
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 189)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
BEXAGLIFLOZIN


Bexagliflozin
THR1442; THR-1442, EGT 0001442; EGT1442
CAS :1118567-05-7
(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2- (cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol
D-Glucitol, 1,5-anhydro-1-C-(4-chloro-3-((4-(2-(cyclopropyloxy)ethoxy)phenyl)methyl)phenyl)-, (1S)-
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
1-[4-Chloro-3-[4-[2-(cyclopropyloxy)ethoxy]benzyl]phenyl]-1-deoxy-beta-D-glucopyranose
1,5-Anhydro-1(S)-[4-chloro-3-[4-[2-(cyclopropyloxy)ethoxy]benzyl]phenyl]-D-glucitol
(1S)-1,5-anhydro-1-C-[4-chloro-3-({4-[2- (cyclopropyloxy)ethoxy]phenyl}methyl)phenyl]-D-glucitol
Chemical Formula: C24H29ClO7
Exact Mass: 464.16018
Mechanism of Action:SGLT2 inhibitor, Sodium-glucose transporter 2 inhibitors
Indication:Type 2 diabetes
FDA APPROVED
| 1/20/2023 |
To improve glycemic control in adults with type 2 diabetes mellitus as an adjunct to diet and exercise
Drug Trials Snapshot
Phase II
Developer:Theracos, Inc.
| Conditions | Phases | Recruitment | Interventions | Sponsor/Collaborators |
|---|---|---|---|---|
| Diabetes Mellitus Type 2 | Phase 2 | Completed | Drug: EGT0001442|Drug: Placebo capsules to match EGT0001442 | Theracos |
| Diabetes Mellitus | Phase 2 | Completed | Drug: EGT0001442|Drug: Placebo | Theracos |
| Type 2 Diabetes Mellitus | Phase 3 | Not yet recruiting | Drug: Bexagliflozin|Drug: Placebo | Theracos |
| Diabetes Mellitus, Type 2 | Phase 2|Phase 3 | Recruiting | Drug: Bexagliflozin tablets | Theracos |

 DIPROLINE COMPLEX
DIPROLINE COMPLEX
Bexagliflozin diproline
RN: 1118567-48-8, C24-H29-Cl-O7.2C5-H9-N-O2
Molecular Weight, 695.2013
L-Proline, compd. with (1S)-1,5-anhydro-1-C-(4-chloro-3-((4-(2-(cyclopropyloxy)ethoxy)phenyl)methyl)phenyl)-D-glucitol (2:1)
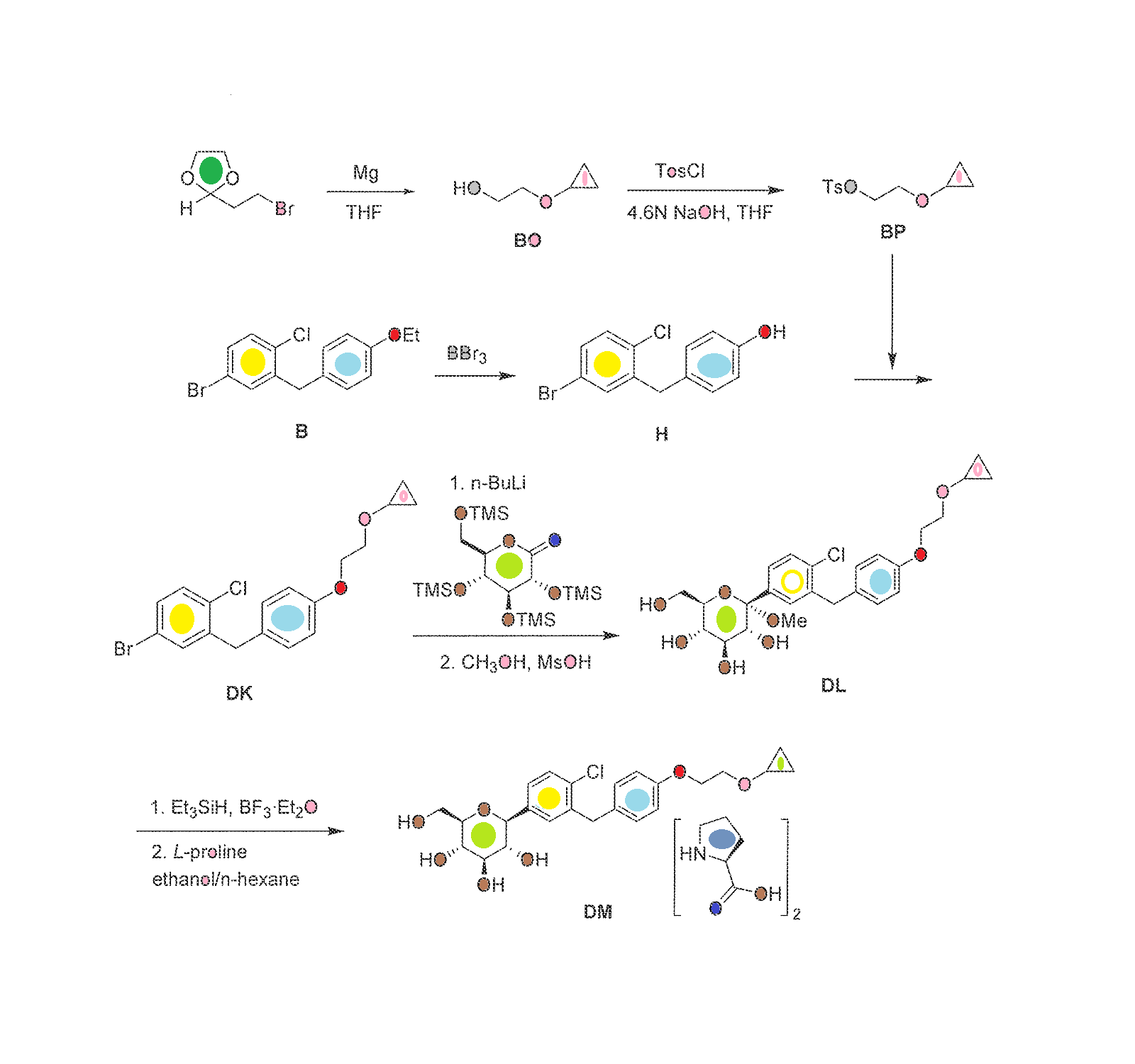

Bexagliflozin [(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2-(cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol] is an orally administered drug for the treatment of Type 2 Diabetes Mellitus (T2DM) and is classified as a Sodium Glucose co-Transporter 2 (SGLT2) Inhibitor. It is in Phase 2b study to evaluate the effect of bexagliflozin tablets in subjects with type 2 diabetes mellitus.
Bexagliflozin, also known as EGT1442, is a potent and selective SGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and prolongs the survival of stroke-prone rats. The IC(50) values for EGT1442 against human SGLT1 and SGLT2 are 5.6μM and 2nM, respectively. In normal rats and dogs a saturable urinary glucose excretion was produced with an ED(50) of 0.38 and 0.09mg/kg, respectively. EGT1442 showed favorable properties both in vitro and in vivo and could be beneficial to the management of type 2 diabetic patients.
One promising target for therapeutic intervention in diabetes and related disorders is the glucose transport system of the kidneys. Cellular glucose transport is conducted by either facilitative (“passive”) glucose transporters (GLUTs) or sodium-dependent (“active”) glucose cotransporters (SGLTs). SGLTl is found predominantly in the intestinal brush border, while SGLT2 is localized in the renal proximal tubule and is reportedly responsible for the majority of glucose reuptake by the kidneys.
Recent studies suggest that inhibition of renal SGLT may be a useful approach to treating hyperglycemia by increasing the amount of glucose excreted in the urine (Arakawa K, et al., Br J Pharmacol 132:578-86, 2001; Oku A, et al., Diabetes 48:1794-1800, 1999).

The potential of this therapeutic approach is further supported by recent findings that mutations in the SGL T2 gene occur in cases of familial renal glucosuria, an apparently benign syndrome characterized by urinary glucose excretion in the presence of normal serum glucose levels and the absence of general renal dysfunction or other disease (Santer R, et al., J Am Soc Nephrol 14:2873-82, 2003). Therefore, compounds which inhibit SGLT, particularly SGL T2, are promising candidates for use as antidiabetic drugs.
Compounds previously described as useful for inhibiting SGLT include C-glycoside derivatives (such as those described in US6414126, US20040138439, US20050209166, US20050233988, WO2005085237, US7094763, US20060009400, US20060019948, US20060035841, US20060122126, US20060234953, WO2006108842, US20070049537 and WO2007136116), O-glycoside derivatives (such as those described in US6683056, US20050187168, US20060166899, US20060234954, US20060247179 and US20070185197), spiroketal-glycoside derivatives (described in WO2006080421), cyclohexane derivatives (such as those described in WO2006011469), and thio- glucopyranoside derivatives (such as those described in US20050209309 and WO2006073197).

PATENT
WO 2009026537……………PRODUCT PATENT
http://www.google.co.in/patents/WO2009026537A1?cl=en


Example 19
[0289] The synthesis of compound BQ within the invention is given below.
[0290] Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (0.87 g, 36.1 mmol) and iodine (catalytic) in THF (4 mL) was added slowly BrCH2CH2Br (4.6 g, 24.5 mmol) in THF (8 mL). The exothermic reaction was cooled in an ice-bath. After complete addition OfBrCH2CH2Br, a solution of 2- (2-bromoethyl)-l,3-dioxolane (1 g, 5.6 mmol) was added dropwise. The reaction mixture was then kept at reflux for 24 h, quenched by addition of aqueous NH4Cl, and extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give crude intermediate BO (400 mg) as yellow oil. [0292] Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
Ts0^°V
To a solution of 2-cyclopropoxyethanol (400 mg, 3.92 mmol) in DCM (10 niL) were added TsCl (821 mg, 4.31 mmol) and Et3N (0.6 mL, 4.31 mmol). The reaction was stirred at room temperature overnight. Then, IN HCl was added, and the reaction was extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give a yellow oil. The oil was purified by preparative TLC to obtain intermediate BP (50 mg) as a yellow oil.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2- cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Compound BQ)

To a solution of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (intermediate Dl) (30 mg, 0.08 mmol) in anhydrous DMF (1 mL) were added 2-cyclopropoxyethyl 4-methylbenzenesulfonate (intermediate BP) (20 mg, 0.08 mmol) and Cs2CO3 (52 mg, 0.16 mmol). The mixture was stirred at room temperature for 12 h. Then the reaction mixture was poured into water, extracted with EA, washed with brine, dried with anhydrous Na2SO4 and concentrated to an oil. The oil was purified by preparative HPLC to obtain compound BQ (11 mg) as a colorless oil. 1H NMR (CD3OD): δ 7.30 (m, 3H), 7.11 (d, J= 8.8 Hz, 2H), 6.82 (d, J= 8.8 Hz, 2H), 4.13 (m, 5H), 3.85 (m, 3H), 3.81 (m, IH), 3.40 (m, 4H), 3.30 (m, IH), 0.52 (m, 4H); MS ESI (m/z) 465 (M+H)+, calc. 464.

Example 33
The synthesis of complex DM within the invention is outlined in FIG. 30, with the details given below.
Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (86.7 g, 3.6 mol) and I2 (catalytic) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) at a rate that maintained the reaction temperature between 40-55° C. A solution of 2-(2-bromoethyl)-1,3-dioxolane (100 g, 0.56 mol) in anhydrous THF (750 mL) was added dropwise, and the reaction mixture was kept at 40-55° C. for 16 h. The reaction was quenched by addition of an aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give intermediate BO (27 g) as yellow oil, which was used in the next step without further purification.
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added crude 2-cyclopropoxyethanol from the previous step (27 g, 0.26 mol) at −5 to 0° C. A solution of p-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise, and the reaction mixture was kept at −5 to 0° C. for 16 h. The reaction mixture was then incubated at room temperature for 30 min, the organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4 and concentrated to get the crude intermediate BP as a yellow oil (53.3 g), which was used for the preparation of intermediate DK below without further purification.
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (Intermediate H)

To a stirred solution of 4-bromo-1-chloro-2-(4-ethoxybenzyl)benzene (intermediate B) (747 g, 2.31 mol) in dichloromethane was added slowly boron tribromide (1.15 kg, 4.62 mol) at −78° C. The reaction mixture was allowed to warm to room temperature. When the reaction was complete as measured by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with an aqueous solution of saturated sodium bicarbonate, then with water, and then with brine, and dried over Na2SO4. The residue was concentrated and then recrystallized in petroleum ether to obtain intermediate H as a white solid (460 g, yield 68%). 1H NMR (CDCl3, 400 MHz): δ 7.23˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Preparation of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (Intermediate DK)

A mixture of 4-(5-bromo-2-chlorobenzyl)phenol (56.7 g, 210 mmol) and Cs2CO3 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 30 min, and then 2-cyclopropoxyethyl 4-methylbenzenesulfonate (crude intermediate BP from the second preceeding step above) (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight, and then diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, then with brine, and dried over Na2SO4. The residue was concentrated and then purified by flash column chromatography on silica gel (eluent PE:EA=10:1) to give intermediate DK as a liquid (51 g, yield 64%). 1H NMR (CDCl3, 400 MHz): δ 7.22˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52 (m, 2H).
Preparation of (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (Intermediate DL)

To a stirred solution of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (213 g) in anhydrous THF/toluene (1:2 v/v, 1.7 L) under argon was added n-BuLi (2.5 M in hexane, 245.9 mL) dropwise at −60±5° C. The mixture was stirred for 30 min, and then transferred to a stirred solution of (3R,4S,5R,6R)-3,4,5-tris(trimethylsilyloxy)-6-((trimethylsilyloxy)methyl)tetrahydro-2H-pyran-2-one (310.5 g) in toluene (1.6 L) at −60±5° C. The reaction mixture was continuously stirred at −60±5° C. for 1 before quenching with an aqueous solution of saturated ammonium chloride (1.5 L). The mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 mL). The combined organic layers were washed with brine (1 L), dried over Na2SO4, and concentrated. The residue was dissolved in methanol (450 mL), and methanesulfonic acid (9.2 mL) was added at 0° C. The solution was allowed to warm to room temperature and stirred for 2.0 h. The reaction was quenched with an aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and then additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated. The crude product was used in the next step without further purification.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex (Complex DM)

To a stirred solution of crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol from the previous step in CH2Cl2/CH3CN (1:1, 1.3 L) at −5° C. was added triethylsilane (28.2 mL, 563 mmol), followed by BF3.Et2O (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm gradually to room temperature. The reaction was quenched by addition of an aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2SO4 and concentrated to give the crude product (230 g, purity 82.3%). To the crude product was added L-proline (113.7 g) in EtOH/H2O (15:1 v/v, 2.09 L), and the mixture was stirred at 80° C. for 1 h until it became a clear solution. Hexane (3.0 L) was added dropwise over 50 min, while the temperature was maintained at about 60° C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/H2O (15:1 v/v, 2×300 mL), hexane (2×900 mL), and dried at 45° C. under vacuum for 10 h to give pure complex DM as a white solid (209 g; HPLC purity 99.2% (UV)). 1H NMR (CD3OD, 400 MHz): δ 7.25˜7.34 (m, 3H), 7.11 (d, J=8.8 Hz, 2H), 6.84 (d, J=8.8 Hz, 2H), 4.03-4.11 (m, 5H), 3.96-4.00 (m, 2H), 3.83-3.90 (m, 3H), 3.68-3.72 (m, 1H), 3.36-3.46 (m, 6H), 3.21-3.30 (m, 3H), 2.26-2.34 (m, 2H), 2.08-2.17 (m, 2H), 1.94-2.02 (m, 4H), 0.56-0.57 (m, 2H), 0.52-0.53 (m, 2H).
Crystalline complex DM was analyzed by X-ray powder diffraction using CuKα1 radiation. The diffraction pattern is shown inFIG. 31 and summarized in Table 1 (only peaks up to 30° in 2θ are listed). The melting point of complex DM was determined by differential scanning calorimetry (DSC) as 151±1° C. (evaluated as onset-temperature; heating from 50° C. to 200° C. at 10° C./min). The DSC spectrum is shown in FIG. 32.
Preparation of (3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate D)
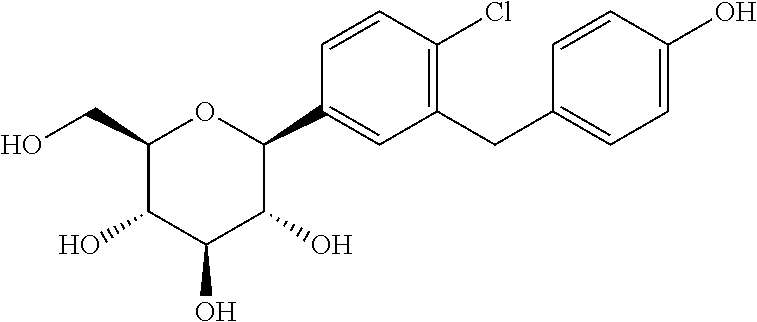
To a stirred solution of (3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate C) (2 g, 5.9 mmol) in dichloromethane was added BBr3 (14.6 mL, 1 M) dropwise at −78° C. After the addition was complete, the mixture was allowed to warm to 0° C. and held at this temperature for 2 h. When LC-MS showed that no starting material remained, the mixture was cooled to −78° C. again, and quenched with water. When the temperature was stable, saturated NaHCO3 solution was added. The mixture was evaporated under reduced pressure, and the residue was extracted with EtOAc. The organic layer was washed with NaHCO3 and brine, dried over Na2SO4, evaporated and purified to obtain intermediate D (0.7 g).
In addition, for use in the synthesis of certain compounds of the invention, the 2S isomer (intermediate D1) and the 2R isomer (intermediate D2) of intermediate D were separated by preparative LC-MS. Intermediate D1: 1H NMR (CD3OD): δ 7.30 (m, 3H), 6.97 (d, 2H, J=6.8 Hz), 6.68 (d, 2H, J=6.8 Hz), 4.56 (s, 1H), 4.16 (s, 1H), 3.91˜4.02 (m, 5H), 3.79 (m, 1H), 3.64 (m, 1H). Intermediate D2: 1H NMR (CD3OD): δ 7.29˜7.33 (m, 3H), 7.00 (d, 2H, J=6.8 Hz), 6.70 (d, 2H, J=6.8 Hz), 4.58 (d, 1H, J=4.0 Hz), 3.96˜4.02 (m, 4H), 3.93˜3.95 (m, 1H), 3.81˜3.85 (m, 1H), 3.64˜3.69 (m, 1H).
PATENT
http://www.google.com/patents/US20130267694

Example 14 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol crystals
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(442-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol bis(L-proline) complex in methanol/water solvent mixture.

(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (1.3 kg) was added to a propylene drum (25 L) and methanol (3.6 kg) and water (1.3 kg) and the mixture was stirred until the solids dissolved. The solution was filtered through filter membrane (Millipore, 0.45 μm) into a clean glass reactor (50 L). The mixture was refluxed for 30 min and water (7.2 kg) was added over 1.0 h while maintaining the temperature between 50 and 65° C. The mixture was slowly cooled to ˜42° C. over 2 h. A suspension of seed crystal (26 g) in cold (−5° C.) mixture of methanol/water (78 mL, 2.8/6.5 (w/w)) and the slow cooling was continued to −5° C. over 12 h. The suspension was stirred for another 5 h and was filtered. The solid was slurried with cold water and filtered (0 to 5° C., 3×2.6 kg). The filter cake was dried under reduced pressure for 24 h until the loss on drying was no more than 0.5% to give a white solid (825 g, 92% yield, 99.3% pure by \HPLC-0001).
Example 15 Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol
This example describes preparation of 4-(2-chloro-5-iodobenzyl)phenol using gaseous hydrobromic acid.

Preparation of (2-chloro-5-iodophenyl)methan-1-ol

A 250 mL of 4-necked flask equipped with thermometer and mechanical stirring was charged with NaBH4 (4.16 g, 0.11 mol) and THF (60 mL) under argon. After cooling to 0˜5° C. with stirring, a solution of iodine in THF (12.7 g I2 in 25 mL THF) was added slowly dropwise over 30 min and the reaction temperature was maintained below 10° C. After the addition was completed, a solution of 2-chloro-5-iodobenzoic acid (15.0 g, 50 mmol) in THF (20 mL) was added dropwise over 30 min and kept the reaction temperature below 10° C. After stirring for another 3 h at 20˜25° C., the reaction mixture was heated to reflux for additional 16 h and monitored by TLC (PE/EA=1:1, Rf=0.2). The mixture was cooled to 20˜25° C. and poured into ice water (100 mL), extracted with ethyl acetate (2×100 mL), washed with water (2×100 mL), brine (100 mL), concentrated and the residue was purified by flash chromatography (PE:EA=20:1 as eluant, 200 mL) to give an off-white solid. Yield: 10.0 g (70%) MS ESI (m/z): 269 [M+1]+.
Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol

A 100 mL of 4-necked flask equipped with thermometer and mechanical stirrer was charged with (2-chloro-5-iodophenyl)methanol (268.5 mg, 1 mmol), anhydrous ZnCl2 (136.3 mg, 1 mmol), dichloromethane (5.0 mL) and n-hexane (29 mL) under argon. After stirring for 10 min at 20 to 25° C., HBr (gas) was bubbled into the mixture for 10 min and a solution of phenol (197.6 mg, 2.1 mmol) in dry dichloromethane (3.0 mL) was added dropwise over 30 min. After bubbling HBr for additional 2 h, the mixture was refluxed for 3 days. The conversion was about 65%. The mixture was quenched with ice water (50 mL), extracted with ethyl acetate (2×30 mL), washed with water (2×30 mL), brine (30 mL), concentrated and the residue was purified by flash chromatography (PE:EA=25:1 as eluant, 200 mL) to give an off-white solid. Yield: 180 mg (52%). 1H NMR (CDCl3, 400 MHz): δ 7.44 (d, J=8.4 Hz, 2H), 7.03˜7.09 (m, 3H), 6.77 (d, J=8.4 Hz, 2H), 4.76 (s, 1H), 3.95 (s, 2H), 3.82 (s, 2H). MS ESI (m/z): 345 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 154.1, 141.4, 139.5, 136.6, 134.2, 131.2, 130.9, 130.1, 115.5, 91.67, 38.07.
Example 16 Preparation of 2-(4-(2-Cyclopropoxyethoxy)Benzyl)-1-Chloro-4-Iodobenzene
This example describes the preparation of 2-(4-(2-cyclopropoxyethoxy)benzyl)-1-chloro-4-iodobenzene via coupling of the 4-(2-chloro-5-iodobenzyl)phenol with 2-cyclopropoxyethyl 4-methylbenzenesulfonate.

Under nitrogen a 500 L glass-lined reactor was charged with acetone (123 kg) with stirring (120 RPM), 4-(2-chloro-5-iodobenzyl)phenol (19.37 kg, 0.056 kmol), 2-cyclopropoxyethyl 4-methylbenzenesulfonate (15.85 kg, 0.062 kmol), cesium carbonate (18.31 kg, 0.0562 kmol) powder, potassium carbonate (23.3 kg, 0.169 kmol) powder and TBAI (4.15 kg, 0.011 kmol). After stirring for 4045 h at 40° C., TLC (PE:EA=4:1, Rf=0.3) showed that starting material was consumed. The mixture was cooled to 20˜25° C.
The reaction mixture was filtered over diatomite (28 kg) and the filter cake was washed with acetone (2×31 kg). The combined filtrates were transferred to a 500 L glass-lined reactor and concentrated. The residue was dissolved in ethyl acetate (175 kg, washed with water (2×97 kg) and concentrated until the volume was about 100 L and was transferred to a 200 L glass-lined reactor and continued to concentrate to get about 22.5 kg of crude material.
The crude material was dissolved in methanol/n-hexane (10:1, 110 kg) under refluxing for 30 min with stirring (100 RPM) until it was a clear solution. The mixture was cooled to 5 to 10° C. and some crystal seeds (20 g) were added. The suspension was stirred for another 5 h at 5 to 10° C. The mixture was filtered at 0 to 5° C. and the filter cake was washed with pre-cooled methanol/n-hexane (10:1, 5° C., 2×11 kg). The filter cake was dried under at 15 to 20° C. for 15 h to give off-white to white solid. Yield: 18.1 kg, 75%. Melting Point: 31° C. (DSC onset). 1H NMR (CDCl3, 400 MHz): δ 7.45˜7.50 (m, 2H), 7.09˜7.12 (m, 3H), 6.88 (d, J=8.8 Hz, 2H), 4.11 (t, J=5.2 Hz, 2H), 3.99 (s, 2H), 3.88 (t, J=5.2 Hz, 2H), 3.40˜3.44 (m, 1H), 0.63˜0.67 (m, 2H), 0.49˜0.54 (m, 1H). MS ESI (m/z): 429 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 157.5, 141.5, 139.5, 136.6, 134.2, 131.2, 130.8, 129.9, 114.9, 91.66, 69.00, 67.13, 53.72, 38.08, 5.63.
Example 9 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex by co-crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol with L-proline in ethanol/water/n-heptane solvent mixture.
The crude (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (2.5 kg) was added to a glass reactor containing ethanol (95%, 16 kg) and L-proline (1.24 kg) and the mixture was refluxed for 1 h. While keeping the temperature above 60° C., n-heptane (8.5 kg) was added over 40 min. The mixture was slowly cooled to 25 to 20° C. and stirred at this temperature for 10 h. The mixture was filtered and the solids were washed with cold (−5° C.) ethanol (95%, 2×2.5 L) and n-heptane (2×5 L) and the solids were dried under reduced pressure at 55 to 65° C. for 20 h to give a white solid (3.03 kg, 81% yield, 99.4% pure by HPLC-0001).
Example 7 Preparation of ((2S,3R,4R,5S,6R)-2-(4-Chloro-3-(4-(2-Cyclopropoxyethoxy)Benzyl)Phenyl)-6-(Hydroxymethyl)Tetrahydro-2H-Pyran-3,4,5-triol
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by removal of the anomeric OH or OMe.

(2S,3R,4S,5S,6R)-2-(4-Chloro-3-(4-(2-Cyclopropoxyethoxy)Benzyl)Phenyl)-6-(Hydroxymethyl)-2-Methoxytetrahydro-2H-Pyran-3,4,5-Triol Solution
A 30 L glass reactor equipped with a thermometer was charged with crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (1.15 kg), DCM (2.3 kg) and acetonitrile (1.4 kg), and the mixture was magnetically stirred until all the solids dissolved under nitrogen sparging. The solution was cooled to ˜−15° C.
Triethylsilane Solution:
BF3.Et2O (1.2 kg) was added to a cold (−20 to −15° C.) solution of triethysilane (1.08 kg) dichloromethane (2.3 kg) and acetonitrile (1.4 kg) with nitrogen sparging.
The cold (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol solution was added to the cold triethylsilane solution at such a rate to maintain the temperature between −20 and −15° C. (˜2 to 3 h).
The reaction mixture was stirred for another 2 to 3 h and then quenched by addition of an aqueous solution of sodium bicarbonate (7.4% w/w, 7.8 kg) and the reaction mixture was stirred for about 15 min. The solvents were removed under reduced pressure (2 h, temperature below 40° C.). The residue was partitioned between ethyl acetate (6.9 kg) and water (3.9 kg). The layers were separated and the aqueous layer was extracted with ethyl acetate (2×3.5 kg). The combined organic layers were washed with brine (2×3.8 kg) and the solvents were removed under reduced pressure. Anhydrous ethanol (2.3 kg) was added and concentrated to give the crude product of the title compound (1 kg, 90% yield, 90% HPLC-0001) as yellow solid.
PATENT
WO 2011153953
https://www.google.com/patents/WO2011153953A1?cl=en
Example 1. Preparation of (2S.iR. R.5S.6R)-2-(4-chloro-3-(4-(2-cvclopropoxyethoxy) benzyl)phenyl)-6-(hvdroxymethyl)tetrahvdro-2H-pyran-3,4,5-triol, bis(X-proline) complex


Example 1A
Preparation of 2-cyclopropoxyethanol (1)

To a suspension of Mg powder (86.7 g, 3.6 mol) and iodine (cat) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) slowly at a rate as to keep the internal temperature between 40-55 °C. After the addition, a solution of 2-(2-bromoethyl)-l,3-dioxolane (lOOg, 0.56 mol) in anhydrous THF (750 mL) was added dropwise. The reaction mixture was kept at 40-55 °C for 16h and was quenched by addition of aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give the title product (27 g) as yellow oil, which was directly used without further purification.
Example IB
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (2)

To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added Example 1A (27 g, 0.26 mol) at -5 to 0 °C. Afterwards, a solution of ji?-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise. The reaction mixture was kept at -5 to 0 °C for 16 h. The reaction mixture was then kept at room temperature for 30 min. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2S04 and concentrated to get the crude product as yellow oil (53.3 g). It was used directly without further purification.
Example 1C
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (3)

To a stirred solution of 4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene (747 g, 2.31 mol) in dichloromethane was added boron tribromide (1.15 kg, 4.62 mol) slowly at -78 °C. The reaction mixture was allowed to rise to room temperature. When the reaction was complete as measure by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with aqueous solution of saturated sodium bicarbonate, water, brine, dried over Na2S04, and concentrated. The residue was recrystallized in petroleum ether to give the title compound as a white solid (460 g, yield 68%). 1H NMR (CDC13, 400MHz): δ 7.23-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Example ID
Preparation of 4-bro -l-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (4)

A mixture of Example 1C (56.7 g, 210 mmol) and Cs2C03 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 0.5 h. Example IB (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight. It was diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, brine, dried over Na2S04, and concentrated. The residue was purified by flash column
chromatography on silica gel eluting with petroleum ether:ethyl acetate (10:1) to give the title compound as liquid (51 g, yield 64%). 1H NMR (CDC13, 400MHz): δ 7.22-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52(m, 2H).
Example IE
Preparation of (25,5R, S,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6-(hydroxymethyl)-2-metlioxytetraliydro-2H-pyran-3,4,5-triol (5)

To a stirred solution of Example ID (213 g) in anhydrous THF/toluene (1 :2 (v/v), 1.7 L) under argon was added n-BuLi (2.5 M hexane, 245.9 mL) drop wise at -60 ± 5 °C. The mixture was stirred for 30 min. before transferred to a stirred solution of 2,3,4,6-tetra-O- trimethylsilyl-P-Z -glucolactone (310.5 g) in toluene (1.6 L) at -60 ± 5 °C. The reaction mixture was continuously stirred at -60 ± 5 °C for 1 h before quenching with aqueous solution of saturated ammonium chloride (1.5 L). Then mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 niL). The combined organic layers were washed with brine (1 L), dried over Na2S04, and concentrated. The residue was dissolved in methanol (450 mL) and methanesulfonic acid (9.2 mL) was added at 0 °C. The solution was allowed to warm to room temperature and stirred for 20 h. It was quenched with aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2S04, concentrated and used directly in the next step without further purification.
Example IF
Preparation of (25,5R, R,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(Z-proline) complex (7)

To stirred solution of Example IE in CH2C12/CH3CN (650 mL:650 mL) at -5 °C was added triethylsilane (28.2 mL, 563 mmol), and followed by BF3-Et20 (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm to room temperature gradually. The reaction was quenched with aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2S04 and concentrated to give the crude product 6 (230 g, purity 82.3%). This product and L-proline (113.7 g) in EtOH/H20 (15:1 v/v, 2.09 L) was stirred at 80 °C for 1 h when it became a clear solution. Hexane (3.0 L) was added dropwise into the above hot solution over 50 min, with the temperature being kept at about 60 °C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/ H20 (15:1 (v/v), 2×300 mL), hexane (2×900 mL), and dried at 45 °C under vacuum for 10 h to give the pure title compound 7 as a white solid (209 g).
Purity (HPLC) 99.2% (UV). 1H NMR (CD3OD, 400 MHz): δ 7.25—7.34 (m, 3H), 7.11 (d, J = 8.8 Hz, 2H), 6.84 (d, J= 8.8 Hz, 2H), 4.03-4.11 (m, 5H), 3.96-4.00 (m, 2H), 3.83-3.90 (m, 3H), 3.68-3.72 (m, 1H), 3.36-3.46 (m, 6H), 3.21-3.30 (m, 3H), 2.26-2.34 (m, 2H), 2.08-2.17 (m, 2H), 1.94-2.02 (m, 4H), 0.56-0.57 (m, 2H), 0.52-0.53(m, 2H).
Example 2. Direct Preparation of Crystalline Compound 8 from Complex 7
This example illustrates the preparation of a crystalline form of (2S, 3R, 4R, 5S, 6R)-2- (4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol.

To a 5.0 L 4-necked flask equipped with a mechanical stirrer was added the starting co-crystal (150.0 g) and methanol (300 mL). The mixture was stirred at room temperature with mechanical stirring (anchor agitator, 2-blades 9 cm) until a cloudy solution/suspension formed, to which distilled water (1500 mL) was added dropwise at a rate of -12.5 mL/min. As the mixture warmed from the exotherm of adding water to methanol, the mixture became clear after adding about 1/5 to 1/3 of the water. After the addition was completed the reaction was stirred continuously at 80 rpm for another 5 h. The reaction mixture was filtered over medium-speed filter paper and the filter cake was washed with distilled water (450 mL and then 300 mL) and dried under vacuum using an oil pump (~6 mm Hg) at 45 °C for 48 hours to give the target product as a white crystalline solid (94.2 g, 93.9% yield, purity (HPLC): 99.3%).
Example 5. Indirect Preparation of Crystalline Compound 8 from Complex 7

[0113] To a 200 L glass lined reactor equipped with a double-tier paddle agitator and a glass condenser was added sequentially complex 7 (7.33 kg), ethyl acetate (67.5 kg) and pure water (74.0 kg). The mixture was heated to reflux and stirred at reflux for 30 min. The reaction mixture was cooled to approximately 50 °C and the organic layer was separated and the aqueous layer was extracted with ethyl acetate (34.0 kg). The combined organic layers were washed with pure water (3×74.0 kg) (IPC test showed that the IPC criteria for L-proline residue was met after three water washes). The mixture was concentrated at 40 °C under vacuum (-15 mmHg) for 3 h until the liquid level dropped below the lower-tier agitator paddle. The mixture (18 kg) was discharged and transferred to a 20L rotary evaporator. The mixture was concentrated under vacuum (40 °C, ~5 mmHg) to a minimum volume. The remaining trace amount of ethyl acetate was removed azeotropically at 40 °C under vacuum with methanol (10 kg). The residue was dried under vacuum of an oil pump (~6 mmHg) at 40 °C for 10 h to give 8 as a white amorphous solid (4.67 kg, purity (HPLC): 99.2%) which was used in the next step without further purification.
The recrystallization was accomplished by the following steps. To a 100 L glass line reactor equipped with a double-tier paddle agitator and a glass condenser was added the above amorphous 8 (4.67 kg) and methanol (18.0 kg). The mixture was refluxed at 70 °C for 30 min until a clear solution formed, to which pure water (45.0 kg) was added over 2 hours. After the addition was completed (the reaction temperature was 41 °C), the reaction mixture was cooled to room temperature and stirred at room temperature for 15 hours. The reaction mixture was filtered and the wet cake was washed with pure water (2×15 kg) and dried under vacuum at 55-60 °C for 12 hours to give the target product as an off-white crystalline solid (3.93 kg, yield: 84% in two steps; purity (HPLC): 99.7%).
Example 6. Direct Preparation of Crystalline Compound 8 from Amorphous 8

A 5 L 4-neck flask was charged with 8 (amorphous), 116 g, and methanol (580 mL). The reaction mixture was heated to 60 C with mechanical stirring and the solution became clear. Water (2320 mL) was added dropwise to the reaction solution at 40 mL/min at 50 °C. The reaction mixture was stirred overnight at room temperature. The reaction mixture was filtered and the filter cake was washed with water (2×200 mL), dried under vacuum at 55 °C for 12 hours, to afford white crystalline 8. Yield is 112.8 g (97.2%).
References:
1. Clinical Trial, A Dose Range Finding Study to Evaluate the Effect of Bexagliflozin Tablets in Subjects With Type 2 Diabetes Mellitus. NCT02390050 (retrieved on 26-03-2015).
| WO2008144346A2 * | May 15, 2008 | Nov 27, 2008 | Squibb Bristol Myers Co | Crystal structures of sglt2 inhibitors and processes for their preparation | |||||||||||||||
| WO2009026537A1 * | Aug 22, 2008 | Feb 26, 2009 | Theracos Inc | Benzylbenzene derivatives and methods of use | |||||||||||||||
| CN1407990A * | Oct 2, 2000 | Apr 2, 2003 | 布里斯托尔-迈尔斯斯奎布公司 | C-aryl glucoside sgltz inhibitors
|
| WO2010022313A2 * | Aug 21, 2009 | Feb 25, 2010 | Theracos, Inc. | Processes for the preparation of sglt2 inhibitors |
////////BEXAGLIFLOZIN, APPROVALS 2023, FDA 2023
c1cc(ccc1Cc2cc(ccc2Cl)[C@H]3[C@@H]([C@H]([C@@H]([C@H](O3)CO)O)O)O)OCCOC4CC4
SYN
https://doi.org/10.1021/acs.jmedchem.4c02079J.Med.Chem.2025,68,2147−2182
Bexagliflozin (Brenzavvy). Bexagliflozin (3) was discoveredanddevelopedbyTheracosBioforthetreatmentof
type2diabetesmellitus.28Bexagliflozinisasodium-dependent glucose cotransporter 2 (SGLT2) inhibitor. Inhibition of SGLT2 reduces blood sugar without stimulating insulin release.29 Bexagliflozin shows >2000-fold selectivity forSGLT2 over SGLT1 and demonstrated improvement inglycemiccontrolwithaoncedaily,20mgdose.28Since 2011, there have been 11 therapeutics targeting
SGLT2.30Thesedrugsexhibit commonstructural features(abiarylmethaneandglycoside)andlikelyfacesimilarsynthetic challenges.31 The medicinal chemistry efforts to identifybexagliflozinweredisclosedintheprimaryliterature.32Apatent fromTheracos, Inc. in2013describedasyntheticapproachto bexagliflozinonmultikilogramscale.33Slightvariations inthe
reactionconditions,yieldandisolationstrategyofintermediates wereincludedinthepatent.Theimplementationoftelescoping intheprocessislikelyduetopoorcrystallinityofintermediates,
whichmaybeacommonchallengetootherSGLT2inhibitors.31
Anotherpatent disclosedbyPiramal Enterprises suggesteda
similarbondformationstrategybut includedanacetylationof bexagliflozinprior tothefinal isolation inorder toprovidea crystallinesolid.34
Bexagliflozinwas assembled by cryogenicmetal halogen exchangeof aryl iodide3.1with turboGrignard(i-PrMgCl·LiCl)andsubsequentadditiontoprotectedgluconolactone3.2
whichwaspreparedbytreatmentofD-(+)-glucono-1,4-lactonewithTMSClandNMMinTHFin94%yield(Scheme4).WhentheGrignardadditionwascomplete,thereactionwasquenchedand a solution of the product inEtOAcwas treatedwith
activated carbon, filtered, concentrated, and diluted with methanol.ThissolutionwastreatedwithconcentratedHCl to remove thesilyl protectinggroupsandprovidecrudemethyl ketal3.3inyields rangingfrom79to95%.Themethyl ketal
functionalitywasreducedusingtriethylsilaneandBF3·Et2Oin DCMandMeCNatcryogenictemperaturestoprovidecrude bexagliflozin (3) as a solid after concentrating the reaction mixture. Alternatively, a larger-scale demonstration of this processinthepatenttelescopedasolutionofcrudebexagliflozin toformabis-L-prolinecomplexinethanol,water,andheptane,
whichwasisolatedasacrystallinesolidin81%yield.Thiswas convertedto the free formin82%yieldbycrystallization in methanolandwater.Arecrystallizationofbexagliflozin(3)was
reported in 92% yield. Details on stereoselectivity of this
approachwerenotdisclosed.
Amilligram-togram-scaleconstructionofthearyliodide3.1 wasalsodisclosedintheTheracospatent from2013(Scheme 5).33First,carboxylicacid3.5wasreducedtoprimaryalcohol
3.6using sodiumborohydride and iodine. Next, the diaryl methanecorewas assembledbyFriedel−Crafts alkylationof phenol with3.6 after activationwithHBr andZnCl2. This reactionwasdemonstratedonmilligramscaleandachieved65% conversion, with 52% isolated yield after chromatographic purification.Analternativeapproachtoabromovariantofaryl iodide3.7waspresentedina2009patentfromTheracos,where Friedel−Craftsacylationprovidedtheanalogousbenzophenone intermediatewhichwas thensubsequentlyreduced.35Finally,alkylationofthephenolwasconductedusingthetosylatedether
3.8toprovidearyl iodide3.1in75%yieldonkilogramscale.A syntheticapproachtothetosylatedetherwasprovidedinthe earlyTheracospatent,35wherecyclopropylether formationin 3.10wasgeneratedviaGrignardformationandrearrangement of 2-(2-bromoethyl)-1,3-dioxolane 3.9 (Scheme 6). The primary alcohol 3.10was protectedas the tosylate3.8and employedinthealkylationstepwithoutpurification.Noyields wereprovided.

(28) Hoy, S. M. Bexagliflozin: first approval. Drugs 2023, 83, 447−
453.
(29) Hsia, D. S.; Grove, O.; Cefalu, W. T. An update on sodium
glucose co-transporter-2 inhibitors for the treatment of diabetes
mellitus. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 73−79.
(30) Guo, Y.-Y.; Zhang, J.-Y.; Sun, J.-F.; Gao, H. A comprehensive
review of small-molecule drugs for the treatment of type 2 diabetes
mellitus: Synthetic approaches and clinical applications. Eur. J. Med.
Chem. 2024, 267, No. 116185.
(31) Aguillón, A. R.; Mascarello, A.; Segretti, N. D.; de Azevedo, H. F.
Z.; Guimaraes, C. R. W.; Miranda, L. S. M.; de Souza, R. O. M. A.
Synthetic strategies toward SGLT2 inhibitors. Org. Process Res. Dev.
2018, 22, 467−488.
(32) Xu, B.; Feng, Y.; Cheng, H.; Song, Y.; Lv, B.; Wu, Y.; Wang, C.;
Li, S.; Xu, M.; Du, J.; et al. C-aryl glucosides substituted at the 4′
position as potent and selective renal sodium-dependent glucose co
transporter 2 (SGLT2) inhibitors for the treatment of type 2 diabetes.
Bioorg. Med. Chem. Lett. 2011, 21, 4465−4470.
(33) Xu, B.; Lv, B.; Xu, G.; Seed, B.; Roberge, J. Y. Process for the
preparation of benzyl-benzene C-glycosides via coupling reaction as
potential SGLT2 inhibitors. US 20130267694, 2013.
(34) Gharpure, M.; Sharma, S. K.; Vishwasrao, S.; Vichare, P.; Varal,
D. Aprocess for the preparation of SGLT2 inhibitor and intermediates
thereof. WO 2018207113, 2018.
(35) Song, Y.; Chen, Y.; Cheng, H.; Li, S.; Wu, Y.; Feng, Y.; Lv, B.; Xu,
B.; Seed, B.; Hadd, M. J.; et al. Preparation of benzylbenzene glycoside
derivatives as antidiabetic agents. WO 2009026537, 2009.
.
European Journal of Medicinal Chemistry
Volume 265, 5 February 2024, 116124
https://doi.org/10.1016/j.ejmech.2024.116124
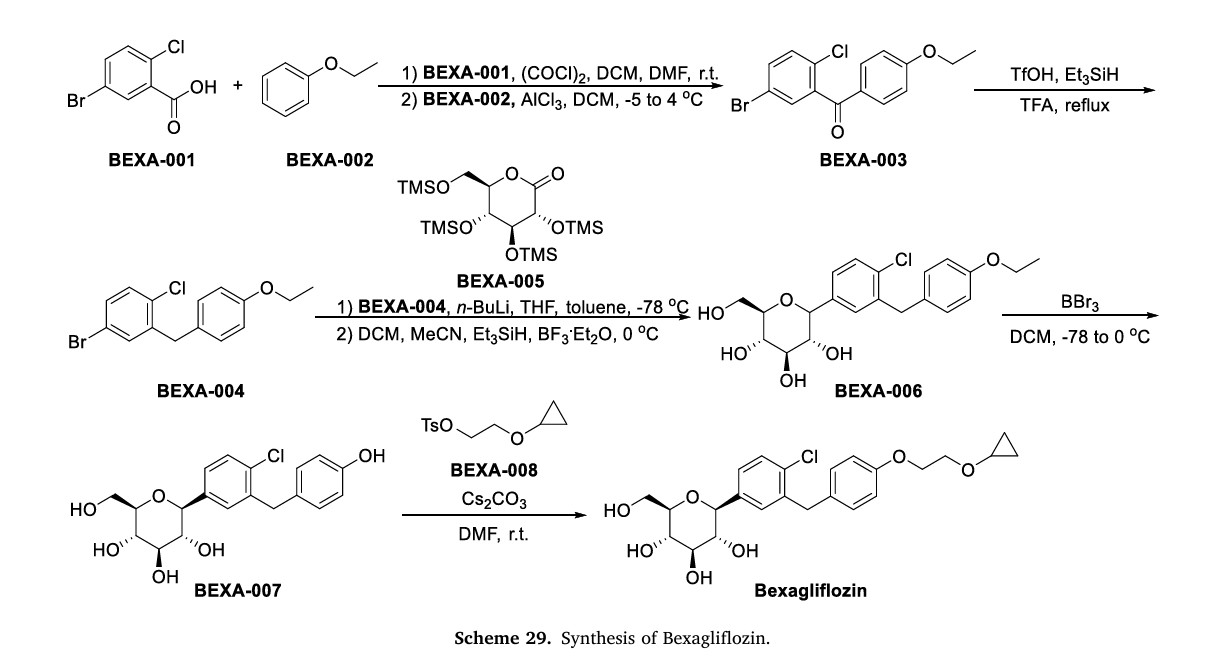
Bexagliflozin (Brenzavvy)
On January 20, 2023, the FDA granted approval to Bexagliflozin, a medication developed by Theracos Inc, for the treatment of type 2 diabetes mellitus (T2DM) [104–106]. The SGLT2 inhibitor Bexagliflozin
can increase energy expenditure, reduce fluid retention, and increase urinary glucose excretion by inhibiting SGLT2 in renal tubular epithelial cells [106]. SGLT2 inhibitors have significant advantages compared to other drugs: (1) they can lower both pre-meal and post-meal blood sugar levels (not all drugs can lower both); (2) they have a lower risk of hypoglycemia as they do not stimulate insulin secretion; (3) they have adiuretic effect due to their primary action on the renal tubules, which
lowers systolic blood pressure; (4) research has shown that SGLT2 in hibitors have therapeutic effects on diabetic kidney disease [107,108].
The process of synthesizing Bexagliflozin started by conducting theFriedel-Crafts acylation of ethoxybenzene (BEXA-002) with 5-bromo-2-chlorobenzoic acid (BEXA-001) (Scheme 29) [109]. This reaction produced ketone BEXA-003. Subsequently, the carbonyl reduction of BEXA-003 was carried out using trifluoromethanesulfonic acid (TfOH),triethylsilane, and TFA. This step yielded BEXA-004. Next, n-butyllithium (n-BuLi) and pyrone BEXA-005 were combined with BEXA-004 at78◦C. This reaction produced an intermediate, which was thenreacted with triethylsilane and BF◦3⋅Et2O at 0C. The final product obtained from this reaction was BEXA-006, which contained a sugar ring.
BEXA-006 underwent dealkylation upon treatment with boron tribromide, resulting in the formation of BEXA-007, which was a phenol.
Subsequently, BEXA-007 was alkylated using 2-cyclopropoxyethyl4-methylbenzenesulfonate (BEXA-008) to yield Bexagliflozin.
[104] S.M. Hoy, Bexagliflozin: first approval, Drugs 83 (2023) 447–453.
[105] W. Zhang, A. Welihinda, J. Mechanic, H. Ding, L. Zhu, Y. Lu, Z. Deng, Z. Sheng,
B. Lv, Y. Chen, J.Y. Roberge, B. Seed, Y.X. Wang, EGT1442, a potent and selectiveSGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and
prolongs the survival of stroke-prone rats, Pharmacol. Res. 63 (2011) 284–293.
[106] O. Azzam, R. Carnagarin, L.M. Lugo-Gavidia, J. Nolde, V.B. Matthews, M.
P. Schlaich, Bexagliflozin for type 2 diabetes: an overview of the data, Expet Opin.
Pharmacother. 22 (2021) 2095–2103.
[107] B.F. Palmer, D.J. Clegg, Kidney-protective effects of SGLT2 inhibitors, Clin. J. Am.
Soc. Nephrol. 18 (2023) 279–289.
[108] M. Singh, A. Kumar, Risks associated with SGLT2 inhibitors: an overview, Curr.
Drug Saf. 13 (2018) 84–91.
[109] Y. Song, Y. Chen, H. Cheng, S. Li, Y. Wu, Y. Feng, B. Lv, B. Xu, B. Seed, M.J. Hadd,
J. Du, C. Wang, J.Y. Roberge, Preparation of Benzylbenzene Glycoside Derivatives
as Antidiabetic Agents, 2009. WO2009026537A1.
.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
Merestinib

1206799-15-6 (Merestinib)
Chemical Formula: C30H22F2N6O3
Exact Mass: 552.17215
N-(3-fluoro-4-((1-methyl-6-(1H-pyrazol-4-yl)-1H-indazol-5-yl)oxy)phenyl)-1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamide
- OriginatorEli Lilly…..Eli Lilly And Company
- ClassAmides; Antineoplastics; Dihydropyridines; Pyrazoles; Small molecules
- Mechanism of ActionMKNK1 protein inhibitors; MKNK2 protein inhibitors; Proto oncogene protein c met inhibitors; ROS1-protein-inhibitors
- 29 Jun 2015Immunocore in collaboration with Eli Lilly plans a phase Ib/II trial for Uveal Melanoma (Metastatic disease, Combination therapy)
- 18 Jun 2015Eli Lilly completes a phase I bioavailability trial in healthy volunteers in USA (NCT02370485)
- 01 Feb 2015Eli Lilly initiates enrolment in a phase I bioavailability trial in healthy volunteers in USA (NCT02370485)
| Company | Eli Lilly and Co. |
| Description | C-Met inhibitor |
| Molecular Target | c-Met receptor tyrosine kinase (c-MET) (MET) (HGFR) (c-Met proto-oncogene) |
| Mechanism of Action | c-Met receptor tyrosine kinase inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase II |
| Standard Indication | Cancer (unspecified) |
| Indication Details | Treat advanced cancer |
 LY2801653 dihydrochloride; UNII-33F79TLF60; LY 2801653 dihydrochloride; LY-2801653 dihydrochloride; 1206801-37-7; 33F79TLF60
LY2801653 dihydrochloride; UNII-33F79TLF60; LY 2801653 dihydrochloride; LY-2801653 dihydrochloride; 1206801-37-7; 33F79TLF60LY2801653 was identified and developed as a novel, potent, and orally active small molecule inhibitor of human c-Met. It demonstrated dose dependent inhibition of c-Met phosphorylation in xenograft tumors with a long lasting PD effect. LY2801653 displayed potent anti-tumor efficacy in a number of non small cell lung, renal, pancreatic, and breast tumor models. Examination of c-Met expression in these tumors by immunohistochemistry (IHC) revealed a good correlation between response and c-Met expression in the tumor tissue. LY2801653 treatment led to increase in functional vessel areas, and decrease in tumor hypoxia. Enhanced anti-tumor efficacy was achieved when Erlotinib was combined with LY2801653. . (source: http://cancerres.aacrjournals.org/cgi/content/meeting_abstract/70/8_MeetingAbstracts/3611).
Patent
MAIN
INTERMEDIATES
https://www.google.com/patents/US8030302
Example 1 N-(3-Fluoro-4-(1-methyl-6-(1H-pyrazol-4-yl)-1H-indazol-5-yloxy)phenyl)-1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamide
To a 100 mL round bottom flask is added tert-butyl 4-(5-(4-amino-2-fluorophenoxy)-1-methyl-1H-indazol-6-yl)-1H-pyrazole-1-carboxylate (1.43 g, 3.38 mmol), 1-(4-flurorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxylic acid (1.25 g, 5.07 mmol), EDCI (1.48 g, 7.6 mmol) and HOBt (776 mg, 5.07 mmol) followed by DMF (15 mL, 193.99 mmol) and then DIPEA (1.47 mL, 8.44 mmol). The mixture is allowed to stir at RT overnight. The reaction mixture is diluted into EtOAc (300 mL) and washed with saturated aqueous sodium chloride (5×100 mL). The combined aqueous solution is extracted with EtOAc (1×100 mL) and then the combined organic solutions are dried over N2SO4, filtered, and concentrated to dryness. The solid is purified on a silica gel column eluting with DCM (A) and a 10% MeOH in a DCM solution (B), gradient from 100% (A) to 80% (A):20% (B) over 70 min to give tert-butyl 4-(5-(2-fluoro-4-(1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamido)phenoxy)-1-methyl-1H-indazol-6-yl)-1H-pyrazole-1-carboxylate as a gold solid (2.20 g, 87% yield). MS (m/z): 653. (M+H), 675 (M+Na).
Example 2 N-(3-Fluoro-4-(1-methyl-6-(1H-pyrazol-4-yl)-1H-indazol-5-yloxy)phenyl)-1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamide

A 12 L round bottom flask is equipped with overhead agitation, a thermocouple, and a N2 purge. tert-Butyl 4-(5-(4-amino-2-fluorophenoxy)-1-methyl-1H-indazol-6-yl)-1H-pyrazole-1-carboxylate (404 g, 954.08 mmol) is dissolved in DMF (2 L) and charged to the flask. DMF (1 L) is used to rinse the flask. 1-(4-Fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxylic acid (259.46 g, 1.05 mol) and EDCI (228.63 g, 1.19 mol) are added and it is rinse in with DMF (500 mL). Then HOBt (189.94 g, 1.24 mol) is added and it is again rinsed in with DMF (500 mL). Finally, DIPEA is slowly added (184.97 g, 1.43 mol). The dark solution is then stirred at RT over the weekend. To a 20 L bottom outlet flask is added DI water (3 L) and DCM (5 L). The reaction mixture is poured in and it is rinsed in with DCM (1 L). The organic layer is separated, washed with DI water (3×3 L), dried over Na2SO4, filtered, rinsed solids with DCM and concentrated the filtrate. EtOAc (2 L) is added to the residue and the solution is stirred for 1 hour. The product crystallizes out. The mixture is concentrated. Another portion of EtOAc (2 L) is added and concentrated to remove all of the DCM. EtOAc (650 mL) and MTBE (3 L) are added to the residue and the solution is stirred in an ice bath for 1 hour. The tan slurry is filtered using a polypropylene pad. The cake is rinsed with MTBE (2×500 mL). The light tan solid is dried overnight in the vacuum oven at 40° C. to give the crude product (553 g). The crude product is purified by silica gel column chromatography eluting with (50% EtOAc (50%):35% DCM (35%): n-heptane (15%)) to give the pure desired product tert-butyl 4-(5-(2-fluoro-4-(1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamido)phenoxy)-1-methyl-1H-indazol-6-yl)-1H-pyrazole-1-carboxylate (424 g, 68%). MS (m/z): 651.0 (M−H).
tert-Butyl 4-(5-(2-fluoro-4-(1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamido)phenoxy)-1-methyl-1H-indazol-6-yl)-1H-pyrazole-1-carboxylate (423.9 g, 649.50 mmol) is dissolved in DCM (4.24 L). HCl in MeOH (5.74 N, 799.99 mL, 4.59 mol) is added and the solution is heated at 30° C. for 1 hour. Then the reaction mixture is heated to 45° C. and DCM (1.5 L) is added. After two hours, the solution is heated to 50° C. and DCM (2 L) is added. After 3 hours, DCM (2 L) is added followed by HCl in MeOH (4.5 N, 721.67 mL, 3.25 mol). After another 45 min, DCM (1 L), HCl in MeOH (4.5 N, 288.67 mL, 1.30 mol), and MeOH (1.5 L) are added. The reaction solution is then heated to 60° C. After 4 hours, MeOH (2 L) is added and 10 min later DCM (1 L) is added followed by HCl in MeOH (4.5 N, 200 mL). After 5 hours, the reaction is complete. The reaction mixture is concentrated to about ⅓ volume. MeOH (2 L) is added and the solution is concentrated to a thick slurry. Again, MeOH (2 L) is added and the mixture is concentrated to a thick slurry. The slurry is cooled to about 10-15° C. and then filtered. The solids are washed with MeOH. The solids are placed in a 55° C. vacuum oven for 2 days to give the desired product N-(3-fluoro-4-(1-methyl-6-(1H-pyrazol-4-yl)-1H-indazol-5-yloxy)phenyl)-1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamide hydrochloride (377 g, 92.8%). MS (m/z): 551.0 (M−H).
To a 22 L round bottom flask equipped with mechanical stirring under nitrogen is added N-(3-fluoro-4-(1-methyl-6-(1H-pyrazol-4-yl)-1H-indazol-5-yloxy)phenyl)-1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamide hydrochloride (367 g, 0.62 mol) followed by DCM (7.34 L) and water (7.34 L). Na2CO3 (181.6 g, 1.71 mol) is added and the mixture is stirred at RT for 30 min. The pH is checked and found to be about 9.4. The mixture is filtered over polypropylene. The solids are collected and placed into a 5 L round bottom flask. A 20% water/MeOH solution (2.6 L) is added and the slurry is stirred for 30 min. The slurry is filtered and the solids are washed with 20% water/MeOH (600 mL). The solids are placed in a vacuum oven at 35° C. overnight. The first weighing indicates 394 g (theoretical yield 324.8 g, about 121% mass recovery). TGA (Thermogravimetric analysis)/DSC (differential scanning calorimetry) shows about 17 wt % free water and 10-11 wt% volatile loss at the melt. The solids are dried at 55° C. in a vacuum oven with a N2 sweep for 3.5 hours (354.7 g, about 109% mass recovery, NMR shows about 9.3 wt % DCM). No free water is present according to TGA/DSC. The material is sent for milling.
The jet mill (Aljet™ 0101) in a glove bag is assembled inside a walk in hood and hooked up to N2 to a 100 lb header. The inlet pusher nozzle is adjusted for maximum draw and max nitrogen flow is introduced into the mill. Pressure readings are noted as 90 psi on pusher nozzle and 85 psi on both grind nozzles. The starting material (353.4 g) is slowly fed to the mill inlet, stopping to empty the receiver sock as needed. The total milling time is 22 min and 25 second. The calculated feed rate is 15.8 g/min (353.4 grams divided by 22.42 min). The milled material (335.7 g, 95%) is obtained with 17.7 g loss. Particle size analysis result of the milled material is d90 of 4.6 microns.
TGA/DSC indicates about 11.4 wt % volatiles at the melt and NMR (DMSO) shows about 9.3 wt % DCM. 1H NMR (DMSO) δ 12.94 (br s, 1 H), 11.88 (s, 1H), 8.44 (d,J=7.47 Hz, 1 H), 8.12 (br s, 1 H), 8.00 (br s, 1 H), 7.96 (s, 1 H), 7.94 (d,J=2.2 Hz, 1 H), 7.91 (d,J=2.6 Hz, 1 H), 7.87 (s, 1H), 7.47-7.37 (m, 5 H), 6.82 (t,J=9.26 Hz, 8.82 Hz, 1 H), 6.65 (d,J=7.49 Hz, 1 H), 4.04 (s, 3 H), 2.03 (s, 3 H). LC/MS: (M+H) 553.1.
Anhydrous Crystal Form Preparation
To 10 mL of EtOH is added 120 mg of the above compound into a 20 mL vial. The sample is heated to 70° C. with stirring. Initially the solids start to dissolve and then a suspension forms followed by a white precipitate. The sample is cooled to RT while being stirred. A small sample of the slurry is taken by pipette and allowed to air dry. This material is highly crystalline and proves to be an ethanol solvate by TGA. To the remaining suspension, 10 mL of heptane is added and then heated to boiling. The measured temperature is monitored at 70.8° C. until the volume has been reduced to 10 mL. When the temperature starts to rise, the heat is removed and the slurry stirred at RT overnight. The solid is isolated by vacuum filtration and dried in a vacuum oven at 45° C. for 3 hours, resulting in 77% recovery. The crystalline form shows a weight loss of 0.17% from 25-238° C. by TGA. The form’s onset of melting is 247.8° C.
PAPER
N-(3-Fluoro-4-((1-methyl-6-(1H-pyrazol-4-yl)-1H-indazol-5-yl)oxy)phenyl)-1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamide (1, LY2801653)
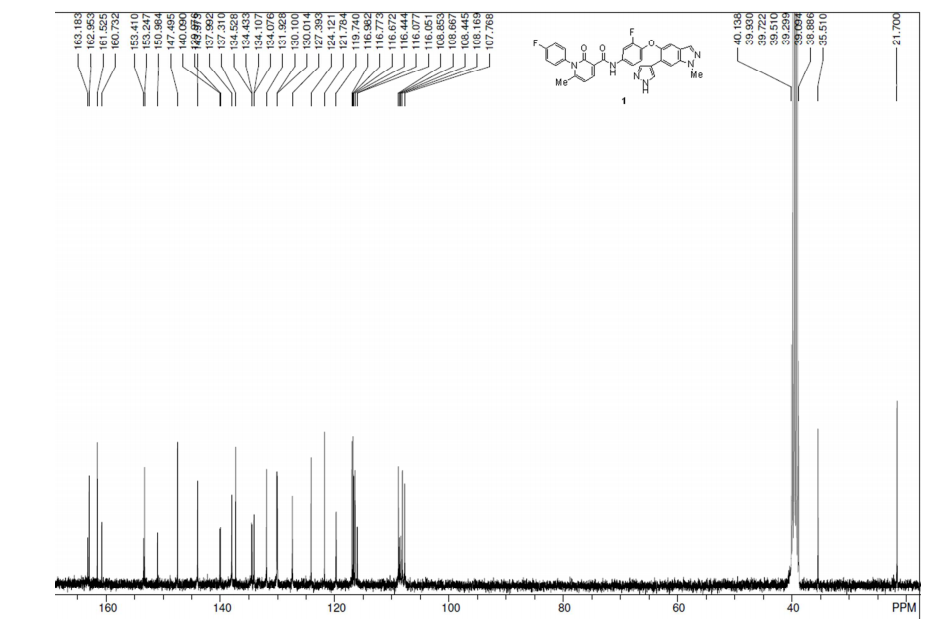

1H NMRof Merestinib
PAPER
 1 MERESTENIB
1 MERESTENIBAn NH4Cl-catalyzed ethoxy ethyl deprotection was developed for the synthesis of merestinib, a MET inhibitor. Alternative reactor technologies using temperatures above the solvent boiling point are combined with this mild catalyst to promote the deprotection reaction. The reaction is optimized for flow and has been used to synthesize over 100 kg of the target compound. The generality of the reaction conditions is also demonstrated with other compounds and protecting groups.
1: 1H NMR (500 MHz, DMSO-d6): δ = 13.00 (s, 1 H), 11.93 (s, 1 H), 8.45 (d, J = 7.5 Hz, 1 H), 8.17 (s, 1 H), 8.05 (s, 1 H), 8.01 (s, 1 H), 7.97 (d, J = 2.2 Hz, 1 H), 7.91 (d, J = 2.6 Hz, 1 H), 7.50–7.46 (m, 2 H), 7.43–7.40 (m, 2 H), 7.27 (s, 1 H), 7.26–7.25 (m, 1 H), 6.86 (t, J = 9.0 Hz, 1 H), 6.67–6.65 (m, 1 H), 4.08 (s, 3 H), 2.03 (s, 3 H) ppm; 13C NMR (125 MHz, DMSO-d6): δ = 163.0, 161.5, 161.0, 153.2, 152.2, 147.5, 144.0, 140.1, 138.0, 137.3, 134.5, 134.1, 132.0, 130.1, 127.4, 124.2, 121.8, 119.8, 117.0, 116.8, 116.6, 116.1, 108.9, 108.6, 108.2, 107.8, 35.5, 21.7 ppm; HR-MS [ESI]: Calcd for C30H23F2N6O3+ [M + H+]: 553.1794, found 553.1793.

……….
WO 2010011538
http://www.google.co.in/patents/WO2010011538A1?cl=en
Example 1 N-(3 -Fluoro-4-( 1 -methyl-6-( lH-pyrazol-4-yl)- lH-indazol-5 -yloxy)phenyl)- 1 -(4- fluorophenyl)-6-methyl-2-oxo- 1 ,2-dihydropyridine-3 -carboxamide

To a 100 mL round bottom flask is added tert-butyl 4-(5-(4-amino-2- fluorophenoxy)-l -methyl- IH- indazol-6-y I)- lH-pyrazole-1-carboxylate (1.43 g, 3.38 mmol), l-(4-fluorophenyl)-6-methyl-2-oxo-l,2-dihydropyridine-3-carboxylic acid (1.25 g, 5.07 mmol), EDCI (1.48 g , 7.6 mmol) and ΗOBt (776 mg, 5.07 mmol) followed by DMF (15 mL, 193.99 mmol) and then DIPEA (1.47 mL, 8.44 mmol). The mixture is allowed to stir at RT overnight. The reaction mixture is diluted into EtOAc (300 mL) and washed with saturated aqueous sodium chloride (5 x 100 mL). The combined aqueous solution is extracted with EtOAc (1 x 100 mL) and then the combined organic solutions are dried over Na2SO4, filtered, and concentrated to dryness. The solid is purified on a silica gel column eluting with DCM (A) and a 10% MeOH in a DCM solution (B), gradient from 100% (A) to 80% (A):20% (B) over 70 min to give tert-butyl 4-(5-(2- fluoro-4-(l-(4-fluoroplienyl)-6-metliy 1-2-oxo- 1,2-dihy dropyridine-3- carboxamido)phenoxy)- 1 -methyl- lH-indazol-6-yl)- lH-pyrazole- 1 -carboxylate as a gold solid (2.20 g, 87% yield). MS (m/z): 653. (M+Η), 675 (M+Na).
To a round bottom flask is added tert-butyl 4-(5-(2-fluoro-4-(l-(4-fluorophenyl)- 6-methyl-2-oxo- 1 ,2-dihydropyridine-3 -carboxamido)phenoxy)- 1 -methyl- lH-indazol-6- yl)-lΗ-pyrazole-l -carboxylate (1.92 g, 2.94 mmol) and DCM (50 mL) followed by triethylsilane (1.88 mL, 11.77 mmol) and TFA (17.8 mL, 235.35 mmol). The reaction mixture is allowed to stir at RT for 1.5 hours. The solvent is removed and diluted into DCM (150 mL) and washed with saturated aqueous NaHCθ3 solution (2 x 100 mL). The organic solution is dried with Na2SO4, and concentrated under reduced pressure to give a solid material. The solid is purified on a silica gel column eluting with DCM (A) and a 10% MeOH in DCM solution (B), gradient from 100% (A) to 75%(A):25%(B) over 70 min, held at this 75:25 ratio for 15 min to give the title compound as an off-white solid. The solid is dissolved in hot EtOH (50 mL) followed by a portion-wise addition of distilled water (250 mL) causing a white solid to precipitate. The solid is filtered over a Buchner funnel and washed with distilled water (3 x 15 mL), air dried, and vacuum dried at 60 0C for 15 hours to give the title compound as an off-white solid (1.27 g, 78% yield). MS (m/z): 552.8 (M+H).
Example 2
N-(3-Fluoro-4-(l-methyl-6-(lH-pyrazol-4-yl)-lH-indazol-5-yloxy)phenyl)-l-(4- fluorophenyl)-6-methyl-2-oxo-l,2-dihydropyridine-3-carboxamide
A 12 L round bottom flask is equipped with overhead agitation, a thermocouple, and a N2 purge. tert-Butyl 4-(5-(4-amino-2-fluorophenoxy)-l -methyl- lH-indazol-6-yl)- lH-pyrazole-1-carboxylate (404 g, 954.08 mmol) is dissolved in DMF (2 L) and charged to the flask. DMF (1 L) is used to rinse the flask. l-(4-Fluorophenyl)-6-methyl-2-oxo- l,2-dihydropyridine-3-carboxylic acid (259.46 g ,1.05 mol) and EDCI (228.63 g , 1.19 mol) are added and it is rinse in with DMF (500 mL). Then ΗOBt (189.94 g, 1.24 mol) is added and it is again rinsed in with DMF (500 mL). Finally, DIPEA is slowly added (184.97 g, 1.43 mol). The dark solution is then stirred at RT over the weekend. To a 20 L bottom outlet flask is added DI water (3 L) and DCM (5 L). The reaction mixture is poured in and it is rinsed in with DCM (1 L). The organic layer is separated, washed with DI water (3 X 3 L), dried over Na2SO4, filtered, rinsed solids with DCM and concentrated the filtrate. EtOAc (2 L) is added to the residue and the solution is stirred for 1 hour. The product crystallizes out. The mixture is concentrated. Another portion of EtOAc (2 L) is added and concentrated to remove all of the DCM. EtOAc (650 mL) and MTBE (3 L) are added to the residue and the solution is stirred in an ice bath for 1 hour. The tan slurry is filtered using a polypropylene pad. The cake is rinsed with MTBE (2 x 500 mL). The light tan solid is dried overnight in the vacuum oven at 40 0C to give the crude product (553 g). The crude product is purified by silica gel column chromatography eluting with (50% EtOAc (50%):35% DCM (35%): n-heptane (15%)) to give the pure desired product tert-butyl 4-(5-(2-fluoro-4-(l-(4-fluorophenyl)-6-methyl-2-oxo-l,2- dihydropyridine-3 -carboxamido)phenoxy)- 1 -methyl- lH-indazol-6-yl)- lH-pyrazole- 1 – carboxylate (424 g, 68%). MS (m/z): 651.0 (M-H). tert-Butyl 4-(5-(2-fluoro-4-(l-(4-fluorophenyl)-6-methyl-2-oxo-l,2- dihydropyridine-3 -carboxamido)phenoxy)- 1 -methyl- lH-indazol-6-yl)- lH-pyrazole- 1 – carboxylate (423.9 g, 649.50 mmol) is dissolved in DCM (4.24 L). HCl in MeOH (5.74 N, 799.99 mL, 4.59 mol) is added and the solution is heated at 30 0C for 1 hour. Then the reaction mixture is heated to 45 0C and DCM (1.5 L) is added. After two hours, the solution is heated to 50 0C and DCM (2 L) is added. After 3 hours, DCM (2 L) is added followed by HCl in MeOH (4.5 N, 721.67 mL, 3.25 mol). After another 45 min, DCM (1 L), HCl in MeOH (4.5 N, 288.67 mL, 1.30 mol), and MeOH (1.5 L) are added. The reaction solution is then heated to 60 0C. After 4 hours, MeOH (2 L) is added and 10 min later DCM (1 L) is added followed by HCl in MeOH (4.5 N, 200 mL). After 5 hours, the reaction is complete. The reaction mixture is concentrated to about 1/3 volume. MeOH (2 L) is added and the solution is concentrated to a thick slurry. Again, MeOH (2 L) is added and the mixture is concentrated to a thick slurry. The slurry is cooled to about 10- 15 0C and then filtered. The solids are washed with MeOH. The solids are placed in a 55 0C vacuum oven for 2 days to give the desired product N-(3-fluoro-4-(l-methyl-6-(lH- pyrazol-4-yl)- lH-indazol-5-yloxy)phenyl)- 1 -(4-fluorophenyl)-6-methyl-2-oxo- 1,2- dihydropyridine-3-carboxamide hydrochloride (377 g, 92.8%). MS (m/z): 551.0 (M-H).
To a 22 L round bottom flask equipped with mechanical stirring under nitrogen is added N-(3-fluoro-4-(l-methyl-6-(lH-pyrazol-4-yl)-lH-indazol-5-yloxy)phenyl)-l-(4- fluorophenyl)-6-methyl-2-oxo-l,2-dihydropyridine-3-carboxamide hydrochloride (367 g, 0.62 mol) followed by DCM (7.34 L) and water (7.34 L). Na2CO3 (181.6 g, 1.71 mol) is added and the mixture is stirred at RT for 30 min. The pΗ is checked and found to be about 9.4. The mixture is filtered over polypropylene. The solids are collected and placed into a 5 L round bottom flask. A 20% water/MeOΗ solution (2.6 L) is added and the slurry is stirred for 30 min. The slurry is filtered and the solids are washed with 20% water/MeOΗ (600 mL). The solids are placed in a vacuum oven at 35 0C overnight. The first weighing indicates 394 g (theoretical yield 324.8 g, about 121% mass recovery).
TGA (Thermogravimetric analysis)/DSC (differential scanning calorimetry) shows about 17 wt % free water and 10-11 wt% volatile loss at the melt. The solids are dried at 55 0C in a vacuum oven with a N2 sweep for 3.5 hours (354.7 g, about 109% mass recovery, NMR shows about 9.3 wt % DCM). No free water is present according to TGA/DSC. The material is sent for milling. The jet mill (AIj et™ 0101) in a glove bag is assembled inside a walk in hood and hooked up to N2 to a 100 Ib header. The inlet pusher nozzle is adjusted for maximum draw and max nitrogen flow is introduced into the mill. Pressure readings are noted as 90 psi on pusher nozzle and 85 psi on both grind nozzles. The starting material (353.4 g) is slowly fed to the mill inlet, stopping to empty the receiver sock as needed. The total milling time is 22 min and 25 second. The calculated feed rate is 15.8 g/min (353.4 grams divided by 22.42 min). The milled material (335.7 g, 95%) is obtained with 17.7 g loss. Particle size analysis result of the milled material is d90 of 4.6 microns.
TGA/DSC indicates about 11.4 wt % volatiles at the melt and NMR (DMSO) shows about 9.3 wt % DCM. 1H NMR (DMSO) δ 12.94 (br s, 1 H), 11.88 (s, IH), 8.44 (d, J= 7.47 Hz, 1 H), 8.12 (br s, 1 H), 8.00 (br s, 1 H), 7.96 (s, 1 H), 7.94 (d, J= 2.2 Hz, 1 H), 7.91 (d, J= 2.6 Hz, 1 H), 7.87 (s, IH), 7.47-7.37 (m, 5 H), 6.82 (t, J= 9.26 Hz, 8.82 Hz, 1 H), 6.65 (d, J= 7.49 Hz, 1 H), 4.04 (s, 3 H), 2.03 (s, 3 H). LC/MS: (M + H) 553.1.
Anhydrous Crystal Form Preparation To 10 mL of EtOH is added 120 mg of the above compound into a 20 mL vial.
The sample is heated to 70 0C with stirring. Initially the solids start to dissolve and then a suspension forms followed by a white precipitate. The sample is cooled to RT while being stirred. A small sample of the slurry is taken by pipette and allowed to air dry. This material is highly crystalline and proves to be an ethanol solvate by TGA. To the remaining suspension, 10 mL of heptane is added and then heated to boiling. The measured temperature is monitored at 70.8 0C until the volume has been reduced to 10 mL. When the temperature starts to rise, the heat is removed and the slurry stirred at RT overnight. The solid is isolated by vacuum filtration and dried in a vacuum oven at 45 0C for 3 hours, resulting in 77% recovery. The crystalline form shows a weight loss of 0.17% from 25-238 0C by TGA. The form’s onset of melting is 247.80C.
|
References |
1: Yan SB, Peek VL, Ajamie R, Buchanan SG, Graff JR, Heidler SA, Hui YH, Huss KL, Konicek BW, Manro JR, Shih C, Stewart JA, Stewart TR, Stout SL, Uhlik MT, Um SL, Wang Y, Wu W, Yan L, Yang WJ, Zhong B, Walgren RA. LY2801653 is an orally bioavailable multi-kinase inhibitor with potent activity against MET, MST1R, and other oncoproteins, and displays anti-tumor activities in mouse xenograft models. Invest New Drugs. 2012 Dec 29. [Epub ahead of print] PubMed PMID: 23275061.
-
Liu, X.; Newton, R. C.; Scherle, P. A. Expert Opin. Invest. Drugs 2011, 20, 1225,DOI: 10.1517/13543784.2011.600687
-
Yan, S. B.; Peek, V. L.; Ajamie, R.; Buchanan, S. G.; Graff, J. R.; Heidler, S. A.; Hui, Y.;Huss, K. L.; Konicek, B. W.; Manro, J. R.; Shih, C.; Stewart, J. A.; Stewart, T. R.; Stout, S. L.; Uhlik, M. T.; Um, S. L.; Wang, Y.; Wu, W.; Yan, L.; Yang, W. J.; Zhong, B.; Walgren, R. A. Invest. New Drugs 2013, 31, 833, DOI: 10.1007/s10637-012-9912-9
-
Kallman, N. J.; Liu, C.; Yates, M. H.; Linder, R. J.; Ruble, J. C.; Kogut, E. F.; Patterson, L. E.; Laird, D. L. T.; Hansen, M. M. Org. Process Res. Dev. 2014, 18, 501,DOI: 10.1021/op400317zKallman, N.J.; Yates, M.H.; Linder, R.J.; Hansen, M.M.
Route design and development of c-Met inhibitor LY2801653
244th Am Chem Soc (ACS) Natl Meet (August 19-23, Philadelphia) 2012, Abst ORGN 212
////////
UPDATE…..

A new synthesis of a key indazole-containing building block for the MET kinase inhibitor merestinib was designed and demonstrated. Crucial to the successful construction of the challenging indazole is an SNAr reaction, which forges the heterocyclic ring. Continuous processing was applied to two of the five steps: nitration of a benzaldehyde and high-temperature hydrolysis of an aniline to phenol. Compared to a highly developed historical route, the new route shows clear benefits in terms of product quality and potentially manufacturability and robustness.
An Alternative Indazole Synthesis for Merestinib
 , Kevin P. Cole† , Jared W. Fennell†, Todd D. Maloney†, David Mitchell†§ , Ramesh Subbiah‡, and Balakumar Ramadas‡
, Kevin P. Cole† , Jared W. Fennell†, Todd D. Maloney†, David Mitchell†§ , Ramesh Subbiah‡, and Balakumar Ramadas‡6-Bromo-5-(2-fluoro-4-nitrophenoxy)-1-methyl-1H-indazole (2)
SILDENAFIL

SILDENAFIL
The chemical name of sildenafil is 5-[2-ethoxy-5-(4-methylpiperazin-1-ylsulfonyl)phenyl]-1- methyl-3-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one and its formula is C22H30N6O4S. The melting point of sildenafil is 189-190oC. Its solubility is 3.5 mg/mL in water.
The 1H NMR data of sildenafil is given below. The abbreviations used are s for singlet, d for doublet, t for triplet and q for quartet. The chemical shifts are given in ppm (parts per million) and are followed by the number of Hydrogens the peaks account for:
| peak (ppm) | integration | multiplicity |
| 0.94 | 3H | t |
| 1.32 | 3H | t |
| 2.15 | 3H | s |
| 2.35 | 4H | broad s |
| 2.76 | 2H | t |
| 2.88 | 4H | broad s |
| 4.14 | 3H | s |
| 4.18 | 2H | q |
| 7.36 | 1H | d |
| 7.80 | 2H | multiplet |
| 12.16 | 1H | broad s |
Sildenafil, sold as Viagra and other trade names, is a medication used to treat erectile dysfunction and pulmonary arterial hypertension.[1] Its effectiveness for treating sexual dysfunction in women has not been demonstrated.[1]
Common side effects include headaches and heart burn, as well as flushed skin. Caution is advised in those who have cardiovascular disease. Rare but serious side effects include prolonged erections, which can lead to damage to the penis, and sudden-onset hearing loss. Sildenafil should not be taken by people who take nitrates such as nitroglycerin, as this may result in a severe and potentially fatal drop in blood pressure.[1]
It acts by inhibiting cGMP-specific phosphodiesterase type 5 (PDE5), an enzyme that promotes degradation of cGMP, which regulates blood flow in the penis.
It was originally discovered by Pfizer scientists Andrew Bell, David Brown, and Nicholas Terrett.[2][3] Since becoming available in 1998, sildenafil has been a common treatment for erectile dysfunction; its primary competitors are tadalafil (Cialis) and vardenafil (Levitra).

EP0463756A,US6469012,WO2008074512A1
Chemical synthesis
Dunn PJ (2005). “Synthesis of Commercial Phosphodiesterase(V) Inhibitors”. Org Process Res Dev 2005 (1): 88–97. doi:10.1021/op040019c.
The preparation steps for synthesis of sildenafil are:[40]
- Methylation of 3-propylpyrazole-5-carboxylic acid ethyl ester with hot dimethyl sulfate
- Hydrolysis with aqueous NaOH to free acid
- Nitration with oleum/fuming nitric acid
- Carboxamide formation with refluxing thionyl chloride/NH4OH
- Reduction of nitro group to amino
- Acylation with 2-ethoxybenzoyl chloride
- Cyclization
- Sulfonation to the chlorosulfonyl derivative
- Condensation with 1-methylpiperazine.

|
The synthesis of sildenafil citrate was first reported in the Bioorganic & Medicinal Chemistry Letters, Vol 6, pp. 1819, 1824, 1996. The reaction scheme is reproduced below. Sildenafil was reported in this journal as “a potent and selective inhibitor of type 5 PDE with utility for the treatment of male erectile dysfunction”. |

| he first step of the synthesis is the reaction of a diketoester (1) and hydrazine to give the pyrazole ring. The regioselective N-methylation of the pyrazole and hydrolysis gives a carboxylic acid (3). Compound (3) is then reacted with HNO3 and H2SO4 to give a nitrated product. This is then followed by a carboxamide formation and the reduction of the nitro group. The compound (4) is then acylated under basic conditions and this produces the pyrazolopyrimidinone (6). (6) is then chlorosulphonylated selectively on the 5′-position of the phenyl ring. This can then couple with an amine to give sildenafil (7). The yield of each step is given on the reaction scheme. |
|
This is the original synthesis which was reported in the literature when the molecule was first synthesised. A variant of the synthesis was published but the changes it involved only consisted in the change of a few reactants, and no major changes were reported. This synthesis appeared in the January 1999 issue of Chemistry in Britain. This journal only reported the original discovery synthesis and said that the synthesis used commercially had not been published. The drug is commercially manufactured by an alternative route. The reaction scheme is described in the patent which was published on 17 decembre 1997. However, the synthesis used in the commercial manufacture could be different to this. The patent was filed by the Pfizer Research and Development Company. The scheme is reproduced below.
The synthesis was described in a lot of detail, including the solvents that were the best to use, however, these details have not been reproduced here. These and further details about the synthesis can be found on the original patent document. The reaction pathway is explained in more detail below. The reagents employed in the reactions can vary, but the following are among the ones recommended by the submitters of the patent: |


|

SCHEME2

Chemical synthesis
…………………………………………………………………………………
SYNTHESIS

……………………………………………

…………………………………………………………………
SYNTHESIS

…………………………………………………………………………..

……………………………
PRECURSORS
…………………………………..
SYNTHESIS

Patents
European Union
Pfizer’s patent on sildenafil citrate expired in some member countries of the EU, Austria, Denmark, France, Germany, Ireland, Italy, The Netherlands, Spain, Sweden, the United Kingdom and Switzerland on 21 June 2013.[53][54][55] A UK patent held by Pfizer on the use of PDE5 inhibitors (see below) as treatment of impotence was invalidated in 2000 because of obviousness; this decision was upheld on appeal in 2002.[56][57]
United States
In 1992, Pfizer filed a patent covering the substance sildenafil and its use to treat cardiovascular diseases.[58] This patent was published in 1993 and expired in 2012. In 1994, Pfizer filed a patent covering the use of sildenafil to treat erectile dysfunction.[59] This patent was published in 2002 and will expire in 2019. Teva sued to have the latter patent invalidated, but Pfizer prevailed in an August 2011 federal district court case.[60]
The patent on Revatio (indicated for pulmonary arterial hypertension rather than erectile dysfunction) expired in late 2012. Generic versions of this low-dose form of sildenafil have been available in the U.S. from a number of manufacturers, including Greenstone, Mylan, and Watson, since early 2013.[61] No legal barrier exists to doctors prescribing this form of sildenafil “off label” for erectile dysfunction, although the dosage typically required for treating ED requires patients to take multiple pills.
Canada
In Canada, Pfizer’s patent 2,324,324 for Revatio (sildenafil used to treat pulmonary hypertension) was found invalid by the Federal Court in June 2010, on an application by Ratiopharm Inc.[62][63]
On November 8, 2012, the Supreme Court of Canada ruled that Pfizer’s patent 2,163,446 on Viagra was invalid from the beginning because the company did not provide full disclosure in its application. The decision, Teva Canada Ltd. v. Pfizer Canada Inc., pointed to section 27(3)(b) of The Patent Act which requires that disclosure must include sufficient information “to enable any person skilled in the art or science to which it pertains” to produce it. It added further: “As a matter of policy and sound statutory interpretation, patentees cannot be allowed to ‘game’ the system in this way. This, in my view, is the key issue in this appeal.”[64]
Teva Canada launched Novo-Sildenafil, a generic version of Viagra, on the day the Supreme Court of Canada released its decision.[65][66][67] To remain competitive, Pfizer then reduced the price of Viagra in Canada.[68] However, on November 9, 2012, Pfizer filed a motion for a re-hearing of the appeal in the Supreme Court of Canada,[69] on the grounds that the court accidentally exceeded its jurisdiction by voiding the patent.[70] Finally, on April 22, 2013, the Supreme Court of Canada invalidated Pfizer’s patent altogether.[71]
India
Manufacture and sale of sildenafil citrate drugs known as “generic viagra” is common in India, where Pfizer’s patent claim does not apply. Trade names include Kamagra (Ajanta Pharma), Silagra (Cipla), Edegra (Sun Pharmaceutical), Penegra (Zydus Cadila), and Zenegra (Alkem Laboratories).
China
Manufacture and sale of sildenafil citrate drugs is common in China, where Pfizer’s patent claim is not widely enforced.
Other countries
Egypt approved Viagra for sale in 2002, but soon afterwards allowed local companies to produce generic versions of the drug, citing the interests of poor people who would not be able to afford Pfizer’s price.[72]
Pfizer’s patent on sildenafil citrate expired in Brazil in 2010.[73]
References
- “Sildenafil Citrate”. The American Society of Health-System Pharmacists. Retrieved Dec 1, 2014.
- “Patent US5250534 – Pyrazolopyrimidinone antianginal agents – Google Patents”.
- Boolell M, Allen MJ, Ballard SA, Gepi-Attee S, Muirhead GJ, Naylor AM, Osterloh IH, Gingell C (June 1996). “Sildenafil: an orally active type 5 cyclic GMP-specific phosphodiesterase inhibitor for the treatment of penile erectile dysfunction”. Int. J. Impot. Res. 8 (2): 47–52. PMID 8858389.
- Vardi M, Nini A (2007). Vardi, Moshe, ed. “Phosphodiesterase inhibitors for erectile dysfunction in patients with diabetes mellitus”. Cochrane Database Syst Rev (1): CD002187. doi:10.1002/14651858.CD002187.pub3. PMID 17253475.
- “FDA Approves Pfizer’s Revatio as Treatment for Pulmonary Arterial Hypertension” (Press release). Pfizer. June 6, 2005. Archived from the original on August 28, 2005.
- Nurnberg HG, Hensley PL, Gelenberg AJ, Fava M, Lauriello J, Paine S (January 2003). “Treatment of antidepressant-associated sexual dysfunction with sildenafil: a randomized controlled trial”. JAMA 289 (1): 56–64. doi:10.1001/jama.289.1.56. PMID 12503977.
- Nurnberg HG, Hensley PL, Lauriello J, Parker LM, Keith SJ (August 1999). “Sildenafil for women patients with antidepressant-induced sexual dysfunction”. Psychiatr Serv 50 (8): 1076–8. PMID 10445658.
- Nurnberg HG, Hensley PL, Heiman JR, Croft HA, Debattista C, Paine S (2008). “Sildenafil Treatment of Women With Antidepressant-Associated Sexual Dysfunction”. JAMA 300 (4): 395–404. doi:10.1001/jama.300.4.395. PMID 18647982.
- Richalet JP, Gratadour P, Robach P, et al. (2005). “Sildenafil inhibits altitude-induced hypoxemia and pulmonary hypertension”. Am. J. Respir. Crit. Care Med. 171 (3): 275–81. doi:10.1164/rccm.200406-804OC. PMID 15516532.
- Perimenis P (2005). “Sildenafil for the treatment of altitude-induced hypoxaemia”. Expert Opin Pharmacother 6 (5): 835–7. doi:10.1517/14656566.6.5.835. PMID 15934909.
- Fagenholz PJ, Gutman JA, Murray AF, Harris NS (2007). “Treatment of high altitude pulmonary edema at 4240 m in Nepal”. High Alt. Med. Biol. 8 (2): 139–46. doi:10.1089/ham.2007.3055. PMID 17584008.
- “Viagra Prescribing Information”. Pfizer. October 2007. Archived from the original (PDF) on 14 November 2012. Retrieved 14 November 2012.
- “Viagra and vision”. VisionWeb. 29 October 2001. Retrieved 2009-02-10.
- “FDA Updates Labeling for Viagra, Cialis and Levitra for Rare Post-Marketing Reports of Eye Problems”. United States Food and Drug Administration. 8 July 2005. Archived from the original on February 23, 2008. Retrieved 2009-02-10.
- Laties AM (2009). “Vision disorders and phosphodiesterase type 5 inhibitors: a review of the evidence to date”. Drug Saf 32 (1): 1–18. doi:10.2165/00002018-200932010-00001. PMID 19132801.
- “FDA Announces Revisions to Labels for Cialis, Levitra and Viagra”. United States Food and Drug Administration. 18 October 2007. Archived from the original on 11 July 2007. Retrieved 10 February 2009.
- “Viagra (sildenafil citrate) tablets” (PDF). page 29: Pzifer. October 2007. Retrieved 2009-10-25.
- Kloner RA (2005). “Pharmacology and drug interaction effects of the phosphodiesterase 5 inhibitors: focus on alpha-blocker interactions”. Am J Cardiol 96 (12B): 42M–46M. doi:10.1016/j.amjcard.2005.07.011. PMID 16387566.
- Cheitlin MD, Hutter AM Jr, Brindis RG, Ganz P, Kaul S, Russell RO Jr, Zusman RM (1999). “ACC/AHA expert consensus document. Use of sildenafil (Viagra) in patients with cardiovascular disease. American College of Cardiology/American Heart Association”. Journal of the American College of Cardiology 33 (1): 273–82. doi:10.1016/S0735-1097(98)00656-1. PMID 9935041.
- Peterson K (2001-03-21). “Young men add Viagra to their drug arsenal”. USAToday.
- Smith KM, Romanelli F (2005). “Recreational use and misuse of phosphodiesterase 5 inhibitors”. J Am Pharm Assoc (2003) 45 (1): 63–72; quiz 73–5. doi:10.1331/1544345052843165. PMID 15730119.
- Sildenafil Will Not Affect Libido – Fact!
- Mondaini N, Ponchietti R, Muir GH, Montorsi F, Di Loro F, Lombardi G, Rizzo M (June 2003). “Sildenafil does not improve sexual function in men without erectile dysfunction but does reduce the postorgasmic refractory time”. Int. J. Impot. Res. 15 (3): 225–8. doi:10.1038/sj.ijir.3901005. PMID 12904810.
- McCambridge J, Mitcheson L, Hunt N, Winstock A (March 2006). “The rise of Viagra among British illicit drug users: 5-year survey data”. Drug Alcohol Rev 25 (2): 111–3. doi:10.1080/09595230500537167. PMID 16627299.
- Eloi-Stiven ML, Channaveeraiah N, Christos PJ, Finkel M, Reddy R (November 2007). “Does marijuana use play a role in the recreational use of sildenafil?”. J Fam Pract 56 (11): E1–4. PMID 17976333.
- “The 2007 Ig Nobel Prize Winners”. Improbable Research. 4 October 2007. Retrieved 2009-02-10.
- Agostino PV, Plano SA, Golombek DA (June 2007). “Sildenafil accelerates reentrainment of circadian rhythms after advancing light schedules”. Proc. Natl. Acad. Sci. U.S.A. 104 (23): 9834–9. doi:10.1073/pnas.0703388104. PMC 1887561. PMID 17519328.
- Teri Thompson, Christian Red, Michael O’Keefffe, and Nathaniel Vinton (10 June 2008). “Source: Roger Clemens, host of athletes pop Viagra to help onfield performance”. Daily News (Daily News). Retrieved 2009-02-10.
- Busbee J (2012-11-28). “Bears’ Brandon Marshall says some NFL players use Viagra … ON THE FIELD”. Yahoo! Sports. Retrieved 2012-11-28.
- Venhuis BJ, de Kaste D. (2006–2012). “Towards a decade of detecting new analogues of sildenafil, tadalafil and vardenafil in food supplements: a history, analytical aspects and health risks”. Journal of Pharmaceutical and Biomedical Analysis 69: 196–208. doi:10.1016/j.jpba.2012.02.014. PMID 22464558.
- Oh SS, Zou P, Low MY, Koh HL. Detection of sildenafil analogues in herbal products for erectile dysfunction (2006). “Detection of sildenafil analogues in herbal products for erectile dysfunction”. Journal of Toxicology and Environmental Health Part A 69 (21): 1951–1958. doi:10.1080/15287390600751355. PMID 16982533.
- Venhuis BJ, Blok-Tip L, de Kaste D (2008). “Designer drugs in herbal aphrodisiacs”. Forensic Science International 177 (2–3): 25–27. doi:10.1016/j.forsciint.2007.11.007. PMID 18178354.
- FDA letter to Libidus distributor
- FDA Warns Consumers About Dangerous Ingredients in “Dietary Supplements” Promoted for Sexual Enhancement
- Hidden Risks of Erectile Dysfunction “Treatments” Sold Online
- R. Baselt, Disposition of Toxic Drugs and Chemicals in Man, 9th edition, Biomedical Publications, Seal Beach, CA, 2011, pp. 1552–1553. http://www.biomedicalpublications.com/dt9.pdf
- Sung, B. J.; Hwang, K.; Jeon, Y.; Lee, J. I.; Heo, Y. S.; Kim, J.; Moon, J.; Yoon, J.; Hyun, Y. L.; Kim, E.; Eum, S.; Park, S. Y.; Lee, J. O.; Lee, T.; Ro, S.; Cho, J. (2003). “Structure of the catalytic domain of human phosphodiesterase 5 with bound drug molecules”. Nature 425 (6953): 98–102. doi:10.1038/nature01914. PMID 12955149.
- Webb, D.J.; Freestone, S.; Allen, M.J.; Muirhead, G.J. (March 4, 1999). “Sildenafil citrate and blood-pressure-lowering drugs: results of drug interaction studies with an organic nitrate and a calcium antagonist”. Am. J. Cardiol. 83 (5A): 21C–28C. doi:10.1016/S0002-9149(99)00044-2. PMID 10078539.
- “Viagra Clinical Pharmacology”. RxList.com. 2008. Retrieved 2008-08-20.
- Dunn PJ (2005). “Synthesis of Commercial Phosphodiesterase(V) Inhibitors”. Org Process Res Dev 2005 (1): 88–97. doi:10.1021/op040019c.
- “Research”. ABM. Abertawe Bro Morgannwg University Health Board. 4 July 2008. Archived from the original on 26 September 2008. Retrieved 6 August 2008.
Our clinicians regularly offer patients the opportunity to take part in trials of new drugs and treatments. Morriston Hospital in Swansea, was the first in the world to trial Viagra!
- Terrett NK, Bell AS, Brown D, Elllis P (1996). “Sildenafil (Viagra), a potent and selective inhibitor of Type 5 cGMP phosphodiesterase with utility for the treatment of male erectile dysfunction”. Bioorg Med Chem Lett 6 (15): 1819–1824. doi:10.1016/0960-894X(96)00323-X.
- Kling J (1998). “From hypertension to angina to Viagra”. Mod. Drug Discov. 1: 31–38. ISSN 1532-4486. OCLC 41105083.
- Viagra sales peak at $1,934m in 2008
- Ciment, J (1999). “Missouri fines internet pharmacy”. BMJ (British Medical Journal) 319 (7221): 1324. doi:10.1136/bmj.319.7221.1324g. PMC 1174637. PMID 10567131.
- Devine, Amy (September 29, 2008). “Chemists plan to sell Viagra on the internet”. Daily Record. Retrieved 2012-04-30.
- Keith A (2000). “The economics of Viagra”. Health Aff (Millwood) 19 (2): 147–57. doi:10.1377/hlthaff.19.2.147. PMID 10718028.
- McGuire S (2007-01-01). “Cialis gaining market share worldwide”. Medical Marketing & Media. Haymarket Media. Archived from the original on 2009-01-03. Retrieved 2009-02-10.
- Mullin, Rick (June 20, 2005). “Viagra”. Chemical & Engineering News 83 (25). Retrieved 2008-08-20.
- Berenson, Alex (December 4, 2005). “Sales of Impotence Drugs Fall, Defying Expectations”. The New York Times. Retrieved 2008-08-20.
- “Over-the-counter Viagra piloted”. BBC News. BBC News. 11 February 2007. Retrieved 2009-02-10.
- “Pfizer to sell Viagra online, in first for Big Pharma: AP”. CBS News. Retrieved 6 May 2013.
- “Actavis Launches Generic Viagra in Europe as Patents Expire”. Retrieved 2013-10-25.
- Jim Edwards (October 21, 2009). “What Will Happen When Viagra Goes Generic?”. AccessRx.com. Retrieved 2011-03-25.
- “Is Viagra about to lose its pulling power in the UK?”. The Guardian. Retrieved 13 June 2013.
- Murray-, Rosie (23 January 2002). “Viagra ruling upsets Pfizer”. London: Telegraph Media Group Limited. Archived from the original on 22 August 2009. Retrieved 10 February 2009.
- “Pfizer Loses UK Battle on Viagra Patent”. UroToday. Thomson Reuters. 17 June 2002. Archived from the original on 25 June 2007. Retrieved 10 February 2009.
- U.S. Patent 5,250,534
- U.S. Patent 6,469,012
- “Pfizer Wins Viagra Patent Infringement Case Against Teva Pharmaceuticals”. Bloomberg. August 15, 2011. Retrieved 2012-04-01.
- “Pfizer’s Revatio Goes Generic”. Zacks Equity Research. November 15, 2012. Retrieved 2013-10-05.
- “Revation patent ruled invalid for lack of sound prediction and obviousness”. Canadian Technology & IP Law. Stikeman Elliott. 2010-06-18. Retrieved 2012-11-14.
- “Pfizer Canada Inc. v. Ratiopharm Inc., 2010 FC 612″. CanLII.
- Teva Canada Ltd. v. Pfizer Canada Inc. 2012 SCC 60 at par. 80 (8 November 2012)
- John Spears (2012-11-08). “Supreme Court ruling could lead to cheaper versions of Viagra”. The [Toronto] Star. Retrieved 2012-11-14.
- Ken Hanly (2012-11-08). “Canadian Supreme court rules Viagra patent invalid”. Digital Journal. Retrieved 2012-11-14.
- “Viagra patent tossed out by Supreme Court: Decision allows generic versions of drug to be produced”. CBC News. 2012-11-08. Retrieved 2012-11-14.
- “Pfizer Canada drops Viagra price after generic versions get Supreme Court green light”. Financial Post. 2012-11-22. Retrieved 2013-02-09.
- “SCC Case Information, Docket No. 33951”. Retrieved 2012-11-14.
- Kirk Makin (2012-11-15). “In rare move, Pfizer asks Supreme Court to reconsider ruling that killed Viagra patent”. The Globe and Mail. Retrieved 2012-11-15.
- Gowling Lafleur Henderson LLP, Hélène D’Iorio (2013-04-22). “The Supreme Court of Canada holds Pfizer’s Viagra patent invalid”. Lexology. Retrieved 2013-12-27.
- Allam, Abeer (October 4, 2002). “Seeking Investment, Egypt Tries Patent Laws”. New York Times. Retrieved April 1, 2013.
External link
Official Viagra Website
- Official Viagra UK Website
- Official Revatio Website
- prescribing information for Viagra and prescribing information for Revatio from Pfizer
- FDA Information
- MedlinePLUS information, including side effects
- U.S. National Library of Medicine: Drug Information Portal – Sildenafil
- Viagra at The Periodic Table of Videos (University of Nottingham)
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
1-[4-ethoxy-3-(6,7-dihydro-1-methyl-
7-oxo-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl) phenylsulfonyl]-4-methylpiperazine |
|
| Clinical data | |
| Trade names | Viagra, Revatio, others |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a699015 |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral, IV |
| Pharmacokinetic data | |
| Bioavailability | 40% |
| Metabolism | Hepatic (mostly CYP3A4, also CYP2C9) |
| Biological half-life | 3 to 4 hours |
| Excretion | Fecal (80%) and renal (around 13%) |
| Identifiers | |
| CAS Registry Number | 139755-83-2 |
| ATC code | G04BE03 |
| PubChem | CID: 5281023 |
| DrugBank | DB00203 |
| ChemSpider | 56586 |
| UNII | 3M7OB98Y7H |
| KEGG | D08514 |
| ChEBI | CHEBI:58987 |
| ChEMBL | CHEMBL1737 |
| PDB ligand ID | VIA (PDBe, RCSB PDB) |
| Chemical data | |
| Formula | C22H30N6O4S |
| Molecular mass | base: 474.6 g/mol |
///////
Defibrotide


Defibrotide sodium is an oligonucleotide mixture with profibrinolytic properties. The chemical name of defibrotide sodium is polydeoxyribonucleotide, sodium salt. Defibrotide sodium is a polydisperse mixture of predominantly single-stranded (ss) polydeoxyribonucleotide sodium salts derived from porcine intestinal tissue having a mean weighted molecular weight of 13-20 kDa, and a potency of 27-39 and 28-38 biological units per mg as determined by two separate assays measuring the release of a product formed by contact between defibrotide sodium, plasmin and a plasmin substrate. The primary structure of defibrotide sodium is shown below.
DEFITELIO (defibrotide sodium) injection is a clear, light yellow to brown, sterile, preservative-free solution in a single-patient-use vial for intravenous use. Each milliliter of the injection contains 80 mg of defibrotide sodium and 10 mg of Sodium Citrate, USP, in Water for Injection, USP. Hydrochloric Acid, NF, and/or Sodium Hydroxide, NF, may have been used to adjust pH to 6.8-7.8.
Defibrotide is the sodium salt of a mixture of single-stranded oligodeoxyribonucleotides derived from porcine mucosal DNA. It has been shown to have antithrombotic, anti-inflammatory and anti-ischemic properties (but without associated significant systemic anticoagulant effects). It is marketed under the brand names Dasovas (FM), Noravid, and Prociclide in a variety of countries, but is currently not approved in the USA. The manufacturer is Gentium.
Defibrotide is used to treat or prevent a failure of normal blood flow (occlusive venous disease, OVD) in the liver of patients who have had bone marrow transplants or received certain drugs such as oral estrogens, mercaptopurine, and many others.
In 2012, an IND was filed in Japan seeking approval of the compound for the treatment of veno-occlusive disease.
Approved 3/30/3016 US FDA, defibrotide sodium, (NDA) 208114

To treat adults and children who develop hepatic veno-occlusive disease with additional kidney or lung abnormalities after they receive a stem cell transplant from blood or bone marrow called hematopoietic stem cell transplantation
Polydeoxyribonucleotides from bovine lung or other mamalian organs with molecular weight between 15,000 and 30,000 Da
CAS 83712-60-1
Defibrotide is a polydisperse mixture of oligonucleotides produced by random, chemical cleavage (depolymerisation) of porcine DNA. It is predominantly single stranded, of varying base sequence, lengths and conformations; unfolded, folded or combined. The mean oligonucleotide length is 50 bases with a mean molecular weight of 17 ± 4 kDa. No individually defined component is at more than femtomolar concentration. The only meaningful scientific information that can be obtained about the biochemical nature of defibrotide (aside from determination of percentage of each nucleobase) is a measurement of its average length and its average percentage double stranded character. Therefore, it can be established that this active substance is of highly heterogenic nature.

Defibrotide (Defitelio, Gentium)[1] is a deoxyribonucleic acid derivative (single-stranded) derived from cow lung or porcine mucosa. It is an anticoagulant with a multiple mode of action (see below).
It has been used with antithrombin III.[2]
Jazz Pharmaceuticals plc announced that the FDA has accepted for filing with Priority Review its recently submitted New Drug Application (NDA) for defibrotide. AS ON OCT 2015
Defibrotide is an investigational agent proposed for the treatment of patients with hepatic veno-occlusive disease (VOD), also known as sinusoidal obstruction syndrome (SOS), with evidence of multi-organ dysfunction (MOD) following hematopoietic stem-cell transplantation (HSCT).
Priority Review status is designated for drugs that may offer major advances in treatment or provide a treatment where no adequate therapy exists. Based on timelines established by the Prescription Drug User Fee Act (PDUFA), FDA review of the NDA is expected to be completed by March 31, 2016.
“The FDA’s acceptance for filing and Priority Review status of the NDA for defibrotide is an important milestone for Jazz and reflects our commitment to bringing meaningful medicines to patients who have significant unmet needs,” said Karen Smith, M.D., Ph.D., Global Head of Research and Development and Chief Medical Officer of Jazz Pharmaceuticals. “We look forward to continuing to work closely with the FDA to obtain approval for defibrotide for patients with hepatic VOD with evidence of MOD in the U.S. as quickly as possible, as there are no other approved therapies for treating this rare, often fatal complication of HSCT.”
The NDA includes safety and efficacy data from three clinical studies of defibrotide for the treatment of hepatic VOD with MOD following HSCT, as well as a retrospective review of registry data from the Center for International Blood and Marrow Transplant Research. The safety database includes over 900 patients exposed to defibrotide in the clinical development program for the treatment of hepatic VOD.
The compound was originally developed under a collaboration between Sanofi and Gentium. In December 2001, Gentium entered into a license and supply agreement with Sigma-Tau Pharmaceuticals, pursuant to which the latter gained exclusive rights to distribute, market and sell the product for the treatment of VOD in the U.S. This agreement was expanded in 2005 to include all of North America, Central America and South America.
Defibrotide was granted orphan drug designations from the FDA in July 1985, May 2003 and January 2007 for the treatment of thrombotic thrombocytopenic purpura (TTP), for the treatment of VOD and for the prevention of VOD, respectively. Orphan drug was also received in the E.U. for the prevention and treatment of hepatic veno-occlusive disease (VOD) in 2004 and for the prevention of graft versus host disease (GvHD) in 2013.
Pharmacokinetics
Defibrotide is available as an oral, intravenous, and intramuscular formulation. Its oral bioavailability is in the range of 58-70% of theparenteral forms. T1/2 alpha is in the range of minutes while T1/2 beta is in the range of hours in studies with oral radiolabelleddefibrotide. These data suggest that defibrotide, in spite of its macromolecular nature, is absorbed well after oral administration. Due to the drug’s short half-life, it is necessary to give the daily dose divided in 2 to 4 doses (see below).
In 2014, Jazz Pharmaceuticals (parent of Gentium) acquired the rights of the product in U.S. and in the Americas
Mode of action
The drug appears to prevent the formation of blood clots and to help dissolve blood clots by increasing levels of prostaglandin I2, E2, and prostacyclin, altering platelet activity, increasing tissue plasminogen activator (tPA-)function, and decreasing activity of tissue plasminogen activator inhibitor. Prostaglandin I2 relaxes the smooth muscle of blood vessels and prevents platelets from adhering to each other. Prostaglandin E2 at certain concentrations also inhibits platelet aggregation. Moreover, the drug provides additional beneficial anti-inflammatory and antiischemic activities as recent studies have shown. It is yet unclear, if the latter effects can be utilized clinically (e.g., treatment of ischemic stroke).
Unlike heparin and warfarin, defibrotide appears to have a relatively mild anticoagulant activity, which may be beneficial in the treatment of patients at high risk of bleeding complications. Nevertheless, patients with known bleeding disorders (e.g., hemophilia A) or recent abnormal bleedings should be treated cautiously and under close medical supervision.
The drug was marketed under the brand names Dasovas (FM), Noravid, and Prociclide in a variety of countries. It is currently not approved in the USA. The manufacturer is Gentium.
Defibrotide also received fast track designation from the FDA for the treatment of severe VOD in recipients of stem cell transplants. In 2011, the compound was licensed to Medison Pharma by Gentium in Israel and Palestine. The license covers the management of named-patient sales program and local registration, authorization, marketing, reimbursement and medical affairs for the treatment of peripheral vascular disease.
Usual indications
Defibrotide is used to treat or prevent a failure of normal blood flow (Veno-occlusive disease, VOD) in the liver of patients having had bone marrow transplants or received certain drugs such as oral estrogens, mercaptopurine, and many others. Without intensive treatment, VOD is often a fatal condition, leading to multiorgan failure. It has repeatedly been reported that defibrotide was able to resolve the condition completely and was well tolerated.
Other indications are: peripheral obliterative arterial disease, thrombophlebitis, and Raynaud’s phenomenon. In very high doses, defibrotide is useful as treatment of acute myocardial infarction. The drug may also be used for the pre- and postoperative prophylaxis of deep venous thrombosis and can replace the heparin use during hemodialytic treatments.
It has been investigated for use in treatment of chronic venous insufficiency.[3]
Potential indications in the future
Other recent preclinical studies have demonstrated that defibrotide used in conjunction with Granulocyte Colony-Stimulating Factor (rhG-CSF) significantly increases the number of Peripheral Blood Progenitor Cells (Stem cells). The benefit of this increase in stem cells may be crucial for a variety of clinical indications, including graft engineering procedures and gene therapy programs. This would expand the clinical usefulness of defibrotide to a complete distinct area.
Very recently (since early 2006) combination therapy trials (phase I/II) with defibrotide plus melphalan, prednisone, and thalidomide in patients with multiple myeloma have been conducted. The addition of defibrotide is expected to decrease the myelosuppressive toxicity of melphalan. However, is too early for any definitive results at that stage.
Cautions and contraindications
- The efficacy of the drug has been reported to be poorer in patients with diabetes mellitus.
- Pregnancy: The drug should not be used during pregnancy, because adequate and well controlled human studies do not exist.
- Lactation: No human data is available. In order to avoid damage to the newborn, the nursing mother should discontinue either the drug or breastfeeding, taking into account the importance of treatment to the mother.
- Known Bleeding Disorders or Bleeding Tendencies having occurred recently: Defibrotide should be used cautiously. Before initiation of treatment, the usual coagulation values should be obtained as baseline and regularly controlled under treatment. The patient should be observed regularly regarding local or systemic bleeding events.
Side-effects
Increased bleeding and bruising tendency, irritation at the injection site, nausea, vomiting, heartburn, low blood pressure. Serious allergic reactions have not been observed so far.
Drug interactions
Use of heparin with defibrotide may increase the aPTT, reflecting reduced ability of the body to form a clot. Nothing is known about the concomitant application of other anticoagulants than heparin and dextran containing plasma-expanders, but it can be anticipated that the risk of serious bleeding will be increased considerably.
PATENT
WO 2001078761
G-CSF (CAS registry number 143011-2-7/Merck Index, 1996, page 4558) is a haematopoietic growth factor which is indispensable in the proliferation and differentiation of the progenitor cells of granulocytes; it is a 18-22 kDa glycoprotein normally produced in response to specific stimulation by a variety of cells, including monocytes, fibroblasts and endothelial cells. The term defibrotide (CAS registry number 83712-60-1) normally identifies a polydeoxyribonucleotide obtained by extraction (US 3,770,720 and US 3,899,481) from animal and/or vegetable tissue; this polydeoxyribonucleotide is normally used in the form of a salt of an alkali metal, generally sodium. Defibrotide is used principally for its anti- thrombotic activity (US 3,829,567) although it may be used in different applications, such as, for example, the treatment of acute renal insufficiency (US 4,694,134) and the treatment of acute myocardial ischaemia (US 4,693,995). United States patents US 4,985,552 and US 5,223,609, finally, describe a process for the production of defibrotide which enables a product to be obtained which has constant and well defined physico-chemical characteristics and is also free from any undesired side-effects
References
- “Jazz Pharma Acquiring Gentium for $1B”. Gen. Eng. Biotechnol. News (paper) 34 (2). January 15, 2014. p. 10.
- Haussmann U, Fischer J, Eber S, Scherer F, Seger R, Gungor T (June 2006). “Hepatic veno-occlusive disease in pediatric stem cell transplantation: impact of pre-emptive antithrombin III replacement and combined antithrombin III/defibrotide therapy”. Haematologica 91 (6): 795–800. PMID 16769582.
- Coccheri S, Andreozzi GM, D’Addato M, Gensini GF (June 2004). “Effects of defibrotide in patients with chronic deep insufficiency. The PROVEDIS study”. Int Angiol 23 (2): 100–7.PMID 15507885.
External links
- Palmer KJ, Goa KL. Defibrotide: a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in vascular disorders. Drugs 1993;45:259-94.
- http://www.globalrx.com/medinfo/Defibrotide.htm
- Fisher J, Holland TK, Pescador R, Porta R, Ferro L (January 1996). “Study on pharmacokinetics of radioactive labelled defibrotide after oral or intravenous administration in rats”. Thromb. Res. 81 (1): 55–63. doi:10.1016/0049-3848(95)00213-8. PMID 8747520.
- http://www.gentium.it/Defibrotide.aspx (information provided by manufacturer)
- “Melphalan: profile and news”. Archived from the original on 2007-09-28. (on cytostatic combination therapy)
- Beşişik SK, Oztürk GB, Calişkan Y, Sargin D (March 2005). “Complete resolution of transplantation-associated thrombotic microangiopathy and hepatic veno-occlusive disease by defibrotide and plasma exchange”. Turk J Gastroenterol 16 (1): 34–7. PMID 16252186.
| WO2003101468A1 * | Jun 2, 2003 | Dec 11, 2003 | Guenther Eissner | Method for the protection of endothelial and epithelial cells during chemotherapy |
| US4985552 | Jul 5, 1989 | Jan 15, 1991 | Crinos Industria Farmacobiologica S.P.A. | Process for obtaining chemically defined and reproducible polydeoxyribonucleotides |
| US5223609 | May 26, 1992 | Jun 29, 1993 | Crinos Industria Farmacobiologica S.P.A. | Process for obtaining chemically defined and reproducible polydeoxyribonucleotides |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1999026639A1 * | 24 Nov 1998 | 3 Jun 1999 | Allegheny University Of The He | Methods for mobilizing hematopoietic facilitating cells and hematopoietic stem cells into the peripheral blood |
| EP0317766A1 * | 20 Oct 1988 | 31 May 1989 | Crinos Industria Farmacobiologica S.p.A. | A method for preventing blood coaguli from being formed in the extra-body circuit of dialysis apparatus and composition useful thereof |
| EP0416678A1 * | 10 Aug 1990 | 13 Mar 1991 | Crinos Industria Farmacobiologica S.p.A. | Topical compositions containing Defibrotide |
| US5199942 * | 26 Sep 1991 | 6 Apr 1993 | Immunex Corporation | Method for improving autologous transplantation |
| US5977083 * | 5 Jun 1995 | 2 Nov 1999 | Burcoglu; Arsinur | Method for using polynucleotides, oligonucleotides and derivatives thereof to treat various disease states |
| Reference | ||
|---|---|---|
| 1 | * | CARLO-STELLA, C. (1) ET AL: “Defibrotide significantly enhances peripheral blood progenitor cell mobilization induced by recombinant human granulocyte colony – stimulating factor ( rhG – CSF.” BLOOD, ( NOVEMBER 16, 2000 ) VOL. 96, NO. 11 PART 1, PP. 553A. PRINT. MEETING INFO.: 42ND ANNUAL MEETING OF THE AMERICAN SOCIETY OF HEMATOLOGY SAN FRANCISCO, CALIFORNIA, USA DECEMBER 01-05, 2000 AMERICAN SOCIETY OF HEMATOLOGY. , XP002176349 |
| 2 | * | GURSOY A: “PREPARATION, CHARACTERIZATION AND ANTI-INFLAMMATORY EFFECT OF DEFIBROTIDE LIPOSOMES” PHARMAZIE,DD,VEB VERLAG VOLK UND GESUNDHEIT. BERLIN, vol. 48, no. 7, 1 July 1993 (1993-07-01), pages 549-550, XP000372658 ISSN: 0031-7144 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2005017160A2 * | 12 Aug 2004 | 24 Feb 2005 | Childrens Hosp Medical Center | Mobilization of hematopoietic cells |
| WO2009115465A1 * | 13 Mar 2009 | 24 Sep 2009 | Gentium Spa | Synthetic phosphodiester oligonucleotides and therapeutical uses thereof |
| EP2103689A1 * | 19 Mar 2008 | 23 Sep 2009 | Gentium S.p.A. | Synthetic phosphodiester oligonucleotides and therapeutical uses thereof |
| US7417026 | 12 Aug 2004 | 26 Aug 2008 | Children’s Hospital Medical Center | Mobilization of hematopoietic cells |
| US7915384 | 5 Jan 2009 | 29 Mar 2011 | Children’s Hospital Medical Center | Chimeric peptides for the regulation of GTPases |
| US8242246 | 28 Feb 2011 | 14 Aug 2012 | Children’s Hospital Medical Center | Chimeric peptides for the regulation of GTPases |
| US8674075 | 13 Aug 2012 | 18 Mar 2014 | Children’s Medical Center Corporation | Chimeric peptides for the regulation of GTPases |
| US8980862 | 12 Nov 2010 | 17 Mar 2015 | Gentium S.P.A. | Defibrotide for use in prophylaxis and/or treatment of Graft versus Host Disease (GVHD) |
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
oral, i.m., i.v. |
| Pharmacokinetic data | |
| Bioavailability | 58 – 70% orally (i.v. and i.m. = 100%) |
| Biological half-life | t1/2-alpha = minutes; t1/2-beta = a few hours |
| Identifiers | |
| CAS Registry Number | 83712-60-1 |
| ATC code | B01AX01 |
| DrugBank | DB04932 |
| UNII | 438HCF2X0M |
| KEGG | D07423 |
///////////Approved, 3/30/3016, US FDA, defibrotide sodium, NDA 208114, FDA 2016
Updates……….
FDA approves first treatment for rare disease in patients who receive stem cell transplant from blood or bone marrow
For Immediate Release
March 30, 2016
Release
The U.S. Food and Drug Administration today approved Defitelio (defibrotide sodium) to treat adults and children who develop hepatic veno-occlusive disease (VOD) with additional kidney or lung abnormalities after they receive a stem cell transplant from blood or bone marrow called hematopoietic stem cell transplantation (HSCT). This is the first FDA-approved therapy for treatment of severe hepatic VOD, a rare and life-threatening liver condition.
HSCT is a procedure performed in some patients to treat certain blood or bone marrow cancers. Immediately before an HSCT procedure, a patient receives chemotherapy. Hepatic VOD can occur in patients who receive chemotherapy and HSCT. Hepatic VOD is a condition in which some of the veins in the liver become blocked, causing swelling and a decrease in blood flow inside the liver, which may lead to liver damage. In the most severe form of hepatic VOD, the patient may also develop failure of the kidneys and lungs. Fewer than 2 percent of patients develop severe hepatic VOD after HSCT, but as many as 80 percent of patients who develop severe hepatic VOD do not survive.
“The approval of Defitelio fills a significant need in the transplantation community to treat this rare but frequently fatal complication in patients who receive chemotherapy and HSCT,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research.
The efficacy of Defitelio was investigated in 528 patients treated in three studies: two prospective clinical trials and an expanded access study. The patients enrolled in all three studies had a diagnosis of hepatic VOD with liver or kidney abnormalities after HSCT. The studies measured the percentage of patients who were still alive 100 days after HSCT (overall survival). In the three studies, 38 to 45 percent of patients treated with Defitelio were alive 100 days after HSCT. Based on published reports and analyses of patient-level data, the expected survival rates 100 days after HSCT would be 21 to 31 percent for patients with severe hepatic VOD who received only supportive care or interventions other than Defitelio.
The most common side effects of Defitelio include abnormally low blood pressure (hypotension), diarrhea, vomiting, nausea and nosebleeds (epistaxis). Serious potential side effects of Defitelio that were identified include bleeding (hemorrhage) and allergic reactions. Defitelio should not be used in patients who are having bleeding complications or who are taking blood thinners or other medicines that reduce the body’s ability to form clots.
The FDA granted the Defitelio application priority review status, which facilitates and expedites the development and review of certain drugs in light of their potential to benefit patients with serious or life-threatening conditions. Defitelio also received orphan drug designation, which provides incentives such as tax credits, user fee waivers and eligibility for exclusivity to assist and encourage the development of drugs for rare diseases.
Defitelio is marketed by Jazz Pharmaceuticals based in Palo Alto, California
Efficacy and Safety of Olive in the Management of Hyperglycemia

Postprandial hyperglycemia indicates the abnormality in glucose turnover leading to the onset of type 2 diabetes. Therefore, correction of postprandial hyperglycemia is crucial in the early stage of diabetes therapy. One of the most effective strategies to control postprandial hyperglycemia is medication combined with intake restriction and an exercise program. However, along with the prevalence of chronic diseases with multi-pathogenic factor, drugs with single chemical composition are usually not effective. In this view, phytotherapy has a promising future in the management of diabetes, considered to have less side effects as compared to synthetic drugs.
The World Health Organization estimates that in developing countries about 80% of the population now still depend on herbal treatment. Olive (Olea europea) (OE) has been used in traditional remedies in Europe and Mediterranean countries as a food and medicine for over 5,000 years especially for the prevention and treatment of chronic diseases such as hypertension, atherosclerosis , cancer and diabetes. In addition, olive is considered as the most important component of the Mediterranean diet with many health benefits.
Several experimental studies have demonstrated the beneficial effect of OE on diabetes. This effect has been demonstrated in the animal models such as streptozotocin-induced diabetic rats, alloxaninduced diabetic rats and obese diabetic sand rats fed a hypercaloric diet. In these models olive extracts have been shown to exhibit a significant reduction on both blood glucose and insulin levels. Few randomized clinical trials have demonstrated the beneficial effect of olive and one study has shown that the subjects treated with olive leaf extract exhibited significantly lower Glycated hemoglobin (HbA1c) and fasting plasma insulin levels.
Another study performed in recent onset type 2 diabetic patients has revealed that OE leaves exhibited antidiabetic activity when it added as a mixture of extract of leaves of Juglans regia, Urtica dioica and Atriplex halimus. The underlying mechanism seems to be the improvement of glucose uptake and no side effect was reported while extracts from OE have been found to exhibit cytotoxic effects only at concentrations higher than 500 μg/ mL in cells from the liver hepatocellular carcinoma cell line (HepG2) and cells from the rat L6 muscle cell line. As far as the phytochemical analysis is concerned, it is now well-established that major fatty acid constituents and minor phenolic components in olives and olive oil exert important health benefits particularly for cardiovascular diseases, metabolic syndrome and inflammatory conditions.
Hydroxytyrosol and oleuropein are considered as major polyphenolic compounds in olive leaf. Oleuropeoside, a phenylethanoid isolated from OE demonstrated a significant hypoglycemic activity in alloxan-induced diabetes and the hypoglycemic activity of this compound may result from both the increased peripheral uptake of glucose and potentiation of glucose-induced insulin secretion. In addition, Maslinic acid (MA), a natural triterpene from OE with hypoglycemic activity is a wellknown inhibitor of glycogen phosphorylase in diabetic rats without affecting hematological, histopathologic and biochemical variables, thus suggesting a sufficient margin of safety for its putative use as a nutraceutical. More recently a study has showed that MA exerts antidiabetic effects by increasing glycogen content and inhibiting glycogen phosphorylase activity in HepG2 cells.

Furthermore, MA was shown to induce the phosphorylation level of insulin-receptor β-subunit, protein kinase B (Akt) and glycogen synthase kinase-3β. MA treatment of mice fed with a high-fat diet reduced the model-associated adiposity, mRNA expression of proinflammatory cytokines and then insulin resistance, and increased the accumulated hepatic glycogen.
Finally, a recent clinical study has revealed that supplementation with olive leaf polyphenols significantly improved insulin sensitivity and pancreatic β-cell secretory capacity in overweight middle-aged men at risk of developing the metabolic syndrome. In conclusion, OE has been and continue to represent a natural source of phytocompounds eliciting a beneficial effect in human health especially in the management of hyperglycemia [1–15].
- Bennani-Kabchi N Fdhil H,Cherrah Y,El Bouayadi F,Kehel L,et al.(2000) Therapeutic effect ofOleaeuropeavar. oleaster leaves on carbohydrate and lipid metabolism in obese and prediabetic sand rats (Psammomysobesus).Ann Pharm Fr 58: 271-7.
- Said O,Fulder S, Khalil K,Azaizeh H, Kassis E, et al. (2008) Maintaining a physiological blood glucose level with ‘glucolevel’, a combination of four anti-diabetesplants used in the traditional arab herbal medicine. Evid Based Complement Alternat Med 5:421-8.
- Bouallagui Z,Han J,Isoda H,Sayadi S. Hydroxytyrosol rich extract from olive leaves modulates cell cycle progression in MCF-7 human breast cancer cells (2011) Food ChemToxicol 49:179-84.
- Nan JN,Ververis K ,Bollu S,Rodd AL,Swarup O,et al. (2014) Biological effects of the olive polyphenol, hydroxytyrosol: An extra view from genome-wide transcriptome analysis.Hell J Nucl Med 17: 62-9.
- Guan T, Qian Y, Tang X, Huang M, Huang L, et al. (2011) Maslinic acid, a natural inhibitor of glycogen phosphorylase, reduces cerebral ischemic injury in hyperglycemic rats by GLT-1 up-regulation. Neurosci Res 89: 1829-39.
- Sánchez-González M, Lozano-Mena G, Juan ME, García-Granados A, Planas JM (2013)Assessment of thesafetyof maslinic acid, a bioactive compound from Oleaeuropaea L.Mol Nutr Food Res 57:339-46.
- Liu J, Wang X, Chen YP, Mao LF, Shang J, et al. (2014) Maslinic acidmodulates glycogen metabolism by enhancing the insulin signaling pathway and inhibiting glycogen phosphorylase.Chin J Nat Med 12: 259-65.
- de Bock M, Derraik JG, Brennan CM, Biggs JB, Morgan PE,(2013) Olive(Oleaeuropaea L.) leaf polyphenols improve insulin sensitivity in middle-aged overweight men: a randomized, placebo-controlled, crossover trial.

Prof. Mohamed Eddouks
Dean, Polydisciplinary Faculty of Errachidia
Moulay Ismail University, Morocco

RESEARCH EXPERIENCE
- Eddouks M, Chattopadhyay D, Zeggwagh NA.Animal models as tools to investigate antidiabetic and anti-inflammatory plants.Evid Based Complement Alternat Med. 2012;2012:142087.
- Zeggwagh NA, Michel JB, Eddouks M.Vascular Effects of Aqueous Extract of Chamaemelum nobile: In Vitro Pharmacological Studies in Rats.Clin Exp Hypertens. 2012.
- Oufni L, Taj S, Manaut B, Eddouks M. 2011.Transfer of uranium and thorium from soil to different parts of medicinal plants using SSNTD. Journal of Radioanalytical and Nuclear Chemistry, 287; 403-411.
- Zeggwagh NA, Moufid A, Michel JB, Eddouks M. Hypotensive effect of Chamaemelum nobile aqueous extract in spontaneously hypertensive rats.Clin Exp Hypertens. 2009.31(5):440-50.
- Zeggwagh NA, Farid O, Michel JB, Eddouks M. Cardiovascular effect of Artemisia herba alba aqueous extract in spontaneously hypertensive rats.Methods Find Exp Clin Pharmacol. 2008. 30(5):375-81.
- Eddouks M, Maghrani M, Louedec L, Haloui M, Michel JB.Antihypertensive activity of the aqueous extract of Retama raetam Forssk. leaves in spontaneously hypertensive rats.J Herb Pharmacother. 2007;7(2):65-77.
- Zeggwagh, N-A., Eddouks, M . Anti-hyperglycaemic and hypolipidemic effects of Ocimum basilicum aqueous extract in diabetic rats. American Journal of Pharmacology and Toxicology. 2(3): 123-129, 2007.
- Lemhadri, A., Burcelin, R., Eddouks, M. Chamaemelum nobile L. aqueous extract represses endogenous glucose production and improves insulin sensitivity in streptozotocin-induced diabetic mice. American Journal of Pharmacology and Toxicology. 2(3): 116-122, 2007.
- Lemhadri, A., Eddouks, M., Burcelin, R. Anti-hyperglycaemic and anti-obesity effects of Capparis spinosa and Chamaemelum nobile aqueous extracts in HFD mice. American Journal of Pharmacology and Toxicology. 2(3): 106-110, 2007.
- Zeggwagh, N.A., Michel, J.B, and Eddouks, M. Acute Hypotensive and Diuretic Activities of Chamaemelum nobile Aqueous Extract in Normal Rats. American Journal of Pharmacology and Toxicology. 2(3): 140-145, 2007.
- Zeggwagh, N-A., Michel, JB., Eddouks, M . Cardiovascular effect of Capapris spinosa aqueous extract in rats Part II: Furosemide-like effect of Capparis spinosa aqueous extract in normal rats. 2(3): 130-134, 2007.
- Zeggwagh, N-A., Michel, JB., Eddouks, M . Cardiovascular effect of Capparis spinosa aqueous extract. Part III: Antihypertensive effect in spontaneously hypertensive rats. American Journal of Pharmacology and Toxicology. 2(3): 111-115, 2007.
- Zeggwagh, N-A., Eddouks, M .Michel, JB. Cardiovascular effect of Capparis spinosa aqueous extract. Part VI: in vitro vasorelaxant effect.American Journal of Pharmacology and Toxicology. 2(3): 135-139, 2007.
- Eddouks, M., Ouahidi, M.L., Farid, O., Moufid, A., Lemhadri, A. The use of medicinal plants in the treatment of diabetes in Morocco. Phytothérapie. 2007, 5, no4, pp.194-203.
- Eddouks M; Khalidi A; Zeggwagh N.-A; Pharmacological approach of plants traditionally used in treating hypertension in Morocco. Phytothérapie. 2009, 7, no2, pp. 122-127.
- Zeggwagh NA, Ouahidi ML, Lemhadri A, Eddouks M. 2006. Study of hypoglycaemic and hypolipidemic effects of Inula viscosa L. aqueous extract in normal and diabetic rats. Journal ofEthnopharmacology. 24; 108(2): 223-7.
- Lemhadri A, Hajji L, Michel JB, Eddouks M. Cholesterol and triglycerides lowering activities of caraway fruits in normal and streptozotocin diabetic rats. Journal ofEthnopharmacology 2006 19; 106(3):321-6.
- Eddouks, M., Maghrani, M, Michel, J-B.Antihypertensive action of Lepidium sativum in SHR rats. In Press. Journal of Herbal Pharmacotherapy.Eddouks, M., Michel, J-B., Mghrani, M. Effect of Lepidium sativum L. On renal glucose reabsorption and urinary TGF B levels in diabetic rats. Phytotherapy Research. 2008 ;22(1):1-5.
- Eddouks M, Maghrani M, Michel JB.2005.Hypoglycaemic effect of Triticum repens P. Beauv. in normal and diabetic rats. Journal of Ethnopharmacology. 2005 ; 102(2):228-32.
- Eddouks, M. 2005. Les plantes anti-diabétiques. Phytothérapie Européenne. 28, 8-12.
- Zhang J, Onakpoya IJ, Posadzki P, Eddouks M. The safety of herbal medicine: from prejudice to evidence. Evid Based Complement Alternat Med. 2015;2015:316706.
- Yakubu MT, Sunmonu TO, Lewu FB, Ashafa AO, Olorunniji FJ, Eddouks M. Efficacy and safety of medicinal plants used in the management of diabetes mellitus. Evid Based Complement Alternat Med. 2014; 2014: 793035.
- Eddouks M, Chattopadhyay D, De Feo V, Cho WC. Medicinal plants in the prevention and treatment of chronic diseases 2013. Evid Based Complement Alternat Med. 2014;2014:180981.
- Eddouks M, Bidi A, El Bouhali B, Hajji L, Zeggwagh NA. Antidiabetic plants improving insulin sensitivity. J Pharm Pharmacol. 2014 Sep;66(9):1197-214.
Efficacy and Safety of Olive in the Management of Hyperglycemia

Eddouks M*
Faculty of Sciences and Techniques Errachidia, Moulay Ismail university, BP 21, Errachidia, 52000, Morocco
MOHAMED EDDOUKS
Professor
Faculty of Sciences and Techniques Errachidia
Moulay Ismail University
Morocco
Dr. Mohamed Eddouks is currently working as a professor at Moulay ismail university, morocco. He worked as assistant professor at faculty of sciences and techniques errachidia (1995) and as head of the department of biology at faculty of sciences and techniques errachidia (2003). He completed his PhD degree in Physiology and Pharmacology from University of Liege, Belgium and Sidi Mohammed Ben Abdellah University. He published many articles in international journals.
Eddouks M
Faculty of Sciences and Techniques Errachidia
Moulay Ismail university, BP 21
Errachidia, 52000, Morocco
Tel: +212535574497
Fax: +212535574485
E-mail: mohamed.eddouks@laposte.net
Citation: Eddouks M (2015) Efficacy and Safety of Olive in the Management of Hyperglycemia. Pharmaceut Reg Affairs 4:e145. doi:10.4172/2167-7689.1000e145




Er Rachidi; Errachidia



………..
Morocco


////////
New FDA Requirements for the Development of Herbal Medicinal Products
DRUG REGULATORY AFFAIRS INTERNATIONAL
The previous FDA guideline for herbal medicinal products from 2004 is supposed to be replaced by a new version. In August 2015, the FDA has presented the draft of the revised guideline. Find out more about the FDA Guideline Botanical Drug Development.
In August 2015, the FDA has published a draft of the guideline “Botanical Drug Development”. This guideline addresses issues arising from the particular nature of herbal medicinal products. After its finalization it is supposed to replace the previous guideline from June 2004.
The general approach in the development of herbal medicinal products remained unchanged since 2004. But due to the better understanding of herbal medicinal products and the experience gained during the review of the approval documents for herbals (NDAs/New Drug Applications and INDs/Investigational New Drug Applications), specific recommendations could be adjusted. Still, new sections will be supplemented to better address the late development phase.
The…
View original post 34 more words
Genotoxic impurities: the new ICH M7 addendum to calculation of compound-specific acceptable intakes
DRUG REGULATORY AFFAIRS INTERNATIONAL

Genotoxic impurities: the new ICH M7 addendum to calculation of compound-specific acceptable intakes
The draft for a guideline ICH M7(R1) published recently supplements the ICH-M7 guideline published last year. Read more about the calculation of compound-specific acceptable intakes of genotoxic impurities.
The final document of the ICH-Guideline M7 was published in June 2014. It describes the procedure for evaluating the genotoxic potential of impurities in medicinal products (see also our news Final ICH M7 Guideline on Genotoxic Impurities published dated 23 July 2014).
An important approach to the risk characterisation of impurities is the TTC concept (TTC = threshold of toxicological concern). According to this approach the exposure to a mutagenic impurity having the concentration of 1.5 µg per adult person per day is considered to be associated with a negligible risk. It can be used as default evaluation approach to most pharmaceuticals for long-term treatment (> 10 years)…
View original post 200 more words
TR 700, TR 701FA, Tedizolid phosphate

“TR-700”
5R)-3-{3-Fluoro-4-[6-(2-methyl-2H-1,2,3,4-tetrazol-5-yl)-pyridin-3-yl]-phenyl}-5-hydroxymethyl-1,3-oxazolidin-2-one
US Patent Publication No. 20070155798, which is hereby incorporated by reference in its entirety, recently disclosed a series of potently anti-bacterial oxazolidinones including

wherein R═H, PO(OH)2, and PO(ONa)2.

(R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate, CAS 856867-55-5

DISODIUM SALT

CAS 856867-39-5
- C17 H16 F N6 O6 P . 2 Na
- 2-Oxazolidinone, 3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5-[(phosphonooxy)methyl]-, sodium salt (1:2), (5R)-
-
- DA 7218, Tedizolid phosphate disodium salt
In addition, improved methods of making the free acid are disclosed in U.S. patent application Ser. No. 12/577,089, which is assigned to Trius Therapeutics, Inc., and which is incorporated herein by reference
crystalline (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate 1 (R═PO(OH)2), was more stable and non-hygroscopic than the salt forms that were tested. In addition, unlike typical crystallizations, where the crystallization conditions, such as the solvent and temperature conditions, determine the particular crystalline form, the same crystalline form of 1 (R═PO(OH)2) was produced using many solvent and crystallization conditions. Therefore, this crystalline form was very stable, was made reproducibly, and ideal for commercial production because it reduced the chances that other polymorphs would form contaminating impurities during production. However, in all preliminary testing, the free acid crystallized as fine particles, making filtering and processing difficult.
To overcome difficulties in filtering and processing crystalline (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate 1 (R═PO(OH)2), processes described herein result in significantly reduced filtering time, avoid more toxic solvents, and significantly increased ease of preparing dosage forms such as tablets. It has been found that implementing various processes can control the particle size distribution of the resulting material, which is useful for making the crystalline form, and for commercial production and pharmaceutical use. Surprisingly, the process for increasing the particle size reduces the amount of the dimer impurity, in comparison to the process for making the free acid disclosed in U.S. patent application Ser. No. 12/577,089. Thus, various methods of making and using the crystalline form are also provided.
In addition, by using methods of making the free acid disclosed in U.S. patent application Ser. No. 12/577,089, which is assigned to the same assignee as in the present application, and by using the crystallization methods described herein, a crystalline free acid having at least 96% purity by weight may be formed that comprises a compound having the following formula:

(hereinafter “the chloro impurity”), i.e., (R)-5-(chloromethyl)-3-(3-fluoro-4-(6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl)phenyl)oxazolidin-2-one in an amount less than 1%.
Similarly, by using methods of making the free acid disclosed in U.S. patent application Ser. No. 12/577,089, which is assigned to the same assignee as in the present application, and by using the crystallization methods described herein, a crystalline free acid having at least 96% purity by weight may be formed that comprises a compound having the following formula:
(hereinafter “TR-700”), i.e., 5R)-3-{3-Fluoro-4-[6-(2-methyl-2H-1,2,3,4-tetrazol-5-yl)-pyridin-3-yl]-phenyl}-5-hydroxymethyl-1,3-oxazolidin-2-one, in an amount less than 1%.
The crystalline free acid may have one or more of the attributes described herein.
In some aspects, a purified crystalline (R)-3-(4-(2-(2-methyltetrazol-5-yl)-pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate, i.e., the free acid, has a purity of at least about 96% by weight. In some embodiments, the crystalline free acid has a median volume diameter of at least about 1.0 μm.
BRIEF DESCRIPTION OF THE DRAWINGS……http://www.google.com/patents/US8426389
FIG. 1 the FT-Raman spectrum of crystalline 1 (R═PO(OH)2).

FIG. 2 shows the X-ray powder pattern of crystalline 1 (R═PO(OH)2).
http://www.google.com/patents/US8426389
FIG. 3 shows the differential scanning calorimetry (DSC) thermogram of crystalline 1 (R═PO(OH)2).
http://www.google.com/patents/US8426389
FIG. 4 shows the 1H NMR spectrum of 1 (R═PO(OH)2).

FIG. 5 depicts the TG-FTIR diagram of crystalline 1 (R═PO(OH)2).
http://www.google.com/patents/US8426389
FIG. 6 is a diagram showing the dynamic vapor sorption (DVS) behavior of crystalline 1 (R═PO(OH)2).
FIG. 7 is a manufacturing process schematic for 1 (R═PO(OH)2) (TR-701 FA) in a tablet dosage form.

FIG. 8 is a manufacturing process schematic for 1 (R═PO(OH)2) (TR-701 FA) Compounding Solution for Lyophilization.

FIG. 9 is a manufacturing process schematic for 1 (R═PO(OH)2) (TR-701 FA) for Injection, 200 mg/vial: sterile filtering, filling, and lyophilization.
FIG. 10 is a representative particle size distribution of crystalline free acid without regard to controlling particle size distribution as also described herein.
FIG. 11 is a representative particle size distribution of crystalline free acid made using laboratory processes to control particle size described herein.
FIG. 12 is a representative particle size distribution of crystalline free acid made using scaled up manufacturing processes to control particle size described herein.
These impurities include

i.e., 5R)-3-{3-Fluoro-4-[6-(2-methyl-2H-1,2,3,4-tetrazol-5-yl)-pyridin-3-yl]-phenyl}-5-hydroxymethyl-1,3-oxazolidin-2-one (“TR-700”) and/or

i.e., (R)-5-(chloromethyl)-3-(3-fluoro-4-(6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl)phenyl)oxazolidin-2-one (“chloro impurity”).
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US4128654 | Feb 10, 1978 | Dec 5, 1978 | E. I. Du Pont De Nemours And Company | 5-Halomethyl-3-phenyl-2-oxazolidinones |
| US4250318 | Aug 9, 1978 | Feb 10, 1981 | Delalande S.A. | Novel 5-hydroxymethyl oxazolidinones, the method of preparing them and their application in therapeutics |
| US4340606 | Oct 23, 1980 | Jul 20, 1982 | E. I. Du Pont De Nemours And Company | 3-(p-Alkylsulfonylphenyl)oxazolidinone derivatives as antibacterial agents |
| US4461773 | Jan 5, 1984 | Jul 24, 1984 | E. I. Dupont De Nemours And Company | P-Oxooxazolidinylbenzene compounds as antibacterial agents |
| US4476136 | Feb 24, 1982 | Oct 9, 1984 | Delalande S.A. | Aminomethyl-5 oxazolidinic derivatives and therapeutic use thereof |
| US4948801 | Jul 29, 1988 | Aug 14, 1990 | E. I. Du Pont De Nemours And Company | Aminomethyloxooxazolidinyl arylbenzene derivatives useful as antibacterial agents |
| US5523403 | May 22, 1995 | Jun 4, 1996 | The Upjohn Company | Tropone-substituted phenyloxazolidinone antibacterial agents |
| US5565571 | Apr 28, 1994 | Oct 15, 1996 | The Upjohn Company | Substituted aryl- and heteroaryl-phenyloxazolidinones |
| US5652238 | Sep 27, 1994 | Jul 29, 1997 | Pharmacia & Upjohn Company | Esters of substituted-hydroxyacetyl piperazine phenyl oxazolidinones |
| US5688792 | Aug 16, 1994 | Nov 18, 1997 | Pharmacia & Upjohn Company | Substituted oxazine and thiazine oxazolidinone antimicrobials |
| US6365751 | Apr 17, 2001 | Apr 2, 2002 | Zeneca Ltd. | Antibiotic oxazolidinone derivatives |
| US6627646 * | Jul 17, 2001 | Sep 30, 2003 | Sepracor Inc. | Norastemizole polymorphs |
| US6689779 | May 18, 2001 | Feb 10, 2004 | Dong A Pharm. Co., Ltd. | Oxazolidinone derivatives and a process for the preparation thereof |
| US7129259 | Dec 1, 2004 | Oct 31, 2006 | Rib-X Pharmaceuticals, Inc. | Halogenated biaryl heterocyclic compounds and methods of making and using the same |
| US7141583 | Apr 23, 2001 | Nov 28, 2006 | Astrazeneca Ab | Oxazolidinone derivatives with antibiotic activity |
| US7144911 | Dec 24, 2003 | Dec 5, 2006 | Deciphera Pharmaceuticals Llc | Anti-inflammatory medicaments |
| US7202257 | Jul 6, 2004 | Apr 10, 2007 | Deciphera Pharmaceuticals, Llc | Anti-inflammatory medicaments |
| US7396847 | Sep 9, 2002 | Jul 8, 2008 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline as antibacterial agents |
| US7462633 | Jun 29, 2004 | Dec 9, 2008 | Merck & Co., Inc. | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US7473699 | Feb 25, 2003 | Jan 6, 2009 | Astrazeneca Ab | 3-cyclyl-5-(nitrogen-containing 5-membered ring)methyl-oxazolidinone derivatives and their use as antibacterial agents |
| US7498350 | Nov 24, 2003 | Mar 3, 2009 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| US7816379 | Dec 17, 2004 | Oct 19, 2010 | Dong-A Pharm. Co., Ltd. | Oxazolidinone derivatives |
| US20020115669 | Aug 29, 2001 | Aug 22, 2002 | Wiedeman Paul E. | Oxazolidinone chemotherapeutic agents |
| US20030166620 | May 18, 2001 | Sep 4, 2003 | Jae-Gul Lee | Novel oxazolidinone derivatives and a process for the preparation thereof |
| US20040180906 | Dec 24, 2003 | Sep 16, 2004 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20050038092 | Jun 29, 2004 | Feb 17, 2005 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US20050107435 | Sep 9, 2002 | May 19, 2005 | Gravestock Michael B. | Oxazolidinone and/or isoxazoline as antibacterial agents |
| US20050288286 | Jul 6, 2004 | Dec 29, 2005 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20060116386 | Nov 24, 2003 | Jun 1, 2006 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| US20060116400 | Nov 24, 2003 | Jun 1, 2006 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline derivatives as antibacterial agents |
| US20060270637 | Feb 24, 2004 | Nov 30, 2006 | Astrazeneca Ab | Hydroxymethyl substituted dihydroisoxazole derivatives useful as antibiotic agents |
| US20070155798 | Dec 17, 2004 | Jul 5, 2007 | Dong-A Pharm. Co., Ltd. | Novel oxazolidinone derivatives |
| US20070185132 | Jun 29, 2004 | Aug 9, 2007 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereo |
| US20070191336 | Dec 23, 2004 | Aug 16, 2007 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20070203187 | Jan 22, 2007 | Aug 30, 2007 | Merck & Co., Inc. | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US20070208062 | May 24, 2005 | Sep 6, 2007 | Astrazeneca Ab | 3-(4-(2-dihydroisoxazol-3-ylpyridin-5-yl)phenyl)-5-triazol-1-ylmethyloxazolidin-2-one derivatives as mao inhibitors for the treatment of bacterial infections |
| US20080021012 | May 24, 2005 | Jan 24, 2008 | Astrazeneca Ab | 3-[4-{6-Substituted Alkanoyl Pyridin-3-Yl}-3-Phenyl]-5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones As Antibacterial Agents |
| US20080021071 | May 24, 2005 | Jan 24, 2008 | Astrazeneca Ab | 3-{4-(Pyridin-3-Yl) Phenyl}-5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones as Antibacterial Agents |
| US20080064689 | May 24, 2004 | Mar 13, 2008 | Astrazeneca Ab | 3-[4-(6-Pyridin-3-Yl)-3-Phenyl] -5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones as Antibacterial Agents |
| US20090018123 | Jun 19, 2006 | Jan 15, 2009 | Milind D Sindkhedkar | Oxazolidinones Bearing Antimicrobial Activity Composition and Methods of Preparation |
| US20090192197 | Jul 30, 2009 | Dong-A Pharm. Co., Ltd. | Novel oxazolidinone derivatives | |
| US20100093669 | Oct 9, 2009 | Apr 15, 2010 | Trius Therapeutics | Methods for preparing oxazolidinones and compositions containing them |
| US20100227839 | Sep 9, 2010 | Trius Therapeutics | Crystalline form of r)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin- 5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate | |
| AU2004299413A1 | Title not available | |||
| AU2009200606A1 | Title not available | |||
| CA2549062A1 | Dec 17, 2004 | Jun 30, 2005 | Dong-A Pharm. Co., Ltd. | Novel oxazolidinone derivatives |
| CN101982468A | Dec 17, 2004 | Mar 2, 2011 | 东亚制药株式会社 | Novel oxazolidinone derivatives and pharmaceutical compositions comprising the derivatives |
| EP0312000A1 | Oct 12, 1988 | Apr 19, 1989 | The Du Pont Merck Pharmaceutical Company | Aminomethyl oxooxazolidinyl aroylbenzene derivatives useful as antibacterial agents |
| EP0352781A2 | Jul 27, 1989 | Jan 31, 1990 | The Du Pont Merck Pharmaceutical Company | Aminomethyloxooxazolidinyl arylbenzene derivatives useful as antibacterial agents |
| EP1699784A1 | Dec 17, 2004 | Sep 13, 2006 | Dong-A Pharmaceutical Co., Ltd. | Novel oxazolidinone derivatives |
| EP2305657A2 | Dec 17, 2004 | Apr 6, 2011 | Dong-A Pharmaceutical Co., Ltd. | Oxazolidinone derivatives |
| EP2435051A1 | May 27, 2010 | Apr 4, 2012 | Trius Therapeutics | Oxazolidinone containing dimer compounds, compositions and methods to make and use |
| IN236862A1 | Title not available | |||
| JPS5799576A | Title not available | |||
| KR20110071107A | Title not available | |||
| NZ547928A | Title not available | |||
| NZ575842A | Title not available | |||
| WO1993009103A1 | Oct 5, 1992 | May 13, 1993 | Upjohn Co | Substituted aryl- and heteroarylphenyloxazolidinones useful as antibacterial agents |
| WO1993023384A1 | Apr 21, 1993 | Nov 25, 1993 | Upjohn Co | Oxazolidinones containing a substituted diazine moiety and their use as antimicrobials |
| WO1995007271A1 | Aug 16, 1994 | Mar 16, 1995 | Michael R Barbachyn | Substituted oxazine and thiazine oxazolidinone antimicrobials |
| WO1995014684A1 | Sep 27, 1994 | Jun 1, 1995 | Michel R Barbachyn | Esters of substituted-hydroxyacetyl piperazine phenyl oxazolidinones |
| WO2001094342A1 | May 18, 2001 | Dec 13, 2001 | Cho Jong Hwan | Novel oxazolidinone derivatives and a process for the preparation thereof |
| WO2002081470A1 | Apr 3, 2002 | Oct 17, 2002 | Astrazeneca Ab | Oxazolidinones containing a sulfonimid group as antibiotics |
| WO2003022824A1 | Sep 9, 2002 | Mar 20, 2003 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline as antibacterial agents |
| WO2003035648A1 | Oct 23, 2002 | May 1, 2003 | Astrazeneca Ab | Aryl substituted oxazolidinones with antibacterial activity |
| WO2003047358A1 | Dec 2, 2002 | Jun 12, 2003 | Vaughan Leslie Crow | Cheese flavour ingredient and method of its production |
| WO2003072575A1 | Feb 25, 2003 | Sep 4, 2003 | Astrazeneca Ab | 3-cyclyl-5-(nitrogen-containing 5-membered ring) methyl-oxazolidinone derivatives and their use as antibacterial agents |
| WO2003072576A2 | Feb 25, 2003 | Sep 4, 2003 | Astrazeneca Ab | Oxazolidinone derivatives, processes for their preparation, and pharmaceutical compositions containing them |
| WO2004048350A2 | Nov 24, 2003 | Jun 10, 2004 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| WO2004083205A1 | Mar 16, 2004 | Sep 30, 2004 | Astrazeneca Ab | Antibacterial 1, 3- oxazolidin -2- one derivatives |
| WO2005005398A2 | Jun 29, 2004 | Jan 20, 2005 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| WO2005051933A1 | Nov 23, 2004 | Jun 9, 2005 | Vijay Kumar Kaul | An improved process for the synthesis of 4-(4-benzyloxy-carbonylamino-2-fluorophenyl)-piperazine-1-carboxylic acid tert-butyl ester, a key intermediate for oxazolidinone antimicrobials and compounds prepared thereby |
| WO2005058886A1 | Dec 17, 2004 | Jun 30, 2005 | Dong A Pharm Co Ltd | Novel oxazolidinone derivatives |
| WO2005116017A1 | May 24, 2005 | Dec 8, 2005 | Astrazeneca Ab | Process for the preparation of aryl substituted oxazolidinones as intermediates for antibacterial agents |
| WO2006038100A1 | Oct 6, 2005 | Apr 13, 2006 | Ranbaxy Lab Ltd | Oxazolidinone derivatives as antimicrobials |
| WO2007023507A2 | Jun 19, 2006 | Mar 1, 2007 | Milind D Sindkhedkar | Oxazolidinones bearing antimicrobial activity composition and methods of preparation |
| WO2007138381A2 | Oct 13, 2006 | Dec 6, 2007 | Delorme Daniel | Phosphonated oxazolidinones and uses thereof for the prevention and treatment of bone and joint infections |
| WO2010042887A2 | Oct 9, 2009 | Apr 15, 2010 | Trius Therapeutics | Methods for preparing oxazolidinones and compositions containing them |
| WO2010091131A1 | Feb 3, 2010 | Aug 12, 2010 | Trius Therapeutics | Crystalline form of r)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate |
| WO2010138649A1 | May 27, 2010 | Dec 2, 2010 | Trius Therapeutics, Inc. | Oxazolidinone containing dimer compounds, compositions and methods to make and use |
Wockhardt, WO 2007023507, N-[[3-[3,5-difluoro-4-[4-(tetrazol-2-yl)piperidin-1-yl]phenyl]-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide


Cas 928156-95-0,
Acetamide, N-[[(5S)-3-[3,5-difluoro-4-[4-(2H-tetrazol-2-yl)-1-piperidinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]-
| C18H21F2N7O3 | |
| Molecular Weight: | 421.401246 g/mol |
|---|
N-[[3-[3,5-difluoro-4-[4-(tetrazol-2-yl)piperidin-1-yl]phenyl]-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide

Example- 14 and 15
(S)-N- { 3- [4-(4-(2H-tetrazol-2-yl)-piperidin- 1 -yl)-3 , 5-difluorophenyl] -2-oxo-oxazolidin-
5-ylmethyl }-acetamide and
(S)-N- { 3- [4-(4-(l H-tetrazol- 1 -yl)-piperidin- 1 -yl)-3 , 5-difluorophenyl] -2-oxo-oxazolidin-
5-ylmethyl }-acetamide

and

A mixture of (S)-N-{3-[4-methanesulphonyloxy piperidin-l-yl)-3,5-difluorophenyl]-2- oxo-oxazolidin-5-ylmethyl}-acetamide (1.12 mM), tetrazole (1.68 mM), and K2CO3 (1.68 mM) in DMF (6 ml) was heated for 22 hrs at 850C. The resulting mixture was poured into ice-water mixture, stirred for 30 min. And the separated solid was purified by column chromatography to obtain two isomeric products in 18% and 12% yields respectively. Isomer A: M.P. 234-2370C; MS(M+1)- 422 ; M.F. C18H21F2N7O3 Isomer B: M.P. 214-2170C; MS(M+1)- 422 ; M.F. C18H2JF2N7O3
WOCKHARDT LIMITED [IN/IN]; D-4, MIDC Area, Chikalthana, Aurangabad 431006 (IN)
Our New Drug Discovery team has developed a number of lead molecules, mainly in the area of anti-infectives; these are currently at various stages of development.
Of these molecules, the most advanced of the New Chemical Entities (NCE) is WCK 771, which has commenced Phase II human clinical trials.
WCK 771 is a broad-spectrum antibiotic, which has proven effective in treating diverse staphylococcal infections like MRSA and VISA.
Other lead molecules at various stages of pre-clinical trials are: WCK 2349, WCK 4873 and WCK 4086.
http://www.wockhardt.com/how-we-touch-lives/new-drug-discover.aspx


///////
Wockhardt, WO 2015136473, sodium (2S, 5R)-6-(benzyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octane-2-carboxylate

WO-2015136473
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015136473&redirectedID=true
WOCKHARDT LIMITED [IN/IN]; D-4, MIDC Area, Chikalthana, Aurangabad 431006 (IN)
Our New Drug Discovery team has developed a number of lead molecules, mainly in the area of anti-infectives; these are currently at various stages of development.
Of these molecules, the most advanced of the New Chemical Entities (NCE) is WCK 771, which has commenced Phase II human clinical trials.
WCK 771 is a broad-spectrum antibiotic, which has proven effective in treating diverse staphylococcal infections like MRSA and VISA.
Other lead molecules at various stages of pre-clinical trials are: WCK 2349, WCK 4873 and WCK 4086.
http://www.wockhardt.com/how-we-touch-lives/new-drug-discover.aspx
WO-2015136473
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015136473&redirectedID=true
Process for the synthesis of sodium (2S, 5R)-6-(benzyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octane-2-carboxylate (disclosed in WO2014135929) is claimed. Used as an intermediate in the synthesis of several antibacterial compounds. For a concurrent filing see WO2015136387, claiming the combination of an antibacterial agent with sulbactam.
In September 2015, Wockhardt’s pipeline lists several antibacterial programs, including WCK-771 and WCK-2349 (both in phase II), WCK-5107 (phase I), and also investigating iv and oral second generation oxazolidinones, WCK-4873, and iv and oral formulation of WCK-4086 (in preclinical stage) for treating the bacterial infection.
For a prior filing see WO2015125031, claiming the combination of an antibacterial agent (eg cefepime or cefpirome) and nitrogen containing bicyclic compound, useful for treating bacterial infection.
A compound of Formula (I), chemically known as sodium (25, 5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylate, can be used as an intermediate in the synthesis of several antibacterial compounds and is disclosed in PCT International Patent Application No. PCT/IB2013/059264. The present invention discloses a process for preparation of a compound of Formula (I).

Scheme 1
Example 1
Synthesis of sodium (25, 5R)-6-(benzyloxy)-7-oxo-l,6-diazabicvclor3.2.11octane-2- carboxylate
Step 1; Preparation of -Γl-Γ(feΓt-butyldimethylsilyl -oxymethyll-5-Γdimethyl(oxido -λ-4-sulfanylidenel-4-oxo-pentyll-carbamic acid tert-butyl ester (III):
To a suspension of trimethylsulfoxonium iodide (180.36 gm, 0.819 mol) in tetrahydrofuran (900 ml), sodium hydride (32.89 g, 0.819 mol, 60% in mineral oil) was charged in one portion at 30°C temperature. The reaction mixture was stirred for 15 minutes and then dropwise addition of dimethylsulphoxide (1.125 ml) was done over a period of 3 hours at room temperature to provide a white suspension. The white suspension was added to a pre-cooled a solution of 2-(feri-butyldimethylsilyl-oxymethyl)-5-oxo-pyrrolidine-l-carboxylic acid tert-buty\ ester (II) (225 g, 0.683 mol, prepared as per J. Org Chem.; 2011, 76, 5574 and WO2009067600) in tetrahydrofuran (675 ml) and triethylamine (123.48 ml, 0.887 mol) mixture at -13°C by maintaining the reaction mixture temperature below -10°C. The resulting suspension was stirred for additional 1 hour at -10°C. The reaction mixture was carefully quenched by addition of saturated aqueous ammonium chloride (1.0 L) at -10°C to 10°C. The reaction was extracted by adding ethyl acetate (1.5 L). The layers were separated and aqueous layer was re-extracted with ethyl acetate (500 ml x 3). The combined organic layer was washed successively with saturated aqueous sodium bicarbonate (1.0 L), water (2.0 L) followed by saturated aqueous sodium chloride solution (1.0 L). Organic layer was dried over sodium sulfate and evaporated under vacuum to provide 265 g of 5-[l-[(ieri-butyldimethylsilyl)-oxymethyl]-5-[dimethyl(oxido)- -4-sulfanylidene]-4-oxo-pentyl]-carbamic acid tert-buty\ ester (III) as an yellow oily mass.
Analysis:
Mass: 422.3 (M+l); for Molecular weight: 421.68 and Molecular Formula: ![]()
1H NMR (CDC13): δ 4.77 (br d, 1H), 4.38 (br s, 1H), 3.58 (br s, 3H), 3.39 (s, 3H), 3.38 (s, 3H), 2.17-2.27 (m, 2H), 1.73-1.82 (m, 2H), 1.43 (s, 9H), 0.88 (s, 9H), 0.01 (s, 3H), 0.04 (s, 3H).
Step 2: Preparation of 5-r4-benzyloxyimino-l-(fert-butyldimethylsilyl-oxymethyl)-5-chloro-pentyll-carbamic acid tert- butyl ester (IV):
To a suspension of 5-[l-[(ieri-butyldimethylsilyl)-oxymethyl]-5-[dimethyl(oxido)- -4-sulfanylidene]-4-oxo-pentyl]-carbamic acid tert-butyl ester (III) (440.0 g, 1.045 mol) in tetrahydrofuran (6.6 L), O-benzhydroxylamine hydrochloride (200.0 g, 1.254 mol) was charged. The reaction mixture was heated to 50°C for 2.5 hours. The reaction mixture was filtered through pad of celite and filtrate was concentrated to provide a residue. The residue was dissolved in ethyl acetate (5.0 L) and washed successively with saturated aqueous sodium bicarbonate (1.5 L), water (1.5 L) and saturated aqueous sodium chloride (1.5 L). Organic layer was dried over sodium sulfate. Solvent was evaporated under vacuum to yield 463.0 g of 5-[4-benzyloxyimino-l-(tert-butyldimethylsilyl-oxymethyl)-5-chloro-pentyl]-carbamic acid tert-butyl ester (IV) as an oily mass.
Analysis:
Mass: 486.1 (M+l); for Molecular weight: 485.4 and Molecular Formula: ![]()
1H NMR (CDCI3): δ 7.26-1 6 (m, 5H), 5.10 (s, 2H), 4.66 (br d, 1H), 3.58-4.27 (m, 2H), 3.56-3.58 (m, 3H), 2.40-2.57 (m, 2H), 1.68-1.89 (m, 2H), 1.44 (s, 9H), 0.89 (s, 9H), 0.02 (s, 3H), 0.04 (s, 3H).
Step 3: Preparation of 5-5-benzyloxyimino-2-(fert-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (V):
To a solution of 5-[4-benzyloxyimino-l-(tert-butyldimethylsilyl-oxymethyl)-5-chloro-pentyl]-carbamic acid tert-butyl ester (IV) (463.0 g 0.954 mol) in tetrahydrofuran (6.9 L), was charged potassium feri-butoxide (139.2 g, 1.241 mol) in portions over a period of 30 minutes by maintaining temperature -10°C. The resulting suspension was stirred for additional 1.5 hours at -10°C to -5°C. The reaction mixture was quenched by addition of saturated aqueous ammonium chloride (2.0 L) at -5°C to 10°C. The organic layer was separated and aqueous layer was extracted with ethyl acetate (1.0 L x 2). The combined organic layer was washed with saturated aqueous sodium chloride solution (2.0 L). Organic layer was dried over sodium sulfate, and then evaporated under vacuum to yield 394.0 g of 5-5-benzyloxyimino-2-(ieri-butyldimethylsilyl-oxymethyl)-piperidine- 1 -carboxylic acid tert-butyl ester (V) as an yellow oily mass.
Analysis:
Mass: 449.4 (M+l) for Molecular weight: 448.68 and Molecular Formula: C24H4oN204Si;
1H NMR (CDC13): δ 7.25-1 3 (m, 5H), 5.04-5.14 (m, 2H), 4.35 (br s, 1H), 3.95 (br s, 1H), 3.63-3.74 (br d, 2H), 3.60-3.63 (m, 1H), 2.70-2.77 (m, 1H), 2.33-2.41 (m, 1H), 1.79-1.95 (m, 2H), 1.44 (s, 9H), 0.88 (s, 9H), 0.03 (s, 3H), 0.04 (s, 3H).
Step 4: Preparation of (25,5R5)-5-benzyloxyamino-2-(tert-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (VI):
To a solution of 5-5-benzyloxyimino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (V) (394.0 g, 0.879 mol) in dichloromethane (5.0 L) and glacial acetic acid (788 ml), was charged sodium cyanoborohydride (70.88 g, 1.14 mol) one portion. The resulting reaction mixture was stirred at temperature of about 25 °C to 30°C for 2 hours. The mixture was quenched with adding aqueous solution of sodium bicarbonate (1.3 kg) in water (5.0 L). The organic layer was separated and aqueous layer was extracted with dichloromethane (2.0 L). The combined organic layer washed successively with water (2.0 L), saturated aqueous
sodium chloride (2.0 L) and dried over sodium sulfate. Solvent was evaporated under vacuum to provide a residue. The residue was purified by silica gel column chromatography to yield 208 g of (25,5i?5)-5-benzyloxyamino-2-(ieri-butyldimethylsilyl-oxymethyl)-piperidine- 1 -carboxylic acid tert-buty\ ester (VI) as pale yellow liquid.
Analysis:
Mass: 451.4 (M+l); for Molecular weight: 450.70 and Molecular Formula: C24H42N204Si;
1H NMR (CDC13): δ 7..26-7.36 (m, 5H), 4.90-5.50 (br s, 1H), 4.70 (dd, 2H), 4.09-4.25 (m, 2H), 3.56-3.72 (m, 2H), 2.55-3.14 (m, 2H), 1.21-1.94 (m, 4H), 1.45 (s, 9H), 0.89 (s, 9H), 0.05 (s, 6H).
Step 5: Preparation of (25,5R5)-5-benzyloxyamino-2-(tert-butyldimethylsilyl-oxymethyl)-piperidine (VII):
To a solution of 5-5-benzyloxyamino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (VI) (208 g, 0.462 mol) in dichloromethane (3.0 L), boron trifluoride diethyletherate complex (114.15 ml, 0.924 mol) was charged in one portion. The resulting reaction mixture was stirred at temperature of about 25°C to 35°C temperature for 2 hours. The reaction mixture was quenched with saturated aqueous sodium bicarbonate (2.0 L). The organic layer was separated and aqueous layer was extracted with dichloromethane (1.5 L x 2). The combined organic layer was washed with saturated aqueous sodium chloride (1.0 L) and dried over sodium sulfate. Solvent was evaporated under vacuum to yield 159 g of (25,5i?5)-5-benzyloxyamino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine (VII) as a yellowish syrup.
Analysis:
Mass: 351.3 (M+l); for Molecular weight: 350.58 and Molecular Formula: C19H34N202Si.
Step-6: Preparation of (25,5R)-6-benzyloxy-2-(fert-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicyclo-r3.2.11octane (VIII):
Part 1; Preparation of (2S,5RS)-6-benzyloxy-2-(fert-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicvclo-r3.2.11octane:
To a solution of (25,5i?5)-5-benzyloxyamino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine (VII) (159.0 g, 0.454 mol) in a mixture of acetonitrile (2.38 L) and diisopropylethylamine (316.5 ml, 1.81 mol) was added triphosgene (59.27 gm, 0.199 mol) dissolved in acetonitrile (760 ml) at -15°C over 30 minutes under stirring. The resulting reaction mixture was stirred for additional 1 hour at -10°C. The reaction mixture was quenched by addition of saturated aqueous sodium bicarbonate (2.0 L) at -5°C to 10°C. Acetonitrile was evaporated from the reaction mixture under vacuum and to the left over aqueous phase, dichloromethane (2.5 L) was added. The organic layer was separated and aqueous layer extracted with dichloromethane (1.5 L x 2). The combined organic layer was washed successively with water (2.0 L), saturated aqueous sodium chloride (2.0 L) and dried over sodium sulfate. Solvent was evaporated under vacuum and the residue was passed through a silica gel bed to yield 83.0 g of diastereomeric mixture (25, 5i?5)-6-benzyloxy-2-(feri-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicyclo-[3.2.1]octane in 50:50 ratio as a yellow liquid.
Part-2: Separation of diastereomers to prepare (25,5R)-6-benzyloxy-2-(fert-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicvclo-r3.2.11octane:
A mixture of diastereomers (2S,5Z?S)-6-benzyloxy-2-(teri-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicyclo-[3.2.1]octane in 50:50 ratio (47.0 gm, 0.125 mol), was dissolved in n-hexane (141 ml) and stirred at temperature of about 10°C to 15°C for 1 hour. Precipitated solid was filtered and washed with n-hexane (47 ml) to provide 12.0 g of diastereomerically pure (25,5i?)-6-benzyloxy-2-(tert-butyl-dimethylsilyl-oxymethyl)-7-oxo- 1,6-diaza-bicyclo-[3.2.1] octane (VIII) as a white crystalline material.
Analysis:
Mass: 377.3 (M+l); for Molecular weight: 376.58 and Molecular Formula: ![]()
1H NMR (CDCI3): δ Ί -Ί.ΑΑ (m, 5H), 4.95 (dd, 2H), 3.76-3.85 (ddd, 2H), 3.37-3.40 (m, 1H), 3.28-3.31 (m, 2H), 2.89 (brd, 1H), 1.90-2.02 (m, 2H), 1.62- 1.74 (m, 2H), 1.56 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H).
Diastereomeric purity as determined by HPLC: 99.85%
Step-7: Preparation of (25,5R)-6-benzyloxy-2-hvdroxymethyl)-7-oxo-l,6-diaza-bicvclo-r3.2.11octane (IX):
To a solution of (25,5i?)-6-benzyloxy-2-(ieri-butyl-dimethylsilyl-oxymethyl)-7-oxo- l,6-diaza-bicyclo-[3.2.1]octane (VIII) ( 12.0 g, 31.9 rnmol) in tetrahydrofuran (180 ml) was charged tetra 7? -butyl ammonium fluoride (38.0 ml, 38 mmol, 1 M in tetrahydrofuran) at room temperature. The reaction mixture was stirred for 2 hours. It was quenched with saturated aqueous ammonium chloride ( 100 ml). The organic layer was separated and aqueous layer extracted with dichloromethane (150 ml x 3). The combined organic layer was washed with saturated aqueous sodium chloride (150 ml), dried over sodium sulfate and evaporated under vacuum to yield 24.0 g of (25,5i?)-6-benzyloxy-2-hydroxymethyl)-7-oxo-l ,6-diaza-bicyclo-[3.2.1]octane (IX) as a yellow liquid. The compound of Formula (IX) was purified by silica gel (60-120 mesh) column chromatography using a mixture of ethyl acetate and hexane as an eluent.
Analysis:
Mass: 263.1 (M+l); for Molecular weight: 262.31 and Molecular Formula: C14H18N203
1H NMR (CDCb): δ 7.34-7.42 (m, 5H), 4.95 (dd, 2H), 3.67-3.73 (m, 1H), 3.53-3.60 (m, 2H), 3.32-3.34 (m, 1H), 2.88-3.01 (m, 2H), 2.09 (brs, 1H), 1.57-2.03 (m, 2H), 1.53- 1.57 (m, 1H), 1.37- 1.40 (m, 1H).
Step 8: Preparation of sodium salt of (25, 5R)-6-benzyloxy-7-oxo-l,6-diaza-bicvclor3.2.11-octane-2-carboxylic acid (I):
Step I:
Compound of Formula (IX) obtained in step 8 above was used without any further purification. To the clear solution of (25,5i?)-6-benzyloxy-2-hydroxymethyl)-7-oxo-l,6-diaza-bicyclo-[3.2.1]octane (IX) (24.0 g, 31.8 mmol) (quantities added based upon theoretical basis i.e 8.3 g ) in dichloromethane (160 ml), was added Dess-Martin reagent (24.1 g, 57.24 mmol) in portions over 15 minutes. The resulting suspension was stirred for 2 hours at 25°C. The reaction was quenched by adding a solution, prepared from saturated aqueous sodium hydrogen carbonate solution (160 ml) and 72.0 g of sodium thiosulfate. Diethyl ether (160 ml) was added to the reaction mixture and it was stirred for 5-10 minutes and filtered through celite. Biphasic layer from filtrate was separated. Organic layer was washed with saturated aqueous sodium hydrogen carbonate solution (160 ml) followed by saturated aqueous sodium chloride solution (160 ml). Organic layer was dried over sodium sulfate and evaporated to dryness at 30°C to obtain 20.0 g of intermediate aldehyde, which was used immediately for the next reaction.
Step II:
To the crude intermediate aldehyde (20.0 g, 31.6 mmol) (quantities added based upon theoretical yield i.e. 8.2 g) obtained as above, was charged i-butyl alcohol (160 ml) and cyclohexene (10.8 ml, 110.6 mmol). The reaction mixture was cooled to temperature of about 10°C to 15°C. To this mixture was added clear solution prepared from sodium hypophosphate (14.8 g, 94.8 mmol) and sodium chlorite (5.7 g, 63.2 mmol) in water (82.0 ml) over a period of 30 minutes by maintaining temperature between 10°C to 15°C. The reaction mixture was further stirred for 1 hour and was quenched with saturated aqueous ammonium chloride solution. The reaction mixture was subjected to evaporation under vacuum at 40°C to remove i-butyl alcohol. Resulting mixture was extracted with dichloromethane (3 x 150 ml). Layers were separated. Combined organic layer was washed with aqueous brine solution, dried over sodium sulfate and evaporated to dryness under vacuum to obtain 16.0 g of crude residue. To this residue was added acetone (83 ml) to provide a clear solution and to it was added dropwise a solution of sodium 2-ethyl hexanoate (4.5 g) in acetone (24 ml). The reaction mixture was stirred for 15 hours at 25°C to 30°C to provide a suspension. To the suspension was added diethyl ether (215 ml) and stirred for 30 minutes. Resulting solid was filtered over suction, and wet cake was washed with cold acetone (83 ml) followed by diethyl ether (83 ml). The solid was dried under vacuum at 40°C to provide 3.6 g of off-white colored, non-hygroscopic sodium salt of (25, 5i?)-6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]-octane-2-carboxylic acid (I).
Analysis:
Mass: 275.2 as M-1 (for free acid) for Molecular Weight: 298 and Molecular Formula: ![]()
NMR (DMSO-d6): δ 7.43-7.32 (m, 5H), 4.88 (q, 2H), 3.48 (s, IH), 3.21 (d, IH), 2.73 (d, IH), 2.04-2.09 (m, IH), 1.77-1.74 (m, IH), 1.65-1.72 (m, IH), 1.55-1.59 (m, IH);
Purity as determined by HPLC: 97.47%;
[a]D25: -42.34° (c 0.5, water).
///////
