



| Patent | Submitted | Granted |
|---|---|---|
| Triphenylalkene derivatives and their use as selective estrogen receptor modulators [US6576645] | 2003-06-10 | |
| Combination therapy for the treatment of estrogen-sensitive disease [US2002119502] | 2002-08-29 | |
| Triphenylalkene derivatives and their use as selective estrogen receptor modulators [US6875775] | 2003-12-04 | 2005-04-05 |
| Combination therapy for the treatment of estrogen-sensitive disease [US2005176691] | 2005-08-11 | |
| Anti-IGFR1 antibody therapeutic combinations [US8017735] | 2005-06-23 | 2011-09-13 |
| Combination therapy for the treatment of estrogen-sensitive disease [US2005228053] | 2005-10-13 | |
| Combination therapy for the treatment of estrogen-sensitive drugs [US2005232862] | 2005-10-20 | |
| Toremifene crystallization method [US7368607] | 2007-04-26 | 2008-05-06 |
| Platinum therapeutic combinations [US2006205810] | 2006-09-14 | |
| Methods and compositions for treating or preventing cancer [US2006233810] | 2006-10-19 |
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Toremifene

Toremifene
2-[4-[(Z)-4-chloro-1,2-diphenylbut-1-enyl]phenoxy]-N,N-dimethylethanamine
(Z)-2-[4-(4-Chloro-1,2-diphenyl-1-butenyl)phenoxy]-N,N-dimethylethanamine
(Z)-4-Chloro-1,2-diphenyl-1-[4-[2-(N,N-dimethylamino)ethoxy]phenyl]-1-butene
(Z)-Toremifene
2-({4-[(1Z)-4-chloro-1,2-diphenylbut-1-en-1-yl]phenyl}oxy)-N,N-dimethylethanamine
4-chloro-1,2-diphenyl-1-[4-[2-(N ,N-dimethylamino)ethoxy]phenyl]-1-butene
Toremifene; Acapodene; Farestone; Z-Toremifene; Toremifeno; Toremifenum
| Molecular Formula: | C26H28ClNO |
|---|---|
| Molecular Weight: | 405.95962 g/mol |
cas 89778-26-7
Launched – 1988.Orion (FI), greast cancer
- Citrate, Toremifene, GTx-006
NK-622 - Fareston
- FC 1157a
- FC-1157a
- FC1157a
- Toremifene
- Toremifene Citrate
- Toremifene Citrate (1:1)
- Toremifene, (E)-Isomer
-
- C26H28ClNO · C6H8O7
- Molecular Weight 598.08
 Toremifene Citrate
Toremifene Citrate
Toremifene is a first generation selective estrogen receptor modulator (SERM). Like TAMOXIFEN, it is an estrogen agonist for bone tissue and cholesterol metabolism but is antagonistic on mammary and uterine tissue.
The company GTx is conducting phase III clinical trials for the prevention of prostate cancer in men who have been diagnosed with high grade prostatic intraepithelial neoplasia (PIN).
Toremifene citrate is an oral selective estrogen receptor modulator (SERM) which helps oppose the actions of estrogen in the body. Licensed in the United States under the brand name Fareston, toremifene citrate is FDA-approved for use in advanced (metastatic)breast cancer. It is also being evaluated for prevention of prostate cancer under the brand name Acapodene.[1]
In 2007 the pharmaceutical company GTx, Inc was conducting two different phase 3 clinical trials; First, a pivotal Phase clinical trial for the treatment of serious side effects of androgen deprivation therapy (ADT) (especially vertebral/spine fractures and hot flashes, lipid profile, and gynecomastia) for advanced prostate cancer, and second, a pivotal Phase III clinical trial for the prevention of prostate cancer in high risk men with high grade prostatic intraepithelial neoplasia, or PIN. Results of these trials are expected by first quarter of 2008[2]
An NDA for the first application (relief of prostate cancer ADT side effects) was submitted in Feb 2009,[3] and in Oct 2009 the FDA said they would need more clinical data, e.g. another phase III trial.[4]
Originally developed at Orion, toremifene was subsequently licensed to Nippon Kayaku in Japan and to Asta Medica (now, part of Meda) in Germany.
Synthesis

……….
PATENT
http://www.google.com/patents/CN104230723A?cl=en
Toremifene (Toremifene), chemical name (Z) -4- chloro-1,2-diphenyl–1- [4- (2- (N, N- dimethylamino) ethoxy yl) phenyl] -1-butene, having the structure I.Toremifene to tamoxifen (Tamoxifen) analogues with anti-estrogenic activity, can be used in the treatment of hormone-dependent breast cancer, and its E-isomer has the presence of estrogenic activity, E isomers toremifene may counteract anti-estrogenic activity, and therefore isomeric purity is essential toremifene.Toremifene was developed in 1983 by the Finnish Famos company, listed in 1996 by the Orion company in the EU, the trade name Fareston, 2002 to enter the country, the trade name of toremifene.

RJ Toivola et al., European Patent EP95875, disclosed in U.S. Patent US4696949A synthetic route toremifene, that following a synthetic route, the synthetic route to phenol as a raw material, by acylation, rearrangement, alkyl group and an addition reaction to give 1,2-diphenyl -1- [4- [2- (N, N- dimethylamino) ethoxyphenyl]] – 1,4-diol (Compound 5) as the key intermediate, further HCl in ethanol or hydrochloric acid elimination reaction occurs, then get toremifene thionyl chloride reaction. The main problem with this approach is that the elimination reaction of the compound 5 in ethanol occurs when hydrochloric acid or concentrated hydrochloric acid, the resulting triaryl alcohol butyrate (Compound 6) having a Z / E configuration, both the ratio of 1: 2 ~ 2: 1, stereo selectivity is not high, and there are 5% of the cyclization by-product; on the Z / E configuration Miyoshi butyric fractional crystallization of alcohol, you can get pure Z-type Miyoshi butyric alcohol , but the yield is only 41%; then, Z-type Miyoshi butyric alcohol chlorination reaction occurs in the action of thionyl chloride, the purified product toremifene.

U.S. Patent US5491173A also reported another synthetic route toremifene namely the following two synthetic routes. The route to the aryl ketone (Compound 7) with a phenyl Grignard reagent addition reaction of ketone carbonyl groups to give triaryl-butanediol (compound 5), which is the elimination of toremifene and chlorinated reaction products happen again.

Chinese Patent Publication No. CN1125716A application reported an efficient synthesis of Z-type Miyoshi butyric alcohol (compound 6) method, US4696949A compared with the US patent, the method mild conditions, reduce the acid concentration and reaction temperature, reaction time, triarylphosphine butanediol (Compound 5) in concentrated hydrochloric acid or concentrated hydrochloric isopropanol or ethanol effect of concentrated hydrochloric acid, can be 60-78% selectivity and 95% yield of type 2 Miyoshi butyric alcohol But after Publication No. 0 02,126,969 attached eight patent applications after the inventor repeated experiments show that the technique disclosed in the patent application programs can not achieve their claimed technical effect.
Publication No. CN102126969A of Chinese patent applications through the intermediate Miyoshi butyric alcohol occurs at a catalytic converter configuration of concentrated hydrochloric acid, while taking advantage of differences in solubility, so E- type Miyoshi butyric alcohol continuously into Z-type Miyoshi butyric alcohol (compound 6) precipitates, thereby undermining the balance, so that one of the E-type Miyoshi butyric alcohol continuously into Z-type Miyoshi butyric alcohol (compound 6) to give the Z-Miyoshi butyric alcohol ( Compound 6) and then get toremifene thionyl chloride after chlorination. Although to some extent, improve the yield, but increased operating procedure, is not conducive to industrial production.
Currently, the key intermediate is patent protected, and z-type Miyoshi butyric alcohol (compound 6) stereoselective low yield and isolated intermediates, to solve this problem, to overcome technical barriers to foreign pharmaceutical companies, urgent need to find a simple process, low cost, easy to separate and viable for large-scale production of synthetic routes.
To achieve the above object, according to one aspect of the present invention, there is provided a method of synthesizing toremifene, synthetic method comprising: a step S1, so that a compound having the structural formula II with a compound B having the structural formula III C occurs Mike Murray to give compound D having the structural formula IV; step S2, the Compound D and Compound E or Compound E of the hydrochloride salt of the formula V having a phenolic hydroxyl group on the occurrence of a selective alkylation reaction, to give a compound having the structural formula VI F; step S3, the compound F is reacted with thionyl chloride to give toremifene, wherein,
Formula II is:

Structural formula III as follows:

Formula IV is

Of formula V is C1CH2CH2N (CH 3) 2; formula VI is

FIG. 1 illustrates the present invention obtained in Example 1 H NMR spectrum of compound D of implementation;

FIG. 2 shows the 1 H NMR spectrum of the present invention, the compound obtained in Example F;
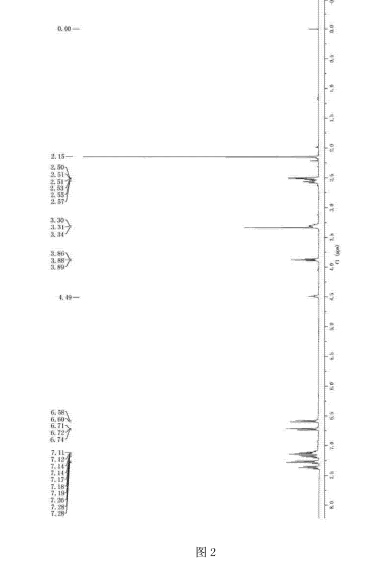
FIG. 3 shows the present invention is a proton nuclear magnetic resonance spectrum of toremifene obtained in Example.
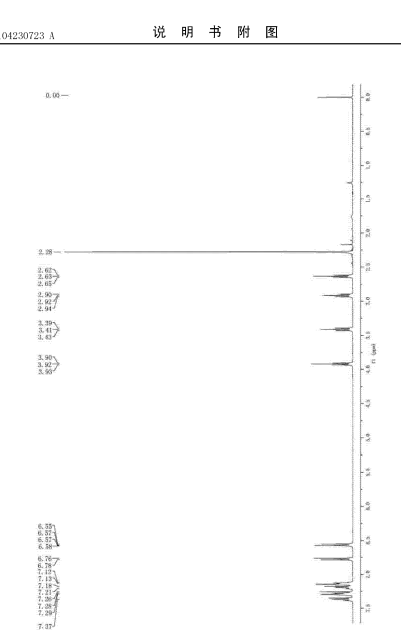
Figure 1, which shows a spectrum of results for Che bandit? (400 cm take, 01 ^ 0) 3 = 9.20 (! 8,1 1), 7.37 (^ = 7.4 to take, 2 1!), 7.30- 7. 23 (m, 3H), 1.22- 7. 15 (m, 2H), 7. 15 – 7. 06 ( m, 3H), 6. 61 (dd, J = 9. 0, 2. 2Hz, 2H), 6. 49 -. 6. 32 (m, 2H), 4 48 (s, 1H), 3 30 (. m, 2H), 2 55 (t, J = 7. 5Hz, 2H);. F proton nuclear magnetic resonance spectrum of the compound attached to the
Figure 2, showing spectrum results Che NMR (400MHz, DMSO) δ = 7. 36 (d, J = 7. 3Hz, 2H), 7. 31 – 7. 25 (m, 3H), 7. 21 – 7. 10 (m, 5H), 6. 75 – 6. 69 (m , 2H), 6. 59 (d, J = 8. 8Hz, 2H), 4. 49 (s, 1H), 3. 88 (t, J = 5. 8Hz, 2H), 3. 31 (d, J = 4. 3Hz, 2H), 2. 57 (t, J = 7.5Hz, 2H), 2.52 (t, J = 4.6Hz, 2H), 2 15 (s, 6H);.
Tommy remifentanil NMR hydrogen spectrum in Figure 3 attached, showing spectrum results Che NMR (400MHz, CDC13) δ = 7. 41 -. 7. 33 (m, 2H), 7 29 (dt, J = 7. 1, 2. 9Hz, 3H), 7. 20 (dd, J = 10. 0, 4. 3Hz, 2H), 7. 13 (dd, J = 7. 1, 4. 3Hz, 3H), 6.87- 6. 72 (m, 2H), 6. 57 (dd, J = 6. 8, 4. 8Hz, 2H), 3. 92 (t, J = 5. 8Hz, 2H), 3. 41 (t, J = 7. 5Hz, 2H), 2. 92 (t, J = 7. 5Hz, 2H), 2. 63 (t, J = 5. 8Hz, 2H), 2. 28 (s, 6H).
The synthetic routes above synthetic method are as follows:

Synthesis of toremifene:
To a 2L reaction flask 1. 1L of toluene, 110g (0. 28mol) obtained in the above step Z configuration compound F, mixed to obtain a sixth system, the cooling system to the sixth mixed 0~5 ° C , was slowly added dropwise 99. 93g (0. 84mol) thionyl chloride addition was complete the formation of the seventh mixed system, the mixed system was slowly warmed to a seventh ll〇 ° C, for 1 hour to obtain a third product system, stop The third product heating and cooling system to 15~25 ° C, the third product system slowly poured into 1L of water, adding NaOH solution to a pH 9~10 and get the second system, the second in and a system for liquid separation, and the resulting aqueous phase to obtain a second solution was extracted with 1L toluene extraction, the organic phase of the second extraction solution and liquid separation were combined and concentrated to give crude toremifene, the crude product was mass ratio of 1 : mixed solvent of ethyl acetate and acetone 1 crystals to give 103. 7g toremifene products.
Synthesis of toremifene:
[0062] To a 5L reaction flask 3. 3L of toluene, 110g (0. 28mol) obtained in the above step Z configuration compound F, mixed to obtain a sixth system, the cooling system to the sixth mixed 0~5 ° C , was slowly added dropwise 33. 31g (0. 28mol) thionyl chloride addition was complete the formation of the seventh mixed system, the mixed system was slowly warmed to a seventh ll〇 ° C, after the reaction for 6 hours to obtain a third product system, stop The third product heating and cooling system to 15~25 ° C, the third product system slowly poured into 1L of water, potassium carbonate solution to a pH 9~10 and get the second system, the second and system for liquid separation, and the resulting aqueous phase to obtain a second solution was extracted with 1L ethyl acetate, the organic phase after the second extraction solution and liquid separation were combined and concentrated to give crude toremifene, the crude product was quality ratio was crystallized from acetone to give 92. 2g toremifene products.
Purity of toremifene following method:
[0107] to take the product, add the mobile phase dissolved and diluted into 1ml of 1. Omg solution containing, according to HPLC octadecylsilane bonded silica as a filler to square 1% trifluoroacetic acetic acid aqueous solution (A) and acetonitrile (B) as the mobile phase gradient elution (T = Omin 10% B; T = lOmin 95% B; T = 12min 100% B; T = 15min 10% B), detection wavelength 210nm; area normalization method to calculate the Z configuration purity compound F, where F Z configuration compound retention time of 6. 76min. The purity of the above calculation or Z configuration detection obtained compound D, compound D Z configuration and E configuration of the weight ratio, toremifene yield and purity are reported in Table 1 below.
……………..
PATENT
http://www.google.com/patents/US5491173
c) 4-chloro-1,2-diphenyl-1-[4-[2-(N ,N-dimethylamino)ethoxy]phenyl]-1-butene (Z and E)
(Z)-isomer: The reaction is performed under dry conditions. 42.4 g of (Z)-1,2-diphenyl-1-[4-[2-(N,N-dimethylamino )ethoxy]phenyl]-1-buten-4-ol are dissolved in 250 ml of chloroform. Then 23.8 g of thionyl chloride areadded dropwise. The mixture is refluxed 3 h. The solvent is evaporated, after which the product is recrystallized from ethyl acetate. The yield ofthe hydrochloride salt is 36.7 g (83%), m.p. 194°-6° C. The base can be liberated from the Salt with 1M sodium carbonate solution, after which the base is extracted in toluene. The toluene solution is dried and the solvent is evaporated. The free base has m.p. 108°-10° C. (from acetone).
1 H-NMR-spectrum (CDCl3): δ 2.27 (6H, s), 2.63 (2H, t), 2.91 (2H, t), 3.41 (2H, t), 3.92 (2H, t), 6.54 (2H, d), 6.79 (2H. d), 7.15(5H, s), 7.31 (5H, s). MS: m/z 405/407 (M+, 7/3), 72 (20), 58 (100).
The citric acid salt can be prepared as follows: 40.6 g of the (Z)-isomer as a free base are dissolved in 175 ml of warm acetone and 24.3 g of citric acid are dissolved in 100 ml of warm acetone. The solutions are combined and the mixture is allowed to cool. The citrate, m.p. 160°-162° C., is collected by filtration.
(E)-isomer: The compound is prepared from (E)-1,2-diphenyl-1-[4-[2-(N ,N-dimethylamino)ethoxy]phenyl]-1-buten-4-ol in the same manner as the corresponding (Z)-isomer. The hydrochloride salt is crystallized from toluene. The yield is 35.8 g (81%) of a product having m.p. 183°-5° C. The base can be liberated from the salt in the same manner as the corresponding (Z)-isomer. It has m.p. 69°-71° C. (from hexane).
1 H-NMR-spectrum (CDCl3): b 2.34 (6H, s), 2.74 (2H, t), 2.97 (2H,t), 3.43 (2H, t), 4.08 (2H, t), 6.80-7.30 (14H, m).
MS: m/z 405/407 (M+, 7/3) 72 (19) 58 (100)
EXAMPLE 4
4-chloro-1,2-diphenyl-1-[4-[2-(N ,N-diethylamino)ethoxy]phenyl ]-1-butene (Z and E)
43.3 g of 1,2-diphenyl-1-[4-[2-(N,N-diethylamino)ethoxy]phenyl]butane-1,4-diol (pureenantiomer pairs or their mixture: m.p. of (RR,SS)-pair is 107°-9° C.)is suspended in 250 ml of toluene, after which 25ml toluene is distilled off to dry the solution. The mixture is cooled to 0° C. with stirring. While stirring and keeping the temperature at 0° C. or a little below, 47.6 g of thionyl chloride of good qualityare added. The mixture is stirred for 1 h at 0° C. and the temperature is then allowed to rise to 22° C. The mixture is stirred at 80° C. until the reaction is completed (about 3 h). After that, water is added to decompose the excess of thionyl chloride followed by 20% sodium hydroxide solution to liberate the product from itshydrochloride salt. The aqueous layer is discarded and the toluene layer iswashed with water. Then the solvent is evaporated to leave a mixture of (Z)- and (E)isomers (Z:E 7:3) as an oil in quantitative yield.
(Z)-isomer: The (Z)-isomer is isolated from the isomer mixture above as thehydrochloride salt because of the low melting point of the free base. The m.p. of the hydrochloride salt is 178°-80° C. The (Z)-isomermay be freed from its salt by any normal method.
1 H-NMR-spectrum (CDCl3): δ 1.01 (6H, t), 2.57 (4H, q), 2.77 (2H, t), 2.91 t), 3.41 (2H, t), 3.90 t), 6.53 (2H, d), 6.78 (2H, d), 7.15 (5H, s), 7.31 (5H, s). (E)-isomer:
1 H-NMR-spectrum (CDCl3): δ 1.07 (6H, t), 2.66 (4H, q), 2.89 (2H, t), 2.97 (2H, t), 3.42 (2H, t), 4.07 (2H, t), 6.90-7.20 (10H, m).
……………….
SEE
http://www.google.co.ug/patents/EP0095875B1?cl=en
………….
References
- Price N, Sartor O, Hutson T, Mariani S. Role of 5a-reductase inhibitors and selective estrogen receptor modulators as potential chemopreventive agents for prostate cancer. Clin Prostate Cancer 2005;3:211-4. PMID 15882476
- “GTx’s Phase III Clinical Development of ACAPODENE on Course Following Planned Safety Review” (Press release). GTx Inc. 2007-07-12. Retrieved 2006-07-14.
- “GTx Announces Toremifene 80 mg NDA Accepted for Review by FDA” (Press release).
- “GTx and Ipsen End Prostate Cancer Collaboration due to Costs of FDA-Requested Phase III Study”. 2 Mar 2011
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-{4-[(1Z)-4-chloro-1,2-diphenyl-but-1-en-1-yl]phenoxy}-N,N-dimethylethanamine
|
|
| Clinical data | |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a608003 |
| Pharmacokinetic data | |
| Protein binding | more than 99.5% |
| Biological half-life | 5 days |
| Identifiers | |
| CAS Registry Number | 89778-26-7 |
| ATC code | L02BA02 |
| PubChem | CID: 3005573 |
| IUPHAR/BPS | 4325 |
| DrugBank | DB00539 |
| ChemSpider | 2275722 |
| UNII | 7NFE54O27T |
| KEGG | D08620 |
| ChEBI | CHEBI:9635 |
| ChEMBL | CHEMBL1655 |
| Chemical data | |
| Formula | C26H28ClNO |
| Molecular mass | 405.959 g/mol |
from PubChem……….https://pubchem.ncbi.nlm.nih.gov/compound/3005573#section=Depositor-Supplied-Patent-Identifiers
///////
What is SBM-TFC-039 an SGLT Inhibitor from Sirona Biochem !!
A new “flozin” seems to me appearing on the horizon in form of SBM-TFC-039 an SGLT Inhibitor from Sirona Biochem, picked up a list from WO 2012160218, from TFChem…….see link , Sirona Biochem Announces SGLT2 Inhibitor and Skin Lightening Patent Granted, 29 Jun 2015, Patent entitled “Family of aryl, heteroaryl, o-aryl and o-heteroaryl carbasugars”
This led me to search, “Family of aryl, heteroaryl, o-aryl and o-heteroaryl carbasugars”
WO 2012160218 A1, IN 2013-DN10635, CN 103649033Tf化学公司
| Applicant | Tfchem |

List above as in http://www.google.com/patents/WO2012160218A1?cl=en
FROM THE ABOVE LIST, SBM-TFC-039 MAY BE PREDICTED/OR AS SHOWN BELOW
COMPD 16 as in/WO2012160218
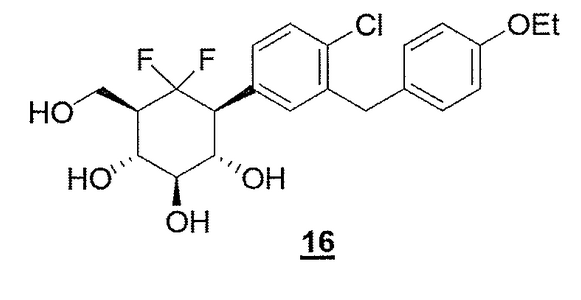
COMPD 16, PREDICTED/LIKELY SBM-TFC-039 has CAS 1413373-30-4, name D-myo-Inositol, 1-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1,2,3-trideoxy-2,2-difluoro-3-(hydroxymethyl)-
Just scrolling through the patent gave me more insight
MORE EVIDENCE….http://www.google.com/patents/WO2012160218A1?cl=en, this patent descibes compd 16 as follows
Compound 16 according to the invention has been compared to Dapaglifozin to underline the improvement of the duration of action, i.e. the longer duration of glucosuria, of the compound when the intracyclic oxygen atom of the glucose moiety is replaced by a CF2 moiety.

This assay has been carried out at a dose of 3 mg/ kg.
The results obtained are presented on Figure 5. It appears thus that 16 (3 mg/kg) triggered glucosuria that lasted beyond 24 hours compared to Dapagliflozin.
• Compound 16 according to the invention has been compared to the compound 9 of WO 2009/1076550 to underline the improvement of the duration of action of the compound when a mimic of glucose bearing a CH-OH moiety instead of the intracyclic oxygen atom is replaced by a mimic of glucose bearing a CF2 in place of the CH-OH moiet .

NOTE=COMPD 9 OF WO 2009/1076550 has CAS 1161430-16-5, D-scyllo-Inositol, 1-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1,3-dideoxy-3-(hydroxymethyl)- and is very similar to the compd under discussion
%7D)
| Company | Sirona Biochem Corp. |
| Description | Sodium-glucose cotransporter 2 (SGLT2) inhibitor |
| Molecular Target | Sodium-glucose cotransporter 2 (SGLT2) |
| Mechanism of Action | Sodium-glucose cotransporter 2 (SGLT2) inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Preclinical |
| Standard Indication | Diabetes |
| Indication Details | Treat Type II diabetes |
| Regulatory Designation | |
| Partner | Shanghai Fosun Pharmaceutical Group Co. Ltd. |
SBM-TFC-039
PATENT
WO 2012160218
http://www.google.com/patents/WO2012160218A1?cl=en
Examples within this first subclass include but are not limited to:

Synthesis of compound 8
C35H34O5 M = 534.64 g.mol“
Mass: (ESI ): 535.00 (M + H); 552.00 (M + H20); 785.87; 1086.67 (2M + H20)

A.

Procedure A:
To a solution of 4 (10.5g, 15.89mmol, leq) in toluene (400mL) were added 18-crown-6 (168mg, 0.64mmol, 0.04eq) and potassium carbonate (6.69g, 48.5mmol, 3.05eq.). The mixture was stirred overnight at room temperature, and then the remising insoluble material was filtered off and washed with toluene. The filtrate and the washings were combined, washed with 2N hydrochloric acid aqueous solution followed by saturated sodium hydrogencarbonate aqueous solution, dried over sodium sulphate, filtered and concentrated under reduced pressure. The residue was purified on silica gel chromatography (cyclohexane/ethyl acetate 98:2 to 80:20) to afford cyclohexenone 8 (4.07g; 48% yield) as yellowish oil.
Procedure B:
A solution of 7 (3.27g, 5.92mmol, leq) in pyridine (14mL) was cooled to 0°C before POCl3 (2.75mL, 29.6mmol, 5eq) was added dropwise. The mixture was stirred at this temperature for 10 min before the cooling bath was removed. The reaction mixture was stirred overnight at room temperature before being re-cooled to 0°C. POCI3 (2.75mL, 29.6mmol, 5eq) was added once again trying to complete the reaction. The mixture was stirred for an additional 20h at room temperature before being diluted with Et20 (20mL) and poured onto crushed ice. 1M HC1 aqueous solution (lOOmL) was added, and the mixture was extracted with Et20 (200mL & l OOmL). The combined organic extracts were washed with brine (lOOmL), dried over sodium sulphate, filtered and concentrated before being purified on silica gel chromatography (cyclohexane / ethyl acetate 98:2 to 80:20) to afford compound 8 (1.46g, 46% yield) as an orange oil. Synthesis of compound 9
C15H12BrC102 M = 339.61 g.moF1
Mass: (GC-MS): 338-340

The synthesis of this product is described in J. Med. Chem. 2008, 51, 1 145—1149.Synthesis of compound 10
C15H14B1CIO M = 325.63 g.mof1

10 The synthesis of this product is described in J. Med. Chem. 2008, 51, 1145-1 149.
Synthesis of compound 11
C50H49CIO6 M = 781.37 g.moF1
Mass: ESI+): 798.20 (M + H20)

Under inert atmosphere, Mg powder (265mg, 10.9mmol, 2.4eq) was charged into a three necked flask, followed by addition of a portion of 1/3 of a solution of the 4- bromo-l-chloro-2-(4-ethylbenzyl)benzene (2.95g, 9.1mmol; 2eq) in dry THF (25mL) and 1 ,2-dibromoethane (10 mol % of Mg; 85mg; 0.45mmol). The mixture was heated to reflux. After the reaction was initiated (exothermic and consuming of Mg), the remaining solution of 2-(4-ethylbenzyl)-4-bromo-l-chlorobenzene in dry TFIF was added dropwise. The mixture was then allowed to react for another one hour under gentle reflux until most of the Mg was consumed.
The above Grignard reagent was added dropwise into the solution of cyclohexenone 8 (2.42g, 4.53mmol, leq) in dry THF (25mL) under inert atmosphere at room temperature (about 25°C), then allowed to react for 3h. A saturated aqueous solution of ammonium chloride was added into the mixture to quench the reaction. The mixture was extracted with Et20, washed with brine, dried over sodium sulphate, filtered and concentrated. The residue was purified on silica gel chromatography (cyclohexane/ethyl acetate 100:0 to 80:20) to afford the target compound 11 as a yellow oil (3.01g, 86%).
Synthesis of compound 12
C5oH49C105 M = 765.37 g.mol“1
+): 782.13 (M + H20)

Triethylsilane (0.210mL, 1.30mmol, 3eq) and boron-trifluoride etherate (48% BF3, O. l lOmL, 0.866mmol, 2eq) were successively added into a solution of alcohol 1 1 (338mg, 0.433mmol, leq) in dichloromethane (5mL) under inert atmosphere at -20°C. After stirring for 2.5h, a saturated aqueous solution of sodium chloride was added to quench the reaction. The mixture was extracted with CH2C12 (10mLx3) and the organic layer was washed with brine, dried over Na2S04, filtrated and concentrated. The residue was purified on silica gel chromatography (cyclohexane/ethyl acetate 9.8:0.2 to 8:2) to afford the target compound 12 as a white powder (278 mg, 0.363mmol, 84%).
Synthesis of compound 13
C5oH5tC106 M = 783.39g.moF1
Mass: (ESI+): 800 (M + H20); 1581 (2M + H20)

Under inert atmosphere, borane-dimethyl sulfide complex (2M in THF, 16.7mL, 33mmol, 10.5eq) was added to a solution of 12 (2.41g; 3.15mmol, leq) in dry THF (lOOmL) cooled to 0°C. The reaction mixture was then refluxed for lh,cooled to 0°C and treated carefully with sodium hydroxide (3M in H20, 10.5mL, 31.5mmol, lOeq), followed by hydrogen peroxide (30% in H20, 3.2mL, 31.5mmol, l Oeq) at room temperature (above 30°C). The mixture was allowed to react overnight at room temperature (~25°C) before a saturated aqueous solution of ammonium chloride was added to quench the reaction. The mixture was extracted with ethyl acetate and the organic layer was washed with brine, dried over Na2S04, filtered, and concentrated. The residue was purified by silica gel chromatography (cyclohexane/ethyl acetate 97:3 to 73:27) to afford the desired compound 13 (1.05g; 43%) as a yellowish oil.
Synthesis of compound 14
C50H49CIO6 M = 781.37g.mol“1
Mass: (ESI+): 798 (M + H20); 1471; 1579 (2M + H20)

13 14
Dess-Martin periodinane (81mg; 1.91mmol; 1.5eq) was added portion wise to a solution of alcohol 13 (l .Og; 1.28mmol, leq) in anhydrous dichloromethane (20mL) at 0°C. The reaction was then stirred overnight at room temperature before being quenched with IN aqueous solution of sodium hydroxide. The organic layer was separated and the aqueous layer was extracted with dichloromethane. The combined organic layers were dried over sodium sulphate, filtered and concentrated. The residue was purified on silica gel chromatography (cyclohexane / ethyl acetate 98:2 to 82: 18), to afford the target ketone 14 (783mg, 79% yield) as a colorless oil. Synthesis of compound 15
C5oH49ClF206 M = 803.37g.moF1
19 F NMR (CDCU, 282.5MHz): -100.3 (d, J=254Hz, IF, CFF); -1 13.3 (td, Jl=254Hz, J2=29Hz, IF, CFF).
Mass: (ESI+): 820.00 (M+H20)

14 15
A solution of ketone 14 (421mg, 0.539mmol, leq) in DAST (2mL, 16.3mmol, 30eq.) was stirred under inert atmosphere at 70°C for 12h. The mixture was then cooled to room temperature and dichloromethane was added. The solution was poured on a mixture of water, ice and solid NaHC03. Agitation was maintained for 30min while reaching room temperature. The aqueous layer was extracted with dichloromethane and the organic phase was dried over Na2S04, filtered and concentrated. The crude product was purified on silica gel chromatography (cyclohexane/ethyl acetate 98:2 to 80:20) to afford the desired compound 15 as a yellowish oil ( 182mg, 42% yield).
Synthesis of compound 16
C22H25CIF2O5 M = 442.88g.mor1
19 F NMR (MeOD, 282.5MHz): -96.7 (d, J=254Hz, IF, CFF); 12.2 (td,
Jl=254Hz, J2=28Hz, IF, CFF).
Mass: (ESI+): 465.3 (M+Na)

o-Dichlorobenzene (0.320mL, 2.82mol, lOeq) followed by Pd/C 10% (0.342g, 0.32mol, l .leq) were added to a solution of 15 (228mg, 0.28mmol, leq) in a mixture of THF and MeOH (2: 1, v/v, 160mL). The reaction was placed under hydrogen atmosphere and stirred at room temperature for 2h. The reaction mixture was filtered and concentrated before being purified on silica gel chromatography (dichloromethane/methanol 100: 1 to 90: 10) to afford compound 16 (105mg, 83% yield).
…………………….
CN 103649033
Sirona Biochem’s SGLT Inhibitor Performs Better Than Johnson and Johnson’s SGLT Inhibitor, According to Study
Vancouver, British Columbia – December 7, 2012 – Sirona Biochem Corp. (TSX-V: SBM), announced its sodium glucose transporter (SGLT) inhibitor for Type 2 diabetes reduced blood glucose more effectively than Johnson and Johnson’s canagliflozin, an advanced SGLT inhibitor being considered for market approval in Europe and the U.S. Studies compared Sirona Biochem’s SGLT Inhibitor, SBM-TFC-039, with canagliflozin and were conducted on Zucker Diabetic Fatty (ZDF) rats.
In the study, SBM-TFC-039 significantly and rapidly reduced blood glucose levels at a dose of 1.0 mg/kg. Six (6) hours after administration, SBM-TFC-039 reduced blood glucose by 44% compared to canagliflozin at 26%. SBM-TFC-039 also had a longer duration of effect than canagliflozin. At 36 and 48 hours after treatment, SBM-TFC-039, at a dose of 1.0 mg/kg, was still effective at reducing blood glucose, whereas canagliflozin lost its effect after 36 hours. Studies were conducted at the Institut Universitaire de Cardiologie et de Pneumologie de Québec (IUCPQ) by Principal Investigator Dr. Denis Richard, Research Chair on Obesity and Professor, Faculty of Medicine, Department of Anatomy & Physiology at Laval University.
“SGLT Inhibitors are a ground-breaking new treatment for Type 2 diabetes and these results demonstrate that SBM-TFC-039 will be a significant competitor for other SGLT Inhibitors,” said Neil Belenkie, Chief Executive Officer of Sirona Biochem. “The first SGLT Inhibitor,Forxiga™, was approved last month by the European Commission. We believe there is tremendous market potential worldwide for SGLT Inhibitors in the treatment of diabetes.”
SBM-TFC-039 is a sodium glucose transporter (SGLT) inhibitor. SGLT inhibitors are a new class of drug candidates for the treatment of diabetes. In the kidneys, SGLT inhibitors reduce the reabsorption of glucose into the bloodstream by eliminating excess glucose into the urine.
About Sirona Biochem Corp.
Sirona Biochem is a biotechnology company developing diabetes therapeutics, skin depigmenting and anti-aging agents for cosmetic use, biological ingredients and cancer vaccine antigens. The company utilizes a proprietary chemistry technique to improve pharmaceutical properties of carbohydrate-based molecules. For more information visit www.sironabiochem.com.
![]()
Laboratory – France
TFChem
Voie de l’innovation
Pharma Parc II
Chaussée du Vexin
27100 Val de Reuil
France
Phone:+33(0)2.32.09.01.16
Fax:+33(0)2.32.25.07.64



……………………………………………………………………………….
Shanghai Fosun Pharmaceutical Group Co. Ltd.

![]()
//////
Fispemifene for hypogonadism

Fispemifene, HM 101
Fispemifene; UNII-3VZ2833V08;
cas 341524-89-8
| Molecular Formula: | C26H27ClO3 |
|---|---|
| Molecular Weight: | 422.94378 g/mol |
2-[2-[4-[(Z)-4-chloro-1,2-diphenylbut-1-enyl]phenoxy]ethoxy]ethanol
Treatment of Hypogonadism
Androgen Decline in the Aging Male (Andropause) in phase 2
Fispemifene is the Z-isomer of the compound of formula (I)

WO 01/36360 describes a group of SERMs, which are tissue-specific estrogens and which can be used in women in the treatment of climacteric symptoms, osteoporosis, Alzheimer’s disease and/or cardiovascular diseases without the carcinogenic risk. Certain compounds can be given to men to protect them against osteoporosis, cardiovascular diseases and Alzheimer’s disease without estrogenic adverse events (gynecomastia, decreased libido etc.). Of the compounds described in said patent publication, the compound (Z)-2-{2-[4-(4-chloro-1,2-diphenylbut-1-enyl)phenoxy]ethoxy}ethanol (also known under the generic name fispemifene) has shown a very interesting hormonal profile suggesting that it will be especially valuable for treating disorders in men. WO 2004/108645 and WO 2006/024689 suggest the use of fispemifene for treatment or prevention of age-related symptoms in men, such as lower urinary tract symptoms and diseases or disorders related to androgen deficiency in men.
Quatrx had been conducting phase II clinical development for the treatment of androgen decline in the aging male. Unlike testosterone replacement therapies that are typically topical or injection therapies, fispemifene is an oral treatment and is not a formulation of testosterone. Fispemifene utilizes the body’s normal feedback mechanism to increase testosterone levels. Originally developed at Hormos, QuatRx gained rights to the drug candidate following a merger of the companies pursuant to which Hormos became a wholly-owned subsidiary of QuatRx.
Known methods for the syntheses of compounds like ospemifene and fispemifene include rather many steps. WO 02/090305 describes a method for the preparation of fispemifene, where, in a first step, a triphenylbutane compound with a dihydroxysubstituted butane chain is obtained. This compound is in a second step converted to a triphenylbutene where the chain is 4-chlorosubstituted. Then the desired Z-isomer is crystallized. Finally, the protecting group is removed to release the ethanol-ethoxy chain of the molecule.
Fispemifene is a selective estrogen receptor modulator (SERM) studied in phase II clinical trials at Forendo Pharma for the treatment low testosterone in men. The compound is also in phase II clinical studies at Apricus for the treatment of men with secondary hypogonadism.
In 2013, Forendo Pharma acquired the drug from Hormos Medical for the treatment of male low testosterone.
In 2014, Apricus Biosciences acquired U.S. rights for development and commercialization
PATENT
https://www.google.com/patents/US7504530
EXAMPLE 2 2-{2-[4-(4-Chloro-1,2-diphenyl-but-1-enyl)-phenoxy]-ethoxy}-ethanol (Compound I)
{2-[4-(4-Chloro-1,2-diphenyl-but-1-enyl)-phenoxy]-ethoxy}-acetic acid ethyl ester was dissolved in tetrahydrofuran at room temperature under nitrogen atmosphere. Lithium aluminium hydride was added to the solution in small portions until the reduction reaction was complete. The reaction was quenched with saturated aqueous ammonium chloride solution. The product was extracted into toluene, which was dried and evaporated in vacuo. The residue was purified with flash chromatography with toluene/triethyl amine (9.5:0.5) as eluent. Yield 68%.
1H NMR (200 MHz, CDCl3):
2.92 (t, 2H, ═CH 2CH2Cl),
3.42 (t, 2H, ═CH2 CH2 Cl),
3.59-3.64 (m, 2H, OCH2CH2O CH2CH 2OH),
3.69-3.80 (m, 4H, OCH2 CH 2OCH 2 CH2OH),
3.97-4.02 (m, 2H, OCH2CH2OCH2CH2OH),
6.57 (d, 2H, aromatic proton ortho to oxygen),
6.78 (d, 2H, aromatic proton meta to oxygen),
7.1-7.43 (m, 10H, aromatic protons).
………….
PATENT
WO 2001036360
https://www.google.com/patents/WO2001036360A1?cl=en
……………
PATENT
WO 2002090305
http://www.google.co.in/patents/WO2002090305A1?cl=en
EXAMPLE
a) [2-(2-chloroethoxy)ethoxymethyl]benzene
is prepared from benzyl bromide and 2-(2-chloroethoxy)ethanol by the method described in literature (Bessodes, 1996).
b) {4-[2-(2-Benzyloxyethoxy)ethoxy]phenyl}phenylmethanone

The mixture of 4-hydroxybenzophenone (16.7 g, 84.7 mmol) and 48 % aqueous sodium hydroxide solution (170 ml) is heated to 80 °C. Tetrabutylammonium bromide (TBABr) (1.6 g, 5.1 mmol) is added and the mixture is heated to 90 °C. [2-(2-Chloroethoxy)ethoxymethyl]benzene (18. g, 84.7 mmol) is added to the mixture during 15 min and the stirring is continued for additional 3.5 h at 115-120 °C. Then the mixture is cooled to 70 °C and 170 ml of water and 170 ml of toluene are added to the reaction mixture and stirring is continued for 5 min. The layers are separated and the aqueous phase is extracted twice with 50 ml of toluene. The organic phases are combined and washed with water, dried with sodium sulphate and evaporated to dryness. Yield 31.2 g.
Another method to prepare {4-[2-(2-benzyloxyethoxy)ethoxy]phenyl}phenyl- methanone is the reaction of 2-(2-benzyloxyethoxy)ethyl mesylate with 4- hydroxybenzophenone in PTC-conditions.
Η NMR (CDCI3): 3.64-3.69 (m, 2H), 3.74-3.79 (m, 2H), 3.90 (dist.t, 2H), 4.22 (dist.t, 2H), 4.58 (s, 2H), 6.98 (d, 2H), 7.28-7.62 (m, 8H), 7.75 (td, 2H), 7.81 (d, 2H).
c) 1- {4-[2-(2-Benzyloxyethoxy)ethoxy]phenyl} – 1 ,2-diphenyl -butane- 1 ,4-diol
 R = BENZYL
R = BENZYL
Lithium aluminum hydride (1.08 g, 28.6 mmol) is added into dry tetrahydrofuran (60 ml) under nitrogen atmosphere. Cinnamaldehyde (6.65 g, 50 mmol) in dry tetrahydrofuran (16 ml) is added at 24-28 °C. The reaction mixture is stirred at ambient temperature for 1 h. {4-[2-(2- Benzyloxyethoxy)ethoxy]phenyl}-phenyl-methanone (14.0 g, 37 mmol) in dry tetrahydrofuran (16 ml) is added at 50-55 °C. The reaction mixture is stirred at 60 °C for 3 h. Most of tetrahydrofuran is evaporated. Toluene (70 ml) and 2 M aqueous hydrogen chloride (50 ml) are added. The mixture is stirred for 5 min and the aqueous layer is separated and extracted with toluene (30 ml). The toluene layers are combined and washed with 2M HC1 and water, dried and evaporated. The product is crystallized from isopropanol as a mixture of stereoisomers (8.8 g, 50 %).
Η NMR (CDCI3 ): 1.75-2.10 (m, 2H), 3.20-4.16 (m, 1 OH), 4.52 and 4.55 (2s, together 2H), 6.61 and 6.88 (2d, together 2H), 6.95-7.39 (m, 15H), 7.49 and 7.57 (2d, together 2H).
d) Z- 1 – {4-[2-(2-Benzyloxyethoxy)ethoxy]phenyl} -4-chloro- 1 ,2-diphenyl-but- 1-ene
 R = BENZYL
R = BENZYL
1 – {4- [2-(2-Benzyloxy-ethoxy)ethoxy]phenyl} – 1 ,2-diphenyl -butane- 1 ,4-diol (10.0 g, 19.5 mmol) is dissolved in toluene (50 ml). Triethylamine (2.17 g, 21.4 mmol) is added to the solution and the mixture is cooled to -10 °C. Thionyl chloride (6.9 g, 58.5 mmol) is added to the mixture at -10 – ±0 °C. The mixture is stirred for 1 hour at 0-5 °C, warmed up to 70 °C and stirred at this temperature for 4 hours. Solvent is evaporated, the residue is dissolved to toluene, washed three times with 1M HC1 solution and twice with water. The Z-isomer of the product is crystallized from isopropanol-ethyl acetate. Yield 3.0 g. The filtrate is purified by flash chromatography to give E-isomer.
Z-isomer: Η NMR (CDCI3): 2.91 (t, 2H), 3.41 (t, 2H), 3.55-3.85 (m, 6H), 3.99 (dist.t, 2H), 4.54 (s, 2H), 6.40 (s, 1H), 6.56 (d, 2H), 6.77 (d, 2H), 7.10- 7.50 (m, 15H)
E-isomer: 1H NMR (CDCI3): 2.97 (t, 2H), 3.43 (t, 2H), 3.65-3.82 (m, 4H), 3.88 (dist.t, 2H), 4.15 (dist.t, 2H), 4.58 (s, 2H), 6.86 -7.45 (m, 19H)
FINAL STEP
e) 2- {2-[4-(4-Chloro- 1 ,2-diphenyl-but- 1 -enyl)phenoxy]ethoxy } ethanol:
Z- 1 – {4-[2-(2-Benzyloxy-ethoxy)ethoxy]phenyl} -4-chloro- 1 ,2-diphenyl -but- 1-ene (3.8 g, 7.4 mmol) is dissolved in ethyl acetate under nitrogen atmosphere , Zn powder (0.12 g, 1.85 mmol) and acetyl chloride (1.27 g, 16.3 mmol) are added and the mixture is stirred at 50 °C for 3 h (Bhar, 1995). The reaction mixture is cooled to room temperature, water (10 ml) is added and stirring is continued for additional 10 min. The aqueous layer is separated and the organic phase is washed with 1 M aqueous hydrogen chloride solution and with water. Ethyl acetate is evaporated and the residue is dissolved in methanol (16 ml) and water (4 ml). The acetate ester of the product is hydrolysed by making the mixture alkaline with sodium hydroxide (1 g) and stirring the mixture at room temperature for 1 h. Methanol is evaporated, water is added and the residue is extracted in ethyl acetate and washed with 1 M hydrogen chloride solution and with water. Ethyl acetate is evaporated and the residue is dissolved in toluene (25 ml), silica gel (0.25 g) is added and mixture is stirred for 15 min. Toluene is filtered and evaporated to dryness. The residue is crystallised from heptane-ethyl acetate (2:1). The yield is 71 %.
Z-isomer: 1H NMR (CDCI3): 2.92 (t, 2H), 3.41 (t, 2H), 3.58-3.63 (m, 2H), 3.69-3.80 (m, 4H), 3.96-4.01 (m, 2H), 6.56 (d, 2H), 6.78 (d, 2H), 7.10-7.40 (m, 10H).
 Z ISOMER IE FISPEMIFENE
Z ISOMER IE FISPEMIFENE
E-2- {2- [4-(4-Chloro- 1 ,2-diphenyl-but- 1 -enyl)phenoxy]ethoxy} ethanol is prepared analogously starting from E-l-{4-[2-(2-benzyloxy- ethoxy)ethoxy]phenyl} -4-chloro- 1,2-diphenyl-but-l-ene. The product is purified by flash chromatography with toluene-methanol (10:0.5) as eluent.
E-isomer: 1H NMR (CDCI3): 2.97 (t, 2H), 3.43 (t, 2H), 3.65-3.79 (m, 4H), 3.85-3.90 (m, 2H), 4.13-4.17 (m, 2H), 6.85-7.25 (m, 2H).
Debenzylation of 1 – {4-[2-(2-benzyloxy-ethoxy)ethoxy]phenyl} -4-chloro- 1 ,2- diphenyl-but- 1-ene is also carried out by hydrogenation with Pd on carbon as a catalyst in ethyl acetate-ethanol solution at room temperature.
………….
PATENT
http://www.google.com/patents/US5491173

| Patent | Submitted | Granted |
|---|---|---|
| Method for the preparation of 2-{2-[4-(4-chloro-1,2-diphenylbut-1-enyl)phenoxy]ethoxy}ethanol and its isomers [US6891070] | 2004-06-17 | 2005-05-10 |
| Formulations of fispemifene [US2007104743] | 2007-05-10 | |
| METHODS FOR THE PREPARATION OF FISPEMIFENE FROM OSPEMIFENE [US7504530] | 2008-09-04 | 2009-03-17 |
| METHOD FOR THE PREPARATION OF THERAPEUTICALLY VALUABLE TRIPHENYLBUTENE DERIVATIVES [US2011015448] | 2011-01-20 | |
| METHOD FOR THE PREPARATION OF THERAPEUTICALLY VALUABLE TRIPHENYLBUTENE DERIVATIVES [US7812197] | 2008-08-28 | 2010-10-12 |
| WO2001036360A1 | 1 Nov 2000 | 25 May 2001 | Pirkko Haerkoenen | Triphenylalkene derivatives and their use as selective estrogen receptor modulators |
| EP0095875A2 | 20 May 1983 | 7 Dec 1983 | Farmos Group Ltd. | Novel tri-phenyl alkane and alkene derivatives and their preparation and use |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2008099059A1 * | 13 Feb 2008 | 21 Aug 2008 | Hormos Medical Ltd | Method for the preparation of therapeutically valuable triphenylbutene derivatives |
| WO2008099060A2 * | 13 Feb 2008 | 21 Aug 2008 | Hormos Medical Ltd | Methods for the preparation of fispemifene from ospemifene |
| CN101636372B | 13 Feb 2008 | 27 Mar 2013 | 霍尔莫斯医疗有限公司 | Method for the preparation of therapeutically valuable triphenylbutene derivatives |
| EP1636159A1 * | 5 May 2004 | 22 Mar 2006 | Hormos Medical Ltd. | Method for the treatment or prevention of lower urinary tract symptoms |
| EP2518039A1 | 13 Feb 2008 | 31 Oct 2012 | Hormos Medical Ltd. | Method for the preparation of therapeutically valuable triphenylbutene derivatives |
| EP2821385A2 | 13 Feb 2008 | 7 Jan 2015 | Hormos Medical Ltd. | Method for the preparation of therapeutically valuable triphenylbutene derivatives |
| US7504530 | 13 Feb 2008 | 17 Mar 2009 | Hormos Medical Ltd. | Methods for the preparation of fispemifene from ospemifene |
| US7812197 | 13 Feb 2008 | 12 Oct 2010 | Hormos Medical Ltd. | Method for the preparation of therapeutically valuable triphenylbutene derivatives |
| US8293947 | 16 Sep 2010 | 23 Oct 2012 | Hormos Medical Ltd. | Method for the preparation of therapeutically valuable triphenylbutene derivatives |
| US8962693 | 19 Aug 2013 | 24 Feb 2015 | Hormos Medical Ltd. | Method for the treatment or prevention of lower urinary tract symptoms |
| WO2002090305A1 | Mar 21, 2002 | Nov 14, 2002 | Hormos Medical Corp | A new method for the preparation of 2-{2-[4-(4-chloro-1,2-diphenylbut-1-enyl)phenoxy]ethoxy}ethanol and its isomers |
| WO2004108645A1 | May 5, 2004 | Dec 16, 2004 | Hormos Medical Corp | Method for the treatment or prevention of lower urinary tract symptoms |
| WO2006024689A1 * | Jul 20, 2005 | Mar 9, 2006 | Taru Blom | Use of a selective estrogen receptor modulator for the manufacture of a pharmaceutical preparation for use in a method for the treatment or prevention of androgen deficiency |
| WO2007099410A2 * | Nov 9, 2006 | Sep 7, 2007 | Hormos Medical Ltd | Formulations of fispemifene |
| WO2014060640A1 | Oct 17, 2013 | Apr 24, 2014 | Fermion Oy | A process for the preparation of ospemifene |
| CN100526277C | May 5, 2004 | Aug 12, 2009 | 霍尔莫斯医疗有限公司 | Method for the treatment or prevention of lower urinary tract symptoms |
| CN102532073A * | Dec 30, 2011 | Jul 4, 2012 | 北京赛林泰医药技术有限公司 | Ethylene derivative serving as selective estrogen receptor modulators (SERMs) |
| EP1786408A1 * | Jul 20, 2005 | May 23, 2007 | Hormos Medical Ltd. | Use of a selective estrogen receptor modulator for the manufacture of a pharmaceutical preparation for use in a method for the treatment or prevention of androgen deficiency |
| EP1951250A2 * | Nov 22, 2006 | Aug 6, 2008 | SmithKline Beecham Corporation | Chemical compounds |
| EP2258360A2 | May 5, 2004 | Dec 8, 2010 | Hormos Medical Ltd. | Method for the treatment or prevention of lower urinary tract symptoms |
| EP2518039A1 | Feb 13, 2008 | Oct 31, 2012 | Hormos Medical Ltd. | Method for the preparation of therapeutically valuable triphenylbutene derivatives |
| EP2821385A2 | Feb 13, 2008 | Jan 7, 2015 | Hormos Medical Ltd. | Method for the preparation of therapeutically valuable triphenylbutene derivatives |
| US6891070 | Mar 21, 2002 | May 10, 2005 | Hormos Medical Corporation | Method for the preparation of 2-{2-[4-(4-chloro-1,2-diphenylbut-1-enyl)phenoxy]ethoxy}ethanol and its isomers |
| US7504530 | Feb 13, 2008 | Mar 17, 2009 | Hormos Medical Ltd. | Methods for the preparation of fispemifene from ospemifene |
| US7560589 | Jul 27, 2004 | Jul 14, 2009 | Smithkline Beecham Corporation | Cycloalkylidene compounds as modulators of estrogen receptor |
| US7569601 | May 14, 2007 | Aug 4, 2009 | Smithkline Beecham Corporation | Cycloalkylidene compounds as modulators of estrogen receptor |
| US7799828 | Jun 8, 2009 | Sep 21, 2010 | Glaxosmithkline Llc | Cycloalkylidene compounds as modulators of estrogen receptor |
| US7812197 | Feb 13, 2008 | Oct 12, 2010 | Hormos Medical Ltd. | Method for the preparation of therapeutically valuable triphenylbutene derivatives |
| US7825107 | May 22, 2007 | Nov 2, 2010 | Hormos Medical Ltd. | Method of treating men suffering from chronic nonbacterial prostatitis with SERM compounds or aromatase inhibitors |
| US8293947 | Sep 16, 2010 | Oct 23, 2012 | Hormos Medical Ltd. | Method for the preparation of therapeutically valuable triphenylbutene derivatives |
| US8299112 | Sep 15, 2011 | Oct 30, 2012 | Aragon Pharmaceuticals, Inc. | Estrogen receptor modulators and uses thereof |
| US8455534 | Sep 13, 2012 | Jun 4, 2013 | Aragon Pharmaceuticals, Inc. | Estrogen receptor modulators and uses thereof |
| US8962693 | Aug 19, 2013 | Feb 24, 2015 | Hormos Medical Ltd. | Method for the treatment or prevention of lower urinary tract symptoms |
| WO1996007402A1 * | Sep 6, 1995 | Mar 14, 1996 | Michael Degregorio | Triphenylethylenes for the prevention and treatment of osteoporosis |
| WO1996035417A1 * | May 10, 1996 | Nov 14, 1996 | Cancer Res Campaign Tech | Combinations of anti-oestrogen compounds and pkc modulators and their use in cancer therapy |
| WO1997032574A1 * | Mar 4, 1997 | Sep 12, 1997 | Degregorio Michael | Serum cholesterol lowering agent |
| WO1999042427A1 * | Feb 19, 1999 | Aug 26, 1999 | Kalapudas Arja | E-2-[4-(4-chloro-1,2-diphenyl-but-1-enyl)phenoxy]ethanol and pharmaceutical compositions thereof |
| WO1999063974A2 * | Jun 10, 1999 | Dec 16, 1999 | Endorecherche Inc | Selective estrogen receptor modulator in combination with denydroepiandrosterone (dhea) or analogues |
| EP0095875A2 * | May 20, 1983 | Dec 7, 1983 | Farmos Group Ltd. | Novel tri-phenyl alkane and alkene derivatives and their preparation and use |
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Linkedin

Join me on Facebook 
Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
 LIONEL MY SON
LIONEL MY SONHe was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
///////
Zydus Cadila Healthcare Ltd, WO 2015102017, lorcaserin












Processes for the preparation of lorcaserin
Zydus Cadila Healthcare Ltd
| Applicants: | CADILA HEALTHCARE LIMITED [IN/IN]; Zydus Tower, Satellite Cross Roads Ahmedabad – 380 015 Gujarat (IN) |
| Inventors: | DWIVEDI, Shriprakash Dhar; (IN). SHAH, Alpeshkumar Pravinchandra; (IN). GAJJAR, Samir Rameshbhai; (IN). KHERA, Brij; (IN) |

On 10 May 2012, after a new round of studies submitted by Arena, an FDA panel voted to recommend lorcaserin with certain restrictions and patient monitoring. The restrictions include patients with a BMI of over 30, or with a BMI over 27 and a comorbidity such as high blood pressure or type 2 diabetes.
On 27 June 2012, the FDA officially approved lorcaserin for use in the treatment of obesity for adults with a BMI equal to or greater than 30 or adults with a BMI of 27 or greater who “have at least one weight-related health condition, such as high blood pressure, type 2 diabetes, or high cholesterol
Useful for treating obesity.
The present invention relates to stable crystalline Form I of Iorcaserin hydrochloride of Formula (IA) and processes for its preparation. The invention also relates to processes for the preparation of lorcaserin and pharmaceutically acceptable salts, solvates and hydrates thereof.
Stable crystalline form I of lorcaserin hydrochloride and its process of preparation are claimed. Represents the first patenting from Cadila on lorcaserin, which was developed and launched by Arena Pharma and Eisai.
In July 2015, Newport Premium™ reported that Cadila is potentially interested in lorcaserin.
Lorcaserin hydrochloride is an agonist of the 5-HT2c receptor and shows effectiveness at reducing obesity in animal models and humans developed by Arena Pharmaceuticals. It is chemically represented as (R)-8-chloro-l -methyl -2,3,4,5-tetrahydro-lH-3-benzazepine hydrochloride having Formula (I) as depicted herein below.

(IA)
U.S. Patent No. 6,953,787 B2 discloses compound of Formula (I) and pharmaceutically acceptable salt, solvates or hydrates thereof and process for preparation thereof.
U.S. Patent No. 8,168,624 B2 discloses (R)-8-chloro-l-methyl-2,3,4,5-tetrahydro-lH-3-benzazepine hydrochloride hemihydrate and process for its preparation. The patent also discloses crystalline Form I, Form II and Form III of (R)-8-chloro-l-methyl-2,3,4,5-tetrahydro-lH-3-benzazepine hydrochloride. The crystalline Form
I and Form II are reported as anhydrous, non-solvated crystal forms. The crystalline Form III displays a dehydration feature calculated as a 3.7% weight loss which is consistent with the theoretical weight loss of 3.7% for a hemihydrate.
The patent discloses that anhydrous Form I and Form II readily converts to a hemihydrate, upon exposure to moisture. The dynamic vapor sorption (DVS) data for each of the three crystal forms reveals the hygroscopic nature of both Forms I and II, which readily adsorb moisture at relative humidity (RH) greater than about 40-60%. In addition, both Forms I and II were calculated to adsorb about 3.8% moisture between about 40 and about 80% RH which is consistent with conversion to the hemihydrate (Form III). X-ray powder diffraction (XRPD) carried out on both Forms I and II after the DVS cycle confirmed this conversion. In contrast, the DVS data in connection with Form III shows that it is substantially non-hygroscopic, adsorbing less than 0.5% water at 90% RH and the XRPD pattern showed no change in crystalline form after the DVS cycle.
International (PCT) Publication Nos. WO 2003/086306 Al, WO 2005/019179 Al, WO 2006/069363 Al, WO 2007/120517 Al, WO 2008/07011 1 Al and WO 2009/1 1 1004 Al disclose various synthetic approaches for the preparation of (R)-8-chloro-l-methyl-2,3,4,5-tetrahydro-lH-3-benzazepine, its related salts, enantiomers, crystalline forms and intermediates.
International (PCT) Publication No. WO 2006/071740 Al discloses combination of (R)-8-chloro-l-methyl-2,3,4,5-tetrahydro-lH-3-benzazepine with other agents. International (PCT) Publication No. WO 2012/030938 Al discloses various salts of lorcaserin with optically active acids.
U.S. PG-Pub No. US 2014/0187538 Al discloses amorphous lorcaserin hydrochloride and amorphous solid dispersion comprising lorcaserin hydrochloride and one or more pharmaceutically acceptable carriers and processes for their preparation.
International (PCT) Publication No. WO 2014/135545 Al discloses solid dispersion comprising amorphous lorcaserin hydrochloride and one or more pharmaceutically acceptable water soluble polymers.

Example-7: Preparation of crystalline Form I of lorcaserin hydrochloride. In a round bottom flask, 560g of methyl ethyl ketone and 40 ml water were taken and 100 g of 8-chloro-l-methyl-2,3,4,5-tetrahydro-lH-3-benzazepine was added and stirred for 10 minutes. The reaction mass heated to 55 to 60°C and 19.3 g of. L-(+)-tartaric acid was added slowly and stirred for one to two hours. The reaction mass was further stirred at 10-15°C for an hour and the product was filtered and washed with a mixture of methyl ethyl ketone and water. The wet cake and 150 ml methyl ethyl ketone were taken in another flask and heated to 75-80°C. 20-25 ml water was, added and stirred for an hour. Further, the reaction mass was stirred for an hour at 0-5°C. The product was filtered and washed with methyl ethyl ketone.
100 g tartrate salt of 8-chloro-l-methyl-2,3,4,5-tetrahydro-lH-3-benzazepine and 300 mL water were taken in another round bottom flask. 200 mL methylene dichloride was added and the reaction mass was cooled to 10-20°C. 17.2 g sodium hydroxide dissolved in 89 ml water was added into the reaction mass at 10-20°C. The reaction mass was stirred for an hour at 25-30°C and the layers were separated. The solvent was removed from the organic layer under vacuum and then 100 mL ethyl acetate was added into that and distilled out. Further, 100 mL ethyl acetate was added and stirred for 15 minutes. The reaction mass was filtered through a hyflow bed and the filtrate was treated with dry HC1 gas till a pH of 1.5 to 2.5 was obtained at 0-10°C and it was stirred for about 30 minutes to an hour. The product was then filtered and washed with ethyl acetate and then dried in a vacuum oven at 50°C to 55°C for 2 hours. The product was further dried at 90°C to 110°C for 20 hours to obtain crystalline Form I of lorcaserin hydrochloride. Yield: 87.5-98.6 %.
Example-8: Preparation of crystalline Form I of lorcaserin hydrochloride

In a round bottom flask, 2.20 g lorcaserin, 30 mL methylene chloride, 17.4 mL of 1M HCI in ether were added and the mixture was stirred for 5-15 minutes at room temperature. The solvent was removed under reduced pressure to give a white solid. This solid was again dissolved in 30 ml methylene chloride, 17.4 mL of 1M HCI solution and stirred for 5-15 minutes at room temperature. The solvent was removed under reduced pressure to give lorcaserin hydrochloride. The product was dried in a vacuum oven at 50°C to 55°C for 2 hours. The product was further dried at 90°C to 110°C for 20 hours to obtain crystalline Form I of lorcaserin hydrochloride.
Example-9: Preparation of crystalline Form I lorcaserin hydrochloride
50 g of lorcaserin hydrochloride hemihydrate and 50 g of hydroxypropylmethyl cellulose (HPMC) 3CPC were mixed in a blender at 25°C to 35°C. The mixture was mixed for 30 minutes and unloaded. The solid thus obtained was dried in a vacuum oven at 50°C to 55°C for 2 hours. The product was further dried at 90°C to 110°C for 20 hours to obtain crystalline Form I of lorcaserin hydrochloride.

Pankaj R. Patel (right), Chairman and Managing Director,
/////////
How flow chemistry can make processes greener.. Case study 1 Methylation with DMC.
How flow chemistry can make processes greener
Case study 1 Methylation with DMC.
Increasing reaction efficiency through access to a wider range of reaction conditions
Efficient utilization of energy and time is fundamental to green chemistry and engineering. These factors are directly related to the rate of a chemical reaction, as a fast reaction will require less operating time. Economical use of space is also important, and fast reactions may allow for a smaller reactor to be utilized, particularly in continuous processes. The most straightforward way to increase reaction rate is with an increase in temperature; however, in a batch reactor, this is generally limited to the atmospheric boiling point of the solvent or reagents. In a flow reactor, pressure and temperature can be safely manipulated far beyond atmospheric conditions. Analogous to microwaves synthesis,1 reactions done in flow are often faster than in the corresponding batch reactions, which gives…
View original post 426 more words
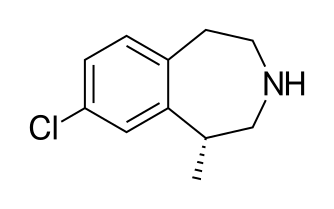
AXITINIB


Axitinib (AG013736; trade name Inlyta) is a small molecule tyrosine kinase inhibitor developed by Pfizer. It has been shown to significantly inhibit growth of breast cancer in animal (xenograft) models[2] and has shown partial responses in clinical trials with renal cell carcinoma (RCC)[3] and several other tumour types.[4] It was approved by the U.S. Food and Drug Administration after showing a modest increase in progression-free survival,[5] though there have been reports of fatal adverse effects.[6]
Axitinib, a small-molecule indazole derivative chemically known as (E)-N-methyl-2-(3-(2-(pyridin-2-yl)-vinyl)-1H-indazol-6-ylthio)benzamide developed by Pfizer, was approved in January 2012 by the U.S. FDA with the trade name Inlyta. It selectively inhibits vascular endothelial growth factor receptors for the treatment of renal cell carcinoma
On January 27, 2012, axitinib was approved with the trade name INLYTA for treatment of patients in the United States with advanced renal cell carcinoma after failure of one prior systemic therapy.
It has received FDA (27 January 2012), EMA (13 September 2012), MHRA (3 September 2012) and TGA (26 July 2012) approval for use as a treatment for renal cell carcinoma.[11][12][13][14]
A study published in 2015[15] showed that axitinib effectively inhibits a mutated gene (BCR-ABL1[T315I]) that is common in chronic myeloid leukemias and adult acute lymphoblastic leukemias which have become resistant to other tyrosine kinase inhibitors likeimatinib. This is one of the first examples of a new indication for an existing drug being discovered by screening known drugs using a patient’s own cells.

The discovery and development of an efficient synthesis route to axinitib is reported. The first-generation route researched by Pfizer implemented two Pd-catalyzed coupling reactions as key steps. In this work, the development of Heck-type and C–S coupling reactions catalyzed by CuI is briefly described, using an economial and practical protocol. Aspects of this route, such as selecting optimal ligands, solvent, and other conditions, are discussed in detail. The scale-up experiment was carried out to provide more than 300 g of active pharmaceutical ingredients of axitinib in Form XLI with 99.9% purity in 39% yield. In short, we provide a new choice of synthesis route to axitinib, through two copper-catalyzed coupling reactions with good yield.
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.5b00123
(E)-N-Methyl-2-(3-(2-(pyridin-2-yl)vinyl)-1H-indazol-6-ylthiol)benzamide (Axitinib) Form XLI (326.4 g in 96% yield with purity 99.91%). Residual Cu content was determined to be 2.2 ppm by atomic absorption spectroscopy: mp 227.7 °C;
1H NMR (300 MHz, DMSO-d6) δ 13.27 (s, 1H), 8.60 (d, J = 4.8 Hz, 1H), 8.29 (d, J = 5.4 Hz, 1H), 8.18 (d, J = 8.5 Hz, 1H), 7.94 (d, J = 16.4 Hz, 1H), 7.81 (t, J = 7.5 Hz, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.63–7.44 (m, 3H), 7.29 (p, J = 7.4, 6.6 Hz, 3H), 7.19 (d, J = 8.5 Hz, 1H), 7.08 (d, J = 7.4 Hz, 1H), 2.78 (d, J = 4.6 Hz, 3H);
13C NMR (75 MHz, DMSO-d6) δ 167.89, 154.86, 149.54, 142.01, 141.86, 136.92, 136.88, 135.67, 132.52, 130.32, 129.99, 129.25, 127.80, 126.15, 125.59, 123.66, 122.68, 122.50, 121.79, 120.29, 114.76, 26.13.
………………………..
Axitinib (Axitinib, AG-013736, CAS: 319460-85-0) is a Pfizer research and development by the United States of new, mainly targeting VEGFR kinase GABA, inhibiting angiogenesis anticancer small molecule drug, trade name Inlyta, for other systems therapy for advanced renal cell carcinoma (Renal Cell Carcinoma, RCC), 2008 has been approved in the domestic clinical, and Pfizer’s cancer drug Sutent another similar imatinib (Sunitinib) , Axitinib also potent and selective multi-targeted tyrosine kinase inhibitor, can inhibit the vascular endothelial growth factor receptor (Vascular EndothelialGrowth Factor Rec India tor, VEGFR), including VEGFl receptor, VECF2 receptors and VECF3 receptor, can inhibit platelet-derived growth factor receptor (Platelet-derived growth factor receptor, PDGFR) and c_KIT. Axitinib is called sunitinib second generation, better than sunitinib adverse reactions.
Axitinib (II) chemical name 6- [2_ (methylcarbamoyl) phenylsulfanyl] -3-E- [2_ (Batch-2-yl) ethenyl] indazole structural formula as follows:

Axitinib (II)
Assi synthesis method for Nepal mainly in the following three ways:
(I) Patent US20060094881 (Agouron Pharmaceuticals), EP2163544 (Pfizer) reported the first synthesis method Axitinib to 3,6-diiodo-indazole as a starting material, first-iodo-6-position is substituted mercapto group, protection of the NH group, then the Heck reaction occurs (pyridine-2-yl) vinyl 3-position, after deprotection Axitinib whole synthesis route is as follows:

Axitinib Scheme I
This method although the synthesis route is shorter, but the catalyst and reagents used relatively expensive and require purified through the column, the total yield is low, is not conducive to industrial production.
[0004] (2) The second method of synthesis Axitinib e.g. W00102369 (Agouron Pharmaceuticals), US6531491 (Agouron Pharmaceuticals) reported in 6-nitro-indazole as a starting material, the 3-position first iodo, followed by the protecting group NH, Suzuki coupling reaction with boronic acid to give 3- styryl styryl-position, a nitro group reduced to an amino group, an amino diazotization reaction was iodo, the 3-position of the styrene-based ozone of the obtained aldehyde, followed by Wittig reaction to give the 3-position (pyridin-2-yl) ethenyl, 6-position is substituted mercapto iodine, alkaline hydrolysis then amidated, and finally deprotection Axitinib, the entire reaction formula as follows:

Axitinib Scheme 2
The method of synthesis route is long, harsh reaction conditions, complex process, the total yield is low, does not apply to industrial production.
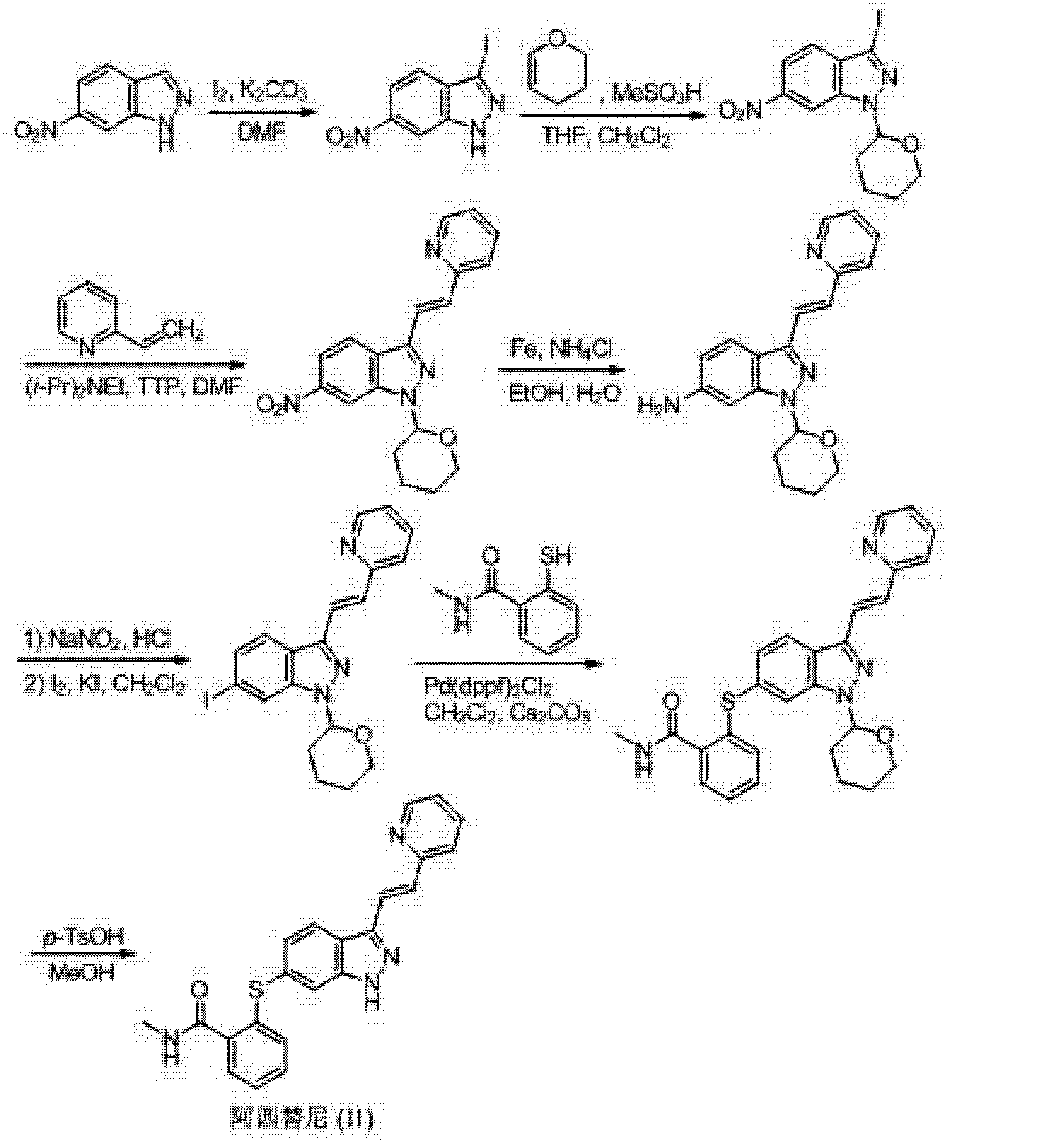
[0005] (3) The third method is W02006048745 (Pfizer) discloses to 6-nitro-indazole as a starting material, the 3-position iodo first, followed by the protecting group NH, 3- bits Heck coupling reaction, a nitro group reduced to an amino group, an amino diazotization reaction was iodo, iodo-6-position is substituted mercapto group, and finally deprotected to give Axitinib, the entire reaction is as follows:
This method has an advantage over the first two methods, it is possible to enlarge the production, but the reaction was not complete in the reaction step, will generate new impurities through the column needs to be purified.
SYNTHESIS

aReagents and conditions: (a) I2, K2CO3, DMF; (b) CH2Cl2, CH3SO3H, dihydrofuran; (c) compound B, i-Pr2EtN, Pd(OAc)2, (o-Tol)3P, DMF; (d) iron, EtOH, NH4Cl; (e) AcOH, NaNO2, CH2Cl2, I2/KI; (f) compound C, Pd(dppf)Cl2, Cs2CO3, DMF; (h) 1, p-TsOH, MeOH; 2, NaHCO3; (i) AcOH, MeOH, Pd removal, recrystallization.
http://www.google.com/patents/WO2006048745A1?cl=en
Example 15: Final deprotectioπ step to produce 6-r2-(methylcarbamoyl)phenylsulfanyll-3-E-f2- (pyridine-2-yl)ethenyllindazole

N-1 THP 6-[2-(methylcarbamoyl)phenylsulfanyl]-3-E-[2-(pyridine-2-yl)ethenyl]indazole (355 g) was suspended in 2,485 ml_ of methanol, after which p-toluenesulfonic acid monohydrate (718 g) was added. The mixture was then heated to 65 0C (hard reflux) for 4 hours under argon while the reaction was monitored by HPLC (gluco method). Heating continued until less than 1% of the N-1 THP protected starting material persisted. The heating was then removed and the reaction was cooled to room temperature. The solid was filtered and the wet cake was washed with methanol (2 volumes, 710 mL) then the solids were rinsed with ethyl acetate (2 volumes, 710 mL). The wet cake was transferred to a reactor containing sodium bicarbonate (126.84 g), deionized water (1800 mL), and ethyl acetate (975 mL), which was then stirred for 2 hours at 2O0C. The solids were filtered and washed with 5 volumes of deionized water (1800 mL), then with 2 volumes of ethyl acetate (760 mL), and then dried in a vacuum oven at 400C for 16 hours. The isolated yield for the reaction was 92.5% (274 g). The isolated material was identified as crystalline Form III free base (0.5 ethyl acetate solvate). 1H NMR, 300 MHz, (DMSO-D6), ppm; 13.35 (1 H, s), 8.60 (1 H, d, J=3.8 Hz), 8.39 (1 H, m), 8.23 (1 H, d, J=8.5 Hz), 7.95 (1 H, d, J=16.4 Hz), 7.82 (1 H, ddd, J=7.7, 7.6, 1.8 Hz), 7.67 (1 H, d, J=7.8 Hz), 7.60 (a H, s), 7.57 (1 H, d, J=16.4 Hz), 7.49 (1 H, dd, J=7.1 , 1.6 Hz), 7.35-7.26 (3 H, m), 7.19 (1 H, d, J=8.4 Hz), 7.04 (1 H, d, J=7.8 Hz), 2.77 (3 H, d, J=4.6 Hz). 13C NMR, 75 MHz, (DMSO-D6) ppm: 168.23, 155.18, 149.81 , 142.35, 142.22, 137.31 , 136.00, 132.89, 130.64, 130.36, 129.51 , 128.14, 126.50, 125.93, 124.08, 123.01 , 122.85, 122.12, 120.642, 115.08, 26.45.
Example 21 : Preparation of 6-F2-(methylcarbamovDphenylsulfanyll-3-Z-r2-(pyridine-2- vDethenyllindazole

To a 100 ml_ 3-neck flask containing a solution of 0.95 g of 6-[2- (methylcarbamoyl)phenylsulfanyl]-3-[2-(pyridine-2-yl)ethynyl]indazole was added 2.5 g of phenyliodide diacetate followed by 1.0 mL of H2NNH2 H2O. After the bubbling had settled, more phenyliodide diacetate and H2NNH2 H2O were added in small portions, until LC/MS indicated the disappearance of 6-[2-(methylcarbamoyl)phenylsulfanyl]-3-[2-(pyridine-2-yl)ethynyl]indazole and the formation of 6-[2-(methylcarbamoyl)phenylsuIfanyl]-3-Z-[2-(pyridine-2-yl)ethenyl]indazole. Example 22: Palladium removal and polymorph control of 6-[2-(methylcarbamoyl)phenylsulfanvn- 3-E-r2-(pyridine-2-vDethenyllindazole

4) MeOH, reflux
Polymorph Form IV
5) HOAc/Xylenes
To a 12 L 3-neck flask, equipped with a mechanical stirrer, was added 160.20 g of 6-[2- (methylc’arbamoyl)phenylsulfanyl]-3-E-[2-(pyridine-2-yl)ethenyl]indazole and 1.6 L of DMA and 1.6 L of THF. After stirring for 20 minutes, the mixture became homogeneous. To the clear solution was added 800.99 g of 10% cysteine-silica and the resulting mixture was allowed to stir at room temperature overnight.
The mixture was filtered through a medium sintered glass fritted funnel, and the cake was washed with a solution of 500 mL of DMA and 500 mL of THF. The cake was further washed with 2.0 L of THF and the filtrate was collected into a separate flask. The volatile parts in the latter filtrate were removed in vacuo and the residue was combined with the main filtrate. The combined filtrate was recharged back into the 12 L flask, followed by 800 g of 10% cysteine-silica. The flask was equipped with a mechanical stirrer and stirred over the weekend at room temperature. The mixture was then filtered through a medium sintered glass fritted funnel and the silica was washed with a mixture of solvents of 500 ml. of DMA and 500 ml_ of THF, followed by 3.0 L of THF. The volatile parts in the filtrate were removed in vacuo and the remaining solution was transferred to a 22 L 3-neck flask and treated with 12 L of water (added over a 20 minute period of time), a thick precipitate formed at this stage. After stirring overnight, the mixture was filtered and the cake was washed with 2.0 L of water and sucked dry.
The cake was charged to a 5 L 3-neck flask, followed by 1.6 L of THF and 160 mL of DMF. The flask was equipped with a mechanical stirrer, a reflux condenser and the mixture was heated at reflux for 8 hours. After cooling overnight, the mixture was filtered through sharkskin filter paper and sucked dry. The cake was charged to a 5 L 3-neck flask and 1.6 L of MeOH was added. The flask was equipped with a mechanical stirrer, a water condenser and the contents were heated at reflux for 6 hours. After cooling overnight, the mixture was filtered through sharkskin filter paper and sucked dry.
The cake was dissolved into 1.6 L of HOAc with the assistance of gentle heating in the water bath of a rotary evaporator. The solution was filtered through #3 filter paper and the total volume of the filtrate was reduced to ~500 mL in volume on the rotary evaporator at 60 °C/60 mmHg. At this stage, the bulk of the mixture remained a yellow solution and a small amount of precipitate formed. To the flask was charged 500 mL of xylenes (precipitate formed) and the total volume was reduced to -500 mL in volume on the rotary evaporator at 60°C/60 mmHg. The process was repeated two more times. After cooling, the mixture was filtered, the cake was washed with 500 mL of xylenes and sucked dry. The cake was transferred to a glass dish and further dried at 80°C/27 inch vacuum overnight.
The cake was off-white in color and weighed 108.38g. X-ray powder diffraction analysis indicated that a crystalline form was present, which was characterized as Form IV by a powder X- ray diffraction pattern comprising peaks at the following approximate diffraction angles (20): 8.9, 12.0, 14.6, 15.2, 15.7, 17.8, 19.2, 20.5, 21.6, 23.2, 24.2, 24.8, 26.2, and 27.5.
While the invention has been illustrated by reference to specific and preferred embodiments, those skilled in the art will recognize that variations and modifications may be made through routine experimentation and practice of the invention. Thus, the invention is intended not to be limited by the foregoing description, but to be defined by the appended claims and their equivalents.
………………………..
Chekal, B. P.; Guinness, S. M.; Lillie, B. M.; McLaughlin, R. W.; Palmer, C. W.; Post, R. J.; Sieser, J. E.; Singer, R. A.; Sluggett, G. W.; Vaidyanathan, R.; Withbroe, G. Org. Process Res. Dev. 2014, 18, 266 http://pubs.acs.org/doi/abs/10.1021/op400088k

The manufacturing process of axitinib (1) involves two Pd-catalyzed coupling reactions, a Migita coupling and a Heck reaction. Optimization of both of these pivotal bond-formation steps is discussed as well as the approach to control impurities in axitinib. Essential to the control strategy was the optimization of the Heck reaction to minimize formation of impurities, in addition to the development of an efficient isolation of crude axitinib to purge impurities.
Babu, S.; Dagnino, R., Jr.; Ouellette, M. A.; Shi, B.; Tian, Q.; Zook, S. E. PCT Int. Appl. WO/2006/048745, 2006.
…………………..

………………………………
http://www.google.com/patents/CN103570696A?cl=en

formula:

A Axitinib intermediate (1) production method, based on 6-nitro-indazole as a starting material, in the first catalyst is reacted with 3,4-dihydro -2H- pyran, bits of NH the protecting group tetrahydro -2H- pyran-2-yl, then the three iodide, to give the key intermediate in high yield 3-iodo-6-nitro-1- (tetrahydro -2H- pyrazol pyran-2-yl) -1H- indazole (I), comprising the following synthetic steps:
(1) 6-nitro-indazole dissolved in an aprotic solvent, and 3,4-dihydro -2H- pyran catalyst, 6-nitro-indazole in the catalyst and the 3,4-dihydro -2H – pyran reaction, the protecting group NH-position, was prepared to give 6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, the reaction equation is:

Wherein the 3,4-dihydro -2H- pyran an amount of 3 equivalents wide;
Aprotic solvent is acetonitrile, ethyl acetate, toluene or xylene;
The catalyst is 2,3-dichloro-5,6-dicyano-p-benzoquinone, p-toluenesulfonic acid or methanesulfonic acid;
The reaction temperature is 7 (T90 ° C, the reaction time is 1 to 4 hours;
(2) 6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole dissolved in a polar aprotic solvent, iodine was added and the acid-binding agent, an inorganic base, to afford 3- iodo-6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (I), the reaction equation is:

Wherein the polar aprotic solvent is N, N- dimethylformamide (DMF), N, N- dimethylacetamide, N, N- diethylformamide, N, N- diethyl-acetamide ;
Inorganic base acid binding agent is potassium carbonate, sodium carbonate, potassium hydroxide, sodium hydroxide, potassium bicarbonate, sodium bicarbonate, cesium carbonate, lithium hydroxide;
The reaction temperature is 2 (T40 ° C, the reaction time is 8 to 20 hours.
[0009] A Axitinib intermediate (1) in preparation for the Nepalese Asif application, based on intermediate (1) and 2-vinyl pyridine Heck coupling reaction, followed sequentially nitro reduction and the diazotization reaction of iodine, and finally with a 2-mercapto–N- methylbenzamide was prepared by deprotection docking axitinib, including the following synthetic steps:
(I) Intermediate (1) and be given 2_ vinylpyridine Jie Heck coupling reaction to give (E) _6_ nitro _3- [2_ (P than-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, the reaction equation is:

(2) (E) -6- nitro-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1Η- nitro indazole group reduction reaction, to give (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, The reaction equation is:

(3) (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1Η- indazole diazo of the iodide to give (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole The reaction equation is:

(4) (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1Η- indazole with 2- mercapto-methylbenzamide reaction -N-, to give (E) -N- methyl-2 – {[3- (2- (pyridin-2-yl) ethenyl) -1- (tetrahydro -2H- pyrazol pyran-2-yl) -1H- indazol-6-yl] thio} benzamide, the reaction equation is:

(5) (E) -N- methyl-2- {[3- (2- (pyridin-2-yl) ethenyl) -1- (tetrahydro -2H- pyran-2-yl) -1H- indazol-6-yl] thio} benzamide deprotected Axitinib (II), the reaction equation is:

Example 1
A Assi intermediates for preparing Nigeria, comprising the steps of:
Synthesis of (I) 6- nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole
A 5L reaction flask was added acetonitrile (2L), followed by addition of 6-nitro-indazole (163.1g, 1.0mol), 3, 4- dihydro -2H- pyran (168.2g, 2.0mol), 2,3- dichloro-5,6-dicyano-p-benzoquinone (22.7g, 0.1mol), was heated to 820C under reflux for 2 hours to complete the reaction, cooled to room temperature, rotary evaporated to dryness, added water and dichloromethane 2L 2L, stirring I hour, delamination, the organic phase washed with brine, dried over anhydrous sodium sulfate, filtered, and rotary evaporated to dryness, and then dissolved in acetonitrile and 2L, stirring ice-salt bath chilled to _5 ° C for 2 hours, suction filtered, the filter cake washed with a small amount of cold acetonitrile, recrystallized from ethanol, 60 ° C and dried in vacuo 12 hours to give an off-white solid, 6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, 236.3 g, yield 95.6%, m.p. 110 ~ 120 ° C, 1Η NMR (CDCl3): δ 1.30-1.83 (m, 6Η, Η3, _Η5,), 3.82-3.93 (m, 2Η, Η6 ‘), 5.86 (m , 1Η, Η2 ‘), 8.10-8.12 (m, 2Η, Η3, Η5), 8.31 (m, 1Η; Η4), 8.55 (s, 1Η, Η7);
The reaction equation is as follows:

(2) 3-iodo-6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (I),
5L reaction flask in DMF 700mL, followed by addition of 6-nitro-_1_ (tetrahydro -2H- pyran-2-yl) -1H- indazole (225.0g, 0.91mol, l.0eq) and potassium carbonate ( 251.6g, 1.82mol, 2.0eq), ice-cooled (10 ° C or less), followed by stirring, iodine (415.8g, 1.64mol, 1.8eq) was dissolved in DMF 300mL, was added dropwise to the reaction system, addition time 2 hours , the reaction system was stirred at 25 ° C for 16 hours to complete the reaction, sodium thiosulfate was added (223.0g, 1.41mol, 1.55eq) and 1.50g of potassium carbonate aqueous solution (1.5L), while maintaining the internal temperature 30 ° C Hereinafter, stirred for 30 minutes at room temperature, water was added with stirring 2L, solid precipitated, stirred for 30 minutes at room temperature, suction filtered, the filter cake was washed with water, 60 ° C and dried in vacuo 12 hours to give a pale yellow solid (Ι), 326.5g, yield 96.2%, m.p. 135 ~ 137 ° C / H NMR (DMS0_d6): δ 1.60-1.61 (m, 2H, H4,, H5 ‘), 1.73-1.76 (m, 1H, H5’), 2.01-2.04 (m, 2H, H3 ‘, H4’), 2.35-2.38 (m, 1H, H3 ‘), 3.81-3.87 (m, 2H, H6’), 6.11-6.14 (dd, 1H, H2 ‘), 7.70-7.72 (d , 1H, H4),
8.05-8.07 (dd, 1H, H5), 8.79 (s, 1H, H7).
The reaction equation is as follows:

A Axitinib intermediate (1) in the preparation for the Nepalese Asif applications, including the following synthetic steps:
Synthesis of (I) (E) -6- nitro-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole
A 5L reaction flask was added DMF (2L), followed by addition of the intermediate (1) (312.0g, 0.84mol), 2- vinylpyridine (127.5g, 1.21mol), N, N- diisopropylethylamine ( 205.3g, 1.59mol), tri-o-tolylphosphine (22.3g, 0.073mol) and palladium chloride (4.9g, 0.028mol), nitrogen, and heated to 100 ° C for 12 hours to complete the reaction, cooled to 45 ° C, isopropanol was added 1L, stirring at 45 ° C for 30 minutes, diluted with water and 5L, stirring at room temperature for I h, suction filtered, washed with water, isopropanol was added to the filter cake 1.2L, stirred at 55 ° C for 30 minutes, then stirred at room temperature for 30 minutes, suction filtered, the filter cake washed with cold isopropanol, 50 ° C and dried under vacuum for 12 hours to give (E) -6- nitro-3- [2- (pyridin-2 – yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, 275.3g, 94.0% yield, m.p. 175 ~ 176 ^, ¾ NMR (DMSO-Cl6): δ 1.63-1.64 (m, 2H, H4 ‘, H5’), 1.79-1.81 (m, 1H, H5 ‘), 2.05-2.07 (m, 2H, H3’, H4 ‘), 2.44-2.50 (m, 1H , H3 ‘), 3.86-3.90 (m, 2H, H6’), 6.15-6.18 (dd, 1H, H2 ‘), 7.30-7.33 (dd, 1H, pyridine H5), 7.65-7.69 (d, 1H, J = 16Hz, vinyl H2), 7.72-7.74 (d, 1H, pyridine H4), 7.82-7.86 (m, 1H, pyridine H3), 7.96-8.00 (d, 1H, J = 16Hz, vinyl HI), 8.07 -8.10 (dd, 1H, H4), 8.44-8.46 (d, 1H, H5), 8.63-8.64 (d, 1H, pyridine H6), 8.77-8.78 (d, 1H, H7);
The reaction equation is as follows:

Synthesis of (2) (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2Η-) -1H- indazole
5L reaction flask in ethanol HOOmLdjC 1000mL and ammonium chloride (300.0g, 5.61mol), was dissolved with stirring, followed by addition of (E) -6- nitro-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (255.0g, 0.73mol), was added iron powder (162.6g, 2.91mol), heated to 50 ° C the reaction was stirred for 2 hours to completion of the reaction, was cooled to 22 ° C, tetrahydrofuran 2L, stirred for I hour at room temperature, filtered through Celite, the filter cake washed with tetrahydrofuran and the filtrate was rotary evaporated to dryness, cooled to room temperature, water was added 2L, stirred for I hour at room temperature, pumping filtered, the filter cake washed with petroleum ether, 50 ° C and dried under vacuum for 12 hours to give a pale yellow solid 206.5g, (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1Η- indazole, yield 88.6%, m.p. 162 ~ 164 ° C / H NMR (CDCl3): δ 1.63-1.77 (m, 2H, H4 ‘, H5 ‘), 2.02-2.06 (m, 1H, H5’), 2.17-2.18 (m, 1H, H4 ‘), 2.55-2.60 (m, 1H, H3’) 3.70-3.72 (m, 2H, H3 ‘, H6 ‘), 3.91 (s, 2H, NH2), 4.04-4.07 (m, 1H, H6’), 5.57-5.60 (dd, 1H, H2 ‘), 6.64-6.66 (dd, 1H, H5), 6.74-6.75 (d, 1H, H7), 7.13-7.16 (dd, 1H, pyridine H5), 7.48-7.50 (d, 1H, pyridine H4), 7.49-7.53 (d, 1H, J = 16Hz, vinyl H2), 7.64 -7.68 (m, 1H, pyridine H3), 7.78-7.82 (d, 1H, J = 16Hz, vinyl Hl), 7.82-7.83 (d, 1H, H4), 8.60-8.61 (d, 1H, pyridine H6) ;
The reaction equation is as follows:

Synthesis of (3) (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1Η- indazole
A 5L reaction flask was added 600mL of water and sodium nitrite (70.2g, 1.02mol), stirred and dissolved, and cooled to (TC, (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl ] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (200.0g, 0.62mol) was dissolved in glacial acetic acid 1.3L, dropwise added to the system dropwise over I h, a solution process maintain an internal temperature of 0 ° C, the same temperature for I hour, dropping HCl solution (concentrated hydrochloric acid 112mL, water 200mL) at O ° C, the dropping time of 10 minutes, with the temperature for I h, TLC plate tracking point diazonium salt formation reaction (PE: EA = 1: 1). dropwise 800mL dichloromethane between 0 ° C, the dropping time of 5 minutes, potassium iodide (207.3g, l.25mol) and iodine (79.2g, 0.31mol) was dissolved water 600mL, in (TC dropwise added to the system at the same temperature for 2 hours to complete the reaction. The reaction mixture was poured into the system to 20% sodium thiosulfate solution (2L) and dichloromethane SOOmL and stirred, layered , the aqueous phase was extracted with dichloromethane frozen (2x800mL), dichloromethane phases were combined burning, 3M sodium hydroxide solution was added dropwise 3.5L, adjust the aqueous phase pH = 9 ~ 12, and water was added ammonia 200mL 400mL, stirred for 30 minutes , separated and the aqueous phase was extracted with dichloromethane (2×1.2L), the organic phases were combined, rotary evaporated to dryness, and purified through silica gel to give (E) -6- iodo-3- [2- (pyridin-2-yl ) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, 176.0g, 65.4% yield, m.p. 142 ~ 143 ° C, 1H NMR (DMS0_d6): δ 1.58- 1.61 (m, 2H, H4 ‘, H5,) 1.72-1.78 (m, 1H, H5,), 1.97-2.04 (m, 2H, H3,, H4,), 2.38-2.44 (m, 1H, H3,) , 3.79-3.81 (m, 1H, H6,), 3.88-3.90 (m, 1H, H6,), 5.91-5.94 (dd, 1H, H2,), 7.29-7.31 (m, 1H, pyridine H5), 7.56 -7.60 (d, 1H ,, J = 16Hz, vinyl H2), 7.57-7.59 (m, 1H, pyridine H4), 7.69-7.71 (d, 1H, pyridine H3), 7.80-7.84 (m, 1H, H4 ), 7.89-7.93 (d, 1H, J = 16Hz, vinyl HI), 8.01-8.03 (d, 1H, H5), 8.25 (s, 1H, H7), 8.61-8.62 (d, 1H, pyridine H6) ;
The reaction equation is as follows:

(4) (E) -N- methyl-2 – {[3- (2- (pyridin-2-yl) ethenyl) _1_ (tetrahydro -2H- pyran-2-yl) -1H- indazole 6-ylthio} benzamide]
A 5L reaction flask was added DMF (1750mL) and (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1H- indazole (175.0g, 0.41mol), nitrogen, was added [1, I, – bis (diphenylphosphino) ferrocene] dichloropalladium dichloromethane complex (14.9g, 0.018mmol ), cesium carbonate (198.3g, 0.61mol) and dichloromethane 20mL, was added 2-mercapto -N- methylbenzamide (84.9g, 0.5Imol), heated to 80 ° C for 16 hours to complete the reaction, spin distilled was removed DMF, cooled to room temperature, ethyl acetate was added 3L, water 4L, stirred for 40 minutes, the organic phase was separated, washed with brine, layered, dried over sodium sulfate, filtered, and rotary evaporated to dryness, to give (E) -N- methyl-2 – {[3- (2- (pyridin-2-yl) ethenyl) -1- (tetrahydro -2H- pyran-2-yl) -1H- indazol-6-yl] thio } benzamide, 165.6g, a yield of 86.7%, the melting point of 142 ~ 143 ° C;
The reaction equation is as follows:

(5) Synthesis of axitinib
In a 2L reaction flask was added (E) -N- methyl-2 – {[3- (2- (pyridin-2-yl) ethenyl) _1_ (tetrahydro -2H- pyran-2-yl) -1H – indazol-6-yl] thio} benzamide (150.0g, 0.32mol), p-toluenesulfonic acid monohydrate (303.2g, 1.59mol), methanol (800mL) and water (150mL), nitrogen, heated to 65 ° C for 4 hours, spin evaporated to dryness and ethanol (800mL), 65 ° C was stirred for I hour, the ethanol was removed by rotary evaporation, then repeated three times, TLC spot plate tracking reaction (petroleum ether: ethyl acetate = 1: 1). Completion of the reaction, cooled to room temperature, rotary evaporated to dryness, water was added 500mL, stirred for I h, filtered, and the filter cake was washed with methanol and ice, and then added to the reaction vessel, ethyl acetate was added 450mL, stirred at 65 ° C 30 minutes. cooled to room temperature, suction filtered, the filter cake washed with ethyl acetate and freeze paint, water paint, 50 ° C and dried under vacuum for 12 hours to give a white solid 117.5g, Axitinib (II), yield 95.4%, HPLC purity 98.8 % / H NMR (DMS0_d6): δ 2.78 (d, 3H, CH3), 7.05 (dd, 1H), 7.19 (dd, 1H), 7.36-7.23 (m, 3H), 7.50 (dd, 1H), 7.58 ( d, 1H), 7.61 (s, 1H), 7.66 (d, 1H), 7.85-7.76 (m, 1H), 7.96 (d, 1H, J = 16Hz), 8.21 (d, 1H), 8.39 (q, 1H), 8.61 (d, 1H), 13.35 (s, 1H).
The reaction equation is as follows:

Example 2
A Assi intermediates for preparing Nigeria, comprising the steps of:
Synthesis of (1) 6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole
A 5L reaction flask was added ethyl acetate (2L), followed by addition of 6-nitro-indazole (163.14g, 1.0mol), 3, 4- dihydro -2H- pyran (210.3g, 2.5mol), toluene acid (20.7g, 0.12mol), heated to 78 ° C under reflux for 3 hours to complete the reaction, cooled to room temperature, rotary evaporated to dryness, added water and dichloromethane 2L 2L, stirred for I hour, stratification, the organic phase was washed with brine, dried over anhydrous sodium sulfate, filtered, and rotary evaporated to dryness, and then dissolved in acetonitrile and 2L, stirring ice-salt bath chilled to _5 ° C for 2 hours, suction filtered, the filter cake washed with a small amount of cold acetonitrile, recrystallized from ethanol , 60 ° C and dried in vacuo 12 hours to give an off-white solid 223.3g, 6- nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, yield 90.3%, m.p. 110 ^ 11 TC;
The reaction equation is as follows:

(2) 3-iodo-6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (I),
5L reaction flask in DMF 700mL, followed by addition of 6-nitro-_1_ (tetrahydro -2H- pyran-2-yl) -1H- indazole (200.0g, 0.81mol, l.0eq) and sodium hydroxide (64.7g, 1.62mol, 2.0eq), ice-cooled (10 ° C or less), followed by stirring, iodine (369.6g, 1.46mol, 1.8eq) was dissolved in DMF 300mL, was added dropwise to the reaction system, addition time 2 hours, the reaction system was stirred at 25 ° C for 12 hours to complete the reaction, sodium thiosulfate was added (198.2g, 1.25mol, 1.55eq) and 1.50g of potassium carbonate aqueous solution (1.5L), while maintaining the temperature of 30 ° C or less, and stirred for 30 minutes at room temperature, water was added with stirring 2L, solid precipitated, stirred for 30 minutes at room temperature, suction filtered, the filter cake was washed with water, 60 ° C and dried in vacuo 12 hours to give a pale yellow solid
(1), 294.3g, 97.5% yield, m.p. 136 ~ 137. . .
[0014] The reaction equation is as follows:

A Axitinib intermediate (1) in the preparation for the Nepalese Asif applications, including the following synthetic steps:
Synthesis (1) (E) -6- nitro-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2Η-) -1H- indazole
A 5L reaction flask was added DMF (2L), followed by addition of the intermediate (1) (312.0g, 0.84mol), 2- vinylpyridine (127.5g, 1.21mol), N, N- diisopropylethylamine ( 205.3g, 1.59mol), tri-o-tolylphosphine (22.3g, 0.073mol) and palladium chloride (4.9g, 0.028mol), nitrogen, and heated to 100 ° C for 12 hours to complete the reaction, cooled to 45 ° C, isopropanol was added 1L, stirring at 45 ° C for 30 minutes, diluted with water and 5L, stirring at room temperature for I h, suction filtered, washed with water, isopropanol was added to the filter cake 1.2L, stirred at 55 ° C for 30 minutes, then stirred at room temperature for 30 minutes, suction filtered, the filter cake washed with cold isopropanol, 50 ° C and dried under vacuum for 12 hours to give (E) -6- nitro-3- [2- (pyridin _2 _-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, 275.3g, 94.0% yield, m.p. 175 ~ 176 ^, ¾ NMR (DMSO-Cl6): δ 1.63-1.64 (m, 2H, H4 ‘, H5’), 1.79-1.81 (m, 1H, H5 ‘), 2.05-2.07 (m, 2H, H3’, H4 ‘), 2.44-2.50 (m, 1H , H3 ‘), 3.86-3.90 (m, 2H, H6’), 6.15-6.18 (dd, 1H, H2 ‘), 7.30-7.33 (dd, 1H, pyridine H5), 7.65-7.69 (d, 1H, J = 16Hz, vinyl H2), 7.72-7.74 (d, 1H, pyridine H4), 7.82-7.86 (m, 1H, pyridine H3), 7.96-8.00 (d, 1H, J = 16Hz, vinyl HI), 8.07 -8.10 (dd, 1H, H4), 8.44-8.46 (d, 1H, H5), 8.63-8.64 (d, 1H, pyridine H6), 8.77-8.78 (d, 1H, H7);
The reaction equation is as follows:

Synthesis of (2) (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole
5L reaction flask in ethanol HOOmLdjC 1000mL and ammonium chloride (300.0g, 5.61mol), was dissolved with stirring, followed by addition of (E) -6- nitro-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (255.0g, 0.73mol), was added iron powder (162.6g, 2.91mol), heated to 50 ° C the reaction was stirred for 2 hours to completion of the reaction, was cooled to 22 ° C, tetrahydrofuran 2L, stirred for I hour at room temperature, filtered through Celite, the filter cake washed with tetrahydrofuran and the filtrate was rotary evaporated to dryness, cooled to room temperature, water was added 2L, stirred for I hour at room temperature, pumping filtered, the filter cake washed with petroleum ether, 50 ° C and dried under vacuum for 12 hours to give a pale yellow solid 206.5g, (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1Η- indazole, yield 88.6%, m.p. 162 ~ 164 ° C / H NMR (CDCl3): δ 1.63-1.77 (m, 2H, H4 ‘, H5 ‘), 2.02-2.06 (m, 1H, H5’), 2.17-2.18 (m, 1H, H4 ‘), 2.55-2.60 (m, 1H, H3’) 3.70-3.72 (m, 2H, H3 ‘, H6 ‘), 3.91 (s, 2H, NH2), 4.04-4.07 (m, 1H, H6’), 5.57-5.60 (dd, 1H, H2 ‘), 6.64-6.66 (dd, 1H, H5), 6.74-6.75 (d, 1H, H7), 7.13-7.16 (dd, 1H, pyridine H5), 7.48-7.50 (d, 1H, pyridine H4), 7.49-7.53 (d, 1H, J = 16Hz, vinyl H2), 7.64 -7.68 (m, 1H, pyridine H3), 7.78-7.82 (d, 1H, J = 16Hz, vinyl Hl), 7.82-7.83 (d, 1H, H4), 8.60-8.61 (d, 1H, pyridine H6) ;
The reaction equation is as follows:

Synthesis of (3) (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1Η- indazole
A 5L reaction flask was added 600mL of water and sodium nitrite (70.2g, 1.02mol), stirred and dissolved, and cooled to (TC, (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl ] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (200.0g, 0.62mol) was dissolved in glacial acetic acid 1.3L, dropwise added to the system dropwise over I h, a solution process maintain an internal temperature of 0 ° C, the same temperature for I hour, dropping HCl solution (concentrated hydrochloric acid 112mL, water 200mL) at O ° C, the dropping time of 10 minutes, with the temperature for I h, TLC plate tracking point diazonium salt formation reaction (PE: EA = 1: 1). dropwise 800mL dichloromethane between 0 ° C, the dropping time of 5 minutes, potassium iodide (207.3g, l.25mol) and iodine (79.2g, 0.31mol) was dissolved water 600mL, in (TC dropwise added to the system at the same temperature for 2 hours to complete the reaction. The reaction mixture was poured into the system to 20% sodium thiosulfate solution (2L) and dichloromethane SOOmL and stirred, layered , the aqueous phase was extracted with dichloromethane frozen (2x800mL), dichloromethane phases were combined burning, 3M sodium hydroxide solution was added dropwise 3.5L, adjust the aqueous phase pH = 9 ~ 12, and water was added ammonia 200mL 400mL, stirred for 30 minutes , separated and the aqueous phase was extracted with dichloromethane (2×1.2L), the organic phases were combined, rotary evaporated to dryness, and purified through silica gel to give (E) -6- iodo-3- [2- (pyridin-2-yl ) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, 176.0g, 65.4% yield, m.p. 142 ~ 143 ° C, 1H NMR (DMS0_d6): δ 1.58- 1.61 (m, 2H, H4 ‘, H5,) 1.72-1.78 (m, 1H, H5,), 1.97-2.04 (m, 2H, H3,, H4,), 2.38-2.44 (m, 1H, H3,) , 3.79-3.81 (m, 1H, H6,), 3.88-3.90 (m, 1H, H6,), 5.91-5.94 (dd, 1H, H2,), 7.29-7.31 (m, 1H, pyridine H5), 7.56 -7.60 (d, 1H ,, J = 16Hz, vinyl H2), 7.57-7.59 (m, 1H, pyridine H4), 7.69-7.71 (d, 1H, pyridine H3), 7.80-7.84 (m, 1H, H4 ), 7.89-7.93 (d, 1H, J = 16Hz, vinyl HI), 8.01-8.03 (d, 1H, H5), 8.25 (s, 1H, H7), 8.61-8.62 (d, 1H, pyridine H6) ;
The reaction equation is as follows:

(4) (E) -N- methyl-2 – {[3- (2- (pyridin-2-yl) ethenyl) _1_ (tetrahydro -2H- pyran-2-yl) -1H- indazole 6-ylthio} benzamide]
A 5L reaction flask was added DMF (1750mL) and (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1H- indazole (175.0g, 0.41mol), nitrogen, was added [1, I, – bis (diphenylphosphino) ferrocene] dichloropalladium dichloromethane complex (14.9g, 0.018mmol ), cesium carbonate (198.3g, 0.61mol) and dichloromethane 20mL, was added 2-mercapto -N- methylbenzamide (84.9g, 0.5Imol), heated to 80 ° C for 16 hours to complete the reaction, spin distilled was removed DMF, cooled to room temperature, ethyl acetate was added 3L, water 4L, stirred for 40 minutes, the organic phase was separated, washed with brine, layered, dried over sodium sulfate, filtered, and rotary evaporated to dryness, to give (E) -N- methyl-2 – {[3- (2- (pyridin-2-yl) ethenyl) -1- (tetrahydro -2H- pyran-2-yl) -1H- indazol-6-yl] thio } benzamide, 165.6g, a yield of 86.7%, the melting point of 142 ~ 143 ° C;
The reaction equation is as follows:

(5) Synthesis of axitinib
In a 2L reaction flask was added (E) -N- methyl-2 – {[3- (2- (pyridin-2-yl) ethenyl) _1_ (tetrahydro -2H- pyran-2-yl) -1H – indazol-6-yl] thio} benzamide (150.0g, 0.32mol), p-toluenesulfonic acid monohydrate (303.2g, 1.59mol), methanol (800mL) and water (150mL), nitrogen, heated to 65 ° C for 4 hours, spin evaporated to dryness and ethanol (800mL), 65 ° C was stirred for I hour, the ethanol was removed by rotary evaporation, then repeated three times, TLC spot plate tracking reaction (petroleum ether: ethyl acetate = 1: 1). Completion of the reaction, cooled to room temperature, rotary evaporated to dryness, water was added 500mL, stirred for I h, filtered, and the filter cake was washed with methanol and ice, and then added to the reaction vessel, ethyl acetate was added 450mL, stirred at 65 ° C 30 minutes. cooled to room temperature, suction filtered, the filter cake washed with ethyl acetate and freeze paint, water paint, 50 ° C and dried under vacuum for 12 hours to give a white solid 117.5g, Axitinib (II), yield 95.4%, HPLC purity 98.8 % / H NMR (DMS0_d6): δ 2.78 (d, 3H, CH3), 7.05 (dd, 1H), 7.19 (dd, 1H), 7.36-7.23 (m, 3H), 7.50 (dd, 1H), 7.58 ( d, 1H), 7.61 (s, 1H), 7.66 (d, 1H), 7.85-7.76 (m, 1H), 7.96 (d, 1H, J = 16Hz), 8.21 (d, 1H), 8.39 (q, 1H), 8.61 (d, 1H), 13.35 (s, 1H).
The reaction equation is as follows:

Example 3
A Assi intermediates for preparing Nigeria, comprising the steps of:
Synthesis of (1) 6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole
5L reaction flask in toluene (2L), followed by addition of 6-nitro-indazole (163.lg, 1.0mol), 3,4- dihydro -2H- pyran (193.5g, 2.3mol), methanesulfonic acid (14.4g, 0.15mol), heated to 85 ° C under reflux for 3.5 hours, to complete the reaction, cooled to room temperature, rotary evaporated to dryness, added water and dichloromethane 2L 2L, stirred for I hour, stratification, the organic phase was washed with brine wash, dried over anhydrous sodium sulfate, filtered, and rotary evaporated to dryness, and then dissolved in acetonitrile and 2L, stirring ice-salt bath chilled to _5 ° C for 2 hours, suction filtered, the filter cake washed with a small amount of cold acetonitrile and paint, and recrystallized from ethanol , 60 ° C and dried in vacuo 12 hours to give an off-white solid, 6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, 234.4g, 94.8% yield, m.p. 111 ~ 112.. ;
The reaction equation is as follows:

(2) 3-iodo-6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (I),
5L reaction flask in DMF 700mL, followed by addition of 6-nitro-_1_ (tetrahydro -2H- pyran-2-yl) -1H- indazole (225.0g, 0.91mol, 1.0eq) and potassium hydroxide ( 102.lg, 1.82mol, 2.0eq), ice-cooled below 10 ° C, with stirring, iodine (415.8g, 1.64mol, 1.8eq) was dissolved in DMF 300mL, was added dropwise to the reaction system dropwise over 2 hours, The reaction system was stirred at 30 ° C for 10 hours to complete the reaction, sodium thiosulfate was added (223.0g, 1.41mol, 1.55eq) and 1.50g of potassium carbonate aqueous solution (1.5L), while maintaining the internal temperature below 30 ° C , stirred for 45 minutes at room temperature, water was added with stirring 2L, solid precipitated, stirred for 45 minutes at room temperature, suction filtered, the filter cake was washed with water, 60 ° C and dried in vacuo 12 hours to give a pale yellow solid
(1), 317.2g, 93.4% yield, m.p. 135 ~ 136 ° C.
The reaction equation is as follows:

A Axitinib intermediate (1) in the preparation for the Nepalese Asif applications, including the following synthetic steps:
Synthesis (1) (E) -6- nitro-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole
A 5L reaction flask was added DMF (2L), followed by addition of the intermediate (1) (312.0g, 0.84mol), 2- vinylpyridine (127.5g, 1.21mol), N, N- diisopropylethylamine ( 205.3g, 1.59mol), tri-o-tolylphosphine (22.3g, 0.073mol) and palladium chloride (4.9g, 0.028mol), nitrogen, and heated to 100 ° C for 12 hours to complete the reaction, cooled to 45 ° C, isopropanol was added 1L, stirring at 45 ° C for 30 minutes, diluted with water and 5L, stirring at room temperature for I h, suction filtered, washed with water, isopropanol was added to the filter cake 1.2L, stirred at 55 ° C for 30 minutes, then stirred at room temperature for 30 minutes, suction filtered, the filter cake washed with cold isopropanol, 50 ° C and dried under vacuum for 12 hours to give (E) -6- nitro-3- [2- (pyridin _2 _-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, 275.3g, 94.0% yield, m.p. 175 ~ 176 ^, ¾ NMR (DMSO-Cl6): δ 1.63-1.64 (m, 2H, H4 ‘, H5’), 1.79-1.81 (m, 1H, H5 ‘), 2.05-2.07 (m, 2H, H3’, H4 ‘), 2.44-2.50 (m, 1H , H3 ‘), 3.86-3.90 (m, 2H, H6’), 6.15-6.18 (dd, 1H, H2 ‘), 7.30-7.33 (dd, 1H, pyridine H5), 7.65-7.69 (d, 1H, J = 16Hz, vinyl H2), 7.72-7.74 (d, 1H, pyridine H4), 7.82-7.86 (m, 1H, pyridine H3), 7.96-8.00 (d, 1H, J = 16Hz, vinyl HI), 8.07 -8.10 (dd, 1H, H4), 8.44-8.46 (d, 1H, H5), 8.63-8.64 (d, 1H, pyridine H6), 8.77-8.78 (d, 1H, H7);
The reaction equation is as follows:

Synthesis of (2) (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole
5L reaction flask in ethanol HOOmLdjC 1000mL and ammonium chloride (300.0g, 5.61mol), was dissolved with stirring, followed by addition of (E) -6- nitro-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (255.0g, 0.73mol), was added iron powder (162.6g, 2.91mol), heated to 50 ° C the reaction was stirred for 2 hours to completion of the reaction, was cooled to 22 ° C, tetrahydrofuran 2L, stirred for I hour at room temperature, filtered through Celite, the filter cake washed with tetrahydrofuran and the filtrate was rotary evaporated to dryness, cooled to room temperature, water was added 2L, stirred for I hour at room temperature, pumping filtered, the filter cake washed with petroleum ether, 50 ° C and dried under vacuum for 12 hours to give a pale yellow solid 206.5g, (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1Η- indazole, yield 88.6%, m.p. 162 ~ 164 ° C / H NMR (CDCl3): δ 1.63-1.77 (m, 2H, H4 ‘, H5 ‘), 2.02-2.06 (m, 1H, H5’), 2.17-2.18 (m, 1H, H4 ‘), 2.55-2.60 (m, 1H, H3’) 3.70-3.72 (m, 2H, H3 ‘, H6 ‘), 3.91 (s, 2H, NH2), 4.04-4.07 (m, 1H, H6’), 5.57-5.60 (dd, 1H, H2 ‘), 6.64-6.66 (dd, 1H, H5), 6.74-6.75 (d, 1H, H7), 7.13-7.16 (dd, 1H, pyridine H5), 7.48-7.50 (d, 1H, pyridine H4), 7.49-7.53 (d, 1H, J = 16Hz, vinyl H2), 7.64 -7.68 (m, 1H, pyridine H3), 7.78-7.82 (d, 1H, J = 16Hz, vinyl Hl), 7.82-7.83 (d, 1H, H4), 8.60-8.61 (d, 1H, pyridine H6) ;
The reaction equation is as follows:

Synthesis of (3) (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1Η- indazole
A 5L reaction flask was added 600mL of water and sodium nitrite (70.2g, 1.02mol), stirred and dissolved, and cooled to (TC, (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl ] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (200.0g,
0.62mol) was dissolved in glacial acetic acid 1.3L, dropwise added to the system dropwise over I hour, added dropwise to maintain the internal temperature process 0 ° C, the same temperature for I h, HCl solution was added dropwise at O ° C (112mL of concentrated hydrochloric acid , water 200mL), was added dropwise for 10 minutes, with the temperature for I h, TLC plate tracking point diazonium salt formation reaction (PE: EA = 1: 1). Solution of methylene chloride at 0 ° C and 800mL, dropping time of 5 minutes, potassium iodide (207.3g, l.25mol) and iodine (79.2g, 0.31mol) dissolved in water 600mL, at (TC dropwise added to the system, same temperature for 2 hours to complete the reaction. The reaction system was poured into a mixture of 20% sodium thiosulfate solution (2L) and dichloromethane SOOmL and stirred, layers were separated, the aqueous phase was extracted with dichloromethane frozen (2x800mL ), methylene chloride phases were combined burning, 3M sodium hydroxide solution was added dropwise 3.5L, adjust the aqueous phase pH = 9 ~ 12, and water was added ammonia 200mL 400mL, stirred for 30 minutes, separated and the aqueous phase extracted with dichloromethane ( 2×1.2L), the organic phases were combined, rotary evaporated to dryness, and purified through silica gel to give (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H – pyran-2-yl) -1H- indazole, 176.0g, 65.4% yield, m.p. 142 ~ 143 ° C, 1H NMR (DMS0_d6): δ 1.58-1.61 (m, 2H, H4 ‘, H5,) 1.72-1.78 (m, 1H, H5,), 1.97-2.04 (m, 2H, H3,, H4,), 2.38-2.44 (m, 1H, H3,), 3.79-3.81 (m, 1H, H6,) , 3.88-3.90 (m, 1H, H6,), 5.91-5.94 (dd, 1H, H2,),
7.29-7.31 (m, 1H, pyridine H5), 7.56-7.60 (d, 1H ,, J = 16Hz, vinyl H2), 7.57-7.59 (m, 1H, pyridine H4), 7.69-7.71 (d, 1H, pyridine H3), 7.80-7.84 (m, 1H, H4), 7.89-7.93 (d, 1H, J = 16Hz, vinyl HI), 8.01-8.03 (d, 1H, H5), 8.25 (s, 1H, H7 ), 8.61-8.62 (d, 1H, pyridine H6); reaction equation is as follows:

(4) (E) -N- methyl-2 – {[3- (2- (pyridin-2-yl) ethenyl) _1_ (tetrahydro -2H- pyran-2-yl) -1H- indazole 6-ylthio} benzamide]
A 5L reaction flask was added DMF (1750mL) and (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1H- indazole (175.0g, 0.41mol), nitrogen, was added [1, I, – bis (diphenylphosphino) ferrocene] dichloropalladium dichloromethane complex (14.9g, 0.018mmol ), cesium carbonate (198.3g, 0.61mol) and dichloromethane 20mL, was added 2-mercapto -N- methylbenzamide (84.9g, 0.5Imol), heated to 80 ° C for 16 hours to complete the reaction, spin distilled was removed DMF, cooled to room temperature, ethyl acetate was added 3L, water 4L, stirred for 40 minutes, the organic phase was separated, washed with brine, layered, dried over sodium sulfate, filtered, and rotary evaporated to dryness, to give (E) -N- methyl-2 – {[3- (2- (pyridin-2-yl) ethenyl) -1- (tetrahydro -2H- pyran-2-yl) -1H- indazol-6-yl] thio } benzamide, 165.6g, a yield of 86.7%, the melting point of 142 ~ 143 ° C;
The reaction equation is as follows:
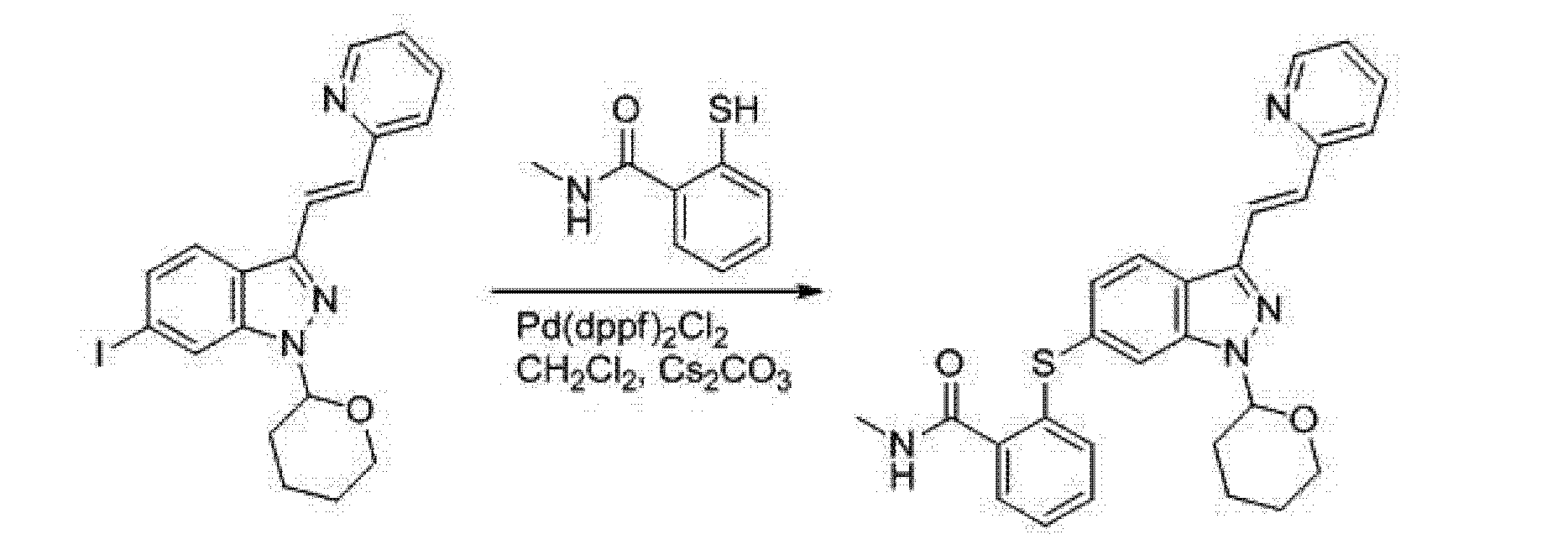
(5) Synthesis of axitinib
In a 2L reaction flask was added (E) -N- methyl-2 – {[3- (2- (pyridin-2-yl) ethenyl) _1_ (tetrahydro -2H- pyran-2-yl) -1H – indazol-6-yl] thio} benzamide (150.0g, 0.32mol), p-toluenesulfonic acid monohydrate (303.2g, 1.59mol), methanol (800mL) and water (150mL), nitrogen, heated to 65 ° C for 4 hours, spin evaporated to dryness and ethanol (800mL), 65 ° C was stirred for I hour, the ethanol was removed by rotary evaporation, then repeated three times, TLC spot plate tracking reaction (petroleum ether: ethyl acetate = 1: 1). Completion of the reaction, cooled to room temperature, rotary evaporated to dryness, water was added 500mL, stirred for I h, filtered, and the filter cake was washed with methanol and ice, and then added to the reaction vessel, ethyl acetate was added 450mL, stirred at 65 ° C 30 minutes. cooled to room temperature, suction filtered, the filter cake washed with ethyl acetate and freeze paint, water paint, 50 ° C and dried under vacuum for 12 hours to give a white solid 117.5g, Axitinib (II),
yield 95.4%, HPLC purity 98.8 % / H NMR (DMS0_d6): δ 2.78 (d, 3H, CH3), 7.05 (dd, 1H), 7.19 (dd, 1H), 7.36-7.23 (m, 3H), 7.50 (dd, 1H), 7.58 ( d, 1H), 7.61 (s, 1H), 7.66 (d, 1H), 7.85-7.76 (m, 1H), 7.96 (d, 1H, J = 16Hz), 8.21 (d, 1H), 8.39 (q, 1H), 8.61 (d, 1H), 13.35 (s, 1H).
The reaction equation is as follows:

…………………….

………………………

SEE NMR……….
http://orgspectroscopyint.blogspot.in/2015/07/axitinib.html
………..
NMR source apexbt
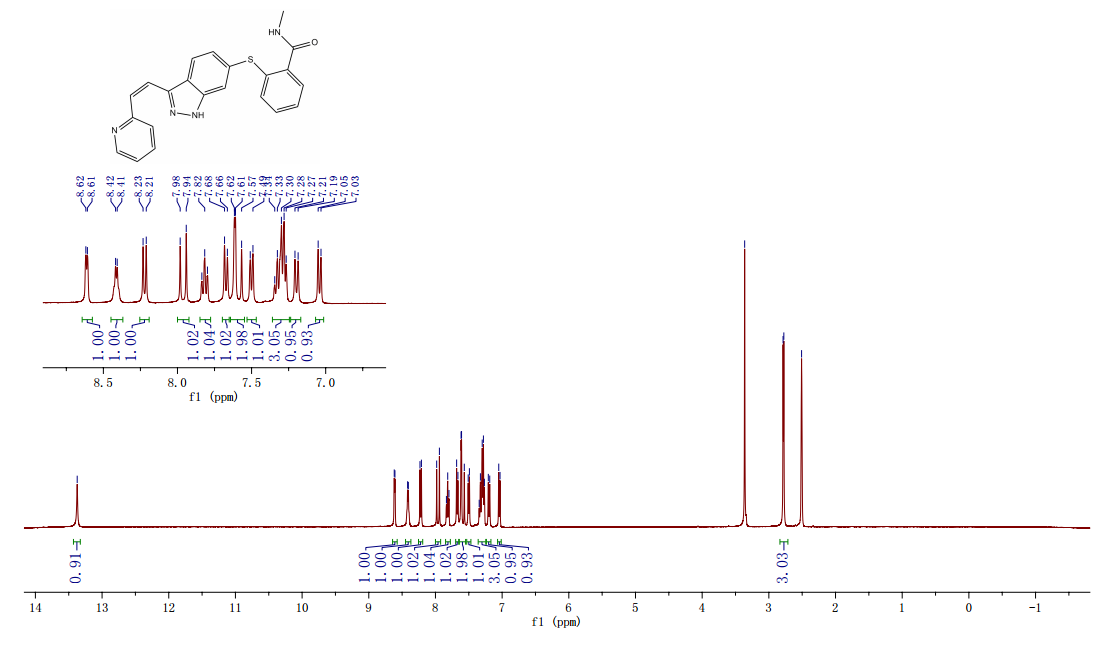

http://dmd.aspetjournals.org/content/suppl/2014/03/07/dmd.113.056531.DC1/Supplemental__Data_Figures_56531.pdf
NMR..SEE……..http://www.abmole.com/download/axitinib-hnmr.pdf

MASS

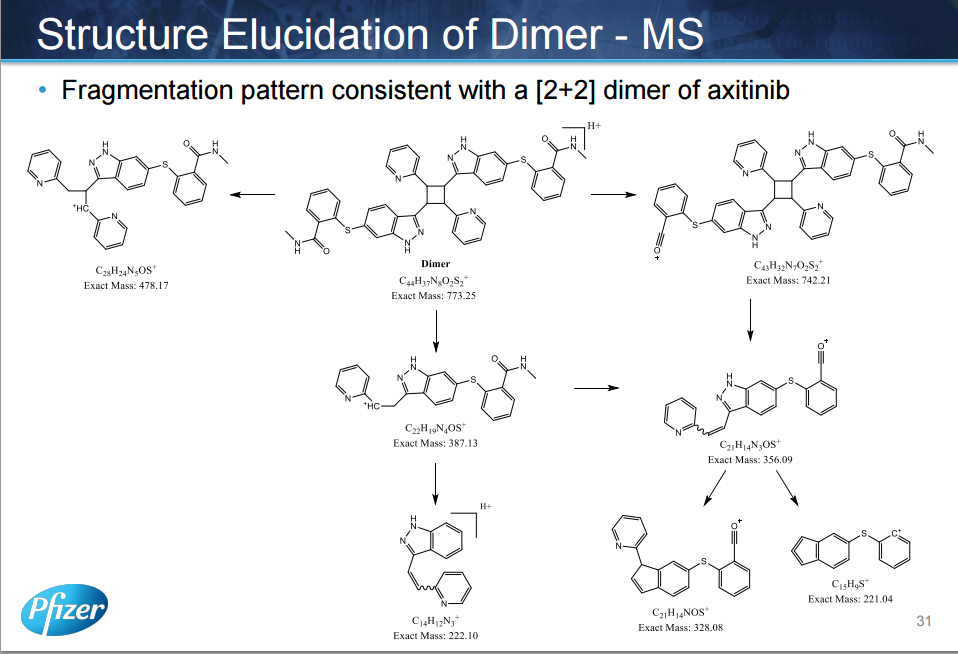

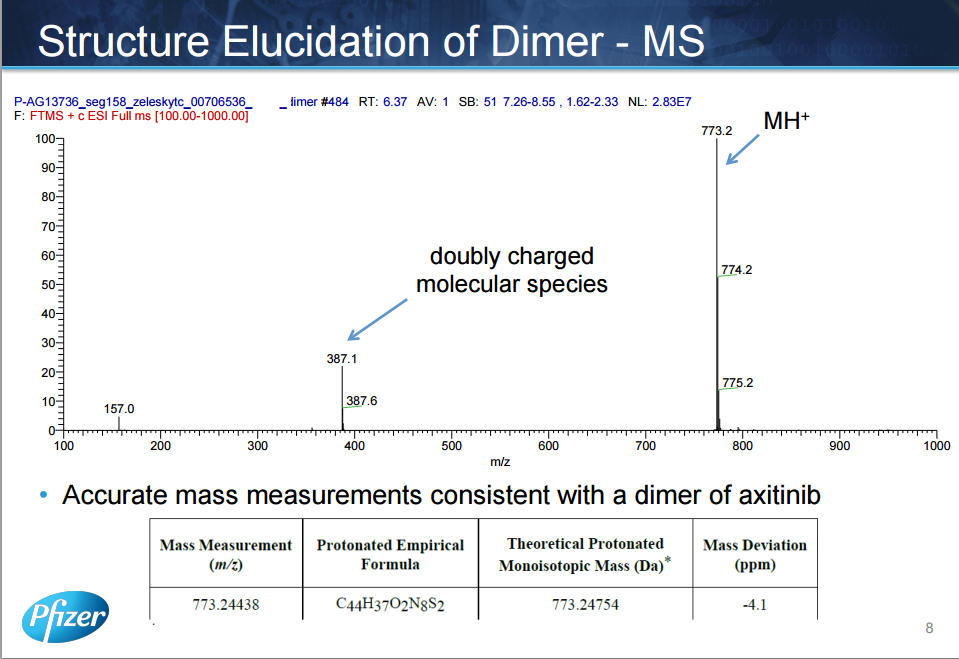

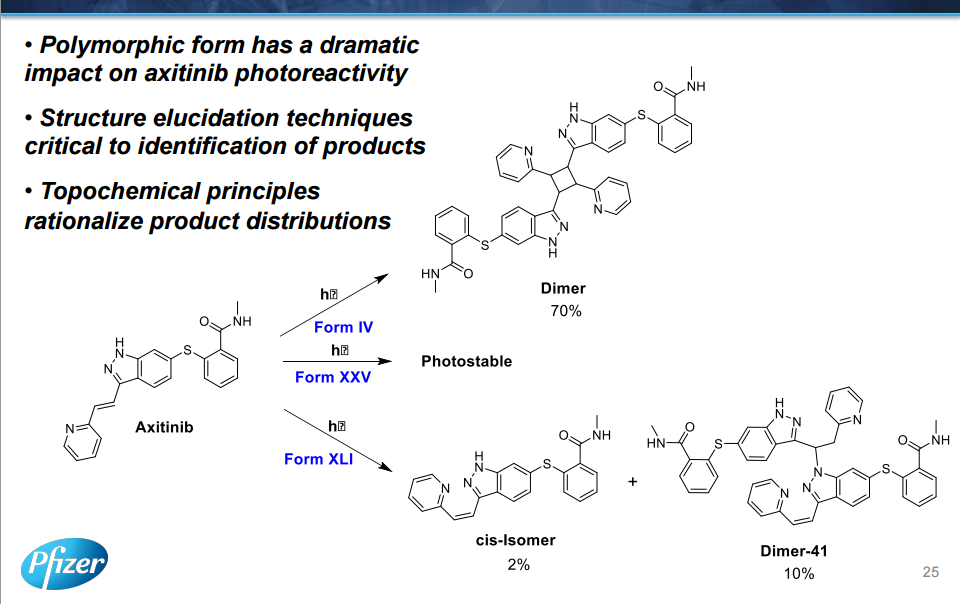
References
- “Inlyta (axitinib) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 25 January 2014.
- Wilmes, LJ; Pallavicini, MG; Fleming, LM; Gibbs, J; Wang, D; Li, KL; Partridge, SC; Henry, RG; Shalinsky, DR; Hu-Lowe, D; Park, JW; McShane, TM; Lu, Y; Brasch, RC; Hylton, NM (April 2007). “AG-013736, a novel inhibitor of VEGF receptor tyrosine kinases, inhibits breast cancer growth and decreases vascular permeability as detected by dynamic contrast-enhanced magnetic resonance imaging”. Magnetic Resonance Imaging 25 (3): 319–27. doi:10.1016/j.mri.2006.09.041. PMID 17371720.
- Rini, B; Rixe, O; Bukowski, R; Michaelson, MD; Wilding, G; Hudes, G; Bolte, O; Steinfeldt, H; Reich, SD; Motzer, R (June 2005). “AG-013736, a multi-target tyrosine kinase receptor inhibitor, demonstrates anti-tumor activity in a Phase 2 study of cytokine-refractory, metastatic renal cell cancer (RCC)”. Journal of Clinical Oncology ASCO Annual Meeting Proceedings 23 (16S): 4509.
- Rugo, HS; Herbst, RS; Liu, G; Park, JW; Kies, MS; Steinfeldt, HM; Pithavala, YK; Reich, SD; Freddo, JL; Wilding, G (August 2005). “Phase I trial of the oral antiangiogenesis agent AG-013736 in patients with advanced solid tumors: pharmacokinetic and clinical results”(PDF). Journal of Clinical Oncology 23 (24): 5474–83. doi:10.1200/JCO.2005.04.192.PMID 16027439.
- “FDA Approves Inlyta for Advanced Renal Cell Carcinoma”. Drugs.com. January 27, 2012.
- John Fauber, Elbert Chu (Oct 27, 2014). “The Slippery Slope: Is a Surrogate Endpoint Evidence of Efficacy?”. Milwaukee Journal Sentinel/MedPage Today.
- Spano, JP; Chodkiewicz, C; Maurel, J; Wong, R; Wasan, H; Barone, C; Létourneau, R; Bajetta, E; Pithavala, Y; Bycott, P; Trask, P; Liau, K; Ricart, AD; Kim, S; Rixe, O (June 2008). “Efficacy of gemcitabine plus axitinib compared with gemcitabine alone in patients with advanced pancreatic cancer: an open-label randomised phase II study”. Lancet 371(9630): 2101–2108. doi:10.1016/S0140-6736(08)60661-3. PMID 18514303.
- “Pfizer pancreatic cancer drug fails, trial halted”. Reuters. January 30, 2009.
- “Pfizer’s Phase III Trial in mRCC Turns Up Positive Results”. 19 Nov 2010.
- “ODAC Unanimously Supports Axitinib for Renal Cell Carcinoma”. 7 Dec 2011.
- “INLYTA (axitinib) tablet, film coated [Pfizer Laboratories Div Pfizer Inc]”. DailyMed. Pfizer Laboratories Div Pfizer Inc. September 2013. Retrieved 25 January 2014.
- “Inlyta : EPAR – Product Information” (PDF). European Medicines Agency. Pfizer Ltd. 17 December 2013. Retrieved 25 January 2014.
- “Inlyta 1 mg 3mg, 5 mg & 7mg film-coated tablets – Summary of Product Characteristics (SPC)”. electronic Medicines Compendium. Pfizer Limited. 5 December 2013. Retrieved25 January 2014.
- “PRODUCT INFORMATION INLYTA (axitinib)” (PDF). TGA eBusiness Services. Pfizer Australia Pty Ltd. 5 July 2013. Retrieved 25 January 2014.
- Tea Pemovska,Eric Johnson,Mika Kontro,Gretchen A. Repasky,Jeffrey Chen,Peter Wells,Ciarán N. Cronin,Michele McTigue,Olli Kallioniemi,Kimmo Porkka,Brion W. Murray & Krister Wennerberg. “Axitinib effectively inhibits BCR-ABL1(T315I) with a distinct binding conformation”. Nature. doi:10.1038/nature14119.
- “FDA Prescribing Information” (PDF). 30 Jan 2012.
- Escudier, B; Gore, M. “Axitinib for the Management of Metastatic Renal Cell Carcinoma” (PDF). Drugs in R&d 11 (2): 113–126. doi:10.2165/11591240-000000000-00000. PMC 3585900. PMID 21679004.
- Zhang Y (Jan 2014). “Screening of kinase inhibitors targeting BRAF for regulating autophagy based on kinase pathways.”. J Mol Med Rep 9 (1): 83–90.doi:10.3892/mmr.2013.1781. PMID 24213221.
- [1] http://www.cancer.gov/cancertopics/druginfo/axitinib[2] http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm289439.htm[3] Kosugi M, Shimizu T, T. Migita, Chemistry Letters , 1978 , pp 13-14.[4] Organic Process Research & Development 2008 , 12, 869? 876.[5] Furstner A. Chem. Commun ., 2008 , 2873? 2875.[6] Thorarensen A. , Synlett , 2010 , 2 pp 219 – 222.
[7] http://en.wikipedia.org/wiki/Heck_reaction – where you can find the reaction mechanism and many other useful information.
[8] Aoyama, T., Synthesis , 2004 , 8 pp 1183-1186.
Raw Material Variation into QbD Risk Assessment
DRUG REGULATORY AFFAIRS INTERNATIONAL
Areas of discussion included how expectations for raw material control are evolving within changing regulatory and business paradigms including quality by design (QbD), counterfeiting, complex supply chains, and sourcing changes. discussed risk assessment and mitigation strategies along with supplier risk management plans.

Regulatory Considerations
the lack of a consistent definition of raw materials in regulations pertaining to the pharmaceutical industry. In its Q7 guideline, the International Conference on Harmonisation of Technical Requirements for the Registration of Pharmaceuticals for Human Use (ICH) defines raw materials as “starting materials, reagents, and solvents intended for use in the production of intermediates or APIs.” However, the term as defined by different speakers could cover a wide range of materials including the following:
• starting or source materials (cell lines, viral or bacterial stocks, media components, chemicals, tissues, serum, water)
• in-process materials (resins, buffers, filters, column housings, tubing, reagents)
• excipients
• packaging components, both…
View original post 3,002 more words
FDA Warning Letter on Data Integrity
DRUG REGULATORY AFFAIRS INTERNATIONAL

The integrity of data is currently in the focus of international authorities In particular the US FDA issued serious violations in Warning Letters to the companies concerned. Read more about the current complaints in a Warning Letter issued to the API manufacturer VUAB Pharma.
For authorities the integrity of data is an essential quality attribute in the manufacture of pharmaceutical products. After some serious deviations international authorities have moved the topic into the centre of their interest. In particular the US FDA issued serious violations in Warning Letters to the companies concerned.

In a current letter to the API manufacturer VUAB Pharma in the Czech Republic the inspector and the authority criticised multiple aspects with regard to “failure to prevent unauthorized access or change to data and to provide controls preventing data omissions”:
- ‘The firm did not retain complete raw data from testing performed to assure the quality…
View original post 408 more words
Development of monographs for Indian pharmacopoeia
DRUG REGULATORY AFFAIRS INTERNATIONAL

Development of monographs for Indian pharmacopoeia
http://www.ipapharma.org/events/speakerpresentation/8-%20Dr%20S%20C%20Mathur.pdf

ALOGLIPTIN
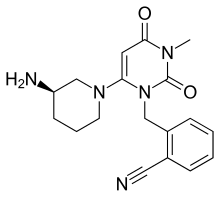
ALOGLIPTIN
Alogliptin is a potent, selective inhibitor of DPP-4 with IC50 of <10 nM, exhibits greater than 10,000-fold selectivity over DPP-8 and DPP-9.
Alogliptin (trade name Nesina in the US[1] and Vipidia in Europe[2]) is an orally administered anti-diabetic drug in the DPP-4 inhibitor class,[3] developed by Syrrx, a company which was acquired by Takeda Pharmaceutical Company in 2005. Like other medications for the treatment of Type 2 diabetes, alogliptin does not decrease the risk of heart attack and stroke. Like other members of the gliptin class, it causes little or no weight gain, exhibits relatively little risk of causing hypoglycemia, and exhibits relatively modest glucose-lowering activity. Alogliptin and other gliptins are commonly used in combination with metformin in patients whose diabetes cannot adequately be controlled with metformin alone.[4]
Clinical study
Alogliptin is a dipeptidyl peptidase-4 inhibitor (DPP-4i) that is designed to slow the inactivation of incretin hormones GLP-1 (glucagon-like peptide-1) and GIP (glucose-dependent insulinotropic peptide). [5]
A randomized clinical trial reporting in 2011 aimed to determine the efficacy and safety of alogliptin versus placebo and vogliboseamong newly diagnosed Type 2 diabetes patients in Japan. The main outcome indicated that alogliptin was statistically superior to both comparitors.[6]
A randomized clinical trial reporting in 2012 aimed to demonstrate that alogliptin was “non-inferior” to a “very low fat/calorie traditional Japanese diet” among newly diagnosed Type 2 diabetes patients in Japan. The outcome indicated that both the drug and dietary treatments comparably impacted indicators of the diabetic condition, such as HbA1c levels and glycemic efficacy. The drug treatment had its impact without changing body mass index (BMI), but the dietary treatment was accompanied by a significant reduction in the BMI.[7]
A randomized clinical trial reporting in 2011 aimed to demonstrate the efficacy of alogliptin as an add-on agent in combination withmetformin and pioglitazone versus simply increasing the dosage of pioglitazone in combination with metformin; in other words, this was a study to look at a three-agent therapy versus a two-agent therapy. The outcome of this study suggested that the addition of alogliptin to metformin and pioglitazone provided superior impact on diabetes biomarkers (e.g. HbA1c) than increasing the dose of pioglitazone in a two agent therapy with metformin.[8]
Reported adverse events
Adverse events appear to be restricted to mild hypoglycemia based on clinical studies.[6][7][8]
Alogliptin is not associated with increased weight, increased risk of cardiovasular events, or heart failure.[9][10]
Market access
In December 2007, Takeda submitted a New Drug Application (NDA) for alogliptin to the United States Food and Drug Adminiistration (USFDA),[11] after positive results from Phase III clinical trials.[1] In September of 2008, the company also filed for approval in Japan,[12] winning approval in April 2010.[11] The company also filed a Marketing Authorization Application (MAA) elsewhere outside the United States, which was withdrawn in June 2009 needing more data.[12] The first USFDA NDA failed to gain approval and was followed by a pair of NDAs (one for alogliptin and a second for a combination of alogliptin and pioglitazone) in July 2011.[11] In 2012, Takeda received a negative response from the USFDA on both of these NDAs, citing a need for additional data.[11]
In 2013 the FDA approved the drug in three formulations: As a stand-alone with the brand-name Nesina. Combined with metforminusing the name Kazano, and when combined with pioglitazone as Oseni.
Diabetes affects millions of people worldwide and is considered one of the main threats to human health in the 21st century. In 2006, the World Health Organization (WHO) estimated that over 180 million people worldwide had diabetes, and the number is projected to double by 2030. Over time, uncontrolled diabetes can damage body systems, including the heart, blood vessels, eyes, kidneys and nerves. According to the WHO, approximately 1.1 million people died from diabetes in 2005, and it is estimated that diabetes-related deaths will increase by more than 50% in the next decade. Globally, the socioeconomic burden of diabetes is substantial.
There are two main types of diabetes, designated type 1 and type 2, with type 2 diabetes accounting for over 90% of all diabetes cases globally. Type 1 diabetes is characterized by insulin deficiency, primarily caused by autoimmune-mediated destruction of pancreatic islet β-cells, and type 2 diabetes is characterized by abnormal insulin secretion and concomitant insulin resistance. To prevent the development of ketoacidosis, people with type 1 diabetes must take exogenous insulin for survival. Although those with type 2 diabetes are not dependent on exogenous insulin as much as subjects with type 1 diabetes, they may require exogenous insulin to control blood glucose levels.
As diabetes has become a global health concern, research interest in the condition has rapidly increased. In addition to studies on prevention, many studies with the aim of developing new interventions for the treatment of diabetes, especially type 2 diabetes, have been conducted. Currently available medications for the treatment and management of type 2 diabetes include metformin, sulfonylureas, thiazolidinediones and insulin. However, these therapies are commonly associated with secondary failure and may cause hypoglycemia. Insulin resistance and progressively worsening hyperglycemia caused by reduced β-cell function are major challenges in managing type 2 diabetes. Evidence suggests that patients with insulin resistance do not develop hyperglycemia until their β-cells are unable to produce enough insulin. New agents that can enhance insulin secretion from islet β-cells in a sustained glucose-dependent manner could therefore hold promise for the treatment of type 2 diabetes.
One promising approach is based on inhibition of the serine protease dipeptidyl- peptidase IV (DPP IV), a postproline dipeptidyl aminopeptidase that belongs to the S9b peptidase family of proteolytic enzymes. It is known that DPP IV plays a key role in maintaining glucose homeostasis by controlling the incretin activity of glucagon-like peptide 1 (GLP-I) and glucose-dependent insulinotropic polypeptide (GIP, also known as gastric inhibitory polypeptide). Inhibition of DPP IV is therefore recognized as a novel therapeutic approach for the treatment of type 2 diabetes.
Recently, a series of DPP IV inhibitors were developed. Among these highly potent compounds, alogliptin benzoate (SYR-322) and its analogs demonstrated encouraging antidiabetic efficacy (EP 1586571 (WO 2005/095381); WO 2008/067465; WO 2007/035379, and US 2004/097510).
Alogliptin benzoate can be prepared as described in EP 1586571 (WO 2005/095381) according to the process set forth in Scheme 1 :

Scheme 1
In accordance with this process, 6-Chlorouracil (1) is alkylated with 2- (bromomethyl)benzonitrile in the presence of NaH and LiBr in a mixture of DMF- DMSO to produce the TV-benzyluracil derivative (2) in 54% yield. Compound (2) is further alkylated with iodomethane and NaH in DMF/THF to give the 1 ,3 disubstituted uracil (3) in 72% yield. Subsequent displacement of chlorouracil (IV) with 3(R)- aminopiperidine dihydrochloride in the presence of either NaHCO3 in hot methanol or K2CO3 in aqueous isopropanol provides alogliptin (4), which is isolated as the corresponding benzoate salt by treatment with benzoic acid in ethanol. The overall yield of this three-stage process is -20-25%. One of the disadvantages of above described process is the difficulty to separate and purify mixtures of solvents with high boiling point (for example, DMF/DMSO) for recycling. Another disadvantage is the usage of hazardous materials such as sodium hydride, which requires anhydrous solvents as a reaction media.
Intermediate 2-((6-chloro-3-methyl-2,4-dioxo-3 ,4-dihydropyrimidin- 1 (2H)-yl)methyl) benzonitrile (3) is alternatively obtained by alkylation of 6-chloro-3 methyluracil with 2-(bromomethyl)benzonitrile by means of diisopropylethylamine in hot NMP (WO 2007/035629). Although this process is more technological than the previously described process (EP 1586571), the overall yield is still moderate (50-55%). The problem of mixed solvents (toluene, NMP, diisopropylethylamine) separation persists for this process as well.

………….
http://www.google.com/patents/EP2410855A1?cl=en
EXAMPLE 1
Preparation of (R)-2-((6-(3 -aminopiperidin-l-yl)-3 -methyl-2,4-dioxo-3 ,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile (alogliptin) via 6-chloro-l-(2- isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (Scheme 3):

Scheme 3
Preparation of l-(2-isocyanobenzyl)-3-methylurea
2-cyanobenzylamine hydrochloride (90 g) and Dichloromethane (800 ml) were taken into a round bottomed (RB) flask. Methyl isocyanate (45.6 g) was added at 5°C. Triethylamine (81 g) in Dichloromethane (300 ml) was added at the same temperature and stirred at room temperature for 16h. Water (1 L) was added and stirred for 30 min. The obtained solid was collected by filtration and dried in oven at 50°C for 12h. The yield is 85% and the purity 99.8%.
Preparation of l-(2-isocyanobenzyl)-3-methyIpyrimidine-2,4,6(lH,3H,5H)-trione
a). To a stirred solution of 0.11 mol of sodium ethanolate in 80 ml of ethanol abs. was added 0.1 mol of l-(2-isocyanobenzyl)-3-methylurea and 0.1 mol diethyl malonate. The mixture was refluxed for 3-5 h. The cooled residue was acidified with 0.1 M hydrochloric acid (60 ml). The solid which separated was filtered off and recrystallized from ethanol or any suitable solvent. The yield is 78-85% and purity >95%.
b). In an alternate embodiment, l-(2-isocyanobenzyl)-3-methylurea (30 g), acetic acid (105 ml) and malonic acid (18 g) were mixed and heated to 60°C. Acetic anhydride (60 ml) was added at 60°C and heating was continued for two hours at 80°C. The reaction mixture was poured over ice water (300 ml) and the obtained solid was filtered, washed with water (1×500 ml) and methyl-tert-butylether (100 ml). The yield is 60% with 93.4% purity.
The compound thus prepared can be used for the next step without purification or purified by crystallization or column chromatography.
Preparation of 6-chloro-l-(2-isocyanobenzyl)-3-methylpyriinidine-2,4(lH,3H)- dione
a). l-(2-isocyanobenzyl)-3-methylpyrimidine-2,4,6(lH,3H,5H)-trione (30 g) was mixed with phosphorus oxychloride (300 ml) and cooled to 0°C. Water (9 ml) was added slowly, stirred for 10 min. and heated to reflux at 110°C for 5h. Progress of the reaction was monitored by TLC (50% Ethyl acetate/Hexane). On completion of the reaction, phosphorus oxychloride was distilled off. The crude compound was dissolved in dichloromethane (500 ml) and poured into ice water (500 ml) by small portions. The layers were separated and the aqueous layer was extracted with dichloromethane (200 ml). The combined organic extracts were washed with water and brine, dried over sodium sulphate and concentrated under reduced pressure. The mixture of two isomers (4-chloro and 6-chloro derivatives = 1:1) was isolated and separated by column chromatography using neutral alumina and eluent – 25-50% of ethylacetate and hexane). The off-white solid was obtained, yield – 37%, purity – 99.8%. 1H NMR corresponds to literature data (J. Med. Chem. 2007, 50, 2297-2300).
b). In an alternate embodiment, a solution of l-(2-isocyanobenzyl)-3-methylpyrimidine- 2,4,6(1 H,3H,5H)-trione (18 mmol), phosphorus oxychloride (85 ml), benzyltriethylammonium chloride (16.5 g, 72 mmol) and phosphorus pentachloride (3.8 g, 18 mol) in acetonitrile (80 ml) was refluxed for 4-5 h with stirring. After evaporation under reduced pressure, the resulting oily residue was mixed with methylene chloride (or chloroform) and the mixture was poured into water and ice (50 ml). The layers were separated and the aqueous layer was extracted with dichloromethane (200 ml). The combined organic extracts were washed with water and brine, dried over sodium sulphate and concentrated under reduced pressure. Crude product was crystallized from THF-hexanes to give desired compound in 70.5% yield.
c). In an alternate embodiment, a solution of l-(2-isocyanobenzyl)-3-methylpyrimidine- 2,4,6(1 H,3H,5H)-trione (13.1 mmol) in POCl3 (30 ml) was refluxed for 1-3 h. The solvent was concentrated and then partitioned with CH2Cl2 (100 ml) and water (100 ml). The organic layer was washed with brine, dried over Na2SO4, and concentrated to give 6-chloro compound as a solid (-95%). Compound can be also precipitated from concentrated methylene chloride solution by hexanes and used as a crude for the next step or purified by reslurring in isopropanol, filtered off, washed with isopropanol, and dried under vacuum at 55-60° C.
Preparation of (R)-tert-butyl l-(3-(2-isocyanobenzyI)-l-methyl-2,6-dioxo-l,2,3,6- tetrahydropyrimidin-4-yl)piperidin-3-yl carbamate
a). 6-chloro- l-(2-isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (13 g), Dimethylformamide (130 ml), Potassium carbonate (13 g) and tert-butyl (R)-piperidin- 3-ylcarbamate (10.4 g) were heated to 80°C for 7 hrs. The mixture was then allowed to come to room temperature and poured over ice water (500 ml). The obtained solid was filtered and washed with cold water (500 ml). The solid thus obtained was taken in Methyl-tert-butylether (50 ml) stirred for 10 min. filtered and washed with Hexane (50 ml), to give the N-tert-butyloxycarbonyl protected compound in -75% yield. b). In an alternate embodiment, a flask charged with a stir bar, 6-chloro-l-(2- isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (4.10 mmol), (Λ)-3- terrtnityloxycarbonylaminopiperidine (4.64 mmol), K2CO3 (1.15 g, 8.32 mmol) and DMF (12 mL) was stirred at 75 °C for 6 h. Then, water was added and the mixture was extracted with methylene chloride. The organic layer was washed with brine, dried over Na2SO4, and concentrated to give the N-ter/butyloxycarbonyl protected compound in -93-96% yield.
Preparation of (R)-2-((6-(3-aminopiperidin-l-yl)-3-methyl-2,4-dioxo-3,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile salts
a). Preparation of (R)-2-((6-(3-aminopiperidin-l-yl)-3-methyl-2,4-dioxo-3,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile hydrochloride
The crude (R)-tert-butyl l-(3-(2-isocyanobenzyl)-l-methyl-2,6-dioxo-l,2,3,6- tetrahydropyrimidin-4-yl)piperidin-3-yl carbamate from previous procedure was dissolved in THF and acidified with 6M hydrochloric acid while maintaining the temperature below 15° C. The resultant slurry was cooled to 0-5° C, stirred at this temperature for 3-5 h and then filtered. The filter cake was washed twice with isopropanol and dried in vacuum at 45-5O0C to provide hydrochloride as a white crystalline solid.
b). Preparation of (R)-2-((6-(3-aminopiperidin-l-yl)-3-methyl-2,4-dioxo-3,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile trifluoroacetate
TFA (ImL) was added into the methylene chloride solution of (R)-tert-butyl l-(3-(2- isocyanobenzyl)- 1 -methyl-2,6-dioxo- 1 ,2,3,6-tetrahydropyrimidin-4-yl)piperidin-3-yl carbamate from the above-mentioned procedure. The solution was stirred at room temperature for 1 h and then the mixture was concentrated in vacuo. The residue was dissolved in a small amount of MeOH or isopropanol and the desired salt was precipitated by addition of diisopropyl ether. The solids were filtered off, washed with diisopropyl ether and dried in vacuum at 45-5O0C to provide trifluoroacetate as an off- white powder. c). Preparation of (R)-2-((6-(3-aminopiperidin-l-yl)-3-methyl-2,4-dioxo-3,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile benzoate (Alogliptin)
The crude (R)-tert-butyl l-(3-(2-isocyanobenzyl)-l-methyl-2,6-dioxo-l,2,3,6- tetrahydropyrimidin-4-yl)piperidin-3-yl carbamate was dissolved in ethanol. A solution of benzoic acid in ethanol was added and the mixture was slowly heated to 65-70°C. The solution was stirred at this temperature for Ih and was then crystallized by cooling to 0-5° C and stirring for 12 hrs. The solution was filtered, washed with alcohol. The wet cake was then conditioned under nitrogen for 2 hours. The cake was dried for 8 hrs at 40-50° C to provide the benzoic acid salt of alogliptin as a white crystalline solid.
EXAMPLE 2:
Preparation of (R)-2-((6-(3-aminopiperidin-l-yl)-3-methyl-2,4-dioxo-3,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile (alogliptin) via 6-amino-l-(2- isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (Scheme 4)


Scheme 4 Preparation of 6-amino-l-(2-isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)- dione
a). l-(2-isocyanobenzyl)-3-methylurea (0.2 mol) and cyanoacetic acid (0.22 mol) were dissolved in acetic anhydride (400 ml), and the mixture was heated at 80°C for 2 hours. Acetic anhydride was distilled off under reduced pressure and water (200 ml) was added. The mixture was cooled to 0-5 0C and 2N NaOH solution (220 ml) was added and stirring was continued for 2 hours. The obtained solids were filtered off, washed with cold methanol and dried under vacuum. The yield of 6-amino-l-(2- isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione was 72 %.
b). Under nitrogen atmosphere, l-(2-isocyanobenzyl)-3-methylurea (98.4 g) and cyanoacetic acid (80.0 g) was added to N,N-dimethylformamide (836 ml). The mixture was stirred at room temperature and methanesulfonyl chloride (72.8 ml) was added dropwise with stirring at this temperature. The mixture was stirred at room temperature for 4 hrs, cooled with water, and water-isopropanol [2:1 (volume ratio), 1670 ml] was added drop wise. The mixture was stirred under water-cooling for 1 hr, and the precipitated crystals were collected by filtration and dried to give 3-(2-cyano-acetyl)-3- methyl-l-(2-isocyanobenzyl)-urea with 68% yield.
To 3-(2-cyano-acetyl)-3-methyl-l-(2-isocyanobenzyl)-urea (120 g) were added water (962 ml) and 2N aqueous sodium hydroxide solution (24.9 ml), and the mixture was stirred with heating at 80° C for 1 hr. After allowing to cool to room temperature, the crystals were collected by filtration and dried to give 6-amino-l-(2-isocyanobenzyl)-3- methylpyrimidine-2,4(lH,3H)-dione in 76% yield.
c). 6-amino-l-(2-isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (0.1 mol) was mixed with (R)-piperidin-3-yl-carbamic acid tert.-butyl ester hydrochloride (0.1 mol) of the appropriate amine hydrochloride and (R)-piperidin-3-yl-carbamic acid tert.-butyl ester (0.1 mol). The mixture was heated at 100°C and bubbling continued for 3 hr. Water was added to the cooled mixture and the mixture was extracted with methylene chloride. The organic layer was washed with brine, dried over Na2SO4, and concentrated to give N-tert-butyloxycarbonyl protected compound in ~93-96% yield.
d). Benzoate salt of alogliptin was prepared as described above. While certain embodiments of the invention have been illustrated and described, it will be clear that the invention is not limited to the embodiments described herein. Numerous modifications, changes, variations, substitutions and equivalents will be apparent to those skilled in the art without departing from the spirit and scope of the present invention as described by the claims, which follow.
………………
Patent EP2410855A1
http://www.google.com/patents/EP2410855A1?cl=en

…………..

NMR
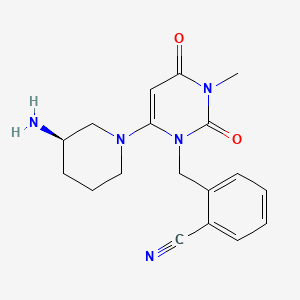
SOURCE APEXBT

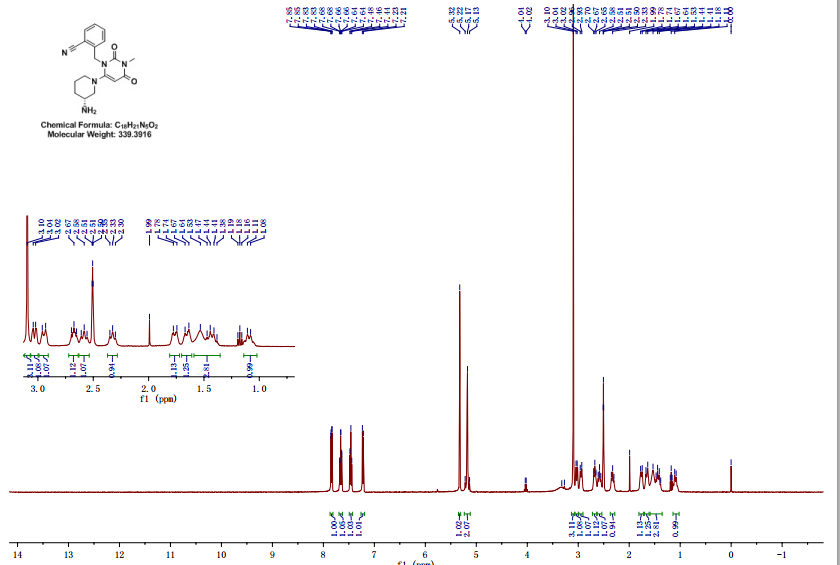
References
- “Takeda Submits New Drug Application for Alogliptin (SYR-322) in the U.S.” (Press release). Takeda Pharmaceutical Company. January 4, 2008. Retrieved January 9, 2008.
- Vipidia: EPAR summary for the public (European Medicines Agency)
- Feng, Jun; Zhang, Zhiyuan; Wallace, Michael B.; Stafford, Jeffrey A.; Kaldor, Stephen W.; Kassell, Daniel B.; Navre, Marc; Shi, Lihong; Skene, Robert J.; Asakawa, Tomoko; Takeuchi, Koji; Xu, Rongda; Webb, David R.; Gwaltney II, Stephen L. (2007). “Discovery of alogliptin: a potent, selective, bioavailable, and efficacious inhibitor of dipeptidyl peptidase IV”. J. Med. Chem.50 (10): 2297–2300.doi:10.1021/jm070104l.PMID 17441705.
- “www.aace.com” (PDF).
- http://www.takeda.com/news/2013/20130618_5841.html
- Seino, Yutaka; Fujita, Tetsuya; Hiroi, Shinzo; Hirayama, Masashi; Kaku, Kohei (September 2011), “Efficacy and safety of alogliptin in Japanese patients with type 2 diabetes mellitus: a randomized, double-blind, dose-ranging comparison with placebo, followed by a long-term extension study (abstract only)”, Current Medical Research and Opinion 27 (9): 1781–1792,doi:10.1185/03007995.2011.599371,PMID 21806314, retrieved April 26,2012
- Kutoh, Eiji; Ukai, Yasuhiro (2012),“Alogliptin as an initial therapy in patients with newly diagnosed, drug naïve type 2 diabetes: a randomized, control trial (abstract only)”, Endocrine(January 17, 2012), doi:10.1007/s12020-012-9596-0, PMID 22249941, retrieved April 26, 2012
- Bosi, Emanuele; Ellis, G.C.; Wilson, C.A.; Fleck, P.R. (October 2011), “Alogliptin as a third oral antidiabetic drug in patients with type 2 diabetes and inadequate glycaemic control on metformin and pioglitazone: a 52-week, randomized, double-blind, active-controlled, parallel-group study”, Diabetes, Obesity and Metabolism (October 27, 2011) 13 (12): 1088–1096, doi:10.1111/j.1463-1326.2011.01463.x, retrieved April 26,2012
- White WB, Cannon CP, Heller SR et al. (October 2013). “Alogliptin after acute coronary syndrome in patients with type 2 diabetes”. N. Engl. J. Med. 369(14): 1327–35.doi:10.1056/NEJMoa1305889.PMID 23992602.
- White WB, Zannad F (January 2014). “Saxagliptin, alogliptin, and cardiovascular outcomes”. N. Engl. J. Med. 370 (5): 484.doi:10.1056/NEJMc1313880.PMID 24482824.
- Grogan, Kevin (April 26, 2012),“FDA wants yet more data on Takeda diabetes drug alogliptin”,PharmaTimes (PharmaTimes), PharmaTimes online, retrieved April 26,2012
- “GEN News Highlights: Takeda Pulls MAA for Type 2 Diabetes Therapy”. Genetic Engineering & Biotechnology News. June 4, 2009.
| EP1083172A1 * | May 26, 1998 | Mar 14, 2001 | Rimma Iliinichna Ashkinazi | N-substituted derivatives of 5-oxyiminobarbituric acid |
| US2598936 * | Apr 13, 1950 | Jun 3, 1952 | Searle & Co | Disubstituted cyanoalkanoylureas and thioureas and methods for their production |
| US6066641 * | Dec 12, 1995 | May 23, 2000 | Euro-Celtique S.A. | Aryl thioxanthines |
| US6248746 * | Jan 7, 1999 | Jun 19, 2001 | Euro-Celtique S.A. | 3-(arylalkyl) xanthines |
| US20080194593 * | Jan 11, 2008 | Aug 14, 2008 | Rao Kalla | A2b adenosine receptor antagonists |
| WO1994003456A1 * | Aug 5, 1993 | Feb 17, 1994 | Boehringer Ingelheim Kg | Asymmetrically substituted xanthine with adenosine-antagonistic properties |
| WO2001029010A1 * | Oct 18, 2000 | Apr 26, 2001 | Amjad Ali | Gram-positive selective antibacterial compounds, compositions containing such compounds and methods of treatment |
| WO2007035629A2 * | Sep 15, 2006 | Mar 29, 2007 | Takeda Pharmaceutical | Process for the preparation of pyrimidinedione derivatives |
| WO2007150011A2 * | Jun 22, 2007 | Dec 27, 2007 | Smithkline Beecham Corp | Prolyl hydroxylase inhibitors |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-({6-[(3R)-3-aminopiperidin-1-yl]-3-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl}methyl)benzonitrile
|
|
| Clinical data | |
| Trade names | Nesina, Vipidia Kazano, Vipidomet (withmetformin) Oseni, Incresync (withpioglitazone) |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 100% |
| Protein binding | 20% |
| Metabolism | Limited, hepatic (CYP2D6– and3A4-mediated) |
| Biological half-life | 12–21 hours |
| Excretion | Renal (major) and fecal (minor) |
| Identifiers | |
| CAS Registry Number | 850649-62-6 |
| ATC code | A10BH04 |
| PubChem | CID: 11450633 |
| IUPHAR/BPS | 6319 |
| ChemSpider | 9625485 |
| UNII | JHC049LO86 |
| KEGG | D06553 |
| ChEBI | CHEBI:72323 |
| ChEMBL | CHEMBL376359 |
| Synonyms | SYR-322 |
| Chemical data | |
| Formula | C18H21N5O2 |
| Molecular mass | 339.39 g/mol |
 Alogliptin benzoate
Alogliptin benzoate
| MF: | C18H21N5O2.C7H6O2 |
| MW: | 461.519 |
| Melting Point: | 185-188°C |
| Optical Rotation: | -56.3° (c=1, MeOH) |
Solubility:Soluble in MeOH; Insoluble in ACN
850649-62-6 CAS
Alogliptin
-
- Synonyms:SYR-322
- ATC:A10BH04
- Use:antidiabetic, DPP-4 inhibitor
- Chemical name:2-[[5-[(3R)-3-amino-1-piperidinyl]-3,4-dihydro-3-methyl-2,4-dioxo-2H-pyrimidin-1(2H)-yl]methyl]benzonitrile
- Formula:C18H21N5O2
- MW:339.40 g/mol
- CAS-RN:850649-61-5
Derivatives
benzoate
- Formula:C19H19NO2
- MW:293.37 g/mol
- CAS-RN:850649-62-6
Substance Classes
Synthesis Path
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 22115-41-9 | C8H6BrN | 2-(bromomethyl)benzonitril | |
| C12H8ClN3O2 | 2-[[6-chloro-3,4-dihydro-2,4-dioxo-1(2H)-pyrimidinyl]methyl]benzonitrile | ||
| C13H10ClN3O2 | 2-[[6-chloro-3,4-dihydro-3-methyl-2,4-dioxo-1(2H)-pyrimidinyl]methyl]benzonitrile | ||
| 4270-27-3 | C4H3ClN2O2 | 6-chloro-2,4(1H,3H)-pyrimidinedione | |
| 74-88-4 | CH3I | methyl iodide | Methane, iodo- |
| 127294-73-9 | C5H12N2 | (3R)-3-piperidinamine |
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| J | Nesina | Takeda ,2010 |
Formulations
- tabl. 12.5 and 25 mg
References
-
- Feng, J. et al.: J. Med. Chem. (JMCMAR) 50, 2297-2300 (2007).
- WO 2 005 095 381 (SYRRX; 13.10.2005; appl. 15.12.2004; USA-prior. 15.3.2004).
- WO 2 010 109 468 (MAPI Pharma; 30.9.2010; appl. 25.3.2010; USA-prior. 26.3.2009).
-
solid preparation comprising Alogliptin and Pioglitazone:
- US 20 100 092 551 (Takeda Pharm.; 15.4.2010; appl. 30.1.2008; J-prior. 1.2.2007).
-
solid preparation comprising Alogliptin and Metformin:
- US 20 200 136 127 (Takeda Pharm.; 3.6.2010; appl. 16.7.2008; J-prior. 19.7.2007).
 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy
