




























| Patent | Submitted | Granted |
|---|---|---|
| NORMALIZATION OF CULTURE OF CORNEAL ENDOTHELIAL CELLS [US2015044178] | 2012-12-27 | 2015-02-12 |
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 181)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA approves first drug to treat a rare enzyme disorder in pediatric and adult patients
![]()
Sebelipase alfa

CAS No. 1276027-63-4
Synageva… innovator
ALEXION
EMA AUG 28 2015
12/08/2015
Today, the U.S. Food and Drug Administration approved Kanuma (sebelipase alfa) as the first treatment for patients with a rare disease known as lysosomal acid lipase (LAL) deficiency.
December 8, 2015
Release
Today, the U.S. Food and Drug Administration approved Kanuma (sebelipase alfa) as the first treatment for patients with a rare disease known as lysosomal acid lipase (LAL) deficiency.
Patients with LAL deficiency (also known as Wolman disease and cholesteryl ester storage disease [CESD]) have no or little LAL enzyme activity. This results in a build-up of fats within the cells of various tissues that can lead to liver and cardiovascular disease and other complications. Wolman disease often presents during infancy (around 2 to 4 months of age) and is a rapidly progressive disease. Patients with Wolman disease rarely survive beyond the first year of life. CESD is a milder, later-onset form of LAL deficiency and presents in early childhood or later. Life expectancy of patients with CESD depends on the severity of the disease and associated complications. Wolman disease affects one to two infants per million births, and CESD affects 25 individuals per million births.
Today’s action involved approvals from two FDA centers. The Center for Veterinary Medicine (CVM) approved an application for a recombinant DNA (rDNA) construct in chickens that are genetically engineered (GE) to produce a recombinant form of human lysosomal acid lipase (rhLAL) protein in their egg whites. The FDA regulates GE animals under the new animal drug provisions of the Federal Food, Drug, and Cosmetic Act, because an rDNA construct introduced into an animal to change its structure or function meets the definition of a drug. The Center for Drug Evaluation and Research (CDER) approved the human therapeutic biologic (Kanuma), which is purified from those egg whites, based on its safety and efficacy in humans with LAL deficiency.
“LAL deficiency is a rare inherited genetic disorder that can lead to serious and life-threatening organ damage, especially when onset begins in infancy,” said CDER Director Janet Woodcock, M.D. “Using this technology, these patients for the first time ever have access to a treatment that may improve their lives and chances of survival.”
The new therapy, Kanuma, provides an rhLAL protein that functions in place of the missing, partially active or inactive LAL protein in the patient. Kanuma is produced by GE chickens containing an rDNA construct responsible for producing rhLAL protein in their egg whites. These egg whites are refined to extract the rhLAL protein that is eventually used to produce Kanuma and treat patients with LAL deficiency. The GE chickens are used only for producing the drug substance, and neither the chicken nor the eggs are allowed in the food supply.
Kanuma is approved for use in patients with LAL deficiency. Treatment is provided via intravenous infusion once weekly in patients with rapidly progressive LAL deficiency presenting in the first six months of life, and once every other week in all other patients.
CDER evaluated the safety and efficacy of Kanuma in an open-label, historically controlled trial in nine infants with rapidly progressive Wolman disease and in a double-blind, placebo-controlled trial in 66 pediatric and adult patients with CESD. In the trial in infants with Wolman disease, six of nine infants (67 percent) treated with Kanuma were alive at 12 months of age, whereas none of the 21 infants in the historical control group survived. In the trial in CESD patients, there was a statistically significant improvement in LDL-cholesterol levels and other disease-related parameters in those treated with Kanuma versus placebo after 20 weeks of treatment.
The most common side effects observed in patients treated with Kanuma are diarrhea, vomiting, fever, rhinitis, anemia, cough, headache, constipation, and nausea.
In its review of the GE chicken application, CVM assessed the safety of the rDNA construct, including the safety of the rDNA construct to the animals, as well as a full review of the construct and its stability in the genome of the chicken over several generations. No adverse outcomes were noted in the chickens. As required by the National Environmental Policy Act and its implementing regulations, CVM evaluated the potential environmental impacts of approval of the sponsor’s GE chickens and determined that the approval does not cause any significant impact on the environment, because the chickens are raised in highly secure indoor facilities.
“We reviewed all of the data to ensure that the hens do produce rhLAL in their egg whites, without suffering any adverse health effects from the introduced rDNA construct. The company has taken rigorous steps to ensure that neither the chickens nor the eggs will enter the food supply, and we have confirmed their containment systems by inspecting the manufacturing facilities,” said CVM Director Bernadette Dunham, D.V.M., Ph.D.
The FDA granted Kanuma orphan drug designation because it treats a rare disease affecting fewer than 200,000 patients in the United States. Orphan drug designation provides financial incentives for rare disease drug development such as clinical trial tax credits, user fee waivers, and eligibility for market exclusivity to promote rare disease drug development. Kanuma was also granted breakthrough therapy designation as it is the first and only treatment available for Wolman disease, the very severe infant form of the disease. The breakthrough therapy designation program encourages the FDA to work collaboratively with sponsors, by providing timely advice and interactive communications, to help expedite the development and review of important new drugs for serious or life-threatening conditions. The Kanuma application was also granted a priority review, which is granted to drug applications that show a significant improvement in safety or effectiveness in the treatment of a serious condition. The manufacturer of Kanuma was granted a rare pediatric disease priority review voucher –– a provision intended to encourage development of new drugs and biologics for the prevention and treatment of rare pediatric diseases.
Kanuma is produced by Alexion Pharmaceuticals Inc., based in Cheshire, Connecticut.


///////// Kanuma, sebelipase alfa, rare disease, lysosomal acid lipase (LAL) deficiency,
WCK ? trans-7-oxo-6-(sulphoxy)-1,6-diazabicvclo[3.2.1]-octane-2- carbonitrile from Wockhardt
 .
.
WCK ?
WATCH OUT FOR THIS POST, THIS MAY BE WCK 4234
Cas 1427462-70-1, 1706523-58-1
| Molecular Formula: | C7H9N3O5S |
|---|---|
| Molecular Weight: | 247.22846 g/mol |
Sulfuric acid, mono[(1R,2S,5R)-2-cyano-7-oxo-1,6-diazabicyclo[3.2.1]oct-6-yl] ester
[(2S,5R)-2-cyano-7-oxo-1,6-diazabicyclo[3.2.1]octan-6-yl] hydrogen sulfate
CAS 1427462-59-6, 1804915-68-1, SODIUM SALT, (2S, 5R)-1,6-DIAZA-BICYCLO [3.2.1]OCTANE-2-CARBONITRILE-7-OXO-6-(SULFOOXY)-MONO SODIUM SALT
1408/MUM/2014 and 1407/MUM/2014 INDIAN PATENT, WO2013038330
trans-7-oxo-6-(sulphooxy)-l,6-diazabicyclo[3.2.1]octane-2-carbonitrile
(2S, 5R)-7-oxo-6-(sulphooxy)-l,6-diazabicyclo [3.2.1]octane-2-carbonitrile
sulphuric acid, mono[(1R,2S,5R)-2-cyano-7-oxo-l,6-diazabicyclo[3.2.1]oct-6-yl] ester
mono[(1R,2S,5R)-2-cyano-7-oxo-1,6- diazabicyclo[3.2.1]oct-6-yl] ester,
trans-7-oxo-6-(sulphoxy)-l,6-diazabicvclo[3.2.1]-octane-2- carbonitrile
Sodium salt (also known as “sodium salt of sulphuric acid, mono[(li?,25,5i?)-2-cyano-7-oxo-l,6- diazabicyclo[3.2.1]oct-6-yl] ester” or “sulphuric acid, mono[(lR,25,5R)-2-cyano-7-oxo-l,6- diazabicyclo[3.2.1]oct-6-yl] ester, sodium salt (1: 1); CAS Registry Number: 1427462-59-6”); CAS 1804915-68-1

(2S, 5R)-1,6-DIAZA-BICYCLO [3.2.1]OCTANE-2-CARBONITRILE-7-OXO-6-(SULFOOXY)-MONO SODIUM SALT
Potassium salt (also known as “potassium salt of sulphuric acid, mono[(li?,25,5i?)-2-cyano-7-oxo- l,6-diazabicyclo[3.2.1]oct-6-yl] ester” or “sulphuric acid, mono[(lR,25,5R)-2-cyano-7-oxo-l,6- diazabicyclo[3.2.1]oct-6-yl] ester, potassium salt (1: 1); CAS Registry Number: 1427462-60-9”); CAS 1804915-69-2
And
Other salts such as “l-butanarninium, Ν,Ν,Ν-tributyl-, (lR,25,5R)-2-cyano-7-oxo-l,6- diazabicyclo[3.2.1]oct-6-yl sulphate (1: 1); CAS Registry Number: 1427462-72-3”.
PATENT
http://google.com/patents/WO2013038330A1?cl=en
Scheme 1

l( M = Na) a: Base,water, RT;b:Boc-anhydride,TEA,DIV1AP, DCIv1 , RT; c:LiOH, acetone; d: PivaloyI chloride, TEA; e. Ammonia(g); f:Trifluoroacetic anhydride,TEA,DC g: TFA, DC ; h: Triphosgene,TEA, D AP, DCM; i:H2, Pd/C; j:S03-DIVlF;
k: Tetrabutyl ammonium acetate, DCM; I: Dowex 50WX8 200 Na+ resin Scheme 2

a: Water, reflux, 24h; b:1-Hydroxybenzotriazole ammonium salt, DCC,D F; c: Boc-anhydride,TEA,D AP,DC ,RT; d:Trifluoroacetic anhydride,TEA, DCM;
e:TMSOI, NaH,DMSO,THF, -10°C 1 hr; f: O-Benzyl hydroxyl amine.HCI, EtOAc 60°C,2.5hr; g: Methane sulphonic acid, ethyl acetate,40°C; h:.KHC03, water, 55 °C;
i: sodium triacetoxy borohydride, STABH, H2S04; j: Triphosgene,TEA,DMAP,DCM;
Scheme-1 : further steps as depicted in scheme-1 Scheme 3

IX
: Water, reflux, 24h; b:1 -Hydroxybenzotriazole ammonium salt, DCC,D F;
: Boc-anhydride,TEA,D AP, DC ,rt; d:T SOI, NaH, D SO,THF, -1 0 °C 1 hr;
: O-Benzyl hydroxyl amine.HCI, EtOAc 60 °C, 2.5hr; f: Methane sulphonic acid, ethyl acetate, 40 °C g:.KHC03, water, 55 °C; g: sodium triacetoxy borohydride,
STABH, H2S04; h: Triphosgene,TEA,DMAP,DCIvl; i: Trifluoroacetic anhydride,
TEA, DCM; Scheme-1 : further steps as depicted in scheme-1
Step 1: Preparation of freebase and – Boc protection

The oxalate salt II (30g, 0.0697moles) was partitioned between water (300ml), and ethyl acetate (300ml) followed by addition of sodium bicarbonate (11.7gm, 0.139moles) under stirring. After lhr the organic layer was separated and the aqueous layer was extracted with ethyl acetate (150ml). The combined organic layer was washed with water (150ml) then brine (150ml), dried (over Na2S04) and the solvent evaporated under reduced pressure to obtain the free base Ila, 24gm.
To a cooled (5-10°C solution of the free base (24g, 0.0705moles) in DCM (240ml) were added triethylamine (19.68ml, 0.141moles), Boc anhydride (17.8ml, 0.0775moles) under stirring. After 30min. was added DMAP (0.86gm, 0.00705moles) and the resulting solution was allowed to warm to room temperature and stirred for a further 16hrs. The reaction mixture was diluted with saturated aqueous ammonium chloride solution (10ml), stirred well and the DCM layer was separated, washed with water (10ml) and finally with brine (10ml). The solvent was evaporated under reduced pressure and the residue chromatographed on a column of silica gel (60-120 mesh). Elution with mixtures of ethyl acetate: hexane 25-50% and concentration of the combined fractions gave the product as a colorless oil, 25gm(yield: 80%).
MS: 439 [M+]; MF: C26H33NO5; MW: 439.
Step 2: Hydrolysis of Benzyl ester ^S | LiOH.Acetone Bn0 HN / ^-
N’^COOBn L JL
J N COOH X
To a solution of the compound lib (25gm, 0.0567moles) in acetone (500ml), at 0 °C, was added lithium hydroxide solution (3.8 lgm, 0.0908moles in mixture of 228.6ml water and 76.2 ml acetone) drop-wise under vigorous stirring. The reaction mixture was allowed to warm to RT and stirring continued further for 5hrs. The resulting mixture was cooled to 0 °C and pH adjusted to 8 to 8.5 with 2N HC1 (~10ml). The reaction mixture was diluted with brine (75ml) and toluene (250ml) under stirring, and after 10 minutes the organic layer was separated. The aqueous layer was re-extracted with toluene (2 X 120ml). The aqueous layer was acidified to pH 3-4 by using 2N HC1 and the solution extracted with ethyl acetate (3X200ml).,The combined organic layer was washed with water (200ml), and brine (200ml), dried (over Na2S04)and the solvent evaporated under reduced pressure to obtain the product as a thick oil, 21g, (quantitative yield).
MS: 349(M+); MF: C19H27NO5; MW: 349
Step 3: Conversion of Acid to Amide

IV V
To a stirred solution of compound IV (21gm, 0.06moles) in DCM (210ml) at 0°C was added TEA (25.12ml, 0.18moles) followed by slow addition of Pivaloyl chloride (11.07ml, 0.09moles). The resulting mixture was stirred further for 1.5hrs. The reaction mixture was cooled to -40°C and dry ammonia gas was bubbled through the reaction mixture for 30 min. The reaction mixture was allowed to warm to RT and the suspended white solid was filtered off. The solvent was evaporated under reduced pressure and the residue chromatographed on a column of silica gel (60-120 mesh). Elution with a mixture of acetone: hexane system (1 :4) and concentration of the combined solvents gave the product, as thick oil, 10.2gm (yield: 49%)
MS: 348[M+] ; MF: C19H28N2O4; MW: 348.
Step 4: Conversion of Amide to Cyano

To a cooled (0°C) and stirred solution of compound VI (10.2gm, 0.0286moles) in DCM (306ml) was added Triethylamine (17.99ml, 1.289moles) and followed by the slow addition of Trifluoro acetic anhydride (12.08gm, 0.0573moles). The resulting solution was allowed to warm to RT and stirred for a further 6h. The reaction mixture was washed water (3* 100ml), Saturated ammonium chloride solution (100ml) and brine (100ml). The organic layer was dried (Na2S04) and the solvent evaporated under reduced pressure. The residue was chromatographed on a column of silica gel (60-120 mesh) using a mixture of Acetone: Hexane (1: 19). Concentration of the combined fractions gave the product, as a white solid, 9.7gm (yield – quantitative). MS: 331(M+); MF: C18H25N3O3; MW: 331
Step 5: Deprotection of Cyano

VI VII
To a chilled (-15°C) and stirred solution of compound VII (6gm,) in DCM (150ml) was added Trifluoro acetic acid (12ml) and the mixture was allowed to warm to RT. The reaction mixture was stirred for a further 4hrs. The solvent was evaporated under reduced pressure at 40± 5°C and the residue diluted with aqueous sat. sodium bicarbonate solution (60ml) and the mixture extracted with DCM (2 X 60ml). The combined extracts were washed with water (60ml), dried (over sodium sulphate) and evaporated under reduced pressure at 35± 5°C to obtain 4.2gm of compound VIII.
Step 6: Formation of bi-cyclic compound

To the cooled (0- 5°C) and stirred solution of compound VIII (4.2gm) in acetonitrile (63ml) was added triethyl amine (5.28ml) followed by a slow addition of a solution of Triphosgene (1.9gm) in Acetonitrile (16.8ml). Stirring was further continued for 30min. followed by addition of Dimethyl amino pyridine (0.178gm). The reaction mixture was allowed to warm to RT and stirred for further 16hrs. A aqueous sat. solution of sodium bicarbonate (33.6ml) was added to the reaction mixture and the resulting mixture stirred for 30min. The mixture was concentrated to l/3rd volume under reduced pressure. The residue was diluted with water (42ml) and the resulting mixture extracted with DCM (2 X 42ml). The solvent was evaporated under reduced pressure and the residue purified over a column of silica-gel (60 -120 mesh). Elution with a 1 :4 mixture of acetone: hexane and concentration of the combined fractions gave the product as white solid, 2.3g (yield: 48%).
MS: 314(M+); MF; Ci6Hi8N403; MW; 314 Step 7: Synthesis of TBA sulfate salt

To a solution of benzyl compound VIII (6 gm, 0.0233 mol) in a 1 : 1 mixture of DCM (30 ml)& DMF (30 ml), was added 1.5 gm of dry 10% Palladium charcoal and the mixture was hydrogenated under 3 kg Hydrogen pressure for 3 hour at 25-30°C.The reaction mixture was filtered through micron filter to remove catalyst and the filtrate concentrated under reduced pressure to obtain the debenzylated compound IX.
The debenzylated compound (IX) was dissolved in Ν,Ν’ -Dimethyl formamide (30 ml) under argon atmosphere and the solution cooled to 0°C. DMF: SO3 (4.26 gm, 0.0278mol) was added to the cooled solution and the stirring continued further for 30 min at 0°C. The mixture was then allowed to warm to RT and stirred for 1 hour. TLC showed complete conversion of N-Hydroxy compound to product X.
The solution containing the sulfate(X) was re-cooled to 0°C and a solution of Tetra butyl ammonium acetate (9 gm, 0.0301mol dissolved in 30ml water) was added to it. The reaction mixture was allowed to warm to 25°C and stirred for 1 hour. The volatiles were removed under reduced pressure and residue was co-evaporated with 2×50 ml Xylene to remove traces of Ν,Ν’ -Dimethyl formamide. The residue was partitioned between a 1: 1 mixture of water and dichloromethane (120ml). The aqueous layer was re-extracted with dichloromethane (30 ml). The combined organic extracts were washed with water (2x30ml), brine (30 ml). And dried over Na2S04 and the solvent evaporated under reduced pressure to obtain the crude TBA sulfate (5.2 gm). Crude compound was triturated with hexane (2×30 ml) & dried on rotavapor under 4mmHg pressure to obtain the TBA salt (XI), 5.0 g, yield-
44%.
Mass: 246 (M-H) of sulfate M.W: 488, M.F: C23H44N4O5S.
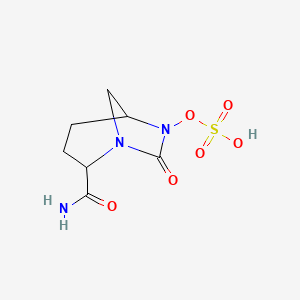
Step 8: Synthesis of Sodium salt of trans-7-oxo-6-(sulphoxy)-l,6-diazabicyclo[3.2.1]- octane-2-carbonitrile I

XI The TBA sulfate (4.4g, 0.009mol) was dissolved in 5% THF in water (2ml) and the solution was passed through column (45cm length and 2.0cm diameter) packed with Dowex 50WX8 200 Na+ resin. The column was eluted with 5% THF-water mixture (100ml). The combined fractions were evaporated under reduced pressure (4 mmHg) to obtain the product as white semi-solid, 1.5 gm, yield: 62%.
MS: 246 (M-H) of sulfate; M.W.: 269; M.F.: CyHgNaOsSNa,
XH NMR (DMSO):8 4.54 (d, 1H), 4.06 (s, 1H), 3.22 (m, 2H), 1.96 (m, 2H), 1.84 (m, 2H).
PATENT
(WO2015159167) PHARMACEUTICAL COMPOSITIONS COMPRISING ANTIBACTERIAL AGENTS
http://google.com/patents/WO2015159167A1?cl=en
PATENT
(2S, 5R)-1,6-DIAZA-BICYCLO [3.2.1]OCTANE-2-CARBONITRILE-7-OXO-6-(SULFOOXY)-MONO SODIUM SALT
Patent
https://www.google.co.in/patents/WO2015114595A1?cl=en

EXAMPLES
Example 1
Synthesis of (25, 5R)-l,6-diaza-bicyclo r3.2.11octane-2-carbonitrile-7-oxo-6-(sulfooxy)- mono sodium salt
Step 1; Synthesis of (25, 5R)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (III):
Method 1:
To a stirred suspension of sodium (25,5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II) (1 g, 0.00335 mol) in dichloromethane (15 ml), triethylamine hydrochloride (0.688 g, 0.00503 mol) was added in small portions at 25°C. After 30 minutes, triethylamine (0.678g, 0.0067 moles) was added, followed by addition of pivaloyl chloride (0.605 g, 0.00502 mol) at 0-5°C under stirring. After 2 hours, the reaction mass was cooled further to -20°C and aqueous ammonia (25% solution, 0.75 ml, 0.01 mol) was added slowly. The completion of the reaction was confirmed after 30 minutes by thin layer chromatography using acetone: hexane (35:65) solvents. The reaction mixture was diluted with water (10 ml) and the mixture was allowed to warm to room temperature. The dichloromethane layer was separated and the aqueous layer was re-extracted with dichloromethane (5 ml). The combined organic layer was dried (over anhydrous sodium sulfate) and the solvent was evaporated under reduced pressure. The residue was purified by re-crystallization from n-butyl chloride to obtain 0.75 g of (25, 5i?)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (III) as an off-white solid in 81 % yield.
Analysis:
Mass: 276.1 (M+l) for Molecular Weight of 275.31 and Molecular Formula of C14H17N303;
1H NMR (400MHz, CDC13): 57.43-7.35 (m, 5H), 6.56 (brs, 1H), 5.58 (brs, 1H), 5.07-4.89 (dd, 2H), 3.95-.393 (d, 1H), 3.31 (s, 1H), 3.04-3.01 (d, 1H), 2.78-2.75 (d, 1H), 2.38-2.32 (m, 1H), 2.03-1.88 (m, 2H), 1.64-1.58(m, 1H);
Purity as determined by HPLC: 98.9%.
Method 2:
To a stirred suspension of sodium (25,5i?)-6-(benzyloxy)-7-oxo- l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II) (5 g, 0.0167 mol) in dimethylformamide (25 ml) pivaloyl chloride (3.03 g, 0.0251 mol) was added drop wise at about 0 – 5°C. After stirring for 3 hours, the resulting mixture was cooled to -20°C and aqueous ammonia (25% solution, 3.75 ml, 0.0501 mol) was added slowly under stirring. The completion of the reaction was confirmed after 30 minutes by thin layer chromatography using acetone: hexane (35:65) solvents. The reaction mixture was diluted with water (125 ml) and dichloromethane (50 ml), and allowed to warm to room temperature. The dichloromethane layer was separated and the aqueous layer extracted with fresh dichloromethane (25 ml). The combined organic layer was dried (over anhydrous sodium sulfate) and the solvent was evaporated under reduced pressure. The residue was purified by re-crystallization using n-butyl chloride to obtain 0.7 g of (25, 5i?)-6-(benzyloxy)-7-oxo-l ,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (III) as an off-white solid in 15 % yield.
Analysis:
Purity as determined by HPLC: 93.9%.
Method 3:
To a stirred suspension of sodium (25,5i?)-6-(benzyloxy)-7-oxo- l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II) (5 g, 0.0167 mol) in tetrahydrofuran (50 ml), 1-methyl-2-pyrrolidinone (7.44 g, 0.0751 mol) and pivaloyl chloride (8.0 g, 0.0668 mol) was added at about 0 – 5°C. After stirring for 3 hours the resulting mixture was cooled to -20°C and aqueous ammonia (25% solution, 6.2 ml, 0.0835 mol) was added slowly under stirring. The completion of the reaction was confirmed after 30 minutes by thin layer chromatography using acetone: hexane (35:65) solvents. The reaction mixture was diluted with water (50 ml) and allowed to warm to room temperature. The tetrahydrofuran layer was separated and the aqueous layer was extracted with dichloromethane (25 ml). The combined organic layer was dried (over anhydrous sodium sulfate) and the solvent evaporated under reduced pressure. The residue was purified by re-crystallization from n-butyl chloride to obtain 2.32 g of (25, 5R)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (III) in 50 % yield.
Analysis:
Purity as determined by HPLC: 91.6%.
Method 4:
To a stirred suspension of sodium (25,5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II) (5 g, 0.0167 mol) in tetrahydrofuran (50 ml), l-methyl-2-pyrrolidine (6.39 g, 0.0751 mol) and pivaloyl chloride (8.0 g, 0.0668 mol) was added at about 0 – 5°C. After stirring for 3 hours, the resulting mixture was cooled to -20°C and aqueous ammonia (25% solution, 6.2 ml, 0.0835 mol) was added slowly under stirring. The completion of the reaction was confirmed after 30 minutes by thin layer chromatography using acetone: hexane (35:65) solvents. The reaction mixture was diluted with water (50 ml) and allowed to warm to room temperature. The tetrahydrofuran layer was separated and the aqueous layer was extracted with dichloromethane (25 ml). The combined organic layer was dried (over anhydrous sodium sulfate) and the solvent was evaporated under reduced pressure. The residue was purified by re-crystallization from n-butyl chloride, to obtain 4.35 g of (25, 5i?)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (III) in 94% yield.
Analysis:
Purity as determined by HPLC: 97.6%.
Analytical data for (25, 5i?)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide obtained from Method 2, 3 and 4 was consistent with that obtained in Method 1.
Step 2: Synthesis of (25, 5R)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (IV):
Trifluoroacetic anhydride (48 ml, 0.340 mol) was added slowly to a solution of (25,5i?)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxamide (III) (47 g, 0.170 mol) in dichloromethane, (1430 ml) containing triethylamine (107 ml, 0.765 mol), under stirring at about -5°C. After 2 hours, the reaction mixture was diluted with water (1450 ml) and the resulting mixture was stirred for further 15 minutes. The dichloromethane layer was separated, washed with aqueous saturated sodium bicarbonate solution (470 ml), brine (470 ml), dried (over anhydrous sodium sulfate) and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel (60-120 mesh) using acetone: hexane (0-15% acetone in hexane) solvents. The combined solvent fractions were concentrated under reduced pressure to obtain 32 g of (25, 5i?)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (IV) as a white solid in 74% yield.
Analysis:
Mass: 258 (M+l) for Molecular Weight of 257 and Molecular Formula of ![]()
1H NMR (400 MHz, DMSO): δ 7.42-7.36 (m, 5H), 5.06-4.88 (dd, 2H), 4.37-4.35 (d, 1H), 3.36-3.35 (m, 1H), 3.29-3.26 (d, 1H), 3.16-3.12 (m, 1H), 2.30-2.25 (m, 1H), 2.13-2.09(m, 1H), 1.90-1.83 (m, 2H);
Purity as determined by HPLC: 100%.
Step 3: Synthesis of (25, 5R)-6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (V):
A solution of (25,5i?)-6-(benzyloxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (IV) (32 g, 0.124 mol) in a mixture of dimethylformamide and dichloromethane (1 : 1, 160 ml: 160 ml) containing 10% palladium on carbon (4.6 g, 50% wet) was hydro genated at 50-55 psi for 2 hours at 25 °C. The resulting mixture was filtered through a celite pad and residue was washed with mixture of dimethylformamide and dichloromethane (1 : 1, 25 ml: 25 ml). The solvent from the combined filtrates was evaporated under reduced pressure to obtain 20.66 g of (25, 5i?)-6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (V) as an oil. The obtained product was used as such for the next reaction without further purification.
Step 4: Synthesis of (25, 5R)-6-(sulfooxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile, tetrabutylammonium salt (VI):
To a solution of (25,5i?)-6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (20.66 g, 0.124 mol) in dimethylformamide (160 ml), sulfur trioxide dimethylformamide complex (22.8 g, 0.149 mol) was added in one portion under stirring at about -5°C. After 60 minutes of stirring, the completion of the reaction was monitored by thin layer chromatography using mixture of chloroform and methanol (9: 1). To the resulting mixture was slowly added a solution of tetrabutylammomum acetate (48.6 g, 0.161 mol) in water (160 ml). After 1 hour of stirring, the solvent was evaporated under reduced pressure to obtain an oily residue. The oily residue was co-evaporated with xylene (2 x 200 ml), to yield a thick mass. This mass was partitioned between dichloromethane (320 ml) and water (320 ml). The organic layer was separated and the aqueous layer re-extracted with dichloromethane (160 ml). The combined organic extracts were washed with water (3 x 160 ml), dried (over anhydrous sodium sulfate) and the solvent was evaporated under reduced pressure at about 35°C. The residual oily mass was triturated with ether (3 xl60 ml), each time the ether layer was decanted and finally the residue was dried under reduced pressure, to obtain 52.5 g of (25, 5i?)-6-(sulfooxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile, tetrabutyl ammonium salt (VI) as an oil in 86% yield.
Analysis:
Mass: 246 (M-l) as free sulfonic acid; for Molecular Weight of 488 and Molecular Formula of C23H44N4O5S;
1H NMR (400 MHz, CDC13): δ 4.39 (brs, 1H), 4.34-4.32 (d, 1H), 3.41-3.33 (m, 2H), 3.27-3.22 (m, 8H), 2.28 (m, 2H), 1.89-1.84 (m, 2H), 1.67-1.59 (m, 8H), 1.47-1.37 (m, 8H), 1.00-0.96 (m, 12H);
Purity as determined by HPLC: 95.24%.
Step 5: Synthesis of (25, 5R)-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile-7-oxo-6-(sulfooxy)-mono sodium salt (I):
A column loaded with activated Amber lite 200 sodium resin (1200 gm) was washed with water followed by 10% tetrahydrofuran in water. A solution of (25,5i?)-6-(sulfooxy)-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile, tetrabutylammomum salt (VI) (51.5 g, 0.105 mol) in tetrahydrofuran (50 ml) was poured over the column. The column was further eluted by using 10% tetrahydrofuran in water. Tetrahydrofuran from the combined fractions was evaporated under reduced pressure and the aqueous layer extracted with ethyl acetate (5 x 250 ml). The aqueous layer was stirred with neutral charcoal (3 g) for 1 hour and then filtered through celite bed and further washed with water (100 ml). The combined filtrate was
evaporated under reduced pressure till free of moisture, to obtain 20.5 g of (25, 5i?)-l,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile-7-oxo-6-(sulfooxy)-mono sodium salt in 72% yield.
Analysis:
Mass: 246 (M-1) as free sulfonic acid; for Molecular Weight of 269 and Molecular Formula of CvHgNsOsSNa;
1H NMR (400 MHz, DMSO): δ 4.56-4.54 (d, 1H), 4.08 (brs, 1H), 3.24-3.18 (m, 2H), 1.97-1.82 (m, 4H); and
Purity as determined by HPLC: 98.46%.
PATENT
WO 2015159265
http://google.com/patents/WO2015159265A1?cl=en
PATENT
WO 2015136387
https://www.google.co.in/patents/WO2015136387A1?cl=en
PATENT
WO 2015059642
http://www.google.com/patents/WO2015059642A1?cl=en
PATENT
http://www.google.com/patents/US20140296526
- Example 1
-

-
The oxalate salt (II) (30 gm, 0.0697 moles) was partitioned between water (300 ml), and ethyl acetate (300 ml) followed by addition of sodium bicarbonate (11.7 gm, 0.139 moles) under stirring. After 1 hour the organic layer was separated and the aqueous layer was extracted with ethyl acetate (150 ml). The combined organic layer was washed with water (150 ml) then brine (150 ml), dried (over sodium sulphate) and the solvent evaporated under reduced pressure to obtain the free base (IIa), 24 gm.
-
To a cooled (5-10° C. solution of the free base (24 gm, 0.0705 moles) in dichloromethane (240 ml) were added triethylamine (TEA) (19.68 ml, 0.141 moles), Boc anhydride ((Boc)2O) (17.8 ml, 0.0775 moles) under stiffing. After 30 minutes was added DMAP (0.86 gm, 0.00705 moles) and the resulting solution was allowed to warm to room temperature and stirred for a further 16 hours. The reaction mixture was diluted with saturated aqueous ammonium chloride solution (10 ml), stirred well and the dichloromethane layer was separated, washed with water (10 ml) and finally with brine (10 ml). The solvent was evaporated under reduced pressure and the residue chromatographed on a column of silica gel (60-120 mesh). Elution with mixtures of ethyl acetate:hexane 25-50% and concentration of the combined fractions gave the product as colorless oil, 25 gm (yield: 80%).
-
Analysis:
-
Mass: 439 [M+]; Molecular Formula: C26H33NO5; Molecular Weight: 439.
- Preparation of Sodium salt of trans-7-oxo-6-(sulphoxy)-1,6-diazabicyclo[3.2.1]-octane-2-carbonitrile IStep 1: Preparation of Freebase and -Boc Protection
Step 2: Hydrolysis of Benzyl Ester
-

-
To a solution of the compound (IIb) (25 gm, 0.0567 moles) in acetone (500 ml), at 0° C., was added lithium hydroxide solution (3.81 gm, 0.0908 moles in mixture of 228.6 ml water and 76.2 ml acetone) drop-wise under vigorous stiffing. The reaction mixture was allowed to warm to room temperature and stiffing continued further for 5 hours. The resulting mixture was cooled to 0° C. and pH adjusted to 8 to 8.5 with 2N HCl (about 10 ml). The reaction mixture was diluted with brine (75 ml) and toluene (250 ml) under stiffing, and after 10 minutes the organic layer was separated. The aqueous layer was re-extracted with toluene (2×120 ml). The aqueous layer was acidified to pH 3-4 by using 2N HCl and the solution extracted with ethyl acetate (3×200 ml). The combined organic layer was washed with water (200 ml), and brine (200 ml), dried (over sodium sulphate) and the solvent evaporated under reduced pressure to obtain the product (III) as a thick oil, 21 gm.
-
Analysis:
-
Mass: 349 (M+); Molecular Formula: C19H27NO5; Molecular Weight: 349.
Step 3: Conversion of Acid to Amide
-

-
To a stirred solution of compound (IV) (21 gm, 0.06 moles) in dichloromethane (210 ml) at 0° C. was added (triethylamine) TEA (25.12 ml, 0.18 moles) followed by slow addition of Pivaloyl chloride (11.07 ml, 0.09 moles). The resulting mixture was stirred further for 1.5 hours. The reaction mixture was cooled to −40° C. and dry ammonia gas was bubbled through the reaction mixture for 30 minutes. The reaction mixture was allowed to warm to room temperature and the suspended white solid was filtered off. The solvent was evaporated under reduced pressure and the residue chromatographed on a column of silica gel (60-120 mesh). Elution with a mixture of acetone: hexane system (1:4) and concentration of the combined solvents gave the product (V), as thick oil, 10.2 gm (yield: 49%)
-
Analysis:
-
Mass: 348[M+]; Molecular Formula: C19H28N2O4; Molecular Weight: 348.
Step 4: Conversion of Amide to Cyano
-

-
To a cooled (0° C.) and stirred solution of compound (VI) (10.2 gm, 0.0286 moles) in dichloromethane (306 ml) was added triethylamine (TEA) (17.99 ml, 1.289 moles) and followed by the slow addition of trifluoroacetic anhydride (12.08 gm, 0.0573 moles). The resulting solution was allowed to warm to room temperature and stirred for a further 6 hours. The reaction mixture was washed with water (3×100 ml), Saturated ammonium chloride solution (100 ml) and brine (100 ml). The organic layer was dried (over sodium sulphate) and the solvent evaporated under reduced pressure. The residue was chromatographed on a column of silica gel (60-120 mesh) using a mixture of Acetone:Hexane (1:19). Concentration of the combined fractions gave the product, as a white solid, 9.7 gm (yield-quantitative).
-
Analysis:
-
Mass: 331(M+); Molecular Formula: C18H25N3O3; Molecular Weight: 331
Step 5: Deprotection of Cyano
-

-
To a chilled (−15° C.) and stirred solution of compound (VII) (6 gm,) in dichloromethane (150 ml) was added trifluoroacetic acid (12 ml) and the mixture was allowed to warm to room temperarture. The reaction mixture was stirred for a further 4 hours. The solvent was evaporated under reduced pressure at 40±5° C. and the residue diluted with aqueous saturated sodium bicarbonate solution (60 ml) and the mixture extracted with dichloromethane (2×60 ml). The combined extracts were washed with water (60 ml), dried (over sodium sulphate) and evaporated under reduced pressure at 35±5° C. to obtain 4.2 gm of compound (VIII).
Step 6: Formation of Bi-Cyclic Compound
-

-
To the cooled (0-5° C.) and stirred solution of compound (VIII) (4.2 gm) in acetonitrile (63 ml) was added triethyl amine (5.28 ml) followed by a slow addition of a solution of Triphosgene (1.9 gm) in Acetonitrile (16.8 ml). Stirring was further continued for 30 minutes followed by addition of Dimethylaminopyridine (DMAP) (0.178 gm). The reaction mixture was allowed to warm to room temperature and stirred for further 16 hours. A aqueous saturated solution of sodium bicarbonate (33.6 ml) was added to the reaction mixture and the resulting mixture stirred for 30 minutes. The mixture was concentrated to ⅓rd volume under reduced pressure. The residue was diluted with water (42 ml) and the resulting mixture extracted with dichloromethane (2×42 ml). The solvent was evaporated under reduced pressure and the residue purified over a column of silica-gel (60-120 mesh). Elution with a 1:4 mixture of acetone: hexane and concentration of the combined fractions gave the product as white solid, 2.3 gm (yield: 48%).
-
Analysis:
-
Mass: 314 (M+); Molecular Formula: C16H18N4O3; Molecular Weight: 314.
Step 7: Synthesis of TBA Sulfate Salt
-

-
To a solution of benzyl compound (VIII) (6 gm, 0.0233 mol) in a 1:1 mixture of dichloromethane (30 ml) and dimethylformamide (30 ml), was added 1.5 gm of dry 10% Palladium charcoal and the mixture was hydrogenated under 3 kg hydrogen pressure for 3 hour at 25-30° C. The reaction mixture was filtered through micron filter to remove catalyst and the filtrate concentrated under reduced pressure to obtain the debenzylated compound IX.
-
The debenzylated compound (IX) was dissolved in N,N′-Dimethyl formamide (30 ml) under argon atmosphere and the solution cooled to 0° C. Dimethylformamide sulfur trioxide complex (DMF: SO3) (4.26 gm, 0.0278 mol) was added to the cooled solution and the stiffing continued further for 30 minutes at 0° C. The mixture was then allowed to warm to room temperature and stirred for 1 hour. Thin layer chromatography showed complete conversion of N-Hydroxy compound to product (X).
-
The solution containing the sulfate (X) was re-cooled to 0° C. and a solution of tetra butyl ammonium acetate (TBAA) (9 gm, 0.0301 mol dissolved in 30 ml water) was added to it. The reaction mixture was allowed to warm to 25° C. and stirred for 1 hour. The volatiles were removed under reduced pressure and residue was co-evaporated with 2×50 ml xylene to remove traces of N,N′-Dimethyl formamide. The residue was partitioned between a 1:1 mixture of water and dichloromethane (120 ml). The aqueous layer was re-extracted with dichloromethane (30 ml). The combined organic extracts were washed with water (2×30 ml), brine (30 ml) and dried over sodium sulphate and the solvent evaporated under reduced pressure to obtain the crude TBA sulfate compound (XI) (5.2 gm). Crude compound was triturated with hexane (2×30 ml) and dried on rotavapor under 4 mm Hg pressure to obtain the TBA salt (XI), 5.0 gm, yield-44%.
-
Analysis:
-
Mass: 246 (M−1) of sulfate; Molecular Weight: 488, Molecular Formula: C23H44N4O5S.
Step 8: Synthesis of Sodium salt of trans-7-oxo-6-(sulphoxy)-1,6-diazabicyclo[3.2.1]-octane-2-carbonitrile (I
-

-
The TBA sulfate compound (XI) (4.4 gm, 0.009 mol) was dissolved in 5% tetrahydrofuran (THF) in water (2 ml) and the solution was passed through column (45 cm length and 2.0 cm diameter) packed with Dowex 50WX8 200 Na+resin. The column was eluted with 5% THF-water mixture (100 ml). The combined fractions were evaporated under reduced pressure (4 mm Hg) to obtain the product (I) as white semi-solid, 1.5 gm, yield: 62%.
-
Analysis:
-
Mass: 246 (M−1) of sulfate; Molecular Weight: 269; Molecular Formula: C7H8N3O5SNa,
-
1H NMR (DMSO): δ 4.54 (d, 1H), 4.06 (s, 1H), 3.22 (m, 2H), 1.96 (m, 2H), 1.84 (m, 2H).
Example 2Preparation of Sodium salt of trans-7-oxo-6-(sulphooxy)-1,6-diazabicyclo[3.2.1]-octane-2-carbonitrile IStep 1: Preparation of (S)-5-oxopyrrolidine-2-carboxamide (III)
-

-
To a stirred solution of L-pyroglutamic acid (II) (75 gm, 0.580 mol, commercially available) in dimethylformamide (750 ml) was added 1-hydroxy benzotriazole ammonium salt (106 gm, 0.696 mol, prepared according the literature procedure described in WO 2006100119) in one lot at 25° C. To this reaction mass, DCC was added in small portions over a period of 30 minutes at 0-5° C. The reaction mixture was allowed to warm to room temperature and stiffing continued further for 2 hours. The precipitates were removed by filtration and the filtrate concentrated under reduced pressure. The residue was treated with ethyl acetate (1000 ml) and stirred for 1 hour. The precipitate formed was filtered under suction and washed with additional ethyl acetate (2×75 ml). The combined filtrate was concentrated under reduced pressure to obtain 73 gm of (S)-5-oxopyrrolidine-2-carboxamide (III) as a white solid in 98% yield. The solid thus obtained was used without further purification in the next step.
-
Analysis:
-
Mass: 129 (M+1) for Molecular Weight: 128.13 and Molecular Formula: C5H8N2O2;
-
1H-NMR (400 MHz, DMSO): δ7.71 (s, 1H), 7.34 (s, 1H), 7.01 (s, 1H), 3.93-3.90 (m, 1H), 2.27-2.14 (m, 1H), 2.12-2.01 (m, 2H), 1.89-1.81 (m, 1H).
Step 2: Preparation of (S)-tert-butyl 2-carbamoyl-5-oxopyrrolidine-1-carboxylate (IV)
-

-
To a cooled (0° C.), stirred solution of (S)-5-oxopyrrolidine-2-carboxamide (70 gm, 0.546 mol) in dimethylformamide (700 ml), triethylamine (TEA) (164.5 gm, 1.6 mol) was added in one lot. After stiffing for 5 minutes Boc anhydride [(Boc)2O] (225 gm, 1.031 mol) was added, followed by the addition of DMAP (6.7 gm, 0.0549 mol). Stirring was continued further for 3 hours, and the completion of the reaction was monitored by thin layer chromatography. The solvent was evaporated under reduced pressure, the residue was leached with diethyl ether (350 ml) and the same procedure repeated with additional diethyl ether (600 ml). The separated solid was filtered under suction and the residue washed with fresh diethyl ether (2×35 ml). The solid was dried at 2 mm Hg, at 45° C. for 2 hour, to obtain 102 gm of (S)-tert-butyl 2-carbamoyl-5-oxopyrrolidine-1-carboxylate as white solid in 82% yield.
-
Analysis:
-
M.P.: 99-102° C.;
-
Mass m/z: 229 (M+H) for MW: 228 and M.F: C10H16N2O4;
-
1H NMR (400 MHz, DMSO): δ 7.60 (s, 1H), 7.15 (s, 1H), 4.42-4.39 (m, 1H), 2.48-2.32 (m, 2H), 2.20-2.15 (m, 1H), 1.77-1.72 (m, 1H), 1.38 (s, 9H).
Step 3: Preparation of (S)-tert-butyl 2-cyano-5-oxopyrrolidine-1-carboxylate (V)
-

-
Trifluoroacetic anhydride (178 gm, 0.845 mol) was added slowly to a stirred solution of (2S)-tert-butyl 2-carbamoyl-5-oxopyrrolidine-1-carboxylate (IV) (97 gm, 0.425 mol), containing triethylamine (TEA) (193 gm, 1.907 mol) in dichloromethane (DCM) (2900 ml) at 0° C. After 2 hours of stirring, reaction mixture was diluted with water (1450 ml) and stirred further for 10 minutes. The organic layer was separated and washed with aqueous saturated solution of sodium hydrogen carbonate solution (500 ml), followed by brine (500 ml). The organic layer was dried over anhydrous sodium sulphate, and the solvent evaporated under reduced pressure. To the residue was added diethyl ether (200 ml), stirred well and the separated solid was filtered under suction to obtain the product. The filtrate was concentrated under reduced pressure and the residue was chromatographed on a column of silica gel using mixtures of ethyl acetate and hexane. The evaporation of the combined fractions gave 64.5 gm of (S)-tert-butyl 2-cyano-5-oxopyrrolidine-1-carboxylate (V) as white solid in 72% yield.
-
Analysis:
-
Melting point: 107-109° C.;
-
1H -NMR (400 MHz, DMSO): δ55.07-5.05 (m, 1H), 2.67-2.2.60 (m, 1H), 2.46-2.36 (m, 2H), 2.20-2.17 (m, 1H), 1.46 (s, 9H).
Step 4: Preparation of Sulfoxonium, [(5S)-5-[[(1,1-dimethylethoxy)carbonyl]amino]-2-oxo-5-cyanopentyl]dimethyl-, inner salt (VI)
-

-
Dimethyl sulfoxide (DMSO) (175 ml) was slowly added to a stirred suspension of sodium hydride (NaH) (7.3 gm, 0.182 mol, 60%) and trimethylsulfoxonium iodide (TMSOI) (40.2 gm, 0.182 mol) in tetrahydrofuran (THF) (140 ml) over a period of 1 hour at 25° C. The stirring was continued further for 1 hour and the resulting suspension cooled to −10° C. This suspension was slowly added to a stirred solution of (S)-tert-butyl-2-cyano-5-oxopyrrolidine-1-carboxylate (V) (35 gm, 0.166 mol, prepared according to the procedure described in step 3) in tetrahydrofuran (105 ml) containing triethylamine (TEA) (30 ml, 0.215 mol), over a period of 30 minutes at −10° C. Stirring was continued further for 1 hour at the same temperature. Saturated aqueous ammonium chloride solution (350 ml) was added to the reaction mass (after completion of the reaction as indicated by thin layer chromatography) and the reaction mixture was allowed to warm to 25° C. The organic layer was separated and the aqueous layer re-extracted by adding ethyl acetate (350 ml). The combined organic layer was washed with aqueous saturated solution of sodium hydrogen carbonate (350 ml) and brine (350 ml). The organic layer was dried over anhydrous sodium sulphate and the solvent evaporated under reduced pressure. To the residual concentrate, diethyl ether (350 ml) was added and the mixture was stirred for 1 hour. The separated solid was filtered, and the residual solid was washed with additional diethyl ether (20 ml). The solid was dried under reduced pressure to provide 35 gm of Sulfoxonium, [(5S)-5[[(1,1-dimethylethoxy)carbonyl]amino]-2-oxo-5-cyanopentyl]dimethyl-, inner salt (VI) as a white solid, in 70% yield.
-
Analysis:
-
Melting Point: 150-153° C.;
-
Mass: 303 (M+1) for Molecular Weight: 302 and Molecular Formula: C13H22N2O4S;
-
1H-NMR (400 MHz, CDCl3): δ 6.04 (br, 1H), 4.55 (br, 1H), 4.45 (s, 1H), 3.40-3.38 (d, 6H), 2.51-2.35 (m, 2H), 2.13-2.03 (m, 2H), 1.44 (s, 9H).
Step 5: Preparation of Carbamic acid, N-[(1S)-5-chloro-1-cyano-4-[(benzyloxy)imino]pentyl, 1,1-dimethylethyl ester (VII)
-

-
To a stirred solution of Sulfoxonium, [(5S)-5-[[(1,1-dimethylethoxy)carbonyl]amino]-2-oxo-5-cyanopentyl]dimethyl-, inner salt (VI) (15 gm, 0.049 mol, prepared according to the procedure described in step 4) in ethyl acetate (EtOAc) (225 ml) was added O-benzyl hydroxylamine hydrochloride (9.5 gm, 0.059 mol) in one lot, at 25° C. The reaction mixture was heated to 60° C. for 2.5 hours. After completion (checked by thin layer chromatography), the reaction mixture was allowed to cool to 25° C. and filtered to remove the precipitates. The filtrate was washed with water (75 ml) and brine (75 ml) and dried over anhydrous sodium sulphate. The solvent was evaporated under reduced pressure to obtain 17.5 gm of Carbamic acid, N-[(1S)-5-chloro-1-cyano-4-[(benzyloxy)imino]pentyl, 1,1-dimethylethyl ester (VII) as an oil in 96% yield.
-
Analysis:
-
Mass: 366 (M+1) for Molecular Weight: 365 and Molecular Formula: C18H24ClN3O3;
-
1H -NMR (400 MHz, CDCl3): δ 7.36-7.7.33 (m, 5H), 5.13 (s, 2H), 4.97 (br, 1H), 4.53 (br, 1H), 4.10 (s, 2H), 2.64-2.50 (m, 2H), 2.15-2.01 (m, 2H), 1.46 (s, 9H).
Step 6: Preparation of (2S)-5-[(benzyloxy)imino]-2-cyanopiperidine (IX)
-

-
Methane sulphonic acid (9 ml, 0.138 mol) was slowly added to a stirred solution of carbamic acid, N-[(1S)-5-chloro-1-cyano-4-[(phenylmethoxy)imino]pentyl, 1,1-dimethylethyl ester (VII) (17 gm, 0.0465 mol, prepared according to the procedure described in step 5) in ethyl acetate (EtOAc) (130 ml), at 25° C. The resulting mixture was heated to 45° C., while monitoring the reaction with thin layer chromatography. After 45 minutes, the reaction mixture was allowed to cool to 25° C. and the resulting reaction mixture (Intermediate VIII) was slowly added to stirred aqueous suspension of potassium hydrogen carbonate (28 gm in 57 ml water). The resulting mixture was stirred and heated to 50-55° C. for 3 hours. The reaction mixture was allowed to cool to 25° C. and the organic layer was separated. The aqueous layer was re-extracted with ethyl acetate (100 ml). The combined organic layer was washed with water (75 ml) and brine (75 ml), dried over anhydrous sodium sulphate and the solvent evaporated under reduced pressure to obtain 11 gm of (2S)-5-[(benzyloxy)imino]-2-cyanopiperidine (IX) as an oil.
-
Analysis:
-
1H-NMR (400 MHz, CDCl3): δ7.36-7.7.33 (m, 5H), 5.09 (s, 2H), 4.14-4.07 (m, 1H), 3.65-3.52 (m, 1H), 3.52-3.45 (m, 1H), 3.16-3.11 (m, 1H), 2.66-2.35 (m, 2H), 2.02-1.89 (m, 2H).
Step 7: Preparation of (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine (X)
-

-
Sulphuric acid (11.7 ml, 0.217 mol) was slowly added to a stirred solution of (2S)-5-[(benzyloxy)imino]-2-cyanopiperidine (IX) (10 gm, 0.0436 mol, prepared according to the procedure described in step 6) in ethyl acetate (150 ml) at −10° C. After 10 minutes of stirring, sodium triacetoxy borohydride (NaHB(OOCCH3)3) (11.7 gm, 0.0519 mol, 95% purity) was added in small portions while maintaining temperature below −5° C. After completion of the addition, stirring was further continued for 2 hour at the same temperature. The pH of the reaction mixture was adjusted to about pH 7 by using 30% aqueous potassium hydrogen carbonate solution. The mixture was allowed to warm to 25° C. and the reaction mixture was filtered under suction. The organic layer was separated and the aqueous layer extracted with fresh ethyl acetate (50 ml). The combined organic layer was washed with water (50 ml) and brine (50 ml), dried over anhydrous sodium sulphate and the solvent evaporated under reduced pressure to obtain 8.88 gm of (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine (X) as an oil, in 88% yield. This was used as such for the next step without further purification.
-
Analysis:
-
Mass: 232 (M+1) for Molecular Weight: 231 and Molecular Formula: C13H17N3O.
Step 8: Preparation of (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine ethanedioate (1:1) (XI)
-

-
A solution of oxalic acid dihydrate (5.28 gm, 0.0418 mol) in a mixture of ethyl acetate:acetone (1:1, 28 ml:28 ml) was slowly added to a stirred solution of (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine (X) (8.8 gm, 0.0380 mol, prepared according to the procedure described in step 7) in ethyl acetate (35 ml) at 25° C. After 3 hour of stirring, the separated solid was filtered under suction, washed with additional 50 ml of v/v mixture of ethyl acetate: acetone solution (1:1, 25 ml: ml) and the solid dried under reduced pressure to obtain 6.7 gm of (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine ethanedioate (1:1) (XI) in 55% yield.
-
Analysis:
-
Mass: 232 (M+1) for Molecular Weight: 321 and Molecular Formula: C13H17N3O.C2H2O4;
-
1H-NMR (400 MHz, DMSO): δ7.25 (m, 5H), 4.59 (s, 2H), 4.22 (br, 1H), 4.07-4.04 (m, 1H), 3.10-3.07 (m, 1H), 2.97-2.83 (m, 1H), 2.61-2.52 (m, 1H), 1.83-1.63 (m, 3H), 1.41-1.25 (m, 1H).
Separation of (2S,5R)-5-[(benzyloxy)amino]-2-cyanopiperidine ethanedioate from two isomeric (1:1) mixture of (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine ethanedioate
-
A suspension of (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine ethanedioate (1:1) (XI) (13 gm, 0.0404 moles) in methanol (260 ml) was heated under reflux, with stirring, for 3 hour. The resulted suspension was allowed to cool to 35° C. and the resulting suspension filtered under suction. The solid was washed with additional methanol (2×13 ml). The solid was dried under reduced pressure (4 mm Hg), to obtain (2S,5R)-5-[(benzyloxy) amino]-2-cyanopiperidine ethanedioate (XIA) as a white solid, 7.3 gm, yield 56%.
-
Analysis:
-
Mass m/z: 232.2 (M+H) for MW: 321 and M.F: C13H17N3O.C2H2O4.
-
1H-NMR (400 MHz, DMSO): δ 7.37-7.24 (m, 5H), 4.57 (s, 2H), 3.92-3.91 (m, 1H), 3.06-3.02 (m, 1H), 2.92-2.88 (m, 1H), 2.56-2.51 (m, 1H), 1.96-1.91 (m, 1H), 1.76-1.55 (m, 2H), 1.44-1.38 (m, 1H).
-
Purity as determined by HPLC: (2S,5R isomer) 88.44% (RT-9.74) and (2S,5S isomer) 5.47% (RT-8.61).
Step 9: Preparation of (2S,5R)-6-(benzyloxy)-2-cyano-7-oxo-1,6-diazabicyclo[3.2.1]octane (XIII) and (2S,5S)-6-(benzyloxy)-2-cyano-7-oxo-1,6-diazabicyclo[3.2.1]octane (XIV)
-

-
To a stirred suspension of (2S)-5-[(benzyloxy) amino]-2-cyanopiperidine ethanedioate (1:1) (XI) (3.7 gm, 0.0115 mol, prepared according to the procedure described in step 8) in ethyl acetate:water (1:1, 37 ml:37 ml) was added solid sodium bicarbonate (1.9 gm, 0.022 mol) at 25° C. After 30 minutes of stirring the organic layer was separated. The aqueous layer was re-extracted with ethyl acetate (20 ml). The combined organic layer was washed with water (20 ml) and brine (20 ml), dried over anhydrous sodium sulphate and concentrated under reduced pressure to obtain 3 gm of ((2S)-5-[(benzyloxy)amino]-2-cyanopiperidine (XII) as an oil. The oily product, (2S)-5-[(benzyloxy)amino]-2-cyanopiperidine (XII) (1 gm, 0.00432 mol, prepared as mentioned above), was dissolved in acetonitrile (ACN) (15 ml), cooled to 10° C., stirred and triethyl amine (1.8 ml, 0.0129 mol) was added in one portion. To this mixture was added slowly a solution of triphosgene (0.564 gm, 0.0019 mol) in acetonitrile (6 ml). After 15 minutes of stirring, DMAP (0.0527 gm, 0.000432 mol) was added and the reaction mixture allowed to warm to 25° C. After 16 hours of stirring, the thin layer chromatography (ethyl acetate:hexane (1:1)) showed the two separable mixture of isomers. A solution of saturated sodium bicarbonate (10 ml) was added to the reaction mass and stirring continued for another 30 minutes. The volatiles were removed under reduced pressure. The residual mass was partitioned between ethyl acetate (10 ml) and water (10 ml). The organic layer was separated and the aqueous layer re-extracted with ethyl acetate (10 ml). The combined organic layer was washed with water (10 ml) and brine (10 ml), dried over anhydrous sodium sulphate and the solvent evaporated under reduced pressure. The resulting mixture was dissolved in dichloromethane (15 ml) and washed with 5% potassium hydrogen sulphate solution (3×10 ml), saturated sodium hydrogen carbonate (10 ml) and water (10 ml). The organic layer was concentrated under reduced pressure, to yield 0.610 gm of crude oily product.
-
[0204]The oily mixture was purified by column chromatography using silica gel (60-120 mesh) by eluting with mixture of ethyl acetate and hexane. The upper spot was eluted out by using 25% ethyl acetate in hexane and the lower spot was eluted out by using 45% ethyl acetate in hexane. The combined pure fractions were concentrated under reduced pressure, to obtain the 0.130 gm of (2S,5R)-6-(benzyloxy)-2-cyano-7-oxo-1,6-diazabicyclo[3.2.1]octane (XIII) and 0.105 gm of (2S,5S)-6-(benzyloxy)-2-cyano-7-oxo-1,6-diazabicyclo[3.2.1]octane (XIV).
-
Analysis for compound of Formula (XIII):
-
Rf: 0.49;
-
Melting Point: 95-99° C.;
-
Mass: 258 (M+1) for Molecular Weight: 257 and Molecular Formula: C14H15N3O2;
-
1H-NMR (400 MHz, CDCl3): δ 7.43-7.35 (m, 5H), 5.06-5.03 (d, 1H), 4.91-4.88 (d, 1H), 4.38-4.36 (d, 1H), 3.36-3.29 (m, 2H), 3.16-3.12 (m, 1H), 2.33-2.10 (m, 2H), 1.90-1.79 (m, 2H).
-
Analysis for compound of Formula (XIV):
-
Rf: 0.12;
-
Melting Point: 115-118° C.
-
Mass: 258 (M+1) for Molecular Weight: 257 and Molecular Formula: C14H15N3O2;
-
1H-NMR (400 MHz, CDCl3): δ7.43-7.33 (m, 5H), 5.06-5.04 (d, 1H), 4.92-4.89 (d, 1H), 3.96-3.92 (dd, 1H), 3.32-3.23 (m, 2H), 2.76-2.73 (m, 1H), 2.29-2.18 (m, 2H), 2.05-1.99 (m, 1H), 1.71-1.63 (m, 1H).
Step 10: Preparation of (2S,5R)-6-hydroxy-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (XIIIa)
-

-
A solution of (2S,5R)-6-(benzyloxy)-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (XIII) (1 gm, 0.00389 mol) in a mixture of ethyl acetate and tetrahydrofuran (THF) (4:6, 4 ml:6 ml) containing 10% palladium over carbon (0.300 gm, 50% wet) was hydrogenated at 50-55 psi, for 6 hours at 25° C. The resulting mixture was filtered through a celite pad and residue was washed with mixture of ethyl acetate and tetrahydrofuran (4:6, 4 ml:6 ml). The solvent from the combined filtrate was evaporated under reduced pressure to obtain 0.649 gm of the titled compound of Formula (XIIIa) as oil, which was used as such for the next reaction without further purification.
Preparation of (2S,5S)-6-hydroxy-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (XIVa)
-

-
A solution of (2S,5S)-6-(benzyloxy)-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (XIV) (545 mg, 2.120 mol) in a mixture of ethyl acetate and tetrahydrofuran (5:5, 8 ml:8 ml) containing 10% palladium over carbon (0.109 gm, 50% wet) was hydrogenated at 50-55 psi, for 45 minutes at 25° C. The resulting mixture was filtered through a celite pad and residue was washed with mixture of dichloromethane and dimethylformamide (5:5, 10 ml:10 ml). The solvent from the combined filtrate was evaporated under reduced pressure to obtain the product as oil, which was triturated with diethyl ether (5 ml). The diethyl ether layer was decanted and the residue was dried under reduced pressure at 40° C. for 15 minutes to obtain 0.343 gm of compound of Formula (XIVa), which was used as such for the next step.
Step 11: Preparation of (2S,5R)-6-(sulfooxy)-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile, tetrabutylammonium salt (XIII b)
-

-
To a stirred solution of (2S,5R)-6-hydroxy-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (XIIIa) (0.649 gm, 0.00389 mol) in a mixture of dichloromethane (5 ml) and dimethylformamide (1 ml), sulfur trioxide dimethylformamide complex (1.07 gm, 0.007 mol) was added in one portion at about 10° C. After 90 minutes, the completion of the reaction was monitored by thin layer chromatography (9:1, chloroform:methanol). To the resulting reaction mass was added tetrabutylammonium hydrogen sulphate (TBAHS) in one portion (2.37 gm, 0.007 mol) under stirring. After 1 hour, water (10 ml) was added and the mixture stirred for 5 minutes. The organic layer was separated and washed with water (2×10 ml), dried (over anhydrous sodium sulphate) and the solvent evaporated under reduced pressure at 35° C. The residual oily mass was triturated with ether (2×10 ml), each time the ether layer was decanted and finally the residue was concentrated under reduced pressure, to obtain 0.6 gm of the titled compound of Formula (XIIIb) in 31% yield.
-
Analysis:
-
Mass: 246 (M−1), for Molecular Weight: 488 and Molecular Formula: C23H44N4O5S;
-
1H NMR (400 MHz, CDCl3): δ4.43 (brs, 1H), 4.35-4.33 (d, 1H), 3.47-3.44 (m, 2H), 3.28-3.24 (m, 8H), 2.33-2.29 (m, 2H), 1.92-1.85 (m, 2H), 1.69-1.61 (m, 8H), 1.48-1.39 (m, 8H), 1.02-0.98 (m, 12H).
-
Purity as determined by HPLC: 95.57%
Preparation of (2S,5S)-6-(sulfooxy)-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile, tetrabutylammonium salt (XIVb)
-

-
To a stirred solution of (2S,5S)-6-hydroxy-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile (XIVa) (343 mg, 2.05 mol) in dimethylformamide (3 ml) sulfur trioxide dimethylformamide complex (390 mg, 2.549 mol) was added in one portion, at 10° C. and stirring continued further. After 60 minutes, thin layer chromatography (9:1, chloroform:methanol) showed the complete conversion. To the resulting reaction mixture was added, slowly, a solution of tetrabutylammonium acetate (TBAA) (831 mg, 2.756 mol) in water (3 ml) under stirring. After 1 hour of stirring, the solvent from the reaction mixture was evaporated under reduced pressure to obtain an oily residue. The oily mass was co-evaporated with xylene (2×10 ml), to yield a thick mass which was partitioned between dichloromethane (10 ml) and water (10 ml). The organic layer was separated and the aqueous layer re-extracted with dichloromethane (10 ml). The combined organic extracts were washed with water (3×10 ml), dried (over anhydrous sodium sulphate) and the solvent evaporated under reduced pressure at 35° C. The residual oily mass was triturated with ether (2×10 ml), each time the ether layer was decanted and finally the residue was dried under reduced pressure, to obtain 634 mg of compound of Formula (XIVb) as an oil in 61% yield.
-
Analysis:
-
Mass: 246 (M−1); for Molecular Weight: 488 and Molecular Formula: C23H44N4O5S;
-
1H NMR (400 MHz, CDCl3): δ4.38 (m, 1H), 3.98-3.93 (dd, 1H), 3.98-3.54 (m, 1H), 3.32-3.28 (m, 8H), 2.43-2.39 (m, 1H), 2.31-2.30 (m, 1H), 2.15-2.01 (m, 2H), 1.76-1.63 (m, 8H), 1.49-1.40 (m, 8H), 1.02-0.99 (m, 12H);
-
Purity as determined by HPLC: 98.22%.
Step 12: Preparation of (2S,5R)-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile-7-oxo-6-(sulfooxy)-mono sodium salt (I)
-

-
An activated Amberlite 200 sodium resin (20 gm) was loaded on a glass column and was washed with de-mineralized water (50 ml) followed by 10% tetrahydrofuran in water (50 ml). A solution of (2S,5R)-6-(sulfooxy)-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile, tetrabutyl ammonium salt (XIIIb) (575 mg, 1.176 mol) in tetrahydrofuran (THF) (1.1 ml) was loaded on column. It was eluted by using 10% tetrahydrofuran in water. The pure fractions were combined and the solvents evaporated under reduced pressure to obtain 280 mg of the compound of Formula (I) in 85% yield.
-
Analysis:
-
Mass: 246 (M−1) as free sulfonic acid, for Molecular Weight: 269 and Molecular Formula:
-
C7H8N3O5SNa;
-
1H NMR (400 MHz, DMSO): δ4.54-4.53 (d, 1H), 4.06 (brs, 1H), 3.20 (m, 2H), 1.96-1.81 (m, 4H);
-
Purity as determined by HPLC: 97.07%.
Preparation of (2S,5S)-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile-7-oxo-6-(sulfooxy)-mono sodium salt (Ia)
-

-
An activated Amberlite 200 sodium resin (20 gm) was loaded on a glass column and was washed with de-mineralized water (100 ml) followed by 10% tetrahydrofuran (THF) in water (100 ml). A solution of (2S,5S)-6-(sulfooxy)-7-oxo-1,6-diaza-bicyclo[3.2.1]octane-2-carbonitrile, tetrabutylammonium salt (XIVb) (475 mg, 0.971 mol) in tetrahydrofuran (1.5 ml) was loaded on column. It was eluted by using 10% tetrahydrofuran in water. The pure fractions were combined and the solvent evaporated under reduced pressure to obtain 242 mg of compound of Formula (Ia) as white solid, in 92% yield.
-
Analysis:
-
Mass: 246 (M−1) as free sulfonic acid, for Molecular Weight: 269 and Molecular Formula: C7H8N3O5SNa;
-
1H NMR (400 MHz, DMSO): δ 4.53-4.50 (dd, 1H), 3.98 (brs, 1H), 3.17-3.02 (dd, 2H), 1.99-1.96 (m, 2H), 1.77-1.75 (m, 2H);
-
Purity as determined by HPLC: 99.59%.
note
Avibactam is

1192500-31-4; SULFURIC ACID, MONO[(1R,2S,5R)-2-(AMINOCARBONYL)-7-OXO-1,6-DIAZABICYCLO[3.2.1]OCT-6-YL] ESTER;
COMPD IS
References
IN 2013MU03308
IN 2011MU02582
| Patent | Submitted | Granted |
|---|---|---|
| Nitrogen containing compounds and their use [US8969334] | 2014-05-04 | 2015-03-03 |
| Nitrogen containing compounds and their use [US8969567] | 2014-05-10 | 2015-03-03 |
| Nitrogen containing compounds and their use [US8754102] | 2012-09-11 | 2014-06-17 |
| WO2013014496A1 * | 4 Oct 2011 | 31 Jan 2013 | Wockhardt Limited | Pharmaceutical compositions comprising sulbactam and beta-lactamase inhibitor |
| WO2013038330A1 * | 11 Sep 2012 | 21 Mar 2013 | Wockhardt Limited | Nitrogen containing compounds and their use |
| WO2013030733A1 * | Aug 24, 2012 | Mar 7, 2013 | Wockhardt Limited | 1,6- diazabicyclo [3,2,1] octan-7-one derivatives and their use in the treatment of bacterial infections |
| WO2013038330A1 * | Sep 11, 2012 | Mar 21, 2013 | Wockhardt Limited | Nitrogen containing compounds and their use |
| WO2013149121A1 * | Mar 29, 2013 | Oct 3, 2013 | Cubist Pharmaceuticals, Inc. | 1,3,4-oxadiazole and 1,3,4-thiadiazole beta-lactamase inhibitors |
| WO2014108872A1 * | Jan 13, 2014 | Jul 17, 2014 | Wockhardt Limited | Compositions and methods for treating bacterial infections |
| CA2874279A1 * | May 30, 2013 | Dec 5, 2013 | Meiji Seika Pharma Co., Ltd. | Novel .beta.-lactamase inhibitor and process for preparing the same |
| US20130289012 * | Mar 29, 2013 | Oct 31, 2013 | Cubist Pharmaceuticals, Inc. | 1,2,4-oxadiazole and 1,2,4-thiadiazole beta-lactamase inhibitors |
////////
C1CC(N2CC1N(C2=O)OS(=O)(=O)O)C#N
or
C1C2CN(C(C1)C#N)C([C@@H]2OS(=O)(=O)O)=O
or
O=S(=O)(O)ON2C(=O)N1C[C@H]2CC[C@H]1C#N
////////
WCK ? NEW ANTIBACTERIALS FROM WOCKHARDT

WCK ?
TRANS-SULFURIC ACID MONO-{2-[5-(2-METHYLAMINO-ETHYL)-[1,3,4]-OXADIAZOL-2-YL]-7-OXO-1,6-DIAZA-BICYCLO [3.2.1]OCT-6-YL} ESTER
Trans-sulfuric acid mono- { 2-[5-(2-methylamino-ethyl)-[l,3,4]-oxadiazol-2-yl]-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl} ester.
CAS 1824664-22-3
MW 347.35, C11 H17 N5 O6 S
|
Beta lactamase inhibitor
|
To treat
|
Bacterial infection
|
Several l,6-diazabicyclo[3.2.1]octan-7-one derivatives have been described as antibacterial agents in PCT International Patent Application No. PCT/IB2012/054296. A compound of Formula (I), chemically known as irans-sulfuric acid mono- {2- [5 -(2-methylamino-ethyl)-[l,3,4]-oxadiazol-2-yl]-7-oxo-l,6-diazabicyclo[3.2.1]oct-6-yl} ester has antibacterial properties and is also disclosed in PCT International Patent Application No. PCT/US2013/034562
PATENT
WO2015173663

(VII) Formula (I)
Scheme 1
Example 1
Synthesis of traras-sulfuric acid mono-{2-[5-(2-methylamino-ethyl)-[l,3,4]-oxadiazol- 2-yl]-7-oxo-l,6-diazabicyclo[3.2.1]oct-6-yl} ester (I)
Step 1; Preparation of tr «s-{3-[N’-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane-2-carbonyl)-hydrazino]-3-oxo-propyl}-methyl-carbamic acid fert-butyl ester (IV):
Sodium salt of 6-benzyloxy-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylic acid (III) (5.9 g, 0.02 mol; prepared using a method disclosed in Indian Patent Application No 699/MUM/2013) was dissolved in water (100 ml) to obtain a clear solution under stirring at 25°C. To the clear solution was added successively, (3-hydrazinocarbonyl-ethyl)-methyl-carbamic acid tert-buty\ ester (II) (4.5 g, 0.02 mol), EDC. HC1 (5.7 g, 1.5 mol), and HOBt (2.7 g, 0.02 mol) followed by water (20 ml) under stirring at 25°C. The reaction mixture was stirred at 30°C for 20 hours. As maximum precipitation was reached, thin layer chromatography (acetone: hexane, 35:65) showed completion of reaction. The suspension was filtered under suction and the wet cake was washed with additional water (100 ml) and dried under vacuum at 45°C to furnish 5.5 g of ir ns-{3-[N’-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonyl)-hydrazino]-3-oxo-propyl}-methyl-carbamic acid tert-buty\ ester (IV) as a white powder in 58% yield.
Analysis:
Mass: 476.4 (M+l); for Molecular Formula: C23H33N5O6 and Molecular Weight:
475.2;
1H NMR (CDCI3): δ 7.43-7.35 (m, 5H), 5.04 (d, 1H), 4.90 (d, 1H), 4.01 (d, 1H), 3.54 (t, 2H), 3.33 (br s, 1H), 3.14-3.07 (m, 2H), 2.85 (s, 3H), 2.53 (br s, 2H), 2.33-2.30 (m, 1H), 2.07-1.94 (m, 2H), 1.64-1.61 (m, 4H), 1.40 (s, 9H), 1.25-1.17 (m, 2H).
Step 2: Preparation of tr «s-{2-[5-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-ethyl}-methyl-carbamic acid tert-butyl ester (V):
To a solution of triphenylphosphine (3.3 g, 0.0126 mol) in dichloromethane (70 ml) at was added iodine (3.2 g, 0.0126 mol) and triethyl amine (7.0 ml, 0.0525 mol) under stirring at 25°C. Separately prepared solution of ir ns-{3-[N’-(6-benzyloxy-7-oxo-1 ,6-diaza-bicyclo[3.2.1 ]octane-2-carbonyl)-hydrazino] -3-oxo-propyl)-methyl-carbamic acid tert-butyl ester (IV) (5.5 g, 0.0105 mol) dissolved in dichloromethane (30 ml) was added to above reaction mixture and the mixture was stirred at 25°C for 30 minutes. The reaction mixture was concentrated and to this ethyl acetate (100 ml) was added. The separated triphenylphosphine oxide was filtered off. The filtrate was concentrated and the residue purified by silica gel column chromatography using mixture of ethyl acetate and hexane, to afford 5 g of the titled compound.
Analysis:
Mass: 458.3 (M+l); for Molecular Formula: C23H31N5O5 and Molecular Weight:
457.53;
1H NMR (CDCI3): δ 7.44-7.35 (m, 5H), 5.04 (d, 1H), 4.93 (d, 1H), 4.70 (t, 1H), 3.62 (br s, 2H), 3.36 (s, 1H), 3.07 (t, 2H), 2.93 (br d, 1H), 2.85 (br s, 4H), 2.32-2.27 (m, 2H), 2.12 (br d, 2H), 1.95 (br s, 1H), 1.40 (s, 9H).
Step 3: Preparation of traras-{2-[5-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-ethyl}-methyl-carbamic acid tert-butyl ester (VI):
To a solution of trans-{2-[5-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-ethyl}-methyl-carbamic acid tert-butyl ester (V) (5 g, 0.0109 mol) in methanol (50 ml) was added 10% palladium on carbon (1.5 g) at 25°C. The reaction mixture was stirred under 1 atmospheric pressure of hydrogen at 35°C for 2 hours. The catalyst was removed by filtering the reaction mixture under suction over a celite bed. The celite bed was washed with methanol (50 ml). The combined filtrate was evaporated under vacuum below 35°C to provide 3.8 g of trans- {2- [5-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-ethyl}-methyl-carbamic acid tert-butyl ester (VI) in 93% yield; it was used as such for the next reaction.
Step 4: Preparation of trans -tetrabutyl ammonium salt-methyl-{2-[5-(7-oxo-6-sulphooxy-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-ethyl}-carbamic acid tert-butyl ester (VII):
A solution of trans-{2-[5-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[1,3,4] oxadiazol-2-yl] -ethyl }-methyl-carbamic acid tert-butyl ester (VI) (3.8 g, 9.8 mmol), in dichloromethane (38 ml) was charged with triethylamine (2.6 ml, 19.7 mmol) under stirring to provide a clear solution. To this clear solution was added sulfur trioxide -pyridine complex (2.35 g, 14.8 mmol) under stirring at 30°C. The reaction mixture was stirred for 3 hours and to this 0.5 M aqueous potassium dihydrogen phosphate (38 ml) was added followed by ethyl acetate (76 ml). The biphasic mixture was stirred for 15 minutes at 30°C. Aqueous layer was separated and re-extracted with dichloromethane and ethyl acetate mixture (1:2 v/v, 76 ml twice). To the aqueous layer was added solid tetrabutyl ammonium hydrogen sulfate (3 g, 8.8 mmol) and stirring was continued for 1
hour at room temperature. The reaction mixture was extracted with dichloromethane (3 x 50 ml). Layers were separated and dichloromethane layer dried over sodium sulfate and then evaporated under vacuum at 35°C to provide 2.8 g of irans-tetrabutyl ammonium salt-methyl- {2-[5-(7-oxo-6-sulphooxy-l,6-diaza-bicyclo[3.2. l]oct-2-yl)-[l, 3, 4]oxadiazol -2-yl] -ethyl} -carbamic acid tert-buty\ ester (VII). This was purified by column chromatography to afford 2.0 g of pure product in 29% yield.
Analysis:
Mass: 446.5 (M-l) as free sulfonic acid; for Molecular Formula:![]()
(C4H9)4 and Molecular Weight: 688.5;
1H NMR (CDC13): δ 4.67 (d, 1H), 4.36 (br s, 1H), 3.33-3.29 (m, 8H), 3.23 (d, 1H), 3.08 (t, 2H), 2.87 (s, 3H), 2.83 (s, 1H), 2.28-2.22 (m, 3H), 2.07-2.00 (m, 8H), 1.50-1.41 (m, 17H), 1.28 (s, 3H), 1.01 (t, 12 H), 1.41-1.52 (m, 10 H).
Step 5: traras-sulfuric acid mono-{2-[5-(2-methylamino-ethyl)-[l,3,4]-oxadiazol-2-yl]-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl} ester:
irans-Tetrabutyl ammonium salt-methyl- {2-[5-(7-oxo-6-sulphooxy- 1 ,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol -2-yl] -ethyl} -carbamic acid tert-butyl ester (VII) (2.0 g, 2.9 mmol) was dissolved in dichloromethane (5 ml) and to the clear solution was slowly added trifluoroacetic acid (5 ml) at 0 to -10 °C. The reaction mixture was stirred at 0 to -10 °C for 1 hour. The solvent and excess trifluoroacetic acid was evaporated under vacuum below 40°C to approximately 1/3 of its original volume to provide pale yellow oily residue. The oily residue was stirred with diethyl ether (100 ml) for 10-15 minutes. The suspension formed was filtered under suction to provide a solid. This process was repeated twice. The solid was charged in a round bottom flask and to it was added dichloromethane (100 ml). The suspension was stirred for 15 minutes and filtered under suction to provide a solid. The obtained solid was dried under vacuum below 40°C to furnish 850 mg of trans- sulfuric acid mono-{2-[5-(2-methylamino-ethyl)-[l,3,4]-oxadiazol-2-yl]-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl} ester as white solid in 85% yield.
Analysis:
Mass: 346.3 (M-1) as a free sulfonic acid; for Molecular Formula: C11H17N5O6S and Molecular Weight: 347.35;
NMR (D20): δ 4.74 (d, IH), 4.16 (br s, IH), 3.45 (t, 2H), 3.31 (t, 2H), 3.15 (d, IH), 2.91 (d, IH), 2.98 (s, 3H), 2.27-2.22 (m, IH), 2.16-2.11 (m, 2H), 1.94-1.91 (m, IH);
Purity as determined by HPLC: 95.56%.
/////////
WCK ? New molecules from Wochkardt to treat bacterial infections

(2S, 5R)-7-OXO-N-[(3S)-PYRROLIDIN-3-YLOXY]-6-(SULFOOXY)-1,6-DIAZABICYCLO [3.2.1]OCTANE-2-CARBOXAMIDE
- (2S,5R)-7-Oxo-N-((3S)-pyrrolidin-3-yloxy)-6-(sulfooxy)-1,6-diazabicyclo[3.2.1]octane-2-carboxamide
- C11 H18 N4 O7 S, 350.35
- Sulfuric acid, mono[(1R,2S,5R)-7-oxo-2-[[[(3S)-3-pyrrolidinyloxy]amino]carbonyl]-1,6-diazabicyclo[3.2.1]oct-6-yl] ester
CAS 1452458-72-8
KEEP WATCHING THIS POST
SODIUM SALT CAS 1629221-44-8
Sulfuric acid, mono[(1R,2S,5R)-7-oxo-2-[[[(3S)-3-pyrrolidinyloxy]amino]carbonyl]-1,6-diazabicyclo[3.2.1]oct-6-yl] ester, sodium salt (1:1)
Patent
http://www.google.com/patents/WO2015110886A1?cl=en

Formula (II) Formula (III) Formula (IV)
Hydrogenolysis

Formula (I)
Scheme – 1

Formula (VII) Formula (VIII)
Hydrazine hydrate

Formula I
Scheme – 2
Example 1
Synthesis of tert-butyl (3S)-2-(aminooxy)pyrrolidine-l-carboxylate (III):
Step 1; Preparation of 3-(R)-hydroxypyrrolidine hydrochloride (VIII):
To a stirred suspension of commercially available (25, 4i?)-4-hydroxy-2-pyrrolidinecarboxylic acid (L-hydroxyproline) (VII) (100 g, 0.762 mol) in anhydrous cyclohexanol (500 ml), was added 2-cyclohexen-l-one (5 ml). The resulting mixture was heated under reflux at about 154°C for about 48 hour. The obtained clear solution was allowed to cool to room temperature and then was cooled further to 10°C. To this, about 15 % solution of hydrochloric acid in ethanol (234 ml) was added and then stirred for 30 minutes. The separated solid was filtered under suction and washed with ethyl acetate (2 x 100 ml). The solid was dried under reduced pressure to obtain 47.5 g of 3-(R)-hydroxypyrrolidine hydrochloride (VIII) in 51 % yield. The solid was used without further purification in the next step.
Analysis:
Mass: 87.8 (M+l) as free base; for Molecular weight of 123.57 and Molecular Formula of C4Hi0ClNO; and
1H NMR (400MHz, DMSO): 5 9.58 – 9.32 (brd, 2H), 5.36 (brs, 1H), 4.36 – 3.39 (brs, 1H), 3.17 (brs, 2H), 3.11-2.96 (dd, 2H), 1.90 – 1.81 (m, 2H).
Step 2: Preparation of (3R)-l-(tert-butoxycarbonyl)-3-hydroxypyrrolidine (IX):
To a stirred suspension of 3-(i?)-hydroxypyrrolidine hydrochloride (VIII) (110 g, 0.9 mol) in dichloromethane (1100 ml), triethylamine (273 g, 2.7 mol) was added at 0-5°C. After 5 minute of stirring di-feri-butyldicarbonate [(Boc)20] (245 g, 1.125 mol) was added to the reaction mixture in small portions, followed by 4-dimethylaminopyridine (10.99 g, 0.09 mol). The reaction mixture was stirred for 2 hour and then poured in to water (1100 ml). The organic layer was separated and washed with saturated ammonium chloride solution (1×1100 ml) and water (1100 ml). The organic layer was dried over anhydrous sodium sulphate and the solvent evaporated under reduced pressure. The residue was purified by silica gel (60-120 mesh) column chromatography using 1-5% mixtures of acetone: hexane as an eluent. The combined fractions were evaporated, to obtain the 118 g of (3i?)-l-(ieri-butoxycarbonyl)-3-hydroxypyrrolidine (IX), as a white solid, in 71 % yield.
Analysis:
Melting point: 55 – 58°C;
Mass: 188 (M+l); for Molecular Weight of 187.24 and Molecular Formula of C9H17N03; and
1H NMR (400MHz, CDC13): 54.428 – 4.424 (s, 1H), 3.46 – 3.43 (m, 2H), 3.37 -3.28 (m, 2H), 2.36 – 2.30 (d, 1H), 2.00 – 1.86 (m, 2H), 1.44 (s, 9H).
Step 3: Preparation of (5)-3-[(l,3-dihydro-l,3-dioxo-isoindol-2-yl)oxy]pyrrolidine-l-carbox lic acid tert- butyl ester (X):
To a stirred solution of di-isopropyl azodicarboxylate (97.17 g, 0.481 mol) in tetrahydrofuran (1200 ml), a solution triphenyl phosphine (125.9 g, 0.481 mol) in tetrahydrofuran (300 ml) was added at temperature below -10°C. The resulting reaction mixture was stirred for further 45 minute at the same condition and a solution of (3i?)-l-(ieri-butoxycarbonyl)-3-hydroxypyrrolidine (IX) (60 g, 0.3204 mol) in tetrahydrofuran (300 ml) was added over a period of 15 minute. After another 45 minute of stirring, N-hydroxy phthalimide (52.4 g, 0.3204mol) was added in one portion to the reaction mass. The reaction mixture was allowed to warm to room temperature and stirred for 16 hour.
The completion of the reaction was monitored by thin layer chromatography. After completion of reaction, the solvent was evaporated under reduced pressure. The residue thus obtained was stirred with di-isopropyl ether (600 ml). The precipitate formed was filtered under suction. The filtrate was concentrated under reduced pressure and the residual mass was purified by silica gel (60-120 mesh) column chromatography using 1-5 % mixtures of acetone: hexane as an eluent. The solvent from the combined fractions was evaporated to obtain 63 g of (5)-3-[(l,3-dihydro-l,3-dioxo-isoindol-2-yl)oxy]pyrrolidine-1-carboxylic acid tert-buty\ ester (X), as a white solid, in 59% yield.
Analysis:
Melting point: 112-115°C;
Mass: 333.2 (M+l); for Molecular Weight of 332.36 and Molecular Formula of ![]()
1H NMR (400 MHz, CDC13): 57.86-7.83 (m, 2H), 7.78-7.75 (m, 2H), 4.99 – 4.94 (d, 1H), 3.80 – 3.68 (m, 2H), 3.60 – 3.53 (m, 2H), 2.28-2.25 (m, 1H), 2.02 (m, 1H), 1.48 (s, 9H).
Step 4: Preparation of tert-butyl (35)-2-(aminooxy)pyrrolidine-l-carboxylate (III):
To a stirred suspension of the (5)-3-[(l,3-dihydro-l,3-dioxo-isoindol-2-yl) oxy]pyrrolidine-l-carboxylic acid tert-buty\ ester (X) (12.68 g, 0.0381 mol) in dichloromethane (200 ml) was added 99% hydrazine hydrate (3.81 g, 0.0762 mol) drop-wise over a period of 30 minutes, at 25°C. After 2 hour of stirring, the separated solid was filtered and washed with dichloromethane (2 x 50 ml). The filtrate and washings were combined and washed with water (2 x 65 ml) and finally with brine (1 x 65 ml). The organic layer was dried over anhydrous sodium sulphate and the solvent was evaporated under reduced pressure to obtain 7.71 g of tert-buty\ (3S)-2-(aminooxy pyrrolidine- 1-carboxylate (III) as pale yellow oil.
Analysis:
Mass: 203 (M+l); for Molecular Weight of 202.26 and Molecular Formula of C9H18N203.
Example 2
Synthesis of (25, 5R)-7-oxo-N-r(35)-pyrrolidin-3-yl-oxyl-6-(sulfooxy)-l,6-diaza bicyclor3.2. lloctane-2-carboxamide (I) :
Step 1: Preparation of fert-butyl-(35)-3-[({[25, 5R)-6-(benzyloxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate (IV):
To a clear, stirred solution of sodium (25, 5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II) (11.38 g, 0.0382 mol) in water (114 ml), was added EDC.HC1 (18.24 g, 0.0955 mol) at 15°C, in small portions. After 10 minutes, a solution of feri-butyl-(35)-3-(aminooxy) pyrrolidine- 1-carboxylate (III, 7.72 g, 0.0382 mol), prepared as per the literature procedure: US5233053, Chemistry Letters, 893-896, (1986) and depicted in scheme 2), in dimethylformamide (24 ml) was added drop wise, to the above stirred solution, at about 10°C. The reaction mass was allowed to warm to 25°C and HOBt (5.15 g, 0.0382 mol) was added in small portions over a period of 15 minutes and the reaction mixture was stirred further at room temperature for 16 hour. After completion of the reaction (monitored by thin layer chromatography using solvent system acetone: hexane (35:65)) the resulting mixture was filtered and the residue was washed with water (120 ml). The residual white solid was suspended in fresh water (120 ml) and the mixture stirred at 50°C, for 3 hour. The resulting suspension was filtered and the residual solid dried under reduced pressure to obtain 16.1 g of tert-buty\ (35)-3-[({ [25,5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino) oxy]pyrrolidine- 1-carboxylate (IV) as off white solid in 92% yield.
Analysis:
Mass: 461.3 (M+l); for Molecular weight of 460.53 and Molecular formula of ![]()
1H NMR (400MHz, CDC13): δ 9.08-9.03 (d, 1H), 7.43-7.36 (m, 5H), 5.06-4.88 (dd, 2H), 4.63-4.57 (d, 1H), 3.97-.396 (d, 1H), 3.64-3.53 (m, 2H), 3.47-3.37 (m, 2H), 3.31 (s, 1H), 3.02-2.99 (d, 1H), 2.75-2.73 (d, 1H), 2.29(m, 2H), 2.18-2.15 (m, 1H), 2.01-1.90 (m, 3H), 1.66 (m, 1H), 1.46 (s, 9H).
Step 2: Preparation of tert-butyl-(35)-3-[({[25,5R)-6-hydroxy-7-oxo-l,6-diazabicylco
[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate (V):
ieri-Butyl-(35)-3-[({ [25,5R)-6-(benzyloxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate (IV) (10 g, 0.02171 mol) was dissolved in a mixture of dimethylformamide and dichloromethane ( 1 : 1 , 50 ml : 50 ml) to obtain a clear solution. To this solution, was added 10% palladium on carbon (2.5 g, 50% wet) catalyst. The suspension was stirred for 4 hour, at 50 psi hydrogen atmosphere, at 25°C. After completion of the reaction (monitored by thin layer chromatography), the resulting mixture was filtered through a celite pad. The residue was washed with dichloromethane (50 ml). The solvent from the combined filtrate was evaporated under reduced pressure to obtain 8.04 g of ieri-butyl(35)-3-[({ [25,5i?)-6-hydroxy-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl
amino)oxy]pyrrolidine-l-carboxylate (V) as oil. This was used as such for the next reaction without further purification.
Analysis:
Mass: 371.2 (M+l); for Molecular Weight of 370.4 and Molecular Formula of
Step 3: Preparation of tert-butyl-(35)-3-[({[25,5R)-6-(sulfooxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate, tetrabutyl ammonium salt (VI):
To a stirred solution of ieri-butyl(35)-3-[({ [25,5i?)-6-hydroxy-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate (V) (8.04 g, 0.0217 mol) in dimethylformamide (50 ml), was added sulfur trioxide dimethyl formamide complex (3.98 g, 0.0260 mol) in one portion, at about 10°C. The stirring was continued further for 30 minute and then the reaction mixture was allowed to warm to room temperature. After 2 hour, a solution of tetrabutylammonium acetate (7.83 g, 0.0260 mol) in water (25.8 ml) was added to the resulting reaction mass under stirring. After additional 2 hour of stirring, the solvent from the reaction mixture was evaporated under reduced pressure to obtain an oily residue. The oily mass was co-evaporated with xylene (2 x 20 ml) to obtain thick mass. This mass was partitioned between dichloromethane (100 ml) and water (100 ml). The organic layer was separated and the aqueous layer re-extracted with dichloromethane (50 ml). The combined organic extracts were washed with water (3 x 50 ml), dried over anhydrous sodium sulphate and the solvent evaporated under reduced pressure. The residual oily mass was triturated with ether (3 x 50 ml), each time the ether layer was decanted and finally the residue was concentrated under reduced pressure to obtain 11.3 g of tert-butyl(3S)-3-[({ [2S,5R)-6-(sulfooxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy] pyrrolidine- 1-carboxylate, tetrabutylammonium salt (VI), as a white foam, in 75 % yield.
Analysis:
Mass: 449.3 (M-l, without TBA); for Molecular weight of 691.94 and Molecular formula of C32H61N5O9S; and
1H NMR (400MHz, CDC13): 59.14-9.10 (d, 1H), 4.63 (s, 1H), 4.35 (s, 1H), 3.94-3.92 (d, 1H), 3.66-3.35 (m, 5H), 3.29-3.27 (m, 8H), 2.83-2.80 (d, 1H), 2.35-2.17 (m, 3H), 1.98-1.87 (m, 2H), 1.73 (m, 1H), 1.70-1.62 (m, 8H), 1.49-1.40 (m, 17H), 1.02-0.99 (t, 12H).
Step 4: Preparation of (25,5R)-7-oxo-iV-[(35)-pyrrolidin-2-yl-oxy]-6-(sulfooxy)-l,6-diazabicyclo [3.2.1]octane-2-carboxamide (I):
To a stirred solution of ieri-butyl(35)-3-[({ [25,5i?)-6-(sulfooxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate tetrabutyl ammonium salt (VI) (11 g, 0.0158 mol) in dichloromethane (55 ml), was added trifluoroacetic acid (55 ml) drop wise at about -10 °C over a period of 1 hour. After 1 hour of stirring, the resulting mixture was poured into hexane (550 ml), stirred well for 30 minute and the separated oily layer was collected. This procedure was repeated one more time and finally the combined oily layer was added to diethyl ether (110 ml) under vigorous stirring, at about 25 °C. The ether layer was removed by decantation from the precipitated solid. This procedure was repeated twice again with diethyl ether (2 x 110 ml). The solid thus obtained was stirred with fresh dichloromethane (110 ml) for 30 minutes and filtered. The residual solid was dried at about 45 °C under reduced pressure to obtain 5.7 g of (25,5i?)-7-oxo-N-[(35)-pyrrolidin-2-yl-oxy]-6-(sulfo-oxy)- l,6-diaza bicyclo[3.2.1] octane-2-carboxamide (I), as a white amorphous solid having XRPD as shown in Figure 1.
Analysis:
Mass: 349.2 (M-l); for Molecular Weight of 350.35 and Molecular Formula of ![]()
1H NMR (400MHz, DMSO-D6): δ 11.44 (brs, 1H), 8.80 (brs, 2H), 4.64-4.63 (m, 1H), 4.00 (s, 1H), 3.78-3.77 (d, 1H), 3.38-3.23 (m, 4H), 3.03-2.93 (dd, 2H), 2.48-2.11 (m, 1H), 2.00- 1.94 (m, 2H), 1.88- 1.86 (m, 1H), 1.71-1.65 (m, 2H).
Example 3
Preparation of Crystalline Form I of (25,5R)-7-oxo-jV-r(35)-pyrrolidin-2-yl-oxyl-6-(sulfooxy)-l,6-diaza bicyclor3.2.11 octane-2-carboxamide:
The solid (5 g) obtained in Step 4 of Example 2 was dissolved in water (30 ml) with stirring. To this solution, Isopropanol (210 ml) was slowly added at 25 °C and stirred for 12 hours. The separated solid was filtered and washed with additional isopropanol ( 10 ml) and dried under reduced pressure to obtain 3.9 g of (25,5i?)-7-oxo-N-[(35)-pyrrolidin-2-yl-oxy]-6-(sulfo-oxy)-l,6-diazabicyclo[3.2.1]octane-2-carboxamide as crystalline Form I, having XRPD as shown in Figure 2, in 78 % yield.
Analysis:
Purity as determined by HPLC: 95.56 %; and
X-ray powder diffraction pattern comprising peak at (2 Theta Values): 10.57 (± 0.2), 12.01 (± 0.2), 13.61 (± 0.2), 15.47 (± 0.2), 17.86 (± 0.2), 18.34 (± 0.2), 19.09 (± 0.2), 19.81 (± 0.2), 22.69 (± 0.2), 24.79 (± 0.2), 27.22 (± 0.2) and 33.41 (± 0.2)
//////
WCK ? , WCK Series by Wockhardt for treating the bacterial infection

(2S,5R)-7-0X0-N-[(2S)-PYRROLLIDIN-2-YL-METHYLOXY]-6-(SULFOOXY)-1,6-DIAZABICYCLO[3.2.1 ]OCTANE-2-CARBOXAMIDE
(2S,5R)-7-Oxo-N-((2S)-pyrrolidin-2-ylmethyloxy)-6-(sulfooxy)-1,6-diazabicyclo[3.2.1]octane-2-carboxamide
Sulfuric acid, mono[(1R,2S,5R)-7-oxo-2-[[[(2S)-2-pyrrolidinylmethoxy]amino]carbonyl]-1,6-diazabicyclo[3.2.1]oct-6-yl] ester
KEEP WATCHING THIS POST
MW 364.37, C12 H20 N4 O7 S
CAS 1452459-04-9 FREE FORM
CAS Na SALT 1572988-44-3
Sulfuric acid, mono[(1R,2S,5R)-7-oxo-2-[[[(2S)-2-pyrrolidinylmethoxy]amino]carbonyl]-1,6-diazabicyclo[3.2.1]oct-6-yl] ester, sodium salt (1:1)
PATENTS, WO 2015079329, WO 2015079389 , WO 2015063714, US 20130225554
Emergence of bacterial resistance to known antibacterial agents is becoming a major challenge in treating bacterial infections. One way forward to treat bacterial infections, and especially those caused by resistant bacteria, is to develop newer antibacterial agents that can overcome the bacterial resistant. Coates et al. (Br. J. Pharmacol. 2007; 152(8), 1147-1154.) have reviewed novel approaches to developing new antibiotics. However, the development of new antibacterial agents is a challenging task. For example, Gwynn et al. (Annals of the New York Academy of Sciences, 2010, 1213: 5-19) have reviewed the challenges in discovery of antibacterial agents.
Several compounds have been described in the prior art for use in treatment of bacterial infections (for example, see Patent Application Nos. PCT/IB2012/054296, PCT/IB2012/054290, US20130225554, PCT/US2010/060923, PCT/EP2010/067647, PCT/US2010/052109, PCT/US2010/048109, PCT/GB2009/050609, PCT/EP2009/056178, PCT/US2009/041200, PCT/US2013/034562, PCT/US2013/034589, PCT/IB2013/053092 and PCT/IB2012054706). However, there remains a need for potent antibacterial agents for preventing and/or treating bacterial infections, including those caused by bacteria that are resistant to known antibacterial agents.

PATENT
https://encrypted.google.com/patents/WO2015079329A2?cl=en



Formula (I)
Scheme -1

Formula (VII) Formula (VIII)

Formula (III) Formula (X) Formula (IX)
Scheme 2
Example 1
Synthesis of fert-butyl (25)-2-r(aminooxy)methyllpyrrolidine-l-carboxylate
Step 1: Synthesis of l-(tert-butoxycarbonyl)-(25)-pyrrolidine-2-carboxylic acid (VII):
To a stirred suspension of (2S)-pyrrolidine-2-carboxylic acid (L-proline) (200 g, 1.73 mol) in 1,4-dioxan and water mixture (1: 1, 1000 ml : 1000 ml) was added a solution of sodium hydroxide (138.97 g, 3.47 mol in 740 ml water) over a period of 20 minutes at 0 °C. Bi-feri-butyl dicarbonate (415.3 ml, 1.9 mol in 400 ml 1,4-dioxan) was added to the resulting clear solution over a period of 30 minutes, at temperature of about 0-5 °C. The reaction mixture was allowed to warm to room temperature and stirred for 16 hours. After completion of the reaction (monitored by thin layer chromatography), the reaction mixture was concentrated to 40 % of the initial volume under reduced pressure at 40-50 °C. The pH of the residual mixture was adjusted to 2 – 2.5 using 30 % aqueous potassium hydrogen sulphate at 15 °C under continuous stirring. The separated solid was filtered under suction and washed with water (2×400 ml) and dried under reduced pressure (4 mm Hg), to obtain 370 g of l-(ieri-butoxycarbonyl)-(25)-pyrrolidine-2-carboxylic acid (VII) as white solid.
Analysis:
Mass: 216 (M+l), for Molecular Weight: 215.24 and Molecular Formula:
1H NMR (400 MHz, CDC13): δ 10.60 (s, 1H), 4.35-4.24 (dd, 1H), 3.54-.3.34 (M, 2H), 2.27-1.91 (unresolved, 4H), 1.47-1.41 (d, 9H);
Purity as determined by HPLC: 99.92 %.
Step 2: Synthesis of tert-iutyl-(25)-2-(hydroxymethyl)-pyrrolidine-l-carboxylate (IX):
N-Methylmorpholine (113 ml, 1.114 mol) was added to the suspension of \-{tert-butoxycarbonyl)-(25)-pyrrolidine-2-carboxylic acid (VII, 30 g, 139 mmol) in tetrahydrofuran (2000 ml) under stirring at temperature of about 0 °C. Ethyl chloroformate (106 ml, 1.114 mol) was added drop- wise to the above obtained clear solution over a period of 30 minutes. After stirring for 1 hour, the resulting suspension was filtered over celite and the residue was washed with tetrahydrofuran (2×200 ml). To the combined filtrate was added dropwise a solution of sodium borohydride (42.1 g, 1.114 mol) in 210 ml water, containing a catalytic amount of sodium hydroxide, at temperature of about -10 °C over a period of 1-2 hours under stirring. The reaction mixture was allowed to warm to room temperature and stirred further for an hour. The reaction mixture was filtered through celite bed and the filtrate concentrated under reduced pressure to yield 180 g of ieri-butyl(25)-2-(hydroxymethyl)-pyrrolidene-l-carboxylate (IX) as colorless oil.
Analysis:
Mass: 202 (M+l), for Molecular Weight: 201.2 and Molecular Formula: C10H19NO3;
1H NMR (400 MHz, CDC13): δ 3.94-.3.92 (m, 1H), 3.80 (board, 1H), 3.63-3.54 (m, 2H), 3.45-3.40 (m, 1H), 3.32-3.28 (m, 1H), 2.01-1.96 (m, 1H), 1.84-1.75 (m, 2H), 1.63 (m, 1H), 1.45 (s, 9H);
Purity as determined by HPLC: 87.7 %.
Step 3: Synthesis of fert-butyl (25)-2-[[(l,3-dihydro-l,3-dioxo-2H-isoindol-2-yl)oxy] methyl] -pyrrolidine-1 -carboxylate (X) :
Triphenylphosphine (328.4 g, 1.253 mol) in tetrahydrofuran (1260 ml) was added to solution of Diisopropyl azodicarboxylate (253.3 g, 1.253 mol) in tetrahydrofuran at temperature of -15 °C under stirring. After stirring for an hour, N-feri-butoxylcarbonyl-L-prolinol (IX) (180 g, 0.895 mol) in tetrahydrofuran (540 ml) was added to the resulting mixture over a period of 15 minutes. After stirring the mixture for 45 minutes, N-Hydroxy phthalimide (146 g, 0.895 mol) was added and the mixture was allowed to warm to room temperature and stirred further for 16 hours. The solvent was evaporated under reduced pressure and residual oil was dissolved in dichloromethane (5000 ml) and washed with an aqueous 5 % sodium hydrogen carbonate solution (2×300 ml). The organic layer was dried over anhydrous sodium sulfate and the solvent evaporated under reduced pressure to obtain viscous oil. Diisopropyl ether (720 ml) was added to the oil, the mixture was stirred well and separated solid was filtered under suction. The filtrate was concentrated under reduced pressure and the residue was further purified by chromatography over a silica gel column (60 -120 mesh) and eluted with mixtures of ethyl acetate and hexane. Upon concentration of the combined eluted fractions, 230 g of teri-butyl (25)-2-[[( l,3-dihydro- l,3-dioxo-2H-isoindol-2-yl)oxy]methyl]-pyrrolidine- l-carboxylate (X) was obtained as yellow oil.
Analysis:
Mass: 347.3 (M+l), for Molecular Weight: 346.39 and Molecular Formula: ![]()
1H NMR (400 MHz, CDCI3): δ 7.80-7.78 (m, 2H), 7.72-7.70 (m, 2H), 4.32 (brs, 1H), 4.05 (brs, 2H), 3.36-3.31 (m, 2H), 2.27-2.25 (m, 1H), 2.08(m, 1H), 1.88-1.87 (m, 2H), 1.43 (s, 9H).
Step 4: Synthesis of fert-butyl (25)-2-[(aminooxy)methyl]pyrrolidine-l-carboxylate (HI):
To a stirred solution of the compound of Formula (X) ( 100 g, 0.288 mol) in dichloromethane (2000 ml) was added 99 % hydrazine hydrate (28.9 g, 0.577 mol) drop-wise over a period of 30 minutes at temperature of about 25 °C. The stirring was continued further for a period of 3 hours. The separated solid was filtered and the solid washed with additional dichloromethane (2 x 500 ml). The combined organic layer was collected and washed with water (2 x 500 ml). The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain 62.4 g of tert-butyl (25)-2-[(aminooxy)methyl]pyrrolidine-l-carboxylate (III) as a colorless oil. This was used as such for the next reaction without further purification.
Analysis:
Mass: 215.1 (M- l), for Molecular Weight: 216.2 and Molecular Formula:
Example 2
Synthesis of (25,5R)-7-oxo-N-r(25)-pyrrolidin-2-yl-methyloxyl-6-(sulfooxy)-l,6- diazabicvclor3.2.11octane-2-carboxamide (I)
Step 1: Synthesis of tert-butyl (25)-2-{[({[25,5R)-6-(benzyloxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]methyl}pyyrolidine-l-carboxylate
(IV):
Sodium(25,5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II, 77.4 g, 0.259 mol; prepared according to the procedure disclosed in Indian patent application No. 699/MUM/2013) was dissolved in water (774 ml) to obtain a clear solution. To the clear solution was added EDC.HC1 (120.8 g, 0.632 mol) at temperature of about 15°C and after 10 minutes a solution of tert-buty\ (25)-2-[(aminooxy)methyl]pyrrolidine-l-carboxylate (III, 62.4 g, 0.288 moles prepared as per the literature procedure depicted in scheme 2) in dimethylformamide (125 ml) was added drop wise under continuous stirring at temperature of about 10 °C. The reaction mass was allowed to warm to temperature of about 25°C and then HOBt (38.96 g, 0.288 mol) was added in small portions over a period of 15 minutes and the resulting mixture was further stirred at room temperature for 16 hours. The reaction progress was monitored using thin layer chromatography using mixture of acetone and hexane (35: 65) as solvent system. The resulting suspension was filtered and the residue was washed with water (200 ml). The residual white solid was suspended in water (200 ml) and the mixture stirred with heating at temperatyre of about 50 °C for 3 hours. The resulting suspension was filtered, the residue dried at atmospheric temperature and then dried under vacuum to obtain 105 g of ierr-Butyl(25)-2- { [( { [25,5R)-6-(benzyloxy)-7-oxo- l,6-diazabicylco[3.2. l]oct-2-yl]carbonyl} amino)oxy]methyl}pyyrolidine-l-carboxylate (IV) as off white solid.
Analysis:
Mass: 475.4 (M+l), for Molecular Weight of 474.56 and Molecular Formula of ![]()
1H NMR (400 MHz, CDCI3): δ 10.16 (br s, 1H), 7.43-7.35 (m, 5H), 5.06-4.88 (dd, 2H), 4.12 (s, 1H), 3.94-.393 (d, 2H), 3.83 (unresolved s, 1H), 3.75-3.73 (m, 1H), 3.37-3.28 (dt, 2H), 3.02-2.86 (dd, 2H), 2.31-2.26 (m, 1H), 2.02-1.84 (m, 6H), 1.71-1.68 (m, 1H), 1.45 (s, 9H).
Step 2: Synthesis of tert-butyl(25)-2-{[({[25,5R)-6-hydroxy-7-oxo-l,6-diazabicylco
[3.2.1]oct-2-yl]carbonyl}amino)oxy]methyl}pyyrolidine-l-carboxylate (V):
tert-butyl(25)-2-{ [({ [25,5R)-6-(benzyloxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl] carbonyl}amino)oxy]methyl}pyyrolidine-l-carboxylate (IV, 85 g, 0.179 mol) was dissolved in a mixture of dimethylformamide and dichloro methane (1: 1, 425 ml : 425 ml) to obtain a clear solution. To this solution was added 10 % Pd-C (17 g, 50 % wet) catalyst. The suspension was stirred for 4 hours under 7 psi hydrogen atmosphere at temperature of about 25 °C. The resulting mixture was filtered through celite under suction. The residue was washed with dichloromethane (170 ml). The solvent from the filtrate was evaporated under reduced pressure to furnish 68.8 g of tert-buty\(2S)-2-{ [( { [25,5i?)-6-hydroxy-7-oxo- l,6-diazabicylco[3.2. l]oct-2-yl]carbonyl} amino)oxy] methyl}pyyrolidine-l-carboxylate (V) as oil. The obtained product was used as such for the next reaction without further purification.
Analysis:
Mass: 385.4 (M+l), for Molecular Weight of 384.4 and Molecular Formula of C17H28N406.
Step 3: Synthesis of tert-butyl(25)-2-{[({[25,5R)-6-(sulfooxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]methyl}pyyrolidine-l-carboxylate, tetra butyl ammonium salt (VI):
To solution of ieri-butyl(25)-2-{ [({ [25,5i?)-6-hydroxy7-oxo-l,6-diazabicylco [3.2.1]oct-2-yl]carbonyl}amino)oxy]methyl}pyyrolidine-l-carboxylate (V, 68.8 g, 0.178 mol) in dimethylformamide, (345 ml) was added sulfur trioxide dimethylformamide complex (30 g, 0.196 mol) under stirring at temperature of about 10 °C. The reaction mass was stirred at the same temperature for 30 minutes and then allowed to warm to room temperature. After 2 hours solution of tetra butyl ammonium acetate (59.09 g, 0.196 mol) in water (178 ml) was added to the reaction mixture under stirring. After 2 hours, the solvent from the reaction mixture was evaporated under reduced pressure to obtain an oily residue. The oily mass was co-evaporated with xylene (2×140 ml) to obtain thick mass. This mass was partitioned between dichloromethane (690 ml) and water (690 ml). The organic layer was separated and the aqueous layer re-extracted with dichloromethane (345 ml). The combined organic extracts were washed with water (3×345 ml) and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure and the resulting oily mass was triturated with ether (3×140 ml), each time the ether layer was decanted and finally the residue was concentrated under reduced pressure to obtain 102 g of ieri-butyl(25)-2- { [({ [25,5i?)-6-(sulfooxy)-7-oxo- l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]methyl}pyyrolidine- l-carboxylate, tetrabutyl ammonium salt (VI) as fluffy material.
Analysis:
Mass: 463.4 (M- l without TBA), for Molecular Weight of 705.96 and Molecular Formula of C33H63N5O9 S;
1H NMR (400 MHz, CDCI3): δ 10.2 (s, 1H), 4.35 (s, 1H), 4.14 (s, 1H), 3.91 -3.92 (d, 2H), 3.74 (m, 1H), 3.36-3.27 (m, 10H), 2.96-2.88 (dd, 2H), 2.31-2.26 (m, 2H), 2.19-1.98 (m, 2H), 1.95-1.70 (m, 4H), 1.68- 1.62 (p, 8H), 1.49- 1.40 (m, 17H), 1.02-0.98 (t, 12H).
Step 4: (25,5R)-7-oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)-l,6-diaza bicyclo [3.2.1]octane-2-carboxamide (I):
feri-butyl(25)-2-{ [( { [25,5i?)-6-(sulfooxy)-7-oxo-l ,6-diazabicylco[3.2.1]oct-2-yl] carbonyl}amino)oxy]methyl}pyyrolidine- l-carboxylate, tetrabutylammonium salt (VI) (50 g, 0.0708 mol) was dissolved in dichloromethane (250 ml) and to the clear solution was slowly added trifluoroacetic acid (250 ml) at temperature of about -10 °C over a period of 1 hour under stirring. After stirring for an hour, the resulting mixture was poured into hexane (2500 ml) and the oily layer was separated. This procedure was repeated one more time and finally the separated oily layer was added to diethyl ether (500 ml) under vigorous stirring at temperature of about 25 °C. The ether layer was removed by decantation from the precipitated solid. This procedure was repeated twice again with diethyl ether (2x500ml). The solid thus obtained was stirred with fresh dichloromethane (500 ml) for 30 minutes and filtered. The residual solid was dried at temperature of about 45 °C under reduced pressure to yield 25 g of (25,5i?)-7-Oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)- l,6-diazabicyclo[3.2.1]octane-2-carboxamide (I) in amorphous form. The XRD of the obtained amorphous form is shown in Figure 1.
Analysis:
Mass: 363.2 (M- l), for Molecular Weight: 364.37 and Molecular Formula: C12H2oN407S;
1H NMR (400 MHz, DMSO-D6): δ 1 1.73 (s, 1H), 8.62-8.83 (d, 2H), 3.88-4.00 (m, 3H), 3.74-3.81 (m, 2H), 3.19 (t, 2H), 2.94-3.04 (dd, 2H), 1.96-2.03 (m, 2H), 1.80-1.92 (m, 3H), 1.54- 1.73 (m, 3H);
Purity as determined by HPLC: 90.30 %.
Example 3
Preparation of Crystalline Form I of (25,5R)-7-oxo-N-r(25)-pyrrolidin-2-yl- methyloxyl-6-(sulfooxy)-l,6-diaza bicvclor3.2.11octane-2-carboxamide
The amorphous solid obtained in the Step 4 of Example 2 was dissolved in water (75 ml) and to this solution isopropanol (200 ml) was slowly added at temperature of about 25 °C. The solution was further stirred for 12 hours. The separated solid thus obtained was filtered and washed with additional isopropanol (25 ml) and dried under reduced pressure to obtain 19 g of (25,5i?)-7-Oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)-l ,6-diazabicyclo[3.2.1]octane-2-carboxamide as crystalline Form I. The XRD of the obtained crystalline Form I is shown in Figure 2.
X-ray powder diffraction pattern comprising peak at (2 Theta Values): 8.08 (± 0.2), 1 1.45 (± 0.2), 16.26 (± 0.2), 17.89 (± 0.2), 18.15 (± 0.2), 19.66 (± 0.2), 21.15 (± 0.2), 23.55 (± 0.2), 24.23 (± 0.2), 24.94 (± 0.2), 25.66 (± 0.2) and 29.41 (± 0.2).
Typical X-ray analysis was performed as follows. Pass the test substance through sieve #100 BSS or gently grind it with a mortar and pestle. Place the test substance uniformly on a sample holder having cavity surface on one side, press the sample and cut into thin uniform film using a glass slide in such a way that the surface of the sample should be smooth and even. Record the X-ray diffractogram using the following instrument parameters:
Instrument : X-Ray Diffractometer
(PANalytical, Model X’Pert Pro
MPD)
Target source : CuK(a)
Antiscattering slit (Incident beam) : 1°
Programmable Divergent slit : 10 mm (fixed)
Anti- scattering slit (Diffracted beam) : 5.5 mm
Step width : 0.02°
Voltage : 40 kV
Current : 40 mA
Time per step : 30 seconds
Scan range : 3 to 40°
Example 4
Preparation of Pure (25,5R)-7-oxo-N-r(25)-pyrrolidin-2-yl-methyloxyl-6-(sulfooxy)- l,6-diazabicyclor3.2.11octane-2-carboxamide
(25,5i?)-7-Oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)- l,6-diazabicyclo [3.2.1] octane-2-carboxamide (5 g) was slowly dissolved in water (50 ml) under stirring until clear solution appears. To this clear solution 350 ml of isopropanol was added drop wise under stirring over the period of 2 hours. Formation of fine white precipitates was observed after the completion of the addition of isopropanol. The resulted fine suspension was stirred at temperature of about 25 °C for 20 hours. The formed white precipitates were filtered and vacuum dried at temperature of about 30-40 °C, under reduced pressure (2 mm Hg) to get 4.4 g of (2S,5i?)-7-oxo-N-[(2S)-pyrroMin-2-yl-methyloxy]-6-(sulfooxy)-l,6-diazabicyclo [3.2.1] octane-2-carboxamide.
The above obtained (25,5i?)-7-oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)-l,6-diazabicyclo[3.2.1]octane-2-carboxamide (3.4 gm) was dissolved in 34 ml of water to get clear solution. To the obtained clear solution 170 ml of isopropanol was added drop wise over a period of 1 hour. Formation of fine oily globules was observed and allowed to stand still for 15 minutes. The upper clear water and isopropanol layer was decanted from the oily mass. The clear decanted solution was allowed to stand at temperature of about 25 °C for 48 hours. Formation of crystals was observed and were collected by filtration. The collected crystals were dried at temperature of about 30-40 °C, under reduced pressure (2 mm Hg) to get 2 g of (2S,5i?)-7-oxo-N-[(2S)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)- 1 ,6-diazabicyclo[3.2.1 ]octane-2-carboxamide which was analyzed for content of various components using HPLC and the results are described in Table 1.
The relative % content of (25,5i?)-7-oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)-l,6-diazabicyclo[3.2.1]octane-2-carboxamide with other substances (Table 1) was determined using HPLC (Agilent 1100 or equivalent). The HPLC column having 250 mm length and 4.6 mm ID packed with 5 μ particles of octa-decyl silane (ODS) was used. Mobile phase A used was a mixture of buffer (0.02 M potassium dihydrogen phosphate in HPLC grade water, pH adjusted to 2.5 with orthophosphoric acid and again readjusted to 7.0 with dilute ammonia), HPLC grade water and acetonitrile in a ratio of 40 : 60 : 0.2; v/v/v. Mobile phase B was mixture of buffer and acetonitrile in a ratio of 40 : 60; v/v. Mobile phase was run in gradient mode. Initially mobile phase A and B was run at 100 : 0 for 15 minutes, slowly ratio of mobile phase B was raised to 100 % in 10 minutes, held for 10 minutes at this concentration and brought back to initial condition in next 5 minutes and held for 10 minutes before next run. Flow rate of mobile phase was maintained at 1.0 ml/min. Column temperature was maintained at temperature of about 30°C. Detection was carried out using UV detector at wavelength 225 nm. Test solutions were prepared in mobile phase A. The method is capable of resolving diastereomers (Table 1, Sr. No. 1 and 2) with resolution of not less than 2.0.

WCK 5222, Wockhardt receives QIDP status for its new drug WCK 5222 from USFDA
WCK 5222
Watch this post as I get to the structure…………..
DEC2015
Wockhardt has received Qualified Infectious Disease Product (QIDP) status for its new drug WCK 5222, a product from its breakthrough New Drug Discovery program in Anti Infectives from the US Food and Drug Administration (FDA).
This is the fourth product from the company to receive this coveted status. During last year, the company has received approval for WCK 771 & WCK 2349 and in early this year approval was received for WCK 4873. The only company globally to receive QIDP status for 4 drugs from US FDA.
Wockhardt is one of the few companies with end to end integrated capabilities for its products, starting with the manufacture of the oral and sterile API’s, the dose forms and marketing through wholly owned subsidiary in the US, enabling the company to capture maximum value.
Ten compounds generally represented by a general Formula (I) were used and are as follows:
(a) Sodium salt of ir ns-7-oxo-6-sulphooxy-l ,6-diazabicyclo[3.2.1]-octane-2-carbonitrile (Compound A);

(b) trans-sulphuric acid mono-[2-(5-carboxamido)-[l ,3,4]-oxadiazol-2-yl)-7-oxo-l,6-diazabicyclo[3.2.1]-octan-6-yl] ester (Compound B);

(c) trans-sulphuric acid mono-[2-(5-(piperidin-4-yl)-[l ,3,4]-oxadiazol-2-yl)-7-oxo-l,6-diazabicyclo[3.2.1]-octan-6-yl] ester (Compound C);

(d) trans-sulphuric acid mono-[2-(5-azetidin-3-ylmethyl-[l ,3,4]-oxadiazol-2-yl)-7-oxo-l,6-diazabicyclo[3.2.1]-octan-6-yl] ester (Compound D);

(e) (25,5i?)-7-Oxo-6-sulphooxy-2-[N’-((i?)-piperidine-3-carbonyl)-hydrazinocarbonyl] -1,6-diaza-bicyclo[3.2.1]octane (Compound E);

(f) (25, 5i?)-7-Oxo-N-[(25)-pyrrolidin-2-ylmethoxy]-6-(sulfooxy)-l,6-diaza bicyclo [3.2.1] octane-2-carboxamide (Compound F);

(g) (25,5i?)-7-Oxo-6-sulphooxy-2-[N’-((i?)-pyrrolidine-3-carbonyl)-hydrazinocarbonyl]-l ,6-diaza -bicyclo[3.2.1]octane (Compound G);

(h) (25,5i?)-7-Oxo-N-[(25)-piperidine-2-ylmethyloxy]-6-(sulfooxy)-l ,6-diazabicyclo
octane-2-carboxamide (Compound H);

(i) trans-sulphuric acid mono-[2-(5-((5)-l-amino-ethyl)-[l ,3,4]-oxadiazol-2-yl)-7-oxo-l,6-diazabicyclo[3.2.1]-octan-6-yl] ester (Compound I); and

j) trans-sulphuric acid mono-[2-(5-((5)-pyrrolidin-2-yl)-[l,3,4]-oxadiazol-2-yl)-7-oxo-l,6-diazabicyclo[3.2.1]-octan-6-yl] ester (Compound J).

////
SEMAPIMOD

Semapimod Mesylate
CPSI-2364, AXD-455, CN-1493, CNI 1493
CAS No. 352513-83-8(Semapimod base)
Cas 164301-51-3 4x HCl
CAS 872830-80-3 (Semapimod mesylate)
MW 1129
CROHNS DISEASE, PHASE 1
N,N’-bis[3,5-bis[(E)-N-(diaminomethylideneamino)-C-methylcarbonimidoyl]phenyl]decanediamide
Decanediamide, N,N’-bis[3,5-bis[1-[(aminoiminomethyl)hydrazono]ethyl]phenyl]-, methanesulfonate
N,N’-Bis(3,5-bis(1-(carbamimidoylhydrazono)ethyl)phenyl)decanediamide
- N,N’-Bis(3-acetylphenyl)decane diamide tetrakis (amidinohydrazone)
- N,N’-bis(3,5-bis{(1E)-N-[amino(imino)methyl]ethanehydrazonoyl}phenyl)decanediamide
- N,N’-bis[3,5-bis[(E)-N-(diaminomethylideneamino)-C-methylcarbonimidoyl]phenyl]decanediamide
- N,N’-bis[3,5-bis[(E)-N-guanidino-C-methyl-carbonimidoyl]phenyl]decanediamide
A nitric oxide synthesis inhibitor and a p38 MAPK inhibitor potentially for the treatment of Crohn’s disease.

Crohn’s disease (CD) is a chronic inflammatory disease involving the upper and lower gastrointestinal tract and characterized by abdominal pain, weight loss, gastrointestinal bleeding and formation of fistulas between loops of bowel and from the bowel to the skin or other organs. Current therapy for active Crohn’s disease consists of symptomatic treatment, nutritional therapy, salicylates and immunosuppressants or surgical management.
Tumor necrosis factor a (TNF-a) plays a central role in the initiation and amplification of the granulomatous inflammatory reaction seen in CD (van Deventer, 1997). Increased TNF-a is present in gut mucosa as well as in stool of patients with active CD (Braegger et al, 1992). CNI-1493 is a synthetic guanylhydrazone compound that is an inhibitor of TNF-a synthesis. A monoclonal antibody to TNF, infliximab, is now approved for treatment of CD, but not all patients respond and many who do respond eventually become refractory to this treatment as well.
CNI-1493 is a synthetic compound which blocks the production of several inflammatory cytokines, including TNF. Because it blocks production of multiple inflammatory mediators, it may be more active than products targeted to a specific cytokine. In addition, as it is not a biologic, it should not cause hypersensitivity reactions or induce formation of antibodies.
The purpose of this trial is to determine if CNI-1493 is safe and effective in treating patients with moderate to severe Crohn’s Disease in a placebo controlled setting………https://clinicaltrials.gov/ct2/show/NCT00038766
Semapimod (INN), formerly known as CNI-1493, is an investigational new drug which has anti-inflammatory,[1] anti-cytokine,[2] immunomodulatory,[3] antiviral[4] and antimalarial[5] properties.
History
Semapimod was developed at the former Picower Institute for Medical Research, and is now licensed to Cytokine PharmaSciences. In 2000, Cytokine PharmaSciences licensed anti-infective applications of semapimod to Axxima Pharmaceuticals, but Axxima became insolvent in Dec. 2004 and its assets were acquired by GPC Biotech, which has recently merged into Agennix AG[1]. Although the disposition of Axxima’s partial rights to semapimod was not specified in these merger announcements, Cytokine PharmaSciences does not currently list any licensees for semapimod on its website.
Mechanism of action
Semapimod was first developed to inhibit nitric oxide synthesis by inflammatory macrophages, via inhibition of the uptake of arginine which macrophages require for nitric oxide synthesis.[1] Subsequently it was found that suppression of nitric oxide synthesis occurred even at semapimod concentrations 10-fold less than required for inhibition of arginine uptake, suggesting that this molecule was a more general inhibitor of inflammatory responses.[2] Further work revealed that semapimod suppressed the translation efficiency of tumor necrosis factor production.[6] Specifically, semapimod was found to be an inhibitor of p38 MAP kinase activation.[7] Surprisingly, however, the primary mode of action in vivo is now thought to be via stimulation of the vagus nerve, thereby down-regulating inflammatory pathways via the recently discovered cholinergic anti-inflammatory pathway.[8][9]
Pharmacology and clinical trials
In a preclinical study in rats, semapimod was found to suppress cytokine-storm induction by the anticancer cytokine interleukin-2 (IL-2) without decreasing its anticancer properties, allow larger doses of IL-2 to be administered.[10] A subsequent phase I trial in humans failed to show an increase in the tolerated dose of IL-2, although indications of pharmacological activity as an inhibitor of tumor necrosis factor production were observed.[11]
In a preliminary clinical trial of semapimod in patients with moderate to severe Crohn’s disease, positive clinical changes were observed, including endoscopic improvement, positive responses in some patients not responding to infliximab, healing of fistulae, and indications for tapering of steroids; no significant adverse effects were observed.[12]
In a small clinical trial against post-ERCP pancreatitis, significant suppression was not observed, although investigators observed a significant reduction of the incidence of hyperamylasemia and the levels of post-ERCP amylase.[13]
In the clinical trials above, semapimod tetrahydrochloride was administered by intravenous injection. This route has drawbacks such as dose-limiting phlebitis.[2] Recently Cytokine PharmaSciences has announced the development of novel salt forms of semapimod which are said to be orally absorbable; a phase I clinical trial of one of these salt forms, CPSI-2364, has been completed, and a phase II trial is planned for 2010.[3][4]
Chemistry
Semapimod is synthesized by reacting 3,5-diacetylaniline[14] with sebacoyl chloride in the presence of pyridine, followed by reaction of the resulting tetraketone with aminoguanidine hydrochloride.[1]
PATENT
-
N,N′-bis(3,5-diacetylphenyl) decanediamide tetrakis (amidinohydrazone) tetrahydrochloride (CNI-1493), which has the following structural formula:
-

SYNTHESIS
The reaction of decanedioyl dichloride (I) with 3,5-diacetylaniline (II) by means of pyridine in dichloromethane gives the corresponding diamide (III), which is condensed with aminoguanidine (IV) in refluxing aqueous ethanol to afford the target tetrakis amidinohydrazone. EP 0746312; EP 1160240; US 5599984; WO 9519767
http://www.google.com/patents/EP0746312A1?cl=en
References
- 1 Bianchi, M.; Ulrich, P.; Bloom, O.; Meistrell m, M. , I. I.; Zimmerman, G. A.; Schmidtmayerova, H.; Bukrinsky, M.; Donnelley, T.; Bucala, R.; Sherry, B.; Manogue, K. R.; Tortolani, A. J.; Cerami, A.; Tracey, K. J. (Mar 1995). “An inhibitor of macrophage arginine transport and nitric oxide production (CNI-1493) prevents acute inflammation and endotoxin lethality”. Molecular Medicine (Cambridge, Mass.) 1 (3): 254–266. ISSN 1076-1551. PMC 2229913. PMID 8529104.
- 2
- Bianchi, M.; Bloom, O.; Raabe, T.; Cohen, P. S.; Chesney, J.; Sherry, B.; Schmidtmayerova, H.; Calandra, T.; Zhang, X.; Bukrinsky, M.; Ulrich, P.; Cerami, A.; Tracey, K. J. (Mar 1996). “Suppression of proinflammatory cytokines in monocytes by a tetravalent guanylhydrazone”. The Journal of Experimental Medicine 183 (3): 927–936. doi:10.1084/jem.183.3.927. ISSN 0022-1007. PMC 2192362. PMID 8642296.
- 3
- Martiney, J.; Rajan, A. J.; Charles, P. C.; Cerami, A.; Ulrich, P. C.; MacPhail, S.; Tracey, K. J.; Brosnan, C. F. (Jun 1998). “Prevention and treatment of experimental autoimmune encephalomyelitis by CNI-1493, a macrophage-deactivating agent” (Free full text). Journal of immunology (Baltimore, Md. : 1950) 160 (11): 5588–5595. ISSN 0022-1767. PMID 9605164.
- 4
- Hauber, I.; Bevec, D.; Heukeshoven, J.; Krätzer, F.; Horn, F.; Choidas, A.; Harrer, T.; Hauber, J. (Jan 2005). “Identification of cellular deoxyhypusine synthase as a novel target for antiretroviral therapy”. The Journal of Clinical Investigation (Free full text) 115 (1): 76–85. doi:10.1172/JCI21949. ISSN 0021-9738. PMC 539192. PMID 15630446.
- 5
- Specht, S.; Sarite, R.; Hauber, I.; Hauber, J.; Görbig, F.; Meier, C.; Bevec, D.; Hoerauf, A.; Kaiser, A. (May 2008). “The guanylhydrazone CNI-1493: an inhibitor with dual activity against malaria-inhibition of host cell pro-inflammatory cytokine release and parasitic deoxyhypusine synthase”. Parasitology research 102 (6): 1177–1184. doi:10.1007/s00436-008-0891-x. ISSN 0932-0113. PMID 18256853.
- 6
- Cohen, P. S.; Nakshatri, H.; Dennis, J.; Caragine, T.; Bianchi, M.; Cerami, A.; Tracey, K. J. (Apr 1996). “CNI-1493 inhibits monocyte/macrophage tumor necrosis factor by suppression of translation efficiency”. Proceedings of the National Academy of Sciences of the United States of America 93 (9): 3967–3971. Bibcode:1996PNAS…93.3967C. doi:10.1073/pnas.93.9.3967. ISSN 0027-8424. PMC 39469. PMID 8632999.
- 7
- Cohen, P. S.; Schmidtmayerova, H.; Dennis, J.; Dubrovsky, L.; Sherry, B.; Wang, H.; Bukrinsky, M.; Tracey, K. J. (May 1997). “The critical role of p38 MAP kinase in T cell HIV-1 replication”. Molecular Medicine (Cambridge, Mass.) 3 (5): 339–346. ISSN 1076-1551. PMC 2230081. PMID 9205949.
- 8
- Tracey, J. (Feb 2007). “Physiology and immunology of the cholinergic antiinflammatory pathway”. The Journal of Clinical Investigation (Free full text) 117 (2): 289–296. doi:10.1172/JCI30555. ISSN 0021-9738. PMC 1783813. PMID 17273548.
- 9
- Oke, L.; Tracey, J. (Mar 2008). “From CNI-1493 to the immunological homunculus: physiology of the inflammatory reflex” (Free full text). Journal of Leukocyte Biology 83 (3): 512–517. doi:10.1189/jlb.0607363. ISSN 0741-5400. PMID 18065685.
- 10
- Kemeny, M. M.; Botchkina, G. I.; Ochani, M.; Bianchi, M.; Urmacher, C.; Tracey, K. J. (1998). “The tetravalent guanylhydrazone CNI-1493 blocks the toxic effects of interleukin-2 without diminishing antitumor efficacy”. Proceedings of the National Academy of Sciences of the United States of America 95 (8): 4561–4566. Bibcode:1998PNAS…95.4561K. doi:10.1073/pnas.95.8.4561. PMC 22529. PMID 9539777.
- 11
- Atkins, M. B.; Redman, B.; Mier, J.; Gollob, J.; Weber, J.; Sosman, J.; MacPherson, B. L.; Plasse, T. (2001). “A phase I study of CNI-1493, an inhibitor of cytokine release, in combination with high-dose interleukin-2 in patients with renal cancer and melanoma”. Clinical Cancer Research 7 (3): 486–492. PMID 11297238.
- 12
- Hommes, D.; Van Den Blink, B.; Plasse, T.; Bartelsman, J.; Xu, C.; MacPherson, B.; Tytgat, G.; Peppelenbosch, M.; Van Deventer, S. (2002). “Inhibition of stress-activated MAP kinases induces clinical improvement in moderate to severe Crohn’s disease”. Gastroenterology 122 (1): 7–14. doi:10.1053/gast.2002.30770. PMID 11781274.
- 13 Vanwesterloo, D.; Rauws, E.; Hommes, D.; De Vos, A.; Van Der Poll, T.; Powers, B.; Fockens, P.; Dijkgraaf, M.; Bruno, M. (2008). “Pre-ERCP infusion of semapimod, a mitogen-activated protein kinases inhibitor, lowers post-ERCP hyperamylasemia but not pancreatitis incidence”. Gastrointestinal Endoscopy 68 (2): 246–254. doi:10.1016/j.gie.2008.01.034. PMID 18455169.
- 14 Ulrich, P.; Cerami, A. (Jan 1984). “Trypanocidal 1,3-arylene diketone bis(guanylhydrazone)s. Structure-activity relationships among substituted and heterocyclic analogues”. Journal of Medical Chemistry 27 (1): 35–40. doi:10.1021/jm00367a007. ISSN 0022-2623. PMID 6690682.
| Patent | Submitted | Granted |
|---|---|---|
| Neural tourniquet [US2005282906] | 2005-12-22 | |
| Guanylhydrazone Salts, Compositions, Processes of Making, and Methods of Using [US2008262090] | 2008-10-23 | |
| Protective role of semapimod in necrotizing enterocolitis [US7795314] | 2007-12-06 | 2010-09-14 |
| METHOD OF TREATING ILEUS BY PHARMACOLOGICAL ACTIVATION OF CHOLINERGIC RECEPTORS [US2011112128] | 2011-05-12 | |
| Method of treating ileus by pharmacological activation of cholinergic receptors [US2007213350] | 2007-09-13 | |
| Pharmaceutically active aromatic guanylhydrazones [US2005171176] | 2005-08-04 | |
| Guanylhydrazone salts, compositions, processes of making and methods of using [US7244765] | 2006-01-19 | 2007-07-17 |
| GUANYLHYDRAZONE SALTS, COMPOSITIONS, PROCESSES OF MAKING, AND METHODS OF USING [US8034840] | 2008-06-19 | 2011-10-11 |
| METHOD FOR TREATING GLIOBLASTOMAS AND OTHER TUMORS [US2014323576] | 2014-03-14 | 2014-10-30 |
| Methods of treatment of fatty liver disease by pharmacological activation of cholinergic pathways [US8865641] | 2012-06-14 | 2014-10-21 |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N,N’-bis[3,5-bis[N-(diaminomethylideneamino)-C-methylcarbonimidoyl]phenyl] decanediamide tetrahydrochloride
|
|
| Identifiers | |
| CAS Number | 164301-51-3 352513-83-8 (base) |
| ATC code | None |
| PubChem | CID: 5745214 |
| UNII | 9SGW2H1K8P |
| ChEMBL | CHEMBL2107779 |
| Chemical data | |
| Formula | C34H56Cl4N18O2 |
| Molecular mass | 890.73984 g/mol |
see………http://worlddrugtracker.blogspot.in/2015/12/semapimod.html
/////////Semapimod Mesylate, CPSI-2364, AXD-455, CN-149, PHASE 1, FERRING, CNI 1493
CC(=NN=C(N)N)C1=CC(=CC(=C1)NC(=O)CCCCCCCCC(=O)NC2=CC(=CC(=C2)C(=NN=C(N)N)C)C(=NN=C(N)N)C)C(=NN=C(N)N)C
Zucapsaicin for osteoarthritis

Zucapsaicin (珠卡赛辛)
cis-Capsaicin; (Z)-Capsaicin
Zucapsaicin; Civamide; Cis-Capsaicin; 25775-90-0; (Z)-Capsaicin; (Z)-N-(4-Hydroxy-3-methoxybenzyl)-8-methylnon-6-enamide;
(Z)-N-[(4-Hydroxy-3-methoxyphenyl)methyl]-8-methylnon-6-enamide
CAS No. 25775-90-0
| MF C18H27NO3 | |
| Molecular Weight: | 305.41188 g/mol |
|---|
WINSTON INNOVATOR
SANOFI
(Zuacta®/Civanex®
A medication used to treat osteoarthritis of the knee and other neuropathic pain.TRPV1 CHANNEL AGONIST



Zucapsaicin (Civanex) is a medication used to treat osteoarthritis of the knee and other neuropathic pain. It is applied three times daily for a maximum of three months. It reduces pain, and improves articular functions. It is the cis-isomer of capsaicin. Civamide, manufactured by Winston Pharmaceuticals, is produced in formulations for oral, nasal, and topical use (patch and cream).[1]
Zucapsaicin has been tested for treatment of a variety of conditions associated with ongoing nerve pain. This includes herpes simplex infections; cluster headaches and migraine; and knee osteoarthritis.[2]
Civanex (zucapsaicin) cream is a TRPV-1 modulator in development for the treatment of signs and symptoms of osteoarthritis of the knee.
Zucapsaicin, the cis-isomer of the natural product capsaicin, is a
topical analgesic that was initially developed by Winston Pharmaceuticals
and approved in Canada in July 2010 for the treatment of
severe pain in adults with osteoarthritis of the knee.
Bronson, J.; Dhar, M.; Ewing, W.; Lonberg, N. In Annual Reports in MedicinalChemistry; John, E. M., Ed.; Academic Press, 2011; Vol. 46, p 433.
The advantagesof zucapsaicin compared with naturally-occurring capsaicin, are reported to be a lesser degree of local irritation (stinging, burning,
erythema) in patients and a greater degree of efficacy in preclinical
animal models of pain.
Bernstein, J. E. U.S. 5063060, 1991.
Bernstein, J. E. U.S. 20050084520 A1, 2005.
The analgesic action of both
zucapsaicin and capsaicin is mediated through the transient receptor
potential vanilloid type 1 (TRPV1) channel, a ligand-gated ion
channel expressed in the spinal cord, brain, and localized on neurons
in sensory projections to the skin, muscles, joints, and
gut.
Westaway, S. M. J. Med. Chem. 2007, 50, 2589.
The scale preparation of zucapsaicin likely parallels the original
approach described by Gannett and co-workers involving the
coupling of vanillylamine with (Z)-8-methylnon-6-enoyl chloride.
Gannett, P. M.; Nagel, D. L.; Reilly, P. J.; Lawson, T.; Sharpe, J.; Toth, B. J. Org.Chem. 1988, 53, 1064.
Orito and co-workers elaborated this original approach in
an effort to prepare both capsaicin and zucapsaicin on gram-scale,
Kaga, H.; Miura, M.; Orito, K. J. Org. Chem. 1989, 54, 3477.
References
- 1 Winston Pharmaceuticals website http://www.winstonlabs.com/productdevelopment/civamide.asp
- 2 Zucapsaicin information from the National Library of Medicine http://druginfo.nlm.nih.gov/drugportal
Janusz, John M.; Buckwalter, Brian L.; Young, Patricia A.; LaHann, Thomas R.; Farmer, Ralph W.; et al. Journal of Medicinal Chemistry, 1993 , vol. 36, # 18 p. 2595 – 2604
Journal of Organic Chemistry, , vol. 53, # 5 p. 1064 – 1071
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(Z)-N-[(4-Hydroxy-3-methoxyphenyl)methyl]-8-methylnon-6-enamide
|
|
| Clinical data | |
| Trade names | Civanex |
| Routes of administration |
Topical |
| Identifiers | |
| CAS Number | 25775-90-0 |
| ATC code | M02AB02 |
| PubChem | CID: 1548942 |
| ChemSpider | 1265956 |
| UNII | 15OX67P384 |
| Synonyms | Civamide; (Z)-Capsaicin; cis-Capsaicin |
| Chemical data | |
| Formula | C18H27NO3 |
| Molecular mass | 305.41188 g/mol |
////Zucapsaicin
Oc1ccc(cc1OC)CNC(CCCC\C=C/C(C)C)=O
see………..http://apisynthesisint.blogspot.in/2015/12/zucapsaicin-for-osteoarthritis.html
Iptakalim Hydrochloride 盐酸埃他卡林

Iptakalim Hydrochloride 盐酸埃他卡林
NDA Filed china
A K(ir) 6.1/SUR2B activator potentially for the treatment of pulmonary arterial hypertension.

179.7, C9H21N.HCl
CAS No. 642407-44-1(Iptakalim)
642407-63-4(Iptakalim Hydrochloride)
N-(1-methylethyl)-2,3-dimethyl-2-butylamine
| Catholic Healthcare West (D/B/A/ St. Joseph’s Hospital And Medical Center) |


Hypertension is a multifactorial disorder, and effective blood pressure control is not achieved in most individuals. According to the most recent report of the American Heart Association, for 2010, the estimated direct and indirect financial burden for managing hypertension is estimated to be $76.6 billion. Overall, almost 75% of adults with cardiovascular diseases/comorbidities have hypertension, which is associated with a shorter overall life expectancy. Alarmingly, rates of prehypertension and hypertension are increasing among children and adolescents due, in part, to the obesity epidemic we currently face. There is also the problem of an aging population and the growing rates of diabetes and obesity in adults, all factors that are associated with high blood pressure.Thus, the need is great for novel drugs that target the various contributing causes of hypertension and the processes leading to end organ damage.
Iptakalim (IPT), chemically 2, 3–dimethyl-N-(1-methylethyl)-2-butanamine hydrochloride, is novel adenosine triphosphate–sensitive potassium (KATP) channel opener. KATP channels are composed of discrete pore-forming inward rectifier subunits (Kir6.1s) and regulatory sulphonylurea subunits (SUR).IPT shows high selectivity for cardiac KATP (SUR2A/Kir6.2) and vascular KATP (SUR2B/Kir6.1 or SUR6B/Kir6.2). Because of this high selectivity, IPT does not exhibit the adverse side effects associated with the older nonspecific K+ channel openers, which limit their use to the treatment of severe or refractory hypertension. IPT produces arteriolar and small artery vasodilatation, with no significant effect on capacitance vessels or large arteries. Vasodilatation is induced by causing cellular hyperpolarization via the opening of K+ channels, which in turn decreases the opening probability of L-type Ca2+ channels. Of particular note, IPT is very effective in lowering the blood pressure of hypertensive humans but not of those with normal blood pressure.
-
The present compd relates generally to a novel method for decreasing a human’s cravings for cigarettes and reducing instances of relapse during detoxification once smoking abstinence has been achieved, and more specifically, to a method for decreasing nicotine use by treating a human with a novel type of nicotinic acetylcholine receptor antagonist, iptakalim hydrochloride (IPT).
-
Cigarette smoking is a prevalent, modifiable risk factor for increased morbidity and mortality in the United States, and perhaps in the world. Smokers incur medical risks attributable to direct inhalation. Bystanders, termed passive smokers, also incur medical risks from second-hand smoke. Society, as a whole, also bears the economic costs associated with death and disease attributable to smoking. Although the majority of smokers have tried repeatedly to quit smoking, eighty percent of smokers return to tobacco in less than two years after quitting. Therefore, tobacco dependence is a health hazard for millions of Americans.
-
Nicotine is the biologically active substance that is thought to promote the use of tobacco products by approximately one-quarter of the world populations. Tobacco-related disease is personally and economically costly to the any nation. Unfortunately, once use of tobacco has begun, it is hard for a smoker to quit because of nicotinic dependence and addiction.
-
The initiation and maintenance of tobacco dependence in a human is due to certain bio-behavioral and neuromolecular mechanisms. Nicotinic acetylcholine receptors (nAChRs) in humans are the initial binding sites for nicotine. The binding of nicotine to nAChRs is thought to modulate the brain’s “reward” function by triggering dopamine release in the human brain. The nAChRs exist as a diverse family of molecules composed of different combinations of subunits derived from at least sixteen genes. nAChRs are prototypical members of the ligand-gated ion channel superfamily of neurotransmitter receptors. nAChRs represent both classical and contemporary models for the establishment of concepts pertaining to mechanisms of drug action, synaptic transmission, and structure and function of transmembrane signaling molecules.
-
Basic cellular mechanisms of nicotinic dependence also involve the functional state changes during repeated nicotinic agonists exposure and receptor changes in the number of receptors during chronic nicotinic exposure. nAChRs can exist in many different functional states, such as resting, activated, desensitized or inactivated The activation and/or desensitization of nAChRs plays an important role in initiating nicotinic tolerance and dependence. Recovery from receptor activation and/or desensitization contributes to nicotinic withdrawal symptoms.
-
The most abundant form of nAChRs in the brain contains α4 and β2 subunits. α4β2-nAChRs bind nicotine with high affinity and respond to levels of nicotine found in the plasma of smokers. α4β2-nAChR also have been implicated in nicotine self-administration, reward, and dependence. Therefore, selective drug action at nAChRs, especially at those containing α4 subunits, is thought to be an ideal way for nicotine cessation and reducing nicotine withdrawal syndrome. Unfortunately, thus far, no optimal compound can meet this purpose. The brain-blood-barrier permeable nAChR antagonist, mecamylamine is popularly used systemically but exhibits much less nAChR subtype selectivity.
-
Although a variety of psychopharmacological effects contribute to drug reinforcement, actions on the mesolimbic dopaminergic pathway is the predominant hypothesis for mechanisms of nicotinic reward. The mesolimbic dopaminergic pathway originates in the ventral tegmental area (VTA) of the midbrain and projects to forebrain structures including the prefrontal cortex and to limbic areas such as the olfactory tubercle, the amygdala, the septal region, and the nucleus accumbens. Many studies have indicated that dopamine release in the nucleus accumbens of the human brain is “rewarding” or signals an encounter with a “reward” from the environment. Other substances, such as alcohol, cocaine, and opiates, operate in the same manner, resulting in a cycle of substance or alcohol abuse.
-
Therefore, a considerable need exists for a novel compound that can selectively block α4 subtypes of nAChRs to prevent smoking-induced “reward”, to limit increasing nicotine-induced dopamine release, and/or to diminish nicotinic withdrawal symptoms.
Patent
https://www.google.com/patents/US20040266822
Example 1
-
Production of N-(1-methylethyl)-2,3-dimethyl-2-butylamine (Compound 1): Method 1. The solution of 7.6 g (0.0745 mole) 2,3-dimethyl-2-butanol in 3.24 mL glacial acetic acid was cooled and maintained at −5 to −8 degree of centigrade (° C.), then was added 7.3 g (0.49 mole) of powdered potassium cyanide in several times under stirring. 32.4 mL concentrated sulfuric acid was added dropwise while keeping the temperatue below 20° C., after which, the reaction mixture was stirred for 3.5 hours below 20° C. and another 6 hours at room temperature, then stood overnight. After poured into ice colded water, the mixture was adjusted to pH10 with 20% aqueous sodium hydroxide solution, and extracted with ether (×4). The extract was dried over anhydrous sodium sulfate. After filtration on the next day, the dessicator was removed, and the filtrate was evaporated off the ether, then distilled in vacuum to give 8.8 g (yield 91.6%) N-[2-(2,3-dimethylbutyl)]-fomide; bp 105-108° C./5 mmHg.
-
To the mixture of 7.7 g (0.0597 mole) N-[2-(2,3-dimethylbutyl)]-formide, 6.2 mL ethanol and 51.6 mL wate, 17.4 mL concentrated hydrochloric acid was added. The reaction mixture was refluxed for 4 hours in the oil bath, then distilled off ethanol in vacuum. The residue was adjusted to above pH12 with 40% aqueous sodium hydroxide solution, and extracted with ether. The extract was dried over anhydrous potassiun carbonate. After recovering the ether, The residue was distilled at atmosphere to give 3.75 g (yield 62.2%) 2,3-dimethyl-2-butylamine, bp 97-104° C.
-
The mixture of 10.6 g (0.15 mole) 2,3-dimethyl-2-butylamine, 6.45 g (0.0524 mole) 2-bromopropane, 3.0 mL glycol and 22.0 mL toluene was added into an autoclave, and heated with stirring for 17 hours at temperature of 170° C., after which, the organic layer was separated and extracted with 6N hydrochloric acid (15 mL×4). The extract was combined and washed once with toluene, then adjusted to pH 12-13 with 4% aqueous sodium hydroxide in the ice bath. The mixture was extracted with ether and then dried over anhydrous potassium carbonate. After recovering the ether, The filtrate was distilled to yield the fraction of bp 135-145° C. (yield 68.8%). The hydrochloride’s Mp is 228-230° C. (1-PrOH-Et2O). Elemental analysis for C9H22ClN(%): Calculated C, 60.14; H, 12.34; N, 7.79, Cl 19.73; Found C, 60.14; H, 12.48; N, 7.31, Cl 19.67.
-
1H-NMR(D2O, ppm) 0.98(d, J=6.75H, 6H), 1.33(s, 6H), 1.37(d, J=6.46, 6H), 2.10(m, 1H), 3.70(m, 1H). MS(m/z) 143 (M+), 100(B).
-
Method 2. To the mixture of 288 mL glacial acetic acid, 412 g (6.86 mole) urea and 288 g (3.43 mole) 2,3-dimethyl-2-butene, the solution of 412 mL concentrated sulfuric acid and 412 mL of glacial acetic acid was added dropwise under stirring, while maintaining the reaction temperature at the range of 45° C. to 50° C., then stirred for 5 hours at the temperature of 50-55° C. The mixture stood overnight. Next day, the mixture was reacted for another 7 hours at the temperature of 50-55° C., then poured into the solution of 1200 g (30 mole) sodium hydroxide in 8L glacial water. The resulting solid was filtered, washed with water (200 mL×5) and dried to give 404 g (yield 81.8%) N-(2,3-dimethyl-2-butyl)urea as white solid, mp 175-176° C. Elemental analysis for C7H16N2O(%): Calculated C 58.30, H 11.18, N 19.42; Found C, 58.70; H, 11.54; N, 19.25, 1H-NMR(CDCl3, ppm) 0.88-0.91(d, 6H, 2×CH3), 1.26(s, 6H, 2×CH3), 2.20-2.26(m, 1H, CH), 4,45(br, 2H), 4.65(br, 1H). MS(m/z) 145.0, 144.0(M+), 143.0, 129.1, 101.0, 86.1, 69.1, 58.0(B).
-
To the mixture of 196 g (1.36 mole) N-(2,3-dimethyl-2-butyl)urea and 392 mL glycol or tri-(ethanol)amine, a solution of 118 g (2.95 mole) sodium hydroxide in 118 mL water was added. The reaction mixture was heated for 8 hours in an oil bath at temperature of 120° C., then distilled at atmosphere to collect the fraction of bp 95-102° C. To the fraction, 75 g anhydrous potassium carbonate and. 40 g sodium hydroxide were added. The resulting mixture was distilled to give 88.5 g (yield 64.3%) 2,3-dimethyl-2-butylamine as colorless liquid, bp 99-101° C.
-
1H-NMR(CDCl3, ppm) 0.88-0.91(d, 6H, 2×CH3), 1.04 (s, 6H, 2×CH3), 1.53(m, 1H, CH).
-
To a 50.0 ml autoclave, 10.6 g (0.15 mole) 2,3-dimethyl-2-butylamine, 6.45 g (0.0524 mol) 2-bromopropane, 3.0 ml glycol and 22.0 ml toluene were added, and heated with stirring for 17 hours at 170° C., after which the organic layer was seperated and extracted with 6N hydrochloric acid (15 ml×4). The extract was combined and washed once with toluene, then adjusted to pH 12-13 with 4% aqueous sodium hydroxide in the ice bath. The mixture was extracted with ether and then dried over anhydrous potassium carbonate the ether was recovered, and distilled to give the fraction of bp 135-145° C. (yield 68.8%). mp of the hydrochloride is 228-230° C., (i-PrOH: Et2O). Elemental analysis for C9H22ClN(%): Calculated C, 60.14; H, 12.34; N, 7.79, Cl 19.73; Found C 60.14, H 12.48, N 7.31, Cl 19.67. 1H-NMR(D2O, ppm) 0.98(d, J=6.75H, 6H), 1.33(s, 6H), 1.37(d, J=6.46, 6H), 2.10(m, 1H), 3.70(m, 1H). MS(m/z) 143 (M+), 100(B).
-
Method 3. a solution of 0.10 mole enamine (prepared from the condensation of methyl iso-propyl ketone and iso-propylamine) in 20 mL hexane was filled with N2 and added dropwise to a solution containing 0.10 mole lithium methide with stirring in ice bath. After the reaction is complete, the mixture was poured into 500 g glacial water, and stirred. The aqueous layer was extracted with ether (×2). The resulting organic layer was concentrated. 3N hydrochloric acid was added to acified the organic layer to pH<1. The mixture was kept minutes and adjusted to pH>11 with 10% aqueous sodium hydroxide, then extracted with ether (×3). The extract was dried over anhydrous potassium carbonate and filtered. The filtrate was distilled at atmosphere to give a fraction of bp 140-145° C. with a yield of 80%.
REF
http://www.google.com/patents/US20060293393
//////Iptakalim Hydrochloride, 盐酸埃他卡林 , K(ir) 6.1/SUR2B activator, pulmonary arterial hypertension, nda
see……….http://apisynthesisint.blogspot.in/2015/12/iptakalim-hydrochloride.html
Tesmilifene , Antagonist of intracellular histamine

Tesmilifene
BMS-217380; BMY-33419; DPPE
CAS No. 98774-23-3(Tesmilifene), 92981-78-7(Tesmilifene hydrochloride)
Tesmilifene
CAS 98774-23-3
N,N-Diethyl-2-[4-(phenylmethyl)phenoxy]ethanamine
DPPE
MFC19H25NO
MW 283.41
Percent Composition: C 80.52%, H 8.89%, N 4.94%, O 5.65%

Hydrochloride
CAS 92981-78-7
BMS-217380-01; BMY-33419
MF C19H25NO.HCl
MF 319.87
Percent Composition: C 71.34%, H 8.19%, N 4.38%, O 5.00%, Cl 11.08%
Properties: White crystals from isopropanol + acetone (3:1), mp 156-158°. pKa 10.9.
Melting point: mp 156-158°
pKa: pKa 10.9
Therap-Cat: Antineoplastic adjunct (chemosensitizer).
Tesmilifene is a novel potentiator of chemotherapy which, when added to doxorubicin, achieved an unexpected and very large survival advantage over doxorubicin alone in a randomized trial in advanced breast cancer.
PHASE 23 FOR An estrogen receptor antagonist potentially for the treatment of advanced breast cancer, gastric cancer
Tesmilifene is a novel agent that augments cytotoxicity of various chemotherapeutic agents both in vitro and in vivo. It binds selectively to the high-affinity microsomal antiestrogen binding site (Ki=50nm) but has no affinity for estrogen receptors. Inhibits concanavalin-A-induced histamine release in mast cells and acts as a novel antagonist of intracellular histamine.
US 4803227


The target product can be prepared by reacting para-benzylphenol (I) with 2-diethylaminoethylchloride hydrochloride (II) either by means of NaOH in H2O or with K2CO3 in DMF/acetone (at 60 C in both cases), followed by treatment with HCl to obtain the corresponding hydrochloride salt.
| EP 0153160; JP 1985190742; US 4803227 |
US 4803227
http://www.google.com/patents/US4803227
Tesmilifene is a small molecule chemopotentiator under development by YM BioSciences, a Candian pharmaceutical company that specialises in the development of cancer treatments. It is indicated for use in combination with standard cytotoxic drugs, such as taxanes and anthracyclines, which are widely used in the treatment of metastatic disease – when cancers spread to distant sites in the body.
Tesmilifene, the company’s lead investigational compound, is currently in phase III development for patients with metastatic breast cancer. At the end of January 2007, an independent safety monitoring board advised the company that its ongoing registration trial should be stopped; it was considered unlikely that significant differences in overall survival (primary endpoint) between treatment arms would emerge over time. The company had hoped that the addition of tesmilifene to standard epirubicin/cyclophosphamide therapy would confer a survival benefit similar to that seen in its earlier phase III trial.
In light of these disappointing results, YM BioSciences plans a detailed analysis of its phase III data in advanced breast cancer to see if it can identify why tesmilifene failed to add clinical benefit in this trial.
DRUG RESISTANCE LIMITS EFFECTIVENESS OF CHEMOTHERAPY
Cytotoxic drugs have proved potent weapons in the fight against malignant tumours and are considered first-line therapy for the treatment of many cancers. However, while patients often respond well to a first course of chemotherapy over time the response to drug treatment diminishes and the tumour may eventually become drug resistant. In some cases resistance can develop across several classes of anti-cancer drugs, leading to multidrug resistance. The development of drug resistance limits the effectiveness of many anti-cancer agents and is an important contributor to cancer deaths.
The development of agents that can overcome drug resistance is seen as one of the most important areas of cancer research and for which there is significant unmet need. Various approaches are being explored to boost the use of cytotoxic agents including chemopotentiators, chemoprotectants and liposomal formulations.
Clearly any agent that can prevent or reverse drug resistance would have a major impact on treatment strategies, enhancing the benefits of standard cytotoxic drugs.
TESMILIFENE MAY BOOST CYTOTOXIC EFFECTS OF ANTHRACYCLINES
Anthracyclines are a class of cytotoxic agents with proven efficacy in the treatment of breast cancer. They include agents such as doxorubicin and epirubicin among others. Because patients with metastatic breast cancer may have received anthracycline therapy for earlier stage breast cancer (adjuvant therapy) or following disease recurrence, there is a risk that they will fail to respond to continued treatment.
A phase III trial in 305 patients with advanced breast cancer has shown that when tesmilifene is combined with doxorubicin it appears to improve survival over treatment with doxorubicin alone. In this trial approximately half the patients were treated with both tesmilifene and doxorubicin, while the other half received doxorubicin alone. Although there were no significant differences in tumour response rates, progression-free survival, or average duration of response between treatment arms at endpoint, overall survival was significantly improved in the combination arm. Among patients treated with tesmilifene and doxorubicin overall survival was 23.6 months compared with 15.6 months for those treated with doxorubicin alone.
Researchers have suggested that tesmilifene may enhance the anti-tumour effects of anthracyclines in several ways:
- Reducing the cancer cell’s ability to become resistant
- Decreasing the metabolism or “break-down” of doxorubicin
- Disrupting the cancer cell’s energy source
TESMILIFENE REGISTRATION TRIAL
In March 2004 YM BioSciences began its pivotal international phase III trial of tesmilifene in metastatic breast cancer. By September 2005, 723 patients had been enrolled in the trial, which was designed once again to compare the efficacy and safety of tesmilifene and an antrhacycline (epirubicin) with epirubicin alone.
“At the end of January 2007, an independent safety monitoring board advised the company that its ongoing registration trial should be stopped.”
Given the survival benefit seen in the earlier trial, which was carried out by the Canadian National Cancer Institute, the company was optimistic about outcome in its pivotal registration trial. However, an interim analysis of 351 events suggested that significant differences in overall survival were unlikely to be seen between the two treatment arms as the data matured and the trial was brought to a premature end.
In addition to its work on anthracyclines, YM BioSciences has also been exploring the potential of tesmilifene to enhance the efficacy of taxanes, also standard chemotherapy for metastatic breast cancer. Other potential applications include:
- Adjuvant therapy for breast cancer, i.e. immediately post-surgery and before the cancer has recurred or metastasised
- Hormone-refractory prostate cancer
- Lung cancer
- Non-Hodgkin’s lymphoma
MARKETING COMMENTARY
Although there have been major advances in the treatment of breast cancer in the last 10 to 15 years, it remains a disease for which improved treatments are still urgently needed. Estimates from the WHO suggest that metastatic breast cancer will claim the lives of over 40,000 patients a year.
Current treatments for metastatic breast cancer are rarely curative but can nonetheless do much to improve patients’ quality of life or duration of survival. . By boosting the cytotoxic effects of standard chemotherapy agents such as anthracyclines, while protecting healthy cells, tesmilifene was thought to have potential to extend the benefits of cytotoxic therapy to more patients. This is now in doubt following premature ending of its pivotal registration trial in advanced breast cancer.
Literature References: Intracellular histamine antagonist with chemopotentiating and cytoprotective activity. Structurally similar to tamoxifen, q.v., although binds anti-estrogen binding site (AEBS) with no affinity for the estrogen receptor.
Prepn: L. J. Brandes, M. W. Hermonat, Biochem. Biophys. Res. Commun. 123, 724 (1984); and use as antineoplastic: eidem, US 4803227 (1989 to Univ. Manitoba); and study of binding affinity: M. Poirot et al., Bioorg. Med. Chem. 8, 2007 (2000). Spectral analysis of interaction with P450 isozymes: L. J. Brandes et al., Cancer Chemother. Pharmacol. 45, 298 (2000).
Clinical evaluation in combination with cyclophosphamide in prostate cancer: L. J. Brandes et al., J. Clin. Oncol. 13, 1398 (1995); in combination with doxorubicin in breast cancer: L. Reyno et al., J. Clin. Oncol. 22, 269 (2004).
Bioorg Med Chem 2000,8(8),2007
Product Literature References
Enhancement of cytotoxicity of natural product drugs against multidrug resistant variant cell lines of human head and neck squamous cell carcinoma and breast carcinoma by tesmilifene.: P. J. Ferguson, et al.; Cancer Lett. 274, 279 (2009), Abstract;
Phase III study of N,N-diethyl-2-[4-(phenylmethyl) phenoxy]ethanamine (BMS-217380-01) combined with doxorubicin versus doxorubicin alone in metastatic/recurrent breast cancer: National Cancer Institute of Canada Clinical Trials Group St: L. Reyno, et al.; J. Clin. Oncol. 22, 269 (2004), Abstract;
Synergy between tamoxifen and cisplatin in human melanoma cells is dependent on the presence of antiestrogen-binding sites.: J.A. Jones, et al.; Cancer Res. 57, 2657 (1997), Abstract;
Influence of DPPE on histamine release from isolated rat mast cells.: N. Grosman; Agents Actions 41, 1 (1994), Abstract;
Histamine is an intracellular messenger mediating platelet aggregation.: S.P: Saxena, et al.; Science 243, 1596 (1989), Abstract;
///////Tesmilifene, Antineoplastic Adjunct, Chemosensitizer, PHASE 3, Tesmilifene hydrochloride, BMY-33419, BMS-217380, DPPE, N,N-DPPE, Antagonist of intracellular histamine
CCN(CC)CCOC1=CC=C(C=C1)CC2=CC=CC=C2
see……….http://apisynthesisint.blogspot.in/2015/12/tesmilifene-antagonist-of-intracellular.html
