
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 285)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Glenmark Kicks Off Monoclonal Antibody Pain Studies

Glenmark Pharmaceuticals S.A., a wholly owned Swiss subsidiary of Glenmark Pharmaceuticals Ltd., announced that GBR 900, a novel monoclonal antibody is entering human trials. GBR 900 targets TrkA, a receptor for nerve growth factor (NGF) involved in chronic pain signaling.
In 2010, Glenmark gained an exclusive worldwide license from Lay Line Genomics S.p.A. (Italy) for anti-TrkA antibodies and their entire intellectual property portfolio in the TrkA field. GBR 900 is the optimized anti-TrkA antibody emerging from this exclusive worldwide license.
read all at
http://www.dddmag.com/news/2014/04/glenmark-kicks-monoclonal-antibody-pain-studies
Glenmark Kicks Off Monoclonal Antibody Pain Studies

Glenmark Pharmaceuticals today said its novel monoclonal antibody for potential treatment of chronic pain is entering human trials.
Tafenoquine…..GSK Launches Phase 3 Malaria Drug Trials

Tafenoquine
N-[2,6-dimethoxy-4-methyl-5-[3-(trifluoromethyl)phenoxy]quinolin-8-yl]pentane-1,4-diamine
Medicines for Malaria Venture
Walter Reed Army Institute (Originator)
April 28, 2014
GlaxoSmithKline (GSK) and Medicines for Malaria Venture (MMV) announced the start of a Phase 3 global program to evaluate the efficacy and safety of tafenoquine, an investigational medicine which is being developed for the treatment and relapse prevention (radical cure) of Plasmodium vivax (P. vivax) malaria.
P. vivax malaria, a form of the disease caused by one of several species of Plasmodium parasites known to infect humans, occurs primarily in South and South East Asia, Latin America and the horn of Africa. Severe anemia, malnutrition and respiratory distress are among the most serious consequences described to be caused by the infection.
The Phase 3 program includes two randomized, double-blind treatment studies to investigate tafenoquine in adult patients with P. vivax malaria. The DETECTIVE study (TAF112582) aims to evaluate the efficacy, safety and tolerability of tafenoquine as a radical cure for P. vivax malaria, co-administered with chloroquine, a blood stage anti-malarial treatment. The GATHER study (TAF116564) aims to assess the incidence of hemolysis and safety and efficacy of tafenoquine compared to primaquine, the only approved treatment currently available for the radical cure of P. vivax malaria.
Tafenoquine is not yet approved or licensed for use anywhere in the world.
“P. vivax malaria can affect people of all ages and is particularly insidious because it has the potential to remain dormant within the body in excess of a year, and causes some patients to experience repeated episodes of illness after the first mosquito bite,” said Nicholas Cammack, head, Tres Cantos Medicines Development Center for Diseases of the Developing World. “Our investigation of tafenoquine for the treatment of P. vivax malaria is part of GSK’s efforts to tackle the global burden of malaria. Working with our partners, including MMV, we are determined to stop malaria in all its forms.”
“One of the big challenges we face in tackling malaria is to have new medicines to prevent relapse, caused by dormant forms of P. vivax,” said Dr. Timothy Wells, MMV’s chief scientific officer. “The Phase 3 program is designed to build upon the promising results of the Phase 2b study which showed that treatment with tafenoquine prevented relapses. If successful, tafenoquine has the potential to become a major contributor to malaria elimination. It’s a great privilege to be working with GSK on this project; they have a clear commitment to changing the face of public health in the countries in which we are working.”


Tafenoquine succinate, Etaquine, SB-252263, WR-238605
in phase 2
Medicines for Malaria Venture
Walter Reed Army Institute (Originator)
Tafenoquine is an 8-aminoquinoline drug manufactured by GlaxoSmithKline that is being investigated as a potential treatment for malaria, as well as for malaria prevention.[1][2]
The proposed indication for tafenoquine is for treatment of the hypnozoite stages of Plasmodium vivax (and also Plasmodium ovale) that are responsible for relapse of these malaria species even when the blood stages are successfully cleared. This is only now achieved by administration of daily primaquine for 14 days. The main advantage of tafenoquine is that it has a long half-life (2–3 weeks) and therefore a single treatment may be sufficient to clear hypnozoites. The shorter regimen has been described as an advantage.[3]
Like primaquine, tafenoquine causes haemolysis in people with G-6-P deficiency.[1] Indeed the long half-life of tafenoquine suggests that particular care should be taken to ensure that individuals with severe deficiency do not receive the drug.
The dose of tafenoquine has not been firmly established, but for the treatment of Plasmodium vivax malaria, a dose of 800 mg over three days has been used.[4]
Synonyms

………………..
US 4431807

Nitration of 1,2-dimethoxybenzene (XXIX) with HNO3/AcOH gives 4,5-dimethoxy-1,2-dinitrobenzene (XXX), which is treated with ammonia in hot methanol to yield 4,5-dimethoxy-2-nitroaniline (XXXI). Cyclization of compound (XXXI) with buten-2-one (XXXII) by means of H3PO4 and H3AsO4 affords 5,6-dimethoxy-4-methyl-8-nitroquinoline (XXXIII), which is selectively mono-demethylated by means of HCl in ethanol to provide 5-hydroxy-6-methoxy-4-methyl-8-nitroquinoline (XXXIV). Reaction of quinoline (XXXIV) with POCl3 gives the corresponding 5-chloro derivative (XXXV), which is condensed with 3-(trifluoromethyl)phenol (IV) by means of KOH to yield the diaryl ether (XXXVI). Finally, the nitro group of (XXXVI) is reduced by means of H2 over PtO2 in THF or H2 over Raney nickel.

Nitration of 2-fluoroanisole (XXXVII) with HNO3/Ac2O gives 3-fluoro-4-methoxynitrobenzene (XXXVIII), which is reduced to the corresponding aniline (XXXIX) with SnCl2/HCl. Reaction of compound (XXXIX) with Ac2O yields the acetanilide (XL), which is nitrated with HNO3 to afford 5-fluoro-4-methoxy-2-nitroacetanilide (XLI). Hydrolysis of (XLI) with NaOH provides 5-fluoro-4-methoxy-2-nitroaniline (XLII), which is cyclized with buten-2-one (XXXII) by means of As2O5 and H3PO4 to furnish 5-fluoro-6-methoxy-4-methyl-8-nitroquinoline (XLIII). Condensation of quinoline (XLIII) with 3-(trifluoromethyl)phenol (IV) by means of K2CO3 gives the diaryl ether (XXXIV), which is finally reduced by means of H2 over PtO2 in THF.
………………..
US 4617394

Reaction of 8-amino-6-methoxy-4-methyl-5-[3-(trifluoromethyl)phenoxy]quinoline (XIV) with phthalic anhydride (XV) affords the phthalimido derivative (XVI), which is oxidized with MCPBA to yield the quinoline N-oxide (XVII). Treatment of compound (XVII) with neutral alumina gives the quinolone derivative (XVIII), which by reaction with POCl3 in refluxing CHCl3 provides the 2-chloroquinoline derivative (XIX). Alternatively, reaction of the quinoline N-oxide (XVII) with POCl3 as before also gives the 2-chloroquinoline derivative (XIX) The removal of the phthalimido group of compound (XIX) by means of hydrazine in refluxing ethanol gives the chlorinated aminoquinoline (XX), which is finally treated with MeONa in hot DMF.
……………….
US 6479660; WO 9713753

Chlorination of 6-methoxy-4-methylquinolin-2(1H)-one (I) with SO2Cl2 in hot acetic acid gives the 5-chloro derivative (II), which is nitrated with HNO3 in H2SO4 to yield the 8-nitroquinolinone (III). Condensation of compound (III) with 3-(trifluoromethyl)phenol (IV) by means of KOH in NMP provides the diaryl ether (V), which is treated with refluxing POCl3 to afford the 2-chloroquinoline (VI). Reaction of compound (VI) with MeONa in refluxing methanol results in the 2,6-dimethoxyquinoline derivative (VII), which is reduced with hydrazine over Pd/C to give the 8-aminoquinoline derivative (VIII). Condensation of aminoquinoline (VIII) with N-(4-iodopentyl)phthalimide (IX) by means of diisopropylamine in hot NMP yields the phthalimido precursor (X), which is finally cleaved with hydrazine in refluxing ethanol.

Reaction of 1,4-dibromopentane (XI) with potassium phthalimide (XII) gives N-(4-bromopentyl)phthalimide (XIII), which is then treated with NaI in refluxing acetone.

Reaction of 4-methoxyaniline (XXI) with ethyl acetoacetate (XXII) by means of triethanolamine in refluxing xylene gives the acetoacetanilide (XXIII), which is cyclized by means of hot triethanolamine and H2SO4 to yield 6-methoxy-4-methylquinolin-2(1H)-one (I), which is treated with refluxing POCl3 to provide 2-chloro-6-methoxy-4-methylquinoline (XXIV). Reaction of compound (XXIV) with SO2Cl2 in hot AcOH affords 2,5-dichloro-6-methoxy-4-methylquinoline (XXV), which is treated with MeONa in refluxing methanol to furnish 5-chloro-2,6-dimethoxy-4-methylquinoline (XXVI). Alternatively, the reaction of compound (XXIV) with MeONa as before gives 2,6-dimethoxy-4-methylquinoline (XXVII), which is treated with SO2Cl2 in hot AcOH to give the already described 5-chloro-2,6-dimethoxy-4-methylquinoline (XXVI). Nitration of compound (XXVI) with KNO3 and P2O5 gives the 8-nitroquinoline derivative (XXVIII), which is condensed with 3-(trifluoromethyl)phenol (IV) by means of KOH in hot NMP to yield the diaryl ether (VII). Finally, the nitro group of compound (VII) is reduced with hydrazine over Pd/C.
//////////////////////
J Med Chem 1989,32(8),1728-32

Synthesis of the intermediate diazepinone (IV) is accomplished by a one-pot synthesis. Condensation of 2-chloro-3-aminopyridine (I) with the anthranilic ester (II) is effected in the presence of potassium tert-butoxide as a catalyst. The resulting anthranilic amide (III) is cyclized under the influence of catalytic amounts of sulfuric acid. Treatment of (IV) with chloroacetylchloride in toluene yields the corresponding choroacetamide (V). The side chain of AQ-RA 741 is prepared starting from 4-picoline, which is alkylated by reaction with 3-(diethylamino)propylchloride in the presence of n-butyllithium. Hydrogenation of (VIII) using platinum dioxide as a catalyst furnishes the diamine (IX), which is coupled with (V) in the presence of catalytic amounts of sodium iodide in acetone leading to AQ-RA 741 as its free base.
- Shanks GD, Oloo AJ, Aleman GM et al. (2001). “A New Primaquine Analogue, Tafenoquine (WR 238605), for prophylaxis against Plasmodium falciparum malaria”. Clin Infect Dis 33 (12): 1968–74. doi:10.1086/324081. JSTOR 4482936.PMID 11700577.
- Lell B, Faucher JF, Missinou MA et al. (2000). “Malaria chemoprophylaxis with tafenoquine: a randomised study”.Lancet 355 (9220): 2041–5. doi:10.1016/S0140-6736(00)02352-7. PMID 10885356.
- Elmes NJ, Nasveld PE, Kitchener SJ, Kocisko DA, Edstein MD (November 2008). “The efficacy and tolerability of three different regimens of tafenoquine versus primaquine for post-exposure prophylaxis of Plasmodium vivax malaria in the Southwest Pacific”. Transactions of the Royal Society of Tropical Medicine and Hygiene 102 (11): 1095–101.doi:10.1016/j.trstmh.2008.04.024. PMID 18541280.
- Nasvelda P, Kitchener S. (2005). “Treatment of acute vivax malaria with tafenoquine”. Trans R Soc Trop Med Hyg 99 (1): 2–5. doi:10.1016/j.trstmh.2004.01.013. PMID 15550254.
- Peters W (1999). “The evolution of tafenoquine–antimalarial for a new millennium?”. J R Soc Med 92 (7): 345–352.PMID 10615272.
- J Med Chem 1982,25(9),1094
|
8-3-2007
|
Methods and compositions for treating diseases associated with pathogenic proteins
|
|
|
12-6-2006
|
Process for the preparation of quinoline derivatives
|
|
|
3-14-2002
|
PROCESS FOR THE PREPARATION OF ANTI-MALARIAL DRUGS
|
|
|
4-2-1998
|
MULTIDENTATE METAL COMPLEXES AND METHODS OF MAKING AND USING THEREOF
|
|
|
4-18-1997
|
PROCESS FOR THE PREPARATION OF ANTI-MALARIAL DRUGS
|
|
|
12-20-1996
|
MULTIDENTATE METAL COMPLEXES AND METHODS OF MAKING AND USING THEREOF
|
|
|
12-15-1993
|
Use of interferon and a substance with an antimalarial activity for the treatment of malaria infections
|
|
|
10-15-1986
|
4-methyl-5-(unsubstituted and substituted phenoxy)-2,6-dimethoxy-8-(aminoalkylamino) quinolines
|
FDA Approves Zykadia, Ceritinib, LDK378 for ALK-Positive NSCLC



Acting 4 months ahead of schedule, the FDA has granted an accelerated approval to ceritinib (Zykadia; LDK378) as a treatment for patients with ALK-positive metastatic non-small cell lung cancer (NSCLC) following treatment with crizotinib (Xalkori), based on a single-arm clinical trial demonstrating a durable improvement in overall response rates (ORR).
The approval for the second-generation ALK inhibitor was supported by results from an analysis of 163 patients treated with single-agent ceritinib at 750 mg daily following progression on crizotinib. In these select patients, the ORR was 54.6% with a 7.4-month median duration of response, according to data submitted to the FDA by Novartis, the company developing the drug. Based on these findings, the FDA granted ceritinib a Breakthrough Therapy designation, Priority Review, and orphan product designation.
“Today’s approval illustrates how a greater understanding of the underlying molecular pathways of a disease can lead to the development of specific therapies aimed at these pathways,” Richard Pazdur, MD, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a statement. “It also demonstrates the FDA’s commitment to working cooperatively with companies to expedite a drug’s development, review and approval, reflecting the promise of the breakthrough therapy designation program.”
In the study that was the basis for the approval, the primary endpoint was ORR by RECIST criteria with a secondary outcome measure of duration of response. Treatment with ceritinib resulted in an ORR of 54.6% by investigator assessment with a median duration of response of 7.4 months. By blinded independent central review, the ORR was 43.6% and the duration of response was 7.1 months.
Earlier this year, results from a dose escalation study that examined ceritinib in 130 patients who were untreated or refractory to crizotinib were published in the New England Journal of Medicine. In this analysis for patients who received doses of at least 400 mg (n = 114), the ORR was 58%. Patients who had progressed on crizotinib (n = 80) experienced an ORR of 56% and those who were crizotinib-naïve (n =34) had an ORR of 62%.
The median progression-free survival was 7.0 months and the median duration of response was 8.2 months (95% CI; 6.9-11.4). Additionally, responses were seen in patients with untreated metastatic brain lesions who progressed on prior therapy with crizotinib, the authors of the study noted.
The most frequent adverse events were nausea (82%), diarrhea (75%), vomiting (65%), fatigue (47%) and increased alanine aminotransferase levels (35%). These adverse events were generally mild and resolved when treatment stopped or the dose was reduced.
The most common grade 3 or 4 drug-related adverse events were increased alanine aminotransferase levels (21%), increased aspartate aminotransferase levels (11%), diarrhea (7%) and increased lipase levels (7%), all of which were reversible upon treatment discontinuation.
“Zykadia represents an important treatment option for ALK-positive NSCLC patients who relapse after starting initial therapy with crizotinib,” Alice Shaw, MD, PhD, of the Massachusetts General Hospital (MGH) Cancer Center, lead author of the report, said in a statement. “This approval will affect the way we manage and monitor patients with this type of lung cancer, as we will now be able to offer them the opportunity for continued treatment response with a new ALK inhibitor.”
Two phase III studies are enrolling patients to further explore the efficacy and safety of ceritinib in patients with ALK-positive NSCLC. These studies will likely act as confirmation for the accelerated approval. In the first, ceritinib will be compared with chemotherapy in untreated patients with ALK-rearranged NSCLC (NCT01828099). The second will compare ceritinib to chemotherapy in ALK-positive patients with NSCLC following progression on chemotherapy and crizotinib (NCT01828112).
“The approval of Zykadia less than three and a half years after the first patient entered our clinical trial exemplifies what is possible with a highly focused approach to drug development and strong collaboration,” Alessandro Riva, MD, president of Novartis Oncology ad interim and global head of Oncology Development and Medical Affairs, said in a statement. “The dedication of clinical investigators, patients, the FDA and others has enabled us to bring this medicine to patients in need as swiftly as possible.”
![]()

Nitration of 2-chloro-4-fluoro-1-methylbenzene with KNO3 in the presence of H2SO4 gives 1-chloro-5-fluoro-2-methyl-4-nitrobenzene , which upon condensation with isopropyl alcohol in the presence of Cs2CO3 in 2-PrOH at 60 °C yields 5-isopropoxy-2-methyl-4-nitrobenzene .
Suzuki coupling of chloride with 4-pyridineboronic acid in the presence of Pd2dba3, K3PO4 and SPhos in dioxane/water at 150 °C (microwave irradiation) provides 4-(5-isopropoxy-2-methyl-4-nitrophenyl)pyridine , which is then subjected to global reduction using H2 over PtO2 in the presence of TFA in AcOH to afford 2-isopropoxy-5-methyl-4-piperidin-4-ylaniline .
N-Protection of piperidine with Boc2O in the presence of Et3N in CH2Cl2 furnishes the corresponding carbamate (VIII), which upon Buchwald-Hartwig cross coupling with 2,5-dichloropyrimidine derivative (prepared by condensation of 2-(isopropylsulfonyl)aniline and 2,4,5-trichloropyrimidine in the presence of NaH in DMSO/DMF) in the presence of Pd(OAc)2, Xantphos and Cs2CO3 in THF affords Boc-protected ceritinib . Finally, removal of Boc-group in compound using TFA in CH2Cl2 furnishes the target compound ceritinib
/////////////////

Anaplastic lymphoma kinase (ALK), a member of the insulin receptor superfamily of receptor tyrosine kinases, has been implicated in oncogenesis in hematopoietic and non- hematopoietic tumors. The aberrant expression of full-length ALK receptor proteins has been reported in neuroblastomas and glioblastomas; and ALK fusion proteins have occurred in anaplastic large cell lymphoma. The study of ALK fusion proteins has also raised the possibility of new therapeutic treatments for patients with ALK-positive malignancies. (Pulford et al., Cell. MoI. Life Sci. 61:2939-2953 (2004)).
Focal Adhesion Kinase (FAK) is a key enzyme in the integrin-mediated outside-in signal cascade (D. Schlaepfer et al., Prog Biophys MoI Biol 1999, 71, 43578). The trigger in the signal transduction cascade is the autophosphorylation of Y397. Phosphorylated Y397 is a SH2 docking site for Src family tyrosine kinases; the bound c-Src kinase phosphorylates other tyrosine residues in FAK. Among them, phsophorylated Y925 becomes a binding site for the SH2 site of Grb2 small adaptor protein. This direct binding of Grb2 to FAK is one of the key steps for the activation of down stream targets such as the Ras-ERK2/MAP kinase cascade.
Zeta-chain-associated protein kinase 70 (ZAP-70), a member of the protein tyrosine kinase family, is of potential prognostic importance in chronic lymphocytic leukemia (CLL). ZAP-70, known to be of importance in T and NK cell signaling but absent in normal peripheral B cells, is expressed in the majority of the poorer prognosis unmutated CLL and absent in most cases with mutated IgVH genes. ZAP-70 is also expressed in a minority of other B cell tumors. (Orchard et al., Leuk. Lymphoma 46:1689-98 (2005)). [0006] Insulin- like growth factor (IGF-I) signaling is highly implicated in cancer, with the IGF-I receptor (IGF-IR) as the predominating factor. IGR-IR is important for tumor transformation and survival of malignant cells, but is only partially involved in normal cell growth. Targeting of IGF-IR has been suggested to be a promising option for cancer therapy. (Larsson et al., Br. J. Cancer 92:2097-2101 (2005)).
Because of the emerging disease-related roles of ALK, FAK, ZAP-70 and IGF-IR, there is a continuing need for compounds which may be useful for treating and preventing a disease which responds to inhibition of ALK, FAK, ZAP-70 and/or IGF-IR
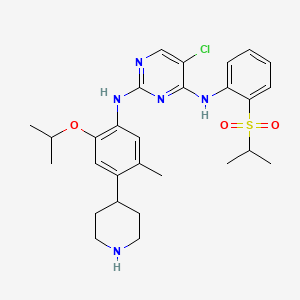
The compound 5-Chloro-N2-(2-isopropoxy-5-methyl-4-piperidin-4-yl-phenyl)-N4-[2- (propane-2-sulfonyl)-phenyl]-pyrimidine-2, 4-diamine, in the form of a free base, of formula

(I)
is an anaplastic lymphoma kinase (ALK) inhibitor, a member of the insulin receptor super family of receptor tyrosine kinases. Compound I was originally described in WO 2008/073687 Al as Example 7, compound 66. WO 2008/073687 Al , however, provides no information about crystalline forms of 5-
Chloro-N2-(2-isopropoxy-5-methyl-4-piperidin-4-yl-phenyl)-N4-[2-(propane-2-sulfonyl)- phenyl]-pyriniidine-2, 4-diamine or its corresponding salts. Crystalline forms of 5-Chloro-N2- (2-isopropoxy-5-methyl-4-piperidin-4-yl-phenyl)-N4-[2-(propane-2-sulfonyl)-phenyl]- pyrimidine-2, 4-diamine have been discovered, which are useful in treating diseases which respond to inhibition of anaplastic lymphoma kinase activity, focal adhesion kinase (FAK), zeta- chain-associated protein kinase 70 (ZAP-70) insulin-like growth factor (IGF-1 or a
combination thereof. The crystalline forms exhibit new physical properties that may be exploited in order to obtain new pharmacological properties, and that may be utilized in the drug product development of 5-Chloro-N2-(2-isopropoxy-5-methyl-4-piperidin-4-yl-phenyl)-N4-[2- (propane-2-sulfonyl)-phenyl]-pyrimidine-2, 4-diamine.
…………………….
WO 2008073687
http://www.google.com/patents/WO2008073687A2?cl=en
Example 7
5-Chloro-N2-(2-isopropoxy-5-methyl-4-piperidin-4-yl-phenyl)-N4-r2-(propane-2-sulfonyl)- phenvH-pyrimidine-2,4-diamine (66)

1 : 4-(5-Isopropoxv-2-methvl-4-nitro-phenyl)-pvridine

[0111] 4-Pyridineboronic acid (147 mg, 1.20 mmol, 1.1 equiv.) is dissolved in a 2:1 v/v mixture of dioxane and H2O (15 mL) and N2 is bubbled through for 5 minutes. Tris(dibenzylidene acetone)dipalladium (0) (100 mg, 0.109 mmol, 0.1 equiv.), 2- dicyclohexylphosphine-2′-6′-dimethoxy biphenyl (112 mg, 0.272 mmol, 0.25 equiv.), 1-chloro- 5-isopropoxy-2-methyl-4-nitro-benzene (Intermediate 4, 250 mg, 1.09 mmol, 1.0 equiv.) and K3PO4 (462 mg, 2.18 mmol, 2.0 equiv.) are added under a N2 blanket. The reaction vessel is sealed and heated with microwave irradiation to 150 0C for 20 min. After cooling to room temperature, the reaction is diluted with ethyl acetate and washed with 1 N aqueous NaOH (2x), the organic layer is then dried over Na2SO4 and filtered. After concentration, the crude product is purified by silica gel chromatography (gradient from hexanes to 30% ethyl acetate in hexanes) to give 4-(5-Isopropoxy-2-methyl-4-nitro-phenyl)-pyridine as a brown solid: ESMS m/z 273.1 (M + H+).
Steps 2 and 3 : 4-(4-Amino-5-isopropoxy-2-methyl-phenyl)-piperidine-l-carboxylic acid tert- butyl ester

[0112] 4-(5-Isopropoxy-2-methyl-4-nitro-phenyl)-pyridine from the previous step(438 mg, 1.61 mmol) dissolved in acetic acid (30 mL) is treated with TFA (0.24 mL, 3.22 mmol) and PtO2 (176 mg, 40% w/w). The reaction mixture is vigorously stirred under 1 atm. H2 for 36 hours. The reaction mixture is filtered and the filtrate is concentrated under vacuum. The resulting residue is diluted with ethyl acetate and washed with 1 N aqueous NaOH (2x), the organic layer is then dried over Na2SO4 and filtered. After concentration, the crude product (391 mg) is dissolved in anhydrous CH2Cl2 (30 mL). TEA is added (0.44 mL, 3.15, 2 equiv.) followed by Boc2O (344 mg, 1.57 equiv, 1 equiv.). The reaction is stirred at room temperature for 30 min. The reaction is concentrated under vacuum. The resulting residue is purified by silica gel chromatography (gradient from hexanes to 30% ethyl acetate in hexanes) to give 4-(4-amino-5- isopropoxy-2-methyl-phenyl)-piperidine-l-carboxylic acid tert-butyl ester as a sticky foam: ESMS m/z 293.1 (M-?Bu+H)+.
Steps 4 and 5
[0113] 4-(4-Amino-5-isopropoxy-2-methyl-phenyl)-piperidine-l-carboxylic acid tert-butyl ester (170 mg, 0.488 mmol) from the previous step, (2,5-Dichloro-pyrimidin-4-yl)-[2-(propane- 2-sulfonyl)-phenyl]-amine (Intermediate 2, 169 mg, 0.488 mmol, 1 equiv.), xantphos (28 mg, 0.049 mmol, 0.1 equiv.), palladium acetate (5.5 mg, 0.024 mmol, 0.05 equiv.), and Cs2CO3 (477 mg, 1.46 mmol, 3 equiv.) are dissolved in anhydrous THF (6 mL). N2 is bubbled through the reaction mixture for 5 minutes and then the reaction vessel is sealed and heated with microwave irradiation to 150 0C for 20 min. The reaction is filtered and the filtrate concentrated under vacuum. After concentration, the crude product is purified by silica gel chromatography (gradient from hexanes to 30% ethyl acetate in hexanes) to give 4-(4-{5-chloro-4-[2-(propane-2- sulfonyl)-phenylamino]-pyrimidin-2-ylamino}-5-isopropoxy-2-methyl-phenyl)-piperidine-l- carboxylic acid tert-butyl ester as a yellow film: ESMS m/z 658.3 (M + H+). This product (105 mg, 0.160 mmol) is dissolved in CH2Cl2 (3 mL) and treated with TFA (3 mL). After 45 min., the reaction is concentrated under vacuum. 1 N HCl in Et2O (5 mL x 2) is added causing the product HCl salt to precipitate. The solvent is removed by decantation. The resulting 5- Chloro-N2-(2-isopropoxy-5-methyl-4-piperidin-4-yl-phenyl)-N4-[2-(propane-2-sulfonyl)- phenyl]-pyrimidine-2,4-diamine (66) is dried under high vacuum, generating an off-white powder: 1H NMR (400 MHz, DMSO-J6+ trace D2O) δ 8.32 (s, IH), 8.27 (d, IH), 7.88 (d, IH), 7.67 (dd, IH), 7.45 (dd, IH), 7.42 (s, IH), 6.79 (s, IH), 4.56-4.48 (m, IH), 3.49-3.32 (m, 3H), 3.10-2.91 (m, 3H), 2.09 (s, 3H), 1.89-1.77 (m, 4H), 1.22 (d, 6H), 1.13 (d, 6H); ESMS m/z 558.1 (M + H+).
 66
66
……………………..
WO 2012082972
http://www.google.com/patents/WO2012082972A1
EXAMPLE 1
Preparation of Form A of 5-chloro-N-(2-isopropoxy-5-methyl-4-(piperidin-4-ylphenyl)-N-2- (isopropylsulfonyl phenyl)-2^-diamine
5-chloro-N-(2-isopropoxy-5-methyl-4-(piperidin-4-ylphenyl)-N-2-(isopropylsulfonyl)phenyl)- 2,4-diamine di-hydrochloride salt
The compound 2-isopropoxy-5-methyl-4-(piperdin-4-yl) aniline dihydrochloride (33.00 g, 85.25 mmol) and 2,5-dichloro-N-(2-(isopropyl sulfonyl )phenyl)pyrimidin-4-amine (32.53 g) was added to a 3 -necked 500-mL round-bottomed flask equipped with mechanical stirring, thermocouple, reflux condenser and N2 inlet-outlet. A solvent, 2-propanol (255.0 g, 325 mL), was added and the mixture to heated to reflux at 82±2 °C and stirred for at least 14 hours. The mixture was cooled to 22±3 °C over 1 hour and stirred at 22±3 °C for 3 hours. The resulting solids were filtered and rinsed with 3 x 40 g (3 x 51 mL) of 2-propanol. The solids were dried at 50±5 °C/10 mbar for 16 hours to yield 44.63 g of 5-chloro-N-(2-isopropoxy-5-methyl-4- (piperidin-4-ylphenyl)-N-2-(isopropylsulfonyl)phenyl)-2,4-diamine di-hydrochloride salt. Chemical Purity (as determined by HPLC): 97.3%. Corrected yield: 71.6%. LOD = 11.60%. The dihydrochloride salt was recrystallized using acetone:water (10:l,v/v). Chemical Purity (as determined by HPLC): 98.8%.
…………….
J Med Chem 2013, 56(14): 5675
http://pubs.acs.org/doi/abs/10.1021/jm400402q

Synthesis of 5-Chloro-N2-(2-isopropoxy-5-methyl-4-piperidin-4-ylphenyl)-N4-[2-(propane-2-sulfonyl)phenyl]pyrimidine-2,4-diamine 15b
|
10-21-2011
|
COMPOUNDS AND COMPOSITIONS AS PROTEIN KINASE INHIBITORS
|
|
|
10-21-2011
|
COMPOUNDS AND COMPOSITIONS AS PROTEIN KINASE INHIBITORS
|
|
|
10-19-2011
|
COMPOUNDS AND COMPOSITIONS AS PROTEIN KINASE INHIBITORS
|
|
|
8-5-2011
|
COMPOUNDS AND COMPOSITIONS AS PROTEIN KINASE INHIBITORS
|
Water molecules control inactivation and recovery of potassium channels

Depiction of simulated potassium channel and surrounding environment. Potassium ions (green) are unable to pass through because water molecules (red and white) are present inside the protein, locking the channel into an inactivated state. Credit: Benoit Roux, University of Chicago
Just 12 molecules of water cause the long post-activation recovery period required by potassium ion channels before they can function again. Using molecular simulations that modeled a potassium channel and its immediate cellular environment, atom for atom, University of Chicago scientists have revealed this new mechanism in the function of a nearly universal biological structure, with implications ranging from fundamental biology to the design of pharmaceuticals. Their findings were published online July 28 in Nature.
“Our research clarifies the nature of this previously mysterious inactivation state. This gives us better understanding of fundamental biology and should improve the rational design of drugs, which often target the inactivated state of…
View original post 339 more words
GSK3β inhibitor, AZD8926

[4-[5-Fluoro-4-[3-tetrahydropyran-4-yl-2-(trifluoromethyl)imidazol-4-yl]-pyrimidin-2-yl]aminophenyl]-(4-methylpiperazin-1-yl)-methanone
[4- [5-Fluoro-4- [3-tetr ahydropy r an-4-yl-2-(trifluor omethyl)imidazol-4-yl] -pyrimidin- 20 2-yl] aminophenyl]-(4-methylpiperazin-l-yl)-methanone
ASTRAZENECA
GSK3β inhibitor
AZD8926 is a potent glycogen synthase kinase-3β (GSK3β) inhibitor which has potential for treating several CNS disorders, such as Alzheimer’s disease (AD), schizophrenia, and chronic as well as acute neurodegenerative diseases


…………………..


Development of a new, safe, and scalable route to the GSK3β inhibitor, AZD8926, is presented.
In brief, the process constitutes of (i) a synthesis of 1-(pyran-4-yl)-2-trifluoromethyl-imidazole, 14; (ii) a Ziegler-type coupling of lithiated 14 with commercially available 2-chloro-5-fluoropyrimidine via 1,2-addition over the 3,4-C–N bond; (iii) a copper-catalyzed dehydrogenative aromatization using oxygen as the stoichiometric oxidant; and (iv) an aromatic C–N bond formation using either a Buchwald–Hartwig coupling or an acid-catalyzed amination. This process circumvents the main issue in the early-phase route, in which serious process safety constraints were associated with the hazardous properties of the structure, formation, and reduction of 5-methyl-4-nitroisoxazole, 2 (4200 J/g). The new process has been demonstrated on a multigram, 2-L scale. The overall yield was improved from 4 to 14%, and the number of steps decreased from 12 to 10
http://pubs.acs.org/doi/full/10.1021/op300365e?prevSearch=triphosgene&searchHistoryKey=
[4-[5-Fluoro-4-[3-tetrahydropyran-4-yl-2-(trifluoromethyl)imidazol-4-yl]-pyrimidin-2-yl]aminophenyl]-(4-methylpiperazin-1-yl)-methanone (9)
 7.25 min) was conducted by HPLC analysis with UV detection at 292 nm. HRMS m/z found 452.1345 [M + H]+, C20H18N5O3F4 requires 452.1346.2.90 min) was conducted by HPLC analysis with UV detection at 292 nm.
7.25 min) was conducted by HPLC analysis with UV detection at 292 nm. HRMS m/z found 452.1345 [M + H]+, C20H18N5O3F4 requires 452.1346.2.90 min) was conducted by HPLC analysis with UV detection at 292 nm.Scheme I:

Scheme 1
Anilines of formula (III) are commercially available compounds, or they are known in the literature, or they are prepared by standard processes known in the art.
Process b). Compounds of formula (IV) and amines of formula (V) may be reacted together under standard Buchwald conditions as described in Process a.
A synthesis of pyrimidines of formula (IV) is described in Scheme 2(RX may be the same or different and is Q.βalkytyT should not be there

Scheme 2
Compounds of formula (V) are commercially available compounds, or they are known in the literature, or they are prepared by standard processes known in the art.
Compounds of formula (VI) in which R6 has the general structure Ra-CH-Rb (wherein Ra and Rb are as defined in formula I and Rx may be the same or different and is C1-6alkyl) and R9 is F may be prepared according to Scheme 3

(VI) (VIf)
Scheme 3
Example 104
[4- [5-Fluoro-4- [3-tetr ahydropy r an-4-yl-2-(trifluor omethyl)imidazol-4-yl] -pyrimidin- 20 2-yl] aminophenyl]-(4-methylpiperazin-l-yl)-methanone hydrochloride

The title compound was prepared in accordance with the general method E and work-up procedure B. The product was purified by flash chromatography (CH2Cl2MeOH 30:1, 20:1 then 15:1). Using 5-fluoro-4-[l-(tetrahydro-2H-pyran-4-yl)-2-(trifluoromethyl)-lH- 25 imidazol-5-yl]pyrimidin-2-amme (obtained from Example 34(d)) (33 mg, 0.1 mmol), l-(4- bromobenzoyl)-4-methylpiperazine (0.027 g, 0.095 mmol), Cs2CO3 (65 mg, 0.2 mmol), Pd2(dba)3 (6.8 mg, 0.0075 mmol) and X-Phos (7 mg, 0.015 mmol), the base of the title compound (35 mg, 70%) was obtained as a solid. The hydrochloride was prepared in accordance with the method described in general method D. 1H NMR (DMSO-<fc, 300 MHz) δ 10.60 (br s, 1 H), 10.11 (s, 1 H), 8.82 (s, 1 H), 7.74 (d, J = 8.4 Hz, 2 H), 7.56 (s, 1 H), 7.42 (d, J= 8.4 Hz, 2 H), 4.80 (t, 1 H), 3.80 (d, J= 8.4 Hz, 2 H), 3.22 (t, J= 11.5 Hz, 2 H), 3.2-3.0 (m, 2 H), 2.78 (s, 3 H), 2.2-2.1 (m, 2 H), 2.0-1.8 (m, 2 H); 6 Hydrogens were not assigned in the region 3.6 -2.2 ppm due to the presence of the water and DMSO peaks in this region; MS (ESI) m/z 534.5 (M+ 1); MS (ESI) m/z 532.5 (M-I).
Example 34 (d) 5-Fluoro-4-[l-(tetrahydro-2H-pyran-4-yl)-2-(trifluoromethyl)-lH- imidazol-5-yl]pyrimidin-2-amine

The title compound was prepared in accordance with the general method B with the exception that guanidine carbonate was used. Using (2Z)-3-dimethylamino-2-fluoro-l-[l- (tetrahydro-2H-pyran-4-yl)-2-trifluoromethyl- lH-imidazol-5 -y l]prop-2-en- 1 -one (0.330 g, 1.0 mmol, obtained from Example 34(c)) and guanidine carbonate (0.45 g, 2.50 mmol). After purification by flash chromatography (heptane/EtOAc 1 :2), the title compound was obtained (0.170 g, 51 %) as a white solid.
1HNMR (CDCl35 300 MHz) δ 8.29 (s, 1 H)5 7.63 (d, J= 2.7 Hz, 1 H)5 5.10 (br.s., 2 H)5 4.88-4.76 (m, 1 H), 4.16-4.07 (m, 2 H)53.53-3.42 (m5 2 H)5 2.80-2.65 (m, 2 H), 1.89-1.81 (m, 2 H); MS (ES) m/z 332 (M+l).
REF………….
(a) Bhat, R. V.; Budd, S. L. Neurosignals 2002, 11, 251
Sphaelactone dimethylamine fumarate


Sphaelactone dimethylamine fumarate
Accenda Tech Co Ltd; Nankai University
Acute leukemia
Protein farnesyltransferase inhibitor; Ras GTPase inhibitor
Accendatech, 天津尚德药缘科技有限公司,Nankai University, 南开大学
crystalline lactone dimethylamine fumaric and method of the present invention belongs to the field of pharmaceutical technology, in particular, relates to a lactone dimethylamine smile crystalline fumarate ship their preparation. Patent CN 101978959 A discloses a lactone and derivatives thereof, pharmaceutical compositions for use in the treatment of cancer, including the formula (π) compounds, lactone dimethylamine. Activity test results show that the compounds of formula (Π) of the test cell comprising: HepG-2, Ec9706, SGC790K SW1116, A498, ASPC-1, H -29, HeLa, GL15, B16F1, T24, SKOV3, SW579, PC -3, are showing strong inhibitory activity; rather stab at 50 μ Μ normal cells, the filaments exhibit significant killing effect.

WO 2011/131103 A1 discloses a formula including the inner (I) compound smile lactone derivative or a salt thereof a pharmaceutical composition, preparation and use for the preparation of anticancer drugs. But no problem about the compound of formula polymorph (I), have not been reported for formula (I) compounds of the crystalline areas. The present invention provides compounds of formula (I) dimethylamine smile lactone crystalline fumarate and its preparation method.

An object of the present invention to provide a compound of formula (I) a lactone compound smile dimethylamine i.e. crystalline fumarate polymorph A. Another object of the present invention to provide a method for preparing crystalline compound of formula (I). A lactone compound of dimethylamine fumaric polymorph A boat characteristics of formula (I):
http://www.google.com/patents/EP2562172A1?cl=en

………………………………………………………………………………………………..
Example 1:
11PH, 13 – two smiling Yue amino lactone (Compound II) (the structural formula of the formula (II)) Preparation of

Smile lactone (106 mg, 0.40 mmol), triethylamine (2.0mL), Yue alcohol (30 mL) was added lOO mL round bottom flask and heated at reflux for 3 hours, concentrated under reduced pressure, cross stone column chromatography (petroleum ether : ethyl acetate: triethylamine = 50: 50: 0.5) to give a white solid 107.4 mg, yield: 86%.
Formula: C 17 H 27 N0 3
Weight: 293
Appearance: white amorphous 4 minutes late
Spectral data:
¾ NMR (CDCl 3 , 400 MHz) delta 3.76 (t, J = 5.0 Hz, 1H), 2.96 (s, 1H), 2.49-2.67 (m, 3H), 2.28-2.34 (m, IH), 2.30-2.34 (m, 2H), 2.18 (s, 6H), 2.09 (br s, 2H), 1.96 (d, J = 11.2, IH), 1.67-1.73 (m, 2H), 1.60 (s, 3H) 1.22 (br s, 3H), 1.18 (br s, 2H); 13 C NMR (CDCl 3 , 100 MHz) delta 177.0, 131.8, 131.3, 84.0, 80.2, 58.3, 58.1, 50.9, 46.0, 44.6, 38.4, 35.3, 30.0, 27.2, 23.7, 22.8.
……………………………………..
A new crystalline form of sphaelactone dimethylamine fumarate and its preparation are claimed. The parent compound is ACT-001 (DMAMCL) which Accenda Tech and Nankai University are investigating for potential oral treatment of acute leukemia. Preclinical studies were completed in October 2012, and an application for clinical trials was planned for 2013. Picks up from WO2013163936, which claims preparation of similar compounds.

Sprifermin offers benefit for cartilage loss from knee osteoarthritis
In a new study in patients with osteoarthritis (OA) of the knee, at 12 months, total femorotibial cartilage thickness loss was reduced in sprifermin (recombinant human fibroblast growth factor 18)-treated knees compared to placebo-treated knees, with effects being significant in the lateral femorotibial compartment but not in the central femorotibial compartment.
Results published in Arthritis & Rheumatology, a journal of the American College of Rheumatology (ACR), showed that sprifermin dosed at 100µg reduced loss of cartilage thickness and volume in the total femorotibial joint and in the lateral knee compartment (outside of the knee).
The 2010 Global Burden of Disease Study estimates that OA affects 150 million people around the world, with the ACR reporting 27 million Americans over 25 years of age diagnosed with the disease. While OA is the most common cause of physical disability in older adults, studies suggest that the average age at diagnosis is…
View original post 321 more words
(S,E)-4-(2-(3-(3-chloro-2-fluoro-6-(1H-tetrazol-1-yl)phenyl)acryloyl)-5-(4-methyl-2-oxopiperazin-1-yl)-1,2,3,4-tetrahydroisoquinoline-1-carboxamido) benzoic acid,


(S,E)-4-(2-(3-(3-chloro-2-fluoro-6-(lH-tetrazol-l-yl)phenyl)acryloyl)-5 -(4-methyl-2-oxopiperazin- 1 -yl)- 1,2,3 ,4-tetrahydroisoquinoline- 1 -carboxamido)benzoic acid
(S,E)-4-(2-(3-(3-chloro-2-fluoro-6-(1H-tetrazol-1-yl)phenyl)acryloyl)-5-(4-methyl-2-oxopiperazin-1-yl)-1,2,3,4-tetrahydroisoquinoline-1-carboxamido) benzoic acid
(S,E)-4-(2-(3-(3-chloro-2-fluoro-6-(lH-tetrazol-l-yl)phenyl)acryloyl)-5-(4-methyl-2- oxopiperazin-l-yl)-l,2,3,4-tetrahydroisoquinoline-l-carboxamido)benzoic acid
4-[[[(1S)-2-[(2E)-3-[3-chloro-2-fluoro-6-(1H– tetrazol-1-yl)phenyl]-1-oxo-2-propen-1-yl]-1,2,3,4- tetrahydro-5-(4-methyl-2-oxo-1-piperazinyl)-1-isoquinolinyl] carbonyl]amino]-Benzoic acid,has cas 1430114-34-3
Benzoic acid,4-[[[(1S)-2-[(2E)-3-[3-chloro-2-fluoro-6-(1H– tetrazol-1-yl)phenyl]-1-oxo-2-propen-1-yl]-1,2,3,4- tetrahydro-5-(4-methyl-2-oxo-1-piperazinyl)-1-isoquinolinyl] carbonyl]amino]-, 2,2,2-trifluoroacetate (1:1) has cas 1430114-35-4
hydrochloride ……1430115-97-1
Bristol-Myers Squibb Company innovator
Acute coronary syndrome; Thromboembolism; Unstable angina Factor XIa antagonist
WO-2014059203 describes Crystalline forms of (S,E)-4-(2-(3-(3-chloro-2-fluoro-6-(1H-tetrazol-1-yl)phenyl)acryloyl)-5-(4-methyl-2-oxopiperazin-1-yl)-1,2,3,4-tetrahydroisoquinoline-1-carboxamido) benzoic acid, and their use for treating thromboembolic disorders eg unstable angina or acute coronary syndrome, are claimed. This compound appears to have emerged as a lead from the factor XIa antagonists claimed in WO2013056060. This compound may be the parenteral factor XIa inhibitor or the oral factor XIa inhibitor which were being investigated by BMS. However both programs were no longer listed on the company website in February 2014. The concurrently published WO2014059202 and ‘214 claim similar compounds.
CRYSTALLINE FORMS OF A FACTOR XIA INHIBITOR (Fri, 18 Apr 2014) The instant invention provides crystalline forms of (S,E)-4-(2-(3-(3-chloro-2-fluoro-6-(1H-tetrazol-1-yl)phenyl)acryloyl)-5-(4-methyl-2-oxopiperazin-1-yl)-1,2,3,4-tetrahydroisoquinoline-1-carboxamido)benzoic acid and its solvates thereof; processes for the production of such crystalline forms; pharmaceutical compositions comprising such crystalline forms; and methods of treating thromboembolic disorders with such crystalline forms or such pharmaceutical compositions. >> read more
http://patentscope.wipo.int/search/en/detail.jsf;jsessionid=510CB24BFD57B0C6EA3C7FAC7EA701D3.wapp1nB?docId=WO2014059203&recNum=1&maxRec=4340&office=&prevFilter=&sortOption=&queryString=EN_ALL%3Anmr+AND+PA%3A%28Bristol-Myers+Squibb%29+&tab=PCTDescription WO 2013/056060, which is herein incorporated by reference, discloses a factor XIa inhibitor, (S,E)-4-(2-(3-(3-chloro-2-fluoro-6-(lH-tetrazol-l-yl)phenyl)acryloyl)-5 -(4-methyl-2-oxopiperazin- 1 -yl)- 1,2,3 ,4-tetrahydroisoquinoline- 1 -carboxamido)benzoic acid, (hereinafter referred to as “Compound (I)”):
(I) which is useful in preventing or treating thromboembolic disorders. Compound (II) is obtained through Ugi reaction (Schuster, I. et al. {Letters in Organic Chemistry, 4(2): 102-108 (2007)). Deprotection of Compound (II) leads to Compound (I).  (II) (I)
(II) (I)
…………………………………………………….
WO 2013056060 http://www.google.com/patents/WO2013056060A1?cl=en
Scheme 1 :

[00223] Scheme 2 describes an alternative method to access compounds of this invention. Reaction of acid le, isocyanide 2a, and imine 2b can give Ugi product 2d (Schuster, I. et al, Letters in Organic Chemistry, 4(2): 102-108 (2007)). Selective oxidation of tetrahydroisoquinoline 2c using known methods such as Mn02 (Aoyama, T. et al, Synlett, 1 :35-36 (1998)) can yield imine 2b, which can then be used via the three component Ugi coupling procedures described above. The Ugi coupling procedures can be used extensively with other imino derived intermediates contained in this invention. Further manipulations of the Ugi derived products can afford compounds of this invention.
Scheme 2:

[00224] Scheme 3 describes methods for preparing the tetrahydroisoquinoline intermediate 3c and 3e. Method A uses Bischler-Napieralski cyclization to access compounds such as intermediate 3c (Al-Hiari, Y. M. et al, Journal of Heterocyclic Chemistry, 42(4): 647-659 (2005)) or 3e (Zalan, Z. et al, Tetrahedron, 62(12): 2883- 2891 (2006)). Method B uses the Friedel-Crafts alkylation reaction to access compounds such as intermediate 3c (Topsom, R. D. et al, Journal of the Chemical Society [Section] D: Chemical Communications, 15:799 (1971)). Alternatively, as described in Method C, cyclization of intermediate 3h and 3-aminopropanol (3i) can afford 3j. Reduction with NaBH4, followed by PCC oxidation gave β-amino aldehyde, which can be converted to 3c under basic conditions (Umetsu, K.; Asao, N., Tetrahedron Letters, 49(17): 2722-2725 (2008)). In Method D, lactam 31 can be synthesized from ketone 3k by the Beckmann rearrangement. Reduction of 31 can afford intermediates such as 3c (Vernier, J. et al, WO 2008024398 (2008)). In Method E, the dihydroisoquinoline carbaldehyde (3m) was converted to 3c under basic conditions (Martin, S. et al, WO 2006134143 (2006)). In Method F, dihydroisoquinolinethione was converted to 3c treating the thione 3o with bromopropene followed by treatment with perchloric acid and sodium borohydride (Mohinder, B, et al, Indian Journal of Chemistry, Section B: Organic Chemistry
Including Medicinal Chemistry, 18B (4); 312-15 (1979)).
Scheme 3:

[00225] Preparation of substituted THQ analogs is shown in Scheme 4. Bromide 4a can be converted to nitrile 4b under lithiation conditions. Hydrolysis under basic conditions should lead to acid 4c, which can be converted to carbamate 4e via Curtius rearrangement. Formation of the THQ intermediate 4f can then be accomplished by treatment with paraformaldehyde in a mixture of acetic and sulfuric acid (Bigge, C. F. et al, Bioorganic & Medicinal Chemistry Letters, 3(1): 39-42 (1993)). Deprotection of carbamate 4f followed by protection with B0C2O should afford intermediate 4h, which can be subjected to the Suzuki cross coupling reaction with an appropriate boronate or boronic acid or the Stille coupling procedures known to those in the art.
Scheme 4:
Isobutyronitrile, DPPA/TEA,
LiHMDS/THF, Toluene, NaH (60%), 0 °C to rt, 0 °C, 1 h, MeOH/THF, 3 h 110 °C, 4 h. 0 °C to rt, 3 h.

4a 4b 4d


[00231] The synthesis was described as Intermediate 1 in PCT International Application, WO 2009/1 14677 published 09/17/09. Intermediate 2: (E)-3-(5-chloro-2-tetrazol-l-yl-phenyl)-acrylic acid

[00232] The synthesis was described as Intermediate IB in PCT International Application, WO 2009/1 14677 published 09/17/09. Intermediate 3: (E)-3-(3-Chloro-2-fluoro-6-tetrazol-l-yl-phenyl)-acrylic acid 2,5-dioxo- pyrrolidin-l-yl ester

[00233] Intermediate 3A: (E)-3-(3-chloro-2-fluoro-6-(lH-tetrazol-l-yl)phenyl)acrylic acid: The synthesis of Intermediate 3A was described as Intermediate 7 in PCTInternational Application, WO 2009/1 14677 published 09/17/09. [00234] Intermediate 3 : To a slightly turbid mixture of Intermediate 3 A (1.0 g, 3.72 mmol) in THF (18.70 mL) and DMF (1.870 mL) was added l-hydroxypyrrolidine-2,5- dione (0.471 g, 4.09 mmol) and DIC (0.638 mL, 4.09 mmol). The reaction was stirred at rt and a white precipitate formed overtime. The solid was collected by suction filtration and washed with MeOH and H20. The crude product was then air-dried and finally dried under vacuum to give Intermediate 3 (0.98 g, 72%), as a white solid. ¾ NMR (500 MHz, DMSO-d6) δ 9.92 (s, 1H), 8.06 (t, J= 8.12 Hz, 1H), 7.72 (d, J= 8.80 Hz, 1H), 7.36 (d, J = 16.23 Hz, 1H), 6.81 (d, J = 16.51 Hz, 1H), 2.84 (s, 4 H) ppm. MS (ESI) m/z: 366.2 (M+H)+. Intermediate 4: (E)-3-(2-acetyl-5-chlorophenyl)acrylic acid

[00235] Intermediate 4A: (E)-tert-butyl 3-(2-acetyl-5-chlorophenyl)acrylate: To a degassed solution of l-(2-bromo-4-chlorophenyl)ethanone (1.0 g, 4.28 mmol), tributylamine (2.041 mL, 8.57 mmol), and tert-butyl acrylate (1.255 mL, 8.57 mmol) in DMF (10 mL) was added palladium on carbon (0.456 g, 0.428 mmol) and palladium (II) acetate (0.096 g, 0.428 mmol). The reaction mixture was warmed to 100 °C. After 16 h, the reaction was cooled to rt and filtered. The solid was rinsed with DMF and the filtrate was diluted with EtOAc and washed with H20 (2x) followed by brine. The crude product was then dried over a2S04, filtered and concentrated. Purification by normal phase chromatography afforded Intermediate 4A (0.760 g, 63%), as a brown oil. MS (ESI) m/z: 225.0 (M-C4H8+H)+. [00236] Intermediate 4: A solution of Intermediate 4A (0.048 g, 0.171 mmol) in 50% TFA/DCM (2 mL) was stirred at rt. After 1 h, the reaction was concentrated to give Intermediate 4 (0.038 g, 100%) as a yellow solid. The material was carried onto the next step without further purification. MS (ESI) m/z: 225.1 (M+H)+. Intermediate 5: (E)-3-(5-chloro-4-fluoro-2-(lH-tetrazol-l-yl)phenyl)acrylic acid


[00312] Example 57 (Table 7): (E)-tert-butyl 4-(2-(3-(3-chloro-2-fluoro-6-(lH- tetrazol- 1 -yl)phenyl)acryloyl)-5 -(4-methyl-2-oxopiperazin- 1 -yl)- 1,2,3,4- tetrahydroisoquinoline-l-carboxamido)benzoate: Intermediate 3A (0.320 g, 1.192 mmol) and Intermediate 22 (0.29 g, 1.192 mmol) were combined in a vial in EtOH (5mL) and after 10 min., Intermediate 6 (0.315 g, 1.550 mmol) in EtOH (3mL) was added and reaction was heated at 55 °C for 24 h. The reaction was concentrated and the residue was purified by silica gel column chromatography followed by reverse phase HPLC and freeze-dried to afford 0.339g (32.6%) of Example 57 (Table 7) as a white solid. ¾ NMR (400 MHz, MeOD) δ: 9.44 (1 H, s), 7.74 – 7.84 (2 H, m), 7.62 – 7.73 (1 H, m), 7.43 – 7.58 (3 H, m), 7.37 (1 H, dd, J= 8.72, 1.64 Hz), 7.31 (1 H, td, J= 7.83, 2.78 Hz), 7.19 (1 H, t, J= 6.82 Hz), 6.98 – 7.1 1 (1 H, m), 6.79 – 6.94 (1 H, m), 5.80 (1 H, s), 3.94 – 4.20 (3 H, m), 3.84 – 3.95 (1 H, m), 3.62 – 3.80 (3 H, m), 3.53 – 3.64 (1 H, m), 2.99 (3 H, s), 2.92 – 2.96 (1 H, m), 2.61 – 2.77 (1 H, m), 1.47 (9 H, d, J= 2.02 Hz) ppm. MS (ESI) m/z: 715.3. Analytical HPLC: RT = 6.82 min. [00313] Example 183 was prepared from Example 57 (Table 7) and isolated as the first eluting peak after chiral HPLC separation using Chiralpak AD-H, 250 X 30 mm, 5μιη, using 60/40 C02/1 : 1 EtOH-IPA-0.1% DEA at 90 mL/min, 150 bar BP, 35 °C followed by deprotection with TFA/DCM and HPLC purification to afford 96.8 mgs (25.8%) of a white solid. XH NMR (400 MHz, MeOD) δ: 9.44 (1 H, s), 7.78 – 7.95 (2 H, m), 7.69 (1 H, td, J=8.08, 2.53 Hz), 7.44 – 7.60 (3 H, m), 7.27 – 7.41 (2 H, m), 7.15 – 7.25 (1 H, m), 6.98 – 7.11 (1 H, m), 6.77 – 6.98 (1 H, m), 5.78 – 5.88 (1 H, m), 3.83 – 4.19 (4 H, m), 3.64 – 3.80 (3 H, m), 3.54 – 3.64 (1 H, m), 3.03 (3 H, s), 2.93 – 3.00 (1 H, m), 2.63 – 2.78 (1 H, m) ppm MS (ESI) m/z: 659.3 (M+H)+. Analytical HPLC: RT = 4.90 min. Example 184: (S,E)-4-(2-(3-(3-chloro-2-fluoro-6-(lH-tetrazol-l-yl)phenyl)acryloyl)-5-(4-methyl-2- oxopiperazin-l-yl)-l,2,3,4-tetrahydroisoquinoline-l-carboxamido)benzoic acid, TFA salt
[00314] Example 184 was isolated as the second eluting enantiomer from Example 57 (Table 7) and deprotected and purified as described in Example 183 to afford 104 mgs (27.7%) of a white solid. ¾ NMR (400 MHz, MeOD) δ: 9.45 (1 H, s), 7.79 – 7.92 (2 H, m), 7.64 – 7.74 (1 H, m), 7.44 – 7.62 (3 H, m), 7.27 – 7.43 (2 H, m), 7.15 – 7.24 (1 H, m), 6.97 – 7.12 (1 H, m), 6.72 – 6.90 (1 H, m), 5.77 – 5.88 (1 H, m), 3.82 – 4.17 (4 H, m), 3.53 – 3.82 (4 H, m), 2.99 – 3.03 (1 H, m), 2.98 (3 H, s), 2.60 – 2.77 (1 H, m) ppm. MS (ESI) m/z: 659.3 (M+H)+. Analytical HPLC: RT = 4.94 min.
GKT-137831 a NOX1 and NOX4 inhibitor from GenKyoTex being developed for diabetic nephropathy
GTK 137831
1218942-37-0
Genkyotex Sa INNOVATOR
1H-Pyrazolo[4,3-c]pyridine-3,6(2H,5H)-dione, 2-(2-chlorophenyl)-4-[3-(dimethylamino)phenyl]-5-methyl-
C21 H19 Cl N4 O2
-
2-(2-Chlorophenyl)-4-(3-(dimethylamino)phenyl)-5-methyl-1H-pyrazolo(4,3-c)pyridine-3,6(2H,5H)-dione
- 394.8601 mw
- in phase 2
- UNII-45II35329V
drug recently advancing to phase II trials is GKT-137831, a NOX1 and NOX4 inhibitor from GenKyoTex being developed for diabetic nephropathy, the leading cause of chronic kidney disease in the US and Europe.
GKT137831 is a selective NOX1/4 inhibitor in Phase II clinical development for the treatment of diabetic nephropathy, one of the complications of diabetes. It is a potent, NOX specific, small molecule with good oral availability.
Data from the Phase 1 programme to assess safety and exposure to single and multiple oral doses of GKT137831 was presented at the ASN Kidney week in San Diego in 2012. More than 100 subjects have been exposed to GKT137831 and the drug was well tolerated with no serious adverse events. In summer 2013, the FDA approved the IND to allow commencement of the Ph2 PoC trial of GKT137831 in diabetic nephropathy. Subsequently, approvals have been received from the competent authorities in Australia, Canada, Germany, Czech Republic and Poland. Enrollment to this study is ongoing and data is expected in H1 2015.
GKT137831 has been found to be effective in a range of preclinical disease models. This work has been conducted by leading academic collaborators in disease models of diabetic nephropathy, atherosclerosis, idiopathic pulmonary fibrosis, liver fibrosis and angiogenesis. GKT137831 has therefore, the potential to treat a wide range of important and poorly managed diseases

PATENT
WO 2010035221
http://www.google.com/patents/WO2010035221A1?cl=en
Scheme 1

R18 = Me Pr, iPr, Bu
G /NH Toluen
Il

G1 as described above G1 = H (Ib) (Ia) VIII
Scheme 2

R18 = Me, Et, Pr, iPr, Bu
Toluene
G^

G1 as described above G1 = H (Ib) (Ia) VIII
Scheme 3

IV R19 = Me, Et, XII
R18 = Me, Et, Pr, iPr, Bu


G1 = H, G3 = CH2NR20R21 (Ia) XIV XIII

G1, G3 as described above (Ib)

-
Genkyotex’s GKT137831 Found to Reverse Fibrosis and Improve Survival in a Model of Persistent Lung Fibrosis
-
Genkyotex, the leading developer of selective NOX enzyme inhibitors, announced today the publication of data showing that GKT137831, a first in class NOX1 and 4 inhibitor, was able to reverse lung fibrosis associated with aging in a new model of idiopathic pulmonary fibrosis. Collaborators led by Professor Victor Thannickal at the University of Alabama at Birmingham published the results in the April 9, 2014 issue of Science Translational Medecine. Genkyotex is investigating GKT137831 in a Phase II trial in patients with diabetic nephropathy, another progressive fibrotic disease.
- 23 MARCH 2014
-
Keystone Symposia Conference 2014
-
March 26th, 2014. Today, Dr. Philippe Wiesel, CMO at Genkoytex presented preclinical data showing the beneficial effect of NOX1/4 inhibitor for the treatment of NASH (Non-Alcoholic Steatohepatitis)
Genkyotex held a breakfast meeting on the 28th on the role of NADPH oxidases in fibrosis
The presentations can be downloaded here
- 11 NOVEMBER 2013
-
Genkyotex NOX Inhibitor GKT137831 Successfully Shown to Halt Diabetic Kidney Disease
-
Genkyotex, the leading developer of selective NOX enzyme inhibitors, announced today that data from a group of academic collaborators demonstrated that NOX4 is an important driver of kidney injury in diabetes and that its novel, first in class NOX 1 and 4 inhibitor, GKT137831, has the potential to prevent or delay the development of diabetic nephropathy. Data were presented at the American Society of Nephrology’s Kidney Week 2013 in Atlanta and have been accepted for publication in the Journal of the American Society of Nephrology (JASN).
- 08 NOVEMBER 2013
-
Genkyotex attended the American Society of Nephrology Annual Meeting during Kidney week in Atlanta GA.
-
November 7th to 10th, 2013. Genkyotex attended the American Society of Nephrology Annual Meeting during Kidney week in Atlanta, GA. Ursula Ney, CEO, Philippe Wiesel, CMO, and the clinical team attended. Presentations from the Ancillary meeting held on 8th November can be found here.
- 05 NOVEMBER 2013
-
Genkyotex Initiates Multinational Phase II Study with First in Class NOX Inhibitor GKT137831 in Diabetic Nephropathy Patients
-
Genkyotex, the leading developer of selective NOX enzyme inhibitors, announced today the initiation of a multinational Phase II clinical study of GKT137831 in patients with diabetic nephropathy. GKT137831 is a first in class inhibitor targeting NOX1 and NOX4 enzymes, both of which play a key role in the development of diabetic complications and chronic kidney disease in particular. In phase I studies in more than 100 subjects, GKT137831 was found to be safe and well tolerated when administered orally once and twice daily.
- 21 OCTOBER 2013
-
Genkyotex Collaborators Elucidate Role of NOX4 in Osteoporosis
-
Genkyotex, the leading developer of NOX enzyme inhibitors, announced today that a group of collaborators have discovered a link between the enzyme NOX4 and development of osteoporosis. These results, published online in the Journal of Clinical Investigationdoi:10.1172/JCI67603), indicate that inhibitors of NOX4, such as GKT137831 developed by Genkyotex could lead to a novel way of treating patients with osteoporosis. GKT137831, the first in class NOX1 and 4 inhibitor, has shown favorable safety and pharmacokinetic profiles in Phase I studies, and following a recently FDA approved IND will enter a Phase II trial in patients with diabetic nephropathy.
- 08 SEPTEMBER 2013
-
Genkyotex Receives FDA IND Approval for Phase II Clinical Study with First in Class NOX Inhibitor GKT137831
-
Genkyotex, the leading developer of NOX enzyme inhibitors, announced today that the U.S. Food and Drug Administration has approved the company’s Investigational New Drug (IND) application to begin a Phase II clinical study of GKT137831 in patients with diabetic nephropathy. GKT137831 is a first in class inhibitor targeting NOX1 and NOX4 enzymes. Enrollment of patients into the multinational Phase II study is expected to begin during Q4, 2013.
- 07 MAY 2013
-
Genkyotex Collaborators Discover Role of NOX in Development of Atherosclerosis in Diabetic Mice
-
Genkyotex, the leading developer of NOX inhibitors to treat oxygen-radical mediated diseases, announced today that its collaborators at the Baker IDI Heart & Diabetes Research Institute, Melbourne (Australia) and Maastricht University (The Netherlands) have elucidated the role of NOX1 in causing atherosclerosis in diabetic mice. The researchers found that NOX1 produces toxic amounts of oxygen radicals in the wall of blood vessels, which along with other inflammatory chemicals led to atherosclerotic plaque development. The researchers also demonstrated that Genkyotex’s selective NOX1 and 4 inhibitor, GKT137831, was able to dramatically reduce development of atherosclerosis. The research and accompanying editorial from Dr. David G. Harrison from Vanderbilt University was published in May 7th issue ofCirculation.
- 17 DECEMBER 2012
-
Genkyotex Issued U.S. Patent Covering Parent NOX Inhibitor Chemical Series
-
Genkyotex, the leading developer of NOX inhibitors to treat oxygen-radical mediated diseases, today announced that the United States Patent and Trademark Office (USPTO) has issued a Notice of Allowance for U.S. Patent Application No. 12/532,336, titled “pyrazolo pyridine derivatives as NADPH oxidase inhibitors”.
- 02 NOVEMBER 2012
-
Genkyotex’s NOX Inhibitor GKT137831 Phase I Data Presented at Kidney Week 2012
-
Genkyotex, the leading developer of NOX inhibitors to treat oxygen-radical mediated diseases, announced today that Phase I studies have demonstrated excellent safety and tolerability following single and multiple oral doses of GKT137831, the first in class NOX 1 and 4 inhibitor. In addition, GKT137831 demonstrated a favourable pharmacokinetic profile in these subjects.
- 15 OCTOBER 2012
-
Genkyotex’s First in Class NOX Inhibitor GKT137831 to be Presented at Kidney Week
-
Genkyotex will present data from single and multiple dose Phase I studies with the NOX 1 and 4 inhibitor, GKT137831, at Kidney Week 2012 (San Diego, October 30 – November 4). The Phase I data will be presented on Friday, November 2, 2012, 10.00 AM -12.00 PM (PosterBoard# FR-PO831; Abstract# 2279).
- 08 AUGUST 2012
-
Genkyotex’s Lead NOX Inhibitor GKT137831 Demonstrates Activity in Models of Liver Fibrosis
-
Genkyotex, with collaborator Professor David Brenner, M.D., Dean, School of Medicine, University of California San Diego, has published data online in Hepatology regarding its lead (NOX) inhibitor, GKT137831, in models of liver fibrosis, a scarring process associated with chronic liver disease that can lead to loss of liver function. The data demonstrates the specificity of GKT137831 and its ability to attenuate development of fibrosis in the liver and production of reactive oxygen species (ROS) in two models of disease, as well as inhibiting messenger RNA expression of fibrotic and NOX genes.
- 09 JULY 2012
-
Genkyotex closes CHF25 million (USD26 million) extension to its Series C financing.
-
Investors in the Series C round, including Eclosion, Edmond de Rothschild Investment Partners, Vesalius Biocapital Partners, MP Healthcare Venture, all participated in the financing extension. The proceeds will be used to advance clinical development of Genkyotex’s lead compound, the NOX1/4 inhibitor GKT137831, through Phase II development for the treatment of diabetic nephropathy.
- 22 JUNE 2012
-
Genkyotex Announces Successful Phase Ia Data with First in Class NOX Inhibitor GKT137831
-
Diabetic Nephropathy First Target Indication for NOX1/4 Inhibitor
- 31 OCTOBER 2011
-
GenKyoTex Starts Phase I Trial with First in Class NOX inhibitor GKT137831
-
GenKyoTex, the leading developer of NOX inhibitors to treat oxygen-radical mediated diseases, announced today that a Phase I study has been initiated with GKT137831, a first in class dual inhibitor of NOX1 and NOX4 enzymes.
-
GenKyoTex raises CHF 18 million in a Series C Venture Financing to develop NOX enzyme inhibitors.
-
Appoints New Management Team & Board
- 02 DECEMBER 2010
-
GKT137831 granted Orphan Drug status for Idiopathic Pulmonary Fibrosis by the EC (EMEA)
-
Genkyotex announced today that its lead clinical candidate GKT137831 has been granted the orphan drug status by the European Commission for the treatment of idiopathic pulmonoary fibrosis.
- 27 SEPTEMBER 2010
-
FDA granting Genkyotex Orphan Drug Designation of GKT137831 for IPF
-
Genkyotex announced today having received a letter from FDA dated of 21st September 2010, granting Genkyotex Orphan Drug Designation of GKT137831 for the treatment of Idiopathic Pulmonary Fibrosis (IPF).
-
Medical Mushrooms – The Future of Cancer Treatment?
Cancer rates are on the rise worldwide, which means that in coming generations more and more people will have their lives turned inside out with a diagnosis, and with having to turn their attention to battling this new plague. The psychological effects of having your world turned on its so quickly can be devastating, and often put people in a depressed, anxious and negative emotional state.
With so many types of cancers affecting people these days, there is no such thing as a single cure for cancer, because each type is different and will respond to different remedies. Finding the miracle cure often requires an intense search, deviation from standard doctor’s recommendations, a huge investment of time and money, and tremendous amount of hope, belief and faith. Not everything works for every cancer, but, some things consistently aid in the struggle with all cancers, like the right diet…
View original post 1,125 more words
