
Home » 2023
Yearly Archives: 2023
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Capivasertib
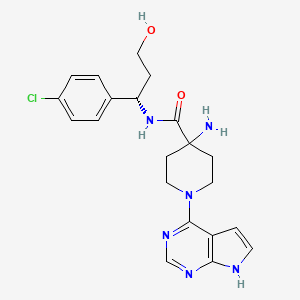
Capivasertib
C21H25ClN6O2
428.915
- 1143532-39-1
AZD 5363
4-amino-N-[(1S)-1-(4-chlorophenyl)-3-hydroxypropyl]-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide
(S)-4-AMINO-N-(1-(4-CHLOROPHENYL)-3-HYDROXYPROPYL)-1-(7H-PYRROLO[2,3-D]PYRIMIDIN-4-YL)PIPERIDINE-4-CARBOXAMIDE
FDA APPROVED 11/16/2023, To treat breast cancer that meets certain disease criteria, Truqap
Capivasertib, sold under the brand name Truqap, is an anti-cancer medication used for the treatment of breast cancer.[1][2]
The most common adverse reactions include diarrhea, cutaneous adverse reactions, increased random glucose, decreased lymphocytes, decreased hemoglobin, increased fasting glucose, nausea, fatigue, decreased leukocytes, increased triglycerides, decreased neutrophils, increased creatinine, vomiting, and stomatitis.[3]
In November 2023, capivasertib was approved in the United States for people with hormone receptor-positive, human epidermal growth factor receptor 2-negative breast cancer when used in combination with fulvestrant.[3][4][5]
Capivasertib is a novel pyrrolopyrimidine derivative, and an orally available inhibitor of the serine/threonine protein kinase AKT (protein kinase B) with potential antineoplastic activity. Capivasertib binds to and inhibits all AKT isoforms. Inhibition of AKT prevents the phosphorylation of AKT substrates that mediate cellular processes, such as cell division, apoptosis, and glucose and fatty acid metabolism. A wide range of solid and hematological malignancies show dysregulated PI3K/AKT/mTOR signaling due to mutations in multiple signaling components. By targeting AKT, the key node in the PIK3/AKT signaling network, this agent may be used as monotherapy or combination therapy for a variety of human cancers.
Medical uses
Capivasertib, used in combination with fulvestrant (Faslodex), is indicated for adults with hormone receptor-positive, human epidermal growth factor receptor 2-negative locally advanced or metastatic breast cancer with one or more PIK3CA/AKT1/PTEN-alterations, as detected by an FDA-approved test, following progression on at least one endocrine-based regimen in the metastatic setting or recurrence on or within twelve months of completing adjuvant therapy.[1][3]
History
Efficacy was evaluated in CAPItello-291 (NCT04305496), a randomized, double-blind, placebo-controlled, multicenter trial in 708 participants with locally advanced or metastatic HR-positive, HER2-negative breast cancer, of which 289 participants had tumors with PIK3CA/AKT1/PTEN-alterations.[3] All participants were required to have progression on aromatase inhibitor-based treatment.[3] Participants could have received up to two prior lines of endocrine therapy and up to one line of chemotherapy for locally advanced or metastatic disease.[3]
PATENT
EXAMPLE 9: (S)-4-AMINO-N-(1-(4-CHLOROPHENYL)-3-HYDROXYPROPYL)-1-(7H-PYRROLO[2,3-D]PYRIMIDIN-4-YL)PIPERIDINE-4-CARBOXAMIDE (E9)
EXAMPLE 9 ALTERNATIVE ROUTE 1: (S)-4-AMINO-N-(1-(4-CHLOROPHENYL)-3-HYDROXYPROPYL)-1-(7H-PYRROLO[2,3-D]PYRIMIDIN-4-YL)PIPERIDINE-4-CARBOXAMIDE
EXAMPLE 9 ALTERNATIVE ROUTE 2: (S)-4-AMINO-N-(1-(4-CHLOROPHENYL)-3-HYDROXYPROPYL)-1-(7H-PYRROLO[2,3-D]PYRIMIDIN-4-YL)PIPERIDINE-4-CARBOXAMIDE


AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
//////////
| Clinical data | |
|---|---|
| Trade names | Truqap |
| Other names | AZD-5363 |
| AHFS/Drugs.com | Truqap |
| License data | US DailyMed: Capivasertib |
| Routes of administration | By mouth |
| Drug class | Threonine kinase inhibitor |
| ATC code | L01EX27 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1143532-39-1 |
| PubChem CID | 25227436 |
| DrugBank | DB12218 |
| ChemSpider | 28189073 |
| UNII | WFR23M21IE |
| KEGG | D11371 |
| ChEMBL | ChEMBL2325741 |
| PDB ligand | 0XZ (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID40150710 |
| ECHA InfoCard | 100.208.066 |
| Chemical and physical data | |
| Formula | C21H25ClN6O2 |
| Molar mass | 428.92 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
- ^ Jump up to:a b c “Truqap- capivasertib tablet, film coated”. DailyMed. 16 November 2023. Archived from the original on 20 November 2023. Retrieved 20 November 2023.
- ^ Turner NC, Oliveira M, Howell SJ, Dalenc F, Cortes J, Gomez Moreno HL, et al. (June 2023). “Capivasertib in Hormone Receptor–Positive Advanced Breast Cancer”. New England Journal of Medicine. 388 (22): 2058–2070. doi:10.1056/NEJMoa2214131. PMID 37256976. S2CID 259002400.
- ^ Jump up to:a b c d e f “FDA approves capivasertib with fulvestrant for breast cancer”. U.S. Food and Drug Administration. 16 November 2023. Archived from the original on 17 November 2023. Retrieved 17 November 2023.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Oncology (Cancer) / Hematologic Malignancies Approval Notifications”. U.S. Food and Drug Administration. 16 November 2023. Archived from the original on 17 November 2023. Retrieved 17 November 2023.
- ^ “Truqap (capivasertib) plus Faslodex approved in the US for patients with advanced HR-positive breast cancer”. AstraZeneca (Press release). 17 November 2023. Archived from the original on 17 November 2023. Retrieved 17 November 2023.
External links
- Clinical trial number NCT04305496 for “Capivasertib+Fulvestrant vs Placebo+Fulvestrant as Treatment for Locally Advanced (Inoperable) or Metastatic HR+/HER2- Breast Cancer (CAPItello-291)” at ClinicalTrials.gov
///////Capivasertib, Truqap, FDA 2023, APPROVALS 2023, AZD 5363
NC1(CCN(CC1)C1=C2C=CNC2=NC=N1)C(=O)N[C@@H](CCO)C1=CC=C(Cl)C=C1
Eplontersen

Eplontersen
AKCEA-TTR-LRx
- ION-682884 FREE ACID
- ISIS-682884 FREE ACID
UNII0GRZ0F5XJ6
CAS number1637600-16-8


Eplontersen, FDA APP, 12/21/2023, To treat polyneuropathy of hereditary transthyretin-mediated amyloidosis, Wainua
AKCEA-TTR-LRx is under investigation in clinical trial NCT04136184 (Neuro-ttransform: A Study to Evaluate the Efficacy and Safety of Akcea-ttr-lrx in Participants With Hereditary Transthyretin-mediated Amyloid Polyneuropathy).
Eplontersen, sold under the brand name Wainua, is a medication used for the treatment of transthyretin-mediated amyloidosis.[1] It is a transthyretin-directed antisense oligonucleotide.[1] It was developed to treat hereditary transthyretin amyloidosis by Ionis Pharmaceuticals and AstraZeneca.[2][3][4][5]
It was approved for medical use in the United States in December 2023.[6][7][8]
Medical uses
Eplontersen is indicated for the treatment of the polyneuropathy of hereditary transthyretin-mediated amyloidosis in adults.[1]
Society and culture
Names
Eplontersen is the international nonproprietary name.[9]

//////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
//////////
| Clinical data | |
|---|---|
| Trade names | Wainua |
| Other names | AKCEA-TTR-LRx |
| AHFS/Drugs.com | Eplontersen |
| License data | US DailyMed: Eplontersen |
| Routes of administration | Subcutaneous |
| ATC code | N07XX21 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 1637600-16-8 |
| DrugBank | DB16199 |
| UNII | 0GRZ0F5XJ6 |
References
- ^ Jump up to:a b c d https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/217388s000lbl.pdf
- ^ “Ionis announces FDA acceptance of New Drug Application for eplontersen for the treatment of hereditary transthyretin-mediated amyloid polyneuropathy (ATTRv-PN)” (Press release). Ionis Pharmaceuticals. 7 March 2023. Archived from the original on 26 September 2023. Retrieved 21 December 2023 – via PR Newswire.
- ^ Coelho, Teresa; Waddington Cruz, Márcia; Chao, Chi-Chao; Parman, Yeşim; Wixner, Jonas; Weiler, Markus; et al. (February 2023). “Characteristics of Patients with Hereditary Transthyretin Amyloidosis-Polyneuropathy (ATTRv-PN) in NEURO-TTRansform, an Open-label Phase 3 Study of Eplontersen”. Neurology and Therapy. 12 (1): 267–287. doi:10.1007/s40120-022-00414-z. PMC 9837340. PMID 36525140.
- ^ Coelho, Teresa; Marques, Wilson; Dasgupta, Noel R.; Chao, Chi-Chao; Parman, Yeşim; França, Marcondes Cavalcante; et al. (October 2023). “Eplontersen for Hereditary Transthyretin Amyloidosis With Polyneuropathy”. The Journal of the American Medical Association. 330 (15): 1448–1458. doi:10.1001/jama.2023.18688. PMC 10540057. PMID 37768671.
- ^ Diep, John K.; Yu, Rosie Z.; Viney, Nicholas J.; Schneider, Eugene; Guo, Shuling; Henry, Scott; et al. (December 2022). “Population pharmacokinetic/pharmacodynamic modelling of eplontersen, an antisense oligonucleotide in development for transthyretin amyloidosis”. British Journal of Clinical Pharmacology. 88 (12): 5389–5398. doi:10.1111/bcp.15468. PMID 35869634. S2CID 250989659.
- ^ “Eplontersen: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 21 December 2023.
- ^ “Wainua (eplontersen) granted regulatory approval in the U.S. for the treatment of adults with polyneuropathy of hereditary transthyretin-mediated amyloidosis”. Ionis Pharmaceuticals, Inc. (Press release). 21 December 2023. Retrieved 22 December 2023.
- ^ “Wainua (eplontersen) granted first-ever regulatory approval in the US for the treatment of adults with polyneuropathy of hereditary transthyretin-mediated amyloidosis”. AstraZeneca US (Press release). 22 December 2023. Retrieved 22 December 2023.
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 85”. WHO Drug Information. 35 (1). hdl:10665/340684.
External links
- Clinical trial number NCT04136184 for “NEURO-TTRansform: A Study to Evaluate the Efficacy and Safety of Eplontersen (Formerly Known as ION-682884, IONIS-TTR-LRx and AKCEA-TTR-LRx) in Participants With Hereditary Transthyretin-Mediated Amyloid Polyneuropathy” at ClinicalTrials.gov
- Clinical trial number NCT01737398 for “Efficacy and Safety of Inotersen in Familial Amyloid Polyneuropathy” at ClinicalTrials.gov
///////////Eplontersen, Wainua, FDA 2023, APPROVALS 2023, ION-682884 FREE ACID, ISIS-682884 FREE ACID
Iptacopan


Iptacopan
1644670-37-0
422.525, C25H30N2O4
- 4-((2S,4S)-4-ethoxy-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl) benzoic acid
- BENZOIC ACID, 4-((2S,4S)-4-ETHOXY-1-((5-METHOXY-7-METHYL-1H-INDOL-4-YL)METHYL)-2-PIPERIDINYL)-
- Iptacopan
- LNP 023
- LNP-023
- LNP023
- NVP-LNP023
- NVP-LNP023-NX
Fda approved, To treat paroxysmal nocturnal hemoglobinuria, 12/5/2023, Fabhalta ‘CHINA 2024
Iptacopan is a small-molecule factor B inhibitor previously investigated as a potential treatment for the rare blood disease paroxysmal nocturnal hemoglobinuria (PNH) by inhibiting the complement factor B.1 Factor B is a positive regulator of the alternative complement pathway, where it activates C3 convertase and subsequently C5 convertase.2 This is of particular importance to PNH, where one of the disease hallmarks is the mutation of the PIGA gene. Due to this mutation, all progeny erythrocytes will lack the glycosyl phosphatidylinositol–anchored proteins that normally anchor 2 membrane proteins, CD55 and CD59, that protect blood cells against the alternative complement pathway.3 Additionally, iptacopan has the benefit of targeting factor B, which only affect the alternative complement pathway, leaving the classic and lectin pathway untouched for the body to still mount adequate immune responses against pathogens.2
On December 6th, 2023, Iptacopan under the brand name Fabhalta was approved by the FDA for the treatment of adults with PNH. This approval was based on favorable results obtained from the phase III APPL-PNH and APPOINT-PNH studies, where 82.3% and 77.5% of patients experienced a sustained hemoglobin improvement without transfusions respectively.5
Iptacopan , sold under the brand name Fabhalta, is a medication used for the treatment of paroxysmal nocturnal hemoglobinuria.[1] It is a complement factor B inhibitor that was developed by Novartis.[1] It is taken by mouth.[1]
Iptacopan was approved by the US Food and Drug Administration (FDA) for the treatment of adults with paroxysmal nocturnal hemoglobinuria in December 2023.[2][3]
Medical uses
Iptacopan is indicated for the treatment of adults with paroxysmal nocturnal hemoglobinuria.[1][4]
Side effects
The FDA label for iptacopan contains a black box warning for the risk of serious and life-threatening infections caused by encapsulated bacteria, including Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae type B.[1]
Research
In a clinical study with twelve participants, iptacopan as a single drug led to the normalization of hemolytic markers in most patients, and no serious adverse events occurred during the 12-week study.[5][6]
Iptacopan is also investigated as a drug in other complement-mediated diseases, like age-related macular degeneration and some types of glomerulopathies.[7]
PATENT
https://patents.google.com/patent/US9682968B2/en
Example-26Example-26a4-((2S,4S)-(4-ethoxy-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl))benzoic acid ((+) as TFA Salt)

A mixture of methyl 4-((2S,4S)-4-ethoxy-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl)benzoate, Intermediate 6-2b peak-1 (tr=1.9 min), (84 mg, 0.192 mmol) and LiOH in H2O (1 mL, 1 mmol) in THF (1 mL)/MeOH (2 mL) was stirred at room temperature for 16 h, and then concentrated. The resulting residue was purified by RP-HPLC (HC-A) to afford the title compound. Absolute stereochemistry was determined by comparison with enantiopure synthesis in Example-26c. 1H NMR (TFA salt, 400 MHz, D2O) δ 8.12 (d, J=8.19 Hz, 2H), 7.66 (br. d, J=8.20 Hz, 2H), 7.35 (d, J=3.06 Hz, 1H), 6.67 (s, 1H), 6.25 (d, J=3.06 Hz, 1H), 4.65 (dd, J=4.28, 11.49 Hz, 1H), 4.04 (d, J=13.00 Hz, 1H), 3.87-3.98 (m, 2H), 3.53-3.69 (m, 5H), 3.38-3.50 (m, 1H), 3.20-3.35 (m, 1H), 2.40 (s, 3H), 2.17-2.33 (m, 2H), 2.08 (br. d, J=15.70 Hz, 1H), 1.82-1.99 (m, 1H), 1.28 (t, J=7.03 Hz, 3H); HRMS calcd. for C26H31N2O3 (M+H)+ 423.2284, found 423.2263.
PATENT
Example 1
PAPER
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.9b01870
The alternative pathway (AP) of the complement system is a key contributor to the pathogenesis of several human diseases including age-related macular degeneration, paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic uremic syndrome (aHUS), and various glomerular diseases. The serine protease factor B (FB) is a key node in the AP and is integral to the formation of C3 and C5 convertase. Despite the prominent role of FB in the AP, selective orally bioavailable inhibitors, beyond our own efforts, have not been reported previously. Herein we describe in more detail our efforts to identify FB inhibitors by high-throughput screening (HTS) and leveraging insights from several X-ray cocrystal structures during optimization efforts. This work culminated in the discovery of LNP023 (41), which is currently being evaluated clinically in several diverse AP mediated indications.



a Reagents and conditions: (a) i PrMgCl·LiCl, Cbz-Cl, THF; (b) Zn, AcOH; (c) LiBH4, THF; (d) TBDPS-Cl, imidazole, DMF; (e) separation of diastereomers by flash chromatography; (f) TBAF, THF; (g) NaH, EtI, DMF; (h) Ba(OH)2, i PrOH, H2O; (i) K2CO3, MeI, DMF; (j) H2, Pd/C, MeOH; (k) (±)-50, DIPEA, DMA; (l) K2CO3, MeOH; then TMS-diazomethane, toluene, MeOH; (m) chiral SFC; (n) LiOH, H2O, MeOH, THF; (o) (2S,4S)-50, NaBH(OAc)3, DCE.
4-((2S,4S)-(4-Ethoxy-1-((5-methoxy-7-methyl-1H-indol-4- yl)methyl)piperidin-2-yl))benzoic Acid (41, LNP023). Step 1: tert-Butyl 4-(((2S,4S)-4-Ethoxy-2-(4-(methoxycarbonyl)phenyl)- piperidin-1-yl)methyl)-5-methoxy-7-methyl-1H-indole-1-carboxylate (58). To a solution of tert-butyl 4-formyl-5-methoxy-7-methyl1H-indole-1-carboxylate (57) (1.5 g, 5.18 mmol) and methyl 4- ((2S,4S)-4-ethoxypiperidin-2-yl)benzoate ((2S,4S)-50) (1.185 g, 4.50 mmol) in DCE (20 mL) was added NaBH(OAc)3 (3 g, 14.1 mmol), and this was stirred at rt for 21.5h. Additional tert-butyl 4-formyl-5- methoxy-7-methyl-1H-indole-1-carboxylate (57) (500 mg, 1.90 mmol) was added, and this was stirred for 20 h. The reaction was diluted with EtOAc, washed successively with 5% aqueous NaHCO3, H2O, and brine, dried over Na2SO4, filtered, and concentrated to provide the title compound (2.415 g, quant) which was used without further purification. MS (ESI+) m/z 537.4 (M + H). The absolutestereochemistry was ultimately determined via cocrystallization of 41 with the catalytic domain of FB. Step 2: 4-((2S,4S)-(4-Ethoxy-1-((5-methoxy-7-methyl-1H-indol-4- yl)methyl)piperidin-2-yl))benzoic Acid (41, LNP023). To a solution of tert-butyl 4-(((2S,4S)-4-ethoxy-2-(4-(methoxycarbonyl)phenyl)- piperidin-1-yl)methyl)-5-methoxy-7-methyl-1H-indole-1-carboxylate (58) (2.415 g, 4.50 mmol) in THF (10 mL) and MeOH (20 mL) was added 1 M LiOH in H2O (15 mL, 15 mmol), and this was stirred at 70 °C for 8 h. The reaction was cooled to rt, diluted with H2O, half saturated aqueous KHSO4 and citric acid, saturated with sodium chloride, then extracted with 9:1 DCM/TFE, dried with Na2SO4, filtered, and concentrated. RP-HPLC-B purification provided the title compound (730 mg, 38% for 2 steps). 1 H NMR (400 MHz, D2O) δ 7.96 (d, J = 8.0 Hz, 2H), 7.58 (d, J = 8.1 Hz, 2H), 7.30 (d, J = 3.2 Hz, 1H), 6.66 (s, 1H), 6.20 (s, 1H), 4.62−4.47 (m, 1H), 4.06 (d, J = 13.2 Hz, 1H), 3.97−3.76 (m, 2H), 3.66−3.48 (m, 5H), 3.43−3.29 (m, 1H), 3.26−3.15 (m, 1H), 2.35 (s, 3H), 2.31−2.11 (m, 2H), 2.00 (d, J = 15.4 Hz, 1H), 1.93−1.74 (m, 1H), 1.25−1.07 (m, 3H). HRMS calcd for C25H31N2O4 (M + H)+ 423.2284, found 423.2263. 4-((2S,4S)-(4-Ethoxy-1-((5-methoxy-7-methyl-1H-indol-4- yl)methyl)piperidin-2-yl))benzoic Acid Hydrochloride (41· HCl). To a solution of 41 (620 mg, 1.47 mmol) in H2O (10 mL) and acetonitrile (3 mL) was added 5 M aqueous HCl (0.5 mL, 2.5 mmol). The mixture was then lyophilized, and the resulting solid was suspended in i PrOH and heated to 70 °C. The mixture turned into a solution after 1.5 h and was then cooled to rt with stirring. After about 5 h, the mixture turned into a suspension and the solid was collected by filtration and dried under high vacuum at 50 °C to provide the title compound as the hydrochloride salt (450 mg, 65%). 1 H NMR (400 MHz, methanol-d4) δ 10.73 (s, 1H), 8.23 (d, J = 8.2 Hz, 2H), 7.74 (d, J = 8.3 Hz, 2H), 7.36−7.31 (m, 1H), 6.77 (s, 1H), 6.42−6.31 (m, 1H), 4.40−4.19 (m, 2H), 3.87−3.80 (m, 1H), 3.76 (s, 3H), 3.68− 3.50 (m, 4H), 3.45−3.38 (m, 1H), 2.51 (s, 3H), 2.30−2.18 (m, 2H), 2.13−1.89 (m, 2H), 1.31 (t, J = 7.0 Hz, 3H). MS (ESI+) m/z 423.3 (M + H).
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Iptacopan (Fabhalta®), a first-in-class oral therapeutic agent discovered by Novartis, specifically targets the complement Factor B protein within the alternative complement system. NMPA granted
marketing authorization in 2024, indicated for complement inhibitor-naïve adult patients diagnosed with paroxysmal nocturnal hemoglobinuria (PNH) [75]. By competitively binding to the catalytic domain of
Factor B, the drug effectively blocks C3 convertase assembly, thereby suppressing downstream cleavage of C3 into its active fragments. This dual inhibitory action addresses both intravascular erythrocyte
destruction and extravascular hemolytic processes characteristic of PNHpathogenesis [76]. Clinical validation emerged from the multinational APPOINT-PNH study (ClinicalTrials.gov identifier NCT04820530), where treatment-naïve participants exhibited sustained hemoglobin
stabilization (≥12 g/dL) in 79.6 % of cases, achieving transfusion in dependence over 24 weeks. Secondary endpoints revealed significant improvements in fatigue scores and health-related quality metrics [77]. Safety monitoring identified encapsulated bacterial infection as critical risks, necessitating mandatory vaccination ≥2 weeks pre-treatment. Common treatment-emergent adverse events comprised transient gastrointestinal disturbances (nausea 18.3 %, diarrhea 14.7 %) and mild
cephalgia (22.1 %), with resolution typically occurring within 4 weeks [78].
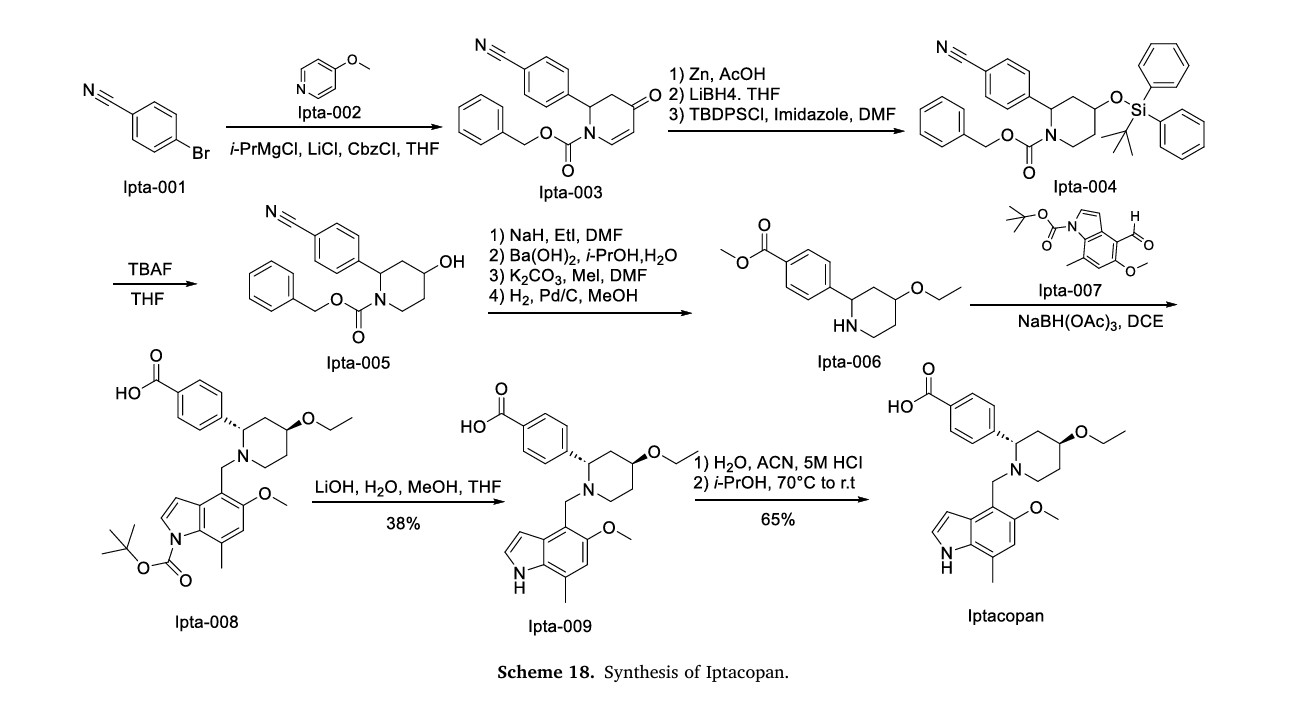
The synthetic pathway of Iptacopan, delineated in Scheme 18, initiates with nucleophilic substitution between Ipta-001 and Ipta-002, followed by Grignard coupling yielding Ipta-003 [79]. This intermedi
ate undergoes NaBH4-mediated reduction and TMSCl-induced silanization to afford Ipta-004. Acid-catalyzed TMS deprotection (HCl/MeOH) delivers Ipta-005, which progresses through sequential alkylation (methyl iodide/K2CO3 catalytic hydrogenation (H)/Pd–C), transesterification (EtONa), and to construct Ipta-006. Condensation with Ipta-007 and subsequent reduction forms Ipta-008. Strategic TFA-mediated Boc cleavage in DCM followed by HCl-induced salt formation in dioxane ultimately furnishes Iptacopan hydrochloride.
75-79
[75] Iptacopan, Drugs and Lactation Database (Lactmed®), National Institute of Child
Health and Human Development, Bethesda (MD), 2006.
[76] J.H. Jang, L. Wong, B.S. Ko, S.S. Yoon, K. Li, I. Baltcheva, P.K. Nidamarthy,
R. Chawla, G. Junge, E.S. Yap, Iptacopan monotherapy in patients with paroxysmal
nocturnal hemoglobinuria: a 2-cohort open-label proof-of-concept study, Blood
Adv 6 (2022) 4450–4460.
[77] A.M. Risitano, C. de Castro, B. Han, A.G. Kulasekararaj, J.P. Maciejewski,
P. Scheinberg, Y. Ueda, S. Vallow, G. Bermann, M. Dahlke, R. Kumar, R. Peffault de
Latour, Patient-reported improvements in patients with PNH treated with
iptacopan from two phase 3 studies, Blood Adv 9 (2025) 1816–1826.
[78] C.M. de Castro, B.J. Patel, Iptacopan for the treatment of paroxysmal nocturnal
hemoglobinuria, Expert Opin Pharmacother 25 (2024) 2331–2339.
[79] N. Mainolfi, T. Ehara, R.G. Karki, K. Anderson, A. Mac Sweeney, S.M. Liao, U.
A. Argikar, K. Jendza, C. Zhang, J. Powers, D.W. Klosowski, M. Crowley,
T. Kawanami, J. Ding, M. April, C. Forster, M. Serrano-Wu, M. Capparelli,
R. Ramqaj, C. Solovay, F. Cumin, T.M. Smith, L. Ferrara, W. Lee, D. Long,
M. Prentiss, A. De Erkenez, L. Yang, F. Liu, H. Sellner, F. Sirockin, E. Valeur,
P. Erbel, D. Ostermeier, P. Ramage, B. Gerhartz, A. Schubart, S. Flohr, N. Gradoux,
R. Feifel, B. Vogg, C. Wiesmann, J. Maibaum, J. Eder, R. Sedrani, R.A. Harrison,
M. Mogi, B.D. Jaffee, C.M. Adams, Discovery of 4-((2S,4S)-4-Ethoxy-1-((5-
methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl)benzoic acid (LNP023), a
factor B inhibitor specifically designed to be applicable to treating a diverse array
of complement mediated diseases, J. Med. Chem. 63 (2020) 5697–5722.
.
//////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
///////////
| Clinical data | |
|---|---|
| Trade names | Fabhalta |
| Other names | LNP023 |
| AHFS/Drugs.com | Fabhalta |
| License data | US DailyMed: Iptacopan |
| Routes of administration | By mouth |
| Drug class | Complement factor B inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 1644670-37-0 |
| PubChem CID | 90467622 |
| DrugBank | DB16200 |
| ChemSpider | 75533872 |
| UNII | 8E05T07Z6W |
| KEGG | D12251D12252 |
| ChEMBL | ChEMBL4594448 |
| PDB ligand | JGQ (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C25H30N2O4 |
| Molar mass | 422.525 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
- ^ Jump up to:a b c d e f “Fabhalta- iptacopan capsule”. DailyMed. 5 December 2023. Archived from the original on 10 December 2023. Retrieved 10 December 2023.
- ^ “Novartis receives FDA approval for Fabhalta (iptacopan), offering superior hemoglobin improvement in the absence of transfusions as the first oral monotherapy for adults with PNH”. Novartis (Press release). Archived from the original on 12 December 2023. Retrieved 6 December 2023.
- ^ “Novel Drug Approvals for 2023”. U.S. Food and Drug Administration (FDA). 6 December 2023. Archived from the original on 21 January 2023. Retrieved 10 December 2023.
- ^ https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2023/218276Orig1s000ltr.pdf Archived 10 December 2023 at the Wayback Machine This article incorporates text from this source, which is in the public domain.
- ^ Jang JH, Wong L, Ko BS, Yoon SS, Li K, Baltcheva I, et al. (August 2022). “Iptacopan monotherapy in patients with paroxysmal nocturnal hemoglobinuria: a 2-cohort open-label proof-of-concept study”. Blood Advances. 6 (15): 4450–4460. doi:10.1182/bloodadvances.2022006960. PMC 9636331. PMID 35561315.
- ^ “Novartis Phase III APPOINT-PNH trial shows investigational oral monotherapy iptacopan improves hemoglobin to near-normal levels, leading to transfusion independence in all treatment-naïve PNH patients”. Novartis (Press release). Archived from the original on 12 December 2023. Retrieved 6 September 2023.
- ^ Schubart A, Anderson K, Mainolfi N, Sellner H, Ehara T, Adams CM, et al. (April 2019). “Small-molecule factor B inhibitor for the treatment of complement-mediated diseases”. Proceedings of the National Academy of Sciences of the United States of America. 116 (16): 7926–7931. Bibcode:2019PNAS..116.7926S. doi:10.1073/pnas.1820892116. PMC 6475383. PMID 30926668.
External links
- Clinical trial number NCT04558918 for “Study of Efficacy and Safety of Twice Daily Oral LNP023 in Adult PNH Patients With Residual Anemia Despite Anti-C5 Antibody Treatment (APPLY-PNH)” at ClinicalTrials.gov
- Clinical trial number NCT04820530 for “Study of Efficacy and Safety of Twice Daily Oral Iptacopan (LNP023) in Adult PNH Patients Who Are Naive to Complement Inhibitor Therapy (APPOINT-PNH)” at ClinicalTrials.gov
///////Iptacopan, fda 2023, approvals, 2023, paroxysmal nocturnal hemoglobinuria, 12/5/2023, Fabhalta , LNP 023, LNP-023, LNP023, NVP-LNP023, NVP-LNP023-NX

NEW DRUG APPROVALS
ONE TIME
$10.00
CAREBASTINE
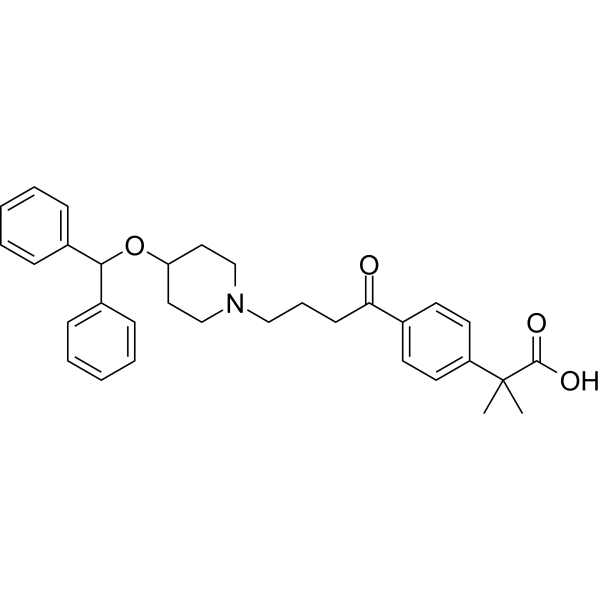
CAREBASTINE
| Molecular Weight | 499.64 |
|---|---|
| Appearance | Solid |
| Formula | C32H37NO4 |
| CAS No. | 90729-42-3 |
Carebastine is the active metabolite of Ebastine. Carebastine is a histamine H1 receptor antagonist. Carebastine inhibits VEGF-induced HUVEC and HPAEC proliferation, migration and angiogenesis in a dose-dependent manner. Carebastine suppresses the expression of macrophage migration inhibitory factor.
Carebastine is the active metabolite of Ebastine. Carebastine is a histamine H1 receptor antagonist. Carebastine inhibits VEGF-induced HUVEC and HPAEC proliferation, migration and angiogenesis in a dose-dependent manner[1]. Carebastine suppresses the expression of macrophage migration inhibitory factor[2].
Literature References: Nonsedating type histamine H1-receptor antagonist. Prepn: J. M. P. Soto et al., EP 134124; eidem, US 4550116 (both 1985 to Fordonal). Metabolized in vivo to carebastine, its active carboxylic acid metabolite.

PATENT
https://patents.google.com/patent/US8067604B2/en
These schemes also illustrate the interrelatedness of the processes and intermediates.










EXAMPLE 1
One gram of 9 was dissolved in 20 mL of DMF and 18 mg of P(tBu)3, 41 mg of Pd(dba)2, 230 mg of ZnF2 and 1.2 g of 5 were added. A mixture was stirred at 80° for 18 hours, cooled to room temperature, diluted with ether and washed with water. The organic layer was dried over sodium sulfate, filtered and stripped in vacuo. The resulting product was flash chromatographed on silica gel using 4:1 hexane ethyl acetate to yield 1.0 g (91%) of 10. A repeat of the reaction on larger scale using 15 g of 9 provided 15.2 g (93%) of 10.
EXAMPLE 2
Five grams of 9 was dissolved in 50 mL of methylene chloride and cooled to 0° C. To the solution was added 5.78 g of trimethylsilyl iodide. The mixture was stirred for 30 minutes and excess sodium bisulfite solution was added with vigorous stirring at room temperature. The layers were separated and the aqueous layer extracted twice with methylene chloride. Combined organic layers were dried, filtered and stripped in vacuo to provide 7.7 g (98%) of 1. The reaction was repeated on a larger scale using 15 g of 9 to produce 22.5 g of 1 (96%) yield.
EXAMPLE 3
Six grams of potassium carbonate, 5.8 g of piperidine 2 and 7.6 g of 1 are combined in 100 mL of DMF. The suspension is stirred at room temperature until TLC in 4:1 hexane-ethyl acetate indicates a complete reaction. The reaction mixture is poured into 400 mL of water and extracted three times with methylene chloride. The combined organic extracts are dried, filtered and reduced in vacuo. The resulting product is flash chromatographed on silica gel using ethyl acetate containing 10% triethylamine to yield 3.
EXAMPLE 4
Seven grams of 3 is dissolved in 100 mL of methanol, cooled to 0° C. and 1.1 g of sodium borohydride is added. The mixture is stirred 1 hour, concentrated and partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The bicarbonate layer is extracted twice with ethyl acetate, the combined organic layers are dried over sodium sulfate and the solution is reduced in vacuo to provide 4.
EXAMPLE 5
Two grams of 4 is dissolved in 30 mL of DMF. To this are added 16.2 mg of P(tBu)3, 36.6 mg of Pd(dba)2, 209 mg of ZnF2 and 1.056 g of 5. The mixture is heated at 80° C., cooled, diluted with ether and worked up as in example 1. The resulting product is flash chromatographed on silica gel using 9:1 ethyl acetate-triethylamine to provide 7.
EXAMPLE 6
One hundred fifty milligrams of 6 is slurried in 5 mL of water and 10 mL of methanol. To the slurry is added 175 mg of sodium hydroxide. The slurry is refluxed for one hour, cooled to room temperature and the methanol removed in vacuo. The resulting aqueous solution is distributed between water and chloroform, the chloroform layer is discarded, the aqueous layer is adjusted to pH 2.3 and extracted with chloroform. The organic layer is dried, filtered and reduced in vacuo to provide carebastine.
EXAMPLE 7
Five grams of 1 was combined with 2.64 g of 2 and 2.0 g of potassium carbonate and 80 mL of DMF. The mixture was stirred at room temperature for two hours, poured into 400 mL of water and extracted three times into methylene chloride. The combined organic layers were dried, filtered and reduced in vacuo. The resulting product was flash chromatographed on silica gel using 9:1 ethyl acetate-triethylamine to provide 2.0 g (54%) of 3.
EXAMPLE 8
One and seven-tenths grams of 3, 90 mg of P(tBu)3, 300 mg of Pd(dba)2, 250 mg of ZnF2 and 1.1 g of 5 were dissolved in 330 mL of DMF under argon. The mixture was heated to 80° for two hours, cooled to room temperature, diluted with ether and worked up as described in example 1. The resulting product was filtered through silica to provide 1.2 g (67.8%) of 6.
EXAMPLE 9
Two grams of 20, 170 mg of P(tBu)3, 560 mg of Pd(acac)2, 474 mg of ZnF2 and 2.0 g of 5 were combined in 50 mL of DMF under argon. The mixture was heated to 80° C. and monitored by HPLC. When reaction was complete, the mixture was cooled to room temperature and 250 mL of water was added. The mixture was extracted three times with ether, dried, filtered and reduced in vacuo. The resulting product was flash chromatographed in 4:1 hexane-ethyl acetate to provide 1.89 g (85%) of 8.
EXAMPLE 10
Two grams of the triflate analog of 20 were reacted as in the foregoing example with 134 mg P(tBu)3, 433 mg of Pd(acac)2, 375 mg of ZnF2 and 1.58 g of 5 to provide 1.56 g (90% yield) of 8.
Example 11
Piperidinol 25 is reacted with chlorodiphenylmethane as described in Fujii et al. Arzneim.-Forsch. 44, 527-538 (1994) to provide 6.
PATENT
WO/2023/213182CAREBASTINE SALT AND USE THEREOF
WIPO – Search International and National Patent Collections
Example 1: Potassium 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionate (carristin potassium salt ) preparation
[0060]
[0061]
Step 1: Preparation of methyl 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionate
[0062]
[0063]
Add 4-(diphenylmethoxy)piperidine hydrochloride (473mg, 1.77mmol), DMAC (4.5ml), K 3 PO 4 (1.13g, 5.3mmol), KI (29mg, 0.177mmol) to a 25ml single-neck bottle. , stir and heat to 100°C. Weigh 2-[4-(4-chloro-1-butyryl)phenyl]-2-methylpropionate methyl ester (600mg, 2.12mmol) and dissolve it in 1ml of DMAC. Add the reaction solution slowly and dropwise, and keep the reaction for 4~ 6h, TLC detects that the raw material reaction is complete. Cool to room temperature, add isopropyl acetate and water, and stir to separate layers. The aqueous phase was then extracted with isopropyl acetate, the organic phases were combined, washed twice with water, dried over anhydrous sodium sulfate, filtered, concentrated, and passed through a silica gel column to obtain 500 mg of the title product, yield 45%, purity: 97.3%.
[0064]
ESI-MS: m/z = 514.3(M+H) +。
[0065]
1H NMR (400 MHz, CDCl 3) δ: 7.93 (d, J=8.3Hz, 2H), 7.47 (m, 4H), 7.42 (d, J=8.3Hz, 2H), 7.30 (m, 4H), 7.18 (m, 2H), 3.64 (s, 3H),2.98 (m, 4H), 2.42 – 2.40 (m, 4H), 1.96 (m, 4H), 1.62 (s, 6H), 1.42 (m, 4H)。
[0066]
Step 2: Preparation of 2-(4-(4-(4-(Diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionic acid (carristin)
[0067]
[0068]
Add (5-methyl-2-oxo-1,3-dioxo-4-yl)methyl-2-(4-(4-(4-(diphenylmethoxy))piperidine-1 to a 25ml three-necked flask) -Methyl)-butyryl)phenyl)-2-methylpropionate (320 mg, 0.62 mmol), 1.5 ml of methanol, 2 ml of 10% NaOH, heated to 60°C for 2 hours, and the TLC raw material reaction was completed. After the reaction is completed, cool to room temperature, concentrate to dryness, add EA, add hydrochloric acid to adjust the pH to 2~3, layer the layers, wash once with water, dry the organic phase, and concentrate to dryness to obtain 300 mg of the title product. Yield: 95%, purity 95.0%.
[0069]
ESI-MS: m/z = 500.3(M+H) +。
[0070]
1H NMR (400 MHz, CDCl 3) δ:7.75-7.63 (m, 2H), 7.57–7.24 (m,12H), 5.48 (s,1H),3.73 (m, 1H), 3.05–3.02 (m, 2H), 2.77–2.66 (m, 6H), 2.20–2.07 (m, 2H), 2.00–1.81 (m,4H), 1.58 (s, 6H)。
[0071]
Step 3: Potassium 2-(4-(4-(4-(Diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionate (Carristine Potassium Salt) Preparation
[0072]
[0073]
Add 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionic acid (499mg, 1mmol) and acetonitrile 3.5 to a 25ml three-necked flask. ml, heated to 60°C, added potassium hydroxide (56 mg, 1 mmol), stirred, cooled down, a white solid precipitated, filtered, and dried to obtain 500 mg of carristine potassium salt, with a yield of 90% and a purity of 98.67%.
[0074]
ESI-MS: m/z = 500.3(M+H) +。
[0075]
1H NMR (400 MHz, CDCl 3) δ:7.75-7.63 (m, 2H), 7.57–7.24 (m,12H), 5.48 (s,1H),3.73 (m, 1H), 3.05–3.02 (m, 2H), 2.77–2.66 (m, 6H), 2.20–2.07 (m, 2H), 2.00–1.81 (m,4H), 1.58 (s, 6H)。
[0076]
Example 2: Sodium 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionate (carristine sodium salt ) preparation
[0077]
[0078]
In this example, the preparation method of 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionic acid is the same as in Example 1.
[0079]
Add 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionic acid (499mg, 1mmol) and acetonitrile 3.5 to a 25ml three-necked flask. ml, heated to 60°C, added sodium hydroxide (40 mg, 1 mmol) and stirred for 1 hour, concentrated to dryness, added methyl tert-butyl ether and stirred, filtered, and dried to obtain 458 mg of carristin sodium salt, yield 85%, purity 96.98 %.
[0080]
ESI-MS: m/z = 500.3(M+H) +。
[0081]
1H NMR (400 MHz, CDCl 3) δ:7.75-7.63 (m, 2H), 7.57–7.24 (m,12H), 5.48 (s,1H),3.73 (m, 1H), 3.05–3.02 (m, 2H), 2.77–2.66 (m, 6H), 2.20–2.07 (m, 2H), 2.00–1.81 (m,4H), 1.58 (s, 6H)。
//////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
///////////

NEW DRUG APPROVALS
ONE TIME
$10.00
VODOBATINIB
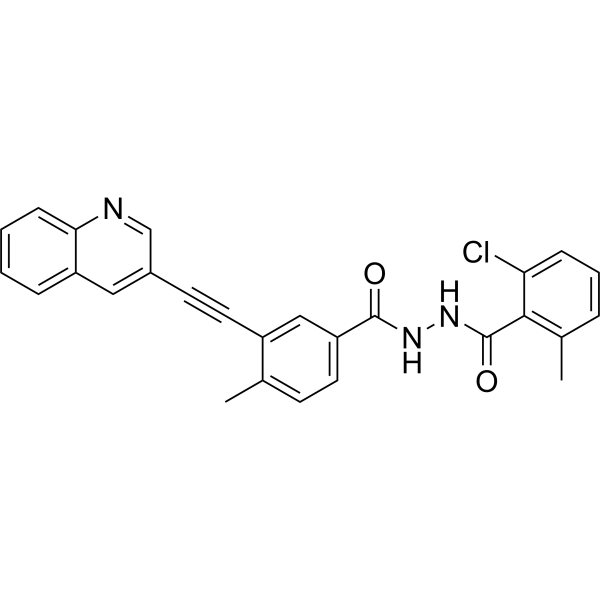
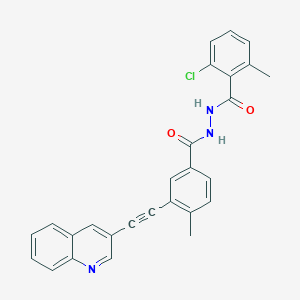
VODOBATINIB
1388803-90-4
| Molecular Weight | 453.92 |
|---|---|
| Appearance | Solid |
| Formula | C27H20ClN3O2 |
- SCO-088
- K0706
- K-0706
2-chloro-6-methyl-N‘-[4-methyl-3-(2-quinolin-3-ylethynyl)benzoyl]benzohydrazide
Vodobatinib (K0706) is a potent, third generation and orally active Bcr-Abl1 tyrosine kinase inhibitor with an IC50 of 7 nM. Vodobatinib exhibits activity against most BCR-ABL1 point mutants, and has no activity against BCR-ABL1T315I. Vodobatinib can be used for chronic myeloid leukemia (CML) research. Vodobatinib is a click chemistry reagent, itcontains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
| Vodobatinib (K0706) is a potent, third generation and orally active Bcr-Abl1 tyrosine kinase inhibitor with an IC50 of 7 nM. Vodobatinib exhibits activity against most BCR-ABL1 point mutants, and has no activity against BCR-ABL1T315I. Vodobatinib can be used for chronic myeloid leukemia (CML) research[1][2]. Vodobatinib is a click chemistry reagent, itcontains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups. |
Brain penetrant kinase inhibitors: Learning from kinase neuroscience discovery
Publication Name: Bioorganic & Medicinal Chemistry Letters
Publication Date: 2018-06-15
PMID: 29752185
DOI: 10.1016/j.bmcl.2018.05.007
PATENT
WO2012098416
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012098416
EXAMPLES
Reƒerence Example 1
Methyl 3-ethynyl-4-methylbenzoate
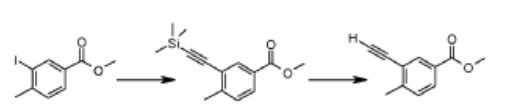

A mixture of methyl 3-iodo-4-methylbenzoate (2.0g, 7mmol), trimethylsilylacetylene (1.2ml, 8mmol), Pd(PPh3)4 (0.42g, 0.3mmol), CuI (0.137g, 0.7mmol) and diisopropylethylamine (2.5ml, 11.4mmol) in THF (20ml) was heated at 50°C for 12hrs under nitrogen atmosphere. The reaction mixture was cooled to ambient temperature and filtered through a Celite® bed. The clear filtrate was concentrated and the residue purified by flash chromatography on silica gel (elution with 2% ethyl acetate in n-hexane) to provide methyl 4-methyl-3-[(trimethylsilyl)ethynyl]benzoate.
To the solution of methyl 4-methyl-3-[(trimethylsilyl)ethynyl]benzoate (2.3g) in THF (40ml) was added tetrabutylammonium fluoride (1.0M in THF, 3.2ml, 1 1mmol) at ambient
temperature and stirred for 15 minutes, concentrated and the residue purified by flash chromatography on silica gel (elution with 2% ethyl acetate in n-hexane) to provide methyl 3 – ethynyl- 4-methylbenzo at e .
1H NMR (500 MHz in DMSO-d6), δ 2.50 (s, 3H), 3.90 (s, 3H), 4.57 (s, 1H), 7.51 (d, J = 8.0 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.99 (s, 1H).
Similarly were prepared the following ester compounds from their corresponding iodo esters:
Methyl 3-ethynyl-4-fluorobenzoate
Methyl 3-ethynyl-4-methoxybenzoate
Reƒerence Example 2
4-Methyl-3-[(quinolin-3-yl)ethynyl]benzoic acid
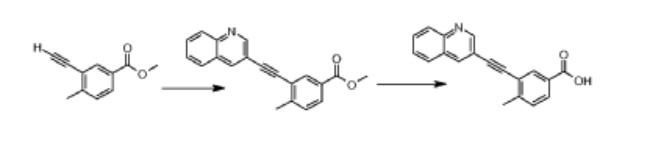

A mixture of methyl 3-ethynyl-4-methylbenzoate (0.341 g, 2mmol), 3-iodoquinoline (0.5g, 2mmol), Pd(PPh3)4 (0.1 1g, 0.01mmol), CuI (0.179g, 0.1mmol) and diisopropylethylamine (0.5ml, 3mmol) in DMF (15ml) was stirred at ambient temperature for 12hrs under an atmosphere of nitrogen. The reaction mixture was concentrated and the crude product was purified by flash chromatography on silica gel (elution with 10% ethyl acetate in n-hexane) to provide methyl 4-methyl-3-[(quinolin-3-yl)ethynyl]benzoate.
Sodium hydroxide (0.15g, 3.71mmol) was added to a solution of the above methyl ester in methanol (20ml) and water (3ml) and stirred at 50°C for 3hrs and then concentrated in vacuo. Water (10ml) was added to the residue, adjusted pH to 4.0-4.5 with citric acid. The solid obtained was filtered, washed successively with water and diethyl ether and dried at ambient temperature to obtain 4-methyl-3-[(quinolin-3-yl)ethynyl]benzoic acid. 1H NMR (500 MHz in DMSO-d6), δ 2.66 (s, 3H), 7.56 (d, J = 8.0 Hz, 1H), 7.75 (t, J; = 15.1 Hz, J2 = 8.2 Hz, 1H), 7.89 (t, J} = 13.7 Hz, J2 = 8.5 Hz, 1H), 7.95 (d, J = 8.0 Hz, 1H), 8.09 (d, J = 8.2 Hz, 1H), 8.12 (d, J = 8.1 Hz, 1H), 8.17 (s, 1H), 8.75 (s, 1H), 9.1 1 (s, 1H), 12.84 (s, 1H).
Reƒerence Example 3
4-Methyl-3-[2-(3-quinolyl)ethynyl]benzohydrazide
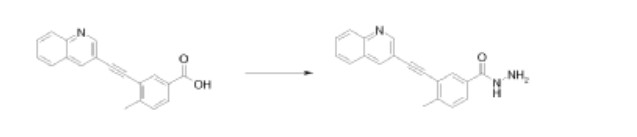

A mixture of 4-methyl-3-[(quinolin-3-yl)ethynyl]benzoic acid (0.15g, 0.5mmol), N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (0.15g, 0.7mmol) and 1-hydroxybenzotriazole (0.1g, 0.7mmol) in N,N-dimethylformamide (15ml) was stirred at room temperature for 1hr. Hydrazine hydrate (1.52ml, 0.5mmol) was then added and the mixture stirred for another 3hrs. Concentration and trituration of the residue with water produced a solid which was filtered, washed successively with water and diethyl ether, and finally dried in vacuo to get the hydrazide as a pale yellow solid.
1H NMR (400 MHz in DMSO-d6), δ 2.63 (s, 3H), 4.79 (s, 2H), 7.51 (d, J = 8.0 Hz, 1H), 7.75 (t, J1 = 14.7 Hz, J2 = 7.6 Hz, 1H), 7.85-7.96 (m, 2H), 8.09-8.13 (m, 3H), 8.73 (s, 1H), 9.09 (s, 1H), 9.91 (s, 1H).
Reƒerence Example 4
N’-(3-iodo-4-methylbenzoyl)-2,4,6-trichlorobenzohydrazide
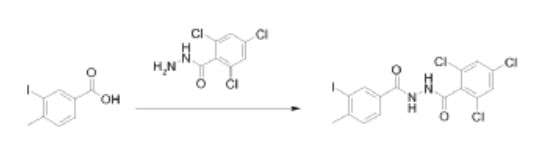

N’-(3-iodo-4-methylbenzoyl)-2,4,6-trichlorobenzohydrazide was prepared by the reaction of 3-iodo-4-methylbenzoic acid with 2,4,6-trichlorobenzohydrazide. The coupling was performed in a manner similar to that described in Reference Example 3.
Example 1.1
2,4,6-Trichloro-N’-[4-methyl-3-[2-(3-quinolyl)ethynyl]benzoyl]benzohydrazide
Method A:
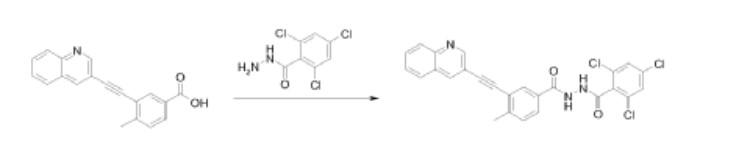

A mixture of 4-methyl-3-[(quinolin-3-yl)ethynyl]benzoic acid (0.15g, 0.5mmol), N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (0.15g, 0.7mmol) and 1-hydroxybenzotriazole (0.1g, 0.7mmol) in N,N-dimethylformamide (15ml) was stirred at ambient temperature for 1hr. 2,4,6-Trichlorobenzohydrazide (0.125g, 0.5mmol) was added and the mixture stirred for 12hrs at ambient temperature. Concentration and trituration of the residue with water produced a solid which was filtered, washed with water and the crude product was purified by flash chromatography on silica gel (elution with 10% methanol in dichloromethane) to get 2,4,6-trichloro-N-[4-methyl-3-[2-(3-quinolyl)ethynyl]benzoyl] benzohydrazide as a white solid.
Method B:
2,4,6-Trichloro-N’-[4-methyl-3-[2-(3-quinolyl)ethynyl]benzoyl] benzohydrazide was also prepared by the reaction of 4-methyl-3-[(quinolin-3-yl)ethynyl]benzoic acid with 2,4,6-trichlorobenzohydrazide in diethyl cyanophosphonate. The condensation reaction was performed in a manner similar to that described in Method A.
Method C:
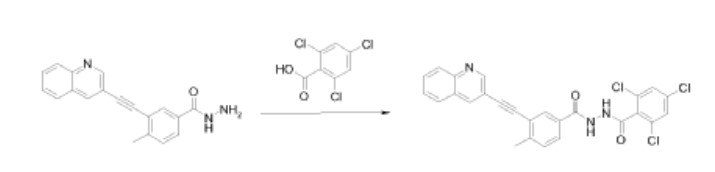

2,4,6-Trichloro-N-[4-methyl-3-[2-(3-quinolyl)ethynyl]benzoyl]benzohydrazide was also prepared by the reaction of 4-methyl-3-[(quinolin-3-yl)ethynyl]benzohydrazide with 2,4,6- trichlorobenzoyl chloride. The condensation reaction was performed in a manner similar to that described in Method A.
The compounds 1.2 to 1.14, 1.21 to 1.34, 1.36 to 1.40, and 1.43 to 1.59 were prepared in a manner similar to Example I.1, by following either of the methods A, B or C, using the appropriate substrates.
PATENT
WO2023214314 VODOBATINIB FOR REDUCING PROGRESSION OF PARKINSON’S DISEASE (wipo.int)
Vodobatinib (N’-(2-chloro-6-methylbenzoyl)-4-methyl-3-[2-(3-quinolyl) ethynyl]-benzohydrazide), a c-Abl inhibitor is represented by Formula I (referred hereinafter interchangeably as vodobatinib or compound of Formula
International Publication Nos. WO 2017/208267A1, WO 2020/250133 Al and WO 2022/024072A1, which are hereby incorporated by reference, disclose methods of use of the compound of Formula I for the treatment of Parkinson’s disease, synucleinopathies and Alzheimer’s disease (AD) respectively.
There is a continuing need for effective and safe methods for the treatment of, and delaying the progression of, neurodegenerative diseases, including in the early-stage of the diseases.
- N’-(2-chloro-6-methylbenzoyl)-4-methyl-3-[2-(3-quinolyl) ethynyl]-benzohydrazide for treatment of alzheimer’s diseasePublication Number: WO-2022024072-A1Priority Date: 2020-07-31
- Compounds for the treatment of covid-19Publication Number: EP-3875078-A1Priority Date: 2020-03-06
- Treatment for synucleinopathiesPublication Number: US-2022257582-A1Priority Date: 2019-06-11
- Novel amorphous dispersion of 4-Methyl-3-quinolin-3-ylethynyl-benzoic acid N’-(2-chloro-6-methyl-benzoyl) hydrazidePublication Number: AU-2018235446-A1Priority Date: 2017-03-15
- Novel amorphous dispersion of 4-methyl-3-quinolin-3-ylethynyl-benzoic acid n’-(2-chloro-6-methyl-benzoyl) hydrazidePublication Number: EP-3596050-A1Priority Date: 2017-03-15
- Novel amorphous dispersion of 4-methyl-3-quinolin-3-ylethynyl-benzoic acid n’-(2-chloro-6-methyl-benzoyl) hydrazidePublication Number: US-2020085751-A1Priority Date: 2017-03-15
- Novel amorphous dispersion of 4-methyl-3-quinolin-3-ylethynyl-benzoic acid n’-(2-chloro-6-methyl-benzoyl) hydrazidePublication Number: WO-2018167802-A1Priority Date: 2017-03-15
- NOVEL AMORPHOUS DISPERSION OF 4-METHYL-3-QUINOLIN-3-YETHYNYL-BENZOIC ACID HYDRAZIDE N- (2-CHLORO-6-METHYL-BENZOYL)Publication Number: WO-2018167802-A9Priority Date: 2017-03-15
- Novel amorphous dispersion of 4-methyl-3-quinolin-3-ylethynyl-benzoic acid n’-(2-chloro-6-methyl-benzoyl) hydrazidePublication Number: US-2021267906-A1Priority Date: 2017-03-15
- Novel amorphous dispersion of 4-Methyl-3-quinolin-3-ylethynyl-benzoic acid N’-(2-chloro-6-methyl-benzoyl) hydrazidePublication Number: AU-2018235446-B2Priority Date: 2017-03-15Grant Date: 2022-04-07
- Amorphous dispersion of 4-methyl-3-quinolin-3-ylethynyl-benzoic acid n’-(2-chloro-6-methyl-benzoyl) hydrazidePublication Number: US-11351123-B2Priority Date: 2017-03-15Grant Date: 2022-06-07
- Treatment of parkinson’s diseasePublication Number: US-2019275017-A1Priority Date: 2016-06-02
- Treatment of Parkinson’s diseasePublication Number: US-10849887-B2Priority Date: 2016-06-02Grant Date: 2020-12-01
- Treatment of Parkinson’s diseasePublication Number: IL-263188-APriority Date: 2016-06-02
- Treatment of Parkinson’s diseasePublication Number: CN-109475539-BPriority Date: 2016-06-02Grant Date: 2021-12-28
- Treatment of Parkinson’s diseasePublication Number: JP-6974357-B2Priority Date: 2016-06-02Grant Date: 2021-12-01
- Treatment for parkinson’s diseasePublication Number: US-2022273632-A1Priority Date: 2016-06-02
- Diarylacetylene hydrazide containing tyrosine kinase inhibitorsPublication Number: AU-2012208388-A1Priority Date: 2011-01-21
- Diarylacetylene hydrazide containing tyrosine kinase inhibitorsPublication Number: AU-2012208388-A2Priority Date: 2011-01-21
- Diarylacetylene hydrazide containing tyrosine kinase inhibitorsPublication Number: EP-2665709-B1Priority Date: 2011-01-21Grant Date: 2016-12-07
- Tyrosine kinase inhibitors containing diarylacetylene hydrazidePublication Number: ES-2608829-T3Priority Date: 2011-01-21Grant Date: 2017-04-17
- Diarylacetylene hydrazide containing tyrosine kinase inhibitorsPublication Number: US-2013296557-A1Priority Date: 2011-01-21
- Diarylacetylene hydrazide containing tyrosine kinase inhibitorsPublication Number: US-9024021-B2Priority Date: 2011-01-21Grant Date: 2015-05-05
//////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
///////////
Ref
- [1]. Orlando Antelope, et al. BCR-ABL1 tyrosine kinase inhibitor K0706 exhibits preclinical activity in Philadelphia chromosome-positive leukemia. Exp Hematol. 2019 Sep;77:36-40.e2. [Content Brief][2]. Phase 1 Trial of Vodobatinib, a Novel Oral BCR-ABL1 Tyrosine Kinase Inhibitor (TKI): Activity in CML Chronic Phase Patients Failing TKI Therapies Including Ponatinib. Session: 632: Chronic Myeloid Leukemia: Therapy: CML: New and Beyond.
///////VODOBATINIB, SCO-088, K0706, K-0706
CC1=C(C(=CC=C1)Cl)C(=O)NNC(=O)C2=CC(=C(C=C2)C)C#CC3=CC4=CC=CC=C4N=C3
Etrasimod

Etrasimod
- APD334
- C26H26F3NO3
- 457.493
1206123-37-6
2-[(3R)-7-{[4-cyclopentyl-3-(trifluoromethyl)phenyl]methoxy}-1H,2H,3H,4H-cyclopenta[b]indol-3-yl]acetic acid

| Etrasimod arginine | MXE5EMA09L | 1206123-97-8 | GVPVVOSNDUAUKM-BPGOJFKZSA-N |
Name: Etrasimod arginine
CAS#: 1206123-97-8 (arginine)
Chemical Formula: C32H40F3N5O5
Exact Mass: 631.30
Molecular Weight: 631.700
FDA APPROVED, To treat moderately to severely active ulcerative colitis in adults,
| 10/12/2023 |
Etrasimod, sold under the brand name Velsipity, is a medication that is used for the treatment of ulcerative colitis (UC).[1] It is a selective sphingosine-1-phosphate (S1P) receptor modulator that modifies the activity of the immune system.[1] It is taken by mouth.[1]
Etrasimod was discovered by Arena Pharmaceuticals, with subsequent development by Pfizer.[2]
Etrasimod is a synthetic next-generation selective Sphingosine 1-phosphate (S1P) receptor modulator that targets the S1P1,4,5 with no detectable activity on S1P2 and S1P3 receptors. S1P receptors are membrane-derived lysophospholipid signaling molecules that are involved in the sequestration of circulating peripheral lymphocytes in lymph nodes.1 Therefore, S1P receptor modulators like etrasimod were investigated in treating immune-mediated diseases like ulcerative colitis where a high level of inflammatory T cells is present in the gastrointestinal tract, thus causing diffuse mucosal inflammation.1 In fact, it has been observed that antigen-activated T cells within peripheral lymphoid organs can transiently downregulate S1P receptor levels to facilitate immune cells trafficking into the intestinal mucosa.2
Etrasimod was approved on October 13, 2023, by the FDA under the brand name VELSIPITY for the treatment of adults with moderately to severely active ulcerative colitis. This approval was based on favorable results obtained from Pfizer’s Elevate UC Phase III registrational program, consisting of the Elevate UC 52 and Elevate UC 12 clinical trials, that investigates the efficacy of a 2-mg daily dose regimen of etrasimod, with a 32% and 26% remission rate observed in UC 52 and UC 12 trials respectively.4
Medical uses
Etrasimod is used for the treatment of moderate to severe ulcerative colitis.[1]
Mechanism of action
It works by causing T cells to become trapped in the lymph nodes, preventing them from entering the bloodstream, from where they would travel to other tissues in the body and mediate inflammation.[3][4][5][6][7][8]
Society and culture
Legal status
Velsipity was approved by the US Food and Drug Administration (FDA) in October 2023.[1][9][10]
Names
Etrasimod is the international nonproprietary name.[11]
SYN
ACS Med. Chem. Lett.2014, 5, 12, 1313–1317
Publication Date:November 4, 2014
https://doi.org/10.1021/ml500389m
APD334 was discovered as part of our internal effort to identify potent, centrally available, functional antagonists of the S1P1 receptor for use as next generation therapeutics for treating multiple sclerosis (MS) and other autoimmune diseases. APD334 is a potent functional antagonist of S1P1 and has a favorable PK/PD profile, producing robust lymphocyte lowering at relatively low plasma concentrations in several preclinical species. This new agent was efficacious in a mouse experimental autoimmune encephalomyelitis (EAE) model of MS and a rat collagen induced arthritis (CIA) model and was found to have appreciable central exposure.

APD334 is the second eluting enantiomer (most retained) with a retention time of 48.4 minutes. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.54-1.75 (m, 4H), 1.79-1.92 (m, 2H), 1.95-2.16 (m, 3H), 2.39 (dd, J = 16.0, 8.8 Hz, 1H), 2.61-2.83 (m, 4H), 3.23-3.34 (m, 1H), 3.45-3.56 (m, 1H), 5.14 (s, 2H), 6.74 (dd, J = 8.7, 2.4 Hz, 1H), 6.92 (d, J = 2.3 Hz, 1H), 7.24 (d, J = 8.8 Hz, 1H), 7.64 (d, J = 8.1 Hz, 1H), 7.72 (d, J = 8.6 Hz, 1H), 7.74 (s, 1H), 10.50 (s, 1H), 12.24 (bs, 1H). 13C APT NMR (100 MHz, DMSO-d6): δ up (C, CH2): 23.1, 25.5, 35.5, 35.6, 68.6, 117.0, 124.7 (q, J = 273 Hz), 124.2, 126.8 (q, J = 28 Hz), 128.7, 136.1, 136.2, 144.6, 147.0, 151.9, 173.4; down (CH, CH3): 35.0, 40.5, 102.1, 110.0, 112.4, 124.1 (q, J = 5.7 Hz), 128.4, 131.7. 19F NMR (400 MHz, DMSO-d6) δ ppm -57.4. LCMS (ESI+): calcd for C26H27F3NO3+ [M+H] 458.19; found, 458.4. HRMS (ESI-): calcd for C26H25F3NO3- [M-H] 456.1792; found, 456.1776.




AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
///////////
| Skeletal formula of etrasimod | |
| Clinical data | |
|---|---|
| Trade names | Velsipity |
| Other names | APD334, APD-334 |
| License data | US DailyMed: Etrasimod |
| Routes of administration | By mouth |
| Drug class | Sphingosine-1-phosphate receptor modulator |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Pharmacokinetic data | |
| Protein binding | 97.9%[medical citation needed] |
| Metabolism | Liver (CYP2C8, 2C9, 3A4)[medical citation needed] |
| Elimination half-life | 30 hours[medical citation needed] |
| Excretion | Feces (82%), kidneys (5%)[medical citation needed] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1206123-37-6as arginine: 1206123-97-8 |
| PubChem CID | 44623998 |
| DrugBank | DB14766as arginine: DBSALT003430 |
| ChemSpider | 52084233as arginine: 57643656 |
| UNII | 6WH8495MMHas arginine: MXE5EMA09L |
| KEGG | D10930as arginine: D10931 |
| ChEMBL | ChEMBL3358920 |
| Chemical and physical data | |
| Formula | C26H26F3NO3 |
| Molar mass | 457.493 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
- ^ Jump up to:a b c d e f Pfizer (12 October 2023). “Velsipity (etrasimod) tablets, for oral use” (PDF). U.S. Food and Drug Administration (FDA). Retrieved 18 October 2023.
- ^ Bayer M (2 May 2023). “Pfizer tosses newly acquired meds out of the Arena”. Fierce Biotech. Retrieved 13 October 2023.
- ^ Atreya R, Neurath MF (April 2023). “The sphingosine-1-phosphate receptor agonist etrasimod in ulcerative colitis”. Lancet. 401 (10383): 1132–1133. doi:10.1016/S0140-6736(23)00228-3. PMID 36871570.
- ^ Sandborn WJ, Vermeire S, Peyrin-Biroulet L, Dubinsky MC, Panes J, Yarur A, et al. (April 2023). “Etrasimod as induction and maintenance therapy for ulcerative colitis (ELEVATE): two randomised, double-blind, placebo-controlled, phase 3 studies”. Lancet. 401 (10383): 1159–1171. doi:10.1016/S0140-6736(23)00061-2. PMID 36871574.
- ^ Dal Buono A, Gabbiadini R, Alfarone L, Solitano V, Repici A, Vetrano S, et al. (July 2022). “Sphingosine 1-Phosphate Modulation in Inflammatory Bowel Diseases: Keeping Lymphocytes Out of the Intestine”. Biomedicines. 10 (7). doi:10.3390/biomedicines10071735. PMC 9313037. PMID 35885040.
- ^ Argollo M, Furfaro F, Gilardi D, Roda G, Allocca M, Peyrin-Biroulet L, et al. (April 2020). “Modulation of sphingosine-1-phosphate in ulcerative colitis”. Expert Opin Biol Ther. 20 (4): 413–420. doi:10.1080/14712598.2020.1732919. PMID 32093531.
- ^ Al-Shamma H, Lehmann-Bruinsma K, Carroll C, Solomon M, Komori HK, Peyrin-Biroulet L, et al. (June 2019). “The Selective Sphingosine 1-Phosphate Receptor Modulator Etrasimod Regulates Lymphocyte Trafficking and Alleviates Experimental Colitis”. J Pharmacol Exp Ther. 369 (3): 311–317. doi:10.1124/jpet.118.254268. PMID 30872391.
- ^ Peyrin-Biroulet L, Christopher R, Behan D, Lassen C (May 2017). “Modulation of sphingosine-1-phosphate in inflammatory bowel disease”. Autoimmun Rev. 16 (5): 495–503. doi:10.1016/j.autrev.2017.03.007. PMID 28279838.
- ^ Brooks M (13 October 2023). “FDA Approves New Drug for Ulcerative Colitis”. Medscape. Retrieved 13 October 2023.
- ^ https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2023/216956Orig1s000ltr.pdf
- ^ World Health Organization (2017). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 78”. WHO Drug Information. 31 (3). hdl:10665/330961.
/////////Etrasimod, APD334, Velsipity, FDA 2023, APPROVALS 2023

NEW DRUG APPROVALS
ONE TIME
$10.00
Motixafortide

Motixafortide
- 4F-benzoyl-TN-14003
- BKT-140
- BKT140
- BL-8040
Protein Chemical FormulaC97H144FN33O19S2
Protein Average Weight2159.6 Da
UNIIDA9G065962CAS number664334-36-5>Motixafortide sequence RRXCYXKKPYRXCR
| Motixafortide acetate | 2639893-42-6 | GMUZYOKQKWMETH-AQDOTSTFSA-N |
fda approved,9/8/2023,
мотиксафортид[Russian]
موتيكسافورتيد[Arabic]
莫替福肽[Chinese]
Motixafortide is a peptide inhibitor of CXCR4 used to mobilize hematopoietic stem cells prior to collection and autologous transplantation in multiple myeloma patients.
To use with filgrastim (G-CSF) to mobilize hematopoietic stem cells to the peripheral blood for collection and subsequent autologous transplantation in patients with multiple myeloma
Motixafortide, sold under the brand name Aphexda, is a medication used for the treatment of multiple myeloma.[1] Motixafortide is a hematopoietic stem cell mobilizer and a CXCR4 antagonist.[1] It is given by subcutaneous injection.[1]
Motixafortide was approved for medical use in the United States in September 2023.[2][3]
Motixafortide is a cyclic peptide hematopoietic stem cell mobilizer used to improve stem cell collection prior to autologous transplantation.3 Hematopoietic stem cell transplantation (HSCT) is commonly employed in the context of hematologic cancers – high-dose chemotherapy regimens destroy cancerous blood cells, which are then replaced via infusion of the patient’s own stem cells (i.e. an autologous transplant).4 Similar in mechanism to the previously approved plerixafor, motixafortide is an inhibitor of C-X-C Motif Chemokine Receptor 4 (CXCR4), a protein that helps to anchor stem cells to bone marrow matrix.3 When administered alongside filgrastim, another agent used to aid in stem cell collection, motixafortide enabled the collection of an adequate number of stem cells in ~92% of patients within two apheresis procedures, compared to ~26% of patients receiving only filgrastim.1
Motixafortide was approved by the FDA in September 2023, in combination with filgrastim, for use in stem cell mobilization prior to autologous stem cell transplant in patients with multiple myeloma.5 It has also been investigated alongside pembrolizumab for the treatment of pancreatic cancer.2
Medical uses
Motixafortide is indicated in combination with filgrastim, a granulocyte-colony stimulating factor (G-CSF), to mobilize hematopoietic stem cells to the peripheral blood for collection and subsequent autologous transplantation in people with multiple myeloma.[1][2]
Society and culture
Names
Motixafortide is the international nonproprietary name.[4]
| Clinical data | |
|---|---|
| Trade names | Aphexda |
| Other names | BL-8040 |
| License data | US DailyMed: Motixafortide |
| Routes of administration | Subcutaneous |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 664334-36-5as acetate: 2639893-42-6 |
| PubChem CID | 91865076 |
| DrugBank | DB14939 |
| ChemSpider | 64854351 |
| UNII | DA9G065962as acetate: 3ZPX60DV8A |
| KEGG | D12281as acetate: D12282 |
| ChEBI | CHEBI:145536 |
| Chemical and physical data | |
| Formula | C97H144FN33O19S2 |
| Molar mass | 2159.55 g·mol−1 |
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
///////////
References
- ^ Jump up to:a b c d e “Aphexda- motixafortide injection, powder, lyophilized, for solution”. DailyMed. 4 September 2023. Archived from the original on 14 September 2023. Retrieved 13 September 2023.
- ^ Jump up to:a b “Aphexda approval letter” (PDF). 8 September 2023. Archived from the original (PDF) on 14 September 2023. This article incorporates text from this source, which is in the public domain.
- ^ “BioLineRx Announces FDA Approval of Aphexda (motixafortide) in Combination with Filgrastim (G-CSF) to Mobilize Hematopoietic Stem Cells for Collection and Subsequent Autologous Transplantation in Patients with Multiple Myeloma” (Press release). BioLineRx Ltd. 11 September 2023. Retrieved 13 September 2023 – via PR Newswire.
- ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 82”. WHO Drug Information. 33 (3). hdl:10665/330879.
Further reading
- Crees ZD, Rettig MP, Jayasinghe RG, Stockerl-Goldstein K, Larson SM, Arpad I, et al. (April 2023). “Motixafortide and G-CSF to mobilize hematopoietic stem cells for autologous transplantation in multiple myeloma: a randomized phase 3 trial”. Nature Medicine. 29 (4): 869–879. doi:10.1038/s41591-023-02273-z. PMC 10115633. PMID 37069359.</ref>
External links
- Clinical trial number NCT03246529 for “A Phase III, Safety, Tolerability and Efficacy of Combination Treatment of BL-8040 and G-GSF as Compared to Placebo and G-CSF for thE MobilizatioN of HematopoiEtic Stem Cells for Autologous TransplantatIon in SubjectS With MM (GENESIS)” at ClinicalTrials.gov
/////fda 2023, approvals 2023, Motixafortide, 4F-benzoyl-TN-14003, BKT-140, BKT140, BL 8040, Aphexda, мотиксафортид, موتيكسافورتيد , 莫替福肽 ,
Evobrutinib
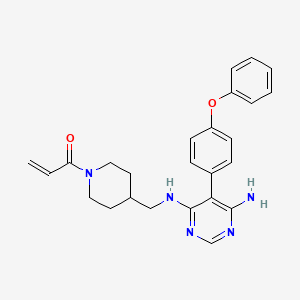
Evobrutinib
429.5 g/mol,C25H27N5O2
- Evobrutinib
- 1415823-73-2
- Evobrutinib [INN]
- 1-(4-(((6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl)amino)methyl)piperidin-1-yl)prop-2-en-1-one
- MSC2364447C
- MSC2364447C
- M-2951
- MSC-2364447C
- ZA45457L1K
- 1-[4-[[[6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl]amino]methyl]piperidin-1-yl]prop-2-en-1-one
- M2951
Evobrutinib is under investigation in clinical trial NCT03934502 (Effect of Meal Composition and Timing on Evobrutinib Bioavailability).
Evobrutinib is an inhibitor of Bruton’s tyrosine kinase (BTK) with potential antineoplastic activity. Upon administration, evobrutinib inhibits the activity of BTK and prevents the activation of the B-cell antigen receptor (BCR) signaling pathway. This prevents both B-cell activation and BTK-mediated activation of downstream survival pathways, which leads to the inhibition of the growth of malignant B-cells that overexpress BTK. BTK, a member of the Src-related BTK/Tec family of cytoplasmic tyrosine kinases, is overexpressed in B-cell malignancies; it plays an important role in B-lymphocyte development, activation, signaling, proliferation and survival.
Evobrutinib is in clinical development to investigate its potential as a treatment for multiple sclerosis (MS). It is an oral, highly selective inhibitor of Bruton’s tyrosine kinase (BTK) which is important in the development and functioning of various immune cells including B lymphocytes and macrophages.
Evobrutinib is designed to inhibit primary B cell responses such as proliferation and antibody and cytokine release, without directly affecting T cells. BTK inhibition is thought to suppress autoantibody-producing cells, which preclinical research suggests may be therapeutically useful in certain autoimmune diseases.
U.S. Patent No. 9073947 discloses a pyrimidine derivative of Evobrutinib which chemically named as l-(4-(((6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl)amino)methyl)
piperidin-l-yl)prop-2-en-l-one and pharmaceutically acceptable salts, solvates and pharmaceutical compositions thereof.
U.S. Patent No. 9073947 and ‘Journal of Medicinal Chemistry 2019, 62(17), 7643-7655’ discloses process for the preparation of Evobrutinib which involves column purifications and lyophilisation methods to provide Evobrutinib with low yield, which is not viable at large scale production.
https://www.frontiersin.org/articles/10.3389/fnume.2021.820235/full
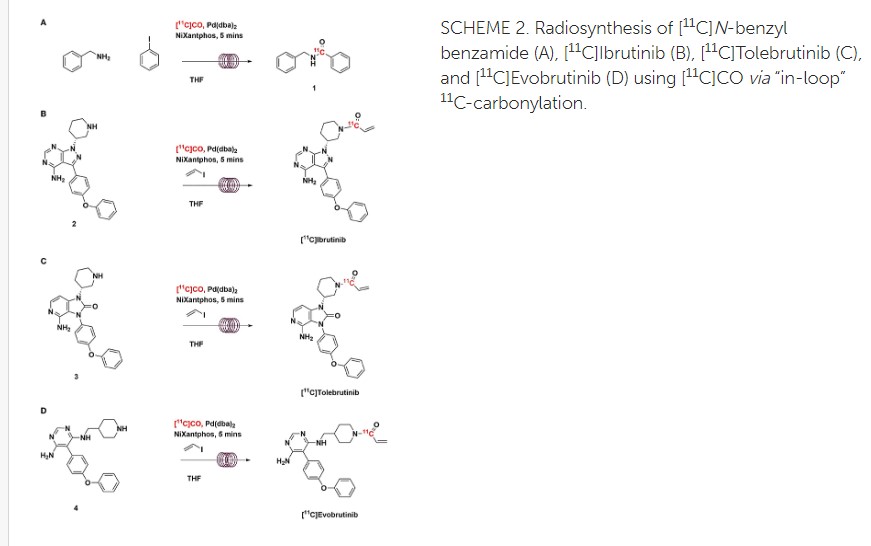

Radiosynthesis of [11C]Evobrutinib. [11C]Evobrutinib was synthesized similarly to the Tolebrutinib example above with the following exceptions. First, the precursor 5-(4-phenoxyphenyl)-N4-(piperidin-4-ylmethyl)pyrimidine-4,6-diamine (4) (1 mg, 2.7 μmol) was used and the crude reaction mixture after the carbonylation reaction was purified by semi-preparative HPLC (column: Luna C18(2), 5 μ (250 x 9.6 mm); mobile phase: 44% MeCN in 200 mM ammonium formate; flow rate: 5 ml/min; UV: 254 nm). The [11C]1-(4-(((6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl)amino)methyl)piperidin-1-yl)prop-2-en-1-one ([11C]evobrutinib) was isolated between the 15.5 and 18 min mark of the chromatogram and this sample was collected into a dilution flask that contained 50 ml of a 2 mg/ml sodium ascorbate aqueous solution. This solution was transferred to an HLB light (30 mg) SPE cartridge. After transfer, the cartridge was eluted with 1 ml of ethanol into the sterile product vial that contained 4 ml of sterile saline. Using this method, 2.2 ± 0.6 GBq (81.4 ± 22.2 mCi) [11C]evobrutinib was isolated (n = 3), and the product was analyzed via reverse phase HPLC using the following methods. Method A described above and Method B (Isocratic and molar activity): column: Luna C18(2) 3-μm (250×4.6 mm); mobile phase Isocratic: 36% acetonitrile in aqueous 0.1% TFA; flow rate: 1.3 ml/min; UV: 254 nm. Method A was used to confirm chemical identity using a co-injection of non-radioactive standard. Radiochemical purity and molar activity were determined by Method B. [11C]Evobrutinib was confirmed by co-injection with a verified non-radioactive reference standard. Am was determined using a 4-point standard curve (analytical HPLC peak area) (Y) vs. standard concentration (X: in nmol) by comparison with an evobrutinib reference standard of known concentration (2.3 mg in 1 ml). The isolated [11C] evobrutinib was co-eluted with a non-radioactive reference standard. The sample was >99% radiochemically pure, >95% chemically pure (HPLC, UV: 254 nm), with a molar activity of 496.5 ± 74 GBq/μmol (13.4 Ci/μmol) The overall synthesis time from the end of cyclotron bombardment was 37–46 min.
Patent
U.S. Patent No. 9073947
PAPER
Journal of Medicinal Chemistry 2019, 62(17), 7643-7655
https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b00794

Step 4
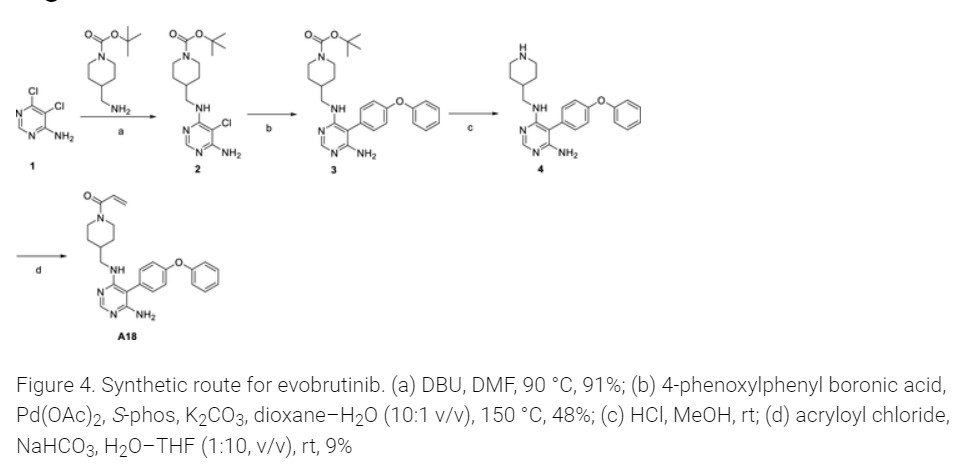
To a 20 mL vial was added 5-(4-phenoxyphenyl)-N-(piperidin-4-ylmethyl)pyrimidine-4,6-diamine (210.00 mg, 0.56 mmol, 1.00 equiv), sodium bicarbonate (70.48 mg, 0.84 mmol, 1.50 equiv), THF (8.00 mL, 98.74 mmol, 176.55 equiv), and water (0.80 mL, 44.41 mmol, 79.40 equiv). The mixture was cooled to 0 °C on an ice bath. Acryloyl chloride (0.15 mL, 1.83 mmol) was then added dropwise. The ice bath was removed, and the reaction was stirred at room temperature for 12 h before it was purified by silica gel chromatography (25 g KPNH silica, 0–100% methanol/ethyl acetate) to afford the title compound (A18) (21 mg, 8.7% yield) was synthesized with a similar protocol to prepared as described in the main body of the article. 1H NMR (DMSO-d6) δ 7.93 (s, 1 H), 7.40–7.08 (m, 9H), 6.76 (dd, J = 4 Hz, 1 H), 6.04 (d, J = 4 Hz, 1 H), 5.61 (d, J = 4 Hz, 1 H), 5.43 (s, 2H), 4.34 (d, J = 12 Hz, 1 H), 3.98 (d, J = 8 Hz, 1 H), 3.12 (m, 2H), 2.95 (m, 1 H), 2.56 (m, 1 H), 1.81 (m, 1 H), 1.59 (m, 2H), 0.92 (m, 2H). [ES-MS] (ESI+): m/z calcd for C25H28N5O2 [M + H]+ 430, found 430.
PATENT

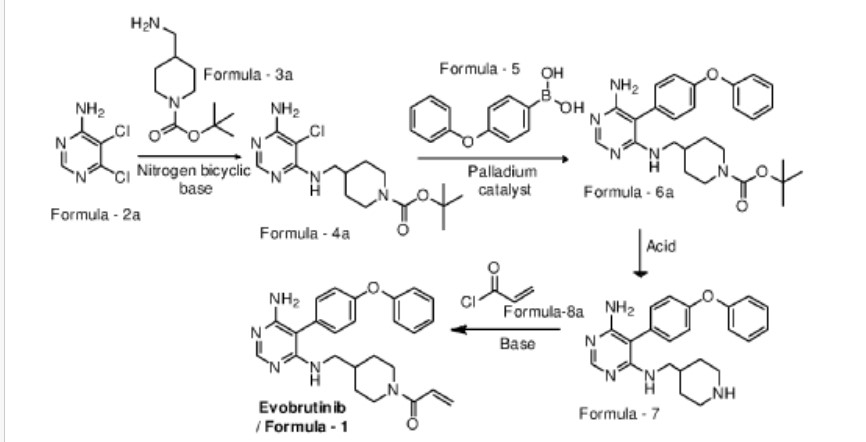

Examples:
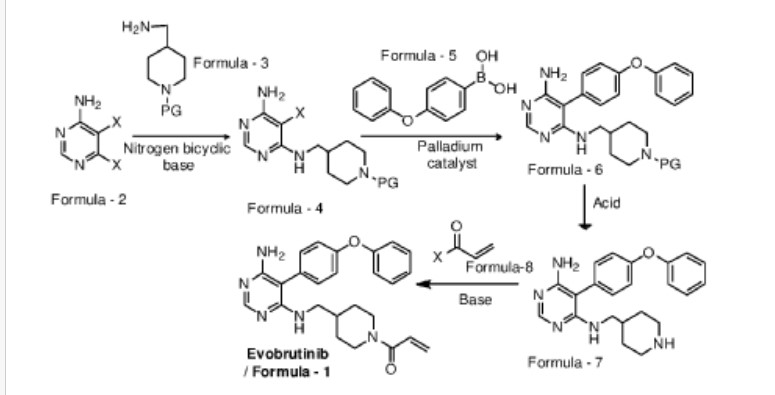
Example-1: Preparation of tert-butyl 4-(((6-amino-5-chloropyrimidin-4-yl)amino) met hy 1 jpiperid ine- 1 -carboxylate
Tert-butyl-4-(aminomethyl)piperidine-l -carboxylate (81 ml) and 1,8-diazabicyclo [5.4.0]undec-7-ene (60.34 g) were added to a mixture of 5,6-dichloropyrimidin-4-amine (50 g) in N,N-dimethylformamide (500 ml) at 25-35°C. Heated the mixture to 90-95°C and stirred for 22 hrs. Cooled the mixture to 25-30°C. Water was added to the mixture at 25-35°C and stirred for 5 hrs. Filtered the precipitated solid, washed with water and n-heptane and dried to get the title compound. Yield: 73.0 gms; Purity by HPLC: 98.7%
Example-2: Preparation of tert-butyl 4-(((6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl) amino)methyl)piperidine-l-carboxylate
(4-Phenoxyphenyl)boronic acid (75.12 g) was added to a mixture of tert-butyl 4-(((6-amino-5-chloropyrimidin-4-yl)amino)methyl)piperidine-l-carboxylate(100 g), 2-di cyclo hexylphosphino-2′,6′-dimethoxybiphenyl (12 g) and potassium carbonate (121.28 g) in 1,4-di oxane (1000 ml) at 25-30°C and stirred for 30 minutes under nitrogen atmosphere. Palladium acetate (1.96 g) was added to the mixture at 25-30°C. Heated the mixture to 100-105°C and stirred for 3 hrs. Cooled the mixture to 25-30°C. Water and ethyl acetate were added to the mixture at 25-35°C and stirred for 30 minutes. Filtered the mixture by using hyflow bed. Organic layer was separated from the filtrate. Organic layer was treated with carbon powder and distilled-off the solvent under reduced pressure, n-heptane (800 ml) was added to the obtained compound. Heated the mixture to 60-65°C and stirred for 90 minutes. Cooled the mixture to 25-30°C and stirred for 2 hrs. Filtered the precipitated solid, washed with n-heptane and dried to get the title compound. Yield: 120 gms, Purity by HPEC: 97.6% Example-3: Preparation of 5-(4-phenoxyphenyl)-N4-(piperidin-4-ylmethyl)pyrimidine-4,6-diamine
Tert-butyl-4-(((6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl)amino)methyl) piperidine- 1 -carboxylate (200 g) in methanol (600 ml) was cooled to 0-5°C. Hydrochloric acid in ethyl acetate (500 ml) was slowly added to the mixture at 0-5°C. Mixture allowed to warm to 25-30°C and stirred for 20 hours. Water was added to the mixture and treated the mixture with aqueous ammonia solution. Dichloromethane was added to the mixture at 25-30°C and stirred for 10 minutes. Layers were separated and distilled-off the organic layer under reduce pressure. Obtained compound was treated with isopropyl ether and dried to get the title compound. Yield: 150 gms, Purity by HPLC: 76.4%
Example-4: Preparation of Evobrutinib
Sodium bicarbonate (23.86 g) and water (301 ml) were added to the mixture of 5-(4-phenoxyphenyl)-N4-(piperidin-4-ylmethyl)pyrimidine-4,6-diamine (70 g) in tetrahydrofuran (2800 ml). Cooled the mixture to 0-5°C. Acryloyl chloride (23.62 g) was slowly added to the mixture. Mixture allowed to warm to 25-30°C and stirred for 20 hrs. Distilled-off the solvent from the mixture under reduced pressure. Ethyl acetate and water were added to the mixture and stirred for 10 minutes. Both the layers were separated. Organic layer was treated with aqueous hydrochloric acid solution and carbon powder. Distilled-off the organic layer under reduced pressure. Isopropyl ether was added to the mixture at 25-30°C and stirred for 14 hrs. Filtered the mixture and washed with isopropyl ether. Dried to get the title compound.
Yield: 41.8 gms, Purity by HPLC: 97.6%
AS ON AUG2023 4,071,221 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
///////////
//////////PHASE 3, MSC2364447C, M-2951, MSC-2364447C, ZA45457L1K, M2951, M 2951, Evobrutinib

NEW DRUG APPROVALS
ONE TIME
$10.00
Zuranolone
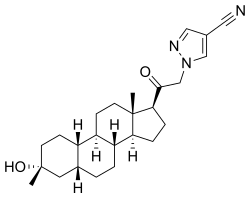
Zuranolone
CAS 1632051-40-1
FDA APPROVED 8/4/2023, To treat postpartum depression
Press Release
WeightAverage: 409.574
Monoisotopic: 409.272927379Chemical FormulaC25H35N3O2
- SAGE 217
- SAGE-217
- SAGE217
Zuranolone, sold under the brand name Zurzuvae, is a medication used for the treatment of postpartum depression.[1][2] It is taken by mouth.[1]
The most common side effects include drowsiness, dizziness, diarrhea, fatigue, nasopharyngitis, and urinary tract infection.[1][2] An orally active inhibitory pregnane neurosteroid, zuranolone acts as a positive allosteric modulator of the GABAA receptor.[6][7][8]
Zuranolone was approved for medical use in the United States for the treatment of postpartum depression in August 2023.[2] It was developed by Sage Therapeutics and Biogen.[9]
Medical uses
Zuranolone is indicated for the treatment of postpartum depression.[1][2]
Adverse effects
The most common side effects include drowsiness, dizziness, diarrhea, fatigue, nasopharyngitis (cold-like symptoms), and urinary tract infection.[2]
The US FDA label contains a boxed warning noting that zuranolone can impact a person’s ability to drive and perform other potentially hazardous activities.[2] Use of zuranolone may cause suicidal thoughts and behavior.[2] Zuranolone may cause fetal harm.[2]
History
Zuranolone was developed as an improvement on the intravenously administered neurosteroid brexanolone, with high oral bioavailability and a biological half-life suitable for once-daily administration.[7][10] Its half-life is around 16 to 23 hours, compared to approximately 9 hours for brexanolone.[4][5]
The efficacy of zuranolone for the treatment of postpartum depression in adults was demonstrated in two randomized, double-blind, placebo-controlled, multicenter studies.[2] The trial participants were women with postpartum depression who met the Diagnostic and Statistical Manual of Mental Disorders criteria for a major depressive episode and whose symptoms began in the third trimester or within four weeks of delivery.[2] In study 1, participants received 50 mg of zuranolone or placebo once daily in the evening for 14 days.[2] In study 2, participants received another zuranolone product that was approximately equal to 40 mg of zuranolone or placebo, also for 14 days.[2] Participants in both studies were monitored for at least four weeks after the 14-day treatment.[2] The primary endpoint of both studies was the change in depressive symptoms using the total score from the 17-item Hamilton depression rating scale (HAMD-17), measured at day 15.[2] Participants in the zuranolone groups showed significantly more improvement in their symptoms compared to those in the placebo groups.[2] The treatment effect was maintained at day 42—four weeks after the last dose of zuranolone.[2]
Society and culture
Zuranolone is the international nonproprietary name.[11]
Legal status
Zuranolone was approved by the US Food and Drug Administration (FDA) for the treatment of postpartum depression in August 2023.[2][12] The FDA granted the application for zuranolone priority review and fast track designations.[2] Approval of Zurzuvae was granted to Sage Therapeutics, Inc.[2]
Zuranolone has also been under development for the treatment of major depressive disorder, but the application for this use was given a Complete Response Letter (CRL) by the FDA due to insufficient evidence of effectiveness.[13]
Research
In a randomized, placebo-controlled phase III trial to assess its efficacy and safety for the treatment of major depressive disorder, subjects in the zuranolone group (50 mg oral zuranolone once daily for 14 days) experienced statistically significant and sustained improvements in depressive symptoms (as measured by HAM-D score) throughout the treatment and follow-up periods of the study.[14]
Other investigational applications include insomnia, bipolar depression, essential tremor, and Parkinson’s disease.[15][6][16]
syn
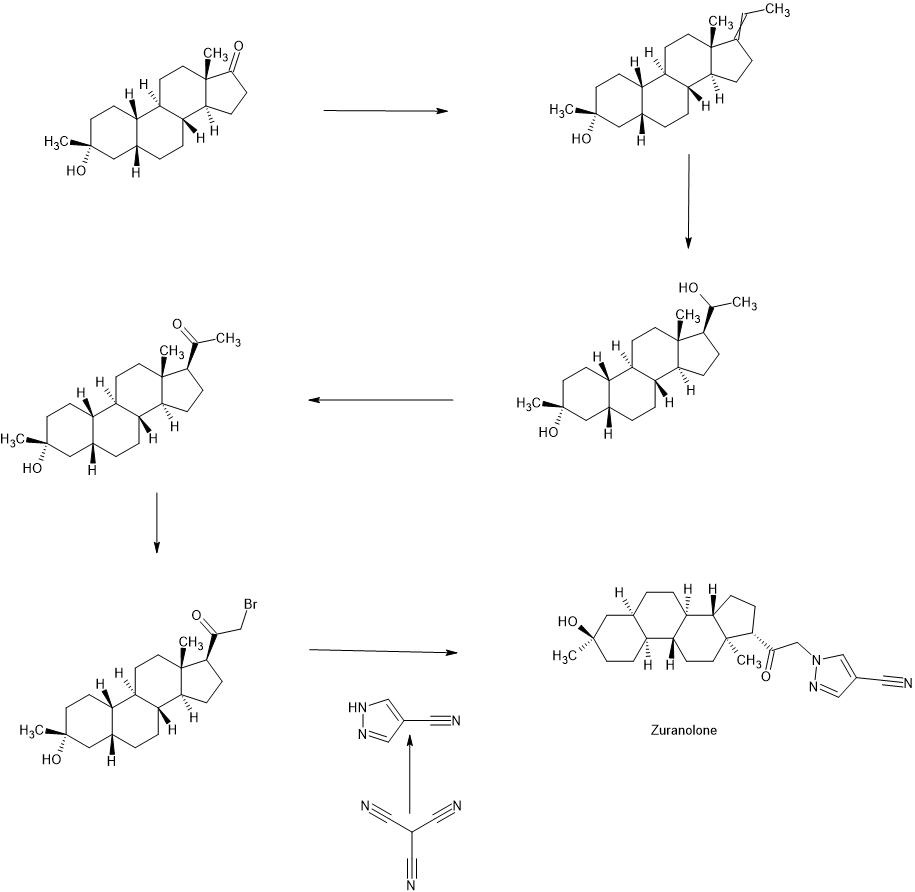

PATENT
WO2022020363
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022020363&_cid=P11-LLRZ9A-38538-1
Example 1. Synthesis of 1-(2-((3R,5R,8R,9R,10S,13S,14S,17S)-3-hydroxy-3,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-2-oxoethyl)-1H-pyrazole-4-carbonitrile (Compound 1).
[00488] To a suspension of K2CO3 (50 mg, 0.36 mmol) in THF (5 mL) was added 1H-pyrazole-4-carbonitrile (100 mg, 0.97 mmol) and 2-bromo-1-((3R,5R,8R,9R,10S,13S,14S,17S)-3-hydroxy-3,13-dimethylhexadecahydro-1H-cyclopenta[ ^]phenanthren-17-yl)ethan-1-one (50 mg, 0.12 mmol). The mixture was stirred at room temperature for 15 hours. The reaction mixture was poured into 5 mL H2O and extracted with ethyl acetate (2×10 mL). The combined organic layers were washed with brine, dried over sodium sulfate, filtered and concentrated. The residue mixture was purified by reverse-phase preparative HPLC to afford Compound 1 as a white solid (9 mg, 17.4% yield).1H NMR (500 MHZ, CDCl3) δ (ppm) 7.87 (1H, s), 7.82 (1H, s), 5.02 (1H, AB), 4.2 (1H, AB), 2.61 (1H, t), 2.16-2.24 (1H, m), 2.05 (1H, dxt), 1.70-1.88 (6H, m), 1.61-1.69 (2H, m), 1.38-1.52 (6H, m), 1.23-1.38 (5H, m), 1.28 (3H, s), 1.06-1.17 (3H, m), 0.67 (3H, s). LCMS: rt=2.24 min, m/z=410.1 [M+H]+.
PAPER
Journal of Medicinal Chemistry (2017), 60(18), 7810-7819
https://pubs.acs.org/doi/10.1021/acs.jmedchem.7b00846
Certain classes of neuroactive steroids (NASs) are positive allosteric modulators (PAM) of synaptic and extrasynaptic GABAA receptors. Herein, we report new SAR insights in a series of 5β-nor-19-pregnan-20-one analogues bearing substituted pyrazoles and triazoles at C-21, culminating in the discovery of 3α-hydroxy-3β-methyl-21-(4-cyano-1H-pyrazol-1′-yl)-19-nor-5β-pregnan-20-one (SAGE-217, 3), a potent GABAA receptor modulator at both synaptic and extrasynaptic receptor subtypes, with excellent oral DMPK properties. Compound 3 has completed a phase 1 single ascending dose (SAD) and multiple ascending dose (MAD) clinical trial and is currently being studied in parallel phase 2 clinical trials for the treatment of postpartum depression (PPD), major depressive disorder (MDD), and essential tremor (ET).

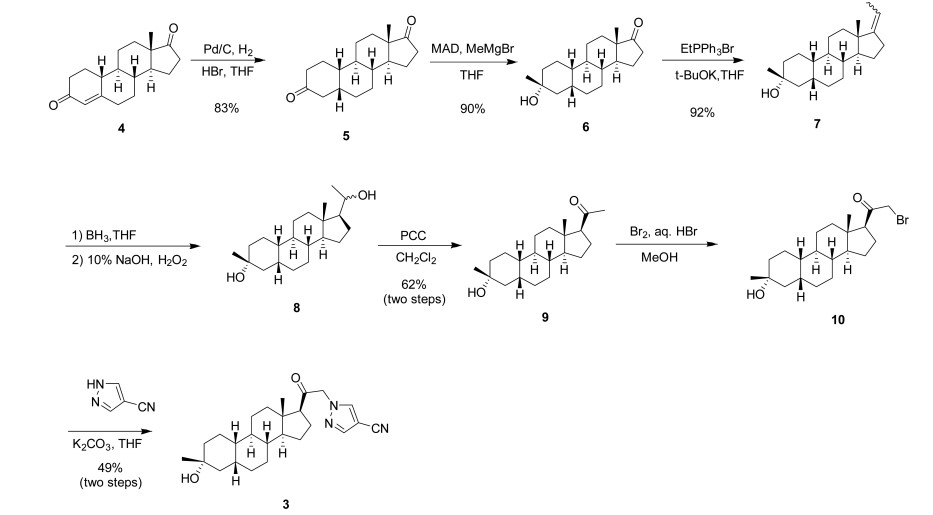

3α-Hydroxy-3β-methyl-21-(4-cyano-1H-pyrazol-1′-yl)-19- nor-5β-pregnan-20-one (3). Yield: 28 g (49%) as an off-white solid. LC-MS: tR = 1.00 min, m/z = 410 (M + 1). 1 H NMR (400 MHz, CDCl3): δ 7.86 (s, 1H), 7.80 (s, 1H), 5.08−4.84 (m, 2H), 2.70−2.55 (m, 1H), 2.25−2.15 (m, 1H), 2.10−2.00 (m, 1H), 1.88−1.59 (m, 7H), 1.53−1.30 (m, 15H), 1.25−1.00 (m, 3H), 0.67 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 13.92 (CH3), 23.20, 24.44, 25.54, 25.78, 26.15 (5 × CH2), 26.69 (CH3), 31.43, 34.61 (2 × CH2), 34.77, 37.71 (2 × CH), 39.26 (CH2), 40.35 (CH), 41.21 (CH2), 41.75 (CH), 45.56 (C), 56.04, 61.24 (2 × CH), 61.78 (CH2), 72.14 (C), 93.25 (C), 113.35 (CN), 136.16, 142.49 (2 × CH), 202.23 (CO). HRMS m/z 410.2803 calcd for C25H36N3O2 + 410.2802
PATENT
WO2014169833
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014169833&_cid=P11-LLRZJ9-40598-1
Synthetic Procedures
The compounds of the invention can be prepared in accordance with methods described in the art (Upasmi et al., J. Med. Chem. 1997, 40:73-84; and Hogenkamp et al., J. Med. Chem. 1997, 40:61- 72) and using the appropriate reagents, starting materials, and purification methods known to those skilled in the art. In some embodiments, compounds described herein can be prepared using methods shown in general Schemes 1-4, comprising a nucleophilic substitution of 19-nor pregnane bromide with a neucleophile. In certain embodiments, the nucleophile reacts with the 19-nor pregnane bromide in the presence of K2CO3 in THF.


Synthesis of compound SA-B. Compound SA (50 g, 184 mmol) and palladium black (2.5 g) in tetrahydrofuran (300 mL) and concentrated hydrobromic acid (1.0 mL) was hydrogenated with 10 atm hydrogen. After stirring at room temperature for 24h, the mixture was filtered through a pad of celite and the filtrate was concentrated in vacuo to afford the crude compound. Recrystallization from acetone gave compound SA-B (42.0 g, yield: 83.4%) as white powder.
1H NMR: (400 MHz, CDCl3) δ 2.45-2.41 (m, 1H), 2.11-3.44 (m, 2H), 3.24 (s, 3H), 2.18-2.15 (m, 1H), 2.01-1.95 (m, 1H), 1.81-1.57 (m, 7H), 1.53-1.37 (m, 7H), 1.29-1.13 (m, 3H), 1.13-0.90 (m, 2H), 0.89 (s, 3H).
Synthesis of compound SA-C. A solution of SA-B (42.0 g, 153.06 mmol) in 600 mL anhydrous toluene was added dropwise to the methyl aluminum bis(2,6-di-tert-butyl-4-methylphenoxide (MAD) (459.19 mmol, 3.0 eq, freshly prepared) solution under N2 at -78°C. After the addition was completed, the reaction mixture was stirred for 1 hr at -78°C. Then 3.0 M MeMgBr (153.06 mL, 459.19 mmol) was slowly added dropwise to the above mixture under N2 at -78°C. Then the reaction mixture was stirred for 3 hr at this temperature. TLC (Petroleum ether/ethyl acetate = 3:1) showed the reaction was completed. Then saturated aqueous NH4Cl was slowly added dropwise
to the above mixture at -78°C. After the addition was completed, the mixture was filtered, the filter cake was washed with EtOAc, the organic layer was washed with water and brine, dried over anhydrous Na2SO4, filtered and concentrated, purified by flash Chromatography on silica gel (Petroleum ether/ ethyl acetate20:1 to 3:1) to afford compound SA-C (40.2 g, yield: 90.4%) as white powder. 1H NMR: (400 MHz, CDCl3) δ 2.47-2.41 (m, 1H), 2.13-2.03 (m, 1H), 1.96-1.74 (m, 6H), 1.70-1.62 (m, 1H), 1.54-1.47 (m, 3H), 1.45-1.37 (m, 4H), 1.35-1.23 (m, 8H), 1.22-1.10 (m, 2H), 1.10-1.01 (m, 1H), 0.87 (s, 3H).
Synthesis of compound SA-D. To a solution of PPh3EtBr (204.52 g, 550.89 mmol) in THF (500 mL) was added a solution of t-BuOK (61.82 g, 550.89 mmol) in THF (300 mL) at 0°C. After the addition was completed, the reaction mixture was stirred for 1 h 60 °C, then SA-C (40.0 g, 137.72 mmol) dissolved in THF (300 mL) was added dropwise at 60°C. The reaction mixture was heated to 60 °C for 18 h. The reaction mixture was cooled to room temperature and quenched with Sat. NH4Cl, extracted with EtOAc (3*500 mL). The combined organic layers were washed with brine, dried and concentrated to give the crude product, which was purified by a flash column chromatography (Petroleum ether/ ethyl acetate50:1 to 10:1) to afford compound SA-D (38.4 g, yield:92%) as a white powder. 1H NMR: (400 MHz, CDCl3) δ 5.17-5.06 (m, 1H), 2.42-2.30 (m, 1H), 2.27-2.13 (m, 2H), 1.89-1.80 (m, 3H), 1.76-1.61 (m, 6H), 1.55-1.43 (m, 4H), 1.42-1.34 (m, 3H), 1.33-1.26 (m, 6H), 1.22-1.05 (m, 5H), 0.87 (s, 3H).
Synthesis of compound SA-E. To a solution of SA-D (38.0 g, 125.62 mmol) in dry THF (800 mL) was added dropwise a solution of BH3.Me2S (126 mL, 1.26 mol) under ice-bath. After the addition was completed, the reaction mixture was stirred for 3 h at room temperature (14-20 °C). TLC (Petroleum ether/ ethyl acetate3:1) showed the reaction was completed. The mixture was cooled to 0 °C and 3.0 M aqueous NaOH solution (400 mL) followed by 30% aqueous H2O2 (30%, 300 mL) was added. The mixture was stirred for 2 h at room temperature (14-20 °C), and then filtered, extracted with EtOAc (3*500 mL). The combined organic layers were washed with saturated aqueous Na2S2O3, brine, dried over Na2SO4 and concentrated in vacuum to give the crude product (43 g , crude) as colorless oil. The crude product was used in the next step without further purification.
Synthesis of compound SA-F. To a solution of SA-E (43.0 g, 134.16 mmol) in dichloromethane (800 mL) at 0 °C and PCC (53.8 g, 268.32 mmol) was added portion wise. Then the reaction mixture was stirred at room temperature (16-22 °C) for 3 h. TLC (Petroleum ether/ ethyl acetate3:1) showed the reaction was completed, then the reaction mixture was filtered, washed with DCM. The organic phase was washed with saturated aqueous Na2S2O3, brine, dried over Na2SO4 and concentrated in vacuum to give the crude product. The crude product was purified by a flash column chromatography (Petroleum ether/ ethyl acetate50:1 to 8:1) to afford compound SA-F (25.0 g, yield:62.5%, over two steps) as a white powder. 1H NMR (SA-F): (400 MHz, CDCl3) δ 2.57-2.50 (m, 1H), 2.19-2.11 (m, 4H), 2.03-1.97 (m, 1H), 1.89-1.80 (m, 3H), 1.76-1.58 (m, 5H), 1.47-1.42 (m, 3H), 1.35-1.19 (m, 10H), 1.13-1.04 (m, 3H), 0.88-0.84 (m, 1H), 0.61 (s, 3H).
Synthesis of compound SA. To a solution of SA-F (10 g, 31.4 mmol) and aq. HBr (5 drops, 48% in water) in 200 mL of MeOH was added dropwise bromine (5.52 g, 34.54 mmol). The reaction mixture was stirred at 17 °C for 1.5 h. The resulting solution was quenched with saturated aqueous NaHCO3 at 0°C and extracted with EtOAc (150 mLx2). The combined organic layers were dried and concentrated. The residue was purified by column chromatography on silica gel eluted with (PE: EA=15:1 to 6:1) to afford compound SA (9.5 g, yield: 76.14%) as a white solid. LC/MS: rt 5.4 mm ; m/z 379.0, 381.1, 396.1.

To a suspension of K2CO3 (50 mg, 0.36mmol) in THF (5 mL) was added ethyl 1H-pyrazole-4-carbonitrile (100 mg, 0.97 mmol ) and SA (50 mg,0.12 mmol). The mixture was stirred at rt for 15h. The reaction mixture was poured in to 5 mL H2O and extracted with EtOAc (2 x 10 mL). The combined organic layers were washed with brine, dried over sodium sulfate, filtered and concentrated. The residue mixture was purified with by reverse-phase prep-HPLC to afford the title compound as a white solid (9mg, 17.4%). 1H NMR (500 MHz, CDCl3), δ (ppm) 7.87 (1H, s),
7.82 (1H, s), 5.02 (1H, AB), 4.92 (1H, AB), 2.61 (1H, t), 2.16-2.24 (1H, m), 2.05 (1H, dXt), 1.70-1.88 (6H, m), 1.61-1.69 (2H, m), 1.38-1.52 (6H, m), 1.23-1.38 (5H, m), 1.28 (3H, s), 1.06-1.17 (3H, m), 0.67 (3H, s). LCMS: rt = 2.24 mm, m/z = 410.1 [M+H]+.
PATENT
WO2020150210
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
///////////
References
- ^ Jump up to:a b c d e “Zurzuvae (zuranolone) capsules, for oral use, [controlled substance schedule pending]” (PDF). Archived (PDF) from the original on 5 August 2023. Retrieved 5 August 2023.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t “FDA Approves First Oral Treatment for Postpartum Depression”. U.S. Food and Drug Administration (FDA) (Press release). 4 August 2023. Retrieved 4 August 2023. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “Zuranolone”. DrugBank Online.
- ^ Jump up to:a b Cerne R, Lippa A, Poe MM, Smith JL, Jin X, Ping X, et al. (2022). “GABAkines – Advances in the discovery, development, and commercialization of positive allosteric modulators of GABAA receptors”. Pharmacology & Therapeutics. 234: 108035. doi:10.1016/j.pharmthera.2021.108035. PMC 9787737. PMID 34793859. S2CID 244280839.
- ^ Jump up to:a b Faden J, Citrome L (2020). “Intravenous brexanolone for postpartum depression: what it is, how well does it work, and will it be used?”. Therapeutic Advances in Psychopharmacology. 10: 2045125320968658. doi:10.1177/2045125320968658. PMC 7656877. PMID 33224470.
- ^ Jump up to:a b “SAGE 217”. AdisInsight. Archived from the original on 29 March 2019. Retrieved 10 February 2018.
- ^ Jump up to:a b Blanco MJ, La D, Coughlin Q, Newman CA, Griffin AM, Harrison BL, et al. (2018). “Breakthroughs in neuroactive steroid drug discovery”. Bioorganic & Medicinal Chemistry Letters. 28 (2): 61–70. doi:10.1016/j.bmcl.2017.11.043. PMID 29223589.
- ^ Martinez Botella G, Salituro FG, Harrison BL, Beresis RT, Bai Z, Blanco MJ, et al. (2017). “Neuroactive Steroids. 2. 3α-Hydroxy-3β-methyl-21-(4-cyano-1H-pyrazol-1′-yl)-19-nor-5β-pregnan-20-one (SAGE-217): A Clinical Next Generation Neuroactive Steroid Positive Allosteric Modulator of the (γ-Aminobutyric Acid)A Receptor”. Journal of Medicinal Chemistry. 60 (18): 7810–7819. doi:10.1021/acs.jmedchem.7b00846. PMID 28753313.
- ^ Saltzman J (4 August 2023). “FDA approves postpartum depression pill from two Cambridge drug firms”. The Boston Globe. Archived from the original on 6 August 2023. Retrieved 5 August 2023.
- ^ Althaus AL, Ackley MA, Belfort GM, Gee SM, Dai J, Nguyen DP, et al. (2020). “Preclinical characterization of zuranolone (SAGE-217), a selective neuroactive steroid GABAA receptor positive allosteric modulator”. Neuropharmacology. 181: 108333. doi:10.1016/j.neuropharm.2020.108333. PMC 8265595. PMID 32976892.
- ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 82”. WHO Drug Information. 33 (3). hdl:10665/330879.
- ^ “FDA Approves Zurzuvae (zuranolone), the First and Only Oral Treatment Approved for Women with Postpartum Depression, and Issues a Complete Response Letter for Major Depressive Disorder” (Press release). Biogen Inc. 4 August 2023. Retrieved 4 August 2023 – via GlobeNewswire.
- ^ McKenzie H. “Sage Hints at Difficult Decisions After Zuranolone’s Rejection in MDD”.
- ^ Clayton AH, Lasser R, Parikh SV, Iosifescu DV, Jung J, Kotecha M, et al. (May 2023). “Zuranolone for the Treatment of Adults With Major Depressive Disorder: A Randomized, Placebo-Controlled Phase 3 Trial”. The American Journal of Psychiatry: appiajp20220459. doi:10.1176/appi.ajp.20220459. PMID 37132201. S2CID 258461851.
- ^ Deligiannidis KM, Meltzer-Brody S, Gunduz-Bruce H, Doherty J, Jonas J, Li S, et al. (2021). “Effect of Zuranolone vs Placebo in Postpartum Depression: A Randomized Clinical Trial”. JAMA Psychiatry. 78 (9): 951–959. doi:10.1001/jamapsychiatry.2021.1559. PMC 8246337. PMID 34190962.
- ^ Bullock A, Kaul I, Li S, Silber C, Doherty J, Kanes SJ (2021). “Zuranolone as an oral adjunct to treatment of Parkinsonian tremor: A phase 2, open-label study”. Journal of the Neurological Sciences. 421: 117277. doi:10.1016/j.jns.2020.117277. PMID 33387701. S2CID 229333842.
External links
- Clinical trial number NCT04442503 for “A Study to Evaluate the Efficacy and Safety of SAGE-217 in Participants With Severe Postpartum Depression (PPD)” at ClinicalTrials.gov
- Clinical trial number NCT02978326 for “A Study to Evaluate SAGE-217 in Participants With Severe Postpartum Depression” at ClinicalTrials.gov
/////////Zuranolone, FDA 2023, APPROVALS 2023, Zurzuvae, postpartum depression , SAGE 217, SAGE-217, SAGE217
[H][C@@]1(CC[C@@]2([H])[C@]3([H])CC[C@]4([H])C[C@](C)(O)CC[C@]4([H])[C@@]3([H])CC[C@]12C)C(=O)CN1C=C(C=N1)C#N
EIDD-2173, ATI-2173, Fosclevudine alafenamide

EIDD-2173; also known as ATI-2173
Fosclevudine alafenamide
Phase 2
| Molecular Weight | 529.45 |
|---|---|
| Formula | C22H29FN3O9P |
| CAS No. | 1951476-79-1 |
Hepatitis B virus (HBV) is an infectious disease that targets the liver resulting in either an acute infection, with symptoms arising in 45 to 160 days, or a chronic infection, which 350 million people worldwide are affected by. Estimates indicate that 600,000 deaths occur each year as a result of consequences related to HBV infection. HBV possesses a 3.2- kb relaxed circular DNA (rcDNA) genome that is used to form covalently closed circular DNA (cccDNA) in a host cell. The cccDNA is then transcribed by RNA polymerase II, a host DNA-dependent RNA polymerase, to produce pregenomic RNA (pgRNA). The pgRNA is then used by the virally encoded reverse transcriptase to form rcDNA. The goals of current treatments for chronic HBV infections are to reduce HBV replication and reduce liver damage.
Current treatments for chronic HBV infections include pegylated alpha interferon and nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs). NRTIs are converted to their corresponding 5 ‘-triphosphate, or diphosphate in the case of phosphonate containing NRTIs, and reduce viral replication by inhibiting the HBV encoded polymerase. Clevudine is an NRTI that is no longer being developed for the treatment of chronic HBV because of drug-related skeletal myopathy that was a result of mitochondrial dysfunction in patients.
Interestingly, clevudine triphosphate has been shown to be a competitive nonsubstrate inhibitor of the HBV encoded polymerase, and due to its long intracellular half-life, is able to suppress HBV replication for an extended period of time after drug withdrawal.
The discovery and synthesis of the (S,S) and (S,R) diastereomers of clevudine phosphoramidate has been previously reported. These studies were undertaken to address the myopathy concerns associated with clevudine. The phosphoramidate moiety was utilized to deliver clevudine, as its 5 ‘-monophosphate, to the liver reducing 1) systemic exposure to clevudine and 2) the possibility of skeletal myopathy. Both phosphoramidates showed anti-HBV activity similar to clevudine with the (S,S) diastereomer being slightly more potent.
SYN
See U.S. Patent No. 10,683,319.
PATENT
WO20 17223421
PATENT


EXAMPLE 11 :
[0374] Preparation of intermediate ATI-2173 from Compound-10.
[0375] Experimental Procedure
[0376] (H-l) Crude Compound-10 in THF (see Example 10) was divided into three aliquots and stirred with 2% HC1 aq. solution at pH 5-6, 4-5, and 3-4; for 16 h at 20-25 °C; the aliquots were combined and stirred at 15-20 °C for 72 h, with no degradation of ATI-2173 observed over the second time period;
[0377] (H-2) the pH of the mixture was adjusted to 7 with 7% NaHCCh;
[0378] (H-3) phase separation was carried out using 2-MeTHF, the organic phase was washed with NA2SO4 aqueous soltion, then concentrated to 1-3 V, and MTBE (5 V) was added; this operation was repeated twice;
[0379] (H-5) ATI-2173 was precipitated gradually upon addition of seed crystal and addition of n-heptane (5 V);
[0380] (H-6) the product was filtered; and
[0381] (H-7) the wetcake was dried, resulting in ATI-2173 in 99.73a% purity and
84.4% yield.
[0382] EXAMPLE 12:
[0383] Crystallization of ATI-2173
[0384] Initial studies examined the use of single or mixed solvent systems to crystalize the amorphous product, ATI-2173. Several solvent conditions were screened, including single solvent and mixed solvent systems, in order to determine the potential for obtaining a crystalline material from the amorphous material. None of the solvents tested worked and all conditions produced an oil product. The results are shown below in Tables 8 and 9.
[0385] Table 8: Single Solvent Systems
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
///////////
////////// EIDD-2173, ATI-2173, EIDD 2173, ATI 2173, Hepatitis B virus, ANTI HBV, Fosclevudine alafenamide, PHASE 2

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}