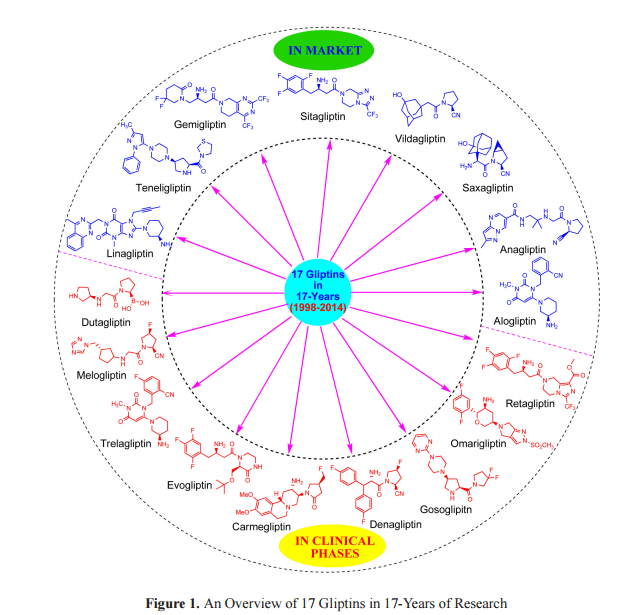
XP 23829 from Xenoport is an interesting molecule and as on 27 July 2014, I did not find conclusive evidence
See some structures below
 Not sure about the structure of XP 23829
Not sure about the structure of XP 23829
OR
 Best fit
Best fit
OR

Not sure?
(N,N-dimethylcarbamoyl)methyl methyl(2E)but-2-ene-1,4-dioate.
OR

I AM NOT SURE ABOUT THIS ONE ALSO????????
As Football worldcup2014 goes on in Brazil

A thought for it is due…………
……………………………………………………
Best fit is probably is as shown below, and there are reasons
(N,N- Diethylcarbamoyl)methyl methyl (2E)but-2-ene-l,4-dioate 
Introduction
(N,N-Diethylcarbamoyl)methyl methyl (2E)-but-2-ene-1,4-dioate
C11 H17 N O5, mw 243.13
M.p.: 53-56 °C.
1 H NMR (CDCI3, 400 MHz): δ 6.99-6.90 (m, 2H), 4.83 (s, 2H), 3.80 (s, 3H), 3.39 (q, J = 1.1 Hz, 2H), 3.26 (q, J = 7.2 Hz, 2H), 1 .24 (t, J = 7.2 Hz, 3H), 1 .14 (t, J = 7.2 Hz, 3H). MS (ESI): m/z 244.13 (M+H)+.
Cas…….1208229-58-6
XP-23829 PROBABLE
For the treatment of moderate-to-severe chronic plaque-type psoriasis.
XP-H-093
US8148414 Basic patent
Basic patent
Xenoport, Inc. Innovator
XenoPort has initiated a phase II trial of XP-23829, a proprietary investigational next-generation fumaric acid product candidate (ClinicalTrials.gov Identifier NCT02173301). The multicenter, randomized, double-blind, placebo-controlled study is designed to assess the efficacy and safety of XP-23829 as a potential treatment of patients with moderate to severe chronic plaque-type psoriasis. XenoPort expects to enroll approximately 200 subjects in this trial, which is being conducted in the U.S. The study will include a screening and washout phase of up to 4 weeks, a 12-week treatment phase and a 4-week post-treatment phase. Eligible study subjects will be randomized to placebo or one of three treatment arms of XP-23829: 400 or 800 mg once daily or 400 mg twice daily. The primary endpoint will examine the percent change in Psoriasis Area and Severity Index (PASI) score from baseline at the end of week 12. Secondary endpoints will include the proportion of subjects who achieve a reduction of 75% or greater from baseline in PASI (PASI75) score and subjects who achieve a Static Physicians Global Assessment score of “clear” or “almost clear.” Topline results are expected in the third quarter of 2015 (XenoPort News Release).
XP23829 — A Prodrug of Monomethyl Fumarate
Our third product candidate, XP23829, is in Phase 1 clinical development. Provided we are able to demonstrate the safety and desired pharmacokinetic, or PK, profile of XP23829 in our Phase 1 trials, we believe that XP23829 could be a potential treatment of patients with RRMS, psoriasis and/or certain other disorders where the mechanism of action of XP23829 may be relevant. For example, we are exploring the potential of XP23829 to protect against neurodegeneration in experimental preclinical models of Parkinson’s disease through a grant from The Michael J. Fox Foundation. We hold a composition-of-matter patent and a formulation patent in the United States on XP23829 and hold patents or pending patent applications directed to the XP23829 methods of synthesis and use in the United States. We have also filed applications directed to the XP23829 composition of matter and methods of synthesis and use in other jurisdictions.
Prodrug Background
XP23829 is a fumaric acid ester compound and a patented prodrug of MMF. Fumaric acid ester compounds have shown immuno-modulatory and neuroprotective effects in cell-based systems and preclinical models of disease. A product containing a combination of fumaric acid ester compounds, known as Fumaderm, is approved in Germany for the treatment of psoriasis. Tecfidera (a formulation of DMF, also known as BG-12) from Biogen Idec Inc. is another fumaric acid ester prodrug that converts to MMF in the body. Phase 3 clinical trials of Tecfidera as a potential treatment for RRMS showed statistically significant benefits of Tecfidera versus placebo. Tecfidera is currently under U.S. regulatory review as a potential treatment for RRMS.
Our Prodrug
XP23829 is a novel prodrug of MMF that we believe may provide improved tolerability and efficacy compared to DMF. In preclinical studies that compared molar equivalent doses of XP23829 to DMF, XP23829 provided higher blood levels of the biologically active molecule MMF and a similar or greater degree of efficacy in MS and psoriasis animal models. Toxicology studies conducted in two species showed that XP23829 caused less stomach irritation when compared to DMF.
Phase 1 Clinical Trial in Healthy Volunteers
In October 2012, we reported favorable preliminary results from our first Phase 1 clinical trial in healthy adults designed to assess the pharmacokinetics, safety and tolerability of single doses of four different formulations of XP23829. The trial was a randomized, double-blind, two-period crossover, food effect comparison clinical trial of XP23829. Sixty subjects were assigned to five cohorts of 12, with each cohort receiving one of four different formulations of XP23829 or placebo. The trial demonstrated that administration of XP23829 resulted in the expected levels of MMF in the blood. As anticipated, the four formulations produced


April 4, 2012
http://investor.xenoport.com/releasedetail.cfm?ReleaseID=708145
XenoPort Awarded U.S. Patent Directed to Composition and Formulations of XP23829, a Novel Fumarate Analog for the Potential Treatment of Relapsing-Remitting Multiple Sclerosis and Psoriasis
SANTA CLARA, Calif.–(BUSINESS WIRE)–Apr. 4, 2012– XenoPort, Inc. (Nasdaq: XNPT) announced today that it was awarded U.S. Patent 8,148,414 for “Prodrugs of Methyl Hydrogen Fumarate, Pharmaceutical Compositions Thereof, and Methods of Use.” The term of the patent extends until 2029, subject to potential Hatch-Waxman patent term extensions.
The patent is directed to the XP23829 compound, analogs thereof and formulations thereof. A related U.S. patent application directed to therapeutic uses of XP23829 is now pending.
XP23829 is a prodrug of methyl hydrogen fumarate, also known as monomethyl fumarate (MMF). In cell- and animal-based models, MMF has been shown to exhibit immuno-modulatory properties and inhibit damage from oxidative stress.
In XenoPort’s preclinical animal studies that compared molar equivalent doses of XP23829 to dimethyl fumarate (DMF), another prodrug of MMF, XP23829 demonstrated a greater degree of efficacy in animal models of both multiple sclerosis (MS) and psoriasis. Toxicology studies conducted in two species showed that XP23829 caused less stomach irritation compared to DMF.
XenoPort intends to file an Investigational New Drug Application (IND) for XP23829 for the treatment of relapsing remitting MS with the U.S. Food and Drug Administration (FDA) in the second quarter of 2012 and expects to initiate human clinical trials later this year.
XenoPort owns all rights to XP23829.
About XenoPort
XenoPort is a biopharmaceutical company focused on developing and commercializing a portfolio of internally discovered product candidates for the potential treatment of neurological disorders. Horizant® (gabapentin enacarbil) Extended-Release Tablets is XenoPort’s first FDA-approved product. GlaxoSmithKline holds commercialization rights and certain development rights for Horizant in the United States. Regnite® (gabapentin enacarbil) is approved for the treatment of moderate-to-severe primary restless legs syndrome in Japan. Astellas Pharma Inc. holds all development and commercialization rights for Regnite in Japan and five Asian countries. XenoPort holds all other world-wide rights and has co-promotion and certain development rights to gabapentin enacarbil in the United States. XenoPort’s pipeline of product candidates includes potential treatments for patients with postherpetic neuralgia, spasticity and Parkinson’s disease.
To learn more about XenoPort, please visit the company Website at http://www.XenoPort.com.
More info about this drug
SEE a patent
WO 2010022177
……………………………………….
WO 2013181451
http://www.google.com/patents/WO2013181451A1?cl=en
Scheme 5:
ONE OUT OF THESE
Example 6: (/V,/V-Diethylcarbamoyl)methyl methyl (2£)but-2-ene-1 ,4-dioate
[0138] Following general procedure A, methyl hydrogen fumarate (MHF) (0.39 g, 3.00 mmol) dissolved in NMP was reacted at about 55 °C with 2-chloro-/V,/V-diethylacetamide (0.44 g, 3.00 mmol) in the presence of CsHC03 (0.69 g, 3.60 mmol) to afford 0.37 g (51 % yield) of the title compound after purification by silica gel column chromatography (Biotage) using a mixture of ethyl acetate (EtOAc) and hexanes (1 :1 ) as eluent. M.p.: 53-56 °C. 1 H NMR (CDCI3, 400 MHz): δ 6.99-6.90 (m, 2H), 4.83 (s, 2H), 3.80 (s, 3H), 3.39 (q, J = 1.1 Hz, 2H), 3.26 (q, J = 7.2 Hz, 2H), 1 .24 (t, J = 7.2 Hz, 3H), 1 .14 (t, J = 7.2 Hz, 3H). MS (ESI): m/z 244.13 (M+H)+.
Example 7: Methyl 2-morpholin-4-yl-2-oxoethyl (2 £)but-2-ene-1 ,4-dioate
[0139] Following general procedure A, methyl hydrogen fumarate (MHF) (0.50 g, 3.84 mmol) dissolved in NMP was reacted at about 55 °C with 4-(chloroacetyl) morpholine (0.75 g, 4.61 mmol) in the presence of CsHC03 (0.89 g, 4.61 mmol) to afford 0.34 g (35% yield) of the title compound as a white solid after purification by mass-guided preparative HPLC and lyophilization. M.p.: 124 to 126°C; 1 H NMR (CDCI3, 400 MHz): δ 6.97-6.91 (m, 2H), 4.84 (s, 2H), 3.82 (s, 3H), 3.72-3.70 (m, 4H), 3.64-3.62 (m, 2H), 3.46-3.41 (m, 2H). MS (ESI): m/z 258.04 (M+H)+. Example 8: A/,A/-Dimethylcarbamoyl)methyl methyl (2E)but-2-ene-1 ,4-dioate

[0140] Following general procedure A, methyl hydrogen fumarate (MHF) (0.50 g, 3.84 mmol) dissolved in NMP was reacted at about 55 °C with /V,/V-dimethyl chloroacetamide (0.56 g, 4.61 mmol) in the presence of CsHC03 (0.89 g, 4.61 mmol). The crude material was precipitated out from a mixture of ethyl acetate (EtOAc) and hexanes (Hxn) (1 :1 ) to provide a white solid. This solid was further dissolved in dichloromethane (DCM) and the organic layer washed with water. After removal of the solvents 0.55 g (67% yield) of the title compound was obtained as a white solid. 1 H NMR (CDCI3, 400 MHz): δ 6.98- 6.90 (m, 2H), 4.84 (s, 2H), 3.80 (s, 3H), 2.99-2.97 (2s, 6H). MS (ESI): m/z 216 (M+H)+.
Example 9: Methyl (2-morpholino-4-ylethyl) fumarate
[0141] Following general Procedure A, methyl hydrogen fumarate (MHF) dissolved in NMP is reacted at about 55 °C with 4-(chloroethyl) morpholine (0.75 g, 4.61 mmol) in the presence of CsHC03 to afford the title compound after purification by mass-guided preparative HPLC and lyophilization. Example 10: Methyl (3-mor holino-4-ylpropyl) fumarate
[0142] Following the procedure of Methyl (2-morpholino-4-ylethyl) fumarate, and replacing 4-(chloroethyl) morpholine with 4-(chloropropyl) morpholine provides the title compound.
Example 11 : Methyl (4-morpholino-4-ylbutyl) fumarate
[0143] Following the procedure of Methyl (2-morpholino-4-ylethyl) fumarate, and replacing 4-(chloroethyl) morpholine with 4-(chlorobutyl) morpholine provides the title compound. Example 12: Methyl 5-morpholino-4-ylpentyl) fumarate
[0144] Following the procedure of Methyl (2-morpholino-4-ylethyl) fumarate, and replacing 4-(chloroethyl) morpholine with 4-(chloropentyl) morpholine provides the title compound. Example 13: (A/-cyclopropyl-W-ethylcarbamoyl)methyl methyl 2(E)but-2-ene-1 ,4-dioate
[0145] Following the general procedure A, methyl hydrogen fumarate (MHF) (38.7 g, 0.297 mol) suspended in toluene (100 mL) was reacted at about 80 °C with 2-chloro-/V-cyclopropyl- N-ethylacetamide (48 g, 0.297 mol) in the presence of W,/V-diisopropylethylamine (DIEA; 42.3 g, 57 mL, 0.327 mol) to afford 50 g (63.3%) of the title compound after recrystallization using methyl ferf-butyl ether. The crystalline compound had a melting point of 92.1 °C. 1 H NMR (CDCI3, 400 MHz): δ 7.01 -6.92 (m, 2H), 4.99 (s, 2H), 3.81 (s, 3H), 3.44 (q, J = 7.2 Hz, 2H), 2.69-2.66 (m, 1 H), 1 .14 (t, J = 7.2 Hz, 3H), 0.94-0.91 (m, 2H), 0.83-0.81 (m, 2H). MS (ESI): m/z 256.2 (M+H)+.
Example 14: (/V-cyclopropyl-/V-methylcarbamoyl)methyl methyl 2(E)but-2-ene-1 , 4- dioate
[0146] Following general procedure A, methyl hydrogen fumarate (MHF) (38.7 g, 0.40 mol) suspended in toluene (100 mL) was reacted at about 80 °C with 2-chloro-/V-cyclopropyl-/V- methylacetamide (60 g, 0.40 mol) in the presence of Ν,Ν-diisopropylethylamine (DIEA; 57.8 g, 78 mL, 0.44 mol) to afford 50 g (50.86%) of the title compound after recrystallization using methyl fe/t-butyl ether. The crystalline compound had a melting point of 93.6 °C. 1 H NMR (CDCI3, 400 MHz): δ 7.01 -6.91 (m, 2H), 5.01 (s, 2H), 3.82 (s, 3H), 2.94 (s, 3H), 2.73-2.68 (m, 1 H), 0.94-0.86 (m, 2H), 0.83-0.78 (m, 2H). MS (ESI): m/z 242.2 (M+H)+.
Example 15: Methyl 2-oxo-2-pyrrolidinylethyl 2(E)but-2-ene-1 ,4-dioate
[0147] Following general procedure A, methyl hydrogen fumarate (MHF) (20.78 g, 0.159 mol) suspended in toluene (60 mL) was reacted at about 80 °C with 2-chloro-1 -pyrrolidin-1 -yl- ethanone (23.5 g, 0.159 mol) in the presence of N,N-diisopropylethylamine (DIEA; 22.69 g, 31 .5 mL, 0.175 mol) to afford 24 g (62.3%) of the title compound after recrystallization using methyl fe/t-butyl ether. The crystalline compound had a melting point of 102.1 °C. 1 H NMR (CDCI3, 400 MHz): δ 7.00-6.92 (m, 2H), 4.75 (s, 2H), 3.81 (s, 3H), 3.53-3.49 (t, J = 6.8 Hz, 2H), 3.42-3.39 (t, J = 6.8 Hz, 2H), 2.20-1 .97 (m, 2H), 1 .91 -1 .82 (m, 2H). MS (ESI): m/z 242 (M+H)+.
………………………………
Patent
http://www.google.co.in/patents/US8148414
(I):
Example 1(N,N-Diethylcarbamoyl)methyl methyl(2E)but-2-ene-1,4-dioate (1)…………. best fit
Following general procedure A, methyl hydrogen fumarate (MHF) (0.39 g, 3.00 mmol) dissolved in NMP was reacted at ca. 55° C. with 2-chloro-N,N-diethylacetamide (0.44 g, 3.00 mmol) in the presence of CsHCO3 (0.69 g, 3.60 mmol) to afford 0.37 g (51% yield) of the title compound (1) after purification by silica gel column chromatography (Biotage) using a mixture of ethyl acetate (EtOAc) and hexanes (1:1) as eluent. M.p.: 53-56° C. 1H NMR (CDCl3, 400 MHz): δ 6.99-6.90 (m, 2H), 4.83 (s, 2H), 3.80 (s, 3H), 3.39 (q, J=7.2 Hz, 2H), 3.26 (q, J=7.2 Hz, 2H), 1.24 (t, J=7.2 Hz, 3H), 1.14 (t, J=7.2 Hz, 3H). MS (ESI): m/z 244.13 (M+H)+.
Example 162-(4-Acetylpiperazinyl)-2oxoethyl methyl(2E)but-2ene-1,4-dioate (16)
Methyl 2-oxo-2-piperazinylethyl(2E)but-2-ene-1,4-dioate hydrochloride (14) (0.20 g, 0.68 mmol) was reacted with acetyl chloride (AcCl) (0.60 mL, 0.66 g, 0.84 mmol) and diisopropylethylamine (0.70 mL, 0.52 g, 4.0 mmol) in dichloromethane (DCM). Following aqueous work-up, the crude product was purified by silica gel flash chromatography to afford 0.12 g (54% yield) of the title compound (16) as a white solid. 1H NMR (CDCl3, 400 MHz): δ 6.98-6.93 (m, 2H), 4.86 (s, 2H), 3.83 (s, 3H), 3.66 3.63 (m, 4H), 3.50-3.40 (m, 4H), 2.14 (s, 3H). MS (ESI): m/z 299.12 (M+H)+.
Example 9N,N-Dimethylcarbamoyl)methyl methyl(2E)but-2-ene-1,4-dioate (9)
Following general procedure A, methyl hydrogen fumarate (MHF) (0.50 g, 3.84 mmol) dissolved in NMP was reacted at ca. 55° C. with N,N-dimethyl chloroacetamide (0.56 g, 4.61 mmol) in the presence of CsHCO3 (0.89 g, 4.61 mmol). The crude material was precipitated out from a mixture of ethyl acetate (EtOAc) and hexanes (Hxn) (1:1) to provide a white solid. This solid was further dissolved in dichloromethane (DCM) and the organic layer washed with water. After removal of the solvents 0.55 g (67% yield) of the title compound (9) was obtained as a white solid. 1H NMR (CDCl3, 400 MHz): δ 6.98-6.90 (m, 2H), 4.84 (s, 2H), 3.80 (s, 3H), 2.99-2.97 (2s, 6H). MS (ESI): m/z 216 (M+H)+.
………………………..
http://www.google.com/patents/WO2014031844A1?cl=en
Compound (1).
Table 1 : Flushing Incidence as a Function of MMF Cmax
*Formulation 2 is the dosage form described in Example 10; Formulation 3 is the dosage form described in Example 3 ; Formulation 4 is the dosage form described in Example 5 ;
** maximum average Concentration; ***average Cmax; Poster (see above); Compound (1) referred to in the above table is an MMF prodrug of Formula (II); (N,N- Diethylcarbamoyl)methyl methyl (2£)but-2-ene-l,4-dioate having the following chemical structure:
Compound (1).
The maximum slope values ( dose and ng) for different dosage treatments are given in Table 2. The Figures 15-16 show plots of maximum MMF slope vs flushing incidence. The curves in the figures were fitted using a Hill Emax model. Table 2
Compound, Flushing
Table 3: Composition of Enteric Coated Sustained Release Tablet (15% HPMC in Core)
Quantity Quantity
Component Manufacturer Role
(mg tablet) (%w/w)
Vertellus (Greensboro,
Triethyl Citrate Plasticizer 1.25 0.42
NC)
Emerson Resources Anti- tacking
PlasAC YL™ T20 2.41 0.80
(Norristown, PA) agent
Total Enteric
27.87 9.30 Coating
Total Tablet 334.69 111.68
[00191] The tablets were made according to the following steps. The core tablets were prepared using a wet granulation process. The granulation was performed in two batches at 456 g per batch. Compound (1) and hydroxypropyl cellulose were passed through a conical mill with a 610 micron round holed screen. Compound (1) and hydroxypropyl cellulose were then combined in a Key KG- 5 granulator bowl and mixed with water addition for approximately 7 minutes. The wet granules were dried in a Glatt GPCG-1 fluid bed dryer at 40 °C. The two portions of dried granules were sized by passing through a conical mill with an approximately 1300 micron grater type screen. The milled granules were blended with the hypromellose 2208, silicon dioxide, and lactose monohydrate for 10 minutes in an 8 quart (7.6 1) V-blender. This blend was passed through an 850 micron mesh screen. The magnesium stearate was passed through a 600 micron mesh screen and blended with the additional core materials in the V-blender for 5 minutes. Core tablets (299.69 mg) were compressed using a GlobePharma Minipress II rotary tablet press with 8.6 mm round concave tooling. The core tablets had a final mean hardness of approximately 12 kp. For the coating, an aqueous suspension was prepared by mixing with an impeller 63.8 g Opadry 03019184 with 770.7 g of purified water. The water contained in the suspension is removed during the film coating process and therefore not included in the final formulation in Table 3. The tablets were coated with the aqueous suspension in an O’ Hara Technologies Labcoat M coater with a 12″ (30.5 cm) diameter perforated pan until the desired weight gain of barrier coat was achieved. The coating process occurred at an inlet temperature of approximately 52 °C and an outlet temperature of 36 °C. After coating, the tablets were dried for 2 hours at 40 °C. An aqueous suspension was prepared by mixing with an impeller 405.1 g methacrylic acid copolymer dispersion, 6.3 g triethyl citrate, 60.6 g PlasACRYL™ T20 with 228.1 g water. The water contained in the methacrylic acid copolymer dispersion and the
PlasACRYL™ T20 is removed during the film coating process and therefore not included in the final formulation in Table 3. The tablets were coated with the aqueous suspension in the O’ Hara Technologies Labcoat M coater until the desired weight gain of enteric film was achieved. The coating process occurred at an inlet temperature of approximately 40 °C and an outlet temperature of 30 °C. After coating, the tablets were dried for 2 hours at 40 °C.
Example 2
In Vitro Dissolution Profile of Example 1 Dosage Form
[00192] A two-stage dissolution method was used to determine the in vitro dissolution profile of dosage forms prepared according to Example 1. The 2-stage dissolution test was used to better approximate the pH conditions experienced by a dosage form after swallowing by a patient, i.e., low pH of the stomach followed by near neutral pH of the intestines. The dosage forms were first placed into a dissolution vessel (USP, Type I, basket) containing 750 mL of 0.1 N hydrochloric acid (pH 1.2). After 2 hours, 250 mL of 200 mM tribasic sodium phosphate was added to the vessel resulting in a pH adjustment from 1.2 to 6.8. The dissolution medium was kept at 37 °C and was agitated at 100 rpm.
[00193] For the Example 1 dosage forms, samples of the dissolution medium were withdrawn after 1 and 2 hours in the low pH stage, and at 0.5, 2, 4, 7, 10, and 14 hours following buffer addition. The released amount of the MMF prodrug in the samples was determined by reverse phase HPLC using a C18 column and a 7 minute gradient method according to Table 4 where Mobile Phase A is water/0.1 ]¾Ρθ4 and Mobile Phase B is water/acetonitrile/H3PC>4 (10/90/0.1 by volume) with UV detection at 210 nm.
Table 4: HPLC Gradient Conditions
[00194] As shown in FIG. 1, for dosage forms prepared according to Example 1, drug release is delayed for approximately 2 hours, followed by sustained release reaching >90 at 12 hours.
Example 3
Preparation of Delayed Sustained Release Dosage Form (Enteric Coated, 15% HPMC in Core, without Barrier Layer) [00195] Delayed sustained release tablets containing compound (1) were made having the ingredients shown in Table 5:
Table 5: Composition of Enteric Coated Sustained Release Tablet (15% HPMC in Core, without Barrier Layer)

[00196] The tablets were made according to the following steps. The core tablets were prepared using a wet granulation process. The granulation was performed in two batches at 463.9 g per batch. Compound (1) and hydroxypropyl cellulose were passed through a conical mill with a 610 micron round holed screen. Compound (1) and hydroxypropyl cellulose were then combined in a Key KG- 5 granulator bowl and mixed with water addition for approximately 10 minutes. The wet granules were dried in a Glatt GPCG-1 fluid bed dryer at 40 °C. The two portions of dried granules were blended with silicon dioxide and sized by passing through a conical mill with an approximately 1300 micron grater type screen. The milled granules were blended with the hypromellose 2208 and lactose monohydrate for 10 minutes in an 8 quart (7.6 1) V-blender. This blend was passed through an 850 micron mesh screen. The magnesium stearate was passed through a 600 micron mesh screen and blended with the additional core materials in the V-blender for 5 minutes. Core tablets (299.68 mg) were compressed using a GlobePharma Minipress II rotary tablet press with 11/32″ round concave tooling. The core tablets had a final mean hardness of approximately 11 kp. For the coating, an aqueous suspension was prepared by mixing with an impeller 578.7 g methacrylic acid copolymer dispersion, 9.0 g triethyl citrate, 86.5 g PlasACRYL™ T20 with 325.8 g water. The water contained in the methacrylic acid copolymer dispersion and the
PlasACRYL™ T20 is removed during the film coating process and therefore not included in the final formulation in Table 4. The tablets were coated with the aqueous suspension in the O’ Hara Technologies Labcoat M coater until the desired weight gain of enteric film was achieved. The coating process occurred at an inlet temperature of approximately 41 °C and an outlet temperature of 31 °C. After coating, the tablets were dried for 2 hours at 40 °C.
…………………………
WO 2014071371
http://www.google.com/patents/WO2014071371A1?cl=en
(N,N-Diethylcarbamoyl)methyl methyl (2E)but-2-ene-1 ,4-dioate has the following chemical structure:
This compound was synthesized in Example 1 of Gangakhedkar et al., U.S. Patent No. 8,148,414. The compound is a prodrug of methyl hydrogen fumarate (MHF) and has a disclosed melting point of between 53 °C and 56 °C.
Cocrystals are crystals that contain two or more non-identical molecules that form a crystalline structure. The intermolecular interactions between the non-identical molecules in the resulting crystal structures can result in physical and chemical properties that differ from the properties of the individual components. Such properties can include, for example, melting point, solubility, chemical stability, mechanical properties and others. Examples of cocrystals may be found in the Cambridge Structural Database and in Etter, et al.,
“The use of cocrystallization as a method of studying hydrogen bond preferences of 2-aminopyridine” J. Chem. Soc, Chem. Commun. (1990), 589-591 ; Etter, et al., “Graph-set analysis of hydrogen-bond patterns in organic crystals” Acta Crystallogr., Sect. B, Struct. Sci. (1990), B46: 256-262; and Etter, et al., “Hydrogen bond directed cocrystallization and molecular recognition properties of diarylureas” J. Am. Chem. Soc. (1990), 1 12: 8415-8426. Additional information relating to cocrystals can be found in: Carl Henrik Gorbotz and Hans-Petter Hersleth,
“On the inclusion of solvent molecules in the crystal structures of organic compounds”; Acta Cryst. (2000), B56: 625-534; and Senthil Kumar, et al., “Molecular Complexes of Some Mono- and Dicarboxylic Acids with trans-1 ,4,-Dithiane-1 ,4-dioxide” American Chemical Society, Crystal Growth & Design (2002) , 2(4) : 313-318.
(N,N-Diethylcarbamoyl)methyl methyl (2E)but-2-ene-1 ,4-dioate is a prodrug of methyl hydrogen fumarate. Once administered, the compound is metabolized in vivo into an active metabolite, namely, methyl hydrogen fumarate (MHF) which is also referred to herein as monomethyl fumarate (MMF). The in vivo metabolism of (N,N-Diethylcarbamoyl)methyl
(N,N-Diethylcarbamoyl)methyl methyl Methyl hydrogen fumarate N ^ diethyl glycolamide
(2E)but-2-ene-1 ,4-dioate
Table 1
As can be seen from the data in Table 1 , the six cocrystals disclosed herein each exhibit a higher melting point than crystalline (N,N-Diethylcarbamoyl)methyl methyl (2E)but-2-ene-1 ,4- dioate.

…………………………….
Steady state pharmacokinetics of formulations of XP23829, a novel prodrug of monomethyl fumarate (MMF), in healthy subjects
66th Annu Meet Am Acad Neurol (AAN) (April 26-May 3, Philadelphia) 2014, Abst P1.188
………………………………….
Lymphocyte and eosinophil responses in healthy subjects dosed with Tecfidera and XP23829, a novel fumaric acid ester (FAE)
66th Annu Meet Am Acad Neurol (AAN) (April 26-May 3, Philadelphia) 2014, Abst P1.201
………………………..
A comparison of XP23829 with DMF, the active ingredient of BG-12
4th Cooperative Meet Consorti Mult Scler Cent (CMSC) Am Comm Treat Res Mult Scler (ACTRIMS) (May 30-June 2, San Diego) 2012, Abst SC03
http://annualmeeting.mscare.org/index.php?option=com_content&view=article&id=174&Itemid=101
…………………………..
Favorable metabolism and pharmacokinetics of formulations of XP23829, a novel fumaric acid ester, in healthy subjects
65th Annu Meet Am Acad Neurol (AAN) (March 16-23, San Diego) 2013, Abst P05.189
…………………………………..
Comparison of the efficacy and tolerability of a novel methyl hydrogenfumarate prodrug with dimethyl fumarate in rodent EAE and GI irritation models
Neurology 2011, 76(9): Abst P05.040
| WO2013119791A1 * |
Feb 7, 2013 |
Aug 15, 2013 |
Xenoport, Inc. |
Morpholinoalkyl fumarate compounds, pharmaceutical compositions, and methods of use |
| US20120034303 * |
Jan 8, 2010 |
Feb 9, 2012 |
Forward Pharma A/S |
Pharmaceutical formulation comprising one or more fumaric acid esters in an erosion matrix |
| US20120095003 * |
Oct 14, 2011 |
Apr 19, 2012 |
Xenoport, Inc. |
Methods of using prodrugs of methyl hydrogen fumarate and pharmaceutical compositions thereof |
| US20120157523 * |
Oct 14, 2011 |
Jun 21, 2012 |
Xenoport, Inc. |
Prodrugs of methyl hydrogen fumarate, pharmaceutical compositions thereof, and methods of use |
| WO2013119791A1 * |
Feb 7, 2013 |
Aug 15, 2013 |
Xenoport, Inc. |
Morpholinoalkyl fumarate compounds, pharmaceutical compositions, and methods of use |
| US20100048651 * |
Aug 19, 2009 |
Feb 25, 2010 |
Xenoport, Inc. |
Prodrugs of methyl hydrogen fumarate, pharmaceutical compositions thereof, and methods of use |
| US8669281 |
20 Sep 2013 |
11 Mar 2014 |
Alkermes Pharma Ireland Limited |
Prodrugs of fumarates and their use in treating various diseases |
| WO2014031894A1 |
22 Aug 2013 |
27 Feb 2014 |
Xenoport, Inc. |
Oral dosage forms of methyl hydrogen fumarate and prodrugs thereof |
| WO2014071371A1 |
5 Nov 2013 |
8 May 2014 |
Xenoport, Inc. |
Cocrystals of (n,n-diethylcarbamoyl)methyl methyl (2e)but-2-ene-1,4-dioate |



































 Philip Kocienski, Professor of Organic Chemistry.
Philip Kocienski, Professor of Organic Chemistry.