WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Edoxaban (DU-176b, trade names Savaysa, Lixiana) is an anticoagulant drug which acts as a direct factor Xa inhibitor. It was developed by Daiichi Sankyo and approved in July 2011 in Japan for prevention of venous thromboembolisms (VTE) following lower-limb orthopedic surgery.[1] It was also approved by the FDA in January 2015 for the prevention of stroke and non–central-nervous-system systemic embolism.[2]
Daiichi Sankyo, APPROVED IN JAPAN as tosylate monohydrate salt in 2011 for the prevention of venous embolism in patients undergoing total hip replacement surgery
Edoxaban (INN, codenamed DU-176b, trade name Lixiana) is an anticoagulant drug which acts as a direct factor Xa inhibitor. It is being developed by Daiichi Sankyo. It was approved in July 2011 in Japan for prevention of venous thromboembolisms (VTE) following lower-limb orthopedic surgery.[1]
In animal studies, edoxaban is potent, selective for factor Xa and has good oral bioavailability.[2]
Daichi Sankyo’s edoxaban tosilate is an orally administered
coagulation factor Xa inhibitor that was approved and launched
in Japan for the preventive treatment of venous thromboembolic
events (VTE) in patients undergoing total knee arthroplasty, total
hip arthroplasty, or hip fracture surgery. Edoxaban has been
shown to have a rapid onset of anticoagulant effect due to short
Tmax (1–2 h) after dosing and sustained for up to 24 h post-dose.
Marketed under the brand name Lixiana, it is currently in phase
III studies in the US for the prevention of stroke and systemic embolic
events in patients with atrial fibrillation (AF) and venous
thromboembolism (VTE).
A large phase III trial showed that edoxaban was non inferior to warfarin in preventing recurrent venous thromboembolic events with fewer episodes of major bleeding.[7]
N1– (5-chloropyridin-2-yl) -N2– ( (IS, 2R/4S) -4- [ (dimethylamino) carbo nyl] -2- { [ ( 5-methyl-4 , 5,6, 7-tetrahydrothiazolo [5 , 4-c] pyridin-2-yl ) carbonyl] amino}eyelohexyl) ethanediamide , represented by the following formula (A) :
(A) The p-toluenesulfonic acid monohydrate salt of compound A is represented b the following formula (B) :
(B)
Edoxaban is known as a compound that exhibits an inhibitory effect on activated blood coagulation factor X (also referred to as activated factor X or FXa) , and is useful as a preventive and/or therapeutic drug for thrombotic diseases.
Several processes are known in the literature for preparing edoxaban for example, U.S. Patent No. 7365205; U.S. Publication No . 20090105491.
U.S. Patent No. 7365205 provides a process for the preparation of edoxaban, wherein the process involves the use of
(IS, 4S, 5S) -4-iodo-6-oxabicyclo [3.2.1] octan-7-one, represented by the following formula (C) :
(C)
as an intermediate.
The present inventors have identified that
(IS, 4S, 5S) -4-bromo-6-oxabicyclo [3.2.1] octan-7-one, represented by the following formula (I) :
( I )
could also be used as an intermediate for the preparation of FXa inhibitory compounds like edoxaban. The present inventors have found that replacement of
(IS, 4S, 5S) -4-iodo-6-oxabicyclo [3.2.1] octan-7-one (C) with
(IS, 4S, 5S) -4-bromo-6-oxabicyclo [3.2.1] octan-7-one (I) has a better atom economy and also an impact on cost.
A method for the synthesis of the
(IS, 4S, 5S) -4 -bromo- 6 -oxabicyclo [3.2.1] octan-7-one (I) was reported in Tetrahedron Letters, 51, (2010) Pages 3433-3435 which involves the reaction of ( IS) -cyclohex-3 -ene- 1-carboxylic acid represented by the following formula (II) :
( Π )
with N-bromosuccinimide in the presence of molecular sieves using dichloromethane as a solvent. However, this reaction is carried out in dark over a period of 7 hours and does not provide a pure product .
Tetrahedron, Vol. 28, Pages 3393 -3399 , 1972 provides a process for the preparation of 4 -bromo- 6 -oxabicyclo [3.2.1] octan-7-one which involves the addition of 20% excess of a 2M solution of bromine in chloroform to a stirred solution of cyclohex- 3 -ene- 1-carboxylic acid (0.04 mol) in chloroform (250 mL) in the absence of a base . Extraction with aqueous sodium bicarbonate followed by acidification gave, after extraction with ether and evaporation of the extract, a mixture of cis & trans 3 , 4-dibromocyclohexanecarboxylic acid (6.7 g) and evaporation of the chloroform layer afforded the bromolactone (0.59 g) . It further provides a process for the preparation of
4 -bromo-6 -oxabicyclo [3.2.1] octan-7-one which involves the treating of cyclohex-3-ene-l-carboxylic acid (0.08 mol) dissolved in chloroform (450 mL) with 20% excess bromine in the presence of an equimolar amount of triethylamine (8.1 g) . After extraction of the amine with 2N hydrochloric acid, and work-up, bromolactone (10.7 g) and a mixture of cis & trans 3 , 4 -dibromocyclohexanecarboxylic acid (6.6 g) were obtained.
Tetrahedron Vol. 48, No. 3, Pages 539-544, 1992 provides a process for the preparation of
(IS, 4S, 5S) -4-bromo-6-oxabicyclo [3.2.1] octan-7-one (I) which involves the addition of 1M solution of bromine in chloroform (30 mL) at 0°C to a solution of ( IS) -cyclohex-3 -ene- 1-carboxylic acid (0.024 mol) of formula (II) in chloroform (600 mL) in the presence of an equimolar amount of triethylamine (3.33 mL) . After work-up, the crude bromolactone obtained was recrystallized from petroleum ether.
However, bromination using bromine does not provide a pure product in good yield.
Heterocycles, Vol. 23, No. 8, Pages 2035-2039, 1985 provides a process for the 4-bromo-6-oxabicyclo [3.2.1] octan-7-one which involves the addition of cyclohex-3-ene-l-carboxylic acid (1.0 mM) in 1 , 2 -dimethoxyethane (2 mL) to a stirred solution of 90% Lead (IV) acetate (1.1 or 2.2 mM) in 1 , 2 -dimethoxyethane (4 mL) followed by the addition of Zinc bromide (2.2 mM) in 1 , 2 -dimethoxyethane (4 mL) and continuing the stirring for 10-30 minutes at 0°C . The reaction mixture was poured into a solution of ice-cold water (30 mL) and 10% hydrochloric acid (10 mL) , and extracted with ether (50 mL X 3) . The combined ether extract was washed successively with saturated sodium hydrogen carbonate solution (20 mL) , 10% sodium thiosulphate solution (5 mL) , and brine (10 mL) , and dried over sodium sulphate. Evaporation of the solvent gave crude lactone which were separated and purified (42% yield) . However, this reaction does not provide a pure product in good yield.
Heterocycles, Vol. 31, No. 6, Pages 987-991, 1990 provides a method for bromolactonization using a
dimethylsulfoxide-trimethylsilyl bromide-amine system. The bromolactonization is carried out for 10 to 72 hours using different solvents and triethylamine or diisopropylethyl amine as base. However, this process does not provide a product in high yield. Further the process afforded the cis isomer exclusively. Journal of the Chemical Society, Perkin Transactions 1:
Organic and Bio-Organic Chemistry (1972-1999) (1994) , (7) , Pages 847-851 provides a method for bromolactonization using a
dimethylsulfoxide-trimethylsilyl bromide-amine system. The bromolactonization is carried out for 12 hours using
dimethylsulfoxide and chloroform solvent system and triethylamine or diisopropylethyl amine as base. However, this process resulted in a low yield of about 55%. Citation List
Patent Literature
PTLl: U.S. Patent No. 7365205
PTL2: U.S. Publication No. 20090105491.
Non Patent Reference
NPLl: Feng Chen et al . , Tetrahedron Letters, 51, (2010) Pages 3433-3435.
NPL2 : G. Belluci et al . , Tetrahedron, Vol. 28, No. 13, Pages 3393-3399, 1972.
NPL3 : Marco Chini et al ., Tetrahedron Vol .48, No. 3, Pages 539-544 , 1992.
NPL4 : Y. Fujimoto et al . , Heterocycles , Vol. 23, No. 8, Pages 2035-2039, 1985.
NPL5: C. Iwata et al . , Heterocycles, Vol. 31, No. 6, Pages 987-991, 1990. –
NPL6 : K. Miyashita et al . , Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999) (1994) , (7) , Pages 847-851.
Summary of Invention
Technical Problem
It is an object of the present invention to solve the problems associated with the prior art, and to provide an improved and efficient method for the preparation of
(IS, 4S, 5S) -4-bromo-6-oxabicyclo [3.2.1] octan-7-one of formula (I).
Solution to Problem As a result of conducting diligent studies to attain the object, the present inventors have found that: surprisingly, the use of N-bromosuccinimide or bromohydantoin (representative is
1, 3-dibromo-5, 5-dimethylhydantoin) as brominating agent in the presence of a base selected from calcium oxide or calcium hydroxide, in specific mole ratios in a solvent selected from the group consisting of dichloromethane , toluene, tetrahydrofuran, ethyl acetate, hexanes, cyclopentyl methyl ether (CPME) or a mixture thereof can efficiently produce a pure
( IS , 4S , 5S) -4 -bromo- 6 -oxabicyclo [3.2.1] octan- 7 -one (I) in better yields. The process provides obvious benefits with respect to economics, convenience to operate at a commercial scale.
(Reference Example 6) N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide (X) (production method described in the pamphlet of International Publication No. WO 2007/032498)
Methanesulfonic acid (66 ml) was added to a suspension of tert-butyl [(1R,2,S,5S)-2-({[(5-chloropyridin-2-yl)amino](oxo)acetyl}amino)-5-(dimethylaminocarbonyl)cyclohexyl]carbamate (5) (95.1 g) in acetonitrile (1900 ml) at room temperature, and the mixture was stirred at this temperature for 2 hours. To the reaction solution, triethylamine (155 ml), 5-methyl-4,5,6,7-tetrahydro[1,3]thiazolo[5,4-c]pyrzdine-2-carboxylic acid hydrochloride (8) (52.5 g), 1-hydroxybenzotriazole (33.0 g), and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (46.8 g) were added under ice cooling, and the mixture was stirred at room temperature for 16 hours. Triethylamine and water were added thereto, and the mixture was stirred for 1 hour under ice cooling. Then, crystals were collected by filtration to obtain the title compound (X) (103.2 g). 1H-NMR (CDCl3) δ : 1.60-1.98 (3H, m), 2.00-2.16 (3H, m), 2.52 (3H, s), 2.78-2.90 (3H, m), 2.92-2.98 (2H, m), 2.95 (3H, s), 3.06 (3H, s), 3.69 (1H, d, J = 15.4 Hz), 3.75 (1H, d, J = 15.4 Hz), 4.07-4.15 (1H, m), 4.66-4.72 (1H, m), 7.40 (1H, dd, J = 8.8, 0.6 Hz), 7. 68 (1H, dd, J = 8.8, 2.4 Hz), 8.03 (1H, d, J = 7.8 Hz), 8.16 (1H, dd, J = 8.8, 0.6 Hz), 8.30 (1H, dd, J = 2. 4, 0.6 Hz), 9.72 (1H, s). MS (ESI) m/z: 548 (M+H)+.
TOSYLATE
(Reference Example 7) N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide mono-p-toluenesulfonate monohydrate (X-a) (production method described in the pamphlet of International Publication No. WO 2007/032498)
N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide (X) (6.2 g) was dissolved in methylene chloride (120 ml). To the solution, a 1 mol/L solution of p-toluenesulfonic acid in ethanol (11.28 ml) was added, and the solvent was distilled off. To the residue, 15% hydrous ethanol (95 ml) was added, and the mixture was dissolved by stirring at 60°C. Then, the mixture was cooled to room temperature and stirred for 1 day. The precipitated crystals were collected by filtration, washed with ethanol, and then dried under reduced pressure at room temperature for 2 hours to obtain the title compound (X-a) (7.4 g). 1H-NMR (DMSO-d6) δ : 1. 45-1. 54 (1H, m), 1.66-1.78 (3H, m), 2.03-2.10 (2H, m), 2.28 (3H, s), 2.79 (3H, s), 2.91-3.02 (1H, m), 2.93 (3H, s), 2.99 (3H, s), 3.13-3.24 (2H, m), 3.46-3.82 (2H, m), 3.98-4.04 (1H, m), 4.43-4.80 (3H, m), 7.11 (2H, d, J = 7.8 Hz), 7.46 (2H, d, J = 8.2 Hz), 8.01 (2H, d, J = 1.8 Hz), 8.46 (1H, t, J = 1.8 Hz), 8.75 (1H, d, J = 6.9 Hz), 9.10-9.28 (1H, br), 10.18 (1H, br), 10.29 (1H, s).
MS (ESI) m/z: 548 (M+H)+.
Anal.: C24H30ClN7O4S·C7H8O3S·H2O
Theoretical: C; 50.43, H; 5.46, N; 13.28, Cl; 4.80, S; 8.69.
Found: C; 50.25, H; 5.36, N; 13.32, Cl; 4.93, S; 8.79. mp (dec.): 245-248°C.
A compound represented by the following formula (X) [hereinafter, also referred to as compound (X)] or a pharmacologically acceptable salt thereof, or a hydrate thereof is a compound that exhibits an FXa inhibitory effect, as disclosed in Patent Literatures 1 to 3, and is useful as a preventive and/or therapeutic drug for thrombotic and/or embolic diseases:
The pamphlet of International Publication No. WO 2007/032498discloses a process for preparing an FXa inhibitor compound (X) or a pharmacologically acceptable salt thereof, or a hydrate thereof. The process for producing compound (X) disclosed therein involves, as shown in [Scheme A] below, azidifying compound (2) to produce azide compound (3), subsequently reducing compound (3) into amino compound (1a), subsequently treating compound (1a) with anhydrous oxalic acid to obtain compound (1), which is then treated with compound (4) (ethyl[5-chloropyridin-2-yl]amino](oxo)acetate hydrochloride) in the presence of a base to produce compound (5), followed by several steps from compound (5). This pamphlet also discloses crystals of the oxalate of compound (1) as a production intermediate.
wherein Boc represents a tert-butoxycarbonyl group.
Citation ListPatent Literatures
Patent Literature 1: International Publication No.WO 2004/058715
Patent Literature 2: International Publication No.WO 2003/016302
Patent Literature 3: International Publication No.WO 2003/000680
Patent Literature 4: International Publication No.WO 2007/032498
Furugohri T, Isobe K, Honda Y, Kamisato-Matsumoto C, Sugiyama N, Nagahara T, Morishima Y, Shibano T (September 2008). “DU-176b, a potent and orally active factor Xa inhibitor: in vitro and in vivo pharmacological profiles”. J. Thromb. Haemost.6 (9): 1542–9. doi:10.1111/j.1538-7836.2008.03064.x. PMID18624979.
Raskob, G.; Cohen, A. T.; Eriksson, B. I.; Puskas, D.; Shi, M.; Bocanegra, T.; Weitz, J. I. (2010). “Oral direct factor Xa inhibition with edoxaban for thromboprophylaxis after elective total hip replacement”. Thrombosis and Haemostasis104 (3): 642–649. doi:10.1160/TH10-02-0142.PMID20589317. edit
Weitz JI, Connolly SJ, Patel I, Salazar D, Rohatagi S, Mendell J, Kastrissios H, Jin J, Kunitada S (September 2010). “Randomised, parallel-group, multicentre, multinational phase 2 study comparing edoxaban, an oral factor Xa inhibitor, with warfarin for stroke prevention in patients with atrial fibrillation”. Thromb. Haemost.104 (3): 633–41. doi:10.1160/TH10-01-0066.
Edoxaban versus Warfarin in Patients with Atrial Fibrillation Robert P. Giugliano, M.D., Christian T. Ruff, M.D., M.P.H., Eugene Braunwald, M.D., Sabina A. Murphy, M.P.H., Stephen D. Wiviott, M.D., Jonathan L. Halperin, M.D., Albert L. Waldo, M.D., Michael D. Ezekowitz, M.D., D.Phil., Jeffrey I. Weitz, M.D., Jindřich Špinar, M.D., Witold Ruzyllo, M.D., Mikhail Ruda, M.D., Yukihiro Koretsune, M.D., Joshua Betcher, Ph.D., Minggao Shi, Ph.D., Laura T. Grip, A.B., Shirali P. Patel, B.S., Indravadan Patel, M.D., James J. Hanyok, Pharm.D., Michele Mercuri, M.D., and Elliott M. Antman, M.D. for the ENGAGE AF-TIMI 48 InvestigatorsDOI: 10.1056/NEJMoa1310907
“Edoxaban versus Warfarin for the Treatment of Symptomatic Venous Thromboembolism”. N. Engl. J. Med. August 2013. doi:10.1056/NEJMoa1306638. PMID23991658.
WO 03/000657 pamphlet WO 03/000680 pamphlet WO 03/016302 pamphlet WO 04/058715 pamphlet WO 05/047296 pamphlet WO 07/032498 pamphlet WO 08/129846 pamphlet WO 08/156159 pamphlet
Ohta T, Komoriya S, Yoshino T, et al. Preparation of N,N’-bis( heterocyclicacyl) cycloalkanediamine and heterocyclediamine derivatives as inhibitors of activated blood coagulation factor X (factor Xa): WO, 2003 000657 [P]. 2003-01-03. (CA 2003, 138: 73271)
[3]
Ohta T, Komoriya S, Yoshino T, et al. Preparation of heterocyclic moiety-containing diamine derivatives as FXa inhibitors: WO, 2003 000680 [P]. 2003-01-03. (CA 2003, 138: 89801)
[4]
Mochizuki A, Nagata T. Triamine derivative: WO, 2006106963 [P]. 2005-03-31. (CA 2006, 145: 419128)
[5]
Kawanami K, Ishikawa H, Shoji M. Process for preparation of optically active (1S,3R,4R)-3-amino-4-hydroxy-N,Ndimethylcyclohexanecarboxamide derivative salt: WO, 2012002538 [P]. 2012-01-05. (CA 2012, 156: 122056)
[6]
Sato K, Kubota K. Process for producing optically active carboxylic acid: WO, 2010067824 [P]. 2010-06-17. (CA 2010, 153: 36882)
[7]
Yoshikawa K, Yokomizo A, Naito H, et al. Design, synthesis, and SAR of cis-1,2-diaminocyclohexane derivatives as potent factor Xa inhibitors. Part I: Exploration of 5-6fused rings
[8]
as alternative S1 moieties [J]. Bioorg Med Chem, 2009, 17(24): 8206-8220.
[9]
Sato K, Kawanami K, Yagi T. Process for the preparation of optically active cyclohexane-1,2-diamine derivative from 7-oxabicyclo[4.1.0]heptane compound: WO, 2007032498
[10]
2007-03-22. (CA 2007, 146: 358502)
[11]
Kawanami K. Method for the preparation of optically active diamine derivative: WO, 2010104106 [P]. 2010-09-16. (CA 2010, 153: 406061)
[12]
Koyama T, Kondo S. Process for the preparation of diamine derivative: WO, 2010104078 [P]. 2010-09-16. (CA 2010, 153: 382938)
[13]
Suzuki T, Ono M. Crystal of diamine derivative and method of producing same: WO, 2011115066 [P]. 2011-09-22. (CA 2011, 155: 467954)
Process for the preparation of (1s,4s,5s)-4-bromo-6-oxabicyclo[3.2.1] octan-7-one
Molecular Formula
C24H30ClN7O4S.C7H7HSO3
Molecular Weight
720.26
CAS Registry Number
480449-71-6 (912273-65-5)
Drug formulation , lixiana, edoxaban tosylate monohydrate, CAS 912273-65-5, C24 H30 Cl N7 O4 S . C7 H8 O3 S . H2 O, 738.274
N1-(5-chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide p-toluenesulfonic acid monohydrate represented by the following formula (A) (hereinafter, also referred to as compound A) :
is known as a compound that exhibits an inhibitory effect on activated blood coagulation factor X (FXa), and is useful as a preventive and/or therapeutic drug for thrombotic diseases (Patent Literature 1 to 8).
For example, a method comprising mixing the free form of compound A represented by the following formula (B) (hereinafter, also referred to as compound B):
with p-toluenesulfonic acid or p-toluenesulfonic acid monohydrate, followed by crystallization from aqueous ethanol, is known as a method for obtaining compound A (Patent Literature 1 to 8). These literature documents do not make any mention about adding p-toluenesulfonic acid or p-toluenesulfonic acid monohydrate in a stepwise manner in the step of obtaining compound A from compound B.
Citation ListPatent Literature
Patent Literature 1: International Publication No. WO 03/000657
Patent Literature 2: International Publication No. WO 03/000680
Patent Literature 3: International Publication No. WO 03/016302
Patent Literature 4: International Publication No. WO 04/058715
Patent Literature 5: International Publication No. WO 05/047296
Patent Literature 6: International Publication No. WO 07/032498
Patent Literature 7: International Publication No. WO 08/129846
Patent Literature 8: International Publication No. WO 08/156159
Edoxaban is reported to be a member of the so-called “Xaban-group” and as such to be a low molecular inhibitor of the enzyme factor Xa, participating in the blood coagulation system. Therefore, edoxaban is classified as an antithrombotic drug and its possible medical indications are reported to be treatment of thrombosis and thrombosis prophylaxis after orthopaedic operations, such as total hip replacement, as well as for stroke prevention in patients with atrial fibrillation, the prophylaxis of the acute coronary syndrome and the prophylaxis after thrombosis and pulmonary embolism.
The IUPAC name for edoxaban is N’-(5-chloropyridin-2-yl)-N-[(15,2^,4S)-4- (dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[l ,3]thiazolo[5,4-c]pyridine-2- carbonyl)amino]cyclohexyl]oxamide. The chemical structure of edoxaban is shown in the formula (1) below:
formula ( 1 ) While Edoxaban is reported to be soluble in strongly acidic aqueous solutions, its solubility is considered to be very low in neutral or alkaline aqueous media. EP 2 140 867 A 1 claims an edoxaban-containing pharmaceutical composition comprising a water-swelling additive and/or a sugar alcohol. Further, it is alleged that compositions comprising lactose or cornstarch do not have good dissolution properties. The claimed pharmaceutical compositions in EP 2 140 867 Al are considered to show good dissolution properties in a neutral aqueous medium as well. Tablets comprising said composition were produced by wet granulation. However, it turned out that prior art pharmaceutical formulations comprising edoxaban being suitable for oral administration are still improvable with regards to dissolution rate and bioavailability. Further, stability and content uniformity of the known formulations could be improved. Further, due to the intolerance of many people to sugar alcohol(s), such as sorbitol, the use of sugar alcohol(s) should be avoided.
(Reference Example 1) 2-amino-5-methyl-4,5,6,7-tetrahydro thiazolone [5,4-c] pyridine (1-n) (The method described in WO 2005/047296 Pamphlet )
[0091]
[Of 35] in 2-PrOH (1.44L) solution was heated to 50 ℃ 1- methyl-4-piperidone (180.0g), 2-PrOH (360mL) solution of cyanamide (67.0g), and sulfur powder (51.0 g) it was added. Pyrrolidine (13.3mL) was added to the reaction mixture, after stirring for 2 hours at 50 ℃, followed by stirring overnight and allowed to cool to room temperature. The reaction mixture was cooled to 10 ℃ less in an ice water bath and stirred for 1 hour at the same temperature. Is filtered and the precipitated crystals were washed with 2-PrOH (540mL), the title compound was dried under reduced pressure at 40 ℃ (209.9g, 78%) was obtained.
[0092]
1 H-NMR (CDCl 3 ) ppm: 4.86 (Br, 2H), 3.47-3.46 (t, 2H, J = 1.9 Hz), 2.78-2.71 (M, 2H), 2.71-2.65 (M, 2H), 2.47 . (s, 3H)
MS (FAB) M / z: 170 (M + H) +
elemental analysis: C 7 H 11 N 3 as S,
theoretical value: C, 49.68; H, 6.55; N, 24.83; S, 18.95
measured value: C, 49.70; H, 6.39; N, 24.91; S, 19.00.
In 10 mL test tube, compound (5-ms) (the compound of Reference Example 8) (100 mg, 0.216 mmol), Compound (1-p2) (81.4 mg, 0.216 mmol), K 3 PO 4 (91.7 mg, 0.432 mmol) and DMF (1 mL) was added, and the mixture was stirred at room temperature conditions for 3 hours. H To the reaction mixture 2 O (2 mL) was added and the resulting slurry was stirred at room temperature overnight, the solid was filtered. The resulting solid H 2 was washed with O (1 mL), was obtained by drying under reduced pressure the title compound (110.0 mg, 92.9%) as a solid.
(Reference Example 6) N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide (X) (production method described in the pamphlet of International Publication No. WO 2007/032498)
Methanesulfonic acid (66 ml) was added to a suspension of tert-butyl [(1R,2,S,5S)-2-({[(5-chloropyridin-2-yl)amino](oxo)acetyl}amino)-5-(dimethylaminocarbonyl)cyclohexyl]carbamate (5) (95.1 g) in acetonitrile (1900 ml) at room temperature, and the mixture was stirred at this temperature for 2 hours. To the reaction solution, triethylamine (155 ml), 5-methyl-4,5,6,7-tetrahydro[1,3]thiazolo[5,4-c]pyrzdine-2-carboxylic acid hydrochloride (8) (52.5 g), 1-hydroxybenzotriazole (33.0 g), and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (46.8 g) were added under ice cooling, and the mixture was stirred at room temperature for 16 hours. Triethylamine and water were added thereto, and the mixture was stirred for 1 hour under ice cooling. Then, crystals were collected by filtration to obtain the title compound (X) (103.2 g).
(Reference Example 7) N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide mono-p-toluenesulfonate monohydrate (X-a) (production method described in the pamphlet of International Publication No. WO 2007/032498)
N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide (X) (6.2 g) was dissolved in methylene chloride (120 ml). To the solution, a 1 mol/L solution of p-toluenesulfonic acid in ethanol (11.28 ml) was added, and the solvent was distilled off. To the residue, 15% hydrous ethanol (95 ml) was added, and the mixture was dissolved by stirring at 60°C. Then, the mixture was cooled to room temperature and stirred for 1 day. The precipitated crystals were collected by filtration, washed with ethanol, and then dried under reduced pressure at room temperature for 2 hours to obtain the title compound (X-a) (7.4 g).
In 10 mL test tube, compound (5-ms) (the compound of Reference Example 8) (100 mg, 0.216 mmol), Compound (1-p2) (81.4 mg, 0.216 mmol), K 3 PO 4 (91.7 mg, 0.432 mmol) and DMF (1 mL) was added, and the mixture was stirred at room temperature conditions for 3 hours. H To the reaction mixture 2 O (2 mL) was added and the resulting slurry was stirred at room temperature overnight, the solid was filtered. The resulting solid H 2 was washed with O (1 mL), was obtained by drying under reduced pressure the title compound (110.0 mg, 92.9%) as a solid.
Yoshiyuki, I., et al. “Biochemical and pharmalogical profile of darexaban, an oral direct Xa inhibitor.” European Journal of Pharmacology (2011): 49-55
Katsung, B., S. Masters and A. Trevor. Basic and Clinical Pharmacology 11th Edition. United States of America: McGraw-Hill, 2009
Turpie AG (January 2008). “New oral anticoagulants in atrial fibrillation”. European Heart Journal29 (2): 155–65. doi:10.1093/eurheartj/ehm575. PMID18096568.
Edoxaban, a factor Xa inhibitor, is supplied as edoxaban tosylate monohydrate. The chemical name is N-(5-Chloropyridin-2-yl)-N’-[(1S,2R,4S)-4-(N,N-dimethylcarbamoyl)-2-(5-methyl- 4,5,6,7-tetrahydro[1,3]thiazolo[5,4-c]pyridine-2-carboxamido)cyclohexyl] oxamide mono (4- methylbenzenesulfonate) monohydrate. Edoxaban tosylate monohydrate has the empirical formula C24H30ClN7O4S•C7H8O3S•H2O representing a molecular weight of 738.27. The chemical structure of edoxaban tosylate monohydrate is:
It is a white to pale yellowish-white crystalline powder. The solubility of edoxaban tosylate (pKa 6.7) decreases with increasing pH. It is slightly soluble in water, pH 3 to 5 buffer, very slightly soluble at pH 6 to 7; and practically insoluble at pH 8 to 9.
SAVAYSA is available for oral administration as a 60 mg, 30 mg, or 15 mg round shaped, film-coated tablet, debossed with product identification markings. Each 60 mg tablet contains 80.82 mg edoxaban tosylate monohydrate equivalent to 60 mg of edoxaban. Each 30 mg tablet contains 40.41 mg edoxaban tosylate monohydrate equivalent to 30 mg of edoxaban. Each 15 tablet contains 20.20 mg edoxaban tosylate monohydrate equivalent to 15 mg of edoxaban.
The inactive ingredients are: mannitol, pregelatinized starch, crospovidone, hydroxypropyl cellulose, magnesium stearate, talc, and carnauba wax. The color coatings contain hypromellose, titanium dioxide, talc, polyethylene glycol 8000, iron oxide yellow (60 mg tablets and 15 mg tablets), and iron oxide red (30 mg tablets and 15 mg tablets).
Entinostat, developed by Syndax Pharmaceuticals, is an oral selective histone deacetylase (HDAC) inhibitor primarily targeting class IHDACs (HDAC1, HDAC2, and HDAC3) . It was later licensed to Jiangsu Hengrui Medicine Co., Ltd., for development and commercialization in China. In 2024, Entinostat has been approved by the NMPA for use in combination with exemestane to treat advanced breast cancer that is HR-positive and HER2-negative.
TOKYO and WALTHAM, Mass., Jan. 7, 2015 /PRNewswire/ — Kyowa Hakko Kirin Co., Ltd., (Headquarters: Chiyoda-ku, Tokyo; president and CEO: Nobuo Hanai, “Kyowa Hakko Kirin”) and Syndax Pharmaceuticals, Inc., (Waltham, Mass.; president and CEO:Arlene M. Morris, “Syndax”) today jointly announced that the companies have entered into a license agreement for the exclusive rights to develop and commercialize entinostat in Japan and Korea. Entinostat is a Class I selective histone deacetylase (HDAC) inhibitor being developed by Syndax in the United States and Europe in combination with hormone therapy for advanced breast cancer and immune therapy combinations in solid tumors.
Entinostat inhibits class I HDAC1 and HDAC3 with IC50 of 0.51 μM and 1.7 μM, respectively.[2]
Entinostat (formerly known as MS-275) is a histone deacetylase (HDAC) inhibitor in phase III clincal trials at Syndax in combination with exemestane for the treatment of advanced HR-positive breast cancer.
Entinostat (MS-275) preferentially inhibits HDAC1 (IC50=300nM) over HDAC3 (IC50=8µM) and has no inhibitory activity towards HDAC8 (IC50>100µM). MS-275 induces cyclin-dependent kinase inhibitor 1A (p21/CIP1/WAF1), slowing cell growth, differentiation, and tumor development in vivo. Recent studies suggest that MS-275 may be particularly useful as an antineoplastic agent when combined with other drugs, like adriamycin.
In September 2013, Syndax Pharmaceuticals entered into a licensing, development and commercialization agreement with Eddingpharm in China and other asian countries. In 2013, a Breakthrough Therapy Designation was assigned to the compound for the treatment of locally recurrent or metastatic estrogen receptor-positive (ER+) breast cancer when added to exemestane in postmenopausal women whose disease has progressed following non-steroidal aromatase inhibitor therapy.
Clinical trials
There is an ongoing phase II trial studying the effect of entinostat on Hodgkin’s lymphoma.[3] It is in other phase II trials for advanced breast cancer (in combination with aromatase inhibitors)[4] and for metastatic lung cancer (in combination with erlotinib).[5] As of September 2013, the Food and Drug Administration is working with the industry to design phase III clinical trials. They seek to evaluate the application of Entinostat for the reduction, or prevention of, treatment resistance to aromatase inhibitors in hormone receptor positive breast cancer.[6] Syndax pharmaceuticals currently holds the rights to Entinostat and recently received $26.6 million in funds to advance treatments of resistant cancers using epigenetic tools.[7]
PHASE 3………..SYNDAX, BREAST CANCER
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Entinostat, developed by Syndax Pharmaceuticals, is an oral selec tive histone deacetylase (HDAC) inhibitor primarily targeting class I HDACs (HDAC1, HDAC2, and HDAC3) [7]. It was later licensed to Jiangsu Hengrui Medicine Co., Ltd., for development and commercial ization in China. In 2024, Entinostat has been approved by the NMPA for use in combination with exemestane to treat advanced breast cancer that is HR-positive and HER2-negative. This approval is specifically for pa tients whose disease has progressed following prior endocrine therapy [8]. Entinostat inhibits HDACs, increasing histone acetylation and reactivating tumor suppressor genes. This mechanism restores sensi tivity to endocrine therapy and prevents cancer cell proliferation [9]. The therapeutic agent exerts its effects by modulating the tumor microenvironment through the suppression of immune regulatory cells, thereby augmenting the immune response. Its clinical efficacy was confirmed in the E2112 trial (NCT02115282), a global Phase III study. When used in combination with exemestane, Entinostat demonstrated the ability to extend PFS in patients with HR-positive, HER2-negative breast cancer [10]. The median PFS was significantly extended to 6.32 months, contrasting with the 3.72 months observed in the control cohort. In terms of safety profile, Entinostat demonstrated favorable tolerability. The frequently encountered adverse events were primarily neutropenia, fatigue, and nausea. Severe neutropenia occurred in 43 % of patients but was manageable with supportive care. Liver function abnormalities were reported but manageable with dose adjustments [11]. The synthetic route of Entinostat is shown in Scheme 2 [12]. Enti-001 is first treated with trifluoroacetic anhydride to afford Enti-002. Reaction of Enti-002 with oxalyl chloride yields the acyl chloride intermediate, which undergoes condensation with Enti-003 to form Enti-004. Subsequent alkaline hydrolysis of Enti-004 produces Enti-005. This compound is activated with CDI followed by reaction with Enti-006 to generate Enti-007. The synthesis concludes with acidic removal of the Boc protecting group from Enti-007, yielding Entinostat
[8] W. Li, Z. Sun, Mechanism of action for HDAC inhibitors-insights from omics approaches, Int. J. Mol. Sci. 20 (2019) 1616. [9] N. Bharathy, N.E. Berlow, E. Wang, J. Abraham, T.P. Settelmeyer, J.E. Hooper, M. N. Svalina, Z. Bajwa, M.W. Goros, B.S. Hernandez, J.E. Wolff, R. Pal, A.M. Davies, A. Ashok, D. Bushby, M. Mancini, C. Noakes, N.C. Goodwin, P. Ordentlich, J. Keck, D.S. Hawkins, E.R. Rudzinski, A. Mansoor, T.J. Perkins, C.R. Vakoc, J.E. Michalek, C. Keller, Preclinical rationale for entinostat in embryonal rhabdomyosarcoma, Skelet Muscle 9 (2019) 12. [10] B. Xu, Q. Zhang, X. Hu, Q. Li, T. Sun, W. Li, Q. Ouyang, J. Wang, Z. Tong, M. Yan, H. Li, X. Zeng, C. Shan, X. Wang, X. Yan, J. Zhang, Y. Zhang, J. Wang, L. Zhang, Y. Lin, J. Feng, Q. Chen, J. Huang, L. Zhang, L. Yang, Y. Tian, H. Shang, Entinostat, a class I selective histone deacetylase inhibitor, plus exemestane for Chinese patients with hormone receptor-positive advanced breast cancer: a multicenter, randomized, double-blind, placebo-controlled, phase 3 trial, Acta Pharm. Sin. B 13 (2023) 2250–2258. [11] E.T. Roussos Torres, W.J. Ho, L. Danilova, J.A. Tandurella, J. Leatherman, C. Rafie, C. Wang, A. Brufsky, P. LoRusso, V. Chung, Y. Yuan, M. Downs, A. O’Connor, S. M. Shin, A. Hernandez, E.L. Engle, R. Piekarz, H. Streicher, Z. Talebi, M.A. Rudek, Q. Zhu, R.A. Anders, A. Cimino-Mathews, E.J. Fertig, E.M. Jaffee, V. Stearns, R. M. Connolly, Entinostat, nivolumab and ipilimumab for women with advanced HER2-negative breast cancer: a phase Ib trial, Nat Cancer 5 (2024) 866–879. [12] T. Suzuki, T. Ando, K. Tsuchiya, T. Nakanishi, A. Saito, S. Yamashita, G. Shiraishi, E. Tanaka, Preparation of Benzamide Derivatives as Anticancer Agents, 1998 JP10152462
In EP 0 847 992 A1 (which co-patent is US 6,794,392) benzamide derivatives as medicament for the treatment of malignant tumors, autoimmune diseases, de- rmatological diseases and parasitism are described. In particular, these derivatives are highly effective as anticancer drugs, preferred for the haematological malignancy and solid tumors. The preparation of N-(2-aminophenyl)-4-[N- (pyridine-3-yl)methoxycarbonylaminomethyl]-benzamide is described on page 57, Example 48. The compound is neither purified by chromatography nor purified by treatment with charcoal. The final step of the process comprises the re- crystallization from ethanol.
Said compound has a melting point (mp) of 159 – 160 0C.
The IR spectrum shows the following bands: IR(KBr) cm“1: 3295, 1648, 1541 , 1508, 1457, 1309, 1183, 742.
The data indicate the Polymorph A form.
In EP 0 974 576 B1 a method for the production of monoacylated phenylenediamine derivatives is described. The preparation of N-(2- aminophenyl)-4-[N-(pyridine-3-yl)methoxycarbonylamino-methyl] benzamide is described on pages 12 to 13, Example 6. The final step of the process comprises the purification of the compound via silica gel column chromatography.
Said compound has a melting point (mp) of 159 – 160 0C.
The IR spectrum shows the following bands: IR(KBr) cm‘1: 3295, 1648, 1541 , 1508, 1457, 1309, 1183, 742.
The data indicate the Polymorph A form. In J. Med. Chem. 1999, 42, 3001-3003, the synthesis of new benzamide derivatives and the inhibition of histone deacetylase (HDAC) is described. The process for the production of N-(2-aminophenyl)-4-[N-(pyridine-3-yl) meth- oxycarbonylaminomethyl] benzamide is described. The final step of the process comprises the purification of the compound via silica gel column chromatography (ethyl acetate).
Said compound has a melting point (mp) of 159 – 160 0C.
The IR spectrum shows the following bands: IR(KBr) cm‘1: 3295, 1648, 1541 , 1508, 1457, 1309, 1183, 742.
The data indicate the Polymorph A form.
In WO 01/12193 A1 a pharmaceutical formulation comprising N-(2- aminophenyl)-4-[N-(pyridine-3-yl)methoxycarbonylamino-methyl]benzamide is described.
In WO 01/16106 a formulation comprising N-(2-aminophenyl)-4-[N-(pyridine-3- yl)methoxycarbonylamino-methyl]benzamide, having an increased solubility and an improved oral absorption for benzamide derivatives, and pharmaceutically acceptable salts thereof are described.
In WO 2004/103369 a pharmaceutical composition is described which comprises histone deacetylase inhibitors. That application concerns the combined use of N-(2-aminophenyl)-4-[N-(pyridine-3-yl)methoxycarbonylamino- methyl]benzamide together with different cancer active compounds. In fact that application is a later application, which is based on the above mentioned matter and thus concerns the Polymorph A form. Finally, JP 2001-131130 (11-317580) describes a process for the purification of monoacylphenylenediamine derivatives. In Reference Example 2, the process for the production of crude N-(2-aminophenyl)-4-[N-(pyridine-3-yl) meth-oxycarbonylaminomethyl] benzamide is described. Said compound has a melting point (mp) of 159 – 160 0C,
The IR spectrum shows the following bands: IR(KBr) cm“1: 3295, 1648, 1541 , 1508, 1457, 1309, 1183, 742.
The data indicate the Polymorph A form.
Moreover, Working Example 1 describes the purification of crude N-(2- aminophenyl)-4-[N-(pyridine-3-yl) methoxycarbonylaminomethyl] benzamide in aqueous acid medium together with carbon The final crystallization is done under aqueous conditions at 40-500C.
Following the description to that example it can be seen from the Comparative Examples 1 – 3 that the crude N-(2-aminophenyl)-4-[N-(pyridine-3-yl) meth- oxycarbonylaminomethyl] benzamide is not purified by dissolution under reflux conditions in either ethanol, methanol or acetonithle followed by a recrystalliza- tion at 2°C. As a result, these recrystallisations do not yield any pure compound.
In addition a “purification” of crude N-(2-aminophenyl)-4-[N-(pyridine-3-yl) methoxycarbonylaminomethyl] benzamide in ethanol under reflux conditions to- gether with carbon is dechbed. After filtering off the carbon the compound is re- crystallized at 2°C. The purification effect of this method is very limited. 1 ,1 % of an impurity remain in the N-(2-aminophenyl)-4-[N-(pyridine-3-yl) methoxycarbonylaminomethyl] benzamide. As a result, this procedure does not yield any pure compound.
None of the state of the art documents refer to a polymorph B of N-(2- aminophenyl)-4-[N-(pyridine-3-yl)methoxycarbonylamino-methyl]benzamide and no physicochemical features of said compound are known. Several biological and clinical studies have been done with N-(2-aminophenyl)- 4-[N-(pyridine-3-yl) meth-oxycarbonylaminomethyl] benzamide. For example, Kummar et al., Clin Cancer Res. 13 (18), 2007, pp 5411-5417 describe a phase I trial of N-(2-aminophenyl)-4-[N-(pyridine-3-yl) meth-oxycarbonylaminomethyl] benzamide in refractory solid tumors. The compound was applied orally.
The crude N-(2-aminophenyl)-4-[N-(pyridine-3-yl)methoxycarbonylaminomethyl]- benzamide of step a) can be produced according to the method described in example 6 of EP 0974 576 B1.
Example 6Synthesis of N-(2-aminophenyl)-4-[N-(pyridin-3-ylmethoxycarbonyl)aminomethyl]benzamide (an example in which after activation with N,N’-carbonyldiimidazole, an acid was added to carry out reaction)
[0082]
7.78 g (48 mmole) of N,N’-carbonyldiimidazole were added to a 1,3-dimethyl-2-imidazolidinone (50 g) suspension including 11.45 g (40 mmole) of 4-[N-(pyridin-3-ylmethoxycarbonyl)aminomethyl]benzoic acid. After stirring at room temperature for 2 hours, 17.30 g (0.16 mole) of 1,2-phenylenediamine were added to the solution. After cooling to 2°C, 9.60 g (0.1 mole) of methanesulfonic acid were added dropwise. After stirring for 2 hours, water was added, and the deposited solid was collected by filtration. Purification was then carried out through silica gel column chromatography to obtain 10.83 g (yield: 72%) of N-(2-aminophenyl)-4-[N-(pyridin-3-ylmethoxycarbonyl)aminomethyl]benzamide. Reaction selectivity based on the result in HPLC
Suzuki et al (Suzuki et al Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives, J Med Chem 1999, 42, (15), 3001-3) discloses benzamide derivatives having histone deacetylase inhibitory activity and methods of making benzamide derivatives having histone deacetylase inhibitory activity. Suzuki et al is hereby incorporated herein by reference in its entirety.
[18] An example of the synthesis method of Suzuki et al to produce MS-275 via a three- step procedure in 50.96% overall yield is outlined in Scheme 3 below.
Scheme 3: Previous Procedure for Synthesis of MS-275 en rt, 4h
(used without purification)
[Overall yield: 0.91 x 0.56 x 100 = 50.96%;
MS-275 [19] In addition to the modest overall yield, the procedure of Suzuki et al has other disadvantages, such as a tedious method for the preparation of an acid chloride using oxalyl chloride and requiring the use of column chromatography for purification.
The synthesis of MS-275 is shown below in Scheme 4 as an example of Applicants invention of a two-step procedure: [37] Scheme 4: Preparation of MS-275
Scheme 4: New Synthesis of MS-275 (4)
Condensation of 3-(hydroxymethyl)pyridine (7) and 4-(aminomethyl)benzoic in the presence of CDI gave 4-[N-(pyridin-3-ylmethoxycarbonyl)aminomethyl]benzoic Acid (8) in 91.0% yield. In the previous method of Suzuki et ah, the carboxylic acid derivative 8 was first converted into acyl chloride hydrochloride by treatment of oxalyl chloride in toluene and then reacted with imidazole to form the acylimidazole intermediate. (Suzuki et al., Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives. J Med Chem 1999, 42, (15), 3001-3.). However, Applicants synthesized the imidazolide of intermediate 8 by treatment with CDI at about 55-60 0C in THF. The imidazolide was cooled to ambient and further reacted in situ with 1,2-phenylenediamine in the presence of TFA to afford MS-275
[63] To a suspension of 4-[N-(Pyridin-3-ylmethoxycarbonyl)aminomethyl]benzoic
Acid (5.0 g, 0.017 mol) in THF (100 mL) was added CDI (3.12 g, 0.019 mol), and the mixture stirred for 3 h at 60 0C. After formation of acylimidazole the clear solution was cooled to room temperature (rt). To this was added 1,2-phenylenediamine (15.11 g, 0.14 mmol) and trifluoroacetic acid (1.2 mL, 0.015 mol) and then stirred for 16 h. The reaction mixture was evaporated to remove THF and crude product was stirred in a mixture of hexane and water (2:5, v/v) for 1 h and filtered and dried. The residue was stirred in dichloromethane twice to afford pure MS-275 (4) as off white powder 5.25 g, 80% yield:
HRMS: calcd 376.1560 (C2iH2oN4θ3), found 376.1558. These spectral and analytical data are as previously reported in J Med Chem 1999, 42, (15), 3001-3.
[64] 4-[7V-(Pyridin-3-ylmethoxycarbonyI)aminomethyl] benzoic Acid (8) may be prepared as follows. To a suspension of l, l’-carbonyldiimidazole (CDI, 25.6 g, 158 mmol) in THF (120 mL) was added 3-pyridinemethanol (7, 17.3 g, 158 mmol) in THF (50 mL) at 10 0C, and the mixture stirred for 1 h at rt. The resulting solution was added to a suspension of 4-(aminomethyl)benzoic acid (22.6 g, 158 mmol), DBU (24.3 g, 158 mmol), and triethylamine (22.2 mL, 158 mmol) in THF (250 mL). After stirring for 5 h at rt, the mixture was evaporated to remove THF and then dissolved in water (300 mL). The solution was acidified with HCl (pH 5) to precipitate a white solid which was collected by filtration, washed with water (300 mL) and methanol (50 mL), respectively, and dried to yield pure 8 (41.1 g, 91% yield):
mp 207-208 0 C;
IR (KBr) 3043, 1718, 1568, 1434, 1266, 1 108, 1037, 984, 756 cm4; 1H NMR (DMSO-^6) δ 4.28 (d, 2H, J= 5.9 Hz), 5.10 (s, 2H), 7.3-7.5 (m, 3H), 7.7-8.1 (m, 4H), 8.5-8.7 (m, 2H). These spectral and analytical data are as previously reported in Suzuki et al, J Med Chem 1999, 42, (15), 3001-3.
N-(2-Aminophenyl)-4-[N-(pyridin-3-ylmethoxycarbonyl)aminomethyl]benzamide (1, MS-275). To a solution of imidazole (0.63 g, 9.2 mmol) in THF (20 mL) was added 3 (1 g, 2.9 mmol), and the mixture stirred for 1 h at room temperature. After imidazole hydrochloride was removed by filtration, 1,2-phenylenediamine (2.52 g, 23.2 mmol) and trifluoroacetic acid (0.2 mL, 2.6 mmol) were added to the filtrate and stirred for 15 h. The reaction mixture was evaporated to remove THF and partitioned between ethyl acetate (500 mL) and water (400 mL). The organic layer was washed with water and dried and then purified by silica gel column chromatography (ethyl acetate) to give 1 (0.62 g, 56% yield):
References: 1. Saito, A. et al. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc Natl Acad Sci USA 96 4592-4597 (1999). 2. Jaboin, J., et al. MS-27-275, an inhibitor of histone deacetylase, has marked in vitro and in vivo antitumor activity against pediatric solid tumors. Cancer Res 62 6108-6115 (2002). 3. Rosato RR, et al. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res 2003; 63: 3637–3645.
For the treatment of patients with predominantly classic subfoveal choroidal neovascularization due to age-related macular degeneration, pathologic myopia or presumed ocular histoplasmosis syndrome. Verteporfin can also be used to destroy tumors.
Verteporfin (trade name Visudyne), a benzoporphyrin derivative, is a medication used as a photosensitizer for photodynamic therapy to eliminate the abnormal blood vessels in the eye associated with conditions such as the wet form of macular degeneration. Verteporfin accumulates in these abnormal blood vessels and, when stimulated by nonthermal red light with awavelength of 693 nm in the presence of oxygen, produces highly reactive short-lived singlet oxygen and other reactive oxygen radicals, resulting in local damage to the endothelium and blockage of the vessels.[1][2]
Verteporfin, otherwise known as benzoporphyrin derivative (trade name Visudyne®), is a medication used as a photosensitizer for photodynamic therapy to eliminate the abnormal blood vessels in the eye associated with conditions such as the wet form of macular degeneration. Verteporfin accumulates in these abnormal blood vessels and, when stimulated by nonthermal red light with a wavelength of 693 nm in the presence of oxygen, produces highly reactive short-lived singlet oxygen and other reactive oxygen radicals, resulting in local damage to the endothelium and blockage of the vessels.
Administration
Verteporfin is given intravenously, 15 minutes before laser treatment.[1]
VISUDYNE® (verteporfin) for Injection is a light activated drug used inphotodynamic therapy. The finished drug product is a lyophilized dark green cake. Verteporfin is a 1:1 mixture of two regioisomers (I and II), represented by the following structures:
The chemical names for the verteporfin regioisomers are:
9-methyl (I) and 13-methyl (II) trans-(±)-18-ethenyl-4,4a-dihydro-3,4-bis(methoxycarbonyl)-4a,8,14,19-tetramethyl-23H, 25H-benzo[b]porphine-9,13-dipropanoate
The molecular formula is C41H42N4O8 with a molecular weight of approximately 718.8. Each mL of reconstituted VISUDYNE contains:
ACTIVE:
Verteporfin, 2 mg
INACTIVES:
Lactose, egg phosphatidylglycerol, dimyristoyl phosphatidylcholine, ascorbyl palmitate and butylated hydroxytoluene
Most common side effects are blurred vision, headache, and local effects at the injection site. Also, photosensitivity; it is advised to avoid exposure to sunlight and unscreened lighting until 48 hours after the injection of verteporfin.[1]
Interactions
None known. Verteporfin has no influence on the liver enzyme CYP3A4, which metabolises many pharmaceutical drugs.[1]
Verteporfin (CAS # 129497-78-5) is a benzoporphyrin derivative which has been used clinically for photodynamic therapy of age related macular degeneration (23).
Verteporfin is photoactivated for photodynamic therapy to eliminate the abnormal blood vessels in the eye associated with conditions such as the wet form of macular
degeneration. Verteporfin accumulates in these abnormal blood vessels and, when stimulated by nonthermal red light with a wavelength of 693 ran in the presence of oxygen, the photoactivated verteporfin produces highly reactive short-lived singlet oxygen and other reactive oxygen radicals, resulting in local damage to the endothelium and blockage of the vessels. Benzoporphoryrins, are described for example, in US patents 5,095,030, 5,214,036, and 6,008,241.
Verteporfin (CAS # 129497-78-5) as used herein may include the two regioisomers as shown below:
and the 2 entantiomers of each of the two regioisomers as shown below:
The verteporfin as disclosed herein contains at least one chiral center and thus may exist in various stereoisomeric forms. If desired, such stereoisomers, including enantiomers, may be separated using techniques standard in the art (for example, chiral columns). However, racemic mixtures or mixtures containing more than one diastereomer may also be used and are contenplated herein. However, the compounds tested herein were in either of the trans entantiomers shown above. The compounds shown in Formulas IA, IB, Tables 1, 2 and Figure 10, are representative of the individual optical isomers, enantiomers or diastereomers as the case may be, as well as mixtures of these individual chiral isomers.
Visudyne™, as used herein, is the liposomal formulation of verteporfin used in humans for photodynamic therapy. Visudyne™ is given intravenously, usually within 15 minutes prior to laser treatment to eliminate the abnormal blood vessels in the eye in the treatment of wet macular degeneration. The verteporfin compound accumulates in these abnormal blood vessels and, when stimulated by a nonthermal red light laser with a wavelength of 693 nm in the presence of oxygen, produces highly reactive short-lived singlet oxygen and other reactive oxygen radicals, resulting in local damage to the endothelium and blockage of the vessels. Patients given Visudyne™ experience photosensitivity and are advised to avoid exposure to sunlight and unscreened lighting for at least 48 hours after the injection of verteporfin.
In contrast to the current use of verteporfin in photodynamic therapy, subjects administered the BPDs described herein, in accordance with the methods and uses described herein, do not require photoactivation of the BPD via nonthermal red light laser with a wavelength of 693 nm or otherwise. The activity of the BPDs to inhibit early stage autophagy is independent of the activity associated with photoactivation and would likely be hindered by photoactivation. Accordingly, a person of skill in the art would appreciate that the precautions associated with photosensitivity should also apply to the present methods and uses (i.e. avoid exposure to sunlight and unscreened lighting for at least 48 hours after the injection of of the BPD).
ICH announces Q&A Document on Q11 Guideline – Main Focus: API Starting Materials
The ICH Q11 Guideline entitled “Development and Manufacture of Drug Substances” from May 2012 has been implemented in the three ICH regions EU, USA and Japan for 2 years now. It describes the approach to developing APIs based on an in-depth understanding of the manufacturing process and adequate strategies to control this process. The document indicates what information should be provided about the quality of the API in Module 3 of the CTD (Common Technical Document) within the framework of a marketing authorisation application.
In the meantime, there has been an accumulation of cases where the applicant and the regulatory authorities adopted different positions with regard to the interpretation of the requirements in this guideline. Particularly, this concerned the definition of starting materials for the manufacture of APIs. The standards according to which regulatory authorities accept a specific compound as a starting material are far from uniform across the 3 ICH regions, and, all the more across Europe. It is thus obvious that this – in the context of global authorisation procedures – costs a lot of time, energy and money.
The ICH has now faced this problem and created an Implementation Working Group (IWG) which has the task of elaborating a Q&A document on API Starting Materials. As is usual in such circumstances (see also our News from 10 December 2014), the ICH has justified the necessity of the Q&A document in a Business Plan and a Concept Paper – both entitled “Q11: Q&As on Selection and Justification of Starting Materials for the Manufacture of Drug Substances”. (Business Plan and Concept Paper are the 1st step of the ICH procedure consisting of 5 steps. The last step always ends with a “Harmonised Tripartite Guideline”.)
The Concept Paper provides further details about the benefits expected of the Q&A document for the industry, authorities and patients:
The selection and justification of starting materials should be extensively harmonised.
The connection between the selection of starting materials and GMP aspects, control strategies, length of the chemical syntheses (number of synthesis steps) and the relevance of the manufacturing steps with regard to the API quality should be clarified.
It should be specified which information has to be provided in the application dossier for the justification of the selection of the starting materials.
The expectations regarding the lifecycle management should be explained.
This Question-&-Answer document is certainly interesting for all those confronted with diverse regulatory expectations regarding starting materials in relation to supra-regional registration and marketing authorisation procedures. Yet, according to the timing indicated in both the Business Plan and the Concept Paper, a first draft of this document (as Step 2a/b) should be released in one year at the earliest, namely in November 2015.
NEWS………….DUBLIN and BUDAPEST, Hungary, Jan. 6, 2015 /PRNewswire/ — Actavis plcand Gedeon Richter Plc. today announced that the U.S. Food and Drug Administration (FDA) has acknowledged receipt of Actavis’ New Drug Application (NDA) resubmission for its atypical antipsychotic cariprazine, a potent dopamine D3/D2 receptor partial agonist with preferential binding to D3 receptors. The Prescription Drug User Fee Act (PDUFA) date is expected to be in the second quarter of 2015…….
The most prevalent side effects for cariprazine include akathisia, insomnia, and weight gain. Cariprazine does not appear to impact metabolic variables or prolactin levles, and unlike many other antipsychotics, does not increase the electrocardiogram (ECG) QT interval. In short term clinical trials extrapyramidal effects, sedation, akathisia, nausea, dizziness, vomiting, anxiety, and constipation were observed. One review characterized the frequency of these events as “not greatly different from that seen in patient treated with placebo”[5] but a second called the incidence of movement-related disorders “rather high”[6][7] .
Pharmacodynamics
Cariprazine acts as an antipsychotic that is effective against the positive and negative symptoms of schizophrenia.[8] Unlike many antipsychotics that are D2 and 5-HT2Areceptor antagonists, cariprazine is a D2 and D3partial agonist. It also has a higher affinity for D3 receptors. The D2 and D3 receptors are important targets for the treatment of schizophrenia, because the overstimulation of dopamine receptors has been implicated as a possible cause of schizophrenia.[9] Cariprazine acts to inhibit overstimulated dopamine receptors (acting as an antagonist) and stimulate the same receptors when the endogenous dopamine levels are low. Cariprazine’s high selectivity towards D3 receptors could prove to reduce side effects associated with the other antipsychotic drugs, because D3receptors are mainly located in the ventral striatum and would not incur the same motor side effects (extrapyramidal symptoms) as drugs that act on dorsal striatum dopamine receptors.[8] Cariprazine also acts on 5-HT1A receptors, though the affinity is considerably lower than the affinity to dopamine receptors (seen in monkey and rat brain studies).[8][10] In the same studies, cariprazine has been noted to produce pro-cognitive effects, the mechanisms of which are currently under investigation. An example of pro-cognitive effects occurred in pre-clinical trials with rats: rats with cariprazine performed better in a scopolamine-induced learning impairment paradigm in a water labyrinth test. This may be due to the selective antagonist nature of D3 receptors, though further studies need to be conducted.[8] This result could be very useful for schizophrenia, as one of the symptoms includes cognitive deficits.
Cariprazine has partial agonist as well as antagonist properties depending on the endogenous dopamine levels. When endogenous dopamine levels are high (as is hypothesized in schizophrenic patients), cariprazine acts as an antagonist by blocking dopamine receptors. When endogenous dopamine levels are low, cariprazine acts more as an agonist, increasing dopamine receptor activity.[11] In monkey studies, the administration of increasing does of cariprazine resulted in a dose-dependent and saturable reduction of specific binding. At the highest dose (300 μg/kg), the D2/D3 receptors were 94 % occupied, while at the lowest dose (1 μg/kg), receptors were 5 % occupied.[10]
Cariprazine has high oral bioavailability and can cross the blood brain barrier easily in humans because it is lipophilic.[2] In rats, the oral bioavailability was 52 % (with a dose of 1 mg/kg).[7]
Example 1 1-(2,3-dichlorophenyl)-[1,4]diazepine (starting material)
2.25 g (10 mmol) 1-bromo-2,3-dichloro-benzene was dissolved in dry toluene (50 ml), 2.3 (11 mmol) of [1 ,4]diazepine-1 -carboxylic acid tert-butylester was added followed by 0.2 g BINAP (2,2-bis(diphenylρhosphino)-1 ,1′-binaphtyl), 85 mg tris(dibenzylideneacetone)dipalladium(0) and 1.2 g (12mmol) sodium-tert-butoxyde. The reaction mixture was refluxed for eight hours and filtered. The organic layer was washed with water, dried and evaporated in vacuo. The residue was purified by chromatography and deprotected at 10 °C using 20 ml ethylacetate saturated with gaseous hydrochloric acid, the precipitate was filtered giving 2.1 g (yield: 75 %) hydrochloride salt of the title compound, melting at 182-3 °C. Example 2 Trans-N-{4-[2-[4-(2,3-dichloro-phenyl)-hexahydro-[1 ,4]diazepin-1-yl]-ethyl]- cyclohexyl}-carbamic acid tert-butylester (intermediate) 0.7 g (2.5 mmol) of 1 -(2,3-dichlorophenyl)-[1 ,4]diazepine hydrochloride and
0.6 g (2.5 mmol) of frat?s-2-{1 -[4-(N-tert-butyloxycarbonyl)amino]cyclohexyl}- acetaldehyde were dissolved in dichloroethane (35 ml), 0.35 ml (2.5 mmol) triethylamine was added, then 0.79 g (3.7 mmol) sodium triacetoxyborohydride was added portionswise and the reaction mixture was stirred for 20 hours at ambient temperature, then 20 % potassium carbonate solution in water (20 ml) was added. The organic layer was separated, dried and evaporated to dryness in vacuo. The precipitate was recrystallized from acetonitrile to give the title compound 1 .0 g (yield: 85.8 %), m.p.: 95-8 °C. Example 3
0.93 g (2.1 mmol) frarjs-N-{4-[2-[4-(2,3-dichloro-phenyl)-hexahydro- [1 ,4]diazepin-1 -yl]-ethyl]-cyclohexyl}-carbamic acid tert-butylester was deprotected at
10 °C using 15 ml ethylacetate saturated with gaseous hydrochloric acid, after 4 hours the precipitate was filtered giving 0.91 g (yield: 98 %) dihydrochloride salt of the title compound, melting at 260-6 °C. Method A
Trans-1-{4-[2-[4-(2,3-dichlorophenyl)-piperazin-1-yi]-ethyl]-cyclohexyl}-3,3- dimethyl-urea (compound 1 ) 1 .39g (3 mmol) trans-4-{2-[4-(2,3-dichlorophenyl)-ρiperazin-1 -yl]-ethyl}- cyclohexyl-amine trihydrochloride was suspended in dichloromethane (100 ml), triethylamine (2.1 ml, 15 mmol) was added followed by 0.30 ml (3.3 mmol) N,N- dimethylcarbamoylchloride. The reaction mixture was stirred for 48 hours at room temperature, filtered. The filtrate was washed with water (2 x 20 ml), dried and evaporated in vacuo. Recrystallizing from methanol gave the title compound (0.83 g, 65 %), melting at 212-4 °C.
Method B
7rans-1-{4-[2-[4-(2,3-dichlorophenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-3-ethyl- urea (compound 2) 0.56g (1.2 mmol) trans-4-{2-[4-(2,3-dichlorophenyl)-piperazin-1-yl]-ethyl}- cyclohexyl-amine was dissolved in dry dichloromethane (20 ml), ethylisocyanate (0.1 ml, 1.3 mmol) was added and the reaction mixture was stirred at room temperature for 4 hours. The solvent was removed in vacuo. The residue was stirred with water, the precipitate was filtered, giving the title compound (0.33 g, 65 %). Melting point:
235-8 °C.
Method C rrans-1-{4-[2-[4-(2,3-dichlorophenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-3,3- dimethyl-urea (compound 1 )
0.56g (1.2 mmol) trans-4-{2-[4-(2,3-dichlorophenyl)-piperazin-1-yl]-ethyl}- cyclohexyl-amine trihydrochloride was suspended in dry dichloromethane (50 ml), triethylamine 0.77 ml, 6 mmol) was added and 0.13g (0.44 mmol) triphosgene dissolved in dichloromethane was dropped in. After one hour stirring at room temperature dimetilamine hydrochloride (0.49 g, 6 mmol) followed by triethylamine (0.84 ml, 6 mmol) was added and the stirring was continued for 20 hours. The mixture was filtered, the filtrate washed with water, dried and evaporated in vacuo. Recrystallizing the product from methanol gave the title compound (0.27 g, 52 %). Melting point: 212-4 °C.
U.S. Patent Publication No. 2006/0229297 discloses (thio)-carbamoyl-cyclohexane derivatives that are D3 and D2 dopamine receptor subtype preferring ligands, having the formula (I):
(I)
wherein R1, R2, X, and n are as defined therein. One particular compound disclosed therein is trans-1{4-[2-[4-(2,3-dichlorophenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-3,3-dimethyl-urea, which is also known as trans-4-{2-[4-(2,3-dichlorophenyl)-piperazin-1-yl]-ethyl}-N,N-dimethylcarbamoyl-cyclohexylamine, the structural formula for which is shown below:
Compounds of formula (I) act as a dopamine receptor antagonists, particularly D3/D2 receptor antagonists, and are useful in the treatment and prevention of pathological conditions which require modulation of dopamine receptors.
In some cases, an appropriate salt of an active may improve certain properties suitable for pharmaceutical compounds (i.e., stability, handling properties, ease of large scale synthesis, etc.). However, selection of a suitable salt for a particular active agent is not always straightforward, since the properties of salts of different compounds formed with the same salt forming agent may differ greatly. Moreover, formation of particular salts of a compound possessing more than one basic centre may be difficult to achieve in high yield due to formation of multiple products.
We have surprisingly found that by reacting trans 4-{2-[4-(2,3-dichlorophenyl)- piperazine-l-yl]-ethyl}-cyclohexylamine of formula (III)
with a carbonic acid derivative of general formula (VI)R-O-CO-Z (VI)
then reacting the compound of general formula (IV) obtained
with an amine derivative of general formula (V)
get the compounds of general formula (I)
EXAMPLES
The invention is illustrated by the following non-limiting examples.
Example 1
Trans N-(4-{2-[4-(2,3-dichlorophenyl)-piperazine-l-yl]-ethyl}-cyclohexyl)-carbamic acid methylester 6.45 g (0.015 mol) of dihydrochloride of compound of formula (III) was added to a mixture of 125 ml dichloromethane and 12.25 ml triethylamine and the thick suspension obtained was stirred at a temperature between 20-25°C for one hour. The so obtained suspension was added to a solution of 2.3 ml (0.03 mol) methyl chloroformate in 25 ml of dichloromethane at a temperature between 5-10°C. The reaction mixture obtained was stirred at a temperature between 20-25°C for 3 hours then extracted with 3×150 ml (150 g) of distilled water. The organic phase was evaporated in vacuum and the residue was recrystallized from methanol. In this manner 4.5 g of the title product was obtained.
Yield: 72 %.
Melting point: 143-147 °C
Example 2
Trans N-(4-{2-[4-(2,3-dichlorophenyl)-piperazine-l-yl]-ethyl}-cyclohexyl)-carbamic acid isopropylester
6.45 g (0.015 mol) of dihydrochloride of compound of formula (III) was added to a mixture of 125 ml dichloromethane and 12.25 ml of triethylamine and the thick suspension obtained was stirred at a temperature between 20-25°C-on for one hour. The suspension was added to a solution of 3.7 g (0.03 mol) of isopropyl chloroformate in 30 ml of toluene at a temperature between 5-10°C. The reaction mixture was stirred at a temperature between 20-25°C for 3 hours and then extracted with 3×150 ml (150 g) of distilled water. The organic phase was evaporated in vacuum and the residue obtained was recrystallized from isopropanole.
In this manner 4,4 g of title compound was obtained. Yield: 67 %.
Melting point: 128-131°C
Example 3
Trans 4-{2-[4-(2,3-dichlorophenyl)-piperazine-l-yl]-ethyl}-N,N-dimethylcarbamoyl- cyclohexylamine
6.45 g (0.015 mol) of dihydrochloride of compound of formula (III) was added to a mixture of 125 ml of dichloromethane and 12.25 ml of triethylamine and the thick suspension obtained was stirred at a temperature between 20-25°C for one hour. The suspension was added to a solution of 4.9 g of bis(trichloromethyl)carbonate in 50 ml of dichloromethane at a temperature between -5-(-10)°C for one hour. The reaction mixture obtained was added to a solution of 13 g dimethylamine in 100 ml isopropyl alcohol (IP A) (40 ml, 0.12 mol) cooled at a temperature between 0-(-10)°C during which the temperature of the reaction mixture was kept under 0°C. After stirring at a temperature between 0-(-5)°C for 30 minutes to the reaction mixture 100 ml of distilled water was added under stirring. Then the pH of the aqueous phase was adjusted to 7-8 by adding concentrated hydrochloric acid and volume of the reaction mixture was concentrated to 130 ml under vacuum. To the reaction mixture obtained additional 70 ml of distilled water was added and the mixture was concentrated to 170 ml under vacuum. The suspension was stirred at 20-25°C for one hour and the product obtained was isolated by filtration.
In this manner 6.6 g of title compound was obtained.
Yield: 95 %
Melting point: 208-211 °C Example 4
Trans 4-{2-[4-(2,3-dichlorophenyl)-piperazine-l-yl]-ethyI}-N,N-dimethylcarbamoyl- cyclohexylamine 4.4 g (0.011 mol) of trans N-(4-{2-[4-(2,3-dichlorophenyl)-piperazine-l-yl]-ethyl}- cyclohexyl)-carbamic acid methylester was dissolved in 120 ml of dichloromethane. The solution obtained was added to a solution of 13 g dimethylamine in 100 ml isopropyl alcohol (IP A) (100 ml, 0.3 mol) cooled at a temperature between 0-(-10)°C during which the temperature of the reaction mixture was kept under 0°C. After stirring at a temperature between 0-(-5)°C for 30 minutes to the reaction mixture 100 ml of distilled water was added under stirring. Then the pH of the aqueous phase was adjusted to 7-8 by adding concentrated hydrochloric acid and volume of the reaction mixture was concentrated to 100 ml under vacuum. To the reaction mixture obtained additional 70 ml of distilled water was added and the mixture was concentrated to 120 ml under vacuum. The suspension was stirred at 20-25°C for one hour and the product obtained was isolated by filtration.
In this manner 4.3 g of title compound was obtained.
Yield: 95 %
Melting point: 208-211 °C
Example 5
Trans 4-{2-[4-(2,3-dichlorophenyl)-piperazine-l-yl]-ethyl}-N,N-dimethylcarbamoyl- cyclohexylamine hydrochloride 6.45 g (0.015 mol) dihydrochloride of formula (III) was added to a mixture of 125 ml of dichloromethane and 12.25 ml of triethylamine and the thick suspension obtained was stirred at a temperature between 20-25°C for one hour. The suspension was added to the solution of 4.9 g of bis(trichloromethyl)carbonate in 50 ml of dichloromethane at a temperature between -5-(-10)°C for one hour. The reaction mixture obtained was added to a solution of 13 g dimethylamine in 100 ml isopropyl alcohol (IP A) (40 ml, 0.12 mol) cooled at a temperature between 0-(-10)°C during which the temperature of the reaction mixture was kept under 0°C. After stirring at a temperature between 0-(-5)°C for 30 minutes 100 ml of distilled water was added to the reaction mixture under stirring. Then the pH of the aqueous phase is adjusted to 2-3 by adding concentrated hydrochloric acid and the reaction mixture was concentrated to 130 ml, additional 70 ml of distilled water was added and the mixture was concentrated to 170 ml. The suspension was stirred at 20-25°C for one hour and the product obtained was isolated by filtration.
In this manner 6.7 g of title compound was obtained.
Yield: 96 %
Melting point: 221-224 °C
Example 6
Trans 4-{2-[4-(2,3-dichlorophenyl)-piperazine-l-yl]-ethyl}-N,N-dimethylcarbamoil- cyclohexylamine hydrochloride 6.72 g (0.015 mol) dihydrochloride monohydrate of compound of formula (III) was added to a mixture of 125 ml of dichloromethane and 12.25 ml of triethylamine and the thick suspension obtained was stirred at a temperature between 20-25 °C for one hour. The suspension was added to the solution of 4.9 g of bis(trichloromethyl)carbonate in 50 ml of dichloromethane at a temperature between -5-(-10)°C for one hour. The reaction mixture obtained was added to a solution of 13 g dimethylamine in 100 ml isopropyl alcohol (IP A) (40 ml, 0,12 mol) cooled at a temperature between 0-(-10)°C during which the temperature of the reaction mixture was kept under 0°C. After stirring at a temperature between 0-(-5)°C for 30 minutes to the reaction mixture 100 ml of distilled water was added and the pH of the aqueous phase was adjusted to 2-3 by adding concentrated hydrochloric acid. The reaction mixture was concentrated to 130 ml under vacuum then additional 70 ml of water was added and the mixture was concentrated to 170 ml. The suspension was stirred at a temperature between 20-25°C for one hour and the product obtained was isolated by filtration.
In this manner 6.7 g of title compound was obtained.
Éva Ágai-Csongor, György Domány, Katalin Nógrádi, János Galambos, István Vágó, György Miklós Keserű, István Greiner, István Laszlovszky, Anikó Gere, Éva Schmidt, Béla Kiss, Mónika Vastag, Károly Tihanyi, Katalin Sághy, Judit Laszy, István Gyertyán, Mária Zájer-Balázs, Larisza Gémesi, Margit Kapás, Zsolt Szombathelyi
Cariprazine, a potential atypical antipsychotic agent has been identified during the optimization of novel series of 4-aryl-piperazine derivatives. The recently available top line results from pivotal clinical trials demonstrated the safety and efficacy of cariprazine in bipolar mania and schizophrenia indications.
Biased agonism offers an opportunity for the medicinal chemist to discover pathway-selective ligands for GPCRs. A number of studies have suggested that biased agonism at the dopamine D2 receptor (D2R) may be advantageous for the treatment of neuropsychiatric disorders, including schizophrenia. As such, it is of great importance to gain insight into the SAR of biased agonism at this receptor. We have generated SAR based on a novel D2R partial agonist, tert-butyl (trans-4-(2-(3,4-dihydroisoquinolin-2(1H)-yl)ethyl)cyclohexyl)carbamate (4). This ligand shares structural similarity to cariprazine (2), a drug awaiting FDA approval for the treatment of schizophrenia, yet displays a distinct bias toward two different signaling end points. We synthesized a number of derivatives of 4 with subtle structural modifications, including incorporation of cariprazine fragments. By combining pharmacological profiling with analytical methodology to identify and to quantify bias, we have demonstrated that efficacy and biased agonism can be finely tuned by minor structural modifications to the head group containing the tertiary amine, a tail group that extends away from this moiety, and the orientation and length of a spacer region between these two moieties.
Using 50 (40 mg, 112 μmol) as the amine, following general procedure F the product was eluted (CHCl3/CH3OH, 20:1 to 10:1) to give the title compound as a white solid (27 mg, 56%).
Following an adapted literature procedure,(38) 10% Pd/C (881 mg, 828 μmol) was carefully added to an orange suspension of 5 (5.00 g, 27.6 mmol) in H2O (150 mL). The reaction mixture was hydrogenated on a Parr shaker at 60 psi at rt for 3 days until the uptake of hydrogen was complete and no starting materials remained by TLC (CHCl3/CH3OH, 1:1). The mixture was filtered through a Celite pad and washed with water (30 mL), and the filtrate evaporated to dryness in vacuo to reveal a white solid. The material was taken up in absolute EtOH (70 mL) to which concentrated HCl (10 mL) was addedm and the mixture was heated at reflux for 2 h. TLC confirmed ethyl ester formation, and the solvents were concentrated in vacuo. The material was basified with a 1 M NaOH solution to pH 14, and a white precipitate emerged. The product was then extracted from the mixture with EtOAc (3 × 30 mL), and the combined organic extracts were washed with brine and then dried over anhydrous Na2SO4. The product was then converted to the HCl salt by the addition of 1 M HCl in Et2O (27.6 mL, 27.6 mmol), and the solvents were concentrated to half volume in vacuo. The solution was then cooled to 0 °C, resulting in fractional crystallization of the trans stereoisomer as a white solid, which was then collected by filtration and washed with cold CH3CN (1.34 g, 22%). mp: 164–166 °C (lit.(J. Med. Chem.1998, 41, 760– 77)
the metabolite of the present invention is selected from:
EXAMPLESThe metabolites of the present invention were synthetized according to the following procedures:Example 1Trans-1-{4-[2-[4-(2,3-dichlorophenyl)-1-oxo-piperazin-1-yl]-ethyl]-cyclohexyl}-3,3-dimethyl-urea (compound D)
0.8 g (1.6 mmol) trans-1-{4-[2-[4-(2,3-dichlorophenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-3,3-dimethyl-urea was dissolved in dichloromethane (60 ml). A solution of 0.54 g (2.4 mmol) 3-chloro-perbenzoic acid in dichloromethane (10 ml) was dropped in and the reaction mixture stirred for 24 hours at room temperature. The reaction was monitored by TLC. The solution was washed twice with saturated NaHCO3 solution, the organic layer dried and evaporated in vacuo. Flash chromatography gave 0.45 g (63.3%) of the title compound melting at 175-8° C.
Example 2Trans-1-{4-[2-[4-(2,3-dichloro-4-hydroxy-phenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-3,3-dimethyl-urea (compound C)
0.92 g (2 mmol) trans-4-{2-[4-(2,3-dichloro-4-methoxy-phenyl)-piperazin-1-yl]-ethyl}-cyclohexyl-amine dihydrochloride was suspended in dichloromethane (60 ml), triethylamine (1.26 ml, 9 mmol) was added followed by 0.21 ml (2.3 mmol) N,N-dimethylcarbamoylchloride. The reaction mixture was stirred for 48 hours at room temperature. The solution was washed with water (2×10 ml), dried and evaporated in vacuo. Purification with flash chromatography gave 0.66 g trans-1-{4-[2-[4-(2,3-dichloro-4-methoxy-phenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-3,3-dimethyl-urea, melting at 196-8° C. This product was dissolved in dichloromethane (60 ml), then 6.4 ml (6.4 mmol) borontribromid solution (1M in CH2Cl2) was dropped in at 5° C. and the mixture stirred at room temperature for 24 hours. The reaction was monitored by TLC. 4 ml methanol was added, followed by 25 ml saturated NaHCO3 solution. After separation the organic layer was dried and evaporated in vacuo. Purification with flash chromatography gave 0.4 g of the title compound, melting at 278-80° C.
Example 3Trans-1-{4-[2-[4-(2,3-dichloro-4-hydroxy-phenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-3-methyl-urea (compound B)
1.38 g (3 mmol) trans-4-{2-[4-(2,3-dichloro-4-methoxy-phenyl)-piperazin-1-yl]-ethyl}-cyclohexyl-amine dihydrochloride was suspended in dry dichloromethane (100 ml), triethylamine (1.72 ml, 12.4 mmol) was added and 0.34 g (1.14 mmol) triphosgene dissolved in dichloromethane was dropped in. After one hour stirring at room temperature methylamine (33% solution in ethanol) was added and the stirring was continued for 20 hours. The mixture was evaporated. 20 ml water was added, the precipitate filtered, washed with water, dried. Recrystallizing the product from methanol gave trans-1-{4-[2-[4-(2,3-dichloro-4-methoxy-phenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-3-methyl-urea (0.86 g, 65%) melting above 250° C. This product was dissolved in dichloromethane (60 ml), then 10 ml (10 mmol) borontribromid solution (1M in CH2Cl2) was dropped in at 5° C. and the mixture stirred at room temperature for 24 hours. The reaction was monitored by TLC. 4 ml methanol was added and the mixture evaporated. 35 ml saturated NaHCO3 solution was added. The precipitate was filtered, washed with water and dried, recrystallized from methanol giving 0.34 g of title compound, melting at 237-41° C.
Example 4Trans-1-{4-[2-[4-(2,3-dichloro-4-hydroxy-phenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-urea (compound A)
1.38 g (3 mmol) trans-4-{2-[4-(2,3-dichloro-4-methoxy-phenyl)-piperazin-1-yl]-ethyl}-cyclohexyl-amine dihydrochloride was suspended in dry dichloromethane (100 ml), triethylamine 1.72 ml, 12.4 mmol) was added and 0.34 g (1.14 mmol) triphosgene dissolved in dichloromethane was dropped in. After one hour stirring at room temperature ammonia (20% solution in methanol) was added and the stirring was continued for 20 hours. The mixture was evaporated. 20 ml water was added, the precipitate filtered, washed with water, dried. Recrystallizing the product from methanol gave 0.86 g trans-1-{4-[2-[4-(2,3-dichloro-4-methoxy-phenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-urea melting above 250° C. This product was dissolved in dichloromethane (60 ml), then 10 ml (10 mmol) borontribromid solution (1M in CH2Cl2) was dropped in at 5° C. and the mixture stirred at room temperature for 24 hours. The reaction was monitored by TLC. 4 ml methanol was added and the mixture evaporated. 35 ml saturated NaHCO3 solution was added. The precipitate was filtered, washed with water and dried, recrystallized from methanol giving 0.37 g of title compound, melting at 195-8° C.
Kiss B; Horváth A; Némethy Z; Schmidt E; Laszlovszky I; Bugovics G; Fazekas K; Hornok K; Orosz S; Gyertyán I; Agai-Csongor E; Domány G; Tihanyi K; Adham N; Szombathelyi Z (2010). “Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile”. The Journal of Pharmacology and Experimental Therapeutics333 (1): 328–340. doi:10.1124/jpet.109.160432. PMID20093397.
Gründer G (2010). “Cariprazine, an orally active D2/D3 receptor antagonist, for the potential treatment of schizophrenia, bipolar mania and depression”. Current Opinion in Investigational Drugs11 (7): 823–832. PMID20571978.
Citrome, L (February 2013). “Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability”. Expert Opinion on Drug Metabolism and Toxicology9 (2): 193–206. doi:10.1517/17425255.2013.759211. PMID23320989.
Citrome L (February 2013). “Cariprazine in schizophrenia: clinical efficacy, tolerability, and place in therapy”. Adv Ther30 (2): 114–26. doi:10.1007/s12325-013-0006-7. PMID23361833.
Veselinović T, Paulzen M, Gründer G (November 2013). “Cariprazine, a new, orally active dopamine D2/3 receptor partial agonist for the treatment of schizophrenia, bipolar mania and depression”. Expert Rev Neurother13 (11): 1141–59. doi:10.1586/14737175.2013.853448. PMID24175719.
Citrome, L (February 2013). “Cariprazine in Schizophrenia: Clinical Efficacy, Tolerability, and Place in Therapy”. Advances in Therapy30 (2): 114–126. doi:10.1007/s12325-013-0006-7.PMID23361833.
Domany, G.
Discovery of novel dopamine D3/D2 ligands for the treatment of schizophrenia
234th ACS Natl Meet (August 19-23, Boston) 2007, Abst MEDI 383
crystalline form of trans-1 {4-[2-[4-(2,3-dichlorophenyl)-piperazin-1-yl]-ethyl]-cyclohexyl}-3,3-dimethyl-urea hydrochloride (Form III) Cariprazine {RGH-188); Dysfunction of the dopaminergic neurotransmitter system is involved in the pathology of several neuropsychiatric and neurodegenerative disorders
I was sitting in the lobby of my accountant’s office, flipping absentmindedly through a magazine when she walked in. I’ve never had a visceral reaction as when I saw her walk through that door. There was just something about her; I felt head over heels… My heart started racing and I had butterflies in my stomach…
This is the amazing time when you are truly love-struck. With an irresistible cocktail of chemicals, our brain entices us to fall in love. But is it really us or is it yet another nature’s trick to keep our species alive?
Scientists agree that there are three stages and processes in love:
Stage 1 – Attraction: Dopamine and Adrenaline
When you fall in love, your brain starts sending signals before you can even blink. Your heart races and palms sweat: adrenaline is getting released from neurons. Then, when you are close to your…
A convenient and simple PdCl2-based hydrogenation catalyst has been developed. The liquid, air, and moisture stable precursor is pumped into the reactor where it is temporarily immobilized and reduced on the channel surface into Pd(0), providing a constant high activity for hydrogenation reaction. The catalyst is leached with time, avoiding any kind of clogging problems during long time runs.
GSK-557296 is being developed in early clinical studies at GlaxoSmithKline for enhancement of embryo and or blastocyst implantation in women undergoing IVF treatment. The product has been in phase II clinical development for the treatment of premature ejaculation.
Preterm labor is a major clinical problem leading to death and disability in newborns and accounts for 10% of all births and causes 70% of all infant mortality and morbidity.
Oxytocin (OT) is a potent stimulant of uterine contractions and is responsible for the initiation of labor via the interaction with the OT receptors in the mammalian uterus. OT antagonists have been shown to inhibit uterine contractions and delay preterm delivery. So there is increasing interest in OT antagonists because of their potential application in the prevention of preterm labor. Although several tocolytics have already been approved in clinical practice, they have harmful maternal or fetal side effects.
The first clinically tested OT antagonist atosiban has a much more tolerable side effect profile and has recently been approved for use in Europe. However, atosiban is a peptide and a mixed OT/vasopressin V1a receptor antagonist that has to be given by iv infusion and is not suitable for long-term maintenance treatment, as it is not orally bioavailable.
Hence there has been considerable interest in overcoming the shortcomings of the peptide OT antagonists by identifying orally active nonpeptide OT antagonists with a higher degree of selectivity toward the vasopressin receptors (V1a, V1b, V2) with good oral bioavailability. Although several templates have been investigated as potential selective OT antagonists, few have achieved the required selectivity for the OT receptor vs the vasopressin receptors combined with the bioavailability and physical chemical properties required for an efficacious oral drug.
Therefore our objective was to design a potent, orally active OT antagonist with high levels of selectivity over the vasopressin receptor with good oral bioavailability in humans that would delay labor safely by greater than seven days and with improved infant outcome, as shown by a reduced combined morbidity score.
A suspension of {(3R,6R)-3-(2,3-dihydro-1H-inden-2-yl)-6-[(1S)-1-methylpropyl]-2,5-dioxo-1-piperazinyl}(2,6-dimethyl-3-pyridinyl)acetic acid hydrochloride (5.0 g, 10.3 mmol) (intermediate 5) in dry dichloromethane (50 ml) was treated with 1,1-carbonyldiimidazole (2.6 g, 16 mmol) and the reaction mixture was stirred under nitrogen for 18 hours. Morpholine (4.8 ml, 55 mmol) was added and the resultant solution was left to stand under nitrogen for 18 hours. The solvent was removed in vacuo and the residue was separated between ethyl acetate and water. The organic phase was washed with brine and dried over anhydrous magnesium sulphate. The solvent was removed in vacuo and the residue was dissolved in dichloromethane. This was applied to a basic alumina cartridge (240 g) and eluted using a gradient of 0-7.5% methanol in diethyl ether (9CV), 7.5-10% methanol in diethyl ether (1CV) and 10% methanol in diethyl ether (1CV). The required fractions were combined and evaporated in vacuo to give (3R,6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione as a white solid (2.4 g, 45%).
This invention relates to novel crystalline forms of (3R, 6R)-3-(2,3-dihydro-1 H- inden-2-yl)-1 -[(1 R)-1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1 S)-1 – methylpropyl]-2,5-piperazinedione benzenesulfonate salt, processes for their preparation, pharmaceutical compositions containing them and to their use in medicine. The benzenesulfonate salt of Compound A is represented by the following structure:
In one aspect, the present invention provides a crystalline form of {3R, 6R)-3- (2,3-dihydro-1 H-inden-2-yl)-1 -[(1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2- oxoethyl]-6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate, wherein said crystalline form provides an X-ray powder diffraction pattern substantially in accordance with Figure 1 .
In another aspect, the invention encompasses a crystalline form of (3R, 6R)-3- (2,3-dihydro-1 H-inden-2-yl)-1 -[(1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2- oxoethyl]-6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate, wherein said crystalline form is characterized by an X-ray powder diffraction pattern comprising the peaks:
In an additional aspect, the invention includes a crystalline form of {3R, 6R)-3- (2,3-dihydro-1 H-inden-2-yl)-1 -[(1 R)-1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2- oxoethyl]-6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate hydrate, wherein said compound is characterized by an X-ray powder diffraction pattern substantially in accordance with Figure 2.
In certain aspects, the invention encompasses a crystalline form of (3R, 6R)-3- (2,3-dihydro-1 H-inden-2-yl)-1 -[(1 R)-1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2- oxoethyl]-6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate hydrate, wherein said compound is characterized by an X-ray powder diffraction pattern substantially in accordance with Figure 2 In one aspect, the invention also provides a crystalline form of {3R, 6R)-3-(2,3- dihydro-1 H-inden-2-yl)-1-[(1 R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]- 6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate hydrate, wherein said crystalline form is characterized by an X-ray powder diffraction pattern comprising the peaks:
Experimental
Process Scheme
Stage 4
Acetone / Water Recrystallisation
Compound A-form I Ste8e 5 Besylate salt
MW 676.83 Acetone / Water
Recrystallisation MW 676.83 Process description for isolation of Compound A-Form 1
Stage 0
methyl d-alloisoleucinate hydrochloride (Compound 2) was charged to ethyl acetate. A solution of potassium carbonate in water was then added. The mixture was then stirred vigorously at room temperature for 1 hour. The two layers were separated and the aqueous layer further extracted with ethyl acetate. The organic layers were combined and washed with brine. The organic layers were then concentrated in vacuo and filtered to yield methyl D-alloisoleucinate (Compound 3) as a pale yellow oil.
Stage 1
2,6-dimethyl-3-pyridinecarbaldehyde (Compound 4) in methanol at ambient temperature was treated with D-alloisoleucinate (Compound 3) in methanol followed by 2,2,2- trifluoroethanol and the reaction mixture was warmed to 40°C. When formation of the intermediate imine (methyl A/-[(2,6-dimethyl-3-pyridinyl)methylidene]-D-alloisoleucine) was complete Compound 5 was added followed by 1-isocyano-2- [(phenylmethyl)oxy]benzene (Compound 6) and the reaction mixture was stirred at 40°C until formation of Compound 7 was deemed complete.
Stage 2
Palladium on carbon catalyst was treated with a solution of Compound 7 in methanol and 2,2,2-trifluoroethanol and diluted with acetic acid. The vessel was purged with nitrogen and the reaction mixture warmed to 50°C and hydrogenated at 4.0-4.5 barg. When the reaction was deemed complete it was cooled to ambient temperature and the catalyst removed by filtration and washed through with methanol. The organic solution of 2- {(3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-6-[(1 S)-1 -methylpropyl]-2,5-dioxo-1-piperazinyl}- 2-(2,6-dimethyl-3-pyridinyl)-/\/-(2-hydroxyphenyl)acetamide (Compound 8) was concentrated at reduced pressure and then diluted with /‘so-propyl acetate and concentrated at reduced pressure.
The residue was diluted with /‘so-propyl acetate and washed with aqueous ammonia. The aqueous phase was separated and extracted into another portion of /‘so-propyl acetate. The combined organic phases were washed with water, concentrated by distillation at reduced pressure, diluted with /‘so-propyl acetate and concentrated by distillation at reduced pressure, to leave a concentrated solution of 2-{(3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-6-[(1 S)-1 -methylpropyl]-2,5-dioxo-1 – piperazinyl}-2-(2,6-dimethyl-3-pyridinyl)-/\/-(2-hydroxyphenyl)acetamide (Compound 8). The product was finally dissolved in 1 ,4-dioxane for the next stage and stored into drums.
Stage 3 Solution of 2-{(3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-6-[(1 S)-1 -methylpropyl]-2,5-dioxo-1 – piperazinyl}-2-(2,6-dimethyl-3-pyridinyl)-/\/-(2-hydroxyphenyl)acetamide (Compound 8) in 1 ,4-dioxane was treated with 1 ,1 ‘-carbonyl diimidazole at ambient temperature to form a solution containing (3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-1 -[1-(2,6-dimethyl-3-pyridinyl)- 2-oxo-2-(2-oxo-1 ,3-benzoxazol-3(2H)-yl)ethyl]-6-[(1 S)-1 -methylpropyl]-2,5- piperazinedione (Compound 9).
In a separate vessel morpholine in 1 ,4-dioxane was heated to 80-85°C. The solution containing (3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-1-[1 – (2,6-dimethyl-3-pyridinyl)-2-oxo-2-(2-oxo-1 ,3-benzoxazol-3(2H)-yl)ethyl]-6-[(1 S)-1- methylpropyl]-2,5-piperazinedione (Compound 9) was slowly added to the morpholine in 1 ,4-dioxane. The reaction mixture was stirred for one hour at 80-85°C and cooled before concentration by distillation at reduced pressure.
The concentrated solution of Compound A was diluted with /‘so-propyl acetate and washed with aqueous sodium hydroxide followed by water. The /so-propyl acetate solution of COMPOUND A was then concentrated by distillation at reduced pressure and cooled to ambient temperature. The concentrated solution of Compound A was then diluted with acetone and treated with benzenesulfonic acid and seed crystals were added and the reaction mixture stirred until crystallisation occurred. The slurry of Compound A besylate was heated to 50°C, a temperature cycle was performed, and finally the slurry was cooled to -10°C and isolated by filtration. The filter cake was washed with cold acetone (-10°C) to give Compound A besylate (intermediate grade) as a wet cake.
Yield: 44% from Compound 5
39% from Compound 5
Stage 4
Compound A besylate (intermediate grade wet cake, Compound A besylate ) was suspended in acetone (17.4 vol including acetone content of wet cake) and heated to 55- 60°C. Water (0.66 vol) was added until dissolution was observed. The reaction mixture was then filtered into another vessel and the lines washed through with acetone (3.2 vol). The temperature of the reaction mixture was adjusted to 45-50°C before the addition of seed crystals (0.00025wt). When crystallisation was complete the reaction mixture was cooled to 20-25°C and stirred at 20-25°C for 30mins.
The reaction mixture was heated to 45-50°C and stirred at 45-50°C for 30mins. The reaction mixture was cooled to 20-25°C and stirred at 20-25°C for 30mins. The reaction mixture was heated to 45-50°C and stirred at 45-50°C for 30mins. The reaction mixture was cooled to -3-2°C over 4.5 h and stirred for at least 1 h before the product was isolated by filtration. The wet cake was washed with acetone at 0°C (3 x 3.1 vol) and blown dry before being unloaded. COMPOUND A besylate was dried at 50°C under vacuum for 3 days. Compound A besylate was then milled. Yield: 66% Stage 5
Compound A besylate (OBU-D-02) was suspended in acetone (8 vol) and water (1 .1 vol) and heated to 48-52°C until dissolution was observed. The reaction mixture was then filtered into another vessel and the lines washed through with acetone (2 vol). The reaction mixture was cooled to 20-25°C before the addition of Form 1 seed crystals (0.0025wt). When crystallisation was complete the reaction mixture was cooled to 0-5°C over 1 h and stirred at 0-5°C for 30mins. The reaction mixture was heated to 20-25°C and stirred at 20-25°C for 30mins. The reaction mixture was cooled to 0-5°C over 1 h and stirred at 0-5°C for 30mins.
The reaction mixture was heated to 20-25°C and stirred at 20-25°C for 30mins. The reaction mixture was cooled to -12— 8°C over 3.5 h and stirred for 15 h before the product was isolated by filtration. The wet cake was washed with acetone at -10°C (2 x 3 vol) and blown dry before being unloaded. Compound A besylate was dried at ambient temperature under vacuum for 6 days with a wet nitrogen bleed to afford Form 1 . Compound A besylate was then milled. Yield: 67%
Recrystallisation of Compound A besylate anhydrate (Form 2)
Besylate salt ………………………………………………………………Besylate salt
C30H38 4O4■ C6H603S C30H38 4O4■
MW 676.83 MW 676.83
COMPOUND A besylate is charged to the vessel and treated with methyl ethyl ketone (MEK) (8vol) and water (0.35vol) and the solution heated until dissolution is observed (ca. 55-60°C). The solution is then filtered and recharged to the vessel. Pressure is then reduced to 650mbar and the reaction mixture heated further to distil out solvent. MEK is added at the same rate as solvent is removed by distillation keeping the reaction mixture volume constant. After 4 volumes of MEK have been added the reaction mixture is treated with Form 2 seed crystals (2%wt) and the distillation continued in the same manner until another 7 volumes of MEK has been added. The vacuum is then released to an atmospheric pressure of nitrogen and the temperature of the reaction mixture adjusted to 65°C. The reaction mixture is then filtered and washed with pre heated MEK (2vol at 65°C). The purified COMPOUND A besylate anhydrate is then sucked dry and dried further in a vacuum oven at 65°C at l OOmbar with a nitrogen bleed. Yield 89%
A six-stage stereoselective synthesis of indanyl-7-(3′-pyridyl)-(3R,6R,7R)-2,5-diketopiperazines oxytocin antagonists from indene is described. SAR studies involving mono- and disubstitution in the 3′-pyridyl ring and variation of the 3-isobutyl group gave potent compounds (pKi > 9.0) with good aqueous solubility. Evaluation of the pharmacokinetic profile in the rat, dog, and cynomolgus monkey of those derivatives with low cynomolgus monkey and human intrinsic clearance gave 2′,6′-dimethyl-3′-pyridyl R–sec-butyl morpholine amide Epelsiban (69), a highly potent oxytocin antagonist (pKi = 9.9) with >31000-fold selectivity over all three human vasopressin receptors hV1aR, hV2R, and hV1bR, with no significant P450 inhibition. Epelsiban has low levels of intrinsic clearance against the microsomes of four species, good bioavailability (55%) and comparable potency to atosiban in the rat, but is 100-fold more potent than the latter in vitro and was negative in the genotoxicity screens with a satisfactory oral safety profile in female rats.
3.78–3.39 and 3.14–2.80 (m, 13H, 8× morpholinyl-H, indanyl-3H, –1H, –2H)),
2.79 and 2.78 (2s, 6H, pyridyl-2Me, -6Me),
1.85–1.74 (m, 1H, CHHMe),
1.59–1.48 (m, 1H, CHHMe),
1.15–1.01 (m, 1H, CHMeCH2),
0.92 (d, J =6.3 Hz, 3H, CHMe),
0.85 (t, J = 7.3 Hz, 3H, CH2Me).
LCMS m/z 519 MH+ single components, tR2.72 min;
circular dichroism (CH3CN) λmax 225.4 nm, dE −15.70, E15086; λmax 276 nm, dE 3.82, E5172.
HRMS calcd for C30H38N4O4 (MH+) 519.2971, found 519.2972.
Anal. (C30H38N4O4·C6H6O3S·3.0H2O) C, H, N, S.
References
Borthwick AD, Liddle J, Davies DE, Exall AM, Hamlett C, Hickey DM, Mason AM, Smith IE, Nerozzi F, Peace S, Pollard D, Sollis SL, Allen MJ, Woollard PM, Pullen MA, Westfall TD, Stanislaus DJ (January 2012). “Pyridyl-2,5-diketopiperazines as potent, selective, and orally bioavailable oxytocin antagonists: synthesis, pharmacokinetics, and in vivo potency”. Journal of Medicinal Chemistry55 (2): 783–96. doi:10.1021/jm201287w. PMID205501.
2 Borthwick, A. D.; Liddle, J. (January 2013). “Retosiban and Epelsiban: Potent and Selective Orally available Oxytocin Antagonists”. In Domling, A. Methods and Principles in Medicinal Chemistry: Protein-Protein Interactions in Drug Discovery. Weinheim: Wiley-VCH. pp. 225–256. ISBN978-3-527-33107-9.
Crystalline forms of (3R, 6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione
Crystalline forms of (3R, 6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione
The WordPress.com stats helper monkeys prepared a 2014 annual report for this blog.
Here’s an excerpt:
The Louvre Museum has 8.5 million visitors per year. This blog was viewed about 350,000 times in 2014. If it were an exhibit at the Louvre Museum, it would take about 15 days for that many people to see it.
Gellért Sipos1, Viktor Gyollai1, Tamás Sipőcz1, György Dormán1, László Kocsis1, Richard V. Jones1, Ferenc Darvas1
1ThalesNano Zahony u. 7 1031 Budapest Hungary
László Kocsis holds a Masters degree in Bioorganic Chemistry from the Eötvös Lóránd University in Budapest, Hungary (2001) and a PhD in Organic Chemistry from the Eötvös Lóránd University in Budapest, Hungary (2008). In 2004 he began working as a research chemist at the Reanal Finechemical Company in Budapest, Hungary. He became the Head of the R&D laboratory in 2007 and a manager of production in 2008. In 2011 he joined ThalesNano Inc. as Head of Chemistry. He has experience in organic chemistry, with emphasis on sythesis of amino acid derivatives and peptides, focusing mainly on the following subjects: structure – relationship studies in opiod peptides, methodological studies in the internal solubilization of the sekf-aggregating peptides, industrial scale sythesis of protected amino acid derivatives, and peptides, heterogeneous catalysis, reactions under continuous flow conditions. He is the co-author of 10 pulications and a member of the European Peptide Society.
Abstract
The atom economy concept is one of the earliest recognition for green and sustainable aspects of organic synthesis. Over the years, novel technologies emerged that made this important feature of reactions into practice. Continuous-flow devices increased the efficiency of the chemical transformations with novel process windows (high T, high p and heterogeneous packed catalysts etc.) and increased safety which turned the attention to reexamine old, industrial processes. Oxidation can be performed under flow catalytic conditions with molecular oxygen; alcohols can be oxidized to carbonyl compounds with high atom economy (AE = 87 %). Using O2 and 1 % Au/TiO2, alcohol oxidation in flow was achieved with complete conversion and >90 % yield. N-alkylation is another good example for achieving high atom economy. Under flow catalytic conditions (Raney Ni), amines were successfully reacted with alcohols directly (AE = 91 %) with >90 % conversion and selectivity. In both examples, the effective residence time was less than 1 min. These two examples demonstrate the significant contribution of flow technology to the realization of key principles in green and sustainable chemistry.
ThalesNano Nanotechnology Inc, GraphisoftPark. Záhony u. 7. H-1031 Budapest HUNGARY

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....








































![N'-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide,4-methylbenzenesulfonic acid,hydrate NMR spectra analysis, Chemical CAS NO. 1229194-11-9 NMR spectral analysis, N'-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide,4-methylbenzenesulfonic acid,hydrate H-NMR spectrum](https://images-blogger-opensocial.googleusercontent.com/gadgets/proxy?url=http%3A%2F%2Fpic11.molbase.net%2Fnmr%2Fnmr_image%2F2015-01-11%2F001%2F717%2F1717235_1h.png&container=blogger&gadget=a&rewriteMime=image%2F*)
![N'-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide,4-methylbenzenesulfonic acid,hydrate NMR spectra analysis, Chemical CAS NO. 1229194-11-9 NMR spectral analysis, N'-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide,4-methylbenzenesulfonic acid,hydrate C-NMR spectrum](https://images-blogger-opensocial.googleusercontent.com/gadgets/proxy?url=http%3A%2F%2Fpic11.molbase.net%2Fnmr%2Fnmr_image%2F2015-01-11%2F001%2F717%2F1717235_13c.png&container=blogger&gadget=a&rewriteMime=image%2F*)



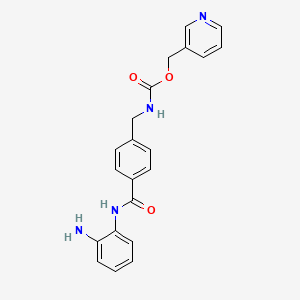





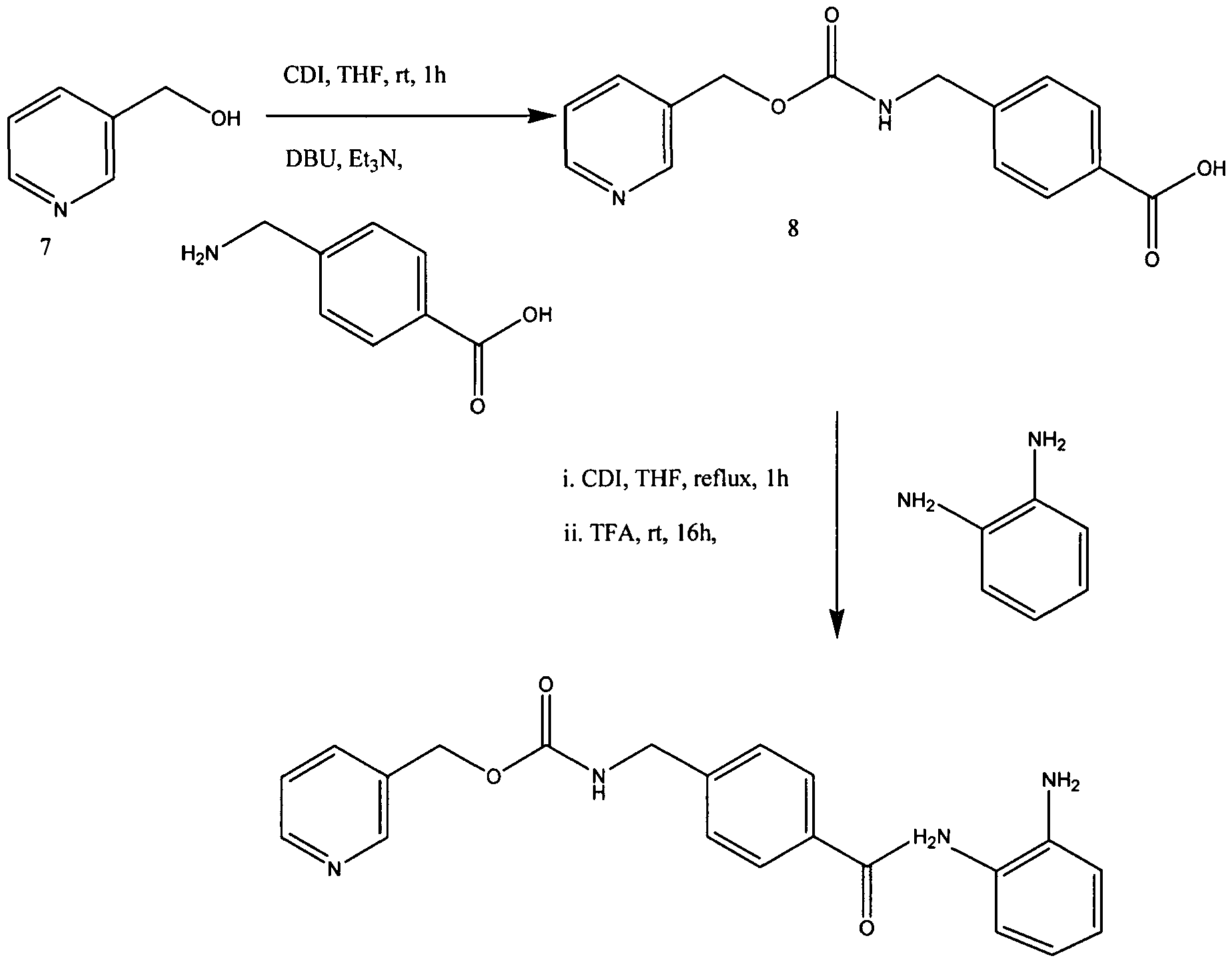


















 NEWS………….DUBLIN and BUDAPEST, Hungary, Jan. 6, 2015 /PRNewswire/ — Actavis plcand Gedeon Richter Plc. today announced that the U.S. Food and Drug Administration (FDA) has acknowledged receipt of Actavis’ New Drug Application (NDA) resubmission for its atypical antipsychotic cariprazine, a potent dopamine D3/D2 receptor partial agonist with preferential binding to D3 receptors. The Prescription Drug User Fee Act (PDUFA) date is expected to be in the second quarter of 2015…….
NEWS………….DUBLIN and BUDAPEST, Hungary, Jan. 6, 2015 /PRNewswire/ — Actavis plcand Gedeon Richter Plc. today announced that the U.S. Food and Drug Administration (FDA) has acknowledged receipt of Actavis’ New Drug Application (NDA) resubmission for its atypical antipsychotic cariprazine, a potent dopamine D3/D2 receptor partial agonist with preferential binding to D3 receptors. The Prescription Drug User Fee Act (PDUFA) date is expected to be in the second quarter of 2015…….










































 Click here to see the complete report.
Click here to see the complete report.








 László Kocsis holds a Masters degree in Bioorganic Chemistry from the Eötvös Lóránd University in Budapest, Hungary (2001) and a PhD in Organic Chemistry from the Eötvös Lóránd University in Budapest, Hungary (2008). In 2004 he began working as a research chemist at the Reanal Finechemical Company in Budapest, Hungary. He became the Head of the R&D laboratory in 2007 and a manager of production in 2008. In 2011 he joined ThalesNano Inc. as Head of Chemistry. He has experience in organic chemistry, with emphasis on sythesis of amino acid derivatives and peptides, focusing mainly on the following subjects: structure – relationship studies in opiod peptides, methodological studies in the internal solubilization of the sekf-aggregating peptides, industrial scale sythesis of protected amino acid derivatives, and peptides, heterogeneous catalysis, reactions under continuous flow conditions. He is the co-author of 10 pulications and a member of the European Peptide Society.
László Kocsis holds a Masters degree in Bioorganic Chemistry from the Eötvös Lóránd University in Budapest, Hungary (2001) and a PhD in Organic Chemistry from the Eötvös Lóránd University in Budapest, Hungary (2008). In 2004 he began working as a research chemist at the Reanal Finechemical Company in Budapest, Hungary. He became the Head of the R&D laboratory in 2007 and a manager of production in 2008. In 2011 he joined ThalesNano Inc. as Head of Chemistry. He has experience in organic chemistry, with emphasis on sythesis of amino acid derivatives and peptides, focusing mainly on the following subjects: structure – relationship studies in opiod peptides, methodological studies in the internal solubilization of the sekf-aggregating peptides, industrial scale sythesis of protected amino acid derivatives, and peptides, heterogeneous catalysis, reactions under continuous flow conditions. He is the co-author of 10 pulications and a member of the European Peptide Society.




{kind=link}