
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 28)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Bezisterim, HE 3286; NE-3107




Bezisterim, HE 3286; NE-3107
CAS 1001100-69-1
(1R,3aS,3bR,4R,7S,9aR,9bS,11aS)-1-ethynyl-9a,11a-dimethyl-1H,2H,3H,3aH,3bH,4H,6H,7H,8H,9H,9aH,9bH,10H,11H,11aH-cyclopenta[a]phenanthrene-1,4,7-triol
- (3β,7β,17α)-Pregn-5-en-20-yne-3,7,17-triol
- 17α-Ethynyl-5-androstene-3β,7β,17β-triol
- 17α-Ethynyl-Δ5-androstene-3β,7β,17β-triol
- 17α-Ethynylandrost-5-ene-3β,7β,17β-triol
- 3β,7β,17β-Trihydroxy-17α-ethynylandrost-5-ene
- Bezisterim
- HE 3286
- NE 3107
- Triolex
(3S,7R,8R,9S,10R,13S,14S,17R)-17-ethynyl-10,13-dimethyl-1,2,3,4,7,8,9,11,12,14,15,16-dodecahydrocyclopenta[a]phenanthrene-3,7,17-triol
| Formula | C21H30O3 |
|---|---|
| Molar mass | 330.468 g·mol−1 |
Q27286562
(3beta,7beta,17alpha)-Pregn-5-en-20-yne-3,7,17-triol
17.ALPHA.-ETHYNYL-5-ANDROSTENE-3.BETA.,7.BETA.,17.BETA.-TRIOL
PREGN-5-EN-20-YNE-3,7,17-TRIOL, (3.BETA.,7.BETA.,17.ALPHA.)-
- (1R,3aS,3bR,4R,7S,9aR,9bS,11aS)-1-ethynyl-9a,11a-dimethyl-1H,2H,3H,3aH,3bH,4H,6H,7H,8H,9H,9aH,9bH,10H,11H,11aH-cyclopenta(a)phenanthrene-1,4,7-triol
- (1R,3aS,3bR,4R,7S,9aR,9bS,11aS)-1-ethynyl-9a,11a-dimethyl-1H,2H,3H,3aH,3bH,4H,6H,7H,8H,9H,9aH,9bH,10H,11H,11aH-cyclopenta[a]phenanthrene-1,4,7-triol
- 17-ethynyl-5-androstene-3, 7, 17-triol

Bezisterim (developmental code names NE3107, HE3286) is a synthetic analogue of androstenetriol that is believed to have anti-inflammatory and insulin-sensitizing effects in the brain.[1] The compound crosses the blood–brain barrier and does not activate any neurotransmitter receptors.[2] It has been tested as a treatment for Alzheimer’s disease,[3][4][5][6] Parkinson’s disease,[1] and traumatic brain injury.[7] The drug is under development for a variety of conditions and its highest developmental phase is phase 3 for Alzheimer’s disease.[1]
- Originator Hollis-Eden Pharmaceuticals
- Developer BioVie; Harbor Therapeutics; National Institutes of Health (USA); NeurMedix
- Class Anti-inflammatories; Antidementias; Antiepileptic drugs; Antifibrotics; Antiglaucomas; Antihyperglycaemics; Antimigraines; Antineoplastics; Antiparkinsonians; Antirheumatics; Hormones; Insulin sensitisers; Nootropics; Obesity therapies; Small molecules
- Mechanism of Action Adiponectin stimulants; Interleukin 23 inhibitors; Interleukin 6 inhibitors; Mitogen-activated protein kinase 1 inhibitors; Mitogen-activated protein kinase 3 inhibitors; NF-kappa B inhibitors; Tumour necrosis factor inhibitors
- Cystic fibrosis
- Phase III Alzheimer’s disease
- Phase II Parkinson’s disease; Traumatic brain injuries
- Preclinical Multiple myeloma; Prostate cancer
- No development reported Drug-induced dyskinesia
- Discontinued Amyotrophic lateral sclerosis; Cognition disorders; Cystic fibrosis; Epilepsy; Glaucoma; Huntington’s disease; Migraine; Myositis; Optic neuritis; Rheumatoid arthritis; Type 1 diabetes mellitus; Type 2 diabetes mellitus; Ulcerative colitis; Uveitis
28 Feb 2025BioVie plans the phase II ADdRESs-LC trial for Post-acute COVID-19 syndrome in USA (PO, Capsule), in February 2025 (NCT06847191)
- 18 Feb 2025Phase-II clinical trials in Parkinson’s disease (Early-stage disease, In the elderly) in USA (PO) (NCT06757010)
- 03 Jan 2025BioVie plans a phase II SUNRISE-PD trial for Parkinsons disease (Early stage disease) in February 2025 (PO) (NCT06757010)
SCHEME

US20100227841
https://patentscope.wipo.int/search/en/detail.jsf?docId=US43352763&_cid=P11-M9JSD6-84971-1
17α-Ethynylandrost-5-ene-3β,7β,17β-triol was prepared as follows
US20100222315 https://patentscope.wipo.int/search/en/detail.jsf?docId=US43344622&_cid=P11-M9JSIE-88638-1
WO2009149392
PATENT’
WO2009149392
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2009149392&_cid=P11-M9JSL7-90448-1

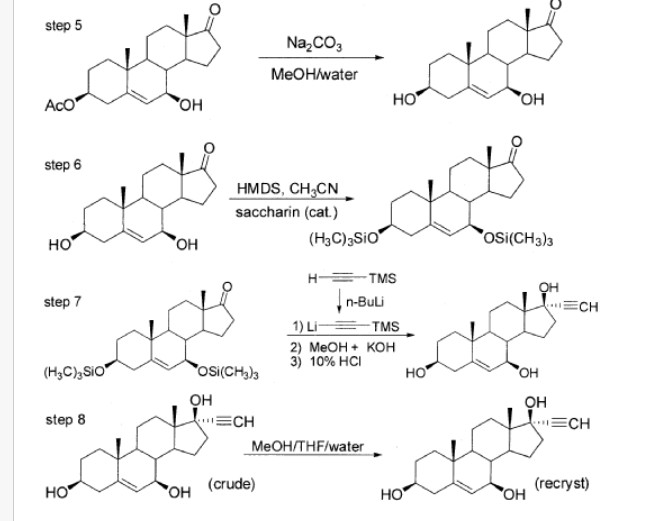
49] Example 7. Synthesis of 3β-acetoxy-androst-5-ene-17,17-ethylenedioxy: A 300L reactor was charged with 36 kg of triethylorthoformate, 20 kg of 3β-acetoxy-5-androsten-17-one, 12.6 kg of ethylene glycol and 400 g of p-toluenesulfonic acid. The mixture was heated to reflux under nitrogen until the reaction was complete (about 2-3 hours). The mixture was then cooled to 60 0C and 16 kg of anhydrous ethanol and 400 ml of pyridine were added. The resulting solution was transferred to a container and refrigerated overnight. The solids that formed were filtered and washed with 80 kg of 50% ethanol and dried at 40-50 0C to afford 18.5-21.0 kg (81.5-92.5%) of the title compound. [50] Example 8. Synthesis of 3β-acetoxy-androst-5-en-7-one-17,17-ethylenedioxy: A 500 L reactor was charged with 200 kg ethyl acetate and 25 kg of 3β-acetoxy-androst-5-en-17,17-ethylenedioxy. The mixture was stirred for 30 minutes whereupon 55 kg of 70% t-butyl peroxide and 9 kg of sodium bicarbonate were added. The reaction mixture was then cooled to 0 0C and 116 kg of 13% sodium perchlorate (aq.) was added over 10 hours so that a reaction temperature below 5 0C and pH between 7.5 and 8.5 were maintained. After the reaction was complete, the organic layer was separated and the aqueous phase was extracted with ethyl acetate (35 kg x 2). The combined organic phase was combined with a solution 33 kg of sodium sulfite in 167 kg of water, and the resulting mixture was stirred at 40 0C for 3 hours. The organic phase was washed with 50 kg of brine and concentrated to 55-60 kg whereupon 50 kg of methanol was added. After refrigeration overnight, a white solid was formed that was filtered and washed with 10 kg of methanol, and dried at 40-50 0C to yield 7.1-7.8 kg (27.4-30.1%) of the title compound.
[51] Example 9. Synthesis of 3β-acetoxy-androst-5-ene-17,17-ethylenedioxy-7β-ol. A 500 L reactor was charged with 48 kg of THF, 10 kg of 3β-acetoxy-androst-5-en-7-one-17,17-ethylenedioxy and a solution of 9.6 kg CeCI3-7H2O in 95 kg methanol. This mixture was cooled to 0 0C whereupon 2.0 kg of NaBH4 was added in batches over 3 hours in order to maintain the temperature below 5 0C. After stirring for 30 more minutes, 28 kg of acetone was added slowly in order to maintain the temperature below 5 0C, with stirring continued for another 30 minutes. To the mixture was added 240 kg water with stirring continued for 1 hour. The organic solvents were removed under vacuum and the residue was extracted with ethyl acetate (100 kg + 50 kg). The combined organic phase was washed with brine. Solvent was then removed to provide 8.6-8.9 kg (85.1-88.1 %) of the title compound. [52] Example 10. Synthesis of 3β-acetoxy-androst-5-en-17-one-7β-ol: A 500 L reactor was charged with 315 kg of acetone and 18 kg of 3β-acetoxy-androst-5-en-17,17-ethylenedioxy-7β-ol. The mixture was cooled to 5 0C and 2.34 kg of p-toluenesulfonic acid was added slowly to maintain the temperature below 10 0C. After stirring the mixture at 8-15 0C for 36-48 hours, 3.0 kg of sodium bicarbonate was added with stirring continued for 1 hour. Acetone was removed under vacuum, and to the residue was added 100 kg of water. The mixture was placed in a refrigerator overnight to give a white precipitate which was filtered to provide 33 kg (wet) of the title compound.
[53] Example 11. Synthesis of androst-5-en-17-one-3β,7β-diol: A 500 L reactor was charged 230 kg methanol, 33 kg (wet) 3β-acetoxy-7β-hydroxy-5-androsten-17-one, 108 kg water and 15 kg NaaCOβ. The mixture was heated to reflux for 3 hours. Methanol was removed under vacuum whereupon 250 kg of water was added to the residue. The mixture was put in refrigerator overnight to give a precipitate. The solids were collected by filtration, then washed with water and dried at 40-50 0C to yield 9.5-10.5 kg (67.9-75.0%) of the title compound as a white solid.
[54] Example 12. Purification of androst-5-en-17-one-3β,7β-diol: A 500 L reactor was charged with 20 kg crude 3β, 7β-dihydroxyandrost-5-en-17-one and 200 kg methanol and heated until all the solid dissolved. The solution was filtered while hot and after the filtrate cooled a white crystalline solid formed. The solids were collected by filtration, washed with small amount of methanol and dried at 40-50 0C. The solid was then refluxed in 50 kg of ethyl acetate for 20 minutes. After cooling the solid was filtered and dried at 40-50 0C under vacuum to provide 15.2 kg (76%) of purified title compound.
[55] Example 13. Synthesis of 3β,7β-bis-(trimethylsiloxy)-5-androsten-17-one: A mixture of 14.87 Kg of androst-5-en-17-one-3β,7β-diol, 23.8 Kg HMDS and 0.7 Kg saccharin catalyst in 100 L acetonitrile was heated to reflux for 8 hours with stirring under a nitrogen atmosphere. Liberated ammonia was purged under slight vacuum. The reaction volume was then reduced by distillation to collect 3OL of distillate (requires about 2 h). The reaction volume was further reduced to half of the original reaction volume by distillation under reduced pressure (700 mmHg), which requires about 2h of heating at 50 0C. The resulting uniform thick slurry is cooled to 5 0C (requires about 3 h), with additional acetonitrile added to maintain a minimum mixing volume, and held at that temperature for 1. The precipitated product was collected by filtration and dried at 45-50 0C under vacuum (29 mmHg) to a loss on drying (LOD) of not more than 1 % (requires 20 h) to provide 16 Kg (81 % yield) of the title compound (95% purity). [56] Example 14. Synthesis of 17α-ethynyl-5-androstene-3β,7β,17β-triol: To 11.02 Kg TMS-acetylene in 56.5 L tetrahydrofuran (THF) at -27 0C under a nitrogen atmosphere was added 8.51 L 10M n-BuLi. The n-butyl lithium was added very slowly to maintain a temperature at -7 to -27 0C (requires about 2 h) and the resulting reaction was stirred 10 min. at approximately 0°C to produce TMS-lithium-acetylide. To the TMS-lithium-acetylide solution was added a solution of 25.41 Kg of 3β,7β-bis-(trimethylsiloxy)-5-androsten-17-one in 95.3 L THF filtered through a 25 μm filter while allowing the reaction temperature to rise to 20-25 0C. After addition was completed, the reaction temperature was increased to 40-45 0C. To quench the reactor contents, 31.8 L of methanol was added over a period of about 1 h followed by 3.81 Kg KOH in 18.4 L of water giving a final reactor temperature of 50 0C. Liberated acetylene is purged under slight vacuum. The reactor contents were then concentrated by distillation at 80 0C for 1 h then under vacuum (175 mmHg) at about 70 0C (with an initial temperature of 25 0C to avoid bumping) to half of the original pot volume. The residue was cooled to about 10 0C and 35.0 Kg of deionized water was added, followed by 16.4 Kg 12N HCI while maintaining a pot temperature of about 10 0C and giving a final pH of 1. Additional 26.0 kg deionized water was added and the resulting mixture was stirred at about 5 0C for 1 h. The resulting slurry was filtered and washed with 75/25 mixture of methanol/water (16.9 L methanol, 5.6 L water). The collected solids were dried under vacuum (28 in Hg) at 45 0C for 12h for a loss on drying of no more than 0.5% to provide 9.6 Kg of the title compound (83% yield).
[57] Example 15. Recrystallization of 17α-ethynyl-5-androstene-3β,7β,17β-triol: Crude 9.6 Kg 17α-ethynyl-5-androstene-3β,7β,17β-triol prepared in
Example 14 was dissolved in refluxing 50/50 methanol/water (4.2 Kg methanol and 5.4 Kg water). To the solution was added 33.4 Kg methanol followed by 37.6 Kg of THF. The mixture was heated to reflux and stirring was continued until all solids have dissolved, whereupon 99.8 Kg of deionized water was added while maintaining a reactor temperature of 60-75 0C. The mixture was cooled to 0-5 0C over a period of 2 h and maintain at that temperature for 1 h while stirring was continued. The solids were recovered by filtration, washed with 9.6 Kg cold 50/50 methanol water and dried under vacuum (28 in Hg) at 50 0C for 8 h to provide 8.2 Kg of 17α-ethynyl-5-androstene-3β,7β,17β-triol. This first recrystallization is used to remove trace colored impurities from the initial product. A second recrystallization was conducted by heating the solid from the first recrystallization in ~10:1 methanohwater (145.8 Kg methanol and 18.2 Kg of water) to 80°C until all the solids have dissolved. The solution at 55-60 0C was filtered through a 25 μm filter to remove particulate impurities, whereupon 2.5 Kg of methanol at 55-60 0C (used to rinse the reactor) was added. Vacuum distillation at 125 mmHg at 70 0C was conducted until 0.9 to 1.2 times the volume of methanol that was added to the reactor was collected as distillate with water added as necessary to permit stirring (about 120-160 Kg water added). Final reaction volume was 200-225 L. The reactor mixture was cooled to 0-5 0C and maintained at that temperature for 1 h. The resulting slurry was filtered and the filter cake rinsed with 10 Kg deionized water and dried under vacuum (28 in Hg) at 50 0C for 12 h to a residual water content of less than 0.5%. This isolation procedure was used to reduce the THF content in the final product. The yield was 8.0 Kg of recrystallized title compound (83% yield).

[59] Example 16. Synthesis of 3β-acetoxy-androst-5-en-7-on-17-oxime: 3β-Acetoxy-androst-5-en-7,17-dione (45 g, 130 mmol) was dissolved in 800 ml_ methanol, 200 ml_ dichloromethane and 14.5g Et3N (144 mmol). To the solution at RT was added a solution of 10 g of hydroxylamine hydrochloride dissolved in 200 ml_ methanol. After stirring overnight, 200 ml_ of water was added followed by removal of volatile organics by evaporation under reduced pressure. To the resulting residue was added an additional 1 L of water to give a while solid that was filtered and washed well with water. Obtained was 45 g of crude title oxime in 95% purity by 1H-NMR, which was used in the next step without further purification.
[60] Example 17. Synthesis of 3β-acetoxy-androst-5-en-17-oxime-7β-ol: To a solution of 44 g of 3β-acetoxy-androst-5-en-7-on-17-oxime (100 mol%) in 800 ml_ methanol and 200 ml_ tetrahydrofuran was added 50 g of cerium chloride heptahydrate (110 mol%) in 20 ml_ of methanol. The resulting mixture was stirred until the solids were completely dissolved. To the solution cooled to about -5 0C was added 7 g sodium borohydride over 30 min. After stirring an additional 1.5 h at -5 0C, the reaction mixture was quenched with acetone (100 mL) and then allowed to warm to room temperature over a 30 min. period. The quenched reaction mixture was concentrated under vacuum to remove volatile organics. To the residue was added 800 mL of water followed by extraction with ethyl acetate (3 x 500 mL). The combined organic extracts were washed with brine, dried over Na2SO4, then concentrated to provide 42 g of the title compound as a white foam, which was used in the next step without further purification.. [61] Example 18. 3β-acetoxy-androst-5-en-17-one-7β-ol: To a solution of 42 g of 3β-acetoxy-androst-5-en-17-oxime-7β-ol (100 mol%) in 200 mL of ethanol was added 100 mL of water followed by 80 g (400 mol%) of sodium dithionite. The reaction was heated at 55 0C and stirred 16 h. After cooling, the reaction was concentrated under reduced pressure. The residue was diluted with 100 mL of water, and the resulting solid was collected by filtration and redissolved in 1 L dichloromethane. To the DCM solution was added 1 g activated carbon. After stirring overnight the mixture was filtered, and the resulting filtrate was washed with water, dried and concentrated to provide 25 g of crude product. Recrystallization from ethyl acetate gave 22g of the title compound. [62] Example 19. Estrogen receptor binding assay: A suitable example system is an estrogen receptor- kit manufactured by PanVera for ERβ, which contains recombinant estrogen receptor β ligand, FLUORMONE™ ES2 (ES2), a fluorescently labeled estrogen ligand, and appropriate buffer. The system was used in a fluorescence polarization competition assay in which a test article, such as a preparation of Compound 1 or a positive control displaces ES2 from its binding site. When bound to ERβ, ES2 tumbles slowly and has a high fluorescence polarization value. Unbound ES2 tumbles quickly and displays a low fluorescence polarization value. The change in polarization value in the presence of test compound then determines relative binding affinity of that test compound for ERβ as expressed by its IC50, which is the concentration of test compound that results in half-maximum shift in polarization. From IC50, K/ was calculated using the Cheng-Prusoff equation [Biochem. Pharmacol. 22: 3099-3108, (1973)]: K, = IC50Z(I + D/Kd) where D is the concentration of ES2 and Kd is the dissociation constant for binding of ES2 to ERβ (Kd = 4 ± 2 nM).
[63] The competition assay was conducted according to the manufacturer’s protocol (Lit. No. L0712, Rev. 10/03). Assay reagents used were bacculovirus expressed, full length human ERβ 4.5 pmol/μL in 50 mM Bis-Tris Propane (pH = 9), 400 mM KCI, 2 mM DTT, 1 mM EDTA, 10% glycerol, ES2 400 nM in methanol and E2 screening buffer consisting of 100 mM potassium phosphate (pH = 7.4), 100 μg/mL BGG, 0.02% NaN3. The ES2-ERβ complex was formed with 20 μL 20 nM ERβ (0.020 pmol/μL) and 20 μl_ 2 nM ES2 (0.002 pmol/μL). Positive control (estrogen) solution was prepared using 20 μL of a 1.0 mM stock solution in DMSO and 80 μL DMSO. In a first dilution, 50 μL of this solution is added to 50 μL of DMSO, which is followed by dilutions in 2-fold increments, to provide for a 14 point dilution curve. In a second dilution, to 4 μL of each DMSO solution from the first dilution is added 400 μL of ES2 screening buffer. To 20 μL of test compound, serially diluted in the manner described immediately above, in a 384 well black flat bottom microtiter plate, was added 20 μL of the ES2-ERβ complex (0.5% final DMSO concentration) followed by incubation in the dark at 20-30 0C for 1-4 h. Test compound was treated similarly except the starting concentration was 10 mM. Fluorescence polarization values are obtained using 485 nm excitation and 530 nm emission interference filters. Binding assay for ERa was conducted as for ERβ except bacculovirus expressed, full length human 2.8 pmol/μL ERa was used as reagent with the ERα-ES2 complex formed from 20 μL 30 nM (0.030 pmol/μL) and 20 μL 2 nM ES2 (0.002 pmol/μL). [64] Example 20. AR, GR and PR receptor binding assays. The AR competition assay was conducted according to the manufacturer’s protocol (Lit. No. L0844, Rev. 05/02) in the manner described for ERβ with the following exceptions. Reagents used were recombinant rat androgen receptor ligand binding domain tagged with His and GST [AR-LBD (His-GST)] 0.38 pmol/μL in buffer containing protein stabilizing agents and glycerol (pH = 7.5), 200 nM FLUORMONE™ AL Green, which is a fluorescently labeled androgen ligand, in 20 mM Tris, 90% methanol and AR screening buffer containing stabilizing agents and glycerol (pH = 7.5) with 2 μL of 1 mM DTT added per mL screening buffer (AR screening buffer 2 mM in added DTT) was used as the reagents. The AL Green-AR complex was formed with 20 μL 50 nM AR (0.050 pmol/μL) and 20 μL 2 nM AL Green (0.002 pmol/μL). K, was calculated using, for the dissociation constant for binding of the fluorophore to receptor, Kd = 20 ± 10 nM. [65] The PR competition assay was conducted according to the manufacturer’s protocol (Lit. No. L0503, Rev. 06/03) in the manner described for ERβ with the following exceptions. Reagents used were recombinant human progesterone receptor ligand binding domain tagged with GST [PR-LBD (GST)] 3.6 pmol/μL in 50 mM Tris (pH = 8.0), 500 mM KCI, 1 M urea, 5 mM DTT, 1 mM EDTA and 50% glycerol, 400 nM FLUORMONE™ PL Green, which is a fluorescently labeled progesterone ligand, in 20 mM Tris 90% methanol (pH = 6.8) and PR screening buffer containing protein stabilizing agents and glycerol (pH = 7.4) with 4 μL of 1 mM DTT added per mL screening buffer (PR screening buffer 4 mM in added DTT). The PL Green-PR complex was formed with 20 μL 80 nM PR (0.080 pmol/μL) and 20 μL 4 nM PL Green (0.004 pmol/μL). K, was calculated using, for the dissociation constant for binding of the fluorophore to receptor, Kd = 40 nM.
[66] The GR competition assay was conducted according to the manufacturer’s protocol (Lit. No. L0304, Rev. 12/01) in the manner described for ERβ with the following exceptions. Reagents used were recombinant full length human glucocorticoid receptor 0.240 pmol/μL in 10 mM phosphate buffer (pH = 7.4), 200 mM Na2MoO4, 0.1 mM EDTA, 5 mM DTT and 10% glycerol, 200 nM FLUORMONE™ GS1 , which is a fluorescently labeled glucocorticoid ligand, in 75% methanol, and GR screening buffer containing 100 mM potassium phosphate (pH = 7.4), 200 mM Na2MoO4, 1 mM EDTA, 20% DMSO with 5 μL of 1 mM DTT per mL screening buffer added (GR screening buffer 5 mM in added DTT), 1 mM GR stabilizing peptide, which is a co-activator related peptide [see Chang, CY. MoI. Cell Biol. 19: 8226-36 (1999)] in 10 mM phosphate buffer (pH = 7.4) and 1 M DTT in water were used as the reagents. To 2.5 mL of the GR screening buffer is added 2.5 mL GR stabilizing peptide solution and 125 μL of 1 M DTT to form the GR stabilizing peptide-glucocorticoid receptor complex. Order of addition to the microtiter plate was 20 μL test compound in 1 % DMSO, 10 μL of 16 nM GR (0.016 pmol/μL) and finally 10 μL of 4 nM GS1 , followed by incubation in the dark at 20-30 0C for 4 h (total experiment time should not exceed 7 h). K, was calculated using, for the dissociation constant for binding of the fluorophore to receptor, Kd = 0.3 ± 0.1 nM.
[67] Example 21. Impurity profiling of 17α-ethynyl-5-androstene-3β,7β,17β- triol (Compound 1) preparations.
[68] Process A: HPLC conditions for Impurity profiling of Compound 1 preparations form Process B are give in Table 1.
[69]
Table 1. HPLC Conditions for Impurity Profiling of Compound 1 Preparations form Process A
PATENT
Hollis-Eden Pharmaceuticals, Inc. WO2008039566
Zhejiang Xianju Junye Pharmaceutical Co., Ltd.; Jiangxi Junye Biopharmaceutical Co., Ltd.CN114478672
Harbor BioSciences, Inc.US20100227841
Harbor BioSciences, Inc. US20100222315 A1
Hollis-Eden Pharmaceuticals, Inc. US20100075937
Neurmedix Inc. US20080153792 A1
Hollis-Eden Pharmaceuticals, Inc.; Harbor Therapeutics, Inc. US20080146532 A1
Harbor Therapeutics, Inc.; Neurmedix, Inc. US20160045516 A1
Harbor Therapeutics, Inc. US8354396 B2
Hollis-Eden Pharmaceuticals, Inc. WO2009149392
| Clinical data | |
|---|---|
| Other names | NE3107; NE-3107; HE3286; HE-3286; 17α-Ethynyl-5-androstene-3β,7β,17β-triol; |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1001100-69-1 |
| PubChem CID | 16739648 |
| DrugBank | DB05212 |
| ChemSpider | 20571043 |
| UNII | PH8858757I |
| KEGG | D12932 |
| ChEMBL | ChEMBL4297284 |
| CompTox Dashboard (EPA) | DTXSID501267252 |
| Chemical and physical data | |
| Formula | C21H30O3 |
| Molar mass | 330.468 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
- ^ Jump up to:a b c “Bezisterim”. AdisInsight. 5 September 2024. Retrieved 26 September 2024.
- ^ Reading, Chris L; Ahlem, Clarence N; Parameswaran, Narayanan (December 2021). “Rationale for an anti-inflammatory insulin sensitizer in a phase 3 Alzheimer’s disease trial”. Alzheimer’s & Dementia. 17 (S9). doi:10.1002/alz.057438.
- ^ Stoiljkovic, Milan; Horvath, Tamas L.; Hajós, Mihály (July 2021). “Therapy for Alzheimer’s disease: Missing targets and functional markers?”. Ageing Research Reviews. 68: 101318. doi:10.1016/j.arr.2021.101318. PMC 8131215. PMID 33711510.
- ^ Balzano, Tiziano; Esteban-García, Noelia; Blesa, Javier (2 January 2023). “Neuroinflammation, immune response and α-synuclein pathology: how animal models are helping us to connect dots”. Expert Opinion on Drug Discovery. 18 (1): 13–23. doi:10.1080/17460441.2023.2160440. PMID 36538833. S2CID 254959175.
- ^ Liu, Ping; Wang, Yunyun; Sun, Yan; Peng, Guoping (April 2022). “Neuroinflammation as a Potential Therapeutic Target in Alzheimer’s Disease”. Clinical Interventions in Aging. 17: 665–674. doi:10.2147/CIA.S357558. PMC 9064449. PMID 35520949.
- ^ Xi, Yilong; Chen, Yun; Jin, Yi; Han, Guochen; Song, Mingjie; Song, Tingting; Shi, Yang; Tao, Ling; Huang, Zewei; Zhou, Jianping; Ding, Yang; Zhang, Huaqing (May 2022). “Versatile nanomaterials for Alzheimer’s disease: Pathogenesis inspired disease-modifying therapy”. Journal of Controlled Release. 345: 38–61. doi:10.1016/j.jconrel.2022.02.034. PMID 35257810. S2CID 247285338.
- ^ “U.S. Clinical Trial: Neurological Associates of West Los Angeles Listed a New Clinical Trial to Study Insulin-sensitizing NE3107 in Improving Sleep and Fatigue in Subjects With Traumatic Brain Injury.” Contify Life Science News, 1 Aug. 2023, p. NA. Gale OneFile: Health and Medicine, link.gale.com/apps/doc/A759542006/HRCA?u=anon~bb46c85&sid=sitemap&xid=0c315c7e. Accessed 14 Dec. 2023.
/////Bezisterim, HE 3286, NE 3107, Triolex, NE3107, NE-3107, HE3286, HE-3286, PHASE 2
Ensartinib



Ensartinib
X396, X 396
- 1370651-20-9
- C26H27Cl2FN6O3,
561.4 g/mol - SMA5ZS5B22
6-amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-N-[4-[(3R,5S)-3,5-dimethylpiperazine-1-carbonyl]phenyl]pyridazine-3-carboxamide
- 3-Pyridazinecarboxamide, 6-amino-5-((1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-N-(4-(((3R,5S)-3,5-dimethyl-1-piperazinyl)carbonyl)phenyl)
- 6-amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-N-[4-[(3R,5S)-3,5-dimethylpiperazine-1-carbonyl]phenyl]pyridazine-3-carboxamide
- 6-Amino-5-((1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)- N-(4-((3R,5S)-3,5-dimethylpiperazine- 1-carbonyl)phenyl)pyridazine-3-carboxamide
FDA 12/18/2024, Ensacove, To treat non-small cell lung cancer
Ensartinib, sold under the brand name Ensacove, is an anti-cancer medication used for the treatment of non-small cell lung cancer.[1] Ensartinib is an Anaplastic lymphoma kinase (ALK) inhibitor used as the salt ensartinib hydrochloride.[1] It is taken by mouth.[1]
The most common adverse reactions include rash, musculoskeletal pain, constipation, cough, pruritis, nausea, edema, pyrexia, and fatigue.[2]
Ensartinib was approved for medical use in the United States in December 2024.[1][2][3][4]
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US90227390&_cid=P11-M9JBTT-36001-1
Synthesis of 6-[bis(tert-butoxycarbonyl)amino]-5-[(1R)-1-(2,6-dichloro-3-fluoro-phenyl)ethoxy]pyridazine-3-carboxylic acid (B)


Synthesis of 6-[bis(tert-butoxycarbonyl)amino]-5-[(1S)-1-(2,6-dichloro-3-fluoro-phenyl)ethoxy]pyridazine-3-carboxylic acid (C)

| Step 1: To a solution of A5 (41.8 g, 200 mmol) in 1,2-dichloroethane (800 mL) was added Boc-L-Pro (26.9 g, 125 mmol) followed by EDCI (31.1 g, 163 mmol) and DMAP (4.12 g, 33.8 mmol) at 0° C. The resulting mixture was stirred at r.t. overnight and then water (350 mL) was added and separated, the water phase was extracted with DCM(150 mL×3), dried over MgSO 4, concentrated and purified by column chromatography to (PE:EA=30:1) to give C1 (13.72 g, yield: 65.6%). |
| Step 2: The procedure from C1 to C was similar to that of B1 to B (9.46 g, yield: 26.4% from C1). |

Medical uses
Ensartinib is indicated for the treatment of adults with anaplastic lymphoma kinase (ALK)-positive locally advanced or metastatic non-small cell lung cancer who have not previously received an ALK-inhibitor.[1][2]
History
Efficacy was evaluated in eXALT3 (NCT02767804), an open-label, randomized, active-controlled, multicenter trial in 290 participants with locally advanced or metastatic ALK-positive non-small cell lung cancer who had not previously received an ALK-targeted therapy.[2] Participants were randomized 1:1 to receive ensartinib or crizotinib.[2]
Society and culture
Legal status
Ensartinib was approved for medical use in the United States in December 2024.[2][3][5]
Name
Ensartinib is the international nonproprietary name.[6]
Ensartinib is sold under the brand name Ensacove.[1][2][3]
References
- ^ Jump up to:a b c d e f g “Ensacove (ensartinib) capsules, for oral use” (PDF). Xcovery Holdings, Inc. U.S. Food and Drug Administration. December 2024.
- ^ Jump up to:a b c d e f g “FDA approves ensartinib for ALK-positive locally advanced or metastatic non-small cell lung cancer”. U.S. Food and Drug Administration (FDA). 18 December 2024. Retrieved 20 December 2024.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 20 December 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ “FDA Approval of Ensartinib for ALK-Positive Locally Advanced or Metastatic Non-Small Cell Lung Cancer (NSCLC)” (Press release). Xcovery Holdings. 19 December 2024. Retrieved 20 December 2024 – via Business Wire.
- ^ World Health Organization (2017). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 77”. WHO Drug Information. 31 (1). hdl:10665/330984.
External links
- “Ensartinib (Code C102754)”. NCI Thesaurus.
- Clinical trial number NCT02767804 for “eXalt3: Study Comparing X-396 (Ensartinib) to Crizotinib in ALK Positive Non-Small Cell Lung Cancer (NSCLC) Patients” at ClinicalTrials.gov
Horn L, Infante JR, Reckamp KL, Blumenschein GR, Leal TA, Waqar SN, Gitlitz BJ, Sanborn RE, Whisenant JG, Du L, Neal JW, Gockerman JP, Dukart G, Harrow K, Liang C, Gibbons JJ, Holzhausen A, Lovly CM, Wakelee HA: Ensartinib (X-396) in ALK-Positive Non-Small Cell Lung Cancer: Results from a First-in-Human Phase I/II, Multicenter Study. Clin Cancer Res. 2018 Jun 15;24(12):2771-2779. doi: 10.1158/1078-0432.CCR-17-2398. Epub 2018 Mar 21. [Article]- FDA Approved Drug Products: ENSACOVETM (ensartinib) capsules, for oral use (Dec 2024) [Link]
- NCI Formulary: Ensartinib (X-396) [Link]
| Clinical data | |
|---|---|
| Trade names | Ensacove |
| Other names | X-396 |
| License data | US DailyMed: Ensartinib |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 1370651-20-9 |
| PubChem CID | 56960363 |
| DrugBank | DB14860 |
| ChemSpider | 58828042 |
| UNII | SMA5ZS5B22 |
| KEGG | D11346 |
| ChEMBL | ChEMBL4113131 |
| ECHA InfoCard | 100.306.918 |
| Chemical and physical data | |
| Formula | C26H27Cl2FN6O3 |
| Molar mass | 561.44 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
\/////////Ensartinib, FDA 2024, APPROVALS 2024, Ensacove, X396, X 396, GLXC-15836, BCP26265, EX-A2941, NSC793150, s8230
Bexicaserin



Bexicaserin
CAS 2035818-24-5
| Formula | C15H19F2N3O |
|---|---|
| Molar mass | 295.334 g·mol−1 |
(3R)-N-(2,2-difluoroethyl)-3-methyl-1,10-diazatricyclo[6.4.1.04,13]trideca-4,6,8(13)-triene-5-carboxamide
- (3R)-N-(2,2-difluoroethyl)-3-methyl-1,10-diazatricyclo[6.4.1.04,13]trideca-4,6,8(13)-triene-5-carboxamide
- (7R)-N-(2,2-difluoroethyl)-7-methyl-1,2,3,4,6,7-hexahydro[1,4]diazepino[6,7,1-hi]indole-8-carboxamide
- Pyrrolo[3,2,1-jk][1,4]benzodiazepine-8-carboxamide, N-(2,2-difluoroethyl)-1,2,3,4,6,7-hexahydro-7-methyl-, (7R)-
Bexicaserin is under investigation in clinical trial NCT05626634 (Open-label, Long-term Safety Study of LP352 in Subjects With Developmental and Epileptic Encephalopathy).
PATENT
Arena Pharmaceuticals, Inc.WO2023172685
Arena Pharmaceuticals, Inc., WO2016176177
https://patents.google.com/patent/WO2016176177A1/en
Example 1: Syntheses of Compounds of Table A Example 1.1: Preparation of N-(2,2-difluoroethyl)-7-methyl-l,2,3,4,6,7-hexahydro- [l,4]diazepino[6,7,l-hi]indole-8-carboxamide (Compound 1)
Step A: Preparation of methyl 3-formyl-lH-indole-4-carboxylate
2M solution of oxalyl dichloride in dichloromethane (DCM) (1.712 ml, 3.425 mmol) was added to DCM (15 mL) cooled down in an ice-water bath. N,N-dimethylformamide (0.250 g, 3.425 mmol) was added dropwise under nitrogen. The reaction mixture was stirred at 0 °C for 30 min. Then methyl lH-indole-4-carboxylate (0.5 g, 2.854 mmol) in DCM (10 mL) was added. The reaction mixture was warmed to room temperature and stirred for 1 h. The solvent was removed. THF (15 mL) and 20% aqueous ammonium acetate were added. The reaction mixture was stirred under reflux (-70 °C) for 30 min. The reaction mixture was then extracted with ethyl acetate. The combined organics (organic phases) were concentrated; the residue was purified by silica gel column chromatography with 90% ethyl acetate/hexanes to give the title compound (551 mg, 95.0 %) as white solid. LCMS m/z = 204.2 [M+H]+; Ή NMR (400 MHz, CDC13) δ ppm 4.00 (s, 3H), 7.34 (t, / = 7.8 Hz, 1H), 7.63 (dd, / = 8.0 and 1.0 Hz, 1H), 7.87 (dd, / = 7.5 and 1.0 Hz, 1H), 8.10 (d, / = 3.2 Hz, 1H), 9.08 (br s, 1H), 10.53 (s, 1H).
Step B: Preparation of methyl 3-methyl-lH-indole-4-carboxylate
To a stirred solution of methyl 3-formyl-lH-indole-4-carboxylate (551 mg, 2.712 mmol) in DMF (8 mL) was added 4-methylbenzenesulfonohydrazide (0.657 g, 3.525 mmol) followed by p- toluenesulfonic acid monohydrate (77.37 mg, 0.407 mmol) and tetramethylene sulfone (sulfolane, 8 mL). The reaction mixture was stirred at 100 °C for 1 h, cooled to room temperature. Sodium cyanoborohydride (0.682 g, 10.85 mmol) was added portionwise. Then the mixture was stirred at 100 °C for 2 h. The reaction mixture was cooled down, diluted with water, and extracted with 50% ethyl acetate in hexanes. The organics were concentrated; the residue was purified by silica gel column chromatography with 20% ethyl acetate hexanes to give the title compound (355 mg, 69.2 %) as off- white solid. LCMS m/z = 190.4 [M+H]+; Ή NMR (400 MHz, CDC13) δ ppm 2.41 (d, / = 1.0 Hz, 3H), 3.96 (s, 3H), 7.07-7.10 (m, 1H), 7.18 (t, / = 7.8 Hz, 1H), 7.50 (dd, / = 8.0 and 1.0 Hz, 1H), 7.64 (dd, J = 7.5 and 1.0 Hz, 1H), 8.12 (bs, 1H).
Step C: Preparation of methyl 3-methylindoline-4-carboxylate
To a solution of methyl 3-methyl-lH-indole-4-carboxylate (1.253 g, 6.622 mmol) in TFA (trifluoroacetic acid) (4.06 mL) in an ice-water bath was added triethylsilane (4.231 ml, 26.49 mmol) drop wise under N2. The reaction mixture was warmed to room temperature and stirred overnight. The mixture was concentrated and added water. After adjusting pH to 8 with saturated aqueous NaHC03 solution, the mixture was extracted with ethyl acetate. The combined organics were concentrated. The residue was purified by silica gel column chromatography with 25% ethyl acetate/hexanes (column prewashed with 0.1% Et3N/hexanes) to give the title compound (1.013 g, 80.0 %) as orange-red oil. LCMS m/z = 192.2 [M+H]+; Ή NMR (400 MHz, CDC13) δ ppm 1.25 (d, J = 6.9 Hz, 3H), 3.25 (dd, J = 8.6 and 1.7 Hz, 1H), 3.68 (t, / = 8.5 Hz, 1H), 3.83-3.92 (m, 1H), 3.90 (s, 3H), 6.78 (dd, / = 7.8 and 1.0 Hz, 1H), 7.07 (t, / = 7.8 Hz, 1H), 7.34 (dd, / = 7.8 and 1.0 Hz, 1H).
Step D: Preparation of 2-tert-butyl 8-methyl 7-methyl-3,4,6,7-tetrahydro- [l,4]diazepino[6,7,l-hi]indole-2,8(lH)-dicarboxylate
A mixture of methyl 3-methylindoline-4-carboxylate (1.013 g, 5.297 mmol) and 2- bromoethanamine hydrobromide (1.302 g, 6.357 mmol) was heated at 115 °C overnight. The residue was dissolved in methanol and purified by preparative HPLC (5-60% CH3CN/H20 with 0.1% TFA over 30 min). The combined fractions were then concentrated to give methyl l-(2-aminoethyl)-3- methylindoline-4-carboxylate. LCMS m/z = 235.4 [M+H]+; Ή NMR (400 MHz, CDC13) δ ppm 1.24 (d, / = 7.0 Hz, 3H), 2.92-3.00 (m, 3H), 3.25 (dd, / = 8.5 and 1.7 Hz, 1H), 3.30-3.40 (m, 2H), 3.80-3.90 (m, 1H), 3.88 (s, 3H), 6.64 (d, / = 7.7 Hz, 1H), 7.11 (t, / = 7.8 Hz, 1H), 7.27 (dd, / = 7.9 and 0.8 Hz, 1H).
Methyl l-(2-aminoethyl)-3-methylindoline-4-carboxylate obtained above was dissolved in methanol (10 mL), 37% formaldehyde in water (1.183 ml, 15.89 mmol) was added, followed by TFA (1.217 ml, 15.89 mmol). The reaction mixture was heated at 80 °C for lh and concentrated. The residue was dissolved in THF (8 mL), and added saturated aqueous NaHC03 (8 mL) solution and di-tert-butyl dicarbonate (0.776 ml, 5.297 mmol). The reaction mixture was stirred at room temperature overnight, diluted with water, and extracted with ethyl acetate. The combined organics were concentrated. The residue was purified by silica gel column chromatography with 25% ethyl acetate/hexanes to give the title compound (1.212 g, 66.0 %) as colorless oil. LCMS m/z = 347.2 [M+H]+; Ή NMR (400 MHz, CDC13) δ ppm rotamers 1.20 (d, / = 6.9 Hz, 3H), 1.35-1.45 (br, 9H), 2.80-2.95 (m, 1H), 3.08-3.18 (m,lH), 3.24-3.35 (m, 2H), 3.35-3.45 (m, 1H), 3.85-3.95 (m, 1H), 3.86 (s, 3H), 3.97-4.08 (m, 2H), 4.62-4.88 (m, 1H), 6.91-7.06 (m, 1H), 7.36 (d, J = 8.0 Hz, 1H).
Step E: Preparation of 2-(tert-butoxycarbonyl)-7-methyl-l,2,3,4,6,7-hexahydro- [l,4]diazepino[6,7,l-hi]indole-8-carboxylic acid
To a solution of 2-tert-butyl 8-methyl 7-methyl-3,4,6,7-tetrahydro-[l,4]diazepino[6,7,l- hi]indole-2,8(lH)-dicarboxylate (1.212 g, 3.499 mmol) in dioxane (10 mL) was added a 1M solution of lithium hydroxide in water (13.99 ml, 13.99 mmol). The reaction mixture was stirred at 80 °C for 2 h. Organic solvent was evaporated. The residue was diluted with water, adjusted pH to 3-4 with aqueous 5% citric acid. The off-white precipitate was collected and dried to give the title compound (1.116 g, 96.0 %) as off-white solid. LCMS m/z = 333.4 [M+H]+.
Step F: Preparation of N-(2,2-difluoroethyl)-7-methyl-l,2,3,4,6,7-hexahydro- [l,4]diazepino[6,7,l-hi]indole-8-carboxamide
To the solution of 2-(tert-butoxycarbonyl)-7-methyl- 1,2, 3,4,6, 7-hexahydro- [l,4]diazepino[6,7,l-hi]indole-8-carboxylic acid (25 mg, 75.21 μιηοΐ), HATU (42.87 mg, 0.113 mmol) and triethylamine (20.97 μί, 0.150 mmol) in DMF (2 mL) was added 2,2-difluoroethanamine (9.146 mg, 0.113 mmol). The reaction was stirred at room temperature overnight. The mixture was purified by semi preparative HPLC (15-85% CH3CN/H20 with 0.1% TFA over 30 min). The combined fractions were lyophilized to give tert-butyl 8-((2,2-difluoroethyl)carbamoyl)-7-methyl-3,4,6,7-tetrahydro- [l,4]diazepino[6,7,l-hi]indole-2(lH)-carboxylate, which was dissolved in dioxane (0.5 mL). A solution of 4M HC1 in dioxane (0.5 mL) was added. The reaction mixture was stirred at room temperature for 4 h and concentrated. The residue was purified by semi preparative HPLC (5-60% CH3CN/H20 with 0.1% TFA over 30 min). The combined fractions were lyophilized to give the title compound as TFA salt (17 mg, 55.2 %). LCMS m z = 296.2 [M+H]+; Ή NMR (400 MHz, CD3OD) δ ppm 1.18 (d, / = 6.9 Hz, 3H), 3.10-3.20 (m, 1H), 3.26-3.40 (m, 2H), 3.40-3.64 (m, 3H), 3.65-3.85 (m, 3H), 4.21 (d, / = 14.9 Hz, 1H), 4.40 (d, J = 14.9 Hz, 1H), 6.00 (tt, J = 56.0 and 3.9 Hz, 1H), 6.99 (d, J = 7.8 Hz, 1H), 7.12 (d, / = 7.9 Hz, 1H).
Example 1.2: Preparation of (S)- N-(2,2-difluoroethyl)-7-methyl-l,2,3,4,6,7-hexahydro- [l,4]diazepino[6,7,l-hi]indole-8-carboxamide (Compound 2) and (R)- N-(2,2-difluoroethyl)-7- methyl-l,2,3,4,6,7-hexahydro-[l,4]diazepino[6,7,l-hi]indole-8-carboxamide (Compound 3)
Enantiomers of N-(2,2-difluoroethyl)-7-methyl- 1,2,3, 4,6, 7-hexahydro-[l, 4]diazepino[6,7,l-hi]indole-8- carboxamide were obtained by chiral HPLC separation using following conditions. Column: Chiralpak IC column 250 x 20 mm (L x I.D.)
Flow: 12 mL/min
Eluent: 12 % ethanol/8 % mTBE/80 % hexanes with 0.1 % Et3N
Detector: UV 254 nm
Retention time: 1st eluting enantiomer 22.0 min, 2nd eluting enantiomer 23.5 min
After separation, both enantiomers were further purified by semi preparative HPLC (5-60%
CH3CN/H20 with 0.1% TFA (trifluoroacetic acid) over 30 min). The combined fractions were lyophilized to give the title compounds as the TFA salt.
SCHEME

Bexicaserin (INNTooltip International Nonproprietary Name; developmental code names LP352 and AN352) is a selective serotonin 5-HT2C receptor agonist which is under development for the treatment of seizures in developmental disabilities such as Dravet syndrome and Lennox-Gastaut syndrome.[1][3][2] It is taken by mouth.[2][1]
The drug is highly selective for the serotonin 5-HT2C receptor, with negligible affinity for the serotonin 5-HT2A and 5-HT2B receptors.[2] Because it does not activate the serotonin 5-HT2B receptor, bexicaserin is not expected to pose a risk of cardiac valvulopathy, unlike the existing agent fenfluramine.[2]
As of October 2024, bexicaserin is in phase 3 clinical trials for treatment of developmental disabilities.[1][3] It is being developed by Longboard Pharmaceuticals.[1][3]
The activation of 5HT2c receptors has been shown to reduce epileptic seizure activity by inhibiting CaV3 calcium channels which mediate the T-type calcium current.[4] CaV3 calcium channels facilitate high frequency burst firing in princible neurons of the subiculum. This firing pattern is upregulated following status epilepticus, with these hyperactive neurons often serving as the initiation point for seizures.[5][6][7]
References
- ^ Jump up to:a b c d e f “Bexicaserin – Longboard Pharmaceuticals”. AdisInsight. 16 October 2024. Retrieved 29 October 2024.
- ^ Jump up to:a b c d e f Dell’isola GB, Verrotti A, Sciaccaluga M, Roberti R, Parnetti L, Russo E, et al. (June 2024). “Evaluating bexicaserin for the treatment of developmental epileptic encephalopathies”. Expert Opinion on Pharmacotherapy. 25 (9): 1121–1130. doi:10.1080/14656566.2024.2373350. PMID 38916481.
- ^ Jump up to:a b c “Delving into the Latest Updates on Bexicaserin with Synapse”. Synapse. 28 October 2024. Retrieved 29 October 2024.
- ^ Petersen AV, Jensen CS, Crépel V, Falkerslev M, Perrier JF (2017). “Serotonin Regulates the Firing of Principal Cells of the Subiculum by Inhibiting a T-type Ca2+ Current”. Frontiers in Cellular Neuroscience. 11: 60. doi:10.3389/fncel.2017.00060. PMC 5339341. PMID 28326015.
- ^ Menendez de la Prida L, Gal B (June 2004). “Synaptic contributions to focal and widespread spatiotemporal dynamics in the isolated rat subiculum in vitro”. The Journal of Neuroscience. 24 (24): 5525–36. doi:10.1523/JNEUROSCI.0309-04.2004. PMC 6729319. PMID 15201325.
- ^ Su H, Sochivko D, Becker A, Chen J, Jiang Y, Yaari Y, et al. (May 2002). “Upregulation of a T-type Ca2+ channel causes a long-lasting modification of neuronal firing mode after status epilepticus”. The Journal of Neuroscience. 22 (9): 3645–55. doi:10.1523/JNEUROSCI.22-09-03645.2002. PMC 6758371. PMID 11978840.
- ^ Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002 Nov 15;298(5597):1418-21. doi: 10.1126/science.1076510. PMID 12434059.
| Clinical data | |
|---|---|
| Other names | LP352; LP-352; AN352; AN-352 |
| Routes of administration | Oral[1] |
| Drug class | Serotonin 5-HT2C receptor agonist[1][2] |
| Pharmacokinetic data | |
| Elimination half-life | 5–7 hours[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2035818-24-5 |
| PubChem CID | 122662787 |
| DrugBank | DB18885 |
| ChemSpider | 129309383 |
| UNII | R8XR1D6SCB |
| KEGG | D13035 |
| ChEMBL | ChEMBL5314507 |
| Chemical and physical data | |
| Formula | C15H19F2N3O |
| Molar mass | 295.334 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////Bexicaserin, PHASE 2, LP 352, LP-352, AN 352, AN-352
Olezarsen


Olezarsen
Olezarsen is an ASO directed inhibitor of Apolipoprotein C-III (apoC-III) mRNA, conjugated to a ligand containing three N-acetyl galactosamine (GalNAc) residues to enable delivery of the ASO to hepatocytes.
TRYNGOLZA contains olezarsen sodium as the active ingredient. Olezarsen sodium is a white to yellow solid and it is freely soluble in water and in phosphate buffer. The molecular formula of olezarsen sodium is C 296H 419N 71O 154P 20S 19Na 20and the molecular weight is 9124.48 daltons. The chemical name of olezarsen sodium is DNA, d(P-thio) ([2′- O-(2-methoxyethyl)] rA-[2′- O-(2-methoxyethyl)] rG-[2′- O-(2-methoxyethyl)] m5rC-[2′- O-(2-methoxyethyl)] m5rU-[2′- O-(2-methoxyethyl)] m5rU-m5C-T-T-G-T-m5C-m5C-A-G-m5C-[2′- O-(2-methoxyethyl)] m5rU-[2′- O-(2-methoxyethyl)] m5rU-[2′- O-(2-methoxyethyl)] m5rU-[2′- O-(2-methoxyethyl)] rA-[2′- O-(2-methoxyethyl)]m5rU), 5′-[26-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]-14,14-bis[[3-[[6-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]hexyl]amino]-3-oxopropoxy]methyl]-8,12,19-trioxo-16-oxa-7,13,20-triazahexacos-1-yl hydrogen phosphate], sodium salt (1:20).


Olezarsen
FDA APPROVED 12/19/2024, Tryngolza, To treat familial chylomicronemia syndrome
Drug Trials Snapshot
- AKCEA-APOCIII-LRX
- ALL-P-AMBO-5′-O-(((6-(5-((TRIS(3-(6-(2-ACETAMIDO-2-DEOXY-.BETA.-D-GALACTOPYRANOSYLOXY)HEXYLAMINO)-3-OXOPROPOXYMETHYL))METHYL)AMINO-5-OXOPENTANAMIDO)HEXYL))PHOSPHO)-2′-O-(2-METHOXYETHYL)-P-THIOADENYLYL-(3′-O->5′-O)-2′-O-(2-METHOXYETHYL)-P-THIOGUANYLYL-(3
- DNA, D(P-THIO)((2′-O-(2-METHOXYETHYL))RA-(2′-O-(2-METHOXYETHYL))RG-(2′-O-(2-METHOXYETHYL))M5RC-(2′-O-(2-METHOXYETHYL))M5RU-(2′-O-(2-METHOXYETHYL))M5RU-M5C-T-T-G-T-M5C-M5C-A-G-M5C-(2′-O-(2-METHOXYETHYL))M5RU-(2′-O-(2-METHOXYETHYL))M5RU-(2′-O-(2-METHOXYETH
- IONIS-APOCIII-LRX
- ISIS-APOCIII-LRX
- ISIS-678354
Olezarsen, sold under the brand name Tryngolza, is a medication used in the treatment of familial chylomicronemia syndrome.[1][2] It is given by injection under the skin.[1]
Olezarsen was approved for medical use in the United States in December 2024.[1][3] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[4]
PATENT
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US9127276 | No | 2015-09-08 | 2034-05-01 |  |
| US9181549 | No | 2015-11-10 | 2034-05-01 | |
| US9593333 | No | 2014-02-14 | 2034-02-14 | |
| US9157082 | No | 2012-04-27 | 2032-04-27 | |
| US9163239 | No | 2014-05-01 | 2034-05-01 | |
Medical uses
Olezarsen is indicated as an adjunct to diet to reduce triglycerides in adults with familial chylomicronemia syndrome.[1]
Pharmacology
Olezarsen is an apolipoprotein C-III-directed antisense oligonucleotide.[1] By binding to apolipoprotein C-III mRNA, it causes its degradation, which in turn increases clearance of plasma triglycerides and very low-density lipoprotein (VLDL).[5]
Adverse effects
In a 66-patient trial, olezarsen was demonstrated to cause following side effects:[5][6]
- injection site reactions
- hypersensitivity reactions (due to immunogenic potential of the medication)
- arthralgia
- thrombocytopenia
- hyperglycemia
- elevation of liver enzymes
History
The US Food and Drug Administration (FDA) granted the application of olezarsen orphan drug designation in February 2024.[7] In August 2024, European Medicines Agency also granted olezarsen this designation.[8]
Society and culture
Legal status
Olezarsen was approved for medical use in the United States in December 2024.[3][9]
Names
Olezarsen is the international nonproprietary name.[10]
Olezarsen is sold under the brand name Tryngolza.[1]
References
^ Jump up to:a b c d e f g “Tryngolza- olezarsen sodium injection, solution”. DailyMed. 19 December 2024. Retrieved 25 January 2025.
- ^ Spagnuolo, Catherine M; Hegele, Robert A (2023). “Recent advances in treating hypertriglyceridemia in patients at high risk of cardiovascular disease with apolipoprotein C-III inhibitors”. Expert Opinion on Pharmacotherapy. 24 (9): 1013–1020. doi:10.1080/14656566.2023.2206015. PMID 37114828.
- ^ Jump up to:a b “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 20 December 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ Jump up to:a b Stroes, Erik S.G.; Alexander, Veronica J.; Karwatowska-Prokopczuk, Ewa; Hegele, Robert A.; Arca, Marcello; Ballantyne, Christie M.; et al. (16 May 2024). “Olezarsen, Acute Pancreatitis, and Familial Chylomicronemia Syndrome”. New England Journal of Medicine. 390 (19): 1781–1792. doi:10.1056/NEJMoa2400201. ISSN 0028-4793.
- ^ Ionis Pharmaceuticals, Inc. (11 December 2024). A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Study of AKCEA-APOCIII-LRx Administered Subcutaneously to Patients With Familial Chylomicronemia Syndrome (FCS) (Report). clinicaltrials.gov.
- ^ “Olezarsen Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). Retrieved 20 December 2024.
- ^ “EU/3/24/2973 – orphan designation for treatment of familial chylomicronaemia syndrome | European Medicines Agency (EMA)”. http://www.ema.europa.eu. 21 August 2024. Retrieved 22 February 2025.
- ^ “Tryngolza (olezarsen) approved in U.S. as first-ever treatment for adults living with familial chylomicronemia syndrome as an adjunct to diet” (Press release). Ionis Pharmaceuticals. 19 December 2024. Retrieved 20 December 2024 – via PR Newswire.
- ^ World Health Organization (2022). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 87”. WHO Drug Information. 36 (1). hdl:10665/352794.
Further reading
Karwatowska-Prokopczuk, Ewa; Tardif, Jean-Claude; Gaudet, Daniel; Ballantyne, Christie M.; Shapiro, Michael D.; Moriarty, Patrick M.; et al. (2022). “Effect of olezarsen targeting APOC-III on lipoprotein size and particle number measured by NMR in patients with hypertriglyceridemia”. Journal of Clinical Lipidology. 16 (5): 617–625. doi:10.1016/j.jacl.2022.06.005. PMID 35902351.
- Tardif, Jean-Claude; Karwatowska-Prokopczuk, Ewa; Amour, Eric St; Ballantyne, Christie M; Shapiro, Michael D; Moriarty, Patrick M; et al. (6 April 2022). “Apolipoprotein C-III reduction in subjects with moderate hypertriglyceridaemia and at high cardiovascular risk”. European Heart Journal. 43 (14): 1401–1412. doi:10.1093/eurheartj/ehab820. PMC 8986458. PMID 35025993.
External links
“Olezarsen (Code C180652)”. NCI Thesaurus.
- Clinical trial number NCT04568434 for “A Study of Olezarsen (Formerly Known as AKCEA-APOCIII-LRx) Administered to Patients With Familial Chylomicronemia Syndrome (FCS) (BALANCE)” at ClinicalTrials.gov
- Tardif JC, Karwatowska-Prokopczuk E, Amour ES, Ballantyne CM, Shapiro MD, Moriarty PM, Baum SJ, Hurh E, Bartlett VJ, Kingsbury J, Figueroa AL, Alexander VJ, Tami J, Witztum JL, Geary RS, O’Dea LSL, Tsimikas S, Gaudet D: Apolipoprotein C-III reduction in subjects with moderate hypertriglyceridaemia and at high cardiovascular risk. Eur Heart J. 2022 Apr 6;43(14):1401-1412. doi: 10.1093/eurheartj/ehab820. [Article]
- Karwatowska-Prokopczuk E, Tardif JC, Gaudet D, Ballantyne CM, Shapiro MD, Moriarty PM, Baum SJ, Amour ES, Alexander VJ, Xia S, Otvos JD, Witztum JL, Tsimikas S: Effect of olezarsen targeting APOC-III on lipoprotein size and particle number measured by NMR in patients with hypertriglyceridemia. J Clin Lipidol. 2022 Sep-Oct;16(5):617-625. doi: 10.1016/j.jacl.2022.06.005. Epub 2022 Jun 23. [Article]
- Hooper AJ, Bell DA, Burnett JR: Olezarsen, a liver-directed APOC3 ASO therapy for hypertriglyceridemia. Expert Opin Pharmacother. 2024 Oct;25(14):1861-1866. doi: 10.1080/14656566.2024.2408369. Epub 2024 Sep 26. [Article]
- Bergmark BA, Marston NA, Prohaska TA, Alexander VJ, Zimerman A, Moura FA, Murphy SA, Goodrich EL, Zhang S, Gaudet D, Karwatowska-Prokopczuk E, Tsimikas S, Giugliano RP, Sabatine MS: Olezarsen for Hypertriglyceridemia in Patients at High Cardiovascular Risk. N Engl J Med. 2024 May 16;390(19):1770-1780. doi: 10.1056/NEJMoa2402309. Epub 2024 Apr 7. [Article]
- FDA News: FDA approves drug to reduce triglycerides in adult patients with familial chylomicronemia syndrome [Link]
- FDA Approved Drug Products: TRYNGOLZA (olezarsen) injection, for subcutaneous use [Link]
| Clinical data | |
|---|---|
| Trade names | Tryngolza |
| Other names | IONIS-APOCIII-LRX |
| License data | US DailyMed: Olezarsen |
| Routes of administration | Subcutaneous |
| Drug class | Antisense oligonucleotide |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2097587-83-02298451-31-5 |
| DrugBank | DB18728 |
| UNII | S3RS2SA30LNSY2BY6PSB |
| KEGG | D13023 |
////Olezarsen, FDA 2024, APPROVALS 2025, Tryngolza, ISIS-678354, ISIS 678354, familial chylomicronemia syndrome
Fitusiran
Fitusiran
1711.0 g/mol, C78H139N11O30
FDA APPROVED 3/28/2025, Qfitlia, To prevent or reduce the frequency of bleeding episodes in hemophilia A or B
Press Release
- CAS 1499251-18-1
- EX-A12034
- DA-53206
- N-[1,3-Bis[3-[3-[5-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxypentanoylamino]propylamino]-3-oxopropoxy]-2-[[3-[3-[5-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxypentanoylamino]propylamino]-3-oxopropoxy]methyl]propan-2-yl]-12-[(2R,4R)-4-hydroxy-2-methylpyrrolidin-1-yl]-12-oxododecanamide
Fitusiran Sodium

43 Sodium salt of duplex of [(2S,4R)-1-{1-[(2-acetamido-2-deoxy-β-D-galactopyranosyl)oxy]-16,16-bis({3-[(3-{5-[(2-acetamido-2-deoxy-β-D-galactopyranosyl)oxy]pentanamido}propyl)amino]-3-oxopropoxy}methyl)-5,11,18-trioxo-14-oxa-6,10,17-triazanonacosan-29-oyl}-4-hydroxypyrrolidin-2-yl]methyl hydrogen all–P–ambo-2′-deoxy-2′-fluoro-P-thioguanylyl-(3’→5′)-2′-O-methyl-P-thioguanylyl-(3’→5′)-2′-deoxy-2′-fluorouridylyl-(3’→5′)-2′-O-methyluridylyl-(3’→5′)-2′-deoxy-2′-fluoroadenylyl-(3’→5′)-2′-O-methyladenylyl-(3’→5′)-2′-deoxy-2′-fluorocytidylyl-(3’→5′)-2′-O-methyladenylyl-(3’→5′)-2′-deoxy-2′-fluorocytidylyl-(3’→5′)-2′-deoxy-2′-fluorocytidylyl-(3’→5′)-2′-deoxy-2′-fluoroadenylyl-(3’→5′)-2′-O-methyluridylyl-(3’→5′)-2′-deoxy-2′-fluorouridylyl-(3’→5′)-2′-O-methyluridylyl-(3’→5′)-2′-deoxy-2′-fluoroadenylyl-(3’→5′)-2′-O-methylcytidylyl-(3’→5′)-2′-deoxy-2′-fluorouridylyl-(3’→5′)-2′-O-methyluridylyl-(3’→5′)-2′-deoxy-2′-fluorocytidylyl-(3’→5′)-2′-O-methyladenylyl-(3’→5′)-2′-deoxy-2′-fluoro-3′-adenylate and all–P–ambo-2′-O-methyl-P-thiouridylyl-(3’→5′)-2′-deoxy-2′-fluoro-P-thiouridylyl-(3’→5′)-2′-O-methylguanylyl-(3’→5′)-2′-deoxy-2′-fluoroadenylyl-(3’→5′)-2′-O-methyladenylyl-(3’→5′)-2′-deoxy-2′-fluoroguanylyl-(3’→5′)-2′-O-methyluridylyl-(3’→5′)-2′-deoxy-2′-fluoroadenylyl-(3’→5′)-2′-O-methyladenylyl-(3’→5′)-2′-deoxy-2′-fluoroadenylyl-(3’→5′)-2′-O-methyluridylyl-(3’→5′)-2′-O-methylguanylyl-(3’→5′)-2′-O-methylguanylyl-(3’→5′)-2′-deoxy-2′-fluorouridylyl-(3’→5′)-2′-O-methylguanylyl-(3’→5′)-2′-deoxy-2′-fluorouridylyl-(3’→5′)-2′-O-methyluridylyl-(3’→5′)-2′-deoxy-2′-fluoroadenylyl-(3’→5′)-2′-O-methyladenylyl-(3’→5′)-2′-deoxy-2′-fluorocytidylyl-(3’→5′)-2′-O-methyl-P-thiocytidylyl-(3’→5′)-2′-O-methyl-P-thioadenylyl-(3’→5′)-2′-O-methylguanosine
C520H636F21N175Na43O309P43S6 : 17193.39
[1609016-97-8]
Fitusiran, sold under the brand name Qfitlia, is a medication used for the treatment of hemophilia.[1] It is an antithrombin-directed small interfering ribonucleic acid.[1] It is given by subcutaneous injection.[1] Fitusiran reduces the amount of a protein called antithrombin.[2]
The most common side effects include viral infection, common cold symptoms (nasopharyngitis) and bacterial infection.[2]
Fitusiran was approved for medical use in the United States in March 2025.[2]
PATENT
https://patents.google.com/patent/WO2023240199A2/en
Medical uses
Fitusiran is indicated for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in people aged twelve years of age and older with hemophilia A or hemophilia B, with or without factor VIII or IX inhibitors (neutralizing antibodies).[1][2]
Adverse effects
The US Food and Drug Administration prescription label for fitusiran contains a boxed warning for thrombotic events (blood clotting) and gallbladder disease (with some recipients requiring gallbladder removal).[2] The label also has a warning about liver toxicity and the need to monitor liver blood tests at baseline and then monthly for at least six months after initiating treatment with fitusiran or after a dose increase of fitusiran.[2]
History
The efficacy and safety of fitusiran were assessed in two multicenter, randomized clinical trials which enrolled a total of 177 adult and pediatric male participants with either hemophilia A or hemophilia B.[2] In one study, participants had inhibitory antibodies to coagulation factor VIII or coagulation factor IX and previously received on-demand treatment with medicines known as “bypassing agents” for bleeding.[2] In the second study, participants did not have inhibitory antibodies to coagulation factor VIII or coagulation factor IX and previously received on-demand treatment with clotting factor concentrates.[2] In the two randomized trials, participants received either a fixed dose of fitusiran monthly or their usual on-demand treatment (bypassing agents or clotting factor concentrates) as needed for nine months.[2] The fixed dose of fitusiran is not approved because it led to excessive clotting in some participants.[2]
The US Food and Drug Administration (FDA) granted the application for fitusiran orphan drug and fast track designations. The FDA granted the approval of Qfitlia to Sanofi.
Society and culture
Legal status
Fitusiran was approved for medical use in the United States in March 2025.[2][3]
Names
Fitusiran is the international nonproprietary name.[4]
Fitusiran is sold under the brand name Qfitlia.[1][2]
References
^ Jump up to:a b c d e f “Qfitlia- fitusiran injection, solution”. DailyMed. 26 March 2025. Retrieved 2 April 2025.
- ^ Jump up to:a b c d e f g h i j k l m “FDA Approves Novel Treatment for Hemophilia A or B, with or without Factor Inhibitors”. U.S. Food and Drug Administration. 28 March 2025. Retrieved 29 March 2025. This article incorporates text from this source, which is in the public domain.
- ^ “Qfitlia approved as the first therapy in the US to treat hemophilia A or B with or without inhibitors”. Sanofi (Press release). 28 March 2025. Retrieved 29 March 2025.
- ^ World Health Organization (2016). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 75”. WHO Drug Information. 30 (1). hdl:10665/331046.
Further reading
Srivastava A, Rangarajan S, Kavakli K, Klamroth R, Kenet G, Khoo L, et al. (May 2023). “Fitusiran prophylaxis in people with severe haemophilia A or haemophilia B without inhibitors (ATLAS-A/B): a multicentre, open-label, randomised, phase 3 trial”. The Lancet. Haematology. 10 (5): e322 – e332. doi:10.1016/S2352-3026(23)00037-6. PMID 37003278.
- Young G, Kavakli K, Klamroth R, Matsushita T, Peyvandi F, Pipe SW, et al. (March 2025). “Safety and efficacy of a fitusiran antithrombin-based dose regimen in people with hemophilia A or B: the ATLAS-OLE study”. Blood. doi:10.1182/blood.2024027008. PMID 40053895.
- Young G, Srivastava A, Kavakli K, Ross C, Sathar J, You CW, et al. (April 2023). “Efficacy and safety of fitusiran prophylaxis in people with haemophilia A or haemophilia B with inhibitors (ATLAS-INH): a multicentre, open-label, randomised phase 3 trial”. Lancet (London, England). 401 (10386): 1427–1437. doi:10.1016/S0140-6736(23)00284-2. PMID 37003287.
External links
- Clinical trial number NCT03417102 for “A Study of Fitusiran (ALN-AT3SC) in Severe Hemophilia A and B Patients With Inhibitors (ATLAS-INH)” at ClinicalTrials.gov
- Clinical trial number NCT03417245 for “A Study of Fitusiran (ALN-AT3SC) in Severe Hemophilia A and B Patients Without Inhibitors” at ClinicalTrials.gov
- Clinical trial number NCT03754790 for “Long-term Safety and Efficacy Study of Fitusiran in Patients With Hemophilia A or B, With or Without Inhibitory Antibodies to Factor VIII or IX (ATLAS-OLE)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Qfitlia |
| Other names | ALN-AT3SC |
| License data | US DailyMed: Fitusiran |
| Routes of administration | Subcutaneous |
| Drug class | Anthithrombin production inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 1499251–18–1 |
| DrugBank | DB15002 |
| UNII | SV9W47ZLE1 |
| KEGG | D11810 |
| Chemical and physical data | |
| Formula | C520H636F21N175Na43O309P43S6 |
| Molar mass | 17193.48 g·mol−1 |
////////Fitusiran, Qfitlia, FDA 2025, APPROVALS 2025, EX-A12034, DA-53206
Tibremciclib


Tibremciclib
cas 2397678-18-9, GTPL12881
CRB7BT5JDQ
518.6 g/mol, C28H32F2N8
N-[5-[(4-ethylpiperazin-1-yl)methyl]pyridin-2-yl]-5-fluoro-4-[(1R)-6-fluoro-1-methyl-1,2,3,4-tetrahydropyrido[1,2-a]benzimidazol-8-yl]pyrimidin-2-amine
Tibremciclib is a CDK4 inhibitor with antineoplastic activity[1].
- Originator Betta Pharmaceuticals Co Ltd
- Class Antineoplastics; Small molecules
- Mechanism of Action Cyclin-dependent kinase 4 inhibitors; Cyclin-dependent kinase 6 inhibitors
- Phase III Breast cancer; Solid tumours
13 Sep 2024 Efficacy and adverse event data from a phase III trial in Breast cancer presented at the 49th European Society for Medical Oncology Congress 2024 (ESMO-2024)
- 30 Jun 2023Phase-III clinical trials in Breast cancer (Metastatic disease, Late-stage disease, Combination therapy, Second-line therapy or greater) in China (PO) (NCT05433480)
- 02 Jun 2023Efficacy, adverse events and PK data from a phase I trial in Solid tumours presented at the 59th Annual Meeting of the American Society of Clinical Oncology (ASCO-2023)
Cyclin-dependent kinases (CDKs) are a class of serine / threonine protein kinases that participate in the regulation of the cell cycle, transcription initiation, and control of certain specific metabolic cascades. Different CDKs and cyclins form CDK-cyclin complexes. If the CDK activity is dysregulated, it will directly or indirectly cause uncontrolled cell proliferation, genomic instability (increased DNA mutation, chromosome deletion, etc.) and chromosomal instability (change in chromosome number). )Wait.
The CDKs family has identified more than 20 subtypes. CDK1, CDK2, CDK4, and CDK6 are involved in cell cycle regulation; CDK7, CDK8, CDK9, and CDK11 are involved in transcription regulation; and other kinases include CDK3 and CDK5. Among them, CDK4 / 6 (cyclin-dependent kinases 4 and 6) is a key factor in regulating the cell cycle. Cancer-related cell cycle mutations mainly exist in the G1 and G1 / S phase transformation. CDK4 / 6 binds to CyclinD A complex with kinase activity is formed and phosphorylation of the tumor suppressor gene Rb product pRb releases the bound transcription factor E2F to initiate transcription of genes related to the S phase, prompting cells to pass the checkpoint and transfer from the G1 phase to the S phase. The specific activation of CDK4 / 6 is closely related to the proliferation of some tumors. About 80% of human tumors have abnormalities in the cyclin D-CDK4 / 6-INK4-Rb pathway. CDK4 / 6 inhibitors block the cell cycle in the G1 phase, thereby inhibiting tumor proliferation.
The development of drugs targeting CDK4 / 6 kinases is a significant area. The advantages of anti-tumor targets are: (1) Most proliferating cells rely on CDK2 or CDK4 / 6 to proliferate, but CDK4 / 6 inhibitors do not show Cytotoxicity of “pan-CDK inhibitors”, such as bone marrow suppression and intestinal response; (2) Preclinical experiments show that if the level of cyclin D or the inactivation of P16INK4a can increase the sensitivity of cells to drugs, due to tumors Compared with normal cells, cells have the above phenomenon, so the targeting of drugs is increased to a certain extent.
PCT International Application PCT / CN2017 / 117950 describes a class of benzimidazole derivatives that are used as CDK4 / 6 protein kinase inhibitors, and most of these compounds effectively inhibit CDK4 and CDK6. Because there are still unmet needs in the treatment options for kinase-mediated diseases, here we further screen the salt forms and crystal forms of benzimidazole derivatives to meet the medical needs of patients.
SCHEME
SIDE CHAIN

SIDE CHAIN

MAIN

Patent
Betta Pharmaceuticals Co., Ltd., WO2019242719
https://patents.google.com/patent/WO2019242719A1/en


Synthesis of 1-A1-01 (Step 1)
In a 50L reactor, add 20L of dichloromethane (DCM), 1-A1-S1 (300g), and triethylamine (390g). While stirring, lower the temperature to below -5 ° C, and add benzyl chloroformate / Cbz- Cl (570 g) was added dropwise for 5 hours, and the temperature was naturally raised to room temperature. TLC (ethyl acetate: n-hexane = 1: 3) was monitored until the reaction was completed. Water (1.5 L) was added, and concentrated hydrochloric acid (80 mL) was slowly added dropwise to control the pH to 1-2. The solution was allowed to stand and separate. The organic phase was washed with 15 L of water, dried over anhydrous sodium sulfate for 0.5 hours, filtered to remove the desiccant, and collected the filtrate. And concentrated to obtain 730 g of light yellow oily liquid, which is crude 1-A1-01, yield 95.4%
Synthesis of 1-A1-02 (Step 2)
720mL of DCM, N, N-dimethylsulfoxide (90g) was added to a 20L reaction flask, protected by nitrogen, and the temperature was lowered below -65 ° C under stirring, and oxalyl chloride (106g) was added dropwise. The addition was completed in 2 hours. Stir for 20 minutes under heat preservation; add 1-A1-01’s dichloromethane solution (143g / 500mL DCM) dropwise. After 40 minutes, the addition is complete and the reaction is held for 15 minutes. Controlled at this temperature, TEA was added dropwise. After the addition was completed for 2 hours, the temperature was naturally raised to -20 ° C. 250 L of water was added to the system. The pH of the system was adjusted to 1-2 with hydrochloric acid. × 2) Washed, dried over anhydrous sodium sulfate, filtered to remove the desiccant, collected the filtrate and concentrated to obtain 432 g of a yellow oily liquid, which is the crude product 1-A1-02, which was directly used in the next reaction.
Synthesis of 1-A1-03 (Step 3)
In a stirred state, 400 mL of tetrahydrofuran (THF) and potassium tert-butoxide (215 g) were sequentially added to a 1 L reaction kettle, the temperature was lowered to 5-15 ° C., and triethyl phosphoryl acetate (430 g) was added dropwise. The dropwise addition was completed in 50 minutes. At a controlled temperature of 15 ° C, a tetrahydrofuran solution of 1-A1-02 (431 g / 100 mL of THF) was added dropwise. After the dropwise addition was completed for 1 hour, TLC (ethyl acetate: n-hexane = 1: 3) was monitored to complete the reaction, and the system was added. Saturated aqueous sodium chloride solution (1.5L), allowed to stand and separate, and collected the tetrahydrofuran phase; the aqueous phase was extracted with dichloromethane (2L), and the organic phases were combined and dried over anhydrous sodium sulfate for 0.5 hours, and the drying agent was removed by filtration. The filtrate was collected and concentrated, and the concentrate was purified by column chromatography to obtain 390 g of a pale yellow oily liquid, which was 1-A1-03 product.
Synthesis of 1-A1-041 (step 4)
In a 5L reactor, an aqueous solution of sodium hydroxide (301 g / 1.5 L of water) was added to a tetrahydrofuran (601 g / 2.3 L of THF) solution of 1-A1-03, and the mixture was heated to reflux for 3-4 hours to stop the reaction. The temperature was lowered to 40-50 ° C, and the layers were left to stand. The organic phase (THF) was collected and concentrated to a large amount of solids; the solids were dissolved by adding water (20L), and the aqueous phase was sequentially treated with methyl tert-butyl ether (2L) and ethyl acetate. Ester (2L), methyl tert-butyl ether (2L) washing; the aqueous phase was adjusted to pH 1-2 with concentrated hydrochloric acid, extracted twice with ethyl acetate (1.5L, 3L), the organic phases were combined, and anhydrous sulfuric acid was used Sodium was dried for 0.5 hours; the desiccant was removed by filtration, and the filtrate was collected and concentrated to a large amount of solids. The solids were added with isopropyl ether (3L) and slurried for 2 hours. The solids were collected by filtration and the solids were rinsed with isopropyl ether (1L). The solid was air-dried at 50 ° C for 3-4 hours to obtain 331 g of a pale yellow solid, which is a 1-A1-041 product with a yield of 52.7%.
Synthesis of 1-051 (step 5)
In a stirred state, 1-A1-041 (600g), methanol (25L), and concentrated sulfuric acid were added to a 50L reactor, and the reaction was heated under reflux for 3-4 hours. After the reaction was completed, the temperature was reduced to room temperature. Dichloromethane (15L) was added to the concentrate, and the pH was adjusted to 9-10 with an aqueous solution of potassium carbonate. The organic phase was collected by stirring, standing, and separating. The organic phase was dried over anhydrous sodium sulfate for 0.5 hours. The desiccant was removed by filtration and the filtrate was collected. And concentrated to obtain 6.37 kg of off-white solid, which is 1-A1-051 product, with a yield of 97.3%.
Synthesis of 1-A1 (step 6)
In a 2L hydrogenation kettle, add 1-A1-051 (500g), methanol (1.8L), and palladium on carbon. The system replaces nitrogen 3 times and hydrogen 3 times in sequence. The system maintains a hydrogen atmosphere, and the temperature is increased to 85 ° C and the pressure is 3.0. The reaction was carried out at Mpa for 3 hours, and the reaction was completed. The temperature was lowered to room temperature, the palladium on carbon was removed by filtration, and the organic phase was collected and concentrated until a large amount of light yellow solid appeared. Isopropyl ether (3L) was added to freeze (-20 ° C) for crystallization, and the solid product was collected by filtration. Ether (500 mL) was rinsed to obtain 234 g of a pale yellow solid, which was a 1-A1 product with a yield of 90.5%.
Synthesis of 1-A2 (Step 7)
In a stirred state, 1-A1 (200g), 4-bromo-2,6-difluoroaniline (410g), and toluene (1.2L) were added to a 50L reactor, and phosphorus oxychloride (413g) was added dropwise to the system. The addition was completed in 1 hour. Triethylamine was added dropwise under an ice bath, and the addition was completed in 1 hour. The temperature was raised to 110 ° C, and the reaction was performed for 1 hour. Reduce the temperature of the system to 2-10 ° C, add 1L of water, adjust the pH = 9-10 with saturated potassium carbonate aqueous solution, extract twice with ethyl acetate (1.5L, 1L), and combine the organic phases with 2L saturated sodium chloride aqueous solution. Wash, dry with anhydrous sodium sulfate for 0.5 hours, remove the desiccant by filtration, collect the filtrate and concentrate to the appearance of a solid product, add isopropyl ether (1L) to beat the solid for 10 minutes, filter, and collect 460 g of a yellow solid as a 1-A2 product.
Synthesis of 1-A3 (step eight)
Under stirring, 1-A2 (450g), N, N-dimethylformamide (2L), and cesium carbonate (700g) were added to the reaction kettle, and the reaction was heated to 110 ° C for 24 hours, and the reaction was detected by TLC. Ethyl acetate (3L) was added to the system, and solid impurities were removed by filtration. The filtrate was washed with a saturated sodium chloride aqueous solution (1L × 5), and the organic phase was dried over anhydrous sodium sulfate for 0.5 hours, concentrated to the appearance of a large amount of solid, Butyl ether (1L × 2) was beaten for 30 minutes, and 382 g of pale yellow solid product was obtained by filtration, that is, 1-A3, and the yield was 90.10%.
Synthesis of 1-01 (step 9)
With stirring, 1-A3 (380 g), pinacol diborate (400 g), potassium acetate (340 g), palladium acetate (6 g), tricyclohexyl phosphorus (7 g), and 1,4-dioxane were sequentially added. The ring was added to the reaction kettle, protected by nitrogen, and heated to 90 ° C for 2 hours. TLC was monitored until the reaction was complete. The temperature was reduced to room temperature, and the filtrate was concentrated to remove a large amount of 1,4-dioxane. The concentrate was purified by n-hexane and dichloromethane column chromatography, and n-hexane (1.2 L) was slurried for 1 hour to obtain 334 g of a gray solid. That is 1-01, and the yield is 70.10%.
Synthesis of 1-02 (step 10)
Under stirring, take 1-01 (128g), 1,4-dioxane (1L), 1-S3 (85g), potassium carbonate (110g), and purified water and add them to a 2L three-necked flask in sequence. [1,1′-Bis (diphenylphosphine) ferrocene] palladium dichloromethane complex (Pd (dppf) Cl 2 .DCM) was added. The temperature was raised to 60 ° C. After 4 hours of reaction, the reaction was complete. The reaction solution was cooled to room temperature, and concentrated under reduced pressure to remove most of 1,4-dioxane. Dichloromethane (1.5 L) and purified water (1.1 L) were added, stirred, and allowed to stand and separate. The layers were separated, and water was added. The phases were extracted with dichloromethane (10 L), the organic phases were combined, washed with 0.5% dilute hydrochloric acid (1 L x 2), saturated aqueous sodium chloride solution (1 L), and the layers were separated. The organic phase was dried over anhydrous sodium sulfate (500 g), filtered to remove the drying agent, and the filtrate was concentrated under reduced pressure. Ethyl acetate (0.5 L) was added to the concentrate and the mixture was stirred for 30 minutes to precipitate a solid. After filtration, the obtained solid was rinsed with ethyl acetate (0.5 L) and dried under vacuum at 45 ° C for 3 hours to obtain 120 g of a yellow solid.
Synthesis of 1-03 (step 11)
Under stirring, take 1-02 (100g), 1,4-dioxane (1L), 1-C2 (80g), and cesium carbonate (163g) into a 2L three-necked bottle in sequence, protected by nitrogen, and add palladium acetate ( 2g) and 4,5-bisdiphenylphosphine-9,9-dimethylxanthracene (Xantphos) (4g), heated to 85 ° C. until the reaction was complete. The reaction solution was cooled to room temperature and filtered to obtain a solid product. The solid was rinsed with ethyl acetate, and then added to a mixed system of dichloromethane (1.5 L) and purified water (1.1 L), stirred, allowed to stand, and separated into layers. The aqueous phase was extracted with dichloromethane (700 mL). The organic phases were combined and washed with purified water (700 mL x 2). The organic phase was dried by adding anhydrous sodium sulfate (700 g), filtered to remove the desiccant, and the filtrate was concentrated. Methanol (0.5 L) was added, heated to 55-65 ° C. and stirred for 0.5 hours, lowered to room temperature, and filtered. The solid product was filtered and rinsed with 500 mL of ethyl acetate. The solid was dried under vacuum at 45 ° C for 8 hours to obtain 111.79 g of a pale yellow solid 1-03.
Synthesis of compound II (step twelve)
Under stirring, take 1-03 (500g) and anhydrous methanol (3.8L), add them to a 10L reactor in sequence, and heat to 65 ° C. After the reaction system is clarified for 0.5 hours, add L-tartaric acid in methanol (150.89) dropwise. g of tartaric acid is dissolved in 500mL of anhydrous methanol), and the dropping time is controlled to be 45 to 60 minutes. After the addition is complete, the reaction is kept at 65 ° C for 4 hours. ), Control the dropwise addition time to 30 to 45 minutes. After the dropwise addition is complete, hold the reaction at 65 ° C for 1 hour. Continue to dropwise add L-tartaric acid in methanol (36.55g of tartaric acid dissolved in 250mL of anhydrous methanol) and control the dropwise addition time to 30. To 45 minutes, the dropwise addition was completed. The temperature was kept at 65 ° C for 1.5 hours, and the heating was stopped. The temperature was naturally lowered to 20-30 ° C, filtered, the filter cake was rinsed with methanol (400mL × 2), and dried at 45 ° C under vacuum for 36 hours. 530.64 g of crystalline powder was Compound II, which was identified by X-ray powder diffraction, and showed that the crystal form was Form A of Compound II.
WO2022199656
WO2023131179
///Tibremciclib, GTPL12881, BETTA, PHASE 3, CANCER
Gepotidacin




Gepotidacin
CAS
1075236-89-3 |
GSK2140944
WeightAverage: 448.527
Monoisotopic: 448.222288786 Chemical FormulaC24H28N6O3
(3R)-3-({4-[({2H,3H,4H-pyrano[2,3-c]pyridin-6-yl}methyl)amino]piperidin-1-yl}methyl)-1,4,7-triazatricyclo[6.3.1.0^{4,12}]dodeca-6,8(12),9-triene-5,11-dione
FDA APPROVED 3/25/2025,Blujepa, To treat uncomplicated urinary tract infections
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Gepotidacin hydrochloride | 30Z5B7ACV6 | 1075235-46-9 | DPAHPKBTWARMFG-FSRHSHDFSA-N |
| Gepotidacin mesylate | 5P7X0H2O6B | 1624306-20-2 | MTLHHQWYERWLIX-RGFWRHHQSA-N |
Gepotidacin, sold under the brand name Blujepa, is an antibiotic medication used for the treatment of urinary tract infection.[1] Gepotidacin is a triazaacenaphthylene bacterial type II topoisomerase inhibitor.[1][2] It is used as the salt gepotidacin mesylate, and is taken by mouth.[1]
Gepotidacin was approved for medical use in the United States in March 2025.[1][3]
SYNTHESIS
Gepotidacin
Gepotidacin (GSK2140944) is a triazaacenaphtylene developed by GSK and belongs to the class of Novel Bacterial Topoisomerase Inhibitors (NBTI). This new antibiotic is currently being investigated in three phase 3 clinical trials.
Gepotidacin is derived from the analogue GSK299423 described by Bax et al. [9], which results from a medicinal chemistry program initiated after an unbiased antibacterial screening [10].
2.2.1 Chemical synthesis
The synthesis of gepotidacin has been described in two patents in 2008 and 2016 and comprises 11 steps (Fig. 2) [11,12]. First, 2-chloro-6-methoxy-3-nitro-pyridine reacts with 2-amino-propane-1,3-diol through nucleophilic aromatic substitution (SNAr). The resulting diol is then protected with 2,2-dimethoxypropane in presence of p-toluenesulfonic acid (PTSA) followed by the reduction of the nitro group with hydrogen and 10% Pd/C. The aniline thus formed is then alkylated with ethyl bromoacetate. Cyclization is performed in basic conditions using sodium hydride, followed by oxidation using manganese dioxide. The acetal is then cleaved and the released diol reacts with methanesulfonic anhydride to form the third cycle of the triazaacenaphtylene core. Substitution with Boc-amino-piperidine, followed by deprotection and subsequent purification by chiral chromatography affords the primary amine derivative, which can be condensed by reductive amination with the corresponding aldehyde to give the free base of gepotidacin. The mono-hydrochloride salt is obtained by reaction with one equivalent of HCl 1 M in diethylether [13].

PATENT
WO2021219637A1
https://patents.google.com/patent/WO2021219637A1/en
Gepotidacin mesylate dihydrate (Form 1)
Example la – Preparation Method 1
Acetone (5 ml) was added to gepotidacin (294.14 mg). To the slurry, methanesulfonic acid (3M solution in water, 1 equivalent) was added over a period of 60 minutes. The slurry was heated to 50°C for 3 hours, cooled slowly to 20°C, left stirring at 20°C for 5 hours and cooled further to 5°C. The slurry was stirred at 5°C overnight. The crystalline solids were filtered under vacuum, washed with acetone and dried in a vacuum oven at 60°C to give crystalline gepotidacin mesylate dihydrate (Form 1) in 72.9% yield.
References:
GLAXO GROUP LIMITED WO2008/128942, 2008, A1Yield:-
Steps:
Multi-step reaction with 12 steps
1.1: ethanol; water / 4 h / 0 °C / Heating / reflux
2.1: toluene-4-sulfonic acid / 20 °C
2.2: 0.33 h
3.1: hydrogen / palladium 10% on activated carbon / 1,4-dioxane / 20 °C / 760.05 Torr
4.1: potassium carbonate / N,N-dimethyl-formamide / 20 °C
5.1: sodium hydride / tetrahydrofuran / 3.25 h / 0 – 20 °C
6.1: manganese(IV) oxide / dichloromethane / 2 h / 20 °C
7.1: hydrogenchloride; water / tetrahydrofuran / 1 h / 20 °C
7.2: pH ~ 8
8.1: triethylamine / chloroform / 4.5 h / Heating / reflux
9.1: pyridine / acetonitrile / 5 h / 50 – 90 °C
10.1: hydrogenchloride / 1,4-dioxane; dichloromethane / 1 h / 20 °C
11.1: isopropylamine / methanol; acetonitrile / Resolution of racemate
12.1: methanol; chloroform / 20 °C
12.2: 0.5 h / 20 °C
Example 10 (lR)-l-({4-[(3,4-Dihydro-2H-pyrano[2,3-c]pyridin-6-ylmethyl)amino]- l-piperidinyl}methyl)-l,2-dihydro-4H,9H-imidazo[l,2,3-//]-l,8-naphthyridine-4,9- dione hydrochloride

A suspension of (\R)- 1 -[(4-amino- 1 -piperidinyl)methyl]- 1 ,2-dihydro-4Η,9Η- imidazo[l,2,3-ij]-l,8-naphthyridine-4,9-dione (for a preparation see Example 5(j)) (51 mg, 0.14 mmol) in chloroform:methanol (9:1, 3 ml) at rt under argon was treated with triethylamine (0.06ml) and stirred at rt for 10 min. The solution was then treated with 1,3- dihydrofuro[3,4-c]pyridine-6-carbaldehyde (for a synthesis see WO2004058144,
Example 126(e)) (21mg, 0.133mmol) and stirred for a further 2h. The solution was then treated with NaBH(OAc)3 (87mg) and stirred at rt for 2h. The reaction was then treated with saturated aqueous NaHCO (10ml) and extracted with 20% methanol/DCM (3 x 50ml). The combined organic extracts were dried (MgSO ), filtered, evaporated and chromatographed (0-20% methanol/DCM) to give the free base of the title compound as a light brown solid (20mg, 32%) MS (ES+) m/z 448 (MH+). δH (CDCl3, 400MHz) 1.15-1.49 (2H, m), 1.61-1.95 (2H, m), 1.99-2.09 (2H, m) 2.20-2.38 (IH, m), 2.45-2.85 (6H, m), 2.92-3.02(1H, m), 3.05-3.15 (IH, m), 3.78 (2H, s), 4.20 (2H, t), 4.30-4.42 (IH, m), 4.52-4.61 (IH, m), 4.95-5.05 (IH, m), 6.23-6.32 (2H, m), 7.00 (IH, s), 7.47-7.50 (2H, m), 8.07 (IH, s).
The free base in DCM was treated with one equivalent IM HCl in diethyl ether and then evaporated to give the title monohydrochloride salt.
PATENT
WO2004058144
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2004058144&_cid=P20-M9AS9E-95245-1
Medical uses
Gepotidacin is indicated for the treatment of females aged twelve years of age and older weighing at least 40 kilograms (88 lb) with uncomplicated urinary tract infections (uUTI) caused by Escherichia coli, Klebsiella pneumoniae, Citrobacter freundii complex, Staphylococcus saprophyticus, and Enterococcus faecalis.[1]
Society and culture
Legal status
In October 2024, gepotidacin was granted priority review by the US Food and Drug Administration (FDA) for the treatment of uncomplicated urinary tract infections.[4]
Gepotidacin was approved for medical use in the United States in March 2025.[1][5]
Names
Gepotidacin is the international nonproprietary name.[6]
Gepotidacin is sold under the brand name Blujepa.[1][5]
Research
Gepotidacin is being studied for the treatment of Neisseria gonorrhoeae (gonorrhea) infection, including multidrug resistant strains.[7][8]
References
- ^ Jump up to:a b c d e f g h “Blujepa- gepotidacin tablet, film coated”. DailyMed. 25 March 2025. Retrieved 2 April 2025.
- ^ Biedenbach DJ, Bouchillon SK, Hackel M, Miller LA, Scangarella-Oman NE, Jakielaszek C, et al. (January 2016). “In Vitro Activity of Gepotidacin, a Novel Triazaacenaphthylene Bacterial Topoisomerase Inhibitor, against a Broad Spectrum of Bacterial Pathogens”. Antimicrobial Agents and Chemotherapy. 60 (3): 1918–1923. doi:10.1128/aac.02820-15. PMC 4776004. PMID 26729499.
- ^ Fick M, Sneha SK, Sunny ME (2025). “FDA approval”. Reuters.
- ^ “GSK’s investigational antibiotic granted FDA priority review for urinary tract infections”. PMLiVE. 18 October 2024. Retrieved 21 October 2024.
- ^ Jump up to:a b “Blujepa (gepotidacin) approved by US FDA for treatment of uncomplicated urinary tract infections (uUTIs) in female adults and pediatric patients 12 years of age and older”. GSK (Press release). 25 March 2025. Retrieved 28 March 2025.
- ^ World Health Organization (2015). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 74”. WHO Drug Information. 29 (3). hdl:10665/331070.
- ^ Scangarella-Oman NE, Hossain M, Dixon PB, Ingraham K, Min S, Tiffany CA, et al. (December 2018). “Microbiological Analysis from a Phase 2 Randomized Study in Adults Evaluating Single Oral Doses of Gepotidacin in the Treatment of Uncomplicated Urogenital Gonorrhea Caused by Neisseria gonorrhoeae“. Antimicrobial Agents and Chemotherapy. 62 (12). doi:10.1128/AAC.01221-18. PMC 6256812. PMID 30249694.
- ^ Jacobsson S, Golparian D, Scangarella-Oman N, Unemo M (August 2018). “In vitro activity of the novel triazaacenaphthylene gepotidacin (GSK2140944) against MDR Neisseria gonorrhoeae“. The Journal of Antimicrobial Chemotherapy. 73 (8): 2072–2077. doi:10.1093/jac/dky162. PMC 6927889. PMID 29796611.
Further reading
- Wagenlehner F, Perry CR, Hooton TM, Scangarella-Oman NE, Millns H, Powell M, et al. (February 2024). “Oral gepotidacin versus nitrofurantoin in patients with uncomplicated urinary tract infection (EAGLE-2 and EAGLE-3): two randomised, controlled, double-blind, double-dummy, phase 3, non-inferiority trials”. Lancet. 403 (10428): 741–755. doi:10.1016/S0140-6736(23)02196-7. PMID 38342126. S2CID 267548740.
External links
- Clinical trial number NCT04020341 for “A Study to Evaluate Efficacy and Safety of Gepotidacin in the Treatment of Uncomplicated Urinary Tract Infection (UTI)” at ClinicalTrials.gov
- Clinical trial number NCT04187144 for “Comparative Study to Evaluate Efficacy and Safety of Gepotidacin to Nitrofurantoin in Treatment of Uncomplicated Urinary Tract Infection (UTI)” at ClinicalTrials.gov
- Ross JE, Scangarella-Oman NE, Flamm RK, Jones RN: Determination of disk diffusion and MIC quality control guidelines for GSK2140944, a novel bacterial type II topoisomerase inhibitor antimicrobial agent. J Clin Microbiol. 2014 Jul;52(7):2629-32. doi: 10.1128/JCM.00656-14. Epub 2014 Apr 23. [Article]
- Oviatt AA, Gibson EG, Huang J, Mattern K, Neuman KC, Chan PF, Osheroff N: Interactions between Gepotidacin and Escherichia coli Gyrase and Topoisomerase IV: Genetic and Biochemical Evidence for Well-Balanced Dual-Targeting. ACS Infect Dis. 2024 Apr 12;10(4):1137-1151. doi: 10.1021/acsinfecdis.3c00346. Epub 2024 Mar 5. [Article]
- GSK Press Release: Blujepa (gepotidacin) approved by US FDA for treatment of uncomplicated urinary tract infections (uUTIs) in female adults and paediatric patients 12 years of age and older [Link]
- FDA Approved Drug Products: Blujepa (gepotidacin) tablets for oral use (March 2025) [Link]
| Clinical data | |
|---|---|
| Trade names | Blujepa |
| Other names | GSK2140944 |
| AHFS/Drugs.com | Blujepa |
| License data | US DailyMed: Gepotidacin |
| Routes of administration | By mouth |
| ATC code | J01XX13 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1075236-89-3 |
| DrugBank | DB12134 |
| ChemSpider | 34982930 |
| UNII | DVF0PR037D5P7X0H2O6B |
| KEGG | D10878D10879 |
| ECHA InfoCard | 100.249.088 |
| Chemical and physical data | |
| Formula | C24H28N6O3 |
| Molar mass | 448.527 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
///////Gepotidacin, FDA 2025, APPROVALS 2025, Blujepa, GSK-2140944, GSK2140944
Vimseltinib


Vimseltinib
1628606-05-2 |
2/14/2025 FDA APPROVED, Romvimza
3-methyl-5-[6-methyl-5-[2-(1-methylpyrazol-4-yl)pyridin-4-yl]oxypyridin-2-yl]-2-(propan-2-ylamino)pyrimidin-4-one
C23H25N7O2, 431.5
- DP-6865
- PX9FTM69BF
- DCC3014
- UNII-PX9FTM69BF
- WHO 11443
DCC-3014- DP-6865
To treat symptomatic tenosynovial giant cell tumor for which surgical resection will potentially cause worsening functional limitation or severe morbidity
Vimseltinib is an orally bioavailable inhibitor of the tyrosine kinase receptor colony stimulating factor 1 receptor (CSF1R; CSF-1R; C-FMS; CD115; M-CSFR), with potential antineoplastic, macrophage checkpoint-inhibitory and immunomodulating activities. Upon administration, vimseltinib targets and binds to CSF1R expressed on monocytes, macrophages, and osteoclasts and inhibits the binding of the CSF1R ligands colony-stimulating factor-1 (CSF-1) and interleukin-34 (IL-34), to CSF1R. This prevents CSF1R activation and CSF1R-mediated signaling in these cells. This blocks the production of inflammatory mediators by macrophages and monocytes and reduces inflammation. By blocking the recruitment to the tumor microenvironment (TME) and activity of CSF1R-dependent tumor-associated macrophages (TAMs), vimseltinib inhibits the immunomodulating activity by macrophages and enhances T-cell infiltration and anti-tumor T-cell immune responses, which inhibits the proliferation of tumor cells. TAMs play key roles in the TME and allow for immune suppression; TAMs promote inflammation, tumor cell proliferation, angiogenesis, invasiveness and survival.
Vimseltinib, sold under the brand name Romvimza, is an anti-cancer medication used for the treatment of tenosynovial giant cell tumor.[1][2] Vimseltinib is a kinase inhibitor.[1][2] Vimseltinib is a macrophage colony-stimulating factor receptor antagonist.[3]
The most common adverse reactions, including laboratory abnormalities, include increased aspartate aminotransferase, periorbital edema, fatigue, rash, increased cholesterol, peripheral edema, face edema, decreased neutrophils, decreased leukocytes, pruritus, and increased alanine aminotransferase.[2]
Vimseltinib was approved for medical use in the United States in February 2025.[2][4]
PATENT
vimseltinib is a c-FMS (CSF-IR) and c-KIT dual inhibitor with anticancer and antiproliferative activities, can excite tyrosine protein kinase activity, influence protooncogene transcription, and is widely applied to research of anticancer drugs as an active molecule.
CN105120864B discloses heating the reaction mixture in a sealed tube at 100 ℃ for 2 days. The mixture was then cooled to room temperature, the solids were removed by filtration and the filtrate was concentrated to dryness and purified by silica gel chromatography to give 2- (isopropylamino) -3-methyl-5- (6-methyl-5- ((2- (1-methyl-1H-pyrazol-4-yl) pyridin-4-yl) oxy) pyridin-2-yl) pyrimidin-4 (3H) -one, amorphous form described.
CN113880812a reports another preparation method of Vimseltinib, and a small amount of target product meeting the requirement is finally obtained through a column chromatography purification process. The preparation method has complicated process and is not beneficial to industrialized mass production. There is no mention in this patent of reports on solid or crystalline forms of the compound of formula (I), and the purification process of column chromatography (EA/meoh=120:1 to 100:1) was repeated to give form a.
CN116283919A
https://patents.google.com/patent/CN116283919A/en
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014145025&_cid=P21-M98JKR-94364-1
Example 2: A solution of Example C2 (0.13 g, 0.309 mmol) in DCM (5 mL) was treated portion-wise with mCPBA (0.09 g, 0.37 mmol), stirred at RT overnight, treated with TEA (0.5 mL) and Ν,Ν-dimethylamine HCl salt (500 mg) and stirred at RT for 2 h. The mixture was treated with satd. NaHCO3, extracted with DCM (2x) and the combined organics were dried over Na2SO4, concentrated to dryness and purified via silica gel chromatography (MeOH/DCM) to obtain 4-methoxy-N,N-dimethyl-5-(6-methyl-5-((2-(1-methyl-1H-pyrazol-4-yl)pyridin-4-yl)oxy)pyridin-2-yl)pyrimidin-2-amine (60 mg, 47%). MS (ESI) m/z: 418.2 (M+H+).
[0199] A solution of 4-methoxy-N,N-dimethyl-5-(6-methyl-5-((2-(1-methyl-1H-pyrazol-4-yl)pyridin-4-yl)oxy)pyridin-2-yl)pyrimidin-2-amine (0.060 g, 0.144 mmol) in acetic acid (5 mL) was treated with HBr (0.065 mL, 0.575 mmol), heated at 90°C for 6 h, cooled to RT and quenched with ice water. The solution was treated with NaHCO3 and NaCl, extracted with 1 : 1 THF/EtOAc (3x) and the combined organics were dried over Na2SO4 and concentrated to dryness. The material was treated with MeCN (1 mL), allowed to stand at RT and the
resulting solid was collected via filtration to afford 2-(dimethylamino)-5-(6-methyl-5-((2-(1-methyl-1H-pyrazol-4-yl)pyridin-4-yl)oxy)pyridin-2-yl)pyrimidin-4(3H)-one (43 mg, 71%). 1H NMR (400 MHz, DMSO-d6): δ 11.23 (s, 1 H), 8.73 (s, 1 H), 8.36 (d, J = 5.7 Hz, 1 H), 8.30 (m, 1H), 8.26 (s, 1 H), 7.97 (s, 1 H), 7.51 (m, 1H), 7.23 (d, J = 2.4 Hz, 1 H), 6.62 (br s, 1 H), 3.85 (s, 3 H), 3.12 (s, 6 H), 2.35 (s, 3 H); MS (ESI) m/z: 404.2 (M+H+).
Example 3: A solution of Example C2 (0.13 g, 0.309 mmol) in DCM (5 mL) was treated portion-wise with mCPBA (0.09 g, 0.37 mmol), stirred at RT overnight, treated with isopropyl amine (0.5 mL) and stirred at RT overnight. The mixture was treated with satd. NaHCO3, extracted with DCM (2x) and the combined organics were dried over Na2SO4, concentrated to dryness and purified via silica gel chromatography (MeOH/DCM) to obtain N-isopropyl-4-methoxy-5-(6-methyl-5-((2-(1-methyl-1H-pyrazol-4-yl)pyridin-4-yl)oxy)pyridin-2-yl)pyrimidin-2-amine (63 mg, 47%). MS (ESI) m/z: 432.2 (M+H+).
PAPER
Discovery of vimseltinib (DCC-3014), a highly selective CSF1R switch-control kinase inhibitor, in clinical development for the treatment of Tenosynovial Giant Cell Tumor (TGCT)
https://www.sciencedirect.com/science/article/pii/S0960894X22004048

Medical uses
Vimseltinib is indicated for the treatment of adults with symptomatic tenosynovial giant cell tumor for which surgical resection will potentially cause worsening functional limitation or severe morbidity.[1][2]
History
The efficacy of vimseltinib was evaluated in MOTION (NCT05059262), a double-blind, multicenter, randomized (2:1), placebo-controlled trial in participants with tenosynovial giant cell tumor for whom surgical resection may cause worsening functional limitation or severe morbidity.[2] Eligible participants had a confirmed diagnosis of tenosynovial giant cell tumor with measurable disease (RECIST v1.1) with at least one lesion having a minimum size of 2 cm.[2] Pp[-[p;articipants were randomized to placebo or vimseltinib, 30 mg twice weekly administered for 24 weeks, during the double-blind period (part 1).[2] During the open-label period (part 2), patients could continue vimseltinib and those receiving placebos could crossover to vimseltinib.[2] Randomization was stratified by tumor location (lower limb versus all other) and region (United States versus Non-US).[2] A total of 123 participants were randomized: 83 to the vimseltinib arm and 40 to placebo during part 1.[2]
The US. Food and Drug Administration (FDA) granted the application for vimseltinib priority review designation.[2]
Society and culture
Legal status
Vimseltinib was approved for medical use in the United States in February 2025.[2][5]
Names
Vimseltinib is the international nonproprietary name.[6]
Vimseltinib is sold under the brand name Romvimza.[1][2]
References
- ^ Jump up to:a b c d e “Romvimza- vimseltinib capsule”. DailyMed. 18 February 2025. Retrieved 3 March 2025.
- ^ Jump up to:a b c d e f g h i j k l m n “FDA approves vimseltinib for symptomatic tenosynovial giant cell tumor”. U.S. Food and Drug Administration (FDA). 14 February 2025. Retrieved 16 February 2025. This article incorporates text from this source, which is in the public domain.
- ^ Caldwell TM, Ahn YM, Bulfer SL, Leary CB, Hood MM, Lu WP, et al. (October 2022). “Discovery of vimseltinib (DCC-3014), a highly selective CSF1R switch-control kinase inhibitor, in clinical development for the treatment of Tenosynovial Giant Cell Tumor (TGCT)”. Bioorganic & Medicinal Chemistry Letters. 74: 128928. doi:10.1016/j.bmcl.2022.128928. PMID 35961460.
- ^ “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 21 February 2025. Retrieved 9 March 2025.
- ^ “U.S. FDA Grants Full Approval of Deciphera’s Romvimza (vimseltinib) for the Treatment of Symptomatic Tenosynovial Giant Cell Tumor (TGCT)” (Press release). Deciphera Pharmaceuticals. 14 February 2025. Retrieved 16 February 2025 – via Business Wire.
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 85”. WHO Drug Information. 35 (1). hdl:10665/340684.
External links
- Clinical trial number NCT05059262 for “Study of Vimseltinib for Tenosynovial Giant Cell Tumor (MOTION)” at ClinicalTrials.gov
- Caldwell TM, Ahn YM, Bulfer SL, Leary CB, Hood MM, Lu WP, Vogeti L, Vogeti S, Kaufman MD, Wise SC, Le Bourdonnec B, Smith BD, Flynn DL: Discovery of vimseltinib (DCC-3014), a highly selective CSF1R switch-control kinase inhibitor, in clinical development for the treatment of Tenosynovial Giant Cell Tumor (TGCT). Bioorg Med Chem Lett. 2022 Oct 15;74:128928. doi: 10.1016/j.bmcl.2022.128928. Epub 2022 Aug 10. [Article]
- Smith BD, Kaufman MD, Wise SC, Ahn YM, Caldwell TM, Leary CB, Lu WP, Tan G, Vogeti L, Vogeti S, Wilky BA, Davis LE, Sharma M, Ruiz-Soto R, Flynn DL: Vimseltinib: A Precision CSF1R Therapy for Tenosynovial Giant Cell Tumors and Diseases Promoted by Macrophages. Mol Cancer Ther. 2021 Nov;20(11):2098-2109. doi: 10.1158/1535-7163.MCT-21-0361. Epub 2021 Aug 25. [Article]
- FDA Approved Drug Products: Romvimza (vimseltinib) capsules for oral use (February 2025) [Link]
- FDA News Release: FDA approves vimseltinib for symptomatic tenosynovial giant cell tumor [Link]
| Clinical data | |
|---|---|
| Trade names | Romvimza |
| License data | US DailyMed: Vimseltinib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1628606-05-2 |
| PubChem CID | 86267612 |
| IUPHAR/BPS | 11190 |
| DrugBank | DB17520 |
| ChemSpider | 95499700 |
| UNII | PX9FTM69BF |
| KEGG | D12238 |
| ChEMBL | ChEMBL5095202 |
| Chemical and physical data | |
| Formula | C23H25N7O2 |
| Molar mass | 431.500 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
//////Vimseltinib, FDA 2025, APPROVALS 2025, Romvimza, DCC-3014, DCC 3014, DP-6865, PX9FTM69BF, C3014, WHO 11443, DCC-3014, DP-6865,
TREOSULFAN



TREOSULFAN
C6H14O8S2 MW 278.29
FDA APPROVED 1/21/2025 Grafapex
CAS
299-75-2 |
299-75-2
Treosulphan
Ovastat
Treosulfano
NSC-39069
- Dihydroxybusulfan
- L-threitol-1,4-dimethanesulfonate
[(2S,3S)-2,3-dihydroxy-4-methylsulfonyloxybutyl] methanesulfonate
Trecondi, Treosulfan was authorized for medical use in the European Union in June 2019
For use in combination with fludarabine as a preparative regimen for allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia and myelodysplastic syndrome
Treosulfan, sold under the brand name Trecondi among others, is an alkylating medication given to people before they have a bone marrow transplant from a donor known as allogeneic hematopoietic stem cell transplantation. It is used as a ‘conditioning’ treatment to clear the bone marrow and make room for the transplanted bone marrow cells, which can then produce healthy blood cells.[9][10] It is used together with another medicine called fludarabine in adults and children from one month of age with blood cancers as well as in adults with other severe disorders requiring a bone marrow transplant.[9] It belongs to the family of drugs called alkylating agents.[9] In the body, treosulfan is converted into other compounds called epoxides which kill cells, especially cells that develop rapidly such as bone marrow cells, by attaching to their DNA while they are dividing.[9]
The most common side effects include infections, nausea (feeling sick), stomatitis (inflammation of the lining of the mouth), vomiting, diarrhea, and abdominal pain (belly ache).[9] Tiredness, febrile neutropenia (low white blood cell counts with fever) and high blood levels of bilirubin (a breakdown product of red blood cells) are also seen in more than 1 in 10 adults, and rash also affects more than 1 in 10 children.[9] The most common adverse reactions include musculoskeletal pain, stomatitis, pyrexia, nausea, edema, infection, and vomiting.[7] Selected grade 3 or 4 nonhematological laboratory abnormalities include increased GGT, increased bilirubin, increased ALT, increased AST, and increased creatinine.[7]
Treosulfan was authorized for medical use in the European Union in June 2019,[9] and approved for medical use in the United States in January 2025.[7][11]
Medical Uses
Treosulfan in combination with fludarabine is indicated as part of conditioning treatment prior to allogeneic haematopoietic stem cell transplantation in adults with malignant and non malignant diseases, and in children older than one month with malignant diseases.[7][9]
History
Two main studies showed that treosulfan is at least as effective as busulfan, another medicine used to prepare people for haematopoietic stem cell transplantation.[9]
In one of the studies, involving 570 adults with acute myeloid leukaemia (a blood cancer) or myelodysplastic syndromes (conditions in which large numbers of abnormal blood cells are produced), 64% of patients given treosulfan (with fludarabine) had a successful transplant and were alive and disease-free after 2 years, compared with 51% of patients given busulfan (with fludarabine).[9]
In an additional study in 70 children with blood cancers, 99% of children given treosulfan (with fludarabine) were alive three months after their transplant.[9]
Efficacy was evaluated in MC-FludT.14/L Trial II (NCT00822393), a randomized active-controlled trial comparing treosulfan to busulfan with fludarabine as a preparative regimen for allogeneic transplantation. Eligible patients included adults 18 to 70 years old with AML or MDS, Karnofsky performance status ≥ 60%, and age ≥ 50 years or hematopoietic cell transplantation comorbidity index [HCTCI] score > 2. There were 570 patients randomized to treosulfan (n=280) or busulfan (n=290).
Society and culture
Legal status
Treosulfan was authorized for medical use in the European Union in June 2019,[9] and approved for medical use in the United States in January 2025.[11][12][13]
The US Food and Drug Administration granted orphan drug designation to treosulfan in 1994, for the treatment of ovarian cancer;[14] and in 2015, for conditioning treatment prior to hematopoietic stem cell transplantation in malignant and non-malignant diseases in adults and pediatric patients.[15]
In February 2004, orphan designation (EU/3/04/186) was granted by the European Commission to medac Gesellschaft fuer klinische Spezialpräparate mbH, Germany, for treosulfan for the conditioning treatment prior to haematopoietic progenitor cell transplantation.[16]
Names
Treosulfan is the international nonproprietary name.[17]
Treosulfan is sold under the brand names Trecondi[9] and Grafapex.[7]
SYN
Treosulfan is an active ingredient of the drug Ovastat . Treosulfan is indicated for the treatment of ovarian cancer and belongs to the class of alkylating agents, which prevents the growth and division of cancerous cells.
US3155702 discloses the preparation of Treosulfan by methanesulphonation of (2S,3S)- l,4-dibromobutane-2,3-diol with excess amount of silver methanesulphonate. The presence of free 2,3-diol in the starting material leads to side reactions and formation of undesired by-products which necessitates an additional purification step and thereby results in lower yields. Further, an additional filtration operation is also required to remove silver bromide salt generated during the process and un-reacted silver methanesulphonate, which makes the process less attractive for commercial manufacturing.
US3246012 discloses the preparation of Treosulfan by protection of hydroxyl group of dialkyl tartrates with corresponding aldehyde, ketone or a reactive derivatives to form corresponding cyclic 2,3-O-acetals and 2,3-O-ketals of butanetetrol esters followed by reduction using lithium aluminium hydride to obtain 2,3-O-acetal or ketal protected butanetetrol, which is further methanesulphonated and treated with acid. The use of highly pyrophoric and hazardous reducing agent renders the above process not ideal for industrial production. Organic Syntheses, Coll. Vol. 10, p. 297, 2004 discloses a similar reaction sequence followed by the final de-protection of methanesulphonated 2,3-O-diisopropylidene-L- threitol in methanesulfonic acid at reflux temperature, which leads to a sluggish reaction mixture and a higher number of impurities due to maintaining the reaction mixture for longer time at higher temperature.
IN 1568/MUM/2012 also discloses similar reaction sequence involving reduction of dimethyl-2,3-0-isopropylidene-L-tartrate by sodium-bis(2-methoxyethoxy) aluminium hydride followed by methanesulphonation and final deprotection with formic acid to yield Treosulfan.
KR101367641 describes reduction using lithium borohydride, which requires about 14 hours to complete the reaction and is further extended due to involvement of column chromatography purification. Tetrahedron, vol. 49, no. 30, p. 6645, 1993 describes reduction using sodium borohydride and lithium chloride, followed by flash chromatography purification. Reduction conditions as per Chem. Pharm. Bull. Vol. 42, No. 3, p. 68, 1994, are again not commercially feasible because of lithium aluminium hydride as reducing agent.
Haberland, M., Weber, S., Sharma, A. K., Upadhyay, S., Dua, H., Musmade, S., Singh, G., Lahiri, S., & Cabri, W. (2019). A process for the preparation of Treosulfan (Patent No. WO2019043587A2).
EXAMPLES Detailed experimental parameters suitable for the preparation of Treosulfan or intermediates according to the present invention are provided by the following examples, which are intended to be illustrative and not limiting.
Reference Example 1 (repetition of Tetrahedron, vol. 46, No. 12, p. 4165, 1990):
A reaction mixture of dimethyl-L-tartrate (10. Og), p-toluene sulfonic acid (0.013g) and p- anisaldehydedimethylacetal (l l.Og) in toluene (150ml) was refluxed and the azeotropical mixture of toluene-methanol was continuously removed from the reaction mixture for 3-5 hours. The reaction mixture was cooled to ambient temperature, diluted with dichloromethane (50ml) and neutralised by addition of potassium carbonate (5.0g) followed by stirring for an hour . The reaction mixture was filtered and filtrate was evaporated to give yellow crude compound, which was further dissolved in dichloromethane (25ml) followed by addition of petroleum ether (100ml) and stirred for an hour at ambient temperature. The solid was filtered, washed with petroleum ether (20ml) and dried under vacuum at 35-40°C for 15-20 hours to obtain 16.63g (72.15%) of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5-dicarboxylate having purity 98.4% by HPLC.
Reference Example 2 (repetition of Synthesis, No. 15, p. 2488-90, 2008):
A reaction mixture of dimethyl-L-tartrate (5.0g), p-toluene sulfonic acid (0.0064g) and p- anisaldehyde dimethylacetal (5.35g) in toluene (25ml) was refluxed and the azeotropical mixture of toluene-methanol was continuously removed from the reaction mixture for 3-5 hours. The reaction mixture was cooled to ambient temperature, diluted with dichloromethane (25ml) and neutralised by addition of potassium carbonate (5.0g) followed by stirring for an hour. The reaction mixture was filtered and filtrate was evaporated to give yellow crude residues. The crude was further re-crystallized in petroleum ether (25ml), filtered the solid and washed with petroleum ether (15ml) followed by drying under vacuum at 35-40°C for 15-20 hours to obtain 7.4g (89.15%) of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5-dicarboxylate having purity 98.8% by HPLC. Example-1: Preparation of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane- 4,5-dicarboxylate
A reaction mixture of dimethyl-L-tartrate (500g), p-toluene sulfonic acid (5.38g) and p- anisaldehyde dimethylacetal (665g) in toluene (2250ml) was refluxed to 110-115°C. The azeotropical mixture of toluene-methanol was continuously removed from the reaction mixture till the completion of the reaction. The reaction mixture was cooled to ambient temperature and quenched with aq. saturated sodium bicarbonate solution (2500ml), layers were separated. Resulting organic layer was washed with water (2500ml x 2) followed by evaporation of organic layer. Isopropyl alcohol (3500ml) was charged to the residue and heated to 60-70°C followed by cooling at ambient temperature. Reaction mixture was stirred at 0-5°C for 1-2 hours and filtered. The solid thus obtained was washed with pre- cooled isopropyl alcohol and dried under vacuum at 35-40°C for 15-20 hours to obtain 767.0g (92.93%) of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- dicarboxylate having purity 99.97% by HPLC.
Example-2: Preparation of (4S,5S)-2-(4-methoxyphenyl)-l 53-dioxo!ane-4,5- diyifdimethanol
Method-l :To a mixture of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- dicarboxylate (765g), Iodine (13. lg) in tetrahydrofuran (3750ml) and water (76ml), sodium borohydride (146.52g) was added at 0-15°C and stirred for 1 -2 hours at ambient temperature. The reaction was quenched with 30% aq. ammonium chloride (6100ml) solution and dichloromethane (7650ml). The layers were separated and the aqueous layer was extracted by dichloromethane (3800ml x 3) followed by washing of combined organic layers with water (3800ml), The resulting organic layer was evaporated at 35-65°C to obtain 525.0g (83.9%) of (4S,5S)-2-(4-methoxyphenyl)-l,3- dioxolane-4,5-diyl]dimethanol having purity 99.72% by HPLC. Method-2: To a mixture of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane- 4,5-dicarboxylate (765g), Iodine (13.10g) in tetrahydrofuran (3750ml) and water (76.5ml), sodium borohydride (146.52g) was added at 0-10°C and stirred for Ihours at 0-5°C and stirred for 3-4 hours at ambient temperature. The reaction was quenched with 30% aq. ammonium chloride (6120ml) solution and dichloromethane (7650ml) at ambient temperature. The layers were separated and the aqueous layer was extracted by dichloromethane (3825m! x 3) followed by washing of combined organic layers with water (3825ml). The resulting organic layer was evaporated at 50-60°C to obtain 525 g (84.7%) of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5-diyl]dirnethaiiol having purity 99.72% by HPLC. Example-3: Preparation of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- diyl]bis(methylene) dimethanesulfonate
Method-l:To a solution of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- diyl]dimethanol (145g) in dichloromethane (2175ml), pyridine (191g) and methanesulphonyl chloride (190. l g) was added at 0-5 °C. The reaction mixture was stirred for 2-3 hours at ambient temperature followed by quenching with water (1450ml). The organic layer was washed with water (1450ml x 4) and evaporated. The resulting residue was added to isopropanol (725ml) and stirred for 1-2 hours at ambient temperature and further for 1-2 hours at 0-5 C. The solid was filtered and washed with pre-cooled isopropanol (145ml). The resulting product was dissolved in acetone (1300ml) followed by addition of isopropanol (2610ml). Resulting reaction mixture was stirred for 1-2 hours at ambient temperature and then cooled at 0-5 °C. The solid thus obtained was filtered and washed with pre-cooled isopropanol (145ml x 2) and dried under vacuum at 30-35°C for 15-20 hours to give 190.8g (79.4%)of (4S,5S)-2-(4- methoxyphenyl)-l,3-dioxolane-4,5-diyl]bis(methylene) dimethanesulfonate. Method-2: To a solution of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- diyl]dimethanol (525, Og) in dichloromethane (7350ml), di-isopropylamine (663. Og) was added at ambient temperature followed by addition of methanesulphonyl chloride solution (624. Og in 525ml dichloromethane) at 0-10°C. The reaction mixture was stirred for 1-2 hours at 0-10 °C followed by stirring for 3-4 hours at ambient temperature. The organic layer was washed with water (2 x 5250ml) and evaporated. The residues were dissolved in acetone (4725ml) followed by addition of isopropanol (9450ml), stirred for about 1-2 hour at ambient temperature and then at 0-5 °C for 1-2 hours. The resulting solid was filtered, washed with pre-cooled isopropanol (525 x 2 ml)and dried under vacuum at 35-45°C for 15-20 hours to give 705.0g (81.45%) of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5-diyl]bis(methylene)
dimethanesulfonate having purity 99.92% by HPLC.
Example-4: Preparation of Treosulfan
Method-1: To a solution of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- diyl]bis(methylene) dimethanesulfonate (745. Og) in methanol (7450ml), concentrated hydrochloric acid (260ml) was added at 15-25°C followed by stirring for 10-15 hours at ambient temperature. The reaction mixture was cooled to 0-5°C and further stirred for 1-2 hours at 0-5°C followed by filtration and washing the solid with pre-cooled methanol (745ml). The solid thus obtained was dissolved in acetone (3725ml) followed by microne filtration. Di-isopropyl ether (7450ml) was added to the filtrate and stirred for 1-2 hours at ambient temperature and then cooled at 0-5°C. The solid thus obtained was filtered and washed with di-isopropyl ether (745ml x 2) followed by drying at 30-35°C for 15-20 hours to obtain 96.5g of Treosulfan having purity 99.9% by HPLC.
XRPD of Treosulfan obtained by above process is shown in Fig. 1. Method-2:To a solution of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- diyl]bis(methylene)dimethanesulfonate (650. Og) in methanol (6500ml), 9N hydrochloric acid (227.5ml) was added at 0-10°C followed by stirring for 6-8 hours at ambient temperature. The reaction mixture was cooled to 0-5°C and further stirred for 1-2 hours followed by filtration and washing the solid with pre-cooled methanol (2 x 650ml). The solid thus obtained was dissolved in acetone (3250ml). Di-isopropyl ether (6500ml) was added to the resulting solution, stirred for 1-2 hours at ambient temperature and then cooled at 0-5°C. The solid thus obtained was filtered and washed with di- isopropyl ether (650ml x 2) followed by drying at 30-35°C for 15-20 hours to obtain 312g (68.4) of Treosulfan having purity 99.81% by HPLC.
PATENT
https://patents.google.com/patent/WO2020064815A1/en
Example 1 – Preparation of form B using water/isopropanol
99.8 mg treosulfan were weighed in a vial (volume 4.0 ml) which was equipped with a PTFE (Polytetrafluoroethylene) sealing and a stirrer. 1.5 ml of a mixture of 80 % by weight water and 20 % by weight isopropanol preheated to 65°C were then added. The resulting solution was completely taken up with a syringe (volume 5 ml) and filtered using a 0.2 pm filter into a second vial (volume 4.0 ml) . The syringe, second vial and filter had been tempered at 65°C before use. The solvents were allowed to evaporate from the open vial at room temperature to dryness which resulted in formation of crystals.
The XRPD pattern of the obtained crystals of form B according to the invention is shown in Figure 1.
PATENT
1568/MUM/2012
Abstract
Abstract: The present invention provides a convenient and cost-effective process for preparation of Treosulfan. The process comprises reduction of dimethyl 2,3-O-isopropylidene-L-tartrate with sodium-bis(2-methoxyethoxy)aluminum hydride to give the alcohol 2,3-O-isopropylidene-L-threitol (III), which on reaction with methanesulfonyl chloride led to 2,3-O-isopropylidene-L-threitol 1,4-bismethanesulfonate of formula (IV) and further treatment of compound (IV) with formic acid gave Treosulfan (I) having desired purity.
Treosulfan (I), chemically known as (2S,3S)-2,3-Dihydroxy-4-memylsidfonyIoxybutylj methanesulfonate is a drug commonly used for treating ovarian cancer. It belongs to the family of anti-cancer medicines called the alkylating agents, which prevent the growth and division of cancerous cells. Treosulfan has been used for bone-marrow ablation before stem-cell transplantation and in the treatment of malignant melanoma and breast cancer.
US 3,155,702 discloses synthesis of Treosulfan by replacement of the halogen function in L-Threitol-l,4-dibromobutane-2,3-diol, by treating with a large excess of an expensive reagent like silver methanesulfonate. Further, the presence of unprotected hydroxyl groups in the starting material inevitably leads to the formation of undesired impurities, which requires additional purification steps for removal of impurities as well for lowering the level of free silver in the active ingredient as per ICH guidelines, which results in lower yields and increases the costs substantially.
Another method reported in US 3,246,012 involves acetal formation of diethyl-L-tartrate with acetone to obtain 2,3-O-isopropylidene-diethyl-L-tartrate, which, when reduced with lithium aluminium hydride gives 2,3-0-methylene-L-threitol. The obtained alcohol was treated with methanesulfonyl chloride to yield the penultimate Treosulfan intermediate, 2,3-O-methylene-L-threitol-1,4-di-(methanesulfonate).
A similar approach which employs tartrate esters in the synthesis of Treosulfan, is disclosed in Organic Syntheses, (1993), Vol.8, p. 155 and Organic .Syntheses, (2004), Coll.Vol.10, p.297. L-tartaric acid is reacted with 2,2-dimethoxypropane in presence of methanol. The resulting methyl ester, dimethyl 2,3-O-isopropylidene-L-tartrate is reduced with lithium aluminium hydride to obtain 2,3-di-O-isopropylidene-L-threitol, which, upon reaction with methanesulfonyl chloride, followed by treatment with methanesulfonic acid yields Treosulfan.
Although these routes involve protection of the diol group and avoid impurities arising out of substitution at those alcohol functionalities, use of a highly pyrophoric, hazardous reagent such as lithium aluminium hydride severely limits their synthetic applicability, especially on commercial scale. Further, the final step involves reaction of 2,3-di-O-isopropylidene-L-threitol with methanesulfonic acid, which is quite sluggish and causes considerable rise in the total number of impurities due to long reaction time.
Thus, there is a need for a convenient, economical process for a commercial scale synthesis of Treosulfan (I), which overcomes the shortcomings of the prior art, does not involve use of hazardous, pyrophoric reagents and yields Treosulfan conforming to regulatory specifications.
The present inventors have developed a novel process for preparation of (2S,3S)-2,3-Dihydroxy-4-methylsulfonyloxybutyl] methanesulfonate (I). The scheme for synthesis comprises reaction of dimethyl 2,3-O-isopropylidene-L-tartrate of formula (II) with sodium-bis(2-methoxyethoxy) aluminum hydride to give the protected diol, 2,3-0-isopropylidene-L-threitoI (III), which on further treatment with methanesulfonyl chloride, followed by reaction of the resultant ester, 2,3-O-isopropyliden-L-threitol 1,4 bismethanesulfonate (IV) with formic acid, yields Treosulfan (I) having desired purity and with impurity levels conforming to ICH guidelines.
Scheme 1; Method embodied in the present invention for the preparation of Treosulfan (I)
In an embodiment, dimethyl 2,3 -O-isopropylidene-L-tartrate of formula (II) was treated with sodium-bis-(2-methoxyethoxy) aluminium hydride in presence of an organic solvent, and in the temperature range of 25 to 80°C, but preferably 60 to 75°C.
The organic solvent was selected from the group of toluene, xylenes, nitrobenzene, hexane, cyclohexane, heptane, N-methyl-2-pyrroIidone, ethers etc.
Upon completion of the reaction, as monitored by TLC, water was carefully added to the reaction mass and the mixture was extracted with a water immiscible organic solvent.
The organic solvent was selected from the group comprising of n-hexane, cyclohexane, heptane, methyl isobutyl ketone, 2-methyl tetrahydrofuran, cyclopentyl methyl ether etc.
The organic layer was separated and concentrated under reduced pressure to give 2,3-0-isopropylidene-L-threitol of formula (III) of desired purity.
It is pertinent to mention that the reaction was quite facile and the desired product was obtained with minimal formation of associated impurities and did not require any subsequent purification.
Further reaction of compound (III) with methanesulfonyl chloride was carried out at 25 to 35°C, in an organic solvent, in presence of an organic base.
The organic solvent was selected from the group comprising of chloroform, ethylene dichloride, dichloromethane, carbon tetrachloride etc., but preferably dichloromethane.
The organic base was selected from triethyl amine, tributyl amine and pyridine.
The reaction mixture was stirred at 25-35°C and after completion of the reaction as monitored by TLC, aqueous solution of sodium bicarbonate was added slowly to the reaction mass. The organic layer was separated, concentrated under reduced pressure and stirred with isopropyl alcohol to obtain the desired compound, 2,3-O-isopropylidene-L-threitol-l,4-bis(methanesulfonate) of formula (IV).
In a further embodiment, compound (TV) was hydrolyzed by treating with formic acid at 25 to 35°C based on TLC. After completion of the reaction, the reaction mass was concentrated and the product Treosulfan (I) was isolated by addition of isopropyl alcohol to the concentrated mass.
It is pertinent to mention that Organic Syntheses (2004), Coll.Vol. 10, p.297 discloses the hydrolysis reaction using methanesulfonic acid in ethanol at reflux temperature. However, the time taken for completion is about ten hours and the procedure is applicable only for laboratory scale reaction. The hydrolysis step disclosed in the present invention is easily scalable and so facile that it takes place at room temperature and within one to two hours. This reduces the time cycle for each batch run and also reduces the possibility of formation of undesired side products.
Dimethyl 2,3-O-isopropylidene-L-tartrate of formula (II) was prepared by the reaction of dimethyl -L-tartrate with acetone by following known synthetic procedures.
The following examples are meant to be illustrative of the present invention. These examples exemplify the invention and are not to be construed as limiting the scope of the invention.
EXAMPLES
Example 1: Synthesis of 2,3-O-isopropylidene-L-threitol (HI)
A solution of dimethyl-2,3-0-isopropylidene-L-tartrate (50.3 g) in toluene (50 ml) was gradually added to the stirred mixture of sodium-bis(2-methoxyethoxy) aluminum hydride (122.8 g) in toluene (50 ml) at 20-40°C. The reaction mixture was heated to 60-80°C, and the reaction was continued till completion, as monitored by TLC. When the reaction was complete, the mass was cooled to 25-3 5°C, quenched with careful addition of water (10ml) and concentrated. Treatment of the resulting residue with methyl tertiary butyl ether, followed by evaporation of the organic layer under reduced pressure afforded 2,3-0-isopropyliden -L-threitol ( III) as pale yellow oil. Yield: 29.8 g (81.2%) [α]D20 + 4.6.°(CHC13, c 5)
Example 2: Synthesis of 2,3-0-isopropylidene-L-threitol-l,4-bis(methanesulfonate)
(IV)
A stirred solution of 2,3-O-isopropylidene-L-threitol (100.2 g), methylene chloride (1250
ml) and pyridine (146.3 g) was cooled to 0-5°C and methanesulfonyl chloride (176.6 g)
was slowly added to it. Temperature of the reaction mixture was raised to 25-35°C and the
reaction was continued at the same temperature till completion of the reaction, as
monitored by HPLC. After completion of the reaction, aqueous sodium bicarbonate
solution was slowly added to the reaction mass and the organic layer was separated.
Aqueous layer from the reaction mixture was extracted with methylene chloride and the
organic layers were combined. Distillation of the organic solvent, optionally followed by
addition of isopropyl alcohol gave the product, 2,3-0-isopropylidene-L-threitol-l,4-
bis(methanesulfonate).
Yield: 160.7 g (79.7%)
[α]D20-21.6°(acetone,c2)
Example 3: Synthesis of Treosulfan (I)
A mixture of formic acid (98%, 1000 ml) and 2,3-0-isopropylidene-L-threitol-l,4-bis(methanesulfonate) (100.5 g) was stirred at room temperature until completion of the desired reaction, as monitored by TLC, When the reaction was complete, the reaction mass was concentrated under reduced pressure..
Treatment of the residue after evaporation with isopropanol yielded the final product Treosulfan, which was optionally subjected to further treatment with acetone and nexanes or petroleum ether, Yield: 74.3 g (85.0%) [α]D20 – 5.3°(acetone, c 2) Purity: > 99 %.
References
- ^ Jump up to:a b “Trecondi APMDS”. Therapeutic Goods Administration (TGA). 11 October 2022. Retrieved 25 January 2025.
- ^ “Updates to the Prescribing Medicines in Pregnancy database”. Therapeutic Goods Administration (TGA). 21 December 2022. Archived from the original on 3 April 2022. Retrieved 2 January 2023.
- ^ “Trecondi (Link Medical Products Pty Ltd T/A Link Pharmaceuticals)”. Therapeutic Goods Administration (TGA). 14 January 2025. Retrieved 25 January 2025.
- ^ “AusPAR: Trecondi”. Therapeutic Goods Administration (TGA). 4 July 2023. Retrieved 25 January 2025.
- ^ “Health product highlights 2021: Annexes of products approved in 2021”. Health Canada. 3 August 2022. Retrieved 25 March 2024.
- ^ “Treosulfan 5g Powder for Solution for Infusion – Summary of Product Characteristics (SmPC)”. (emc). Archived from the original on 20 May 2022. Retrieved 21 April 2020.
- ^ Jump up to:a b c d e f “Grafapex- treosulfan injection, powder, lyophilized, for solution”. DailyMed. 31 January 2025. Retrieved 2 April 2025.
- ^ “Trecondi Product Information” (PDF). European Medicines Agency (EMA). 21 April 2020.
- ^ Jump up to:a b c d e f g h i j k l m “Trecondi EPAR”. European Medicines Agency (EMA). 11 December 2018. Archived from the original on 16 March 2023. Retrieved 21 April 2020. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Romański M, Wachowiak J, Główka FK (October 2018). “Treosulfan Pharmacokinetics and its Variability in Pediatric and Adult Patients Undergoing Conditioning Prior to Hematopoietic Stem Cell Transplantation: Current State of the Art, In-Depth Analysis, and Perspectives”. Clinical Pharmacokinetics. 57 (10): 1255–1265. doi:10.1007/s40262-018-0647-4. PMC 6132445. PMID 29557088.
- ^ Jump up to:a b “FDA approves treosulfan with fludarabine as a preparative regimen for alloHSCT in adult and pediatric patients with AML or MDS”. U.S. Food and Drug Administration (FDA). 6 February 2025. Retrieved 8 March 2025. This article incorporates text from this source, which is in the public domain.
- ^ “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 21 February 2025. Retrieved 9 March 2025.
- ^ “Medexus Announces FDA Approval of Grafapex (treosulfan) for Injection and Provides Business Update” (Press release). Medexus Pharmaceuticals. 22 January 2025. Retrieved 25 January 2025 – via Newsfile.
- ^ “Treosulfan Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 16 May 1994. Retrieved 9 March 2025.
- ^ “Treosulfan Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 8 April 2015. Retrieved 9 March 2025.
- ^ “EU/3/04/186”. European Medicines Agency (EMA). 17 September 2018. Archived from the original on 16 October 2019. Retrieved 21 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ World Health Organization (1972). “International nonproprietary names for pharmaceutical substances (INN). recommended INN: list 12”. WHO Chronicle. 26 (10).
External links
- “Treosulfan”. National Cancer Institute.
- [1]
- Clinical trial number NCT00822393 for “Clinical Phase III Trial Treosulfan-based Conditioning Versus Reduced-intensity Conditioning (RIC)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Trecondi, others |
| Other names | 1,2,3,4-Butanetetrol, 1,4-dimethanesulfonate, Threitol 1,4-dimethanesulfonate, Threitol 1,4-bismethanesulfonate; L-Threitol 1,4-bis(methanesulfonate); Threosulphan; Treosulphan; Tresulfan |
| AHFS/Drugs.com | International Drug Names |
| License data | US DailyMed: Treosulfan |
| Pregnancy category | AU: D[1][2] |
| Routes of administration | By mouth, intravenous |
| ATC code | L01AB02 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)[1][3]<[4]CA: ℞-only[5]UK: POM (Prescription only)[6]US: ℞-only[7]EU: Rx-only[8]In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 299-75-2 |
| PubChem CID | 9882105 |
| DrugBank | DB11678 |
| ChemSpider | 8057780 |
| UNII | CO61ER3EPI |
| KEGG | C19557D07253 |
| ChEBI | CHEBI:82557 |
| CompTox Dashboard (EPA) | DTXSID0026173 |
| ECHA InfoCard | 100.005.529 |
| Chemical and physical data | |
| Formula | C6H14O8S2 |
| Molar mass | 278.29 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 101.5 to 105 °C (214.7 to 221.0 °F) |
| showSMILES | |
| showInChI | |
- Romanski M, Baumgart J, Bohm S, Glowka FK: Penetration of Treosulfan and its Active Monoepoxide Transformation Product into Central Nervous System of Juvenile and Young Adult Rats. Drug Metab Dispos. 2015 Dec;43(12):1946-54. doi: 10.1124/dmd.115.066050. Epub 2015 Oct 1. [Article]
- EMA Summary of Product Characteristics: Trecondi (treosulfan) powder for solution for infusion [Link]
- FDA Approved Drug Products: GRAFAPEX (treosulfan) for injection, for intravenous use [Link]
- EMC Summary of Product Characteristics: Treosulfan 5g Powder for Solution for Infusion [Link]
- NIH LiverTox: Alkylating Agents [Link]
- FDA News Release: FDA approves treosulfan with fludarabine as a preparative regimen for alloHSCT in adult and pediatric patients with AML or MDS [Link]
////////TREOSULFAN, Treosulphan, Ovastat, Treosulfano, Grafapex, acute myeloid leukemia, myelodysplastic syndrome, NSC-39069, Dihydroxybusulfan, L-threitol-1,4-dimethanesulfonate, Trecondi, FSA 2025, APPROVALS 2025, EMA 2019, EU 2019
CS(=O)(=O)OC[C@H](O)[C@@H](O)COS(C)(=O)=O
Bevemipretide




Bevemipretide,
CAS 2356106-71-1 FREE BASE
CAS SBT-272 Trihydrochloride, 2589640-11-7
607.7 g/mol, C31H45N9O4 F553HAL9V8
- SBT-272
- (2R)-2-amino-N-[(2S)-1-[[(1S)-5-amino-1-(3-benzyl-1,2,4-oxadiazol-5-yl)pentyl]amino]-3-(4-hydroxy-2,6-dimethylphenyl)-1-oxopropan-2-yl]-5-(diaminomethylideneamino)pentanamide
- L-Tyrosinamide, D-arginyl-N-[(1S)-5-amino-1-[3-(phenylmethyl)-1,2,4-oxadiazol-5-yl]pentyl]-2,6-dimethyl-
- (2R)-2-amino-N-[(2S)-1-[[(1S)-5-amino-1-(3-benzyl-1,2,4-oxadiazol-5-yl)pentyl]amino]-3-(4-hydroxy-2,6-dimethylphenyl)-1-oxopropan-2-yl]-5-(diaminomethylideneamino)pentanamide
- L-Tyrosinamide, D-arginyl-N-[(1S)-5-amino-1-[3-(phenylmethyl)-1,2,4-oxadiazol-5-yl]pentyl]-2,6-dimethyl-
- (2R)-2-amino-N-[(1S)-1-{[(1S)-5-amino-1-(3-benzyl-1,2,4-oxadiazol-5-yl)pentyl]carbamoyl}-2-(4-hydroxy-2,6-dimethylphenyl)ethyl]-5-carbamimidamidopentanamide
- Originator Stealth BioTherapeutics
- ClassAntidementias; Antiparkinsonians; Neuroprotectants; Peptidomimetics
- Mechanism of Action Adenosine triphosphatase stimulants; Cardiolipin modulators; Reactive oxygen species inhibitors
- Orphan Drug Status Yes – Amyotrophic lateral sclerosis
Phase IAmyotrophic lateral sclerosis
- Preclinical Dry age-related macular degeneration; Frontotemporal dementia; Parkinson’s disease
- No development reported Multiple system atrophy
18 Sep 2024Pharmacodynamics data from a preclinical trial in dry age related macular degenration released by Stealth BioTherapeutics
- 18 Sep 2024Preclinical trials in Dry age-related macular degeneration in USA (Opthalmic)
- 18 Sep 2024Stealth Biotherapeutics plans clinical trial for Dry age related macular degeneration (Topical)
The present technology relates generally to compounds (i.e. peptidomimetics), compositions (e.g. medicaments) and methods for treating, preventing, inhibiting, amelioration or delaying the onset of ophthalmic diseases, disorders or conditions in a mammalian subject. In some embodiments, the ophthalmic disease, disorder or condition is associated with deterioration of the integrity of the ellipsoid zone of one or more eyes of the mammalian subject. For example, the present technology may relate to administering one or more mitochondrial-targeting peptidomimetics (alone, as formulated and/or in combination with other active pharmaceutical ingredients) in effective amounts to treat, prevent, inhibit, ameliorate or delay the onset of ophthalmic diseases, disorders or conditions (e.g., macular degeneration (including (wet or dry) age-related macular degeneration), dry eye, diabetic retinopathy, diabetic macular edema, cataracts, autosomal dominant optic atrophy (DOA), Leber hereditary optic neuropathy (LHON), pigmentary retinopathy, retinitis pigmentosa, glaucoma, ocular hypertension, uveitis, chronic progressive external ophthalmoplegia (often referred to as CPEO or just PEO, e.g., Kearns-Sayre syndrome), and/or Leber congenital amaurosis (LCA)), in mammalian subjects
[0003] The following introduction is provided to assist the understanding of the reader. None of the information provided, or references cited, is admitted as being prior art to the present technology.
[0004] Diseases, disorders and degenerative conditions of the optic nerve and retina are the leading causes of blindness in the world. Many ophthalmic diseases disorders or conditions result from, or are associated with, mitochondrial dysfunction.
[0005] A significant degenerative condition of the retina is age-related macular degeneration (AMD). AMD is the most common cause of blindness in people over the age of 50 in the United States and its prevalence increases with age. AMD is classified as either wet (neovascular) or dry (non-neovascular). The dry form of the disease is more common.
Macular degeneration occurs when the central retina has become distorted and thinned. This change is usually associated with age but also characterized by intra-ocular inflammation and angiogenesis (wet AMD only) and/or intra-ocular infection. The subsequent generation of free radicals, resulting in oxidative tissue damage, local inflammation and production of growth factors (such as VEGF and FGF) and inflammatory mediators, can lead to inappropriate neovascularization in common with the wet form of AMD. Mitochondrial dysfunction is believed to play a role in age-related disorders such as AMD. (Liu et al., Appl. Sci. (2021) 11: 7385). Pieramici & Ehlers have reported that: “RPE mitochondria in AMD eyes undergo more pronounced degenerative changes, with lower mitochondrial density, organelle area and cristae number.” (Pieramici & Ehlers, Presentation at 54th Annual Retina Society Meeting, Sept.30, 2021, slide 3).
[0006] Retinopathy is a leading cause of blindness in type I diabetes and is also common in type II diabetes. The degree of retinopathy depends on the duration of diabetes, and generally begins to occur ten or more years after onset of diabetes. Diabetic retinopathy may be classified as non-proliferative, where the retinopathy is characterized by increased capillary permeability, edema and exudates, or proliferative, where the retinopathy is characterized by neovascularization extending from the retina to the vitreous, scarring, deposit of fibrous tissue and the potential for retinal detachment. Diabetic retinopathy is believed to be caused by the development of glycosylated proteins due to high blood glucose and leads to damage in small blood vessels in the eye. Diabetic retinopathy (often if left untreated) can progress to diabetic macular edema. Diabetic macular edema involves damage to the blood vessels in the retina that progress to a point where they leak fluid into the macula thereby causing the macula to swell and this results in blurred vision. Mitochondrial dysfunction has been linked to the pathogenesis of diabetic retinopathy. (Wu et al. Hindawi Oxidative Medicine and Cellular Longevity, Volume 2018, Article 3420187)
[0007] Glaucoma is made up of a collection of eye diseases that cause vision loss by damage to the optic nerve and retinal ganglion cells (RGCs). An intraocular pressure (IOP) of over 21 mmHg without optic nerve damage is known as ocular hypertension. Elevated IOP due to inadequate ocular drainage is the primary cause of glaucoma. Lowering IOP reduces the risk of progressive RGC loss in glaucoma; however, no currently available treatments directly prevent RGC damage. Glaucoma often develops as the eye ages, or it can occur as the result of an eye injury, inflammation, tumor or in advanced cases of cataract or diabetes. It can also be caused by the increase in IOP caused by treatment with steroids. Drug therapies that are proven to be effective in glaucoma reduce IOP either by decreasing vitreous humor production or by facilitating ocular draining. Such agents are often vasodilators and as such act on the sympathetic nervous system and include adrenergic antagonists. It has been stated that: “… mitochondrial dysfunction plays an important role in the pathogenesis of neurodegenerative diseases…” and “… mitochondrial damage may provide potential strategies for the treatment of glaucoma….” (Liu et al., Appl. Sci. (2021) 11: 7385).
[0008] Autosomal dominant optic atrophy (DOA) is a genetic X-linked neuro-ophthalmic condition characterized by bilateral degeneration of optic nerves. It affects approximately 1 in 10,000 (Denmark) to 1 in 30,000 (worldwide) persons. The nerve damage causes visual loss. It generally begins to manifest itself during the first decade of life and progresses thereafter. The disease itself affects primarily the retinal ganglion nerves. Mutations in the genes known as OPA1 and OPA3, which encode inner mitochondrial membrane proteins (resulting in mitochondrial dysfunction), are generally associated with DOA.
[0009] Leber Hereditary Optic Neuropathy (LHON) is a genetically-based inherited disease that generally starts to manifest itself between the ages of 15 and 35. In LHON, mitochondrial mutations affect complex I subunit genes in the respiratory chain leading to selective degeneration of retinal ganglion cells (RGCs) and optic atrophy generally within a year of disease onset. LHON is caused by mutations in the MT-NDI1, MT-ND4, MT-ND4L and MT-ND6 genes; all of which are associated with mitochondrial genome coding. LHOH affects approximately 1 in 50,000 people worldwide. It generally starts in one eye and progresses quickly to the other eye. Subjects with LHON may eventually become legally or totally blind, often before they turn 50. LHON affects vision needed for tasks such as reading, driving and recognizing others.
[0010] Retinitis pigmentosa (RP) is a group of hereditary retinal degenerative disorders characterized by progressive vision loss. RP is a leading cause of inherited blindness in the developed world. Clinically, RP is manifested by night vision difficulties due to the death of rod photoreceptors followed by the progressive loss of peripheral vision eventually leading to central vision impairment from the secondary loss of cone photoreceptors. RP is caused by mutations of at least 87 genes. The pathogenesis of RP is not well understood. However, mitochondrial dysfunction and oxidative damage are believed to play a key role in the pathogenesis of photoreceptor cell death in RP. (Gopalakrishnan et al., Scientific Reports (2020) 10: 20382)
[0011] Pigmentary retinopathy (PR) is a frequent feature of retinitis pigmentosa.
Pigmentary retinopathy is a non-specific finding that may be found in several mitochondrial diseases, such as Neurogenic weakness, Ataxia, and Retinitis Pigmentosa (NARP). PR is an inherited degenerative disorder of the retina, characterized by progressive photoreceptor damage. The damage leads to atrophy and cell death of the photoreceptors. Patients with PR can follow an autosomal-dominate, autosomal recessive or X-linked recessive pattern. The prevalence is about one in about three to four thousand individuals. Symptoms of the disease include nyctalopia (night blindness), peripheral visual field constriction, and sometimes loss of the central visual acuity or visual field.
[0012] Uveitis is array of intraocular inflammatory diseases of the eye that often results in irreversible visual loss. Uveitis is responsible for an estimated 30,000 new cases of legal blindness annually in the USA. It is believed that this disease is at least in part due to retinal tissue damage caused excessive mitochondrial oxidative stress that triggers a damaging immune response.
[0013] Chronic progressive external ophthalmoplegia (CPEO) is a condition characterized mainly by a loss of the muscle functions including in eye and eyelid movement. The condition typically appears in adults between ages 18 and 40 and slowly worsens over time. CPEO can be caused by genetic changes in any of several genes, which may be located in mitochondrial DNA or nuclear DNA. CPEO can occur as part of other underlying conditions, such as ataxia neuropathy spectrum and Kearns-Sayre syndrome. These conditions may not only involve CPEO, but various additional features that are not shared by most individuals with CPEO.
[0014] Kearns-Sayre syndrome is a condition that affects many parts of the body, especially the eyes. The features of Kearns-Sayre syndrome usually appear before age 20, and the condition is diagnosed by a few characteristic signs and symptoms. People with Kearns-Sayre syndrome have progressive external ophthalmoplegia. Affected individuals also have an eye condition called pigmentary retinopathy, which results from breakdown (degeneration) of the retina that gives it a speckled and streaked appearance.
[0015] Leber congenital amaurosis (LCA) is a rare genetic eye disorder that affects infants. The infants are often blind at birth. LCA can be associated with mitochondrial dysfunction. (Castro-Gago et al., J. Child Neurol. (1996) 11(2):108-11) Children born with LCA have light-gathering cells (rods and cones) of the retina that do not function properly. LCA has been estimated to be 1-2/100,000 births. This disorder affects males and females in equal numbers.
[0016] Drusen are small yellow or white spots between the retinal pigment epithelium and Bruch’s membrane in the retina that can be detected by an ophthalmologist during a dilated eye exam or with retinal photography. Drusen can also be imaged and monitored by optical coherence tomography (OCT). Drusen are made up of lipids and proteins. Drusen are a defining feature of macular degeneration. Drusen can be hard or soft. Larger numbers of drusen, as well as drusen of larger size, indicate higher risk for some vision loss in the future. “Hard” drusen are small and indicate lower risk of future vision loss than “soft” drusen. “Soft” drusen are larger, cluster together, and have edges that are not as clearly defined. Soft drusen are more likely to lead to vision loss.
[0017] Geometric Atrophy (GA) is generally considered part of the later stage of age-related macular degeneration (AMD) and refers to progression of the disease to a point where in regions of the retina, cells begin to waste away and die (i.e. atrophy).
[0018] Best corrected visual acuity (BCVA) is a measure of the best possible vision an eye can achieve with the use of glasses or corrective lenses. It is typically measured using Snellen lines on an eye chart. Repeated testing of the BCVA over time can be used to determine if a subject’s vision is stable, improving or deteriorating.
[0019] Low luminance visual acuity (LLVA) involves standard visual acuity testing under low-light conditions. This is often achieved by adding a neutral density filter in front of the testing eye. It is a useful visual function marker in those with geographic atrophy (GA) and neovascular age-related macular degeneration. Repeated testing of the LLVA over time can be used to determine if a subject’s vision, under low light conditions, is stable, improving or deteriorating.
[0020] Optical coherence tomography (OCT) is a non-invasive imaging method used to generate a picture of the back of the eye (i.e. the retina). OCT uses a low-powered laser to create pictures of the layers of the retina and optic nerve. The cross-sectional images are three-dimensional and color-coded. OCT can measure the thickness of the retina and optic nerve. OCT can be used to diagnose and manage Glaucoma, AMD, diabetes-related retinopathy, cystoid macular edema, macula pucker and macular hole.
[0021] Spectral domain optical coherence tomography (SDOCT) is an interferometric technique that provides depth-resolved tissue structure information encoded in the magnitude and delay of the back-scattered light by spectral analysis of the interference fringe pattern. SDOCT increases axial resolution 2- to 3-fold and scan speed 60- to 110-fold vs conventional (TD) OCT.
[0022] The ellipsoid zone can be mapped using SCOCT and the integrity of (or changes in) the ellipsoid zone can be determined from such mapping/scanning activity. (Itoh et al., Br J Ophthalmol. (2016) 100(3): 295-299). The technology is capable of evaluating the structures of the external limiting membrane (ELM), ellipsoid zone (EZ), interdigitation zone (IZ) and the retinal pigment epithelium (RPE). Id. Use of this technology is capable of accessing EZ integrity and EZ-RPE alterations. Id. The EZ and ELM, in particular, have been linked to visual outcomes and prognosis in numerous macular conditions, such as age-related macular degeneration (AMD) Id. Itoh et al. suggest that the utility of SDOCT as an assessment tool for EZ integrity for clinical trials and disease prognostication/management may prove particularly useful.
[0023] Swept source OCT (SS-OCT) and OCT angiography (OCTA) are relatively new techniques that are capable of better resolution of the retinal pigment epithelium (RPE), Bruch’s membrane (BM) and choriocapillaris (CC) structures. (Zhou et al. Biomedical Optics Express (2020) 11(4): 1834-1850) Using this technology it is possible to generate relative distance and thickness maps of the RPE-BM-CC complex. Id. Use of these techniques may provide a better understanding of the CC in three dimensions, and further
investigate potential functional relationships between RPE, BM and CC, and their involvement in age-related ocular diseases. Id.
[0024] The ellipsoid zone (EZ) of the eye is a mitochondrial rich tissue (Ball et al., Sci. Adv.8, eabn2070 (2022)). The ellipsoid zone can be imaged using optical coherence tomography (Fujita et al., Scientific Reports (2019) 9:12433). The integrity of the EZ can be quantified. (Fugita et al.). There is a clear relationship between the integrity of the ellipsoid zone and visual function. (Fugita et al., Figure.3). Ball et al. suggest that tightly packed mitochondria in the ellipsoid “focus” light for entry into the outer segment and that healthy mitochondria structure (including cristae structure) might be important for producing a Stiles-Crawford effect (SCE) and maintaining visual resolution in mammals. Pieramici & Ehlers describe mapping the ellipsoid zone to thereby observe the ellipsoid zone and possibly monitor changes in the integrity of the ellipsoid zone. (Pieramici & Ehlers, Presentation at 54th Annual Retina Society Meeting, Sept.30, 2021). Pieramici & Ehlers further described the use of Sub-RPE compartment maps as a means to find and monitor drusen formation and RPE atrophy in a subject. In the study being described (which described results from a P2 clinical trial involving treatments with elamipretide), Pieramici & Ehlers concluded, inter alia, that: (i) “Average BCVA and LLVA in NCGA and HRD patients improved significantly at 24 weeks [of treatment with elamipretide]” and (ii) “Baseline higher order OCT parameters, such as EZ integrity, correlated with improved LLVA in Elamipretide-treated eyes” (Pieramici & Ehlers at slide 15).
[0025] In brief, there are many ophthalmic diseases for which there remains a need for treatments/therapies or improved treatments/therapies. For example, there remains a need for treatments/therapies, or improved treatments/therapies, to address ophthalmic diseases, disorders or conditions such as macular degeneration (including (wet or dry) age-related macular degeneration), dry eye, diabetic retinopathy, diabetic macular edema, cataracts, autosomal dominant optic atrophy (DOA), Leber hereditary optic neuropathy (LHON), pigmentary retinopathy, retinitis pigmentosa, glaucoma, ocular hypertension, uveitis, chronic progressive external ophthalmoplegia (e.g., Kearns-Sayre syndrome), and/or Leber congenital amaurosis (LCA). This forgoing discussion addresses these needs.
SCHEME

MAIN

PATENT
WO2023069255
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023069255&_cid=P22-M93M8P-34013-1

Synthesis of (R)-2-amino-N-((S)-1-(((S)-5-amino-1-(3-benzyl-1,2,4-oxadiazol-5- yl)pentyl)amino)-3-(4-hydroxy-2,6-dimethylphenyl)-1-oxopropan-2-yl)-5- guanidinopentanamide (D-Arg-DMT-NH((S)-5-amino-1-(3-benzyl-1,2,4-oxadiazol-5- yl)pent-1-yl), 7a (a.k.a. ((Formula IIa)):
[0134] In some embodiments, Compound 7a (a.k.a. Formula IIa) may be synthesized as illustrated in Scheme 5, below (Also see WO2019/118878, incorporated herein by reference), wherein compound 12a can be prepared as illustrated in Scheme 6, below
[0135] Step a: Synthesis of benzyl (S)-2-((R)-2-((tert-butoxycarbonyl)amino)-5- guanidinopentanamido)-3-(4-hydroxy-2,6-dimethylphenyl)propanoate (3a). To a suspension of 2,6-Dmt-OBn.HCl (2a, 45.0 g, 134 mmol) in ACN (800 mL), NMM (32.7 mL, 298 mmol) was added at 00C. The reaction mixture was stirred until the reaction mixture became transparent. Then Boc-D-Arg-OH.HCl (1a, 46.3 g, 149 mmol) and HOBt.H2O (9.11 g, 59.5 mmol) were added to reaction mixture and stirred for 15 min. Finally, EDC.HCl (38.5 g, 201 mmol) was added and mixture was stirred at 00C for 4 h. Then EtOAc (450 mL), 1N HCl in brine (300 mL) were added. The combined organic extracts were washed with 1N HCl in brine (7×150 mL), NaHCO3/brine (300 mL and until pH of aqueous layer is about pH=6 to 7), dried over Na2SO4, filtered and concentrated to afford 86.0 g (97%) of Boc-D-Arg-DMT- OBn (3a) that was used without further purification.1H-NMR (400 MHz, Methanol-d4) δ 7.33 – 7.18 (m, 5H), 6.43 (s, 2H), 5.06 (s, 2H) 4.71 (t, J=7.8Hz, 1H), 4.07 (t, J=6.7Hz,1H), 3.19 – 3.09 (m, 3H), 3.03-2.97 (m, 1H), 2.23 (s, 6H), 1.72 – 1.65 (m, 1H), 1.54 – 1.43 (m, 3H), 1.45 (s, 9H).
[0136] Step b: Synthesis of (S)-2-((R)-2-((tert-butoxycarbonyl)amino)-5-guanidinopentanamido)-3-(4-hydroxy-2,6-dimethylphenyl)propanoic acid (4a). To a solution of Boc-D-Arg-DM-Tyr-OBn (3a, 84.0 g, 142 mmol) in MeOH (1000 mL) Pd/C (10% w/w, 14.0 g) was added. The hydrogen was purged in reaction mixture at room temperature for 4h. Then reaction mixture was filtrated through filter paper and washed with MeOH (150 mL). The solvent was removed by evaporation. White foam product 4a was obtained (74.0 g, 93%) and used without further purification.1H-NMR (400 MHz, Methanol-d4) δ 6.44 (s, 2H), 4.68 (t, J = 7.2 Hz, 1H), 4.04 (t, J = 6.8 Hz, 1H), 3.15 – 3.09 (m, 3H), 3.02 – 2.94 (m, 1H), 2.29 (s, 6H), 1.74 – 1.59 (m, 1H), 1.54 – 1.43 (m, 1H), 1.45 (s, 9H).
[0137] Step c: Synthesis of tert-butyl ((6R,9S,12S)-1-amino-12-(3-benzyl-1,2,4-oxadiazol-5-yl)-9-(4-hydroxy-2,6-dimethylbenzyl)-1-imino-20,20-dimethyl-7,10,18-trioxo-19-oxa-2,8,11,17-tetraazahenicosan-6-yl)carbamate (6a). DMF (200 mL) was added to 4a (11.17 g, 24 mmol) and stirred at r.t. for 15 min. To the resulting suspension, 12a (10.65 g, 20 mmol) was added and stirred at r.t. for 20 min. After addition of HOBt (612 mg, 4.00 mmol), the suspension was cooled in ice bath. EDC . HCl (5.38 g, 28 mmol) was added in one portion, and the reaction mixture was stirred while cooled in ice bath for 2.5 h and then, for 4.5 h at r.t. The nearly homogeneous reaction mixture was quenched with EtOAc (1500 mL) and the resulting solution was washed for 10 times with brine/aq.0.5 M HCl (1:1; 400 mL). During the 6th and 9th washings, gel in the aqueous phase was formed. After addition of iPrOH (40 mL in each case) and repeated shaking the layers went clear again. Afterwards, the organic phase was washed for 6 times with brine/sat. aq. NaHCO3 (9:1; 400 mL). During the 4th washing, gel in the aqueous phase was formed. After addition of iPrOH (40 mL) and repeated shaking the layers were separated easily. The organic phase was washed with brine (200 mL) and water (100 mL) and the solvent was removed under reduced pressure. No vigorous shaking was performed upon washing with water to avoid difficulties in phase separation. As a result, 16.8 g of the crude product were obtained (6a, 97.0 % purity by HPLC, white amorphous solid).1H-NMR (300 MHz, Methanol-d4) ppm: δ = 7.33–7.16 (m, 5H), 6.38 (s, 2H), 5.18-5.07 (m, 1H), 4.64-4.55 (m, 1H), 4.10 – 3.92 (m, 3H), 3.18-2.77 (m, 6H), 2.20 (s, 6H), 1.97-1.76 (m, 2H), 1.75-1.14 (m, 8H), 1.43 (s, 9H), 1.41 (s, 9H).
[0138] Step d: Synthesis of (R)-2-amino-N-((S)-1-(((S)-5-amino-1-(3-benzyl-1,2,4-oxadiazol-5-yl)pentyl)amino)-3-(4-hydroxy-2,6-dimethylphenyl)-1-oxopropan-2-yl)-5-guanidinopentanamide (7a, but also referred to as (IIa – the tri-hydrochloride salt of Compound I) herein). After 6a (16.8 g) was dissolved in DCM (100 mL) and cooled to 0°C, TFA (20 mL) was added dropwise and the solution was allowed to stir at 0 °C for 10 min, and then at r.t. for 3 h (LC/MS shows no starting material). Then reaction mixture was evaporated (at 0–5 °C) and additionally re-evaporated from DCM (100 mL, at 0–5 °C). The purification by flash chromatography on reverse phase (cartridge C-18, 120G) was performed on crude material divided in 4 parts. Then all solvents were evaporated at reduced pressure at <40oC. White foam was dissolved in isopropanol (100 mL) and 5 mL of HCl in isopropanol (5-6M) was added at 0 oC and evaporated under reduced pressure. This step was repeated 3 times. Additionally, 100 mL of ACN was added and suspension was evaporated one more time. As a result, white powder of 7a was obtained as the tri-hydrochloride salt.1H-NMR (300 MHz, Methanol-d4) δ 7.36 – 7.14 (m, 5H), 6.40 (s, 2H), 5.15 (dd, J = 8.5, 6.3 Hz, 1H), 4.68 (dd, J = 8.7, 7.5 Hz, 1H), 4.07 (s, 2H), 3.97 (t, J = 6.3 Hz, 1H), 3.18 (t, J = 6.9 Hz, 2H), 3.11 (dd, J = 14.2, 8.8 Hz, 1H), 2.95 – 2.84 (m, 3H), 2.22 (s, 6H), 2.02 – 1.59 (m, 6H), 1.57 – 1.28 (m, 4H). MS: EI-MS: m/z 608.4 [M+1].


Synthesis of (S)-1-(3-Benzyl-1,2,4-oxadiazol-5-yl)-5-((tert-Butoxycarbonyl)amino)pentan-1-Aminium 4-Methylbenzenesulfonate (12a)
step a: NH2OH; step b: T3P, NaHCO3; step c: TEA; step d: PTSA
[0139] Step a: Synthesis of N-hydroxy-2-phenylacetimidamide (9a). To a solution of nitrile 8a (1.0 mol) in EtOH (1.2 L) was added NH2OH (50% aqueous solution, 130 g, 2.0 mol).
The solution was heated to reflux and stirred for 12 hours (hrs.). After completion, the reaction mixture was concentrated under reduced pressure. The resulting residue was re-dissolved in EtOH (350 mL) and concentrated under reduced pressure again (this procedure was repeated three times). The resulting solid was triturated in hexane (350 mL), filtered, washed with hexane (100 mL), and then dried to give the desired product 9a as white solid. (10.5 kg; KF = 1295) with good results (purity by HPLC, > 98.9 A%; Assay = 22.2 w%, yield = 91%).1H NMR (300 MHz, DMSO-d6): δ 8.90 (s, 1H), 7.28-7.18 (m, 5H), 5.40 (s, 2H), 3.25 (s, 2H) ppm. MS: (M+H)+: m/z = 151.1
[0140] Step b: Synthesis of (9H-Fluoren-9-yl)methyl tert-Butyl (1-(3-Benzyl-1,2,4-oxadiazol-5-yl)pentane-1,5-diyl) (S)-Dicarbamate (11a). To a solution of protected enantiomerically pure N2-(((9H-fluoren-9-yl)methoxy)carbonyl)-N6-(tert-butoxycarbonyl)-L-lysine (10a, 4.31 kg, 9.2 mol) and hydroxyimidamide 9a (1.1 equivalents “equiv.” or “eq.”) in ethyl acetate was added NaHCO3 (3.0 equiv.). The mixture was stirred at 25 oC for 20 minutes (min.). Then, propane phosphonic acid anhydride (T3P, 50% solution in ethyl acetate, 3.0 equivalents (equiv.)) was added and the reaction mixture was heated to 80 oC and stirred for 4 hrs. (about 60% conversion of compound 10a based on HPLC). Then compound 9a (1.1 equiv.) was added and the reaction mixture was stirred at 80oC for another 20 hr. (about 10% compound 10a remained). The reaction mixture was cooled to room temperature, saturated aqueous NaHCO3 (2.0 L) was added, the mixture was then extracted with ethyl acetate (3x 1.0 L). The combined organic layers were then washed with brine (1 L), dried over anhydrous Na2SO4, filtered and concentrated to give a crude residue, which was generally purified by silica gel column chromatography (Petroleum ether (PE):EtOAc = 5: 1) to give crude product, (9H-fluoren-9-yl)methyl tert-butyl (1-(3-benzyl-1,2,4-oxadiazol-5-yl)pentane-1,5-diyl) (S)-dicarbamate (11a), solution in ACN (19.7 kg, assay = 20%, chiral HPLC purity = 99.12 A%,yield = 73%).1H-NMR (300 MHz, CDCl3): δ 7.78 (d, J = 7.5 Hz, 2H), 7.61 (d, J = 6.3 Hz, 2H), 7.42 (t, J = 7.5 Hz, 2H), 7.35-7.30 (m, 7H), 5.52 (br, 1H), 5.09-5.05 (m, 1H), 4.56-4.37 (m, 3H), 4.22 (t, J = 6.6 Hz, 1H), 4.08 (s, 2H), 1.95-1.86 (m, 2H), 1.48-1.42 (m, 11H) ppm. MS: (M-100+H)+: m/z = 483.2.
[0141] Step c: Synthesis of tert-Butyl (S)-(5-Amino-5-(3-Benzyl-1,2,4-oxadiazol-5-yl)pentyl)-carbamate (5a). To a solution of compound (9H-fluoren-9-yl)methyl tert-butyl (1-(3-benzyl-1,2,4-oxadiazol-5-yl)pentane-1,5-diyl) (S)-dicarbamate (11a) was added TEA (2.5 eq.). The mixture was kept stirring with mechanical stirrer at 20~ 25 °C for 15 h. The reaction mixture was diluted by tap water and MTBE. Separated, aqueous layer was extracted by MTBE for one time. Both MTBE layers were combined, and then washed by NH4Cl. Then anhydrous Na2SO4 was added and that solution stirred for least 2 h, then filtered and washed with MTBE to afford tert-butyl (S)-(5-amino-5-(3-benzyl-1,2,4-oxadiazol-5-yl)pentyl)-carbamate (5a) solution in MTBE (32.9 kg, assay = 6.5%, yield = 88%).1H-NMR (300 MHz, DMSO-d6): δ 7.33-7.25 (m, 5H), 6.78 (br, 1H), 5.09-5.05 (m, 1H), 4.56-4.37 (m, 3H), 4.06 (s, 2H), 3.98 (t, J = 6.6 Hz, 1H), 2.87-2.84 (m, 2H), 2.10 (s, 2H), 1.38-1.34 (m, 2H), 1.24 (s, 9H), 1.20-1.15 (m, 2H) ppm. MS: (M+H)+: m/z = 361.1.
[0142] Step d: Synthesis of (S)-1-(3-Benzyl-1,2,4-oxadiazol-5-yl)-5-((tert-Butoxycarbonyl)-amino)pentan-1-Aminium 4-Methylbenzenesulfonate (12a). p-toluenesulfonic acid (PTSA) was added to solution of crude tert-butyl (S)-(5-amino-5-(3-benzyl-1,2,4-oxadiazol-5-yl)pentyl)-carbamate (5a) in MTBE to afford (S)-1-(3-benzyl-1,2,4-oxadiazol-5-yl)-5-((tert-butoxycarbonyl)amino)pentan-1-aminium 4-methylbenzenesulfonate (12a) (2.7 kg, yield = 85 %, HPLC purity > 99%, ee > 99%) as white solid.1H-NMR (400 MHz, DMSO-d6): δ 8.74 (br, 3H), 7.48 (d, J = 8.0 Hz, 2H), 7.37-7.26 (m, 5H), 7.11 (d, J = 8.0 Hz, 2H), 6.77 (t, J = 5.2 Hz, 1H), 4.82 (t, J = 6.8 Hz, 1H), 4,17 (s, 2H), 2.90-2.86 (m, 2H), 2.29 (s, 3H), 1.39-1.36 (m, 11H), 1.35-1.28 (m, 2H) ppm. MS: (M-172+H)+: m/z = 361.1.
PATENT WO2021016462
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021016462&_cid=P22-M93MJV-41323-1

WO2019118878
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019118878&_cid=P22-M93MNH-43976-1
/////////Bevemipretide, SBT-272 Trihydrochloride, SBT 272, ORHAN DRUG, Stealth BioTherapeutics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}