
http://www.google.com/patents/US8299060
formula III below

with
Preparation 10 (Trans)-methyl 4-(((S)-5-((3,5-bis(trifluoromethyl)benzyl)(2-methyl-2H-tetrazol-5-yl)amino)-7,9-dimethyl-2,3,4,5-tetrahydro-1H-benzo[b]azepin-1-yl)methyl)cyclohexanecarboxylate (12)
Charge a flask equipped with an overhead stirrer, temperature probe, nitrogen inlet with (S)—N-(3,5-bis(trifluoromethyl)benzyl)-7,9-dimethyl-N-(2-methyl-2H-tetrazol-5-yl)-2,3,4,5-tetrahydro-1H-benzo[b]azepin-5-amine (5 g, 10.03 mmoles) and sodium triacetoxyborohydride (3.19 g, 15.05 mmoles) and acetonitrile (40 mL). Immerse the flask in an ice bath to cool the slurry to below about 5° C., then add (trans)-methyl 4-formylcyclohexanecarboxylate (2.99 g, 17.57 mmoles, prepared essentially according to the procedures in Houpis, I. N. et al, Tetrahedron Let. 1993, 34(16), 2593-2596 and JP49048639) dissolved in THF (10 mL) via a syringe while maintaining the reaction mixture at or below about 5° C. Allow the reaction to warm to RT and stir overnight. Add NH4Cl (25 mL, 50% saturated aqueous solution) and separate the aqueous layer from the organic layer. The pH of the organic layer should be about 5.5. Warm the organic layer to about 45° C. and add water (16 mL). Add a seed crystal of the titled compound and cool to about 35° C. Collect the resulting solid by filtration and rinse with ACN. Dry to provide 5.80 g of the title compound.
………….
Evacetrapib
http://www.platinummetalsreview.com/article/56/4/229-235/

…………………….paper
 THE ESTER OF EVACETRAPIB
THE ESTER OF EVACETRAPIB
http://pubs.acs.org/doi/abs/10.1021/op500025v
Development of a Hydrogenative Reductive Amination for the Synthesis of Evacetrapib: Unexpected Benefits of Water

References
- Cao G, Beyer TP, Zhang Y, et al. (December 2011). “Evacetrapib is a novel, potent, and selective inhibitor of cholesteryl ester transfer protein that elevates HDL cholesterol without inducing aldosterone or increasing blood pressure”. J. Lipid Res. 52 (12): 2169–76.doi:10.1194/jlr.M018069. PMID 21957197.
- Joy T, Hegele RA (July 2009). “The end of the road for CETP inhibitors after torcetrapib?”. Curr. Opin. Cardiol. 24 (4): 364–71.doi:10.1097/HCO.0b013e32832ac166. PMID 19522058.
- Nicholls SJ, Brewer HB, Kastelein JJ, Krueger KA, Wang MD, Shao M, Hu B, McErlean E, Nissen SE (2011). “Effects of the CETP inhibitor evacetrapib administered as monotherapy or in combination with statins on HDL and LDL cholesterol”. JAMA 306 (19): 2099–109.doi:10.1001/jama.2011.1649.
(Z)-5-((1-(4-Chloro-2-(trifluoromethyl)benzyl)-1H-indazol-5-yl)methylene)-3-((3R,4R)-3-fluoro-1-methylpiperidin-4-yl)thiazolidine-2,4-dione for the treatment of hyperglycemia in patients with type 2 diabetes mellitus.


Estrogen Related Receptor alpha (ERR-a) modulators useful for treating, ameliorating, or inhibiting the progression of disease states, disorders, and
conditions mediated by ERR-a activity. BACKGROUND OF THE INVENTION
Nuclear receptors are members of a superfamily of transcription factors.
The members of this family share structural similarities and regulate a diverse set of biological effects (Olefsky, J. M. J. Biol. Chem. 2001 , 276(40), 36863-36864). Ligands activate or repress these transcription factors that control genes involved in metabolism, differentiation and reproduction (Laudet, V. and H. Gronmeyer. The Nuclear Receptor Factbooks. 2002, San Diego: Academic Press). Presently, the human genome project has identified about 48 members for this family and cognate ligands have been identified for about 28 of them (Giguere, V. Endocrine Rev. 1999, 20(5), 689-725). This protein family is composed of modular structural domains that can be interchanged within the members of the family without loss of function. A typical nuclear receptor contains a hypervariable N-terminus, a conserved DNA binding domain (DBD), a hinge region, and a conserved ligand- binding domain (LBD). The function of the DBD is targeting of the receptor to specific DNA sequences (Nuclear Hormone Receptor (NHR) response elements or NREs), and the function of the LBD is recognition of its cognate ligand. Within the sequence of the nuclear receptor there are regions involved in transcriptional activation. The Activation Function 1 (AF-1 ) domain is situated at the N-terminus and constitutively activates transcription (Rochette-Egly, C. et al. Cell 1997, 90, 97-107; Rochette-Egly, C. et al. Mol. Endocrinol. 1992, 6, 2197-2209), while the Activation Function 2 (AF-2) domain is embedded within the LBD and its transcriptional activation is ligand dependent (Wurtz, J.M. et al. Nat. Struct. Biol. 1996, 3, 87-94). Nuclear receptors can exist as monomers, homodimers or heterodimers and bind to direct or inverted nucleotide repeats (Laudet and
Gronmeyer, 2002; Aranda, A. and A. Pascual. Physiol. Rev. 2001 , 81 (3), 1269- 1304).
The members of this family exist either in an activated or repressed basal biological state. The basic mechanism of gene activation involves ligand dependent exchange of co-regulatory proteins. These co-regulatory proteins are referred to as co-activators or co-repressors (McKenna, L.J. et al. Endocrine Rev. 1999, 20, 321 -344). A nuclear receptor in the repressed state is bound to its DNA response element and is associated with co-repressor proteins that recruit histone de-acetylases (HDACs) (Jones, P.L. and Y.B. Shi. Curr. Top. Microbiol. Immunol. 2003, 274, 237-268). In the presence of an agonist there is an exchange of co- repressors with co-activators that in turn recruit transcription factors that assemble into an ATP dependent chromatin-remodeling complex. Histones are hyper- acetylated, causing the nucleosome to unfold, and repression is alleviated. The AF-2 domain acts as the ligand dependent molecular switch for the exchange of co-regulatory proteins. In the presence of an agonist the AF-2 domain undergoes a conformational transition and presents a surface on the LBD for interaction with co-activator proteins. In the absence of an agonist or in the presence of an antagonist the AF-2 domain presents a surface that promotes interactions with co- repressor proteins. The interaction surfaces on the LBD for both co-activators, and co-repressors overlap and provide a conserved molecular mechanism for gene activation or repression that is shared by the members of this family of transcription factors (Xu, H.E. et al. Nature 2002, 415 (6873), 813-817).
Natural ligands that modulate the biological activity of nuclear receptors have been identified for only approximately one half of known nuclear receptors. Receptors for which no natural ligand has been identified are termed “orphan receptors.” The discovery of ligands or compounds that interact with an orphan receptor will accelerate the understanding of the role of the nuclear receptors in physiology and disease and facilitate the pursuit of new therapeutic approaches. Estrogen related receptors (ERRs) constitutes a sub-class of these receptors where no ligand has been identified.
ERR-a (also known as ERR-1 ), an orphan receptor, is the first of the three identified members of the estrogen receptor related subfamily of orphan nuclear receptors (ERR-a, β, γ). The ERR subfamily is closely related to the estrogen receptors (ER-a and ER-β). ERR-a and ERR-β were first isolated by a low stringency hybridization screen (Giguere, V. et al. Nature 1988, 331 , 91 -94) followed later with the discovery of ERR-γ (Hong, H. et al. J. Biol. Chem. 1999, 274, 22618-22626). The ERRs and ERs share sequence similarity with the highest homology observed in their DBDs, approximately 60%, and all interact with the classical DNA estrogen response element. Recent biochemical evidence suggested that the ERRs and ERs share target genes, including pS2, lactoferin, aromatase and osteopontin, and share co-regulator proteins (Giguere, V. Trends in Endocrinol. Metab. 2002, 13, 220-225; Vanacker, J.M. et al. EMBO J. 1999, 18, 4270-4279; Kraus, R.J. et al. J. Biol. Chem. 2002, 272, 24286-24834; Hong et al., 1999; Zhang, Z. and C.T. Teng. J. Biol. Chem. 2000, 275, 20387-20846).
Therefore, one of the main functions of ERR is to regulate the response of estrogen responsive genes. The effect of the steroid hormone estrogen is primarily mediated in the breast, bone and endometrium. Thus, the identification of compounds that will interact with ERRs should provide a benefit for the treatment of bone related disease, breast cancer and reproduction.
ERR-a is shown to be present both in normal and breast cancer tissue (Ariazi, E.A. et al. Cancer Res. 2002, 62, 6510-6518). It has been reported that the main function of ERR-a in normal breast tissue is that of a repressor for estrogen responsive genes. In breast cancers or cell lines that are non-estrogen responsive (ER-a negative), ERR-a has been reported to be in an activated state (Ariazi et al., 2002). Therefore, compounds that will interact with ERR-a may be useful agents for the treatment of breast cancer that is ER-a negative and non- responsive to classical anti-estrogenic therapy, or may be used as an adjunct agent for anti-estrogen responsive breast cancers. These agents may act as antagonists by reducing the biological activity of ERR-a in these particular tissues.
Many post-menopausal women experience osteoporosis, a condition that is a result of the reduction of estrogen production. Reduction of estrogen levels results in an increase of bone loss (Turner, R.T. et al. Endocrine Rev. 1994, 15(3), 275-300). An anabolic effect on bone development has been observed on the administration of estrogens to postmenopausal patients with osteoporosis (Pacifici, R. J. Bone Miner. Res. 1996, 1 1 (8), 1043-1051 ) but the molecular mechanism is unknown since ER-a and ER-β knock-out animals have minor skeletal defects, where the action of estrogens is typically mediated (Korach, K. S. Science 1994, 266, 1524-1527; Windahl, S.H. et al. J. Clin. Invest. 1999, 104(7), 895-901 ). Expression of ERR-a in bone is regulated by estrogen (Bonnelye, E. et al. Mol. Endocrin. 1997, 1 1 , 905-916; Bonnelye, E. et al. J. Cell Biol. 2001 , 153, 971 -984). ERR-a is maintained throughout osteoblast differentiation stages.
Over-expression of ERR-a in rat calvaria osteoblasts, an accepted model of bone differentiation, results in an increase of bone nodule formation, while treatment of rat calvaria osteoblasts with ERR-a antisense results in a decrease of bone nodule formation. ERR-a also regulates osteopontin, a protein believed to be involved in bone matrix formation. Therefore compounds that will modulate ERR-a by increasing its activity can have an anabolic effect for the regeneration of bone density and provide a benefit over current approaches that prevent bone loss, but have no anabolic effect. Such compounds can enhance the activity of the receptor by two possible mechanisms: i) enhancing the association of the receptor with proteins that enhance its activity or improve the stability of the receptor; and ii) increasing the intracellular concentrations of the receptor and consequently increasing its activity. Conversely, with respect to bone diseases that are a result of abnormal bone growth, compounds that will interact with ERR-a and decrease its biological activity may provide a benefit for the treatment of these diseases by retarding bone growth. Antagonism of the association of the receptor with co- activator proteins decreases the activity of the receptor.
ERR-a is also present in cardiac, adipose, and muscle tissue and forms a transcriptional active complex with the PGC-1 co-activator family, co-activators implicated with energy homeostasis, mitochondria biogenesis, hepatic
gluconeogenesis and in the regulation of genes involved in fatty acid beta- oxidation (Kamei, Y. et al. Proc. Natl. Acad. Sci. USA 2003, 100(21 ), 12378- 12383). ERR-a regulates the expression of the medium chain acyl-CoA
dehydrogenase promoter (MCAD). Medium chain acyl-CoA dehydrogenase is a gene involved in the initial reaction in fatty acid beta-oxidation. It is believed that in the adipose tissue ERR-a regulates energy expenditure through the regulation of MCAD (Sladek, R. et al. Mol. Cell. Biol. 1997, 17, 5400-5409; Vega, R.B. and D.P. Kelly. J. Biol. Chem. 1997, 272, 31693-31699). In antisense experiments in rat calvaria osteoblasts, in addition to the inhibition of bone nodule formation, there was an increase in adipocyte differentiation markers including aP2 and PPAR-γ (Bonnelye, E. et al. Endocrinology 2002, 143, 3658-3670). Recently an ERR-a knockout model has been described that exhibited reduced fat mass relative to the wild type and DNA chip analysis data indicated alteration of the expression levels of genes involved in adipogenesis and energy metabolism (Luo, J. et al. Mol. Cell. Biol. 2003, 23(22), 7947-7956). More recently it has been shown that ERR-a regulates the expression of endothelial nitric oxide synthase, a gene that has a protective mechanism against arteriosclerosis (Sumi, D. and L.J. Ignarro. Proc Natl. Acad. Sci. 2003, 100, 14451 -14456). The biochemical evidence supports the involvement of ERR-a in metabolic homeostasis and differentiation of cells into adipocytes. Therefore, compounds interacting with ERR-a can affect energy homeostasis and may therefore provide a benefit for the treatment of obesity and metabolic syndrome related disease indications, including arteriosclerosis and diabetes (Grundy, S.M. et al. Circulation 2004, 109(3), 433-438).
There is a continuing need for new ERR-a inverse agonists. There is also a need for ERR-a inverse agonists useful for the treatment of conditions including but not limited to ankylosing spondylitis, artherosclerosis, arthritis (such as rheumatoid arthritis, infectious arthritis, childhood arthritis, psoriatic arthritis, reactive arthritis), bone-related diseases (including those related to bone formation), breast cancer (including those unresponsive to anti-estrogen therapy), cardiovascular disorders, cartilage-related disease (such as cartilage injury/loss, cartilage degeneration, and those related to cartilage formation),
chondrodysplasia, chondrosarcoma, chronic back injury, chronic bronchitis, chronic inflammatory airway disease, chronic obstructive pulmonary disease, diabetes, disorders of energy homeostasis, gout, pseudogout, lipid disorders, metabolic syndrome, multiple myeloma, obesity, osteoarthritis, osteogenesis imperfecta, osteolytic bone metastasis, osteomalacia, osteoporosis, Paget’s disease, periodontal disease, polymyalgia rheumatica, Reiter’s syndrome, repetitive stress injury, hyperglycemia, elevated blood glucose level, and insulin resistance.
Scheme 1



Scheme 2


Scheme 3


Scheme 4


Scheme 5



Scheme 6

Scheme 7

Scheme 8

Scheme 9

without methyl
Example 199
(5Z)-5-({1 -[4-Chloro-2-(trifluoromethyl)benzyl]-1 /-/-indazol-5-yl}methylidene)-3-(c/‘s- 4-fluoropiperidin-3-yl)-1 ,3-thiazolidine-2,4-dione
 note………..this is without methyl
note………..this is without methyl(A) 1 ,1 -Dimethylethyl c/‘s-3-[(5Z)-5-[(1 -[4-chloro-2-(trifluoromethyl)benzyl]-1 H- indazol-5-yl)methylidene]-2,4-dioxo-1 ,3-thiazolidin-3-yl]-4-fluoropiperidine- 1 -carboxylate was prepared from (5Z)-5-({1 -[2-chloro-4-
(trifluoromethyl)benzyl]-1 /-/-indazol-5-yl}methylidene)-2,4-dioxo-1 ,3- thiazolidine (from Example 1 ) and 1 ,1 -dimethylethyl frans-3-hydroxy-4- fluoropiperidine-1 -carboxylate (prepared as described in US 2007/249589) following General Procedure W.
(B) (5Z)-5-({1 -[4-Chloro-2-(trifluoromethyl)benzyl]-1 H-indazol-5- yljmethylidene)- 3-(c/s-4-fluoropiperidin-3-yl)-1 ,3-thiazolidine-2,4-dione was prepared from 1 ,1 -dimethylethyl c/s-3-[(5Z)-5-[(1 -[4-chloro-2- (trifluoromethyl)benzyl]-1 /-/-indazol-5-yl)methylidene]-2,4-dioxo-1 ,3- thiazolidin-3-yl]-4-fluoropiperidine-1 -carboxylate following General
Procedure M.
1 H NMR (400 MHz, CDCI3): δ 8.21 (s, 1 H), 7.95 (s, 1 H), 7.72 (d, 1 H), 7.65 (s, 1 H), 7.45 – 7.50 (m, 1 H), 7.30 – 7.38 (m, 2H), 6.66 (d, 1 H), 5.80 (s, 2H), 4.83 – 5.04 (m, 2H), 4.08 – 4.20 (m, 2H), 3.99 – 4.08 (m, 1 H), 3.81 – 3.91 (m, 1 H), 2.27 – 2.40 (m, 1 H), 2.02 – 2.13 (m, 1 H).
LC/MS: mass calcd. for C24Hi9CIF4N4O2S: 538.08, found 539.5 [M+1 ]+
Example 201
(5Z)-5-({1 -[4-Chloro-2-(trifluoromethyl)benzyl]-1 /-/-indazol-5-yl}methylidene)-3-(c/‘s- 3-fluoropiperidin-4-yl)-1 ,3-thiazolidine-2,4-dione
 note. this is without methyl
note. this is without methyl(A) 1 ,1 -Dimethylethyl c/‘s-4-[(5Z)-5-[(1 -[4-chloro-2-(trifluoromethyl)benzyl]-1 H- indazol-5-yl)methylidene]-2,4-dioxo-1 ,3-thiazolidin-3-yl]-3-fluoropiperidine- 1 -carboxylate was prepared from (5Z)-5-({1 -[2-chloro-4- (trifluoromethyl)benzyl]-1 /-/-indazol-5-yl}methylidene)-2,4-dioxo-1 ,3- thiazolidine (from Example 1 ) and 1 ,1 -dimethylethyl frans-4-hydroxy-3- fluoropiperidine-1 -carboxylate (prepared as described in US 2007/249589) following General Procedure J.(B) (5Z)-5-({1 -[4-Chloro-2-(trifluoromethyl)benzyl]-1 H-indazol-5- yl}methylidene)-3-(c/s-3-fluoropiperidin-4-yl)-1 ,3-thiazolidine-2,4-dione was prepared from 1 ,1 -dimethylethyl c/‘s-4-[(5Z)-5-[(1 -[4-chloro-2- (trifluoromethyl)benzyl]-1 /-/-indazol-5-yl)methylidene]-2,4-dioxo-1 ,3- thiazolidin-3-yl]-3-fluoropiperidine-1 -carboxylate following General
Procedure M.
1 H NMR (400 MHz, CDCI3): δ 8.22 (s, 1 H), 8.00 (s, 1 H), 7.96 (s, 1 H), 7.72 (d, 1 H), 7.48 – 7.54 (m, 1 H), 7.36 (s, 1 H), 7.34 (s, 1 H), 6.68 (d, 1 H), 5.80 (s, 2H), 4.57 – 4.75 (m, 1 H), 4.40 – 4.56 (m, 1 H), 3.25 – 3.46 (m, 2H), 3.18 (qd, 1 H), 2.83 – 3.03 (m, 1 H), 2.72 (t, 1 H), 1 .88 (br. s., 1 H), 1 .72 (d, 1 H).
LC/MS: mass calcd. for C2 H19CIF4N4O2S: 538.08, found 539.5 [M+1 ]+
Example 273
(5Z)-5-({1 -[4-Chloro-2-(trifluoromethyl)benzyl]-1 /-/-indazol-5-yl}methylidene)-3- (frans-3-fluoropiperidin-4-yl)-1 ,3-thiazolidine-2,4-dione
 note——- this is without methyl but precursor to desired compd
note——- this is without methyl but precursor to desired compdPreparation 1 :
(A) To the solution of 1 ,1 -dimethylethyl frans-4-(2,4-dioxo-1 ,3-thiazolidin-3-yl)- 3-hydroxypiperidine-1 -carboxylate (from Example 270, 0.68 mmol) in DCM (5 ml_) in a plastic bottle was added bis(2-methoxyethyl)aminosulfur trifluoride (3 equiv) and a drop of ethanol. After stirring at rt for 3 h, the reaction was concentrated and the resultant residue was purified by silica gel chromatography (hexane/EtOAc) to provide 1 ,1 -dimethylethyl trans-4- (2,4-dioxo-1 ,3-thiazolidin-3-yl)-3-fluoropiperidine-1 -carboxylate as a pale yellow solid.
(B) (5Z)-5-({1 -[4-Chloro-2-(trifluoromethyl)benzyl]-1 H-indazol-5-yl}methylidene)- 3-[frans-3-fluoropiperidin-4-yl]-1 ,3-thiazolidine-2,4-dione was prepared from [4-chloro-2-(trifluoromethyl)benzyl]-1 H-indazol-5-carbaldehyde (from
Example 1 ) and 1 ,1 -dimethylethyl frans-4-(2,4-dioxo-1 ,3-thiazolidin-3-yl)-3- fluoropiperidine-1 -carboxylate following General Procedure F.
Preparation 2:
(A) A mixture of 1 ,1 -dimethylethyl 7-oxa-3-azabicyclo[4.1 .0]heptane-3- carboxylate (from Example 270; 47.7 mmol), [(5Z)-5-({1 -[4-chloro-2- (trifluoromethyl)benzyl]-1 /-/-indazol-5-yl}methylidene)-2,4-dioxo-1 ,3- thiazolidine (from Example 1 ; 31 .8 mmol) and magnesium perchlorate (23.9 mmol) in DMF (70 mL) was heated at 1 15 °C for 2-4 h. After cooling to rt, the mixture was slowly poured into water (300 mL) with vigorous stirring, and the resultant precipitate was filtered, thoroughly washed with water and dried to afford a mixture of 1 ,1 -dimethylethyl frans-4-{(5Z)-5-[(1 –
{[4-chloro-2-(trifluoromethyl)phenyl]methyl}-1 /-/-indazol-5-yl)methylidene]- 2,4-dioxo-1 ,3-thiazolidin-3-yl}-3-hydroxypiperidine-1 -carboxylate and the corresponding regioisomer, 1 ,1 -dimethylethyl frans-3-{(5Z)-5-[(1 -{[4-chloro- 2-(trifluoromethyl)phenyl]methyl}-1 /-/-indazol-5-yl)methylidene]-2,4-dioxo- 1 ,3-thiazolidin-3-yl}-4-hydroxypiperidine-1 -carboxylate in ratio of ~ 3.3 : 1 .
(B) To an ice-cooled solution of the above mixture of 1 ,1 -dimethylethyl frans- 4-{(5Z)-5-[(1 -{[4-chloro-2-(trifluoromethyl)phenyl]methyl}-1 /-/-indazol-5- yl)methylidene]-2,4-dioxo-1 ,3-thiazolidin-3-yl}-3-hydroxypiperidine-1 – carboxylate and the regioisomer, 1 ,1 -dimethylethyl frans-3-{(5Z)-5-[(1 -{[4- chloro-2-(trifluoromethyl)phenyl]methyl}-1 H-indazol-5-yl)methylidene]-2,4- dioxo-1 ,3-thiazolidin-3-yl}-4-hydroxypiperidine-1 -carboxylate in DCM (350 mL) was slowly added bis(2-methoxyethyl)aminosulfur trifluoride (47.7 mmol). After stirring for 1 h, the solution was allowed to warm to rt and stir overnight. The reaction was then quenched with sat’d aq. NaHCO3 and after separating phases, the organic phase was dried (Na2SO4) and concentrated to ~ 40 mL. The solution was loaded onto a silica gel column (Analogix, 200g) and eluted with heptanes/DCM/EtOAc (40:57:3).
Product-containing fractions were combined and concentrated to afford a crude product mixture as a pale yellow foam. Treatment of this foam with ether (~ 20 mL) led to product precipitation; additional ether (200 mL) was added portionwise with stirring and after cooling to ~ 5 °C, the mixture was filtered through a glass fiber filter and washed with cold ether to afford 1 ,1 – dimethylethyl frans-4-{(5Z)-5-[(1 -{[4-chloro-2-(trifluoromethyl)phenyl]- methyl}-1 H-indazol-5-yl)methylidene]-2,4-dioxo-1 ,3-thiazolidin-3-yl}-3- fluoropiperidine-1 -carboxylate as an essentially white powder. (C) (5Z)-5-({1 -[4-Chloro-2-(trifluoromethyl)benzyl]-1 H-indazol-5- yl}methylidene)-3-[frans-3-fluoropiperidin-4-yl]-1 ,3-thiazolidine-2,4-dione was prepared from 1 ,1 -dimethylethyl frans-4-{(5Z)-5-[(1 -{[4-chloro-2- (trifluoromethyl)phenyl]methyl}-1 H-indazol-5-yl)methylidene]-2,4-dioxo-1 ,3- thiazolidin-3-yl}-3-fluoropiperidine-1 -carboxylate following General
Procedure M.
1 H NMR (400 MHz, CDCI3): δ 8.22 (s, 1 H), 8.02 (s, 1 H), 7.96 (s, 1 H), 7.72 (d, 1 H), 7.47 – 7.56 (m, 1 H), 7.36 (s, 1 H), 7.34 (s, 1 H), 6.68 (d, 1 H), 5.80 (s, 2H), 5.10 – 5.33 (m, 1 H), 4.40 – 4.55 (m, 1 H), 3.52 (d, 1 H), 3.14 (d, 1 H), 2.68 (br. s., 2H), 2.43 (qd, 1 H), 1 .70 – 1 .90 (m, 2H).
LC/MS: mass calcd. for C2 H2oCIF4N4O2S: 538.09, found 539.3 [M+1 ]+
main compd
Example 277
(5Z)-5-({1-[4-Chloro-2-(trifluoromethyl)benzyl]-1H-indazol-5-yl}methylidene)-3- (frans-3-fluoro-1-methylpiperidin-4-yl)-1 ,3-thiazolidine-2,4-dione
 desired compd
desired compd(5Z)-5-({1-[4-Chloro-2-(trifluoromethyl)benzyl]-1H-indazol-5-yl}methylidene)- 3-[ trans -3-fluoro-1-methylpiperidin-4-yl]-1,3-thiazolidine-2,4-dione was prepared from (5Z)-5-({1 -[4-chloro-2-(trifluoromethyl)benzyl]-1 H-indazol-5- yl}methylidene)-3-[ trans -3-fluoropiperidin-4-yl]-1 ,3-thiazolidine-2,4-dione (Example 273) and formaldehyde following General Procedure R.
1 H NMR (400 MHz, CDCI3): δ 8.22 (s, 1 H), 8.01 (s, 1 H), 7.96 (s, 1 H), 7.72 (s, 1 H), 7.51 (d, 1 H), 7.36 (s, 1 H), 7.34 (s, 1 H), 6.68 (d, 1 H), 5.80 (s, 2H), 5.25 – 5.48 (m, 1 H), 4.28 – 4.42 (m, 1 H), 3.24 – 3.36 (m, 1 H), 2.85 – 2.96 (m,
1 H), 2.56 (qd, 1 H), 2.37 (s, 3H), 2.07 – 2.17 (m, 2H), 1 .77 (dd, 1 H).
LC/MS: mass calcd. for C25H2iCIF4N4O2S: 552.1 , found 553.3 [M+1 ]+

The development of a reproducible process for multihundred gram production of (Z)-5-((1-(4-chloro-2-(trifluoromethyl)benzyl)-1H-indazol-5-yl)methylene)-3-((3R,4R)-3-fluoro-1-methylpiperidin-4-yl)thiazolidine-2,4-dione (26), a potent and selective inhibitor of estrogen-related receptor 1 (ERR1), is described. This multihundred gram synthesis was achieved via magnesium perchlorate-catalyzed regioselective epoxide ring-opening of tert-butyl 7-oxa-3-azabicyclo[4.1.0]heptane-3-carboxylate (9) with thiazolidine-2,4-dione (6, TZD) to form a diastereomeric mixture tert-butyl 4-(2,4-dioxothiazolidin-3-yl)-3-hydroxypiperidine-1-carboxylate (17), of which the 3-hydroxyl group was functionally transformed to 3-fluoro derivative 19 after treatment with Deoxo-Fluor. Chiral separation of 19 provided the desired diastereomer (3R,4R)-21 that was converted to the secondary amine 23 TFA salt. Reductive amination of 23 produced the key intermediate N-methyl 24. Knoevenagel condensation of24 with 1-(4-chloro-2-(trifluoromethyl)benzyl)-1H-indazole-5-carbaldehyde (5) produced the final product 26 in 10% overall yield (99.7% HPLC area% with ≥99.5% de) after a convergent eight synthetic steps with the only column purification being the chiral HPLC separation of 3R,4R–21 from 3S,4S–22.
Citations
- Bignan, G; WO 2011149841 2011
- Li, X; 246th American Chemical Society National Meeting 2013
- Slade, D; J Org Chem 2009, 74, 6331
- Collot, V; Tetrahedron 1999, 55, 6917
- Patta, S; Indian J Chem 2005, 2404
- Maccari, R; Bioorg Med Chem 2005, 13, 2809
- Corona, J; Org Process Res Dev 2010, 14, 712
- Chen, S; Bioorg Med Chem Lett 2007, 17, 2134
- Boto, A; Eur J Org Chem 2005, 673
- Saavedra, J; J Org Chem 1979, 44, 4516
- Bosmans, J; WO 2005000838 2005
- Kratzel, M; Heterocycles 1995, 41, 897
- Daly, A; Tetrahedron Lett 1999, 40, 3617
- Cresswell, A; Org Lett 2010, 12, 2936
- Ready, J; Angew Chem, Int Ed 2002, 41, 1394
- Tandon, V; Tetrahedron Lett 1993, 34, 4403
- Zhao, S; Heterocycles 1994, 39, 163
- Imanishi, T; Synth Comm 1978, 8, 99
- White, J; J Org Chem 2004, 69, 2573
- Lal, G; Chem Commun 1999, 215
- Singh, R; Synthesis 2002, 17, 2561
- Singh, R; J Fluorine Chem 2002, 116, 23
- Shaw, S; J Org Chem 2013, 78, 8892
- Grunewald, G; J Med Chem 2001, 44, 2849
Afatinib

Afatinib
439081-18-2
850140-73-7 dimaleate
Tovok, BIBW2992, Tomtovok
An irreversible EGFR/HER2 inhibitor
| Molecular Weight: | 485.94 |
| Molecular Formula: | C24H25ClFN5O3 |
N-[4-[(3-Chloro-4-fluorophenyl)amino]-7-[[(3S)-tetrahydro-3-furanyl]oxy]-6-quinazolinyl]-4(dimethylamino)-2-butenamide
4 – [(3-chloro-4-fluorophenyl) amino] -6 – {[4 – (N, N-dimethylamino)-1-oxo-2-buten-1-yl] – amino} -7 – ((S )-tetrahydrofuran-3-yloxy)-quinazoline
(E)-4-Dimethylamino-but-2-enoic acid {4-(3-chloro-4-fluoro- phenylanimo)-7-[(S)-(tetrahydro-furan-3-yl) oxy]-quinazolin-6-yl} -amide
4 – [(3_ chloro-4 – fluorophenyl) amino] -6 – {[4_ (N, N-dimethylamino)-buten-1-oxo-_2_ – yl] amino}-7 – ((S) – tetrahydrofuran-3 – yloxy) – quinazoline
The endorsement for Giotrif (afatinib) covers the drug’s use in the treatment of adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) who have the epidermal growth factor receptor (EGFR) gene mutation, which is present in about 10 per cent of people with NSCLC.
It caps a good month for Boehringer, which won US approval for the drug under the brand name Gilotrif two weeks ago, adding to the company’s list of therapy areas, which so far include chronic obstructive pulmonary disease (COPD), anticoagulation, HIV, Parkinson’s disease and diabetes.
In the US, the drug is approved alongside a companion diagnostic to help determine if a patient’s lung cancer cells express the EGFR mutations, whereas the EMA recommendation just includes the requirement that Giotrif be initiated and supervised by a physician experienced in the use of anti-cancer therapies.
http://www.pmlive.com/pharma_news/boehringers_first_cancer_drug_leads_ema_recommendations_493051
GILOTRIF tablets contain afatinib, a tyrosine kinase inhibitor which is a 4-anilinoquinazoline. Afatinib is presented as the dimaleate salt, with the chemical name 2-butenamide, N-[4-[(3-chloro-4-fluorophenyl)amino]7-[[(3S)-tetrahydro-3-furanyl]oxy]-6-quinazolinyl]-4-(dimethylamino)-,(2E)-, (2Z)-2-butenedioate (1:2). Its structural formula is:
 |
Afatinib dimaleate is a white to brownish yellow powder, water soluble and hygroscopic, with an empirical formula of C32H33ClFN5O11, and a molecular weight of 718.1 g/mol.
GILOTRIF tablets for oral administration are available in 40 mg, 30 mg, or 20 mg of afatinib (equivalent to 59.12 mg, 44.34 mg, or 29.56 mg afatinib dimaleate, respectively). The inactive ingredients of GILOTRIF are the following: Tablet Core: lactose monohydrate, microcrystalline cellulose, crospovidone, colloidal silicon dioxide, magnesium stearate. Coating: hypromellose, polyethylene glycol, titanium dioxide, talc, polysorbate 80, FD&C Blue No. 2 (40 mg and 30 mg tablets only).

Afatinib (BIBW2992) is an irreversible EGFR/Neu inhibitor with an IC50 of 14 nM. Afatinib is a potent inhibitor of EGFR phosphorylation. Afatinib showed positive results in assays against a variety of human cancer cell lines, including A431, murine NIH-3T3 cells, and breast cancer cell line BT-474.
Afatinib[2] (INN; trade name Gilotrif in the US and Giotrif in Europe, previously Tomtovok and Tovok[3]) is a drug approved inmuch of the world (including the United States, Canada, the United Kingdom and Australia) for the treatment of metastatic non-small cell lung carcinoma (NSCLC), developed by Boehringer Ingelheim.[4][5][6] It acts as an angiokinase inhibitor.
Quinazoline derivatives, such as afatinib, are described in WO2002050043. This document also describes certain favourable pharmacological properties of this compound. The dimaleate salt and its crystalline form are described in WO2005037824.
It is known in the W002/50043, which describes the pharmacological properties has important compounds include in particular their pharmacological properties mediated by the tyrosine kinase inhibitory effect and the signal transmission through the skin growth factor receptor (EGF-R) signal transduction mediated inhibitory effect. Therefore, this type of compounds are useful in the treatment of diseases, in particular for the treatment of tumor diseases, lung and gastrointestinal and respiratory tract and gall bladder and bile duct disease.
W002/50043 discloses a method for preparing a compound wherein the amino crotonic group (IV), such as 4_ [(3 – chloro-4 – fluorophenyl) amino] -6 – {[4 – (N, N-two methyl-amino)-oxo-2-1_ – buten-1 – yl] amino} -7 – ((S) – tetrahydrofuran-3 – yloxy) – quinazoline in the one-pot reaction from the corresponding aniline component (II), bromo crotonic acid (III), oxalyl chloride and a secondary amine prepared (see Figure 1).
Figure 1:

In the method, the yield was 50% at most. In addition, the implementation typically purified by column chromatography. Therefore Preparation of 4 – [(3_ chloro-4 – fluorophenyl) amino] -6 – {[4 – (N, N-dimethylamino)-l-oxo-2 – buten-1 – yl] amino} -7 – ((S) – tetrahydrofuran-3 – yloxy) – quinazoline of the method is not for large-scale industrial production. Moreover, the method is not drawback bromo crotonate purchased by a large number of commercial sources, and the corresponding bromo-methyl crotonate only be obtained in a purity of about 80%.These methods are used in this case is also 4 – [(3 – chloro-4 – fluorophenyl) amino] -6 – {[4 – (N, N-dimethylamino) -1 – oxo – butene-1 – yl] amino} -7 – (⑶ – tetrahydrofuran-3 – yloxy) – quinazoline industrialized production adversely affect the applicability.
In the above-mentioned drawbacks of known production methods, the present invention is to provide a produce aminocrotonate aryl amides, in particular 4 – [(3 – chloro-4 – fluorophenyl) amino] -6 – {[4 – (N, N-dimethylamino)-buten-1-oxo-_2_ – yl] amino} -7 – ((S) – tetrahydrofuran-3 – yloxy) – quinazoline The method of the method can be easily obtained using high purity starting materials and does not require the use of any material technology. Thus, the new method should be applicable on an industrial scale synthesis grade and therefore suitable for commercial applications.
This task is according to the present invention for preparing 4 – [(3 – chloro-4 – fluorophenyl) amino] -6 – {[4 – (N, N-dimethylamino) -1 – oxo-2 – buten-1 – yl] amino} -7 – (⑶ – tetrahydrofuran-3 – yloxy) – quinazoline, and other amino crotonic method based compound. In addition to high yield industrially embodiment, the synthesis method according to the present invention also has a very good purity and less than 0.1 of the advantages of a low cis content.
According to Figure 2, in the method according to the present invention, an aryl group corresponding amino compound (V) with two – (Ch-ware yl) _ phosphono acetic acid, preferably with diethyl phosphonoacetate, by After appropriate activation, in a suitable reaction solvent, preferably for the use of the active 1,1 – carbonyldiimidazole, 1,1 – carbonyldiimidazole – triazole or propane phosphonic acid anhydride, is preferred for the use of 1, 1 – carbonyl diimidazole. The solvent used may be, for example, tetrahydrofuran (THF), dimethylformamide (DMF) or ethyl acetate.
The amide may be connected through any possible approach for activation, i.e., for example, 1,1 _ carbonyldiimidazole, 1,1 – carbonyldiimidazole – triazole, DCC (N, N-dicyclohexyl carbodiimide ), EDC (N ‘_ (dimethylaminopropyl)-N-ethylcarbodiimide), TBTU (0 – (benzotriazol-1 – yl)-N, N, N’, N ‘ – pan tetramethyluronium tetrafluoroborate), thiazolidine-2 – thione, or through the use of thionyl chloride may be converted to the corresponding acyl chloride. If desired, activation may be used an organic base such as triethylamine or pyridine embodiment, and can additionally added DMAP (dimethylaminopyridine). Suitable solvents include DMF, THF, ethyl acetate, toluene, chlorinated hydrocarbons or mixtures thereof.

http://www.google.com/patents/CN1867564B?cl=en
Example 1
{[4 – (3 – chloro-4 – fluoro – phenylamino) -7 – (⑶ – tetrahydrofuran _3_-yloxy) – quinazoline _6_ yl carbamoyl] methyl}-_ _ Diethyl

A 3. 58kg of 1,1 _ carbonyldiimidazole (22.16 mol) was placed in 12.8 l of tetrahydrofuran, and at a temperature of 40 ° C was dissolved in it with 6.5 l of tetrahydrofuran, 4. 52kg (22. 16 mol) of diethyl phosphono acetic acid mixture. Temperature at 40 ° C the mixture was stirred for 30 minutes. The resulting solution was referred to as Solution A.
A 6. 39kg (17. 05 moles) of N4-(3_ _4_ chloro fluoro – phenyl) _7_ (tetrahydrofuran _3_ yloxy) quinazoline-4, 6 – diamine Add 26 5 of tetrahydrofuran at 40 ° C and the solution A were mixed and stirred at a temperature 30 ° C for 2 hours.To the suspension was added 64 l tert-butyl methyl ether and, after cooling to 20 ° C, the precipitate was removed by centrifugation. Using 16 liters of tetrahydrofuran and 16 l of a mixture of tert-butyl methyl ether, washed, and then washed with 32 liters of water and dried at 50 ° C.
Yield: 6. 58kg (69. 8%) of white crystals, the content = HPLC 99. IFl%
Example 2
(E) -4 – dimethylamino – D -2 – acid – [4 – (chloro-3_ _4_ fluoro – phenylamino) _7_ (⑶ – tetrahydrofuran-3 – yloxy) – quinoline yl-6 – yl] – amide

A 5.6 l of 30% hydrochloric acid (53.17 mol) was added to 4.4 liters of water. Then the temperature is under 30 ° C was added dropwise over 20 minutes 4. 28kg 95% of (dimethylamino) _ acetaldehyde – diethyl acetal (26.59 mol).Temperature at 35 ° C the reaction solution was stirred for 8 hours was cooled to 5 ° C and kept under argon. This solution is called Solution B.
A 4. 55kg (68. 06 mol) of potassium hydroxide dissolved in 23.5 liters of water and cooled to _5 ° C. This solution is called Solution C.
A 5. 88kg (10. 63 mol) ((4_ (3_ _4_ chloro fluoro – phenylamino) _7_ (tetrahydrofuran _3_-yloxy) – quinazolin-6 – yl carbamoyl) – methyl)-phosphonic acid diethyl ester and 0.45kg _ lithium chloride (10.63 moles) was placed in 23.5 l of tetrahydrofuran and cooled to -7 ° C. Was added over 10 minutes a cold solution of C. Then _7 ° C temperature of the solution was added over 1 hour B. At _5 ° C temperature for 1 hour under stirring the reaction mixture was heated to 20 ° C and mixed with 15 liters of water. After cooling to; TC temperature, the suspension was suction filtered, the precipitate was washed with water and dried. Yield: 5.21kg The crude product, 100%, water content: 6.7%.
Using Titanium Dioxide / methyl cyclohexane embodiment the crystallization of the crude product.
Yield: 78%, purity: HPLC99. 4F1%, water content: 5.4%
Example 3
(E) -4 – dimethylamino – D -2 – acid – (4 – (chloro-3_ _4_ fluoro – phenylamino) ~ 7 ~ ((S) – tetrahydrofuran-3 – yl oxy) – quinazolin-6 – yl) – amide dimaleate
A 6. Okg (12. 35 mol) of (E_) _4_ _2_ dimethylamino acid _ D – (4_ (3_ _4_ chloro fluoro – phenylamino) -7 – ((S) – tetrahydrofuran-3 – yloxy) – quinazolin-6 – yl) – amide into 84 liters of ethanol and heated to 70 ° C, and dissolved in 36 l of ethanol and 2.94kg (25.31 moles) of maleic acid was mixed . At the beginning of crystallization, the first mixture was cooled to 20 ° C and stirred for 2 hours and then at 0 ° C temperature for 3 hours. Precipitate was suction filtered, washed with 19 l of ethanol at a temperature of 40 ° C in vacuo.
Yield: 8. Ilkg (91. 5%)
Melting point: 178 ° C
[0096] 1H-NMR (CD3OD): δ = 2. 47 + 2. 27 (m + m, 2H), 2. 96 (s, 6H), 4. 03 (m, 2Η), 4. 07 +3 . 92 (m + m, 2Η), 4. 18 +4. 03 (m + m, 2Η), 5. 32 (m, 1Η), 6. 26 (s, 4H), 6. 80 (m, 1H ), 6. 99 (m, 1H), 7 · 27 (s, 1Η), 7 · 30 (t, 1Η), 7 · 66 (m, 1Η), 7 · 96 (dd, 1Η), 8 · 62 (s, 1Η), 9 · 07 (s, 1Η) ppm
13
PATENT
Examples:
Example 1
{[4 – (3-chloro-4-fluoro-phenylamino) -7 – ((S)-tetrahydrofuran-3-yloxy)-quinazolin-6-ylcarbamoyl]-methyl)-phosphonic acid diethyl ester

3.58 kg 1 ,1-carbonyldiimidazole (22.16 mole) were placed in 12.8 liters of tetrahydrofuran at 40 ° C with 4.52 kg (22.16 mol) diethylphosphonoacetic acid, dissolved in 6.5 liters of tetrahydrofuran, . The mixture is stirred for 30 minutes at 40 ° C. The solution thus obtained is referred to as solution A.
6.39 kg (17.05 mol) of N 4 – (3-chloro-4-fluoro-phenyl) -7 – (tetrahydrofuran-3-yloxy) quinazolin-4,6-diamine in 26.5 liters of tetrahydrofuran and submitted to 40 ° C and mixed with the solution A and stirred at 30 ° C for 2 hours. To 64 liters of suspension of tert -. Added butyl methyl ether and, after cooling to 20 ° C., the precipitate is removed by centrifugation. It is dried with a mixture of 16 liters and 16 liters of tetrahydrofuran tert-butyl methyl ether and then washed with 32 liters of water at 50 ° C. Yield: 6.58 kg (69.8%) of white crystals Assay: HPLC 99.1 area% Example 2
(E)-4-dimethylamino-but-2-enoic acid [4 – (3-chloro-4-fluoro-phenylamino) -7 – ((S) – tetrahvdrofuran-3-yloxy)-quinazolin-6yl1 amide

5.6 liters to 4.4 liters of water are added 30% hydrochloric acid (53.17 mol). Then 4.28 kg 95% pure (dimethylamino) acetaldehyde diethyl acetal (26.59 mol) at 30 ° C was added dropwise over 20 minutes. The reaction solution is stirred for 8 hours at 35 ° C, cooled to 5 ° C and kept under argon. This solution is referred to as solution B.
4.55 kg (68.06 mol) of potassium hydroxide are dissolved in 23.5 liters of water and cooled to -5 ° C. This solution is called solution C..
5.88 kg (10.63 mol) of ((4 – (3-chloro-4-fluoro-phenylamino) -7 – (tetrahydrofuran-3-yloxy) – quinazolin-6-ylcarbamoyl)-methyl)-phosphonic acid diethyl ester, and 0.45 kg lithium chloride (10.63 mole) were placed in 23.5 liters of tetrahydrofuran and cooled to -7 ° C. The cold solution C is added within 10 minutes. The solution B is added at -7 ° C over 1 hour. After stirring for one hour at -5 ° C, the reaction mixture is heated to 20 ° C and mixed with 15 liters of water. After cooling to 3 ° C, the suspension is filtered with suction, the precipitate washed with water and dried. Yield: 5.21 kg raw 100% Water content: 6.7%
The crystallization of the raw product is butyl acetate / methylcyclohexane yield: 78% HPLC purity 99.4 area%, water content 5.4% Example 3
(E)-4-dimethylamino-but-2-enoic acid (4 – (3-chloro-4-fluoro-pheny hvdrofuran-3-yloxy)-quinazolin-6YL) amide dimaleate
6.0 kg (12.35 mol) of (E)-4-dimethylamino-but-2-enoic acid (4 – (3-chloro-4-fluoro-phenyl-amino) -7 – ((S)-tetrahydrofuran- 3-yloxy) quinazolin-6YL)-amide are in 84 liters
Submitted ethanol and heated to 70 ° C and a solution of 2.94 kg (25.31 mol) of maleic acid in 36 liters of ethanol added.Following the onset of crystallization is first cooled to 20 ° C. and stirred for 2 hours, then 3 hours at 0 ° C. The precipitate is filtered off, washed with 19 liters of ethanol and dried in vacuum at 40 ° C.
Yield: 8.11 kg (91, 5%)
Mp: 178 ° C.
1 H NMR (CD 3 OD): δ = 2.47 + 2.27 (m + m, 2H), 2.96 (s, 6H), 4.03 (m, 2H), 4.07 + 3 , 92
(M + m, 2H), 4.18 + 4.03 (m + m, 2H), 5.32 (m, 1 H), 6.26 (s, 4H), 6.80 (m, 1 H ), 6.99 (m, 1 H), 7.27 (s, 1 H), 7.30 (t, 1 H), 7.66 (m, 1 H), 7.96 (dd, 1 H ), 8.62 (s, 1 H), 9.07 (s, 1H) ppm
…………..

U.S. Patent No. : 8,426,586 patent expires : October 10, 2029
WO200250043A1 (compound);
WO2003094921A2 (anticancer purposes);
WO2003066060A2 (anti-inflammatory purposes);
US2005085495A1 (process);
WO2005037824A2 (process);
WO2007085638A1 (process);
US2011207932A1 (process);
WO2011084796A2 (deuterated);
WO2012121764A1 (crystalline);
WO2013052157A1 (crystalline)
Chinese patents : CN1867564
CN101402631
UPDATE…………………
(WO2015186065) PROCESS FOR THE PREPARATION OF 4-DIMETHYLAMINOCROTONIC ACID
SUN PHARMACEUTICAL INDUSTRIES LIMITED [IN/IN]; Sun House, Plot No. 201 B/1 Western Express Highway Goregaon (E) Mumbai, Maharashtra 400 063 (IN)
VERMA, Shyam Sunder; (IN).
SINGH, Shravan Kumar; (IN).
SINGH, Kaptan; (IN).
PRASAD, Mohan; (IN)
Afatinib is a tyrosine kinase inhibitor disclosed in U.S. Patent Nos. RE43,431 and
6,251,912. Afatinib is depicted by Formula la:

Formula la
Afatinib is presented as the dimaleate salt and is chemically designated as 2-butenamide, N-[4-[(3-chloro-4-fluorophenyl)amino]-7-[[(35)-tetrahydro-3-furanyl]oxy]-6-quinazolinyl]-4-(dimethylamino)-,(2£)-,(2Z)-2-butenedioate (1 :2) having the structure depicted by Formula I:

Formula I
Processes for the preparation of 4-dimethylaminocrotonic acid or its salts are disclosed in U.S. Patent No. 7,126,025 and U.S. Publication No. 2012/0046494.
U.S. Patent No. 7,126,025 discloses a process for the preparation of 4-dimethylaminocrotonic acid or its salts by reacting but-2-enoic acid with
chlorotrimethylsilane in pyridine to obtain trimethylsilylcrotonate, which is brominated with a brominating agent under free radical conditions and in the presence of methylene chloride, acetonitrile, 1,2-dichloroethane, carbon tetrachloride, or ethyl acetate to give trimethylsilyl-4-bromocrotonate. The bromocrotonate compound is treated with dimethylamine in tetrahydrofuran to provide the 4-dimethylaminocrotonic acid.
U.S. Patent No. 7,126,025 also discloses a process for the preparation of 4-dimethylaminocrotonic acid by treating methyl or ethyl 4-bromocrotonate with dimethylamine to provide methyl or ethyl 4-dimethylaminocrotonate, which is hydrolyzed to provide the 4-dimethylaminocrotonic acid.
U.S. Publication No. 2012/0046494 discloses a process for the preparation of 4-dimethylaminocrotonic acid or its salts by converting alkyl 4-chloro-3 -hydroxy butyrate to alkyl 4-hydroxy crotonate, which is brominated to obtain alkyl 4-bromo crotonate. The alkyl 4-bromo crotonate is treated with dimethyl amine to provide alkyl 4-dimethylaminocrotonate, which is hydrolyzed to get the 4-dimethylaminocrotonic acid.
The use of pyridine or carbon tetrachloride is toxic to humans and therefore their use for the manufacture of a drug substance is not advisable. The bromocrotonate compounds, being lachrymatory in nature, are difficult to handle on an industrial scale.
The present invention provides a faster, more efficient, and industrially feasible process for the preparation of 4-dimethylaminocrotonic acid of Formula II, which is used as an intermediate for the preparation of afatinib or its salts.
A first aspect of the present invention provides a process for the preparation of 4-dimethylaminocrotonic acid of Formula II or its salts,

Formula II
comprising the steps of:
i) converting 2,2-diethoxy-N,N-dimethylethanamine of Formula III

Formula III
to ethyl-4-(dimethylamino)crotonate of Formula IV; and

Formula IV
ii) hydrolyzing the ethyl-4-(dimethylamino)crotonate of Formula IV.
A second aspect of the present invention provides a process for the preparation of afatinib of Formula la or its salts,

Formula la
comprising the steps of:
i) converting 2,2-diethoxy-N,N-dimethylethanamine of Formula III

Formula III
to ethyl-4-(dimethylamino)crotonate of Formula IV;

Formula IV
ii) hydrolyzing the ethyl -4-(dimethylamino)crotonate of Formula IV to obtain 4- dimethylaminocrotonic acid of Formula II or its salts; and

Formula II
iii) converting the 4-dimethylaminocrotonic acid of Formula II or its salts to afatinib of Formula la or its salts.
EXAMPLES
Example 1 : Preparation of ethyl-4-(dimethylamino)crotonate (Formula IV)
In a round bottom flask, 2,2-diethoxy-N,N-dimethylethanamine (Formula III, 200 g) and deionized water (100 mL) were added at about 20°C to about 25°C. To the solution, concentrated hydrochloric acid (240 mL) was added at about 25°C to about 50°C. The temperature of the reaction mixture was raised to about 70°C. The reaction mixture was stirred at about 60°C to about 70°C for about 12 hours. The reaction mixture was cooled to about 0°C. To the reaction mixture, about 200 mL of aqueous potassium hydroxide (240 g in 250 mL water) was added at about 0°C to about 10°C to attain a pH of 9.0. To the reaction mixture, ethyl(diethoxyphosphoryl) acetate (200 g) and 2-methyltetrahydrofuran (600 mL) were added at about 0°C to about 5°C. Further, 50 mL of aqueous potassium hydroxide was added to the reaction mixture at about -5°C to about 0°C to attain a pH of about 13.5. The reaction mixture was stirred at about -5°C to about 0°C for about 1 hour. The reaction mixture was filtered, and then the filtrate was recovered under vacuum at about 45°C to about 50°C to obtain ethyl-4-(dimethylamino)crotonate as an oily mass.
Yield: 89%
Example 2: Preparation of 4-dimethylaminocrotonic acid hydrochloride (Formula ID
In a round bottom flask, ethyl -4-(dimethylamino)crotonate (Formula IV, 120 g) and ethanol (480 mL) were added at about 25°C to about 35°C. To the solution, aqueous sodium hydroxide (30.5 g in 60 mL water) was added at about 10°C to about 20°C. The temperature of the reaction mixture was raised to about 50°C. The reaction mixture was stirred at about 50°C to about 55°C for about 1 hour. The reaction mixture was cooled to about 5°C. To the reaction mixture, concentrated hydrochloric acid (120 mL) was added to attain a pH of 1.5. The reaction mixture was filtered on Celite® and washed with ethanol (50 mL). The filtrate was recovered under vacuum at about 55°C to about 60°C to obtain a crude mass. Ethanol (240 mL) was added to the crude mass, and then the reaction mixture was stirred at about 55°C to about 60°C for about 15 minutes to obtain a solution. In the solution, sodium chloride was obtained as a byproduct. The solution was filtered to discard sodium chloride. The filtrate was recovered under vacuum at about 55°C to about 60°C to obtain a residue. To the residue, isopropanol (400 mL) was added, and then the reaction mixture was stirred at about 55°C to about 60°C to obtain a clear solution. The solution was gradually cooled to about 25°C to about 30°C. The solution was further stirred at the same temperature for about 2 hours. The solid obtained was filtered, and then washed with isopropanol (50 mL). The solid was dried under vacuum at about 55°C to about 60°C to provide 4-dimethylaminocrotonic acid hydrochloride.
Yield: 63%
|
5-30-2012
|
Amide derivative for inhibiting the growth of cancer cells
|
|
|
6-15-2011
|
PROCESS FOR PREPARING AMINOCROTONYLAMINO-SUBSTITUTED QUINAZOLINE DERIVATIVES
|
|
|
12-25-2009
|
METHOD FOR TREATING CANCER HARBORING EGFR MUTATIONS
|
|
|
12-11-2009
|
QUINAZOLINE DERIVATIVES FOR THE TREATMENT OF CANCER DISEASES
|
|
|
12-11-2009
|
COMBINATION TREATMENT OF CANCER COMPRISING EGFR/HER2 INHIBITORS
|
|
|
9-12-2008
|
Multi-Functional Small Molecules as Anti-Proliferative Agents
|
|
|
4-22-2005
|
Process for preparing amino crotonyl compounds
|
//////
Afatinib
-
- Synonyms:BIBW 2992
- ATC:L01XE13
- Use:anticancer; tyrosine kinase inhibitor
- Chemical name:N-[4-[(3-chloro-4-fluorophenyl)amino]-7-[[(3S)-tetrahydro-3-furanyl]oxy]-6-quinazolinyl]-4-(dimethylamino)-2-butenamide; N-[(3-chloro-4-fluorophenyl)amino]-6-{[4-(N,N-dimethylamino)-1-oxo-2-buten-1-yl]amino}-7-((S)-tetrahydrofuran-3-yloxy)-quinazoline
- Formula:C24H25ClFN5O3
- MW:485.9 g/mol
- CAS-RN:439081-18-2; 850140-72-6
Derivatives
dimaleate
- Formula:C32H33ClFN5O11
- MW:718.1 g/mol
- CAS-RN:850140-73-7
Substance Classes
Synthesis Path
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 446-32-2 | C7H6FNO2 | 4-fluoro-anthranilic acid | |
| 162012-70-6 | C8H3ClFN3O2 | 4-chloro-7-fluoro-6-nitroquinazoline | |
| 367-21-5 | C6H5ClFN | 3-chloro-4-fluoroaniline | |
| 86087-23-2 | C4H8O2 | (S)-(+)-3-hydroxytetrahydrofuran | |
| 314771-76-1 | C18H16ClFN4O2 | N-(3-chloro-4-fluorophenyl)-7-((tetrahydrofuran-3-yl)oxy)quinazoline-4,6-diamine | |
| 13991-36-1 | C4H5BrO2 | bromocrotonic acid | |
| 3095-95-2 | C6H13O5P | diethylphophonoacetic acid | |
| 618061-76-0 | C24H27ClFN4O6P | Diethyl-{[4-((3-chloro-4-fluorophenyl)amino)-7-(((S)-tetrahydro- furan-3-yloxy)quinazolin-6-yl)carbamoyl]-methyl}phosphonate |
|
| 3616-56-6 | C8H19NO2 | (dimethylamino)-acetaldehyde diethylacetate |
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| USA | Gilotrif | Boehringer Ingelheim, 2013 | |
| EU | Giotrif | Boehringer Ingelheim, 2013 |
Formulations
- tabs.; 20, 30 and 40 mg
References
-
- a US 6 251 912 (American Cyanamid; 26.6.2001; appl. 29.7.1998; USA-prior. 1.8.1997).
- WO 0 250 043 (Boehringer Ingelheim; 27.6.2002; appl. 12.12.2001; DE-prior. 20.12.2000).
- US RE 43431 (Boehringer Ingelheim; 29.5.2012; appl. 18.8.2009; DE-prior. 20.12.2000).
- b US 8 426 586 (Boehringer Ingelheim; 1.2.2007; appl. 14.7.2006; DE-prior. 17.10.2003).
-
crystalline forms of Afatinib di-maleate:
- Solca, F. et al., J. Pharmacol. Exp. Ther., (2012) 343(2), 342-350.
- WO 2013 052157 (Ratiopharm/Teva; 11.4.2013; appl. 25.4.2012; USA-prior. 6.10.2011).
Teva Pharmaceutical has been given a green light by the European Commission (EC) for Lonquex, a rival to Amgen’s blockbuster Neulasta.

lipegfilgrastim
C 864 H 1369 N 225 O 258 S 9 [C 2 H 4 O] N
pegylated granulocyte colony stimulating factor; O3.133-[N5-(N-{[ω-methoxypoly (oxyethylene)] carbonyl} glycyl)-α-neuraminyl-(2 → 6)-α-D-galactopyranosyl]-L-methionyl -des-1-L-alanine-des-37-L-valine-des-38-L-serine-des-39-L-glutamic acid-human granulocyte colony-stimulating factor (G-CSF, pluripoietin)
Lonquex (lipegfilgrastim) has been approved to reduce the duration of neutropaenia (low white blood cell counts) and febrile neutropaenia in patients undergoing cytotoxic chemotherapy for cancer, and is given as a single subcutaneous dose per cycle of chemotherapy.
Like Neulasta (pegfilgrastim), Lonquex is a long-acting recombinant granulocyte colony-stimulating factor (G-CSF) and is dosed at the same frequency as Amgen’s drug.
http://www.pmlive.com/pharma_news/neulasta_rival_from_teva_cleared_in_eu_495953
OMARIGLIPTIN. MK 3102 IN PHASE 3 FOR TYPE 2 DIABETES
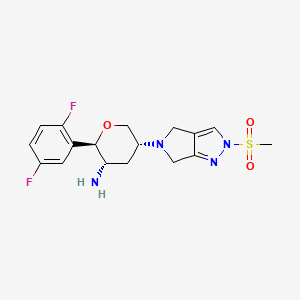
OMARIGLIPTIN. MK 3102
cas 1226781-44-7
Approved in japan SEPT 28 2015
(2R,3S,5R)-2-(2,5-difluorophenyl)-5-[2-(methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl]tetrahydro-2H-pyran-3-amine
(2R,3S,5R)-2-(2,5-difluorophenyl)-5-(2-methylsulfonyl-4,6-dihydropyrrolo[3,4-c]pyrazol-5-yl)oxan-3-amine
(2R,3S,5R)-2-(2,5-Difluorophenyl)-5-[2-(methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl]tetrahydro-2H-pyran-3-amine
IN PHASE 3

CLICK ON IMAGES FOR CLARITY VIEW


PAPER

In our effort to discover DPP-4 inhibitors with added benefits over currently commercially available DPP-4 inhibitors, MK-3102 (omarigliptin), was identified as a potent and selective dipeptidyl peptidase 4 (DPP-4) inhibitor with an excellent pharmacokinetic profile amenable for once-weekly human dosing and selected as a clinical development candidate. This manuscript summarizes the mechanism of action, scientific rationale, medicinal chemistry, pharmacokinetic properties, and human efficacy data for omarigliptin, which is currently in phase 3 clinical development.
Omarigliptin (MK-3102) (2R,3S,5R)-2-(2,5-difluorophenyl)-5-[2-(methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl]tetrahydro-2H-pyran-3-amine, is a 2,3,5-substituted tetrahydropyran analogue currently in phase 3 clinical trial for type 2 diabetes mellitus (T2DM).
is a 2,3,5-substituted tetrahydropyran analogue currently in phase 3 clinical trial for type 2 diabetes mellitus (T2DM).
(2R,3S,5R)-2-(2,5-Difluorophenyl)-5-[2-(methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl]tetrahydro-2H-pyran-3-amine, Omarigliptin
Crystallization from ethyl acetate gave a compound with greater than 99% purity.
Optical rotation [α]D20 −12.0° (c 1.0, CH3OH).
1H NMR (CD3OD, 500 MHz) δ = 1.71 (q, 1H, J = 12 Hz), 2.56–2.61 (m, 1H), 3.11–3.18 (m, 1H), 3.36–3.40 (m, 1H), 3.48 (t, 1H, J = 12 Hz), 3.88–3.94 (m, 4H), 4.30–4.35 (m, 1H), 4.53 (d, 1H, J = 12 Hz), 7.14–7.23 (m, 2H), 7.26–7.30 (m, 1H), 7.88 (s, 1H).
LC–MS: 399.04 (M + 1).
PATENT
http://www.google.com.tr/patents/US20100120863?hl=tr&cl=ja
Example 1

(2R,3S,5R)-2-(2,5-Difluorophenyl)-5-[2-(methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl]tetrahydro-2H-pyran-3-amineStep A: tert-Butyl {(2R,3S,5R)-2-(2,5-difluorophenyl)-5-[2-(methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5 (4H)-yl]tetrahydro-2H-pyran-3-yl}carbamate
A mixture of Intermediate 2 (26.3 g, 80 mmol) and 2-(methylsulfonyl)-2,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole (Intermediate 5) (15.07 g, 80 mmol) in anhydrous methanol (1.5 L) was stirred at room temperature for 2 h. To the resulting white suspension was added decaborane (2.95 g, 24.15 mmol) and the mixture was stirred at room temperature overnight. Methanol was removed and the residue was purified on two 65i Biotage™ columns eluting with 5-50% ethyl acetate in dichloromethane to afford the title compound as a white solid. LC-MS: 499.10 (M+1).
Step B: (2R,3S,5R)-2-(2,5-Difluorophenyl)-5-[2-(methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl]tetrahydro-2H-pyran-3-amine
Removal of the BOC group in the product from Step A (13.78 g, 27.67 mmol) was accomplished with trifluoroacetic acid (100 ml) in dichloromethane (200 mL) at room temperature. After stirring for 2 h, the reaction was concentrated and neutralized with 25% MeOH and 2.5% ammonium hydroxide in dichloromethane. Solvents were removed under reduced pressure and the resulting crude material was purified on a 65i Biotage™ column eluting with 1.25-5% MeOH and 0.125-0.5% ammonium hydroxide in dichloromethane. The isolated material was further purified by recrystallization from 5:1 EtOAc/CH2Cl2 at 60° C. The crystalline product was washed with cold 2:1EtOAc/hexanes to give the title compound as a light brown solid. 1H NMR (500 MHz, CD3OD): 1.71 (q, 1H, J=12 Hz), 2.56-2.61 (m, 1H), 3.11-3.18 (m, 1H), 3.36-3.40 (m, 1H), 3.48 (t, 1H, J=12 Hz), 3.88-3.94 (m, 4H), 4.30-4.35 (m, 1H), 4.53 (d, 1H, J=12 Hz), 7.14-7.23 (m, 2H), 7.26-7.30 (m, 1H), 7.88 (s, 1H). LC-MS: 399.04 (M+1).
Intermediate 2

tert-Butyl[(2R,3S)-5-oxo-2-(2,5-difluorophenyl)tetrahydro-2H-pyran-3-yl]carbamate Step A: 1-(2,5-Difluorophenyl)-2-nitroethanol
To sodium hydroxide (1N, 3L) and methanol (1500 mL) at 5° C. was added a solution of 2,5-difluorobenzaldehyde (350 g, 2.46 mol) and nitromethane (157 mL, 2.9 mol) in methanol (350 mL) dropwise over a period of 1 h. The reaction mixture was then neutralized with glacial acetic acid (165 mL). Diethyl ether (1500 mL) was added and the layers separated. The organic layer was washed successively with saturated aqueous sodium carbonate solution (1000 mL), and saturated aqueous brine (1000 mL). The organic layer was dried over anhydrous magnesium sulfate, filtered and concentrated to afford 1-(2,5-difluorophenyl)-2-nitroethanol that was used without further purification in Step B.
Step B: 2-Nitro-1-(2,5-difluorophenyl)ethanone
A solution of Dess-Martin periodinane (125 g) in dichloromethane (600 mL) was added to a solution of the nitroalcohol made in Step A (46.3 g) at 10° C. over a period of 30 min. Stirring was continued for 2 h, and the reaction mixture was then poured onto a mixture of sodium bicarbonate (300 g) and sodium thiosulfate (333 g) in water (3 L). The desired product was extracted with methyl t-butyl ether (MTBE) (2 L). The aqueous layer was neutralized with HCl (2N, 1.5 L) and extracted with MTBE (3 L). The combined organic layers were dried over anhydrous magnesium sulfate, filtered, evaporated and the residue was purified by chromatography (silica gel, eluting with dichloromethane) to yield the desired nitroketone.
Step C: 3-Iodo-2-(iodomethyl)prop-1-ene
A mixture of 3-chloro-2-(chloromethyl)prop-1-ene (1.0 g, 8 mmol) and sodium iodide (6.6 g, 44 mmol) in acetone (60 mL) was stirred at room temperature for 20 h, evaporated under reduced pressure and partitioned between dichloromethane (150 mL) and water (50 mL). The organic layer was dried over sodium sulfate, filtered and evaporated to yield 3-iodo-2-(iodomethyl)prop-1-ene as a reddish oil.
Step D: 3-Methylene-5-nitro-6-(2,5-difluorophenyl)-3,4-dihydro-2H-pyran
N,N-diisopropylethylamine (184 mL) was added to a solution of 2-nitro-1-(2,5-difluorophenyl)ethanone (92.7 g, 461 mmol) in N,N-dimethylformamide (1000 mL) and 3-iodo-2-(iodomethyl)prop-1-ene (156 g, 507 mmol). The mixture was heated at 60° C. for 2 h, evaporated and purified by chromatography (silica gel, gradient 0-30% dichloromethane in hexane) to yield 3-methylene-5-nitro-6-(2,5-difluorophenyl)-3,4-dihydro-2H-pyran.
Step E: (2R,3S)-5-Methylene-3-nitro-2-(2,5-difluorophenyl)tetrahydro-2H-pyran
This compound was made by following the same method described in Intermediate 1, Step D by using 3-methylene-5-nitro-6-(2,5-trifluorophenyl)-3,4-dihydro-2H-pyran.
Step F: (2R,3S)-5-Methylene-2-(2,5-difluorophenyl)tetrahydro-2H-pyran-3-amine
This compound was made by following the same method described in Intermediate 1, Step E by using (2R,3S)-5-Methylene-3-nitro-2-(2,5-difluorophenyl)tetrahydro-2H-pyran.
Step G: tert-Butyl[(2R,3S)-5-methylene-2-(2,5-difluorophenyl)tetrahydro-2H-pyran-3-yl]carbamate
This compound was made by following the same method described in Intermediate 1, Step F by using (2R,35)-5-methylene-2-(2,5-difluorophenyl)tetrahydro-2H-pyran-3-amine.
Step H: tert-Butyl[(2R,3S)-5-hydroxy-5-(hydroxymethyl)-2-(2,5-difluorophenyl)tetrahydro-2H-pyran-3-yl]carbamate
This compound was made by following the same method described in Intermediate 1, Step G by using tert-butyl[(2R,35)-5-methylene-2-(2,5-difluorophenyl)tetrahydro-2H-pyran-3-yl]carbamate.
Step I: tert-Butyl[(2R,3S)-5-oxo-2-(2,5-difluorophenyl)tetrahydro-2H-pyran-3-yl]carbamate
To a solution of tert-butyl[(2R,3S)-5-hydroxy-5-(hydroxymethyl)-2-(2,5-trifluorophenyl)tetrahydro-2H-pyran-3-yl]carbamate (10.5 g) in methanol (100 mL) at 0° C. was added pyridine (7.8 mL) and lead tetraacetate (21.7 g). The reaction mixture was stirred for 20 min. Aqueous work-up with ethyl acetate gave crude product which was purified by chromatography (silica, 0-50% ethyl acetate/heptane) to yield tert-butyl[(2R,35)-5-oxo-2-(2,5-difluorophenyl)tetrahydro-2H-pyran-3-yl]carbamate as white solid.
Intermediate 3

Step A: tert-Butyl (3Z)-3-[(dimethylamino)methylene]-4-oxopyrrolidine-1-carboxylate
A solution of tert-butyl 3-oxopyrrolidine-1-carboxylate (40 g, 216 mmol) was treated with DMF-DMA (267 g, 2241 mmol) and heated at 105° C. for 40 min. The solution was cooled and evaporated under reduced pressure and the resulting orange solid was treated with hexane (200 mL) and cooled in a refrigerator for 3 days. The resulting brownish-yellow solid obtained as such was collected by filtration, dried and used in the next step without further purification.
Step B: 1,4,5,6-Tetrahydropyrrolo[3,4-c]pyrazole
A solution of hydrazine (3 mL) and tert-butyl (3Z)-3-[(dimethylamino)methylene]-4-oxopyrrolidine-1-carboxylate (19.22 g) in ethanol (40 mL) was heated at 85° C. in a sealed tube for 4 h. Solvent was removed under reduced pressure, and the residue was triturated with dichloromethane (160 mL) and ethyl acetate (15 mL). The resulting solid was filtered. The filtrate was concentrated and the resulting solid was triturated again and filtered. The combined solids were treated with 4N hydrochloric acid (250 mL) in methanol and stirred for 6 h. The reaction mixture was concentrated and dried. The resulting solid was treated again for 6 h with 4N hydrochloric acid (250 mL) in methanol. After concentration and drying, the resulting hydrochloride salt was treated with ammonia in methanol (2N, 300 mL) and ammonium hydroxide solution in water (28%, 30 mL) and concentrated to dryness. The solid obtained was treated with methanol (70 mL) and water (5 mL) and purified in three batches on Biotage Horizon® system (silica, gradient 5-17% methanol containing 10% concentrated ammonium hydroxide in ethyl acetate) to yield 1,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole. 1H NMR (500 MHz, CD3OD): δ 4.04 (d, 4H); 7.39 (s, 1H).
Intermediate 5

2-(Methylsulfonyl)-2,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole Step A: tert-Butyl 1-(methylsulfonyl)]-4,6-dihydropyrrolo[3,4-c]pyrazole-5(1H)-carboxylate (A) and tent-butyl 2-(methylsulfonyl)]-2,6-dihydropyrrolo[3,4-c]pyrazole-5(4H)-carboxylate (B)
A suspension of N-Boc-pyrazolopyrrolidine (Intermediate 3, Step B) (27.16 g, 130 mmol) in anhydrous acetonitrile (1.0 L) was charged in a 2.0 L three-neck flask fitted with a thermometer and an addition funnel and then treated with sodium hydride (60% dispersion in oil, 6.23 g, 156 mmol) while under nitrogen atmosphere in one portion. The reaction mixture was stirred at room temperature for 2 h. The resulting white suspension was then cooled in an ice bath and methanesulfonyl chloride (25.2 mL, 324 mmol) was slowly added via addition funnel The ice bath was then removed and the mixture was stirred 1 h at room temperature. The reaction mixture was quenched with water (500 mL) and the layers were separated. The aqueous layer was then extracted with 2×500 mL of dichloromethane. The combined organic layers were dried over sodium sulfate and concentrated under reduced pressure to give a mixture of products A and B as colorless syrups. NMR in CD3OD indicated a 1:1 mixture of two products, in which the proton on the pyrazole ring in product A appeared at 7.70 ppm while the proton in product B appeared at 7.95 pm. LC-MS: 288.08 (M+1).
Step B: 2-(Methylsulfonyl)-2,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole
Trifluoroacetic acid (200 mL) was added slowly to a solution containing intermediates A and B prepared in the previous step (48.4 g, 168 mmol) in dichloromethane (400 mL) at 0° C. After addition, the cooling bath was removed and the reaction was allowed to stir at room temperature for 2 h. Solvent was removed under reduced pressure and the resulting trifluoroacetate salt was then neutralized with 500 mL of 25% methanol and 2.5% ammonium hydroxide in dichloromethane. After removal of solvent, the desired Intermediate 5 was obtained after chromatography on a Biotage™ column (2×340 g) eluting with 2.5-12.5% methanol and 0.25-1.25% ammonium hydroxide in dichloromethane. LC-MS: 109.85 (M+1).
PATENT
below patent caution…………….similar not same….examples below will help you in synthesis similarities
http://www.google.com/patents/WO2014018355A1?cl=en

Step 1 2 Step 2

Example 1
Synthesis of 3: (Step 1 & 2)
Dimethyldisulfide 1 (5 g, 53 mmol) and acetic acid (6 mL, 106 mmol) were mixed under nitrogen atmosphere and cooled to – 20 °C. Sulfuryl chloride (13 mL, 159 mmol) was added dropwise with stirring. The mixture was then stirred for 1 hour at -20 °C and afterwards allowed to come to room temperature and continued for another two hours. Acetyl chloride was distilled off from the reaction mixture. Crude methanesulfinyl chloride 2 obtained was used in the next step without further purification.
To a solution of chloramine T (14.95 g, 53 mmol) in dry toluene (220 mL) was added a solution of methanesulfinyl chloride 2 (5.2 g, 53 mmol) in dry toluene (10 mL) at 0 °C. The resulting suspension was heated at 80 °C for 2 hours with stirring. After cooling, the solid was filtered off and washed with dry toluene (100 mL). The filtrate was evaporated in vacuo and the crude mixture was purified through silica gel chromatography to obtain 3 as off white solid. XH NMR (300 MHz, CDC13): δ 7.85 – 7.91 (m, J= 8.42 Hz, 2H), 7.31 – 7.38 (m, J= 8.23 Hz, 2H), 3.78 (s, 3H), 2.45 (s, 3H).
Synthesis of 4: (Step 3)
To a solution of Ml (1.0 g, 2.2 mmol) in THF (10 mL) and DMF (10 mL) under nitrogen atmosphere at 0 °C was added Et3N (0.92 mL, 6.6 mmol) followed by B0C2O (0.48 g, 2.2 mmol). The reaction mixture was allowed to come to room temperature and continued the stirring for over night. The reaction mixture was diluted with water (100 mL) and extracted with CH2CI2 (3 x 100 mL). Combined organics were dried over Na2S04, filtered, concentrated under vacuum and purified by silica gel chromatography afforded 4 as a off white solid.
XH NMR (400 MHz, CDC13): δ 7.27 – 7.35 (m, 1H), 4.44 – 4.54 (m, 4H), 1.52 (s, 9H).
Synthesis of 5: (Step 4)
To a suspension of NaH (0.30 g, 7.5 mmol) in dry THF (5 mL) under nitrogen atmosphere at 0 °C was added a solution of 4 (0.78 g, 3.7 mmol) in dry THF (30 mL). The reaction mixture was allowed to come to room temperature and continued the stirring for 2 hours. Reaction mixture was again cooled to 0 °C. A solution of 3 (2.0 g, 7.4 mmol) in THF (25 mL) was added to the reaction mixture and continued the stirring for another 1 hour. The reaction mixture was quenched with water (100 mL) and extracted with EtOAc (3 x 200 mL). Combined organics were dried over Na2S04, filtered, concentrated under vacuum and purified by silica gel chromatography afforded 5 as an off-white solid.
XH NMR (400 MHz, CDC13): δ 7.84 – 7.88 (m, 1H), 7.78 (t, J= 8.27 Hz, 2H), 7.23 – 7.30 (m, 2H), 4.39 – 4.49 (m, 4H), 3.53 (d, J= 2.40 Hz, 3H), 2.42 (s, 3H), 1.53 (s, 9H).; Molecular Formula: Ci8H24N405S2; LCMS purity: 98.18%; Expected: 440.1 ; Observed: 341.0 (M-99).
Synthesis of 6: (Step 5)
To a solution of 5 (0.47 g, 1.06 mmol) in dry CH2CI2 (1 1 mL) under nitrogen atmosphere at 0 °C was added TFA (3 mL). The reaction mixture was allowed to come to room temperature and continued the stirring for 2 hours. Solvent was removed under vacuum and solid mass was washed with Et20 (3 x 10 mL) to get amine TFA salt as white solid.
XH NMR (300 MHz, CD3OD): δ 7.78 (s, 1H), 7.63 – 7.70 (m, J= 8.11 Hz, 2H), 7.26 – 7.35 (m, J = 8.33 Hz, 2H), 3.93 (s, 2H), 3.86 (s, 2H), 3.34 (s, 3H), 2.42 (s, 3H).
The amine TFA salt was dissolved in minimum volume of MeOH:CHCi3 (1 : 1) and passed through a column [Orochem 5 g, 10 ml, Amino (N¾)] using MeOH as eluent.
Organics were concentrated under vacuum to get free 6.
Synthesis of 7: (Step 6) To a stirred solution of 6 (0.34 g, 0.95 mmol) and M2 (0.26 g, 0.79 mmol) in DMAc (6.78 mL) under nitrogen atmosphere for 10 minutes was added AcOH (0.067 mL, 1.19 mmol). The reaction mixture was stirred for further 5 minutes and cooled to 0 °C. NaBH(OAc)3 (0.20 g, 0.95 mmol) was added to the reaction mixture and allowed to stirrer at room temperature for overnight. NH4OH (2 mL) was added to the reaction mixture and heated at 50 °C for 1 hour followed by water (3.39 mL) and again heated at 50 °C for another hour. Reaction mixture was cooled to room temperature and filtered. The solid residue was washed with water (4 x 100 mL) and the crude residue was purified by silica gel chromatography to afford 7.
XH NMR (300 MHz, CDC13): δ 7.80 (d, J= 6.95 Hz, 3H), 7.25 – 7.29 (m, 2H), 7.22 (br. s., 1H), 6.92 – 7.02 (m, 2H), 4.52 (d, J= 9.33 Hz, 1H), 4.24 – 4.40 (m, 2H), 3.85 (br. s., 5H), 3.48 (s, 3H), 3.39 – 3.47 (m, 1H), 3.07 (br. s., 1H), 2.52 (d, J= 10.25 Hz, 1H), 2.44 (s, 3H), 1.61 (br. s., 1H), 1.28 (s, 9H).; Molecular Formula: C29H35F2N506S2; LCMS purity: 99.08%; Expected: 651.2; Observed: 652.0 (M+l). Synthesis of Example 1: (Step 7)
To a solution of 7 (20 mg, 0.03 mmol) in dry CH2CI2 (2 mL) under nitrogen atmosphere at 0 °C was added TFA (0.5 mL). The reaction mixture was allowed to come to room temperature and continued the stirring for 2 hours. Solvent was removed under vacuum and solid mass was washed with Et20 to get amine di-TFA salt Example 1 as white solid. Unless otherwise noted the IC50 values were determined using the assay discussed earlier.
XH NMR (400 MHz, CD3OD): δ 8.05 (s, 1H), 7.73 (d, J= 8.03 Hz, 2H), 7.36 (d, J= 8.28 Hz, 2H), 7.29 – 7.34 (m, 1H), 7.20 – 7.27 (m, 2H), 4.71 (d, J= 10.04 Hz, 1H), 4.40 – 4.53 (m, 5H), 3.72 – 3.82 (m, 2H), 3.68 (s, 3H), 3.59 – 3.65 (m, 1H), 2.77 – 2.85 (m, 1H), 2.44 (s, 3H), 2.00 – 2.14 (m, 1H).; Molecular Formula: C24H27F2 504S2; HPLC purity: 99.74%; LCMS Expected: 551.2; Observed: 552.2 (M+l).
SCHEME 2

Example 2: Synthesis of Compound 1 & 2 (Step 1):
To a suspension of M2 (0.95 g, 2.8 mmol) in water (8.67 mL) was added sodium metabisulfite (0.55 g, 2.8 mmol) and stirred a room temperature for lhour. A solution of M3* (0.52 g, 2.8 mmol) in ethanol (8.67 mL) was added to the above reaction mixture and continued the stirring for further 4 hours. Neat aCN (0.14 g, 2.8 mmol) was added to the above reaction mixture in one portion and heated the reaction mixture at 50 °C for 2 days. Reaction mixture was concentrated under vacuum to remove most of the ethanol. The crude mixture was extracted with CHCI3 (50 x 3 mL). The combined organic layer was washed with water, dried over a2S04, filtered, concentrated and purified by flash chromatography to obtain 1 and 2 as solids.
Compound 1: ‘H NMR (300 MHz, CDC13): δ 7.77 (s, 1H), 7.26 – 7.35 (m, 1H), 7.00 (t, J= 5.76 Hz, 2H), 4.57 (t, J= 9.88 Hz, 2H), 4.32 – 4.39 (m, 1H), 3.85 – 4.09 (m, 5H), 3.60 (d, J= 11.34 Hz, 1H), 3.34 (s, 3H), 2.63 – 2.74 (m, 1H), 2.02 – 2.15 (m, 1H), 1.31 (s, 9H).
Compound 2: XH NMR (300 MHz, CDC13): δ 7.28 – 7.36 (m, 2H), 7.00 (t, J= 5.85 Hz, 2H), 4.55 (d, J= 8.97 Hz, 2H), 4.37 (dd, J= 2.65, 11.25 Hz, 1H), 3.88 – 4.07 (m, 5H), 3.60 (d, J = 1 1.34 Hz, 1H), 2.71 (td, J= 3.45, 12.49 Hz, 1H), 1.97 – 2.12 (m, 1H), 1.31 (s, 9H).; Molecular Formula: C22H25F2 503; LCMS purity: 94.48%; Expected: 445.2; Observed: 446.0 (M+l). (*Preparation of M3: M3.PI1SO3H (1.0 g, 2.8 mmol) was dissolved in minimum volume of MeOH:CHCl3 (1 : 1) and passed through a column [Orochem 5 g, 10 ml, Amino (NH2)] using MeOH as eluent. Organics were concentrated under vacuum to get free M3, which was used directly without further purification.) Synthesis of compound 3 (Step 2):
To a solution of compound 2 (0.40 g, 0.89 mmol) in THF (5 mL) under 2 atmosphere at -78 °C was added a solution of MeMgBr (0.89 mL, 2.6 mmol, 3M in Et20). The reaction mixture was allowed to attain room temperature over 1 hour. TLC shows complete conversion. The reaction mixture was again cooled to -10 °C and quenched with saturated aq. NH4CI solution (10 mL). The reaction mixture was extracted with CH2CI2 (50 x 3 mL).
Combined organics were dried over Na2S04, filtered, concentrated and purified by reversed phase chromatography to obtain 3 as di-TFA salt.
Molecular Formula: C22H28F2 4O3; LCMS purity: 88.82%; Expected: 434.2; Observed: 435.2 (M+l).
Synthesis of Example 2 (Step 3):
To a solution of compound 3 (35 mg, 0.053 mmol) in CH2CI2 (2 mL) was added TFA (0.5 mL) dropwise at 0 °C. Reaction mixture was allowed to attain room temperature over 2 hours time. TLC shows complete conversion. Reaction mixture was concentrated to dryness. The solid residue was washed with Et20 (10 x 3 mL) and dried under vacuum to obtain Example 2 as tri-TFA salt.
XH NMR (400 MHz, CD3OD): δ 7.60 (s, 1H), 7.37 (dd, J= 5.02, 8.03 Hz, 1H), 7.22 – 7.31 (m, 2H), 4.70 (d, J= 10.04 Hz, 1H), 4.48 – 4.61 (m, 4H), 4.17 (dd, J= 2.26, 11.29 Hz, 1H), 3.91 (d, J = 11.04 Hz, 1H), 3.73 – 3.83 (m, 1H), 2.54 – 2.62 (m, 1H), 2.22 (t, J= 12.05 Hz, 1H), 1.71 (s, 3H).; Molecular Formula: C17H20F2 4O; HPLC purity: 94.98%; Expected: 334.2; Observed: 335.2 (M+l).
SCHEME 3

Example 3
Synthesis of 1 & 2: (Step 1)
To a suspension of M2 (0.95 g, 2.8 mmol) in water (8.67 mL) was added sodium metabisulfite (0.55 g, 2.8 mmol) and stirred a room temperature for lhour. A solution of M3* (0.52 g, 2.8 mmol) in ethanol (8.67 mL) was added to the above reaction mixture and continued the stirring for further 4 hours. Neat aCN (0.14 g, 2.8 mmol) was added to the above reaction mixture in one portion and heated the reaction mixture at 50 °C for 2 days. Reaction mixture was concentrated under vacuum to remove most of the ethanol. The crude mixture was extracted with CHCI3 (50 x 3 mL). The combined organic layer was washed with water, dried over a2S04, filtered, concentrated and purified by flash chromatography to obtain 1 and 2 as solids.
Compound 1: ‘H NMR (300 MHz, CDC13): δ 7.77 (s, 1H), 7.35 – 7.26 (m, 1H), 7.00 (t, J= 5.76 Hz, 2H), 4.57 (t, J= 9.88 Hz, 2H), 4.39 – 4.32 (m, 1H), 4.09 – 3.85 (m, 5H), 3.60 (d, J= 1 1.34 Hz, 1H), 3.34 (s, 3H), 2.74 – 2.63 (m, 1H), 2.15 – 2.02 (m, 1H), 1.31 (s, 9H).
Compound 2: XH NMR (300 MHz, CDC13): δ 7.36 – 7.28 (m, 2H), 7.00 (t, J= 5.85 Hz, 2H), 4.55 (d, J= 8.97 Hz, 2H), 4.37 (dd, J= 2.65, 11.25 Hz, 1H), 4.07 – 3.88 (m, 5H), 3.60 (d, J= 1 1.34 Hz, 1H), 2.71 (td, J= 3.45, 12.49 Hz, 1H), 2.12 – 1.97 (m, 1H), 1.31 (s, 9H).; Molecular Formula: C22H25F2 503; LCMS purity: 94.48%; Expected: 445.2; Observed: 446.0 (M+l).
(*Preparation of M3: M3.PI1SO3H (1.0 g, 2.8 mmol) was dissolved in minimum volume of MeOH:CHCl3 (1 : 1) and passed through a column [Orochem 5 g, 10 ml, Amino (NH2)] using MeOH as eluent. Organics were concentrated under vacuum to get free M3, which was used directly without further purification.) Synthesis of compound 3 (Step 2):
To a solution of 2 (0.40 g, 0.89 mmol) in THF (5 niL) under 2 atmosphere at -78 °C was added a solution of MeMgBr (0.89 mL, 2.6 mmol, 3M in Et20). The reaction mixture was allowed to attain room temperature over 1 hour. TLC shows complete conversion. The reaction mixture was again cooled to -10 °C and quenched with saturated aq. NH4CI solution (10 mL). The reaction mixture was extracted with CH2CI2 (50 x 3 mL). Combined organics were dried over Na2S04, filtered, concentrated and purified by reversed phase chromatography to obtain 3 (0.05 g, 8.4%) as di-TFA salt.
Molecular Formula: C22H28F2 4O3; LCMS purity: 88.82%; Expected: 434.2; Observed: 435.2 (M+l).
Synthesis of compound 4 (Step 3):
To a suspension of NaH (22 mg, 0.55 mmol) in dry THF (0.1 mL) under nitrogen atmosphere at 0 °C was added a solution of 3 (120 mg, 0.27 mmol) in dry THF (4.8 mL). The reaction mixture was allowed to come to room temperature and continued the stirring for 2 hours. Reaction mixture was again cooled to 0 °C. Methanesulfonyl chloride (0.42 mL, 0.55 mmol) was added to the reaction mixture and continued the stirring for another 1 hour. The reaction mixture was quenched with water and extracted with EtOAc (3 x 50 mL). Combined organics were dried over Na2S04, filtered, concentrated under vacuum and purified by silica gel chromatography afforded 4 as off white solid.
Molecular Formula: C23H30F2N4O5S; LCMS purity: 95.64%; Expected: 512.2; Observed: 513.2 (M+l). Synthesis of Example 3: (Step 4)
To a stirred solution of compound 4 (9.0 mg, 0.017 mmol) in CH2CI2 (2.0 mL) was added TFA (0.2 mL) dropwise at 0 °C. Reaction mixture was allowed to attain room temperature over 2 hours time. TLC shows complete conversion. Reaction mixture was concentrated to dryness. The solid residue was washed with Et20 (2 x 10 mL) and dried under vacuum. The solids were once again washed with a mixture of CH2CI2 (0.1 mL) and Et20 (5.0 mL) to obtain Example 3 (8.0 mg, 72.7%) as di-TFA salt. The IC50 value of Example 3 is 4nM. ¾ NMR (400MHz ,CD3OD): δ 7.96 (s, 1 H), 7.41 – 7.31 (m, 1 H), 7.30 – 7.19 (m, 2 H), 4.68 – 4.60 (m, 1 H), 4.22 – 4.07 (m, 4 H), 4.01 (d, J= 11.0 Hz, 1 H), 3.77 (d, J= 11.0 Hz, 1 H), 3.74 – 3.63 (m, 1 H), 3.39 (s, 3 H), 2.43 (d, J= 10.8 Hz, 1 H), 2.04 (t, J= 11.9 Hz, 1 H), 1.51 (s, 3 H).; Molecular Formula: C18H22F2 4O3S; HPLC purity: 95.01%; LCMS mass Expected: 412.2;
Observed: 413.0 (M+l).

PAPER
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00267

Development of a convergent synthesis of omarigliptin (MK-3102) suitable for commercial manufacture is described. The target molecule is assembled through a diastereoselective reductive amination of a highly functionalized pyranone with a mesylated pyrazole followed by deprotection of a Boc group. The synthesis of the pyranone relies on three Ru-catalyzed reactions: (1) a DKR reduction of a rac-α-aminoketone to set the two contiguous stereogenic centers, (2) a cycloisomerization of a bis-homopropargylic alcohol to a dihydropyran, and, finally, (3) a Ru-catalyzed oxidation of a pyranol to the desired pyranone. The regioselective synthesis of a N-Boc-1-mesyl pyrazole fragment was achieved via base-promoted mesyl group isomerization to afford 30:1 selectivity. A highlight of the endgame process development is telescoping a Boc deprotection and reductive amination followed by direct crystallization of the penultimate from the reaction mixture. This avoids handling of an unstable, mutagenic 1-mesylpyrazole BSA salt used in the earlier multikilogram deliveries and improves the overall diastereoselectivity and efficiency of the route.


Tesfaye Biftu et al, Omarigliptin (MK-3102): A Novel Long-Acting DPP-4 Inhibitor for Once-Weekly Treatment of Type 2 Diabetes;Journal of Medicinal Chemistry, Articles ASAP, March 24, 2014,DOI: 10.1021/jm401992e
Zacuto, Michael J. et al, Process for preparing chiral dipeptidyl peptidase-IV inhibitors;PCT Int. Appl., WO2013003250
Biftu, Tesfaye et al, Novel tetrahydropyran analogs as dipeptidyl peptidase IV inhibitors: Profile of clinical candidate (2R,3S,5R)-2-(2,5-difluorophenyl)-5-[2-(methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl]tetrahydro-2H-pyran-3-amine, Bioorganic & Medicinal Chemistry Letters, 23(19), 5361-5366; 2013
Biftu, Tesfaye et al, Preparation of aminotetrahydropyrans as dipeptidyl peptidase IV inhibitors for the treatment or prevention of diabetes,PCT Int. Appl., WO2011028455
Biftu, Tesfaye et al, Preparation of aminotetrahydropyrans as dipeptidyl peptidase IV inhibitors for treatment or prevention of diabetes,U.S. Pat. Appl. Publ., US20100120863
Biftu, Tesfaye et al, Preparation of aminotetrahydropyrans as dipeptidyl peptidase IV inhibitors for treatment or prevention of diabetes,U.S. Pat. Appl. Publ., US20100120863
Xu, Feng et al, Process for preparation of chiral trans-2,3-disubstituted 5-oxotetrahydropyrans from ethyl N-(diphenylmethylene)glycinate and propargyl besylate, U.S. Pat. Appl. Publ., US20090187028
Ru(p-cymene)-N-sulfonyl-l,2-diphenylethylenediamine (DPEN) catalyst
R. Noyori, et al., J. Org. Chem., 66: 7931-7944 (2001)
B. Mohar, et al., Chem. Commun., 2572-2573 (2001)
The rhodium-catalyzed cycloisomerization
B. Trost etal., J.Amer. Chem.Soc., 125:7482-7483 (2003).
The ruthenium-catalyzed cycloisomerization
B. Trost, et al., J. Amer. Chem. Soc., 124: 2528-2533 (2002)
Gantz, I.; Chen, M.; Mirza, A.; Suryawanshi, S.; Davies, M. J.; Goldstein, B. J. Effect of MK-3102, a novel once-weekly DPP-4 inhibitor, over 12 weeks in patients with type 2 diabetes mellitus. Presented at the 48th Annual Meeting of the European Association for the Study of Diabetes (EASD), Berlin, Germany, October 2012; Abstract 101 (Clinical Research, Metabolism, Merck Research Laboratories).
Enzyme revealed as promising target to treat asthma and cancer
In experiments with mice, Johns Hopkins Kimmel Cancer Center scientists have identified an enzyme involved in the regulation of immune system T cells that could be a useful target in treating asthma and boosting the effects of certain cancer therapies.
In research described online April 6 in Nature Immunology, the investigators show that mice without the enzyme SKG1 were resistant to dust mite-induced asthma. And mice with melanoma and missing the enzyme, developed far fewer lung tumors—less than half as many—than mice with SKG1.
“If we can develop a drug that blocks the enzyme in a way that mimics what happens when the enzyme is missing, we would not only have a treatment to inhibit asthma, but also a drug that could be used in conjunction with other experimental therapies aimed at helping the immune system fight cancer,” said Jonathan D. Powell, M.D., professor of oncology at the…
View original post 286 more words
New pain relief targets discovered

The immune cell enters the nerve. Credit: Dr. Marzia Malcangio, King’s College London
Scientists have identified new pain relief targets that could be used to provide relief from chemotherapy-induced pain. BBSRC-funded researchers at King’s College London made the discovery when researching how pain occurs in nerves in the periphery of the body.
Dr Marzia Malcangio said: “We have been investigating and identifying mechanisms underlying pain generation and our findings could help chemotherapy patients who suffer pain related side effects.”
One potential side effect of some chemotherapy drugs (such as vincristine) is damage to nerves. This is particularly prominent in hands and feet as the drugs affect nerves in the periphery of the body. This causes pain which doctors treat with painkillers. However, some people find that the pain persists.
Dr Malcangio’s team investigated why the chemotherapy drugs were causing pain in hope to solve the problem. The used mice in…
View original post 257 more words
lei gong teng ,:雷公藤, Tripterygium wilfordii Hook F. for Rheumatoid Arthritis
A Chinese herb called thunder god vine works better than a widely-prescribed pharmaceutical drug at easing rheumatoid arthritis, a new study has found.

The herb has long been used in China to treat this potentially crippling autoimmune disease, which typically strikes hand and foot joints. It is known in Mandarin as ‘lei gong teng’ and to botanists as Tripterygium wilfordii Hook F.
Extracts of the herb have already fired the interest of drug laboratories as they contain hundreds of compounds, including intriguing molecules called diterpenoids which are believed to ease inflammation and immune response.
read at
http://lyranara.me/2014/04/16/chinese-herb-beats-drug-at-treating-rheumatoid-arthritis/

Researchers at the Johns Hopkins School of Medicine have discovered that a natural constituent isolated from a traditional Chinese medicinal herb, Triptergium wilfordii Hook F. (雷公藤, Lei Gong Teng, Thunder God Vine), used for hundreds of years to treat many conditions, works well by blocking gene control machinery in the cell. Thunder God Vine (Lei Gong Teng) is regarded as toxic and used externally to treat rheumatoid arthritis and sciatica. This report, published as a cover story of the March issue of Nature Chemical Biology, suggests that the natural constituent could be a starting point for developing new anti-cancer drugs.

The extracts of Triptergium wilfordii have been used to treat a whole host of conditions and highly lauded for anti-inflammatory, immunosuppressive, contraceptive and anti-tumor activities. The researchers have known about the active compound, triptolide, which can stop cell growth, since 1972, but only now have they figured out what it does.

Triptolide, the active ingredient purified from Tripterygium wilfordii, has been shown in animal models to be effective against cancer, arthritis, and skin graft rejection. In fact, triptolide has been shown to block the growth of all 60 U.S. National Cancer Institute cell lines at very low doses, and even causes some of those cell lines to die. Other experiments have suggested that triptolide interferes with proteins known to activate genes, which gives the researchers an entry point into their research. Using information already known about these proteins and testing the rest to see if triptolide would alter their behaviors, the research team finally found that triptolide directly binds to and blocks the enzymatic activity of a protein.
Triptolide’s general ability to stop enzymatic activity explains its anti-inflammatory and anticancer effects. And its behavior has important additional implications for circumventing the resistance that some cancer cells develop to certain anti-cancer drugs. The researchers are eager to study it further to see what it can do for future cancer therapy.
Source:
http://www.physorg.com/news/2011-03-traditional-chinese-medicine-mystery.html
Tripterygium wilfordii, or léi gōng téng (Mandarin) (Chinese:雷公藤, Japanese: raikōtō), sometimes called thunder god vine but more properly translated thunder duke vine, is a vine used in traditional Chinese medicine for treatment of fever, chills, edema and carbuncle.
Tripterygium wilfordii recently has been investigated as a treatment for a variety of disorders including rheumatoid arthritis, cancer, chronichepatitis, chronic nephritis, ankylosing spondylitis, polycystic kidney disease as well as several skin disorders. It is also under investigation for its apparent antifertility effects, which it is speculated, may provide a basis for a Male oral contraceptive.[1]
Triptolide, a diterpene triepoxide, is a major active component of extracts derived from Tripterygium wilfordii. Triptolide has multiple pharmacological activities including anti-inflammatory, immune modulation, antiproliferative and proapoptotic activity.[2]
The Chinese herb, Lei Gong Teng, comes from the roots, leaves and flowers of the tripterygium wilfordii Hook. f. It is collected during summer and autumn. Tripterygium wilfordii Hook is a deciduous climbing vine growing to 12 meters, with brown, angular, downy twigs. The leaves are light green, smooth on top, and pale gray with light hairs underneath. They have crenate margins and pointed apexes, and are ovate to elliptic, 5-15 cm long, 2.5 – 7 cm wide. The scented hermaphroditic (having male and female organs) flowers, which bloom in September, are small and whitish with five petals and are about 9 mm across, in terminal panicles in July. The fruit is 3-winged, and brownish red, about 1.5 cm long. The plant can grow in light (sandy), medium (loamy) and heavy (clay) soils. It can survive in acid, neutral and basic (alkaline) soil. It can grow in semi-shade (light woodland) or no shade. It requires moist soil.
Source: The whole plant of Triptergium wilfordil Hook. f., family Celastraceae.
Reduction of male fertility
The plant contains many active compounds, at least six of which have male anti-fertility effect (triptolide, tripdiolide, triptolidenol, tripchlorolide, 16-hydroxytriplide and a compound known as T7/19, whose structure is unpublished). The mechanism by which they affect fertility is not yet understood. What is known is that daily doses of these compounds reduce sperm counts and also severely affect the formation and maturation of sperm, causing them to be immotile.
Scientific research into medical effects
Contraception
Certain extracts from Tripterygium wilfordii, as well as from Tripterygium hypoglaucum (now considered identical to T. regelii) and Tripterygium regelii, were discovered in the 1980s to have temporary antifertility effects, which has led to research on its potential as a contraceptive.
“Tripterygium wilfordii Hook.f., known as Leigongteng (Thunder God Vine) in traditional Chinese medicine, has attracted much attention for its applications in relievingautoimmune disorders such as rheumatoid arthritis and systemic lupus erythematosus, and for treating cancer. Molecular analyses of the ITS and 5S rDNA sequences indicate that T. hypoglaucum and T. doianum are not distinct from T. wilfordii, while T. regelii should be recognized as a separate species. The results also demonstrate potential value of rDNA sequence data in forensic detection of adulterants derived from Celastrus angulatus in commercial samples of Leigongteng.”[3]
Not enough is known about T. wilfordii to actually test it as a contraceptive. Research thus far has dealt with establishing the mechanism by which the plant affects fertility, and investigating toxicity and side effects. What has been learned is encouraging, however: in both animals and humans, low doses of various Tripterygium extractscan produce significantly lowered sperm density and motility indices without major side effects. When the treatment was ended in the various trials, all indices returned to normal within months.
T. wilfordii could be an effective pharmaceutical alternative to contraceptives based on hormonal manipulation.
Kidney function
As of 2012 The Nanjing University School of Medicine is conducting a clinical trial of Tripterygium wilfordii to determine its possible beneficial effects on kidney volume and kidney function for polycystic kidney disease (PKD) patients.[4] It should report in late 2013.[dated info]
Immunosuppression
A small molecule Triptolide derived from T. wilfordii has been shown to disrupt mitochondrial function in cells and is under investigation as an anti-tumor agent or to suppress auto-immune disorders.
Rheumatoid arthritis
In China Tripterygium wilfordii has an established history of use in the treatment of rheumatoid arthritis. The herb shows immunosuppressive, cartilage protective, and anti-inflammatory effects.[5][6] The National Center for Complementary and Alternative Medicine has noted that one systematic review of the literature found that Tripterygium wilfordii may improve some RA symptoms, though another systematic review has stated that the serious side effects occur frequently enough to make the risks of taking this herbal supplement too high for the possible benefits.[7]
Pancreatic cancer
Two compounds, the diterpenoid epoxide triptolide and the quinone triterpene celastrol found in the plant may have potential as antitumor drugs.[8]
Drugs derived from the plant also show potential for reduction and elimination of pancreatic tumors in mice. Clinical trials may soon begin for the development of a drug for use in humans.[9]
At medicinal doses, T. wilfordii extract does have significant side effects, including immunosuppression. However, this may not apply to contraceptive use. Many of the side effects are caused by the other active compounds found in the plant, and do not appear when a pure extraction of its compounds with anti-fertility effect is used. In addition, the dose required to lower fertility is significantly lower than the standard medicinal dose.
In August 2011, the UK Medicines and Healthcare products Regulatory Agency (MHRA) published a drug safety bulletin advising consumers not to use medicines containing Lei Gong Teng. This was due to concerns over potentially serious side effects.
Baidu Baike cautions do not take internally; China State Food and Drug Administration issued a warning in April 2012 about this medicine, urging caution.[10]
However, a recent review stated that although Tripterygium wilfordii has toxic potential, careful extraction gives an acceptable frequency of adverse reactions, which are largely related to the gastrointestinal tract and amenorrhea. The review found that T. wilfordii extract is useful remedy for postmenopausal rheumatoid arthritis.[11]
The Beijing TV series of China Medicine has shown people being treated successfully with the herb in a formula for rheumatoid arthritis. and outlined some practice to alleviate problems of using the herb. As often the case of TCM, formulations need to to be adjusted for individual’s physiology for best result.
Composition:
1. Saponins
(1). Wilforgine, wilforgine-B,wilfordine, wilfornine, wilfortrine, wilfortrine-D, wilforzine, wilformine, wilfordinic acid, hydroxywilfordii acid ,wilfornine , neowilforine.
(2). Celacinnine, celafurine, celabenzine, celallocinnine.
(3). Triptofordinine A-1, A-2, triptofordin D-1, D-2, E , triptofordin A, B, C-1 C-2 , triptofordin F-1, F-2, F-3, F-4.
2. Diterpene group
(1). Triptolide, tripdiolide, triptonide,tripterolide.
(2.). Triptolidenol, tripnolide, neotriptophenolide, triptophenolide methyl ether , isoneotrip-tophenolide, hypolide methyl ether.
(3). Triptonoterpene, triptonoterpene methyl ether, triptonoterpenol 12-ydroxy-abieta-8, 11, 13 -trien-3-one, 11-hydroxy-14-methoxy-abieta-8, 11-hydroxy-14-methoxy-abieta-8, 11, 13-trien-3-one.
3. Tetra-triterpene group
(1). Wilforlide A, wilforlide B.
(2). Tritotriterpenoid lactone, tretotriterpenic acid A, tritotriterpenic acid B, tritotriterpenic acid C, 3-epikatonic acid, polpunonic acid, triptodihydroxy acid methyl ester, tripterine.
(3). 3,24-dioxofridelan-29-oic acid, salaspermic acid.
4. Wilfornide
5. 1,8-dihydroxy-4-hydroxymethyl anthraquinone
6. Syringareisno
7 Other Chemicals: dulcitol, glucose, tannin.
8. Trace mineral: iron, manganese, zinc, copper, selenium etc.
Pharmacology
PG490-88 (14-succinyl triptolide sodium salt) is a semisynthetic compound derived from the diterpene triepoxide, triptolide (PG490). PG490 was first isolated and structurally characterized in 1972 when it was extracted from the Chinese medicinal herb, Tripterygium wilfordii Hook F (TWHF), a member of the Celastraceae family. Historically, extracts of TWHF have been used for centuries in traditional Chinese medicine but in the 1970s, they were identified as being effective in the treatment of inflammatory/autoimmune disorders such as rheumatoid arthritis. Since then, more rigorous attempts were made to better identify biologically active constituents of TWHF responsible for its various clinical properties. We now know, for example, that diterpenoid components of TWHF, especially PG490, exert their anti-inflammatory and immunosuppressant effects by inhibition of cytokine production (e.g. , IL-2, IL-4, IFN) by T lymphocytes. These effects of PG490 have also been explored in mouse models where it was shown that PG490 prevents graft versus host disease (GVHD) and prolongs skin, heart, and kidney allograft survival.
The isolation of PG490 has also led to studies supporting its potential development as an antineoplastic agent. Shamon et al., for example, showed that PG490 inhibited growth of several human cancer-derived cell lines (including breast, prostate, and lung) grown in culture. PG490 was also shown to induce apoptosis of human promyelocytic leukemia, T-cell lymphoma, and hepatocellular carcinoma cell lines grown in culture. Interestingly, the inhibitory effects of PG490 on the growth of tumor cells in culture were enhanced in the presence of other inducers of apoptosis such as tumor necrosis factor- (TNF) and chemotherapeutic agents. When combined with chemotherapeutic drugs, PG490 enhanced apoptosis through signaling pathways involving both p53 and p21.
Data on the effects of PG490 on tumor cell growth in vivo , however, are limited. Previous reports have shown that PG490 inhibits tumor development in a hamster model of cholangiocarcinoma and in a murine breast cancer model. These beneficial effects of PG490, however, were counterbalanced by toxicity that was observed at high doses. In the present studies, we further examined the role of PG490 in inhibition of tumor cell growth both in vitroand in a tumor xenograft model. We show that PG490-88, a water-soluble prodrug of PG490, suppresses tumor cell growth in vivo without toxicity. We also show that PG490 acts in synergy with chemotherapy. Our results suggest a potential role of PG490-88 alone and in combination with chemotherapy as a novel antineoplastic regimen for the treatment of patients with solid tumors
The molecular target(s) for PG490 is currently unknown. Clues to the cellular target, however, are emerging from its effect on transcriptional activity. For example, we have shown along with Qiu et al. , that PG490 blocks transcriptional activation of NF- B by blocking transcriptional activation of p65 but without affecting DNA binding by p65. Additionally, we have found that PG490 blocks transcriptional activation by AP-1 and p53 without affecting DNA binding by Jun/Fos or p53. Recent studies show that the transcriptional activity of AP-1, NF-B, and p53 is regulated by a chromatin structure that is controlled, in part, by histone acetylation. In support of this, a recent study showed that p65 interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate NF- B transcriptional activity. Also, silencing mediator of retinoic acid and thyroid hormone receptors (SMRT) was shown to inhibit transactivation of AP-1, NF-B, and serum response factor (SRF) by binding to their cognate transcription factors. Recent studies also show that p53-mediated transcriptional activity is regulated by histone acetylation. However, we have not observed an effect of PG490 on histone acetyltransferase (HAT) activity or histone acetylation.
PG490 at doses of 5–10 ng/ml does not repress basal transcriptional activity mediated by AP-1, NF-B, and p53 but it does block induction of NF-B by TNF and p53 transcriptional activity induced by chemotherapy. Also, PG490 does not affect topoisomerase I or II activity or increase topoisomerase cleavage complexes. Therefore, its synergy with chemotherapy may in large part be due to its inhibition of p21 mediated growth arrest, which activates an apoptotic pathway.
The treatment of solid tumors is evolving to more targeted treatments that may be helped by genetic profiling of tumors and targeting tumor-specific angiogenic and growth factor pathways. Also, several recent studies have shown that disrupting checkpoints in tumors drives tumor cells into apoptosis by abrogating checkpoint arrest. Here we show that PG490-88, a water-soluble derivative of PG490, reduces tumor growth, induces marked regression, or completely eradicates human tumor xenografts. Moreover, PG490-88 is a potent and well-tolerated antitumor agent that acts in synergy with DNA damaging agents and is effective in a clinically relevant dosing schedule. PG490-88 is now in phase I clinical trials for patients with solid tumors. A recent study showing that PG490 inhibits metastasis of solid tumors coupled with our findings that PG490-88 markedly enhances the cytotoxicity of DNA damaging agents suggests that PG490 or PG490-88 alone or in combination with chemotherapy may become an effective therapy for patients with solid tumors. Also, our finding that PG490 sensitizes tumor cells to TNF by blocking NF-B suggests a role for the combination in treating patients with TNF sensitive tumors such as melanoma. Identification of the target of PG490 and its mechanism of action will complement the ongoing clinical trials, and will provide insight into potential mechanisms of toxicity and the design of compounds that may be more selective and more potent.
- Zhen QS, Ye X, Wei ZJ (February 1995). “Recent progress in research on Tripterygium: a male antifertility plant”. Contraception 51 (2): 121–9. doi:10.1016/0010-7824(94)00018-R.PMID 7750290.
- Liu Q. (2011). “Triptolide and its expanding multiple pharmacological functions”. International Immunopharmacology 11 (3): 377–383. doi:10.1016/j.intimp.2011.01.012.PMID 21255694.
- Law et al (2010), p. 21. Source undefined
- Randomized Clinical Trial of Triptolide Woldifii for Autosomal Dominant Polycystic Kidney Disease
- Bao J., Dai S.-M. “A Chinese herb Tripterygium wilfordii Hook F in the treatment of rheumatoid arthritis: mechanism, efficacy, and safety” Rheumatology International 2011 (1-7)
- Moudgil K.D., Venkatesha S.H., Rajaiah R., Berman B.M. “Immunomodulation of autoimmune arthritis by herbal CAM” Evidence-based Complementary and Alternative Medicine 2011 2011 Article Number 986797
- “Rheumatoid Arthritis and Complementary Health Approaches”. National Center for Complementary and Alternative Medicine. Retrieved 21 April 2013.
- Liu Z, Ma L, Zhou GB. (2011). “The main anticancer bullets of the Chinese medicinal herb, thunder god vine.”. Molecules 16 (6): 5283–97. doi:10.3390/molecules16065283.PMID 21701438.
- Drug From Chinese ‘Thunder God Vine’ Slays Tumors in Mice. 17 Oct 2012
- 中医・我が愛しの上海へ/理想の中医学・漢方を求めて-
- Bao J., Dai S.-M., (September 2011). “A Chinese herb Tripterygium wilfordii Hook F in the treatment of rheumatoid arthritis: Mechanism, efficacy, and safety.”. Rheumatology International 31 (9): 1123–1129. doi:10.1007/s00296-011-1841-y. PMID 21365177.
References
- Downloadable PDF – “Molecular analyses of the Chinese herb Leigongteng (Tripterygium wilfordii Hook.f.)” (2010). Sue Ka-Yee Law et al. Phytochemistry 72 (2011) 21–26, Elsevier.[1]
- Adv Exp Med Biol. 2007;599:139-46.
- Journal of Andrology 1998; vol 19 no 4, pp 479–486.
- Contraception 1995; vol 51, pp 121–129.
- Contraception 1986; vol 36 no 3, pp 335–345.
- MHRA safety bulletin
