
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 220)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Automation of Process Control within the Pharmaceutical Industry
![]()
Automation of Process Control within the Pharmaceutical Industry
While most pharmaceutical businesses have adopted process automation in one format or another, the technology has evolved considerably over the past few years, leading to improvements in design, efficiency and reliability.
One of the major drivers for businesses to increase levels of automation is legislation, but the need to compete in the market place and reduce production costs has also played a significant part.
Within the pharmaceutical industry, the key to finding the best automation solution is a thorough analysis of each individual part of the plant or installation.
By carrying out an in-depth analysis of the application, it can be determined if a centralized control system using non-intelligent nodes, will deliver the required performance, or if the sheer size of the system means that the control has to be decentralised using a fieldbus system working with field controls, intelligent valves and actuators.
Download to find out more.
Available Downloads
- Automation of Process Control within the Pharmaceutical Industry
Download

Elemental Impurity Analysis in Pharmaceuticals.free download from Butterworth labs
![]()
Elemental Impurity Analysis in Pharmaceuticals
A method to identify the presence of heavy metals in pharmaceuticals was introduced in the United States Pharmacopeia more than 100 years ago.
Pharmaceutical companies are still using essentially the same method, known as the USP Heavy Metals Limit Test.
This paper will provide an overview of current method limitations, considerations for the new methodology and risk-based assessments being carried out by manufacturers.
 see……
see……Boswellia serrata, -The cure for osteoarthritis in ayurveda, Shallaki,

in Kinnerasani Wildlife Sanctuary,Andhra Pradesh, India.
Boswellia serrata, -The cure for osteoarthritis in ayurveda, Shallaki,
Shallaki-Boswellia serrata
In degenerative and inflammatory pathologies invoving joints, there is no other drug as useful as Guggulu. Many international companies today use shallaki for the manufacture of drugs, ayurvedic and allopathic alike.
Family : Berseraceae
Scientific name : Boswellia serrata
Nomenclature in other languages :
Sanskrit : Shallaki, Susrava, Gajabhakshya
Hindi : Salei
Gujarathi : Dhoopa
Bengali : Salei
Tamil : Olibana
English : Indian Olibanum
Distribution : Gujarat, Rajasthan, Bihar are most commonly the residence of this plant.
Botanical description : It’s a resinous tree that grows to a height of 12m. A tree of moderate height , its bark are grey in colour. Upon time the bark sheds off like scales of a snake. The younger branches and leaflets of this tree are very smooth. The leaves which are compound(pinnate) in nature are 20-37 cm long. The leaflets are 2-5cm long and 1-2.5cm wide. The leaflets are oval shaped. The leaves contains 8 pairs or more of the leaflets . The margins of leaflets are serrated. Flowers are many and the inflorescence is terminal raceme, with it seen in the axilla of the leaf and stem. The petals and sepals are hairy and five in number. The stamen are 10 in number, they are diercted inwards. The fruits are seen in 3-4 numbers and are seen as drupes along with cones. The flowering season in April-May.



C hemical constituents and action
The bark contains carbohydrates, glycosides, beta-sitosterol. The resin contains ditrepene alcohol. This is knownn by the name sitosterol. In addition to that 11-keto-b-boswellic acid also has been extracted from the resin.
Ayurvedic Pharmacoepia
Rasa : kashaya, tikta, madhura
Guna : laghu, rooksha
Veerya : sheeta
Vipaka : katu
Medicinal properties :
Alleiviates vata kapha disorders. Also cures chronic skin lesions of all kinds infective and inflammatory, ulcers, wounds, piles, diseases of mouth, diarhhoea, hepatic disorders etc.
Useful parts : Bark, Resin
Therapeutic uses :
-1gm of resin taken in tablet form daily three times cures rheumatic, neurologic complaints and rheumatic fever.
-for gangrenes in diabetes the resin of this palnt may be applied externally and it taken internally as pills regularly
-the resin of this plant when chewed cures bad odour of mouth and mouth ulcers.
Medical uses
In Ayurvedic medicine Indian frankincense (Boswellia serrata) has been used for hundreds of years for treating arthritis.
Extracts of Boswellia serrata have been clinically studied for osteoarthritis and joint function, particularly for osteoarthritis of the knee, with the research showing a slight improvement of both pain and function compared to a placebo. Positive effects of Boswellia in some chronic inflammatory diseases including rheumatoid arthritis, bronchial asthma, osteoarthritis, ulcerative colitis and Crohn’s disease have been reported. A Boswellia extract marketed under the name Wokvel has undergone human efficacy, comparative, pharmacokinetic studies. Some see Boswellia serrata as a promising alternative to NSAIDs, warranting further investigation in pharmacological studies and clinical trials.

Topical application
Boswellia serrata has been recently developed for topical use in a patent-pending formula in Sano Relief Gel. Boswellia serrata is used in the manufacture of the supposed anti-wrinkle agent “Boswelox”,which has been criticised as being ineffective.
Potential for anti-cancer activity
Boswellic acid, an extract from Boswellia serrata, has been studied for anti-neoplastic activity, especially in experimental primary and secondary brain tumors, indicating potential efficacy from in vitro and limited clinical research. Boswellic acid is also undergoing an early-stage clinical trial at the Cleveland Clinic.
Active constituents
Boswellic acid and other pentacyclic triterpene acids are present. Beta-boswellic acid is the major constituent.
Mechanism of action
Animal studies performed in India show ingestion of a defatted alcoholic extract of Boswellia decreased polymorphonuclear leukocyte infiltration and migration, decreased primary antibody synthesis and almost totally inhibited the classical complement pathway.
Properties
Shallaki has potent analgesic and anti-inflammatory effects that can reduce the pain and inflammation of joints.
Frankincense ‘can ease arthritis’ researches have suggested |
|||
Extracts from Boswellia serrata, a similar species to the variety famous for its role in the Christian nativity, were tested on dozens of patients. Those who received it reported better movement and less pain and stiffness. The herb has been used for thousands of years in Indian Ayurvedic medicine, reports the journal Arthritis Research and Therapy. Current treatments carry a great many adverse effects, and scientists have been hunting for an alternative. The investigation into the properties of Boswellia serrata was led by Dr Siba Raychaudhuri at the University of California, Davis. Eventually they tested an extract of the plant enriched with the chemical – AKBA – thought to be its active ingredient. Some of the 70 patients with severe arthritis in their knees recruited into the trial were given a low-dose capsule, some a higher dose capsule, and the remainder were given a dummy pill with no active ingredients. In as little as seven days, patients taking the frankincense drug reported improvements in their pain and stiffness levels compared with the placebo group, and these continued until the 90-day mark, when the study ended. Alternative therapies Tests of the fluid within affected joints also revealed falls in levels of enzymes linked to the condition. Dr Raychaudhuri said: “We have shown that B. serrata enriched with AKBA can be an effective treatment for osteoarthritis of the knee.” However, UK experts urged caution. Professor Philip Conaghan, from Leeds University, and a spokesman for the Arthritis Research Campaign, said: “Certainly osteoarthritis is in need of new safe analgesics, although many effective therapies that reduce pain such as muscle strengthening exercises, shock-absorbing footwear and weight loss have very few bad side-effects. “This report on treating knee pain with a chemical derivative of B. serrata is interesting but the patient numbers are small, there were some problems with the reported trial design and we need more information on its medium to long-term safety.” |
|||
Boswellia serrata: an overall assessment of in vitro, preclinical, pharmacokinetic and clinical data.
Non-steroidal anti-inflammatory drug (NSAID) intake is associated with high prevalence of gastrointestinal or cardiovascular adverse effects. All efforts to develop NSAIDs that spare the gastrointestinal tract and the cardiovasculature are still far from achieving a breakthrough. In the last two decades, preparations of the gum resin of Boswellia serrata (a traditional ayurvedic medicine) and of other Boswellia species have experienced increasing popularity in Western countries. Animal studies and pilot clinical trials support the potential of B. serrata gum resin extract (BSE) for the treatment of a variety of inflammatory diseases like inflammatory bowel disease, rheumatoid arthritis, osteoarthritis and asthma. Moreover, in 2002 the European Medicines Agency classified BSE as an ‘orphan drug’ for the treatment of peritumoral brain oedema. Compared to NSAIDs, it is expected that the administration of BSE is associated with better tolerability, which needs to be confirmed in further clinical trials. Until recently, the pharmacological effects of BSE were mainly attributed to suppression of leukotriene formation via inhibition of 5-lipoxygenase (5-LO) by two boswellic acids, 11-keto-β-boswellic acid (KBA) and acetyl-11-keto-β-boswellic acid (AKBA). These two boswellic acids have also been chosen in the monograph of Indian frankincense in European Pharmacopoiea 6.0 as markers to ensure the quality of the air-dried gum resin exudate of B. serrata. Furthermore, several dietary supplements advertise the enriched content of KBA and AKBA. However, boswellic acids failed to inhibit leukotriene formation in human whole blood, and pharmacokinetic data revealed very low concentrations of AKBA and KBA in plasma, being far below the effective concentrations for bioactivity in vitro. Moreover, permeability studies suggest poor absorption of AKBA following oral administration. In view of these results, the previously assumed mode of action – that is, 5-LO inhibition – is questionable. On the other hand, 100-fold higher plasma concentrations have been determined for β-boswellic acid, which inhibits microsomal prostaglandin E synthase-1 and the serine protease cathepsin G. Thus, these two enzymes might be reasonable molecular targets related to the anti-inflammatory properties of BSE. In view of the results of clinical trials and the experimental data from in vitro studies of BSE, and the available pharmacokinetic and metabolic data on boswellic acids, this review presents different perspectives and gives a differentiated insight into the possible mechanisms of action of BSE in humans. It underlines BSE as a promising alternative to NSAIDs, which warrants investigation in further pharmacological studies and clinical trials.
Reference :
http://www.ncbi.nlm.nih.gov/pubmed/21553931
http://en.wikipedia.org/wiki/Boswellia_serrata
http://news.bbc.co.uk/2/hi/health/7535733.stm

Olaparib オラパリブ 奥拉帕尼 (AZD-2281, trade name Lynparza) AZ’ first-in-class PARP inhibitor wins EU nod


Olaparib
オラパリブ
奥拉帕尼
Women suffering from advanced relapsed BRCA-mutated ovarian cancer could gain access to a new treatment option after European regulators waved through AstraZeneca’s Lynparza (olaparib).
The European Commission has approved the first-in-class PARP inhibitor for the maintenance treatment of adults with platinum-sensitive relapsed BRCA-mutated high-grade serous epithelial ovarian, fallopian tube, or primary peritoneal cancer, who are in complete response or partial response to platinum-based chemotherapy.


4-[[3-[4-(cyclopropanecarbonyl)piperazine-1-carbonyl]-4-fluorophenyl]methyl]-2H-phthalazin-1-one, cas 763113-22-0
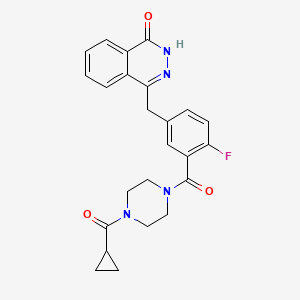
KU-0059436
KU-59436
Olaparib (AZD-2281, trade name Lynparza) is an experimental chemotherapeutic agent, developed by KuDOS Pharmaceuticalsand later by AstraZeneca, that is currently undergoing clinical trials. It is an inhibitor of poly ADP ribose polymerase (PARP), an enzyme involved in DNA repair.[1] It acts against cancers in people with hereditary BRCA1 or BRCA2 mutations, which includes many ovarian, breast and prostate cancers.
Olaparib is an oral poly-ADP-ribose polymerase (PARP) enzyme inhibitor developed by AstraZeneca. The product is awaiting registration in the E.U. and US as a maintenance treatment of patients with BRCA mutated platinum-sensitive relapsed serous ovarian cancer. In 2014, positive opinion was received in the E.U. recommending Lynparza approval for the maintanance treatment of BRCA mutated platinum-sensitive relapsed serous ovarian cancer.
An oral poly (ADP ribose) polymerase (PARP) inhibitor being investigated by British drug company AstraZeneca, is seeking approval from the U.S. Food and Drug Administration (FDA) for the treatment of BRCA mutated platinum-sensitive relapsed ovarian cancer. AstraZeneca filed the US regulatory submission for olaparib in February 2014. Olaparib, one of several cancer drugs AstraZeneca flagged as having strong potential in its defense of a $118 billion take-over bid by Pfizer,was accepted for priority review on April 30, 2014 by the U.S. Food and Drug Administration (FDA). The NDA filing was based on Phase II study 19 data, a randomized, double-blind, placebo-controlled, Phase II study.
On June 25, 2014, FDA Oncologic Drugs Advisory Committee (ODAC), an advisory panel to the U.S. Food and Drug Administration (FDA), voted 11 to two against the accelerated approval of the PARP inhibitor olaparib as a maintenance therapy for women with platinum-sensitive relapsed ovarian cancer who have the germline BRCA (gBRCA) mutation, and who are in complete or partial response to platinum-based chemotherapy. By voting no, the committee recommended waiting for results from the larger confirmatory phase III SOLO-2 trial, which began enrolling in September 2013. According to clincialtrials.gov, the SOLO-2 study (NCT01874353) is slated to wrap in July 2015.
In terms of clinical development, phase III trials are ongoing at AstraZeneca for the treatment of gastric cancer and metastatic breast cancer. Olaparib is also in phase II clinical studies for several indications, including breast cancer, pancreatic cancer and castration-resistant prostate cancer. In March 2014, a phase II was also initiated in GB for the treatment of patients with stage IIIB or stage IV NSCLC that is not amenable to curative therapy. A phase I clinical trial for the treatment of melanoma has been completed. Phase II clinical trials are ongoing at General Hospital Corp. for the treatment of sarcoma. The drug had been in phase II clinical trials for the treatment of colorectal cancer; however no recent developments have been reported.
Discovered by KuDOS Pharmaceuticals, has experienced several twists and turns during its clinical development. Promising results for the drug were reported at the 2011 ASCO Annual Meeting, based on impressive early phase II results, only to have clinical development discontinued later that year after disappointing phase II trial results in a more generalized group of ovarian cancer patients. However, a re-analysis of the data in BRCA-positive patients – coupled with a reformulation of the drug – convinced the British drugmaker to think again and keep it going. AstraZeneca initiates Phase III clinical studies (SOLO 1 and SOLO 2) for olaparib in the U.S. in September 2013. AstraZeneca has filed Marketing Authorisation Application (MAA) for olaparib in EU in September 2013 based on Phase II study 19 data. The U.S. Food and Drug Administration has already granted olaparib orphan drug status for ovarian cancer and will hold an advisory panel hearing on the company’s application on June 25, 2014.
In 2013, orphan drug designation in the U.S. was assigned to the compound for the treatment of ovarian cancer. The compound was originally developed by Kudos Pharmaceuticals, which was acquired by AstraZeneca in 2006.
Early Phase I trials were promising, and olaparib underwent Phase II trials. However, in December 2011, AstraZeneca announced following interim analysis of a phase-II study which indicated that the previously reported progression free survival benefit was unlikely to translate into an overall survival benefit, that it would not progress into Phase III development for the maintenance treatment of serous ovarian cancer,[2] and took a charge of $285 million. The decision to discontinue development of the drug was reversed in 2013,[3] with AstraZeneca posting a new Phase III trial of Olaparib for patients with BRCA mutated ovarian cancer in April 2013.[4]

Mechanism of action
Olaparib acts as an inhibitor of the enzyme Poly ADP ribose polymerase (PARP) and is one of the first PARP inhibitors. Patients with BRCA1/2 mutations may be genetically predisposed to developing some forms of cancer, and are often resistant to other forms of cancer treatment, but this also sometimes gives their cancers a unique vulnerability, as the cancer cells have increased reliance on PARP to repair their DNA and enable them to continue dividing. This means that drugs which selectively inhibit PARP may be of significant benefit in patients whose cancers are susceptible to this treatment.[5][6][7][8][9][10]

Trial results
Phase I clinical trials, in patients with BRCA-mutated tumors including ovarian cancer, were encouraging.[11] In one of these studies, it was given to 19 patients with inherited forms of advanced breast, ovarian and prostate cancers caused by mutations of the BRCA1 and BRCA2 genes. In 12 of the patients, none of whom had responded to other therapies, tumours shrank or stabilised.[12] One of the first patients to be given the treatment (who had castration-resistant prostate cancer) was as of July 2009 still in remission after two years.
In 2009 Phase II clinical trials examining the efficacy of Olaparib in treating breast, ovarian and colorectal cancer were initiated.[13][14] A phase II trial that included 63 cases of ovarian cancer concluded that olaparib is promising for women with ovarian cancer. [7 responses in 17 patients with BRCA1 or BRCA2 mutations and 11 responses in the 46 who did not have these mutations.][15]
Side effects
Olaparib is generally well tolerated, the side effects consist mainly of fatigue, somnolence, nausea, loss of appetite and thrombocytopenia.

………………………

…………….
| LOU Xi-yu, YANG Xuan, DING Yi-li, WANG Jian-jun, YAN Qing-yan, HUANG Xian-gui, GUO Yang-hui, WANG Xiang-jing, XIANG Wen-sheng Synthesis of Olaparib Derivatives and Their Antitumor Activities  |
| 2013 Vol. 29 (2): 231-235 [摘要] ( 390 ) [HTML 1KB] [PDF 0KB] ( 22 ) doi: 10.1007/s40242-013-2448-5 |
……………………….

…………………
4-[3-(4-Cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: A novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1
J Med Chem 2008, 51(20): 6581
…………………………..
http://www.google.co.in/patents/WO2004080976A1?cl=en
Synthesis of Key Intermediates
3- (4-0x0-3 , 4-dihydrophthalazin-l -ylmethyl) benzoic a cid (A)

A mixture of 27% sodium methoxide solution in methanol (400 g, 2 mol) and methanol (150 ml) was added dropwise between ambient temperature and 30°C over 15 minutes to a stirred mixture of phthalide (67 g, 0.5 mol), 3-formylbenzonitrile (65.5 g, 0.5 mol) and ethyl propionate (250 ml) , the mixture was stirred at ambient temperature for 40 minutes and at reflux temperature for 1 hour, then it was allowed to cool to ambient temperature. The resulting red solid was collected by filtration, washed with ethyl acetate (2 x 50 ml) and dissolved in water (1800 ml) . The solution was acidified by the addition of acetic acid (60 ml) and the resulting red solid was collected by filtration, washed with water (2 x 200 ml) and dried in vacuo to give 3- (1,3- dioxoindan-2-yl) benzonitrile (83.2 g) as a dark red solid, m.pt. 179- 182°C, m/z (M+H)+‘ 248, which was used without further purification.
3- (1, 3-Dioxoindan-2-yl) benzonitrile (74.18 g, 0.3 mol) was added in portions to a solution of sodium hydroxide (36 g, 0.9 mol) in water (580 ml), the resulting dark red suspension was stirred at reflux temperature for 5 hours, then it was cooled to ambient temperature and washed with ethyl acetate (3 x 300 ml) . The aqueous solution was acidified by the dropwise addition of concentrated hydrochloric acid (110 ml), the mixture was stirred at ambient temperature for 1 hour, then the resulting solid was collected by filtration, washed with water (2 x 200 ml) and dried in vacuo to give a 1:1 mixture of 3- (1,3- dioxoindan-2-yl)benzoic acid, (M+H)+” 267, and 2- [2- (3- carboxyphenyl) acetyl] benzoic acid, (M+H)+‘ 285, (69.32 g) , which was used without further purification.
The mixture obtained in the previous step (52.8 g) was added to a solution of triethylamine (37.55 g, 0.372 mol) in industrial methylated spirit (500 ml) and the resulting cloudy solution was filtered through a pad of filter-aid to give a clear solution. Hydrazine monohydrate (9.3 g, 0.186 mol) was added in one portion at ambient temperature, the stirred mixture was heated under reflux for 1 hour, then it was concentrated in vacuo to approximately 250 ml and added to a solution of sodium acetate (41 g, 0.5 mol) in water (500 ml) . The mixture was brought to pH 7 by the dropwise addition of concentrated hydrochloric acid, then it was stirred at ambient temperature for 3 hours. The resulting solid was collected by filtration, washed with water (50 ml) and dried in va cuo to give a white solid (15.62 g) . The combined filtrate and washings were acidified to pH 6 by the addition of hydrochloric acid, then the mixture was stirred at ambient temperature for 3 hours. The resulting solid was collected by filtration, washed with water (50 ml) and dried in va cuo to give a second crop of off-white solid (17.57 g) . The combined filtrate and washings from the second crop were readjusted to pH 6 and treated as before to give a third crop of pale orange solid (6.66 g) . The three crops were combined to give essentially pure 3- (4-oxo-3, 4-dihydrophthalazin-l-ylmethyl) benzoic acid (A), (M+H)+‘ 281, δH 4.4 (2H, s), 7.2-7.4 (IH, m) , 7.5-7.6 (IH, ) , 7.7-8.0 (5H, m) , 8.1- 8.2 (IH, m) , 12.6 (IH, s)
b . 2-Fluoro-5- (4-oxo-3 , 4-dihydro-phthalazin -l -ylmethyl) benzoi c a cid (B)

Dimethyl phosphite (22.0 g, 0.2 mol) was added drop-wise to a solution of sodium methoxide (43.0 g) in methanol (100 ml) at 0°C. 2- Carboxybenzaldehyde (21.0 g, 0.1 mol) was then added portion-wise to the reaction mixture as a slurry in methanol (40 ml), with the temperature kept below 5°C. The resulting pale yellow solution was warmed to 20°C over 1 hour. Methanesulphonic acid (21.2 g, 0.22 mol) was added to the reaction drop-wise and the resulting white suspension was evaporated in va cuo . The white residue was quenched with water and extracted into chloroform (3 x 100 ml) . The combined organic extracts were washed with water (2 x 100 ml) , dried over MgS04, and evaporated in va cuo to yield (3-oxo-l, 3-dihydro-isobenzofuran-l-yl) phosphonic acid dimethyl ester as a white solid (32.0 g, 95 %, 95 % purity) . This was then used without further purification in the next stage.
To a mixture of (3-oxo-l, 3-dihydro-isobenzofuran-l-yl) phosphonic acid dimethyl ester (35.0 g, 0.14 mol) in tetrahydrofuran (200 ml) and 2- fluoro-5-formylbenzonitrile (20.9 g, 0.14 mol) in tetrahydrofuran (130 ml) was added triethylamine (14 ml, 0.14 mol) drop-wise over 25 min, with the temperature kept below 15°C. The reaction mixture was warmed slowly to 20°C over 1 hour and concentrated in vacuo . The white residue was slurried in water (250 ml) for 30 minutes, filtered, washed with water, hexane and ether, and dried to yield 2-fluoro-5- (3- oxo-3H-isobenzofuran-l-ylidenemethyl) benzonitrile as a 50:50 mixture of E and Z isomers (37.2 g, 96 %); m/z [M+l]+ 266 (98 % purity) To a suspension of 2-fluoro-5- (3-oxo-3H-isobenzofuran-l- ylidenemethyl) benzonitrile in water (200 ml) was added aqueous sodium hydroxide (26.1 g in 50 ml water) solution and the reaction mixture was heated under nitrogen to 90 °C for 30 minutes. The reaction mixture was partially cooled to 70°C, and hydrazine hydrate (100 ml) was added and stirred for 18 hours at 70°C. The reaction was cooled to room temperature and acidified with 2M HC1 to pH 4. The mixture was stirred for 10 min and filtered. The resulting solid was washed with water, hexane, ether, ethyl acetate and dried to yield 2-fluoro-5- (4-oxo-3, 4- dihydrophthalazin-l-ylmethyl)benzoic acid as a pale pink powder (30.0 g, 77 %) . m/z [M+l]+ 299 (96 % purity), δH 4.4 (2H, s) , 7.2-7.3 (IH, m) , 7.5-7.6 (IH, m) , 7.8-8.0 (4H, m) , 8.2-8.3 (IH, m) , 12.6 (IH, s).
c . 1 – [3- (4-Oxo-S , 4-dihydrophthalazin-l -ylmethyl) benzoyl]piperidine-4- carboxylic a cid (C)
 undesried????????
undesried????????(A) (C)
3- (4-Oxo-3, 4-dihydrophthalazin-l-ylmethyl)benzoic acid (A) (7.0 g, 0.25 mol), ethyl isonipecotate (5 ml, 0.32 mol), 2- (lH-benzotriazol-1-yl) – 1, 1, 3, 3-tetramethyluronium hexafluorophosphate (HBTU) (12.3 g, 0.32 mol) and N, N, -diisopropylethylamine (10.0 ml, 0.55 mol) were added to dimethylacetamide (40 ml) and stirred for 18 h. Water (100 ml) was added to the reaction mixture and the product was extracted into dichloromethane (4 x 50 ml) . The combined organic layers were washed with water (3 x 100 ml), dried over MgS0, filtered and evaporated in va cuo to yield an oil. To a solution of the oil in tetrahydrofuran (100 ml) was added 10 % aqueous sodium hydroxide solution (20 ml) and the reaction was stirred for 18 hours. The reaction was concentrated, washed with ethyl acetate (2 x 30 ml) and acidified with 2M HCl to pH 2. The aqueous layer was extracted with dichloromethane (2 x 100 ml), then the extracts were dried over MgS04, filtered and evaporated to yield 1- [3- (4-oxo-3, 4-dihydrophthalazin-l-ylmethyl)benzoyl]piperidine- 4-carboxylic acid (C) as a yellow solid (7.0 g, 65 %), m/z [M+l]+ 392
(96 % purity), δH 1.3-1.8 (5H, m) , 2.8-3.1 (4H, m) , .4 (2H, s), 7.2- 7.3 (IH, m) , 7.3-7.4 (IH, ) , 7.7-8.0 (5H, m) , 8.2-E 3 (IH, m) , 12.6 (IH, s) .
d . 1 – [2-Fluoro-5- (4 -oxo-3 , 4-dihydrophthala zin-l – ylmethyl) benzoyl]piperidine-4~carboxylic a cid (D)

(B) (D)
2-Fluoro-5- ( -oxo-3, 4-dihydrophthalazin-l-ylmethyl) benzoic acid (B) (3.1 g, 0.14 mol), ethyl isonipecotate (1.7 ml, 0.11 mol), 2-(lH- benzotriazol-1-yl) -1,1,3, 3-tetramethyluronium hexafluorophosphate (HBTU) (5.1 g, 0.13 mol) and N,N, -diisopropylethylamine (10.0 ml, 0.55 mol) were added to dimethylacetamide (15 ml) and stirred for 18 hours. Water (100 ml) was added to the reaction mixture and the product was extracted into dichloromethane (4 x 50 ml) . The combined organic layers were, filtered, washed with water (3 x 100 ml), dried over MgS04, filtered and evaporated in vacuo to yield an orange oil. The oil was purified by flash chromatography (ethyl acetate) to yield l-[2- fluoro-5- (4-oxo-3, 4-dihydrophthalazin-l-ylmethyl) benzoyl] piperidine-4- carboxylic acid as the methyl ester (1.5 g, 33 %, 96 % purity) . To a solution of the methyl ester in tetrahydrofuran: water (2:1, 40 ml) was added sodium hydroxide (0.3 g, 0.075 mol) and the reaction was stirred for 18 h. The reaction was concentrated, washed with ethyl acetate (2 x 20 ml) and acidified with 2M HC1 to pH 2. The aqueous layer was extracted with dichloromethane (2 x 20 ml) , and the combined extracts were dried over MgS04 and evaporated to yield 1- [3- ( 4-oxo-3, 4- dihydrophthalazin-1-ylmethyl) benzoyl] piperidine- -carboxylic acid (D) as a yellow solid (0.6 g, 65 %), m/z [M+l]+ 392 (96 % purity) Example 1 – Synthesis of Key Compounds
a. Synthesis of 4- [3- (piperazine-1-carfoonyl)benzyl] -2H-phthalasin-l- one (1)
 undesired????????
undesired????????(A) (1)
3- (4-0xo-3, 4-dihydrophthalazin-l-ylmethyl) benzoic acid (A) (5.0g, 0.17mol), tert-butyl 1-piperazinecarboxylate (3.9 g, 0.21 mol), 2-(lH- benzotriazol-1-yl) -1,1,3, 3-tetramethyluronium hexafluorophosphate (HBTU) (8.6 g, 0.22 mol) and N, , -diisopropylethylamine (6.7 ml, 0.38 mol) were added to dimethylacetamide (40 ml) and stirred for 18 hours. Water (100 ml) was added and the reaction mixture was heated to 100°C for 1 hour. The suspension was cooled to room temperature, filtered and dried to yield a white solid. The solid was dissolved in a solution of 6M HC1 and ethanol (2:1, 50 ml) and stirred for 1 hour. The reaction was concentrated, basified with ammonia to pH 9, and the product was extracted into dichloromethane (2 x 50 ml). The combined organic layers were washed with water (2 x 50 ml), dried over MgS04, and evaporated in va cuo to yield 4- [3- (piperazine-1-carbonyl) benzyl] – 2H-phthalazin-l-one (1) as a yellow crystalline solid (4.0 g, 77 %); m/z [M+l]+ 349 (97 % purity), δH 2.6-3.8 (8H, ) , 4.4 (2H, s), 7.2-7.5 (4H, m) , 7.7-8.0 (3H, m) , 8.2-8.3 (IH, m) , 12.6 (IH, s)
b . Synthesis of 4 – [4-Fluoro-3- (piperazine-1 -carbonyl) benzyl ] -2H- phthala zin ~l -one (2)
 desired……
desired……(β) (2)
The synthesis was carried out according to the method described in (a) above using 2-fluoro-5- (4-oxo-3, -dihydrophthalazin-l-ylmethyl) benzoic acid (B) to yield 4- [4-fluoro-3- (piperazine-1-carbonyl) benzyl] -2H- phthalazin-1-one (2) as a white crystalline solid (4.8 g, 76 %); m/z [M+l]+ 367 (97 % purity), δH 2.6-3.8 (8H, m) , 4.4 (2H, s), 7.2-7.5 (3H, m) , 7.7-8.0 (3H, m) , 8.2-8.3 (IH, m) , 12.6 (IH, s) .
…………………………..
US 8183369
http://www.google.co.in/patents/US8183369
4-[3-(4-Cyclopropanecarbonyl-piperazine-1-carbonyl)-4-fluoro-benzyl]-2H-phthalazin-1-one (compound A) disclosed in WO 2004/080976:

is of particular interest.
A crystalline form of compound A (Form A) is disclosed in co-pending applications, which claim priority from U.S. 60/829,694, filed 17 Oct. 2006, entitled “Phthalazinone Derivative”, including U.S. Ser. No. 11/873,671 and WO 2008/047082.
Form A

References(a) 4-[3-(4-Cyclopropanecarbonyl-piperazine-1-carbonyl)-4-fluoro-benzyl]-2H-phthalazin-1-one (Compound A)
2-Fluoro-5-[(4-oxo-3,4-dihydrophthalazin-1-yl)methyl]benzoic acid (D)(15.23 g, 51.07 mmol) was suspended with stirring under nitrogen in acetonitrile (96 ml). Diisopropylethylamine (19.6 ml, 112.3 mmol) was added followed by 1-cyclopropylcarbonylpiperazine (I)(9.45 g, 61.28 mmol) and acetonitrile (1 ml). The reaction mixture was cooled to 18° C. 0-Benzotriazol-1-yl-tetramethyluronium hexafluorophosphate (25.18 g, 66.39 mmol) was added over 30 minutes and the reaction mixture was stirred for 2 hours at room temperature. The reaction mixture was cooled to 3° C. and maintained at this temperature for 1 hour, before being filtered. The filter cake was washed with cold (3° C.) acetonitrile (20 ml) before being dried in vacuo at up to 40° C. to give the title compound as a pale yellow solid (20.21 g).
Mass Spectrum: MH+ 435
1H NMR (400 MHz, DMSO-d6) δ: 0.70 (m, 4H), 1.88 (br s, 1H), 3.20 (br s, 2H), 3.56 (m, 6H), 4.31 (s, 2H), 7.17 (t, 1H), 7.34 (dd, 1H), 7.41 (m, 1H), 7.77 (dt, 1H), 7.83 (dt, 1H), 7.92 (d, 1H), 8.25 (dd, 1H), 12.53 (s, 1H).
………………………..
http://www.google.co.in/patents/US8247416
4-[3-(4-Cyclopropanecarbonyl-piperazine-1-carbonyl)-4-fluoro-benzyl]-2H-phthalazin-1-one (compound A) disclosed in WO 2004/080976:

is of particular interest.
In WO 2004/080976, compound A was synthesised as one of a number of library compounds from 4-[4-fluoro-3-(piperazine-1-carbonyl)-benzyl]-2H-phthalazin-1-one (compound B):

by the addition of cyclopropanecarbonyl chloride:

to a solution of (B) in dichloromethane, followed by Hünig’s base (N,N-diisopropylethyl amine). This reaction is carried out with stirring at room temperature for 16 hours, and the resulting compound being purified by preparative HPLC.
The piperazine derivative (B) was prepared by deprotecting 4-[2-fluoro-5-(4-oxo-3,4-dihydro-phthalazin-1-ylmethyl)-benzoyl]-piperazine-1-carboxylic acid tert-butyl ester (compound C):

by the use of 6M HCl and ethanol for 1 hour, followed by basification with ammonia to pH 9, and extraction into dichloromethane.
The Boc-protected piperazine derivative (C) was prepared from 2-fluoro-5-(4-oxo-3,4-dihydro-phthalazin-1-ylmethyl)-benzoic acid (compound D):

by the addition of piperazine-1-carboxylic acid tert-butyl ester:

2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and N,N,-diisopropylethylamine in dimethylacetamide, followed by stirring for 18 hours.
In WO 2004/080976, the following route to compound D is disclosed:

The method of synthesising compound D may further comprise the step of:
(c) synthesising 2-fluoro-5-[(4-oxo-3,4-dihydrophthalazin-1-yl)methyl]benzonitrile (ED):

from compound E by reaction with hydrazine hydrate; and
(d) synthesising compound D from compound ED by reaction with sodium hydroxide.
Step (c) may be achieved by using between 1.1 and 1.3 equivalents of hydrazine hydrate in tetrahydrofuran followed by neutralisation of the excess hydrazine hydrate using acetic acid.
A sixth aspect of the present invention provides the compound ED:

and its use in the synthesis of compound D.
EXAMPLES
Example 1Synthesis of Compound A

Starting material (D) was synthesised by the method disclosed in WO 2004/080976
Methods
Preparative HPLC
Samples were purified with a Waters mass-directed purification system utilising a Waters 600 LC pump, Waters Xterra C18 column (5 μm 19 mm×50 mm) and Micromass ZQ mass spectrometer, operating in positive ion electrospray ionisation mode. Mobile phases A (0.1% formic acid in water) and B (0.1% formic acid in acetonitrile) were used in a gradient; 5% B to 100% over 7 min, held for 3 min, at a flow rate of 20 ml/min.
Analytical HPLC-MS
Analytical HPLC was carried out with a Spectra System P4000 pump and Jones Genesis C18 column (4 μm, 50 mm×4.6 mm). Mobile phases A (0.1% formic acid in water) and B (acetonitrile) were used in a gradient of 5% B for 1 min rising to 98% B after 5 min, held for 3 min at a flow rate of 2 ml/min. Detection was by a TSP UV 6000LP detector at 254 nm UV and range 210-600 nm PDA. The Mass spectrometer was a Finnigan LCQ operating in positive ion electrospray mode.
(a) 4-[2-Fluoro-5-(4-oxo-3,4-dihydro-phthalazin-1-ylmethyl)-benzoyl]-piperazine-1-carboxylic acid tert-butyl ester (C)
To a stirred solution of the starting material D (850 g) in dimethylacetamide (DMA) (3561 ml) at room temperature under nitrogen was added HBTU (2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate) (1402 g) in one portion. Hünig’s base (iPr2NEt, 1096 ml) was then added with the temperature kept between 15 to 25° C. followed by a solution of 1-Boc-piperazine (637 g) in DMA (1428 ml) with the temperature kept between 15 to 25° C.
The solution was stirred at room temperature for 2 hours and sampled for completion (HPLC). Upon completion the solution was added to vigorously stirred water (17085 ml) with the temperature kept between 15 to 25° C. and the solid filtered off, washing with water (2×7131 ml), hexane (2×7131 ml) and methyl tert-butyl ether (MTBE) (2×3561 ml). The solid was then dried overnight and then sampled for water content and chemical purity.
This reaction was then repeated, see table:
| Purity | Water Content | |||
| Batch | Yield (g) | (HPLC Area %) | (K.F.) | Corrected yield |
| 1 | 1571.3 | 86.80 | 24.3 | 1032.5 g (78%) |
| 2 | 2781.6 | 85.00 | 40.3 | 1411.5 g (106%) |
| a. Greater than 100% yield attributed to non-representative sampling | ||||
(b) 4-[4-Fluoro-3-(piperazine-1-carbonyl)-benzyl]-2H-phthalazin-1-one (B)
To a stirred solution of industrial methylated spirits (IMS) (2200 ml) and concentrated HCl (4400 ml) was added compound C (2780.2 g) in portions at room temperature under nitrogen, the foaming was controlled by the addition rate. The solution was then stirred at 15 to 25° C. for 30 minutes and sampled for completion (HPLC).
Upon completion the solution was evaporated to remove any IMS and the aqueous extracted with CH2Cl2 (2×3500 ml) before the pH was adjusted to >8 using concentrated ammonia. The resultant slurry was then diluted with water (10000 ml) and extracted with CH2Cl2 (4×3500 ml), washed with water (2×2000 ml), dried over MgSO4 (250 g) and evaporated. The crude product was then slurried in CH2Cl2 (3500 ml) and added to MTBE (5000 ml). The resultant suspension was filtered and dried at 50° C. overnight yielding 611.0 g (58.5% yield) of material with a purity of 94.12%
(c) 4-[3-(4-Cyclopropanecarbonyl-piperazine-1-carbonyl)-4-fluoro-benzyl]-2H-phthalazin-1-one (A)
To a stirred suspension of compound B (1290 g) in CH2Cl2 (15480 ml) under nitrogen was added a pre-mixed solution of triethylamine (470 ml) and cyclopropane carbonyl chloride (306 ml) in CH2Cl2 (1290 ml) dropwise with the temperature kept below 20° C. The solution was then stirred at 10-15° C. for 15 minutes and sampled for completion. The reaction mixture was found to contain only 1.18% of starting material B and so the reaction was deemed complete and the batch was then worked-up.
The reaction mixture was washed with water (7595 ml), 5% citric acid solution (7595 ml), 5% sodium carbonate solution (7595 ml) and water (7595 ml). The organic layer was then dried over magnesium sulfate (500 g).
The CH2Cl2 containing product layer was then isolated, filtered through Celite and charged to a 251 vessel. CH2Cl2 (8445 ml) was then distilled out at atmospheric pressure and ethanol (10000 ml) added. Distillation was then continued with every 4000 ml of distillate that was removed being replaced with ethanol (4000 ml) until the head temperature reached 73.7° C. The reaction volume was then reduced (to 7730 ml) by which time the head temperature had reached 78.9° C. and the solution was allowed to cool to 8° C. overnight. The solid was then filtered off, washed with ethanol (1290 ml) and dried at 70° C. overnight. Yield=1377.3 g (90%). HPLC purity (99.34% [area %]). Contained 4.93% ethanol and 0.45% CH2Cl2 by GC.
(d) Water Treatment of Compound A
A suspension of compound A (1377.0 g), as produced by the method of Example 1, in water (13770 ml) was heated to reflux for 4 hours, cooled to room temperature and filtered. The solid was washed with water (2754 ml) and dried at 70° C. overnight. Yield=1274.8 g (92.6%). HPLC purity (99.49% [area %]). Contained 0.01% ethanol and 0.01% CH2Cl2 by GC.
1H NMR spectrum of compound A (DMSO-d6) following the water treatment is shown in FIG. 1.

The powder XRD pattern of Compound A following the water treatment is shown in FIG. 2, which shows the compound is as Form A.
Example 2
Alternative Synthesis of Compound A Using 1-(cyclopropylcarbonyl) piperazine

Methods (also for Examples 3 & 4)
NMR
1H NMR spectra were recorded using Bruker DPX 400 spectrometer at 400 MHz. Chemical shifts were reported in parts per million (ppm) on the δ scale relative to tetramethylsilane internal standard. Unless stated otherwise all samples were dissolved in DMSO-d6.
Mass Spectra
Mass spectra were recorded on an Agilent XCT ion trap mass spectrometer using tandem mass spectrometry (MS/MS) for structural confirmation. The instrument was operated in a positive ion elctrospray mode.
(a) 4-[3-(4-Cyclopropanecarbonyl-piperazine-1-carbonyl)-4-fluoro-benzyl]-2H-phthalazin-1-one (Compound A)
2-Fluoro-5-[(4-oxo-3,4-dihydrophthalazin-1-yl)methyl]benzoic acid (D)(15.23 g, 51.07 mmol) was suspended with stirring under nitrogen in acetonitrile (96 ml). Diisopropylethylamine (19.6 ml, 112.3 mmol) was added followed by 1-cyclopropylcarbonylpiperazine (1)(9.45 g, 61.28 mmol) and acetonitrile (1 ml). The reaction mixture was cooled to 18° C. O-Benzotriazol-1-yl-tetramethyluronium hexafluorophosphate (25.18 g, 66.39 mmol) was added over 30 minutes and the reaction mixture was stirred for 2 hours at room temperature. The reaction mixture was cooled to 3° C. and maintained at this temperature for 1 hour, before being filtered. The filter cake was washed with cold (3° C.) acetonitrile (20 ml) before being dried in vacuo at up to 40° C. to give the title compound as a pale yellow solid (20.21 g).
Mass Spectrum: MH+435
1H NMR (400 MHz. DMSO-d6) δ: 0.70 (m, 4H), 1.88 (br s, 1H), 3.20 (br s, 2H), 3.56 (m, 6H), 4.31 (s, 2H), 7.17 (t, 1H), 7.34 (dd, 1H), 7.41 (m, 1H), 7.77 (dt, 1H), 7.83 (dt, 1H), 7.92 (d, 1H), 8.25 (dd, 1H), 12.53 (s, 1H).
Example 3Alternative Synthesis of Compound A Using 1-(cyclopropylcarbonyl) piperazine HCl salt

(a) 1-(Cyclopropylcarbonyl)piperazine HCl salt (I′)
Acetic acid (700 ml) was treated with piperazine (50.00 g, 0.581 mol) portionwise over 15 minutes with stirring under nitrogen The reaction mixture was warmed to 40° C. and maintained at this temperature until a complete solution was obtained. Cyclopropanecarbonyl chloride 59.2 ml, 0.638 mol) was added over 15 minutes. The reaction mixture was stirred at room temperature overnight. The reaction mixture was filtered and the filtrate distilled under reduced pressure until ˜430 ml of distillates had been collected. Toluene (550 ml) was charged to the reaction mixture and reduced pressure distillation continued until a further 400 ml of distillates were collected. A further charge of toluene (550 ml) was added and reduced pressure distillation continued until 350 ml of distillates were collected. The resulting slurry was diluted with toluene (200 ml) and stirred overnight. Further toluene (500 ml) was added in order to mobilise the slurry. The slurry was filtered, washed with toluene (100 ml) and dried in vacuo at 40° C. to give the title compound as an off white solid (86.78 g).
Mass Spectrum: MH+155
1H NMR (400 MHz. D2O) δ: 0.92 (m, 4H), 1.98 (m, 1H), 3.29 (m, 2H), 3.38 (m, 2H), 3.84 (m, 2H), 4.08 (m, 2H).
(b) Compound A
2-Fluoro-5-[(4-oxo-3,4-dihydrophthalazin-1-yl)methyl]benzoic acid (D)(0.95 g, 3.19 mmol) was suspended with stirring under nitrogen in acetonitrile (4 ml). 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) (1.45 g, 3.83 mmol) was added followed by 1-cyclopropylcarbonylpiperazine HCl salt (I′)(0.73 g, 3.83 mmol). Diisopropylethylamine (1.39 ml, 7.98 mmol) was added over 3 minutes and the reaction mixture was stirred for overnight at room temperature. The reaction mixture was cooled to 5° C. and maintained at this temperature for 1 hour, before being filtered. The filter cake was washed with cold (3° C.) acetonitrile (2 ml) before being dried in vacuo at up to 40° C. to give the title compound as a pale yellow solid (0.93 g).
- “Olaparib, a PARP Inhibitor”. Health and Life.
- “AZ updates on olaparib and TC5214”. 20 December 2011.
- http://uk.reuters.com/article/2013/09/04/astrazeneca-cancer-idUKL6N0H00KN20130904
- http://www.clinicaltrials.gov/ct2/show/NCT01844986
- New cancer drug ‘shows promise’ BBC News 24 June 2009
- Olaparib for the treatment of ovarian cancer.
- Vasiliou S, Castaner R, Bolos J. Olaparib. Drugs of the Future. 2009; 34(2): 101.
- Menear KA, Adcock C, Boulter R, Cockcroft XL, Copsey L, Cranston A, Dillon KJ, Drzewiecki J, Garman S, Gomez S, Javaid H, Kerrigan F, Knights C, Lau A, Loh VM, Matthews IT, Moore S, O’Connor MJ, Smith GC, Martin NM (October 2008). “4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1”. Journal of Medicinal Chemistry 51 (20): 6581–91. doi:10.1021/jm8001263. PMID 18800822.
- Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M, Zevenhoven J, Lau A, Boulter R, Cranston A, O’Connor MJ, Martin NM, Borst P, Jonkers J (November 2008). “High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs”. Proceedings of the National Academy of Sciences of the United States of America 105 (44): 17079–84. doi:10.1073/pnas.0806092105. PMC 2579381. PMID 18971340.
- Hay T, Matthews JR, Pietzka L, Lau A, Cranston A, Nygren AO, Douglas-Jones A, Smith GC, Martin NM, O’Connor M, Clarke AR (May 2009). “Poly(ADP-ribose) polymerase-1 inhibitor treatment regresses autochthonous Brca2/p53-mutant mammary tumors in vivo and delays tumor relapse in combination with carboplatin”. Cancer Research 69 (9): 3850–5. doi:10.1158/0008-5472.CAN-08-2388. PMID 19383921.
- http://www.ncri.org.uk/ncriconference/archive/2007/abstracts/pdf/LB57.pdf “A Phase I trial of AZD2281 (KU-0059436), a PARP inhibitor with single agent anticancer activity in patients with BRCA deficient tumours, particularly ovarian cancer”
- Fong PC, Boss DS, Yap TA, et al. (July 2009). “Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers”. N. Engl. J. Med. 361 (2): 123–34.doi:10.1056/NEJMoa0900212. PMID 19553641.
- http://www.cancercompass.com/cancer-news/1,15869,00.htm “Phase II Trials Investigating Oral PARP Inhibitor, Olaparib, In BRCA-Deficient Advanced Breast And Ovarian Cancer” June 2009
- http://clinicaltrials.gov/ct2/show/NCT00912743 Efficacy and Safety of Olaparib in Pretreated Patients With Measurable Colorectal Cancer, Stratified by Microsatellite Instability (MSI) Status
- “Olaparib Looks Promising in Treatment of Non-BRCA Ovarian Cancer”. 26 Aug 2011.
| Patent | Submitted | Granted |
|---|---|---|
| Phthalazinone Derivatives [US2012010204] | 2012-01-12 | |
| PARP1 TARGETED THERAPY [US2012035244] | 2012-02-09 | |
| Phthalazinone derivatives [US7449464] | 2005-03-17 | 2008-11-11 |
| 4- [3- (4-CYCLOPROPANECARBONYL-PIPERAZINE-I-CARBONYL) -4 -FLUORO-BENZYL] -2H-PHTHALAZ IN-1-ONE [US8183369] | 2010-11-11 | 2012-05-22 |
| PHTHALAZINONE DERIVATIVES [US7692006] | 2008-06-19 | 2010-04-06 |
| PHTHALAZINONE DERIVATIVES [US7981889] | 2008-08-21 | 2011-07-19 |
| PHARMACEUTICAL FORMULATION 514 [US2010098763] | 2010-04-22 | |
| PHTHALAZINONE DERIVATIVE [US8247416] | 2009-10-29 | 2012-08-21 |
| WO2002036576A1 * | 25 Oct 2001 | 10 May 2002 | Kudos Pharm Ltd | Phthalazinone derivatives |
| WO2002090334A1 * | 30 Apr 2002 | 14 Nov 2002 | Kudos Pharm Ltd | Isoquinolinone derivatives as parp inhibitors |
| WO2003093261A1 * | 29 Apr 2003 | 13 Nov 2003 | Kudos Pharm Ltd | Phthalazinone derivatives |
extras…………..
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 4-[(3-[(4-cyclopropylcarbonyl)piperazin-4-yl]carbonyl) -4-fluorophenyl]methyl(2H)phthalazin-1-one | |
| Clinical data | |
| Trade names | Lynparza |
| Legal status |
|
| Routes | Oral |
| Identifiers | |
| CAS number | 763113-22-0 |
| ATC code | None |
| PubChem | CID 23725625 |
| ChemSpider | 23343272 |
| UNII | WOH1JD9AR8 |
| ChEMBL | CHEMBL521686 |
| Chemical data | |
| Formula | C24H23FN4O3 |
| Mol. mass | 435.08 g/mol |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
REFERENCES
nmr
 CAS NO. 763113-22-0, olaparib H-NMR spectral analysis |
 CAS NO. 763113-22-0, olaparib C-NMR spectral analysis |
Butoconazole

1-(4-(4-chlorophenyl)-2-(2,6-dichlorophenylthio)-n-butyl)-1H-imidazole
64872-77-1 NITRATE ,
64872-76-0 (free base)
Butoconazole nitrate, RS-35887-00-10-3, RS-35887, Gynomyk, Gynazole-1, Femstat

Brief background information
| Salt | ATC | Formula | MM | CAS |
|---|---|---|---|---|
| – | G01AF15 | C 19 H 17 Cl 3 N 2 S | 411.78 g / mol | 64872-76-0 |
| mononitrate | G01AF15 | C 19 H 17 Cl 3 N 2 S ⋅ HNO 3 | 474.80 g / mol | 64872-77-1 |
No Exclusivity found
| Drug Name | Femstat 3 (from Drugs@FDA) |
|---|---|
| Active Ingredient | Butoconazole nitrate |
| Dosage Form | Cream |
| Route | Vaginal |
| Strength | 2% |
| Market Status | Over the Counter |
| Company | Bayer |
- 64872-77-1 , butoconazole nitrate
- CAS Number: 64872-77-1
- Molecular Weight: 474.79
- Molecular Formula: C19H17Cl3N2S ·HNO3
| Patent No | Patent Expiry | |
|---|---|---|
| 5993856 | Nov 17, 2017 |
Laszlo Czibula, Laszlo Dobay, Eva Werkne Papp, Judit Nagyne Bagdy, Ferenc Sebok, “High Purity Butoconazole Nitrate with Specified Particle Size and a Process for the Preparation Thereof.” U.S. Patent US20080221190, issued September 11, 2008.

 |
|
| Systematic (IUPAC) name | |
|---|---|
| 1-[4-(4-Chlorophenyl)-2-(2,6-dichlorophenyl)sulfanylbutyl]imidazole | |
| Clinical data | |
| Trade names | Gynazole-1, Mycelex-3 |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a682012 |
| Pregnancy cat. |
|
| Legal status | |
| Routes | Vaginal cream |
| Identifiers | |
| CAS number | 67085-13-6 |
| ATC code | G01AF15 |
| PubChem | CID 47472 |
| DrugBank | DB00639 |
| ChemSpider | 43192 |
| UNII | 0Q771797PH |
| KEGG | D00880 |
| ChEBI | CHEBI:3240 |
| ChEMBL | CHEMBL1295 |
| Chemical data | |
| Formula | C19H17Cl3N2S |
| Mol. mass | 411.776 g/mol |
Use
-
an antifungal agent for topical use
Classes substance
-
Eter chlorothiophenol
-
Imidazoles
-
Synthesis pathway
| Synthesis of a) |
|---|
  |
Trade names
| Country | Trade name | Manufacturer |
|---|---|---|
| France | Ginomik | Cassenne |
| USA | Femstat | Syntex |
| Ukraine | Gіnofort | BAT “Gideon Rіhter” Ugorschina |
Formulations
-
2% vaginal cream
Reference for syn
-
Synthesis of a)
-
Walker, KAM et al .: J. Med. Chem. (JMCMAR) 21, 840 (1978).
-
US 4,078,071 (Syntex; USA-prior. 28.7.1975).
-
DOS 2,800,755
-


………………………
Patent
http://www.google.com/patents/EP1709005A1?cl=en
Butoconazole nitrate (chemical name: l-[4-(4-chlorophenyl)-2-(2,6-dichloro- -phenylthio)-n-butyl]-imidazol nitrate) is a compound of the formula (I),

(I)
it belongs among the aryl-ethylimidazole compounds, has fungicidal activity and may be used for the treatment of vaginal infections caused primarily by Candida albicans. Azoles exert their antifungal effect via modifying the ergosterol synthesis of fungus cells; more particularly, imidazoles inhibit the 14α-demethylase enzyme, thereby bringing about an increased level of 14α-methyl sterols which, in turn, causes an alteration of cell membrane permeability leading to the destruction of the fungus cells (Tetrahedron: Asymmetry Vol 4, No. 7, pp. 1521-1526, 1993). The first process for the preparation of the butoconazole nitrate is a multistep synthesis disclosed in the US 4,078,071 patent specification. Here two reaction routes are given for the preparation of the key intermediate of the formula (TV) (l-[4-(4-chlorophenyl)-2-hydroxy-n- -butyl] -imidazole) .

(IN)
According to one of them first an epoxy compound is prepared from an aromatic aldehyde or from an olefinic compound having a terminal double bond; then the epoxy compound is reacted with imidazole to yield the key intermediate. The aromatic aldehyde (VIII)

(VIII)
is treated with expensive and hazardous reagents (trimethylsulfoxonium iodide and sodium hydride) in dry dimethyl sulfoxide and the epoxide formed in the reaction is isolated after a complicated work-up. The epoxide so obtained is converted to the imidazole derivate in a time consuming reaction in the presence of dimethylformamide, then the key intermediate of the formula (IN) (l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) is isolated and purified in an additional step. From the compounds having terminal double bond (Nil)

(Nil) the epoxide is obtained via a highly explosive peracidic oxidation step and the epoxide is then converted into (l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) (IV) in a manner described above. In the other reaction route a poisoning aromatic α-halo-keto compound is used as starting material which is reacted with imidazole to give the corresponding keto-imidazole which, in turn, is reduced with a complex metal hydride – a reagent with potential hazards – to yield the key intermediate (IN). The reaction mixture is worked up in an involved manner. The synthesis way described in J. Med. Chem., 1978, Vol. 21, No. 8, pp 840-843 is as follows: l-chloro-4-chlorophenyl-2-butanol (II)

(II) is treated with the imidazole (III)

(HI)
in the presence of sodium hydride reagent in dimethylformamide solvent. This substitution reaction takes a long time and gives the (l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]- imidazole) (IN) with a poor yield (51.7 %). In the next step of the butoconazole nitrate synthesis
(l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) (IN) is treated with thionyl chloride (which is at once a reagent and a solvent) at 65-70 °C to yield l-[4-(4-chlorophenyl)-2-chloro- -n-butyl] -imidazole of the formula (N).

(V)
The reaction mixture is then evaporated to dryness. The removal of the excess thionyl chloride, a highly corrosive substance, requires special equipment; the same applies to waste treatment, an operation which also involves an environmental risk. The residue is dissolved in dichloromethane, the solution is made alkaline by adding aqueous potassium carbonate solution. Phases are separated, the organic layer is washed with water, dried on magnesium sulphate and evaporated to give l-[4-(4-chlorophenyl)-2-chloro-n-butyl]-imidazole (N), as a gum. Said gum is dissolved in acetone and reacted with 2,6-dichlorothiophenol in the presence of potassium carbonate with a long reaction time. After the reaction has been finished, the inorganic salts are removed by filtration, the solvent is evaporated, and the residue is partitioned between water and ether. Butoconazole nitrate is precipitated with nitric acid from the ethereal layer. The end-product crystals in white plates from a mixture of acetone and ethyl acetate (yield: 84 %). Our aim was to provide a process by which the active agent can be prepared in high purity via reaction steps producing good yields and besides that said steps require neither solvents that are highly flammable and explosive (ether), carcinogenic (dimethylformamide) or corrosive (thionylchloride), nor reagents (e. g. sodium hydride) that are highly flammable or explosive. We have surprisingly found that when the starting material l-chloro-4-chlorophenyl-2-
-butanol (II) is reacted with the imidazole (III) in a mixture of toluene and aqueous sodium hydroxide solution in the presence of a phase transfer catalyst, the
(l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) (IN) key intermediate is obtained with short reaction time and excellent yield (95 %). Next we studied alternative solvents to replace the thionyl chloride in solvent function in the reaction step converting (l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) (IN) into (l-[4-(4-chlorophenyl)-2-chloro-n-butyl]-imidazole) (N). In the inert solvents which could be taken into account such as dichloromethane, toluene, chlorobenzene and dimethylformamide, the chlorinating reaction yielded a sticky reaction mixture which couldn’t be processed. We have surprisingly found, however that when (l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) (IN) is dissolved in 1 ,2-dichloroethane and reacted with approximately equimolar amount of thionyl chloride reagent in the presence of catalytic amount of dimethylformamide at 30-35 °C temperature, a crystal suspension is obtained which is easy-to-stir during the whole reaction time, resulting in that chlorination proceeds completely giving l-[4-(4-chlorophenyl)-2-chloro-n-butyl]-imidazole (N) in quantitative yield. Being the compound sufficiently pure, it is not isolated, but separated by extraction and reacted directly with 2,6-dichlorothiophenol in methyl isobutyl ketone to give 1 -[4-(4-chlorophenyl)-2-(2,6-dichlorophenylthio)-n-butyl]-imidazole (VI) (butoconazole).

(NI)
Example 1. Preparation of (1 4-(4-chlorophenyl)-2-hvdroxy-n-butyll-imidazole) (IV) To a solution of 56.7 g (0.26 mol) of l-chloro-4-chloroρhenyl-2-butanol (J. of Medicinal Chemistry, 1978. Nol. 21. No. 8. p. 842) in 200 ml of toluene 36.2 g (0.9 mol) of sodium hydroxide dissolved in 100 ml of water, 6.4 g (0.028 mol) of benzyltriethyammom‘um chloride and 35.2 g (0.51 mol) of imidazole (III) are added. The reaction mixture is heated at 93-95 °C for one hour then the temperature is returned to about 60 °C, the phases are separated and to the organic layer water (100 ml) is added. The mixture is first stirred at 22-25 °C for 1 hour then at 0-5 °C for two hours. The crystals are separated by filtration, washed with water (2 x 35 ml) of 0-5 °C to yield 74 g of wet (l-[4-(4-chloroρhenyl)-2-hydroxy-n-butyl]-imidazole) which is dried at maximum 50 °C in vacuo to give 61.6 g (95 %) of the product. Recrystallization from ethyl acetate gives 52.4 g (85 %) of dry product melting at 104-106 °C.
Example 2. Preparation of l-[4-(4-chlorophenvπ-2-(2,6-(McMorophenyl o)-n-butyl1-ϊmidazole nitrate (I) 25 g (0.1 mol) of l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole (IN) is suspended in 1,2-dichloroethane (125 ml), to this suspension dimethylformamide (1 ml) and thionyl chloride (13.6 g; 0.11 mol) are added at 30-32 °C and the reaction mixture is kept at 35-38 °C for 1.5 hour under stirring. After the chlorination has been finished the homogenous solution is cooled to 15-18 °C, the excess of thionyl choride is decomposed with water (10 ml) then again water (80 ml) is added to the solution. After stirring at 20-22 °C for 0.5 hour the phases are separated and the organic layer is extracted with water (30 ml). To the aqueous solution methyl isobutyl ketone (250 ml) is added and the pH of the mixture is adjusted to 8.5 – 9 with 15 g (0.14 mol) of sodium carbonate dissolved in water (70 ml). The mixture is stirred at 22-25 °C for 0.5 hour, phases are separated, from the organic layer an 50 ml portion is distilled off to remove water and to the remaining solution 26.8 g (0.15 mol) of 2,6-dichloro-thiophenol and 40 g (0.29 mol) of dry potassium carbonate are added. The suspension is stirred at 105 – 108 °C under nitrogen for 3-4 hours. After the reaction has been finished the inorganic salts are removed by filtration at 22-25 °C, the filtrate is washed and clarified with activated carbon and the pH of the clear solution is adjusted to 3 – 3.5 by adding about 8 – 9 ml of 65 % nitric acid. The solution is stirred at the same temperature for 1 hour then the temperature is lowered to 8 – 12 °C. The crystals obtained are filtered and washed to give 48 g of wet l-[4-(4-chlorophenyl)-2-(2,6-dichlorophenylthio)-n-butyl]- -imidazole nitrate corresponding to 42.6 g (90 %) of dry product.
HPLC
Details of the HPLC method: Type of the apparatus: Spectra System/TSP (manufacturer: Thermo Separation Products, USA) Column: LiChrospher RP-18, 250×4.0 mm ID., 5 μm (Merck, Germany, Cat. No. : 1.50983) Mobile phase: methanol : buffer = 8:2 Bujfer: 2.18 g KH2PO4 + 4.18 g K2HPO4-3H2O dissolved in 1000 ml of distilled water; MeOH (HPLC Gradient grade, Merck, Germany, Cat. No.: 1.06007.2500) KH2PO4 (p.a., Merck, Germany, Cat. No.: 1.04877.1000) K2HPO4-3H2O (p.a., Merck, Germany, Cat. No.: 1.05099.1000) Flow rate: 1.0 ml/min Temperature: 40 °C Detection: UN 229 nm Solvent for sampling: eluent Sample concentration: 1.0 mg/ml Injected volume: 10 μl Duration of analysis: 40 min Evaluation: area normalization method. Approximative retention time: 11.9 min B. Particle size: Particle size was determined by sieve analysis using an Alpine sieve operated by a jet of air.
……………………..
WALKER K A M ET AL: “1-[4-(4-Chlorophenyl)-2-(2,6-dichloro phenylthio)-n-butyl]-1H-imidazole nitrate, a new potent antifungal agent” JOURNAL OF MEDICINAL CHEMISTRY, vol. 21, no. 8, August 1978 (1978-08), pages 840-843,
http://pubs.acs.org/doi/pdf/10.1021/jm00206a028
1- [4-(4-chlorophenyl)-2-(2,6-dichlorophenylthio)-n-b~-
tyll-lH-imidazole nitrate (I).
I as colorless blades
(9.6 g, 84%): mp 162-163 “C (foaming). Anal. (C19H18C13N303S)
C, H, N. The free base prepared by neutralization of a suspension
of 1 in ether with aqueous potassium carbonate and recrystallization
from cyclohexane had mp 68-70.5 “C (foaming).
……………….
FULL SYNTHESIS
SEE
http://www.chemdrug.com/databases/8_0_yyfgohllmfsvfvsx.html

The chlorohydrin (II) is obtained by the reaction of p-chlorobenzylmagnesium chloride (I) with epichlorohydrin (A) in ether. This is then converted to the crystalline alcohol (III) by reaction with sodium imidazole (B) in DMF. On treatment with thionyl chloride is converted to the corresponding chloro compound (IV). When (IV) is reacted with 2,6-dichloro thiophenol (C) in the presence of anhydrous potassium carbonate in acetone, the free base of butoconazole is formed. Neutralization of the free base (V) with nitric acid gives butoconazole.
References
- Seidman, L. S.; Skokos, C. K. (2005). “An evaluation of butoconazole nitrate 2% site release vaginal cream (Gynazole-1) compared to fluconazole 150 mg tablets (Diflucan) in the time to relief of symptoms in patients with vulvovaginal candidiasis”. Infectious diseases in obstetrics and gynecology 13 (4): 197–206. doi:10.1080/10647440500240615. PMC 1784583. PMID 16338779.
- Butoconazole monograph
Literature References:
Imidazole derivative with antifungal properties. Prepn: K. A. M. Walker, US 4078071 (1978 to Syntex).
Prepn, toxicity, activity vs Candida albicans in mice: K. A. M. Walker et al., J. Med. Chem. 21, 840 (1978).
In vitro comparison with other antifungal agents: F. C. Odds et al., J. Antimicrob. Chemother. 14, 105 (1984).
Clinical trials in treatment of vulvovaginal candidiasis: W. Droegemueller et al., Obstet. Gynecol. 64, 530 (1984); J. B. Jacobson et al., Acta Obstet. Gynecol. Scand. 64, 241 (1985).
Comparison with miconazole, q.v.: C. S. Bradbeer et al., Genitourin. Med. 61, 270 (1985).
FDA issues Guidance for a clear Identification of pharmaceutical Companies
DRUG REGULATORY AFFAIRS INTERNATIONAL

| FDA issues Guidance for a clear Identification of pharmaceutical Companies |
| In November the US FDA has issued a Guidance for a clear identification of pharmaceutical companies. The authority now definitely prefers the DUNS system. Get more information. |
In our GMP News from September 2013 you learned about a draft of a FDA Guidance for Industry entitled “Specification of the Unique Facility Identifier (UFI) System for Drug Establishment Registration”. This document’s goal was to clearly identify pharmaceutical sites. The draft comprised (manageable) five pages – including the cover page. And in terms of volume this didn’t change. However, some of the alternatives still mentioned in the draft, are not stated any longer – as one can find out when contacting the authority in these cases. The method now wanted is a registration by a D-U-N-S- (Data Universal Numbering System) number. This number – which is a 9-digit code – is…
View original post 32 more words
Ranbaxy to introduce malarial treatment Synriam in African nations

Ranbaxy to introduce malarial treatment Synriam in African nations
Ranbaxy Laboratories has obtained regulatory approval to introduce India’s first new chemical entity (NCE) Synriam (arterolane maleate 150mg and piperaquine phosphate 750mg drug) in seven African countries.
read at
Synriam is a new age therapy recommended to treat uncomplicated Plasmodium falciparum malaria in adults. It was launched in India in April 2012.
The product was also launched in Uganda and is set to be introduced in Nigeria, Senegal, Cameroon, Guinea, Kenya and Ivory Coast by the end of January 2015.

Arterolane

Arterolane, also known as OZ277 or RBx 11160,is a substance being tested for antimalarial activity[1] by Ranbaxy Laboratories.[2] It was discovered by US and European scientists who were coordinated by the Medicines for Malaria Venture (MMV).[3] Its molecular structure is uncommon for pharmacological compounds in that it has both an ozonide group and an adamantane substituent.[4]
Phase III clinical trials of arterolane, in combination with piperaquine, began in India in 2009.[5] When clinical trial results were disappointing, the MMV withdrew support[2] and Ranbaxy continued developing the drug combination on its own.

Ranbaxy launched India’s first new drug, SynriamTM, treating Plasmodium falciparummalaria in adults. The drug provides quick relief from most malaria-related symptoms, including fever, and has a high cure rate of over 95 %.
Just one tablet per day is required, for three days, instead of two to four tablets, twice daily, for three or more days with other medicines. The drug is independent of dietary restrictions for fatty foods or milk.
Ranbaxy developed Synriam as a fixed-dose combination of arterolane maleate and piperaquine phosphate, where arterolane is the new chemical entity (NCE) that was developed as an alternative to artemisinin. It is the first recently developed antimalarial not based on artemisinin, one of the most effective treatments for malaria, which has shown problems with resistance in recent years. Arterolane was discovered by a collaborative drug discovery project funded by the Medicines for Malaria Venture. Since SynriamTM has a synthetic source, unlike artemisinin-based drugs, production can be scaled up whenever required and a consistent supply can be maintained at a low cost.

The new drug, has been approved by the Drug Controller General of India (DCGI) for marketing in India and conforms to the recommendations of the World Health Organization (WHO) for using combination therapy in malaria. Ranbaxy is also working to make it available in African, Asian and South American markets where Malaria is rampant. SynriamTM trials are ongoing for Plasmodium vivax malaria and a paediatric formulation.
Derek Lowe of the famous In the Pipeline blog had written about arterolane in 2009. At the time it was in Phase III trial, which I assumed were the trials that Ranbaxy was conducting. But it turned out that arterolane was developed by a collaboration between researchers in the US, the UK, Switzerland and Australia who were funded by the World Health Organization and Medicines for Malaria Venture (a Swiss non-profit).
They published this work in Nature in 2004 and further SAR (Structure Activity Relationship) studies in J Med Chem in 2010. So Ranbaxy did not develop the drug from scratch? But the press release quotes Arun Sawhney, CEO and Managing Director of Ranbaxy which misleads people to think so: “It is indeed gratifying to see that Ranbaxy’s scientists have been able to gift our great nation its first new drug, to treat malaria, a disease endemic to our part of the world.
This is a historic day for science and technology in India as well as for the pharmaceutical industry in the country. Today, India joins the elite and exclusive club of nations of the world that have demonstrated the capability of developing a new drug”. So Ranbaxy mixes a known active compound (piperaquine) with a new compound that someone else found to be active (arterolane) and claims that they developed a new drug?
In an interview in LiveMint, Sawhney says, “Ranbaxy spent around $30 million on Synriam and the contribution from DST [India’s Department of Science & Technology] was Rs.5 crore.
The drug went through several phases of development since the project began in 2003. We did not look at this as a commercial development. Instead, this is a CSR [Corporate Social Responsibility] venture for us.” That’s a give away because developing a new drug from scratch has to cost more than $30 million + Rs.50 million.
- Ranbaxy Laboratories Limited (Ranbaxy), India
![]() now taken over by sun
now taken over by sun
SynriamTM
|
|
Description SynriamTM is a fixed dose combination of two antimalarial active ingredients arterolane maleate and piperaquine phosphate.
Arterolane maleate is a synthetic trioxolane compound. The chemical name of arterolane maleate is cis-adamantane-2-spiro-3’-8’-[[[(2’-amino-2’ methylpropyl) amino] carbonyl] methyl] 1’,2’,4’-trioxaspiro [4.5] decane hydrogen maleate. The molecular formula is C26H40N2O8 and molecular weight is 508.61. The structural formula is as follows:

| MALARIA |
| Malaria is one of the most prevalent and deadly parasitic diseases in the world. Up to 289 million cases of malaria may have occurred in 2010, causing between 660,000 and 1.25 million deaths, mainly in Africa and mostly of children younger than 5 years. |
| (WHO: http://www.who.int/malaria/publications/world_malaria_report_2012/en/index.html; Fidock, D. A. Eliminating Malaria. Science 2013, 340, 1531-1533.) |
|


Malaria, the most common parasitic disease of humans, remains a major health and economic burden in most tropical countries. Large areas of Central and South America, Hispaniola (Haiti and the Dominican Republic), Africa, the Middle East, the Indian subcontinent, Southeast Asia, and Oceania are considered as malaria-risk areas. It leads to a heavy toll of illness and death, especially amongst children and pregnant women.
According to the World Health Organization, it is estimated that the disease infects about 400 million people each year, and around two to three million people die from malaria every year. There are four kinds of malaria parasites that infect human: Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale and Plasmodium malariae.
Malaria spreads from one person to another by the bite of mosquito, Anopheles gambiae, which serves as vector. When a mosquito sucks the blood of human, sporozoites are transfused into the human body together with saliva of the mosquito. The sporozoites enter into the hepatocytes, reproduce asexually and finally enter into the blood stream. The parasites continue to multiply inside the red blood cells, until they burst and release large number of merozoites.
This process continues, destroying a significant number of blood cells and causing the characteristic paroxysm (“chills and fever”) associated with the disease. In the red blood cells, some of the merozoites become male or female gametocytes. These gametocytes are ingested by the mosquito when it feeds on blood. The gametocytes fuse in the vector’s gut; sporozoites are produced and are migrated to the vector’s salivary glands.
The clinical symptoms of malaria are generally associated with the bursting of red blood cells causing an intense fever associated with chills that can leave the infected individual exhausted and bedridden. More severe symptoms associated with repeat infections and/or infection by Plasmodium falciparum include anaemia, severe headaches, convulsions, delirium and, in some instances, death.
Quinine, an antimalarial compound that is extracted from the bark of cinchona tree, is one of the oldest and most effective drugs in existence. Chloroquine and mefloquine are the synthetic analogs of quinine developed in 1940’s, which due to their effectiveness, ease of manufacture, and general lack of side effects, became the drugs of choice. The downside to quinine and its derivatives is that they are short-acting and have bitter taste.
Further, they fail to prevent disease relapses and are also associated with side effects commonly known as “Chinchonism syndrome” characterized by nausea, vomiting, dizziness, vertigo and deafness. However, in recent years, with the emergence of drug- resistant strains of parasite and insecticide-resistant strains of vector, the treatment and/or control of malaria is becoming difficult with these conventional drugs.
Malarial treatment further progressed with the discovery of Artemisinin
(qinghaosu), a naturally occurring endoperoxide sesquiterpene lactone isolated from the plant Artemisia annua (Meshnick et al., Microbiol. Rev. 1996, 60, p. 301-315; Vroman et al., Curr. Pharm. Design, 1999, 5, p. 101-138; Dhingra et al., 2000, 66, p. 279-300), and a number of its precursors, metabolites and semi-synthetic derivatives which have shown to possess antimalarial properties. The antimalarial action of artemisinin is due to its reaction with iron in free heme molecules of the malaria parasite, with the generation of free radicals leading to cellular destruction. This initiated a substantial effort to elucidate its molecular mechanism of action (Jefford, dv. Drug Res. 1997, 29, p. 271-325; Cumming et al., Adv. Pharmacol. 1997, 37, p. 254-297) and to identify novel antimalarial peroxides (Dong and Vennerstrom, Expert Opin. Ther. Patents 2001, 1 1, p. 1753-1760).
Although the clinically useful artemisinin derivatives are rapid acting and potent antimalarial drugs, they have several disadvantages including recrudescence,
neurotoxicity, (Wesche et al., Antimicrob. Agents. Chemother. 1994, 38, p. 1813-1819) and metabolic instability (White, Trans. R. Soc. Trop. Med. Hyg., 1994, 88, p. 41-43). A fair number of these compounds are quite active in vitro, but most suffer from low oral activity (White, Trans. R. Soc. Trop. Med. Hyg., 1994, 88, p. 41-43 and van Agtmael et al., Trends Pharmacol. Sci., 1999, 20, p. 199-205). Further all these artemisinin derivatives are conventionally obtained from plant source and are therefore expensive.
As the cultivation of the plant material is dependent on many factors including the weather conditions, the supply source thus becomes finite and there are chances of varying yield and potency. This leads to quality inconsistencies and supply constraints. As malaria is more prevalent in developing countries, a switch to cheaper and effective medicine is highly desirable.
Thus there exists a need in the art to identify new peroxide antimalarial agents, especially those which are not dependent on plant source and can be easily synthesized, are devoid of neurotoxicity, and which possess improved solubility, stability and pharmacokinetic properties.
Following that, many synthetic antimalarial 1 ,2,4-trioxanes (Jefford, Adv. Drug Res. 1997, 29, p. 271-325; Cumming et al., Adv. Pharmacol. 1997, 37, p. 254-297), 1,2,4,5-tetraoxanes (Vennerstrom et al., J. Med. Chem., 2000, 43, p. 2753-2758), and other endoperoxides have been prepared. Various patents/applications disclose means and method for treating malaria using Spiro or dispiro 1,2,4-trioxolanes for example, U.S.
Patent Application No. 2004/0186168 and U.S. Patent Nos. 6,486, 199 and 6,825,230. The present invention relates to solid dosage forms of the various spiro or dispiro 1 ,2,4- trioxolanes antimalarial compounds disclosed in these patents/applications and are incorporated herein by reference.
Active compounds representing various Spiro and dispiro 1 ,2,4-trioxolane derivatives possess excellent potency, efficacy against Plasmodium parasites, and a lower degree of neurotoxicity, in addition to their structural simplicity and ease of synthesis. Furthermore, these compounds have half-lives which are believed to permit short-term treatment regimens comparing favorably to other artemisinin-like drugs. In general, the therapeutic dose of trioxolane derivative may range between about 0.1-1000 mg/kg/day, in particular between about 1-100 mg/kg/day. The foregoing dose may be administered as a single dose or may be divided into multiple doses. For malaria prevention, a typical dosing schedule could be, for example, 2.0-1000 mg/kg weekly beginning 1-2 weeks prior to malaria exposure, continued up to 1-2 weeks post-exposure.
Monotherapy with artemisinin (natural or synthetic) class of drugs might cure the patients within 3 days, however perceiving the potential threat of the malarial parasite developing resistance towards otherwise very potent artemisinin class of drugs, WHO had strictly called for an immediate halt to the provision of single-drug artemisinin malaria pills. Combination therapy in case of malaria retards the development of resistance, improve efficacy by lowering recrudescence rate, provides synergistic effect, and increase exposure of the parasite to the drugs.
Artemsinin based combinations are available in the market for a long time.
Artemether-lumafentrine (Co-artem®) was the first fixed dose antimalarial combination containing an artemisinin derivative and has been known since 1999. This combination has passed extensive safety and efficacy trials and has been approved by more than 70 regulatory agencies. Co-artem® is recommended by WHO as the first line treatment for uncomplicated malaria.
Other artemisinin based combinations include artesunate and amodiaquine (Coarsucam®), and dihydroartemisin and piperaquine (Eurartesim®). Unfortunately, all the available artemisinin based combinations have complicated dosage regimens making it difficult and inconvenient for a patient to comply completely with the total prescribed duration. For example, the dosage regimen of Co-artem®for an adult having body weight of more than 35 kg includes 6 doses over three days.
The first dose comprises four tablets initially, the second dose comprises four tablets after eight hours, the third to sixth doses comprise four tablets twice for another two days; making it a total of 24 tablets. The dosage regimen of Coarsucam® for an adult having body weight of more than 36 kg or age above 14 years includes three doses over three days; each dose comprises two tablets; making it a total of six tablets. The dosage regimen of Eurartesim® for an adult having body weight between 36 kg – 75 kg includes 3 doses over three days, each dose comprises of three tablets, making it a total of nine tablets.
It is evident that the available artemisinin-based combinations have a high pill burden on patients as they need to consume too many tablets. As noted above, this may increase the possibility of missing a few doses, and, consequently, could result in reduced efficacy due to non-compliance and may even lead to development of resistance for the drug. Therefore, there is an urgent and unmet need for anti-malarial combinations with a simplified daily dosing regimen that reduces the pill burden and would increase patient compliance.
Apart from simplifying the regimen, there are certain limitations for formulators developing formulations with trioxolones, the first being their susceptibility to degradation in presence of moisture that results in reduced shelf lives. Another is their bitter taste, which can result in poor compliance of the regimen or selection of another, possibly less effective, therapeutic agent.
……………………..
PATENT
http://www.google.st/patents/US6906205

……………………
PATENT
http://www.google.st/patents/WO2013008218A1?cl=en
structural Formula II.

Formula II
Active compound includes one or more of the various spiro and dispiro trioxolane derivatives disclosed in U.S. Application No. 2004/0186168 and U.S. Patent Nos.
6,486,199 and 6,825,230, which are incorporated herein by reference. These trioxolanes are relatively sterically hindered on at least one side of the trioxolane heterocycle which provides better in vivo activity, especially with respect to oral administration. Particularly, spiro and dispiro 1,2,4-trioxolanes derivatives possess excellent potency and efficacy against Plasmodium parasites, and a lower degree of neurotoxicity.
The term “Active compound I” herein means cis-adamantane-2-spiro-3′-8′-[[[(2′- amino-2′-methylpropyl)amino]carbonyl]-methyl]- 1 ‘,2′,4′-trioxaspiro[4.5]decane hydrogen maleate. The Active compound I may be present in an amount of from about 5% to about 25%, w/w based on the total dosage form.

………………
PATENT
http://www.google.st/patents/WO2007138435A2?cl=en
A synthetic procedure for preparing compounds of Formula I, salts of the free base c«-adamantane-2-spiro-3′-8′-[[[(2′-amino-2′-methyl propyl) amino] carbonyl] methyl]- 1 ‘, 2′, 4′-trioxaspiro [4.5] decane has been disclosed in U.S. 6,906,205.

The process for the preparation of compounds of Formula I wherein a compound of Formula II (wherein R is lower alkyl) is reacted with a compound of Formula III (wherein R is lower alkyl) to obtain compound of Formula IV;


Formula Formula IV
followed by hydrolysis of the compounds of Formula IV to give a compound of Formula V;

Formula V followed by the reaction of the compound of Formula V with an activating agent, for example, methyl chloroformate, ethyl chloroformate, propyl chloro formate, n-butyl chloro formate, isobutyl chloroformate or pivaloyl chloride leads to the formation of mixed anhydride, which is reacted in situ reaction with 1 ,2-diamino-2-methyl propane to give a compound of Formula VI; and

Formula Vl reacting the compound of Formula VI with an acid of Formula HX (wherein X can be the same as defined earlier) to give compounds of Formula I.
Example 1 : Preparation of O-methyl-2-adamantanone oxime
To a solution of 2-adamantanone (50 g, 0.3328 mol, 1 equiv.) in methanol (0.25 lit), sodium hydroxide solution (15 g, 0.3761mol, 1.13 equiv, in 50 mL water) was added followed by methoxylamine hydrochloride (37.5 g x 81.59% Purity= 30.596 g, 0.366 mol, 1.1 equiv) at room temperature under stirring. The reaction mixture was stirred at room temperature for 1 to 2 h. The reaction was monitored by HPLC. The reaction mixture was concentrated at 40- 45°C under vacuum to get a thick residue. Water (250 mL) was added at room temperature and the reaction mixture was stirred for half an hour. The white solid was filtered, washed with water (50 mL), and dried at 40 to 45°C under reduced pressure. O-methyl 2- adamantanone oxime (57 g, 95 % yield) was obtained as a white solid.
(M++l) 180, 1HNMR (400 MHz, CDCl3 ): δ 1.98 – 1.79 (m, 12H), 2.53 (s, IH), 3.46 ( s, IH), 3.81 (s, 3H).
Example 2: Preparation of 4-(methoxycarbonvmethvPcvclohexanone A high pressure autoclave was charged with a mixture of methyl (4- hydroxyphenyl)acetate (50 g, 0.30 mol), palladium ( 5g) (10 %) on carbon (50 % wet) and O- xylene (250 mL). The reaction mixture was stirred under 110 to 115 psi of hydrogen pressure for 7 to 8 h at 1400C. The reaction was monitored by HPLC. The reaction mixture was then cooled to room temperature, and the catalyst was filtered off. Filtrate was concentrated under reduced pressure to get 4-(methoxycarbonylmethyl)cyclohexanone as light yellow to colorless oily liquid (48.7 g, 97.4 %).
(M++!) 171, ‘ HNMR (400 MHz, CDCl 3): δ 1.48 – 1.51 ( m, 2H), 2.1 1-2.07 (m, 2H), 2.4- 2.23 (m, 7H), 3.7 (s, 3H).
Example 3: Preparation of methyl (Is, 4s)-dispiro [cyclohexane-l, 3′-f 1,2,4] trioxolane-5′, 2″-tricvclor3.3.1.13–71decan1-4-ylacetate
A solution of O-methyl-2-adamantanone oxime (example 1) (11.06 g, 61.7 mmol, 1.5 equiv.) and 4-(methoxycarbonymethyl)cyclohexanone (example 2) (7.0 g, 41.1 mmol, 1 equiv.) in cyclohexane ( 200ml) and dichloromethane (40 mL) was treated with ozone (ozone was produced with an OREC ozone generator [0.6 L/min. O2, 60 V] passed through an empty gas washing bottle that was cooled to -780C). The solvent was removed after the reaction was complete. After removal of solvents, the crude product was purified by crystallization from 80% aqueous ethanol (200 mL) to afford the title compound as a colorless solid. Yield: 10.83 g, 78%, mp: 96-980C; 1HNMR (500 Hz3CDCl3): δ 1.20-1.33 (m, 2H), 1.61-2.09 (m, 5 21H), 2.22 (d, J = 6.8Hz, 2H), 3.67(s,3H).
Example 4: Preparation of (Is, 4s)-dispiro [cyclohexane-1, 3′-[l,2,4] trioxolane-5′, 2″- tricvclo [3.3.1.13‘7] decanl-4-ylacetic acid
Sodium hydroxide (3.86 g, 96.57 mmol, 3 equiv.) in water (80 mL) was added to a solution of methyl (\s, 4s)-dispiro [cyclohexane-1, 3′-[l,2,4] trioxolane-5′, 2″-tricyclo
10 [3.3.1.I3‘7] decan]-4-ylacetate (example 3) (10.83 g, 32.19 mmol, 1 equiv.) in 95% ethanol (150 mL). The mixture was stirred at 500C for about 4 h, cooled to O0C, and treated with IM hydrochloric acid (129ml, 4 equiv). The precipitate was collected by filtration, washed with 50 % aqueous ethanol (150 mL) and dried in vacuum at 40 0C to give the title compound as colorless solid. Yield: 9.952 g, 96%, mp: 146-1480C ( 95% ethanol), 1HNMR (500 Hz,
15 CDCl3): δ 1.19-1.41 (m,2H), 1.60-2.05 (m,21H), 2.27 (d, J=6.8 Hz,2H).
Example 5: Preparation of c?s-adamantane-2-spiro-3′-8′-[[[(2′-amino-2′-methyl propyl) amino] carbonyl] methyl]-! ‘, T , 4′-trioxaspiro [4.5] decane
Method A:
(Is, 4s)-dispiro[cyclohexane- 1 ,3 ‘-[ 1 ,2,4]trioxolane-5 ‘,2 ‘ ‘-tricyclo[3.3.1.13‘7]decan]-4-
.0 ylacetic acid (example 4) (5 g ,15.5mmol, 1 equiv) was mixed with triethylamine (2.5 g , 24.8 mmol, 1.6 equiv) in 100ml of dichloromethane. The reaction mixture was cooled to – 1O0C to 00C. Ethyl chloro formate (1.68 g, 17 mmol, 1.0 equiv) in 15 mL dichloromethane was charged to the above reaction mixture at – 100C to 00C. The reaction mixture was stirred at the same temperature for 10 to 30 minutes. The resulting mixed anhydride reaction mixture
15 was added dropwise to a previously prepared solution of l,2-diamino-2-methylpropane (1.64 g, 18.6 mmol, 1.2 equiv), in 100 mL dichloromethane at -100C to O0C. The temperature of reaction mixture was raised to room temperature. The reaction mixture was stirred at the same temperature till the reaction was complete. Reaction monitoring was done by thin layer chromatography using 5 to 10% methanol in dichloromethane. The reaction was complete
>0 within 2 h. Nitrogen atmosphere was maintained throughout the reaction. Water (50 mL) was charged, organic layer was separated and washed with 10% sodium bicarbonate solution (50 mL) and water (50 mL) at room temperature. The organic layer was dried over sodium sulphate and the solvent was removed at 25 to 4O0C under reduced pressure. Hexane (50ml) was added to obtain residue under stirring at room temperature. The mixture was filtered and washed with 5 mL of chilled hexane. The solid was dried under reduced pressure at room 5 temperature.
Yield: 5.2 g (85.4 %), (M++l) 393, 1HNMR (400 MHz, DMSO-J6 ): δ 0.929 ( s, 6H), 1.105 – 1.079 (m, 2H), 1.887-1.641 (m, 21H), 2.030-2.017 (d, 2H), 2.928 (d, 2H).
Method B:
(Is, 4s)-dispiro [cyclohexane-1, 3′-[l,2,4] trioxolane-5′, 2″-tricyclo [3.3.1.I3‘7]
10 decan]-4-ylacetic acid (example 4) (10 g, 31mmol, 1 equiv) was treated with isobutyl chloroformate (4.5 g, 33mmol, 1.1 equiv) in presence of organic base like triethyl amine (5 g, 49.6mmol, 1.6 equiv) at 00C to 7°C in 250ml of dichloromethane. The solution was stirred at O0C to 7°C for aboutlO to 30 minutes. To the above reaction mixture, previously prepared solution of l,2-diamino-2-methylpropane (3.27 g, 37 mmol, 1.2 equiv), in 50 mL of
15 dichloromethane was added at O0C to 7°C in one lot. The temperature of reaction mixture was raised to room temperature. The reaction mixture was stirred at the room temperature till reaction was over. Reaction monitoring was done by thin layer chromatography using 5 to 10% methanol in dichloromethane. Reaction was complete within 2 h. The reaction nitrogen atmosphere was maintained throughout the reaction. Water (250 mL) was charged, organic
20 layer was separated and washed with 10% sodium bicarbonate solution (200 mL) and water (100 mL) at room temperature and the solvent was removed at 25 to 4O0C under reduced pressure. Hexane (100ml) was added to the residue, under stirring, at room temperature. The mixture was filtered and washed with chilled hexane (10 mL). The resultant solid was dried under reduced pressure at room temperature. Yield: 10.63 g (87%), (M++l) 393, 1HNMR
>5 (400 MHz, DMSO-J6 ) :δ 0.928 ( s, 6H), 1.102 – 1.074 (m, 2H), 1.859-1.616 (m, 21H), 2.031- 2.013 (d, 2H), 2.94-2.925 (d, 2H). Method C:
(\s, 4s)-dispiro[cyclohexane-l,3′-[l,2,4]trioxolane-5′,2″-tricyclo[3.3.1.13>7]decan]-4- ylacetic acid (example 4) (5 g, 15.5mmol, 1 equiv) was treated with pivaloyl chloride (1.87 g, 15.5 mmol, 1 equiv) and triethylamine (2.5gm, 24.8mmol, 1.6 equiv) at -15°C to -100C in dichloromethane (125 mL). The solution was stirred at -150C to -100C for aboutlO to 30 minutes. It resulted in the formation of mixed anydride. To the above reaction mixture, previously prepared solution of 1 ,2-diamino-2-methylpropane (1.64 g, 18.6 mmol, 1.2 equiv) in 25 mL dichloromethane was added at -15°C to -100C. The temperature of reaction mixture was raised to room temperature. The reaction mixture was stirred at the room temperature till reaction was over. Reaction monitoring was done by thin layer chromatography using 5 to 10% methanol in dichloromethane. The reaction was complete within 2 h. Nitrogen atmosphere was maintained throughout the reaction. Water (125 mL) was charged, organic layer was separated and washed with 50 mL of 10% sodium bicarbonate solution and 125 mL of water, respectively at room temperature. Finally solvent was removed at 25 to 4O0C under reduced pressure. 50 mL of 5% Ethyl acetate – hexane solvent mixture was added to the residue under stirring at room temperature. The mixture was filtered and washed with 5 mL of chilled hexane. Solid was dried under reduced pressure at room temperature. Yield: 5.03 g (83 %), (M++l) 393, 1JINMR (400 MHz, OMSO-d6 ):δ 0.93 ( s, 6H), 1.113 – 1.069 (m, 2H), 1.861-1.644 (m, 21H), 2.033-2.015 (d, 2H), 2.948-2.933 (d, 2H).
Example 6: Preparation of c/s-adamantane-2-spiro-3′ -8 ‘-πT(2′-amino-2′ -methyl propyl) amino! carbonyl] methyli-l ‘, 2\ 4′-U-JoXaSpJrQ [4.51 decane maleate To a solution of c/s-adamantane-2-spiro-3′-8′-[[[(2′-amino-2′-methyl propyl) amino] carbonyl] methyl]-! ‘, 2′, 4′-trioxaspiro [4.5] decane (example 5) (60 g, 0.153 moles) in ethanol (150 mL) was added a solution of maleic acid (17.3 g, 0.15 moles, 0.98 equiv. in ethanol 90 mL) and the reaction mixture was stirred for about 1 h. To this clear solution, n- heptane (720 mL) was added at room temperature in 1 h and the reaction mixture was stirred for 3 h. It was then cooled to 0 to 100C and filtered. The cake was washed with n-heptane (60 mL) and dried under vacuum at 40-450C.
Yield: 67 g, 77.4%, mp: 1490C (decomp), (M++l) 393.5, 1HNMR (300 MHz, DMSO-^ ): δ 1.05-1.11 (2H,m), 1.18 (6H,s), 1.64-1.89 (21H,m), 2.07(2H,d), 3.21 (2H,d), 6.06 (2H,d), 7.797 (2H, bs), 8.07 (IH, t).



References
- Dong, Yuxiang; Wittlin, Sergio; Sriraghavan, Kamaraj; Chollet, Jacques; Charman, Susan A.; Charman, William N.; Scheurer, Christian; Urwyler, Heinrich et al. (2010). “The Structure−Activity Relationship of the Antimalarial Ozonide Arterolane (OZ277)”. Journal of Medicinal Chemistry 53 (1): 481–91. doi:10.1021/jm901473s. PMID 19924861.
- Blow to Ranbaxy drug research plans at LiveMint.com, Sep 21 2007
- Vennerstrom, Jonathan L.; Arbe-Barnes, Sarah; Brun, Reto; Charman, Susan A.; Chiu, Francis C. K.; Chollet, Jacques; Dong, Yuxiang; Dorn, Arnulf et al. (2004). “Identification of an antimalarial synthetic trioxolane drug development candidate”. Nature 430 (7002): 900–4.doi:10.1038/nature02779. PMID 15318224.
- In the Pipeline: “Ozonides As Drugs: What Will They Think Of Next?”, by Derek Lowe, November 23, 2009, at Corante.com
- Indian company starts Phase III trials of synthetic artemisinin, May 4 2009, at the WorldWide Antimalarial Resistance Network
- http://www.nature.com/nature/journal/v430/n7002/full/nature02779.html
|
5-27-2011
|
PROCESS FOR THE PREPARATION OF DISPIRO 1,2,4-TRIOXOLANE ANTIMALARIALS (OZ277)
|
|
|
2-13-2009
|
STABLE DOSAGE FORMS OF SPIRO AND DISPIRO 1,2,4-TRIOXOLANE ANTIMALARIALS
|
|
|
6-15-2005
|
Spiro and dispiro 1,2,4-trioxolane antimalarials
|
|
|
11-31-2004
|
Spiro and dispiro 1,2,4-trixolane antimalarials
|
ANTIMALARIALS




http://www.rsc.org/chemistryworld/2013/03/new-antimalarial-drug-class-resistance-elq-300-quinolone

http://www.nature.com/nrd/journal/v9/n11/full/nrd3301.html
Structure of NITD609; the 1R,3Sconfiguration is fundamental for its antimalarial activity
FDA gives green light to Novartis acromegaly drug Pasireotide

Pasireotide, Signifor; SOM 320; HY-16381; 396091-73-9
Cyclo[4(R)-[N-(2-aminoethyl)carbamoyloxy]-L-prolyl-L-phenyl-glycyl-D-tryptophyl-L-lysyl-(4-O-benzyl)-L-tyrosyl-L-phenylalanyl]bis(L-aspartic acid)
Regulators in the USA has approved a long-acting release of Novartis’ Signifor as a treatment for acromegaly.
Read more at: http://www.pharmatimes.com/Article/14-12-16/FDA_gives_green_light_to_Novartis_acromegaly_drug.aspx#ixzz3M8Ibn14Q
clinical…..https://clinicaltrials.gov/search/intervention=Pasireotide+OR+SOM-230
Pasireotide (SOM230, trade name Signifor[1]) is an orphan drug approved in the U.S. and Europe for the treatment of Cushing’s disease in patients who fail or are ineligible for surgical therapy.[2][3] It was developed by Novartis. Pasireotide is a somatostatinanalog which has a 40-fold increased affinity to somatostatin receptor 5 than other somatostatin analogs.
The drug showed therapeutical potential in a recent study (PASPORT-CUSHINGS B2305) where 162 patients were treated with either 2x 600 µg or 2x 900 µg pasireotide s.c. daily.[4] The effectiveness of the treatment was checked by the UFC-value (urinary free cortisol) after six months of treatment. The mean reduction of UFC after six months was 47.9%, which also lead to amelioration of clinical symptoms such as blood pressure, cholesterol value, and weight loss.[5]
Pasireotide was approved by the EMEA in October 2009[6] and by the FDA in December 2012.[7]
At present, it is in phase III clinical trials at Novartis for the treatment of carcinoid tumors and symptoms that are not adequately controlled by somatostatin analogues (Sandostatin). Phase II clinical development is also under way at the company for the treatment of gastric dumping syndrome, metastatic carcinoid tumors, meningioma and pituitary adenoma and for the treatment of hepatocellular carcinoma in combination with everolimus. Early clinical trials are also ongoing for the treatment of patients with metastatic melanoma or Merkel cell carcinoma. A phase I clinical trial for the treatment of alcoholic cirrhosis has been completed. The company intends to file for approval in 2007 for these indications. Novartis and Thomas Jefferson University are conducting phase II clinical trials for the treatment of prostate cancer, alone or in combination with everolimus. The Mayo Clinic is conducting phase II clinical trials for the treatment of polycystic liver disease. Phase III clinical trials had been ongoing for the reduction of post-pancreatectomy fistula, leak, and abscess; however, in 2010 these trials were suspended. In 2004, orphan drug designation was assigned in the E.U. for the treatment of functional gastroenteropancreatic endocrine tumors. In 2009, orphan drug designation was received in the U.S. and the E.U. for the treatment of Cushing’s disease and acromegaly. The designation for the treatment of Cushing’s disease was assigned in Australia in 2011 and in Japan in 2012. In 2013, orphan drug designation was assigned in Australia for the treatment of acromegaly.

SIGNIFOR (pasireotide diaspartate) injection is prepared as a sterile solution of pasireotide diaspartate in a tartaric acid buffer for administration by subcutaneous injection. SIGNIFOR is a somatostatin analog. Pasireotide diaspartate, chemically known as (2-Aminoethyl) carbamic acid (2R,5S,8S,11S,14R,17S,19aS)-11-(4-aminobutyl)-5-benzyl-8-(4-benzyloxybenzyl)-14-(1H-indol-3ylmethyl)-4,7,10,13,16,19-hexaoxo-17-phenyloctadecahydro-3a,6,9,12,15,18hexaazacyclopentacyclooctadecen-2-yl ester, di[(S)-2-aminosuccinic acid] salt, is a cyclohexapeptide with pharmacologic properties mimicking those of the natural hormone somatostatin.
The molecular formula of pasireotide diaspartate is C58H66N10O9 • 2C4H7NO4 and the molecular weight is 1313.41. The structural formula is:
 SIGNIFOR is supplied as a sterile solution in a single-dose, 1 mL colorless glass ampule containing pasireotide in 0.3 mg/mL, 0.6 mg/mL, or 0.9 mg/mL strengths for subcutaneous injection.
SIGNIFOR is supplied as a sterile solution in a single-dose, 1 mL colorless glass ampule containing pasireotide in 0.3 mg/mL, 0.6 mg/mL, or 0.9 mg/mL strengths for subcutaneous injection.
Each glass ampule contains:
| 0.3 MG | 0.6 MG | 0.9 MG | |
| Pasireotide diaspartate | 0.3762* | 0.7524* | 1.1286* |
| Mannitol | 49.5 | 49.5 | 49.5 |
| Tartaric acid | 1.501 | 1.501 | 1.501 |
| Sodium hydroxide | ad pH 4.2 | ad pH 4.2 | ad pH 4.2 |
| Water for injection | ad 1ml | ad 1ml | ad 1ml |
| * corresponds to 0.3/0.6/0.9 mg pasireotide base Note: Each ampule contains an overfill of 0.1ml to allow accurate administration of 1 ml from the ampule. |
|||
 |
|
| Systematic (IUPAC) name | |
|---|---|
| [(3S,6S,9S,12R,15S,18S,20R)-9-(4-aminobutyl)-3-benzyl-12-(1H-indol-3-ylmethyl)-2,5,8,11,14,17-hexaoxo-15-phenyl-6-[(4-phenylmethoxyphenyl)methyl]-1,4,7,10,13,16-hexazabicyclo[16.3.0]henicosan-20-yl] N-(2-aminoethyl)carbamate | |
| Clinical data | |
| Trade names | Signifor |
| Licence data | EMA:Link |
| Legal status |
|
| Routes | Subcutaneous |
| Identifiers | |
| CAS number | 396091-73-9 |
| ATC code | H01CB05 |
| PubChem | CID 9941444 |
| UNII | 98H1T17066 |
| Synonyms | SOM230 |
| Chemical data | |
| Formula | C58H66N10O9 |
| Mol. mass | 1107.26 g/mol |
Pasireotide is a multiligand somatostatin analogue with high binding affinity to somatostatin receptors sst1, sst2, sst3 and sst5. Novartis Oncology, a division of Novartis, filed for approval in the E.U. for the treatment of Cushing’s syndrome in 2010. A positive opinion was granted in 2011 and final approval was obtained in 2012. The E.U.’s first launch took place in Germany in June 2012. Also in 2011, Novartis filed an NDA in the U.S. seeking approval of the compound for the treatment of Cushing’s syndrome; however, the application was withdrawn the same year due to an issue related to chemistry, manufacturing and controls. In November 2012, the product was recommended for approval in the U.S. for Cushing’s syndrome. In December 2012, final FDA approval was granted. Phase III clinical trials are ongoing in Japan for this indication. In 2014, the product was approved in the E.U and the U.S. for the treatment of adult patients with acromegaly for whom surgery is not an option or has not been curative and who are inadequately controlled on treatment with a first-generation somatostatin analogue (SSA).
- http://www.google.com/patents/EP2310042B1?cl=en
-
The present invention relates to a new use of Somatostatin (SRIF) peptidomimetics (also referred to as Somatostatin- or SRIF-analogs).
-
Somatostatin is a tetradecapeptide having the structure

-
The somatostatin class is a known class of small peptides comprising the naturally occurring somatostatin-14 and analogues having somatostatin related activity, e.g. as disclosed by A.S. Dutta in Small Peptides, Vol.19, Elsevier (1993). By “somatostatin analog” as used herein is meant any straight-chain or cyclic polypeptide having a structure based on that of the naturally occurring somatostatin-14 wherein one or more amino acid units have been omitted and/or replaced by one or more other amino radical(s) and/or wherein one or more functional groups have been replaced by one or more other functional groups and/or one or more groups have been replaced by one or several other isosteric groups. In general, the term covers all modified derivatives of the native somatostatin-14 which exhibit a somatostatin related activity, e.g. they bind to at least one of the five somatostatin receptor (SSTR), preferably in the nMolar range.
-
Natural somatostatin binds and activates all 5 somatostatin receptors (SSTR1-5) with nmol efficacy and thus causes its multiple physiological effects.
-
Synthetically available somatostatin analogs differ in their binding affinity to the different somatostatin receptor subtypes and often bind selectively to one or few subtypes with significantly higher affinity.
-
Somatostatin analogs of particular interest according to the present invention have a high binding affinity to human SSTR1,2,3,5 and have been described e.g. in WO 97/01579 , the contents of which being incorporated herein by reference. Said somatostatin analogs comprise the amino acid sequence of formula I-(D/L)Trp-Lys-X1 -X2 - Iwherein X1 is a radical of formula (a) or (b)

wherein R1 is optionally substituted phenyl, wherein the substituent may be halogen, methyl, ethyl, methoxy or ethoxy,
R2 is -Z1-CH2-R1, -CH2-CO-O-CH2-R1,
wherein Z1 is O or S, and
X2 is an α-amino acid having an aromatic residue on the Cα side chain, or an amino acid unit selected from Dab, Dpr, Dpm, His,(Bzl)HyPro, thienyl-Ala, cyclohexyl-Ala and t-butyl-Ala, the residue Lys of said sequence corresponding to the residue Lys9 of the native somatostatin-14. -
Somatostatin analogs of particular interest which have a high binding affinity to human SSTR1,2,3,5 have also been described e.g. inWO02/10192. Said somatostatin analogs comprise the compound of formula

also called cyclo[{4-(NH2-C2H4-NH-CO-O-)Pro}-Phg-DTrp-Lys-Tyr(4-Bzl)-Phe] or pasireotide, as well as diastereoisomers and mixtures thereof, in free form, in salt or complex form or in protected form. Phg means -HN-CH(C6H5)-CO- and Bzl means benzyl.




…………………
http://www.google.com/patents/WO2002010192A2?cl=en
Example 1 : Cyclo[{4-(NH2-C2H4-NH-CO-O-
a) Synthesis of Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH
L-hydroxyproline methylester hydrochloride is reacted with Fmoc-OSu in aqueous 1.0 N sodium carbonate/THF at room temperature. After completion of the reaction, Fmoc-Pro(4- OH)-OMe is isolated by precipitation. Fmoc-Pro(4-OH)-OMe is then added dropwise into a solution of trisphosgene (0.6 eq.) in THF to give a chlorocarbonate intermediate. After 1 h dimethylaminopyridine (1.0 eq.) and N-Boc-diaminoethane (6.0 eq.) are added and the reaction is stirred at room temperature. After completion of the reaction, the solvent is removed in vacuo and the resulting Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OMe is extracted from a two phase system of ethyl acetate/0.1 M HCI to give crude product (MH+ = 554) which is purified by crystallization from ethyl acetate. The methyl ester is then cleaved to the free acid by treatment with 1 N NaOH in dioxane/water and the product Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH is purified on silica gel, [(M+Na)]+= 562).
b) H-Phe-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-Phg-DTrp(Boc)-Lys(Boc)-Tyr(Bzl)-OH Commercially available Fmoc-Tyr(Bzl)-O-CH2-Ph(3-OCH3)-O-CH2-Polystyrene resin (SASRIN-resin, 2.4 mM) is used as starting material and carried through a standard protocol consisting of repetitive cycles of Nα-deprotection (Piperidine/DMF, 2:8), repeated washings with DMF and coupling (DIPCI: 4.8 mM/HOBT: 6mM, DMF). The following amino acid- derivatives are sequentially coupled: Fmoc-Lys(Boc)-OH, Fmoc-DTrp(Boc)-OH, Fmoc-Phg- OH, Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH, Fmoc-Phe-OH. Couplings (2 eq. amino acids) are continued or repeated until completion, i.e. until complete disappearance of residual amino groups which is monitored by a negative ‘Kaiser* Ninhydrin test. Before cleavage of the completely assembled protected linear peptide from its resin support the Nα-Fmoc protection from the last residue is removed.
c) H-Phe-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-Phg-DTrp(Boc)-Lys(Boc)-Tyr(Bzl)-OH After washings with CH2CI2) the peptide-resin is transferred into a column or a stirred suction filter and the peptide fragment is cleaved and eluted with a short treatment with 2% TFA in CH2CI2 within 1 h. The eluate is immediately neutralized with a saturated NaHCO3 solution. The organic solution is separated and evaporated and the side chain protected precursor (MH+ = 1366) is cyclized without further purification.
d) cyclo[-Pro(4-OCO-NH-CH2-CH2-NH2)-Phg-DT -Lys-Tyr(Bzl)-Phe-], trifluoroacetate The above linear fragment is dissolved in DMF (4 mM), cooled to minus 5°C and treated with 2 eq. DIPEA then 1.5 eq. of DPPA and stirred until completion (ca. 20h) at 0-4°C. The solvent was almost completely removed in vacuo; the concentrate is diluted with ethyl acetate, washed with NaHCO3, water, dried and evaporated in vacuo.
For deprotection the residue is dissolved at 0°C in TFA H2O 95:5 (ca.50 mM) and stirred in the cold for 30 min. The product is then precipitated with ether containing ca. 10 eq. HCI, filtered, washed with ether and dried. In order to completely decompose remaining Indole-N carbaminic acid the product is dissolved in 5% AcOH and lyophilized after 15 h at ca. 5°C. Preparative RP-HPLC is carried out on a C-18 10 μm STAGROMA column (5-25 cm) using a gradient of 0.5% TFA to 0.5% TFA in 70% acetonitrile. Fractions containing the pure title compound are combined, diluted with water and lyophilized. The lyophilisate is dissolved in water followed by precipitation with 10% Na2CO3 in water. The solid free base is filtered of, washed with water and dried in vacuum at room temperature. The resulting white powder is directly used for the different salts.
Example 2: Cyclo[{4-(NH2-C2H4-NH-CO-O-)Pro}-Phg-DTrp-Lys-Tyr(4-Bzl)-Phe] in salt form a. Acetate
Conversion to the acetate salt form is carried out using an ion-exchange resin (e.g. AG 3- X4). MS (ESI): m/z 524.5 [M+2H]2+ [α]D 20= -42°, c=0.26 in AcOH 95%
b. Aspartate
Conversion to the mono- or di-aspartate is obtained by reacting 1 equivalent of the compound of Example 1 with 1 or 2 equivalent of aspartic acid in a mixture of acetonitrile/water 1 :3. The resulting mixture is frozen and lyophilized. The di-aspartate may also be obtained by dissolving the compound of Example 1 in water/acetonitrile 4:1, filtering, loading on a an ion-exchange resin, e.g. BioRad AG4X4 column, and eluting with water/acetonitrile 4:1. The eluate is concentrated, frozen and lyophilized. [ ]D 20= -47.5°, c= 2.5mg/ml in methanol
……………..
WO2013/174978 A1
http://www.google.im/patents/WO2013174978A1?cl=ru
………………………..
WO2013/131879 A1,
………………………..
WO2005/53732 A1,
http://www.google.com/patents/WO2005053732A1?cl=en
……………………………
Journal of Medicinal Chemistry, 2003 , vol. 46, 12 pg. 2334 – 2344
http://pubs.acs.org/doi/abs/10.1021/jm021093t

A rational drug design approach, capitalizing on structure−activity relationships and involving transposition of functional groups from somatotropin release inhibitory factor (SRIF) into a reduced size cyclohexapeptide template, has led to the discovery of SOM230 (25), a novel, stable cyclohexapeptide somatostatin mimic that exhibits unique high-affinity binding to human somatostatin receptors (subtypes sst1−sst5). SOM230 has potent, long-lasting inhibitory effects on growth hormone and insulin-like growth factor-1 release and is a promising development candidate currently under evaluation in phase I clinical trials.

| residue | group | δ 1H [ppm] | δ 13C [ppm] | residue | group | δ 1H [ppm] | δ 13C [ppm] |
| 1 | l-phenylglycine |
| 1 | NH | 9.73 | 1 | α-CH | 6.47 | 59.3 | |||
| 1 | 2/6-CH | 8.02 | 127.3 | 1 | CO | 169.6 | |||
| 1 | 3/5-CH | 7.41 | 129.1 | 1 | 1-C | 141.0 |
| 1 | 4-CH | 7.21 | 128.0 | |
| 2 | d-tryptophane |
| 2 | 1‘-NH | 12.20 | 2 | α-CH | 5.28 | 55.6 | |||
| 2 | NH | 10.34 | 2 | β-CH2 | 3.72 | 3.30 | 28.5 | ||
| 2 | 7-CH | 7.65 | 112.0 | 2 | CO | 173.9 | |||
| 2 | 4-CH | 7.43 | 119.2 | 2 | 8-C | 137.5 | |||
| 2 | 2-CH | 7.28 | 124.7 | 2 | 9-C | 128.3 | |||
| 2 | 6-CH | 7.23 | 121.6 | 2 | 3-C | 110.3 |
| 2 | 5-CH | 6.96 | 119.2 | |
| 3 | l-lysine |
| 3 | NH | 10.10 | 3 | δ-CH2 | 1.41 | 1.32 | 31.5 | ||
| 3 | α-CH | 4.62 | 55.2 | 3 | γ-CH2 | 0.89 | 23.5 | ||
| 3 | ε-CH2 | 2.80 | 41.0 | 3 | CO | 171.9 | |||
| 3 | β-CH2 | 1.87 | 1.32 | 31.6 | 3 | NH3+ | a |
| 4 | (4-O-benzyl)-l-tyrosine | |||
| 4 | NH | 7.99 | 4 | 7-CH2 | 4.92 | 69.9 | |||
| 4 | 2‘/6‘-CH | 7.46 | 128.0 | 4 | β-CH2 | 3.46 | 3.10 | 39.7 | |
| 4 | 3‘/5‘-CH | 7.37 | 128.9 | 4 | CO | 171.8 | |||
| 4 | 4‘-CH | 7.30 | 128.2 | 4 | 4-C | 157.9 | |||
| 4 | 2/6-CH | 7.21 | 131.5 | 4 | 1‘C | 137.9 | |||
| 4 | 3/5-CH | 6.85 | 114.7 | 4 | 1-C | 129.8 |
| 4 | α-CH | 5.23 | 53.1 | |
| 5 | l-phenylalanine |
| 5 | NH | 9.82 | 5 | α-CH | 4.42 | 53.9 | |||
| 5 | 2/6-CH | 7.38 | 130.0 | 5 | β-CH2 | 3.23 | 3.06 | 37.8 | |
| 5 | 3/5-CH | 7.27 | 129.3 | 5 | CO | 171.2 | |||
| 5 | 4-CH | 7.16 | 127.6 | 5 | 1-C | 136.3 |
| 6 | (γ-O-diaminoethylcarbamate)-l-hydroxyproline | ||||||
| 6 | 2-NH | 8.04 | 6 | 4-CH2 | 2.95 | 42.4 | |||
| 6 | γ-CH | 5.23 | 70.9 | 6 | β-CH2 | 2.63 | 1.25 | 37.0 | |
| 6 | α-CH | 4.22 | 60.6 | 6 | CO | 170.7 | |||
| 6 | δ-CH2 | 4.12 | 51.4 | 6 | 1-CO | 156.7 | |||
| 6 | 3-CH2 | 3.42 | 44.5 | 6 | 4-NH3+ | a |
| A | acetate |
| A | CH3 | 2.20 | 22.1 | A | CO | 174.3 |
a The NH3+ protons are part of the water peak at 5.82 ppm.
References
- Signifor® (pasireotide) Official Website for healthcare professionals outside the US http://www.signifor.com/
- “Novartis drug Signifor® approved in the EU as the first medication to treat patients with Cushing’s disease”. Retrieved 2012-07-08.
- Mancini et al. Therapeutics and Clinical Risk Management 2010;6:505-516
- Colao et al. Pasireotide (SOM230) provides clinical benefit in patients with Cushing’s disease: results from a large, 12-month, randomized-dose, double-blind, Phase III study, Abstract OC1.7. European Neuroendocrine Association (ENEA) 14th Congress, 2010:62-63
- U.S. National Library of Medicine: Treatment of pituitary-dependent Cushing’s disease with the multireceptor ligand somatostatin analog pasireotide (SOM230): a multicenter, phase II trial. http://www.ncbi.nlm.nih.gov/pubmed/18957506?dopt=Abstract
- EMEA Approval for Pasireotide
- “FDA Approves Pasireotide for Cushing’s Disease”.
| WO2005117830A1 | 6 Jun 2005 | 15 Dec 2005 | Camurus Ab | Liquid depot formulations |
| WO2006075124A1 * | 9 Dec 2005 | 20 Jul 2006 | Camurus Ab | Somatostatin analogue formulations |
| WO2006131730A1 | 6 Jun 2006 | 14 Dec 2006 | Camurus Ab | Glp-1 analogue formulations |
| WO2007096055A1 * | 7 Feb 2007 | 30 Aug 2007 | Novartis Ag | Combination of somatostatin-analogs with different selectivity for human somatostatin receptor subtypes |
| WO2010003939A1 * | 7 Jul 2009 | 14 Jan 2010 | Novartis Ag | Use of pasireotide for the treatment of endogenous hyperinsulinemic hypoglycemia |
| US20090155193 * | 9 Dec 2005 | 18 Jun 2009 | Fredrik Joabsson | Topical Bioadhesive Formulations |
LEXIPAFANT

GR-167089
ISV-611
………………………………
Cadila banks on diabetes drug, Lipaglyn, Saroglitazar
(2S)-2-Ethoxy-3-[4-(2-{2-methyl-5-[4-(methylsulfanyl)phenyl]-1H-pyrrol-1-yl}ethoxy)phenyl]propanoic acid
(αS)-α-Ethoxy-4-[2-[2-methyl-5-[4-(methylthio)phenyl]-1H-pyrrol-1-yl]ethoxy]benzenepropanoic Acid
- alpha-ethoxy-4-(2-(2-methyl-5-(4-methylthio)phenyl))-1H-pyrrol-1-yl)ethoxy))benzenepropanoic acid
- alpha-ethoxy-4-(2-(2-methyl-5-(4-methylthio)phenyl))-1H-pyrrol-1-yl)ethoxy))benzenepropanoic acid magnesium salt
- saroglitazar
- ZYH1 compound
- E0YMX3S4JD
- cas no 495399-09-2
Saroglitazar, Lipaglyn
| Molecular Weight | 439.56706 g/mol |
|---|---|
| Molecular Formula | C25H29NO4S |
 |
| Zydus Cadila chairman and MD Pankaj R. Patel (centre) and deputy managing director Sharvil P. Patel (left) in Mumbai on Wednesday. (PTI)JUNE 5, 2013 |
Cadila banks on diabetes drug
Calcutta Telegraph
It generally takes around 10-15 years for a drug to be developed from the time of its discovery In the case of Lipaglyn, the molecule was identified in 2001, and Phase III clinical trials was completed around four years ago. While Zydus has not yet …http://www.telegraphindia.com/1130606/jsp/business/story_16976915.jsp

Mumbai, June 5: Cadila Healthcare will launch a homegrown drug against diabetes by the third quarter of this year.
The Drug Controller General of India has approved its drug — Lipaglyn —…
View original post 2,491 more words
