
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Iodofalan (131I)
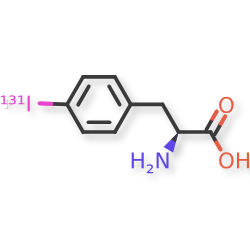
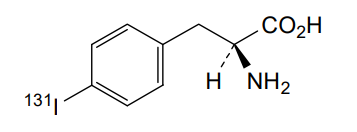
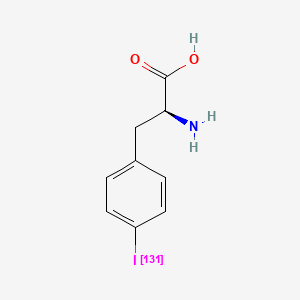
Iodofalan (131I)
CAS 76641-05-9
MFC9H10131INO2
Molecular FormulaC9H10INO2
Molecular Weight295.09
4-(131I)iodo-L-phenylalanine
(2S)-2-amino-3-(4-iodophenyl)propanoic acid
radiopharmaceutical, antineoplastic, Phase 2, Glioblastoma, 606VTF676Y, 131I-TLX-101, ACD 101
- 4-Iodophenylalanine I-131
- 4-(131I)Iodo-L-phenylalanine
- 4-Iodo-L-phenylalanine-131I
- ACD-101
- L-Phenylalanine, 4-(iodo-131I)-
- OriginatorTherapeia
- DeveloperTelix Pharmaceuticals; Therapeia
- ClassAmino acids; Antineoplastics; Radioisotopes; Radiopharmaceutical diagnostics; Radiopharmaceuticals; Small molecules
- Mechanism of ActionApoptosis stimulants; Positron-emission tomography enhancers
- Orphan Drug StatusYes – Glioblastoma
- Phase IIGlioblastoma
- 14 Oct 2025Telix Pharmaceuticals receives IND approval for TLX 101 in Glioblastoma
- 27 Jul 2025Telix Pharmaceuticals plans a phase III IPAX BrIGHT trial for Glioblastoma (Monotherapy, Combination therapy, Recurrent, Second-line therapy or greater) in Australia(IV) (NCT07100730)(EudraCT2025-521785-10) in September 2025
- 16 Apr 2025Telix has submitted for ethics approval a registration-enabling study of TLX101 in recurrent glioblastoma.
Iodofalan (131I) is a radiopharmaceutical that has garnered significant attention in oncological research due to its targeted therapeutic potential. This compound, which includes the radioactive isotope Iodine-131, has been explored for its efficacy in treating certain types of cancers, particularly those associated with the thyroid. Various research institutions worldwide have been studying Iodofalan (131I) to better understand its clinical benefits, optimize its usage, and minimize potential side effects. As a drug type, Iodofalan (131I) is categorized as a targeted radiopharmaceutical therapy, which leverages the properties of radioactive isotopes to destroy cancer cells with precision. Currently, its primary indications include differentiated thyroid cancer and non-resectable metastatic thyroid cancer, among other investigational uses.
Iodofalan (131I) Mechanism of Action
The mechanism of action for Iodofalan (131I) centers on the properties of Iodine-131, a beta-emitting isotope. When administered, Iodofalan (131I) is selectively absorbed by thyroid cells. This selectivity is due to the thyroid gland’s natural ability to uptake iodine, a key element required for the production of thyroid hormones. Cancerous thyroid tissues retain this ability, making them ideal targets for Iodofalan (131I) therapy.
Once absorbed by the thyroid cancer cells, the radioactive decay of Iodine-131 begins. This decay process emits beta particles, which possess sufficient energy to destroy nearby cells. The radiation from these beta particles causes direct DNA damage, leading to cell death. Additionally, the gamma radiation emitted by Iodine-131 can be used diagnostically to track the distribution and uptake of the compound in the body via imaging techniques such as SPECT (Single Photon Emission Computed Tomography).
The dual role of Iodofalan (131I) in both treatment and diagnostic contexts underscores its importance in managing thyroid cancers. By delivering a localized radiation dose to thyroid cancer cells, Iodofalan (131I) minimizes damage to surrounding healthy tissues, which is a significant advantage over traditional external beam radiotherapy.
What is the indication of Iodofalan (131I)?
The primary indication for Iodofalan (131I) is the treatment of differentiated thyroid cancer, a category that includes papillary and follicular thyroid cancers. These subtypes are characterized by their ability to absorb iodine, making them particularly amenable to radioiodine therapy. Iodofalan (131I) is typically used in cases where the thyroid cancer is not amenable to surgical removal or has metastasized to other parts of the body. In such scenarios, the radiopharmaceutical offers a non-invasive therapeutic option that can target and destroy cancer cells even in distant metastatic sites.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US42129729&_cid=P21-MHE8B5-15309-1
EXAMPLE 1
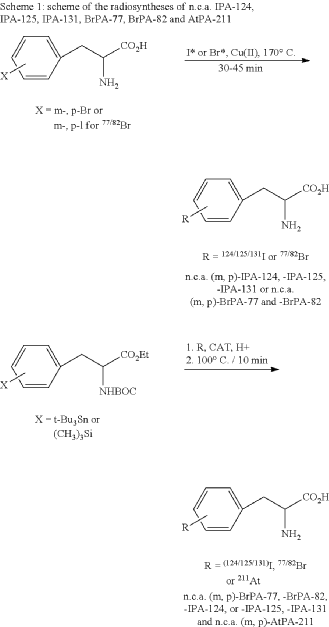
EXAMPLE 2
General synthesis of 3,4-[124I]iodo-L-phenylalanine (m, p-IPA-124), 3,4-[125I]iodo-L-phenylalanine (m,p-IPA-125) and 3,4-[131I]iodo-L-phenylalanine (m,p-IPA-131) by non-isotopic radioiodo-debromination
PAT
- Pharmaceutical combinations and uses thereofPublication Number: US-2024197715-A1Priority Date: 2022-11-18
- Pharmaceutical combinations and uses thereofPublication Number: WO-2024105610-A1Priority Date: 2022-11-18
- Iodine-labeled homoglutamic acid and glutamic acid derivativesPublication Number: US-2013034497-A1Priority Date: 2009-11-17
- MALIGNAS NEOPLASIAS THERAPY.Publication Number: ES-2341575-T3Priority Date: 2005-11-25Grant Date: 2010-06-22
- Therapy of malignant neoplasiasPublication Number: US-2007128108-A1Priority Date: 2005-11-18
- Therapy of malignant neoplasias
- Publication Number: US-9682158-B2
- Priority Date: 2005-11-18
- Grant Date: 2017-06-20



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
//////////Iodofalan (131I), radiopharmaceutical, antineoplastic, Phase 2, Glioblastoma, 606VTF676Y, 131I-TLX-101, ACD 101
Inlexisertib
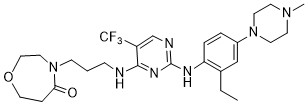
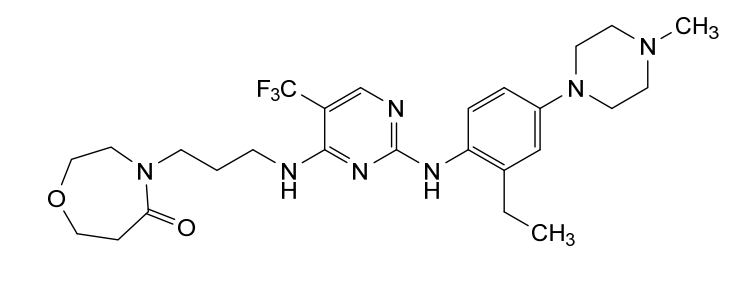
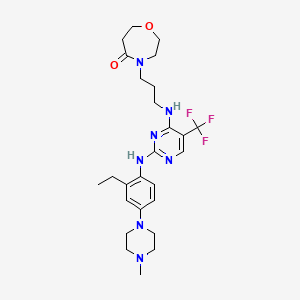
Inlexisertib
CAS 2543673-19-2
MF C26H36F3N7O2, 535.62
4-(3-((2-((2-ethyl-4-(4-methylpiperazin-1-yl)phenyl)amino)-5-(trifluoromethyl)pyrimidin-4-yl)amino)propyl)-1,4-oxazepan-5-one
4-[3-[[2-[2-ethyl-4-(4-methylpiperazin-1-yl)anilino]-5-(trifluoromethyl)pyrimidin-4-yl]amino]propyl]-1,4-oxazepan-5-one

serine/ threonine kinase inhibitor, antineoplastic, DCC 3116, JM2ZTM8S7S
Inlexisertib is an orally bioavailable inhibitor of the serine/threonine-protein kinase ULK 1 and 2, with potential antineoplastic activity. Upon oral administration, inlexisertib targets and binds to ULK1/2. This inhibits cancer autophagy, which mutant RAS cancer cells use for their survival, and results in tumor cell death. ULK1/2 mediates the autophagocytotic process and is often upregulated in cancers, especially in mutant RAS cancers. Autophagy plays a key role in a tumor cell proliferation and survival, and mediates tumor cell resistance.
- A Study of Inlexisertib (DCC-3116) in Combination With Anticancer Therapies in Participants With Advanced MalignanciesCTID: NCT05957367Phase: Phase 1/Phase 2Status: RecruitingDate: 2025-06-05
- A Phase 1/2 Study of Inlexisertib (DCC-3116) in Patients With RAS/MAPK Pathway Mutant Solid TumorsCTID: NCT04892017Phase: Phase 1/Phase 2Status: RecruitingDate: 2025-05-06
SYN
https://patents.google.com/patent/US11530206B2/en
PAT
Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereof
Publication Number: JP-7593947-B2
Priority Date: 2019-05-10
Grant Date: 2024-12-03
- PHENYLAMINOPYRIMIDINE AMIDE INHIBITORS OF AUTOPHAGY AND METHODS OF THEIR APPLICATIONPublication Number: HR-P20231730-T1Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: US-12071432-B2Priority Date: 2019-05-10Grant Date: 2024-08-27
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: CN-118878519-APriority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: CN-118878520-APriority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: CN-118930524-APriority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: AU-2020275392-B2Priority Date: 2019-05-10Grant Date: 2023-09-14
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: CN-114127057-BPriority Date: 2019-05-10Grant Date: 2024-07-12
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: EP-3966207-B1Priority Date: 2019-05-10Grant Date: 2023-11-01
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: EP-4342469-A2Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: ES-2966807-T3Priority Date: 2019-05-10Grant Date: 2024-04-24
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: EP-3966207-A1Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: KR-20220008873-APriority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitor and how to use itPublication Number: JP-2022531801-APriority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: US-11530206-B2Priority Date: 2019-05-10Grant Date: 2022-12-20
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: US-2023039712-A1Priority Date: 2019-05-10
- Combination of dcc-3116 and mapkap pathway inhibitors for use in the treatment of cancerPublication Number: WO-2024050351-A1Priority Date: 2022-09-02
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: US-2020354352-A1Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: WO-2020231806-A1Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitors and methods of use thereofPublication Number: AU-2020275392-A1Priority Date: 2019-05-10
- Phenylaminopyrimidine amide autophagy inhibitor and method of usePublication Number: CN-114127057-APriority Date: 2019-05-10
PAT
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020231806&_cid=P12-MHCSWS-98394-1

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////Inlexisertib, serine/ threonine kinase inhibitor, antineoplastic, DCC 3116, JM2ZTM8S7S
Imocitrelvir
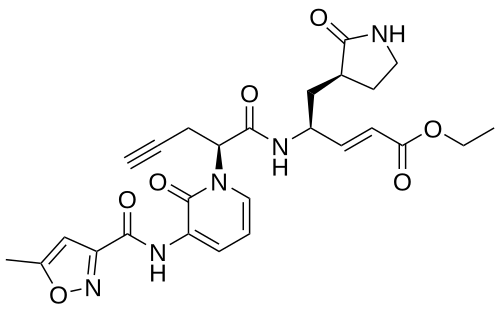
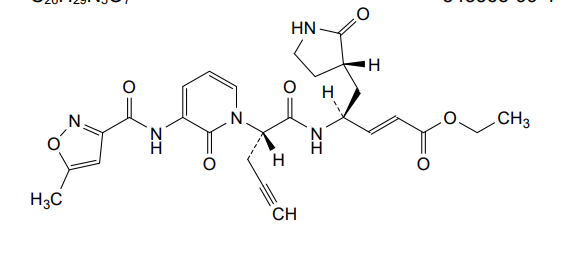
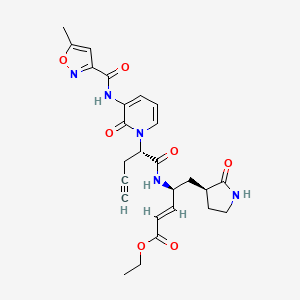
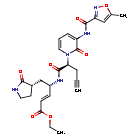
Imocitrelvir
CAS 343565-99-1
MFC26H29N5O7 MW523.5 g/mol
ethyl (2E,4S)-4-{(2S)-2-[3-(5-methyl-1,2-oxazole-3-carboxamido)-2-oxopyridin-1(2H)-yl]pent-4-ynamido}-5-[(3S)-2-oxopyrrolidin-3-yl]pent-2-enoate
ethyl (E,4S)-4-[[(2S)-2-[3-[(5-methyl-1,2-oxazole-3-carbonyl)amino]-2-oxo-1-pyridinyl]pent-4-ynoyl]amino]-5-[(3S)-2-oxopyrrolidin-3-yl]pent-2-enoate
protease inhibitor, antiviral, AG-7404, V-7404, AG 7404, V 7404, VQ1AN3OO42
Imocitrelvir is an investigational new drug that is being evaluated for the treatment of viral infections. It is a 3C protease inhibitor in picornaviruses. Originally developed by Pfizer for treating human rhinovirus infections,[1] this small molecule has shown promise against a broader range of viruses, including polioviruses.[2][3]
SYN
Publication Name: Journal of Medicinal Chemistry
Publication Date: 2003-09-17
PMID: 14521419
DOI: 10.1021/jm030166l
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016044656&_cid=P21-MHBDH2-20719-1
PAT
PAT
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2001040189&_cid=P21-MHBDI9-21481-1
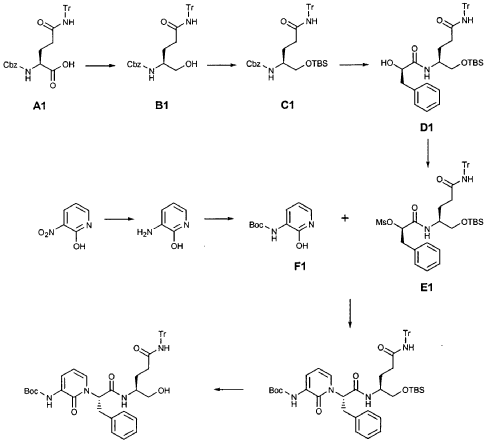
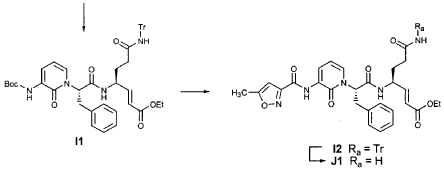
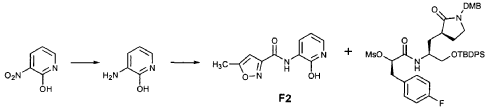
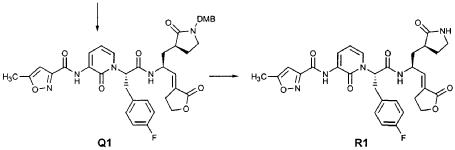
EXAMPLE 21
Preparation of Compound 22: tra«5-(4S,3″”S)-4-(2′-{3″-[(5′”-Methylisoxazole-3′”-carbonyl)amino]-2″-oxo-2″H-pyridin- 1 “-yl} acetylamino)-5-(2″”-oxopyrrilidin-3″”-yl)pent-2-enoic Acid Ethyl Ester
Preparation of Intermediate {3-[(5′-Methylisoxazole-3′-carbonyl)amino]-2-oxo-2H-pyridin-l-yl} acetic Acid tert-Butyl Ester
To a solution of 5-methylisoxazole-3-carboxylic acid (2′-hydroxy-4′-methylpyridin-3′-yl)amide (F2, Example 19) (0.520 g, 2.37 mmol, 1 equiv) in TΗF (20 mL) at 0 °C was added NaΗ (0.095 g, 2.37 mmol, 1.0 equiv). The resulting mixture was stirred at 0 °C for 20 min, and then t-butyl bromoacetate (0.385 mL, 2.61 mmol, 1.1 equiv) was added. The reaction mixture was stirred and warmed to room temperature for 30 min, then was partitioned between 0.5 N ΗC1 (100 mL) and EtOAc (2 x 100 mL). The combined organic layers were dried over Na2SO and were concentrated. Purification of the residue by flash column chromatography (30% EtOAc in hexanes) provided the title intermediate (0.628 g, 79%) as a white solid: IR (cm-1) 3343, 1743, 1651, 1581, 1156; Η NMR (CDC13) δ 1.52 (s, 9H), 2.53 (s, 3H), 4.65 (s, 2H), 6.32 (t, 1H, 7= 7.2), 6.51 (s, IH), 7.01 (dd, 1H, 7= 6.9, 1.8), 8.50 (dd, 1H, 7= 7.5, 1.8), 9.63 (s, br. IH); Anal. C16H19N3O5: C, H, N.
Preparation of Compound 22
The preceding intermediate was transformed into Compound 22 by a process that was analogous to that described in Example 25 for the transformation of V3 to product R3: mp = 102-106 °C; IR (cm”1) 3336, 1684, 1534, 1457; JH NMR (CDCI3) δ 1.27 (t, 3H, 7= 7.2), 1.67-1.75 (m, IH), 1.98-2.09 (m, IH), 2.37-2.49 (m, IH), 2.53 (s, 3H), 2.55-2.61 (m, IH), 3.34-3.46 (m, 2H), 3.51-3.52 (m, IH), 4.17 (q, 2H, 7= 7.2), 4.61-4.78 (m, 3H), 5.98 (dd, IH, 7 = 15.6, 1.5), 6.20 (s, br. IH), 6.35 (t, 1H, 7= 7.8), 6.51 (s, IH), 6.85 (dd, IH, 7= 15.6, 5.1), 7.17 (d, IH, 7= 7.2), 8.33 (d, IH, 7= 7.2), 8.49 (d, IH, 7= 7.5), 9.57 (s, br. IH); Anal.
C23H27N5O7: C, H, N.
EXAMPLE 24
Preparation of Compound 25: trans-(2’S,3″”‘S,4S)-4-(3,-(4″-Fluorophenyl)-2′-{3″‘-[(5″”-methylisoxazole-3″”-carbonyl)amino]-2′”-oxo-2′”H-pyridin- “-yl}propionylamino)-5-(2″ oxopyrrolidin-3′””-yl)pent-2-enoic Acid Ethyl Ester
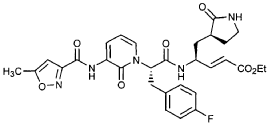
The title compound was prepared from F2 (Example 19) in a manner analogous to that described for the conversion of U2 to 13 in Example 23 utilizing intermediate Y2 (Example 25) where appropriate: IR (cm-1) 3331, 1690, 1590, 1531, 1455; !H NMR (CDCI3) δ 1.30 (t, 3H, 7= 7.0), 1.45-1.55 (m, IH), 1.64-1.75 (m, IH), 2.03-2.31 (m, 3H), 2.49 (s, 3H), 3.10 (dd, IH, 7= 13.7, 7.9), 3.20-3.46 (m, 3H), 4.20 (q, 2H, 7= 7.0), 4.36-4.47 (m, IH), 5.67 (dd, IH, 7 = 15.7, 1.4), 5.85-5.92 (m, IH), 6.29 (t, 1H, 7= 7.2), 6.45 (s, IH), 6.70 (dd, IH, 7= 15.7, 5.7), 6.86 (s, IH), 6.90-6.97 (m, 2H), 7.10-7.16 (m, 2H), 7.60 (dd, IH, 7= 7.2, 1.6), 8.37 (dd, IH, 7 = 7.2, 1.6), 8.51 (d, IH, 7= 6.6), 9.47 (s, IH).
EXAMPLE 25
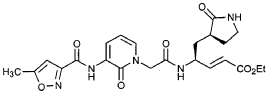
Preparation of Compound 26: tr_.«5-(2’S,3″”S,4S)-4-(2′-{3″-[(5″‘-Methyl-isoxazole-3′”-carbonyl)amino]-2″-oxo-2″H-pyridin-l”-yl}butyrylamino)-5-(2″”-oxopyrrolidin-3″”-yl)pent-2-enoic Acid Ethyl Ester (R3)
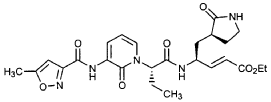
Preparation of Intermediate (2R)-2-Trifluoromethanesulfonyl-oxybutyric acid tert-butyl ester (U3)
Commercially available T3 (0.575 g, 3.59 mmol, 1 equiv) was dissolved in CH2CI2 (25 mL) and cooled in an ice bath. 2,6-Lutidine (0.836 mL, 7.18 mmol, 2 equiv) and trifluoromethanesulfonic anhydride (1.15 mL, 6.84 mmol, 1.9 equiv) were added and the reaction mixture was stirred 30 min. It was then diluted with MTBE (400 mL), washed with a mixture of brine and 1 N HCl (2:1, 100 mL) and brine (100 mL), dried over Na2SO4 and evaporated to provide the title intermediate which was used without further purification.
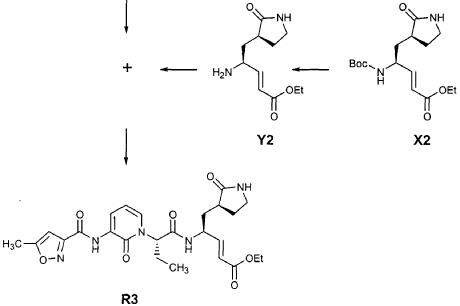
Preparation of Intermediate (2S)-2- {3′-[(5″-Methylisoxazole-3″-carbonyl)amino]-2′-oxo-2’H-pyri din- l’-yl} butyric Acid tert-Butyl Ester (V3)
Intermediate F2 from above (0.200 g, 0.912 mmol, 1.1 equiv) was suspended in TΗF (6 mL). Sodium hydride (60% dispersion in mineral oil, 0.0332 g, 0.830 mmol, 1 equiv) was added in one portion. After stirring 30 min, a solution of intermediate U3 (0.830 mmol, 1 equiv, based on T3) in TΗF (7 mL) was added dropwise. The resulting mixture was stirred 2 hours, then diluted with EtOAc (200 mL) and washed with brine (2 x 50 mL). The organic phase was dried over MgSO4 and evaporated. The residue was purified by flash column chromatography (25% EtOAc in hexanes) to provide the title intermediate (0.178 g, 59%) as an oil: R/= 0.30 (25% EtOAc in hexanes); IR (cm”1) 3331, 1731, 1690, 1649, 1602, 1531 ; *Η NMR (CDCI3) δ 0.93 (t, 3H, 7= 7.3), 1.45 (s, 9H), 1.83-2.01 (m, IH), 2.17-2.31 (m, IH), 2.50 (s, 3H), 5.44-5.51 (m, IH), 6.32 (t, IH, 7= 7.2), 6.48 (s, IH), 7.10 (dd, IH, 7= 7.2, 1.8), 8.45 (dd, 1H, 7= 7.2, 1.8), 9.64 (s, IH); Anal. C18H23N3O5: C, H, N.
Preparation of Intermediate (2S)-2- {3′-[(5″-Methylisoxazole-3″-carbonyl)amino]-2′-oxo-2’H-pyridin-l’-yl}butyric Acid (W3)
Intermediate V3 from above (0.143 g, 0.397 mmol, 1 equiv) was stirred for 1 h in a solution of TFA (2 mL) in CΗ2CI2 (3 mL). The volatiles were evaporated. The residue was suspended in toluene (10 mL) and concentrated to dryness, providing the title intermediate which was used without further purification.
Preparation of Intermediate trα«5-(3’S,4S)-4-Amino-5-(2′-oxopyrrolidin-3′-yl)pent-2-enoic Acid Ethyl Ester (Y2)
Intermediate X2, prepared according to the method disclosed in the co-pending application, U.S. Provisional Patent Application No. 60/150,358, filed August 24, 1999(0.130 g, 0.398 mmol, 1 equiv), was stirred for 30 min in a solution of TFA (2 mL) in CH2CI2 (3 mL). The volatiles were evaporated. The residue was suspended in toluene (10 mL) and concentrated to dryness, providing the title intermediate which was used without further purification.
Preparation of Product R3 (Compound 26)
Intermediates W3 and Y2 (as prepared above) were combined in CH2CI2 (7 mL) and cooled in an ice bath. HOBt (0.064 g, 0.47 mmol, 1.2 equiv), iP^NEt (0.484 mL, 2.78 mmol, 7 equiv) and EDC (0.084 g, 0.44 mmol, 1.1 equiv) were added sequentially. The reaction mixture was allowed to warm to 23 °C overnight, then diluted with EtOAc (500 mL) and washed with 5% KHSO4 , half saturated NaHCO3, and brine (100 mL each). The organic phase was dried over MgSO4 and evaporated. The residue was purified by flash column chromatography (gradient elution, 2→3% CH3OH in CH2CI2) to provide the title intermediate (0.119 g, 58%) as a white foam: IR (cm”1) 3331, 1684, 1649, 1590, 1531; JH NMR (CDCI3) δ 0.92 (t, 3H, J = 7.3), 1.29 (t, 3H, J = 7.1), 1.47-1.58 (m, IH), 1.62-1.77 (m, IH), 1.85-2.00 (m, IH), 2.08-2.33 (m, 4H), 2.49 (s, 3H), 3.25-3.42 (m, 2H), 4.19 (q, 2H, J = 7.1), 4.39-4.50 (m, IH), 5.73 (dd, IH, J = 8.8, 6.8), 5.97 (dd, IH, J = 15.7, 1.4), 6.34 (t, IH, J = 7.2), 6.46 (s, IH), 6.86 (dd, IH, J = 15.7, 5.9), 7.18 (s, IH), 7.59 (dd, IH, J = 7.2, 1.8), 8.42 (dd, IH, J = 7.2, 1.8), 8.58-8.62 (m, IH), 9.56 (s, 1); Anal. C25H31N5O7O.5OH2O: C, H, N.
PAT
- Treatment of infection by human enterovirus d68Publication Number: US-2020016243-A1Priority Date: 2014-09-17
- Treatment of infection by human enterovirus d68Publication Number: WO-2016044656-A1Priority Date: 2014-09-17
- Treatment of infection by human enterovirus d68Publication Number: US-2021052708-A1Priority Date: 2014-09-17
- Treatment of infection by human enterovirus D68Publication Number: US-11191817-B2Priority Date: 2014-09-17Grant Date: 2021-12-07
- Therapeutic compounds and methodsPublication Number: US-2025051283-A1
- Protease Inhibitors for Treatment or Prevention of Coronavirus DiseasePublication Number: US-2023192660-A1Priority Date: 2020-05-08
- Composition and combined medication method for treating enterovirus infectionPublication Number: US-2019030027-A1Priority Date: 2016-01-29
- Composition and combined medication method for treating enterovirus infectionPublication Number: US-10864210-B2Priority Date: 2016-01-29Grant Date: 2020-12-15
- Treatment of infection by human enterovirus D68Publication Number: US-10328128-B2Priority Date: 2014-09-17Grant Date: 2019-06-25
- Treatment of infection by human enterovirus d68Publication Number: US-2017290893-A1Priority Date: 2014-09-17
- Nucleotide and nucleoside therapeutic compositions, combinations and related uses thereofPublication Number: CN-117881402-APriority Date: 2021-05-05
- Nucleotide and nucleoside therapeutic compositions, combinations and uses related theretoPublication Number: EP-4333859-A1Priority Date: 2021-05-05
- Nucleotide and nucleoside therapeutic compositions, combinations, and related usesPublication Number: JP-2024517807-APriority Date: 2021-05-05
- Nucleotide and nucleoside therapeutic compositions, combinations and uses related theretoPublication Number: WO-2022235874-A1Priority Date: 2021-05-05
- Protease inhibitors for treatment or prevention of coronavirus diseasePublication Number: EP-4146267-A1Priority Date: 2020-05-08
- 4′-substituted nucleosides and nucleotides as antiviral agentsPublication Number: WO-2024227159-A2Priority Date: 2023-04-28
- Therapeutic compoundsPublication Number: WO-2024206284-A2Priority Date: 2023-03-27
- Antibody molecules binding to sars-cov-2Publication Number: WO-2024168061-A2Priority Date: 2023-02-07
- Predictive model for variants associated with drug resistance and theranostic applications thereofPublication Number: WO-2023172635-A1Priority Date: 2022-03-08
- Nucleotide and nucleoside therapeutic compositions, combinations and uses related theretoPublication Number: CA-3216679-A1Priority Date: 2021-05-05
LIT
- Structure and inhibition of SARS-CoV-1 and SARS-CoV-2 main proteases by oral antiviral compound AG7404Publication Name: Antiviral ResearchPublication Date: 2022-12PMCID: PMC9632241PMID: 36336176DOI: 10.1016/j.antiviral.2022.105458
- Bioisosteres of the Phenyl Ring: Recent Strategic Applications in Lead Optimization and Drug DesignPublication Name: Journal of Medicinal ChemistryPublication Date: 2021-09-30PMID: 34591488DOI: 10.1021/acs.jmedchem.1c01215
- In Vitro Antiviral Activity of New Oxazoline Derivatives as Potent Poliovirus InhibitorsPublication Name: Journal of Medicinal ChemistryPublication Date: 2018-12-04PMCID: PMC9169555PMID: 30512950DOI: 10.1021/acs.jmedchem.8b01482
- A Novel Series of Highly Potent Small Molecule Inhibitors of Rhinovirus ReplicationPublication Name: Journal of Medicinal ChemistryPublication Date: 2017-06-15PMID: 28581749DOI: 10.1021/acs.jmedchem.7b00175
- Anti-poliovirus activity of protease inhibitor AG-7404, and assessment of in vitro activity in combination with antiviral capsid inhibitor compoundsPublication Name: Antiviral ResearchPublication Date: 2013-05PMID: 23499651DOI: 10.1016/j.antiviral.2013.03.003
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | AG-7404, V-7404 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 343565-99-1 |
| PubChem CID | 5280053 |
| IUPHAR/BPS | 13223 |
| UNII | VQ1AN3OO42 |
| ChEMBL | ChEMBL141157 |
| Chemical and physical data | |
| Formula | C26H29N5O7 |
| Molar mass | 523.546 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “Imocitrelvir”. PatSnap.
- Xie H, Rhoden EE, Liu HM, Ogunsemowo F, Mainou BA, Burke RM, et al. (November 2024). “Antiviral Development for the Polio Endgame: Current Progress and Future Directions”. Pathogens. 13 (11). Basel, Switzerland: 969. doi:10.3390/pathogens13110969. PMC 11597170. PMID 39599522.
- Bandyopadhyay AS, Burke RM, Hawes KM (June 2024). “Polio Eradication: Status, Struggles and Strategies”. The Pediatric Infectious Disease Journal. 43 (6): e207-211. doi:10.1097/INF.0000000000004330. PMID 38564755.
////////Imocitrelvir, protease inhibitor, antiviral, AG-7404, V-7404, AG 7404, V 7404, VQ1AN3OO42
Ilantimod
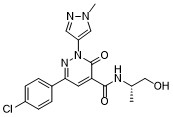
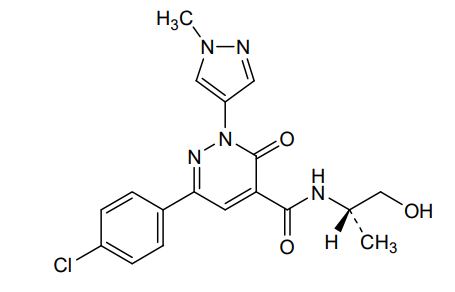
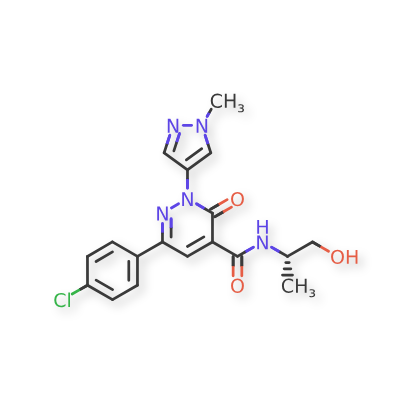
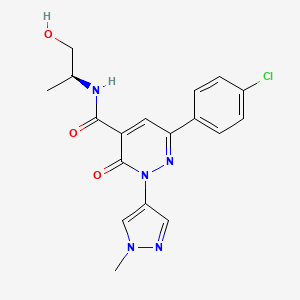
Ilantimod
CAS 2242464-44-2
MF C18H18ClN5O3 MW 387.82
6-(4-chlorophenyl)-N-[(2S)-1-hydroxypropan-2-yl]-2-(1-methyl-1H-pyrazol-4-yl)-3-oxo-2,3-dihydropyridazine-4-carboxamide
(S)-6-(4-chlorophenyl)-N-(1-hydroxypropan-2-yl)-2-(1-methyl-1H-pyrazol-4-yl)-3-oxo-2,3-dihydropyridazine-4-carboxamide
immunomodulator, BAY-2416964, BAY 2416964, Y87V4WXQ4Z
Ilantimod is an orally available formulation containing a small molecule antagonist of the aryl hydrocarbon receptor (AhR; class E basic helix-loop-helix protein 76; bHLHe76) with potential immunomodulating and antineoplastic activities. Upon oral administration, ilantimod specifically binds to AhR, inhibits AhR activation, and prevents AhR-mediated signaling. Abrogation of AhR activation prevents the activation of immune-tolerant dendritic cells (DCs) and regulatory T-cells (Tregs) in the tumor microenvironment (TME). This may restore the immune response against tumor cells. AhR, a member of the basic helix-loop-helix/Per-Arnt-Sim (bHLH/PAS) family of transcription factors, has important roles in regulating immunity and cellular differentiation. AhR can exhibit both pro-oncogenic and tumor suppressor-like functions depending on the tumor type; therefore, its expression may serve as a negative or positive prognostic factor.
- A Study to Learn How Safe the Study Drug BAY 2416964 (AhR Inhibitor) in Combination With the Treatment Pembrolizumab is, How This Combination Affects the Body, the Maximum Amount That Can be Given, How it Moves Into, Through and Out of the Body and Its Action Against Advanced Solid Cancers in AdultsCTID: NCT04999202Phase: Phase 1Status: TerminatedDate: 2025-02-10
- A First-in-Humans Dose Finding Study for an Aryl Hydrocarbon Receptor Inhibitor (AhRi) in Patients With Advanced CancerCTID: NCT04069026Phase: Phase 1Status: CompletedDate: 2024-03-06
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018146010&_cid=P11-MHAFJG-41587-1
Example 17
6-(4-Chlorophenyl)-/V-[(2S)-1 -hydroxypropan-2-yl]-2-(1 -methyl-1 H-pyrazol-4-yl)-3-oxo-2,3-dihydropyridazine-4-carboxamide
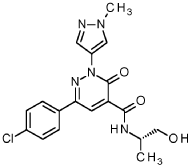

A solution of 80 mg intermediate 1 1 , 29.1 mg (2S)-2-aminopropan-1 -ol, 1 10 mg HATU and 0.1 mL ethyldiisopropylamine in 5 mL of DMF was stirred at room temperature for 14 hours. Then the reaction was quenched by water, and the mixture was extracted with dichloromethane two times. The combined organic phases were dried over sodium sulfate and evaporated to dryness. The residue was subjected to RP-HPLC ((column: X-Bridge C18 5μηι 100x30mm, mobile phase: acetonitrile / water (0.1 vol% formic acid)-gradient)) to yield 50 mg 6-(4-chlorophenyl)-/V-[(2S)-1 -hydroxypropan-2-yl]-2-(1 -methyl-1 H-pyrazol-4-yl)-3-oxo-2,3-dihydropyridazine-4-carboxamide
1H-NMR (400 MHz, CDC ): δ [ppm] = 1.34 (d, 3H); 2.73-2.82 (m, 1 H); 3.66-3.73 (m, 1 H); 3.77-3.84 (m, 1 H); 3.98 (s, 3H); 4.26-4.36 (m, 1 H); 7.49 (d, 2H); 7.87 (d, 2H); 8.12 (s, 1 H); 8.33 (s, 1 H); 8.69 (s, 1 H); 9.82 (bd, 1 H).
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US438191125&_cid=P11-MHAFQQ-47913-1
SEE EX 17
PAT
- 2-Heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamide for the treatment of cancerPublication Number: KR-102627266-B1Priority Date: 2017-02-09Grant Date: 2024-01-24
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: US-11795164-B2Priority Date: 2017-02-09Grant Date: 2023-10-24
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: US-2024294505-A1Priority Date: 2017-02-09
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamidesPublication Number: TW-I770113-BPriority Date: 2017-02-09Grant Date: 2022-07-11
- 2-Heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: CN-110678459-BPriority Date: 2017-02-09Grant Date: 2023-04-04
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: US-2023121195-A1Priority Date: 2017-02-09
- 2-Heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: CN-116531380-APriority Date: 2017-02-09
- 2-Heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: CN-116554152-APriority Date: 2017-02-09
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: WO-2018146010-A1Priority Date: 2017-02-09
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: EP-3580211-B1Priority Date: 2017-02-09Grant Date: 2020-12-02
- 2-HETEROARYL-3-OXO-2,3-DIHYDROPYRIDAZINE-4-CARBOXAMIDES FOR THE TREATMENT OF CANCERPublication Number: HR-P20210143-T1Priority Date: 2017-02-09
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: AU-2018217860-B2Priority Date: 2017-02-09Grant Date: 2021-07-08
- 2-Troaril-3-oxo-3,2-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: IL-268469-APriority Date: 2017-02-09
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: CA-3052718-A1Priority Date: 2017-02-09
- 2-Heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamide for the treatment of cancerPublication Number: CN-110678459-APriority Date: 2017-02-09
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: EP-3580211-A1Priority Date: 2017-02-09
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamide for the treatment of cancerPublication Number: KR-20190115460-APriority Date: 2017-02-09
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamidePublication Number: TW-201840549-APriority Date: 2017-02-09
- Combination of an ahr-inhibitor and an pd1-inhibitor antibody and its use in the treatment of cancerPublication Number: EP-4076462-A1Priority Date: 2019-12-16
- Combinations of AHR inhibitors and PD1 inhibitor antibodies and their use in the treatment of cancerPublication Number: JP-2023505907-APriority Date: 2019-12-16
- Combinations of AHR-inhibitors and PD1-inhibitor antibodies and their use in the treatment of cancerPublication Number: KR-20220128622-APriority Date: 2019-12-16
- Combination of an ahr-inhibitor and an pd1-inhibitor antibody and its use in the treatment of cancerPublication Number: US-2023084899-A1Priority Date: 2019-12-16
- 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancerPublication Number: AU-2018217860-A1Priority Date: 2017-02-09
- Methods and compositions for treating inflammatory and fibrotic pulmonary disordersPublication Number: US-2021401987-A1Priority Date: 2020-03-20
- Methods and compositions for treating inflammatory and fibrotic pulmonary disordersPublication Number: EP-4121111-A1Priority Date: 2020-03-20
- Combination of an ahr-inhibitor and an pd1-inhibitor antibody and its use in the treatment of cancerPublication Number: WO-2021122434-A1Priority Date: 2019-12-16
- Combination of an AhR-inhibitor and an PD1-inhibitor antibody and its use in the treatment of cancerPublication Number: AU-2020403801-A1Priority Date: 2019-12-16
- Combinations of AHR inhibitor and PD1 inhibitor antibodies and their use in cancer therapyPublication Number: CN-114786674-APriority Date: 2019-12-16
- Compositions and methods for treating myelin deficiency by rejuvenating glial progenitor cellsPublication Number: US-2023190961-A1Priority Date: 2021-10-20
- Prophylactic or therapeutic agent for severe pulmonary hypertension, refractory pulmonary hypertension, or drug-induced pulmonary hypertensionPublication Number: WO-2022149605-A1Priority Date: 2021-01-08
- Deuterated 2-arylheterocycle-3-oxo-2,3-dihydropyridazine-4-carboxamide inhibitor and preparation method therefor and application thereofPublication Number: EP-4253374-A1Priority Date: 2020-11-27
- Heteroaromatic ahr inhibitorPublication Number: WO-2022078356-A1Priority Date: 2020-10-15
- Methods and compositions for treating inflammatory and fibrotic pulmonary disordersPublication Number: WO-2021188849-A1Priority Date: 2020-03-20
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Ilantimod, immunomodulator, BAY-2416964, BAY 2416964, Y87V4WXQ4Z
Icovamenib
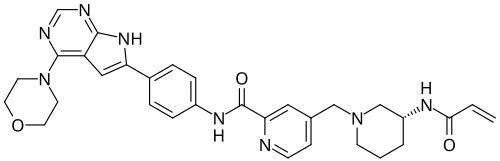
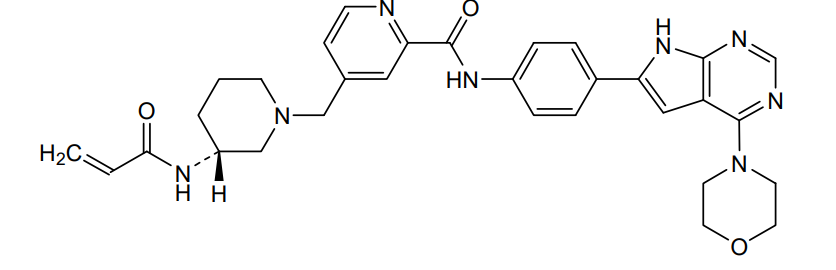
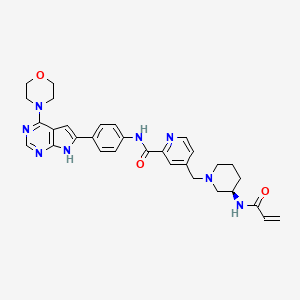
Icovamenib
CAS 2448172-22-1
MF C31H34N8O3 MW 566.7 g/mol
N-{4-[4-(morpholin-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl}-4-{[(3R)-3-(prop-2-enamido) piperidin-1-yl]methyl}pyridine-2-carboxamide
N-[4-(4-morpholin-4-yl-7H-pyrrolo[2,3-d]pyrimidin-6-yl)phenyl]-4-[[(3R)-3-(prop-2-enoylamino)piperidin-1-yl]methyl]pyridine-2-carboxamide
menin-MLL (mixed-lineage leukemia) protein interaction inhibitor,
antineoplastic, BMF-219, BMF 219, 2Z737MY35A, Menin-MLL inhibitor 21
Icovamenib is an investigational irreversible covalent inhibitor of menin. It is developed by Biomea Fusion for diabetes, lymphoma, leukemia, and multiple myeloma.[1][2][3]
Icovamenib is an orally bioavailable, irriversible inhibitor of menin, an essential co-factor of oncogenic menin-mixed lineage leukemia (MLL; myeloid/lymphoid leukemia; KMT2A) fusion proteins, with potential antineoplastic activity. Upon oral administration, icovamenib specifically targets and binds to menin, thereby preventing the interaction between the two proteins menin and MLL and the formation of the menin-MLL complex. This reduces the expression of downstream target genes, such as MYC and Bcl2, and results in an inhibition of the proliferation of MLL-rearranged tumor cells. Menin, an essential transcriptional regulator, plays a key role in oncogenic signaling in cancers driven by oncogenic MLL-fusions.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US299042443&_cid=P20-MH9YDY-31032-1
Example 9
Synthesis of Compound 10
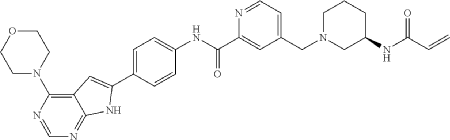
Compound 10
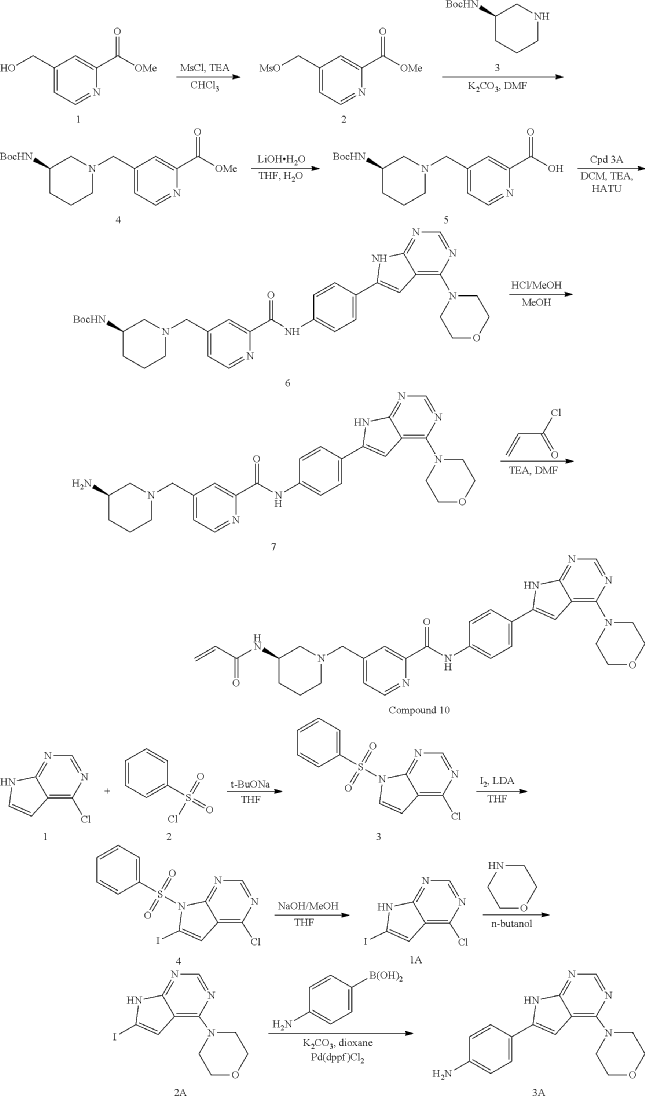
General Procedure for Preparation of Intermediate 2
| 1H NMR: CDCl 3 400 MHz 8.80 (d, J=4.85 Hz, 1H), 8.15 (d, J=0.66 Hz, 1H), 7.53 (dt, J=4.91, 0.85 Hz, 1H), 5.27-5.34 (m, 2H), 4.00-4.08 (m, 3H), 3.11 (s, 3H) |
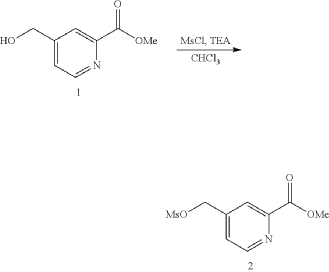
General Procedure for Preparation of Intermediate 5—
| To a solution of Intermediate 4 (1.50 g, 4.29 mmol, 1 eq) in THF (7.00 mL) was added LiOH.H 2O (540.3 mg, 12.8 mmol, 3 eq) in H 2O (7.00 mL). The mixture was stirred at 25° C. for 3 h. TLC (Dichloromethane:Methanol=10:1, R f=0) showed the reaction was complete. The mixture was poured into H 2O (20.0 mL) and extracted with DCM (10.0 mL×3). Then the organic phases dried over Na 2SO 4, filtered and concentrated under vacuum. The crude without purification. Give the Intermediate 5 (1.20 g, crude) as a yellow solid. |
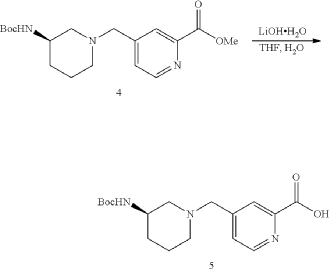
General Procedure for Preparation of Intermediate 6—
| To a solution of Intermediate 5 (0.80 g, 2.39 mmol, 1 eq), Intermediate 3A (704.4 mg, 2.39 mmol, 1 eq), TEA (1.69 g, 16.7 mmol, 2.32 mL, 7 eq) in DCM (10.0 mL) was added HATU (1.36 g, 3.58 mmol, 1.5 eq). The mixture was stirred at 20° C. for 12 h. LCMS showed the reaction was complete. The mixture was poured into H 2O (40.0 mL) and extracted with DCM (20.0 mL×3). Then the organic phases were washed with brine (50.0 mL) dried over Na 2SO 4, filtered and concentrated under vacuum. The crude for next step without purification. Give the Intermediate 6 (0.60 g, crude) as a yellow solid. |
General Procedure for Preparation of Intermediate 7—
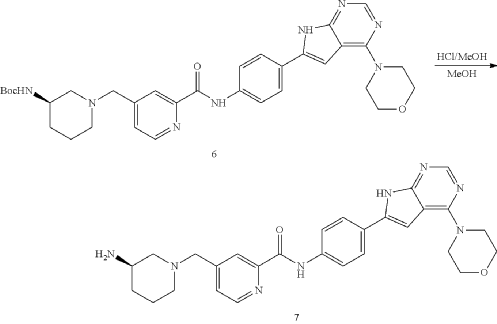
| To a solution of Intermediate 6 (0.50 g, 816.0 umol, 1 eq) in MeOH (5.00 mL) was added HCl/MeOH (4 M, 5.00 mL, 24.51 eq). The mixture was stirred at 20° C. for 12 h. LCMS showed the reaction was complete. The mixture was concentrated under vacuum. The crude for next step without purification. Give the Intermediate 7 (0.50 g, crude, HCl) as a yellow solid. |
General Procedure for Preparation of Compound 10—
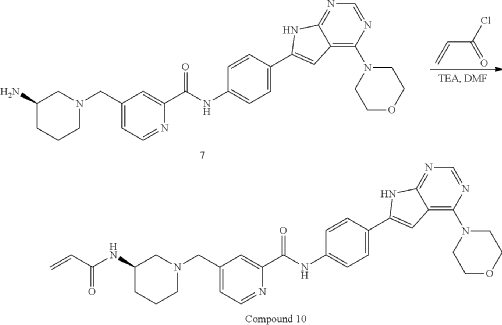
| To a solution of Intermediate 3 (0.50 g, 910.6 umol, 1 eq, HCl) in DMF (10.0 mL) was added TEA (645.0 mg, 6.37 mmol, 887.2 uL, 7 eq) and prop-2-enoyl chloride (82.4 mg, 910.6 umol, 74.2 uL, 1 eq). Then the mixture was stirred at 20° C. for 12 h. LCMS showed the reaction was complete. The mixture was poured into H 2O (50.0 mL), then was filtered and filter cake was concentrated in vacuum. The crude product was purified by reversed-phase HPLC (column: Phenomenex Luna C18 200*40 mm*10 um; mobile phase: [water(0.05% HCl)-ACN]; B %: 10%-30%, 10 min) and (column: Xtimate C18 150*25 mm*5 um; mobile phase: [water(10 mM NH 4HCO 3)-ACN]; B %: 30%-60%, 10 min). Give the Intermediate Compound 10 (20.0 mg, 35.0 umol, 3.85% yield, 99.3% purity) as a yellow solid. |
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024172911&_cid=P20-MH9YNT-37455-1
PAT
- Substituted pyridines as irreversible inhibitors of menin-MLL interactionPublication Number: US-11702421-B2Priority Date: 2018-12-31Grant Date: 2023-07-18
- Irreversible inhibitors of menin-mll interactionPublication Number: US-2023227458-A1Priority Date: 2018-12-31
- N-[4-[4-(4-MORPHOLINYL)-7H-PYRROLO[2,3-d]PYRIMIDIN-6-YL]PHENYL]-4-[[3(R)-[(1-OXO-2-PROPEN-1-YL)AMINO]-1-PIPERIDINYL]METHYL]-2-PYRIDINE CARBOXAMIDE AND USES THEREOFPublication Number: US-2024376112-A1Priority Date: 2018-12-31
- Covalent inhibitors of menin-mll interaction for diabetes mellitusPublication Number: WO-2023018825-A1Priority Date: 2021-08-11
- Covalent inhibitors of Mennin-MLL interaction for diabetesPublication Number: CN-118076357-APriority Date: 2021-08-11
- Irreversible inhibitors of menin-mll interactionPublication Number: US-2020223853-A1Priority Date: 2018-12-31
- Substituted pyridines as irreversible inhibitors of menin-MLL interactionPublication Number: US-11084825-B2Priority Date: 2018-12-31Grant Date: 2021-08-10
- Irreversible inhibitors of menin-mll interactionPublication Number: US-2022169627-A1Priority Date: 2018-12-31
- Crystalline forms of an irreversible inhibitor of menin-mll interactionPublication Number: US-2023086137-A1Priority Date: 2021-08-20
- Crystalline form of n-[4-[4-(4-morpholinyl)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-4-[[3(r)-[(1-oxo -2-propen-1-yl)amino]-1-piperidinyl]methyl]-2-pyridinecarboxamide, an irreversible menin-mll inhibitor for the treatment of cancerPublication Number: EP-4387972-A1Priority Date: 2021-08-20
- Crystalline forms of N-[4-[4-(4-morpholinyl)-7H-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-4-[[3(r)-[(1-oxo-2-propen-1-yl)amino]-1-piperidinyl]methyl]-2-pyridinecarboxamide as an irreversible inhibitor of menin-MLL interactionPublication Number: US-12018032-B2Priority Date: 2021-08-20Grant Date: 2024-06-25
- Crystalline form of n-[4-[4-(4-morpholinyl)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-4-[[3(r)-[(1-oxo -2-propen-1-yl)amino]-1-piperidinyl]methyl]-2-pyridinecarboxamide, an irreversible menin-mll inhibitor for the treatment of cancerPublication Number: WO-2023022912-A1Priority Date: 2021-08-20
- Crystalline forms of an irreversible inhibitor of menin-mll interactionPublication Number: US-2024343731-A1Priority Date: 2021-08-20
- Synthetic methods for preparing a pyridinecarboxamide compoundPublication Number: WO-2024011450-A1Priority Date: 2022-07-13
- Menin-mll inhibitors and compositions for proliferation of beta cellsPublication Number: WO-2024006391-A1Priority Date: 2022-06-28
- Flt3 combination therapy for cancer and compositions thereforPublication Number: WO-2023225005-A1Priority Date: 2022-05-17
- Treatment of cancer with menin inhibitors and immuno-oncology agentsPublication Number: WO-2023172925-A1Priority Date: 2022-03-08
- Treatment of hematological malignancies with menin inhibitors and p-glycoprotein inhibitorsPublication Number: WO-2023150635-A1Priority Date: 2022-02-04
- Crystalline forms of N[4[4-(4-Morpholinyl)-7H-Pyrrolo[2-3-D]Pyrimidin-6-yl]Phenyl]-4-[[3(R)-[(1-Oxo-2-Protein-1-yl)Amino]-1-Piperidinyl]Methyl]2-Pyridinecarboxamide]Publication Number: US-12215113-B2Priority Date: 2023-01-18Grant Date: 2025-02-04
- CRYSTALLINE FORMS OF N-[4-[4-(4-MORPHOLINYL)-7H-PYRROLO[2,3-d]PYRIMIDIN-6-YL]PHENYL]-4-[[3(R)-[(1-OXO-2-PROPEN-1-YL)AMINO]-1-PIPERIDINYL]METHYL]-2-PYRIDINECARBOXAMIDE AS IRREVERSIBLE INHIBITORS OF MENIN-MLL INTERACTIONPublication Number: US-2024417404-A1Priority Date: 2023-01-18
- Crystalline forms of n-[4-[4-(4-morpholinyl)-7h-pyrrolo[2,3-d]pyrimidin-6- yl]phenyl]-4-[[3(r)-[(l-oxo-2-propen-l-yl)amino]-l-piperidinyl]methyl]-2-pyridinecarboxamide as a covalent inhibitor of menin-mll interactionPublication Number: WO-2024155710-A1Priority Date: 2023-01-18
- Crystalline forms of n-[4-[4-(4-morpholinyl)-7h-pyrrolo[2,3-d]pyrimidin-6-yl]phenyl]-4-[[3(r)-[(l-oxo-2-propen-l-yl)amino]-l-piperidinyl]methyl]-2- pyridinecarboxamide as a covalentinhibitor of menin-mll interactionPublication Number: WO-2024155719-A1Priority Date: 2023-01-18
- Combinations of lsd1 inhibitors and menin inhibitors for treating cancerPublication Number: WO-2024110649-A1Priority Date: 2022-11-24
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Rodriguez, Jose E.; Abitbol, Alexander; Abuzgaya, Fathi; Perez, Cesar; Mourya, Sanchita; Munneke, Brian; Morris, Stephan W.; Butler, Thomas (20 June 2023). “91-LB: COVALENT-111, a Phase 1/2 Trial of BMF-219, a Covalent Menin Inhibitor, in Patients with Type 2 Diabetes Mellitus—Preliminary Results”. Diabetes. 72 (Supplement_1) 91-LB. doi:10.2337/db23-91-LB. S2CID 259444592.
- Ravandi-Kashani, F.; Kishtagari, A.; Carraway, H.; Schiller, G.; Curran, E.; Yadav, B.; Cacovean, A.; Morris, S.; Butler, T.; Lancet, J. (23 June 2022). “P587: Covalent-101: A Phase 1 Study of BMF-219, A Novel Oral Irreversible Menin Inhibitor, in Patients with Relapsed/Refractory Acute Leukemia, Diffuse Large B-Cell Lymphoma, and Multiple Myeloma”. HemaSphere. 6: 486–487. doi:10.1097/01.HS9.0000845236.32931.83.
- Somanath, Priyanka; Lu, Daniel; Law, Brian; Archer, Tenley C.; Cacovean, Alexandru; Palmer, James T.; Kinoshita, Taisei; Butler, Thomas (5 November 2021). “Novel Irreversible Menin Inhibitor, BMF-219, Shows Potent Single Agent Activity in Clinically Relevant DLBCL Cells”. Blood. 138 (Supplement 1): 4318. doi:10.1182/blood-2021-148045.
| Clinical data | |
|---|---|
| Other names | BMF-219 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2448172-22-1 |
| PubChem CID | 154988914 |
| ChemSpider | 115037287 |
| UNII | 2Z737MY35A |
| Chemical and physical data | |
| Formula | C31H34N8O3 |
| Molar mass | 566.666 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////Icovamenib, antineoplastic, BMF-219, BMF 219, 2Z737MY35A, Menin-MLL inhibitor 21
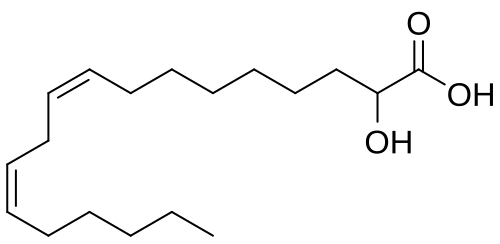
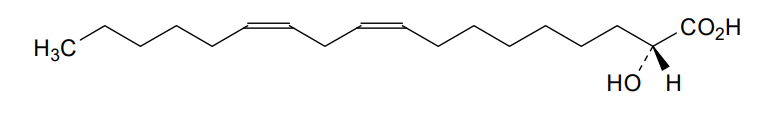
Ibrilatazar
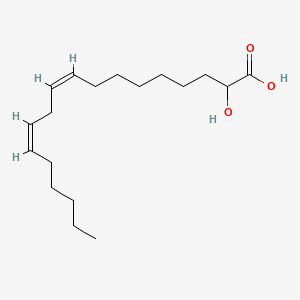
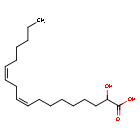
Ibrilatazar
CAS 57818-44-7
MF C18H32O3 MW 296.4 g/mol
rac-(2R)-(9Z,12Z)-2-hydroxyoctadeca-9,12-dienoic acid
(9Z,12Z)-2-hydroxyoctadeca-9,12-dienoic acid
peroxisome proliferator activated receptor (PPAR) alpha and gamma agonist, antineoplastic, ABILITY PHARMA, ABTL 0812, alpha-Hydroxylinoleic acid, ABTL0812
- alpha-Hydroxylinoleic acid
- ABTL0812
- 2-hydroxylinoleic acid
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| ABTL-0812 Sodium | X1840C8161 | Not Available | VFXKYDDSDQXKLC-NBTZWHCOSA-M |
Ibrilatazar also known as α-hydroxylinoleic acid is a small-molecule, experimental cancer drug being developed by Ability Pharmaceuticals.[1]
Ibrilatazar is an orally bioavailable, lipid analogue and inhibitor of raptor-mammalian target of rapamycin (mTOR) (mTOR complex 1; mTORC1), rictor-mTOR (mTOR complex 2; mTORC2) and dihydrofolate reductase (DHFR) with potential antineoplastic activity. Upon oral administration, ibrilatazar binds to and inhibits both mTORC1 and mTORC2, which may result in apoptosis and a decrease in proliferation in mTORC1/2-expressing tumor cells. mTOR is a serine/threonine kinase that is upregulated in some tumors; it plays an important role in the PI3K/Akt/mTOR signaling pathway which is often deregulated in cancer cells. In addition, ibrilatazar inhibits DHFR, an enzyme that reduces dihydrofolic acid to tetrahydrofolic acid, thereby blocking tetrahydrofolate synthesis, and resulting in both the depletion of nucleotide precursors and the inhibition of DNA, RNA and protein synthesis. This induces autophagy-induced cell death and further inhibition of cell proliferation.
- A Study of ABTL0812 in Pancreatic CancerCTID: NCT03417921Phase: Phase 1/Phase 2Status: SuspendedDate: 2024-07-31
- ABTL0812 in Combination With FOLFIRINOX for First-line Treatment of Metastatic Pancreatic StudyCTID: NCT04431258Phase: Phase 1/Phase 2Status: CompletedDate: 2024-03-18
- Phase I/Ib Clinical Trial of ABTL0812 in Advanced Cancer PatientsCTID: NCT02201823Phase: Phase 1Status: CompletedDate: 2015-07-02
- Microbiological production method of γ- and δ-lactonesPublication Number: JP-H03187387-APriority Date: 1989-08-04
- Process for the microbiological production of gamma- and delta-lactonesPublication Number: US-5168054-APriority Date: 1989-08-04Grant Date: 1992-12-01
- Degradation control of environmentally degradable disposable materialsPublication Number: US-2002123546-A1Priority Date: 1988-08-08
- Degradation control of environmentally degradable disposable materialsPublication Number: US-6323307-B1Priority Date: 1988-08-08Grant Date: 2001-11-27
- Degradation control of environmentally degradable disposable materialsPublication Number: US-6740731-B2Priority Date: 1988-08-08Grant Date: 2004-05-25
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US38087288&_cid=P12-MH8IQK-97634-1
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
History
In 2015, Ability announced that it had received orphan drug designation (ODD) for pediatric cancer neuroblastoma from the European Medical Agency (EMA) and the US Food and Drug Administration (FDA).[1] Also in 2016 a preclinical study confirmed that ABTL0812 was well tolerated.[2] In December 2016 the company announced Ibrilatazar has received an Orphan Drug Designation for the treatment of pancreatic cancer.[1]
Mechanism of action
One mechanism of action is the activation of the PPAR-alpha and PPAR-gamma receptors which in turn up-regulate the expression of the TRIB3 gene, leading to inhibition of the PI3K/AKT/mTOR pathway. This pathway is excessively activated in most human cancers, supporting tumor growth. It is a principal target of various new anti-tumour drugs. Tumor cells are killed via autophagic cell death, rather than apoptosis.[3][4]
ABTL0812 activates the PPAR receptors, inducing TRIB3 over-expression. TRIB3 binds to the Akt oncogene and inhibits the Akt/mTOR axis.[3]
Clinical trials
ABTL0812 showed efficacy in Phase I clinical trials in patients with advanced cancer, with low toxicity and high tolerability.[3]
References
- “Ability Pharmaceuticals Announces Orphan Drug Designation in the US for ABTL0812 in Pancreatic Cancer”. Ability Pharmaceuticals SL.
- “Ability Pharmaceuticals Announces Positive Phase 1 1b Study Results Of ABTL0812 In Cancer Patients With Advanced Solid Tumors”. http://www.biospace.com.
- “New mechanism of antitumor action identified”. Medical Xpress. 25 January 2016.
- Erazo T, Lorente M, López-Plana A, Muñoz-Guardiola P, Fernández-Nogueira P, García-Martínez JA, et al. (May 2016). “The New Antitumor Drug ABTL0812 Inhibits the Akt/mTORC1 Axis by Upregulating Tribbles-3 Pseudokinase”. Clinical Cancer Research. 22 (10): 2508–19. doi:10.1158/1078-0432.ccr-15-1808. hdl:2445/207600. PMID 26671995.
| Clinical data | |
|---|---|
| Other names | α-Hydroxylinoleic acid; 2-Hydroxylinoleic acid; ABTL-0812 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 57818-44-7 |
| PubChem CID | 21158511 |
| ChemSpider | 20118100 |
| UNII | 0DE74TJ7EZ |
| ChEBI | CHEBI:136927 |
| CompTox Dashboard (EPA) | DTXSID301258077 |
| Chemical and physical data | |
| Formula | C18H32O3 |
| Molar mass | 296.451 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////Ibrilatazar, peroxisome proliferator activated receptor (PPAR) alpha and gamma agonist, antineoplastic, ABILITY PHARMA, ABTL 0812, alpha-Hydroxylinoleic acid, ABTL0812
Glovadalen



Glovadalen
CAS 2576359-31-2
MF C24H27Cl2N3O3 MW 476.4 g/mol
2-(3,5-dichloro-1-methyl-1H-indazol-4-yl)-1-[(1S,3R)-3-(hydroxymethyl)-5-(2-hydroxypropan-2-yl)-1-methyl-3,4-dihydroisoquinolin-2(1H)-yl]ethan-1-one,
2-(3,5-dichloro-1-methylindazol-4-yl)-1-[(1S,3R)-3-(hydroxymethyl)-5-(2-hydroxypropan-2-yl)-1-methyl-3,4-dihydro-1H-isoquinolin-2-yl]ethanone
dopamine D1 receptor positive allosteric modulator, Phase 2, Parkinson’s disease, UCB-0022, UCB 0022, H8T5VKH4CZ
- OriginatorUCB Biopharma
- ClassAlcohols; Antiparkinsonians; Benzene derivatives; Chlorinated hydrocarbons; Isoquinolines; Ketones; Neuroprotectants; Propanols; Pyrazoles; Small molecules
- Mechanism of ActionDopamine D1 receptor modulators
- Phase IIParkinson’s disease
- 27 Aug 2025Chemical structure information added.
- 21 May 2025UCB Biopharma SRL initiate a phase I trial in healthy volunteers (PO) (NCT06970301)
- 11 Apr 2025UCB Pharma completes a phase-II ATLANTIS trial in Parkinson’s disease (In adults, In the elderly, Adjunctive treatment) in USA (PO) (NCT06055985)
Glovadalen (developmental code name UCB-0022) is a dopamine D1 receptor positive allosteric modulator which is under development for the treatment of Parkinson’s disease.[1][2][3][4][5][6] It has been found to potentiate the capacity of dopamine to activate the D1 receptor by 10-fold in vitro with no actions on other dopamine receptors.[5][6] As of May 2024, glovadalen is in phase 2 clinical trials for this indication.[1][2][5] The drug is under development by UCB Biopharma.[1][4][5] It is described as an orally active, centrally penetrant small molecule.[1][5][6]
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021001288&_cid=P21-MH738G-96748-1

1. Preparation of intermediate of formula (ID- 2-(3,5-dichloro-1-methyl-indazol-4- vDacetic acid

1.1. Preparation of intermediate (Xlb) -1-methyl-5-nitro-indazole
5-Nitro-1H-indazole (Xla) (3.00 kg, 18.4 mol) and DMF (30.0 L) are charged into a 50 L three-neck round-bottom flask at 15-30°C. KOH (2.06 Kg, 36.7 mol) is added in one portion into the reactor at 0-5°C. The mixture is stirred at 0-50°C for 1h. Methyl iodide (2.87 kg, 20.2 mol) is then added at 0-5°C and the mixture is stirred for 3h at 15-30°C. The reaction mixture is added into water (30 L) at 0-10°C and the mixture is stirred for 10 min then filtered. The filter cake is washed with water (5 L) and dried. This overall procedure is carried out on 4 batches of the same size in parallel. The solids obtained from the four batches are combined to give 1-methyl-5-nitro-indazole (Xlb) as a brown solid (10.0 kg, 42.3 mol, 75% purity (LC/MS), 57.5% yield) which is used in the next step without further purification.
1H NMR (400 MHz, CDCIs) d 8.65 (s, 1H), 8.21 (d, J = 9.17 Hz, 1 H), 8.13 (s, 1 H), 7.39 (d, J = 9.17 Hz, 1 H), 4.08 (s, 3 H).
1.2. Preparation of intermediate (Xa)- tert-butyl 2-(1-methyl-5-nitro-indazol-4- yl)acetate
t-BuOK (4.43 kg, 39.5 mol) and THF (30 L) are charged into a 50 L three-neck round-bottom flask and the mixture is cooled to -45 / -35°C under nitrogen and stirring. 1-Methyl-5-nitro-indazole (Xlb) (3.50 kg, 19.7 mol) is then added in portions at -45 / -35°C. Tert-butyl 2-chloroacetate (3.57 kg, 23.7 mol) is added dropwise at the same temperature and the mixture is stirred at 1h. The mixture is warmed up to 15-30°C and stirred for 5h. The reaction is quenched by the addition of a saturated ammonium chloride solution (9 L) and water (2 L) is added. The organic layer is separated and the aqueous layer is extracted with ethyl acetate (2 x 5 L). The organic phases are combined, washed with brine (2 L), dried over Na2SC>4, filtered and concentrated under vacuum. The crude product is purified by recrystallization with ethyl acetate (5 L). This overall procedure is carried out on 2 batches of the same size in parallel. The solids obtained from the two batches are combined and dried together to give tert-butyl 2-(1-methyl-5-nitro-indazol-4-yl)acetate as a yellow solid (Xa) (5.30 kg, 17.7 mol, 97.6% purity (LC/MS), 44.9% yield).
1H NMR (400 MHz, CDCIs) d 8.18-8.20 (m, 2H), 7.37 (d, J = 9.21 Hz, 1 H), 4.27 (s, 2 H), 4.14 (s, 3 H), 1.44 (s, 9 H).
1.3. Preparation of intermediate (Xb) – tert-butyl 2-(5-amino-1-methyl-indazol-4- yl)acetate
Tert-butyl 2-(1-methyl-5-nitro-indazol-4-yl)acetate (Xa) (7.30 kg, 25.0 mol) and MeOH (76 L) are charged into a reactor. Argon is purged and Pd/C (50%, 760 g) is added. Hydrogen is added three times and the mixture is stirred at 50°C under hydrogen atmosphere (50 psi) for 3h. The reaction mixture is filtered and the solid is washed with MeOH (5 L). The mixture is concentrated to give tert-butyl 2-(5-amino-1-methyl-indazol-4-yl)acetate (Xb) as a brown oil (6.50 kg, 23.9 mol, 96.2% purity (LC/MS), 95.4% yield) which is used in the next step without further purification.
1H NMR (400 MHz, CDCI3) d 7.72 (s, 1H), 7.27 (d, J = 8.80 Hz, 1 H), 6.91 (d, J = 8.80 Hz, 1 H), 4.60 (s, 2 H), 3.93 (s, 3 H), 3.68 (s, 2H), 1.38 (s, 9 H).
1.4. Preparation of intermediate (Xc)- 2-(5-chloro-1-methyl-indazol-4-yl)acetic acid
Tert-butyl 2-(5-amino-1-methyl-indazol-4-yl)acetate (Xb) (2.00 kg, 7.65 mol) and concentrated HCI (10.0 L, 12M) are charged into a 50 L three-neck round bottom flask and the mixture is cooled to -10/-5°C and stirred. A water solution (5 L) of sodium nitrite (686 g, 9.95 mol) is added dropwise at -10/-5°C and stirred for 30 min. CuCI (833 g, 8.42 mol) and concentrated HCI (10.0 L, 12M) are charged into a 20 L three-neck round bottom flask and the mixture is stirred for 30 min. at -10/-5°C, then added into the other reactor. The mixture is stirred at -10/-5°C for 1 h, then at 10-30°C for 16h. The reaction mixture is filtered and the solid washed with water. This overall procedure is carried out on 3 batches of the same size in parallel. The solids obtained from the three batches are combined and dried together to give 2-(5-chloro-1-methyl-indazol-4-yl)acetic acid (Xc) as a yellow solid (4.00 kg, 16.3 mol, 92% purity (LC/MS), 71.3% yield) which is used in the next step without further purification.
1.5. Preparation of 2-(3,5-dichloro-1-methyl-indazol-4-yl)acetic acid (II)
2-(5-Chloro-1-methyl-indazol-4-yl)acetic acid (Xc) (1.30 kg, 5.79 mol) and DMF (6.5 L) are charged into a 50 L three-neck round bottom flask at 20°C. N-Chlorosuccinimide (772 g, 5.79 mol) is added portionwise at 20°C and the mixture is stirred at 20°C for 2h. The reaction mixture is poured into water (25 L) and filtered. The crude product is triturated with isopropyl etherethyl acetate (3:1) (7.0 L) at 20°C for 2h then filtered and dried. This overall procedure is carried out on 3 batches of the same size in parallel. The solids obtained from the three batches are combined to give 2-(3,5-dichloro-1-methyl-indazol-4-yl)acetic acid (II) (2.1 kg, 7.9 mol, 97.5% purity (LC/MS), 46% yield).
1H NMR (400 MHz, CDCI3) d 12.67 (s, 1 H), 7.68 (d, J = 9.05 Hz, 1 H), 7.53 (d, J = 9.05 Hz, 1 H), 4.20 (s, 2 H), 4.02 (s, 3 H).
2. Preparation of compound of formula (I)
2-(3,5-dichloro-1-methyl-indazol-4-yl)-1-[(1S,3R)-3-(hydroxymethyl)-5-(1-hydroxy-1- methyl-ethyl)-1-methyl-3,4-dihydro-1H-isoquinolin-2-yl]ethanone

2.1. Preparation of intermediate (IX).
(2R)-2-amino-3-(2-bromophenyl)propan-1-ol – a6
(2R)-2-amino-3-(2-bromophenyl)propanoic acid a5 (34.0 kg, 139 mol) and THF (238 L) are charged into a reactor. Sodium borohydride (15.6 kg, 413 mol) is added slowly at 20-30°C. A solution of iodine (35.3 kg, 139 mol) in dry THF (20.0 L) is added slowly at 0-10°C and the reaction mixture is stirred at 70°C for 12h. The reaction was quenched with methanol (70.0 L) at 0°C and heated to 80°C for 30 min. The mixture was cooled down, concentrated under vacuum and the residue was suspended in NaOH (30.0 L, 2N), then filtered. The filter cake was dried under vacuum to give (2R)-2-amino-3-(2-bromophenyl)propan-1-ol a6 as a white solid (31.0 kg, 135 mol, 96.7% yield) which is used in the next step without further purification. 1H NMR (400 MHz, CDCIs) d 7.57 (d, J = 7.7 Hz, 1H), 7.21 – 7.29 (m, 2H), 7.07 – 7.15 (m, 1H), 3.66 (dd, J = 10.5, 3.6 Hz, 1 H), 3.41 (dd, J = 10.5, 7.2 Hz, 1 H), 3.18 – 3.29 (m, 1 H), 2.95 (dd, J = 13.5, 5.5 Hz, 1 H), 2.70 (dd, J = 13.5, 8.2 Hz, 1H), 1.51 – 1.91 (m, 3H).
2.2. Preparation of intermediate of formula (VIII).
(4R)-4-[(2-bromophenyl)methyl]oxazolidin-2-one – a7
(2R)-2-amino-3-(2-bromophenyl)propan-1-ol a6 (31.0 kg, 135 mol) and dichloromethane (220 L) are charged into a reactor. Triphosgene (13.9 kg, 47.1 mol) is added at room temperature then N,N-diisopropylethylamine (39.1 kg, 303 mol) is slowly added at 0-10°C. The reaction mixture is stirred at 0-10°C for 1h then washed with water (50.0 L) twice, dried with anhydrous sodium sulfate and filtered to give (4R)-4-[(2-bromophenyl)methyl]oxazolidin-2-one a7 as a solution in dichloromethane which is used directly in the next step.
2.3. Preparation of intermediate (VII).
(10aR)-9-bromo-1 ,5, 10, 10a-tetrahydrooxazolo[3,4-b]isoquinolin-3-one a8
A solution of (4R)-4-[(2-bromophenyl)methyl]oxazolidin-2-one a7 (135 mol) in dichloromethane (220 L) is charged into a reactor and cooled down to 0-5°C. Trimethylsilyl triflate (35.9 kg, 162 mol) and paraformaldehyde (13.3 kg, 148 mol) are added at 0-5°C, then stirred for 2h at 15-20°C. Water (170 L) is added into the mixture which is then extracted twice with dichloromethane (50.0 L). the organic layer is dried with anhydrous sodium sulfate, filtered and concentrated under vacuum. A mixture of petroleum etherethyl acetate (1 :1, 45.0 L) is added and the mixture is stirred at room temperature for 6h and filtered. The solid was dried to get (10aR)-9-bromo-1,5,10,10a-tetrahydrooxazolo[3,4-b]isoquinolin-3-one a8 as an off-white solid (29.0 kg, 80.2% yield).
1H NMR (400 MHz, CDCI3) d 7.45 – 7.52 (m, 1H), 7.08 – 7.14 (m, 2H), 4.83 (d, J = 17.0 Hz, 1H), 4.62 (t, J = 8.4 Hz, 1H), 4.36 (d, J = 17.0 Hz, 1H), 4.21 (dd, J = 8.6, 4.9 Hz, 1 H), 3.91 -3.99 (m, 1H), 3.25 (dd, J= 16.3, 4.2 Hz, 1 H), 2.67 (dd, J = 16.1 , 11.0 Hz, 1H).
2.4. Preparation of intermediates (VI)
2.4.1. [(3R)-5-bromo-1,2,3,4-tetrahydroisoquinolin-3-yl]methanol a9
Ethanol (120 L) and water (60.0 L) are mixed into a reactor. (10aR)-9-bromo-1,5,10,10a-tetrahydrooxazolo[3,4-b]isoquinolin-3-one a8 (29.7 kg, 111 mol) is added then sodium hydroxide (13.3 kg, 332 mol) is slowly added at 15-20°C. The reaction mixture is stirred at 90°C for 2h then cooled down to room temperature. Water (300 L) is added into the mixture which is centrifugated. The centrifugal cake is dried in circulation oven to give [(3R)-5-bromo- 1,2,3,4-tetrahydroisoquinolin-3-yl]methanol a9 as a white solid (23.7 kg, 88.3% yield) which is used in the next step without further purification.
1H NMR (400 MHz, CDCIs) d 7.37 – 7.47 (m, 1H), 6.95 – 7.08 (m, 2H), 4.00 – 4.10 (m, 2H), 3.85 (dd, J = 10.9, 3.7 Hz, 1 H), 3.57 (dd, J = 10.9, 7.9 Hz, 1 H), 3.06 (ddt, J = 11.3, 7.6, 4.1 , 4.1 Hz, 1H), 2.79 (dd, J= 17.1, 4.4 Hz, 1H), 2.40 (dd, J= 17.1, 10.9 Hz, 1H), 1.93 (br s, 2H).
2.4.2. [(3R)-5-bromo-1 ,2,3,4-tetrahydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl- silane a10
[(3R)-5-bromo-1,2,3,4-tetrahydroisoquinolin-3-yl]methanol a9 (23.7 kg, 97.8 mol) and dichloromethane (240 L) are charged into a reactor. DMAP (120 g, 0.98 mol) and imidazole (13.3 kg, 196 mol) are added. Tert-butyldimethylsilyl chloride (TBSCI) (17.7 kg, 117 mol) is slowly added at 15-20°C and the mixture is stirred for 12h. Ammonium chloride (100 L) is added into the mixture. The organic phase was separated, washed with water (50.0 L), dried with anhydrous sodium sulfate, filtered and concentrated under vacuum to give [(3R)-5-bromo-1 ,2,3,4-tetrahydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl-silane a10 as a yellow oil (37.6 kg, 86% purity, 93% yield) which is used in the next step without further purification.
1H NMR (400 MHz, CDCI3) d 7.36 – 7.45 (m, 1H), 7.01 (d, J = 4.6 Hz, 1H), 4.01 – 4.13 (m, 2H), 3.84 (dd, J = 9.9, 3.7 Hz, 1 H), 3.64 (dd, J = 9.8, 7.2 Hz, 1 H), 2.96 – 3.08 (m, 1 H), 2.75 (dd, J = 17.0, 4.2 Hz, 1 H), 2.44 (dd, J = 17.0, 10.8 Hz, 1H), 1.76 – 2.20 (m, 2H), 0.89 – 0.97 (m, 9H), 0.08 – 0.14 (m, 6H).
2.5. Preparation of intermediate (V).
[(3R)-5-bromo-3,4-dihydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl-silane a11
[(3R)-5-bromo-1 ,2,3,4-tetrahydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl-silane a10 (3.42 kg, 8.31 mol) and THF (30.0 L) are charged into a reactor. N-Chlorosuccinimide (NCS) (1.17 kg, 8.73 mol) is slowly added at room temperature and the mixture is stirred at 25°C for 30 min. A solution of KOH (1.52 kg, 27.1 mol) in dry methanol (7.00 L) is slowly added at room temperature and the reaction is stirred at 25°C for 1h. The reaction is quenched with water (10.0 L) and extracted with petroleum etherethyl acetate (1:2, 5.00 L). The organic layer is separated, washed with brine (10.0 L), dried with anhydrous sodium sulfate and filtered. This overall procedure is carried out on 10 batches of the same size in parallel and the 10 reaction filtrates are combined and concentrated under vacuum to give [(3R)-5-bromo-3,4-dihydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl-silane a11 as a brown oil (28.0 kg, crude) which is used in the next step without further purification.
1H NMR (400 MHz, CDC ) d 8.24 (d, J = 2.6 Hz, 1H), 7.58 (dd, J = 7.8, 1.2 Hz, 1 H), 7.12 -7.25 (m, 2H), 4.03 (dd, J = 9.5, 4.0 Hz, 1 H), 3.67 – 3.77 (m, 2H), 3.07 (dd, J = 17.0, 6.2 Hz, 1H), 2.68 (dd, J = 17.1, 10.9 Hz, 1 H), 0.88 – 0.91 (m, 9H), 0.07 (d, J= 1.5 Hz, 6H).
2.6. Preparation of intermediates of formula (IV)
2.6.1. [(1S,3R)-5-bromo-1-methyl-1,2,3,4-tetrahydroisoquinolin-3-yl]methoxy-tert-butyl- dimethyl-silane (IVa)
[(3R)-5-bromo-3,4-dihydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl-silane a11 (3.10 kg, 8.75 mol) and THF (20.0 L) are charged into a reactor. The mixture is cooled down to 0°C and methylmagnesium chloride (3M, 11.6 L) is added. The mixture is stirred at 20°C for 12h. The reaction is quenched with a saturated solution of ammonium chloride. The phases are separated and the aqueous layer is extracted twice with petroleum ether: ethyl acetate (3:1, 5.00 L). The combined organic phases are washed with brine (10.0 L), dried over anhydrous sodium sulfate and filtered. This overall procedure is carried out on 9 batches of the same size in parallel and the nine reaction filtrates are combined and concentrated under vacuum. The crude mixture is purified by silica gel chromatography with petroleum ether : ethyl acetate (10:1) to give [(1S,3R)-5-bromo-1 -methyl-1, 2, 3, 4-tetrahydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl-silane (IVa) as a brown oil (4.60 kg, 99.7% purity, 15.7% yield).
1H NMR (400 MHz, DMSO-de) d 7.41 (dd, J=7.7, 0.9 Hz, 1H), 7.12 – 7.18 (m, 1H), 7.03 – 7.11 (m, 1H), 4.12 (q, J= 6.8 Hz, 1H), 3.62 (d, J= 5.7 Hz, 2H), 3.07 – 3.17 (m, 1H), 2.67 – 2.76 (m, 1H), 2.26 (dd, J=16.9, 10.0 Hz, 1H), 2.12 (br s, 1 H), 1.32 (d, J= 6.8 Hz, 3H), 0.84 – 0.93 (m, 9H), 0.07 (d, J=0.9 Hz, 6H).
2.6.2. tert-butyl (1S,3R)-5-bromo-3-[[tert-butyl(dimethyl)silyl]oxymethyl]-1-methyl-3,4- dihydro-1 H-isoquinoline-2-carboxylate (IVb)
[(1S,3R)-5-bromo-1-methyl-1,2,3,4-tetrahydroisoquinolin-3-yl]methoxy-tert-butyl-dimethyl-silane (IVa) (1.85 kg, 4.99 mol) and dichloromethane (13.0 L) are charged in a reactor. N,N-diisopropylethylamine (1.94 kg, 14.9 mol) and di-tert-butyl dicarbonate (1.14 kg, 5.24 mol) are added at room temperature and the mixture is stirred for 12h. The reaction mixture is washed twice with a saturated ammonium chloride solution (10.0 L), the organic layer is dried with anhydrous sodium sulfate and filtered. This overall procedure is carried out on 2 batches of the same size in parallel and the two reaction filtrates are combined and concentrated under vacuum. The crude mixture is purified by silica gel chromatography with petroleum ether ethyl acetate (30:1) to give tert-butyl (1S,3R)-5-bromo-3-[[tert-butyl(dimethyl)silyl]oxymethyl]-1 -methyl-3, 4-dihydro-1 H-isoquinoline-2-carboxylate (IVb) as a yellow oil (4.00 kg, 99.5% purity, 85.2% yield).
1H NMR (400 MHz, DMSO-de) d 7.50 (d, J = 7.9 Hz, 1 H), 7.22 (br d, J = 6.7 Hz, 1 H), 7.06 -7.18 (m, 1 H), 4.84 (br s, 1 H), 4.12 (br s, 1H), 3.46 (br d, J = 15.4 Hz, 2H), 2.94 (br dd, J = 15.8, 5.2 Hz, 1H), 2.71 (br t, J = 9.5 Hz, 1 H), 1.45 (s, 9 H), 1.28 (br s, 3H), 0.81 (s, 9H), -0.08 (s, 6H).
2.6.3. tert-butyl (1S,3R)-3-[[tert-butyl(dimethyl)silyl]oxymethyl]-5-(1-hydroxy-1 -methyl- ethyl)-1 -methyl-3, 4-dihydro-1H-isoquinoline-2-carboxylate (IVc)
A solution of tert-butyl (1S,3R)-5-bromo-3-[[tert-butyl(dimethyl)silyl]oxymethyl]-1-methyl-3,4-dihydro-1 H-isoquinoline-2-carboxylate (IVb) (42.5 g, 90.3 mmol) in dry THF (0.5 M solution) and a commercial solution of n-Buthylithium in Hexanes (1.6 M solution) were pumped at respectively 6.0 ml/min (1.0 equiv) and 2.46 mL/min (1.3 equiv.) and were mixed in a glass microchip cooled at -40°C. The mixed flow stream was pumped through the reaction zone 1 of the microchip (0.3 ml_) and was then combined with a solution of dry acetone (13.5 M) pumped at 6.0 mL/min (27 equiv.). The resulting stream was then passed through the reaction zone 2 of the microchip (0.7 ml_) at -40 °C. Finally, the global flow stream exiting the reactor was collected and quenched at room temperature in a saturated solution of aqueous ammonium chloride. When all the feed solutions were consumed, a bilayer reaction mixture was obtained. The aqueous layer was separated from the organic layer, and then extracted twice with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous sodium sulfate and concentrated under vacuum. A yellow oil was obtained (46.5 g) and was purified by SFC chromatography on a GreenSep Nitro column (10m, 5×22.3 using CO298 %/EtOH 2% eluent). The solvent was removed under vacuum to yield to a white solid, tert-butyl (1S,3R)-3-[[tert-butyl(dimethyl)silyl]oxymethyl]-5-(1-hydroxy-1-methyl-ethyl)-1-methyl-3, 4-dihydro-1H-isoquinoline-2-carboxylate (IVc) (25 g, 56 mmol, 62 % yield).
UPLC_MS basic 1 pic @ 3.83 min (ES+): 350 (M-Boc+H)+, 332 (M-Boc-H20+H)+, 100 % purity.
1H NMR (400 MHz, DMSO-de) d 7.44 (d, J = 7.9 Hz, 1H), 7.19 (dt, J = 8.1 , 5.2 Hz, 1 H), 7.09 (t, J = 9.0 Hz, 1H), 4.99 (s, 1 H), 4.87 (dq, J = 13.4, 6.4 Hz, 1 H), 4.11 (s, 1H), 3.96 (t, J = 14.9 Hz, 1 H), 3.48 (dd, J = 9.4, 4.1 Hz, 1H), 2.98 (dd, J = 16.5, 5.0 Hz, 1H), 2.89 (t, J = 9.6 Hz, 1H), 1.65 (s, 3H), 1.58 (s, 3H), 1.55 (d, J = 2.5 Hz, 9H), 1.34 (dd, J = 20.5, 6.6 Hz, 3H), 0.90 (s, 9H), 0.08 (d, J = 7.2 Hz, 3H), -0.00 (s, 3H).
2.7. Preparation of intermediate (III) 2-[(1S,3R)-3-(hydroxymethyl)-1-methyl- 1.2.3.4-tetrahydroisoquinolin-2-ium-5-yl]propan-2-ol chloride
2.7.1. tert-butyl-dimethyl-[[(1S,3R)-1-methyl-5-(1-methyl-1-trimethylsilyloxy-ethyl)- 1.2.3.4-tetrahydroisoquinolin-3-yl]methoxy]silane- a15
Tert-butyl (1S,3R)-3-[[tert-butyl(dimethyl)silyl]oxymethyl]-5-(1-hydroxy-1-methyl-ethyl)-1-methyl-3, 4-dihydro-1H-isoquinoline-2-carboxylate (IVc) (148 g, 87% purity, 287 mmol) is dissolved in 1000 ml_ dichloromethane and transferred to a 2 liter double walled reactor. 2,6-Lutidine (100 ml_, 860 mmol) is added and the jacket temperature is set at-2°C. Trimethylsilyl trifluoromethanesulfonate (154 g, 129 ml_, 692 mmol) is added over 40 min via an addition funnel. Two hours after the start of addition, the reaction is quenched by adding 650 ml_ of an aqueous citric acid solution (1M) and the temperature of the mixture is brought back to 20°C. One hour after the start of the quench, the layers are separated. The organic layer is washed twice with 350 ml_ of an aqueous solution of citric acid (1M). The organic layer is stirred with 750 ml_ of aqueous sodium carbonate (10% w/w) for 10 min before separation of the layers. The organic layer is dried over anhydrous sodium sulfate. The organic layer is then filtered and the filtrate is concentrated under vacuum at 40°C providing a yellow oil (128 g) of tert-butyl-dimethyl-[[(1S,3R)-1-methyl-5-(1-methyl-1-trimethylsilyloxy-ethyl)-1 ,2,3,4-tetrahydroisoquinolin-3-yl]methoxy]silane a15 which is used in the next step without further purification.
1H NMR (400 MHz, CDC ) d 7.19 (d, J = 7.7 Hz, 1 H), 7.07 (t, J = 7.7 Hz, 1 H), 7.00 (d, J = 7.6 Hz, 1 H), 4.24 (q, J = 6.8 Hz, 1 H), 3.75 (dd, J = 9.7, 4.4 Hz, 1H), 3.60 (dd, J = 9.7, 7.0 Hz, 1H), 3.54 (dd, J = 16.3, 3.5 Hz, 1H), 3.15 (ddt, J = 10.9, 7.4, 4.0 Hz, 1 H), 2.52 (dd, J = 16.3, 10.9 Hz, 1H), 1.66 (d, J = 14.6 Hz, 6H), 1.52 – 1.43 (m, 3H), 0.92 (q, J = 1.2 Hz, 9H), 0.14 (q, J = 1.2 Hz, 2H), 0.09 (d, J = 1.1 Hz, 6H), 0.00 (q, J = 1.2, 0.8 Hz, 9H).
2.7.2. 2-[(1S,3R)-3-(hydroxymethyl)-1-methyl-1,2,3,4-tetrahydroisoquinolin-2-ium-5- yl]propan-2-ol chloride Intermediate (III)
In a three-neck round bottom flask equipped with a mechanical stirrer, tert-butyl-dimethyl-[[(1S,3R)-1-methyl-5-(1-methyl-1-trimethylsilyloxy-ethyl)-1,2,3,4-tetrahydroisoquinolin-3-yljmethoxyjsilane a15 (20.0 g, 47.4 mmol) is dissolved in 220 ml_ of isopropanol. To this solution, 42.3 ml_ of hydrochloric acid in iso-propanol (5-6 M, around 5 eq.) are added. 45 min after addition of hydrochloric acid, a 100 mg of seeds of the desired product are introduced. After 7 hours at room temperature, the reaction mixture is filtered over a sintered glass filter. The filtercake is washed with 40 ml_ isopropanol and dried under vacuum at room temperature overnight. 11.1 g of 2-[(1S,3R)-3-(hydroxymethyl)-1 -methyl-1 , 2,3,4-tetrahydroisoquinolin-2-ium-5-yl]propan-2-ol chloride (III) are obtained as a pinkish solid. The yield over the two deprotection steps is 91%.
1H NMR (400 MHz, CD3OD) d 7.46 (dd, J = 7.8, 1.3 Hz, 1H), 7.28 (t, J = 7.8 Hz, 1H), 7.21 (dd, J = 7.8, 1.3 Hz, 1H), 4.63 (q, J = 6.9 Hz, 1H), 3.97 (dd, J = 11.7, 3.8 Hz, 1 H), 3.88 (dd, J = 17.2, 4.3 Hz, 1H), 3.78 (dd, J = 11.8, 6.1 Hz, 1H), 3.66 – 3.56 (m, 1 H), 3.14 (dd, J = 17.2, 11 .4 Hz, 1 H), 1 .73 (d, J = 6.8 Hz, 3H), 1 .64 (d, J = 4.8 Hz, 6H). OH and NH protons are not observed.
2.8. Preparation of compound of formula (I).
2-(3,5-dichloro-1 -methyl-indazol-4-yl)-1 -[(1 S,3R)-3-(hydroxymethyl)-5-(1 – hydroxy-1 -methyl-ethyl)-1 -methyl-3, 4-dihydro-1 H-isoquinolin-2-yl]ethanone
In a 100 ml. Easymax reactor equipped with a mechanical stirrer, 2-(3,5-dichloro-1 -methyl-indazol-4-yl)acetic acid (II) (4.00 g, 15.4 mmol), 2-[(1S,3R)-3-(hydroxymethyl)-1-methyl-1 ,2,3,4-tetrahydroisoquinolin-2-ium-5-yl]propan-2-ol chloride (III) (4.46 g, 16.4 mmol) and 48 mL of DMF are charged. The suspension is stirred at 20°C and then cooled by setting the jacket temperature to -2°C. Once the temperature of the mixture is below 3°C, N,N-diisopropylethylamine (9.5 mL, 54 mmol) is added. (2-(1 H-benzotriazol-1 -yl)-1 , 1 ,3,3-tetramethyluronium hexafluorophosphate (6.4 g, 17 mmol) is added in four portions over 1 hour. The mixture is stirred for 1 h 45 before setting the jacket temperature at 15°C. 16 mL of water are then added over the course of a few minutes. 15 min later, 30 mg of solid product are added as seeds to initiate the crystallization. The jacket temperature is set at 20°C. Half an hour later, 16 mL of water are added over 17 min. Stirring of the suspension is pursued for 2 h 15 at 20°C before being filtered on sintered glass. The filtercake is washed with two portions of 20 mL of water and then dried at 50°C overnight under vacuum yielding 6.03 g of 2-(3,5-dichloro-1-methyl-indazol-4-yl)-1-[(1 S,3R)-3-(hydroxymethyl)-5-(1 -hydroxy-1 -methyl-ethyl)-1 -methyl-3, 4-dihydro-1 H-isoquinolin-2-yl]ethanone (I) (crude material).
A recristallization is carried out on 5.00 g of the crude material obtained by first suspending in 50 mL acetonitrile. The jacket temperature is set to 70°C. Once the solid has dissolved and the mass temperature has reached 66°C, 720 mI of water are added. The mass temperature is then cooled to 59°C and 125 mg of solid product is added as seeding material. The mass temperature is then decreased to 55°C over 25 min at which stage crystallization is occurring. The jacket temperature is then decreased over two hours from 58°C down to 20°C. After 50 min, the suspension is filtered and the filtercake is washed with 7.5 mL acetonitrile. The filtercake is then dried under vacuum at 45°C overnight and 2 hours at 50°C providing 4.04 g of 2-(3,5-dichloro-1 -methyl-indazol-4-yl)-1-[(1S,3R)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl-ethyl)-1 -methyl-3, 4-dihydro-1 H-isoquinolin-2-yl]ethanone (I) as an off-white powder (hydrate form) Yield = 64%.
1H NMR (400 MHz, DMSO-cfe) d 7.65 (dd, J = 9.0, 2.2 Hz, 1H), 7.52 (dd, J = 9.0, 2.1 Hz, 1 H), 7.37 (ddd, J = 19.6, 7.6, 1 .7 Hz, 1 H), 7.25 – 7.03 (m, 2H), 5.30 (q, J = 6.5 Hz, 0.3H), 5.16 -4.99 (m, 1 .7H), 4.99 – 4.84 (m, 0.7H), 4.63 – 4.30 (m, 3.3H), 4.17 – 3.93 (m, 4H), 3.28 (dt, J = 10.5, 5.1 Hz, 1.3H), 3.10 – 2.85 (m, 1.7H), 1.56 (dd, J = 13.2, 6.9 Hz, 6.7H), 1.24 (d, J = 6.5 Hz, 2.3H).
PAT
- A Substituted Tetrahydroisoquinoiline Derivative as a D1 Positive Allosteric ModulatorPublication Number: US-2022259179-A1Priority Date: 2019-07-01
- Substituted tetrahydroisoquinoline derivatives as D1 positive allosteric modulatorsPublication Number: CN-113993857-BPriority Date: 2019-07-01Grant Date: 2024-01-02
- Substituted tetrahydroisoquinoline derivatives as D1 positive allosteric modulatorsPublication Number: CN-117700395-APriority Date: 2019-07-01
- Substituted tetrahydroisoquinoline derivatives as D1 positive allosteric modulatorsPublication Number: JP-7510444-B2Priority Date: 2019-07-01Grant Date: 2024-07-03
- Substituted Tetrahydroisoquinoline Derivatives as Positive Allosteric Modulators of D1Publication Number: CN-113993857-APriority Date: 2019-07-01
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: EP-3993794-A1Priority Date: 2019-07-01
- D1 Substituted tetrahydroisoquinoline derivatives as positive allosteric modulatorsPublication Number: KR-20220029686-APriority Date: 2019-07-01
- A SUBSTITUTED TETRAHYDROISOQUINOLINE DERIVATIVE AS A POSITIVE ALOSTERIC MODULATOR OF D1Publication Number: PE-20221020-A1Priority Date: 2019-07-01
- Substituted Tetrahydroisoquinoline Derivatives as D1 Positive Allosteric ModulatorsPublication Number: JP-2022539152-APriority Date: 2019-07-01
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: WO-2021001288-A1Priority Date: 2019-07-01
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: TW-202115010-APriority Date: 2019-07-01
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: WO-2021001288-A9Priority Date: 2019-07-01
- A substituted tetrahydroisoquinoline derivative as a D1 positive allosteric modulatorPublication Number: AU-2020299953-A1Priority Date: 2019-07-01
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: CA-3139571-A1Priority Date: 2019-07-01
- A Substituted Tetrahydroisoquinoline Derivative As A D1 Positive Allosteric ModulatorPublication Number: US-2024059665-A1Priority Date: 2020-12-18
- Prodrugs of 2-(3,5-Dichloro-1-methyl-indazol-4-yl)-1-[(1S,3R)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl-ethyl)-1-methyl-3,4-dihydro-1H-isoquinolin-2-yl]ethanonePublication Number: US-2024083925-A1Priority Date: 2020-12-18
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: AU-2021403603-A9Priority Date: 2020-12-18
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: EP-4263517-B1Priority Date: 2020-12-18Grant Date: 2024-10-02
- Prodrugs of 2-(3,5-dichloro-1-methyl-indazol-4-yl)-1-[(1s,3r)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl-ethyl)-1-methyl-3,4-dihydro-1h-isoquinolin-2-yl]ethanonePublication Number: EP-4263519-B1Priority Date: 2020-12-18Grant Date: 2024-10-02
- Prodrugs of 2-(3,5-dichloro-1-methyl-indazol-4-yl)-1-[(1s,3r)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl-ethyl)-1-methyl-3,4-dihydro-1h-isoquinolin-2-yl]ethanonePublication Number: EP-4263519-A1Priority Date: 2020-12-18
- amorphous solid dispersionPublication Number: JP-2023553457-APriority Date: 2020-12-18
- Substituted tetrahydroisoquinoline derivatives as D1-positive allosteric modulatorsPublication Number: JP-2023553671-APriority Date: 2020-12-18
- 2-(3,5-dichloro-1-methyl-indazol-4-yl)-1-[(1S,3R)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl-ethyl)- Prodrug of 1-methyl-3,4-dihydro-1H-isoquinolin-2-yl]ethanonePublication Number: JP-2024500391-APriority Date: 2020-12-18
- Amorphous solid dispersionsPublication Number: US-2024000769-A1Priority Date: 2020-12-18
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: IL-303693-APriority Date: 2020-12-18
- Substituted tetrahydroisoquinoline derivatives as D1 positive allosteric modulatorsPublication Number: KR-20230121849-APriority Date: 2020-12-18
- amorphous solid dispersionPublication Number: KR-20230121867-APriority Date: 2020-12-18
- Amorphous solid dispersionsPublication Number: EP-4262756-A1Priority Date: 2020-12-18
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: EP-4263517-A1Priority Date: 2020-12-18
- Prodrugs of 2-(3,5-dichloro-1-methyl-indazol-4-yl)-1-[(1s,3r)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl-ethyl)-1-methyl-3,4-dihydro-1h-isoquinolin-2-yl]ethanonePublication Number: CA-3203281-A1Priority Date: 2020-12-18
- Substituted tetrahydroisoquinoline derivatives useful as D1 positive allosteric modulatorsPublication Number: CN-116583280-APriority Date: 2020-12-18
- 2-(3,5-Dichloro-1-methyl-indazol-4-yl)-1-[(1S,3R)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl Prodrug of -ethyl)-1-methyl-3,4-dihydro-1H-isoquinolin-2-yl]ethanonePublication Number: CN-116601161-APriority Date: 2020-12-18
- Amorphous Solid DispersionPublication Number: CN-116685308-APriority Date: 2020-12-18
- Amorphous solid dispersionsPublication Number: IL-303688-APriority Date: 2020-12-18
- Amorphous solid dispersionsPublication Number: WO-2022129267-A1Priority Date: 2020-12-18
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: WO-2022129268-A1Priority Date: 2020-12-18
- Prodrugs of 2-(3,5-dichloro-1-methyl-indazol-4-yl)-1-[(1s,3r)-3-(hydroxymethyl)-5-(1-hydroxy-1-methyl-ethyl)-1-methyl-3,4-dihydro-1h-isoquinolin-2-yl]ethanonePublication Number: WO-2022129356-A1Priority Date: 2020-12-18
- Amorphous solid dispersionsPublication Number: AU-2021401128-A1Priority Date: 2020-12-18
- A substituted tetrahydroisoquinoline derivative as a d1 positive allosteric modulatorPublication Number: AU-2021403603-A1Priority Date: 2020-12-18
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | UCB-0022; UCB0022 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2576359-31-2 |
| PubChem CID | 155460962 |
| IUPHAR/BPS | 13232 |
| UNII | H8T5VKH4CZ |
| Chemical and physical data | |
| Formula | C24H27Cl2N3O3 |
| Molar mass | 476.40 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “UCB 0022”. AdisInsight. Springer Nature Switzerland AG. 28 May 2024. Retrieved 10 August 2024.
- “Delving into the Latest Updates on Glovadalen with Synapse”. Synapse. 8 August 2024. Retrieved 10 August 2024.
- McFarthing K, Buff S, Rafaloff G, Fiske B, Mursaleen L, Fuest R, et al. (2023). “Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2023 Update”. Journal of Parkinson’s Disease. 13 (4): 427–439. doi:10.3233/JPD-239901. PMC 10357160. PMID 37302040.
Our analysis of dopaminergic therapies shows a continued emphasis on DA agonists and levodopa reformulation. These include Cerevel’s tavapadon, a D1/D5 receptor partial agonist and UCB0022, a positive allosteric modulator of the D1 receptor, as well as approaches to sub-cutaneously deliver levodopa/carbidopa such as Abbvie’s ABBV-951 and Neuroderm’s ND0612.
- “Glovadalen”. IUPHAR/BPS Guide to PHARMACOLOGY. Retrieved 10 August 2024.
- “UCB0022”. ALZFORUM. 3 May 2024. Retrieved 10 August 2024.
- Vermeiren C, Ates A, Bouzom F, Delaunois A, Gillard M, Kenda B, et al. (7 September 2022). “Preclinical characterization of UCB0022, an oral, brain penetrant, selective, clinical-stage positive allosteric modulator of the dopamine 1 receptor (D1 PAM)”. Movement Disorders. 37 (Suppl 2 [2022 International Congress September 15-18, 2022. Madrid, Spain]). Retrieved 10 August 2024.
////////Glovadalen, dopamine D1 receptor positive allosteric modulator, Phase 2, Parkinson’s disease, UCB-0022, UCB 0022, H8T5VKH4CZ
Girocitinib
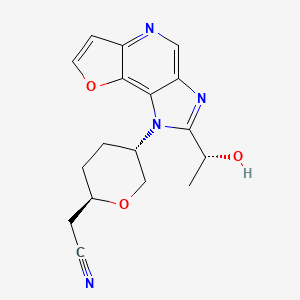
Girocitinib
CAS 2222137-79-1
MFC17H18N4O3 MW 326.36
2-[(2R,5S)-5-[4-[(1R)-1-hydroxyethyl]-12-oxa-3,5,8-triazatricyclo[7.3.0.02,6]dodeca-1,4,6,8,10-pentaen-3-yl]oxan-2-yl]acetonitrile
[(2R,5S)-5-{2-[(1R)-1-hydroxyethyl]-1H-furo[3,2-b]imidazo[4,5-d]pyridin-1-yl}oxan-2-yl]acetonitrile
2-((2R,5S)-5-(2-((R)-1-hydroxyethyl)-1H-furo[3,2-b]imidazo[4,5-d]pyridin-1-yl)tetrahydro-2H-pyran-2-yl)acetonitrile
Janus kinase inhibitor, anti-inflammatory, A0IES9T8GO
In an era where targeted therapies are redefining the landscape of medical treatment, Girocitinib emerges as a beacon of hope for many. This innovative drug, developed by leading pharmaceutical research institutions, primarily targets specific proteins involved in disease progression. Classified as a tyrosine kinase inhibitor (TKI), Girocitinib has shown significant promise in the treatment of various cancers, particularly non-small cell lung cancer (NSCLC). The drug is currently in the advanced stages of clinical trials, with researchers optimistic about its potential to provide a more effective and less toxic treatment option compared to conventional therapies.
Girocitinib is designed to interfere with the signaling pathways that promote cancer cell growth and survival. It does this by inhibiting the activity of tyrosine kinases, enzymes that play a key role in the activation of many proteins by signaling pathways within the cell. Tyrosine kinases are often overactive in cancer cells, leading to unchecked proliferation and survival. By targeting these enzymes, Girocitinib effectively disrupts these malign processes, thereby slowing down or even halting the progression of the disease.
The primary indication for Girocitinib is non-small cell lung cancer (NSCLC), which accounts for approximately 85% of all lung cancer cases. NSCLC is notoriously difficult to treat, especially in its advanced stages, and current treatments often come with significant side effects. Clinical trials have shown that Girocitinib can significantly improve progression-free survival in patients with specific genetic mutations that make them more responsive to TKI therapy. These mutations can be identified through genetic testing, allowing for a more personalized treatment approach that increases the likelihood of success.
In addition to NSCLC, researchers are exploring the potential of Girocitinib to treat other types of cancer, including colorectal cancer and certain forms of leukemia. Early-stage trials have shown encouraging results, suggesting that Girocitinib could become a versatile tool in the oncology arsenal. Its ability to target specific molecular pathways makes it a promising candidate for combination therapies, which aim to enhance treatment efficacy while minimizing resistance and adverse effects.
The development of Girocitinib is a testament to the power of modern science and technology in addressing some of the most challenging health issues of our time. The drug’s journey from the laboratory to clinical trials has been marked by rigorous research and collaboration among scientists, healthcare professionals, and patients. As we await the results of ongoing studies, there is a palpable sense of anticipation in the medical community, as Girocitinib holds the promise of transforming cancer treatment for many patients.
In conclusion, Girocitinib represents a significant advancement in the field of targeted cancer therapy. Its mechanism of action, which involves the inhibition of tyrosine kinases, offers a more precise and potentially less harmful treatment option for patients with NSCLC and possibly other cancers. As research progresses, Girocitinib may well become a cornerstone in the fight against cancer, providing hope and improved outcomes for countless individuals around the world.
PDT PAT
WO2018067422
SYN
https://patents.google.com/patent/US10738060B2/en?oq=US10738060
Example 4: Synthesis of 2-[(2R,5S)-5-[2-[(R)-1-Hydroxyethyl]furo[3,2-b]imidazo[4,5-d]pyridin-1-yl]tetrahydropyran-2-yl] acetonitrile (4)
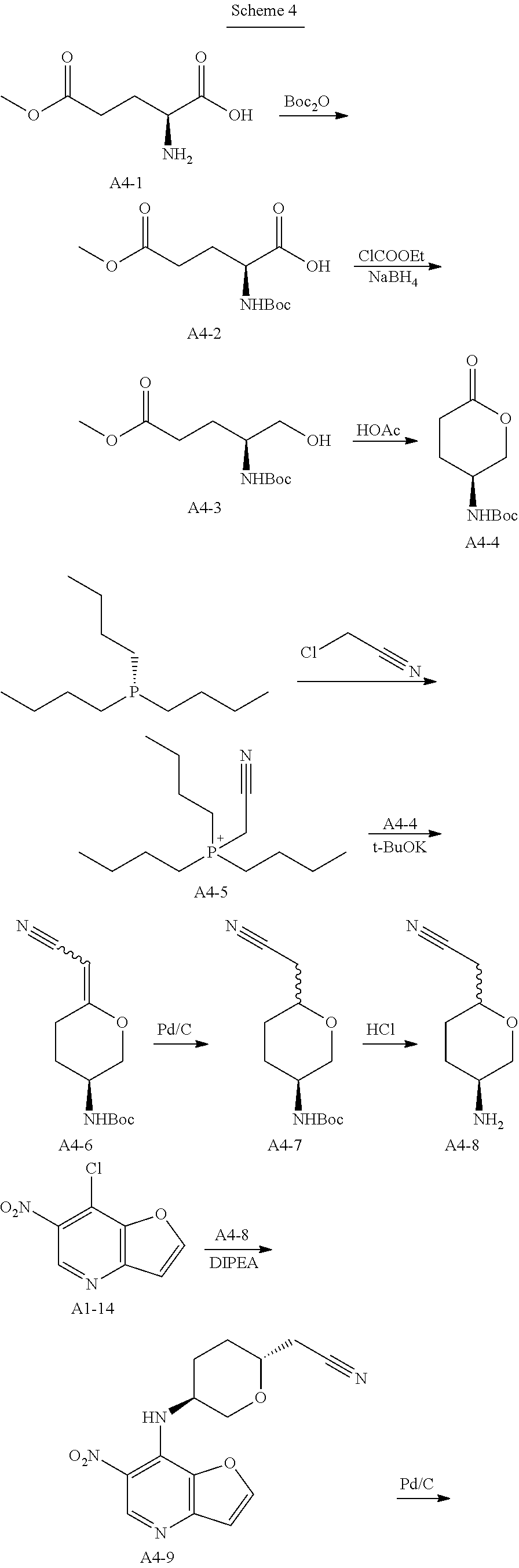
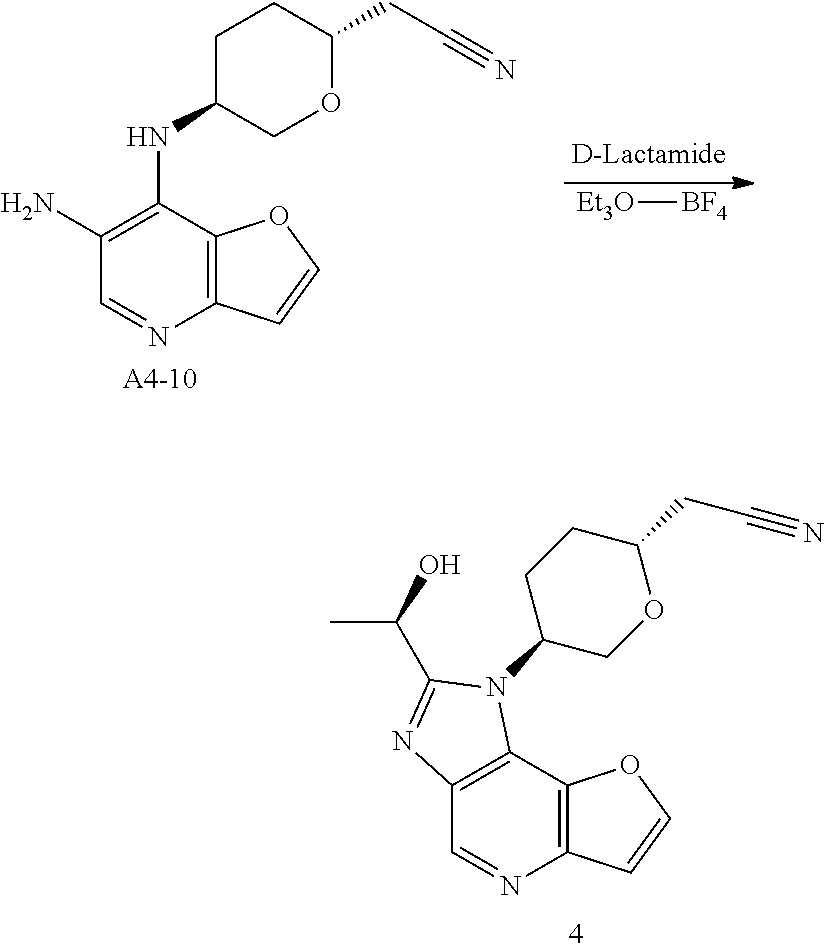
Step 1. In a round bottom flask, triethylamine (188 g, 1.86 mol, 1.0 eq) was added dropwise to a stirred solution of di-tert-butyl dicarbonate (162 g, 0.744 mol, 1.2 eq) and compound A4-1 (100 g, 0.62 mol, 1.0 eq) in water (500 mL) and 1,4-dioxane (500 mL). After stirring for 18 hrs at room temperature, the solution was extracted with MTBE (500 mL*2) and the aqueous phase was cooled on ice and carefully acidified to pH 3 by slow addition of 10% citric acid solution. The urethane was then extracted twice with ethyl acetate, and the combined extracts was washed with brine, dried over anhydrous sodium sulfate, and concentrated to give compound A4-2 as clear viscous oil (180 g, yield 100%). MS-ESI:[M+1]+: 262.1
Step 2. A solution of compound A4-2 (40 g, 0.153 mmol, 1.0 eq) in THF (600 mL) was treated with 4-methylmorpholine (17 g, 0.168, 1.1 eq) at room temperature. The resulting mixture was cooled to 0° C. before being treated with isobutyl chloroformate (22.7 g, 0.166 mmol, 1.08 eq) dropwise. The resulting reaction mixture was stirred at 0° C. for an addition 20 mins before being filtered and washed with THF. Then the clear filtrate solution was cooed to 0° C., and treated with a solution of NaBH4 (11.2 g, 0.295 mol, 1.93 eq) in water (100 mL). The resulting mixture was stirred overnight at room temperature, and then quenched with an aqueous HCl solution (1.0 mol/L,200 mL) dropwise, The mixture was extracted with ethyl acetate, and the combined extracts was washed with brine, dried over anhydrous sodium sulfate, concentrated to give compound A4-3 as a yellow oil (25 g, yield 66%). MS-ESI:[M+1]+: 248.1
Step 3. A solution of compound of A4-3 (25 g, 0.1 mol, 1.0 eq) in toluene (300 mL) and acetic acid (150 mL) was heated to reflux for 5 hrs and then cooled, concentrated under vacuum. The residual was added saturated sodium bicarbonate solution to pH 7-8 in ice-bath. Then the mixture was extracted three times with ethyl acetate, and the combined extracts was washed with brine, dried over anhydrous sodium sulfate, concentrated and recrystallized by ethyl acetate and PE to give compound A4-4 as a white powder (8.0 g, yield 37.2%). GC-MS: 215
Step 4. A solution of tributyl phosphine (72.9 g, 0.36 mol, 1.0 eq) in nitromethane (500 mL), was added dropwise chloroacetonitrile (27.2 g, 0.36 mol, 1.0 eq) in nitrogen atmosphere. The resulting reaction mixture was stirred for 16 hrs at room temperature, then concentrated. The residual oil solidified when a small amount of ethyl acetate was added. The solid was recrystallized by ethyl acetate and DCM to afford compound A4-5 as a white powder (95 g, yield 95%).
Step 5. To a solution of dry compound A4-5 (8.3 g, 30 mmol, 3.0 eq) in N,N-dimethylacetamide (30 mL) in nitrogen atmosphere, was added solid Potassium tert-butoxide (3.1 g, 28 mmol, 2.8 eq) in portions at 0° C. The resulting mixture was gradually warmed to 30° C. and stirred for 2 hrs. The resulting ylide solution was then treated with compound A4-4 (2.15 g, 10 mmol, 1.0 eq), and stirred overnight at 70° C. After cooled to room temperature, the resulting slurry was poured into the mixture of ice-water (100 mL) and saturated sodium bicarbonate solution (100 mL). The mixture was extracted twice with ethyl acetate, and the combined extracts was washed three times with brine, dried over anhydrous sodium sulfate, concentrated to give compound A4-6 as yellow oil without purification (7.5 g, yield 100%). MS-ESI:[M+1]+: 239.1
Step 6. To a solution of compound A4-6 (7.5 g, 10 mmol, 1.0 eq) in methanol (200 mL), was added 10% Pd/C (0.5 g,50% wet). Hydrogenation was carried out under atmospheric pressure at room temperature until hydrogen uptake ceased. The catalyst was filtered and washed by methanol. The filtrates was concentrated under vacuum, and purified by silica gel column chromatography to give compound A4-7 as off-white powder (1.6 g, yield 66.7%). MS-ESI:[M+1]+: 241.1
Step 7. To a solution of compound A4-7 (1.6 g, 6.67 mmol, 1.0 eq) in DCM (20 mL), was added TFA (10 g, 88.5 mmol, 13.2 eq). The reaction mixture was stirred for 2 hrs at room temperature until TLC showed the reaction was complete, then concentrated under vacuum. Water (20 mL) was added and the solution was treated with aqueous sodium hydroxide solution (4 mol/L) to pH 10. Then the aqueous phase was extracted six times with DCM/methanol (10/1). The combined extracts was dried over anhydrous sodium sulfate, concentrated to give compound A4-8 as light-brown oil (950 mg, yield 100%). MS-ESI:[M+1]+: 141.1
Step 8. To a solution of compound A1-14 (prepared as step 4 to 12 in example 1) (600 mg, 3.0 mmol, 1.0 eq) in n-butanol (15 mL), was added compound A4-8 (950 mg, 6.7 mmol, 2.26 eq) and DIPEA (1.36 g, 10.5 mmol, 3.5 eq). The reaction mixture was stirred for 1 hr at 135° C., concentrated and purified by silica gel column chromatography to give compound A4-9 (2R,5S) as light-yellow powder (254 mg, yield 28.0%).MS-ESI: [M+1]+: 303.1.
1H NMR (300 MHz, d6-DMSO): 9.063 (s, 1H), 8.503 (d, 1H), 9.326 (d, 1H), 7.176 (d, 1H), 4.431-4.513 (m, 1H), 4.128-4.156 (m, 1H), 3.633-3.659 (m, 1H), 3.448-3.518 (m, 1H), 2.775-2.841 (m, 2H), 2.205-2.312 (m, 1H), 1.829-1.859 (m, 2H), 1.501-1.521 (m, 1H).
Step 9. To a solution of compound A4-9 (254 g, 0.84 mmol, 1.0 eq) in methanol (20 mL), was added 10% Pd/C (0.15 g,50% wet). Hydrogenation was carried out under atmospheric pressure at room temperature until hydrogen uptake ceased. The catalyst was filtered and washed by methanol. The filtrates was concentrated under vacuum, and compound A4-10 was obtained as yellow oil (230 mg, yield 100%). MS-ESI:[M+1]+: 273.1
Step 10. A solution of D-Lactamide (388 mg, 4.2 mmol, 5.0 eq) and Et3O—BF4 (1.3 g, 6.72 mmol, 8.0 eq) in THF (10 mL) was stirred for 30 mins at room temperature in nitrogen atmosphere. Then the above solution was added to the mixture of compound A4-10 (230 mg, 0.84 mmol, 1.0 eq) in ethanol (10 mL). After stirring for 3 hrs at 85° C. until HPLC showed the reaction was complete, the mixture was concentrated, added water and extracted four times with ethyl acetate. The organic phases was discarded and the aqueous phase was treated with saturated sodium bicarbonate solution to pH 8, extracted twice with ethyl acetate. The second organic phases was dried over anhydrous sodium sulfate, concentrated and purified by silica gel column chromatography to give the title compound as light-yellow powder (120 mg, yield 43.8%). MS-ESI: [M+1]+: 327.6,
1H NMR (300 MHz, CDCl3): 9.039 (s, 1H), 7.939 (d, 1H), 7.196 (d, 1H), 5.235-5.336 (m, 1H), 4.806-4.973 (m, 1H), 4.403-4.483 (t, 1H), 4.096-6.116 (m, 2H), 2.700-2.807 (m, 4H), 2.105-2.312 (m, 2H), 1.830-1.852 (d, 3H).
SYN
US2022227777
https://patents.google.com/patent/US20220227777A1
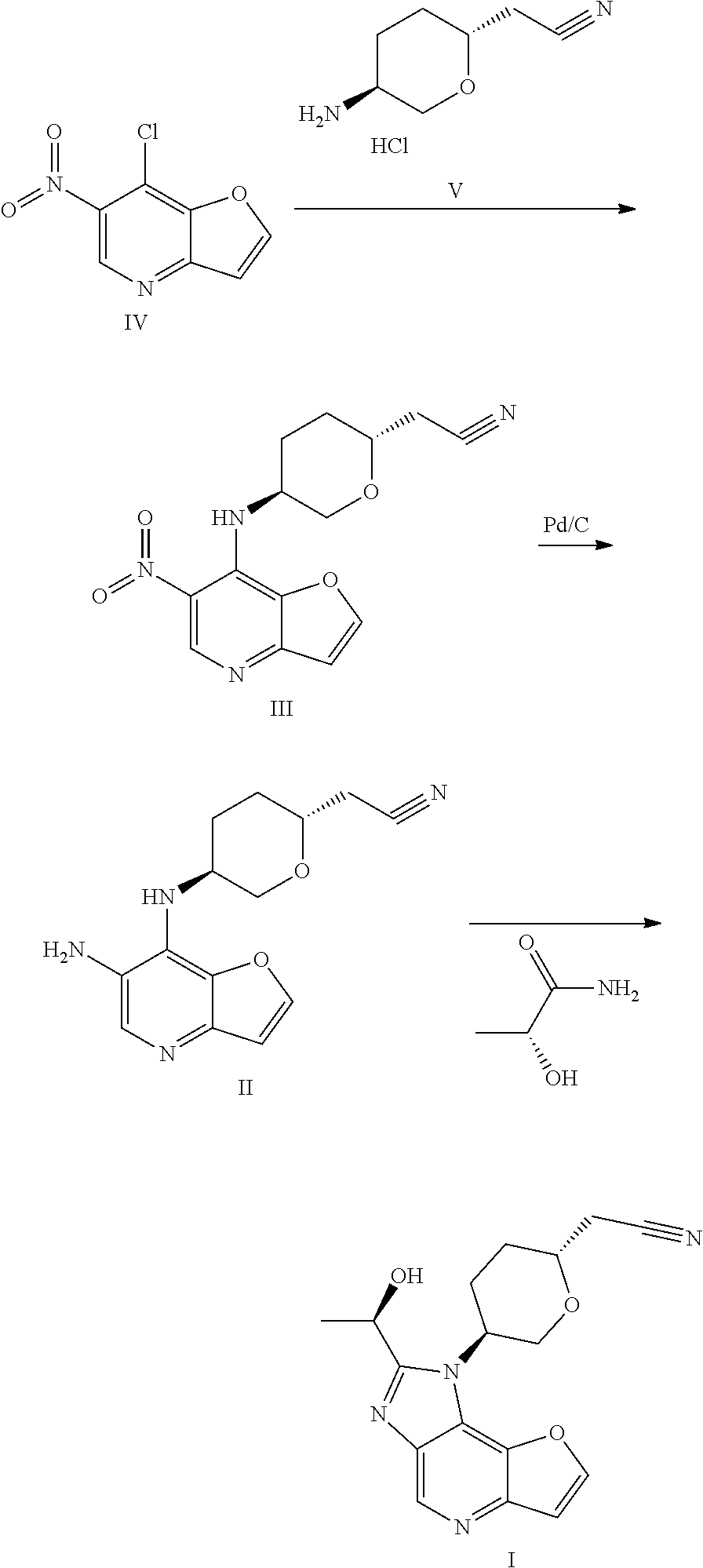
International patent application WO2018067422A1 discloses 1H-furo[3,2-b]imidazo[4,5-d]pyridine derivatives as selective JAK1 kinase inhibitors and preparation methods thereof, wherein compound I and its preparation method is disclosed.
Preparation of a Compound of Formula I
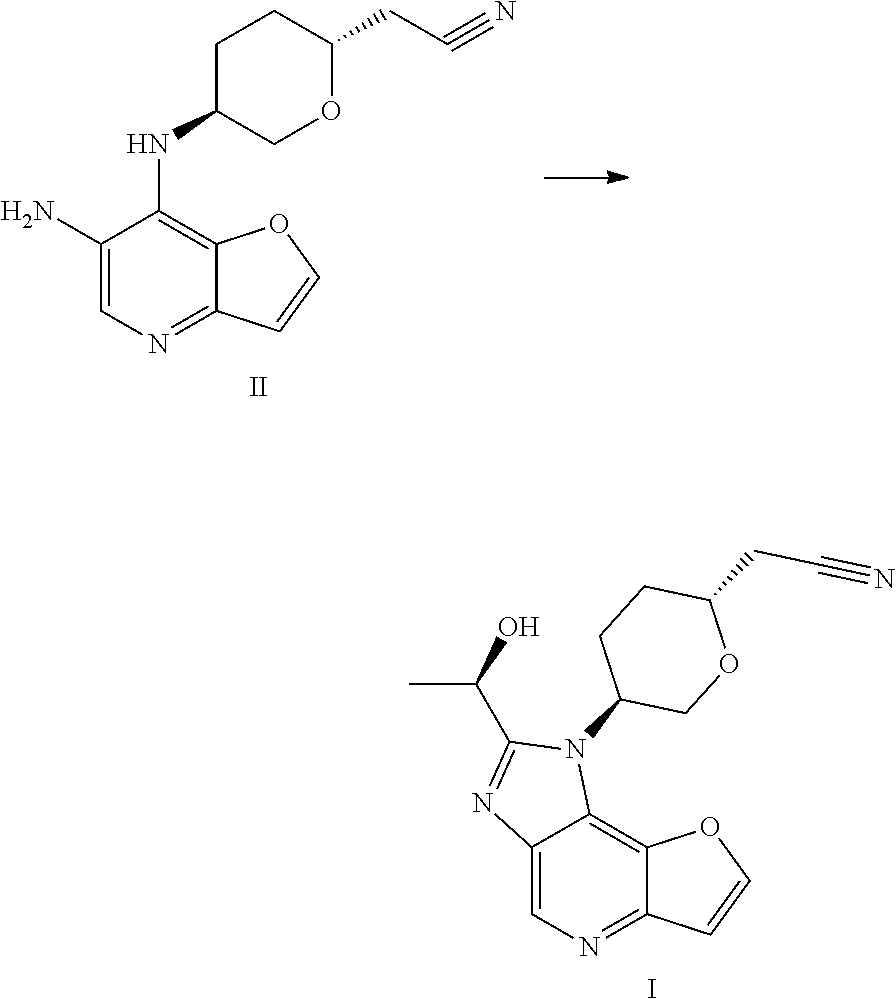
- [0204]THF (60 mL, 12 V), (R)-lactamide (6.6 g, 4.0 eq) and Et3O—BF4 (13.9 g, 4.0 eq) were added to a 250 mL three-necked flask #1, the system was stirred; the materials in three-necked flask #1 were stirred under nitrogen protection for later use; a compound of formula II (5.0 g, 1.0 eq) and ethanol (80 mL, 16 V) were added to another 250 mL three-necked flask #2; the system was heated to 70±5° C. under nitrogen protection; the materials in three-necked flask #1 were added to three-necked flask #2 with a syringe dropwise within 10-20 minutes; the system was heated to 85±5° C. (internal temperature was in the range of 72-75° C.) under nitrogen protection for reacting for 2 hours; the system was cooled to room temperature; the reaction liquid was concentrated with a rotary evaporator until there was basically no fraction flowing out; 1M HCl (80 mL) was added to the residual concentrated liquid, the pH was about 1 (determined with a pH test paper); the system was extracted four times with DCM (50 mL×4); the pH of the aqueous phase was adjusted to 7-8 with saturated sodium bicarbonate solution; the system was stirred at room temperature for 0.5 hour, then was filtered, the filter cake was washed with water (60 mL) and EA (10 mL), respectively; the filter cake was dried under vacuum at 50° C. for 16 hours; 4.3 g of faint yellow solid was obtained, with a purity of 95.0%; the solid was dissolved with methanol (30 mL); 4.1 g of silicon based metal eliminator and 1.0 g of activated carbon were added, the system was heated to 50° C. and stirred for 1 hour, then was cooled, filtered, washed with methanol (30 mL); the filtrate was concentrated with rotary evaporator until there was basically no fraction flowing out; methanol (10 mL) and MTBE (25 mL) were added to the residue, the system was heated to 50° C., and was stirred for 0.5 hour, then was cooled, the system was cooled to 10±5° C. and stirred for 0.5 hour; filtered, the filter cake was washed with MTBE (25 mL); the filter cake was dried under vacuum at 50° C. for 16 hours, 3.2 g of faint yellow solid was obtained, with a purity of 97.9%.
- [0205]MS-ESI: [M+1]+: 327.6
- [0206]1H NMR (400 MHz, CDCl3): 8.988 (s, 1H), 7.922 (d, 1H), 7.175 (d, 1H), 5.200-5.265 (m, 1H), 4.859-4.942 (m, 1H), 4.350-4.406 (t, 1H), 4.020-4.108 (m, 2H), 3.067 (d, 1H), 2.619-2.779 (m, 3H), 2.108-2.269 (m, 2H), 1.790-1.895 (m, 3H).
- [0207]THF (650 mL, 12 V), (R)-lactamide (70.6 g, 4.0 eq) and Et3O—BF4 (150.6 g, 4.0 eq) were added to a 1000 mL three-necked flask #1, the system was stirred; the materials in three-necked flask #1 were stirred under nitrogen protection for later use; a compound of formula II (54 g, 1.0 eq) and ethanol (860 mL, 16 V) were added to another 2000 mL three-necked flask #2; the system was heated to 70±5° C. under nitrogen protection; the materials in three-necked flask #1 were slowly added to three-necked flask #2 dropwise within 1 hour; the system was heated to 85±5° C. (internal temperature was in the range of 72-75° C.) under nitrogen protection for reacting for 2 hours; the system was cooled to room temperature; the reaction liquid was concentrated with a rotary evaporator until there was basically no fraction flowing out; 1M HCl (450 mL) was added to the residual concentrated liquid, the pH was about 1 (determined with a pH test paper); the system was extracted four times with DCM (270 mL×4); the pH of the aqueous phase was adjusted to 7-8 with saturated sodium bicarbonate solution; the system was stirred at room temperature for 0.5 hour, then was filtered, the filter cake was washed with water (540 mL); MTBE (270 mL) was added to the filter cake, the system was stirred at room temperature for 0.5 hour, filtered, the filter cake was washed with MTBE (108 mL); the filter cake was dried under vacuum at 50° C. for 16 hours; 49.2 g of light yellow solid was obtained, with an HPLC purity of 94.2%; the solid was dissolved with methanol (380 mL); silicon based metal eliminator (44 g) and activated carbon (5.4 g) were added, the system was heated to 50° C. and stirred for 1 hour, then was cooled, filtered, washed with methanol (430 mL); the filtrate was concentrated with a rotary evaporator to (80-110 mL, 1.5 V-2 V); MTBE (540 mL) was added to the residue, the system was heated to 50° C., and was stirred for 1 hour, then was cooled to 10±5° C. and stirred for 0.5 hour; filtered, the filter cake was washed with MTBE (270 mL); 42.4 g of filter cake was obtained, with an HPLC purity of 96.9%; the filter cake was dried under vacuum at 50° C. for 16 hours, 41.0 g of light yellow solid was obtained, with an HPLC purity of 96.7%, a yield of 63.3%.
- [0208]Purification of a Compound of Formula I:
- [0209]A compound of formula I (41 g) was dissolved with methanol; silica gel (50 g) was added to the solution, the system was concentrated to dryness for later use; silica gel (200 g) was added to the chromatographic column, the column was compacted with an air pump; a compound of formula I mixed with silica gel was added to the chromatographic column, the column was compacted with an air pump; the chromatographic column was eluted with an eluent (VMeOH:VDCM=1:100-1:30); qualified components were collected, concentrated to dryness; the product was dried under vacuum at 50° C. for 16 hours; 36 g of off-white solid was obtained, with an HPLC purity of 98.5%.
- [0210]The MS-ESI and 1H NMR data are consistent with example 21.
- [0211]THF (60 mL, 6 V), (R)-lactamide (13.2 g, 4.0 eq) and Et3O—BF4 (27.9 g, 4.0 eq) were added to a 100 mL three-necked flask #1, the system was stirred; the materials in #1 were stirred under nitrogen protection for later use; a compound of formula II (10 g, 1.0 eq) and ethanol (100 mL, 10 V) were added to another 250 mL three-necked flask #2; the system was heated to 70±5° C. under nitrogen protection; the materials in three-necked flask #1 were slowly added to three-necked flask #2 dropwise within 20 minutes; the system was heated to 80±5° C. (internal temperature was in the range of 72-75° C.) under nitrogen protection for reacting for 0.5 hour; the system was cooled to room temperature 20-30° C.; the reaction liquid was concentrated to about 50-80 mL with a rotary evaporator between 30-40° C.; water (100 mL, 10 V) was added to the system, then the system was concentrated with a rotary evaporator between 30-40° C. until there was basically no fraction flowing out; the system was cooled to 20-30° C.; the temperature of the system was controlled at 20-30° C., 12M HCl (5.5 g) was used to adjust the pH of the system to 2-3, the system was extracted with ethyl acetate (50 mL×2, 5V×2); the organic phase was discarded, and the aqueous phase was transferred to a flask; the temperature of the system was controlled at 20-30° C., the pH of the system was adjusted to 8-9 with saturated potassium carbonate solution (23 g); the temperature of the system was controlled at 20-25° C., the system was stirred for 2 hours, then was filtered, the filter cake was washed with water (50 mL) and MTBE (50 mL); the filter cake was dried with an air blower at 50° C. for 24 hours, 18 g of earth yellow solid was obtained, with an HPLC purity of 93.5%.
- [0212]The MS-ESI and 1H NMR data are consistent with example 21.
- [0213]THF (120 mL, 12 V), (R)-lactamide (13.2 g, 4.0 eq) and Et3O—BF4 (27.8 g, 4.0 eq) were added to a 250 mL three-necked flask #1, the system was stirred; the materials in #1 were stirred under nitrogen protection for later use; a compound of formula II (10 g, 1.0 eq) and ethanol (140 mL, 14 V) were added to another 500 mL three-necked flask #2; the system was heated to 40-45° C. (internal temperature) under nitrogen protection; the materials in three-necked flask #1 were added to three-necked flask #2 dropwise within 1 hour; the system was maintained at 40-45° C. (internal temperature) under nitrogen protection for reacting for 4.5 hours; the system was cooled to room temperature, and water (20 mL, 2V) was added; the system was concentrated with a rotary evaporator at 30-40° C. until there was basically no fraction flowing out; the system was cooled to 20-30° C.; the temperature of the system was controlled at 20-30° C., 12M HCl (3 mL) was used to adjust the pH of the system to 2-3, the system was extracted with ethyl acetate (50 mL×2, 5V×2); the organic phase was discarded, and the aqueous phase was transferred to a flask; the temperature of the system was controlled at 20-30° C., the pH of the system was adjusted to 8-9 with 50% potassium carbonate solution (15 mL); the temperature of the system was controlled at 20-25° C., the system was stirred for 2 hours, then was filtered, the filter cake was washed with water (50 mL) and acetone (50 mL); the crude product was triturated and stirred with water (50 mL) at 20-25° C. for 1 hour; the system was filtered, the filter cake was washed with water (50 mL) and acetone (50 mL); the filter cake was dried with an air blower at 50° C. for 24 hours, 17.8 g of khaki solid was obtained, with an HPLC purity of 95.3%.
- [0214]The MS-ESI and 1H NMR data are consistent with example 21.
- [0215]THF (60 mL, 12 V), (R)-lactamide (6.6 g, 4.0 eq) and Et3O—BF4 (13.9 g, 4.0 eq) were added to a 250 mL three-necked flask #1, the system was stirred; the materials in three-necked flask #1 were stirred under nitrogen protection for later use; a compound of formula II (5 g, 1.0 eq) and ethanol (70 mL, 14 V) were added to another 250 mL three-necked flask #2; the system was heated to 40-45° C. (internal temperature) under nitrogen protection; the materials in three-necked flask #1 were added to three-necked flask #2 dropwise within 20 minutes; the system was maintained at 40-45° C. (internal temperature) under nitrogen protection for reacting for 3 hours; the system was cooled to room temperature and was filtered, the filter cake was washed with THF (10 mL); water (10 mL, 2V) was added to the filtrate; the filtrate was concentrated with a rotary evaporator to 10-20 mL (2V-4V), the concentrated residue was exchanged with ethyl acetate (25 mL×2) and concentrated to 10-20 mL (2V-4V); water (50 mL, 10V) was added to the concentrated residue; the internal temperature was controlled at 20-25° C., 12M HCl (4.1 g) was used to adjust the pH of the system to 1-2; activated carbon (0.5 g) was added to the system, and the system was stirred at room temperature for 2 hours, and was filtered, the filter cake was washed with water (10 mL) and 1M HCl (10 mL); the combined filtrate was extracted with ethyl acetate (25 mL×2), the organic phase was discarded; the internal temperature was controlled at 20-25° C., the pH of the system was adjusted to 9-10 with saturated potassium carbonate solution (15 g); the internal temperature was controlled at 15-20° C., the system was stirred for 1 hour, and was filtered, the filter cake was washed with water (10 mL); the filter cake was triturated with acetone aqueous solution (50 mL, V/V=1:1) for 1 hour; the system was filtered, the filter cake was washed with acetone aqueous solution (10 mL, V/V=1:1); the filter cake was dried with an air blower at 50° C. for 24 hours; 5.0 g of pale gray solid was obtained, with an HPLC purity of 95.6%, and a yield of 83.5%;
- [0216]Purification of a Compound of Formula I:
- [0217]5.0 g of the obtained solid and methanol (40 mL) were added to a flask, and were stirred for 10 minutes at room temperature, the materials were basically dissolved and the solution was clear; activated carbon (0.5 g) and silica gel (4.0 g) were added to the system; the system was heated to 50-55° C., the temperature was maintained and the system was stirred for 2 hours, then was filtered with silica gel (5 g), the filter cake was washed with methanol (50 mL); the filtrate was concentrated with a rotary evaporator to 5-10 mL; MTBE (50 mL) was added to the concentrated residue; the system was heated to reflux, and was allowed for reflux for 1 hour; the system was cooled to 5-10° C., the temperature was maintained and the system was stirred for 1 hour and was filtered, the filter cake was washed with MTBE; the filter cake was dried with a drying oven under vacuum at 50° C. for 16 hours; 3.0 g of off-white solid was obtained, with a yield of 60% and a purity of 97.9%; the filtrate was concentrated to dryness to obtain 1.4 g of yellow solid.
- [0218]The MS-ESI and 1H NMR data are consistent with example 21.
PAT
- NEW SELECTIVE JAK1 INHIBITORS AND THEIR USEPublication Number: HR-P20211965-T1Priority Date: 2016-10-03
- Novel JAK1 selective inhibitors and uses thereofPublication Number: KR-102399848-B1Priority Date: 2016-10-03Grant Date: 2022-05-19
- Novel Jak1-selective inhibitors and their usesPublication Number: CN-113214278-BPriority Date: 2016-10-03Grant Date: 2022-10-28
- Jak1 selective inhibitors and uses thereofPublication Number: US-RE49834-EPriority Date: 2016-10-03Grant Date: 2024-02-13
- Novel jak1 selective inhibitors and uses thereofPublication Number: US-2019256523-A1Priority Date: 2016-10-03
- JAK1 selective inhibitors and uses thereofPublication Number: US-10738060-B2Priority Date: 2016-10-03Grant Date: 2020-08-11
- Novel Jak1-selective inhibitors and their usesPublication Number: CN-108366994-BPriority Date: 2016-10-03Grant Date: 2021-10-01
- Novel Jak1-selective inhibitors and their usesPublication Number: CN-113214278-APriority Date: 2016-10-03
- Novel jak1 selective inhibitors and uses thereofPublication Number: EP-3509591-B1Priority Date: 2016-10-03Grant Date: 2021-11-17
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereof.Publication Number: MX-2024006688-APriority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: US-12195476-B2Priority Date: 2019-06-06Grant Date: 2025-01-14
- Novel jak1 selective inhibitors and uses thereofPublication Number: CA-3039178-A1Priority Date: 2016-10-03
- Novel jak1 selective inhibitors and uses thereofPublication Number: EP-3509591-A1Priority Date: 2016-10-03
- Novel JAK1 selective inhibitors and uses thereofPublication Number: JP-2019537559-APriority Date: 2016-10-03
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: EP-4248967-A3Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: JP-2023089169-APriority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compounds, crystal forms of furoimidazopyridine compounds and crystal forms of their saltsPublication Number: CN-113906035-BPriority Date: 2019-06-06Grant Date: 2023-11-10
- Synthesis method of furoimidazopyridine compounds, crystal forms of furoimidazopyridine compounds and crystal forms of their saltsPublication Number: CN-117327083-APriority Date: 2019-06-06
- METHOD OF SYNTHESIS OF FUROIMIDAZOPYRIDINE COMPOUND, CRYSTAL FORM OF FUROIMIDAZOPYRIDINE COMPOUND, AND CRYSTAL FORM OF ITS SALT.Publication Number: MX-2024004146-APriority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: US-2022227777-A1Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: AU-2020289149-B2Priority Date: 2019-06-06Grant Date: 2023-05-11
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: EP-4248966-A2Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: EP-4248966-A3Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: EP-4248967-A2Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: WO-2020244348-A1Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: AU-2020289149-A1Priority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound and crystal form of salt thereofPublication Number: CN-113906035-APriority Date: 2019-06-06
- Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereofPublication Number: EP-3981771-A1Priority Date: 2019-06-06
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Girocitinib, Janus kinase inhibitor, anti-inflammatory, A0IES9T8GO
Gildeuretinol
Gildeuretinol
CAS118139-35-8
MF C20H272H3O, MW 289.5 g/mol
(2E,4E,6E,8E)-3-(2H3)methyl-7-methyl-9-(2,6,6-trimethylcyclohex-1-en-1-yl)nona-2,4,6,8-tetraen-1-ol; (20,20,20-2H3)retinol
(2E,4E,6E,8E)-7-methyl-3-(trideuteriomethyl)-9-(2,6,6-trimethylcyclohexen-1-yl)nona-2,4,6,8-tetraen-1-ol
vitamin A analogue, Orphan Drug, Stargardt disease, breakthrough therapy, Pediatric Rare Disease designations, ALK-001, KL-49, ALK 001, KL 49
- OriginatorColumbia University
- DeveloperAlkeus Pharmaceuticals
- ClassEye disorder therapies; Retinoids; Vitamins
- Mechanism of ActionDimerisation inhibitors; Vitamin A replacements
- Orphan Drug StatusYes – Stargardt disease
- Phase II/IIIDry age-related macular degeneration
- Phase IIStargardt disease
- No development reportedRetinal dystrophies
- 08 Sep 2025Gildeuretinol – Alkeus Pharmaceuticals receives Orphan Drug status for Stargardt disease in European Union
- 09 Jan 2025Alkeus Pharmaceuticals announces intention to submit an NDA to US FDA for Stargardt disease in 2025
- 09 Jan 2025Efficacy and adverse event data from phase II trial for Stargardt disease released by Alkeus Pharmaceuticals
Gildeuretinol is an investigational new drug being developed by Alkeus Pharmaceuticals, Inc. for the treatment of retinal diseases, particularly Stargardt disease and geographic atrophy secondary to age-related macular degeneration (AMD). Stargardt disease is caused by a defect in the ABCA4 gene that clears toxic byproducts resulting from the dimerization of vitamin A. Gildeuretinol is new molecular entity designed to reduce the dimerization of vitamin A in the eye without affecting the visual cycle.[1]
Gildeuretinol has received breakthrough therapy, orphan drug and Pediatric Rare Disease designations from the U.S. Food and Drug Administration.[2]
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Zaydon YA, Tsang SH (July 2024). “The ABCs of Stargardt disease: the latest advances in precision medicine”. Cell & Bioscience. 14 (1) 98. doi:10.1186/s13578-024-01272-y. PMC 11282698. PMID 39060921.
- Fitch J (22 November 2024). “Gildeuretinol for Stargardt disease receives Rare Pediatric Disease, Fast Track Designations”. Contemporary Pediatrics.
| Clinical data | |
|---|---|
| Other names | ALK-001, KL-49 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 118139-35-8 |
| PubChem CID | 169490774 |
| UNII | PSZ7W5NR24 |
| KEGG | D12713 |
| ChEMBL | ChEMBL5314606 |
| Chemical and physical data | |
| Formula | C20H30D3O |
| Molar mass | 292.500 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////Gildeuretinol, vitamin A analogue, Orphan Drug, Stargardt disease, breakthrough therapy, Pediatric Rare Disease designations, ALK-001, KL-49, ALK 001, KL 49, PSZ7W5NR24
Frevecitinib
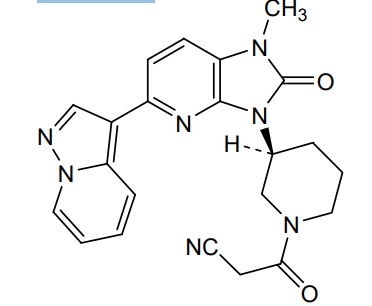
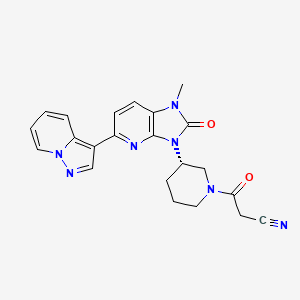
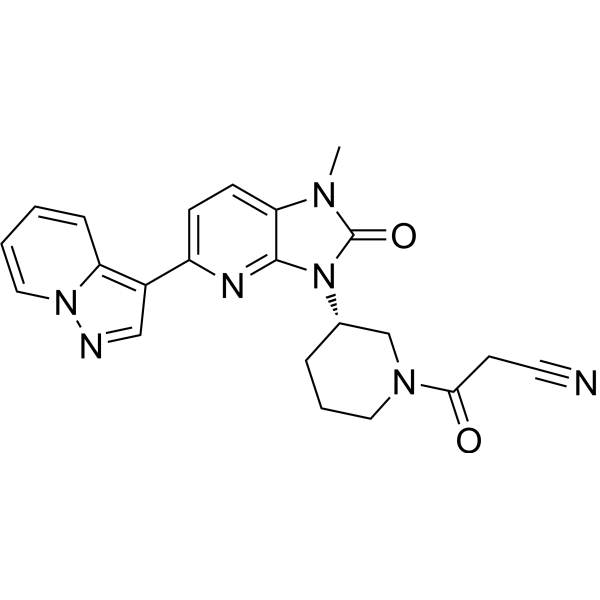
Frevecitinib
CAS 1299417-07-4
MF C22H21N7O2 MW 415.4 g/mol
3-[(3S)-3-(1-methyl-2-oxo-5-pyrazolo[1,5-a]pyridin-3-ylimidazo[4,5-b]pyridin-3-yl)piperidin-1-yl]-3-oxopropanenitrile
3-{(3S)-3-[1-methyl-2-oxo-5-(pyrazolo[1,5-a]pyridin-3-
yl)-1,2-dihydro-3H-imidazo[4,5-b]pyridin-3-yl]piperidin1-yl}-3-oxopropanenitrile
Janus kinase inhibitor, anti-inflammatory, 5N5L287M8T, KN 002, KN-002
Single and Multiple Ascending Dose Study of KN-002
CTID: NCT05006521
Phase: Phase 1
Status: Completed
Date: 2024-08-07
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011157397&_cid=P11-MH2TVG-48083-1
SYN
It has now been found that a drug substance disclosed in WO2011/051452, namely the compound (S)-3-(3-(1-methyl-2-oxo-5-(pyrazolo[1,5-a]pyridine-3-yl)-1H-imidazo[4,5-b]pyridine-3(2H)-yl)piperidin-1-yl)-3-oxopropanenitrile having the structure shown below and known herein as compound (I) can be prepared in different polymorphic forms. Surprisingly one form exists as a polymorph with particularly advantageous stability properties. Compound (I) as prepared following the process in WO2011/051452 is known as Form I herein.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US76222175&_cid=P11-MH2U0A-51623-1
PAT
- N-containing heteroaryl derivatives as jak3 kinase inhibitorsPublication Number: KR-101675614-B1Priority Date: 2009-10-29Grant Date: 2016-11-11
- N-containing heteroaryl derivatives as jak3 kinase inhibitorsPublication Number: US-2012245140-A1Priority Date: 2009-10-29
- N-containing heteroaryl derivatives as jak3 kinase inhibitorsPublication Number: US-2013131038-A9Priority Date: 2009-10-29
- N-containing heteroaryl derivatives as JAK3 kinase inhibitorsPublication Number: US-8501735-B2Priority Date: 2009-10-29Grant Date: 2013-08-06
- N-containing heteroaryl derivatives as JAK3 kinase inhibitorsPublication Number: US-8946257-B2Priority Date: 2009-10-29Grant Date: 2015-02-03
- Heteroaryl imidazolone derivatives as jak inhibitorsPublication Number: EP-2582703-A1Priority Date: 2010-06-15
- Heteroaryl Imidazolone Derivatives as Jap InhibitorsPublication Number: KR-20130113331-APriority Date: 2010-06-15
- Heteroaryl imidazolone derivatives as jak inhibitorsPublication Number: US-2013089512-A1Priority Date: 2010-06-15
- Heteroaryl imidazolone derivatives as jak inhibitorsPublication Number: WO-2011157397-A1Priority Date: 2010-06-15
- N-containing heteroaryl derivatives as jak3 kinase inhibitorsPublication Number: EP-2493895-B1Priority Date: 2009-10-29Grant Date: 2017-04-26
- Novel polymorphsPublication Number: US-2018016284-A1Priority Date: 2015-02-05
- Novel polymorphsPublication Number: US-2019031687-A1Priority Date: 2015-02-05
- Novel polymorphsPublication Number: WO-2016124464-A1Priority Date: 2015-02-05
- Heteroaryl imidazolone derivatives as jak inhibitorsPublication Number: CA-2802588-A1Priority Date: 2010-06-15
- Heteroaryl imidazolone derivatives as JAK inhibitorsPublication Number: CN-102933583-APriority Date: 2010-06-15
- Novel polymorphsPublication Number: EP-3053927-A1Priority Date: 2015-02-05
- Novel polymorphsPublication Number: EP-3253769-B1Priority Date: 2015-02-05Grant Date: 2019-03-13
- New polymorphPublication Number: JP-2018502929-APriority Date: 2015-02-05
- New polymorphPublication Number: JP-6685326-B2Priority Date: 2015-02-05Grant Date: 2020-04-22
- PolymorphsPublication Number: US-10087196-B2Priority Date: 2015-02-05Grant Date: 2018-10-02
- Crystalline form of a JAK3 kinase inhibitorPublication Number: US-10155757-B2Priority Date: 2015-03-10Grant Date: 2018-12-18
- Crystalline form of a jak3 kinase inhibitorPublication Number: US-2018044336-A1Priority Date: 2015-03-10
- Crystalline form of a jak3 kinase inhibitorPublication Number: WO-2016142201-A1Priority Date: 2015-03-10
- Polymorphic forms of (s)-3-(3(1-methyl-2-oxo-5-(pyrazolo[1,5-a]pyridine-3(2h)-yl)piperidin-1-yl)-3-oxopropanenitrilePublication Number: CA-2972977-CPriority Date: 2015-02-05Grant Date: 2019-04-09
- polymorphPublication Number: CN-107207533-BPriority Date: 2015-02-05Grant Date: 2019-04-16
- Formulation of a pan-jak inhibitorPublication Number: TW-202440105-APriority Date: 2022-12-02
- Formulation of a pan-jak inhibitorPublication Number: US-2024261224-A1Priority Date: 2022-12-02
- Formulation of a pan-jak inhibitorPublication Number: WO-2024119058-A2Priority Date: 2022-12-02
- Formulation of a pan-jak inhibitorPublication Number: WO-2024119058-A3Priority Date: 2022-12-02
- Crystalline form of a jak3 kinase inhibitorPublication Number: EP-3268364-A1Priority Date: 2015-03-10
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Frevecitinib, Janus kinase inhibitor, anti-inflammatory, 5N5L287M8T, KN 002, KN-002

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}