
Home » 2020 (Page 3)
Yearly Archives: 2020
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
RALOXIFENE
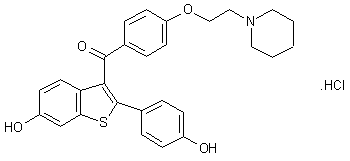
Keoxifene hydrochloride, Raloxifene hydrochloride, LY-139481(free base), LY-156758, Optruma, Loxifen, EvistaTitle: RaloxifeneCAS Registry Number: 84449-90-1CAS Name: [6-Hydroxy-2-(4-hydroxyphenyl)benzo[b]thien-3-yl][4-[2-(1-piperidinyl)ethoxy]phenyl]methanoneAdditional Names: keoxifeneManufacturers’ Codes: LY-139481Molecular Formula: C28H27NO4SMolecular Weight: 473.58Percent Composition: C 71.01%, H 5.75%, N 2.96%, O 13.51%, S 6.77%Literature References: Nonsteroidal, selective estrogen receptor modulator (SERM). Prepn: C. D. Jones, EP62503; idem,US4418068 (1982, 1983 both to Lilly); idemet al.,J. Med. Chem.27, 1057 (1984). Review of pharmacology and toxicology: J. Buelke-Sam et al.,Reprod. Toxicol.12, 217-221 (1998); of clinical pharmacology and pharmacokinetics: D. Hochner-Celnikier, Eur. J. Obstet. Gynecol. Reprod. Biol.85, 23-29 (1999); of clinical efficacy in osteoporosis: D. Agnusdei, ibid. 43-46. Clinical effect on risk of breast cancer: S. R. Cummings et al.,J. Am. Med. Assoc.281, 2189 (1999); on reduction of fracture risk: B. Ettinger et al.,ibid.282, 637 (1999).Properties: Crystals from acetone, mp 143-147°. uv max (ethanol): 290 nm (e 34000).Melting point: mp 143-147°Absorption maximum: uv max (ethanol): 290 nm (e 34000)
Derivative Type: HydrochlorideCAS Registry Number: 82640-04-8Manufacturers’ Codes: LY-156758Trademarks: Evista (Lilly)Molecular Formula: C28H27NO4S.HClMolecular Weight: 510.04Percent Composition: C 65.94%, H 5.53%, N 2.75%, O 12.55%, S 6.29%, Cl 6.95%Properties: Crystals from methanol/water, mp 258°. uv max (ethanol): 286 nm (e 32800).Melting point: mp 258°Absorption maximum: uv max (ethanol): 286 nm (e 32800)
Therap-Cat: Antiosteoporotic.Keywords: Antiosteoporotic; Selective Estrogen Receptor Modulator (SERM).
Raloxifene, sold under the brand name Evista among others, is a medication used to prevent and treat osteoporosis in postmenopausal women and those on glucocorticoids.[4] For osteoporosis it is less preferred than bisphosphonates.[4] It is also used to reduce the risk of breast cancer in those at high risk.[4] It is taken by mouth.[4]
Common side effects include hot flashes, leg cramps, swelling, and joint pain.[4] Severe side effects may include blood clots and stroke.[4] Use during pregnancy may harm the baby.[4] The medication may worsen menstrual symptoms.[5] Raloxifene is a selective estrogen receptor modulator (SERM) and therefore a mixed agonist–antagonist of the estrogen receptor (ER).[4] It has estrogenic effects in bone and antiestrogenic effects in the breasts and uterus.[4]
Raloxifene was approved for medical use in the United States in 1997.[4] It is available as a generic medication.[4][6] A month supply in the United Kingdom costs the NHS about 3.50 £ as of 2019.[6] In the United States the wholesale cost of this amount is about $16.[7] In 2017, it was the 330th most commonly prescribed medication in the United States, with more than 900 thousand prescriptions.[8
Medical uses
Raloxifene is used for the treatment and prevention of osteoporosis in postmenopausal women.[9] It is used at a dosage of 60 mg/day for both the prevention and treatment of osteoporosis.[10] In the case of either osteoporosis prevention or treatment, supplemental calcium and vitamin D should be added to the diet if daily intake is inadequate.[11]
Raloxifene is used to reduce the risk of breast cancer in postmenopausal women. It is used at a dosage of 60 mg/day for this indication.[10] In the Multiple Outcomes of Raloxifene (MORE) clinical trial, raloxifene decreased the risk of all types of breast cancer by 62%, of invasive breast cancer by 72%, and of invasive estrogen receptor-positive breast cancer by 84%.[12] Conversely, it does not reduce the risk of estrogen receptor-negative breast cancer.[12] There were no obvious differences in effectiveness of raloxifene in the MORE trial for prevention of breast cancer at a dosage of 60 mg/m2/day relative to 120 mg/m2/day.[12] In the Study of Tamoxifen and Raloxifene (STAR) trial, 60 mg/day raloxifene was 78% as effective as 20 mg/day tamoxifen in preventing non-invasive breast cancer.[13] Women with undetectable levels of estradiol (<2.7 pg/mL) have a naturally low risk of breast cancer and, in contrast to women with detectable levels of estradiol, do not experience significant benefit from raloxifene in terms of reduction of breast cancer risk.[12]
Contraindications
Raloxifene is contraindicated in lactating women or women who are or who may become pregnant.[14] It also may be of concern to women with active or past history of venous thromboembolic events, including deep vein thrombosis, pulmonary embolism, and retinal vein thrombosis.[15]
Side effects
Common side effects of raloxifene include hot flashes (25–28% vs. 18–21% for placebo),[12] vaginal dryness, and leg cramps (generally mild; 5.5% vs. 1.9% for placebo).[14][1][16] Raloxifene does not cause breast tenderness, endometrial hyperplasia, menstrual bleeding, or endometrial cancer.[17] It does not appear to affect cognition or memory.[15][12] Raloxifene is a teratogen; i.e., it can cause developmental abnormalities such as birth defects.
Raloxifene may infrequently cause serious blood clots to form in the legs, lungs, or eyes.[1] Other reactions experienced include leg swelling/pain, trouble breathing, chest pain, and vision changes. Black box warnings were added to the label of raloxifene in 2007 warning of increased risk of death due to stroke for postmenopausal women with documented coronary heart disease or at increased risk for major coronary events, as well as increased risk for deep vein thrombosis and pulmonary embolism.[14] The risk of venous thromboembolism with raloxifene is increased by several-fold in postmenopausal women (RR = 3.1).[18][12] Raloxifene has a lower risk of thromboembolism than tamoxifen.[13] In the MORE trial, raloxifene caused a 40% decrease in risk of cardiovascular events in women who were at increased risk for coronary artery disease, although there was no decrease in cardiovascular events for the group as a whole.[12]
A report in September 2009 from Health and Human Services’ Agency for Healthcare Research and Quality suggests that tamoxifen and raloxifene, used to treat breast cancer, significantly reduce invasive breast cancer in midlife and older women, but also increase the risk of adverse side effects.[19]
A recent human case report in July 2016 suggests that raloxifene may in fact, at some point, also stimulate breast cancer growth leading to a reduction of advanced breast cancer disease upon the withdrawal of the drug.[20]
Unlike other SERMs, such as tamoxifen, raloxifene has no risk of uterine hyperplasia or endometrial cancer (RR = 0.8).[1][18][13]
Raloxifene does not increase the incidence of breast pain or tenderness in postmenopausal women.[16][21]
Overdose
Raloxifene has been studied in clinical trials across a dosage range of 30 to 600 mg/day, and was well-tolerated at all dosages.[16]
Pharmacology
Pharmacodynamics
Mechanism of action
Raloxifene is a selective estrogen receptor modulator (SERM) and hence is a mixed agonist and antagonist of the estrogen receptor (ER) in different tissues.[4] It has estrogenic activity in some tissues, such as bone and the liver, and antiestrogenic activity in other tissues, such as the breasts and uterus.[4] Its affinity (Kd) for the ERα is approximately 50 pM, which is similar to that of estradiol.[16] Relative to estradiol, raloxifene has been reported to possess about 8 to 34% of the affinity for the ERα and 0.5 to 76% of the affinity for the ERβ.[22][23] Raloxifene acts as a partial agonist of the ERα and as a pure antagonist of the ERβ.[24][25] In contrast to the classical ERs, raloxifene is an agonist of the G protein-coupled estrogen receptor (GPER) (EC50 = 10–100 nM), a membrane estrogen receptor.[26][27]
Clinical effects
Raloxifene has antiestrogenic effects in the mammary glands in preclinical studies.[16] In accordance, raloxifene reduces breast density in postmenopausal women, a known risk factor for breast cancer.[28] It does not stimulate the uterus in postmenopausal women, and results in no increase in risk of endometrial thickening, vaginal bleeding, endometrial hyperplasia, or endometrial cancer.[29][16][21] At the same time, raloxifene has minimal antiestrogenic effect in the uterus in premenopausal women.[29] This may possibly be due to inadequate tissue exposure of the uterus to raloxifene in these estrogen-rich individuals.[29]
In premenopausal women, raloxifene increases levels of follicle-stimulating hormone (FSH) and estradiol.[12] Conversely, in postmenopausal women, raloxifene has been found to reduce levels of the gonadotropins, luteinizing hormone (LH) and FSH, while not affecting levels of estradiol.[12][29] Raloxifene also decreases prolactin levels in postmenopausal women.[29] In men, raloxifene has been found to disinhibit the hypothalamic–pituitary–gonadal axis (HPG axis) and thereby increase total testosterone levels.[30][31][32][33] Due to the simultaneous increase in sex hormone-binding globulin (SHBG) levels however, free testosterone levels often remain unchanged in men during therapy with raloxifene.[30]
Raloxifene has estrogenic effects on liver protein synthesis.[12] It increases SHBG levels in both pre- and postmenopausal women as well as in men.[12][30] The medication decreases levels of total and low-density lipoprotein (LDL) cholesterol, C-reactive protein, apolipoprotein B, and homocysteine.[12][29] Conversely, it has little effect on levels of triglycerides and high-density lipoprotein (HDL).[12] Raloxifene has been shown to inhibit the oxidation of LDL cholesterol in vitro.[16] The medication has been found to decrease insulin-like growth factor 1 (IGF-1) levels in pre- and postmenopausal women as well as in men.[31] It has also been found to increase insulin-like growth factor binding protein 3 (IGFBP-3) levels in pre- and postmenopausal women.[12] Due to activation of estrogen receptors in the liver, raloxifene has procoagulatory effects, such as decreasing levels of fibrinogen and influencing levels of other coagulation factors.[12][29][16] For these reasons, raloxifene increases the risk of thrombosis.[12][29]
Raloxifene increases bone mineral density in postmenopausal women but decreases it in premenopausal women.[12] In the MORE trial, the risk of vertebral fractures was decreased by 30%, and bone mineral density was increased in the spine (by 2.1% at 60 mg, 2.4% at 120 mg) and femoral neck (2.6% at 60 mg, 2.7% at 120 mg).[18] It has been found to possess estrogenic effects in adipose tissue in postmenopausal women, promoting a shift from an android fat distribution to a gynoid fat distribution.[34][35] The medication has been found to increase levels of leptin, an adipokine.[12]
AbsorptionPharmacokinetics
The absorption of raloxifene is approximately 60%.[1][2] However, due to extensive first-pass metabolism, the absolute bioavailability of raloxifene is only 2.0%.[1][2] Raloxifene is rapidly absorbed from the intestines upon oral administration.[1] Peak plasma levels of raloxifene occur 0.5 to 6 hours after an oral dose.[1][2]
Distribution
Raloxifene is widely distributed throughout the body.[1] There is extensive distribution of raloxifene into the liver, serum, lungs, and kidneys.[1] The volume of distribution of raloxifene with a single 30 to 150 mg oral dose is approximately 2348 L.[1][36] Both raloxifene and its metabolites show high plasma protein binding (>95%), including to both albumin and α1 acid glycoprotein, but not to sex hormone-binding globulin.[1][2]
Metabolism
Raloxifene is metabolized in the liver and undergoes enterohepatic recycling.[2] It is metabolized exclusively by glucuronidation and is not metabolized by the cytochrome P450 system.[1][2] Less than 1% of radiolabeled material in plasma comprises unconjugated raloxifene.[2] The metabolites of raloxifene include several glucuronides.[1] The elimination half-life of raloxifene after a single dose is 27.7 hours (1.2 days), whereas its half-life at steady state at a dosage of 60 mg/day is 15.8 to 86.6 hours (0.7–3.6 days), with an average of 32.5 hours (1.4 days).[1][2] The extended half-life of raloxifene is attributed to enterohepatic recirculation and its high plasma protein binding.[1] Raloxifene and its glucuronide conjugates are interconverted by reversible metabolism and enterohepatic recycling, which prolongs the elimination half-life of raloxifene with oral administration.[2] The medication is deconjugated into its active form in a variety of tissues, including liver, lungs, spleen, bone, uterus, and kidneys.[1]
Elimination
Raloxifene is mainly excreted in bile and is eliminated in feces.[1][2] Less than 0.2% of a dose is excreted unchanged in urine and less than 6% of a dose is excreted in urine as glucuronide conjugates.[2]
Chemistry
See also: List of selective estrogen receptor modulators and Benzothiophene
Raloxifene hydrochloride has the empirical formula C28H27NO4S•HCl, which corresponds to a molecular weight of 510.05 g/mol. Raloxifene hydrochloride is an off-white to pale-yellow solid that is slightly soluble in water.[14]
Raloxifene is a benzothiophene derivative and is structurally distinct from the triphenylethylene SERMs like tamoxifen, clomifene, and toremifene.[37] It is the only benzothiophene SERM to have been marketed.[37] A benzothiophene SERM that was not marketed is arzoxifene (LY-353381).[38] Bazedoxifene (Duavee, Viviant) and pipendoxifene (ERA-923) are structurally related to raloxifene but are technically not benzothiophenes and instead are indoles.[38]
History
Raloxifene was approved in the United States for the prevention of postmenopausal osteoporosis in 1997, the treatment of postmenopausal osteoporosis in 1999, and to prevent or reduce the risk of breast cancer in certain postmenopausal women in 2007.[39][40][41][42] It received orphan designation in 2005.[39]
Society and culture

A bottle of raloxifene.
Names
Raloxifene is the generic name of the drug and its INN and BAN, while raloxifène is its DCF and raloxifene hydrochloride is its USAN, BANM, and JAN.[43][44][45][46] It has also been known by the name keoxifene.[43][44][46]
Raloxifene is sold mainly under the brand name Evista and to a lesser extent the brand name Optruma.[46][44] It is also sold under a variety of other brand names in various countries.[46]
Availability
Raloxifene is available widely throughout the world, including in the United States, Canada, the United Kingdom, Ireland, elsewhere throughout Europe, Australia, New Zealand, South Africa, Latin America, Southern, Eastern, and Southeastern Asia, and elsewhere in the world such as in Israel and Egypt.[46][44]
Raloxifene is provided in the form of 60 mg oral tablets.[10]
Controversy
An editorial in Lancet Oncology criticized the way that research about the medication for breast cancer prevention was released.[47]
Research
Clinical studies of raloxifene for metastatic breast cancer in women have been conducted but found little effectiveness at 60 mg/day in those previously treated with tamoxifen, though modest effectiveness has been observed at higher doses.[12][48] In contrast to tamoxifen, raloxifene is not approved for the treatment of breast cancer.[49]
Raloxifene has been studied in men for a variety of uses, such as for treatment of schizophrenia, prostate cancer, and osteoporosis.[50][51][52][53][54][33][32][55][56][57][58] It has been studied in combination with castration and bicalutamide, a nonsteroidal antiandrogen, for the treatment of prostate cancer.[58][55]
Raloxifene has been studied as an adjunct in the treatment of schizophrenia in postmenopausal women.[59] A 2017 meta-analysis concluded that it was safe and effective for this indication, although further studies with larger sample sizes are needed for confirmation.[59] It may be effective in women with less severe symptoms.[59]
A tissue-selective estrogen-receptor complex (TSEC) of estradiol and raloxifene has been studied in postmenopausal women.[60]
Raloxifene (60 mg/day) was reported to be effective in the treatment of pubertal gynecomastia in adolescent boys in a small retrospective chart review.[61][62][63] Other SERMs are also known to be effective in the treatment of gynecomastia.[64]
Raloxifene has been reported to augment the antidepressant effects of selective serotonin reuptake inhibitors (SSRIs).[65]
June 18th 2020, Exscalate4CoV, the private-public consortium supported by the EU’s Horizon 2020 programme for research and innovation, led by Dompé farmaceutici and currently representing 18 partners (including Fraunhofer Institute, CINECA, Chelonia Applied Science, Swiss Institute of Bioinformatics and others) has requested access to clinical trials for the use of Raloxifene in Covid 19 patients. Raloxifene, already proven effective against Mers and Sars in precliinical tests, has been indicated as effective against Sars-Cov2 by the “in-silico” research conducted by the consortium which has shown efficacy in countering the replication of the virus in cells. The IP for its use against Sars-Cov2 has already been protected on May 6 2020 in the name Dompé farmaceutici, Fraunhofer Institute and KU Leuven, to facilitate the largest possible access. Raloxifene would be used in mildly symptomatic Covid19 patients to halt the spread of infection. This result emerged from the first virtual (in silico) screening conducted on the Consortium’s supercomputers of more than 400.000 molecules (safe-in-man drugs and natural products) made available by Dompé farmaceutici and the partner Fraunhofer (IME) to the Consortium. The molecules were prioritized if in clinical stage or already on the market. 7.000 molecules with certain promising characteristics were tested.
SYN

Jones, Charles D.; Jevnikar, Mary G.; Pike, Andrew J.; Peters, Mary K.; Black, Larry J.; Thompson, Allen R.; Falcone, Julie F.; Clemens, James A. (1984). “Antiestrogens. 2. Structure-activity studies in a series of 3-aroyl-2-arylbenzo[b]thiophene derivatives leading to [6-hydroxy-2-(4-hydroxyphenyl)benzo[b]thien-3-yl]-[4-[2-(1-piperidinyl)ethoxy]phenyl]methanone hydrochloride (LY 156758), a remarkably effective estrogen antagonist with only minimal intrinsic estrogenicity”. Journal of Medicinal Chemistry 27 (8): 1057–66.doi:10.1021/jm00374a021. PMID 6431104.
syn 1
EP 0062053; GB 2097788
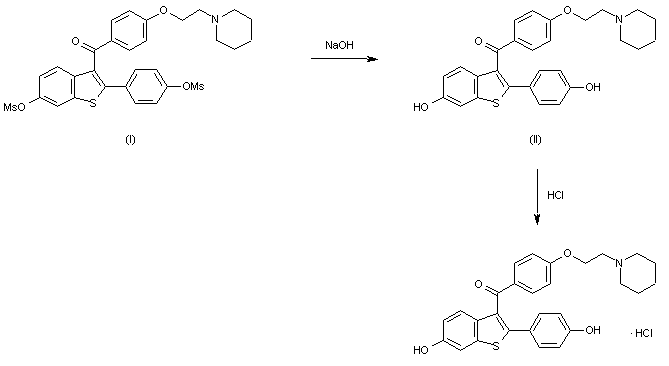
Keoxifene has been synthesized using the following process: A portion of 6-methanesulfonyloxy-2-(4-methanesulfonyloxyphenyl)-3-[4-(2-pipendinoethoxy)benzoyl]benzo[b]thiophene hydrochloride (I) was combined with denatured alcohol and 5N sodium hydroxide, and stirred under a nitrogen atmosphere. The reaction mixture was evaporated to dryness under vacuum, and the residue dissolved in water and washed with diethyl ether. The water layer was degassed under vacuum, and then nitrogen was bubbled through it to remove all traces of ether. The mixture was then acidified with 1N hydrochloric acid, and then made basic with excess sodium bicarbonate The precipitate was collected by filtration and washed with cold water to obtain crude product, which was purified on a column of silica gel. The column was eluted first with 700 ml of 5% methanol in chloroform, followed by 1l of 10% methanol in chloroform. The impurities came off first, and the product-containing fractions were combined and evaporated under vacuum to obtain a yellow oil. The oil was dissolved in acetone seeded and chilled in a freezer to obtain the purified product.
syn2
J Label Compd Radiopharm 1995,36(1),43

The synthesis of radiolabeled raloxifene has been reported: The esterification of 3,5-dibromo-4-hydroxybenzoic acid (I) with methanol/HCl gives the corresponding methyl ester (II), which is condensed with 1-(2-chloroethyl)piperidine (III) by means of K2CO3 in DMF yielding 3,5-dibromo-4-[2-(1-piperidyl)ethoxy]benzoic acid methyl ester (IV). The hydrolysis of (IV) with NaOH in methanol affords the corresponding free acid (V), which by treatment of SOCl2 in toluene is converted to the acyl chloride (VI). The Friedel-Crafts condensation of (VI) with 6-methoxy-2-(4-methoxyphenyl)benzo[b]thiophene (VII) by means of AlCl3 in dichloromethane gives [3,5-dibromo-4-[2-(1-piperidinyl)ethoxy]phenyl]-[6-methoxy-2-(4-methoxy phenyl)benzo[b]thien-3-yl]methanone (VIII), which is demethylated with AlCl3 and ethylmercaptane to dibromoraloxifene (IX). Finally, this compound is submitted to hydrogenolysis with tritium over Pd/C in methanol.
syn 3
Bioorg Med Chem Lett 1997,7(8),993
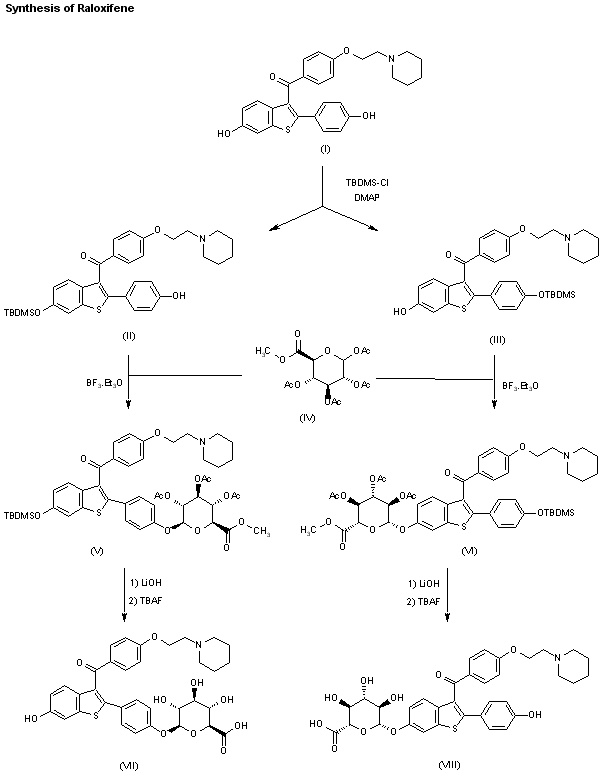
The two major metabolites of raloxifene, the glucuronide conjugates (VI) and (VIII) are synthesized as follows: The partial silylation of raloxifene (I) with tert-butyldimethylsilyl chloride (TBDMS-Cl) by means of dimethylaminopyridine (DMAP) in THF/DMF gives a mixture of the monosilylated compounds (II) and (III), which are separated by chromatography. Compounds (II) and (III) are independently condensed with methyl 1,2,3,4-tetra-O-acetyl-D-glucuronate (IV) by means of BF3.OEt2 in dichloromethane yielding protected glucuronides (V) and (VII), respectively. Finally, both compounds are deprotected by a treatment first with LiOH in dioxane to hydrolyzed the ester groups, and then with tetrabutylammonium fluoride in THF to eliminate the silyl groups, thus obtaining the desired metabolites (VI) and (VIII), respectively.
syn 4
Tetrahedron Lett 1999,40(28),5155
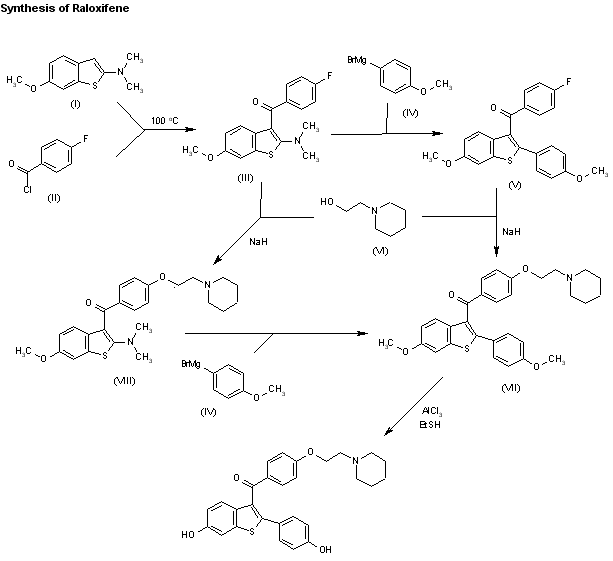
Two related new syntheses of raloxifene have been described: 1) The acylation of N-(6-methoxy-1-benzothiophen-2-yl)-N,N-dimethylamine (I) with 4-fluorobenzoyl chloride (II) by heating at 100 C in chlorobenzene gives the 3-acyl derivative (III), which is condensed with 4-methoxyphenylmagnesium bromide (IV) in THF yielding 3-(4-fluorobenzoyl)-6-methoxy-2-(4-methoxyphenyl)-1-benzothiophene (V). The condensation of (V) with 1-(2-hydroxyethyl)piperidine (VI) by means of NaH in DMF affords the ether (VII), which is finally demethylated with AlCl3 and ethanethiol. 2) The intermediate (III) can also be condensed first with 1-(2-hydroxyethyl)piperidine (VI) by means of NaH as before giving the piperidinoethyl ether (VIII), which is then condensed with the Grignard reagent (IV) affording the previously reported ether (VII).
syn
Org Chem Ind J, Volume: 14( 3)
A GREEN PROCESS FOR DEMETHYLATION REACTION IN SYNTHESIS OF RALOXIFENE HYDROCHLORIDEAuthors : Ramadas Chavakula *, Chakradhar Saladi J S, Narayana Rao Mutyalaa , Vijaya Raju Maddalaa and Raghu Babu Kb

A green process for demethylation reaction in synthesis of raloxifene hydrochloride by using aluminium chloride and odorless decanethiol as demethylation agent instead of aluminium chloride and ethanethiol (foul smell) under normal conditions is described.
Raloxifene hydrochloride [1], is an estrogen agonist/antagonist, commonly referred to as a Selective Estrogen Receptor Modulator (SERM) [1,2] that belongs to the benzothiophene class of compounds. Raloxifene decreases the resorption of bone and reduces the biochemical markers of bone turnover to the premenopausal range [3–5]. Raloxifene hydrochloride may also lower the chance of developing a certain type of breast cancer (invasive breast cancer) in post-menopausal women [6,7]. It can be synthesized [3] directly from aroylation of 6-methoxy-2-(4-methoxyphenyl)benzo[b]thiophene [2] by the acid chloride(4) of 4-[2-(1-piperidinyl)ethoxy]benzoic acid hydrochloride [3] in the presence of AlCl3 followed by addition of ethanethiol (FIG. 1).
Experimental Section
4-[2-(1-Piperidinyl)ethoxy]benzoic acid hydrochloride [3] and 6-methoxy-2-(4-methoxyphenyl) benzo[b] thiophene [2] were prepared by procedures reported previously [3]. Decanethiol was from commercial source. All melting points are uncorrected and were determined in capillary tubes on an Electothermal melting point apparatus. 1H NMR spectra were recorded on a Brucker ADVANCE 400 MHz spectrometer, using DMSO-d6 as solvent and TMS as internal standard. Electrospray ionization mass spectroscopy was performed using an ion trap mass spectrometer (Model 6310 Agilent). All reactions were monitored and checked by Thin Layer Chromatography (TLC) using methanol and spots examined by a UV lamp.
Preparation of [6-hydroxy-2-(4-hydroxyphenyl)benzo[b]thiophen-3-yl][4-[2-(1-piperidyl)ethoxy]phenyl] methanone hydrochloride (Raloxifene hydrochloride) [1]
To a solution of 4-[2-(1-piperidinyl)ethoxy]benzoic acid hydrochloride (3) (14.3 g, 0.05 mol) in methylene dichloride (400 mL) and pyridine (0.5 mL) at 25ºC to 35ºC, thionyl chloride (23.8 g, 0.20 mol) was added dropwise under argon for 15-30 minute. The reaction mixture was stirred for 2 hr. at 40ºC to 45ºC. Excess thionyl chloride and solvent were removed in vacuum at 40◦C to afford 15.0 g of the crude acid chloride hydrochloride salt [4]. The crude solid acid chloride hydrochloride [4] was dissolved in methylene dichloride (150 mL), cooled to 0ºC to 10ºC, 6-methoxy-2-(4-methoxyphenyl)benzo[b] thiophene [2] (10.8 g, 0.04 mol) was added. Then, anhydrous aluminium chloride (37.0 g, 0.28 mol) was added portion wise over a period of 30 min and then the mixture was allowed to warm to 30ºC and stirred for 2 hr at 25-35ºC. Then decanethiol (28.0 g, 0.16 mol) was added and stirred for 2 hr. at 25-35ºC. The reaction mixture was quenched with mixture of methanol (100 mL), ice (200 g) and Conc. HCl (15 mL) and stirred for 1 hr. at 25-35ºC. The precipitated solid was collected, washed with water (100 mL X 2) and dried at 65ºC for 4 h to afford 20.0 g of crude compound 1, which was crystallized from methanol/water (23/1, vol/vol) to yield 13.6 g of compound 1 (53.3 %yield) as a white solid, MP 258-260°C, liter 3, 258°C ; 1H NMR: δ 1.34, 1.72 [2H, m, (CH2CH2)2CH2], 1.76 [4H, m, N(CH2CH2)2], 2.96 (2H, m, N-CH2), 3.43 [4H, m, N(CH2CH2)2], 4.44 (2H, m, O-CH2), 6.67 (2H, d, Ar), 6.85 (1H, d, Ar), 6.95 (2H, d, Ar), 7.18 (2H, d, Ar), 7.25 (1H, d, Ar), 7.35 (1H, s, Ar), 7.70 (2H, d, Ar), 9.77 (1H, s, OH), 9.82 (1H, s, OH), 10.16 (1H, brs, NH), MS (ESI): m/z 474.6 (M +H). “This procedure has been scaled up using 250g of compound 1.”
Results and Discussion
Commonly used thiols like ethanethiol and benzyl mercaptan in demethylation reactions have a foul smell making them difficult and unpleasant to use in the laboratory without fume hoods. The problem becomes even worse in industry on a large scale. Odorless substitutes are therefore always required. Few papers [8,9] discuss the use of long chain thiols to minimize odor, so we used this work as a basis for choosing a long chain thiol for our demethylation reaction. We now report a new, highly active demethylation reagent, an aluminum chloride and decanethiol, characterized by rapid action under mild conditions, easy workup of the reaction product, and high yield (FIG. 2.).

Figure 2: Synthesis of Raloxifene hydrochloride.
Conclusion
In conclusion, we have found that decanethiol is odorless thiol compared to ethanethiol. We believe that removing the foul-smelling thiols and use of these odorless thiols will greatly improve the greenchemistry.
References
- Grese TA, Dodge JA. Selective Estrogen Receptor Modulators (SERMs). Curr Pharm Des. 1998;4:71-92.
- Bryant HU, Dere WH. Selective estrogen receptor modulators: an alternative to hormone replacement therapy. Proc Soc Exp Biol Med. 1998;217:45-52.
- Jones CD, Jevnikar MG, Pike AJ, et al. Antiestrogens. 2. Structure-activity studies in a series of 3-aroyl-2-arylbenzo [b] thiophene derivatives leading to [6-hydroxy-2-(4-hydroxyphenyl) benzo [b] thien-3-yl]-[4-[2-(1-piperidinyl) ethoxy] phenyl] methanone hydrochloride (LY 156758), a remarkably effective estrogen antagonist with only minimal intrinsic estrogenicity. J Med Chem. 1984;27:1057-66.
- Sato M, Grese TA, Dodge JA, et al. Emerging therapies for the prevention or treatment of postmenopausal osteoporosis. J Med Chem. 1999;42:1-24.
- Draper MW, Flowers DE, Huster WJ, et al. A controlled trial of raloxifene (LY139481) HCl: impact on bone turnover and serum lipid profile in healthy postmenopausal women. J Bone Miner Res. 1996;11:835-42.
paper
https://www.sciencedirect.com/science/article/abs/pii/S0223523412001122





syn
https://www.tandfonline.com/doi/abs/10.1080/00397911.2014.943348?journalCode=lsyc20
Piperidine Nucleophilic Substitution Without Solvent: An Efficient Synthesis of RaloxifeneYewei Yang,Tao Zhang,Wenhai Huang &Zhenrong Shen Pages 3271-3276 |
Mild and high-yielding synthesis is described for raloxifene via piperdine nucleophilic substitution of a new raloxifene intermediate 3-aroyl-2-aryl-substituted benzo[b]thiophenes, which is obtained by acylation of para-substituted benzoyl chlorides and 2-arylbenzo[b]thiophenes. The key step is solvent free and offers valuable advantages, such as low cost, and is suitable for industrial production.

Keywords: Friedel–Crafts acylation, green chemistry, nucleophiles, raloxifene, SERM

The improved synthesis of raloxifene 1 was accomplished as shown in Scheme 2. Methyl p-hydroxybenzoate 2, 1-bromo-2-chloroethane, and K2CO3 were refluxed in acetone, yielding compound 3 in 94% yield. Without prior purification, 3 was hydrolyzed to the corresponding p-substituted benzoyl acids 4 in 100% yield. The application of general reaction conditions of methanol as solvent and hydrochloric as acid would afford the substitution impurity 4-(2-methoxyethoxy)-benzoic acid. To control this impurity during reaction, various solvents such as alcohol, ethyl acetate, acetone, and tetrahydrofuran (THF) were screened, and THF gave the best result from the view of impurity formation and yield. Compound 4 is a solid and was easily isolated from THF by adding water. Then 4 was transferred to acid chlorides 5 and substantially reacted with benzothiophene 6 using AlCl3 in dichloromethane at 50 C to afford aroylated benzothiophene 7 in two steps, with yield of 95% (79% from method A[8] and 65.5% from method B[3]). With the requisite 7 in hand, we next examined piperidine nucleophilic substitution to produce the desired beno[b]thien-3-yl ketones 8. In general using reaction conditions A (acetone, NaI, K2CO3, reflux, 70%) and B (acetonitrile, NaI, K2CO3, reflux, 85%), impurity formation was observed from the beginning of the reaction. We screened various conditions and were delighted to found that using excess piperidine at reflux temperature gave negligible impurity formation. Piperidine was not only reagent but also solvent. The isolated product 8 was stable and was converted into the desired raloxifene 1 as reported. In conclusion, we have developed a viable alternative route for the synthesis of raloxifene. The new synthesis would have been better able to support the increase in bulk demand for this drug for the chemoprevention of breast cancer and novel formulations. Our synthetic route has several advantages: the use of difunctionalized coumpunds 5 as key intermediate makes Friedel–Crafts acylation and nucleophilic substitution highly efficient. The using of piperine as reagent and solvent avoids the large waste streams derived from neutralization reaction of sodium hydride. The cost of the new route is less than the current route of manufacture.
Preparation of [4-(2-Chloro-ethoxy)-phenyl]-[6-methoxy-2- (4-methoxy-phenyl)-benzo[b]thiophen-3-yl]-methanone (7) Under an N2 atmosphere, 5 was added to a mixture of 6 (20.25 g, 75 mmol) and AlCl3 (13.30 g, 100 mmol) in DCM (2 mL), and the mixture was stirred for 12 h. The reaction was monitored by TLC (n-hexane/EtOAc, 4:1). After the reaction was judged complete, the reaction mixture was allowed to cool. The crude mixture was poured into H2O and extracted with EtOAc. The organic layer was separated and concentrated. The residue was crystallized from EtOAc to give the product 7 (32.26 g, 95%): yellow solid crystals; mp 119–120 C; IR (KBr) nmax: 2960, 2835, 1647, 1599, 1472, 1251, 1169, 1032, 830 cm1 ; 1 H NMR (400 MHz, CDCl3) d 7.76 (d, J ¼ 8.8 Hz, 2H), 7.53 (d, J ¼ 8.8 Hz, 1H), 7.32 (d, J ¼ 8.4 Hz, 2H), 7.31 (s, 1H), 6.95 (dd, J ¼ 8.4, 2.4 Hz, 1H), 6.75 (dd, J ¼ 9.2, 7.2 Hz, 4H), 4.20 (t, J ¼ 4.0 Hz,2H), 3.87 (s, 3H), 3.78 (t, J ¼ 6.0 Hz, 2H), 3.74 (s, 3H); 13C NMR (100 MHz, CDCl3) d 193.1, 162.1, 159.7, 157.6, 142.7, 139.9, 133.8, 132.3, 130.9, 130.2, 130.1, 125.9, 123.9, 114.8, 114.1, 113.9, 104.4, 67.8, 55.6, 55.2, 41.5; MS (EI) m/z (%):452 (Mþ, 100.0), 437 (13.0), 297 (25.0), 183 (39.0), 121 (44.0). HRMS m/z (EI) calcd. for C25H22ClO4S: (MþH) þ: 453.0927; found: 453.0933.
Preparation of [6-Methoxy-2-(4-methoxy-phenyl)-benzo[b] thiophen-3-yl]-[4-(2-piperidin-1-yl-ethoxy)-phenyl]-methanone (8) Under an N2 atmosphere, a mixture of 7 (8.50 g, 19 mmol) and piperdine (30 ml) was stirred under reflux for 12 h. The reaction was monitored by TLC (n-hexane/EtOAc, 4:1). After the reaction was judged complete, the reaction mixture was allowed to cool. The mixture was concentrated for recovery of piperidine. EtOAc was added and the residue was washed with saturated NaHCO3 aqueous solution. The organic layer was separated and concentrated to give the product 8 (8.80 g, 94%): yellow viscous oil; IR (KBr) nmax: cm1 2933, 1645, 1597, 1535, 1501, 1470, 1249, 1164, 1030, 827; 1 H NMR (400 MHz, CDCl3) d7.76 (d, J ¼ 8.8 Hz, 2H), 7.52 (d, J ¼ 8.8 Hz, 1H), 7.33 (d, J ¼ 8.8 Hz, 2H), 7.30 (d, J ¼ 2.4 Hz, 1H), 6.94 (dd, J ¼ 8.8, 2.0 Hz, 1H), 6.75 (dd, J ¼ 7.2, 5.2 Hz, 4H), 4.08 (t, J ¼ 6.0 Hz, 2H), 3.86 (s, 3H), 3.73 (s, 3H), 2.71 (t, J ¼ 6.0 Hz, 2H), 2.46 (s, 4H), 1.60–1.54 (m, 4H), 1.43–1.41 (m, 2H).13C NMR (100 MHz, CDCl3) d 193.2, 163.0, 159.7, 157.6, 142.4, 140.1, 133.9, 132.3, 130.6, 130.4, 130.2, 126.0, 124.0, 114.8, 114.2, 114.1, 104.5, 66.3, 57.7, 55.6, 55.2, 55.1, 25.9, 24.1. MS (EI) m/z (%): 501 (Mþ, 100.0), 452 (12.0), 402 (21.0), 297 (24.0), 98 (100.0). HRMS m/z (EI) calcd. for C30H32NO4S: (MþH) þ: 502.2052; found: 502.2055.REFERENCES 1. Clemett, D.; Spencer, C. M. Drugs 2000, 60 (2), 379–411. 2. Land, S. R. JAMA 2007, 298 (9), 973–973. 3. Dadiboyena, S. Eur. J. Med. Chem. 2012, 51, 17–34. 4. Schmid, C. R.; Sluka, J. P.; Duke, K. M. Tetrahedron Lett. 1999, 40 (4), 675–678. 5. Bradley, D. A.; Godfrey, A. G.; Schmid, C. R. Tetrahedron Lett. 1999, 40 (28), 5155–5159. 6. Shinde, P. S.; Shinde, S. S.; Renge, A. S.; Patil, G. H.; Rode, A. B.; Pawar, R. R. Lett. Org. Chem. 2009, 6 (1), 8–10.7. Sach, N. W.; Richter, D. T.; Cripps, S.; Tran-Dube, M.; Zhu, H. C.; Huang, B. W.; Cui, J.; Sutton, S. C. Org. Lett. 2012, 14 (15), 3886–889. 8. Jones, C. D.; Jevnikar, M. G.; Pike, A. J.; Peters, M. K.; Black, L. J.; Thompson, A. R.; Falcone, J. F.; Clemens, J. A. J. Med. Chem. 1984, 27 (8), 1057–1066. 9. Grese, T. A.; Cho, S.; Finley, D. R.; Godfrey, A. G.; Jones, C. D.; Lugar, C. W.; Martin, M. J.; Matsumoto, K.; Pennington, L. D.; Winter, M. A.; Adrian, M. D.; Cole, H. W.; Magee, D. E.; Phillips, D. L.; Rowley, E. R.; Short, L. L.; Glasebrook, A. L.; Bryant, H. U. J. Med. Chem. 1997, 40 (2), 146–167.
synChapter 2 – 1-Substituted PiperidinesAuthor links open overlay panelRubenVardanyan
https://doi.org/10.1016/B978-0-12-805157-3.00002-8Piperidine-Based Drug DiscoveryHeterocyclic Drug Discovery2017, Pages 83-1011-Substituted Piperidines
Ruben Vardanyan, in Piperidine-Based Drug Discovery, 2017
Raloxifene (7685)
Raloxifene (Evista) (1.3.4) is a second-generation selective estrogen receptor modulator that functions as an estrogen antagonist on breast and uterine tissues, and an estrogen agonist on bone. Raloxifene is an antiresorptive agent, a new representative of a class of drugs that prevent the loss of bone mass, i.e., used to treat osteoporosis and similar diseases in postmenopausal women and those postmenopausal women at increased risk of invasive breast cancer [41–53].
It was shown that raloxifene can have some affect on cognition, mental health, sleep, and sexual function in menopausal women [54]. Raloxifene was used also as an adjuvant treatment in postmenopausal women with schizophrenia [55].
The first reported synthesis of the raloxifene scaffold consists in Friedel-Crafts aroylation in 1,2-dichloroethane and using AlCl3 as a catalyst by coupling of 4-(2-(piperidin-1-yl)ethoxy)benzoyl chloride (2.3.15) with benzothiophene derivative (2.3.16) followed by alkaline hydrolysis of mesyl groups, which give the desired raloxifene (2.3.4) [56–58] (Scheme 2.9).

The key intermediate – 6-methoxy-2-(4-methoxyphenyl)benzo[b]thiophene (2.3.16) – was prepared by the cyclization-rearrangement of 1-(4-methoxyphenyl)-2-((3-methoxyphenyl)thio)ethan-1-one (2.3.20) induced by polyphosphoric acid (PPA). This rearrangement (Kost rearrangement [59]) is general for 3-(R-substituted)indoles, -benzofurans, and -benzothiophenes, which are converted to the corresponding 2-isomers by heating with PPA.
The synthesis started from thiophenol (2.3.18) and bromoketone (2.3.19), which were coupled in presence of KOH in ethanol/water solution. Obtained (2.3.20) was heated with PPA to give a mixture that is easily separable by crystallization isomeric 2-phenylbenzo[b]thiophenes (2.3.21) and (2.3.22), where preferable, isomer (2.3.22) predominates. Cleavage of the methoxy groups in (2.3.22) was done conveniently with pyridine hydrochloride to give (2.3.23), which was easily converted to mesylate (2.3.16) with methanesulfonyl chloride in pyridine and 4-dimethylaminopyridine as a catalyst (Scheme 2.10).

The second reagent—4-(2-(piperidin-1-yl)ethoxy)benzoyl chloride (2.3.15)—was prepared starting with 4-hydroxybenzoate (2.3.24), which with 1-(2-chloroethyl)piperidine (2.3.25) in anhydrous DMF, and K2CO3 or sodium hydride, gave methyl 4-(2-(piperidin-1-yl)ethoxy)benzoate (2.3.26) hydrolyzed in MeOH/water NaOH solution. The acid (2.3.26) was converted to its chloride (2.3.15) with SOCl2 in 1,2-dichloroethane and a catalytic amount of DMF (Scheme 2.11).

Another novel convenient synthesis of raloxifene (2.3.4) have been proposed [60]. According to this method anisaldehyde (2.3.28) was transformed to corresponding cyanohydrin (2.3.29) using a mixture of sodium cyanide ethanol containing triethylamine through which HCl gas was passed over 30 minutes at 5–10°C.
Gaseous HCl was added to the solution of prepared cyanohydrin (2.3.29) in ethanol at room temperature over 30 minutes in order to give p-methoxybenzaldehyde cyanohydrin iminoether hydrochloride (2.3.30). Then, hydrogen sulfide was bubbled into a solution of the methyl imidate (2.3.30) and triethylamine in methanol at 0°C to give α-(4-methoxy phenyl)-α-hydroxy-N,N dimethylthioacetamide (2.3.31).
To the obtained α-hydroxythioamide (2.3.31) dissolved-in-methylene chloride methanesulfonic acid was slowly added, which transformed the starting material to 2-N,N-dimethylamino-6-methoxy benzo[β]thiophene (2.3.32).
The obtained 2-dimethylaminobenzothiophene (2.3.32) and known 4-(2-piperidinoethoxy)-benzoyl chloride (2.3.15) were partially dissolved in chlorobenzene and the mixture was warmed in a 100–105°C to give 2-(4-methoxyphenyl)-6-methoxy-3-[4-(piperidinoethoxy)benzoyl]-benzo[β]thiophene (2.3.33). 4-Methoxyphenylmagnesium bromide (2.3.34) in THF was added to chilled to 0°C prepared compound (2.3.33) in THF, which gave 2-(4-methoxyphenyl)-6-methoxy-3-[4-(piperidinoethoxy)benzoyl] benzo[β] thiophene (2.3.35). To the prepared benzothiophene (2.3.35) suspended in chlorobenzene was added AlCl3, followed by the addition of n-propanethiol, and the mixture was heated at 35°C. After the workup with aqueous HCl, the desired raloxifene (2.3.4) was separated [60] (Scheme 2.12).

There exist plenty of modifications for these two approaches, as reviewed in [61,62].
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w Morello, Karla C.; Wurz, Gregory T.; DeGregorio, Michael W. (2003). “Pharmacokinetics of Selective Estrogen Receptor Modulators”. Clinical Pharmacokinetics. 42 (4): 361–372. doi:10.2165/00003088-200342040-00004. ISSN 0312-5963. PMID 12648026. S2CID 13003168.
- ^ Jump up to:a b c d e f g h i j k l m n o p q Hochner-Celnikier D (1999). “Pharmacokinetics of raloxifene and its clinical application”. Eur. J. Obstet. Gynecol. Reprod. Biol. 85 (1): 23–9. doi:10.1016/s0301-2115(98)00278-4. PMID 10428318.
- ^ Jeong, Eun Ju; Liu, Yong; Lin, Huimin; Hu, Ming (2005-03-15). “Species- and Disposition Model-Dependent Metabolism of Raloxifene in Gut and Liver: Role of UGT1A10”. Drug Metabolism and Disposition. ASPET. 33 (6): 785–794. doi:10.1124/dmd.104.001883. PMID 15769887. S2CID 24273998.
- ^ Jump up to:a b c d e f g h i j k l m “Raloxifene Hydrochloride Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 22 March 2019.
- ^ Yang, Z. D.; Yu, J.; Zhang, Q. (August 2013). “Effects of raloxifene on cognition, mental health, sleep and sexual function in menopausal women: a systematic review of randomized controlled trials”. Maturitas. 75 (4): 341–348. doi:10.1016/j.maturitas.2013.05.010. ISSN 1873-4111. PMID 23764354.
- ^ Jump up to:a b British national formulary : BNF 76 (76 ed.). Pharmaceutical Press. 2018. pp. 736–737. ISBN 9780857113382.
- ^ “NADAC as of 2019-02-27”. Centers for Medicare and Medicaid Services. Retrieved 3 March 2019.
- ^ “Raloxifene Hydrochloride – Drug Usage Statistics”. ClinCalc. Retrieved 11 April 2020.
- ^ “Raloxifene: MedlinePlus Drug Information”. medlineplus.gov. Retrieved 2018-11-07.
- ^ Jump up to:a b c Mosby (5 March 2013). Mosby’s Drug Reference for Health Professions – E-Book. Elsevier Health Sciences. pp. 1379–. ISBN 978-0-323-18760-2.
- ^ Ohta, Hiroaki; Hamaya, Etsuro; Taketsuna, Masanori; Sowa, Hideaki (January 2015). “Quality of life in Japanese women with postmenopausal osteoporosis treated with raloxifene and vitamin D: post hoc analysis of a postmarketing study”. Current Medical Research and Opinion. 31 (1): 85–94. doi:10.1185/03007995.2014.975339. ISSN 1473-4877. PMID 25299349. S2CID 24671531.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t Fabian CJ, Kimler BF (March 2005). “Selective estrogen-receptor modulators for primary prevention of breast cancer”. J. Clin. Oncol. 23 (8): 1644–55. doi:10.1200/JCO.2005.11.005. PMID 15755972.
- ^ Jump up to:a b c Kirby I. Bland; Edward M. Copeland; V. Suzanne Klimberg; William J Gradishar (29 June 2017). The Breast E-Book: Comprehensive Management of Benign and Malignant Diseases. Elsevier Health Sciences. pp. 231–. ISBN 978-0-323-51187-2.
- ^ Jump up to:a b c d Raloxifene label Last updated 09/2007]
- ^ Jump up to:a b Gizzo S, Saccardi C, Patrelli TS, Berretta R, Capobianco G, Di Gangi S, Vacilotto A, Bertocco A, Noventa M, Ancona E, D’Antona D, Nardelli GB (2013). “Update on raloxifene: mechanism of action, clinical efficacy, adverse effects, and contraindications”. Obstet Gynecol Surv. 68 (6): 467–81. doi:10.1097/OGX.0b013e31828baef9. PMID 23942473. S2CID 9003157.
- ^ Jump up to:a b c d e f g h Goldstein, S. R. (2000). “A pharmacological review of selective oestrogen receptor modulators”. Human Reproduction Update. 6 (3): 212–224. doi:10.1093/humupd/6.3.212. ISSN 1355-4786. PMID 10874566.
- ^ Seeman E (2001). “Raloxifene”. J. Bone Miner. Metab. 19 (2): 65–75. doi:10.1007/s007740170043. PMID 11281162.
- ^ Jump up to:a b c Park, W (2002). “Selective estrogen receptor modulators (SERMS) and their roles in breast cancer prevention”. Trends in Molecular Medicine. 8 (2): 82–88. doi:10.1016/S1471-4914(02)02282-7. ISSN 1471-4914. PMID 11815274.
- ^ Agency for Healthcare Research and Quality, Rockville, MD. (September 2009). “Medications Effective in Reducing Risk of Breast Cancer But Increase Risk of Adverse Effects”. Retrieved 2009-09-14.
- ^ Lemmo, W (2016). “Anti-Estrogen Withdrawal Effect With Raloxifene? A Case Report”. Integrative Cancer Therapies. Published Online Before Print July 13 (3): 245–249. doi:10.1177/1534735416658954. PMC 5739193. PMID 27411856.
- ^ Jump up to:a b Haskell, Sally G. (2003). “Selective Estrogen Receptor Modulators”. Southern Medical Journal. 96 (5): 469–476. doi:10.1097/01.SMJ.0000051146.93190.4A. ISSN 0038-4348. PMID 12911186. S2CID 40607634.
- ^ Weatherman, Ross V; Clegg, Nicola J; Scanlan, Thomas S (2001). “Differential SERM activation of the estrogen receptors (ERα and ERβ) at AP-1 sites”. Chemistry & Biology. 8(5): 427–436. doi:10.1016/S1074-5521(01)00025-4. ISSN 1074-5521. PMID 11358690.
- ^ Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, Nicolas JC, Cavaillès V, Balaguer P (May 2006). “Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta”. Biochem. Pharmacol. 71 (10): 1459–69. doi:10.1016/j.bcp.2006.02.002. PMID 16554039.
- ^ Greene, G. L.; Shiau, A. K.; Nettles, K. W. (2004). “A Structural Explanation for ERα/ERβ SERM Discrimination”. New Molecular Mechanisms of Estrogen Action and Their Impact on Future Perspectives in Estrogen Therapy (46): 33–45. doi:10.1007/978-3-662-05386-7_3. ISBN 978-3-662-05388-1. PMID 15248503.
- ^ Barkhem, Tomas; Carlsson, Bo; Nilsson, Yvonne; Enmark, Eva; Gustafsson, Jan-Åke; Nilsson, Stefan (1998). “Differential Response of Estrogen Receptor α and Estrogen Receptor β to Partial Estrogen Agonists/Antagonists”. Molecular Pharmacology. 54 (1): 105–112. doi:10.1124/mol.54.1.105. ISSN 0026-895X. PMID 9658195.
- ^ Prossnitz ER, Arterburn JB (July 2015). “International Union of Basic and Clinical Pharmacology. XCVII. G Protein-Coupled Estrogen Receptor and Its Pharmacologic Modulators”. Pharmacol. Rev. 67 (3): 505–40. doi:10.1124/pr.114.009712. PMC 4485017. PMID 26023144.
- ^ Petrie WK, Dennis MK, Hu C, Dai D, Arterburn JB, Smith HO, Hathaway HJ, Prossnitz ER (2013). “G protein-coupled estrogen receptor-selective ligands modulate endometrial tumor growth”. Obstet Gynecol Int. 2013: 472720. doi:10.1155/2013/472720. PMC 3863501. PMID 24379833.
- ^ Jeon-Hor, Chen; et al. (September 15, 2003). “Reduction of Breast Density Following Tamoxifen Treatment Evaluated by 3-D MRI: Preliminary Study”. Magn Reson Imaging. 29 (1): 91–8. doi:10.1016/j.mri.2010.07.009. PMC 3005955. PMID 20832226.
- ^ Jump up to:a b c d e f g h Draper, Michael W.; Chin, William W. (2003). “Molecular and Clinical Evidence for the Unique Nature of Individual Selective Estrogen Receptor Modulators”. Clinical Obstetrics and Gynecology. 46 (2): 265–297. doi:10.1097/00003081-200306000-00008. ISSN 0009-9201. PMID 12808380. S2CID 5132467.
- ^ Jump up to:a b c Corona G, Rastrelli G, Ratrelli G, Maggi M (February 2015). “The pharmacotherapy of male hypogonadism besides androgens”. Expert Opin Pharmacother. 16 (3): 369–87. doi:10.1517/14656566.2015.993607. PMID 25523084. S2CID 8891640.
- ^ Jump up to:a b Duarte FH, Jallad RS, Bronstein MD (November 2016). “Estrogens and selective estrogen receptor modulators in acromegaly”. Endocrine. 54 (2): 306–314. doi:10.1007/s12020-016-1118-z. PMID 27704479. S2CID 10136018.
- ^ Jump up to:a b Birzniece V, Sutanto S, Ho KK (April 2012). “Gender difference in the neuroendocrine regulation of growth hormone axis by selective estrogen receptor modulators”. J. Clin. Endocrinol. Metab. 97 (4): E521–7. doi:10.1210/jc.2011-3347. PMID 22319035.
- ^ Jump up to:a b Uebelhart B, Herrmann F, Pavo I, Draper MW, Rizzoli R (September 2004). “Raloxifene treatment is associated with increased serum estradiol and decreased bone remodeling in healthy middle-aged men with low sex hormone levels”. J. Bone Miner. Res. 19 (9): 1518–24. doi:10.1359/JBMR.040503. PMID 15312253. S2CID 36104038.
- ^ Xu, Beibei; Lovre, Dragana; Mauvais-Jarvis, Franck (2016). “Effect of selective estrogen receptor modulators on metabolic homeostasis”. Biochimie. 124: 92–97. doi:10.1016/j.biochi.2015.06.018. ISSN 0300-9084. PMID 26133657.
In healthy postmemopausal women, raloxifene treatment for one year prevented body weight gain and abdominal adiposity by promoting a shift from an android to gynoid fat distribution [46].
- ^ Francucci, C. M.; Pantaleo, D.; Iori, N.; Camilletti, A.; Massi, F.; Boscaro, M. (2014). “Effects of raloxifene on body fat distribution and lipid profile in healthy post-menopausal women”. Journal of Endocrinological Investigation. 28 (9): 623–631. doi:10.1007/BF03347261. ISSN 0391-4097. PMID 16218045. S2CID 28467435.
These results […] suggest, for the first time, that RLX promotes the shift from android to gynoid fat distribution, and prevents the uptrend of abdominal adiposity and body weight compared with untreated women.
- ^ Snyder KR, Sparano N, Malinowski JM (September 2000). “Raloxifene hydrochloride”. Am J Health Syst Pharm. 57 (18): 1669–75, quiz 1676–8. doi:10.1093/ajhp/57.18.1669. PMID 11006795.
- ^ Jump up to:a b Eric S. Orwoll; Michael Bliziotes (2 August 2002). Osteoporosis: Pathophysiology and Clinical Management. Springer Science & Business Media. pp. 320–. ISBN 978-1-59259-278-4.
- ^ Jump up to:a b Stuart Silverman; Bo Abrahamsen (29 December 2015). The Duration and Safety of Osteoporosis Treatment: Anabolic and Antiresorptive Therapy. Springer. pp. 24–. ISBN 978-3-319-23639-1.
- ^ Jump up to:a b Institute of Medicine; Board on Health Sciences Policy; Committee on Accelerating Rare Diseases Research and Orphan Product Development (3 April 2011). Rare Diseases and Orphan Products: Accelerating Research and Development. National Academies Press. pp. 113–. ISBN 978-0-309-15806-0.
- ^ Reducing Breast Cancer Risk with Drugs. Am Cncl on Science, Health. pp. 10–. GGKEY:CBEALLAHP8W.
- ^ Sydney Lou Bonnick (10 November 2007). Bone Densitometry for Technologists. Springer Science & Business Media. pp. 277–. ISBN 978-1-59259-992-9.
- ^ Jie Jack Li; Douglas S. Johnson (27 March 2013). Modern Drug Synthesis. John Wiley & Sons. pp. 2–. ISBN 978-1-118-70124-9.
- ^ Jump up to:a b J. Elks (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 1063–. ISBN 978-1-4757-2085-3.
- ^ Jump up to:a b c d Index Nominum 2000: International Drug Directory. Taylor & Francis. 2000. pp. 909–. ISBN 978-3-88763-075-1.
- ^ I.K. Morton; Judith M. Hall (31 October 1999). Concise Dictionary of Pharmacological Agents: Properties and Synonyms. Springer Science & Business Media. pp. 245–. ISBN 978-0-7514-0499-9.
- ^ Jump up to:a b c d e “Raloxifene”.
- ^ Thelancetoncology (2006). “A STARring role for raloxifene?”. Lancet Oncol. 7 (6): 443. doi:10.1016/S1470-2045(06)70701-X. PMID 16750489.
- ^ Provinciali N, Suen C, Dunn BK, DeCensi A (October 2016). “Raloxifene hydrochloride for breast cancer risk reduction in postmenopausal women”. Expert Rev Clin Pharmacol. 9 (10): 1263–1272. doi:10.1080/17512433.2016.1231575. PMID 27583816. S2CID 26047863.
- ^ James F. Holland; Raphael E. Pollock (2010). Holland-Frei Cancer Medicine 8. PMPH-USA. pp. 743–. ISBN 978-1-60795-014-1.
- ^ Blum A, Hathaway L, Mincemoyer R, Schenke WH, Csako G, Waclawiw MA, Panza JA, Cannon RO (2000). “Hormonal, lipoprotein, and vascular effects of the selective estrogen receptor modulator raloxifene in hypercholesterolemic men”. Am. J. Cardiol. 85 (12): 1491–4, A7. doi:10.1016/s0002-9149(00)00802-x. PMID 10856400.
- ^ Doran PM, Riggs BL, Atkinson EJ, Khosla S (2001). “Effects of raloxifene, a selective estrogen receptor modulator, on bone turnover markers and serum sex steroid and lipid levels in elderly men”. J. Bone Miner. Res. 16 (11): 2118–25. doi:10.1359/jbmr.2001.16.11.2118. PMID 11697809. S2CID 28216610.
- ^ Dimaraki EV, Symons KV, Barkan AL (2004). “Raloxifene decreases serum IGF-I in male patients with active acromegaly”. Eur. J. Endocrinol. 150 (4): 481–7. doi:10.1530/eje.0.1500481. PMID 15080777.
- ^ Duschek EJ, Gooren LJ, Netelenbos C (2004). “Effects of raloxifene on gonadotrophins, sex hormones, bone turnover and lipids in healthy elderly men” (PDF). Eur. J. Endocrinol. 150 (4): 539–46. doi:10.1530/eje.0.1500539. PMID 15080785.
- ^ Smith MR, Fallon MA, Lee H, Finkelstein JS (2004). “Raloxifene to prevent gonadotropin-releasing hormone agonist-induced bone loss in men with prostate cancer: a randomized controlled trial”. J. Clin. Endocrinol. Metab. 89 (8): 3841–6. doi:10.1210/jc.2003-032058. PMID 15292315.
- ^ Jump up to:a b Ho TH, Nunez-Nateras R, Hou YX, Bryce AH, Northfelt DW, Dueck AC, Wong B, Stanton ML, Joseph RW, Castle EP (2017). “A Study of Combination Bicalutamide and Raloxifene for Patients With Castration-Resistant Prostate Cancer”. Clin Genitourin Cancer. 15 (2): 196–202.e1. doi:10.1016/j.clgc.2016.08.026. PMID 27771244. S2CID 19043552.
- ^ Khodaie-Ardakani MR, Khosravi M, Zarinfard R, Nejati S, Mohsenian A, Tabrizi M, Akhondzadeh S (2015). “A Placebo-Controlled Study of Raloxifene Added to Risperidone in Men with Chronic Schizophrenia”. Acta Med Iran. 53 (6): 337–45. PMID 26069170.
- ^ Weickert TW, Weinberg D, Lenroot R, Catts SV, Wells R, Vercammen A, O’Donnell M, Galletly C, Liu D, Balzan R, Short B, Pellen D, Curtis J, Carr VJ, Kulkarni J, Schofield PR, Weickert CS (2015). “Adjunctive raloxifene treatment improves attention and memory in men and women with schizophrenia”. Mol. Psychiatry. 20 (6): 685–94. doi:10.1038/mp.2015.11. PMC 4444978. PMID 25980345.
- ^ Jump up to:a b Fujimura, Tetsuya; Takayama, Kenichi; Takahashi, Satoru; Inoue, Satoshi (2018). “Estrogen and Androgen Blockade for Advanced Prostate Cancer in the Era of Precision Medicine”. Cancers. 10 (2): 29. doi:10.3390/cancers10020029. ISSN 2072-6694. PMC 5836061. PMID 29360794.
- ^ Jump up to:a b c Wang Q, Dong X, Wang Y, Li X (2017). “Raloxifene as an adjunctive treatment for postmenopausal women with schizophrenia: a meta-analysis of randomized controlled trials”. Arch Womens Ment Health. 21 (1): 31–41. doi:10.1007/s00737-017-0773-2. PMID 28849318. S2CID 4524617.
- ^ Carneiro AL, de Cassia de Maio Dardes R, Haidar MA (July 2012). “Estrogens plus raloxifene on endometrial safety and menopausal symptoms–semisystematic review”. Menopause. 19 (7): 830–4. doi:10.1097/gme.0b013e31824a74ce. PMID 22549172. S2CID 196380398.
- ^ Nordt CA, DiVasta AD (2008). “Gynecomastia in adolescents”. Curr. Opin. Pediatr. 20 (4): 375–82. doi:10.1097/MOP.0b013e328306a07c. PMID 18622190. S2CID 205834072.
- ^ Leung KC, Leung AC (2017). “Gynecomastia in Infants, Children, and Adolescents”. Recent Pat Endocr Metab Immune Drug Discov. 10 (2): 127–137. doi:10.2174/1872214811666170301124033. PMID 28260521.
- ^ Lawrence SE, Faught KA, Vethamuthu J, Lawson ML (July 2004). “Beneficial effects of raloxifene and tamoxifen in the treatment of pubertal gynecomastia”. J. Pediatr. 145 (1): 71–6. doi:10.1016/j.jpeds.2004.03.057. PMID 15238910.
- ^ Kanakis, G. A.; Nordkap, L.; Bang, A. K.; Calogero, A. E.; Bártfai, G.; Corona, G.; Forti, G.; Toppari, J.; Goulis, D. G.; Jørgensen, N. (2019). “EAA clinical practice guidelines—gynecomastia evaluation and management”. Andrology. 7 (6): 778–793. doi:10.1111/andr.12636. ISSN 2047-2919. PMID 31099174.
- ^ Sugiyama, Nobuhiro; Barros, Rodrigo P.A.; Warner, Margaret; Gustafsson, Jan-Åke (2010). “ERβ: recent understanding of estrogen signaling”. Trends in Endocrinology & Metabolism. 21 (9): 545–552. doi:10.1016/j.tem.2010.05.001. ISSN 1043-2760. PMID 20646931. S2CID 43001363.
Further reading
- Barrett-Connor E (2001). “Raloxifene: risks and benefits”. Ann N Y Acad Sci. 949 (1): 295–303. Bibcode:2001NYASA.949..295B. doi:10.1111/j.1749-6632.2001.tb04036.x. PMID 11795366. S2CID 41412601.
- Heringa M (2003). “Review on raloxifene: profile of a selective estrogen receptor modulator”. Int J Clin Pharmacol Ther. 41 (8): 331–45. doi:10.5414/cpp41331. PMID 12940590.
- Sporn MB, Dowsett SA, Mershon J, Bryant HU (2004). “Role of raloxifene in breast cancer prevention in postmenopausal women: clinical evidence and potential mechanisms of action”. Clin Ther. 26 (6): 830–40. doi:10.1016/s0149-2918(04)90127-0. PMID 15262454.
- Vogel VG (2009). “The NSABP Study of Tamoxifen and Raloxifene (STAR) trial”. Expert Rev Anticancer Ther. 9 (1): 51–60. doi:10.1586/14737140.9.1.51. PMC 2785111. PMID 19105706.
- Wickerham DL, Costantino JP, Vogel VG, Cronin WM, Cecchini RS, Ford LG, Wolmark N (2009). “The use of tamoxifen and raloxifene for the prevention of breast cancer”. Recent Results Cancer Res. Recent Results in Cancer Research. 181: 113–9. doi:10.1007/978-3-540-69297-3_12. ISBN 978-3-540-69296-6. PMC 5110043. PMID 19213563.
- Vogel VG (2011). “Update on raloxifene: role in reducing the risk of invasive breast cancer in postmenopausal women”. Breast Cancer: Targets and Therapy. 3: 127–37. doi:10.2147/BCTT.S11288. PMC 3846694. PMID 24367182.
- Yang ZD, Yu J, Zhang Q (2013). “Effects of raloxifene on cognition, mental health, sleep and sexual function in menopausal women: a systematic review of randomized controlled trials”. Maturitas. 75 (4): 341–8. doi:10.1016/j.maturitas.2013.05.010. PMID 23764354.
External links
- “Raloxifene”. Drug Information Portal. U.S. National Library of Medicine.
///////Keoxifene hydrochloride, Raloxifene hydrochloride, LY-139481, LY 156758, Optruma, Loxifen, Evista
TUCATINIB

Tucatinib
ツカチニブ;
N6-(4,4-dimethyl-4,5-dihydro-1,3-oxazol-2-yl)-N4-(3-methyl-4-{[1,2,4]triazolo[1,5-a]pyridin-7-yloxy}phenyl)quinazoline-4,6-diamine
| Formula | C26H24N8O2 |
|---|---|
| CAS | 937263-43-9 |
| Mol weight | 480.5212 |
To treat advanced unresectable or metastatic HER2-positive breast cancer
Drug Trials Snapshot
FDA APPROVED 4/17/2020 Tukysa
- ARRY 380
- ARRY-380
- ONT 380
- ONT-380
Tucatinib (INN),[1] sold under the brand name Tukysa, is a small molecule inhibitor of HER2 for the treatment of HER2-positive breast cancer.[2][3] It was developed by Array BioPharma and licensed to Cascadian Therapeutics (formerly Oncothyreon, subsequently part of Seattle Genetics).[4]
Common side effects are diarrhea, palmar-plantar erythrodysesthesia (burning or tingling discomfort in the hands and feet), nausea, fatigue, hepatotoxicity (liver damage), vomiting, stomatitis (inflammation of the mouth and lips), decreased appetite, abdominal pain, headache, anemia and rash.[5][6] Pregnant or breastfeeding women should not take Tucatinib because it may cause harm to a developing fetus or newborn baby.[5]
Tucatinib was approved for medical use in Australia in August 2020.[7]
Medical uses
Tucatinib is a kinase inhibitor indicated in combination with trastuzumab and capecitabine for treatment of adults with advanced unresectable or metastatic HER2-positive breast cancer, including those with brain metastases, who have received one or more prior anti-HER2-based regimens in the metastatic setting.[8]
Clinical trials
Two early stage clinical trials have reported encouraging results, both of which had options to enroll subjects with central nervous system (CNS) metastases.[2][9][10][11][12][10] HER2CLIMB is a Phase 2 randomized, double-blinded, placebo-controlled study of tucatinib in combination with trastuzumab and capecitabine in patients with pretreated, unresectable locally advanced or metastatic HER2-positive breast cancer.[13]
History
In April 2020, the U.S. Food and Drug Administration (FDA) approved tucatinib in combination with chemotherapy (trastuzumab and capecitabine) for the treatment of adults with advanced forms of HER2-positive breast cancer that can’t be removed with surgery, or has spread to other parts of the body, including the brain, and who have received one or more prior treatments.[5][6][14]
The FDA collaborated with the Australian Therapeutic Goods Administration (TGA), Health Canada, Health Sciences Authority (HSA, Singapore) and Swissmedic (SMC, Switzerland) on the review.[5] This was the first Project Orbis partnership between the FDA, HSA and Swissmedic.[5] As of 17 April 2020, the application is still under review at the other agencies.[5]
Tucatinib is a kinase inhibitor meaning it blocks a type of enzyme (kinase) and helps prevent the cancer cells from growing.[5] Tucatinib is approved for treatment after adults have taken one or more anti-HER2-based regimens in the metastatic setting.[5] The FDA approved tucatinib based on the results of the HER2CLIMB trial (NCT02614794) enrolling 612 subjects who had HER2-positive advanced unresectable or metastatic breast cancer and had prior treatment with trastuzumab, pertuzumab and ado-trastuzumab emtansine (T-DM1).[5][6] Subjects with previously treated and stable brain metastases, as well as those with previously treated and growing or untreated brain metastases, were eligible for the clinical trial, and 48% of enrolled subjects had brain metastases at the start of the trial.[5]
Subjects received either tucatinib 300 mg twice daily plus trastuzumab and capecitabine (tucatinib arm, n=410) or placebo plus trastuzumab and capecitabine (control arm, n=202).[6] The primary endpoint was progression-free survival (PFS), or the amount of time when there was no growth of the tumor, assessed by a blinded independent central review, evaluated in the initial 480 randomized patients.[5][6] The median PFS in subjects who received tucatinib, trastuzumab, and capecitabine was 7.8 months (95% CI: 7.5, 9.6) compared to 5.6 months (95% CI: 4.2, 7.1) in those subjects who received placebo, trastuzumab, and capecitabine (HR 0.54; 95% CI: 0.42, 0.71; p<0.00001).[5][6] Overall survival and PFS in subjects with brain metastases at baseline were key secondary endpoints.[5] The median overall survival in subjects who received tucatinib, trastuzumab, and capecitabine was 21.9 months (95% CI: 18.3, 31.0) compared to 17.4 months (95% CI: 13.6, 19.9) in subjects who received placebo, trastuzumab, and capecitabine (HR: 0.66; 95% CI: 0.50, 0.87; p=0.00480).[5][6] The median PFS in subjects with brain metastases at baseline who received tucatinib, trastuzumab and capecitabine was 7.6 months (95% CI: 6.2, 9.5) compared to 5.4 months (95% CI: 4.1, 5.7) in subjects who received placebo, trastuzumab and capecitabine (HR: 0.48; 0.34, 0.69; p<0.00001).[5][6]
The FDA granted the application for tucatinib priority review, breakthrough therapy, fast track, and orphan drug designations.[5][6][15] The FDA granted approval of Tukysa to Seattle Genetics, Inc.[5]
SYN
Recently, the Mao team reported a new route for the efficient synthesis of Tucatinib.
The results were published on Synthesis (DOI: 10.1055/s-0037-1610706).
Previously, the synthesis report route of Tucatinib was published by Array BioPharma in a patent document (WO 2007059257, 2007). The synthetic route reported in the patent is shown in the figure below:

Using 4-nitro-2-cyanoaniline as the raw material, the first step is to condense with DMF-DMA to prepare imine 3 (yield 87%); subsequent catalytic hydrogenation of palladium on carbon to reduce the nitro group to obtain the amine 4 (90% yield); followed by 1,1&39;-thiocarbonyldiimidazole (TCDI) and The amino alcohol undergoes condensation to prepare the thiourea derivative 5 (yield is only 34%); further with the intermediate 6 to undergo ring-closure reaction to obtain the key intermediate 7 (yield 62%) ; Finally, under the action of p-toluenesulfonic acid, intramolecular dehydration and ring closure to form oxazoline, complete the synthesis of the target compound tucatinib.
Reverse synthesis analysis
The author broke the bond of Tucatinib from two points a and b and split them into three fragments. : Thioether oxazoline 17, nitrobenzene 3 and the key fragment of the original research route 6.
Preparation of key fragment 6
4-nitro-3-methylphenol 8 as a starting point The material, with pyridine derivative 9, undergoes aromatic affinity substitution reaction to prepare aryl ether 10 (yield 64%); then it is condensed with DMF-DMA, and then treated with hydroxylamine hydrochloride. The step yield was 81% to obtain the oxime derivative 12; subsequently, the ring was closed under the treatment of trifluoroacetic anhydride, the mostAfter palladium-catalyzed hydrogenation to reduce the nitro group, the key aniline triazole 6 was successfully prepared, with a total yield of 32.8%.
aromatic ring skeleton construction
fragment 3 was synthesized according to the method reported in the literature. The estimated aromatic ring fragment was then constructed with the aniline triazole 6 prepared above:
Compound 6 and fragment 3 were cyclized in acetic acid , 14 was successfully prepared, and finally the nitro group was reduced by palladium-catalyzed hydrogenation to obtain the key arylamine 15 with a two-step yield of 76.4%.
Fragment 17 and Tucatinib synthesis
amino alcohol and 1,1&39;-thiocarbonyl diimidazole (TCDI) The ring is closed to obtain 16, which is then treated with methyl trifluoromethanesulfonate to obtain oxazoline 17, with a total yield of 67.23% in the two steps.
oxazoline17 and arylamine 15 in the presence of cesium carbonate, heated in DMF for 20 hours, and finally completed the synthesis of Tucatinib with a yield of 76%.
Comparison of the new route and the patent route
The yield of the last step of the patent is unknown, starting with key intermediates 3 and 6, total income The rate is less than 19%.
The overview of the new route is as follows:
Correspondingly, starting from the intermediate 3 and 6, the total yield of the new route There is a significant improvement to 39%. Moreover, the purity of the product and other aspects also meet the requirements of API.
Comment
Tucatinib (Tukysa) Tucatinib/Tucatinib as a small-molecule oral tyrosine kinase (TKI) inhibitor for HER2 Positive breast cancer has highly specific targeting selectivity. The study of the new synthetic route
effectively improves the production efficiency in terms of ensuring the purity of the compound, and the raw materials used are relatively simple and easy to obtain.
Medicinal chemists have completed the research and development and synthesis of compounds (from 0 to 1), while process chemists have optimized the synthetic routes and processes, so that the compounds can be prepared more simply, efficiently, economically and environmentally.
SYN PATENT
CN 111825604
PAPER
Synthesis (2019), 51(13), 2660-2664
Abstract
A new and improved synthetic route to tucatinib is described that involves three key intermediates. The first of these, 4-([1,2,4]triazolo[1,5-a]pyridin-7-yloxy)-3-methylaniline, was prepared on a 100 g scale in 33% yield over five steps and 99% purity. Next, N 4-(4-([1,2,4]triazolo[1,5-a]pyridin-7-yloxy)-3-methylphenyl)quinazoline-4,6-diamine was isolated in 67% yield over three steps and >99% purity. Then, 4,4-dimethyl-2-(methylthio)-4,5-dihydrooxazole trifluoromethanesulfonate was prepared under mild conditions in 67% yield over two steps. Finally, tucatinib was obtained in 17% yield over nine steps and in >99% purity (HPLC). Purification methods used to isolate the product and the intermediates involved in the route are also reported.

References
- ^ World Health Organization (2016). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 75”. WHO Drug Information. 30 (1): 161. hdl:10665/331046.
- ^ Jump up to:a b “ONT-380 Active Against CNS Mets in HER2-Positive Breast Cancer”. Cancer Network. 15 December 2015. Retrieved 17 April 2020.
- ^ Martin M, López-Tarruella S (October 2018). “Emerging Therapeutic Options for HER2-Positive Breast Cancer”. American Society of Clinical Oncology Educational Book. American Society of Clinical Oncology. Annual Meeting. 35 (36): e64–70. doi:10.1200/EDBK_159167. PMID 27249772.
- ^ “Tucatinib” (PDF). Statement on a Nonproprietary Name Adopted by the USAN Council.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “FDA Approves First New Drug Under International Collaboration, A Treatment Option for Patients with HER2-Positive Metastatic Breast Cancer”. U.S. Food and Drug Administration (FDA) (Press release). 17 April 2020. Retrieved 17 April 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e f g h i “FDA approves tucatinib for patients with HER2-positive metastatic brea”. U.S. Food and Drug Administration (FDA). 17 April 2020. Retrieved 20 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Tukysa”. Therapeutic Goods Administration (TGA). 21 August 2020. Retrieved 22 September 2020.
- ^ “Tukysa (tucatinib) tablets, for oral use” (PDF). Seattle Genetics. Retrieved 17 April2020.
- ^ “Oncothyreon Inc. Announces Data For ONT-380 In HER2-Positive Breast Cancer Patients With And Without Brain Metastases At The San Antonio Breast Cancer Symposium”. BioSpace (Press release). 9 December 2015. Retrieved 18 April 2020.
- ^ Jump up to:a b Borges VF, Ferrario C, Aucoin N, Falkson CI, Khan QJ, Krop IE, et al. “Efficacy results of a phase 1b study of ONT-380, a CNS-penetrant TKI, in combination with T-DM1 in HER2+ metastatic breast cancer (MBC), including patients (pts) with brain metastases”. Journal of Clinical Oncology. 2016 ASCO Annual Meeting.
- ^ “SABCS15: Promising phase 1 results lead to phase 2 for ONT-380 in HER2+ breast cancer”. Colorado Cancer Blogs. Retrieved 10 June 2016.
- ^ “A Study of Tucatinib (ONT-380) Combined With Capecitabine and/or Trastuzumab in Patients With HER2+ Metastatic Breast Cancer”. ClinicalTrials.gov. 31 December 2013. Retrieved 18 April 2020.
- ^ “A Study of Tucatinib vs. Placebo in Combination With Capecitabine & Trastuzumab in Patients With Advanced HER2+ Breast Cancer (HER2CLIMB)”. ClinicalTrials.gov. Retrieved 18 April 2020.
- ^ “Tukysa: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 20 April 2020.
- ^ “Tucatinib Orphan Drug Designation and Approval”. U.S. Food and Drug Administration(FDA). 24 December 1999. Retrieved 20 April 2020.
External links
- “Tucatinib”. Drug Information Portal. U.S. National Library of Medicine.
- “Tucatinib”. National Cancer Institute.
- Clinical trial number NCT02614794 for “A Study of Tucatinib vs. Placebo in Combination With Capecitabine & Trastuzumab in Patients With Advanced HER2+ Breast Cancer (HER2CLIMB)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Tukysa |
| Other names | ONT-380, ARRY-380 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620032 |
| License data | US DailyMed: Tucatinib |
| Pregnancy category | AU: DUS: N (Not classified yet) |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | AU: S4 (Prescription only)US: ℞-only |
| Identifiers | |
| CAS Number | 937263-43-9 |
| PubChem CID | 51039094 |
| DrugBank | DB11652 |
| ChemSpider | 34995558 |
| UNII | 234248D0HH |
| KEGG | D11141 |
| ChEMBL | ChEMBL3989868 |
| Chemical and physical data | |
| Formula | C26H24N8O2 |
| Molar mass | 480.532 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]CC1=C(C=CC(=C1)NC2=NC=NC3=C2C=C(C=C3)NC4=NC(CO4)(C)C)OC5=CC6=NC=NN6C=C5 | |
| InChI[hide]InChI=1S/C26H24N8O2/c1-16-10-17(5-7-22(16)36-19-8-9-34-23(12-19)28-15-30-34)31-24-20-11-18(4-6-21(20)27-14-29-24)32-25-33-26(2,3)13-35-25/h4-12,14-15H,13H2,1-3H3,(H,32,33)(H,27,29,31)Key:SDEAXTCZPQIFQM-UHFFFAOYSA-N |
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Tukysa | Tablet | 150 mg/1 | Oral | Seattle Genetics, Inc. | 2020-04-17 | Not applicable |  | |
| Tukysa | Tablet | 150 mg | Oral | Seattle Genetics, Inc. | 2020-08-27 | Not applicable |  | |
| Tukysa | Tablet | 50 mg/1 | Oral | Seattle Genetics, Inc. | 2020-04-17 | Not applicable | | |
| Tukysa | Tablet | 50 mg | Oral | Seattle Genetics, Inc. | 2020-10-08 | Not applicable | |
Showing 1 to 4 of 4 entries
///////tucatinib, FDA 2020, TUKSYA, 2020 APROVALS, ARRY 380, ONT 380, ツカチニブ ,
Ripretinib

Ripretinib
リプレチニブ;
| Formula | C24H21BrFN5O2 |
|---|---|
| CAS | 1442472-39-0 |
| Mol weight | 510.3582 |
Antineoplastic, Receptor tyrosine kinase inhibitor
US FDA APPROVED 2020/5/15 QUINLOCK
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Qinlock | Tablet | 50 mg | Oral | Deciphera Pharmaceuticals. Llc | Not applicable | Not applicable | | |
| Qinlock | Tablet | 50 mg/1 | Oral | Deciphera Pharmaceuticals, LLC | 2020-05-15 | Not applicable | |
SYN

Ripretinib, sold under the brand name Qinlock, is a medication for the treatment of adults with advanced gastrointestinal stromal tumor (GIST), a type of tumor that originates in the gastrointestinal tract.[3] It is taken by mouth.[3] Ripretinib is a kinase inhibitor, meaning it works by blocking a type of enzyme called a kinase, which helps keep the cancer cells from growing.[3]
The most common side effects include alopecia (hair loss), fatigue, nausea, abdominal pain, constipation, myalgia (muscle pain), diarrhea, decreased appetite, palmar-plantar erythrodysesthesia syndrome (a skin reaction in the palms and soles) and vomiting.[3][4] Alopecia is a unique side effect to ripretinib, which is not seen with other tyrosine kinase inhibitors used to treat GISTs.
Ripretinib was approved for medical use in the United States in May 2020,[3] and in Australia in July 2020.[1] Ripretinib is the first new drug specifically approved in the United States as a fourth-line treatment for advanced gastrointestinal stromal tumor (GIST).
Medical uses
Ripretinib is indicated for the treatment of adults with advanced gastrointestinal stromal tumor (GIST), a type of tumor that originates in the gastrointestinal tract, who have received prior treatment with three or more kinase inhibitor therapies, including imatinib.[3] GIST is type of stomach, bowel, or esophagus tumor.[4]
Adverse effects
The most common side effects include alopecia (hair loss), fatigue, nausea, abdominal pain, constipation, myalgia (muscle pain), diarrhea, decreased appetite, palmar-plantar erythrodysesthesia syndrome (a skin reaction in the palms and soles) and vomiting.[3][4]
Ripretinib can also cause serious side effects including skin cancer, hypertension (high blood pressure) and cardiac dysfunction manifested as ejection fraction decrease (when the muscle of the left ventricle of the heart is not pumping as well as normal).[3][4]
Ripretinib may cause harm to a developing fetus or a newborn baby.[3][4]
History
Ripretinib was approved for medical use in the United States in May 2020.[3][5][6][4]
The approval of ripretinib was based on the results of an international, multi-center, randomized, double-blind, placebo-controlled clinical trial (INVICTUS/NCT03353753) that enrolled 129 participants with advanced gastrointestinal stromal tumor (GIST) who had received prior treatment with imatinib, sunitinib, and regorafenib.[3][7] The trial compared participants who were randomized to receive ripretinib to participants who were randomized to receive placebo, to determine whether progression free survival (PFS) – the time from initial treatment in the clinical trial to growth of the cancer or death – was longer in the ripretinib group compared to the placebo group.[3] During treatment in the trial, participants received ripretinib 150 mg or placebo once a day in 28-day cycles, repeated until tumor growth was found (disease progression), or the participant experienced intolerable side effects.[3][7] After disease progression, participants who were randomized to placebo were given the option of switching to ripretinib.[3][7] The trial was conducted at 29 sites in the United States, Australia, Belgium, Canada, France, Germany, Italy, the Netherlands, Poland, Singapore, Spain, and the United Kingdom.[4]
The major efficacy outcome measure was progression-free survival (PFS) based on assessment by blinded independent central review (BICR) using modified RECIST 1.1 in which lymph nodes and bone lesions were not target lesions and a progressively growing new tumor nodule within a pre-existing tumor mass must meet specific criteria to be considered unequivocal evidence of progression.[7] Additional efficacy outcome measures included overall response rate (ORR) by BICR and overall survival (OS).[7] The trial demonstrated a statistically significant improvement in PFS for participants in the ripretinib arm compared with those in the placebo arm (HR 0.15; 95% CI: 0.09, 0.25; p<0.0001).[7]
The U.S. Food and Drug Administration (FDA) granted the application for ripretinib priority review and fast track designations, as well as breakthrough therapy designation and orphan drug designation.[3][8] The FDA granted approval of Qinlock to Deciphera Pharmaceuticals, Inc.[3]
The FDA collaborated with the Australian Therapeutic Goods Administration (TGA) and Health Canada on the review of the application as part of Project Orbis.[3][7] The FDA approved ripretinib three months ahead of schedule.[3][7] As of May 2020, the review of the applications was ongoing for the Australian TGA and for Health Canada.[3][7]
Names
Ripretinib is the International nonproprietary name (INN) and the United States Adopted Name (USAN).[9][10]
| PATENT NUMBER | PEDIATRIC EXTENSION | APPROVED | EXPIRES (ESTIMATED) | |
|---|---|---|---|---|
| US8940756 | No | 2012-06-07 | 2032-06-07 | |
| US8461179 | No | 2012-06-07 | 2032-06-07 | |
| US8188113 | No | 2010-07-27 | 2030-07-27 | |
PATENT
US 8461179
PATENT
WO 2013184119
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2013184119
[0125] Example A13: A mixture of Example C5 (2.191 g, 7.94 mmol), Example Bl (1.538 g, 8.33 mmol) and KF on alumina (40 wt%) (9.22 g, 63.5 mmol) in DMA (40 mL) was sonicated for 2 h. The mixture was filtered through a shallow bed of silica gel and rinsed well with EtOAc. The filtrate was washed with satd. NaHC03 (lx), 5% LiCl (2x), then brine (lx), dried (MgS04), and concentrated to dryness to afford 3-(5-amino-2-bromo-4-fluorophenyl)-7-chloro-l -ethyl- l,6-naphthyridin-2(lH)-one (2.793 g, 89% yield) as a brown solid. 1H NMR (400 MHz, DMSO-<¾): δ 8.77 (s, 1 H), 8.00 (s, 1 H), 7.74 (s, 1 H), 7.37 (d, 1 H), 6.77 (d, 1 H), 5.45 (s, 2 H), 4.27 (q, 2 H), 1.20 (t, 3 H); MS (ESI) m z: 398.0 [M+H]+.
[0126] Example A14: A suspension of Example A13 (1.50 g, 3.78 mmol) in dioxane (15 mL) was treated with methylamine (40% in water) (26.4 mL, 303 mmol) in a pressure tube and heated to 100°C overnight. The mixture was cooled to RT, treated with a large amount of brine, then diluted with EtOAc until all of the solids dissolved. The layers were separated, the aqueous layer extracted with additional EtOAc (lx) and the combined organics were washed with satd. NaHC03 (lx), dried (MgS04) and concentrated to dryness. The resulting solid was suspended in MeCN/H20, frozen and lyophilized to afford 3-(5-amino-2-bromo-4-fluorophenyl)-l-ethyl-7-(methylamino)-l,6-naphthyridin-2(lH)-one (1.32g, 89% yield) as a light brown solid. 1H NMR (400 MHz, DMSO-<¾): δ 8.37 (s, 1 H), 7.62 (s, 1 H), 7.30 (d, 1 H), 6.99 (q, 1 H), 6.73 (d, 1 H), 6.21 (s, 1 H), 5.33 (s, 2 H), 4.11 (q, 2 H), 2.84 (d, 3 H), 1.19 (t, 3 H); MS (ESI) m/z: 393.0 [M+H]+.
[0263] Example 31: A mixture of Example A14 (0.120 g, 0.307 mmol) and TEA (0.043 mL, 0.307 mmol) in THF (3.0 mL) was treated with phenyl isocyanate (0.040 g, 0.337 mmol) and stirred at RT for 4 h. Over the course of the next 4 days the mixture was treated with additional phenyl isocyanate (0.056 mL) and stirred at RT. The resulting solid was filtered, rinsed with THF, then triturated with MeOH to afford l-(4-bromo-5-(l-ethyl-7-(methylamino)-2-oxo- 1 ,2-dihydro- 1 ,6-naphthyridin-3 -yl)-2-fluorophenyl)-3 -phenylurea (101 mg, 64.5% yield) as a bright white solid. 1H NMR (400 MHz, DMSO-<¾): δ 9.09 (s, 1 H), 8.68 (s, 1 H), 8.41 (s, 1 H), 8.17 (d, 1 H), 7.70 (s, 1 H), 7.65 (d, 1 H), 7.41 (d, 2 H), 7.27 (m, 2 H), 7.03 (m, 1 H), 6.96 (t, 1 H), 6.23 (s, 1 H), 4.13 (q, 2 H), 2.86 (d, 3 H), 1.20 (t, 3 H); MS (ESI) m/z: 510.1 [M+H]+.
References
- ^ Jump up to:a b c “Qinlock Australian Prescription Medicine Decision Summary”. Therapeutic Goods Administration (TGA). 21 July 2020. Retrieved 17 August 2020.
- ^ “Ripretinib (Qinlock) Use During Pregnancy”. Drugs.com. 10 August 2020. Retrieved 17 August 2020.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t “FDA Approves First Drug for Fourth-Line Treatment of Advanced Gastrointestinal Stromal Tumors”. U.S. Food and Drug Administration (FDA) (Press release). 15 May 2020. Retrieved 15 May 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e f g “Drug Trial Snapshot: Qinlock”. U.S. Food and Drug Administration (FDA). 15 May 2020. Retrieved 2 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ “FDA Grants Full Approval of Deciphera Pharmaceuticals’ Qinlock (ripretinib) for the Treatment of Fourth-Line Gastrointestinal Stromal Tumor”. Deciphera Pharmaceuticals, Inc. (Press release). 15 May 2020. Retrieved 15 May 2020.
- ^ “Qinlock: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 15 May 2020.
- ^ Jump up to:a b c d e f g h i “FDA approves ripretinib for advanced gastrointestinal stromal tumor”. U.S. Food and Drug Administration (FDA). 15 May 2020. Retrieved 18 May 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Ripretinib Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 2 October 2014. Retrieved 15 May 2020.
- ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 81”. WHO Drug Information. 33 (1): 106. hdl:10665/330896. License: CC BY-NC-SA 3.0 IGO.
- ^ “Ripretinib” (PDF). United States Adopted Name (USAN) Drug Finder. Retrieved 17 May 2020.
Further reading
- Schneeweiss M, Peter B, Bibi S, et al. (May 2018). “The KIT and PDGFRA switch-control inhibitor DCC-2618 blocks growth and survival of multiple neoplastic cell types in advanced mastocytosis”. Haematologica. 103 (5): 799–809. doi:10.3324/haematol.2017.179895. PMC 5927976. PMID 29439183.
External links
- “Ripretinib”. Drug Information Portal. U.S. National Library of Medicine.
- “Ripretinib”. NCI Drug Dictionary. National Cancer Institute.
- “Ripretinib”. National Cancer Institute.
- Clinical trial number NCT03353753 for “Phase 3 Study of DCC-2618 vs Placebo in Advanced GIST Patients Who Have Been Treated With Prior Anticancer Therapies (invictus)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Pronunciation | rip re’ ti nib |
| Trade names | Qinlock |
| Other names | DCC-2618 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620035 |
| License data | US DailyMed: Ripretinib |
| Pregnancy category | AU: D[1]US: N (Not classified yet)[2]Use should be avoided |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | AU: S4 (Prescription only) [1]US: ℞-only [3] |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 1442472-39-0 |
| PubChem CID | 71584930 |
| DrugBank | DB14840 |
| ChemSpider | 67886378 |
| UNII | 9XW757O13D |
| KEGG | D11353 |
| ChEMBL | ChEMBL4216467 |
| Chemical and physical data | |
| Formula | C24H21BrFN5O2 |
| Molar mass | 510.367 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]CCN1C(=O)C(=CC2=C1C=C(NC)N=C2)C1=C(Br)C=C(F)C(NC(=O)NC2=CC=CC=C2)=C1 | |
| InChI[hide]InChI=1S/C24H21BrFN5O2/c1-3-31-21-12-22(27-2)28-13-14(21)9-17(23(31)32)16-10-20(19(26)11-18(16)25)30-24(33)29-15-7-5-4-6-8-15/h4-13H,3H2,1-2H3,(H,27,28)(H2,29,30,33)Key:CEFJVGZHQAGLHS-UHFFFAOYSA-N |
////////////Ripretinib, QINLOCK, リプレチニブ , 2020 APPROVALS, FDA 2020
REPROXALAP
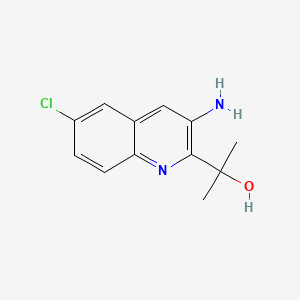
REPROXALAP
レプロキサラップ;
ADX-102
2-(3-amino-6-chloroquinolin-2-yl)propan-2-ol
C12H13ClN2O, 236.7 g/mol
CAS 916056-79-6
UNII-F0GIZ22IJH
2-(3-amino-6-chloroquinolin-2-yl)propan-2-ol
Phase 3 Clinical
Aldeyra Therapeutics is developing reproxalap, which binds and traps free aldehydes, formulated using Captisol technology licensed from Ligand Pharmaceuticals as an eye drop formulation, for treating acute noninfectious anterior uveitis, allergic conjunctivitis and dry eye syndrome.

PATENT
product case, WO2006127945 ,
EU states until 2026
expire US in 2029 with US154 extension.
PATENTS
WO2018170476
United States patent application serial number US 13/709,802, filed December 10, 2012 and published as US 2013/0190500 on July 25, 2013 (“the ‘500 publication,” the entirety of which is hereby incorporated herein by reference), describes certain aldehyde scavenging compounds. Such compounds include com ound A:
[0036] Compound A, (6-chloro-3-amino-2-(2-hydroxypropyl)-l-azanaphthalene), is designated as compound A in the ‘500 publication and the synthesis of compound A is described in detail at Example 5 of the ‘500 publication, and is reproduced herein for ease of reference.
Example A – General Preparation of Compound A
Compound A
[00436] The title compound was prepared according to the steps and intermediates (e.g., Scheme 1) described below and in the ‘500 publication, the entirety of which is incorporated herein by reference.
Step 1: Synthesis of Intermediate A- 1
[00437] To a 2 L round bottom flask was charged ethanol (220 mL), and pyridine (31 g, 392 mmol) and the resulting solution stirred at a moderate rate of agitation under nitrogen. To this solution was added ethyl bromopyruvate (76.6 g, 354 mmol) in a slow, steady stream. The reaction mixture was allowed to stir at 65±5° C. for 2 hours.
Step 2: Synthesis of Intermediate A-2
[00438] Upon completion of the 2-hour stir time in example 1, the reaction mixture was slowly cooled to 18-22° C. The flask was vacuum-purged three times at which time 2-amino-5-chloro-benzaldehyde (ACB) (50.0 g, 321 mmol) was added directly to the reaction flask as a solid using a long plastic funnel. Pyridine (64.0 g, 809 mmol) was added followed by an EtOH rinse (10 mL) and the reaction mixture was heated at 80±3° C. under nitrogen for about 16 hours (overnight) at which time HPLC analysis indicated that the reaction was effectively complete.
Step 3: Synthesis of Intermediate A-3
[00439] The reaction mixture from example 2 was cooled to about 70° C. and morpholine (76.0 g, 873 mmol)) was added to the 2 L reaction flask using an addition funnel. The reaction mixture was heated at 80±2° C. for about 2.5 hours at which time the reaction was considered complete by HPLC analysis (area % of A-3 stops increasing). The reaction mixture was cooled to 10-15° C. for the quench, work up, and isolation.
Step 4: Isolation of Intermediate A-3
[00440] To the 2 L reaction flask was charged water (600 g) using the addition funnel over 30-60 minutes, keeping the temperature below 15° C. by adjusting the rate of addition and using a cooling bath. The reaction mixture was stirred for an additional 45 minutes at 10-15° C. then the crude A-3 isolated by filtration using a Buchner funnel. The cake was washed with water (100 mLx4) each time allowing the water to percolate through the cake before applying a vacuum. The cake was air dried to provide crude A-3 as a nearly dry brown solid. The cake was returned to the 2 L reaction flask and heptane (350 mL) and EtOH (170 mL) were added and the mixture heated to 70±3° C. for 30-60 minutes. The slurry was cooled to 0-5° C. and isolated by filtration under vacuum. The A-3 was dried in a vacuum drying oven under vacuum and 35±3° C. overnight (16-18 hours) to provide A-3 as a dark green solid.
Step 5: Synthesis of Compound A
[00441] To a 2 L round bottom flask was charged methylmagnesium chloride (200 mL of 3.0 M solution in THF, 600 mmol). The solution was cooled to 0-5° C. using an ice bath.
[00442] A 500 mL flask (magnetic stirring) was charged with 22.8 grams A-3 from example 4 and THF (365 mL), stirred to dissolve then transferred to an addition funnel on the 2 L Reaction Flask. The A-3 solution was added drop-wise to the reaction flask over 5.75 hours, keeping the temperature of the reaction flask between 0-5° C throughout the addition. At the end of the addition the contents of the flask were stirred for an additional 15 minutes at 0-5° C. then the cooling bath was removed and the reaction was allowed to stir overnight at ambient temperature.
[00443] The flask was cooled in an ice bath and the reaction mixture was carefully quenched by adding EtOH (39.5 g, 857 mmol) drop-wise to the reaction mixture, keeping the temperature of the reaction mixture below 15° C. during the course of the addition. An aqueous solution of H4C1 (84.7 g H4C1 in 415 mL water) was then carefully added and the mixture stirred under moderate agitation for about 30 minutes then transferred to a separately funnel to allow the layers to separate. Solids were present in the aqueous phase so HO Ac (12.5 g) was added and the contents swirled gently to obtain a nearly homogeneous lower aqueous phase. The lower aqueous layer was transferred back to the 2 L reaction flask and stirred under moderate agitation with 2-methylTHF (50 mL) for about 15 minutes. The original upper organic layer was reduced in volume to approximately 40 mL using a rotary evaporator at≤40° C. and vacuum as needed. The phases in the separatory funnel were separated and the upper 2-MeTHF phase combined with the product residue, transferred to a 500 mL flask and vacuum distilled to an approximate volume of 25 mL. To this residue was added 2-MeTHF (50 mL) and distilled to an approximate volume of 50 mL. The crude compound A solution was diluted with 2-MeTHF (125 mL), cooled to 5-10° C. and 2M H2S04 (aq) (250 mL) was slowly added and the mixture stirred for 30 minutes as the temperature was allowed to return to ambient. Heptane (40 mL) was charged and the reaction mixture stirred for an additional 15 minutes then transferred to a separatory funnel and the layers were allowed to separate. The lower aqueous product layer was extracted with additional heptane (35 mL) then the lower aqueous phase was transferred to a 1 L reaction flask equipped with a mechanical stirrer and the mixture was cooled to 5-10° C. The combined organic layers were discarded. A solution of 25% NaOH(aq) was prepared (NaOH, 47 g, water, 200 mL) and slowly added to the 1 L reaction flask to bring the pH to a range of 6.5-8.5.
[00444] EtOAc (250 mL) was added and the mixture was stirred overnight. The mixture was transferred to a separatory funnel and the lower phase discarded. The upper organic layer was washed with brine (25 mL) then the upper organic product layer was reduced in volume on a rotary evaporator to obtain the crude compound A as a dark oil that solidified within a few minutes. The crude compound A was dissolved in EtOAc (20 mL) and filtered through a plug of silica gel (23 g) eluting with 3/1 heptane/EtOAc until all compound A was eluted (approximately 420 mL required) to remove most of the dark color of compound A. The solvent was removed in vacuo to provide 14.7 g of compound A as a tan solid. Compound A was taken up in EtOAc (25 mL) and eluted through a column of silica gel (72 g) using a mobile phase gradient of 7/1 heptane/EtOAc to 3/lheptane/EtOAc (1400 mL total). The solvent fractions containing compound A were stripped, compound A diluted with EtOAc (120 mL) and stirred in a flask with Darco G-60 decolorizing carbon (4.0 g) for about 1 hour. The mixture was filtered through celite using a fitted funnel, rinsing the cake with EtOAc (3 x 15 mL). The combined filtrates were stripped on a rotary evaporator and compound A dissolved in heptane (160 mL)/EtOAc(16 mL) at 76° C. The
homogeneous solution was slowly cooled to 0-5° C, held for 2 hours then compound A was isolated by filtration. After drying in a vacuum oven for 5 hours at 35° C. under best vacuum, compound A was obtained as a white solid. HPLC purity: 100% (AUC).
Example 1 – Preparation of Free Base Forms A and B of Compound A
Compound A
[00445] Compound A is prepared according to the method described in detail in Examples 1-5 of the ‘500 publication, the entirety of which is hereby incorporated herein by reference.
PATENT
example 5 [WO2018039197A1]
https://patents.google.com/patent/WO2018039197A1/en
Exam le 5: Synthesis of NS2

NS2
[00190] 2-(3-amino-6-chloroquinolin-2-yl)propan-2-ol. To a 2 L round bottom flask was charged methylmagnesium chloride (200 mL of 3.0 M solution in THF, 600 mmol). The solution was cooled to 0-5 °C using an ice bath.
[00191] A 500 mL flask (magnetic stirring) was charged with 22.8 grams A-3a from Example 4 and THF (365 mL), stirred to dissolve, and then transferred to an addition funnel on the 2 L reaction flask. The A-3a solution was added drop-wise to the reaction flask over 5.75 hours, keeping the temperature of the reaction flask between 0-5 °C throughout the addition. At the end of the addition the contents of the flask were stirred for an additional 15 minutes at 0-5 °C, then the cooling bath was removed and the reaction was allowed to stir overnight at ambient temperature.
[00192] The flask was cooled in an ice bath and the reaction mixture was carefully quenched by adding EtOH (39.5 g, 857 mmol) drop-wise to the reaction mixture, keeping the temperature of the reaction mixture below 15 °C during the course of the addition. An aqueous solution of H4CI (84.7 g H4CI in 415 mL water) was then carefully added and the mixture stirred under moderate agitation for about 30 minutes then transferred to a separatory funnel to allow the layers to separate. Solids were present in the aqueous phase so HOAc (12.5 g) was added and the contents swirled gently to obtain a nearly homogeneous lower aqueous phase. The lower aqueous layer was transferred back to the 2 L reaction flask and stirred under moderate agitation with 2-methyl-tetrahydrofuran (2-MeTHF) (50 mL) for about 15 minutes. The original upper organic layer was reduced in volume to approximately 40 mL using a rotary evaporator at < 40 °C under vacuum as needed. The phases in the separatory funnel were separated and the upper 2-MeTHF phase combined with the product residue was transferred to a 500 mL flask and vacuum distilled to an approximate volume of 25 mL. To this residue was added 2-MeTHF (50 mL) and the mixture again distilled to an approximate volume of 50 mL. The crude compound NS2 solution was diluted with 2-MeTHF (125 mL), cooled to 5-10 °C, and 2 M H2S04 (aq) (250 mL) was slowly added and the mixture stirred for 30 minutes as the temperature was allowed to return to ambient. Heptane (40 mL) was charged and the reaction mixture stirred for an additional 15 minutes then transferred to a separatory funnel, and the layers were allowed to separate. The lower aqueous product layer was extracted with additional heptane (35 mL), then the lower aqueous phase was transferred to a 1 L reaction flask equipped with a mechanical stirrer, and the mixture was cooled to 5-10 °C. The combined organic layers were discarded. A solution of 25% NaOH (aq) was prepared (NaOH, 47 g, water, 200 mL) and slowly added to the 1 L reaction flask to bring the pH to a range of 6.5 – 8.5.
[00193] EtOAc (250 mL) was added and the mixture was stirred overnight. The mixture was transferred to a separatory funnel and the lower phase discarded. The upper organic layer was washed with brine (25 mL), then the upper organic product layer was reduced in volume on a rotary evaporator to obtain a obtain the crude compound NS2 as a dark oil that solidified within a few minutes. The crude compound NS2 was dissolved in EtOAc (20 mL) and filtered through a plug of silica gel (23 g) eluting with 3/1 heptane/EtOAc until all compound NS2 was eluted (approximately 420 mL required) to remove most of the dark color of compound NS2. The solvent was removed in vacuo to provide 14.7 g of compound NS2 as a tan solid. Compound NS2 was taken up in EtOAc (25 mL) and eluted through a column of silica gel (72g) using a mobile phase gradient of 7/1 heptane/EtOAc to 3/1 heptane/EtOAc (1400 mL total). The solvent fractions containing compound NS2 were evaporated. Compound NS2 was diluted with EtOAc (120 mL) and stirred in a flask with Darco G-60 decolorizing carbon (4.0 g) for about 1 hour. The mixture was filtered through celite using a firtted funnel, rinsing the cake with EtOAc (3 x 15 mL). The combined filtrates were evaporated on a rotary evaporator and compound NS2 dissolved in heptane (160 mL)/EtOAc (16 mL) at 76 °C. The homogeneous solution was slowly cooled to 0-5 °C, held for 2 hours, then compound NS2 was isolated by filtration. After drying in a vacuum oven for 5 hours at 35 °C under best vacuum, compound NS2 was obtained as a white solid. HPLC purity: 100% (AUC); HPLC (using standard conditions): A-2: 7.2 minutes; A-3 : 11.6 minutes.
Preparation of ACB

[00194] After a N2 atmosphere had been established and a slight stream of N2 was flowing through the vessel, platinum, sulfided, 5 wt. % on carbon, reduced, dry (9.04 g, 3.0 wt. % vs the nitro substrate) was added to a 5 L heavy walled pressure vessel equipped with a large magnetic stir-bar and a thermocouple. MeOH (1.50 L), 5-chloro-2-nitrobenzaldehyde (302.1 g, 1.63 mol), further MeOH (1.50 L) and Na2C03 (2.42 g, 22.8 mmol, 0.014 equiv) were added. The flask was sealed and stirring was initiated at 450 rpm. The solution was evacuated and repressurized with N2 (35 psi), 2x. The flask was evacuated and repressurized with H2 to 35 psi. The temperature of the solution reached 30 °C w/in 20 min. The solution was then cooled with a water bath. Ice was added to the water bath to maintain a temperature below 35 °C. Every 2h, the reaction was monitored by evacuating and repressurizing with N2 (5 psi), 2x prior to opening. The progress of the reaction could be followed by TLC: 5-Chloro-2-nitrobenzaldehyde (Rf = 0.60, CH2CI2, UV) and the intermediates (Rf = 0.51, CH2CI2, UV and Rf = 0.14, CH2CI2, UV) were consumed to give ACB (Rf = 0.43, CH2CI2, UV). At 5 h, the reaction had gone to 98% completion (GC), and was considered complete. To a 3 L medium fritted funnel was added celite (ca. 80 g). This was settled with MeOH (ca. 200 mL) and pulled dry with vacuum. The reduced solution was transferred via cannula into the funnel while gentle vacuum was used to pull the solution through the celite plug. This was chased with MeOH (4 x 150 mL). The solution was transferred to a 5 L three-necked round-bottom flask. At 30 °C on a rotavap, solvent (ca. 2 L) was removed under reduced pressure. An N2 blanket was applied. The solution was transferred to a 5L four-necked round-bottomed flask equipped with mechanical stirring and an addition funnel. Water (2.5 L) was added dropwise into the vigorously stirring solution over 4 h. The slurry was filtered with a minimal amount of vacuum. The collected solid was washed with water (2 x 1.5 L), 2-propanol (160 mL) then hexanes (2 x 450 mL). The collected solid (a canary yellow, granular solid) was transferred to a 150 x 75 recrystallizing dish. The solid was then dried under reduced pressure (26-28 in Hg) at 40°C overnight in a vacuum-oven. ACB (> 99% by HPLC) was stored under a N2 atmosphere at 5°C.
PATENT
WO-2020223717
Process for preparing reproxalap as acetaldehyde dehydrogenase inhibitor useful for treating ocular diseases and cancer.
PATENT
WO-2020223685
Novel crystalline forms of reproxalap (compound 1; designated as Forms A and B) as acetaldehyde dehydrogenase inhibitor useful for treating ocular diseases and cancer.
PATENT
WO 2020123730
//////////REPROXALAP, レプロキサラップ , ADX-102, Phase 3 Clinical
CC(C)(C1=C(C=C2C=C(C=CC2=N1)Cl)N)O
Lercanidipine
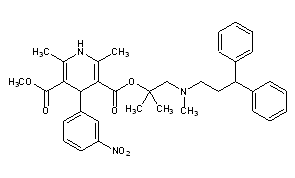
LercanidipineCAS Registry Number: 100427-26-7CAS Name: 1,4-Dihydro-2,6-dimethyl-4-(3-nitrophenyl)-3,5-pyridinedicarboxylic acid 2-[(3,3-diphenylpropyl)methylamino]-1,1-dimethylethyl methyl esterAdditional Names: methyl 1,1,N-trimethyl-N-(3,3-diphenylpropyl)-2-aminoethyl 1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)pyridine-3,5-dicarboxylate; methyl 1,1-dimethyl-2-[N-(3,3-diphenylpropyl)-N-methylamino]ethyl 2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate; masnidipineMolecular Formula: C36H41N3O6Molecular Weight: 611.73Percent Composition: C 70.68%, H 6.76%, N 6.87%, O 15.69%Literature References: Dihydropyridine calcium channel blocker. Prepn: D. Nardi et al.,EP153016; eidem,US4705797 (1985, 1987 both to Recordati). Pharmacology: G. Bianchi et al.,Pharmacol. Res.21, 193 (1989). Clinical evaluation in hypertension: E. Rimoldi et al.,Acta Ther.20, 23 (1994).
Derivative Type: HydrochlorideCAS Registry Number: 132866-11-6Manufacturers’ Codes: Rec-15-2375; R-75Trademarks: Lerdip (Recordati); Zanidip (Napp)Molecular Formula: C36H41N3O6.HClMolecular Weight: 648.19Percent Composition: C 66.71%, H 6.53%, N 6.48%, O 14.81%, Cl 5.47%Properties: Prepd as the hemihydrate, mp 119-123°. LD50 in mice (mg/kg): 83 i.p.; 657 orally (Nardi).Melting point: mp 119-123°Toxicity data: LD50 in mice (mg/kg): 83 i.p.; 657 orally (Nardi)
Therap-Cat: Antihypertensive.Keywords: Antihypertensive; Dihydropyridine Derivatives; Calcium Channel Blocker; Dihydropyridine Derivatives.
Masnidipine hydrochloride, Lercanidipine hydrochloride, TJN-324, Rec-15/2375, Lercan, Cardiovasc, Lerzam, Zanidip, Lerdip, Lercadip, Zanedip
Syn 1
EP 0153016; JP 60199874; US 4772621; US 4968832
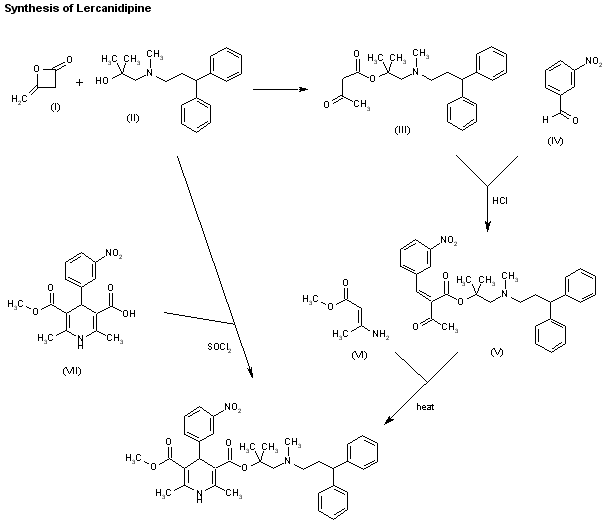
Two new related ways for the synthesis of lercanidipine have been reported: 1) The condensation of diketene (I) with the aminoalcohol (II) gives the corresponding acetoacetate ester (III), which is allowed to react with 3-nitrobenzaldehyde (IV) by means of HCl in chloroform yielding the expected benzylidene derivative (V). Finally, this compound is cyclized with methyl 3-aminocrotonate (VI) in refluxing isopropanol. 2) By esterification of 2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylic acid monomethyl ester (VIII) with alcohol (II) by means of SOCl2 in DMF/dichloromethane.
PATENT
https://patents.google.com/patent/WO2007054969A2/en

PATENT
https://patents.google.com/patent/EP1860102A1/en

PATENT

SPINOSAD

Spinosad
Spinosyn A: The chemical name is: 1H-as-Indaceno[3,2- d]oxacyclododecin-7,a5-dione, 2-[(6-deoxy-2,3,4-tri-O-methyl-alphaL-mannopyranosyl)oxy]-13-[[2R,5S,6R)-5-(dimethylamino) tetrahydro-6-methyl-2H-pyran-2-yl]oxy]-9-ethyl2,3,3a,5a,5b,6,9,10,11,12,13,14,16a,16b-tetradecahydro-14-metyl-, (2R,3aS,5aR,5bS,9S,13S,14R,16aS,16bR)-
Spinosyn D: The chemical name is: 1H-as-Indaceno[3,2- d]oxacyclododecin-7,15-dione, 2-[(6-deoxy-2,3,4-tri-O-methyl-alphaL-mannopyranosyl)oxy]-13-[[2R,5S,6R)-5-(dimethylamino) tetrahydro-6-methyl-2H-pyran-2-yl]oxy]-9-ethyl2,3,3a,5a,5b,6,9,10,11,12,13,14,16a,16b-tetradecahydro-4,14-dimetyl-, (2S,3aSR,5aS,5bS,9S,13S,14R,16aS,16bS)-
168316-95-8
- Molecular FormulaC83H132N2O20
- Average mass1477.938 Da
- Comfortis
- Conserve
- EC 434-300-1
- Natroba
- NaturaLyte
- Spinosad
- Tracer
- Tracer Naturalyte
- UNII-XPA88EAP6V
- XDE 105
Natroba (Spinosad) Suspension 0.9% ParaPro Pharma
New Drug Application (NDA): 022408 appr 01/18/2011
spinosad, is a new molecular entity, and a fermentation product produced by the actinomycete, Saccharopolyspora spinosa. Spinosad contains two components, spinosyn A and D. T

Figure 1. Structure of spinosyn A and DTitle: SpinosynsCAS Registry Number: 131929-60-7Literature References: Class of fermentation derived 12 membered macrocyclic lactones in a unique tetracyclic ring. At least 20 spinosyns have been isolated from Saccharopolyspora spinosa; variations in the two sugars account for most of the structural and insecticidal activity differences. Isolation and biological activity: L. D. Boeck et al.,EP375316 (1990 to Lilly); eidem,US5496931 (1996 to DowElanco); and structure determn: H. A. Kirst et al.,Tetrahedron Lett.32, 4839 (1991). Soil degradation: K. A. Hale, D. E. Portwood, J. Environ. Sci. HealthB31, 477 (1996). HPLC determn in vegetables: L.-T. Yeh et al.,J. Agric. Food Chem.45, 1746 (1997); in soil and water: S. D. West, ibid. 3107. Uptake and metabolism in larvae: T. C. Sparks et al.,Proc. Beltwide Cotton Conf.2, 1259 (1997). Mode of action study: V. L. Salgado et al.,Pestic. Biochem. Physiol.60, 103 (1998). Review of physical and biological properties: C. V. DeAmicis et al.,ACS Symp. Ser.658, 144-154 (1997). Review: G. D. Crouse, T. C. Sparks, Rev. Toxicol.2, 133-146 (1998).
Derivative Type: Spinosyn Acas 131929-60-7CAS Name: (2R,3aS,5aR,5bS,9S,13S,14R,16aS,16bR)-2-[(6-Deoxy-2,3,4-tri-O-methyl-a-L-mannopyranosyl)oxy]-13-[[(2R,5S,6R)-5-(dimethylamino)tetrahydro-6-methyl-2H-pyran-2-yl]oxy]-9-ethyl-2,3,3a,5a,5b,6,9,10,11,12,13,14,16a,16b-tetradecahydro-14-methyl-1H-as-indaceno[3,2-d]oxacyclododecin-7,15-dioneAdditional Names: lepicidin AManufacturers’ Codes: A-83543A; LY-232105Molecular Formula: C41H65NO10Molecular Weight: 731.96Percent Composition: C 67.28%, H 8.95%, N 1.91%, O 21.86%Literature References: Total synthesis: L. A. Paquette et al.,J. Am. Chem. Soc.120, 2553 (1998).Properties: White, odorless crystalline solid, mp 118°. pKa 8.1. uv max (methanol): 243 nm (e 11000). [a]27436 -262.7° (methanol). Vapor pressure: 2.4 ´ 10-10. Soly in water (ppm): 290 (pH 5), 235 (pH 7), 16 (pH 9), distilled 20. Soly (w/v%): methanol 19, acetone 17, dichloromethane >50, hexane 0.45%. LD50 in rats (mg/kg): 3783-5000 orally (Crouse).Melting point: mp 118°pKa: pKa 8.1Optical Rotation: [a]27436 -262.7° (methanol)Absorption maximum: uv max (methanol): 243 nm (e 11000)Toxicity data: LD50 in rats (mg/kg): 3783-5000 orally (Crouse)
Derivative Type: Spinosyn DCAS Registry Number: 131929-63-0Manufacturers’ Codes: A-83543DMolecular Formula: C42H67NO10Molecular Weight: 745.98Percent Composition: C 67.62%, H 9.05%, N 1.88%, O 21.45%Properties: Odorless, white crystalline solid. mp 169°. pKa 7.8. uv max (methanol): 243 nm (e 11000). [a]27436 -297.5° (methanol). Vapor pressure: 2.0 ´ 10-10. Soly in water (ppm): 28 (pH 5), 0.329 (pH 7), 0.04 (pH 9), distilled 1.3. Soly (w/v%): methanol 0.25, acetone 1.0, dichloromethane 45, hexane 0.07%.Melting point: mp 169°pKa: pKa 7.8Optical Rotation: [a]27436 -297.5° (methanol)Absorption maximum: uv max (methanol): 243 nm (e 11000)
Derivative Type: SpinosadCAS Registry Number: 168316-95-8Manufacturers’ Codes: XDE-105; DE-105Trademarks: Conserve (Dow AgroSci.); Justice (Dow AgroSci.); Naturalyte (Dow AgroSci.); SpinTor (Dow AgroSci.); Success (Dow AgroSci.); Tracer (Dow AgroSci.)Literature References: Mixture of spinosyns A and D. Effect on beneficial insects: D. Murray, R. Lloyd, Australian Cottongrower18, 62 (1997).Properties: Light grey to white crystals (tech). LD50 in rats, mallard ducks, quail (mg/kg): >3600, >2000, >2000 orally (Crouse).Toxicity data: LD50 in rats, mallard ducks, quail (mg/kg): >3600, >2000, >2000 orally (Crouse)
Use: Insecticide.(2R,3aS,5aR,5bS,9S,13S,14R,16aS,16bR)-13-{[(2R,5S,6R)-5-(Dimethylamino)-6-methyltetrahydro-2H-pyran-2-yl]oxy}-9-ethyl-14-methyl-7,15-dioxo-2,3,3a,5a,5b,6,7,9,10,11,12,13,14,15,16a,16b-hexadecahydro-1H ;-as-indaceno[3,2-d]oxacyclododecin-2-yl 6-deoxy-2,3,4-tri-O-methyl-α-L-mannopyranoside – (2S,3aR,5aS,5bS,9S,13S,14R,16aS,16bS)-13-{[(2R,5S,6R)-5-(dimethylamino)-6-methyltetrahydro-2H-pyran-2-yl]ox y}-9-ethyl-4,14-dimethyl-7,15-dioxo-2,3,3a,5
1H-as-Indaceno[3,2-d]oxacyclododecin-7,15-dione, 2-[(6-deoxy-2,3,4-tri-O-methyl-α-L-mannopyranosyl)oxy]-13-[[(2R,5S,6R)-5-(dimethylamino)tetrahydro-6-methyl-2H-pyran-2-yl]oxy]-9-ethyl-2,3,3a,5a,5b ,6,9,10,11,12,13,14,16a,16b-tetradecahydro-14-methyl-, (2R,3aS,5aR,5bS,9S,13S,14R,16aS,16bR)-, compd. with (2S,3aR,5aS,5bS,9S,13S,14R,16aS,16bS)-2-[(6-deoxy-2,3,4-tri-O-methyl-α-L-mannopyranosyl)o xy]-13-[[(2R,5S,6R)-5-(dimethylamino)tetrahySpinosad[USAN] [Wiki]168316-95-8 [RN]1H-as-Indaceno[3,2-d]oxacyclododecin-7,15-dione, 2-[(6-deoxy-2,3,4-tri-O-methyl-α-L-mannopyranosyl)oxy]-13-[[2R,5S,6R)-5-(dimethylamino) tetrahydro-6-methyl-2H-pyran-2-yl]oxy]-9-ethyl-2,3,3a,5a,5b,6,9,10,11,12,13,14,16a,16b-tetradecahydro-4,14-dimetyl-,(2S,3aSR,5aS,5bS,9S,13S,14R,16aS,16bS)-1H-as-Indaceno[3,2-d]oxacyclododecin-7,a5-dione, 2-[(6-deoxy-2,3,4-tri-O-methyl-α-L-mannopyranosyl)oxy]-13-[[2R,5S,6R)-5-(dimethylamino) tetrahydro-6-methyl-2H-pyran-2-yl]oxy]-9-ethyl-2,3,3a,5a,5b,6,9,10,11,12,13,14,16a,16b-tetradecahydro-14-metyl-, (2R,3aS,5aR,5bS,9S,13S,14R,16aS,16bR)-NAF-144Spinosad|spinosyn A and D (mixture)spinosyn A and D (mixture)
Spinosad is an insecticide based on chemical compounds found in the bacterial species Saccharopolyspora spinosa. The genus Saccharopolyspora was discovered in 1985 in isolates from crushed sugarcane. The bacteria produce yellowish-pink aerial hyphae, with bead-like chains of spores enclosed in a characteristic hairy sheath.[1] This genus is defined as aerobic, Gram-positive, nonacid-fast actinomycetes with fragmenting substrate mycelium. S. spinosa was isolated from soil collected inside a nonoperational sugar mill rum still in the Virgin Islands. Spinosad is a mixture of chemical compounds in the spinosyn family that has a generalized structure consisting of a unique tetracyclic ring system attached to an amino sugar (D-forosamine) and a neutral sugar (tri-Ο-methyl-L-rhamnose).[2] Spinosad is relatively nonpolar and not easily dissolved in water.[3]
Spinosad is a novel mode-of-action insecticide derived from a family of natural products obtained by fermentation of S. spinosa. Spinosyns occur in over 20 natural forms, and over 200 synthetic forms (spinosoids) have been produced in the lab.[4] Spinosad contains a mix of two spinosoids, spinosyn A, the major component, and spinosyn D (the minor component), in a roughly 17:3 ratio.[1
Mode of action
Spinosad is highly active, by both contact and ingestion, in numerous insect species.[5] Its overall protective effect varies with insect species and life stage. It affects certain species only in the adult stage, but can affect other species at more than one life stage. The species subject to very high rates of mortality as larvae, but not as adults, may gradually be controlled through sustained larval mortality.[5] The mode of action of spinosoid insecticides is by a neural mechanism.[6] The spinosyns and spinosoids have a novel mode of action, primarily targeting binding sites on nicotinic acetylcholine receptors (nAChRs) of the insect nervous system that are distinct from those at which other insecticides have their activity. Spinosoid binding leads to disruption of acetylcholine neurotransmission.[2] Spinosad also has secondary effects as a γ-amino-butyric acid (GABA) neurotransmitter agonist.[2] It kills insects by hyperexcitation of the insect nervous system.[2] Spinosad so far has proven not to cause cross-resistance to any other known insecticide.[7]
Use
Spinosad has been used around the world for the control of a variety of insect pests, including Lepidoptera, Diptera, Thysanoptera, Coleoptera, Orthoptera, and Hymenoptera, and many others.[8] It was first registered as a pesticide in the United States for use on crops in 1997.[8] Its labeled use rate is set at 1 ppm (1 mg a.i./kg of grain) and its maximum residue limit (MRL) or tolerance is set at 1.5 ppm. Spinosad’s widespread commercial launch was deferred, awaiting final MRL or tolerance approvals in a few remaining grain-importing countries. It is considered a natural product, thus is approved for use in organic agriculture by numerous nations.[5] Two other uses for spinosad are for pets and humans. Spinosad has recently been used in oral preparations (as Comfortis) to treat C. felis, the cat flea, in canines and felines; the optimal dose set for canines is reported to be 30 mg/kg.[2]
Spinosad is sold under the trade names, Comfortis, Trifexis, and Natroba.[9][10] Trifexis also includes milbemycin oxime. Comfortis and Trifexis brands treat adult fleas on pets; the latter also prevents heartworm disease. Natroba is sold for treatment of human head lice. Spinosad is also commonly used to kill thrips.[11][12][13]
Spinosyn A
Spinosyn A does not appear to interact directly with known insecticidal-relevant target sites, but rather acts via a novel mechanism.[6] Spinosyn A resembles a GABA antagonist and is comparable to the effect of avermectin on insect neurons.[4] Spinosyn A is highly active against neonate larvae of the tobacco budworm, Heliothis virescens, and is slightly more biologically active than spinosyn D. In general, spinosyns possessing a methyl group at C6 (spinosyn D-related analogs) tend to be more active and less affected by changes in the rest of the molecule.[7] Spinosyn A is slow to penetrate to the internal fluids of larvae; it is also poorly metabolized once it enters the insect.[7] The apparent lack of spinosyn A metabolism may contribute to its high level of activity, and may compensate for the slow rate of penetration.[7]
Safety and ecotoxicology
Spinosad has high efficacy, a broad insect pest spectrum, low mammalian toxicity, and a good environmental profile, a unique feature of the insecticide compared to others currently used for the protection of grain products.[5] It is regarded as natural product-based, and approved for use in organic agriculture by numerous national and international certifications.[8] Spinosad residues are highly stable on grains stored in bins, with protection ranging from 6 months to 2 years.[5][clarification needed] Ecotoxicology parameters have been reported for spinosad, and are:[14]
- in rat (Rattus norvegicus Bergenhout, 1769), acute oral: LD50 >5000 mg/kg (nontoxic)
- in rat (R. norvegicus), acute dermal: LD50 >2000 mg/kg (nontoxic)
- in California quail (Callipepla californica Shaw, 1798), oral toxicity: LD50 >2000 mg/kg (nontoxic)
- in duck (Anas platyrhynchos domestica Linnaeus, 1758), dietary toxicity: LC50 >5000 mg/kg (nontoxic)
- in rainbow trout (Oncorhynchus mykiss Walbaum, 1792), LC50-96h = 30.0 mg/l (slightly toxic)
- in Honeybee (Apis mellifera Linnaeus, 1758), LD50 = 0.0025 mg/bee (highly toxic if directly sprayed on and of dried residues).
Chronic exposure studies failed to induce tumor formation in rats and mice; mice given up to 51 mg/kg/day for 18 months resulted in no tumor formation.[15] Similarly, administration of 25 mg/kg/day to rats for 24 months did not result in tumor formation.[16]
syn
EP 0375,316 (1994, to DowElanco)
US 5496931 (1996 to DowElanco)
PATENT
CN 102190694
https://patents.google.com/patent/CN102190694A/en
Pleocidin compounds (spinosyns) is soil actinomycete thorn many armfuls of bacterium Saccharopolysporaspinosa of sugar secondary metabolites behind aerobic fermentation under developing medium.Pleocidin belongs to macrolides compound, it comprises one a plurality of chiral carbon tetracyclic ring systems (Macrolide tetracycle), big ring is gone up the 9-hydroxyl and is being linked two different hexa-atomic sugar respectively with the 17-hydroxyl, wherein that 17 connections is an aminosugar (Forosamine sugar), and that connect on the 9-position is a rhamnosyl (Rhamnose sugar).Tetracyclic ring system is by one 5,, 6,5-is suitable-and anti–anti–three-loop system condenses one 12 membered macrolide to be formed, and wherein contains an alpha, beta-unsaturated ketone and an independently two key.When 6 on ring is pleocidin A when being substituted by hydrogen, in mixture, account for 85-90%, when ring 6 bit substituents when connecing methyl, be pleocidin D then, in mixture, account for about 10-15%.Up to the present B, C, D, E, F, G, K, L, M, N, O, P, Q, R, S, T, U, more than 20 derivative such as V, W etc. have been found and have isolated it to comprise Spinosyn A.
The commercialization kind has pleocidin Spinosyns (mixture of pleocidin A and pleocidin D) at present, the s-generation pleocidin insecticides Spinetoram. latter is got through semisynthesis by the thick product pleocidin L of biological method preparation and the mixture of J, promptly by 5 of pleocidin J, 6 two key selective reductions, reach 3 ‘ O-ethylization of rhamnosyl and obtain its major ingredient, ethylizing by 3 ‘ O-of pleocidin L rhamnosyl obtains its minor consistuent.
The pleocidin compound can be controlled lepidopteran, Diptera and Thysanoptera insect effectively.It can prevent and treat the pest species of some blade of eating in a large number in Coleoptera and the Orthoptera well.Pleocidin has very high activity to lepidopterous larvaes such as Heliothis virescens, bollworm, beet armyworm, prodenia litura, cabbage looper, small cabbage moth and rice-stem borers, and they are suitable environmental protection, have interesting toxicology character.
U.S. Patent No. 5362634 discloses the derivative that natural pleocidin is replaced by methyl or ethyl on C-21, U.S. Patent application No.60/153513 has disclosed the natural butenyl pleocidin derivative that the 3-4 carbochain replaces on C-21.Pleocidin derivative (John Daeuble, ThomasC.Sparks, Peter Johnson, Paul R.Graupner, the Bioorganic ﹠amp that can prepare C-21 position different substituents by replacement(metathesis)reaction; Medicinal Chemistry17 (2009) 4197-4205).U.S. Patent No. 6001981A, WO 9700265A have openly opened the chemosynthesis of pleocidin compound and have modified, and comprise aminosugar and rhamnosyl and the big chemically modified that encircles in the structure.
PATENT
https://patents.google.com/patent/WO2002077005A1/en
Spinosyns (A83543) are produced by derivatives of Saccharopolyspora spinosa NRRL18395 including strains NRRL 18537, 18538, 18539, 18719, 18720, 18743 and 18823 and derivatives thereof. A more preferred nomenclature for spinosyns is to refer to the pseudoaglycones as spinosyn A 17-Psa, spinosyn D 17-Psa, etc., and to the reverse pseudoaglycones as spinosyn A 9-Psa, spinosyn D 9-Psa, etc. (see Kirst et al., 1991). The known members of this family have been referred to as factors or components, and each has been given an identifying letter designation. These compounds are hereinafter referred to as spinosyn A, B, etc. The spinosyn compounds are useful for the control of arachnids, nematodes and insects, in particular Lepidoptera and Diptera species, and they are quite environmentally friendly and have an appealing toxicological profile. [0004] U.S. Patent No. 5,362,634 and corresponding European Patent Application No. 375316 Al disclose spinosyns A, B, C, D, E, F, G, H, and J. WO 93/09126 discloses spinosyns L, M, N, Q, R, S, and T. WO 94/20518 and US 5,6704,486 disclose spinosyns K,
O, P, U, V, W, and Y, and derivatives thereof. A large number of synthetic modifications to spinosyn compounds have been made, as disclosed in U.S. Patent No. 6,001,981 and WO
97/00265.
PAPER
J. Am. Chem. Soc. 120, 2553 (1998).
Further reading
- The non‐target impact of spinosyns on beneficial arthropods
- Spinosad toxicity to pollinators and associated risk
References
- ^ Jump up to:a b Mertz, Frederick; Raymond C. Yao (Jan 1990). “Saccharopolyspora spinosa sp. nov. Isolated from soil Collected in a Sugar Mill Rum Still”. International Journal of Systematic Bacteriology. 40 (1): 34–39. doi:10.1099/00207713-40-1-34.
- ^ Jump up to:a b c d e Qiao, Meihua; Daniel E. Snyder; Jeffery Meyer; Alan G. Zimmerman; Meihau Qiao; Sonya J. Gissendanner; Larry R. Cruthers; Robyn L. Slone; Davide R. Young (12 September 2007). “Preliminary Studies on the effectiveness of the novel pulicide, spinosad, for the treatment and control of fleas on dogs”. Veterinary Parasitology. 150 (4): 345–351. doi:10.1016/j.vetpar.2007.09.011. PMID 17980490.
- ^ Crouse, Gary; Thomas C Sparks; Joseph Schoonover; James Gifford; James Dripps; Tim Brue; Larry L Larson; Joseph Garlich; Chris Hatton; Rober L Hill; Thomas V Worden; Jacek G Martynow (27 September 2000). “Recent advances in the chemistry of spinosyns”. Pest Manag Sci. 57 (2): 177–185. doi:10.1002/1526-4998(200102)57:2<177::AID-PS281>3.0.CO;2-Z. PMID 11455648.
- ^ Jump up to:a b Watson, Gerald (31 May 2001). “Actions of Insecticidal Spinosyns on gama-Aminobutyric Acid Responses for Small-Diameter Cockroach Neurons”. Pesticide Biochemistry and Physiology. 71: 20–28. doi:10.1006/pest.2001.2559.
- ^ Jump up to:a b c d e Hertlein, Mark; Gary D. Thompson; Bhadriraju Subramanyam; Christos G. Athanassiou (12 January 2011). “Spinosad: A new natural product for stored grain protection”. Stored Products. 47 (3): 131–146. doi:10.1016/j.jspr.2011.01.004. Retrieved 3 May 2012.
- ^ Jump up to:a b Orr, Nailah; Andrew J. Shaffner; Kimberly Richey; Gary D. Crouse (30 April 2009). “Novel mode of action of spinosad: Receptor binding studies demonstrating lack of interaction with known insecticidal target sites”. Pesticide Biochemistry and Physiology. 95: 1–5. doi:10.1016/j.pestbp.2009.04.009.
- ^ Jump up to:a b c d Sparks, Thomas; Gary D crouse; Gregory Durst (30 March 2001). “Natural products as insecticides: the biology, biochemistry and quantitative structure-activity relationships of spinosyns and spinosoids”. Pest Manag Sci. 57 (10): 896–905. doi:10.1002/ps.358. PMID 11695182.
- ^ Jump up to:a b c Sparks, Thomas; James E. Dripps; Gerald B Watson; Doris Paroonagian (6 November 2012). “Resistance and cross-resistance to the spinosyns- A review and analysis”. Pesticide Biochemistry and Physiology. 102: 1–10. doi:10.1016/j.pestbp.2011.11.004. Retrieved 17 November 2011.
- ^ “Spinosad international brands”. Drugs.com. 3 January 2020. Retrieved 30 January2020.
- ^ “Spinosad US brands”. Drugs.com. 3 January 2020. Retrieved 30 January 2020.
- ^ “Spinosad – brand name list from”. Drugs.com. Retrieved 2012-10-20.
- ^ “UC Davis School of Vet Med”. Vetmed.ucdavis.edu. Retrieved 2012-10-20.
- ^ “Safer Flea Control | Insects in the City”. Citybugs.tamu.edu. Retrieved 2012-10-20.
- ^ “Codling Moth and Leafroller Control Using Chemicals” (PDF). Entomology.tfrec.wsu.edu. Retrieved 2012-10-20.
- ^ Stebbins, K. E. (2002). “Spinosad Insecticide: Subchronic and Chronic Toxicity and Lack of Carcinogenicity in CD-1 Mice”. Toxicological Sciences. 65 (2): 276–287. doi:10.1093/toxsci/65.2.276. PMID 11812932. Retrieved 2015-03-08.
- ^ Yano, B. L. (2002). “Spinosad Insecticide: Subchronic and Chronic Toxicity and Lack of Carcinogenicity in Fischer 344 Rats”. Toxicological Sciences. 65 (2): 288–298. doi:10.1093/toxsci/65.2.288. PMID 11812933. Retrieved 2015-03-08.
External links
- “Spinosad”. Drug Information Portal. U.S. National Library of Medicine.
- Monograph
| Spinosyn A | |
| Spinosyn D | |
| Identifiers | |
|---|---|
| CAS Number | 168316-95-8 (A)131929-60-7 (D) |
| ChEBI | CHEBI:9230 (A) CHEBI:9232 (D) |
| ChEMBL | ChEMBL1615373 |
| ChemSpider | 16736513 |
| ECHA InfoCard | 100.103.254 |
| PubChem CID | 183094 (A)443059 (D) |
| CompTox Dashboard (EPA) | DTXSID7032478 |
| InChI[show] | |
| Properties | |
| Chemical formula | C41H65NO10 (A) C42H67NO10 (D) |
| Pharmacology | |
| ATCvet code | QP53BX03 (WHO) |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). |
//////////Spinosad
Dr. Darrin Lew https://www.drdarrinlew.us/insect-control/production-of-spinosad.html
Production of Spinosad
Last Updated on Tue, 29 Oct 2019 | Insect Control
Spinosad is produced directly from the fermentation of a strain of Saccharo-polyspora spinosa. Production strains of S. spinosa have been selected for increased titers of spinosyns A and D, however, no genetic engineering techniques have been used in this process and no genetically-modified organisms are used in the production process. After fermentation, the spinosyn A and D mixture is extracted from the fermentation broth, precipitated and dried to create technical spinosad, which is then formulated into end-use products. Spinosad technical material is also produced under pharmaceutical manufacturing guidelines to be used as a flea control agent in companion animals.
5.9.2 Production of Spinetoram
Production of spinetoram begins with the fermentation of a mutant strain of Saccharopolyspora spinosa that produces primarily spinosyns J and L, rather than spinosyns A and D. This strain was generated through mutagenesis of S. spinosa. However, like the spinosad-producing strains, no genetic engineering techniques were used in this process and no genetically-modified organisms are used in the production process. After fermentation, the spinosyn J and L mixture is extracted from the fermentation broth and precipitated in preparation for the two chemical synthesis steps required to produce spinetoram. The solvents used in extracting and precipitating the spinosyn J and L mixture are recycled.
Spinosyns J and L, unlike spinosyns A and D, have a free hydroxyl group at the 30-position on the rhamnose sugar, which allows for chemical manipulation of this site (see Figure 5.10). In the first synthetic step, the free hydroxyl at the 30-position in spinosyn J and spinosyn L is ethylated to yield a mixture of 30-O-ethyl spinosyn J and 30-O-ethyl spinosyn L. This material is then hydrogenated to yield a mixture of spinetoram-J (30-O-ethyl-5,6-dihydro spinosyn J; see Figure 5.2, structure 5.5) and spinetoram-L (30-O-ethyl spinosyn L; see Figure 5.2, structure 5.6). The hydrogenation conditions are selective and reduce only the disubstituted double bond between C5 and C6 in the 30-O-ethyl spinosyn J intermediate, leaving the 30-O-ethyl spinosyn L unchanged. The material is crystallized from the reaction mixture and dried to create technical spinetoram, which is then formulated into end-use products.
5.9.3 Formulation Attributes of the Spinosyns
To meet a variety of market needs, spinosad and spinetoram products span a very wide range of formulation types (see Table 5.8).
The range of possible formulations for any pesticide is determined by the physical and chemical properties of the active ingredient. Three primary properties determine the formulation characteristics of the spinosyns: (1) both Figure 5.10 Chemical synthesis steps in spinetoram manufacturing.
Figure 5.10 Chemical synthesis steps in spinetoram manufacturing.
Table 5.8 Spinosyn product formulation types and associated uses.
Formulation type
Use pattern
Suspension concentrate
Emulsifiable concentrate Wettable granule Wettable powder Dustable powder Sprayable bait Granular bait Bait stations Granules Tablets
Chewable tablets Gel, paste Creme rinse
Crops, ornamentals, forestry, stored grain, animal health, public health, turf, home and garden Public health Crops
Crops, ornamentals, seed treatment
Stored grain, crops
Crops
Crops, animal health, urban pests
Urban pests
Public health
Public health
Animal health
Urban pests
Public health are fermentation-derived mixtures; (2) both are weak bases; and (3) both have significant solubility in organic solvents.
As fermentation-derived products, spinosad and spinetoram are mixtures composed primarily of two similar, but not identical molecules. In terms of physical properties, a significant difference between the major and minor components of both spinosad and spinetoram is the presence or absence of a methyl group at C6 on the tetracycle (see Table 5.9). With regard to components of spinosad, spinosyn D (methyl group at C6) has a melting point 71 °C higher than that of spinosyn A (hydrogen at C6), and the water solubility of spinosyn D (at pH 7) is almost 1000-fold lower than that of spinosyn A. With regard to the components of spinetoram, spinetoram-L (methyl group at C6) has a melting point 72 °C lower than that of spinetoram-J (hydrogen at C6), and the water solubility of spinetoram-L (at pH 7) is four-fold higher than that of spinetoram-J. The melting points and water solubilities of the mixtures that constitute technical spinosad and technical spinetoram are determined by the relative ratios of the major and minor components.
The predominant components of both spinosad and spinetoram all have pKa values of about 8 (see Table 5.9). As a weak base, the solubility of spinosyns in water increases as the pH is reduced. From a formulation perspective, at pH level above 5, the spinosyns behave like high-melting solids with little water solubility, which results in the predominant agricultural formulations being suspension concentrates and wettable granule formulations composed of milled crystalline particles. Acid salts of spinosyns can be produced and are used in animal health formulations. The basic nature of the spinosyns is also a consideration when combining multiple active ingredients into the same formulation.
The spinosyns have significant solubility in organic solvents (see Table 5.9). This property is relatively rare in high-melting solids with limited water solubility, and has proven to be useful in a number of formulations for
Table 5.9 Selected physical properties of spinosyn A, spinosyn D, spinetoram-
J, and spinetoram-L.
Table 5.9 Selected physical properties of spinosyn A, spinosyn D, spinetoram-
J, and spinetoram-L.
| Property | Spinosyn A133 | Spinosyn D133 | Spinetoram-J134 | Spinetoram-L134 |
| Melting point, °C | 84-99.5a | 161.6-170a | 143.4b | 70.8b |
| Water solubility, | 235 | 0.332 | 11.3 | 46.7 |
| mg/lc’d’e | ||||
| pKaf | 8.10e | 7.87e | 7.86g | 7.59g |
| Solubility in organic solvents, mg/Lc | ||||
| Acetone | 168 000 | 10100 | >250000 | >250000 |
| Ethyl acetate | 194000 | 19 000 | >250000 | >250000 |
| w-Heptane | 12 400 | 300 | 23 900 | >250000 |
| Methanol | 190000 | 2520 | 163 000 | >250000 |
| Xylene | > 250 000 | 64000 | >250000 | >250000 |
“Visual determination. bDiffential scanning calorimetry. cShake flask. ^Buffered to pH 7. eAt 20 °C.
fCapillary zone electrophoresis. gAt 25 °C.
“Visual determination. bDiffential scanning calorimetry. cShake flask. ^Buffered to pH 7. eAt 20 °C.
fCapillary zone electrophoresis. gAt 25 °C.
non-agricultural markets, such as mosquito control and animal health. It is also a consideration when combining the spinosyns with other active ingredients.
////////
https://aem.asm.org/content/82/18/5603

Triheptanoin

Triheptanoin
Approved US FDA 30/6/2020 Dojolvi UX 007
Triheptanoin is a source of heptanoate fatty acids, which can be metabolized without the enzymes of long chain fatty acid oxidation.4 In clinical trials, patients with long chain fatty acid oxidation disorders (lc-FAODs) treated with triheptanoin are less likely to develop hypoglycemia, cardiomyopathy, rhabdomyolysis, and hepatomegaly.1,2 Complications in lc-FAOD patients are reduced from approximately 60% to approximately 10% with the addition of triheptanoin.2
Triheptanoin was granted FDA approval on 30 June 2020.4
Triheptanoin, sold under the brand name Dojolvi, is a medication for the treatment of children and adults with molecularly confirmed long-chain fatty acid oxidation disorders (LC-FAOD).[1][2][3]
The most common adverse reactions include abdominal pain, diarrhea, vomiting, and nausea.[1][2][3]
Triheptanoin was approved for medical use in the United States in June 2020.[4][2][3]
Triheptanoin is a triglyceride that is composed of three seven-carbon (C7:0) fatty acids. These odd-carbon fatty acids are able to provide anaplerotic substrates for the TCA cycle. Triheptanoin is used clinically in humans to treat inherited metabolic diseases, such as pyruvate carboxylase deficiency and carnitine palmitoyltransferase II deficiency. It also appears to increase the efficacy of the ketogenic diet as a treatment for epilepsy.
Since triheptanoin is composed of odd-carbon fatty acids, it can produce ketone bodies with five carbon atoms, as opposed to even-carbon fatty acids which are metabolized to ketone bodies with four carbon atoms. The five-carbon ketones produced from triheptanoin are beta-ketopentanoate and beta-hydroxypentanoate. Each of these ketone bodies easily crosses the blood–brain barrier and enters the brain.
Medical uses
Dojolvi is indicated as a source of calories and fatty acids for the treatment of children and adults with molecularly confirmed long-chain fatty acid oxidation disorders (LC-FAOD).[1][2]
History
Triheptanoin was designated an orphan drug by the U.S. Food and Drug Administration (FDA) in 2006, 2008, 2014, and 2015.[5][6][7][8] Triheptanoin was also designated an orphan drug by the European Medicines Agency (EMA).[9][10][11][12][13][14][15][16]
Triheptanoin was approved for medical use in the United States in June 2020.[4][2]
The FDA approved triheptanoin based on evidence from three clinical trials (Trial 1/NCT018863, Trial 2/NCT022141 and Trial 3/NCT01379625).[3] The trials enrolled children and adults with LC-FAOD.[3] Trials 1 and 2 were conducted at 11 sites in the United States and the United Kingdom, and Trial 3 was conducted at two sites in the United States.[3]
Trial 1 and Trial 2 were used to evaluate the side effects of triheptanoin.[3] Both trials enrolled children and adults diagnosed with LC-FAOD.[3] In Trial 1, participants received triheptanoin for 78 weeks.[3] Trial 2 enrolled participants from other trials who were already treated with triheptanoin (including those from Trial 1) as well as participants who were never treated with triheptanoin before.[3] Trial 2 is still ongoing and is planned to last up to five years.[3]
The benefit of triheptanoin was evaluated in Trial 3 which enrolled enrolled children and adults with LC-FAOD.[3] Half of the participants received triheptanoin and half received trioctanoin for four months.[3] Neither the participants nor the investigators knew which treatment was given until the end of the trial.[3] The benefit of triheptanoin in comparison to trioctanoin was assessed by measuring the changes in heart and muscle function.[3]
Names
Triheptanoin is the international nonproprietary name.[17]
SYN
https://onlinelibrary.wiley.com/doi/abs/10.1002/ejlt.201100425




References
- ^ Jump up to:a b c d “Dojolvi- triheptanoin liquid”. DailyMed. 30 June 2020. Retrieved 24 September2020.
- ^ Jump up to:a b c d e “Ultragenyx Announces U.S. FDA Approval of Dojolvi (UX007/triheptanoin), the First FDA-Approved Therapy for the Treatment of Long-chain Fatty Acid Oxidation Disorders”. Ultragenyx Pharmaceutical. 30 June 2020. Retrieved 30 June 2020 – via GlobeNewswire.
- ^ Jump up to:a b c d e f g h i j k l m n o “Drug Trials Snapshots: Dojolvi”. U.S. Food and Drug Administration. 30 June 2020. Retrieved 16 July 2020.
- ^ Jump up to:a b “Dojolvi: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 30 June 2020.
- ^ “Triheptanoin Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 26 May 2006. Retrieved 30 June 2020.
- ^ “Triheptanoin Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 1 February 2008. Retrieved 30 June 2020.
- ^ “Triheptanoin Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 21 October 2014. Retrieved 30 June 2020.
- ^ “Triheptanoin Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 15 April 2015. Retrieved 30 June 2020.
- ^ “EU/3/12/1081”. European Medicines Agency (EMA). Retrieved 30 June 2020.
- ^ “EU/3/12/1082”. European Medicines Agency (EMA). Retrieved 30 June 2020.
- ^ “EU/3/15/1495”. European Medicines Agency (EMA). Retrieved 30 June 2020.
- ^ “EU/3/15/1508”. European Medicines Agency (EMA). Retrieved 30 June 2020.
- ^ “EU/3/15/1524”. European Medicines Agency (EMA). Retrieved 30 June 2020.
- ^ “EU/3/15/1525”. European Medicines Agency (EMA). Retrieved 30 June 2020.
- ^ “EU/3/15/1526”. European Medicines Agency (EMA). Retrieved 30 June 2020.
- ^ “EU/3/16/1710”. European Medicines Agency (EMA). Retrieved 30 June 2020.
- ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 82”. WHO Drug Information. 33 (3): 694. hdl:10665/330879. License: CC BY-NC-SA 3.0 IGO.
Further reading
- de Almeida Rabello Oliveira M, da Rocha Ataíde T, de Oliveira SL, de Melo Lucena AL, de Lira CE, Soares AA, et al. (March 2008). “Effects of short-term and long-term treatment with medium- and long-chain triglycerides ketogenic diet on cortical spreading depression in young rats”. Neurosci. Lett. 434 (1): 66–70. doi:10.1016/j.neulet.2008.01.032. PMID 18281154. S2CID 7754768.
- Mochel F, DeLonlay P, Touati G, Brunengraber H, Kinman RP, Rabier D, et al. (April 2005). “Pyruvate carboxylase deficiency: clinical and biochemical response to anaplerotic diet therapy”. Mol. Genet. Metab. 84 (4): 305–12. doi:10.1016/j.ymgme.2004.09.007. PMID 15781190.
- Borges K, Sonnewald U (July 2012). “Triheptanoin–a medium chain triglyceride with odd chain fatty acids: a new anaplerotic anticonvulsant treatment?”. Epilepsy Res. 100 (3): 239–44. doi:10.1016/j.eplepsyres.2011.05.023. PMC 3422680. PMID 21855298.
External links
- “Triheptanoin”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT01379625 for “Study of Triheptanoin for Treatment of Long-Chain Fatty Acid Oxidation Disorder (Triheptanoin)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Dojolvi |
| Other names | UX007 |
| AHFS/Drugs.com | Professional Drug Facts |
| License data | US DailyMed: Triheptanoin |
| Pregnancy category | US: N (Not classified yet) |
| Routes of administration | By mouth |
| Drug class | Glycerolipids |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 620-67-7 |
| PubChem CID | 69286 |
| DrugBank | DB11677 |
| ChemSpider | 62497 |
| UNII | 2P6O7CFW5K |
| KEGG | D11465 |
| ChEMBL | ChEMBL4297585 |
| CompTox Dashboard (EPA) | DTXSID40862306 |
| ECHA InfoCard | 100.009.681 |
| Chemical and physical data | |
| Formula | C24H44O6 |
| Molar mass | 428.610 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]CCCCCCC(=O)OCC(COC(=O)CCCCCC)OC(=O)CCCCCC | |
| InChI[hide]InChI=1S/C24H44O6/c1-4-7-10-13-16-22(25)28-19-21(30-24(27)18-15-12-9-6-3)20-29-23(26)17-14-11-8-5-2/h21H,4-20H2,1-3H3 Key:PJHKBYALYHRYSK-UHFFFAOYSA-N |
//////////Triheptanoin, Dojolvi, UX 007, FDA 2020, 2020 APPROVALS
Prescription Products
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Dojolvi | Liquid | 0.96 g/1mL | Oral | Ultragenyx Pharmaceutical Inc. | 2020-07-01 | Not applicable | |
MILVEXIAN
MILVEXIAN
ミルベクシアン;
Molecular Formula,C28-H23-Cl2-F2-N9-O2
Molecular Weight, 626.4441
BMS-986177, JNJ-70033093; JNJ-3093, WHO 11401
CAS 1802425-99-5
(5R,9S)-9-(4-(5-Chloro-2-(4-chloro-1H-1,2,3-triazol-1-yl)phenyl)-6-oxopyrimidin-1(6H)-yl)-21-(difluoromethyl)-5-methyl-21H-3-aza-1(4,2)-pyridina-2(5,4)-pyrazolacyclonaphan-4-one
Prevention and Treatment of Thromboembolic Disorders
Milvexian, also known as BMS-986177, is a blood coagulation factor XIa inhibitor.Bristol-Myers Squibb , in collaboration with Janssen , is developing milvexian (BMS-986177, JNJ-70033093; JNJ-3093), an antithrombotic factor XIa (FXIa) inhibitor, for the oral prevention and treatment of thrombosis.
PATENT
WO-2020210629
Process for preparing milvexian as FXIa and/or plasma kallikrein inhibitors useful for treating deep vein thrombosis, stroke, and atherosclerosis.
(9i?,13ri)-13-{4-[5-chloro-2-(4-chloro- 1 //- 1 2.3-triazol- 1 -yl)phenyl |-6-o\o- 1 6-dihydropyri midin- 1 -yl }-3-(difluoromethyl)-9-methyl-3,4,7,15-tetraazatricyclo[12.3.1.02 (5]octadeca-l(18),2(6),4,14,16-pentaen-8-one, has the structure of Formula (I):
PATENT
WO2020210613
PATENT
WO2016053455
PATENT
product case WO2016053455 novel macrocyclic compounds are FXIa and/or plasma kallikrein inhibitors.
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016053455
Scheme 1
4M HCI or TFA
1c 1a
Scheme 2
2d
Scheme 3
EXAMPLES
Example 1. Preparation of (9i?,135)-13-{4-[5-chloro-2-(4-chloro-lH-l,2,3-triazol-l-yl)phenyl]-6-oxo- 1 ,6-dihydropyrimidin- 1 -yl} -3-(difluoromethyl)-9-methyl-3,4,7, 15-tetraazatricyclo[ 12.3.1.02‘6] -8-one trifluoroacetate
1A. Preparation of l-(difluoromethyl)-4-nitro-lH-pyrazole
CS2CO3 (14.41 g, 44.2 mmol) was suspended in a solution of 4-nitro-lH-pyrazole (5.00 g, 44.2 mmol) and DMF (40 mL). After heating to 120 °C for 5 min, solid sodium 2-chloro-2,2-difluoroacetate (13.48 g, 88 mmol) was added in 10 equal portions over 20 min. The reaction was complete after 10 min of additional heating. The mixture was added to a separatory funnel containing 100 mL water and extracted with Et20 (2 x 50 mL). The combined organic layers were concentrated. Purification by normal-phase chromatography eluting with a gradient of hexanes/EtOAc yielded l-(difluoromethyl)-4-nitro-lH-pyrazole (6.99 g, 42.9 mmol, 97% yield) as a clear, colorless oil. 1H NMR (500MHz, CDCI3) δ 8.58 (s, 1H), 8.22 (s, 1H), 7.39 – 7.05 (t, J= 60 Hz, 1H).
IB. Preparation of (S)-tert-butyl (l-(4-(l-(difluoromethyl)-4-nitro-lH-pyrazol-5-yl)pyridin-2-yl)but-3 -en- 1 -yl)carbamate
To a N2 flushed, 500 mL RBF was added {S)-tert-bvXy\ (l-(4-chloropyridin-2-yl)but-3-en-l-yl)carbamate, prepared as described in Example 3, (10 g, 35.4 mmol), 1-(difluoromethyl)-4-nitro-lH-pyrazol (6.34 g, 38.9 mmol) and dioxane (100 mL). The solution was bubbled with N2 for 5 min. Then Pd(OAc)2 (0.40 g, 1.7 mmol),
di(adamantan-l-yl)(butyl)phosphine (1.27 g, 3.5 mmol), K2CO3 (14.7 g, 106 mmol) and PvOH (1.08 g, 10.61 mmol) were added. The reaction mixture was bubbled with N2 for 5 min then the reaction mixture was heated to 100 °C for 3 h. After this time, the solution was cooled to rt and water (200 mL) was added. The reaction mixture was then extracted with EtOAc (2 x 200 mL). The combined organic extracts were washed with water (200 mL), brine (200 mL), dried over Na2S04, filtered and concentrated in vacuo. Purification by normal phase chromatography eluting with a gradient of hexanes/EtOAc afforded (S)-tert-butyl ( 1 -(4-( 1 -(difluoromethyl)-4-nitro- lH-pyrazol-5 -yl)pyridin-2-yl)but-3 -en- 1 -yl)carbamate (12.91 g, 31.5 mmol, 89% yield) as a slightly yellow oil. MS(ESI) m/z: 410.4 [M+H]+. 1H NMR (400MHz, CDC13) δ 8.80 (dd, J=5.1, 0.7 Hz, 1H), 8.36 (s, 1H), 7.34 (s, 1H), 7.31 (dd, J=5.1, 1.5 Hz, 1H), 7.27 – 6.91 (t, J=58 Hz, 1H), 5.79 – 5.63 (m, 1H), 5.16 – 5.03 (m, 2H), 4.92 (d, J=5.9 Hz, 1H), 2.67 (t, J=6.4 Hz, 2H), 1.46 (br. s., 9H).
1C. Preparation of
(l-(4-(4-amino-l -(difluoromethyl)- lH-pyrazol-5-yl)pyridin-2-yl)but-3 -en- 1 -yl)carbamate
To a 100 mL, 3-necked RBF was added a solution of (S)-tert-butyl (l-(4-(l-(difluoromethyl)-4-nitro-lH-pyrazol-5-yl)pyridin-2-yl)but-3-en-l-yl)carbamate (0.78 g, 1.90 mmol) in MeOH (12 mL) and a solution of NH4C1 (1.02 g, 19 mmol) in water (3 mL). To the solution was added Fe (0.53 g, 9.49 mmol). The reaction mixture was heated to 65 °C for 3 h. Water (50 mL) was added. After cooling to rt, the mixture was filtered through a CELITE® pad and rinsed with MeOH (200 mL). The filtrate was concentrated in vacuo. The residue was partitioned between EtOAC (100 mL) and water (100 mL). The organic phase was separated, washed with water (100 mL), brine (100 mL), dried over Na2S04, filtered and concentrated in vacuo. Purification by normal phase chromatography eluting with a gradient of DCM/MeOH yielded (S)-tert-butyl (l-(4-(4-amino- 1 -(difluoromethyl)- lH-pyrazol-5 -yl)pyridin-2-yl)but-3 -en- 1 -yl)carbamate (0.585 g, 1.54 mmol, 81% yield) as an oil. MS(ESI) m/z: 380.1 [M+H]+. 1H NMR (400MHz,
CDC13) δ 8.70 (dd, J=5.0, 0.7 Hz, 1H), 7.43 (s, 1H), 7.36 (s, 1H), 7.32 (dd, J=5.1, 1.5 Hz, 1H), 7.28 – 6.97 (t, J=58 Hz, 1H), 5.80 – 5.66 (m, 1H), 5.65 – 5.53 (m, 1H), 5.13 – 5.03 (m, 2H), 4.87 (br. s., 1H), 3.22 (br. s., 2H), 2.65 (t, J=6.5 Hz, 2H), 1.52 – 1.37 (m, 9H).
ID. Preparation of tert-butyl ((5)-l-(4-(l-(difiuoromethyl)-4-((i?)-2-methylbut-3-enamido)- lH-pyrazol-5-yl)pyridin-2-yl)but-3-en- 1 -yl)carbamate
To a N2 flushed, 3 -necked, 250 mL RBF was added a solution of {S)-tert-bvXy\ (1-(4-(4-amino-l-(difluoromethyl)-lH-pyrazol-5-yl)pyridin-2-yl)but-3-en-l-yl)carbamate (5 g, 13.18 mmol) and EtOAc (50 ml). The solution was cooled to -10 °C and (R)-2-methylbut-3-enoic acid, as prepared in Example 2, (1.72 g, 17.13 mmol), pyridine (4.26 ml, 52.7 mmol). and T3P® (23.54 ml, 39.5 mmol) were added. The cooling bath was removed and the solution was allowed to warm to rt and then stir over a period of 20 h. Water (30 mL) and EtOAc (30 mL) were added and the mixture was stirred for 30 min. The organic phase was separated and the aqueous layer was extracted with EtOAc (30 mL). The combined organic extracts were washed with brine (50 mL), dried over
Na2SC”4, filtered and concentrated in vacuo. Purification by normal phase
chromatography eluting with a gradient of hexanes/EtOAc gave tert-butyl ((5)-l-(4-(l-(difluoromethyl)-4-((i?)-2-methylbut-3-enamido)-lH-pyrazol-5-yl)pyridin-2-yl)but-3-en-l-yl)carbamate (5.69 g, 12.33 mmol, 94% yield). MS(ESI) m/z: 462.2 [M+H]+. 1H NMR (400MHz, CDC13) δ 8.75 (dd, J=5.0, 0.6 Hz, 1H), 8.37 (s, 1H), 7.32 (t, J=59 Hz, 1H), 7.28 (br. s., 1H), 7.20 (s, 1H), 5.97 – 5.85 (m, 1H), 5.78 – 5.65 (m, 1H), 5.56 – 5.44 (m, 1H), 5.28 – 5.19 (m, 2H), 5.12 (d, J=2.0 Hz, 2H), 4.91 – 4.82 (m, 1H), 3.20 – 3.11 (m, 1H), 2.72 – 2.62 (m, 2H), 1.48 – 1.43 (s, 9H), 1.33 (d, J=6.8 Hz, 3H).
IE. Preparation of tert-butyl N-[(9i?,10E,135)-3-(difluoromethyl)-9-methyl-8-oxo-3,4,7,15-tetraazatricyclo[12.3.1.02‘6]octadeca-l(18),2(6),4,10,14,16-hexaen-13-yl] carbamate
To a N2 flushed, 2 L, 3 -necked, RBF was added a solution of tert-butyl ((S)-l-(4-(1 -(difluoromethyl)-4-((i?)-2-methylbut-3 -enamido)- lH-pyrazol-5 -yl)pyridin-2-yl)but-3 -en-l-yl)carbamate (3 g, 6.50 mmol) in EtOAc (1300 ml). The solution was sparged with argon for 15 min. Grubbs II (1.38 g, 1.63 mmol) was added in one portion. The reaction mixture was heated to reflux for 24 h. After cooling to rt, the solvent was removed and the residue was purified by normal phase chromatography eluting with a gradient of DCM/MeOH to yield tert-butyl N-[(9R, 10E, 135)-3-(difluoromethyl)-9-methyl-8-oxo-3,4,7,15-tetraazatricyclo[12.3.1.02‘6]octadeca-l(18),2(6),4,10,14,16-hexaen-13-yl]carbamate (2.13 g, 4.91 mmol, 76% yield) as a tan solid. MS(ESI) m/z: 434.4 [M+H]+. 1H NMR (400MHz, CDC13) δ 8.71 (d, J=5.1 Hz, 1H), 7.78 (s, 1H), 7.44 – 7.40 (m, 1H), 7.36 (br. s., 1H), 7.27 (t, J=58 Hz, 1H), 6.87 (s, 1H), 6.49 – 6.39 (m, 1H), 5.78 (s, 1H), 4.80 (br. s., 2H), 3.18 – 3.08 (m, 1H), 3.08 – 2.98 (m, 1H), 2.06 – 1.93 (m, 1H), 1.51 (s, 9H), 1.19 (d, J=6.6 Hz, 3H).
IF. Preparation of tert-butyl N-[(9i?,135)-3-(difluoromethyl)-9-methyl-8-oxo-3,4,7,15-tetraazatricyclo[12.3.1.02‘6]octadeca-l(18),2(6),4,14,16-pentaen-13-yl]carbamate
Pd/C (0.60 g, 0.570 mmol) was added to a 250 mL Parr hydrogenation flask containing a solution of tert-butyl N-[(9i?,10E,135)-3-(difluoromethyl)-9-methyl-8-oxo-3,4,7,15-tetraazatricyclo[12.3.1.02‘6]octadeca-l(18),2(6),4,10,14,16-hexaen-13-yljcarbamate (2.46 g, 5.68 mmol) in EtOH (100 mL). The flask was purged with N2 and pressurized to 55 psi of H2 allowed to stir for 18 h. The reaction was filtered through CELITE® and concentrated to yield tert-butyl N-[(9i?,135)-3-(difluoromethyl)-9-methyl-8-oxo-3,4,7,15-tetraazatricyclo[12.3.1.02‘6]octadeca-l(18),2(6),4,14,16-pentaen-13-yl]carbamate (2.17 g, 88% yield) as a tan solid. MS(ESI) m/z: 436.3 [M+H]+. 1H NMR (400MHz, DMSO-d6) δ 9.32 (s, 1H), 8.71 (d, J=5.0 Hz, 1H), 7.96 (t, J=58 Hz, 1H), 7.43 (s, 1H), 7.32 (d, J=4.8 Hz, 1H), 7.22 (d, J=7.3 Hz, 1H), 4.66 (d, J=8.3 Hz, 1H), 2.62 (br. s., 1H), 1.88 (d, J=12.8 Hz, 1H), 1.77 – 1.59 (m, 2H), 1.42 – 1.28 (m, 9H), 1.15 (d, J=18.2 Hz, 2H), 0.83 (d, J=7.0 Hz, 3H).
I G. Preparation of (9R, 13S)-l 3-amino-3-(difiuoromethyl)-9-methyl-3,4,7, 15-tetraazatricyclo[ 12.3.1.02‘6]octadeca- 1(18),2(6),4, 14,16-pentaen-8-one
4 N HC1 in dioxane (3.88 mL, 15.5 mmol) was added to a solution of tert-butyl N-[(9R, 13S)-3-(difluoromethyl)-9-methyl-8-oxo-3,4,7, 15-tetraazatricyclo[12.3.1.02‘6] octadeca-l(18),2(6),4,14,16-pentaen-13-yl]carbamate (2.25 g, 5.2 mmol) in MeOH (10 mL). The reaction was allowed to stir at rt for 2 h. The reaction was cooled in an ice bath, and 7 N NH3 in MeOH (13.3 mL, 93.0 mmol) was added. After 5 min, the reaction was diluted with CH2C12 (80 mL) and the solid that formed was filtered. The filtrate was concentrated to yield (9i?,135)-13-amino-3-(difluoromethyl)-9-methyl-3,4,7,15-tetraazatricyclo[12.3.1.02‘6]octadeca-l(18),2(6),4,14,16-pentaen-8-one (1.3 g, 3.88 mmol, 75% yield). MS(ESI) m/z: 336.3 [M+H]+. 1H NMR (400MHz, DMSO-d6) δ 9.33 (s, 1H), 8.71 (d, J=5.0 Hz, 1H), 7.94 (t, J=58 Hz, 1H), 7.85 (s, 1H), 7.40 (s, 1H), 7.32 (d, J=5.0 Hz, 1H), 4.01 (dd, J=10.2, 5.1 Hz, 1H), 2.63 – 2.53 (m, 1H), 1.90 – 1.69 (m, 2H), 1.53 -1.36 (m, 2H), 1.16 – 1.00 (m, 1H), 0.85 (d, J=7.0 Hz, 3H).
1H. Preparation of (9i?,135)-13-{4-[5-chloro-2-(4-chloro-lH-l,2,3-triazol-l-yl)phenyl]-6-oxo- 1 ,6-dihydropyrimidin- 1 -yl} -3-(difluoromethyl)-9-methyl-3 ,4,7, 15-tetraazatricyclo [12.3.1.02‘6]octadeca- 1 ( 18),2(6),4, 14,16-pentaen-8-one.
To a 100 mL flask containing a white suspension of 6-(5-chloro-2-(4-chloro-lH-l,2,3-triazol-l-yl)phenyl)pyrimidin-4-ol (0.83 g, 2.7 mmol), as prepared in Example 4 in ACN (36 mL) was added HATU (1.12 g, 3.0 mmol) and DBU (0.53 mL, 3.5 mmol). The resulting clear, yellow solution was stirred at rt. After 5 min, (9i?,135)-13-amino-3-(difluoromethyl)-9-methyl-3,4,7,15-tetraazatricyclo[12.3.1.02‘6]octadeca-l(18),2(6),4,14,16-pentaen-8-one (0.9 g, 2.68 mmol) was added and the resulting suspension was stirred at rt for 3 h. The reaction was then concentrated and purified by normal phase silica gel chromatography, eluting with a gradient of 0% to 100% EtOAc in hexanes to yield (9i?,135)-13-{4-[5-chloro-2-(4-chloro-lH-l,2,3-triazol-l-yl)phenyl]-6-oxo- 1 ,6-dihydropyrimidin- 1 -yl} -3-(difluoromethyl)-9-methyl-3 ,4,7, 15-tetraazatricyclo [12.3.1.02‘6]octadeca-l(18),2(6),4,14,16-pentaen-8-one (0.87 g, 50% yield) as a white solid. MS(ESI) m/z: 626.2 [M+H]+. 1H NMR (500MHz, CD3OD) δ 8.91 – 8.83 (m, 1H), 8.78 – 8.71 (m, 1H), 8.33 (s, 1H), 7.88 (d, J=2.5 Hz, 1H), 7.74 (s, 2H), 7.69 – 7.67 (m, 1H), 7.65 (s, 1H), 7.63 (t, J=58 Hz, 1H), 7.52 – 7.50 (m, 1H), 6.36 (d, J=0.8 Hz, 1H),
6.06 – 5.95 (m, 1H), 2.76 – 2.65 (m, 1H), 2.36 – 2.21 (m, 1H), 2.08 – 1.93 (m, 2H), 1.63 -1.53 (m, 1H), 1.53 – 1.42 (m, 1H), 0.99 (d, J=6.9 Hz, 3H). Analytical HPLC (Method A): RT = 8.87 min, purity = 99.7%.
///////////MILVEXIAN, BMS 986177, JNJ 70033093, JNJ 3093, WHO 11401, ミルベクシアン ,
C[C@@H]1CCC[C@H](N2C=NC(=CC2=O)c3cc(Cl)ccc3n4cc(Cl)nn4)c5cc(ccn5)c6c(NC1=O)cnn6C(F)F
Cetuximab sarotalocan sodium
Cetuximab Sarotalocan Sodium (Genetical Recombination)
.png)
Cetuximab Sarotalocan Sodium is an antibody-drug-conjugate (molecular weight: 156,000-158,000) consisting of tetrasodium salt of Sarotalocan (6-({[3-({(OC-6-13)-bis({3-[bis(3-sulfopropyl)(3-sulfonatopropyl)azaniumyl]propyl}dimethylsilanolato-κO,κO‘)[(phtalocyaninato(2-)κN29,κN30,κN31,κN32)-1-yl]silicon}oxy)propoxy]carbonyl}amino)hexanoyl (C70H96N11O24S6Si3; molecular weight: 1,752.22)) attached to an average of 2-3 Lys residues of Cetuximab.
[2166339-33-7 , Cetuximab sarotalocan]
Cetuximab sarotalocan sodium
Enarodustat
Enarodustat
エナロデュスタット
JTZ 951
| Formula | C17H16N4O4 |
|---|---|
| CAS | 1262132-81-9 |
| Mol weight | 340.3333 |
PMDA 2020/9/25 APPROVED ENAROY
Anti-anemic, Hypoxia inducible factor-prolyl hydroxylase (HIF-PH) inhibitor
Originator Japan Tobacco
Developer Japan Tobacco; JW Pharmaceutical
Class Acetic acids; Amides; Antianaemics; Pyridones; Small molecules; Triazoles
Mechanism of Action Hypoxia-inducible factor-proline dioxygenase inhibitors
Preregistration Anaemia
27 Dec 2019 Japan Tobacco and SalubrisBio enter into a development and marketing agreement for enarodustat (JTZ 951) in China, Hong Kong, Macau and Taiwan for Anaemia
29 Nov 2019 Preregistration for Anaemia in Japan (PO)
31 Oct 2019 Phase I development in Anaemia is ongoing in USA
Enarodustat is a potent and orally active factor prolyl hydroxylase inhibitor, with an EC50 of 0.22 μM. Enarodustat has the potential for renal anemia treatment
PATENT
WO 2011007856
PAPER
ACS Medicinal Chemistry Letters (2017), 8(12), 1320-1325
https://pubs.acs.org/doi/10.1021/acsmedchemlett.7b00404
Abstract

Inhibition of hypoxia inducible factor prolyl hydroxylase (PHD) represents a promising strategy for the discovery of a next generation treatment for renal anemia. We identified several 5,6-fused ring systems as novel scaffolds of the PHD inhibitor on the basis of pharmacophore analysis. In particular, triazolopyridine derivatives showed potent PHD2 inhibitory activities. Examination of the predominance of the triazolopyridines in potency by electrostatic calculations suggested favorable π–π stacking interactions with Tyr310. Lead optimization to improve the efficacy of erythropoietin release in cells and in vivo by improving cell permeability led to the discovery of JTZ-951 (compound 14), with a 5-phenethyl substituent on the triazolopyridine group, which increased hemoglobin levels with daily oral dosing in rats. Compound 14 was rapidly absorbed after oral administration and disappeared shortly thereafter, which could be advantageous in terms of safety. Compound 14 was selected as a clinical candidate.

(7-Hydroxy-5-phenethyl-[1,2,4]triazolo[1,5-a]pyridine-8-carbonyl)glycine (14)
To a solution of SI-5 (2.28 g, 6.19 mmol) in EtOH (9.1 mL) was added 2N NaOH aq. (12.4 mL, 24.8 mmol) at room temperature. After stirring at 90 °C for 2 h, 6N HCl aq. (4.1 mL, 24.6 mmol). This was allowed to gradually cool with stirring and crystals were precipitated. The crystals were collected by filtration to give the title compound 14 (2.16 g, 103% yield). 1H NMR (400 MHz, DMSO-D6) δ: 14.22 (s, 1H), 12.98 (br s, 1H), 9.84 (t, J = 5.6 Hz, 1H), 8.58 (s, 1H), 7.33– 7.18 (m, 5H), 6.80 (s, 1H), 4.22 (d, J = 5.6 Hz, 2H), 3.40 (t, J = 7.7 Hz, 2H), 3.12 (t, J = 7.7 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ: 170.28, 167.70, 165.32, 152.95, 148.53, 146.49, 140.05, 128.33, 128.20, 126.17, 106.72, 95.56, 41.00, 31.95, 31.72. HRMS m/z: [M+H]+ calcd for C17H17N4O4, 341.1244; found, 341.1243. Anal. (C17H16N4O4) calcd C 59.99%, H 4.74%, N 16.46%; found C 60.02%, H, 4.78%, N, 16.42%. Melting point: 186 °C Purity: 100.0%.
PATENT
WO 2018097254
PATENT
US 20200017492
/////////////Enarodustat, 2020 APPROVALS, JAPAN 2020, エナロデュスタット , JTZ 951, ENAROY, 2020 APPROVALS,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}