
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Edelinontrine



Edelinontrine
CRD740, PF04447943, cas 1082744-20-4
| Molecular Weight | 395.46 |
|---|---|
| Formula | C20H25N7O2 |
6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-1-(oxan-4-yl)-5H-pyrazolo[3,4-d]pyrimidin-4-one
- 7N969W8Y4O
- 6-((3S,4S)-4-Methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl)-1-(tetrahydro-2H-pyran-4-yl)-1,5-dihydro-4H-pyrazolo(3,4-d)pyrimidin-4-one
- 6-[(3S,4S)-4-METHYL-1-(PYRIMIDIN-2-YLMETHYL)PYRROLIDIN-3-YL]-1-(OXAN-4-YL)-5H-PYRAZOLO[3,4-D]PYRIMIDIN-4-ONE
- 6-((3S,4S)-4-Methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl)-1-(tetrahydro-2H-pyran-4-yl)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one
Edelinontrine (PF-04447943) is a potent inhibitor of human recombinant PDE9A (IC50=12 nM) with >78-fold selectivity, respectively, over other PDE family members (IC50>1000 nM).

PF-04447943 is a potent, selective brain penetrant PDE9 inhibitor (Ki of 2.8, 4.5 and 18 nM) for human, rhesus and rat recombinant PDE9 respectively and high selectivity for PDE9 versus PDEs1-8 and 10-11. PF-04447943 was being developed by Pfizer for the treatment of cognitive disorders. PF-04447943 attenuates a scopolamine-induced deficit in a novel rodent attention task. PF-04447943 enhances synaptic plasticity and cognitive function in rodents. PF-04447943 has completed Phase II clinical trials in subjects with mild to moderate AD in 2013 but this research was discontinued. Pfizer completes a phase I trial in Sickle cell anaemia.
SCHEME
SUDECHAIN

MAIN

Patent
WO2023114995 PFIZER
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023114995&_cid=P20-MB07JE-44537-1
PAPER
Journal of Medicinal Chemistry (2012), 55(21), 9045-9054
PATENT
WO2008139293
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2008139293&_cid=P20-MB07LY-46583-1
EXAMPLE 111
6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyiτolidin-3-yl]-1-αetrahydro-2H- pyran-4-yl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one

///////Edelinontrine, CRD740, PF04447943, CRD 740, PF 04447943, PHASE 1
Sulbactam



Sulbactam
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Sulbactam benzathine | 49MU89FVBV | 83031-43-0 | YSEPFTSCLHUBNH-HFKSPEPWSA-N |
| Sulbactam sodium | DKQ4T82YE6 | 69388-84-7 | NKZMPZCWBSWAOX-IBTYICNHSA-M |
WeightAverage: 233.242
Monoisotopic: 233.035793157
Chemical FormulaC8H11NO5S
(2S,5R)-3,3-dimethyl-4,4,7-trioxo-4λ6-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid
Betamaze, Penicillanic Acid Sulfone, Sulbactamum, CP 45899, CP-45899, CP45899
FDA 2023, Xacduro, 5/23/2023, To treat hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia caused by susceptible isolates of Acinetobacter baumannii-calcoaceticus complex
Press Release
Drug Trials Snapshots
Sulbactam is a β-lactamase inhibitor. This drug is given in combination with β-lactam antibiotics to inhibit β-lactamase, an enzyme produced by bacteria that destroys the antibiotics.[1]
It was patented in 1977 and approved for medical use in 1986.[2]
Sulbactam is a beta (β)-lactamase inhibitor and a derivative of the basic penicillin nucleus. When given in combination with β-lactam antibiotics, sulbactam produces a synergistic effect as it blocks the enzyme responsible for drug resistance by hydrolyzing β-lactams.
PATENT
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US9309245 | No | 2016-04-12 | 2033-04-02 |  |
| US9623014 | No | 2017-04-18 | 2033-04-02 | |
| US9968593 | No | 2018-05-15 | 2035-11-17 | |
| US10376499 | No | 2019-08-13 | 2035-11-17 | |
doi:10.1016/S0040-4039(00)89275-8

SYN
European Journal of Medicinal Chemistry
Volume 265, 5 February 2024, 116124
https://doi.org/10.1016/j.ejmech.2024.116124
On May 23, 2023, the FDA granted approval to Xacduro for the treatment of Baumannii-sensitive strains causing hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia in
patients aged 18 years or older [4]. Xacduro consists of Sulbactam and Durlobactam. Sulbactam, a medication with a similar structure to Penicillin, has the ability to eliminate Acinetobacter baumannii. On the other hand, Durlobactam shields Sulbactam from being broken down by enzymes that may be produced by Acinetobacter baumannii [5].
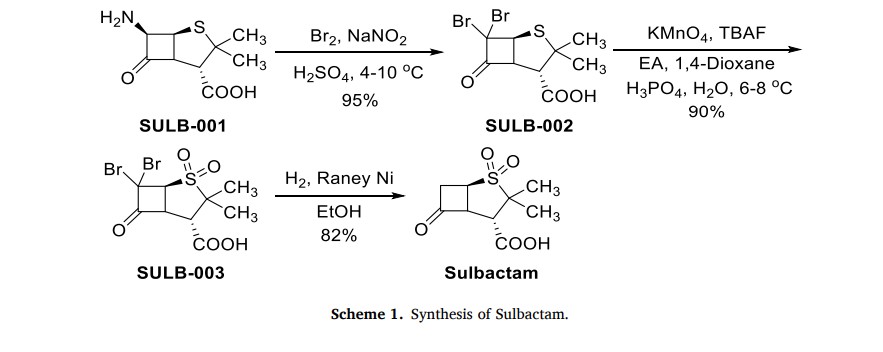
The production process of Sulbactam started with 6-aminopenicilanic acid (6-APA) (SULB-001) as the starting material (Scheme 1) [6]. It underwent bromination reaction with sodium nitrite and bromine
in the presence of sulfuric acid. Then, SULB-002 was oxidized by potassium permanganate to obtain sulfone SULB-003. Finally, the sulfonewas catalytically hydrogenated and dehalogenated in the presence of Raney nickel to get Sulbactam
[5] A. El-Ghali, A.J. Kunz Coyne, K. Caniff, C. Bleick, M.J. Rybak, Sulbactamdurlobactam: a novel β-lactam-β-lactamase inhibitor combination targeting
carbapenem-resistant Acinetobacter baumannii infections, Pharmacotherapy 43
(2023) 502–513.
[6] Z.M. Song, W. Liu, J. Yang, Y. Sun, Improvement on the synthetic process of
sulbactam, Chin. J
PATENT
https://patents.google.com/patent/CN101967155A/en
Embodiment 1
In the four-hole boiling flask of 2000ML, add 600ML methylene dichloride and 180ML2.5N sulfuric acid, stirring is cooled to below 0 ℃, add 28ML bromine and 25g Sodium Nitrite, 0 ± 0.2 ℃, gradation adds 40g 6-APA, and controlled temperature is lower than 5 ℃, stirring reaction 1h, be cooled to then below 0 ℃, 20% aqueous solution of sodium bisulfite of dropping below 0 ℃ leaves standstill phase-splitting to the color fade of bromine, water 100ML dichloromethane extraction 3 times, merge organic phase, with 100ML saturated sodium-chloride water solution washing 2 times, obtain 6, the 6-dibromo penicillanic acid;
To go up 6, the 6-dibromo penicillanic acid changes in the 2000ML beaker mutually. and add 250ML distilled water and stir, be cooled to below 5 ℃, drip 4NNaHCO 3The aqueous solution leaves standstill phase-splitting to pH=7, organic phase extracts 3 times with the 80ML deionized water, merge water, water changes in the 2000ML four-hole boiling flask, stirring is cooled to 0 ℃, and beginning dropping oxidizing agent (44g KMn04+10.8ML H3P04+700MLH20 stirring and dissolving) dripped in 30 minutes, controlled temperature is lower than 10 ℃ in the dropping process, keep 0~5 ℃ then, stirring reaction 1h adds the 500ML ethyl acetate, drip 6N sulfuric acid to pH=1.25, be cooled to 0 ℃, slowly add the color fade of 27.5% hydrogen peroxide (about 45g) to KMn04, during continue to keep pH=1.25 with 6N sulfuric acid, controlled temperature is lower than 10 ℃, reaction 10mi n filters, and adds sodium-chlor in the filtrate to no longer dissolving, leave standstill the branch phase of anhydrating, water 250ML ethyl acetate extraction 4 times merge organic phase, and wash 2 times with the 100ML saturated sodium-chloride water solution, organic phase contains 6, the acid of 6-dibromo sulbactam;
To go up 6,6-dibromo sulbactam acid organic phase changes in the 2000ML four-hole boiling flask, adds 350ML water, is cooled to below 5 ℃, uses 4N NaHCO 3The aqueous solution is transferred pH to 5.0, and add 25ML methyl alcohol, add the 26g zinc powder in batches, and drip 6N sulfuric acid maintenance pH:4.5~5.5, after adding zinc powder, keep stirring reaction 4h below 5 ℃, keep pH=4.5~5.5 with 6N sulfuric acid simultaneously, filter, with 25ML ethyl acetate and 25ML water washing, merging filtrate is transferred pH to 2.0 with 6N sulfuric acid, add sodium-chlor to water insoluble till, leave standstill the branch phase of anhydrating, water merges organic phase with 150ML ethyl acetate extraction 4 times, washs to redness with the 50ML-100ML5% potassium permanganate solution earlier at organic layer and does not take off, again with 150ML saturated sodium-chloride water solution washing 2 times, layering, organic layer add the 2g activated carbon decolorizing, the 15g anhydrous magnesium sulfate drying, suction filtration, be evaporated to feed liquid and be creamy white, cool to 0 ℃ after centrifuging, after the oven dry product Sulbactam (sulbactam acid) 32g, the product yield is 74%, the product colour pure white was placed 30 days the color no change under the room temperature.
Embodiment 2
In the reactor of 2000L, add 600L methylene dichloride and 180L2.5N sulfuric acid, stirring is cooled to below 0 ℃, add 28L bromine and 25Kg Sodium Nitrite, 0 ± 0.2 ℃, gradation adds 40Kg 6-APA, and controlled temperature is lower than 5 ℃, stirring reaction 1h, be cooled to then below 0 ℃, 20% aqueous solution of sodium bisulfite of dropping below 0 ℃ leaves standstill phase-splitting to the color fade of bromine, water 100L dichloromethane extraction 3 times, merge organic phase, with 100L saturated sodium-chloride water solution washing 2 times, obtain 6, the 6-dibromo penicillanic acid;
Will on obtain 6, the 6-dibromo penicillanic acid changes in the 2000L reactor mutually. add the 250L tap water and stir, be cooled to below 5 ℃, drip 4NNaHCO 3The aqueous solution leaves standstill phase-splitting to pH=7, organic phase extracts 3 times with the 80L deionized water, merge water, water changes in the 2000L reactor, stirring is cooled to 0 ℃, and beginning dropping oxidizing agent (44Kg KMn04+10.8L H3P04+700LH20 stirring and dissolving) dripped in 30 minutes, controlled temperature is lower than 10 ℃ in the dropping process, keep 0~5 ℃ then, stirring reaction 1h adds the 500L ethyl acetate, drip 6N sulfuric acid to pH=1.25, be cooled to 0 ℃, slowly add 28% hydrogen peroxide (about 44Kg) color fade to KMn04, during continue to keep pH=1.25 with 6N sulfuric acid, controlled temperature is lower than 10 ℃, reaction 10mi n filters, and adds sodium-chlor in the filtrate to no longer dissolving, leave standstill the branch phase of anhydrating, water 250L ethyl acetate extraction 4 times merge organic phase, and wash 2 times with the 100L saturated sodium-chloride water solution, organic phase contains 6, the acid of 6-dibromo sulbactam;
To go up organic phase and contain 6, the acid of 6-dibromo sulbactam changes in the 2000L reactor, adds 350L water, is cooled to below 5 ℃, uses 4N NaHCO 3The aqueous solution is transferred pH to 5.0, and add 25L methyl alcohol, add the 26Kg zinc powder in batches, and drip 6N sulfuric acid maintenance pH:4.5~5.5, after adding zinc powder, keep stirring reaction 4h below 5 ℃, keep pH=4.5~5.5 with 6N sulfuric acid simultaneously, filter, with 25L ethyl acetate and 25L water washing, merging filtrate is transferred pH to 2.0 with 6N sulfuric acid, add sodium-chlor to water insoluble till, leave standstill the branch phase of anhydrating, water merges organic phase with 150L ethyl acetate extraction 4 times, washs to redness with the 30-50L10% potassium permanganate solution earlier at organic layer and does not take off, again with 150L saturated sodium-chloride water solution washing 2 times, layering, organic layer add the 2Kg activated carbon decolorizing, the 15Kg anhydrous magnesium sulfate drying, suction filtration, be evaporated to feed liquid and be creamy white, cool to 0 ℃ after centrifuging, after the oven dry product Sulbactam (sulbactam acid) 31.5Kg, the product yield is 72.8%, the product colour pure white was placed 30 days the color no change under the room temperature.
Embodiment 3
In the four-hole boiling flask of 1000ML, add 300ML methylene dichloride and 90ML2.5N Hydrogen bromide, stirring is cooled to below 0 ℃, add 14ML bromine and 12.5g Sodium Nitrite, 0 ± 0.2 ℃, gradation adds 20g 6-APA, and controlled temperature is lower than 5 ℃, stirring reaction 1h, be cooled to then below 0 ℃, 20% aqueous solution of sodium bisulfite of dropping below 0 ℃ leaves standstill phase-splitting to the color fade of bromine, water 50ML dichloromethane extraction 3 times, merge organic phase, with 50ML saturated sodium-chloride water solution washing 2 times, obtain 6, the 6-dibromo penicillanic acid;
To go up 6, the 6-dibromo penicillanic acid changes in the 1000ML beaker mutually. and add 125ML distilled water and stir, be cooled to below 5 ℃, drip 4NNaHCO 3The aqueous solution leaves standstill phase-splitting to pH=7, organic phase extracts 3 times with the 40ML deionized water, merge water, water changes in the 1000ML four-hole boiling flask, stirring is cooled to 0 ℃, and beginning dropping oxidizing agent (22g KMn04+5.4ML H3P04+300MLH20 stirring and dissolving) dripped in 30 minutes, controlled temperature is lower than 10 ℃ in the dropping process, keep 0~5 ℃ then, stirring reaction 1h adds the 250ML ethyl acetate, drip 6N sulfuric acid to pH=1.25, be cooled to 0 ℃, slowly add 25% hydrogen peroxide (about 29g) color fade to KMn04, during continue to keep pH=1.25 with 6N sulfuric acid, controlled temperature is lower than 10 ℃, reaction 10mi n filters, and adds sodium-chlor in the filtrate to no longer dissolving, leave standstill the branch phase of anhydrating, water 125ML ethyl acetate extraction 4 times merge organic phase, and wash 2 times with the 50ML saturated sodium-chloride water solution, organic phase contains 6, the acid of 6-dibromo sulbactam.
To go up organic phase and contain 6, the acid of 6-dibromo sulbactam changes in the 1000ML four-hole boiling flask, adds 175ML water, is cooled to below 5 ℃, uses 4N NaHCO 3The aqueous solution is transferred pH to 5.0, and add 12.5ML methyl alcohol, add the 13g zinc powder in batches, and drip 6N sulfuric acid maintenance pH:4.5~5.5, after adding zinc powder, keep stirring reaction 4h below 5 ℃, keep pH=4.5~5.5 with 6N sulfuric acid simultaneously, filter, with 12.5ML ethyl acetate and 12.5ML water washing, merging filtrate is transferred pH to 2.0 with 6N sulfuric acid, add sodium-chlor to water insoluble till, leave standstill the branch phase of anhydrating, water merges organic phase with 75ML ethyl acetate extraction 4 times, washs to redness with the 15ML-35ML7% potassium permanganate solution earlier at organic layer and does not take off, again with 75ML saturated sodium-chloride water solution washing 2 times, layering, organic layer add the 1g activated carbon decolorizing, the 7.5g anhydrous magnesium sulfate drying, suction filtration, be evaporated to feed liquid and be creamy white, cool to 0 ℃ after centrifuging, after the oven dry product Sulbactam (sulbactam acid) 15.9g, the product yield is 73.5%, the product colour pure white was placed 30 days the color no change under the room temperature.
PATENT
https://patents.google.com/patent/US4420426A/en
Medical uses
The combination ampicillin/sulbactam (Unasyn) is available in the United States.[3]
The combination cefoperazone/sulbactam (Sulperazon) is available in many countries but not in the United States.[4]
The co-packaged combination sulbactam/durlobactam was approved for medical use in the United States in May 2023.[5]
Mechanism
Sulbactam is primarily used as a suicide inhibitor of β-lactamase, shielding more potent beta-lactams such as ampicillin.[6] Sulbactam itself contains a beta-lactam ring, and has weak antibacterial activity by inhibiting penicillin binding proteins (PBP) 1 and 3, but not 2.[7]
References
- ^ Totir MA, Helfand MS, Carey MP, Sheri A, Buynak JD, Bonomo RA, Carey PR (August 2007). “Sulbactam forms only minimal amounts of irreversible acrylate-enzyme with SHV-1 beta-lactamase”. Biochemistry. 46 (31): 8980–8987. doi:10.1021/bi7006146. PMC 2596720. PMID 17630699.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 492. ISBN 9783527607495.
- ^ “Unasyn- ampicillin sodium and sulbactam sodium injection, powder, for solution”. DailyMed. U.S. National Library of Medicine. 29 March 2023. Retrieved 25 May 2023.
- ^ “Sulperazon”. drugs.com.
- ^ “FDA Approves New Treatment for Pneumonia Caused by Certain Difficult-to-Treat Bacteria”. U.S. Food and Drug Administration (Press release). 24 May 2023. Retrieved 24 May 2023.
- ^ Crass RL, Pai MP (February 2019). “Pharmacokinetics and Pharmacodynamics of β-Lactamase Inhibitors”. Pharmacotherapy. 39 (2): 182–195. doi:10.1002/phar.2210. PMID 30589457. S2CID 58567725.
- ^ Penwell WF, Shapiro AB, Giacobbe RA, Gu RF, Gao N, Thresher J, et al. (March 2015). “Molecular mechanisms of sulbactam antibacterial activity and resistance determinants in Acinetobacter baumannii”. Antimicrobial Agents and Chemotherapy. 59 (3): 1680–1689. doi:10.1128/AAC.04808-14. PMC 4325763. PMID 25561334.
Further reading
Singh GS (January 2004). “Beta-lactams in the new millennium. Part-II: cephems, oxacephems, penams and sulbactam”. Mini Reviews in Medicinal Chemistry. 4 (1): 93–109. doi:10.2174/1389557043487547. PMID 14754446.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| MedlinePlus | a693021 |
| Routes of administration | Intravenous, intramuscular |
| ATC code | J01CG01 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only) |
| Pharmacokinetic data | |
| Protein binding | 29% |
| Elimination half-life | 0.65–1.20 hrs |
| Excretion | Mainly kidneys (41–66% within 8 hrs) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 68373-14-8 |
| PubChem CID | 130313 |
| ChemSpider | 115306 |
| UNII | S4TF6I2330 |
| KEGG | D08533 |
| ChEBI | CHEBI:9321 |
| ChEMBL | ChEMBL403 |
| CompTox Dashboard (EPA) | DTXSID1023605 |
| ECHA InfoCard | 100.063.506 |
| Chemical and physical data | |
| Formula | C8H11NO5S |
| Molar mass | 233.24 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 148 to 151 °C (298 to 304 °F) |
| showSMILES | |
| showInChI | |
//////////Sulbactam, Xacduro, FDA 2023, APPROVALS 2023, Betamaze, Penicillanic Acid Sulfone, Sulbactamum, CP 45899, CP-45899, CP45899
Eclitasertib
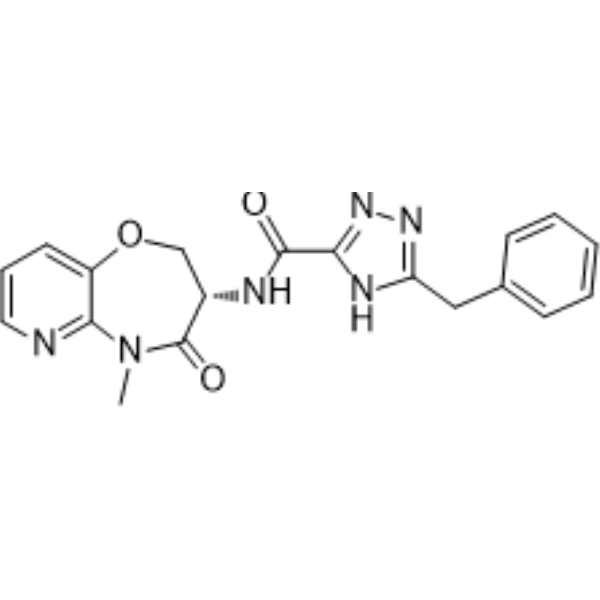
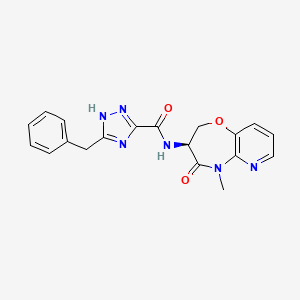
Eclitasertib
CAS 2125450-76-0
5-benzyl-N-[(3S)-5-methyl-4-oxo-2,3-dihydropyrido[3,2-b][1,4]oxazepin-3-yl]-1H-1,2,4-triazole-3-carboxamide
- DNL-758
- SAR-443122
- Eclitasertib (DNL-758) is a potent receptor-interacting protein kinase 1 (RIPK1) inhibitor with an IC50 of 0.0375 µΜ.
- UNII-975AT1P9J6
| Molecular Weight | 378.38 |
|---|---|
| Formula | C19H18N6O3 |
- OriginatorHarvard University
- DeveloperDenali Therapeutics Inc; Sanofi
- Class2 ring heterocyclic compounds; Amides; Anti-inflammatories; Antipsoriatics; Antirheumatics; Oxazepines; Pyridines; Skin disorder therapies; Small molecules; Triazoles
- Mechanism of ActionRIPK1 protein inhibitors
- Phase IIUlcerative colitis
- DiscontinuedCutaneous lupus erythematosus; Psoriasis; Rheumatoid arthritis; SARS-CoV-2 acute respiratory disease
- 12 Mar 2024Discontinued – Phase-I for Psoriasis (In volunteers) in USA (unspecified route) (Denali pipeline, February 2024)
- 12 Mar 2024Discontinued – Phase-I for Rheumatoid arthritis (In volunteers) in USA (unspecified route) (Denali pipeline, February 2024)
- 27 Feb 2024Efficacy and adverse events data from phase II trial in Cutaneous lupus erythematosus released by Sanofi
SAR443122, was investigated in several clinical trials to evaluate its safety and efficacy. NCT04469621 was studied in severe COVID-19 patients, while NCT05588843 is currently recruiting participants with ulcerative colitis. Additionally, NCT04781816, which was completed with results, focused on patients with cutaneous lupus erythematosus.
Eclitasertib is an orally bioavailable, small-molecule inhibitor of receptor-interacting serine/threonine-protein kinase 1 (RIPK1; receptor-interacting protein 1; RIP1), with potential anti-inflammatory and immunomodulatory activities. Upon oral administration, eclitasertib disrupts RIPK1-mediated signaling, and may attenuate inflammation and the resulting tissue damage. RIPK1, a signaling protein in the tumor necrosis factor (TNF) receptor pathway, plays a key role in inflammation and cell death in response to tissue damage and pathogen recognition.
SCHEME
SIDE CHAIN
MAIN
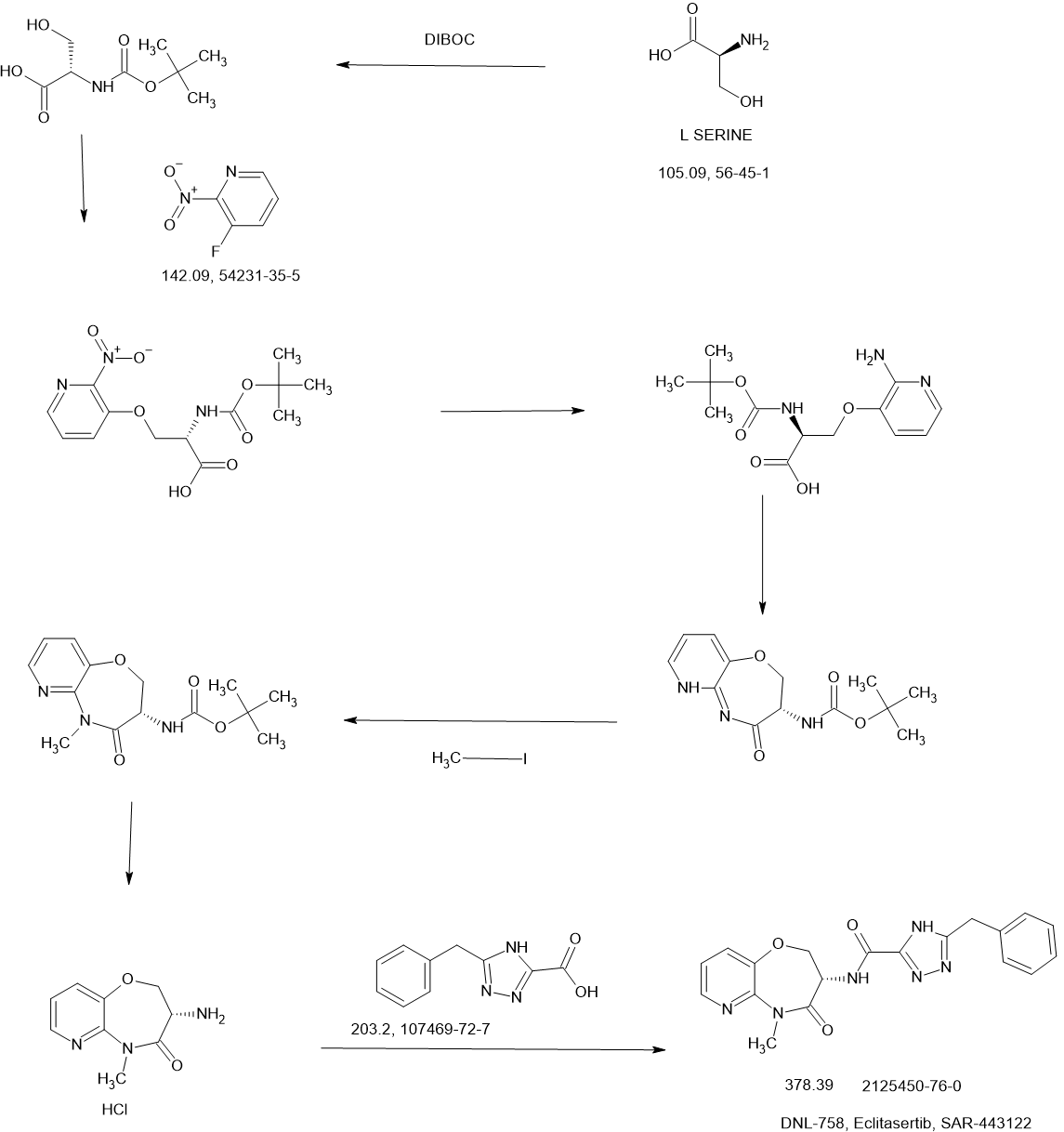
REF
WO2017136727
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017136727&_cid=P22-MAYSGO-11421-1
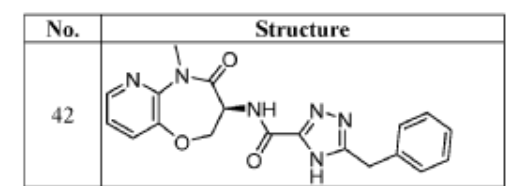
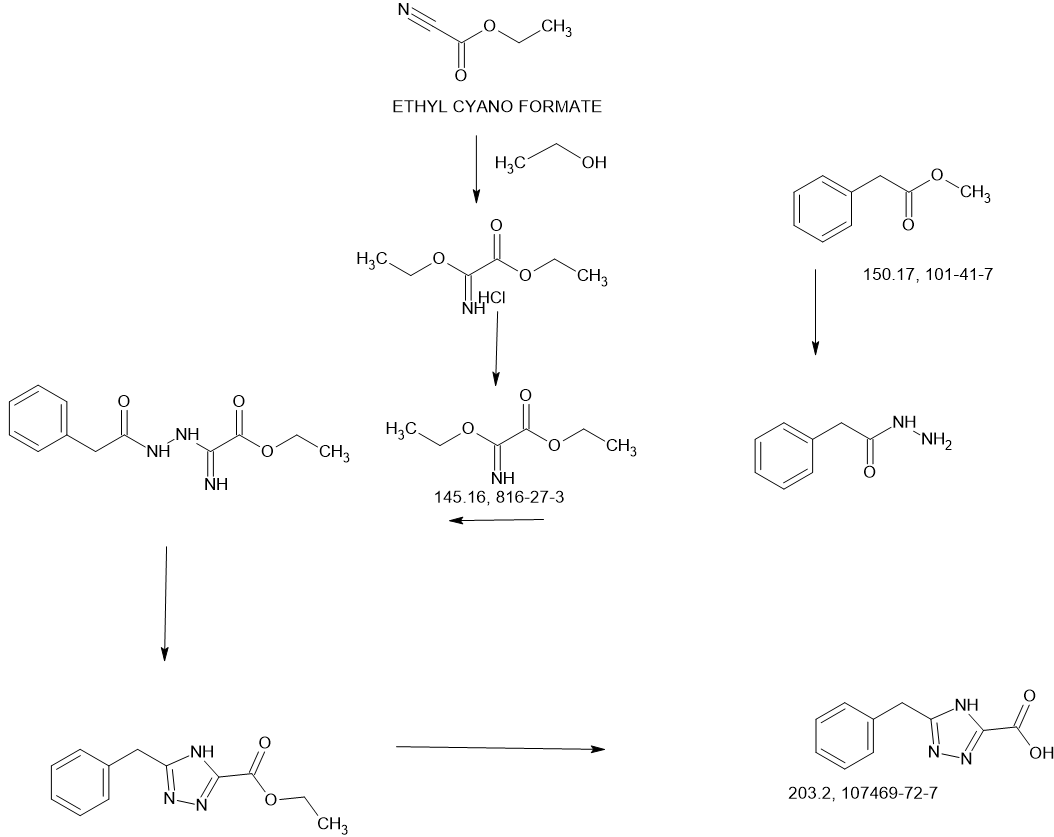
Example 42: (S)-5-benzyl-N-(5-methyl-4-oxo-2,3,4,5-tetrahydropyrido[3,2-b][1,4]oxazepin-3-yl)- 4H-1,2,4-triazole-3-carboxamide
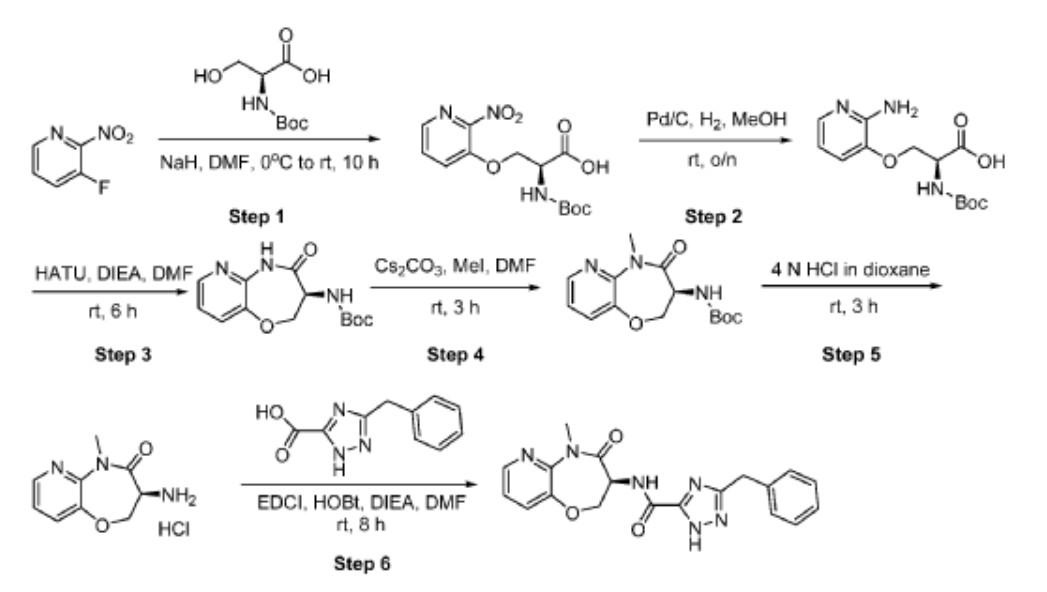
Step 1: Preparation oƒ (2S)-2-(((tert-butoxy)carbonyl)amino)-3-((2-nitropyridin-3-yl)oxy)propanoic acid
[0535] Sodium hydride (60%, 2 g, 50 mmol) was added into a stirring solution of (2S)-2-(tert-butoxycarbonylamino)-3-hydroxypropanoic acid (5 g, 25.0 mmol) in N,N-dimethylformamide (100 mL). The resulting mixture was stirred at 0 °C for 2 hours. 3-Fluoro-2-nitropyridine (3.6 g, 25.3 mmol) was added and the reaction mixture was stirred at room temperature for an additional 8 hours before quenching with hydrochloric acid (3 N, 5 mL). After adjusting the pH to 3-4 with hydrochloric acid (3 N, 20 mL), the resulting mixture was extracted with ethyl acetate (3 x 100 mL). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by reversed phase chromatography with a RP-C18 column (acetonitrile/water, 1/2) to afford the title compound (3.2 g, 39%) as a light yellow oil. LC-MS (Method C): m/z = 272.1 [M+H-(t-BuO)]+, 1.269 min.
Step 2: Preparation oƒ (2S)-3-((2-aminopyridin-3-yl)oxy)-2-(((tert-butoxy)carbonyl)amino)propanoic acid
[0536] (2S)-2-(((tert-butoxy)carbonyl)amino)-3-((2-nitropyridin-3-yl)oxy)propanoic acid (0.45 g, 1.4 mmol) in methanol (20 mL) was aged overnight at room temperature in the presence of palladium on carbon (10%, 0.5 g) under hydrogen atmosphere (2-3 atm). The reaction mixture was filtered through Celite and the filtrate was concentrated under reduced pressure to afford the title compound (0.32 g, 78%) as a yellow oil. LC-MS (Method C): m/z = 298.1 [M+H]+, 0.982 min.
Step 3: Preparation oƒ tert-butyl N-((3S)-4-oxo-2H,3H,4H,5H-pyrido[3,2-b][1,4]oxazepin-3-yl)carbamate
[0537] N,N,N’,N’-tetramethyl-O-(7-azabenzotriazol-1-yl)uronium hexafluorophospate (0.73 g, 1.92 mmol) and N,N-diisopropylethylamine (0.25 g, 1.93 mmol) were added to a stirring solution of (2S)-3-((2-aminopyridin-3-yl)oxy)-2-(((tert-butoxy)carbonyl)amino)propanoic acid (0.45 g, 1.51 mmol) in N,N-dimethylformamide (5 mL). After stirring for 6 hours at room temperature, the reaction mixture was quenched by the addition of water (20 mL), and extracted with ethyl acetate (3 x 100 mL). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. The resulting residue was purified by column chromatography (methanol/dichloromethane, 1/10) to afford the title compound (0.11 g, 26%) as a white solid. LC-MS (Method C): m/z = 280.1 [M+H]+, 1.248 min.
tep 4: Preparation oƒ tert-butylN-((3S)-5-methyl-4-oxo-2H,3H,4H,5H-pyrido[3,2-b][1,4]oxazepin-3-yl)carbamate
[0538] Iodomethane (50 mg, 0.35 mmol) was added dropwise to a stirring solution of tert-butyl N-((3S)-4-oxo-2H,3H,4H,5H-pyrido[3,2-b][1,4]oxazepin-3-yl)carbamate (100 mg, 0.36 mmol) and cesium carbonate (120 mg, 0.36 mmol) in N,N-dimethylformamide (5 mL). After stirring for 3 hours at room temperature, the reaction mixture was diluted with water (20 mL), and extracted with ethyl acetate (3 x 100 mL). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (methanol/dichloromethane, 1/10) to afford the title compound (90 mg, 86%) as a white solid. LC-MS (Method C): m/z = 294.1 [M+H]+, 1.333 min.
Step 5: Preparation oƒ (3S)-3-amino-5-methyl-2H,3H,4H,5H-pyrido-[3,2-b][1,4]oxazepin-4-one hydrochloride
[0539] tert-butyl N-((3S)-5-methyl-4-oxo-2H,3H,4H,5H-pyrido[3,2-b][1,4]oxazepin-3-yl)carbamate (90 mg, 0.31 mmol) was added to a solution of hydrogen chloride in dioxane (4 M, 10 mL). The reaction mixture was stirred for 3 hours at room temperature and concentrated under reduced pressure to afford the title compound (65 mg, 93%) as a white solid, which was used directly in the next step without further purification. LC-MS (Method C): m/z = 194.1 [M+H]+, 0.847 min.
Step 6: Preparation oƒ (S)-5-benzyl-N-(5-methyl-4-oxo-2,3,4,5-tetrahydropyrido[3,2-b][1,4]oxazepin-3-yl)-4H-1,2,4-triazole-3-carboxamide
[0540] A solution of (3S)-3-amino-5-methyl-2H,3H,4H,5H-pyrido-[3,2-b][1,4]oxazepin-4-one hydrochloride (55 mg, 0.24 mmol) in N,N-dimethylformamide (1 mL) was added to a stirring solution of 5-benzyl-2H-1,2,4-triazole-3-carboxylic acid (80 mg, 0.40 mmol), 1-hydroxy-benzotrizole (70 mg, 0.53 mmol), N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (100 mg, 0.52 mmol) and N,N-
diisopropylethylamine (160 mg, 1.21 mmol) in N,N-dimethylformamide (2 mL). After stirring for 8 hours at room temperature, the reaction mixture was quenched by the addition of water (20 mL), and extracted with ethyl acetate (3 x 50 mL). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The resulting residue was purified by Prep-HPLC with the following conditions: Column, XBridge Shield RP18 OBD Column, 5 μm, 19 x 150 mm; mobile phase, water (0.1% formic acid) and ACN (30.0% ACN to 60.0% over 7 min); Detector, UV 254 & 220 nm to afford the title compound. 1H NMR (300 MHz, DMSO-d6) δ 14.45 (s, 1H), 8.67 (d, J= 7.2 Hz, 1H), 8.37 (dd, J= 4.8, 1.8 Hz, 1H), 7.71 (dd, J= 7.8, 1.5 Hz, 1H), 7.37-7.21 (m, 6H), 4.92-4.82 (m, 1H), 4.73 (dd, J= 11.4, 9.6 Hz, 1H), 4.53 (dd, J= 9.6, 7.5 Hz, 1H), 4.14 (s, 2H), 3.37 (s, 3H). LC-MS (Method D): m/z = 379.1 [M+H]+, 1.611 min.
PATENT
WO2023182512
WO2023137035
WO2022208262
WO2021211919
WO2021209740
WO2021205298
WO2021205296
, WO2017136727
PAPER
European Journal of Medicinal Chemistry (2021), 220, 113484
Structure-based bioisosterism design of thio-benzoxazepinones as novel necroptosis inhibitors
Publication Name: European Journal of Medicinal Chemistry
Publication Date: 2021-08-05
PMID: 33930803
DOI: 10.1016/j.ejmech.2021.113484
PATENT
WO2021203011
- [1]. Anthony A. ESTRADA, et al. Compounds, compositions and methods. WO2017136727A2.[2]. Darwish I, et al., Rip1k inhibitors. WO2021203011
//////////Eclitasertib, DNL-758, SAR-443122, DNL 758, SAR 443122, UNII-975AT1P9J6, Phase 2, Ulcerative colitis
Perfluorhexyloctane



WeightAverage: 432.269
Monoisotopic: 432.112266666Chemical FormulaC14H17F13
Perfluorhexyloctane
- 133331-77-8
- MIEBO
- Tetradecane, 1,1,1,2,2,3,3,4,4,5,5,6,6-tridecafluoro-
- 7VYX4ELWQM
- NOV03, NOV 03
- 1-(perfluorohexyl)octane
- F6H8
- NOV03
- Perfluorohexyloctane
1,1,1,2,2,3,3,4,4,5,5,6,6-tridecafluorotetradecane
FDA APPROVED 8/16/2023, Sohonos, To reduce the volume of new heterotopic ossification in adults and pediatric patients (aged 8 years and older for females and 10 years and older for males) with fibrodysplasia ossificans progressiva
Drug Trials Snapshot
Perfluorohexyloctane is a fluoroalkane that is tetradecane in which all of the hydrogen atoms at positions 1, 2, 3, 4, 5, and 6 have been replaced by fluorine atoms. It is an ophthalmic solution used to treat the signs and symptoms of dry eye disease. It has a role as an ophthalmology drug and a nonionic surfactant. It is a fluorohydrocarbon and a fluoroalkane. It derives from a hydride of a tetradecane.
Perfluorohexyloctane (branded as Evotears, Miebo,[a] and Novatears, among others) is a medication used for the treatment of dry eye disease.[4] It is a semifluorinated alkane.[4]
Perfluorohexyloctane has been available in multiple markets since 2015 under the brand names Evotears and Novatears,[5] and was additionally approved for medical use in the United States in May 2023 under the brand name Miebo.[4][6] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[7]
PATENT
Show 102550100 entries
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US11357738 | No | 2022-06-14 | 2036-09-29 | |
| US10058615 | No | 2018-08-28 | 2033-09-12 | |
| US10369117 | No | 2019-08-06 | 2033-09-12 | |
| US10449164 | No | 2019-10-22 | 2033-09-12 | |
| US10507132 | No | 2019-12-17 | 2037-06-21 | |
| US10576154 | No | 2020-03-03 | 2033-09-12 | |
SYN
https://www.scientificupdate.com/process-chemistry-articles/not-a-dry-eye-in-the-house



Medical uses
Perfluorohexyloctane is indicated for the treatment of the signs and symptoms of dry eye disease.[4][8][9]
Availability
Perfluorohexyloctane is sold as an over-the-counter medication under the brand names Evotears and Novatears in multiple countries,[10] costing around NZ$34.00, A$30, and €30 for a one-month supply.
In the US, perfluorohexyloctane is sold under the brand name Miebo; a prescription is required.
Notes and references
- ^ “Notice: Multiple additions to the Prescription Drug List (PDL) [2024-10-18]”. Health Canada. 18 October 2024. Retrieved 25 October 2024.
- ^ “Miebo product information”. Health Canada. 4 September 2024. Retrieved 27 December 2024.
- ^ “Regulatory Decision Summary for Miebo”. Drug and Health Products Portal. 4 September 2024. Retrieved 27 December 2024.
- ^ Jump up to:a b c d e “Miebo- perfluorohexyloctane solution”. DailyMed. 18 May 2023. Retrieved 8 June 2023.
- ^ “URSAPHARM GmbH and Novaliq GmbH Announce European Partnership Agreement” (Press release). Retrieved 15 February 2024.
- ^ “Bausch + Lomb and Novaliq Announce FDA Approval of Miebo (Perfluorohexyloctane Ophthalmic Solution) for the Treatment of the Signs and Symptoms of Dry Eye Disease” (Press release). Bausch + Lomb Corporation. 18 May 2023. Retrieved 8 June 2023 – via Business Wire.
- ^ New Drug Therapy Approvals 2023 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original on 10 January 2024. Retrieved 9 January 2024.
- ^ Ballesteros-Sánchez A, De-Hita-Cantalejo C, Sánchez-González MC, Jansone-Langine Z, de Sotomayor MA, Culig J, et al. (October 2023). “Perfluorohexyloctane in dry eye disease: A systematic review of its efficacy and safety as a novel therapeutic agent”. The Ocular Surface. 30: 254–262. doi:10.1016/j.jtos.2023.10.001. hdl:11441/151762. PMID 37813152. S2CID 263802332.
- ^ Sheppard JD, Evans DG, Protzko EE (November 2023). “A review of the first anti-evaporative prescription treatment for dry eye disease: perfluorohexyloctane ophthalmic solution”. The American Journal of Managed Care. 29 (14 Suppl): S251 – S259. doi:10.37765/ajmc.2023.89464. PMID 37930231. S2CID 265032840.
- ^ “In Australia, NovaTears Eye Drops Are Available on the Pharmaceutical Benefits Scheme (PBS) from Now On” (Press release). Retrieved 15 February 2024.
Further reading
- Azhar A, Taimuri MA, Oduoye MO, Sumbal A, Sheikh A, Iqbal A, et al. (September 2024). “MEIBO (perfluorohexyloctane): a novel approach to treating dry eye disease”. Annals of Medicine and Surgery (2012). 86 (9): 5292–5298. doi:10.1097/MS9.0000000000002322. PMC 11374244. PMID 39239035.
| Clinical data | |
|---|---|
| Trade names | Evotears Miebo (/ˈmaɪboʊ/ MY-bow) Novatears |
| Other names | NOV03; 1-(perfluorohexyl)octane |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a623054 |
| License data | US DailyMed: Perfluorohexyloctane |
| Routes of administration | Eye drops |
| ATC code | None |
| Legal status | |
| Legal status | CA: ℞-only[1][2][3]US: ℞-only[4] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 133331-77-8 |
| PubChem CID | 10477896 |
| DrugBank | DB17823 |
| ChemSpider | 8653305 |
| UNII | 7VYX4ELWQM |
| KEGG | D12604 |
| ChEBI | CHEBI:229658 |
| CompTox Dashboard (EPA) | DTXSID20440585 |
| Chemical and physical data | |
| Formula | C14H17F13 |
| Molar mass | 432.269 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////////Perfluorhexyloctane, Sohonos, APPROVALS 2023, FDA 2023, NOV03, NOV 03, MIEBO, 1-perfluorohexyl)octane, F6H8, NOV03, Perfluorohexyloctane
DIMDAZENIL


DIMDAZENIL
CAS 308239-86-3
WeightAverage: 372.81
Monoisotopic: 372.1101515
Chemical FormulaC17H17ClN6O2
EVT-201, 308239-86-3, EVT201, 6J8AF7CLE4, EVT 201
7-Chloro-3-[5-[(dimethylamino)methyl]-1,2,4-oxadiazol-3-yl]-4,5-dihydro-5-methyl-6H-imidazo[1,5-a]
[1,4]benzodiazepin-6-one
7-chloro-3-[5-[(dimethylamino)methyl]-1,2,4-oxadiazol-3-yl]-5-methyl-4H-imidazo[1,5-a][1,4]benzodiazepin-6-one
EVT 201 is a novel partial positive allosteric modulator of the GABAA receptor complex which is being developed as a treatment for insomnia. It is being developed by Evotec Inc.
- OriginatorRoche
- DeveloperEvotec SE; Zhejiang Jingxin Pharmaceutical
- ClassBenzodiazepines; Chlorobenzenes; Dimethylamines; Imidazoles; Ketones; Oxadiazoles; Sleep disorder therapies; Small molecules
- Mechanism of ActionGABA A receptor modulators
- RegisteredInsomnia
- 29 Nov 2023Registered for Insomnia in China (PO) – First global approval
- 24 Oct 2023Efficacy and adverse events data from a phase III trial in Insomnia released by Zhejiang Jingxin Pharmaceutical
- 21 Oct 2023Efficacy and adverse events data from a phase II trial in Insomnia released by Zhejiang Jingxin Pharmaceutical
Dimdazenil, sold under the brand name Junoenil, is a medication used in the treatment of insomnia in China.[1] It is a benzodiazepine derivative and a partial positive allosteric modulator of the GABAA receptor[2] with two- to four-fold higher functional affinity for the α1 subunit relative to the α2, α3, and α5 subunits.
Medical use
Dimdazenil shows effectiveness in the treatment of insomnia, but has less intrinsic activity in comparison to currently-marketed benzodiazepines and the Z-drugs;[3] however, it is thought that the lower efficacy may result in fewer side effects, such as motor incoordination.[3] In China, dimdazenil is approved for short-term treatment of insomnia.[4]
History
Dimdazenil was originally developed by Roche, based on preclinical data, as a non-sedating anxiolytic, but was found to produce sedation in humans in phase I clinical trials. For this reason, it was subsequently licensed to Evotec, which is now developing it for the treatment of insomnia.[3] By 2007, dimdazenil completed phase II clinical trials for this indication, with positive findings reported.[5] In China, the drug was developed by Zhejiang Jingxin Pharmaceutical.
SCHEME

PATENT
CN111620834
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN306317338&_cid=P10-MAWAJX-84923-1
| Example 16 |
| |
| 1M lithium bis(trimethylsilyl)amide (320 mL, 0.32 mol, 3 eq, 1 Mol/liter) was added to the flask, nitrogen was passed through, the temperature was lowered to -15°C, and the compound K1 (22.6 g, 0.11 mol, 1 eq) obtained in Example 11 was added dropwise. After the addition, the mixture was kept warm at -15°C to -5°C for 2 hours. After the addition, the compound b (26 g, 0.11 mol, 1 eq) obtained by the method of Example 15 was added dropwise. The mixture was kept warm at -15°C to -5°C for 2 hours. After the addition, the mixture was naturally heated to room temperature, and glacial acetic acid was slowly added dropwise. The temperature was controlled to be below 35°C. After completion, the temperature was raised to 55-60°C, and the reaction was kept warm for 2 hours. Then, the mixture was transferred to a rotary evaporator, and the mixture was concentrated under reduced pressure at 45-50°C in batches. The temperature was lowered to 25-30°C, and water and dichloromethane were added in batches. The layers were stirred and separated, and the organic layer was collected. The aqueous layer was extracted once more with dichloromethane, and the organic layers were combined. The layers were washed with a saturated aqueous solution of sodium bicarbonate and water. After washing, the organic layers were collected and transferred to a rotary evaporator for concentration to obtain a solid. The solid was slurried with ethanol at -15°C to -5°C for 15 minutes, filtered, rinsed with cold ethanol, and dried under reduced pressure at 55-60°C to obtain a compound of formula I (36 g, 96.6%), MS: M ++ 1=373.1, HPLC purity 99.85%. |
| 1 H-NMR data: 1 H NMR (400 MHz, DMSO-d 6 δ8.57(s,1H),7.69(d,J=1.9Hz,3H),4.60(d,J=3.7Hz,2H),3.61(s,2H),3.05(s,3H),2.16(s,6H). |
| 13 C-NMR data: 13 C NMR (101 MHz, DMSO) δ 163.35, 163.25, 161.50, 138.88, 134.17, 133.15, 132.81, 130.95, 128.29, 122.67, 114.56, 110.52, 61.10, 46.6 (2), 41.77, 34.48. |
PATENT
WO2000069858
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2000069858&_cid=P10-MAWAOS-90001-1


EXAMPLE
a) 6-Chloro-3,4-dihydro-4-methyl-2H-l,4-benzodiazepine-2,5(lH)-dione (III).
25.0 g 6-chloro-isatoic anhydride (II) and 12.4 g sarcosine were suspended under stirring and argon atmosphere in 100 ml p-xylene and heated at reflux for two hours. The suspension was cooled to room temperature and further stirred 1 hour, then filtered off. The precipitate was washed with 25 ml p-xylene twice and dried at 50°C under vacuum. The solid so obtained (6-chloro-3,4-dihydro-4-methyl-2H-l,4-benzodiazepine-2,5( lH)-dione (II)) was digested in 75 ml deionized water at 0°C for 1 hour, filtered off, washed with 25 ml deionized water and dried under vacuum 18 hours at 80°C. Crude product: 25.2 g as a beige powder, m.p. 230-232°C
b) Ethyl 7-chloro-5,6-dihydro-5-methyl-6-oxo-4H- imidazo[ l,5-a] [ 1,4] benzodiazepine- 3-carboxylate (V).
25.0 g 6-Chloro-3,4-dihydro-4-methyl-2H-l,4-benzodiazepine-2,5( lH)-dione (III) were suspended under stirring and argon atmosphere in 200 ml toluene and 32.1 ml N,N-dimethyl-p-toluidine. The suspension was heated to 100°C and 11.2 ml phosphorus oxychloride were added over 30 minutes and stirring was pursued two and an half hours at 100°C. The dark-orange solution was cooled to 40°C and toluene was removed under reduced pressure to give 82 g of a dark-orange oil.
Meanwhile, 81.2 ml hexamethyldisilazane and 265 ml tetrahydrofuran were mixed and cooled to -35°C. 229.5 ml Butyllithium were added over 45 minutes and, after stirring 30 minutes at -35°C, a solution of 35.2 g ethyl(dimethylamino-methylenamino)acetate in 70.4 ml tetrahydrofuran was added over 30 minutes. The orange solution obtained was stirred one more hour at -35°C and a solution of the crude iminochloride in 100 ml
tetrahydrofuran was added over 1 hour at -15°C. The dark red solution was stirred one hour at -15°C, then 18 hours at room temperature (r.t.). 75 ml Acetic acid were added in 10 minutes, then 75 ml deionized water were added in one portion and the orange suspension was heated at reflux for two hours. Tetrahydrofuran was removed under reduced pressure and the residue was partitioned between 200 ml dichloromethane and 100 ml deionized water. The phases were separated and the organic phase was washed with 100 ml aqueous HC1 IN twice and with 100 ml deionized water. The aqueous phases were extracted twice with 100 ml dichloromethane. The combined organic extracts were dried (Na2S04) and evaporated. The residue was digested in 200 ml n-heptane 30 minutes at r.t. and filtered off. The sticky crystals obtained were digested at reflux for 30 minutes in 213.5 ml ethanol, then stirred 3 hours to r.t. and 2 hours at -20°C. The precipitate (ethyl 7-chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[ l,5-a] [ l,4]benzodiazepine-3-carboxylate (V)) was filtered off, washed three times with 20 ml ethanol and dried under reduced pressure 16 hours at 60°C. Crude product: 23.4 g as a beige powder, m.p. 225.5-226.5 °C c) 7-Chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo [ 1 ,5-a] [ 1 ,4]benzodiazepine-3- carboxamide (VI).
22.8 g Ethyl 7-chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[ l,5-a] [l,4]- benzodiazepine-3-carboxylate (V)were suspended under stirring and argon atmosphere in 91.2 ml 1 ,4-dioxane. 14.1 ml Formamide and 13.9 ml sodium methanolate were successively added to yield a clear light-orange solution, which turned to a white suspension after 10 minutes. This suspension was stirred two hours at 30°C. 200 ml deionized water were added in one portion and 1,4-dioxane was distilled off at 40°C under reduced pressure. The remaining white suspension was stirred two hours at 0°C and filtered. The precipitate (7-chloro-5,6-dihydro-5-methyl-6-oxo-4H- imidazo[ l,5-a] [ l,4]benzodiazepine-3-carboxamide (VI)) was washed with 50 ml deionized water three times and dried under reduced pressure for 18 hours at 80°C. Crude product: 19.43 g as a white powder. m.p.>250°C
d) 7-Chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[l,5-a] [l,4]benzodiazepine-3- carbonitrile (VII).
19.0 g 7-Chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[ l,5-a] [ l,4]benzodiazepine-3- carboxamide (VI) were suspended under stirring and argon atmosphere in 95 ml 1,4- dioxane and 6.58 phosphorous oxychloride were added in one portion. The reaction mixture was heated to reflux for one hour giving a yellow solution, which was concentrated at 50°C under reduced pressure. The residue was digested in 100 ml deionized water for two hours at r.t.. The precipitate (7-chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[l,5- a] [ l ,4]benzodiazepine-3-carbonitrile (VII)) was filtered off, washed three times with 30 ml deionized water and dried under vacuum at 80°C for 18 hours. Crude product: 17.3 g as a light yellow powder, m.p. 238.5-239.5°C
_ e) 7-Chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[l,5-a] [l,4]benzodiazepine-3- carboxamidoxime (VIII).
16.8 g 7-Chloro-5,6-dihydro-5-methyl-6-oxo-4H-imidazo[ l,5-a] [ l,4]benzodiazepine-3- carbonitrile (VIII) were suspended under stirring and argon atmosphere in 101 ml N,N- dimethylformamide and 13.48 g hydroxylamine hydrochloride was added in one portion. 34.2 ml Sodium methanolate were then added over 60 minutes to the yellow suspension, which turned to a colorless suspension. It was stirred one more hour at r.t., then cooled to 0-2°C and 202 ml deionized water were added over 30 minutes. After stirring one more hour at 0°C, the precipitate (7-chloro-5,6-dihydro-5-methyl-6-oxo-4H- imidazo[l,5-a] [l,4]benzodiazepine-3-carboxamidoxime (VIII) was filtered off, washed twice with 40 ml deionized water and dried under vacuum at 70°C for 18 hours Crude product 17.84 g as a white powder m.p.>250°C
f) 7-Chloro-3- (5-chloromethyl- [ 1 ,2,4] oxadiazol-3-yl)-5-methyl-4,5-dihydro- imidazo [ 1 ,5-a] [ 1 ,4] benzodiazepin-6-one (IX).
8.0 g 7-chloro-5,6-dιhydro-5-methyl-6-oxo-4H-ιmιdazo[ 1,5-a] [ l,4]benzodιazepιne-3-carboxamidoxime (VIII) and 1.0 g magnesium oxide were suspended under stirring and argon atmosphere in 160 ml 1,4-dioxane. 2 7 ml Chloracetyl chloride were added in one portion and the white thick gel obtained was stirred 4 hours at r.t. and then 17 hours at reflux to give a lightly orange fluid suspension 100 ml Dioxane were distilled off and the reaction mixture was cooled to room temperature. 180 ml Deionized water were added within 15 minutes and the suspension was stirred 1 hour at r.t . The precipitate was filtered off, washed with 50 ml deionized water twice and dried under vacuum at 80°C for 18 hours Crude product: 8.3 g as a light pink powder. This crude product was dissolved in 120 ml tetrahydrofuran at reflux and 0.83 g active charcoal Darco G 60 were added. The system was refluxed 1 hour, then filtered on 25 g Dicaht-Speedex and the filter cake was washed with three portions of 50 ml warm tetrahydrofuran. The filtrate was concentrated at 40°C under reduced pressure The residue was digested in 80 ml ethanol 1 hour at reflux, then stirred 16 hours at r.t. and finally 2 hours at 2°C. The precipitate (7-chloro-3-(5-chloromethyl- [ l,2,4]oxadιazol-3-yl)-5-methyl-4,5-dιhydro-ιmιdazo [ 1,5-a] [ l,4]benzo-dιazepιn-6-one (IX)) was filtered off, washed with 2 portions of 25 ml cold tert-butyl ethvl- ether and dried under vacuum 5 hours at 80°C Crude product: 7.6 g as a light beige powder, m p. 234-238°C
g) 7-Chloro-3-(5-dimethylaminomethyl-[l,2,4]oxadiazol-3-yl)-5-methyl-4,5- dιhydro-imidazo[l,5-a] [l,4]benzodιazepin-6-one (I).
7.0 g 7-Chloro-3-(5-chloromethyl- [ l,2,4]oxadιazol-3-yl)-5-methyl-4,5-dιhydro-ιmιdazo-[ 1,5-a] [ l,4]benzodιazepιn-6-one (IX) were suspended under stirring and argon
atmosphere in 70 ml 1,4-dioxane and 25.7 ml dimethylamine (33% in ethanol) were added over 60 minutes The reaction mixture was stirred one more hour at r.t. and then the solvents were removed under reduced pressure at 35°C. The residue was partitioned between 50 ml dichloromethane and 20 ml deionized water. The phases were separated and the organic phase was washed twice with 20 ml deionized water. The aqueous phases were extracted separately with the same portion of 25 ml dichloromethane, twice. The combined organic extracts were dried (Na2SO4) and the solvent was removed under reduced pressure Crude product: 8.0 g as a light yellow foam Purification
The crude product was dissolved in 40 ml ethanol at reflux and 400 mg active charcoal Darco G 60 were added. The system was stirred 1 hour at reflux, then filtered on a hot pad of Dicalit Speedex, which was washed with two portions of 40 ml hot ethanol. The filtrate was concentrated to 14 g under reduced pressure, heated to reflux and at this temperature and 40 ml terf-butyl-methylether were added over 5 minutes. The suspension was cooled slowly to r.t., stirred 16 hours, further cooled to 2°C. After stirring 1 hour at 2°C, the precipitate was filtered off, washed with 20 ml tert-butyl-methylether and dried 1 hour at 60°C under vacuum. The so obtained powder was dissolved at reflux in 26 ml ethyl acetate. 6.5 ml Ethyl acetate were then distilled off and the turbid solution obtained was slowly cooled to r.t., then to 0°C. After 1 hour stirring at 0°C, the precipitate was filtered off, washed with 10 ml cold tert-butyl-methylether and dried under vacuum at 60°C for 16 hours. The so obtained powder (7-chloro-3-(5-dimethylaminomethyl-[ 1,2,4] oxadiazol-3-yl)-5-methyl-4,5-dihydro-imidazo[ 1,5-a] [l,4]benzodiazepin-6-one (I)) was crystallized a second time in 24.3 ml ethyl acetate according to the procedure described above. Product: 5.5 g as a white powder, m.p. 151.5-153°C
7-Chloro-3-(5-dimethylaminomethyl-[l,2,4]oxadiazol-3-yl)-5-methyl-4,5-dihydro-imidazo [ 1 ,5-a] [ 1 ,4] benzodiazepin-6-one maleate (1:1)
373 mg 7-Chloro-3-(5-dimethylaminomethyl-[ l,2,4]oxadiazol-3-yl)-5-methyl-4,5-dihydro-imidazo[ 1,5-a] [ l,4]benzodiazepin-6-one (I) and 116 mg maleic acid were dissloved in 3 ml hot ethanol. The salt crystalized on cooling. The suspension was stirred for 10 min at 0°C. Filtration and drying afforded 460 mg 7-Chloro-3-(5-dimethylaminomethyl-[ l,2,4]oxadiazol-3-yl)-5-methyl-4,5-dihydro-imidazo[l,5-a] [ l,4]benzodiazepin-6-one maleate (1:1) as a white solid, m.p. 182-184°C
References
- ^ Huang Z, Zhan S, Chen C, Zhang R, Zhou Y, He J, et al. (February 2024). “Efficacy and safety of Dimdazenil in adults with insomnia disorder: results from a multicenter, randomized, double-blind, placebo-controlled phase III trials”. Sleep. 47 (2). doi:10.1093/sleep/zsad272. PMC 10851846. PMID 37875349.
- ^ Guilleminault C (2010). Sleep Medicine. Elsevier Health Sciences. pp. 574–. ISBN 978-1-4377-1836-2.
- ^ Jump up to:a b c Monti JM, Pandi-Perumal SR, Möhler H (28 September 2010). GABA and Sleep: Molecular, Functional and Clinical Aspects. Springer Science & Business Media. pp. 50–51. ISBN 978-3-0346-0226-6.
- ^ Syed YY (March 2024). “Dimdazenil: First Approval”. Drugs. doi:10.1007/s40265-024-02020-9. PMID 38546956.
- ^ Plunkett JW (September 2007). Plunkett’s Biotech & Genetics Industry Almanac 2008: Biotech & Genetics Industry Market Research, Statistics, Trends & Leading Companies. Plunkett Research, Ltd. pp. 311–. ISBN 978-1-59392-087-6.
External links
| Clinical data | |
|---|---|
| Trade names | Junoenil |
| Other names | EVT-201; EVT201 |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 308239-86-3 |
| PubChem CID | 9885841 |
| DrugBank | DB05721 |
| ChemSpider | 8061514 |
| UNII | 6J8AF7CLE4 |
| ChEMBL | ChEMBL5095096 |
| CompTox Dashboard (EPA) | DTXSID301032055 |
| Chemical and physical data | |
| Formula | C17H17ClN6O2 |
| Molar mass | 372.81 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
//////////DIMDAZENIL, EVT-201, 308239-86-3, EVT201, 6J8AF7CLE4, EVT 201, CHINA 2023, INSOMNIA
Digadoglucitol




Digadoglucitol
DA-52534
CAS 2098944-37-5
μ-[2,2′,2”,2”’,2””,2””’-({[(2S,3R,4R,5R)-2,3,4,5,6- pentahydroxyhexyl]azanediyl}bis{[2-(hydroxy-κO)propane3,1-diyl]-1,4,7,10-tetraazacyclododecane-10,1,4,7-tetraylκ4 N1 ,N4 ,N7 ,N10})hexa(acetato-κO)]digadolinium diagnostic agent
C40H69Gd2N9O19
MW 1,294.536
F2Q2ZU6CAU
- Digadoglucitol free acid
- USL2PB6YFS
- 1-[Bis[2-hydroxy-3-[4,7,10-tris(carboxymethyl)-1,4,7,10-tetraazacyclododec-1-yl]propyl]amino]-1-deoxy-D-glucitol
- 2098944-28-4 FREE ACID
- D-Glucitol, 1-[bis[2-hydroxy-3-[4,7,10-tris(carboxymethyl)-1,4,7,10-tetraazacyclododec-1-yl]propyl]amino]-1-deoxy-

SCHEME

PATENT
Bracco Imaging SpA
WO2017098044
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017098044&_cid=P12-MAUH6W-49653-1



PATENT
WO2023006722
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023006722&_cid=P12-MAUHGF-56290-1
PATENT
WO2022023240
.///////////Digadoglucitol, F2Q2ZU6CAU, X RAY CONTRAST AGENT, DA-52534, DA 52534
Tofersen

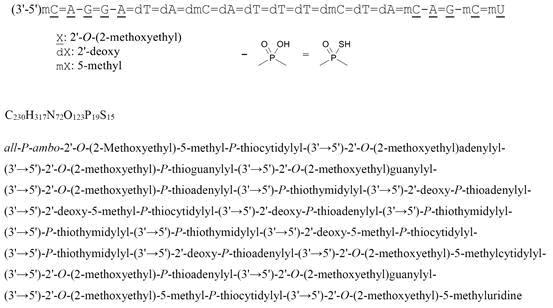
all-P-ambo-2′-O-(2-Methoxyethyl)-5-methyl-P-thiocytidylyl-(3’→5′)-2′-O-(2-methoxyethyl)adenylyl-(3’→5′)-2′-O-(2-methoxyethyl)-P-thioguanylyl-(3’→5′)-2′-O-(2-methoxyethyl)guanylyl-(3’→5′)-2′-O-(2-methoxyethyl)-P-thioadenylyl-(3’→5′)-P-thiothymidylyl-(3’→5′)-2′-deoxy-P-thioadenylyl-(3’→5′)-2′-deoxy-5-methyl-P-thiocytidylyl-(3’→5′)-2′-deoxy-P-thioadenylyl-(3’→5′)-P-thiothymidylyl-(3’→5′)-P-thiothymidylyl-(3’→5′)-P-thiothymidylyl-(3’→5′)-2′-deoxy-5-methyl-P-thiocytidylyl-(3’→5′)-P-thiothymidylyl-(3’→5′)-2′-deoxy-P-thioadenylyl-(3’→5′)-2′-O-(2-methoxyethyl)-5-methylcytidylyl-(3’→5′)-2′-O-(2-methoxyethyl)-P-thioadenylyl-(3’→5′)-2′-O-(2-methoxyethyl)guanylyl-(3’→5′)-2′-O-(2-methoxyethyl)-5-methyl-P-thiocytidylyl-(3’→5′)-2′-O-(2-methoxyethyl)-5-methyluridine
C230H317N72O123P19S15 : 7127.86
[2088232-70-4]

Tofersen
CAS 2088232-70-4
FDA APPROVED 4/25/2023, Qalsody
- BIIB 067
- BIIB067
- Formula
C230H317N72O123P19S15
Molar mass
7127.85 g·mol−1
- Antisense Oligonucleotide Inhibitor Of The Expression Of Superoxide Dismutase 1 Gene
- DNA, D((2′-O-(2-METHOXYETHYL))M5RC-SP-(2′-O-(2-METHOXYETHYL))RA-(2′-O-(2-METHOXYETHYL))RG-SP-(2′-O-(2-METHOXYETHYL))RG-(2′-O-(2-METHOXYETHYL))RA-SP-T-SP-A-SP-M5C-SP-A-SP-T-SP-T-SP-T-SP-M5C-SP-T-SP-A-SP-(2′-O-(2-METHOXYETHYL))M5RC-(2′-O-(2-METHOXYETHYL))R
- IONIS SOD1Rx
To treat amyotrophic lateral sclerosis in adults who have a SOD1 gene mutation
Drug Trials Snapshot
A nucleic acid-based drug indicated for the treatment of a specific type of amyotrophic lateral sclerosis.
Tofersen, sold under the brand name Qalsody, is a medication used for the treatment of amyotrophic lateral sclerosis (ALS).[3] Tofersen is an antisense oligonucleotide that targets the production of superoxide dismutase 1, an enzyme whose mutant form is commonly associated with amyotrophic lateral sclerosis. It is administered as an intrathecal injection.[3]
The most common side effects include fatigue, arthralgia (joint pain), increased cerebrospinal (brain and spinal cord) fluid white blood cells, and myalgia (muscle pain).[3]
Tofersen was approved for medical use in the United States in April 2023,[3][6] and in the European Union in May 2024.[4] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[7]
| Clinical data | |
|---|---|
| Trade names | Qalsody |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a623024 |
| License data | US DailyMed: Tofersen |
| Routes of administration | Intrathecal |
| ATC code | N07XX22 (WHO) |
| Legal status | |
| Legal status | CA: ℞-only[1]US: ℞-only[2][3]EU: Rx-only[4][5] |
| Identifiers | |
| CAS Number | 2088232-70-4 |
| DrugBank | DB14782 |
| UNII | 2NU6F9601K |
| KEGG | D11811 |
| Chemical and physical data | |
| Formula | C230H317N72O123P19S15 |
| Molar mass | 7127.85 g·mol−1 |
References
- ^ “Register of Innovative Drugs”. Health Canada. 3 November 2006. Retrieved 17 April 2025.
- ^ “Qalsody- tofersen injection”. DailyMed. 25 April 2023. Archived from the original on 8 May 2023. Retrieved 10 June 2023.
- ^ Jump up to:a b c d e f g h i j k l “FDA approves treatment of amyotrophic lateral sclerosis associated with a mutation in the SOD1 gene” (Press release). U.S. Food and Drug Administration (FDA). 25 April 2023. Archived from the original on 25 April 2023. Retrieved 25 April 2023.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d “Qalsody EPAR”. European Medicines Agency (EMA). 22 February 2024. Retrieved 24 February 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “Qalsody PI”. Union Register of medicinal products. 3 June 2024. Retrieved 7 September 2024.
- ^ “FDA Grants Accelerated Approval for Qalsody (tofersen) for SOD1-ALS, a Major Scientific Advancement as the First Treatment to Target a Genetic Cause of ALS” (Press release). Biogen. 25 April 2023. Archived from the original on 25 April 2023. Retrieved 25 April 2023 – via GlobeNewswire.
- ^ Jump up to:a b New Drug Therapy Approvals 2023 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original on 10 January 2024. Retrieved 9 January 2024.
- ^ Liu A (1 May 2019). “Biogen’s antisense ALS drug shows promise in early clinical trial”. FierceBiotech. Archived from the original on 2 February 2023. Retrieved 25 April 2023.
- ^ Langreth R (22 March 2023). “Biogen’s ALS Drug Gets Partial Backing From FDA Panel”. Bloomberg News. Retrieved 25 April 2023.
- ^ “FDA approves drug which helps to slow progression of rare form of MND”. http://www.sheffield.ac.uk. 28 April 2023. Retrieved 16 May 2024.
- ^ Berdyński M, Miszta P, Safranow K, Andersen PM, Morita M, Filipek S, et al. (January 2022). “SOD1 mutations associated with amyotrophic lateral sclerosis analysis of variant severity”. Scientific Reports. 12 (1): 103. Bibcode:2022NatSR..12..103B. doi:10.1038/s41598-021-03891-8. PMC 8742055. PMID 34996976.
- ^ Constantino A (25 April 2023). “FDA grants accelerated approval for Biogen ALS drug that treats rare form of the disease”. CNBC. Archived from the original on 25 April 2023. Retrieved 25 April 2023.
- ^ Constantino A (22 March 2023). “FDA advisors vote against effectiveness of Biogen’s ALS drug for rare and aggressive form of the disease”. CNBC. Archived from the original on 10 April 2023. Retrieved 25 April 2023.
- ^ Robins R (25 April 2023). “F.D.A. Approves Drug for Rare Form of A.L.S.” The New York Times. Archived from the original on 25 April 2023. Retrieved 25 April 2023.
- ^ “New treatment for rare motor neuron disease recommended for approval”. European Medicines Agency (EMA) (Press release). 23 February 2024. Retrieved 24 February 2024.
////////////tofersen, Qalsody, FDA 2023, APPROVALS 2023, EU 2024, EMA 2024, BIIB 067, BIIB067, IONIS SOD1Rx
Deupsilocin



Deupsilocin, Psilocin-d10
Psilocin-D10- Deupsilocin
- Psilocine-d10
| Molecular Formula | C12H16N2O |
| Molecular Weight | 214.3299 |
CAS 1435934-64-7
3-[2-[Di(methyl-d3)amino]ethyl-1,1,2,2–d4]-1H-indol-4-ol
3-[2-[bis(trideuteriomethyl)amino]-1,1,2,2-tetradeuterioethyl]-1H-indol-4-ol
| 1H-Indol-4-ol, 3-[2-[di(methyl-d3)amino]ethyl-1,1,2,2-d4]- |
Many mental health disorders, as well as neurological disorders, are impacted by alterations, dysfunction, degeneration, and/or damage to the brain’s serotonergic system, which may explain, in part, common endophenotypes and comorbidities among neuropsychiatric and neurological diseases. Many therapeutic agents that modulate serotonergic function are commercially available, including serotonin reuptake inhibitors, selective serotonin reuptake inhibitors, antidepressants, monoamine oxidase inhibitors, and, while primarily developed for depressive disorders, many of these therapeutics are used across multiple medical indications including, but not limited to, depression in Alzheimer’s disease and other neurodegenerative disease, chronic pain, existential pain, bipolar disorder, obsessive compulsive disorder, anxiety disorders and smoking cessation. However, in many cases, the marketed drugs show limited benefit compared to placebo, can take six weeks to work and for some patients, and are associated with several side effects including trouble sleeping, drowsiness, fatigue, weakness, changes in blood pressure, memory problems, digestive problems, weight gain and sexual problems.
The field of psychedelic neuroscience has witnessed a recent renaissance following decades of restricted research due to their legal status. Psychedelics are one of the oldest classes of psychopharmacological agents known to man and cannot be fully understood without reference to various fields of research, including anthropology, ethnopharmacology, psychiatry, psychology, sociology, and others. Psychedelics (serotonergic hallucinogens) are powerful psychoactive substances that alter perception and mood and affect numerous cognitive processes. They are generally considered physiologically safe and do not lead to dependence or addiction. Their origin predates written history, and they were employed by early cultures in many sociocultural and ritual contexts. After the virtually contemporaneous discovery of (5R,8R)-(+)-lysergic acid-N,N-diethylamide (LSD) and the identification of serotonin in the brain, early research focused intensively on the possibility that LSD and other psychedelics had a serotonergic basis for their action. Today there is a consensus that psychedelics are agonists or partial agonists at brain serotonin 5-hydroxytryptamine 2 A (5-HT2A) receptors, with particular importance on those expressed on apical dendrites of neocortical pyramidal cells in layer V, but also may bind with lower affinity to other receptors such as the sigma-1 receptor. Several useful rodent models have been developed over the years to help unravel the neurochemical correlates of serotonin 5-HT2A receptor activation in the brain, and a variety of imaging techniques have been employed to identify key brain areas that are directly affected by psychedelics.
Psychedelics have both rapid onset and persisting effects long after their acute effects, which includes changes in mood and brain function. Long lasting effects may result from their unique receptor affinities, which affect neurotransmission via neuromodulatory systems that serve to modulate brain activity, i.e., neuroplasticity, and promote cell survival, are neuroprotective, and modulate brain neuroimmune systems. The mechanisms which lead to these long-term neuromodulatory changes are linked to epigenetic modifications, gene expression changes and modulation of pre- and post-synaptic receptor densities. These, previously under-researched, psychedelic drugs may potentially provide the next-generation of neurotherapeutics, where treatment resistant psychiatric and neurological diseases, e.g., depression, post-traumatic stress disorder, dementia and addiction, may become treatable with attenuated pharmacological risk profiles.
Although there is a general perception that psychedelic drugs are dangerous, from a physiologic safety standpoint, they are one of the safest known classes of CNS drugs. They do not cause addiction, and no overdose deaths have occurred after ingestion of typical doses of classical psychotic agents, such as LSD, psilocybin, or mescaline (Scheme 1). Preliminary data show that psychedelic administration in humans results in a unique profile of effects and potential adverse reactions that need to be appropriately addressed to maximize safety. The primary safety concerns are largely psychologic, rather than physiologic, in nature. Somatic effects vary but are relatively insignificant, even at doses that elicit powerful psychologic effects. Psilocybin, when administered in a controlled setting, has frequently been reported to cause transient, delayed headache, with incidence, duration, and severity increased in a dose-related manner [Johnson et al., Drug Alcohol Depend, 2012, 123 (1-3):132-140]. It has been found that repeated administration of psychedelics leads to a very rapid development of tolerance known as tachyphylaxis, a phenomenon believed to be mediated, in part, by 5-HT2A receptors. In fact, several studies have shown that rapid tolerance to psychedelics correlates with downregulation of 5-HT2A receptors. For example, daily LSD administration selectively decreased 5-HT2 receptor density in the rat brain [Buckholtz et al., Eur. J. Pharmacol., 1990, 109:421-425. 1985; Buckholtz et al., Life Sci. 1985, 42:2439-2445].
SCHEME

PATENT
Mindset Pharma Inc., US11591353
https://patentscope.wipo.int/search/en/detail.jsf?docId=US376433397&_cid=P10-MARMO8-36145-1
PATENT
WO2021155470
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021155470&_cid=P10-MARMST-39096-1
PATENT
Cybin IRL Limited, WO2023247665
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023247665&_cid=P10-MARMVV-41020-1
PATENT
WO2023078604
WO2022195011
| Classic psychedelics and dissociative psychedelics are known to have rapid onset antidepressant and anti-addictive effects, unlike any currently available treatment. Randomized clinical control studies have confirmed antidepressant and anxiolytic effects of classic psychedelics in humans. Ketamine also has well established antidepressant and anti-addictive effects in humans mainly through its action as an NMDA antagonist. Ibogaine has demonstrated potent anti-addictive potential in pre-clinical studies and is in the early stages of clinical trials to determine efficacy in robust human studies [Barsuglia et al., Prog Brain Res, 2018, 242:121-158; Corkery, Prog Brain Res, 2018, 242:217-257]. |
/////////Deupsilocin, Psilocin-d10, KXD3HS8D6X, Psilocin-D10, Deupsilocin, Psilocine-d10
Demannose


Demannose
CAS 530-26-7,
3458-28-4
180.16 g/mol
- D-Mannopyranose
- Carubinose
- Seminose
- mannopyranose
- (3S,4S,5S,6R)-6-(hydroxymethyl)oxane-2,3,4,5-tetrol
- C6H12O6
D-mannopyranose congenital glycosylation disorders
D-mannopyranose is d-Mannose in its six-membered ring form. It has a role as a metabolite. It is a D-aldohexose, a D-mannose and a mannopyranose.
SCHEME

LIT
Tetrahedron Letters (1987), 28(31), 3569-72
///////////Demannose, D-Mannopyranose, Carubinose, Seminose, mannopyranose
Leniolisib



Leniolisib
CAS 1354690-24-6
WeightAverage: 450.466
Monoisotopic: 450.199108558
Chemical FormulaC21H25F3N6O2
CDZ-173-NX- CDZ173
- CDZ173-NX
1-[(3S)-3-({6-[6-methoxy-5-(trifluoromethyl)pyridin-3-yl]-5H,6H,7H,8H-pyrido[4,3-d]pyrimidin-4-yl}amino)pyrrolidin-1-yl]propan-1-one
FDA APPROVED Joenja, 3/24/2023, To treat activated phosphoinositide 3-kinase delta syndrome
Drug Trials Snapshot
Leniolisib (INN[3][4]), sold under the brand name Joenja, is a medication used for the treatment of activated phosphoinositide 3-kinase delta syndrome (APDS).[2][5] It is a kinase inhibitor[2][6] that is taken by mouth.[2]
The most common side effects include headache, sinusitis, and atopic dermatitis.[5]
Leniolisib was approved for medical use in the United States in March 2023.[5][7][8] It is the first approved medication for the treatment of activated PI3K delta syndrome.[5] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[9]
PATENT
https://patents.google.com/patent/US8653092B2/en
PATENT
https://patentscope.wipo.int/search/en/WO2012004299
Example 67 was prepared according the general procedure described in scheme 4
Example 67: 1 -{(S)-3-[6-(6-Methoxy-5-trifluoromethyl-pyridin-3-yl)-5,6,7,8-tetrahydro-pyrido[4,3-d]pyrimidin-4-ylamino]-pyrrolidin-1-yl}-propan-1-one
To a solution of (S)-3-[6-(6-methoxy-5-trifluoromethyl-pyridin-3-yl)-5,6,7,8-tetrahydro-pyrido[4,3-d]pyrimidin-4-ylamino]-pyrrolidine-1 -carboxylic acid tert-butyl ester (intermediate 24) (13.4 g, 27.1 mmol) in CH2CI2 (100 mL), was added TFA (41 .8 mL) and the mixture stirred at rt for 1 h. Concentrated in vacuo and partitioned between 2M NaOH(aq) (300 mL) and CH2CI2 (200 mL). The organic phase was separated and the aqueous phase extracted with CH2CI2 (2 x 200 mL). The organic phases were combined, dried (MgS04) and
evaporated in vacuo to give a brown foam. The foam was dissolved in CH2CI2 (50 mL) and was added simultaneously portionwise with sat.NaHC03(aq) (50 mL) to a vigourously stirring solution of propionyl chloride (2.63 g, 28.5 mmol) in CH2CI2 (50 mL) at rt. The resulting biphasic mixture was stirred at rt for 1 h. Further propionyl chloride (0.566g, 6.12 mmol) was added and continued stirring vigorously for 20 min. The organic layer was separated and the aqueous layer extracted with CH2CI2 (100 mL). The organic layers were combined, dried (MgS04) and concentrated in vacuo to give a brown gum. The gum was stirred in EtOAc (100 mL) and the resulting solid filtered (9.4 g). The mother liquors were concentrated in vacuo and purified by column chromatography through a Biotage® amino silica gel eluting with EtOAc / MeOH, 100/0 to 90/10 to give a yellow foam which was then stirred in EtOAc (20 mL) and the resulting solid filtered (870 mg). Both batches of solids were combined and stirred in refluxing EtOAc (50 mL) for 1 h. Filtered to give 1-{(S)-3-[6-(6-methoxy-5-trifluoromethyl-pyridin-3-yl)-5,6,7,8-tetrahydro-pyrido[4,3-d]pyrimidin-4-ylamino]-pyrrolidin-1 -yl}-propan-1 -one as a colourless solid (9.42 g, 76% yield). 1 H NMR (400 MHz, DMSO-d6, 298K) δ ppm 0.95-1.05 (m, 3H) 1 .87-2.32 (m, 4H) 2.77-2.86 (m, 2H) 3.25-3.88 (m, 6H) 3.93 (s, 3H) 3.98 (s, 2H) 4.55-4.80 (m, 1 H) 6.70-6.80 (m, 1 H, N-H) 7.86-7.92 (m, 1 H) 8.27-8.33 (m, 1 H) 8.33-8.37 (m, 1 H) LCMS: [M+H]+=451.0, Rt (6)= 1.49 min.
Alternative synthesis for example 67
A solution of (S)-3-[6-(6-methoxy-5-trifluoromethyl-pyridin-3-yl)-5,6,7,8-tetrahydro-pyrido[4,3-d]pyrimidin-4-ylamino]-pyrrolidine-1-carboxylic acid tert-butyl ester (intermediate 24) (29.04 g, 58.73 mmol) in 2-Me-THF (100 mL) was dropwise added into aqueous HCI solution (150 mL, 31 %) over 15 min. The reaction mixture was partitioned between water (300 mL) and isopropyl acetate (100 mL) and the upper organic phase was discarded. The aqueous phase was partitioned between 25% NaOH (aq) (200 g) and 2-Me-THF (200 mL), and the organic phase was collected and dried. Triethylamine (16.32 mL, 1 17.48 mmol) was added into the organic phase followed by dropwise addition of propionyl chloride (6.0 g, 64.6 mmol) at 0 °C. The resulting mixture was stirred at 0 °C for 1 h. The reaction mixture was washed with water (1 10 mL) and the resulting organic phase was concentrated in vacuo to give a brown gum.
The residue was recrystallized with isopropanol and methyl tert-butyl ether to give 1 -{(S)-3- [6-(6-methoxy-5-trifluoromethyl-pyridin-3-yl)-5,6,7,8-tetrahydro-pyrido[4,3-d]pyrimidin-4- ylamino]-pyrrolidin-1-yl}-propan-1 -one as a colourless solid (17.2 g, 65% yield).
Crystallization of Example 67 by heating in acetonitrile/water
2.0 g of Example 67 (4.440 mol) were dissolved in 10 mL of acetonitrile and 0.5 mL of water at 75°C. The solution was allowed to cool down to rt within 30 min resulting in a suspension. The mixture was stirred for 16 h at rt. The crystals were collected by filtration. The filter cake was washed 2 times with 1 mL of acetonitrile and afterwards dried for 16 h at 24°C and ca. 10 mbar vacuum. Elementary analysis of the material showed a waterless form.
PAPER
https://pubs.acs.org/doi/10.1021/acsmedchemlett.7b00293
ACS Medicinal Chemistry Letters
Cite this: ACS Med. Chem. Lett. 2017, 8, 9, 975–980
https://doi.org/10.1021/acsmedchemlett.7b00293
The predominant expression of phosphoinositide 3-kinase δ (PI3Kδ) in leukocytes and its critical role in B and T cell functions led to the hypothesis that selective inhibitors of this isoform would have potential as therapeutics for the treatment of allergic and inflammatory disease. Targeting specifically PI3Kδ should avoid potential side effects associated with the ubiquitously expressed PI3Kα and β isoforms. We disclose how morphing the heterocyclic core of previously discovered 4,6-diaryl quinazolines to a significantly less lipophilic 5,6,7,8-tetrahydropyrido[4,3-d]pyrimidine, followed by replacement of one of the phenyl groups with a pyrrolidine-3-amine, led to a compound series with an optimal on-target profile and good ADME properties. A final lipophilicity adjustment led to the discovery of CDZ173 (leniolisib), a potent PI3Kδ selective inhibitor with suitable properties and efficacy for clinical development as an anti-inflammatory therapeutic. In vitro, CDZ173 inhibits a large spectrum of immune cell functions, as demonstrated in B and T cells, neutrophils, monocytes, basophils, plasmocytoid dendritic cells, and mast cells. In vivo, CDZ173 inhibits B cell activation in rats and monkeys in a concentration- and time-dependent manner. After prophylactic or therapeutic dosing, CDZ173 potently inhibited antigen-specific antibody production and reduced disease symptoms in a rat collagen-induced arthritis model. Structurally, CDZ173 differs significantly from the first generation of PI3Kδ and PI3Kγδ-selective clinical compounds. Therefore, CDZ173 could differentiate by a more favorable safety profile. CDZ173 is currently in clinical studies in patients suffering from primary Sjögren’s syndrome and in APDS/PASLI, a disease caused by gain-of-function mutations of PI3Kδ.
Synthesis and full characterization of (S)-1-(3-((6-(6-methoxy-5-(trifluoromethyl)pyridin-3-yl)-
5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-yl)amino)pyrrolidin-1-yl)propan-1-one (3h, CDZ173,
leniolisib)
TFA (41.8 mL) was added to a solution of (S)-3-[6-(6-methoxy-5-trifluoromethyl-pyridin-3-yl)-5,6,7,8-
tetrahydro-pyrido[4,3-d]pyrimidin-4-ylamino]-pyrrolidine-1-carboxylic acid tert-butyl ester (13.4 g,
27.1 mmol) in CH2Cl2 (100 mL), and the mixture was stirred at RT for 1 h. After that time, the mixture
was concentrated under reduced pressure, and the residue was partitioned between NaOH (aqu., 2M,
300 mL) and CH2Cl2 (200 mL). The organic phase was separated, and the aqueous phase was extracted
with CH2Cl2 (2 x 200 mL). The combined organic phases were dried (MgSO4) and concentrated under
reduced pressure. The resulting brown foam was dissolved in CH2Cl2 (50 mL) and added simultaneously with a NaHCO3 solution (aqu., saturated) (50 mL) to a vigorously stirring solution of propionyl chloride (2.63 g, 28.5 mmol) in CH2Cl2 (50 mL) at RT. The resulting biphasic mixture was stirred at RT for
1h. Additional propionyl chloride (0.566 g, 6.12 mmol) was added, and vigorous stirring was continued
for 20 min. The organic layer was separated and the aqueous layer extracted with CH2Cl2 (100 mL). The
combined organic layers were dried (MgSO4) and concentrated under reduced pressure. The resulting
brown gum was stirred in EtOAc (100 mL) and the resulting solid was filtered (9.4 g). The mother liquors were concentrated under reduced pressure and purified by column chromatography through a Biotage®
amino silica gel eluting with EtOAc / MeOH, 100/0 to 90/10. After concentration under reduced
pressure, the resulting yellow foam was stirred in EtOAc (20 mL) and the resulting solid was filtered
(870 mg). Both batches of solids were combined and stirred in refluxing EtOAc (50 mL) for 1h. The
resulting solid was filtered to give the title compound as a colorless solid (9.42 g, 76%). 1H NMR (400
MHz, DMSO-d6, 298K, ca. 1:1 mixture of rotamers) δ ppm 8.35 (m, 1H) 8.30 (m, 1H) 7.89 (m, 1H)
6.80-6.70 (m, 1H, N-H) 4.80-4.55 (m, 1H) 3.93 (s, 3H) 3.98 (s, 2H) 3.88-3.25 (m, 6H) 2.86-2.75 (m,
2H) 2.32-1.87 (m, 4H) 1.05-0.95 (m, 3H); 13C-NMR (150 MHz, DMSO-d6, 298K, ca. 1:1 mixture of
rotamers, data given for cis-isomer): δ ppm 171.3, 158.6, 158.1, 155.5, 153.6, 141.3, 138.0, 125.7,
123.3, 111.1, 109.7, 53.7, 50.8, 49.4, 45.8, 45.8, 44.3, 31.2, 29.7, 26.6, 8.97; LCMS method 1: Rt 1.49
min, calcd for C21H26F3N6O2 [M+H]+
451.2, found 451.0, HRMS (ESI+) calcd for C21H26F3N6O2
[M+H]+ 451.20693, found 451.20642
REF

REF
https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2024.1337436/full

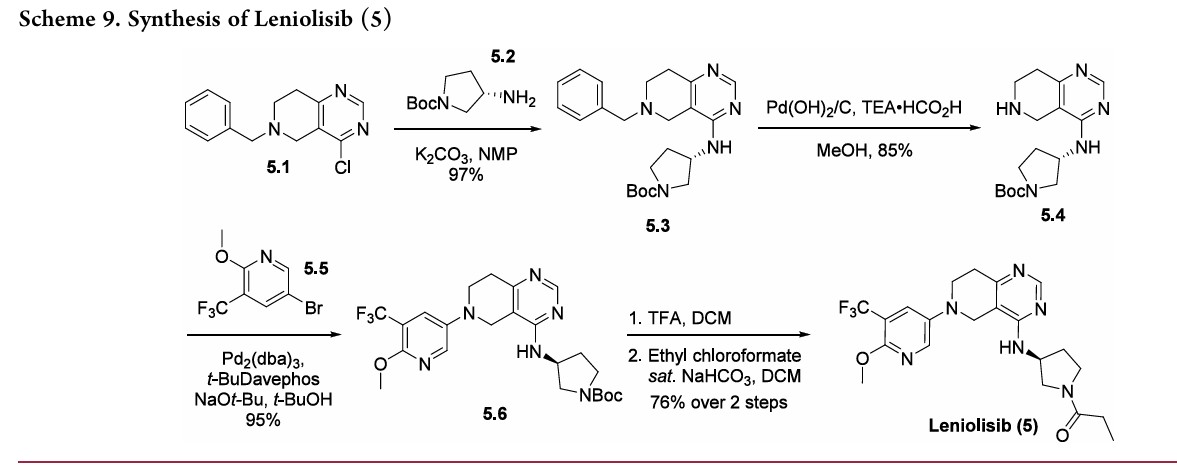
-Benzyl-4-chloro-5,6,7,8-tetrahydro-pyrido[4,3-d]pyrimidine (compound 1) is coupled with (S)-tert-butyl 3-aminopyrrolidine-1-carboxylate (compound 2) in the presence of triethylamine at 120 °C for 42 h to give compound 3 a 93% yield. The benzyl group is deprotected with 20% palladium hydroxide on carbon and ammonium formate in methanol at 65 °C for 2 h to give compound 4 a 66% yield. Compound 4 is coupled with 5-bromo-2-methoxy-3-(trifluoromethyl)pyridine (compound 5) in the presence of sodium –tert-butoxide, tris(dibenzylideneacetone)dipalladium(0), 2-di-t-butylphosphino-2′-(N,N-dimethylamino)biphenyl in tert-butanol at 100 °C for 5 h to give compound 6 a 74% yield. Deprotection of the Boc group in DCM/TFA, followed by coupling with propionyl chloride in the presence of sodium bicarbonate in DCM at room temperature for 1 h gives the final compound 7 (leniolisib) a 76% yield.
REF
https://www.sciencedirect.com/science/article/abs/pii/S0223523424000047

REF
J. Med. Chem. 2025, 68, 2147−2182
Leniolisib (Joenja). Leniolisib (5), is a twice-daily, orally available selective phosphoinositide 3-kinase-delta
(PI3Kδ) inhibitor developed by Novartis and in-licensed by Pharming Group NV for the treatment of activated phosphoinositide 3-kinase-delta syndrome (APDS). APDS is a primary immunodeficiency caused by mutations in PI3Kδ catalytic (PIK3CD) or regulatory (PIK3R1) subunits. The loss or gain of function of these subunits results in hyperactivity of the PI3Kδ pathway which can result in infections, lymphoprolif
eration, autoimmunity, increased risk of malignant lymphoma and early mortality. 44−46
Current treatment strategies include immunosuppressives such as corticosteroids, antiviral, and antibiotic therapies, stem cell transplantation, and immunoglobulin replacement therapy. However, none of these therapeutic strategies treats the underlying hyperactivity of the PI3Kδpathway. Thus, the approval of leniolisib by the USFDA in March 2023 provided a significant breakthrough therapy for patients 12 years and older.47 48
A concise synthetic route to leniolisib has been disclosed by Novartis,beginning with commercially available tetrahydropyridopyrimidine 5.1 (Scheme 9). An SNAr reaction with amine 5.2 furnished intermediate 5.3 in good yield. Transfer hydrogenation with Pd(OH)2 on carbon to remove the benzyl
group gave free amine 5.4, setting up the system for a Buchwald−Hartwig amination with bromide 5.5 to produce 5.6 in good yield. Protecting group removal and subsequent acylation with ethyl chloroformate provided leniolisib (5) in 76% yield over two steps.
(44) Hoegenauer, K.; Soldermann, N.; Stauffer, F.; Furet, P.;
Graveleau, N.; Smith, A. B.; Hebach, C.; Hollingworth, G. J.; Lewis,
I.; Gutmann, S.; et al. Discovery and pharmacological characterization
of novel quinazoline-based PI3K delta-selective inhibitors. ACS Med.
Chem. Lett. 2016, 7, 762−767.
(45) Hoegenauer, K.; Soldermann, N.; Zécri, F.; Strang, R. S.;
Graveleau, N.; Wolf, R. M.; Cooke, N. G.; Smith, A. B.; Hollingworth,
G. J.; Blanz, J.; et al. Discovery of CDZ173 (Leniolisib), representing a
structurally novel class of PI3K delta-selective inhibitors. ACS Med.
Chem. Lett. 2017, 8, 975−980.
(46) Duggan, S.; Al-Salama, Z. T. Leniolisib: first approval. Drugs
2023, 83, 943−948.
(47) Pharming announces US FDA approval of Joenja® (leniolisib) as
the first and only treatment indicated for APDS. Pharming, March 24, 2023 https://www.pharming.com/news/pharming-announces-us-fda
approval-joenja-leniolisib-first-and-only-treatment-indicated-apds (ac
cessed February 2024).
(48) Fernandes Gomes dos Santos, P. A.; Hogenauer, K.;
Hollingworth, G.; Soldermann, N.; Stowasser, F.; Tufilli, N.; Zecri, F.
Solid forms and salts of tetrahydro-pyrido-pyrimidine derivatives. WO
2013001445 A1, 2013
Ref
https://www.sciencedirect.com/science/article/abs/pii/S0223523424000047
Leniolisib developed by Novartis Pharma AG, was approved on March 24, 2023, making it the first treatment drug for APDS [3]. APDS is an immunodeficiency disorder that primarily occurs due to mutations in the gene responsible for encoding phosphotidylinsitol-3-kinase δ(PI3Kδ). These mutations enhance the function of PI3Kδ, resulting in impaired immune response and heightened vulnerability to infections.
Leniolisib is capable of inhibiting the hyperactive PI3Kδ enzyme by obstructing the active binding site within the p110δ subunit [54]. Inisolated enzyme assays conducted without cells, the selectivity of PI3K-δ
was found to be higher compared to PI3Kα(28-fold), PI3K-β (43-fold),and PI3K-γ (257-fold), as well as other enzymes in the kinome [54].Leniolisib demonstrated the ability to decrease phosphoinositide-3-kinase/protein kinase B (pAKT) pathway activity and suppress the growth and activation of B and T cell subsets in cell-based experiments. Leniolisib effectively blocks the signaling pathways responsible for the excessive production of phosphatidylinositol 3,4,5-trisphosphate (PIP3), overactivation of the downstream
mammalian target of rapamycin (mTOR)/protein kinase B (AKT) pathway, and the imbalanced functioning of B and T cells [55].
One representative approach of Leniolisib is depicted in Scheme 15 [55]. Pyrrolidine LENI-003 was obtained by nucleophilic substitution of aminopyrrolidine LENI-001 and the 4-Cl of LENI-002 under alkaline conditions, and LENI-004 was obtained by debenzylation of LENI-003 under palladium hydroxide/carbon. LENI-004 and 5-bromo-2-methox y-3-(trifluoromethyl)pyridine (LENI-005) were coupled to obtain LENI-006. LENI-006 was deprotected by TFA and further condensed with acyl chloride to obtain Leniolisib.
54] V.K. Rao, S. Webster, V. Dalm, A. ˇ Sediv´ a, P.M. van Hagen, S. Holland, S.
D. Rosenzweig, A.D. Christ, B. Sloth, M. Cabanski, A.D. Joshi, S. de Buck,
J. Doucet, D. Guerini, C. Kalis, I. Pylvaenaeinen, N. Soldermann, A. Kashyap,
G. Uzel, M.J. Lenardo, D.D. Patel, C.L. Lucas, C. Burkhart, Effective “activated
PI3Kδ syndrome”-targeted therapy with the PI3Kδ inhibitor leniolisib, Blood 130
(2017) 2307–2316.
[55] K. Hoegenauer, N. Soldermann, F. Z´ ecri, R.S. Strang, N. Graveleau, R.M. Wolf, N.
G. Cooke, A.B. Smith, G.J. Hollingworth, J. Blanz, S. Gutmann, G. Rummel,
A. Littlewood-Evans, C. Burkhart, Discovery of CDZ173 (Leniolisib), representing
a structurally novel class of PI3K delta-selective inhibitors, ACS Med. Chem. Lett.
8 (2017) 975–980.[55] K. Hoegenauer, N. Soldermann, F. Z´ ecri, R.S. Strang, N. Graveleau, R.M. Wolf, N.
G. Cooke, A.B. Smith, G.J. Hollingworth, J. Blanz, S. Gutmann, G. Rummel,
A. Littlewood-Evans, C. Burkhart, Discovery of CDZ173 (Leniolisib), representing
a structurally novel class of PI3K delta-selective inhibitors, ACS Med. Chem. Lett.
8 (2017) 975–980.

,
References
- ^ “Joenja (Ballia Holdings Pty Ltd)”. Therapeutic Goods Administration (TGA). 16 April 2025. Retrieved 3 May 2025.
- ^ Jump up to:a b c d e f “Joenja- leniolisib tablet, film coated”. DailyMed. 29 March 2023. Retrieved 20 June 2023.
- ^ World Health Organization (2016). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 76”. WHO Drug Information. 30 (3). hdl:10665/331020.
- ^ World Health Organization (2017). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 77”. WHO Drug Information. 31 (1). hdl:10665/330984.
- ^ Jump up to:a b c d e f g h i j “FDA approves first treatment for activated phosphoinositide 3-kinase delta syndrome”. U.S. Food and Drug Administration (FDA) (Press release). 24 March 2023. Retrieved 24 March 2023. This article incorporates text from this source, which is in the public domain.
- ^ Duggan S, Al-Salama ZT (July 2023). “Leniolisib: First Approval”. Drugs. 83 (10): 943–948. doi:10.1007/s40265-023-01895-4. PMID 37256490. S2CID 258989663.
- ^ Jump up to:a b “US FDA approves Pharming’s immune disorder drug”. Reuters. Archived from the original on 24 March 2023. Retrieved 24 March 2023.
- ^ “Pharming announces US FDA approval of Joenja (leniolisib) as the first and only treatment indicated for APDS” (PDF). Pharming Group N.V. (Press release). 24 March 2023. Retrieved 25 March 2023.
- ^ New Drug Therapy Approvals 2023 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original on 10 January 2024. Retrieved 9 January 2024.
![]() This article incorporates text from this source, which is in the public domain: Bing Chat output modified to create the initial revision of this article. 25 March 2023. – via Microsoft
This article incorporates text from this source, which is in the public domain: Bing Chat output modified to create the initial revision of this article. 25 March 2023. – via Microsoft
External links
Clinical trial number NCT02435173 for “Study of Efficacy of CDZ173 in Patients With APDS/PASLI” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Joenja |
| Other names | CDZ173 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a623016 |
| License data | US DailyMed: Leniolisib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | L03AX22 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)[1]US: ℞-only[2] |
| Identifiers | |
| CAS Number | 1354690-24-6as salt: 1354691-97-6 |
| DrugBank | DB16217 |
| ChemSpider | 52083264 |
| UNII | L22772Z9CP |
| KEGG | D11158as salt: D11159 |
| ChEMBL | ChEMBL3643413 |
| PDB ligand | 9NQ (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C21H25F3N6O2 |
| Molar mass | 450.466 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
//////////leniolisib, Joenja, FDA 2023, APPROVALS 2023, CDZ-173-NX, CDZ173, CDZ173-NX



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}