

| Record ID | Title | Status | Phase |
|---|---|---|---|
| NCT03041311 | Carboplatin, Etoposide, and Atezolizumab With or Without Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Extensive Stage Small Cell Lung Cancer (SCLC) | Recruiting | 2 |
| NCT02978716 | Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Combination With Gemcitabineand Carboplatin in Metastatic Triple Negative Breast Cancer (mTNBC) | Recruiting | 2 |
| NCT02514447 | Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Patients With Previously Treated Extensive Stage SCLC Receiving Topotecan Chemotherapy | Recruiting | 2 |
| NCT02499770 | Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Combination With Etoposide and Carboplatin in Extensive Stage Small Cell Lung Cancer (SCLC) | Active, not recruiting | 2 |
Home » 2018 (Page 22)
Yearly Archives: 2018
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
BICTEGRAVIR, NEW PATENT, WO 2018005328, CONCERT PHARMA
![]()

BICTEGRAVIR, NEW PATENT, WO 2018005328, CONCERT PHARMA
WO2018005328) DEUTERATED BICTEGRAVIR
CONCERT PHARMACEUTICALS, INC.
TUNG, Roger, D.; (US)

Concert CEO Roger Tung
Novel deuterated forms of bictegravir is claimed. Gilead Sciences is developing the integrase inhibitor bictegravir as an oral tablet for the treatment of HIV-1 infection.
This invention relates to deuterated forms of bictegravir, and pharmaceutically acceptable salts thereof. In one aspect, the invention provides a compound of Formula (I) or a pharmaceutically acceptable salt thereof, wherein each of Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, Y11a, and Y11b is independently hydrogen or deuterium; provided that if each Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, and Y11 is hydrogen, then Y11b is deuterium.

Many current medicines suffer from poor absorption, distribution, metabolism and/or excretion (ADME) properties that prevent their wider use or limit their use in certain indications. Poor ADME properties are also a major reason for the failure of drug candidates in clinical trials. While formulation technologies and prodrug strategies can be employed in some cases to improve certain ADME properties, these approaches often fail to address the underlying ADME problems that exist for many drugs and drug candidates. One such problem is rapid metabolism that causes a number of drugs, which otherwise would be highly effective in treating a disease, to be cleared too rapidly from the body. A possible solution to rapid drug clearance is frequent or high dosing to attain a sufficiently high plasma level of drug. This, however, introduces a number of potential treatment problems such as poor patient compliance with the dosing regimen, side effects that become more acute with higher doses, and increased cost of treatment. A rapidly metabolized drug may also expose patients to undesirable toxic or reactive metabolites.
[3] Another ADME limitation that affects many medicines is the formation of toxic or biologically reactive metabolites. As a result, some patients receiving the drug may experience toxicities, or the safe dosing of such drugs may be limited such that patients receive a suboptimal amount of the active agent. In certain cases, modifying dosing intervals or formulation approaches can help to reduce clinical adverse effects, but often the formation of such undesirable metabolites is intrinsic to the metabolism of the compound.
[4] In some select cases, a metabolic inhibitor will be co-administered with a drug that is cleared too rapidly. Such is the case with the protease inhibitor class of drugs that are used to treat HIV infection. The FDA recommends that these drugs be co-dosed with ritonavir, an inhibitor of cytochrome P450 enzyme 3A4 (CYP3A4), the enzyme typically responsible for their metabolism (see Kempf, D.J. et al., Antimicrobial agents and chemotherapy, 1997, 41(3): 654-60). Ritonavir, however, causes adverse effects and adds to the pill burden for HIV patients who must already take a combination of different drugs. Similarly, the
CYP2D6 inhibitor quinidine has been added to dextromethorphan for the purpose of reducing rapid CYP2D6 metabolism of dextromethorphan in a treatment of pseudobulbar affect. Quinidine, however, has unwanted side effects that greatly limit its use in potential combination therapy (see Wang, L et al., Clinical Pharmacology and Therapeutics, 1994, 56(6 Pt 1): 659-67; and FDA label for quinidine at http://www.accessdata.fda.gov).
[5] In general, combining drugs with cytochrome P450 inhibitors is not a satisfactory strategy for decreasing drug clearance. The inhibition of a CYP enzyme’s activity can affect the metabolism and clearance of other drugs metabolized by that same enzyme. CYP inhibition can cause other drugs to accumulate in the body to toxic levels.
[6] A potentially attractive strategy for improving a drug’s metabolic properties is deuterium modification. In this approach, one attempts to slow the CYP-mediated metabolism of a drug or to reduce the formation of undesirable metabolites by replacing one or more hydrogen atoms with deuterium atoms. Deuterium is a safe, stable, non-radioactive isotope of hydrogen. Compared to hydrogen, deuterium forms stronger bonds with carbon. In select cases, the increased bond strength imparted by deuterium can positively impact the ADME properties of a drug, creating the potential for improved drug efficacy, safety, and/or tolerability. At the same time, because the size and shape of deuterium are essentially identical to those of hydrogen, replacement of hydrogen by deuterium would not be expected to affect the biochemical potency and selectivity of the drug as compared to the original chemical entity that contains only hydrogen.
[7] Over the past 35 years, the effects of deuterium substitution on the rate of metabolism have been reported for a very small percentage of approved drugs (see, e.g., Blake, MI et al, J Pharm Sci, 1975, 64:367-91; Foster, AB, Adv Drug Res 1985, 14:1-40 (“Foster”); Kushner, DJ et al, Can J Physiol Pharmacol 1999, 79-88; Fisher, MB et al, Curr Opin Drug Discov Devel, 2006, 9:101-09 (“Fisher”)). The results have been variable and unpredictable. For some compounds deuteration caused decreased metabolic clearance in vivo. For others, there was no change in metabolism. Still others demonstrated increased metabolic clearance. The variability in deuterium effects has also led experts to question or dismiss deuterium modification as a viable drug design strategy for inhibiting adverse metabolism (see Foster at p.35 and Fisher at p.101).
[8] The effects of deuterium modification on a drug’s metabolic properties are not predictable even when deuterium atoms are incorporated at known sites of metabolism. Only by actually preparing and testing a deuterated drug can one determine if and how the rate of metabolism will differ from that of its non-deuterated counterpart. See, for example, Fukuto et al. (J. Med. Chem.1991, 34, 2871-76). Many drugs have multiple sites where metabolism is possible. The site(s) where deuterium substitution is required and the extent of deuteration necessary to see an effect on metabolism, if any, will be different for each drug.
Exemplary Synthesis
[72] Deuterium-modified analogs of bictegravir can be synthesized by means known in the art of organic chemistry. For instance, using methods described in US Patent No.9,216,996 (Haolun J. et al., assigned to Gilead Sciences, Inc. and incorporated herein by reference), using deuterium-containing reagents provides the desired deuterated analogs.
[73] Such methods can be carried out utilizing corresponding deuterated and optionally, other isotope-containing reagents and/or intermediates to synthesize the compounds delineated herein, or invoking standard synthetic protocols known in the art for introducing isotopic atoms to a chemical structure.
[74] A convenient method for synthesizing compounds of Formula I is depicted in the Schemes below.
 [75] A generic scheme for the synthesis of compounds of Formula I is shown in Scheme 1 above. In a manner analogous to the procedure described in Wang, H. et al. Org. Lett.2015, 17, 564-567, aldol condensation of compound 1 with appropriately deuterated compound 2 affords enamine 3. Enamine 3 is then reacted with primary amine 4 to afford enamine 5, which then undergoes cyclization with dimethyl oxalate followed by ester hydrolysis to provide carboxylic acid 7.
[75] A generic scheme for the synthesis of compounds of Formula I is shown in Scheme 1 above. In a manner analogous to the procedure described in Wang, H. et al. Org. Lett.2015, 17, 564-567, aldol condensation of compound 1 with appropriately deuterated compound 2 affords enamine 3. Enamine 3 is then reacted with primary amine 4 to afford enamine 5, which then undergoes cyclization with dimethyl oxalate followed by ester hydrolysis to provide carboxylic acid 7.
[76] In a manner analogous to the procedure described in US 9,216,996, acetal deprotection of carboxylic acid 7 followed by cyclization with appropriately deuterated aminocyclopentanol 9 provides carboxylic acid intermediate 10. Amide coupling with appropriately deuterated benzylamine 11 followed by deprotection of the methyl ether ultimately affords a compound of Formula I in eight overall steps from compound 1.
[77] Use of appropriately deuterated reagents allows deuterium incorporation at the Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, Y11a, and Y11bpositions of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at any Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, Y11a, and/or Y11b.
[78] Appropriately deuterated intermediates 2a and 2b, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents as exemplified in Scheme 2 below.
S h 2 S th i f C d 2 d 2b

[79] Synthesis of compound 2a (wherein Y3=H) by acetal formation of N,N-dimethylformamide (DMF) with dimethylsulfate has been described in Mesnard, D. et. al. J. Organomet. Chem.1989, 373, 1-10. Replacing DMF with N,N-dimethylformamide-d1 (98-99 atom % D; commercially available from Cambridge Isotope Laboratories) in this reaction would thereby provide compound 2b (wherein Y3=D).
[80] Use of appropriately deuterated reagents allows deuterium incorporation at the Y3 position of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at Y3.
[81] Appropriately deuterated intermediates 4a-4d, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents as exemplified in Scheme 3 below.

[82] As described in Malik, M. S. et. al. Org. Prep. Proc. Int.1991, 26, 764-766, acetaldehyde is converted to alkylhalide 14a via reaction with chlorine gas and subsequent acetal protection with CaCl2 in methanol. As described in CN 103739506, reaction of 14a with aqueous ammonia and then sodium hydroxide provides primary amine 4a (wherein Y9=Y10a=Y10b=H). Replacing acetaldehyde with acetaldehyde-d1, acetaldehyde-2,2,2-d3, or acetaldehyde-d4 (all commercially available from CDN Isotopes with 98-99 atom % D) in the sequence then provides access to compounds 4b (Y9=D, Y10a=Y10b=H), 4c (Y9=H,
Y10a=Y10b=D) and 4d (Y9=Y10a=Y10b=D) respectively (Schemes 3b-d).
[83] Use of appropriately deuterated reagents allows deuterium incorporation at the Y9, Y10a, and Y10b positions of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at any Y9, Y10a, and/or Y10b.
[84] Appropriately deuterated intermediates 9a-9d, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents as exemplified in Scheme 4 below.
 [85] Following the procedures described by Gurjar, M. et. al. Heterocycles, 2009, 77, 909-925, meso-diacetate 16a is prepared in 2 steps from cyclopentadiene. Desymmetrization of 16a is then achieved enzymatically by treatment with Lipase as described in Specklin, S. et. al. Tet. Lett.201455, 6987-6991, providing 17a which is subsequently converted to aminocyclopentanol 9a (wherein Y4a=Y4b=Y5a=Y5b=Y6=Y7a=Y7b=Y8=H) via a 3 step sequence as reported in WO 2015195656.
[85] Following the procedures described by Gurjar, M. et. al. Heterocycles, 2009, 77, 909-925, meso-diacetate 16a is prepared in 2 steps from cyclopentadiene. Desymmetrization of 16a is then achieved enzymatically by treatment with Lipase as described in Specklin, S. et. al. Tet. Lett.201455, 6987-6991, providing 17a which is subsequently converted to aminocyclopentanol 9a (wherein Y4a=Y4b=Y5a=Y5b=Y6=Y7a=Y7b=Y8=H) via a 3 step sequence as reported in WO 2015195656.
[86] As depicted in Scheme 4b, aminocyclopentanol 9b (Y4a=Y4b=Y5a=Y5b=Y6=Y7a=Y7b= Y8=D) is obtained through an analogous synthetic sequence using cyclopentadiene-d6 and performing the penultimate hydrogenation with D2 in place of H2. Cyclopentadiene-d6 is prepared according to the procedure described in Cangoenuel, A. et. al. Inorg. Chem.2013, 52, 11859-11866.
[87] Alternatively, as shown in Scheme 4c, the meso-diol obtained in Scheme 4a is oxidized to the diketone following the procedure reported by Rasmusson, G.H. et. al. Org. Syn.1962, 42, 36-38. Subsequent mono-reduction with sodium borodeuteride and CeCl3 then affords the D1-alcohol in analogy to the method described in WO 2001044254 for the all-protio analog using sodium borohydride. Reduction of the remaining ketone using similar conditions provides the meso-D2-diol in analogy to the method reported in Specklin, S. et. al. Tet. Lett.2014, 55, 6987-6991 for the all protio analog using sodium borohydride. The meso-D2-diol is then converted to 9c (Y4a=Y4b=Y5a=Y5b=Y7a=Y7b=H, Y6=Y8=D) following the same procedures outlined in Scheme 4a.
[88] Likewise, the meso-diol obtained in Scheme 4b may be converted to 9d
(Y4a=Y4b=Y5a=Y5b=Y7a=Y7b=D, Y6=Y8=H) in an analogous manner as depicted in Scheme 4d. The use of deuterated solvents such as D2O or MeOD may be considered to reduce the risk of D to H exchange for ketone containing intermediates.
[89] Use of appropriately deuterated reagents allows deuterium incorporation at the Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, and Y8 positions of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at any Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, and/or Y8.
[90] Appropriately deuterated intermediates 11a-11d, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents exemplified in Scheme 5 below.
Scheme 5. Synthesis of Benzylamines 11a-11d

//////////////////
TRILACICLIB, G1T28
Trilaciclib
update 2021/2/12 US FDA APPROVED COSELA
- Molecular FormulaC24H30N8O
- Average mass446.548 Da
- G1T 28
CAS 1374743-00-6
2′-{[5-(4-Methyl-1-piperazinyl)-2-pyridinyl]amino}-7′,8′-dihydro-6’H-spiro[cyclohexane-1,9′-pyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyrimidin]-6′-one
G1T28, SHR 6390
Spiro[cyclohexane-1,9′(6’H)-pyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyrimidin]-6′-one, 7′,8′-dihydro-2′-[[5-(4-methyl-1-piperazinyl)-2-pyridinyl]amino]-
- 7′,8′-Dihydro-2′-[[5-(4-methyl-1-piperazinyl)-2-pyridinyl]amino]spiro[cyclohexane-1,9′(6’H)-pyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyrimidin]-6′-one
- 2′-[[5-(4-Methylpiperazin-1-yl)pyridin-2-yl]amino}-7′,8′-dihydro-6’H-spiro[cyclohexane-1,9′-pyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyrimidin]-6′-one
UNII:U6072DO9XG
Reduction of Chemotherapy-Induced Myelosuppression
Trilaciclib dihydrochloride
1977495-97-8
In phase II clinical development as a chemoprotectant at G1 Therapeutics for first- or second-line treatment in patients with metastatic triple negative breast cancer, in combination with gemcitabine and carboplatin
![]()
PATENT, WO 2014144326, Compound 89 (also referred to as Compound T)
| WO2014144847A3 | |
| Inventors | Norman E. Sharpless, Jay Copeland Strum, John Emerson Bisi, Patrick Joseph Roberts, Francis Xavier Tavares |
| Applicant | G1 Therapeutics, Inc. |
| Norman Sharpless | |
|---|---|
 |
|
| Born | Norman Edward Sharpless September 20, 1966 Greensboro, North Carolina |
| Nationality | American |
| Other names | Ned Sharpless |
| Occupation | Director, Lineberger Comprehensive Cancer Center Founder, G1 Therapeutics ($GTHX) |
| Notable work | Wellcome Distinguished Professor, American Society of Clinical Investigation Member, Association of American Cancer Institute board of directors, |
NCI Director Dr. Norman E. Sharpless

NCI Director Dr. Norman E. Sharpless, Credit: National Institutes of Health
Norman E. “Ned” Sharpless, M.D., was officially sworn in as the 15th director of the National Cancer Institute (NCI) on October 17, 2017. Prior to his appointment, Dr. Sharpless served as the director of the University of North Carolina (UNC) Lineberger Comprehensive Cancer Center, a position he held since January 2014.
Dr. Sharpless was a Morehead Scholar at UNC–Chapel Hill and received his undergraduate degree in mathematics. He went on to pursue his medical degree from the UNC School of Medicine, graduating with honors and distinction in 1993. He then completed his internal medicine residency at the Massachusetts General Hospital and a hematology/oncology fellowship at Dana-Farber/Partners Cancer Care, both of Harvard Medical School in Boston.
After 2 years on the faculty at Harvard Medical School, he joined the faculty of the UNC School of Medicine in the Departments of Medicine and Genetics in 2002. He became the Wellcome Professor of Cancer Research at UNC in 2012.
Dr. Sharpless is a member of the Association of American Physicians as well as the American Society for Clinical Investigation (ASCI), the nation’s oldest honor society for physician–scientists, and served on the ASCI council from 2011 to 2014. Dr. Sharpless was an associate editor of Aging Cell and deputy editor of the Journal of Clinical Investigation. He has authored more than 150 original scientific papers, reviews, and book chapters, and is an inventor on 10 patents. He cofounded two clinical-stage biotechnology companies: G1 Therapeutics and HealthSpan Diagnostics.
In addition to serving as director of NCI, Dr. Sharpless continues his research in understanding the biology of the aging process that promotes the conversion of normal self-renewing cells into dysfunctional cancer cells. Dr. Sharpless has made seminal contributions to the understanding of the relationship between aging and cancer, and in the preclinical development of novel therapeutics for melanoma, lung cancer, and breast cancer.
Synthesis
WO 2016040858


Trilaciclib (G1T28)
Trilaciclib is a potential first-in-class short-acting CDK4/6 inhibitor in development to preserve hematopoietic stem cells and enhance immune system function during chemotherapy. Trilaciclib is administered intravenously prior to chemotherapy and has the potential to significantly improve treatment outcomes.
G1 is currently evaluating trilaciclib in four Phase 2 clinical trials: three studies in patients with small-cell lung cancer (SCLC), and one study in patients with triple-negative breast cancer (TNBC). Preliminary data from the SCLC trials were presented at the American Society of Clinical Oncology 2017 Annual Meeting and at the 2016 World Conference on Lung Cancer.
Data from a Phase 1 trial in healthy volunteers were presented at the American Society of Clinical Oncology 2015 Annual Meeting and published in Science Translational Medicine. Trilacicilib has been extensively studied in animals; these preclinical data have been presented at several scientific meetings and published in Molecular Cancer Therapeutics, Science Translational Medicine, and Cancer Discovery.
Trilaciclib is a small molecule, competitive inhibitor of cyclin dependent kinases 4 and 6 (CDK4/6), with potential antineoplastic and chemoprotective activities. Upon intravenous administration, trilaciclib binds to and inhibits the activity of CDK4/6, thereby blocking the phosphorylation of the retinoblastoma protein (Rb) in early G1. This prevents G1/S phase transition, causes cell cycle arrest in the G1 phase, induces apoptosis, and inhibits the proliferation of CDK4/6-overexpressing tumor cells. In patients with CDK4/6-independent tumor cells, G1T28 may protect against multi-lineage chemotherapy-induced myelosuppression (CIM) by transiently and reversibly inducing G1 cell cycle arrest in hematopoietic stem and progenitor cells (HSPCs) and preventing transition to the S phase. This protects all hematopoietic lineages, including red blood cells, platelets, neutrophils and lymphocytes, from the DNA-damaging effects of certain chemotherapeutics and preserves the function of the bone marrow and the immune system. CDKs are serine/threonine kinases involved in the regulation of the cell cycle and may be overexpressed in certain cancer cell types. HSPCs are dependent upon CDK4/6 for proliferation.
Trilaciclib (G1T28) is a CDK4/6 inhibitor in phase II clinical development as a chemoprotectant at G1 Therapeutics for first- or second-line treatment in patients with metastatic triple negative breast cancer, in combination with gemcitabine and carboplatin. Also, phase II trials are ongoing in newly diagnosed, treatment-naive small-cell lung cancer patients, in combination with carboplatin, etoposide, and atezolizumab and phase I trials in previously treated small-cell lung cancer patients, in combination with topotecan.
U.S. Patent Nos. 8,822,683; 8,598,197; 8,598,186, 8,691,830, 8,829,102, 8,822,683, 9, 102,682, 9,499,564, 9,481,591, and 9,260,442, filed by Tavares and Strum and assigned to Gl Therapeutics describe a class of N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amine cyclin dependent kinase inhibitors including those of the formula with variables as defined therein):

U.S. Patent Nos. 9,464,092, 9,487,530, and 9,527,857 which are also assigned to Gl Therapeutics describe the use of the above pyrimidine-based agents in the treatment of cancer.
These patents provide a general synthesis of the compounds that is based on a coupling reaction of a fused chloropyrimidine with a heteroaryl amine to form the central disubstituted amine. Such coupling reactions are sometimes referred to as Buchwald coupling (see WO Ί56 paragraph 127; reference WO 2010/020675). The lactam of the fused chloropyrimidine, for example, a 2-chloro-spirocyclo-pyrrolo[2,3-d]pyrimidine-one such as Intermediate K as shown below can be prepared by dehydration of the corresponding carboxylic acid. The reported process to prepare intermediate IK requires seven steps.

(Intermediate IK; page 60, paragraph 215 of WO Ί56)
WO 2013/148748 (U.S. S.N. 61/617,657) entitled “Lactam Kinase Inhibitors” filed by Tavares, and also assigned to Gl Therapeutics likewise describes the synthesis of N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines via the coupling reaction of a fused chloropyrimidine with a heteroaryl amine to form the central disubstituted amine.
WO 2013/163239 (U.S. S.N. 61/638,491) “Synthesis of Lactams” describes a method for the synthesis of this class of compounds with the variation that in the lactam preparation step, a carboxylic acid can be cyclized with a protected amine in the presence of a strong acid and a dehydrating agent, which can be together in one moiety as a strong acid anhydride. The purported improvement is that cyclization can occur without losing the protecting group on the amine before cyclization. The typical leaving group is “tBOC” (t-butoxycarbonyl). The application teaches (page 2 of WO 2013/163239) that the strong acid is, for example, trifluoroacetic acid anhydride, tribromoacetic acid anhydride, trichloroacetic acid anhydride or mixed anhydrides. An additional step may be necessary to take off the N-protecting group. The dehydrating agent can be a carbodiimide-based compound such as DCC (Ν,Ν-dicyclohexylcarbodiimide), EDC (l-ethyl-3-(3-dimethylaminopropyl)carbodiimide, or DIC (Ν,Ν-diisopropylcarbodiimide). DCC and DIC are in the same class of reagents-carbodiimides. DIC is sometimes considered better because it is a liquid at room temperature, which facilitates reactions.
WO 2015/061407 filed by Tavares and licensed to Gl Therapeutics also describes the synthesis of these compounds via the coupling of a fused chloropyrimidine with a heteroaryl amine to form the central disubstituted amine. WO ‘407 focuses on the lactam production step and in particular describes that the fused lactams of these compounds can be prepared by treating the carboxylic acid with an acid and a dehydrating agent in a manner that a leaving group on the amine is not removed during the amide-forming ring closing step.
Other publications that describe compounds of this general class include the following. WO 2014/144326 filed by Strum et al. and assigned to Gl Therapeutics describes compounds and methods for protection of normal cells during chemotherapy using pyrimidine based CDK4/6 inhibitors. WO 2014/144596 filed by Strum et al. and assigned to Gl Therapeutics describes compounds and methods for protection of hematopoietic stem and progenitor cells against ionizing radiation using pyrimidine based CDK4/6 inhibitors. WO 2014/144847 filed by Strum et al. and assigned to Gl Therapeutics describes HSPC-sparing treatments of abnormal cellular proliferation using pyrimidine based CDK4/6 inhibitors. WO2014/144740 filed by Strum et al. and assigned to Gl Therapeutics describes highly active anti -neoplastic and anti-proliferative pyrimidine based CDK 4/6 inhibitors. WO 2015/161285 filed by Strum et al. and assigned to Gl Therapeutics describes tricyclic pyrimidine based CDK inhibitors for use in radioprotection. WO 2015/161287 filed by Strum et al. and assigned to Gl Therapeutics describes analogous tricyclic pyrimidine based CDK inhibitors for the protection of cells during chemotherapy. WO 2015/161283 filed by Strum et al. and assigned to Gl Therapeutics describes analogous tricyclic pyrimidine based CDK inhibitors for use in HSPC-sparing treatments of RB-positive abnormal cellular proliferation. WO 2015/161288 filed by Strum et al. and assigned to Gl Therapeutics describes analogous tricyclic pyrimidine based CDK inhibitors for use as anti -neoplastic and anti-proliferative agents. WO 2016/040858 filed by Strum et al. and assigned to Gl Therapeutics describes the use of combinations of pyrimidine based CDK4/6 inhibitors with other anti-neoplastic agents. WO 2016/040848 filed by Strum et al. and assigned to Gl Therapeutics describes compounds and methods for treating certain Rb-negative cancers with CDK4/6 inhibitors and topoisomerase inhibitors.
Other biologically active fused spirolactams and their syntheses are described, for example, in the following publications. Griffith, D. A., et al. (2013). “Spirolactam-Based Acetyl-CoA Carboxylase Inhibitors: Toward Improved Metabolic Stability of a Chromanone Lead Structure.” Journal of Medicinal Chemistry 56(17): 7110-7119, describes metabolically stable spirolactams wherein the lactam resides on the fused ring for the inhibition of acetyl-CoA carboxylase. WO 2013/169574 filed by Bell et al. describes aliphatic spirolactams as CGRP receptor antagonists wherein the lactam resides on the spiro ring. WO 2007/061677 filed by Bell et al. describes aryl spirolactams as CGRP receptor antagonists wherein the lactam resides on the spiro ring. WO 2008/073251 filed by Bell et al. describes constrained spirolactam compounds wherein the lactam resides on the spiro ring as CGRP receptor antagonists. WO 2006/031606 filed by Bell et al. describes carboxamide spirolactam compounds wherein the spirolactam resides on the spiro ring as CGRP receptor antagonists. WO 2006/031610, WO 2006/031491, and WO 2006/029153 filed by Bell et al. describe anilide spirolactam compounds wherein the spirolactam resides on the spiro ring; WO 2008/109464 filed by Bhunai et al. describes spirolactam compounds wherein the lactam resides on the spiro ring which is optionally further fused.
Given the therapeutic activity of selected N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines, it would be useful to have additional methods for their preparation. It would also be useful to have new intermediates that can be used to prepare this class of compounds.
PATENT
WO 2014144596
PATENT
Compound 89 (also referred to as Compound T)
| WO2014144847A3 | |
| Inventors | Norman E. Sharpless, Jay Copeland Strum, John Emerson Bisi, Patrick Joseph Roberts, Francis Xavier Tavares |
| Applicant | G1 Therapeutics, Inc. |
EXAMPLES
Intermediates B, E, K, L, 1A, IF and 1CA were synthesized according to US 8,598,186 entitled CDK Inhibitors to Tavares, F.X. and Strum, J.C..
The patents WO 2013/148748 entitled Lactam Kinase Inhibitors to Tavares, F.X., WO 2013/163239 entitled Synthesis of Lactams to Tavares, F.X., and US 8,598,186 entitled CDK Inhibitors to Tavares, F.X. and Strum, J.C. are incorporated by reference herein in their entirety. Example 1
Synthesis of tert-butyl N- [2- [(5-bromo-2-chloro-pyrimidin-4yl)amino] ethyl] carbamate, Compound 1

To a solution of 5-bromo-2,4-dichloropyrimidine (3.2 g, 0.0135 mol) in ethanol (80 mL) was added Hunig’s base (3.0 mL) followed by the addition of a solution of N-(tert- butoxycarbonyl)-l,2-diaminoethane (2.5 g, 0.0156 mole) in ethanol (20 mL). The contents were stirred overnight for 20 hrs. The solvent was evaporated under vacuum. Ethyl acetate (200 mL) and water (100 mL) were added and the layers separated. The organic layer was dried with magnesium sulfate and then concentrated under vacuum. Column chromatography on silica gel using hexane/ethyl acetate (0- 60%) afforded tert-butyl N-[2-[(5-bromo-2-chloro-pyrimidin-4- yl)amino]ethyl]carbamate. 1HNMR (d6-DMSO) δ ppm 8.21 (s, 1H), 7.62 (brs, 1H), 7.27 (brs, 1H), 3.39 (m, 2H), 3.12 (m, 2H), 1.34 (s, 9H). LCMS (ESI) 351 (M + H).
Example 2
Synthesis of tert-butyl N-[2-[[2-chloro-5-(3,3-diethoxyprop-l-ynyl)pyrimidin-4- yl] amino] ethyl] carbamate, Compound 2

To tert-butyl N-[2-[(5-bromo-2-chloro-pyrimidin-4-yl)amino]ethyl]carbamate (1.265 g, 6 mmol) in THF (10 mL) was added the acetal (0.778 mL, 5.43 mmol), Pd(dppf)CH2Cl2 (148 g), and triethylamine (0.757 mL, 5.43 mmol). The contents were degassed and then purged with nitrogen. To this was then added Cul (29 mg). The reaction mixture was heated at reflux for 48 hrs. After cooling, the contents were filtered over CELITE™ and concentrated. Column chromatography of the resulting residue using hexane/ethyl acetate (0- 30%) afforded tert-butyl N- [2- [ [2-chloro-5 -(3 ,3 -diethoxyprop- 1 -ynyl)pyrimidin-4-yl]amino] ethyl] carbamate. 1HNMR (d6-DMSO) δ ppm 8.18 (s, 1H), 7.63 (brs, 1H), 7.40 (brs, 1H), 5.55 (s, 1H), 3.70 (m, 2H), 3.60 (m, 2H), 3.42 (m, 2H), 3.15 (m, 2H), 1.19 – 1.16 (m, 15H). LCMS (ESI) 399 (M + H).
Example 3
Synthesis of tert-butyl N-[2-[2-chloro-6-(diethoxymethyl)pyrrolo[2,3-d]pyrimidin-7- yl] ethyl] carbamate, Compound 3

To a solution of the coupled product (2.1 g, 0.00526 mole) in THF (30 mL) was added TBAF solid (7.0 g). The contents were heated to and maintained at 65 degrees for 2 hrs. Concentration followed by column chromatography using ethyl acetate/hexane (0-50%) afforded tert-butyl N-[2-[2-chloro-6-(diethoxymethyl)pyrrolo[2,3-d]pyrimidin-7-yl]ethyl]carbamate as a pale brown liquid (1.1 g). 1FiNMR (d6-DMSO) δ ppm 8.88 (s, 1H), 6.95 (brs, 1H), 6.69 (s, 1H), 5.79 (s, 1H), 4.29 (m, 2H), 3.59 (m, 4H), 3.34 (m, 1H), 3.18 (m, 1H), 1.19 (m, 9H), 1.17 (m, 6H). LCMS (ESI) 399 (M + H).
Example 4
Synthesis of tert-buty\ N-[2-(2-chloro-6-formyl-pyrrolo [2,3-d] pyrimidin-7- yl)ethyl] carbamate, Compound 4

To the acetal (900 mg) from the preceeding step was added AcOH (8.0 mL) and water
(1.0 mL). The reaction was stirred at room temperature for 16 hrs. Cone, and column chromatography over silica gel using ethyl acetate/hexanes (0- 60%) afforded tert-butyl N-[2-(2- chloro-6-formyl-pyrrolo[2,3-d]pyrimidin-7-yl)ethyl]carbamate as a foam (0.510 g). 1HNMR (d6-DMSO) δ ppm 9.98 (s, 1H), 9.18 (s, 1H), 7.66 (s, 1H), 6.80 (brs, 1H), 4.52 (m, 2H), 4.36 (m, 2H), 1.14 (s, 9H). LCMS (ESI) 325 (M + H).
Example 5
Synthesis of 7- [2-(teri-butoxycarbonylamino)ethyl] -2-chloro-pyrrolo [2,3-d] pyrimidine-6- carboxylic acid, Compound 5

To the aldehyde (0.940 g) from the preceeding step in DMF (4 mL) was added oxone (1.95 g, 1.1 eq). The contents were stirred at room temp for 7 hrs. Silica gel column chromatography using hexane/ethyl acetate (0- 100%) afforded l-\2-(tert- butoxycarbonylamino)ethyl]-2-chloro-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid (0.545 g). 1HNMR (d6-DMSO) δ ppm 9.11 (s, 1H), 7.39 (s, 1H), 4.38 (m, 2H), 4.15 (m, 2H), 1.48 (m, 9H). LCMS (ESI) 341(M + H).
Example 6
Synthesis of methyl 7-[2-(teri-butoxycarbonylamino)ethyl]-2-chloro-pyrrolo[2,3- d]pyrimidine-6-carboxylate, Compound 6

To a solution of 2-chloro-7-propyl-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid (0.545 g, 0.00156 mole) from the preceeding step in toluene (3.5 mL) and MeOH (1 mL) was added TMS- diazomethane (1.2 mL). After stirring overnight at room temperature, the excess of TMS- diazomethane was quenched with acetic acid (3 mL) and the reaction was concentrated under vacuum. The residue was purified by silica gel column chromatography with hexane/ethyl acetate (0- 70%) to afford methyl 7-[2-(tert-butoxycarbonylamino)ethyl]-2-chloro-pyrrolo[2,3- d]pyrimidine-6-carboxylate as an off white solid (0.52 g). 1HNMR (d6-DMSO) δ ppm 9.10 (s, 1H), 7.45 (s, 1H), 6.81 (brs, 1H) 4.60 (m, 2H), 3.91 (s, 3H), 3.29 (m, 2H), 1.18 (m, 9H) LCMS (ESI) 355 (M + H).
Example 7
Synthesis of Chloro tricyclic amide, Compound 7

To methyl 7- [2-(tert-butoxycarbonylamino)ethyl] -2-chloro-pyrrolo [2,3 -d]pyrimidine-6- carboxylate (0.50 g, 0.0014 mole) from the preceeding step in dichloromethane (2.0 mL) was added TFA (0.830 mL). The contents were stirred at room temperature for 1 hr. Concentration under vacuum afforded the crude amino ester which was suspended in toluene (5 mL) and Hunig’s base (0.5 mL). The contents were heated at reflux for 2 hrs. Concentration followed by silica gel column chromatography using hexane/ethyl acetate (0- 50%) afforded the desired chloro tricyclic amide (0.260 g). 1HNMR (d6-DMSO) δ ppm 9.08 (s, 1H), 8.48 (brs, 1H), 7.21 (s, 1H) 4.33 (m, 2H), 3.64 (m, 2H). LCMS (ESI) 223 (M + H).
Example 8
Synthesis of chloro-N-methyltricyclic amide, Compound 8

To a solution of the chloro tricycliclactam, Compound 7, (185 mg, 0.00083 mole) in DMF (2.0 mL) was added sodium hydride (55% dispersion in oil, 52 mg). After stirring for 15 mins, methyl iodide (62 μί, 1.2 eq). The contents were stirred at room temperature for 30 mins. After the addition of methanol (5 mL), sat NaHCOs was added followed by the addition of ethyl acetate. Separation of the organic layer followed by drying with magnesium sulfate and concentration under vacuum afforded the N-methylated amide in quantitative yield. 1FiNMR (d6-DMSO) δ ppm 9.05 (s, 1H), 7.17 (s, 1H) 4.38 (m, 2H), 3.80 (m, 2H), 3.05 (s, 3H). LCMS (ESI) 237 (M + H). Example 9
Synthesis of l-methyl-4-(6-nitro-3-pyridyl)piperazine, Compound 9

To 5-bromo-2-nitropyridine (4.93 g, 24.3 mmole) in DMF (20 mL) was added N- methylpiperazine (2.96 g, 1.1 eq) followed by the addition of DIPEA (4.65 mL, 26.7 mmole). The contents were heated at 90 degrees for 24 hrs. After addition of ethyl acetate (200 mL), water (100 mL) was added and the layers separated. Drying followed by concentration afforded the crude product which was purified by silica gel column chromatography using (0-10%) DCM/Methanol. 1HNMR (d6-DMSO) δ ppm 8.26 (s, 1H), 8.15 (1H, d, J = 9.3 Hz), 7.49 (1H, d, J = 9.4 Hz), 3.50 (m, 4H), 2.49 (m, 4H), 2.22 (s, 3H).
Example 10
Synthesis of 5-(4-methylpiperazin-l-yl)pyridin-2-amine, Compound 10

To l-methyl-4-(6-nitro-3-pyridyl)piperazine (3.4 g) in ethyl acetate (100 mL) and ethanol (100 mL) was added 10%> Pd/C (400 mg) and then the reaction was stirred under hydrogen (10 psi) overnight. After filtration through CELITE™, the solvents were evaporated and the crude product was purified by silica gel column chromatography using DCM/ 7N ammonia in MeOH (0- 5%) to afford 5-(4-methylpiperazin-l-yl)pyridin-2-amine (2.2 g). 1HNMR (d6-DMSO) δ ppm 7.56 (1H, d, J = 3 Hz), 7.13 (1H, m), 6.36 (1H, d, J = 8.8 Hz), 5.33 (brs, 2H), 2.88 (m, 4H), 2.47 (m, 4H), 2.16 (s, 3H).
Example 11
Synthesis of tert-butyl 4-(6-amino-3-pyridyl)piperazine-l-carboxylate, Compound 11

This compound was prepared as described in WO 2010/020675 Al .
Synthesis of Compound 89 (also referred to as Compound T)

Compound 89 was synthesized in a similar manner to that described for compound 78 and was converted to an HCl salt. 1HNMR (600 MHz, DMSO-d6) δ ppm 1.47 (br. s., 6 H) 1.72 (br. s., 2 H) 1.92 (br. s., 2 H) 2.77 (br. s., 3 H) 3.18 (br. s., 2 H) 3.46 (br. s., 2 H) 3.63 (br. s., 2 H) 3.66 (d, J=6.15 Hz, 2 H) 3.80 (br. s., 2 H) 7.25 (s, 1 H) 7.63 (br. s., 2 H) 7.94 (br. s., 1 H) 8.10 (br. s., 1 H) 8.39 (br. s., 1 H) 9.08 (br. s., 1 H) 11.59 (br. s., 1 H). LCMS (ESI) 447 (M + H)
PATENT
WO 2014144740
PATENT
Preparation of Active Compounds
Syntheses
The disclosed compounds can be made by the following general schemes:

Scheme 1
In Scheme 1, Ref-1 is WO 2010/020675 Al; Ref-2 is White, J. D.; et al. J. Org. Chem. 1995, 60, 3600; and Ref-3 Presser, A. and Hufher, A. Monatshefte fir Chemie 2004, 135, 1015.

Scheme 2
In Scheme 2, Ref-1 is WO 2010/020675 Al; Ref-4 is WO 2005/040166 Al; and Ref-5 is Schoenauer, K and Zbiral, E. Tetrahedron Letters 1983, 24, 573.

92


93 
3) Pd/C/H2 ![]()
Scheme 6

Scheme 7
NHfOH

Scheme 8
In Scheme 8, Ref-1 is WO 2010/020675 Al; Ref-2 is WO 2005/040166 Al; and Ref-3 is Schoenauer, K and Zbiral, E. Tetrahedron Letters 1983, 24, 573.
Alternatively, the lactam can be generated by reacting the carboxylic acid with a protected amine in the presence of a strong acid and a dehydrating agent, which can be together in one moiety as a strong acid anhydride. Examples of strong acid anhydrides include, but are not limited to, trifluoroacetic acid anhydride, tribromoacetic acid anhydride, trichloroacetic acid anhydride, or mixed anhydrides. The dehydrating agent can be a carbodiimide based compound such as but not limited to DCC (Ν,Ν-dicyclohexylcarbodiimide), EDC (l-ethyl-3-(3-
dimethylaminopropyl)carbodiimide or DIC (Ν,Ν-diisopropylcarbodiimide). An additional step may be necessary to take off the N-protecting group and the methodologies are known to those skilled in the art.
Alternatively, the halogen moiety bonded to the pyrimidine ring can be substituted with any leaving group that can be displaced by a primary amine, for example to create an intermediate for a final product such as Br, I, F, SMe, SO2Me, SOalkyl, SO2alkyl. See, for Exmaple PCT /US2013/037878 to Tavares.
Other amine intermediates and final amine compounds can be synthesized by those skilled in the art. It will be appreciated that the chemistry can employ reagents that comprise reactive functionalities that can be protected and de-protected and will be known to those skilled in the art at the time of the invention. See for example, Greene, T.W. and Wuts, P.G.M., Greene’s Protective Groups in Organic Synthesis, 4th edition, John Wiley and Sons.

Scheme 9
CDK4/6 Inhibitors of the present invention can be synthesized according to the generalized Scheme 9. Specific synthesis and characterization of the Substituted 2-aminopyrmidines can be found in, for instance, WO2012/061156.
Compounds T, Q, GG, and U were prepared as above and were characterized by mass spectrometry and NMR as shown below:
Compound T
1H NMR (600 MHz, DMSO- d6) ppm 1.47 (br. s., 6 H) 1.72 (br. s., 2 H) 1.92 (br. s., 2 H) 2.77 (br. s., 3 H) 3.18 (br. s., 2 H) 3.46 (br. s., 2 H) 3.63 (br. s., 2 H) 3.66 (d, J=6.15 Hz, 2 H) 3.80 (br. s., 2 H) 7.25 (s, 1 H) 7.63 (br. s., 2 H) 7.94 (br. s., 1 H) 8.10 (br. s., 1 H) 8.39 (br. s., 1 H) 9.08 (br. s., 1 H) 11.59 (br. s., 1 H). LCMS ESI (M + H) 447.
PATENT
Synthesis of N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines. The application appears to be particularly focused on methods for the preparation of trilaciclib and an analog of it. Trilaciclib is the company’s lead CDK4/6 inhibitor presently in phase II trials against small-cell lung cancer and triple negative breast cancer. Interestingly, the company is working on a second CDK4/6 inhibitor, G1T38 , which is in a phase II trial against breast cancer.
GENERAL METHODS
The structure of starting materials, intermediates, and final products was confirmed by standard analytical techniques, including NMR spectroscopy and mass spectrometry. Unless otherwise noted, reagents and solvents were used as received from commercial suppliers. Proton nuclear magnetic resonance spectra were obtained on a Bruker AVANCE 500 at 500 MHz in DMSO-dis. HPLC analyses were performed on a Waters HPLC using the below HPLC method.
HPLC Method
Column: Atlantis T3 (150 χ 4.6, 3 μιη)
Column Temperature: 40°C
Flow Rate: 1 mL/min
Detection: UV @ 275 nm
Analysis Time: 36 min
Mobile Phase A: Water (with 0.1% Trifluoroacetic Acid)
Mobile Phase B : Acetonitrile (with 0.1% Trifluoroacetic Acid)
Sample preparation: dissolve PC sample, wet or dry solid (~1 mg of active compound) in acetonitrile/water (1/1) to achieve complete dissolution.
HPLC Method Gradient

Example 1. General Routes of Synthesis

Scheme 1-1 : Starting from an appropriately substituted halo pyrimidine, compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a substituted spirolactam. In Step 2 the appropriately substituted spirolactam is protected with a group selected from R2. In Step 3 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 3, Step 4, Step 5, or Step 6. Oxidation prior to Step 3 results in undesired byproducts. In Step 4 the hydroxyl group of the fused spirolactam is converted to a leaving group.
In Step 5 the leaving group is dehydrated to afford a compound of Formula IV. In Step 6 the compound of Formula IV is optionally deprotected.

Scheme 1-2: Starting from an appropriately substituted halo pyrimidine compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a substituted spirolactam. In Step 2 the appropriately substituted spirolactam is protected with a group selected from R2. In Step 3 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam of Formula IV. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 3 or Step 4. Oxidation prior to Step 3 results in undesired byproducts. In Step 4 the compound of Formula IV is optionally deprotected.

Scheme 1-3 : Starting from an appropriately substituted alkyl glycinate, compounds of the present invention can be prepared. In Step 1 the appropriately substituted alkyl glycinate is subjected to cyclohexanone and TMSCN in the presence of base to afford a cyano species. In Step 2 the appropriately substituted cyanospecies is reduced and subsequently cyclized to afford a compound of Formula I.
Scheme 1-4

Scheme 1-4: Starting from an appropriately substituted l-(aminomethyl)cyclohexan-l-amine, compounds of the present invention can be prepared. In Step 1 the appropriately substituted l-(aminomethyl)cyclohexan-l -amine is reductively aminated with an aldehyde. In Step 2 the appropriately substituted cyclohexane amine is optionally deprotected (i.e.: the group selected from R2 if not H is optionally replaced by H). In Step 3 the cyclohexane amine is cyclized to afford a compound of Formula I. In Step 4 the compound of Formula I is optionally protected.
1-5

Conversion

Scheme 1-5: Starting from an appropriately substituted halo pyrimidine, compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a
substituted spirolactam. In Step 2 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 2, Step 3, Step 4, or Step 5. Oxidation prior to Step 2 results in undesired byproducts. In Step 3 the hydroxyl group of the fused spirolactam is converted to a leaving group. In Step 4 the leaving group is dehydrated to afford a compound of Formula IV. In Step 5 the compound of Formula IV is optionally deprotected.
S

Scheme 1-6: Starting from an appropriately substituted halo pyrimidine compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a substituted spirolactam. In Step 2 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam of Formula IV. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 2 or Step 3. Oxidation prior to Step 2 results in undesired byproducts. In Step 3 the compound of Formula IV is optionally deprotected.

Scheme 1-7: Starting from compound of Formula IV a CDK4/6 inhibitor can be prepared. In Step 1 a heteroaryl amine is subjected to a base and a compound of Formula IV is added slowly under chilled conditions to afford a nucleophilic substitution reaction. The compound of Formula IV can previously be prepared as described in the schemes herein.
Example 2. Representative Routes of Synthesis
Scheme 2-1

quant, yield 2 steps
isolated

70% yield 2 steps 75% yield 95% yield
isolated isolated isolated
Scheme 2-1 : An ester route is one embodiment, of the present invention. Ideally, the best synthesis scheme would afford crystalline intermediates to provide material of consistent purity without column chromatography, and high yielding steps while using safe and cost effective reagents when possible.
The first step in the ester route is a SNAr nucleophilic substitution of CI group in commercially available ester 3 using spirolactam 4. Due to low reactivity of 4, a reaction temperature of 85-95 °C was required. Because of the temperature requirements, DIPEA and dimethylacetamide were selected as the base and solvent, respectively. The reaction follows second-order kinetics and usually stalls after -85% conversion. Therefore, the reaction was typically stopped after 60 hours by first cooling it to room temperature at which point solid formation was observed. The mixture was then partitioned between MTBE and water and product was filtered with excellent purity with -53% yield of the desired product 5. The obtained
compound 5 was protected with a Boc group using Boc anhydride and DMAP as the catalyst and dichloromethane as the solvent. The intermediate 6 was obtained in a quantitative yield. Due to the semi-solid nature of compound 6, the material was taken to the next step without further purification. The Dieckmann condensation was initially performed with strong bases such as LiHMDS and tBuOK. A similar result to the aldehyde route (Scheme 2-2) was obtained: a partial deprotection of Boc group was observed that required column chromatography. However, the best results were obtained when DBU was used as base and THF as solvent. The reaction outcome was complete, clean conversion of 6 to 7. Moreover, the product crystallized from the reaction mixture upon seeding, and a quantitative yield was obtained for the two steps.
The hydroxyl group of 7 was removed via a two-step procedure. First, compound 7 was converted completely into triflate 8 using triflic anhydride and triethylamine in dichloromethane. The reaction was found to proceed well at 0°C. Due to the potential instability of the triflate intermediate, it was not isolated. It was immediately taken to the next step and reduced with triethylsilane and palladium tetrakis to afford the product 9 after ethyl acetate crystallization in -70% yield. The Boc group of 9 was removed using trifluoroacetic acid in dichloromethane to afford 10. Intermediate 10 was converted into the final sulfone 11 using Oxone™ in acetonitrile/water solvent system.
The obtained sulfone 11 was use-tested in the coupling step and was found to perform well. In conclusion, the route to sulfone 11 was developed which eliminated the use of column chromatography with good to excellent yields on all steps.
Scheme 2-2

Molecular Weight: 421 
Scheme 2-2: The first step of Scheme 2-2 consistently afforded product 13 contaminated with one major impurity found in substantial amount. Thorough evaluation of the reaction impurity profile by LC-MS and 2D MR was performed, which showed the impurity was structurally the condensation of two aldehyde 12 molecules and one molecule of lactam 4. Therefore, column chromatography was required to purify compound 13, which consistently resulted in a modest 30% yield. A solvent screen revealed that sec-butanol, amyl alcohol, dioxane, and tert-butanol can all be used in the reaction but a similar conversion was observed in each case. However, tert-butanol provided the cleanest reaction profile, so it was selected as a solvent for the reaction. Assessing the impact of varying the stoichiometric ratio of 4 and 12 on the reaction outcome was also investigated. The reaction was performed with 4 equivalents of amine 4 in an attempt to disrupt the 2: 1 aldehyde/amine composition of the impurity. The result was only a marginal increase in product 13 formation. The temperature impact on the reaction outcome was evaluated next. The coupling of aldehyde 12 and 4 was investigated at two different temperatures: 50 °C and 40 °C with 1 : 1 ratio of aldehyde/amine. Reactions were checked at 2 and 4 hours and then every 12 hours. The reaction progress was slow at 50°C and was accompanied by growth of other impurities. The reaction at 40°C was much cleaner; however the conversion was lower in the same time period. The mode of addition of the reagents was investigated as well at 80°C with a slow addition (over 6 hours) of either aldehyde 12 or amine 4 to the reaction mixture. The product distribution did not change and an about 1 to 1 ratio was observed between product and impurity when amine 4 was added slowly to the reaction mixture containing aldehyde 12 and
DIPEA at reflux. The product distribution did change when aldehyde 12 was added slowly to the mixture of amine 4 and DIPEA. However, the major product of the reaction was the undesired impurity. Other organic bases were tried as well as different ratios of DIPEA. No product was observed when potassium carbonate was used as a base. The results of the experiments are presented in Table 1 below.
Table 1

Compound 13 was successfully formed in three cases: triethylamine, 2,6-lutidine and DIPEA, with the DIPEA result being the best. The use of Boc protected spirolactam 4 had no effect on the impurity formation as well. Its utilization was speculated to be beneficial in performing the coupling step together with the following step, preparation of compound 14.
The major impurity formed during Step 1 of Scheme 2-2 is:

Chemical Formula:€2)Η;Μ(¾ 6( 2ί>2
Molecular Weight: 527.4903
The second step (Boc protection of the free lactam) proceeded well using DMAP as a catalyst in dichloromethane at room temperature. The product 14 is a thick oil, and, therefore, cannot be purified by crystallization. The Boc protected intermediate 14 was cyclized successfully into the desired pentacyclic structure 10 upon treatment with a strong base such as LiHMDS or tBuOK. Surprisingly, the Boc group was partially removed during the reaction. The level of deprotection was independent from the internal reaction temperature and was positively correlated with excess of base used. Therefore the mixture of the desired product 10 and 10-Boc compound was treated with acid to completely deprotect Boc group. The conversion of methyl sulfide into the final sulfone 11 was carried out with Oxone™. Initially a mixture of methanol and water was used for the reaction. As the result, a partial displacement of sulfone by methoxy group was detected. The methanol was replaced with acetonitrile and the sulfone displacement was eliminated.
In summary, the ester route (Scheme 2-1) is preferred because:
1. Formation of the impurity during the first step of Scheme 2-2 was unavoidable and resulted in yields of < 35%.
2. Column purification was required to isolate intermediate 14.
3. The aldehyde starting material was not commercially available and required two synthetic steps from the corresponding ester.

Scheme 2-3 : Starting with cyclohexanone, compounds of the present invention can be prepared. In Step 1 the methyl glycinate is subjected to cyclohexanone and TMSCN in the presence of tri ethyl amine in DCM to afford 15. In Step 2 15 hydrogenated with hydrogen gas in the presence of catalytic platinum oxide and subsequently undergoes an intramolecular cyclization to afford compound 16 which is used in the schemes above.

Scheme 2-4: Starting with compound 17, compounds of the present invention can be prepared. In Step 1 compound 17 is subjected to ethyl 2-oxoacetate in the presence platinum on carbon and hydrogen gas to afford compound 18. In Step 2 compound 18 is Boc-deprotected with hydrochloric acid. In Step 3 compound 18 is cyclized to afford compound 16 which is used in the schemes above.
Scheme 2-5

11 19
Scheme 2-5: Starting from compound 11 the CDK 4/6 inhibitor 19 can be prepared. In Step 1 5-(4-methylpiperazin-l-yl)pyridin-2-amine is subjected to LiHMDS and compound 11 is added slowly under chilled conditions to afford a nucleophilic substitution reaction and compound 19. Compound 11 can be prepared as described in the schemes herein.

Scheme 2-6: Starting from compound 11 the CDK 4/6 inhibitor 20 can be prepared. In Step 1 5-(4-isopropylpiperazin-l-yl)pyridin-2-amine is subjected to LiHMDS and compound 11 is added slowly under chilled conditions to afford a nucleophilic substitution reaction and compound 20. Compound 11 can be prepared as described in the schemes herein.
Preparation of Compound 5:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet, and reflux condenser was charged with ethyl 4-chloro-2-(methylthio)pyrimidine-5-carboxylate 3 (49.2 g, 0.21 mol, 1.00 equiv.), spirolactam 4 (39.2 g, 0.23 mol, 1.10 equiv.), DIPEA (54.7 g, 0.42 mol, 2.00 equiv.), and DMAc (147.6 mL, 3 vol). The batch was heated to 90-95 °C, and after 60 h, IPC confirmed -14% (AUC) of ethyl 4-chloro-2-(methylthio)pyrimidine-5-carboxylate remained. The batch was cooled to RT, and precipitate formation was observed. The suspension was diluted with MTBE (100 mL, 2 vol) and water (442 mL, 9 vol) and stirred for 2 h at RT. The product was isolated by vacuum filtration and washed with MTBE (49 mL, 1 vol). The solid cake was conditioned for 1 h and dried under vacuum at 40 °C for 16 h to afford compound 5 [41.0 g, 53% yield] as an off-white solid with a purity of >99% AUC. ¾ MR (CDCh): δ 8.76 (d, J = 2.0 Hz, 1H), 6.51-6.29 (br, 1H), 4.33 (q, J = 7.0 Hz, 2H), 3.78 (s, 2H), 3.58 (s, 2H), 2.92 (s, 2H), 2.53 (s, 3H), 1.63-1.37 (m, 12H). LCMS (ESI, m/z = 365.3 [M+H]).
Preparation of Compound 6:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 5 [41.0 g, 0.11 mol, 1.00 equiv.], Boc-anhydride (36.8 g, 0.17 mol, 1.50 equiv.), DMAP (1.37 g, 0.01 mol, 0.10 equiv.), and dichloromethane (287 mL, 7 vol). The batch was stirred for 3 h at RT. IPC confirmed no starting material remained (AUC). The batch was concentrated into a residue under reduced pressure and taken to the next step (a quantitative yield is assumed for this step). An aliquot (200 mg) was purified by column chromatography (heptanes/ethyl acetate 0 to 100%) to afford compound 6. 1H MR (CDCh): δ 8.64 (s, 1H), 4.31 (q, J = 7.0 Hz, 2H), 4.07 (s, 2H), 3.83 (S, 2H), 3.15 (m, 2H), 2.56 (s, 3H), 172 (m, 3H), 1.59 (m, 15H), 1.42 (t, J= 7.0 Hz, 3H). LCMS (ESI, m/z = 465.2 [M+H]).
Preparation of Compound 7:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with compound 6 [residue from a previous step, quantitative yield assumed, 52.2 g, 0.11 mol, 1.00 equiv.], and THF (261 mL, 5 vol). The batch was cooled to 0°C and 1,8-diazabicyclo[5.4.0]un-dec-7-ene (17.1 g, 0.11 mmol, 1.00 equiv.) was added keeping the internal temperature in 0-10°C range. After the addition was complete, the cooling bath was removed and the reaction mixture was allowed to warm up to RT and after 2 h, IPC confirmed no starting material remained. The batch was seeded with the product (1.0 g) and was cooled to 0°C. The slurry was stirred at 0°C for 2 h. The product was isolated by vacuum filtration and washed with cold (0°C) THF (50 mL, 1 vol). The solid cake was conditioned for 1 h and dried under vacuum at 40°C for 16 h to afford 7 [47 g, quantitative yield] as a light orange solid with a purity of >99% AUC. The color of the product changed into yellow once the batch was exposed to air for an extended period of time (~ 1 day). Material was isolated with substantial amount DBU, according to proton NMR. However, it did not interfere with the next step. 1H MR (CDCh): δ 8.71 (s, 1H), 4.03 (s, 2H), 2.57 (s, 3H), 1.85 (m, 10H), 1.51 (s, 9H). LCMS (ESI, m/z = 419.2 [M+H]).
Preparation of Compound 8:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 7 [40.8 g, 0.10 mol, 1.00 equiv.], triethylamine (31.5 g, 0.31 mol, 3.20 equiv.), and dichloromethane (408 mL, 10 vol). The batch was purged with N2 for 15 min and was cooled to 0°C. Triflic anhydride (44.0 g, 0.16 mol, 1.60 equiv.) was added keeping the
internal temperature in 0-10°C range. The batch was stirred at 0°C and after 3 h, IPC confirmed -7.0% (AUC) of 7 remained. [It was speculated that the product was hydrolyzing back into starting material during the analysis.] Once the reaction was deemed complete, the batch was transferred to a 1 L, separatory funnel and was washed with 50% saturated sodium bicarbonate (200 mL, 5 vol). [It was prepared by mixing saturated sodium bicarbonate (100 mL) with water (100 mL)).] The aqueous layer was separated and was extracted with DCM (2×40 mL, 1 vol). The organic layers were combined and concentrated into a residue under reduced pressure and taken to the next step. LCMS (ESI, m/z = 551.6 [M+H]).
Preparation of Compound 9:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with compound 8 [residue from a previous step, quantitative yield assumed, 53.7 g, 0.10 mol, 1.00 equiv.], and THF (110 mL, 2 vol). The solvent was removed under vacuum distillation and the procedure was repeated two times. The flask was charged with triethylsilane (22.7 g, 0.20 mol, 2.00 equiv.), and DMF (268 mL, 5 vol). The batch was degassed by five cycles of evacuation, followed by backfilling with nitrogen. The flask was charged with tetrakis(triphenylphosphine)palladium(0) (11.3 g, 0.01 mol, 0.1 equiv.). The batch was heated to 45-50°C, and after 14 h, IPC confirmed no starting material remained. The batch was transferred to a 500 mL, separatory funnel while still warm. The reaction was partitioned between water (5 vol) and ethyl acetate (5 vol). The aqueous layer was extracted with ethyl acetate (3 x3 vol). The organic layers were combined and concentrated down to 2 volumes. The precipitate was filtered and washed with ethyl acetate (2x 1 vol). The solid cake was conditioned for 1 h and dried under vacuum at 40°C for 16 h to afford 9 [27.5 g, 70% yield] as a yellow solid with a purity of -98% AUC. Proton NMR showed some triphenylphosphine oxide present. ¾ NMR (DMSO-i¾):5 9.01 (s, 1H), 7.40 (s, 1H), 4.30 (s, 2H), 2.58 (m, 2H), 2.58 (s, 3H), 1.81 (m, 5H), 1.51 (s, 9H). LCMS (ESI, m/z = 403.4 [M+H]).
Preparation of Compound 10 from the Scheme 2-1 route:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged 9 (12.8 g, 31.8 mmol, 1.00 equiv.) and dichloromethane (64 mL, 5 vol). Trifluoroacetic acid (18.2 g, 159 mmol, 5.00 equiv.) was added over 20 min and the solution was stirred for 2 h at RT. IPC confirmed reaction was complete. The batch was transferred to a 500 mL, separatory funnel and washed with saturated sodium bicarbonate (200 mL). The aqueous layer was extracted with dichlorom ethane (3 x3 vol). The organic layers were combined and concentrated down to 1 volume. The precipitate was filtered and conditioned for 1 h and dried under vacuum at 40 °C for 16 h to afford 9 [6.72 g, 70% yield] as an off-white solid with a purity of 99.1% AUC. ¾ NMR (DMSO-dis): δ 8.95 (s, 1H), 8.32 (s, 1H), 7.15 (s, 1H), 3.68 (d, J = 1.0 Hz, 2H), 2.86 (m, 2H), 2.57 (s, 3H), 1.92 (m, 2H), 1.73 (m, 3H), 1.39 (m, 3H). LCMS, ESI, m/z = 303.2 [M+H]).
Preparation of Compound 10 from Scheme 2-2 route:
A 50 mL, three-neck flask equipped with a magnetic stirring bar, thermocouple, N2 inlet was charged 14 (680 mg, 1.62 mmol, 1.00 equiv.) and THF (6.8 mL, 10 vol). A I M solution of potassium tert-butoxide (3.2 mL, 3.24 mmol, 2.00 equiv.) in THF was added over 10 min and the solution was stirred for 2 h at RT. IPC confirmed reaction was complete. The batch was acidified with 4 N hydrogen chloride solution in dioxane (2.4 mL, 9.72 mmol, 6.00 equiv.) and stirred for additional 1 h. The batch was transferred to a 500 mL, separatory funnel and washed with saturated sodium bicarbonate (100 mL). The aqueous layer was extracted with ethyl acetate (3 x20 vol). The organic layers were combined and concentrated down to 3volumes and product precipitated. The precipitate was filtered and conditioned for 1 h and dried under vacuum at 40 °C for 16 h to afford 9 [489 mg, quantitative yield] as an off-white solid.
Preparation of Compound 11 :
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 10 (9.00 g, 29.8 mmol, 1.00 equiv.), and acetonitrile (180 mL, 20 vol). A solution of Oxone™ (45.9 g, 0.15 mol, 5.00 equiv.) in water (180 mL, 20 vol) was added to the batch over 20 min. The batch was stirred for 2 h and IPC confirmed the reaction was complete. The batch was concentrated down to ½ of the original volume and was extracted with dichloromethane DCM (4x 10 vol). The organic layers were combined; polish filtered and concentrated down to -10 vol of DCM. The product was slowly crystallized out by addition of heptanes (-30 vol). The mixture was cooled to 0°C and the product was filtered and dried under vacuum at 40 °C for 16 h to afford 11 [9.45 g, 95% yield] as an off-white solid with a purity of >99% AUC. ¾ NMR (CDCb): 5 9.24 (s, 1H), 7.78 (s, 1H), 7.46 (s, 1H), 3.89 (d, J= 2.0 Hz, 2H), 3.43 (s, 3H), 2.98 (m, 2H), 2.10 (m, 2H), 1.86 (m, 3H), 1.50 (m, 3H). LCMS (ESI, m/z = 335.2 [M+H]).
Preparation of Compound 13:
A 250 mL, single-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet, and reflux condenser was charged with 4-chloro-2-(methylthio)pyrimidine-5-carbaldehyde (2.00 g, 10.6 mmol, 1.00 equiv.), spirolactam 4 (1.96 g, 11.7 mmol, 1.10 equiv.), DIPEA (2.74 g, 21.2 mmol, 2.00 equiv.), and fert-butanol (20 mL, 10 vol). The batch was heated to 80-85 °C, and after 24 h, IPC confirmed no aldehyde 12 remained. The batch was cool to RT and concentrated into a residue, which was loaded on silica gel column. The product was eluted with ethyl acetate/heptanes (0% to 100%). The product containing fractions were pulled out and concentrated to afford 13 [0.98 g, 29% yield] as an off-white solid.
Preparation of Compound 14:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 13 [0.98 g, 3.00 mmol, 1.00 equiv.], Boc-anhydride (4.90 g, 21.5 mmol, 7.00 equiv.), DMAP (36 mg, 0.30 mmol, 0.10 equiv.), and dichloromethane (7 mL, 7 vol). The batch was stirred for 3 h at RT. IPC confirmed no starting material remained. The batch was cool to RT and concentrated into a residue, which was loaded on silica gel column. The product was eluted with ethyl acetate/heptanes (0% to 100%). The product containing fractions were pulled out and concentrated to afford 14 [0.98 g, 29% yield] as an off-white solid.
Preparation of Compound 15:
To a suspension of methyl glycinate (500 g, 3.98 mol, 1 eq) in DCM (10 L) was added
TEA dropwise at rt under nitrogen atmosphere, followed by the addition of cyclohexanone (781 g, 7.96 mol, 2 eq) dropwise over 15 min. To the resulting mixture was added TMSCN (591 g, 5.97 mol, 1.5 eq) dropwise over 1 hour while maintaining the internal reaction temperature below 35
°C. After stirred at rt for 2 hrs, the suspension became a clear solution. The progress of the reaction was monitored by H- MR.
When the methyl glycinate was consumed completely as indicated by H-NMR analysis, the reaction was quenched by water (5 L). The layers were separated. The aqueous layer was extracted with DCM (1 L). The combined organic phase was washed with water (5 L X 2) and
dried over Na2S04 (1.5 Kg). After filtration and concentration, 1.24 Kg of crude 15 was obtained as oil.
The crude 15 was dissolved in IPA (4 L). The solution was treated with HC1/IPA solution (4.4 mol/L, 1.1L) at RT. A large amount of solid was precipitated during the addition. The resulting suspension was stirred for 2 hrs. The solid product was collected by vacuum filtration and rinsed with MTBE (800 mL). 819 g of pure 15 was obtained as a white solid. The yield was 88.4%. ¾- MR (300 MHz, CD3OD) 4.20 (s, 2H), 3.88 (s, 3H), 2.30-2.40 (d, J = 12 Hz, 2H), 1.95-2.02 (d, J = 12 Hz, 2H), 1.55-1.85 (m, 5H), 1.20-1.40 (m, 1H).
Preparation of Compound 16:
To a solution of 15 (10 g, 43 mmol) in MeOH (100 mL) was added methanolic hydrochloride solution (2 .44 mol/L, 35.3 mL, 2 eq) and Pt02 (0.5 g, 5 wt %). The reaction suspension was stirred with hydrogen bubble at 40 °C for 6 hours. H- MR analysis showed consumption of 15. To the reaction mixture was added K2CO3 (15 g, 108 mmol, 2.5 eq) and the mixture was stirred for 3 hrs. The suspension was filtered and the filtrate was concentrated to dryness. The residual oil was diluted with DCM (100 mL) and resulting suspension was stirred for 3 hrs. After filtration, the filtrate was concentrated to provide crude 16 (6.6 g) as an oil. The crude 16 was diluted with EtOAc/hexane (1 : 1, 18 mL) at rt for 2 hrs. After filtration, 16 (4 g) was isolated. The obtained 16 was dissolved in DCM (16.7 mL) and hexane (100 mL) was added dropwise to precipitate the product. After further stirred for 1 h, 2.8 g of the pure 16 was isolated as a white solid. The yield was 39%. HPLC purity was 98.3%; MS (ESI): 169.2 (MH+); 1 H-NMR (300 MHz, D2O) 3.23 (s, 3H), 3.07 (s, 3H), 1.37-1.49 (m, 10H).
Preparation of compound 19:
5-(4-methylpiperazin-l-yl)pyridin-2-amine (803.1 g; 3.0 equivalents based on sulfone 11) was charged to a 22 L flask. The flask was blanketed with N2 and anhydrous THF added (12.4 kg). The resulting black-purple solution was cooled in an ice bath to < 5°C. 1M LiHMDS (4.7 L; 1.2 equivalents based on sulfone 11) was added via an addition funnel in three equal additions to keep the temperature below 10°C. Upon the completion of the addition, the reaction mixture was warmed to 16°C. The sulfone 11 (455.1 g; 1.00 equivalents) was added in five additions. Reaction proceeded until HPLC analysis of an IPC sample indicated less than 3% of sulfone 11 remained.
To quench the reaction, the contents of the 22L flask were transferred to a 100 L flask containing water. After stirring for 30 minutes at <30°C, the crude product was collected by filtration, washed with water and dried to afford 19 (387 g, 99.1% purity, 63.7% yield).
Preparation of compound 20:
5-(4-isopropylpiperazin-l-yl)pyridin-2-amine (1976.2 g; 3.0 equivalents based on sulfone 11) was charged to a 50 L flask. The flask was blanketed with N2 and anhydrous THF added (10.7 kg). The resulting black-purple solution was cooled in an ice bath to < 5°C. 1M LiHMDS (9.6 kg; 3.6 equivalents based on sulfone) was added via an addition funnel at a rate to keep the temperature below 10°C. Upon the completion of the addition, the reaction mixture was warmed to 16°C over 120 minutes by removing the ice bath. The sulfone (1000 g; 1.00 mol) was added in five additions. The reaction proceeded until HPLC analysis of an IPC sample indicated less than 1% of sulfone 11 remained. After completion of the reaction, ammonium chloride was added to the reaction mixture. The mixture stirred at < 32°C for at least 30 minutes and the solids collected by filtration to afford 20 (900 g, 99.1% purity, 64.2% yield).
Alternate Route to Spirolactam via cyclohexanone:
Scheme 2-7

26
In one embodiment the spirolactam is made via the synthetic scheme above. By reducing the nitrile group before addition of the glycinate group the reaction sequence proceeds in higher yield. The chemistry used in Step 1 is described in the literature (J. Org. Chem. 2005, 70,8027-8034), and was performed on a kilogram scale. The chemistry to convert Compound 24 into the
spirolactam was also demonstrated on kilogram scale. The Boc protection of Compound 23, is carried out at -70°C in order to limit formation of the di-Boc protected product. Experimental details of a 200 g pilot run are described below.
Step 1

200 g of cyclohexanone 21 was converted to 22 using Ti(Oi-Pr)4 /TMSCN/NH3. After work-up, 213 g of 22 was obtained. The H- MR was clean. The yield was 84%. The titanium salts were removed by vacuum filtration. In one embodiment, the titanium salts are removed by centrifugation or Celite filtration.
Step 2

190 g of 22 was mixed with LAH (2 eq) in MTBE for 30 minutes at 45°C. After work-up, 148 g of crude 23 was obtained.
Step 3

136 g of the crude 23 from step 2 was converted to 24 with 0.9 eq of B0C2O at -70°C. The reaction was completed and worked up. After concentration, 188 g of 24 was obtained. The yield was 86%. The H-NMR and C-NMR spectra confirmed that the compound was pure.
Step 4

188 g of 24 was subjected to methyl 2-bromoacetate and K2CO3 in acetonitrile to afford 25. 247 g of crude 25 was obtained.
Step 5

247 g of 25 was subjected to TFA in DCE heated to reflux to afford 26. After work-up, 112 g of 6 as TFA salt was obtained. H- MR was clean.
Step 6

26 27
Compound 26 was stirred in EtOH in the presence at room temperature overnight to afford 27. In one embodiment DCM is used as the solvent instead of EtOH.
Example 3. Purge of residual palladium from Step 5 Scheme 2-1:
Since palladium was used in Step 5 of Scheme 2-1, the levels of residual Pd present in the subsequent synthetic steps was determined. Table 2 below and Figure 3 show the palladium levels in the isolated solids.
Table 2

The material after Step 5 was isolated containing 1.47% (14700 ppm) of residual palladium. This data represents the highest level of palladium in the worst case scenario. The workup conditions of the latter steps purged nearly all of the palladium and the final product, 19 bis HC1 salt, contained 14 ppm of Pd, which is below the standard 20 ppm guidline. The Pd levels will likely be even lower once the catal st loading is optimized in Step 5.

19
The process developed in this route was a significant improvement over the one used for the first generation synthesis. Overall, the scheme consists of seven steps with five isolations, all by crystallization. No silica column chromatography is employed in the synthesis, which makes the process highly scalable. The process workup conditions can successfully purge the 1.47% of residual palladium after step 5 of Scheme 2-1.
///////////////TRILACICLIB, G1T28, G1T 28, SHR 6390, PHASE 2, G1 Therapeutics, Inc.
CN1CCN(CC1)C2=CN=C(C=C2)NC3=NC=C4C=C5C(=O)NCC6(N5C4=N3)CCCCC6
WO-2018001353, APREMILAST, NEW PATENT, ZHEJIANG HUAHAI PHARMACEUTICAL CO., LTD
![]()

WO-2018001353, APREMILAST, NEW PATENT, ZHEJIANG HUAHAI PHARMACEUTICAL CO., LTD
(WO2018001353) METHOD FOR PREPARING APREMILAST
ZHEJIANG HUAHAI PHARMACEUTICAL CO., LTD
DU, Xiaoqiu; (CN).
ZHOU, Lianchao; (CN).
LIU, Jiegen; (CN)
EN)Method one: (S) -1- (3-ethoxy-4-methoxyphenyl) -2- (methylsulfonyl) ethylamine N-acetyl-L-leucine salt of formula II is reacted with 3-acetylaminophthalic anhydride of formula III in an aprotic solvent to produce the compound of formula I; method two: (S) -1- (3-ethoxy-4-methoxyphenyl) -2- (methylsulfonyl) ethylamine N-acetyl-L- leucine salt of formula II is reacted with 3-acetylaminophthalic anhydride of formula III in an organic solvent in the presence of an organic alkaline or an alkali metal hydride to produce the compound of formula I. The method for preparing apremilast requires inexpensive raw materials and reagents , is suitable for industrialized production, and has great economic effects.
Apremilast is a PDE4 inhibitor developed by Celgene. Currently, there are clinical indications such as rheumatoid arthritis, psoriatic arthritis, Behcet’s disease and ulcerative colitis. March 21, 2014 FDA approves first indication – adult active psoriatic arthritis (PsA). Name of Product:  (FDA, as a post-marketing requirement, will evaluate the effect of this drug on pregnant women through a pregnancy registry study.) Three clinical trials evaluated the safety and efficacy of Asprate in the treatment of PsA, The response rates to ACR20 in the prest and placebo groups were 32-41% and 18-19%, respectively.
(FDA, as a post-marketing requirement, will evaluate the effect of this drug on pregnant women through a pregnancy registry study.) Three clinical trials evaluated the safety and efficacy of Asprate in the treatment of PsA, The response rates to ACR20 in the prest and placebo groups were 32-41% and 18-19%, respectively.
Aspast’s oral anti-rheumatic drug, a new mechanism of action, distinguishes itself from currently available anti-TNF monoclonal antibodies. Thomson Pharma predicts rapid sales growth of 201.2 million U.S. dollars in 2015 with sales of US $ 516 million in 2015 . Upstall’s sales are expected to reach a maximum of 2 billion U.S. dollars. Compared with its counterparts, Actuate has the following advantages: It inhibits the production of various proinflammatory mediators (PDE-4, TNF-α, IL-2, interferon γ, leukotriene, NO synthase) Inflammation; selective inhibitor of phosphodiesterase 4 (PDE4), approved for use in psoriatic arthritis in September 2014 FDA approved mid-to-severe treatment of plaque psoriasis for phototherapy or systemic therapy Patient, the first and only PDE4 inhibitor approved for the treatment of plaque psoriasis; clinical trials have shown that OTEZLA reduces erythema, thickening and scaling in patients with moderate to severe plaque psoriasis; clinical trials have demonstrated Painstrept was well tolerated and had minimal adverse reactions. Patients in the Otezla-treated and placebo clinical trials showed signs and symptoms of PsA improvement including tenderness, joint swelling and physical function.
The original patent CN 101683334A reports the synthesis of (S) -1- (3-ethoxy-4-methoxyphenyl) -2- (methylsulfonyl) ethylamine N-acetyl- ) And 3-acetylaminophthalic anhydride (3) Prepared with acetic acid as solvent (1), and the synthetic route is as follows:
The method has low yield, needs lower than 50 DEG C to distill the high-boiling acetic acid, and produces one deacetyl impurity (4) during the reflux reaction and the acetic acid distillation, which affects the product purity. Acetic acid will corrode the equipment at high temperatures. Distillation of high-boiling acetic acid will also increase plant production time. Acetic acid, which is not distilled away, consumes a large amount of lye to neutralize and increases the amount of wastes and production costs, which is not conducive to industrialized production.
Example one
10.0 g (0.0224 mol) of (S) -1- (3-ethoxy-4-methoxyphenyl) -2- (methylsulfonyl) ethylamine N-acetyl- 4.6g (0.0224mol) 3-acetamidophthalic anhydride into a 250mL three-necked flask, then add 50mL of acetonitrile, heating 75 ~ 80 ℃, the reaction incubated for 18 hours and cooled to room temperature. After the reaction mixture was evaporated to dryness, 60 mL of methylene chloride was added, 25 g of 10% sodium carbonate solution was added thereto and the mixture was stirred for 10 to 30 minutes. The mixture was allowed to stand for further delamination and then 25 mL of water was added to the organic layer and stirred for 10-30 minutes. The layers were evaporated to dryness to give a light yellow solid, then add 30mL absolute ethanol, evaporated again. The mixture was hot beaten with ethanol, cooled to 0-5 ° C, stirred for 1-2 hours, filtered and drained. The filter cake was vacuum dried to give 9.4 g of a white powder in 91.2% yield. HPLC: 99.9% ) Has an HPLC area of 0.03%.
Example two
10.0 g (0.0224 mol) of (S) -1- (3-ethoxy-4-methoxyphenyl) -2- (methylsulfonyl) ethylamine N-acetyl- A solution of 4.6 g (0.0224 mol) of 3-acetylaminophthalic anhydride in a 250 mL three-necked flask was charged with 80 mL of toluene and 10 mL of N, N-dimethylformamide. The mixture was heated to 100 ° C and the reaction was incubated for 12 hours and then cooled to room temperature. After the reaction solution was evaporated to dryness, 80 mL of methylene chloride was added, 25 g of 10% sodium carbonate solution was added thereto and the mixture was stirred for 10 to 30 minutes. The mixture was allowed to stand for further delamination and then 50 mL of water was added to the organic layer and stirred for 10 to 30 minutes. Evaporated to a pale yellow solid, then add 30mL of absolute ethanol, evaporated again. Cooled to 0 ~ 5 ℃ and stirred for 1 ~ 2 hours, filtered and drained, the filter cake was dried in vacuo to give 9.2g white powder, yield 89.2%, HPLC: 99.9%, wherein the deacetyl impurities (4 ) Has an HPLC area of 0.03%.
Example three:
10.0 g (0.0224 mol) of (S) -1- (3-ethoxy-4-methoxyphenyl) -2- (methylsulfonyl) ethylamine N-acetyl- To a 250 mL three-necked flask was added 4.6 g (0.0224 mol) of 3-acetamidophthalic anhydride followed by 50 mL of ethyl acetate and 1.81 g (0.8 eq) of triethylamine. The mixture was heated at 75-80 ° C and incubated for 18 hours. The reaction was stopped, 100 mL of ethyl acetate was further added and the mixture was cooled to 20-30 ° C. The reaction solution was added 30g of 8% sodium carbonate solution, stirred for 10 to 30 minutes, allowed to stand layered, the organic layer was added 30mL of water, stirred for 10 to 30 minutes, allowed to stand layered, the organic layer was added 30mL of water, stirred 10 ~ 30 minutes, standing stratification, the organic layer was evaporated to dryness to a pale yellow solid, then add 30mL of absolute ethanol, evaporated again. The mixture was heated to 0-5 ° C for 1 to 2 hours, filtered and drained. The filter cake was vacuum dried to give 9.8 g of a white powder in 95.1% yield. HPLC: 99.9% ) Had an HPLC area of 0.04%.
Example 4:
10.0 g (0.0224 mol) of (S) -1- (3-ethoxy-4-methoxyphenyl) -2- (methylsulfonyl) ethylamine N-acetyl- (0.0224mol) 3-acetamidophthalic anhydride into a 250mL three-necked flask, followed by the addition of 120mL of isopropyl acetate and 30mL of acetonitrile and 1.81g (0.8eq) of triethylamine, heating 75 ~ 80 ℃, incubated reaction 16 hours. Stop the reaction, cooled to 20 ~ 30 ℃. The reaction solution was added 30g of 8% sodium carbonate solution, stirred for 10 to 30 minutes, allowed to stand layered, the organic layer was added 30mL of water, stirred for 10 to 30 minutes, allowed to stand layered, the organic layer was added 30mL of water, stirred 10 ~ 30 minutes, standing stratification, the organic layer was evaporated to dryness to a pale yellow solid, then add 30mL of absolute ethanol, evaporated again. The mixture was hot beaten with ethanol, cooled to 0-5 ° C, stirred for 1-2 hours, filtered and drained. The filter cake was vacuum dried to give 9.6 g of a white powder in 93.1% yield. HPLC: 99.9% ) Has an HPLC area of 0.03%.
Comparative Example:
According to the preparation example of Compound A in original patent CN 101683334A, 10.0 g (0.0224 mol) of (S) -1- (3-ethoxy-4- methoxyphenyl) -2- (methylsulfonyl) N-acetyl-L-leucinate and 4.6 g (0.0224 mol) of 3-acetylaminophthalic anhydride were placed in a 250 mL three-necked flask and 50 mL of acetic acid was added thereto. The mixture was heated at 75 to 80 ° C and the reaction was incubated for 18 hours. The reaction mixture was cooled to 40-50 ° C and the temperature of the water bath was controlled to 40-50 ° C. The reaction mixture was vortexed to glacial acetic acid without any significant fraction. 150 mL of ethyl acetate was added and the mixture was stirred to dissolve. 100 mL of water was added and the mixture was stirred 10 ~ 30 minutes, standing stratification, the organic layer was added 100mL water, stirred for 10 to 30 minutes, allowed to stand for stratification, the organic layer was added 100g 8% sodium bicarbonate solution, stirred for 10 to 30 minutes, The organic layer was added with 100g of 8% sodium bicarbonate solution and stirred for 10-30 minutes. The layers were separated and the organic layer was added with 100 mL of water. The mixture was stirred for 10-30 minutes, and the layers were separated. The organic layer was further added with 100 mL of water and stirred 10 ~ 30 minutes, standing stratification, the organic layer was evaporated to dryness to a pale yellow solid, then add 30mL of absolute ethanol, evaporated again. 68mL of anhydrous ethanol and 34mL of acetone were added to the solid, heated to 60-65 ° C, stirred to make it fully dissolved, and then cooled to 0-5 ° C and stirred for 1 to 2 hours, filtered and drained, and the filter cake was dried under vacuum to give 8.6 Class g white powder, yield 83.4%, HPLC: 99.7% with an HPLC area of deacetylated impurity (4) of 0.22%.
////////////WO 2018001353, APREMILAST, NEW PATENT, ZHEJIANG HUAHAI PHARMACEUTICAL CO., LTD
ELAGOLIX

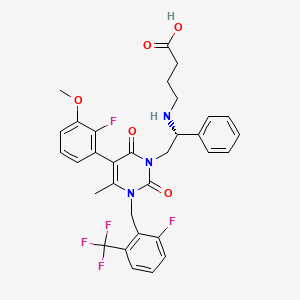
ELAGOLIX
- Molecular FormulaC32H30F5N3O5
- Average mass631.590 Da
NBI56418, ABT 620
UNII:5B2546MB5Z
4-({(1R)-2-[5-(2-Fluoro-3-methoxyphenyl)-3-[2-fluoro-6-(trifluoromethyl)benzyl]-4-methyl-2,6-dioxo-3,6-dihydro-1(2H)-pyrimidinyl]-1-phenylethyl}amino)butanoic acid
834153-87-6 FREE ACID
SODIUM SALT 832720-36-2
Acide 4-({(1R)-2-[5-(2-fluoro-3-méthoxyphényl)-3-[2-fluoro-6-(trifluorométhyl)benzyl]-4-méthyl-2,6-dioxo-3,6-dihydro-1(2H)-pyrimidinyl]-1-phényléthyl}amino)butanoïque
Butanoic acid, 4-[[(1R)-2-[5-(2-fluoro-3-methoxyphenyl)-3-[[2-fluoro-6-(trifluoromethyl)phenyl]methyl]-3,6-dihydro-4-methyl-2,6-dioxo-1(2H)-pyrimidinyl]-1-phenylethyl]amino]-
GNRH antagonist, Endometriosis
Endometriosis PREREGISTERED
Phase III Uterine leiomyoma
| Inventors | Yun-Fei Zhu, Chen Chen, Fabio C. Tucci, Zhiqiang Guo, Timothy D. Gross, Martin Rowbottom, R. Scott Struthers, |
| Applicant | Neurocrine Biosciences, Inc. |
WO 2005007165 PDT PATENT

| Inventors | Zhiqiang Guo, Yongsheng Chen, Dongpei Wu, Chen Chen, Warren Wade, Wesley J. Dwight, Charles Q. Huang, Fabio C. Tucci, |
| Applicant | Neurocrine Biosciences, Inc. |
- Originator Icahn School of Medicine at Mount Sinai
- Developer AbbVie; Neurocrine Biosciences
- Class Antineoplastics; Fluorinated hydrocarbons; Pyrimidines; Small molecules
- Mechanism of Action LHRH receptor antagonists
- Highest Development Phases
- Preregistration Endometriosis
- Phase III Uterine leiomyoma
- Discontinued Benign prostatic hyperplasia; Prostate cancer
- Most Recent Events
- 23 Nov 2017 AbbVie plans a phase III trial for Endometriosis (Monotherapy, Combination therapy) in USA in November 2017 (NCT03343067)
- 01 Nov 2017 Updated efficacy and adverse events data from two phase III extension trials in Endometriosis released by AbbVie
- 27 Oct 2017 Elagolix receives priority review status for Endometriosis in USA
SYN


Elagolix is a specific highly potent non-peptide, orally active antagonist of the GnRH receptor. This compound inhibits pituitary luteinizing hormone (LH) secretion directly, potentially preventing the several week delay and flare associated with peptide agonist therapy.

In 2010, elagolix sodium was licensed to Abbott by Neurocrine Biosciences for worldwide development and commercialization for the treatment of endometriosis. In January 2013, Abbott spun-off its research-based pharmaceutical business into a newly-formed company AbbVie.
AbbVie , following its spin-out from Abbott in January 2013, under license from Neurocrine , is developing elagolix, the lead from a series of non-peptide gonadotropin-releasing hormone antagonists, for treating hormone-dependent diseases, primarily endometriosis and uterine fibroids.
Elagolix sodium is an oral gonadotropin releasing hormone (GnRH) antagonist in development at Neurocrine Biosciences and Abbvie (previously Abbott). In 2017, Abbvie submitted a New Drug Application (NDA) in the U.S. for the management of endometriosis with associated pain. The candidate is being evaluated in phase III trials for the treatment of uterine fibroids.
Elagolix (INN, USAN) (former developmental code names NBI-56418, ABT-620) is a highly potent, selective, orally-active, short-duration, non-peptide antagonist of the gonadotropin-releasing hormone receptor (GnRHR) (KD = 54 pM) which is under development for clinical use by Neurocrine Biosciences and AbbVie.[2][3] As of 2017, it is in pre-registration for the treatment of endometriosis and phase III clinical trials for the treatment of uterine leiomyoma.[1][4] The drug was also under investigation for the treatment of prostate cancer and benign prostatic hyperplasia, but development for these indications was ultimately not pursued.[4] Elagolix is the first of a new class of GnRH inhibitors that have been denoted as “second-generation”, due to their non-peptide nature and oral bioavailability.[1]
Because of the relatively short elimination half-life of elagolix, the actions of gonadotropin-releasing hormone (GnRH) are not fully blocked throughout the day.[1][5] For this reason, gonadotropin and sex hormone levels are only partially suppressed, and the degree of suppression can be dose-dependently adjusted as desired.[1][5] In addition, if elagolix is discontinued, its effects are rapidly reversible.[1][5] Due to the suppression of estrogen levels by elagolix being incomplete, effects on bone mineral density are minimal, which is in contrast to first-generation GnRH inhibitors.[6][7] Moreover, the incidence and severity of menopausal side effects such as hot flashes are also reduced relative to first-generation GnRH inhibitors.[1][5]
Elagolix sodium is a non-peptide antagonist of the gonadotropin-releasing hormone receptor and chemically known as sodium;4-[[(lR)-2-[5-(2-fluoro-3-methoxyphenyl)-3-[[2-fluoro-6-(trifluoromethyl)phenyl]methyl] -4-methyl-2,6-dioxopyrimidin- 1 -yl] -1 -phenylethyl] amino] butanoate as below.

The US patent number 7056927 B2 discloses, elagolix sodium salt as a white solid and process for its preparation in Example-1; Step-IH.
The US patent number 8765948 B2 discloses a process for preparation of amorphous elagolix sodium by spray drying method and solid dispersion of amorphous elagolix sodium with a polymer.
The US patent number 7056927 B2 discloses a process for preparation of elagolix sodium salt in Example -1 as given in below scheme -I.

Scheme -I
The US patent number 8765948 B2 describes a process for preparation of elagolix sodium in example- 1 and 4 as given below scheme-II:

(1c) (1e) (4a)
Scheme-II
Further, the US patent number 8765948 B2 discloses an alternate process for the preparation of compound of formula (le) as mentioned below scheme-Ill.


Scheme -III
PATENT
| WO2001055119A2 * | Jan 25, 2001 | Aug 2, 2001 | Neurocrine Biosciences, Inc. | Gonadotropin-releasing hormone receptor antagonists and methods relating thereto |
PATENT
WO 2005007165
https://encrypted.google.com/patents/WO2005007165A1?cl=en
EXAMPLE 1
3-[2(R)-{HYD OXYCARBONYLPROPYL-AMINθ} -2-PHENYLETHYL]-5-(2-FLUORO-3- METHOXYPHENYL)-l-[2-FLUORO-6-(TRIFLUOROMETHYL)BENZYL]-6-METHYL- PYRIMIDINE-2,4(lH,3H)-DIONE

Step IA: Preparation of 2-fluoro-6-(trifluoromethyl)benzylamine la To 2-fluoro-6-(trifluoromethyl)benzonitrile (45 g, 0.238 mmol) in 60 mL of TΗF was added 1 M BΗ3:TΗF slowly at 60 °C and the resulting solution was refluxed overnight. The reaction mixture was cooled to ambient temperature. Methanol (420 mL) was added slowly and stirred well. The solvents were then evaporated and the residue was partitioned between EtOAc and water. The organic layer was dried over Na2SO4. Evaporation gave la as a yellow oil (46 g, 0.238 mmol). MS (C\) m/z 194.0 (MH+).
Step IB: Preparation of N-|“2-fluoro-6-(trifluoromethyl)benzyl‘|urea lb To 2-fluoro-6-(trifluoromethyl)benzylamine la (51.5 g, 0.267 mmol) in a flask, urea (64 g, 1.07 mmol), HC1 (cone, 30.9 mmol, 0.374 mmol) and water (111 mL) were added. The mixture was refluxed for 6 hours. The mixture was cooled to ambient temperature, further cooled with ice and filtered to give a yellow solid. Recrystallization with 400 mL of EtOAc gave lb as a white solid (46.2 g, 0J96 mmol). MS (CI) m/z 237.0 (MH+).
Step 1C: Preparation of l-[2-fluoro-6-(trifluoromethyl)benzyl]-6- methylpyrimidine-2.4(lH.3H)-dione lc Nal (43.9 g, 293 mmol) was added to N-[2-fluoro-6- (trifluoromethyl)benzyl]urea lb (46.2 g, 19.6 mmol) in 365 mL of acetonitrile. The resulting mixture was cooled in an ice-water bath. Diketene (22.5 mL, 293 mmol) was added slowly via dropping funnel followed by addition of TMSCl (37.2 mL, 293 mmol) in the same manner. The resulting yellow suspension was allowed to warm to room temperature slowly and was stirred for 20 hours. LC-MS showed the disappearance of starting material. To the yellow mixture 525 mL of water was added and stirred overnight. After another 20 hours stirring, the precipitate was filtered via Buchnner funnel and the yellow solid was washed with water and EtOAc to give lc as a white solid (48.5 g, 16 mmol). 1H ΝMR (CDC13) δ 2.15 (s, 3Η), 5.37 (s, 2H), 5.60 (s, 1H), 7.23-7.56 (m, 3H), 9.02 (s, 1H); MS (CI) m/z 303.0 (MH+).
Step ID: Preparation of 5-bromo-l -[2-fluoro-6-(trifluoromethyl)benzyl“|-6- methylpyrimidine-2.4(lH.3H)-dione Id Bromine (16.5 mL, 0.32 mmol) was added to l-[2-fluoro-6-
(trifluoromethyl)benzyl]-6-methylpyrimidine-2,4(lHJH)-dione lc (48.5 g, 0J6 mol) in 145 mL of acetic acid. The resulting mixture became clear then formed precipitate within an hour. After 2 hours stirring, the yellow solid was filtered and washed with cold EtOAc to an almost white solid. The filtrate was washed with sat. ΝaΗCO3 and dried over Na2SO4. Evaporation gave a yellow solid which was washed with EtOAC to give a light yellow solid. The two solids were combined to give 59.4 g of Id (0J56 mol) total. Η NMR (CDC13) δ 2.4 (s, 3H), 5.48 (s, 2H), 7.25-7.58 (m, 3H), 8.61 (s, 1H); MS (CI) m/z 380.9 (MH+). 5-Bromo-l-[2, 6-difluorobenzyl]-6-methylpyrimidine-2,4(lHJH)-dione ld.l was made using the same procedure.
Step IE: Preparation of 5-bromo-l -r2-fluoro-6-(trifluoromethyl)benzyll-6- methyl-3-[2(R)-tert-butoxycarbonylamino-2-phenylethyll-pyrimidine-2.4(lHJH)-dione le To 5-bromo- 1 -[2-fluoro-6-(trifluoromethyl)benzyl]-6-methylpyrimidine- 2,4(lHJH)-dione Id (15 g, 39.4 mmol) in 225 mL of TΗF were added N-t-Boc-D- phenylglycinol (11.7 g, 49.2 mmol) and triphenylphosphine (15.5 g, 59J mmol), followed by addition of di-tert-butyl azodicarboxylate (13.6 g, 59J mmol). The resulting yellow solution was stirred overnight. The volatiles were evaporated and the residue was purified by silica gel with 3:7 EtOAc Ηexane to give le as a white solid (23.6 g, 39.4 mmol). MS (CI) m/z 500.0 (MΗ+-Boc).
Step IF: Preparation of 3-[2(R)-amino-2-phenylethyll-5-(2-fluoro-3- methoxyphenyl)-l-[2-fluoro-6-(trifluoromethyl)benzyll-6-methyl-pyrimidine- 2.4(lH.3H)-dione If To 5-bromo-l-[2-fluoro-6-(trifluoromethyl)benzyl]-6-methyl-3-[2(R)- tert-butoxycarbonylamino-2-phenylethyl]-pyrimidine-2,4(lH,3H)-dione le (15 g, 25 mmol) in 30 mL/90 mL of Η2O/dioxane in a pressure tube were added 2-fluoro-3- methoxyphenylboronic acid (4.25 g, 25 mmol) and sodium carbonate (15.75 g, 150 mmol). N2 gas was bubbled through for 10 min.
Tetrakis(triphenylphosphine)palladium (2.9 g, 2.5 mmol) was added, the tube was sealed and the resulting mixture was heated with stirring at 90 °C overnight. After cooling to ambient temperature, the precipitate was removed by filtration. The volatiles were removed by evaporation and the residue was partitioned between EtOAc/sat. NaHCO3. The organic solvent was evaporated and the residue was chromatographed with 2:3 EtOAc/Hexane to give 13.4 g (20.8 mmol, 83 %) yellow solid. This yellow solid (6.9 g, 10.7 mmol) was dissolved in 20 mL/20 mL CH2C12/TFA. The resulting yellow solution was stirred at room temperature for 2 hours. The volatiles were evaporated and the residue was partitioned between EtOAc/ sat. NaHCO3. The organic phase was dried over Na2SO4. Evaporation gave If as a yellow oil (4.3 g, 7.9 mmol, 74%). Η NMR (CDC13) δ 2.03 (s, 3H), 3.72-4.59 (m, 6H), 5.32-5.61 (m, 2H), 6.74-7.56 (m, 11H); MS (CI) m/z 546.0 (MH+). 3-[2(R)-amino-2-phenylethyl]-5-(2-fluoro-3-methoxyphenyl)-l-[2,6- difluorobenzyl]-6-methyl-pyrimidine-2,4(lH,3H)-dione lf.l was made using the same procedure described in this example.
Step 1G: Preparation of 3-[2(R)- {ethoxycarbonylpropyl-amino} -2-phenylethyll-5-
(2-fluoro-3 -methoxyphenyl)- 1 -[2-fluoro-6-(trifluoromethyl)benzyl“|-6-methyl- pyrimidine-2,4(lHJH)-dione lg To compound 3-[2(R)-amino-2-phenylethyl]-5-(2-fluoro-3- methoxyphenyl)-l-[2-fluoro-6-(trifluoromethyl)benzyl]-6-methyl-pyrimidine- 2,4(lH,3H)-dione If (5 g, 9.4 mmol) in 100 mL of acetonitrile were added ethyl 4- bromobutyrate (4 mL, 28.2 mmol) and Ηunig’s base (1.6 mL, 9.4 mmol). After reflux at 95 °C overnight, the reaction mixture was cooled to ambient temperature and the volatiles were removed. The residue was chromatographed with 10:10: 1 EtOAc/Ηexane/Et3N to give lg as a yellow oil (3.0 g, 4.65 mmol). MS (CI) m/z 646.2 (MH+).
Step 1H: Preparation of 3-[2(R)- {hydroxycarbonylpropyl-amino} -2-phenylethyl]- 5-(2-fluoro-3-methoxyphenyl)-l- 2-fluoro-6-(trifluoromethyl)benzyl1-6-methyl- pyrimidine-2,4(lHJH)-dione 1-1 Compound 3-[2(R)- {ethoxycarbonylpropyl-amino} -2-phenylethyl]-5-(2- fluoro-3-methoxyphenyl)-l-[2-fluoro-6-(trifluoromethyl)benzyl]-6-methyl-pyrimidine- 2,4(lH,3H)-dione lg (2.6 g, 4.0 mmol) was dissolved in 30 mL/30 mL of TΗF/water. Solid NaOΗ (1.6 g, 40 mmol) was added and the resulting mixture was heated at 50 °C overnight. The mixture was cooled to ambient temperature and the volatiles were evaporated. Citric acid was added to the aqueous solution until pΗ = 3. Extraction with EtOAc followed by evaporation of solvent gave 1.96 g of a white gel. The gel was passed through a Dowex MSC-1 macroporous strong cation-exchange column to convert to sodium salt. Lyopholization gave white solid 1-1 as the sodium salt (1.58 g, 2.47 mmol). Η NMR (CD3OD) δ 1.69-1.77 (m, 2H), 2.09 (s, 3H), 2.09-2.19 (t, J = 7.35 Hz, 2H), 2.49-2.53 (t, J = 735 H, 2H), 3.88 (s, 3H), 4.15-4.32 (m, 3H), 5.36-5.52 (m, 2H), 6.60-7.63 (m, 1 IH); HPLC-MS (CI) m/z 632.2 (MH+), tR = 26.45, (method 5)
PATENT
WO 2017221144
Process for the preparation of elagolix sodium and its polymorph forms and intermediates is claimed. Represents first filing from Dr. Reddy’s Laboratories Limited and the inventors on this API.
n a seventh aspect, the present invention provides a process for preparation of compound of formula (VII)

(VII)
wherein R is alkyl such as methyl, ethyl, propyl, isopropyl and the like,
comprising;
a) reacting the compound of formula (II) with compound of formula (III) to obtain the compound of formula (IV)

wherein t-BOC is tertiary butoxycarbonyl group; R is as described above
b) reacting the compound of formula (IV) with the compound of formula (V) to obtain the compound formula (VI), and

c) N-deprotection of the compound of formula (VI) to obtain the compound of formula
(VII)

(VI) (VII)
The reaction of compound of formula (II) with compound of formula (III) to obtain the compound of formula (IV) is carried in the presence of triarylphosphine such as triphenyl phosphine and the like and azodicarboxylates such as diethyl azodicarboxylate, diisopropyl azodicarboxylate and di-tert-butyl azodicarboxylate (DIAD) and the like.
The seventh aspect of the present invention is depicted below scheme-IV.

Scheme-IV
The eighth aspect of the present invention is depicted below scheme-IV.

R=alkyl
Scheme-IV
Example 11: Preparation of ethyl (R)-4-((2-hydroxy-l-phenylethyl)amino)butanoate (Ilia; R is ethyl)
R-(-)-2-phenylglycinol (10 g), DMAP (0.17 g) were added in THF (80 ml) at room temperature under nitrogen atmosphere. Triethylamine (30.48 ml) was added to the reaction mixture and stirred for five minutes. Ethyl-4-bromo butyrate (15.64 ml) was added and the reaction mixture heated to 80°C then stirred for 16 hours. Water (20 volumes) followed by ethyl acetate (200 ml) were added to separate the aqueous and organic layer. The organic layer was washed with IN HC1 (100 ml) followed by neutralize the resulting aqueous layer with saturated sodium carbonate solution then extract with ethyl acetate (100 ml) and the organic layer was dried over anhydrous sodium sulfate then evaporated below 50°C under reduced pressure to obtain the title compound. Yield: 14.50 g. Purity: 94.75% (by HPLC). ¾ NMR (400 MHz, DMSO-d6): δ 7.17-7.30 (m, 5H), 4.83 (m, 1H), 3.99 (q, 2H), 3.58 (dd, 1H, J = 8.8, 4.4 Hz), 3.88 (m, 1H ), 3.27 (m, 1H), 2.38 (m, 1H), 2.26 (m, 3H), 2.10 (s, 1H), 1.61 (m, 2H), 1.12 (t, 3H); m/z: 252 (MH )
Example 12: Preparation of ethyl (R)-4-((tert-butoxycarbonyl)(2-hydroxy-l-phenylethyl) amino)butanoate (III; R is ethyl)
Ethyl (R)-4-((2-hydroxy-l-phenylethyl)amino)butanoate (14 g) was added to THF (140 ml) at room temperature. The reaction mixture was cooled to 0-5 °C. Triethylamine (16.9 mL) was added to the reaction mixture followed by Di-tert-butyl dicarbonate (13.37 g) was added to reaction mixture at 0-5 °C. The reaction mixture was heated to room temperature and stirred for 16 hours. Water (300 mL) and ethyl acetate (300 mL) were added and the layers were separated. The organic layer was washed with sodium chloride then died over sodium sulfate followed by evaporation at 45°C to obtain the crude compound. The crude compound was purified by silica gel (60/120 mesh) withl5-20% EtOAc/Hexane to obtain the title compound as a pale yellow syrup. Yield: 9.5 g. Purity: 95.42% (by HPLC). ¾ NMR (400 MHz, CDC13): δ 7.24-7.34 (m, 5H), 5.08 (m, 1H), 4.09 (m, 4H), 3.10 (m, 2H), 3.00 (s, 1H), 2.21(m, 2H), 1.82 (m, 2H), 1.46 (s, 9H), 1.23 (t, 3H). m/z: 352.20 (MH )
Example 13: Preparation of ethyl (R)-4-((2-(5-bromo)-3-(2-fluoro-6-trifluoromethyl)benzyl)-4-methyl-2,6-dioxo-3,6-dihydropyrimidin-l(2H)-yl)-l-phenylethyl)(tert-butoxycarbonyl) amino)butanoate (IV; R is ethyl)
Ethyl (R)-4-((tert-butoxycarbonyl)(2-hydroxy-l -phenyl ethyl) amino)butanoate (III; R is ethyl) (1.0 g), 5-bromo-l-(2-fluoro-6-trifluoromethyl)benzyl-6-methylpyrimidine-2,4 (1H, 3H)-dione (II) (1.08 g), Triphenyl phosphine (1.49 g) were added to THF (30 mL) at room temperature under nitrogen atmosphere. DIAD (1.11 mL) was added to the reaction mixture and stirred for 16 hours at room temperature. Water (60 volume) was added to the reaction mixture followed by ethylacetate (60 mL) was added then the layers were separated. The organic layer was dried over sodium sulfate and evaporated below 50°C under reduced pressure to obtain the crude compound. The crude compound was purified by silica gel (60/120 mesh) withl5-20% EtOAc/Hexane to obtain the title compound. Yield (1.3 g). Purity: 68.87% (by HPLC); l NMR (DMSO-d6) δ 1.15-2.0 (11H), 2.43-2.48 (4H), 3.9 (2H), 4.71-4.8 (5H), 5.3 -5.4 (3H), 7.28-7.3 (8H), 8.4 (2H); m/z: 616 (M-BOC)+
Example 14: Preparation of ethyl (R)-4-((tert-butoxycarbonyl)-2-(5-(2-fluoro-3-methoxyphenyl)-3-(2-fluoro-6-trifluoromethyl)benzyl)-4-methyl-2,6-dioxo-3,6-dihydropyrimidin-l(2H)-yl)-l-phenylethyl)amino)-butanoate (VI; R is ethyl)
Ethyl (R)-4-((2-(5-bromo)-3-(2-fluoro-6-trifluoromethyl)benzyl)-4-methyl-2,6-dioxo-3,6-dihydropyrimidin-l(2H)-yl)-l-phenylethyl)(tert-butoxycarbonyl) amino)butanoate (IV; R is ethyl) (0.9 g), 2-fluoro-3-methoxy phenyl boronic acid (V) (0.214 g) and sodium carbonate (0.797 g) were added to the mixture of 1,4-dioxane (9 mL) and water (3.06 mL) at room temperature under nitrogen atmosphere. Argon gas was bubbled through for 30 minutes. Tetrakis (triphenylphosphine)palladium (0.145 g) was added to the reaction mixture at room temperature then heated to 90-95 °C and stirred for 5 hours. The reaction mixture cooled to room temperature and filtered through celite bed then the filtrate washed with ethylacetate (9 mL) and water (36 mL) was added and stirred for 30 minutes at room temperature. Ethylacetate (36 mL) was added and the separated organic layer washed with brine and dried over sodium sulfate followed by evaporation at 45°C to obtain the crude compound. The crude compound was purified by silica gel (60/120 mesh) with 20-25% EtOAc/Hexane to obtain the title compound as yellow solid. Yield: 0.5 g; Purity: 75.1% (by HPLC); m/z: 660 (M-BOC)+.
Example 15: Preparation of ethyl (R)-4-((2-(5-(2-fluoro-3-methoxyphenyl)-3-(2-fluoro-6-trifluoromethyl)benzyl)-4-methyl-2,6-dioxo-3,6-dihydropyrimidin-l(2H)-yl)-l-phenylethyl)amino)-butanoate (VII; R is ethyl)
Ethyl(R)-4-((tert-butoxycarbonyl)-2-(5-(2-fluoro-3-methoxyphenyl)-3-(2-fluoro-6-trifluoro methyl)benzyl)-4-methyl-2,6-dioxo-3,6-dihydropyrimidin-l(2H)-yl)-l-phenylethyl)amino)-butanoate (VI; R is ethyl) (0.4 g) was added to dichloromethane (4 mL) at room temperature. The reaction mixture was cooled to 0-5 °C then trifluoroacetic acid (2 mL) was added and stirred for five hours at 0-5 °C. Saturated sodium bicarbonate solution (40 mL) was added to the reaction mixture followed by dichloromethane (40 mL) was added. The organic layer was washed with brine then dried over sodium sulfate and evaporated at 35°C to obtain the crude compound. The crude compound purified by silica gel (60/120 mesh) with 30-35% EtOAc/Hexane to obtain the title compound as yellow solid. Yield: 160 mg; Purity: 88.6% (by HPLC). ‘H NMR (400 MHz, DMSO-d6): δ 7.64 (m, 1H), 7.54 (m, 2H), 7.15-7.27 (m, 6H), 6.85 (m, 2H), 5.31 (s, 2H), 3.99 (m, 3H), 3.87 (m, 2H), 3.83 (s, 3H), 2.30-2.16 (m, 4H), 2.10 (s, 3H), 1.50 (m, 2H), 1.10 (t, 3H). m/z: 660 (MH )
PAPER
Discovery of sodium R-(+)-4-(2-(5-(2-fluoro-3-methoxyphenyl)-3-(2-fluoro-6-(trifluoromethyl-)benzyl)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-yl)-1-phenylethamino)butyrate (elagolix), a potent and orally available nonpeptide antagonist of the human gonadotropin-releasing hormone receptor
J Med Chem 2008, 51(23): 7478
Discovery of Sodium R-(+)-4-{2-[5-(2-Fluoro-3-methoxyphenyl)-3-(2-fluoro-6-[trifluoromethyl]benzyl)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-yl]-1-phenylethylamino}butyrate (Elagolix), a Potent and Orally Available Nonpeptide Antagonist of the Human Gonadotropin-Releasing Hormone Receptor
Department of Medicinal Chemistry, Department of Endocrinology, and Department of Preclinical Development, Neurocrine Biosciences, Inc., 12790 El Camino Real, San Diego, California 92130
J. Med. Chem., 2008, 51 (23), pp 7478–7485
DOI: 10.1021/jm8006454
* To whom correspondence should be addressed. Phone: 1-858-617-7600. Fax: 1-858-617-7925. E-mail: cchen@neurocrine.com, sstruthers@neurocrine.com., †
Department of Medicinal Chemistry., ‡ Department of Endocrinology., § Department of Preclinical Development.

The discovery of novel uracil phenylethylamines bearing a butyric acid as potent human gonadotropin-releasing hormone receptor (hGnRH-R) antagonists is described. A major focus of this optimization was to improve the CYP3A4 inhibition liability of these uracils while maintaining their GnRH-R potency. R-4-{2-[5-(2-Fluoro-3-methoxyphenyl)-3-(2-fluoro-6-[trifluoromethyl]benzyl)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-yl]-1-phenylethylamino}butyric acid sodium salt, 10b (elagolix), was identified as a potent and selective hGnRH-R antagonist. Oral administration of 10b suppressed luteinizing hormone in castrated macaques. These efforts led to the identification of 10b as a clinical compound for the treatment of endometriosis.
NA SALT
(R)-4-{2-[5-(2-Fluoro-3-methoxyphenyl)-3-(2-fluoro-6-[trifluoromethyl]benzyl)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-yl]-1-phenylethylamino}butyric Acid Sodium Salt
sodium salt as a white solid (1.58 g, 2.47 mmol, 62%). HPLC purity: 100% (220 and 254 nm). 1H NMR (CD3OD): 1.72 (m, 2H), 2.08 (s, 3H), 2.16 (t, J = 6.9 Hz, 2H), 2.50 (t, J = 6.9 Hz, 2H), 3.86 (s, 3H), 4.24 (m, 3H), 5.40 (d, J = 9.0 Hz, 1H), 5.46 (d, J = 9.0 Hz, 1H), 6.62 and 6.78 (m, 1H), 7.12 (m, 2H), 7.34 (m, 5H), 7.41 (m, 1H), 7.56 (m, 1H), 7.61 (d, J = 8.0 Hz, 1H). MS: 632 (M − Na + 2H+). Anal. (C32H29F5N3O5Na·0.75H2O): C, H, N, Na.
PATENT
CN 105218389
PATENT
“Elagolix” refers to 4-((R)-2-[5-(2-fluoro-3-methoxy-phenyl)-3-(2- fluoro-6 rifluoromethyl-benzyl)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-l-yl]-l- phenyl-ethylamino)-butyric acid or a pharmaceutically acceptable salt thereof. Elagolix is an orally active, non-peptide GnRH antagonist and is unlike other GnRH agonists and injectable (peptide) GnRH antagonists. Elagolix produces a dose dependent suppression of pituitary and ovarian hormones in women. Methods of making Elagolix and a pharmaceutically acceptable salt thereof are described in WO 2005/007165, the contents of which are herein incorporated by reference.
References
- ^ Jump up to:a b c d e f g Ezzati, Mohammad; Carr, Bruce R (2015). “Elagolix, a novel, orally bioavailable GnRH antagonist under investigation for the treatment of endometriosis-related pain”. Women’s Health. 11(1): 19–28. doi:10.2217/whe.14.68. ISSN 1745-5057.
- Jump up^ Chen C, Wu D, Guo Z, Xie Q, Reinhart GJ, Madan A, Wen J, Chen T, Huang CQ, Chen M, Chen Y, Tucci FC, Rowbottom M, Pontillo J, Zhu YF, Wade W, Saunders J, Bozigian H, Struthers RS (2008). “Discovery of sodium R-(+)-4-{2-[5-(2-fluoro-3-methoxyphenyl)-3-(2-fluoro-6-[trifluoromethyl]benzyl)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-yl]-1-phenylethylamino}butyrate (elagolix), a potent and orally available nonpeptide antagonist of the human gonadotropin-releasing hormone receptor”. J. Med. Chem. 51 (23): 7478–85. doi:10.1021/jm8006454. PMID 19006286.
- Jump up^ Thomas L. Lemke; David A. Williams (24 January 2012). Foye’s Principles of Medicinal Chemistry. Lippincott Williams & Wilkins. pp. 1411–. ISBN 978-1-60913-345-0.
- ^ Jump up to:a b AdisInsight: Elagolix.
- ^ Jump up to:a b c d Struthers RS, Nicholls AJ, Grundy J, Chen T, Jimenez R, Yen SS, Bozigian HP (2009). “Suppression of gonadotropins and estradiol in premenopausal women by oral administration of the nonpeptide gonadotropin-releasing hormone antagonist elagolix”. J. Clin. Endocrinol. Metab. 94 (2): 545–51. doi:10.1210/jc.2008-1695. PMC 2646513
 . PMID 19033369.
. PMID 19033369. - Jump up^ Diamond MP, Carr B, Dmowski WP, Koltun W, O’Brien C, Jiang P, Burke J, Jimenez R, Garner E, Chwalisz K (2014). “Elagolix treatment for endometriosis-associated pain: results from a phase 2, randomized, double-blind, placebo-controlled study”. Reprod Sci. 21 (3): 363–71. doi:10.1177/1933719113497292. PMID 23885105.
- Jump up^ Carr B, Dmowski WP, O’Brien C, Jiang P, Burke J, Jimenez R, Garner E, Chwalisz K (2014). “Elagolix, an oral GnRH antagonist, versus subcutaneous depot medroxyprogesterone acetate for the treatment of endometriosis: effects on bone mineral density”. Reprod Sci. 21 (11): 1341–51. doi:10.1177/1933719114549848. PMC 4212335 . PMID 25249568.
External links
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014143669A1 | Mar 14, 2014 | Sep 18, 2014 | AbbVie Inc . | Compositions for use in treating heavy menstrual bleeding and uterine fibroids |
| EP2881391A1 | Dec 5, 2013 | Jun 10, 2015 | Bayer Pharma Aktiengesellschaft | Spiroindoline carbocycle derivatives and pharmaceutical compositions thereof |
| US8084614 | Apr 4, 2008 | Dec 27, 2011 | Neurocrine Biosciences, Inc. | Gonadotropin-releasing hormone receptor antagonists and methods relating thereto |
| US8263588 | Apr 4, 2008 | Sep 11, 2012 | Neurocrine Biosciences, Inc. | Gonadotropin-releasing hormone receptor antagonists and methods relating thereto |
| US8481738 | Nov 10, 2011 | Jul 9, 2013 | Neurocrine Biosciences, Inc. | Gonadotropin-releasing hormone receptor antagonists and methods relating thereto |
| US8507536 | Aug 10, 2012 | Aug 13, 2013 | Neurocrine Biosciences, Inc. | Gonadotropin-releasing hormone receptor antagonists and methods relating thereto |
| US8952161 | Jun 5, 2013 | Feb 10, 2015 | Neurocrine Biosciences, Inc. | Gonadotropin-releasing hormone receptor antagonists and methods relating thereto |
| US9034850 | Nov 19, 2010 | May 19, 2015 | Sk Chemicals Co., Ltd. | Gonadotropin releasing hormone receptor antagonist, preparation method thereof and pharmaceutical composition comprising the same |
| US9422310 | Jan 8, 2015 | Aug 23, 2016 | Neurocrine Biosciences, Inc. | Gonadotropin-releasing hormone receptor antagonists and methods relating thereto |
 |
|
| Clinical data | |
|---|---|
| Synonyms | NBI-56418; ABT-620 |
| Routes of administration |
By mouth |
| Drug class | GnRH analogue; GnRH antagonist; antigonadotropin |
| Pharmacokinetic data | |
| Biological half-life | 2.4–6.3 hours[1] |
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C32H30F5N3O5 |
| Molar mass | 631.590 g/mol |
| 3D model (JSmol) | |
///////////////ELAGOLIX, NBI 56418, UNII:5B2546MB5Z, ABT 620, priority review status, PHASE 3, AbbVie, Neurocrine Biosciences, Endometriosis
CC1=C(C(=O)N(C(=O)N1CC2=C(C=CC=C2F)C(F)(F)F)CC(C3=CC=CC=C3)NCCCC(=O)O)C4=C(C(=CC=C4)OC)F
Drug Patents International

All about Patents and Intellectual property by DR ANTHONY MELVIN CRASTO, worlddrugtracker, Ph.D ( ICT, Mumbai) , INDIA 30 Yrs Exp. in the feld of Organic Chemistry, Serving chemists around the world.
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India
https://drugpatentsint.blogspot.in/
/////////////////
