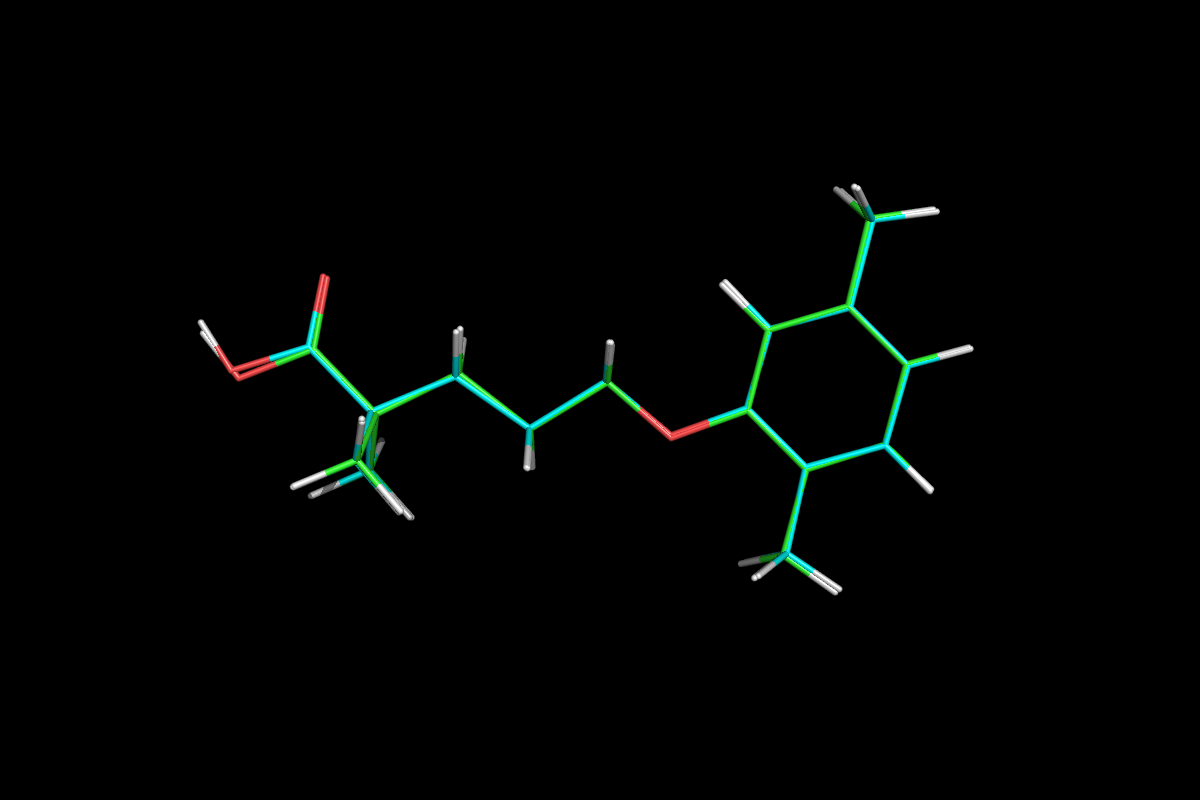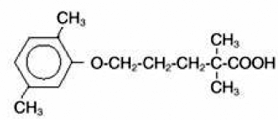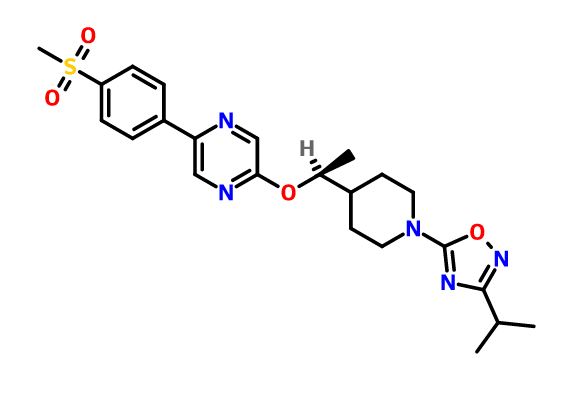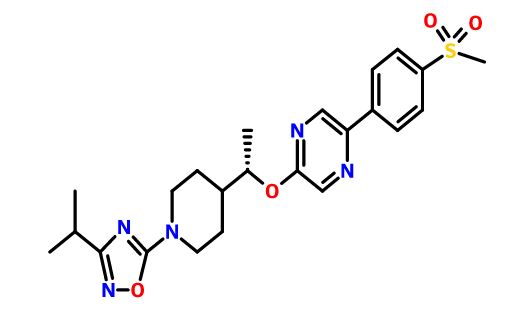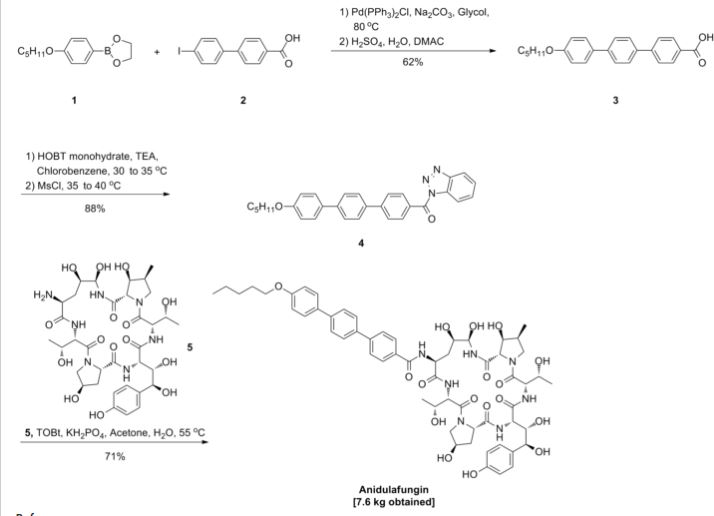

GSK-2041706A
[2-([(1S)-1-(1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl)ethyl]oxy)-5-[4-(methylsulfonyl)phenyl]pyrazine]
2-[((1S)-1-{1-[3-(1-Methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}ethyl)oxy]-5-[4-(methylsulfonyl)phenyl]pyrazine
Potent GPR119 Receptor Agonists
CAS 1032824-43-3
| Molecular Formula: |
C23H29N5O4S |
| Molecular Weight: |
471.57246 g/mol |
G protein-coupled receptor 119 (GPR119) is a G protein-coupled receptor expressed predominantly in pancreatic β-cells and gastrointestinal enteroendocrine cells. Metformin is a first-line treatment of type 2 diabetes, with minimal weight loss in humans. In this study, we investigated the effects of GSK2041706 [2-([(1S)-1-(1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl)ethyl]oxy)-5-[4-(methylsulfonyl)phenyl]pyrazine], a GPR119 agonist, and metformin as monotherapy or in combination on body weight in a diet-induced obese (DIO) mouse model. Relative to vehicle controls, 14-day treatment with GSK2041706 (30 mg/kg b.i.d.) or metformin at 30 and 100 mg/kg b.i.d. alone caused a 7.4%, 3.5%, and 4.4% (all P < 0.05) weight loss, respectively. The combination of GSK2041706 with metformin at 30 or 100 mg/kg resulted in a 9.5% and 16.7% weight loss, respectively. The combination of GSK2041706 and metformin at 100 mg/kg caused a significantly greater weight loss than the projected additive weight loss of 11.8%. This body weight effect was predominantly due to a loss of fat. Cumulative food intake was reduced by 17.1% with GSK2041706 alone and 6.6% and 8.7% with metformin at 30 and 100 mg/kg, respectively. The combination of GSK2041706 with metformin caused greater reductions in cumulative food intake (22.2% at 30 mg/kg and 37.5% at 100 mg/kg) and higher fed plasma glucagon-like peptide 1 and peptide tyrosine tyrosine levels and decreased plasma insulin and glucose-dependent insulinotropic polypeptide levels compared with their monotherapy groups. In addition, we characterized the effect of GSK2041706 and metformin as monotherapy or in combination on neuronal activation in the appetite regulating centers in fasted DIO mice. In conclusion, our data demonstrate the beneficial effects of combining a GPR119 agonist with metformin in the regulation of body weight in DIO mice.
Diabetes mellitus is an ever-increasing threat to human health. For example, in the United States current estimates maintain that about 16 million people suffer from diabetes mellitus.
Type I diabetes, also known as insulin-dependent diabetes mellitus (IDDM), is caused by the autoimmune destruction of the insulin producing pancreatic β-cells, and necessitates regular administration of exogenous insulin. Without insulin, cells cannot absorb sugar (glucose), which they need to produce energy. Symptoms of Type I diabetes usually start in childhood or young adulthood. People often seek medical help because they are seriously ill from sudden symptoms of high blood sugar (hyperglycemia).
Type II diabetes, also known as non-insulin-dependent diabetes mellitus (NIDDM), manifests with an inability to adequately regulate blood-glucose levels. Type II diabetes may be characterized by a defect in insulin secretion or by insulin resistance, namely those that suffer from Type II diabetes have too little insulin or cannot use insulin effectively. Insulin resistance refers to the inability of body tissues to respond properly to endogenous insulin. Insulin resistance develops because of multiple factors, including genetics, obesity, increasing age, and having high blood sugar over long periods of time. Type II diabetes, sometimes called mature or adult onset diabetes, can develop at any age, but most commonly becomes apparent during adulthood. The incidence of Type II diabetes in children, however, is rising
In diabetics, glucose levels build up in the blood and urine causing excessive urination, thirst, hunger, and problems with fat and protein metabolism. If left untreated, diabetes mellitus may cause life-threatening complications, including blindness, kidney failure, and heart disease.
Type II diabetes accounts for approximately 90-95% of diabetes cases, killing about 193,000 U.S. residents each year. Type II diabetes is the seventh leading cause of all deaths. In Western societies, Type II diabetes currently affects 6% of the adult population with world-wide frequency expected to grow by 6% per annum.
Although there are certain inheritable traits that may predispose particular individuals to developing Type II diabetes, the driving force behind the current increase in incidence of the disease is the increased sedentary lifestyle, diet, and obesity now prevalent in developed countries. About 80% of diabetics with Type II diabetes are significantly overweight. As noted above, an increasing number of young people are developing the disease. Type II diabetes is now internationally recognized as one of the major threats to human health in the 21stcentury.
Type II diabetes currently is treated at several levels. A first level of therapy is through the use of diet and/or exercise, either alone or in combination with therapeutic agents. Such agents may include insulin or pharmaceuticals that lower blood glucose levels. About 49% of individuals with Type II diabetes require oral medication(s), about 40% of individuals require insulin injections or a combination of insulin injections and oral medication(s), and about 10% of individuals may use diet and exercise alone.
Current therapies for diabetes mellitus include: insulin; insulin secretagogues, such as sulphonylureas, which increase insulin production from pancreatic-cells; glucose-lowering effectors, such as metformin which reduce glucose production from the liver; activators of the peroxisome proliferator-activated receptor—(PPAR-), such as the thiazolidinediones, which enhances insulin action; and α-glucosidase inhibitors which interfere with gut glucose production. There are, however, deficiencies associated with currently available treatments, including hypoglycemic episodes, weight gain, loss in responsiveness to therapy over time, gastrointestinal problems, and edema.
There are several areas at which research is being targeted in order to bring new, more effective, therapies to the marketplace. For example, on-going research includes exploring a reduction in excessive hepatic glucose production, enhancing the pathway by which insulin transmits its signal to the cells such that they take up glucose, enhancing glucose-stimulated insulin secretion from the pancreatic-cells, and targeting obesity and associated problems with fat metabolism and accumulation.
One particular target is GPR119. GPR119 is a member of the rhodopsin family of G-protein-coupled receptors. In addition to the “GPR119” identifier, several other identifiers exist, including but not limited to RUP 3, Snorf 25, 19 AJ, GPR 116 (believed to be erroneous), AXOR 20, and PS1. GPR119 is expressed in human gastrointestinal regions and in human islets. Activation of GPR119 has been demonstrated to stimulate intracellular cAMP and lead to glucose-dependent GLP-1 and insulin secretion. See, T. Soga et al., Biochemical and Biophysical Research Communications 326 (2005) 744-751, herein incorporated by reference with regard to a background understanding of GPR119.
In type 2 diabetes the action of GLP-1 on the β-cell is maintained, although GLP-1 secretion, itself, is reduced. More recently, therefore, much research has been focused on GLP-1. Studies show glucose-lowering effects in addition to GLP-1’s ability to stimulate glucose-dependent insulin secretion including, but not limited to, an inhibition of the release of the hormone glucagon following meals, a reduction in the rate at which nutrients are absorbed into the bloodstream, and a reduction of food intake. Studies demonstrate that treatments to increase GLP-1, therefore, may be used for a variety of conditions and disorders including but not limited to metabolic disorders, gastrointestinal disorders, inflammatory diseases, psychosomatic, depressive, and neuropsychiatric disease including but not limited to diabetes mellitus (Type 1 and Type 2), metabolic syndrome, obesity, appetite control and satiety, weight loss, stress, inflammation, myocardial ischemia/reperfusion injury, Alzheimer’s Disease, and other diseases of the central nervous system.
The use of exogenous GLP-1 in clinical treatment is severely limited, however, due to its rapid degradation by the protease DPP-IV. There are multiple GLP-1 mimetics in development for type 2 diabetes that are reported in the literature, all are modified peptides, which display longer half-lives than endogenous GLP-1. For example, the product sold under the tradename BYETTA® is the first FDA-approved agent of this new class of medications. These mimetics, however, require injection. An oral medication that is able to elevate GLP-1 secretion is desirable. Orally available inhibitors of DPP-IV, which result in elevation in intact GLP-1, are now available, such as sitagliptin, marketed under the brand name JANUVIA®. Nevertheless, a molecule which may stimulate GLP-1 secretion would provide a therapeutic benefit. A molecule which could stimulate both GLP-1 secretion and insulin secretion through effects on the L-cell and direct effects on the β-cell would hold much promise for type 2 diabetes therapy.
The present invention identifies agonists of GPR119 which increase glucose-disposal in part through elevation of GIP, GLP-1, and insulin. Moreover, studies demonstrate that GPR119 agonists such as the compounds of the present invention can stimulate incretins independently of glucose. GIP and GLP-1 are peptides, known as incretins, secreted from enteroendocrine K and L cells, respectively, in response to ingestion of nutrients, and have a wide variety of physiological effects that have been described in numerous publications over the past two decades. See, for example, Bojanowska, E. et al.,Med. Sci. Monit., 2005, August 11(8): RA271-8; Perry, T. et al., Curr. Alzheimer Res., 2005, July 2(3): 377-85; and Meier, J. J. et al.,Diabetes Metab. Res. Rev., 2005, March-April; 21(2); 91-117 (each herein incorporated by reference with regard to a background understanding of incretins). Moreover, although the mechanisms regulating GLP-1 secretion remain unclear, the initial rapid rise in GLP-1 following a meal may be a result of hormonal stimulation of neuronal afferents involving GIP. See, for example, J. N. Roberge and P. L. Brubaker, Endocrinology 133 (1993), pp. 233-240 (herein incorporated by reference with regard to such teaching). Furthermore, later increases in GLP-1 may involve direct activation of L-cells by nutrients in the distal small-intestine and the colon. GIP and GLP-1 are potent stimulators of the body’s ability to produce insulin in response to elevated levels of blood sugar. In Type 2 diabetes, patients display a decreased responsiveness to GIP but not GLP-1, with respect to its ability to stimulate insulin secretion. The mechanism behind the decreased responsiveness to GIP remains unclear since type 2 diabetics retain sensitivity to a bolus administration of GIP but not to a continuous infusion (Meier et al. 2004 Diabetes 53 S220-S224). Moreover recent studies with a long-acting fatty-acid derivative of GIP showed beneficial effects on glucose homeostasis in ob/ob mice following 14 days of treatment (Irwin N. et al. (2006) J. Med. Chem. 49, 1047-1054.)
Agonists to GPR119 may be of therapeutic value for diabetes and associated conditions, particularly type II diabetes, obesity, glucose intolerance, insulin resistance, metabolic syndrome X, hyperlipidemia, hypercholesterolemia, and atherosclerosis.
NMR
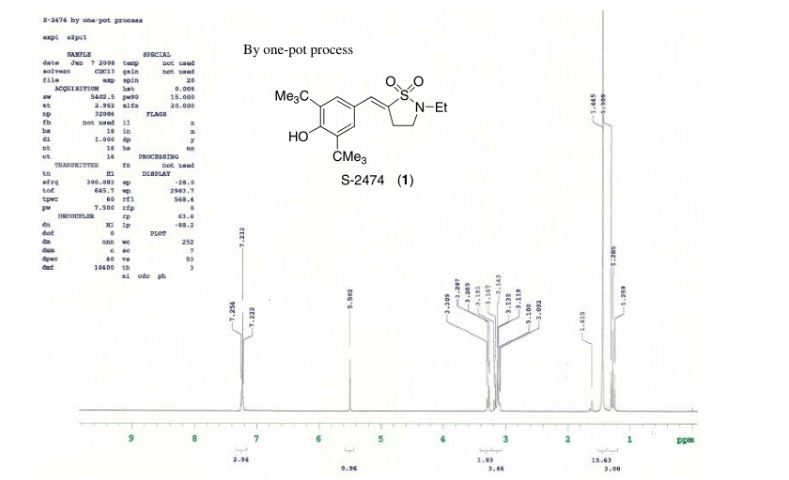
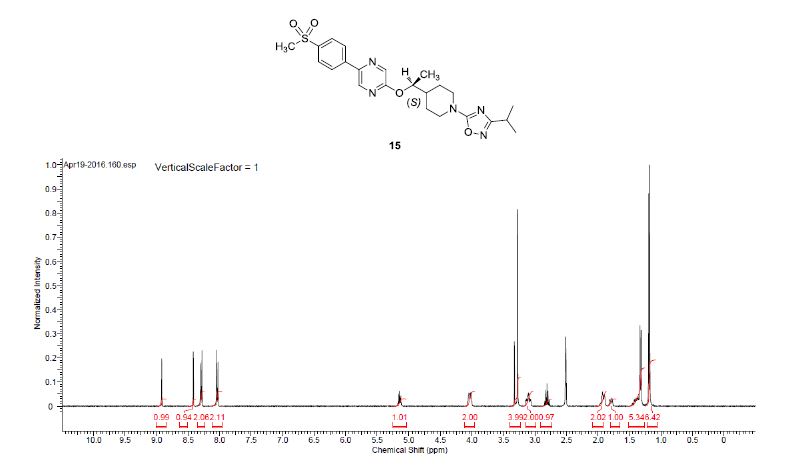
1H NMR (400 MHz, DMSO-d6) δ 8.91 (bs, 1H), 8.40 (bs, 1 H), 8.28 (d, J = 8.5 Hz, 2H), 8.02 (d, J = 8.5 Hz, 2H), 5.17–5.09 (m, 1H), 4.09–3.95 (m, 2H), 3.27 (s, 3H), 3.16–2.99 (m, 2H), 2.80 (q, J = 6.9 Hz, 1H), 1.98–1.85 (m, 2H), 1.83–1.70 (m, 1H), 1.47–1.33 (m, 2H), 1.31 (d, J = 6.3 Hz, 3H), 1.17 (d, J = 6.8 Hz, 6H).
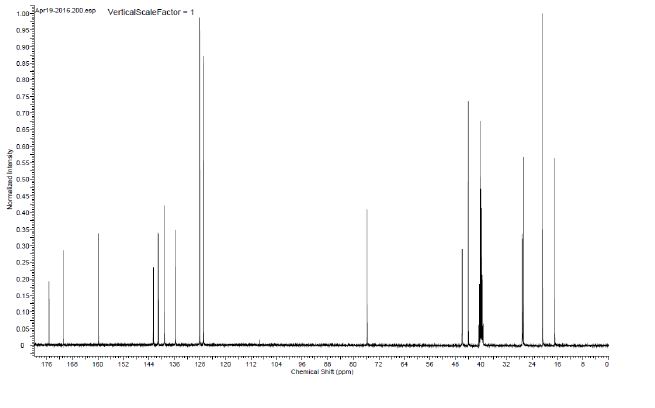
13C NMR (100.6 MHz, DMSO-d6) 175.3, 170.9, 159.8, 142.6, 141.2, 141.0, 139.1, 135.7, 128.1, 126.9, 75.7, 46.0, 45.9, 44.0, 40.2, 27.1, 27.0, 26.7, 20.7, 16.9.
HRMS calcd for C23H30N5O4S (M + H)+ 472.2013, found, 472.2009.

PATENT
Jing Fang, Jun Tang, Andrew J. Carpenter,Gregory Peckham, Christopher R. Conlee,Kien S. Du, Subba Reddy Katamreddy,
http://www.google.co.ug/patents/US20120077812
Example 156(±)-2-[(1-{1-[3-(1-Methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}ethyl)oxy]-5-[4-(methylsulfonyl)phenyl]pyrazine
Step 1: A solution of 3-(1-methylethyl)-5-(trichloromethyl)-1,2,4-oxadiazole (prepared as in Example 158, Alternative synthesis, Step 3, 179 g, 0.78 mol) in MeOH (300 mL) was treated with 4-piperidinemethanol (108 g, 0.94 mol) and stirred and heated at 50° C. overnight. The solvent was removed and the residue was purified by flash chromatography on a silica gel column to give {1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methanol (60 g, 34%) as a pale yellow oil.
Step 2: A solution of {1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methanol (1.50 g, 6.66 mmol) in CH2Cl2 (50 mL) at 0° C. was treated with Dess-Martin periodinane (2.91 g, 6.66 mmol). The reaction mixture was warmed to ambient temperature and stirred overnight. The reaction was quenched with aqueous 20% Na2S2O3(100 mL) and aqueous saturated NaHCO3 (100 mL) and then stirred for 10 minutes. The CH2Cl2 layer was separated and washed with brine, dried over Na2SO4, filtered, and the filtrate was concentrated to give the crude product as a cloudy colorless oil. The crude product was dissolved in 100 mL of 1:1 EtOAc/hexanes, filtered through a pad of silica gel, washed with 200 mL of 1:1 EtOAc/hexanes. The filtrate was concentrated to give 1.07 g (72%) of 1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinecarbaldehyde as a clear colorless oil, which was used without further purification. 1H NMR (400 MHz, CDCl3): δ 9.68 (s, 1H), 4.15-4.00 (m, 2H), 3.30-3.20 (m, 2H), 2.86 (septet, 1H, J=7.0 Hz), 2.55-2.45 (m, 1H), 2.10-1.95 (m, 2H), 1.80-1.65 (m, 2H), 1.26 (d, 6H, J=6.8 Hz).
Step 3: (±)-1-{1-[3-(1-Methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}ethyl methanesulfonate (0.74 g, 49%) was prepared as a light brown oil from 1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinecarbaldehyde (1.07 g, 4.79 mmol) and methylmagnesium bromide (3M in Et2O, 3.51 mL, 10.54 mmol) then methanesulfonyl chloride (0.22 mL, 2.81 mmol) and Et3N (0.66 mL, 4.68 mmol) in a manner similar to Example 139, Steps 1-2. The crude product was used without further purification. 1H NMR (400 MHz, CDCl3): δ 4.70-4.60 (m, 1H), 4.30-4.15 (m, 2H), 3.10-2.95 (m, 5H), 2.87 (septet, 1H, J=7.0 Hz), 1.95-1.70 (m, 3H), 1.55-1.35 (m, 5H), 1.26 (d, 6H, J=6.8 Hz).
Step 4: The title compound (0.212 g, 26%) was prepared as a white foam from 5-[4-(methylsulfonyl)phenyl]-2-pyrazinol (and tautomers thereof) (prepared as in Example 145, Steps 1-2, 0.43 g, 1.72 mmol), (±)-1-{1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}ethyl methanesulfonate (0.74 g, 2.32 mmol) and K2CO3 (0.48 g, 3.44 mmol) in DMF (15 mL) in a manner similar to Example 152, Steps 3. The crude product was purified by chromatography on an ISCO silica gel column using 0 to 25% EtOAc/CH2Cl2, followed by chromatography on a silica gel column eluted with 50% EtOAc/hexanes to give (±)-2-[(1-{1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}ethypoxy]-5-[4-(methylsulfonyl)phenyl]pyrazine as a white solid. 1H NMR (400 MHz, CDCl3): δ 8.53 (s, 1H), 8.25 (s, 1H), 8.10 (d, 2H, J=8.5 Hz), 8.02 (d, 2H, J=8.5 Hz), 5.20-5.10 (m, 1H), 4.35-4.20 (m, 2H), 3.15-3.00 (m, 5H), 2.91 (septet, 1H, J=7.0 Hz), 2.00-1.80 (m, 3H), 1.60-1.40 (m, 2H), 1.34 (d, 3H, J=6.1 Hz), 1.28 (d, 6H, J=7.1 Hz); LRMS (ESI), m/z 472 (M+H).
Example 1572-[((1R)-1-{1-[3-(1-Methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}ethyl)oxy]-5-[4-(methylsulfonyl)phenyl]pyrazin
The racemic 2-[(1-{1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}ethyl)oxy]-5-[4-(methylsulfonyl)phenyl]pyrazine (prepared as in Example 156) was subjected to Chiral HPLC [column: AS-H, column mobile phase: 70% CO2: 30% MeOH (2 mL/min), pressure 140 bar, temperature 40° C., 215 nm] analysis and then separated to give two (R and S) enantiomers. The title compound was isolated as an off-white solid with Tr of 23.42 min (first eluting peak). The (R) absolute stereochemistry was assigned by Ab initio VCD analysis.
Example 158
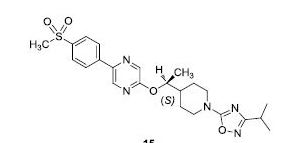
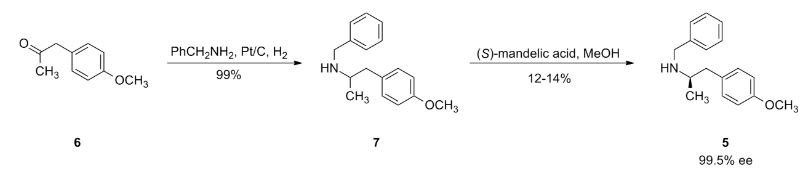
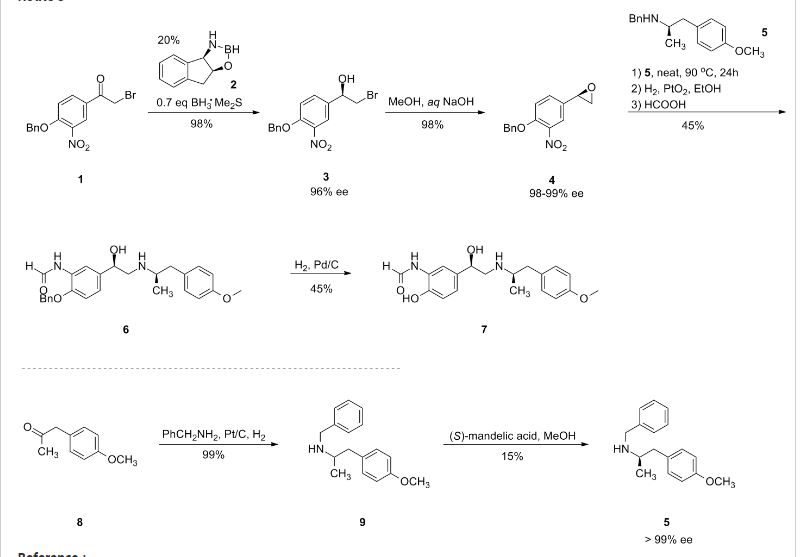
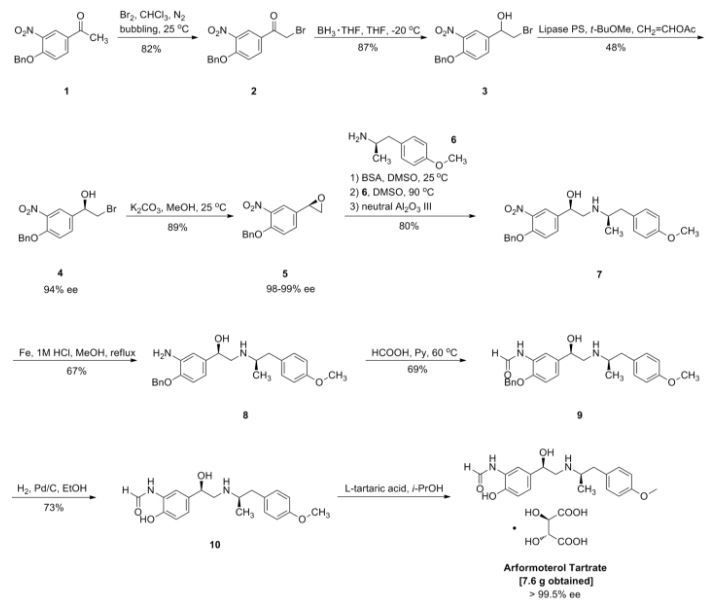
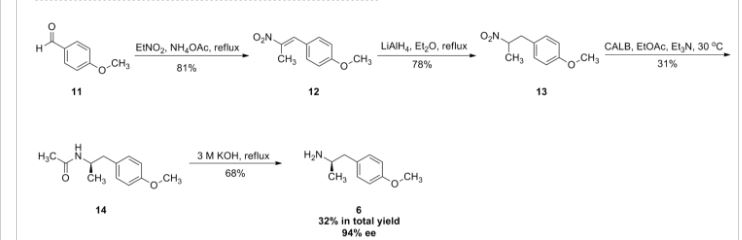
2-[((1S)-1-{1-[3-(1-Methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}ethyl)oxy]-5-[4-(methylsulfonyl)phenyl]pyrazine
The racemic 2-[(1-{1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}ethyl)oxy]-5-[4-(methylsulfonyl)phenyl]pyrazine (prepared as in Example 156) was subjected to Chiral HPLC [column: AS-H, column mobile phase: 70% CO2: 30% MeOH (2 mL/min), pressure 140 bar, temperature 40° C., 215 nm] analysis and then separated to give two (R and S) enantiomers. The title compound was isolated as an off-white solid with Tr of 25.83 min (second eluting peak). The (S) absolute stereochemistry was assigned by Ab initio VCD analysis. Alternative preparation from enantiomerically enriched material:
Step 1: Triethylamine (315 mL, 2.26 mol) was added dropwise to formic acid (150 mL, 3.91 mol) with overhead stirring while maintaining the internal temperature below 60° C. with ice-bath cooling. Neat 4-acetylpyridine (100 mL, 0.904 mol) was then added rapidly while maintaining the temperature below 50° C. Following this addition, the reaction was allowed to cool to 28° C. and the chiral ruthenium catalyst [N-[(1R,2R)-2-(amino-N)-1,2-diphenylethyl]-2,4,6-trimethylbenzenesulfonamidato-N]chloro[(1,2,3,4,5,6-n)-1-methyl-4-(1-methylethyl)benzene]ruthenium (CAS#177552-91-9; for catalyst preparation, see: Uematsu, N.; Fujii, A.; Hashiguchi, S.; Ikariya, T.; Noyori, R.; J. Am. Chem. Soc. 1996, 118, 4916-4917) (3 g, 4.46 mmol) was added. The mixture was stirred under house vacuum for 4 h and then overnight under an atmosphere of nitrogen. The reaction mixture was added dropwise to a stirred solution of 10% Na2CO3 (4 L) and then extracted with EtOAc (3×1 L). The combined EtOAc layers were washed once with brine (1 L), treated with MgSO4 and Darco G-60 decolorizing charcoal and filtered through a 100 g plug of silica gel washing with 10% MeOH/EtOAc (1 L). The filtrate was concentrated to provide a dark oil that crystallized upon standing. The solid was dissolved in warm t-butyl methyl ether (250 mL) and the warm solution was filtered to remove a small amount of insoluble material. The filtrate was allowed to stir with cooling to room temperature and then to −15° C. The solids were collected by filtration, washing with cold t-butyl methyl ether and heptane, and then dried under high vacuum to yield (1R)-1-(4-pyridinyl)ethanol as a dark beige solid (62 g, 52.9% yield). This solid material was 96% ee based on chiral HPLC(HPLC conditions: AS-H column, 5% MeOH/CO2, 40° C., 140 bar, 2 mL/min). The filtrate was combined with the insoluble solid from the crystallization and concentrated in vacuo to yield additional (1R)-1-(4-pyridinyl)ethanol as a dark oil (37.5 g, 32% yield). This oily material was 78% ee based on chiral HPLC (see HPLC conditions above). 1H NMR (400 MHz, DMSO-d6): δ 8.47-8.43 (m, 2H), 7.32-7.28 (m, 2H), 5.37 (d, 1H, J=4.4 Hz), 4.72-4.64 (m, 1H), 1.44 (d, 3H, J=6.6 Hz).
Step 2: A solution of (1R)-1-(4-pyridinyl)ethanol (37 g, 0.3 mol, 78% ee) in MeOH (2 L) was charged with PtO2 (5 g) under nitrogen atmosphere followed by acetic acid (19 mL). The mixture was evacuated and purged with hydrogen several times and then stirred under an atmosphere of hydrogen for 2 d at room temperature. The mixture was filtered to remove catalyst and the filtrate was concentrated in vacuo and triturated with EtOAc to yield a cream-colored solid which was collected by filtration. The filter cake was dissolved in MeOH (500 mL) and 50% NaOH (15.8 g) was added. The resulting solution was stirred at 25° C. for 30 min and concentrated. The resulting solid was triturated with Et2O (700 mL) and stirred at 25° C. for 30 min, the solids were removed by filtration and the filtrate was dried over MgSO4 and filtered again. The final filtrate was concentrated to yield (1R)-1-(4-piperidinyl)ethanol (22 g, 57% yield) as a light beige solid. 1H NMR (400 MHz, CDCl3): δ 3.50 (quint, 1H, J=6.3 Hz), 3.13-3.01 (m, 2H), 2.61-2.47 (m, 2H), 1.88 (br, 2H), 1.84-1.73 (m, 1H), 1.63-1.52 (m, 1H), 1.41-1.27 (m, 1H), 1.23-1.05 (m, 2H), 1.13 (d, 3H, J=6.2 Hz).
Step 3: A stirred solution of N-hydroxy-2-methylpropanimidamide (16.33 g, 160 mmol) in pyridine (16.81 mL, 208 mmol) and dichloromethane (165 mL) at −15° C. was treated with trichloroacetyl chloride (19.63 mL, 176 mmol) over 40 min. The reaction was allowed to warm to ambient temperature and stirred for 42 h. Water (100 mL) was added and the reaction was stirred for 30 min. The dichloromethane was removed and the residue was diluted with water (50 mL) and extracted with ether (300 mL). The ether layer was washed with water, dried over MgSO4 and concentrated to afford 3-(1-methylethyl)-5-(trichloromethyl)-1,2,4-oxadiazole (28.0 g, 76% yield) as an orange liquid.1H NMR (400 MHz, CDCl3): δ 3.13 (septet, 1H, J=7.0 Hz), 1.36 (d, 6H, J=7.0 Hz).
Step 4: A solution of 3-(1-methylethyl)-5-(trichloromethyl)-1,2,4-oxadiazole (25.8 g, 112 mmol) and (1R)-1-(4-piperidinyl)ethanol (13.4 g, 104 mmol) in MeOH (15 mL) was stirred at ambient temperature under a stream of nitrogen for 7 days. The reaction was diluted with MeOH (40 mL), cooled in an ice bath and 1N NaOH (25 mL) was added. The mixture was allowed to warm to ambient temperature and stir for 1 h. The reaction was partitioned in EtOAc (300 mL)/1N NaOH (75 mL) and the layers were separated. The aqueous layer was saturated with NaCl and extracted with EtOAc (200 mL). The combined EtOAc layers were dried over MgSO4, concentrated and placed under high vacuum for 18 h to afford (1R)-1-{1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}ethanol (16.75 g, 68%) as an orange oil. 1H NMR (400 MHz, CDCl3): δ 4.14 (m, 2H), 3.57 (quint, 1H, J=6.3 Hz), 2.98 (m, 2H), 2.83 (septet, 1H, J=7.0 Hz), 1.90 (m, 1H), 1.86 (br, 1H), 1.67 (m, 1H), 1.45 (m, 1H), 1.33 (m, 2H), 1.23 (d, 6H, J=7.0 Hz), 1.16 (d, 3H, J=6.3 Hz); LRMS (ESI), m/z 240 (M+H).
Step 5: A solution of (1R)-1-{1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}ethanol (1.68 g, 7.0 mmol) in dichloromethane (100 mL) at 0° C. was treated with Et3N (1.98 mL, 14.0 mmol) followed by methanesulfonyl chloride (0.66 mL, 8.4 mmol). The mixture was stirred at 0° C. for 1 h, then at room temperature for 2 h. The mixture was diluted with dichloromethane (50 mL), washed with 1M NaH2PO4 (75 mL×2) and brine, and dried over Na2SO4 and concentrated to give (1R)-1-{1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}ethyl methanesulfonate (2.23 g, 7.0 mmol, 100% yield) as a brown oil, which was used without further purification.
Step 6: A mixture of 5-[4-(methylsulfonyl)phenyl]-2-pyrazinol (and tautomers thereof) (prepared as in Example 145, Step 2, 1.3 g, 5.19 mmol), (1R)-1-{1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}ethyl methanesulfonate (2.23 g, 7.0 mmol, 70% ee) and K2CO3 (1.45 g, 10.4 mmol) in DMF (35 mL) was stirred at 100° C. in a preheated oil bath overnight. The mixture was cooled to ambient temperature, treated with water, and the mixture was extracted with EtOAc (75 mL×2). The combined organic extracts were washed with water, brine and dried over Na2SO4, filtered, and the filtrate was concentrated to a brown oil, which was by chromatography on a silica gel column eluted with 50% EtOAc/hexanes followed by chromatography on an ISCO silica gel column using 0 to 60% EtOAc/hexanes to give 2-[((1S)-1-{1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}ethyl)oxy]-5-[4-(methylsulfonyl)phenyl]pyrazine (0.73 g, 70% ee, 30%) as a white solid. The solid was subjected to chiral separation (similar to conditions used above for Example 158) to yield 0.30 g of the title compound as a white solid. 1H NMR (400 MHz, CDCl3): δ 8.53 (d, 1H, J=1.3 Hz), 8.25 (d, 1H, J=1.3 Hz), 8.10 (d, 2H, J=8.3 Hz), 8.02 (d, 2H, J=8.5 Hz), 5.20-5.10 (m, 1H), 4.35-4.20 (m, 2H), 3.15-3.00 (m, 5H), 2.90 (septet, 1H, J=7.0 Hz), 2.00-1.80 (m, 3H), 1.60-1.40 (m, 2H), 1.34 (d, 3H, J=6.3 Hz), 1.28 (d, 6H, J=6.9 Hz); LRMS (ESI), m/z 472 (M+H).
Paper
Development of Large-Scale Routes to Potent GPR119 Receptor Agonists
Richard T. Matsuoka*†, Eric E. Boros#, Andrew D. Brown†, Kae M. Bullock†, Will L. Canoy‡, Andrew J. Carpenter#, Jeremy D. Cobb†, Shannon E. Condon†, Nicole M. Deschamps†, Vassil I. Elitzin†, Greg Erickson†,Jing M. Fang#, David H. Igo§, Biren K. Joshi‡, Istvan W. Kaldor#, Mark B. Mitchell†, Gregory E. Peckham#, Daniel W. Reynolds‡, Matthew C. Salmon†, Matthew J. Sharp†, Elie A. Tabet#, Jennifer F. Toczko†, Lianming Michael Wu‡, and Xiao-ming M. Zhou†
†API Chemistry Department, ‡Analytical Science & Development Department, #Medicinal Chemistry Department, and§Particle Sciences and Engineering Department, GlaxoSmithKline, 709 Swedeland Road, King of Prussia, Pennsylvania 19406, United States
Org. Process Res. Dev., Article ASAP
Publication Date (Web): July 13, 2016
Copyright © 2016 American Chemical Society
Abstract
Practical and scalable syntheses were developed that were used to prepare multikilogram batches of GSK1292263A (1) and GSK2041706A (15), two potent G protein-coupled receptor 119 (GPR119) agonists. Both syntheses employed relatively cheap and readily available starting materials, and both took advantage of an SNAr synthetic strategy.
/////////////GSK2041706A, GSK 2041706A, GSK-2041706A, GSK2041706, GSK 2041706, GSK-2041706
O=S(c4ccc(c3cnc(OC(C2CCN(c1nc(C(C)C)no1)CC2)C)cn3)cc4)(C)=O

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....










































 .
.