
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
An easier, safer, and more accurate treatment for pancreatic cancer

(a) A single axial slice of the pancreas from the pre-treatment CT scans is overlaid with computed contours of light fluence levels around the fiber location. This was simulated using blood content information for tissue absorption from contrast CT. (b) A volume rendering of the blood vessels around the pancreas overlaid with the light dose map in the fiber location, in the same patient. Please see supplementary material at stacks.iop.org/PMB/59/1911/mmedia. Credit: NCCC
Using CT scans with contrast enhancement, Dartmouth researchers measured treatment response to pancreatic cancer photodynamic therapy (PDT) according to a paper published in Physics in Medicine and Biology.
The research team at Dartmouth set out to reduce the imaging obstacles for PDT, a minimally invasive and nontoxic treatment for cancer. “This study implies that treatment response can be reliably predicted using contrast CT. This would represent a major breakthrough in PDT for pancreas cancer that allows…
View original post 466 more words
New treatment could ‘protect against any strain of the flu’

The new biologic (green) binding to the surface of cells (blue nuclei), protecting the cells from invasion by the influenza virus.
Scots scientists have developed a novel treatment that could protect against any strain of the flu.
It is hoped that the new development, led by researchers at the University of St Andrews, has the potential to guard against current, future and even pandemic strains of the virus.
In an international effort, the scientists involved say that the preventative treatment could be used as a ‘frontline defence’ before an effective flu vaccine is developed. Leading influenza experts say the new development is ‘very exciting and potentially of great importance in this era’.
The BBSRC and MRC-funded research was led by Professor Garry Taylor and Dr Helen Connaris in the Biomedical Sciences Research Complex at St Andrews. They said “We have developed an alternative host-targeted approach to prevent influenza by synthesising…
View original post 464 more words
New method to analyse how cancer cells die

A team from The University of Manchester – part of the Manchester Cancer Research Centre – have found a new method to more efficiently manufacture a chemical used to monitor cancer cells.
The technique could lead to clearer and better quality images on PET scans.
The number of cells within tissue is controlled through apoptosis – a process where cells shrink and their components break up, also known as programmed cell death. Cancer is often characterised by a disruption to the normal process of this cell death.
Being able to study this process accurately would allow doctors to more effectively diagnose and monitor cancer and to test and develop new treatments designed to kill cancer cells.
Ideally, cell death would be measured non-invasively to avoid surgery and current methods are focused on using radioactive tracers – molecules that are taken up in regions of tissue where cells are…
View original post 281 more words
RG 7388 is a MDM2 inhibitor with superior potency and selectivity in phase 1 trials

- RG-7388
- Hoffmann-La Roche, Inc. , INNOVATOR
- 4-((2R,3S,4R,5S)-3-(3-chloro-2-fluorophenyl)-4-(4-chloro-2-fluorophenyl)-4-cyano-5-neopentylpyrrolidine-2-carboxamido)-3-methoxybenzoic acid
- 4-{[(2R,3S,4R,5S)-4-(4-Chloro-2-fluoro-phenyl)-3-(3-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carbonyl]-amino}-3-methoxy-benzoic acid
- 4-[[(3S,4R,5S)-3-(3-Chloro-2-fluorophenyl)-4-(4-chloro-2-fluorophenyl)-4-cyano-5-(2,2-dimethylpropyl)-D-prolyl]amino]-3-methoxybenzoic acid
- CAS Number:1229705-06-9
- Mol. Formula:C31H29Cl2F2N3O4
- MW:616.5

- RG-7388 is an MDM2 (hdm2) inhibitor in early clinical trials at Roche for the oral treatment of solid tumors and hematologic cancer.

- INTRO
- RG7388 is a MDM2 inhibitor with superior potency and selectivity
- RG7388 is an oral, selective, small molecule MDM2 antagonist that inhibits binding of MDM2 to p53.
RG7388 is the second generation inhibitor of P53-MDM2 interaction. It is orally active, potently and selectively antagonizing the P53-MDM2 interaction with Ki at low nM. It is designed to selectively target MDM2, a key negative regulator of the p53 tumor suppressor protein. Blocking this essential interaction may lead to apoptosis via activation of p53 in tumor cells with functional p53 signaling. It is currently in clinical evaluation.
Description:
Value IC50: 30 nM (IC50 Average of three wt-p53 SJSA1 Cancer cell lines, RKO, HCT116)
. RG7388 is an Oral, Selective, small molecule antagonist that inhibits binding of MDM2 to p53 MDM2 Blocking the MDM2-p53 Interaction stabilizes p53 and activates p53-mediated cell death and inhibition of cell Growth.
RG7388 Showed all the Characteristics expected of an MDM2 inhibitor in terms of speci? c binding to the target, mechanistic outcomes Resulting from Activation of the p53 pathway, and in vivo ?. Although e cacy Mechanism of Action of the cellular is identical to that of RG7388 RG7112, it is much More potent and Selective.
Tumor suppressor p53 is a powerful growth suppressive and pro-apoptotic protein that plays a central role in protection from tumor development.A potent transcription factor, p53 is activated following cellular stress and regulates multiple downstream genes implicated in cell cycle control, apoptosis, DNA repair, and senescence.While p53 is inactivated in about 50% of human cancers by mutation or deletion, it remains wild-type in the remaining cases but its function is impaired by other mechanisms. One such mechanism is the overproduction of MDM2, the primary negative regulator of p53, which effectively disables p53 function.An E3 ligase, MDM2 binds p53 and regulates p53 protein levels through an autoregulatory feedback loop. Stabilization and activation of wild-type p53 by inhibition of MDM2 binding has been explored as a novel approach for cancer therapy.
//////////////

Restoration of p53 activity by inhibition of the p53–MDM2 interaction has been considered an attractive approach for cancer treatment. However, the hydrophobic protein–protein interaction surface represents a significant challenge for the development of small-molecule inhibitors with desirable pharmacological profiles. RG7112 was the first small-molecule p53–MDM2 inhibitor in clinical development. Here, we report the discovery and characterization of a second generation clinical MDM2 inhibitor, RG7388, with superior potency and selectivity.
http://pubs.acs.org/doi/suppl/10.1021/jm400487c/suppl_file/jm400487c_si_001.pdf …………..for exptal section
//////////////////
US20100152190
http://www.google.com/patents/US20100152190
(Scheme 4).


In a 25 mL round-bottomed flask, (2R,3S,4R,5S)-3-(3-chloro-2-fluorophenyl)-4-(4-chloro-2-fluorophenyl)-4-cyano-5-neopentylpyrrolidine-2-carboxylic acid (250 mg, 535 μmol), was combined with CH2Cl2 (5 ml). DIPEA (277 mg, 374 μl, 2.14 mmol) and dipenylphospenic chloride (380 mg, 306 μl, 1.6 mmol) were added and the reaction was stirred at RT for 20 minutes. Methyl 4-amino-3-methoxybenzoate (100 mg, 552 μumol) was added and the reaction mixture was stirred at RT overnight.
The crude reaction mixture was concentrated in vacuum. The crude material was purified by flash chromatography (silica gel, 40 g, 5% to 25% EtOAc/Hexanes) to give the desired product as a white solid (275 mg, 81% yield).
Example 448 Preparation of 4-{[(2R,3S,4R,5S)-4-(4-Chloro-2-fluoro-phenyl)-3-(3-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carbonyl]-amino}-3-methoxy-benzoic acid

In a 25 mL round-bottomed flask, methyl 4-((2R,3S,4R,5S)-3-(3-chloro-2-fluorophenyl)-4-(4-chloro-2-fluorophenyl)-4-cyano-5-neopentylpyrrolidine-2-carboxamido)-3-methoxybenzoate (150 mg, 238 μmol, Eq: 1.00) was combined with CH2Cl2 (2 ml) to give a colorless solution. Aluminum bromide (Aldrich, 254 mg, 952 μmol, Eq: 4) and dimethyl sulfide (1.69 g, 2 mL, 27.2 mmol, Eq: 114) were added. The reaction mixture was stirred for overnight.
The reaction mixture was diluted with CH3CN (6 ml), EtOAc (10 ml) and water (10 ml), stirred and layers separated. The aqueous layer was extracted with EtOAc (2×10 mL). The organic layers were combined, washed with saturated NaCl (1×15 mL), dried over MgSO4 and concentrated in vacuum.
The crude material was dissolved in DMSO (4 ml) and was purified by preparative HPLC (70-100% ACETONITRILE/water). The fractions were combined, concentrated and freeze dried to give a white powder as desired product (75 mg, 51% yield). (ES+) m/z Calcd: [(M+H)+]: 616, found: 616.
Alternatively, the title compound could be prepared by the following method.
In a 500 mL round-bottomed flask, methyl 4-((2R,3S,4R,5S)-3-(3-chloro-2-fluorophenyl)-4-(4-chloro-2-fluorophenyl)-4-cyano-5-neopentylpyrrolidine-2-carboxamido)-3-methoxybenzoate (3.74 g, 5.93 mmol, Eq: 1.00) was combined with THF (140 ml) and MeOH (160 ml) at 50° C. to give a colorless solution. 1 N NaOH (23.7 ml, 23.7 mmol, Eq: 4) was added. The reaction mixture was stirred at 40° C. for 18 hrs.
The reaction mixture was concentrated to remove about ½ of the solvent, filtered to removed the insoluble, acidified with 1N HCl to PH=4-5 and the resulting solid was collected by filtration and was washed with water, small amount of MeOH and diethyl ether. It was then dried in vacuum oven (60° C.) overnight. Obtained was a white solid as the desired product (2.96 g, 80.5% yield). H1NMR and LC/MASS data were the same as that in the above procedure.

In a manner similar to the method described in Example 1b, 4-chloro-2-fluorophenylacetonitrile (5 g, 30 mmol) was reacted with 3-chloro-2-fluorobenzaldehyde (5 g, 32 mmol), methanolic solution (25 wt %) of sodium methoxide (21 mL, 92 mmol) in methanol (200 mL) at 45° C. for 5 h to give (Z)-3-(3-chloro-2-fluoro-phenyl)-2-(4-chloro-2-fluoro-phenyl)-acrylonitrile as a white powder (9 g, 97%).
Example 52b Preparation of intermediate rac-(2R,3S,4R,5S)-3-(3-chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid tert-butyl ester

In a manner similar to the method described in Example 1c, [3-methyl-but-(E)-ylideneamino]-acetic acid tert-butyl ester prepared in Example 1a (2.3 g, 11 mmol) was reacted with (Z)-3-(3-chloro-2-fluoro-phenyl)-2-(4-chloro-2-fluoro-phenyl)-acrylonitrile (2.5 g, 8 mmol) prepared in Example 52a, AgF (0.7 g, 5.5 mmol), and triethylamine (2.9 g, 29 mmol) in dichloromethane (200 mL) at room temperature for 18 h to give rac-(2R,3S,4R,5S)-3-(3-Chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid tert-butyl ester as a white foam (3 g, 64%).
Example 52c Preparation of intermediate rac-(2R,3S,4R,5S)-3-(3-chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid trifluoroacetic acid

In a manner similar to the method described in Example 25a, rac-(2R,3S,4R,5S)-3-(3-chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid tert-butyl ester prepared in Example 52b (0.4 g, 0.8 mmol) was reacted with trifluoroacetic acid in dichloromethane at room temperature to give rac-(2R,3S,4R,5S)-3-(3-chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid trifluoroacetic acid as a white solid (0.5 g, 100%).
HRMS (ES+) m/z Calcd for C23H22Cl2F2N2O2+H [(M+H)+]: 467.1099, found: 467.1098.

In a manner similar to the method described in Examples 1e, rac-(2R,3S,4R,5S)-3-(3-chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid trifluoroacetic acid prepared in Example 52c (0.5 g, 0.86 mmol) was reacted with a dioxane solution (0.5 M) of ammonia (2 mL, 1 mmol), HATU (0.38 g, 1 mmol) and iPr2NEt (0.6 g, 4.6 mmol) in CH2Cl2 at room temperature for 20 h to give rac-(2R,3S,4R,5S)-3-(3-chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid amide as a white solid (0.3 g, 75%).
HRMS (ES+) m/z Calcd for C23H23Cl2F2N3O+H [(M+H)+]: 466.1259, found: 466.1259.
REFERENCES
1 Discovery of RG7388, a Potent and Selective p53-MDM2 Inhibitor in Clinical Development. By Ding, Qingjie; Zhang, Zhuming; Liu, Jin-Jun; Jiang, Nan; Zhang, Jing; Ross, Tina M.; Chu, Xin-Jie; Bartkovitz, David; Podlaski, Frank; Janson, Cheryl; et al From Journal of Medicinal Chemistry (2013), 56(14), 5979-5983.
2. Pyrrolo[1,2-c]imidazolone derivatives as inhibitors of MDM2-p53 interactions and their preparation and use for the treatment of cancer. By Chu, Xin-Jie; Ding, Qingjie; Jiang, Nan; Liu, Jin-Jun; Ross, Tina Morgan; Zhang, Zhuming From U.S. Pat. Appl. Publ. (2012), US 20120065210 A1 20120315.
3. Pyrrolidine-2-carboxamide derivatives and their preparation and use as anticancer agents. By Chu, Xin-Jie; Ding, Qingjie; Jiang, Nan; Liu, Jin-Jun; Ross, Tina Morgan; Zhang, Zhuming. From U.S. Pat. Appl. Publ. (2012), US 20120010235 A1 20120112.
4. Preparation of substituted pyrrolidine-2-carboxamides as anticancer agents. By Bartkovitz, David Joseph; Chu, Xin-Jie; Ding, Qingjie; Jiang, Nan; Liu, Jin-Jun; Ross, Tina Morgan; Zhang, Jing; Zhang, Zhuming
From PCT Int. Appl. (2011), WO 2011098398 A1 20110818.
5. Preparation of substituted pyrrolidine-2-carboxamides as anticancer agents. By Bartkovitz, David Joseph; Chu, Xin-Jie; Ding, Qingjie; Jiang, Nan; Liu, Jin-Jun; Ross, Tina Morgan; Zhang, Jing; Zhang, Zhuming
From U.S. Pat. Appl. Publ. (2010), US 20100152190 A1 20100617.
6 B. Higgins, et al, Antitumor Activity of the MDM2 Antagonist RG7388, Mol Cancer Ther 2013;12(11 Suppl):B55
Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development
J Med Chem 2013, 46(14): 5979
Erectile dysfunction can be reversed without medication
Men suffering from sexual dysfunction can be successful at reversing their problem, by focusing on lifestyle factors and not just relying on medication, according to new research at the University of Adelaide.
In a new paper published in the Journal of Sexual Medicine, researchers highlight the incidence of erectile dysfunction and lack of sexual desire among Australian men aged 35-80 years.
Over a five-year period, 31% of the 810 men involved in the study developed some form of erectile dysfunction.
“Sexual relations are not only an important part of people’s wellbeing. From a clinical point of view, the inability of some men to perform sexually can also be linked to a range of other health problems, many of which can be debilitating or potentially fatal,” says Professor Gary Wittert, Head of the Discipline of Medicine at the University of Adelaide and Director of the University’s Freemasons Foundation Centre for…
View original post 254 more words
How the body fights against viruses

Illustration “structure”: This is a model of the RNA-binding domain of ADAR1 (green), bound to double-stranded RNA (yellow). Transportin1, which mediates the nuclear transport of ADAR1, is depicted in gray. The structural model reveals that ADAR1 cannot enter the nucleus when bound to RNA, as RNA (yellow) and Transportin1 (gray) clash. Credit: PNAS
Scientists of the Max F. Perutz Laboratories of the University of Vienna and the Medical University of Vienna, together with colleagues of the ETH Zurich, have now shown how double stranded RNA, such as viral genetic information, is prevented from entering the nucleus of a cell. During the immune response against viral infection, the protein ADAR1 moves from the cell nucleus into the surrounding cytoplasm. There it modifies viral RNA to inhibit reproduction of the virus. But how is the human genome protected from inadvertent import of viral RNA into the nucleus? The current study of the research…
View original post 602 more words
A Way To Halt Atherosclerosis
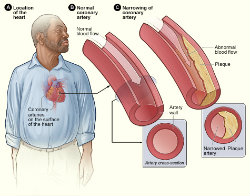
- Johns Hopkins scientists have halted the development of atherosclerotic heart disease in animals by blocking the activity of a sugar-and-fat molecule residing in the membranes of cells.
- Using a widely available man-made compound called D-PDMP, the researchers prevented the buildup of fatty plaque and calcium deposits inside the blood vessels of mice and rabbits fed a high-fat, cholesterol-laden diet.
- Treatment with D-PDMP appears to work by altering a range of biological glitches that affect the body’s ability to properly use, transport and purge itself of cholesterol — the fatty substance that accumulates inside vessels and fuels heart disease.
D-PDMP, which is already widely used in basic research to experimentally block and study cell growth and other basic cell functions, is deemed safe in animals, the investigators say. For example, animals in the current study had no side effects even when given D-PDMP doses 10 times higher than the minimum effective…
View original post 29 more words
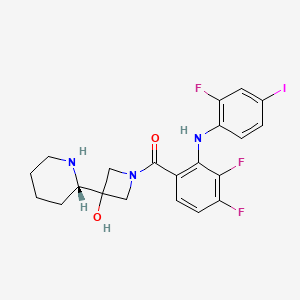
Cobimetinib in phase 3 for metastatic melanoma


Cobimetinib
934660-93-2 cas ………….(S )enantiomer desired
[3,4-Difluoro-2-(2-fluoro-4-iodoanilino)phenyl]{3-hydroxy-3-[(2S)-piperidin-2-yl]azetidin-1-yl} methanone
l-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2S)-piperidin-2- yl]azetidin-3-ol
1-[3,4-Difluoro-2-(2-fluoro-4-iodophenylamino)phenyl]-1-[3-hydroxy-3-[2(S)-piperidinyl]azetidin-1-yl]methanone
 Cobimetinib (racemate)
Cobimetinib (racemate) Cobimetinib (R-enantiomer)
Cobimetinib (R-enantiomer)CAS No: 934660-94-3
cobimetinib fumarate [USAN]
Molecular Formula: C46H46F6I2N6O8
Average mass: 1178.692261 Da
(2E)-2-Butendisäure –{3,4-difluor-2-[(2-fluor-4-iodphenyl)amino]phenyl}{3-hydroxy-3-[(2S)-2-piperidinyl]-1-azetidinyl}methanon (1:2) [German] [ACD/IUPAC Name]
{3,4-Difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}{3-hydroxy-3-[(2S)-2-piperidinyl]-1-azetidinyl}methanone (2E)-2-butenedioate (2:1) [ACD/IUPAC Name]
1369665-02-0 [RN]
Acide (2E)-2-butènedioïque – {3,4-difluoro-2-[(2-fluoro-4-iodophényl)amino]phényl}{3-hydroxy-3-[(2S)-2-pipéridinyl]-1-azétidinyl}méthanone (1:2) [French][ACD/IUPAC Name]
Bis({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}{3-hydroxy-3-[(2S)-piperidin-2-yl]azetidin-1-yl}methanone) (2E)-but-2-enedioate
Methanone, [3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl][3-hydroxy-3-[(2S)-2-piperidinyl]-1-azetidinyl]-, (2E)-2-butenedioate (2:1) (salt
Click to access cobimetinib-fumarate.pdf
Cobimetinib (GDC-0973, XL-518) is a MEK inhibitor being developed by Exelixis and Roche. It is being studied in combination withvemurafenib to treat several cancers, including melanoma.
Cobimetinib is an inhibitor of MEK kinase, which is an enzyme that the mitogen-activated protein kinase (MAPK). This compound was originated by Exelixis and is being developed by Genentech. Currently, Cobimetinb is in phase III trials at Genentech for the treatment of metastatic melanoma and in phase I clinical trials for the treatment of solid tumors. Cobimetinib has received an orphan drug designation in the U.S. for treatment of stage IIb, IIc, III, and IV melanoma with BRAFV600E mutation.
GDC-0973 (XL-518; GDC 0973) is a selective inhibitor of MEK GDC-0973 is also known as mitogen activated protein kinase kinase (MAPKK), is a key component of the RAS / RAF / MEK / ERK pathway, which. is frequently activated in human tumors.
Inappropriate activation of the MEK / ERK pathway promotes cell growth in the absence of exogenous growth factors.
The ERK/MAP kinase cascade is a key mechanism subject to dysregulation in cancer and is constitutively activated or highly upregulated in many tumor types. Mutations associated with upstream pathway components RAS and Raf occur frequently and contribute to the oncogenic phenotype through activation of MEK and then ERK. Inhibitors of MEK have been shown to effectively block upregulated ERK/MAPK signaling in a range of cancer cell lines and have further demonstrated early evidence of efficacy in the clinic for the treatment of cancer. Guided by structural insight, a strategy aimed at the identification of an optimal diphenylamine-based MEK inhibitor with an improved metabolism and safety profile versus PD-0325901 led to the discovery of development candidate 1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2S)-piperidin-2-yl]azetidin-3-ol (XL518, GDC-0973) (1). XL518 exhibits robust in vitro and in vivo potency and efficacy in preclinical models with sustained duration of action and is currently in early stage clinical trials.
Process for the preparation of MEK inhibitors, relates cobimetinib. Genentech and its parent company Roche, under license from Exelixis, are developing cobimetinib, for the treatment of solid tumors, including melanoma, which is in phase 3 trials as of April 2014. The drug was originally disclosed in WO2007044515. For a previous filing on MEK inhibitors, see WO2008124085.
patent
http://www.google.com/patents/WO2007044515A1?cl=en
EXAMPLE 22(a) and 22(b) l-({3,4-difluoro-2-[(2-fluoro-4-iodophenyϊ)amino]phenyl}carbonyI)-3-[(2R)-piperidin-2- yl]azetidin-3-ol

l-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2S)-piperidin-2- yl]azetidin-3-ol
 desired
desired[00334] To a solution of 1 , 1 -dimethylethyl 2-(3 -hydroxy- 1 –
{[(phenylmethyl)oxy]carbonyl}azetidin-3-yl)piperidine-l-carboxylate (368 mg, 0.94 mmol), prepared using procedures similar to those described in Reference 5, in dichloromethane (5 mL) was added DMAP (115 mg, 0.94 mmol) and the resulting solution was cooled to O0C. (i?)-(-)-α-Methoxy-α-trifluoromethylphenylacetyl chloride (105 μL, 0.56 mmol) was added to the solution by syringe and the mixture was allowed to warm to room temperature then stirred an additional 12 hours. The solution was then partitioned with saturated aqueous soldium bicarbonate and the organic phase dried over anhydrous magnesium sulfate then filtered and concentrated to an oily residue.
Silica gel flash chromatography using hexanes:ethyl acetate 3:1 as eluent afforded the less polar 1,1 -dimethyl ethyl (2R)-2-(l- {[(phenylmethyl)oxy]carbonyl}-3-{[(2i?)-3,3,3-trifluoro-2-(methyloxy)-2- phenylpropanoyl]oxy}azetidin-3-yl)piperidine-l-carboxylate (27.5 mg, 5% yield), the more polar 1 , 1 -dimethylethyl (2S)-2-(l -{ [(phenylmethyl)oxy]carbonyl} -3-{ [(2i?)-3,3,3-trifluoro-2- (methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-l-carboxylate (105 mg, 19% yield) and starting material (253 mg, 69% recovery).
[00335] The starting material thus recovered was taken into dichloromethane (3 mL) followed by addition of DMAP (115 mg, 0.94 mmol) and (i?)-(-)-α-methoxy-α- trifluoromethylphenylacetyl chloride (105 μL, 0.56 mmol) and the mixture was allowed to stir at room temperature over 12 hours. Proceeding as before afforded combined 1,1- dimethylethyl (2R)-2-(l-{[(phenylmethyl)oxy]carbonyl}-3-{[(2i?)-3,3,3-trifluoro- 2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-l-carboxylate (46.6 mg, 8% yield), the more polar 1,1 -dimethylethyl (25)-2-(l-{[(phenylmethyl)oxy]carbonyl}-3-{[(2i?)- 3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-l-carboxylate (228 mg, 41% yield) and starting material (100.8 mg, 27% recovery). [00336] The starting material thus recovered was taken into tetrahydrofuran: dichloromethane (1 :1, 2 mL) followed by addition of DMAP (47 mg, 0.39 mmol) and (R)-(-)-α-methoxy-α-trifluoromethylphenylacetyl chloride (80 μL, 0.43 mmol) and the mixture was heated to 60 0C over 12 hours.
Proceeding as before afforded combined less polar 1,1-dimethylethyl (2i?)-2-(l-{[(phenylmethyl)oxy]carbonyl}-3-{[(2i?)-3,3,3- trifluoro-2-(methyloxy)-2-phenylpropanoyl] oxy } azetidin-3 -yl)piperidine- 1 -carboxylate ( 144 mg, 26 % yield). The chiral ester derivatives thus obtained were again subject to silica gel flash chromatography using hexanes:ethyl acetate 3:1 as eluent to give the pure less polar 1,1-dimethylethyl (2i?)-2-(l-{[(phenylmethyl)oxy]carbonyl}-3-{[(2i?)-3,3,3-trifluoro-2-
(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-l-carboxylate (122.8 mg, 22% yield) and the more polar 1,1-dimethylethyl (2£)-2-(l-{[(phenylmethyl)oxy]carbonyl}-3- { [(2R)-3 ,3 ,3 -trifluoro-2-(methyloxy)-2-phenylpropanoyl] oxy } azetidin-3-yl)piperidine- 1 – carboxylate {111.6 mg, 32% yield) both as colorless amorphous residues. [00337] l,l-Dimethylethyl (2i?)-2-(l-{[(phenylmethyl)oxy]carbonyl}-3-{[(2i?)-3,3,3- trifluoro-2-(methyloxy)-2-phenylρropanoyl]oxy}azetidin-3-yl)piperidine-l-carboxylate (122.8 mg, 0.21 mmol) was taken into methanol (4 mL) followed by addition of IM aqueous sodium hydroxide (1 mL) and the resulting solution was stirred for one hour at room temperature.
The solution was then partitioned with ethyl acetate and IN aqueous hydrochloric acid. The organic layer was washed with brine, dried over anhydrous magnesium sulfate then filtered and concentrated. The residue was purified by silica gel flash chromatography using hexanes:ethyl acetate 2:1 to give 1,1-dimethylethyl (2i?)-2-(3- hydroxy- 1 – { [(phenylmethyl)oxy] carbonyl } azetidin-3 -yl)piperidine- 1 -carboxylate (60.8 mg, 81% yield) a colorless amorphous solid. 1,1-dimethylethyl (2ιS)-2-(3-hydroxy-l- {[(phenylmethyl)oxy]carbonyl}azetidin-3-yl)piperidine-l-carboxylate (87.4 mg, 75% yield) was prepared analogously.
[00338] 1 , 1 -Dimethylethyl (2i?)-2-(3-hydroxy- 1 – { [(phenylmethyl)oxy] carbonyl } azetidin- 3 -yl)piperidine-l -carboxylate (60.8 mg, 0.16 mmol) and 10% Pd/C (30 mg) were taken into methanol (2 mL) and the mixture hydrogenated at ambient pressure for one hour. The suspension was then filtered through a celite pad and concentrated then dried in vacuo to a colorless solid. The solid amine was taken into THF (1 mL) followed by addition of DIPEA (42 μL, 0.24 mmol) and 3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]benzoyl fluoride (63 mg, 0.16 mmol), prepared using procedures similar to those described in Reference 1, and the mixture stirred at room temperature for 30 minutes.
The reaction mixture was partitioned with ethyl acetate and 1 N aqueous hydrochloric acid and the organic layer washed with brine, dried over anhydrous magnesium sulfate then filtered and concentrated. Purification of the residue by silica gel flash chromatography using hexanes: ethyl acetate 3:2 as eluent afforded 1,1-dimethylethyl (2i?)-2-[l-({3,4-difluoro-2-[(2-fluoro-4- iodophenyl)amino]phenyl}carbonyl)-3-hydroxyazetidin-3-yl]piperidine-l -carboxylate (74.9 mg, 74% yield) as an amorphous solid. 1,1 -Dimethylethyl (2i?)-2-[l-({3,4-difluoro-2-[(2- fluoro~4-iodophenyl)amino]phenyl}carbonyl)-3-hydroxyazetidin-3-yl]piperidine-l- carboxylate 1R NMR (400 MHz, CDCl3): 8.53 (br s, 0.5H), 8.40 (br s, 0.5H), 7.41-7.38 (dd, IH), 7.34-7.31(dt, IH), 7.17-7.14 (m, IH), 6.86-6.79 (m, IH), 6.63-6.587 (m, IH), 4.24-3.90 (m, 4H), 3.37-3.23 (m, IH), 2.90-2.80 (m, IH), 1.85-1.54 (m, 7H), 1.43 (s, 9H); MS (EI) for C26H29F3IN3O4: 576 (M-C4H9 4).
[00339] 1 , 1 -dimethylethyl (2R)-2-[l -({3,4-difluoro-2-[(2-fluoro-4- iodopheny^aminojphenylJcarbonyO-S-hydroxyazetidin-S-yljpiperidine-l-carboxylate (74.9 mg, 0.12 mmol) was taken into methanol (1 mL) followed by addition of 4 N HCl in dioxane (1 mL) and the solution was stirred at room temperature for one hour. The solution was then concentrated and the residue partitioned with chloroform and saturated aqueous sodium bicarbonate.
The organic layer was washed with brine, dried over anhydrous sodium’ sulfate then filtered and concentrated. Purification of the residue by silica gel flash chromatography using ethyl acetate then concentrated aqueous ammonia in chloroform and methanol (0.1 :10:1) as eluents afforded l-({3,4-difluoro-2-[(2-fluoro-4- iodophenyl)amino]phenyl}carbonyl)-3-[(2i?)-piperidin-2-yl]azetidin-3-ol (57.3 mg) as a colorless amorphous solid.
The free base was taken into methanol (1 mL) then brought to about pH 1 by addition of 4 N HCl in dioxane and the solution concentrated. The residue was triturated with ethyl ether to afford a suspension. The solid was collected by filtration to afford l-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2i?)- piperidin-2-yl]azetidin-3-ol hydrochloride salt (49 mg, 72% yield) as a colorless solid. 1H NMR (400 MHz, CDCl3): 8.43-8.39 (d, IH), 7.41-7.38 (dd, IH), 7.33-7.31(dt, IH), 7.14- 7.10 (m, IH), 6.84-6.80 (m, IH), 6.63-6.57 (m, IH), 4.12-3.99 (m, 4H), 3.10-3.08 (d, IH), 2.72-2.69 (d, IH), 2.64-2.62 (m, IH), 1.61-1.58 (m, 2H), 1.36-1.16 (m, 4H); MS (EI) for C21H2IF3IN3O2: 532 (MH+).

………………….
http://www.google.com/patents/WO2014027056A1?cl=en
[3,4-difluoro-2-[(2-fluoro-4- iodophenyl)amino]phenyl][3-hydroxy-3-[(2S)-2-piperidinyl]-l-azetidinyl]methanone.
GDC-0973 has the chemical structure:

[00156] Compound II may be prepared following the methods described in
US2009/0156576 (the contents of which are hereby incorporated by reference). Compound II has the following CAS Registry Number: 934660-93-2 .
http://www.google.com/patents/US20090156576
Example 22(a) and 22(b) 1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2R)-piperidin-2-yl]azetidin-3-ol

and 1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2S)-piperidin-2-yl]azetidin-3-ol

To a solution of 1,1-dimethylethyl 2-(3-hydroxy-1-{[(phenylmethyl)oxy]carbonyl}azetidin-3-yl)piperidine-1-carboxylate (368 mg, 0.94 mmol), prepared using procedures similar to those described in Reference 5, in dichloromethane (5 mL) was added DMAP (115 mg, 0.94 mmol) and the resulting solution was cooled to 0° C. (R)-(−)-α-Methoxy-α-trifluoromethylphenylacetyl chloride (105 μL, 0.56 mmol) was added to the solution by syringe and the mixture was allowed to warm to room temperature then stirred an additional 12 hours. The solution was then partitioned with saturated aqueous sodium bicarbonate and the organic phase dried over anhydrous magnesium sulfate then filtered and concentrated to an oily residue. Silica gel flash chromatography using hexanes:ethyl acetate 3:1 as eluent afforded the less polar 1,1-dimethylethyl (2R)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (27.5 mg, 5% yield), the more polar 1,1-dimethylethyl (2S)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (105 mg, 19% yield) and starting material (253 mg, 69% recovery).
The starting material thus recovered was taken into dichloromethane (3 mL) followed by addition of DMAP (115 mg, 0.94 mmol) and (R)-(−)-α-methoxy-α-trifluoromethylphenylacetyl chloride (105 μL, 0.56 mmol) and the mixture was allowed to stir at room temperature over 12 hours. Proceeding as before afforded combined 1,1-dimethylethyl (2R)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (46.6 mg, 8% yield), the more polar 1,1-dimethylethyl (2S)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (228 mg, 41% yield) and starting material (100.8 mg, 27% recovery).
The starting material thus recovered was taken into tetrahydrofuran:dichloromethane (1:1, 2 mL) followed by addition of DMAP (47 mg, 0.39 mmol) and (R)-(−)-α-methoxy-α-trifluoromethylphenylacetyl chloride (80 μL, 0.43 mmol) and the mixture was heated to 60° C. over 12 hours. Proceeding as before afforded combined less polar 1,1-dimethylethyl (2R)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (144 mg, 26% yield). The chiral ester derivatives thus obtained were again subject to silica gel flash chromatography using hexanes:ethyl acetate 3:1 as eluent to give the pure less polar 1,1-dimethylethyl (2R)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (122.8 mg, 22% yield) and the more polar 1,1-dimethylethyl (2S)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (177.6 mg, 32% yield) both as colorless amorphous residues.
1,1-Dimethylethyl (2R)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (122.8 mg, 0.21 mmol) was taken into methanol (4 mL) followed by addition of 1M aqueous sodium hydroxide (1 mL) and the resulting solution was stirred for one hour at room temperature. The solution was then partitioned with ethyl acetate and 1N aqueous hydrochloric acid. The organic layer was washed with brine, dried over anhydrous magnesium sulfate then filtered and concentrated. The residue was purified by silica gel flash chromatography using hexanes:ethyl acetate 2:1 to give 1,1-dimethylethyl (2R)-2-(3-hydroxy-1-{[(phenylmethyl)oxy]carbonyl}azetidin-3-yl)piperidine-1-carboxylate (60.8 mg, 81% yield) a colorless amorphous solid. 1,1-dimethylethyl (2S)-2-(3-hydroxy-1-{[(phenylmethyl)oxy]carbonyl}azetidin-3-yl)piperidine-1-carboxylate (87.4 mg, 75% yield) was prepared analogously.
1,1-Dimethylethyl (2R)-2-(3-hydroxy-1-{[(phenylmethyl)oxy]carbonyl}azetidin-3-yl)piperidine-1-carboxylate (60.8 mg, 0.16 mmol) and 10% Pd/C (30 mg) were taken into methanol (2 mL) and the mixture hydrogenated at ambient pressure for one hour. The suspension was then filtered through a celite pad and concentrated then dried in vacuo to a colorless solid. The solid amine was taken into THF (1 mL) followed by addition of DIPEA (42 μL, 0.24 mmol) and 3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]benzoyl fluoride (63 mg, 0.16 mmol), prepared using procedures similar to those described in Reference 1, and the mixture stirred at room temperature for 30 minutes. The reaction mixture was partitioned with ethyl acetate and 1 N aqueous hydrochloric acid and the organic layer washed with brine, dried over anhydrous magnesium sulfate then filtered and concentrated. Purification of the residue by silica gel flash chromatography using hexanes:ethyl acetate 3:2 as eluent afforded 1,1-dimethylethyl (2R)-2-[1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-hydroxyazetidin-3-yl]piperidine-1-carboxylate (74.9 mg, 74% yield) as an amorphous solid. 1,1-Dimethylethyl (2R)-2-[1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-hydroxyazetidin-3-yl]piperidine-1-carboxylate 1H NMR (400 MHz, CDCl3): 8.53 (br s, 0.5H), 8.40 (br s, 0.5H), 7.41-7.38 (dd, 1H), 7.34-7.31 (dt, 1H), 7.17-7.14 (m, 1H), 6.86-6.79 (m, 1H), 6.63-6.587 (m, 1H), 4.24-3.90 (m, 4H), 3.37-3.23 (m, 1H), 2.90-2.80 (m, 1H), 1.85-1.54 (m, 7H), 1.43 (s, 9H); MS (EI) for C26H29F3IN3O4: 576 M-C4H9 +).
1,1-dimethylethyl (2R)-2-[1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-hydroxyazetidin-3-yl]piperidine-1-carboxylate (74.9 mg, 0.12 mmol) was taken into methanol (1 mL) followed by addition of 4 N HCl in dioxane (1 mL) and the solution was stirred at room temperature for one hour. The solution was then concentrated and the residue partitioned with chloroform and saturated aqueous sodium bicarbonate. The organic layer was washed with brine, dried over anhydrous sodium sulfate then filtered and concentrated. Purification of the residue by silica gel flash chromatography using ethyl acetate then concentrated aqueous ammonia in chloroform and methanol (0.1:10:1) as eluents afforded 1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2R)-piperidin-2-yl]azetidin-3-ol (57.3 mg) as a colorless amorphous solid. The free base was taken into methanol (1 mL) then brought to about pH 1 by addition of 4 N HCl in dioxane and the solution concentrated. The residue was triturated with ethyl ether to afford a suspension. The solid was collected by filtration to afford 1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2R)-piperidin-2-yl]azetidin-3-ol hydrochloride salt (49 mg, 72% yield) as a colorless solid. 1H NMR (400 MHz, CDCl3): 8.43-8.39 (d, 1H), 7.41-7.38 (dd, 1H), 7.33-7.31 (dt, 1H), 7.14-7.10 (m, 1H), 6.84-6.80 (m, 1H), 6.63-6.57 (m, 1H), 4.12-3.99 (m, 4H), 3.10-3.08 (d, 1H), 2.72-2.69 (d, 1H), 2.64-2.62 (m, 1H), 1.61-1.58 (m, 2H), 1.36-1.16 (m, 4H); MS (EI) for C21H21F3IN3O2: 532 (MH+).
………………………………..
Novel carboxamide-based allosteric MEK inhibitors: Discovery and optimization efforts toward XL518 (GDC-0973)
ACS Med Chem Lett 2012, 3(5): 416
http://pubs.acs.org/doi/abs/10.1021/ml300049d
http://pubs.acs.org/doi/abs/10.1021/ml300049d
|
8-17-2011
|
Methods of using MEK inhibitors
|
|
|
3-30-2011
|
Azetidines as MEK Inhibitors for the Treatment of Proliferative Diseases
|
|
|
9-29-2010
|
Azetidines as MEK Inhibitors for the Treatment of Proliferative Diseases
|
|
|
3-26-2010
|
Methods of Using PI3K and MEK Modulators
|
ACT-280778 is a L/T calcium channel blocker potentially indicated for the treatment of hypertension and angina pectoris

ACT-280778
(1R,2R,4R)-2-(2-((3-(4,7-Dimethoxy-1H-benzo[d]imidazol-2-yl)propyl)(methyl)amino)ethyl)-5-phenylbicyclo[2.2.2]oct-5-en-2-yl Isobutyrate
Propanoic acid, 2-methyl-, (1R,2R,4R)-2-[2-[[3-(4,7-dimethoxy-1H-benzimidazol-2-yl)propyl]methylamino]ethyl]-5-phenylbicyclo[2.2.2]oct-5-en-2-yl ester
isobutyric acid (1R,2R,4R)-2-(2-{[3-(4,7-dimethoxy-1H-benzoimidazol-2-yl)-propyl]-methyl-amino}-ethyl)-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl ester
Actelion Pharmaceuticals Ltd innovator
C33 H43 N3 O4
1075744-31-8
bis-MALEATE SALT 1537197-53-7
Chiral bicyclic benzimidazole 1 (ACT-280778) is a L/T calcium channel blocker potentially indicated for the treatment of hypertension and angina pectoris
Many cardiovascular disorders have been associated with a ‘calcium overload’ resulting from an abnormal elevated calcium influx through the plasma membrane of cardiac and vascular smooth muscle cells. There are 3 major pathways through which extracellular calcium can enter these cells: 1 ) receptor-activated calcium channels, 2) ligand-gated calcium channels and 3) voltage-operated calcium channels (VOCs). 0 VOCs have been classified into 6 main categories: L (Long-lasting), T (Transient), N (Neuronal), P (Purkinje cells), Q (after P) and R (Remaining or Resistant).
L-type calcium channels are responsible for the inward movement of calcium that initiates contraction in cardiac and smooth muscle cells suggesting a putative application for blockers of these channels in the cardiovascular field. In this view, L-type calcium channel blockers5 have been used in clinic since the early 60s and are now recommended as a first line of treatment for systolic-diastolic hypertension and angina pectoris.
T-type calcium channels are found in various tissues such as coronary and peripheral vasculature, sinoatrial node and Purkinje fibres, brain, adrenal glands and in the kidney. This broad distribution suggests a T-type channel blocker to have a putative cardiovascular0 protection, to have en effect on sleep disorders, mood disorders, depression, migraine, hyperaldosteroneemia, preterm labor, urinary incontinence, brain aging or neurodegenerative disorders such as Alzheimers disease.
Mibefradil (Posicor®), the first L-type and T-type calcium channels blocker demonstrated a superior effect over calcium channel blockers, which target the L channel predominantly. Mibefradil was used for the treatment of hypertension and angina without showing negative side-effects often seen by L channel blockers like inotropy, reflex tachycardia, vasoconstrictive hormone release or peripheral edema. Additionally, mibefradil showed a potentially cardioprotective effect (Villame, Cardiovascular Drugs and Therapy 15, 41-28, 2001 ; Ramires, J MoI Cell Cardiol 1998, 30, 475-83), a renal protective effect (Honda, Hypertension 19, 2031-37, 2001 ), and showed a positive effect in the treatment of heart failure (Clozel, Proceedings Association American Physicians 1999, 1 11 , 429-37).
Despite the enormous demand for a compound of this profile, mibefradil was withdrawn from the market in 1998 (one year after its launch), due to unacceptable CYP 3A4 drug interactions. Moreover, ECG abnormalities (i.e. QT prolongations) and interaction with the MDR-1 mediated digoxin efflux were also reported (du Souich, Clin Pharmacol Ther 67, 249- 57, 2000; Wandel, Drug Metab Dispos 2000, 28, 895-8).
It has now been found that crystalline salt forms of COMPOUND (isobutyric acid (1 R*,2R*,4R*)-2-(2-{[3-(4,7-dimethoxy-1 H-benzoimidazol-2-yl)-propyl]-methyl-amino}-ethyl)- 5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl ester) may under certain conditions be found. Said crystalline salt forms of COMPOUND are novel and may have advantageous properties, especially compared to the free base (WO2008/132679) or the di-hydrochloride salt of COMPOUND. Such advantages may include better flow properties, better solubility, less hygroscopicity, better reproducibiliy in manufacturing (for example better filtration parameters, better reproducibility of formation, better sedimentation),
………………………
http://www.google.com/patents/WO2009130679A1?cl=en
Scheme 1


……………
http://www.google.com/patents/CN102186828A?cl=en
http://www.google.com/patents/EP2344461A1?cl=en
The preparation of COMPOUND is known from WO2008/132679: Preparation of intermediates
General procedures for the preparation of key intermediates K: Key intermediates K1A and K2A which are bicyclo[2.2.2]oct-5-en-2-yl or bicyclo[3.2.2]non-8- en-6-yl derivatives are obtained as a mixture between the major racemate having the relative configuration (R*, R*, R*) (i.e. the bridge -(CH2)2– of the cyclohexene moiety is cis to the group -OR2 being hydroxy) and the minor racemate having the relative configuration (R*, S*, R*) (i.e. the bridge -(CH2)2– of the cyclohexene moiety is trans to the group -OR2 being hydroxy). The major and the minor racemates can be separated as described for key intermediate K1A in procedure A1.5. The major racemate is isolated and used in the preparation of the examples below.
K1 A: rac-(1 R*,2R*,4R*)-(2-Hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid tert. -butyl ester K1 A.1 (Procedure A1.1 ): rac-(1 R*.4R*VBicvclor2.2.21octane-2.5-dione
25 ml. of 2-(trimethylsilyloxy)-1 ,3-cyclohexadiene and 13 ml. of α-acetoxyacrylonitrile were mixed and heated at 1500C in a closed vessel for 22 h. The obtained dark orange viscous oil was dissolved in 200 ml. of MeOH. After dropwise addition of a solution of 2.2 g of sodium methoxide in 150 ml. of MeOH the reaction mixture was stirred for 3 h at rt, poured into ice/water and extracted with DCM. The organic phases were concentrated in vacuo and the crude residue was purified by CC with EtOAc-Hept (1 :2) to yield 7.9 g of rac-(1 R*,4R*)- bicyclo[2.2.2]octane-2,5-dione. LC-MS: tR = 0.44 min.
K1A.2 (Procedure A1.2): rac-(1 R*.4R*VSpirorbicvclor2.2.2loctane-2.2′-ri .3ldioxolanl-5-one To 4.0 g of rac-(1 R*,4R*)-bicyclo[2.2.2]octane-2,5-dione (intermediate K1A.1 ), dissolved in 120 ml. of toluene, 1.7 ml. of ethylene glycol and 0.27 g of TsOH were added and the solution was heated under vigorous stirring to reflux for 3.5 h. The reaction mixture was cooled to rt, quenched with saturated aq. NaHCO3, extracted with Et2O, and the organic phase was evaporated. The crude product was purified by CC with Hex-EtOAc (7:3) to yield 2.41 g of rac-(1 R*,4R*)-spiro[bicyclo[2.2.2]octane-2,2′-[1 ,3]dioxolan]-5-one as yellow oil. LC-MS: tR = 0.64 min; [M+H+CH3CN]+: 224.35. K1A.3 (Procedure A1.3): Mixture of rac-(7R*.8R*.10R*V and rac-(7R*.8S*.10R*V7.10-(1.2- Ethylen)-8-phenyl-1 ,4-dioxa-spiror4.5ldecan-8-ol
To a solution of 2.41 g of rac-(1 R*,4R*)-spiro[bicyclo[2.2.2]octane-2,2′-[1 ,3]dioxolan]-5-one
(intermediate K1A.2) in 80 ml. Et2O, 14.5 ml. phenylmagnesium bromide solution (1 M in Et2O) was added dropwise over 10 min. The reaction mixture was stirred for 4 h at rt. Then, the mixture was quenched carefully with ice, 8 ml. 2N HCI were added and the phases were separated. The organic phase was evaporated and the crude product was purified by CC with Hept-EtOAC (7:3) to give 0.37 g of 7,10-(1 ,2-ethylen)-8-phenyl-1 ,4-dioxa- spiro[4.5]decan-8-ol as colorless oil. (Separation of the diastereomers by CC is possible but was not performed here.)
LC-MS: tR = 0.84 min; [M-H2O+H]+: 243.34.
K1A.4 (Procedure A1.4): rac-(1 R*,4R*)-5-Phenyl-bicvclor2.2.2loct-5-en-2-one
To a solution of 0.54 g of 7,10-(1 ,2-ethylen)-8-phenyl-1 ,4-dioxa-spiro[4.5]decan-8-ol (intermediate K1A.3) in 20 ml. acetone was added 200 mg of TsOH and then the mixture was stirred for 2 d at rt. The reaction mixture was quenched with sat. aq. NaHCO3, extracted with EtOAC and the organic phase was evaporated. The crude product was purified by CC with Hept-EtOAC (7:3) to give 0.34 g of rac-(1 R*,4R*)-5-phenyl-bicyclo[2.2.2]oct-5-en-2-one as colorless oil. LC-MS: tR = 0.93 min; [M+H+CH3CN]+: 240.1 1. K1A.5 (Procedure A1.5): rac-(1 R*.2R*.4R*H2-Hvdroxy-5-phenyl-bicvclor2.2.2loct-5-en-2-vn- acetic acid tert.-butyl ester and rac-(1 R*,2S*,4R*H2-hvdroxy-5-phenyl-bicvclor2.2.2loct-5-en- 2-yl)-acetic acid tert.-butyl ester
To a solution of 0.51 mL of DIPA in 0.5 mL THF 2.2 mL of n-butyllithium (1.6M in Hex) were added dropwise at -200C. After 10 min, 0.5 mL of toluene were added and the solution was stirred for 30 min. The mixture was cooled to -500C, 0.73 mL of tert.-butyl acetate were added and stirring was continued for 1 h at -500C. Then 0.32 g of rac-(1 R*,4R*)-5-phenyl- bicyclo[2.2.2]oct-5-en-2-one (intermediate K1A.4) dissolved in 1 mL of THF was added and the solution was stirred at -50 to -200C over 2.5 h. The reaction mixture was poured on ice/aq. HCI, the organic phase was separated, washed and evaporated. The crude reaction product was purified by CC with Hept-EtOAc (9:1 ) to yield 0.30 g of the major racemate, rac- (1 R*,2R*,4R*)-2-hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid tert.-butyl ester, as white solid and 0.07 g of the minor racemate, rac-(1 R*,2S*,4R*)-2-hydroxy-5-phenyl- bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid tert.-butyl ester, as colorless oil. LC-MS (major racemate): tR = 1.06 min; [M-(CH3)3-H2O+H]+: 241.1 1. LC-MS (minor racemate): tR = 1.05 min; [M+H]+: 315.18. K1A.6: (1 S.2S.4SV(2-Hvdroxy-5-Dhenyl-bicvclor2.2.2loct-5-en-2-vn-acetic acid tert.-butyl ester and (1 R,2R,4R)-(2-Hvdroxy-5-phenyl-bicvclor2.2.2loct-5-en-2-yl)-acetic acid tert.-butyl ester rac-(1 R*,2R*,4R*)-(2-Hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid tert.-butyl ester was separated into the respective enantiomers using prep, chiral HPLC (column: Daicel ChiralPak AD-H, 20×250 mm, 5 μm; Hex/ EtOH 95:5, flow 16 mL/min) Chiral analytic HPLC (Daicel ChiralPak AD-H, 4.6×250 mm, 5 μm; Hex/ EtOH 95:5, flow 0.8 mL/min):
(1 R,2R,4R)-(2-Hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid tert.-butyl ester: Enantiomer A: tR = 6.70 min.
(1S,2S,4S)-(2-Hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid tert.-butyl ester: Enantiomer B: tR = 7.93 min.
BB. [3-(4,7-Dimethoxy-1 H-benzoimidazol-2-yl)-propyl]-methyl-amine
BB.1 3,6-Dimethoxy-benzene-1 ,2-diamine 3, 6-Dimethoxy-benzene-1 ,2-diamine was synthesized by dissolving 6.0 g of 1 ,4-dimethoxy- 2,3-dinitro-benzene (Eur.J.Org.Chem. 2006, 2786-2794) in 220 mL EtOH, evacuating 3 times with N2 and adding 600 mg of 10wt% Pd/C. The reaction was stirred under a H2 atmosphere (balloon). Another 300 mg of 10wt% Pd/C were added after 2 days and the mixture was stirred for another 24 h. Filtration over a pad of celite and washing with EtOH and EtOAc yielded after concentration in vacuo 4.3 g of 3, 6-dimethoxy-benzene-1 ,2-diamine as black solid. LC-MS: tR = 0.48 min; [M+H]+: 169.09.
BB.2 r3-(2-Amino-3,6-dimethoxy-phenylcarbamoyl)-propyll-methyl-carbamic acid benzyl ester To a solution of 3.1 g of 4-(benzyloxycarbonyl-methyl-amino)-butyric acid in 80 mL DCM were added 6.5 mL of DIPEA, 1.8 g of HOBt, 2.6 g of EDC and 154 mg of DMAP. After stirring for 10 min, 2.1 g of 3, 6-dimethoxy-benzene-1 ,2-diamine, dissolved in 20 mL DCM, were added and the mixture was stirred at rt overnight. The reaction was quenched with sat. aq. NaHCO3, the phases were separated and the organic phase was washed with brine, dried over MgSO4 and concentrated in vacuo to yield the crude title compound as black oil. LC-MS: tR = 0.88 min; [M+H]+: 402.06.
BB.3 [3-(4,7-Dimethoxy-1 H-benzoimidazol-2-yl)-propyl1-methyl-carbamic acid benzyl ester
To a mixture of the above crude 3-(2-amino-3,6-dimethoxy-phenylcarbamoyl)-propyl]-methyl- carbamic acid benzyl ester in 16 mL toluene were added 4 mL of DMF and 1.9 g of TsOH and the reaction was heated to 1500C for 2 h in the microwave. Sat. aq. NaHCO3 was added and the phases were separated. The organic phase was washed with brine, dried over MgSO4, concentrated in vacuo, filtered over a short pad of silica gel with EtOAc and concentrated again. Purification by CC with EtOAc yielded 2.7 g of 3-(4,7-dimethoxy-1 H- benzoimidazol-2-yl)-propyl]-methyl-carbamic acid benzyl ester as brown resin. LC-MS: tR = 0.85 min; [M+H]+: 384.62.
BB.4 r3-(4,7-Dimethoxy-1 H-benzoimidazol-2-yl)-propyll-methyl-amine
A solution of 2.6 g of 3-(4,7-dimethoxy-1 H-benzoimidazol-2-yl)-propyl]-methyl-carbamic acid benzyl ester in 60 ml. EtOH was evacuated 3 times with N2 before 260 mg of 10 wt% Pd/C were added. The reaction mixture was then stirred under a H2atmosphere (balloon) for 5 h at rt. Filtration over a pad of celite and washing with EtOH yielded after concentration in vacuo 1.7 g of 3-(4,7-dimethoxy-1 H-benzoimidazol-2-yl)-propyl]-methyl-amine as brown foam. LC-MS: tR = 0.57 min; [M+H]+: 250.13.
Preparation of COMPOUND Reference Example 1A: rac-lsobutyric acid (1 R*,2R*,4R*)-2-(2-{[3-(4,7-dimethoxy-1H- benzoimidazol-2-yl)-propyl]-methyl-amino}-ethyl)-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl ester
1.1 (Procedure P1.1 V rac-(1 R*.2R*.4R*H2-Hvdroxy-5-phenyl-bicvclor2.2.2loct-5-en-2-vn- acetic acid To a solution of 4.0 g of rac-(1 R*,2R*,4R*)-(2-hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)- acetic acid tert.-butyl ester in 25 mL EtOH were added 2.1 g of LiOH-H2O, 8 mL H2O and 22 mL MeOH. The reaction mixture was stirred at rt for 3 d and then concentrated. The residue was partitioned between water and Et2O. The aq. layer was separated and acidified with 1 N HCI resulting in the formation of a white solid. The solid was filtrated, washed with 5 mL aq. HCI and dried in vacuo to obtain 3.2 g of rac-(1 R*,2R*,4R*)-(2-hydroxy-5-phenyl- bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid as white solid. LC-MS: tR = 0.86 min; [M-H2O+H]+: 241.28.
1.2 (Procedure P1.2): rac-(1 R*,2R*,4R*)-N-r3-(4,7-Dimethoxy-1 H-benzoimidazol-2-yl)- propyl1-2-(2-hvdroxy-5-phenyl-bicvclo[2.2.21oct-5-en-2-yl)-N-methyl-acetamide To a solution of 280 mg of rac-(1 R*,2R*,4R*)-(2-hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2- yl)-acetic acid in 7 mL THF were added 0.58 mL of DIPEA, 175 mg of HOBt and 250 mg of EDC at rt. After stirring for 10 min, 270 mg of 3-(4,7-dimethoxy-1 H-benzoimidazol-2-yl)- propyl]-methyl-amine were added and the reaction mixture was stirred at rt overnight. The reaction mixture was quenched with sat. aq. NaHCO3, the phases were separated and the organic phase was washed with water and brine, dried over MgSO4 and concentrated in vacuo. Purification by CC using EtOAc-MeOH (5:1 to 2:1 ) yielded 475 mg of rac- (1 R*,2R*,4R*)-N-[3-(4,7-dimethoxy-1 H-benzoimidazol-2-yl)-propyl]-2-(2-hydroxy-5-phenyl- bicyclo[2.2.2]oct-5-en-2-yl)-N-methyl-acetamide as white foam. LC-MS: tR = 0.91 min; [M+H]+: 490.06.
1.3 (Procedure P1.3): rac-(1 R*.2R*.4R*V2-(2-fr3-(4.7-Dimethoxy-1 H-benzoimidazol-2-ylV propyll-methyl-amino}-ethyl)-5-phenyl-bicvclor2.2.21oct-5-en-2-ol
To a solution of 310 mg of rac-(1 R*,2R*,4R*)-N-[3-(4,7-dimethoxy-1 H-benzoimidazol-2-yl)- propyl]-2-(2-hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)-N-methyl-acetamide in 8 mL toluene were added dropwise 0.77 ml. of a Red-AI solution (65% in toluene) at 00C. After stirring for 10 min at 00C, the cooling bath was removed and stirring was continued for 3 h at rt. The reaction mixture was then carefully poured onto a mixture of 1 M NaOH/ice and stirred for 10 min. The aq. phase was extracted with toluene, the combined organic phases were washed with brine, dried over MgSO4 and concentrated in vacuo. Purification by CC using EtOAc-MeOH (2:1 ) yielded 230 mg of rac-(1 R*,2R*,4R*)-2-(2-{[3-(4,7-dimethoxy-1 H- benzoimidazol^-y^-propyll-methyl-aminoj-ethy^-δ-phenyl-bicyclop^^loct-δ-en^-ol as white foam. LC-MS: tR = 0.79 min; [M+H]+: 476.13. 1.4: rac-lsobutyric acid (1 R*.2R*.4R*‘)-2-(2-fr3-(4.7-dimethoxy-1 H-benzoimidazol-2-vn- propyll-methyl-amino}-ethyl)-5-phenyl-bicvclor2.2.21oct-5-en-2-yl ester
To a solution of 199 mg of rac-(1 R*,2R*,4R*)-2-(2-{[3-(4,7-dimethoxy-1 H-benzoimidazol-2- yO-propyO-methyl-aminoJ-ethy^-δ-phenyl-bicycloP^^loct-δ-en^-ol in 4 mL DCM were added 0.2 mL of NEt3 and 0.1 mL of isobutyrylchloride at 0°C. The reaction mixture was stirred overnight allowing the temperature to reach slowly rt. The reaction was quenched with sat. aq. NaHCO3, the phases were separated and the water phase was re-extracted with DCM. The combined organic phases were washed with brine, dried over MgSO4 and concentrated in vacuo. The residue was redissolved in 3 mL EtOAc, silica gel and 1.5 mL MeOH were added and the mixture was stirred vigorously for 7 d. The mixture was filtered, thouroughly washed with EtOAc-MeOH (2:1 ) and evaporated. Purification by CC using EtOAc-MeOH (5:1 to 3:1 + 0.1 % NEt3) yielded 186 mg of rac-isobutyric acid (1 R*,2R*,4R*)-2- (2-{[3-(4,7-dimethoxy-1 H-benzoimidazol-2-yl)-propyl]-methyl-amino}-ethyl)-5-phenyl- bicyclo[2.2.2]oct-5-en-2-yl ester as beige foam. LC-MS: tR = 0.90 min; [M+H]+: 546.23. Reference Example 2A: lsobutyric acid (1S,2S,4S)-2-(2-{[3-(4,7-dimethoxy-1H- benzoimidazol-2-yl)-propyl]-methyl-amino}-ethyl)-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl ester
2.1 : (1S.2S.4SV(2-Hvdroxy-5-Dhenyl-bicvclor2.2.2loct-5-en-2-vn-acetic acid Prepared according to procedure P1.1 in Reference Example 1A using enantiomer B of rac- (1 R*,2R*,4R*)-(2-hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid tert.-butyl ester (see K1A.6). LC-MS: tR = 0.91 min; [M-H2CHH]+: 241.10.
2.2: (1S.2S.4SV2-(2-fr3-(4.7-Dimethoxy-1 H-benzoimidazol-2-ylVDroDyll-methyl-amino>- ethyl)-5-phenyl-bicvclor2.2.21oct-5-en-2-ol
Prepared according to procedures P1.2 to P1.3 in Reference Example 1A using the above (2-hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid. LC-MS: tR = 0.78 min; [M+H]+: 476.09.
2.3: Isobutyric acid (1S,2S,4S)-2-(2-{[3-(4J-dimethoxy-1 H-benzoimidazol-2-yl)-propyl1- methyl-amino}-ethyl)-5-phenyl-bicvclor2.2.21oct-5-en-2-yl ester
Prepared according to procedure P1.4 in Reference Example 1A using the above 2-(2-{[3-
(4,7-dimethoxy-1 H-benzoimidazol-2-yl)-propyl]-methyl-amino}-ethyl)-5-phenyl- bicyclo[2.2.2]oct-5-en-2-ol.
LC-MS: tR = 0.89 min; [M+H]+: 546.19. Reference Example 3A: lsobutyric acid (1 R,2R,4R)-2-(2-{[3-(4,7-dimethoxy-1H- benzoimidazol-2-yl)-propyl]-methyl-amino}-ethyl)-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl ester
3.1 : (1 R.2R.4RH2-Hvdroxy-5-phenyl-bicvclor2.2.21oct-5-en-2-vn-acetic acid
Prepared according to procedure P1.1 in Reference Example 1 using enantiomer A of rac- (1 R*,2R*,4R*)-(2-hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid tert.-butyl ester (see K1A.6). LC-MS: tR = 0.91 min; [M-H2CHH]+: 241.16.
3.2: (1 R,2R,4R)-2-(2-{[3-(4,7-Dimethoxy-1 H-benzoimidazol-2-yl)-propyl1-methyl-amino}- ethvD-δ-phenyl-bicvclo^^^loct-δ-en^-ol Prepared according to procedures P1.2 to P1.3 in Reference Example 1 using the above (2- hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid. LC-MS: tR = 0.79 min; [M+H]+: 476.09. 3.3: Isobutyric acid (1 R.2R.4RV2-(2-{r3-(4.7-dimethoxy-1 H-benzoimidazol-2-ylVpropyll- methyl-amino}-ethyl)-5-phenyl-bicvclor2.2.21oct-5-en-2-yl ester
Prepared according to procedure P1.4 in Reference Example 1A using the above 2-(2-{[3- (4,7-dimethoxy-1 H-benzoimidazol-2-yl)-propyl]-methyl-amino}-ethyl)-5-phenyl- bicyclo[2.2.2]oct-5-en-2-ol.
LC-MS: tR = 0.89 min; [M+H]+: 546.11. Optical rotation: alpha D (c = 10 mg/mL EtOH) = -21.5°.
1 H NMR (MeOD, 400 MHz) δ 7.39-7.37 (m, 2H), 7.30 (t, J = 6.4 Hz, 2H), 7.24-7.20 (m, 1 H), 6.60 (s, 2 H), 6.43 (br d, J = 7.6 Hz, 1 H), 3.91 (s, 6H), 3.27-3.23 (m, 1 H), 3.18-3.15 (m, 1 H), 2.87 (t, J = 7.6 Hz, 2H), 2.54 (sept, J = 7.0 Hz, 1 H), 2.47-2.37 (m, 4H), 2.21 (s, 3H), 2.19- 2.12 (m, 1 H), 2.01-1.92 (m, 5H), 1.75-1.65 (m, 2H), 1.48-1.38 (m, 1 H), 1.27-1.19 (m, 1 H), 1.16 (d, J = 7.0 Hz, 6H).
Example S5: Preparation and characterization of the di-maleic acid salt of COMPOUND
Maleic acid (256 g, 2.2 mol, 2.1 eq), dissolved in MeOH (630 ml_, 1.1 volumes) was added to a refluxing solution of COMPOUND (682 g, 84% w/w (NMR assay), 1.05 mol) in EtOAc (6.3 L, 11 volumes). The resulting mixture was stirred under reflux for 15 minutes and was then cooled to 65-68°C within 30 minutes and seeded with 0.04% w/w of seeding crystals of di- maleic acid salt of COMPOUND (Seeding crystals were obtained after careful crystallisation using the same protocol.). The mixture was then cooled from 65-68°C to 400C within 3 h. The obtained suspension was then cooled down to 200C over 1 h, filtered under 0.2 bar of nitrogen and rinsed with EtOAc (1500 ml. 2.6 volumes). The obtained white solid was then dried under 1 atmosphere of nitrogen for 24 hours to yield 715 g (88%) of the di-maleic acid salt of COMPOUND.
Table S5: Characterisation data for the di-maleic acid salt of COMPOUND

………….
paper
A scalable access to 1 (ACT-280778), a potent L/T calcium channel blocker, has been developed. The synthesis, amenable to kilogram manufacturing, comprises 10 chemical steps from enantiomerically pure 5-phenylbicyclo[2.2.2]oct-5-en-2-one (3) and 1,4-dimethoxybenzene with a longest linear sequence of 7 steps. Key to the success of this fit-for-purpose approach are a robust and atom-efficient access to benzimidazole 4, the substrate-controlled diastereoselective enolate addition toward carboxylic acid 2 that was isolated by simple crystallization with high dr (>99:1), the convenient selective N-deacylation of intermediate 10, and the identification of a suitable solid form of 1 as the bis-maleate salt (1·2 C4H4O4). As an illustration of the robustness of this process, 14 kg of drug substance, suitable for human use, was produced with an overall yield of 38% over the longest linear sequence (7 steps).




LC-MS were run using the following conditions: Finnigan Navigator with HP 1 100 Binary Pump and DAD, column: 4.6×50 mm, Zorbax SB-AQ, 5 μm, 120 A, gradient: 5-95% acetonitrile in water, 1 min, with 0.04% trifluoroacetic acid, flow: 4.5 mL/min, tR is given in min.
Compounds are purified by preparative HPLC (column: X-terra RP18, 50×19 mm, 5 μm, gradient: 10-95% acetonitrile in water containing 0.5 % of formic acid) or by column chromatography on silica gel. Racemates can be separated into their enantiomers by preparative HPLC (preferred conditions: Daicel, ChiralCel OD 20×250 mm, 10 μm, 4% ethanol in hexane, flow 10-20 mL/min).
-
this work was preliminarily disclosed: Funel, J.-A. In Practical Synthesis of L/T Calcium Channel Blocker ACT-280778, 30th SCI Process Development Symposium, Cambridge, UK, December 5–7, 2012; Funel, J.-A.; In Practical Synthesis of 5-Phenylbicyclo[2.2.2]oct-5-en-2-one toward L/T Calcium Channel Blocker ACT-280778. Application of the Diels–Alder Reaction on kg-Scale, 1st Smart Synthesis and Advanced Purification Conference, April 21–23, 2013; Lyon, FR.
-
(a) Hilpert, K., Hubler, F., and Renneberg, D. WO/2008/132679A1, 2008.
(b) Hubler, F.,Hilpert, K., and Renneberg, D. WO/2009/130679A1, 2009.
-
Funel, J.-A.; Schmidt, G.; Abele, S. Org. Process Res. Dev. 2011, 15, 1420– 1427
-
Abele, S.; Schwaninger, M.; Fierz, H.; Schmidt, G.; Funel, J.-A.; Stoessel, F. Org. Process Res. Dev. 2012, 16, 2015– 2020
-
(a) Abele, S.; Inauen, R.; Funel, J.-A.; Weller, T. Org. Process Res. Dev. 2012, 16, 129–140
(b) Abele, S.; Funel, J.-A. WO/2012/052943A1, 2012.
(c) Abele,S.; Funel, J.-A. WO/2012/052939A2, 2012.
-
Abele, S.; Inauen, R.; Spielvogel, D.; Moessner, C. J. Org. Chem. 2012, 77, 4765– 4773
Antimalarials………….Arterolane from Ranbaxy

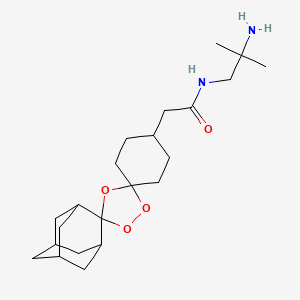
Arterolane

Arterolane, also known as OZ277 or RBx 11160,is a substance being tested for antimalarial activity[1] by Ranbaxy Laboratories.[2] It was discovered by US and European scientists who were coordinated by the Medicines for Malaria Venture (MMV).[3] Its molecular structure is uncommon for pharmacological compounds in that it has both an ozonide group and an adamantane substituent.[4]
Phase III clinical trials of arterolane, in combination with piperaquine, began in India in 2009.[5] When clinical trial results were disappointing, the MMV withdrew support[2] and Ranbaxy continued developing the drug combination on its own.

Ranbaxy launched India’s first new drug, SynriamTM, treating Plasmodium falciparummalaria in adults. The drug provides quick relief from most malaria-related symptoms, including fever, and has a high cure rate of over 95 %.
Just one tablet per day is required, for three days, instead of two to four tablets, twice daily, for three or more days with other medicines. The drug is independent of dietary restrictions for fatty foods or milk.
Ranbaxy developed Synriam as a fixed-dose combination of arterolane maleate and piperaquine phosphate, where arterolane is the new chemical entity (NCE) that was developed as an alternative to artemisinin. It is the first recently developed antimalarial not based on artemisinin, one of the most effective treatments for malaria, which has shown problems with resistance in recent years. Arterolane was discovered by a collaborative drug discovery project funded by the Medicines for Malaria Venture. Since SynriamTM has a synthetic source, unlike artemisinin-based drugs, production can be scaled up whenever required and a consistent supply can be maintained at a low cost.

The new drug, has been approved by the Drug Controller General of India (DCGI) for marketing in India and conforms to the recommendations of the World Health Organization (WHO) for using combination therapy in malaria. Ranbaxy is also working to make it available in African, Asian and South American markets where Malaria is rampant. SynriamTM trials are ongoing for Plasmodium vivax malaria and a paediatric formulation.
Derek Lowe of the famous In the Pipeline blog had written about arterolane in 2009. At the time it was in Phase III trial, which I assumed were the trials that Ranbaxy was conducting. But it turned out that arterolane was developed by a collaboration between researchers in the US, the UK, Switzerland and Australia who were funded by the World Health Organization and Medicines for Malaria Venture (a Swiss non-profit). They published this work in Nature in 2004 and further SAR (Structure Activity Relationship) studies in J Med Chem in 2010. So Ranbaxy did not develop the drug from scratch? But the press release quotes Arun Sawhney, CEO and Managing Director of Ranbaxy which misleads people to think so: “It is indeed gratifying to see that Ranbaxy’s scientists have been able to gift our great nation its first new drug, to treat malaria, a disease endemic to our part of the world. This is a historic day for science and technology in India as well as for the pharmaceutical industry in the country. Today, India joins the elite and exclusive club of nations of the world that have demonstrated the capability of developing a new drug”. So Ranbaxy mixes a known active compound (piperaquine) with a new compound that someone else found to be active (arterolane) and claims that they developed a new drug? In an interview in LiveMint, Sawhney says, “Ranbaxy spent around $30 million on Synriam and the contribution from DST [India’s Department of Science & Technology] was Rs.5 crore. The drug went through several phases of development since the project began in 2003. We did not look at this as a commercial development. Instead, this is a CSR [Corporate Social Responsibility] venture for us.” That’s a give away because developing a new drug from scratch has to cost more than $30 million + Rs.50 million.
- Ranbaxy Laboratories Limited (Ranbaxy), India
![]() now taken over by sun
now taken over by sun
SynriamTM
|
|
Description SynriamTM is a fixed dose combination of two antimalarial active ingredients arterolane maleate and piperaquine phosphate.
Arterolane maleate is a synthetic trioxolane compound. The chemical name of arterolane maleate is cis-adamantane-2-spiro-3’-8’-[[[(2’-amino-2’ methylpropyl) amino] carbonyl] methyl] 1’,2’,4’-trioxaspiro [4.5] decane hydrogen maleate. The molecular formula is C26H40N2O8 and molecular weight is 508.61. The structural formula is as follows:

| MALARIA |
| Malaria is one of the most prevalent and deadly parasitic diseases in the world. Up to 289 million cases of malaria may have occurred in 2010, causing between 660,000 and 1.25 million deaths, mainly in Africa and mostly of children younger than 5 years. |
| (WHO: http://www.who.int/malaria/publications/world_malaria_report_2012/en/index.html; Fidock, D. A. Eliminating Malaria. Science 2013, 340, 1531-1533.) |
|


Malaria, the most common parasitic disease of humans, remains a major health and economic burden in most tropical countries. Large areas of Central and South America, Hispaniola (Haiti and the Dominican Republic), Africa, the Middle East, the Indian subcontinent, Southeast Asia, and Oceania are considered as malaria-risk areas. It leads to a heavy toll of illness and death, especially amongst children and pregnant women.
According to the World Health Organization, it is estimated that the disease infects about 400 million people each year, and around two to three million people die from malaria every year. There are four kinds of malaria parasites that infect human: Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale and Plasmodium malariae.
Malaria spreads from one person to another by the bite of mosquito, Anopheles gambiae, which serves as vector. When a mosquito sucks the blood of human, sporozoites are transfused into the human body together with saliva of the mosquito. The sporozoites enter into the hepatocytes, reproduce asexually and finally enter into the blood stream. The parasites continue to multiply inside the red blood cells, until they burst and release large number of merozoites. This process continues, destroying a significant number of blood cells and causing the characteristic paroxysm (“chills and fever”) associated with the disease. In the red blood cells, some of the merozoites become male or female gametocytes. These gametocytes are ingested by the mosquito when it feeds on blood. The gametocytes fuse in the vector’s gut; sporozoites are produced and are migrated to the vector’s salivary glands.
The clinical symptoms of malaria are generally associated with the bursting of red blood cells causing an intense fever associated with chills that can leave the infected individual exhausted and bedridden. More severe symptoms associated with repeat infections and/or infection by Plasmodium falciparum include anaemia, severe headaches, convulsions, delirium and, in some instances, death.
Quinine, an antimalarial compound that is extracted from the bark of cinchona tree, is one of the oldest and most effective drugs in existence. Chloroquine and mefloquine are the synthetic analogs of quinine developed in 1940’s, which due to their effectiveness, ease of manufacture, and general lack of side effects, became the drugs of choice. The downside to quinine and its derivatives is that they are short-acting and have bitter taste. Further, they fail to prevent disease relapses and are also associated with side effects commonly known as “Chinchonism syndrome” characterized by nausea, vomiting, dizziness, vertigo and deafness. However, in recent years, with the emergence of drug- resistant strains of parasite and insecticide-resistant strains of vector, the treatment and/or control of malaria is becoming difficult with these conventional drugs.
Malarial treatment further progressed with the discovery of Artemisinin
(qinghaosu), a naturally occurring endoperoxide sesquiterpene lactone isolated from the plant Artemisia annua (Meshnick et al., Microbiol. Rev. 1996, 60, p. 301-315; Vroman et al., Curr. Pharm. Design, 1999, 5, p. 101-138; Dhingra et al., 2000, 66, p. 279-300), and a number of its precursors, metabolites and semi-synthetic derivatives which have shown to possess antimalarial properties. The antimalarial action of artemisinin is due to its reaction with iron in free heme molecules of the malaria parasite, with the generation of free radicals leading to cellular destruction. This initiated a substantial effort to elucidate its molecular mechanism of action (Jefford, dv. Drug Res. 1997, 29, p. 271-325; Cumming et al., Adv. Pharmacol. 1997, 37, p. 254-297) and to identify novel antimalarial peroxides (Dong and Vennerstrom, Expert Opin. Ther. Patents 2001, 1 1, p. 1753-1760).
Although the clinically useful artemisinin derivatives are rapid acting and potent antimalarial drugs, they have several disadvantages including recrudescence,
neurotoxicity, (Wesche et al., Antimicrob. Agents. Chemother. 1994, 38, p. 1813-1819) and metabolic instability (White, Trans. R. Soc. Trop. Med. Hyg., 1994, 88, p. 41-43). A fair number of these compounds are quite active in vitro, but most suffer from low oral activity (White, Trans. R. Soc. Trop. Med. Hyg., 1994, 88, p. 41-43 and van Agtmael et al., Trends Pharmacol. Sci., 1999, 20, p. 199-205). Further all these artemisinin derivatives are conventionally obtained from plant source and are therefore expensive. As the cultivation of the plant material is dependent on many factors including the weather conditions, the supply source thus becomes finite and there are chances of varying yield and potency. This leads to quality inconsistencies and supply constraints. As malaria is more prevalent in developing countries, a switch to cheaper and effective medicine is highly desirable.
Thus there exists a need in the art to identify new peroxide antimalarial agents, especially those which are not dependent on plant source and can be easily synthesized, are devoid of neurotoxicity, and which possess improved solubility, stability and pharmacokinetic properties.
Following that, many synthetic antimalarial 1 ,2,4-trioxanes (Jefford, Adv. Drug Res. 1997, 29, p. 271-325; Cumming et al., Adv. Pharmacol. 1997, 37, p. 254-297), 1,2,4,5-tetraoxanes (Vennerstrom et al., J. Med. Chem., 2000, 43, p. 2753-2758), and other endoperoxides have been prepared. Various patents/applications disclose means and method for treating malaria using Spiro or dispiro 1,2,4-trioxolanes for example, U.S.
Patent Application No. 2004/0186168 and U.S. Patent Nos. 6,486, 199 and 6,825,230. The present invention relates to solid dosage forms of the various spiro or dispiro 1 ,2,4- trioxolanes antimalarial compounds disclosed in these patents/applications and are incorporated herein by reference.
Active compounds representing various Spiro and dispiro 1 ,2,4-trioxolane derivatives possess excellent potency, efficacy against Plasmodium parasites, and a lower degree of neurotoxicity, in addition to their structural simplicity and ease of synthesis. Furthermore, these compounds have half-lives which are believed to permit short-term treatment regimens comparing favorably to other artemisinin-like drugs. In general, the therapeutic dose of trioxolane derivative may range between about 0.1-1000 mg/kg/day, in particular between about 1-100 mg/kg/day. The foregoing dose may be administered as a single dose or may be divided into multiple doses. For malaria prevention, a typical dosing schedule could be, for example, 2.0-1000 mg/kg weekly beginning 1-2 weeks prior to malaria exposure, continued up to 1-2 weeks post-exposure.
Monotherapy with artemisinin (natural or synthetic) class of drugs might cure the patients within 3 days, however perceiving the potential threat of the malarial parasite developing resistance towards otherwise very potent artemisinin class of drugs, WHO had strictly called for an immediate halt to the provision of single-drug artemisinin malaria pills. Combination therapy in case of malaria retards the development of resistance, improve efficacy by lowering recrudescence rate, provides synergistic effect, and increase exposure of the parasite to the drugs.
Artemsinin based combinations are available in the market for a long time.
Artemether-lumafentrine (Co-artem®) was the first fixed dose antimalarial combination containing an artemisinin derivative and has been known since 1999. This combination has passed extensive safety and efficacy trials and has been approved by more than 70 regulatory agencies. Co-artem® is recommended by WHO as the first line treatment for uncomplicated malaria.
Other artemisinin based combinations include artesunate and amodiaquine (Coarsucam®), and dihydroartemisin and piperaquine (Eurartesim®). Unfortunately, all the available artemisinin based combinations have complicated dosage regimens making it difficult and inconvenient for a patient to comply completely with the total prescribed duration. For example, the dosage regimen of Co-artem® for an adult having body weight of more than 35 kg includes 6 doses over three days. The first dose comprises four tablets initially, the second dose comprises four tablets after eight hours, the third to sixth doses comprise four tablets twice for another two days; making it a total of 24 tablets. The dosage regimen of Coarsucam® for an adult having body weight of more than 36 kg or age above 14 years includes three doses over three days; each dose comprises two tablets; making it a total of six tablets. The dosage regimen of Eurartesim® for an adult having body weight between 36 kg – 75 kg includes 3 doses over three days, each dose comprises of three tablets, making it a total of nine tablets.
It is evident that the available artemisinin-based combinations have a high pill burden on patients as they need to consume too many tablets. As noted above, this may increase the possibility of missing a few doses, and, consequently, could result in reduced efficacy due to non-compliance and may even lead to development of resistance for the drug. Therefore, there is an urgent and unmet need for anti-malarial combinations with a simplified daily dosing regimen that reduces the pill burden and would increase patient compliance.
Apart from simplifying the regimen, there are certain limitations for formulators developing formulations with trioxolones, the first being their susceptibility to degradation in presence of moisture that results in reduced shelf lives. Another is their bitter taste, which can result in poor compliance of the regimen or selection of another, possibly less effective, therapeutic agent.
……………………..
http://www.google.st/patents/US6906205

……………………
http://www.google.st/patents/WO2013008218A1?cl=en
structural Formula II.

Formula II
Active compound includes one or more of the various spiro and dispiro trioxolane derivatives disclosed in U.S. Application No. 2004/0186168 and U.S. Patent Nos.
6,486,199 and 6,825,230, which are incorporated herein by reference. These trioxolanes are relatively sterically hindered on at least one side of the trioxolane heterocycle which provides better in vivo activity, especially with respect to oral administration. Particularly, spiro and dispiro 1,2,4-trioxolanes derivatives possess excellent potency and efficacy against Plasmodium parasites, and a lower degree of neurotoxicity.
The term “Active compound I” herein means cis-adamantane-2-spiro-3′-8′-[[[(2′- amino-2′-methylpropyl)amino]carbonyl]-methyl]- 1 ‘,2′,4’-trioxaspiro[4.5]decane hydrogen maleate. The Active compound I may be present in an amount of from about 5% to about 25%, w/w based on the total dosage form.

………………
http://www.google.st/patents/WO2007138435A2?cl=en
A synthetic procedure for preparing compounds of Formula I, salts of the free base c«-adamantane-2-spiro-3′-8′-[[[(2′-amino-2′-methyl propyl) amino] carbonyl] methyl]- 1 ‘, 2′, 4’-trioxaspiro [4.5] decane has been disclosed in U.S. 6,906,205.

The process for the preparation of compounds of Formula I wherein a compound of Formula II (wherein R is lower alkyl) is reacted with a compound of Formula III (wherein R is lower alkyl) to obtain compound of Formula IV;


Formula Formula IV
followed by hydrolysis of the compounds of Formula IV to give a compound of Formula V;

Formula V followed by the reaction of the compound of Formula V with an activating agent, for example, methyl chloroformate, ethyl chloroformate, propyl chloro formate, n-butyl chloro formate, isobutyl chloroformate or pivaloyl chloride leads to the formation of mixed anhydride, which is reacted in situ reaction with 1 ,2-diamino-2-methyl propane to give a compound of Formula VI; and

Formula Vl reacting the compound of Formula VI with an acid of Formula HX (wherein X can be the same as defined earlier) to give compounds of Formula I.
Example 1 : Preparation of O-methyl-2-adamantanone oxime
To a solution of 2-adamantanone (50 g, 0.3328 mol, 1 equiv.) in methanol (0.25 lit), sodium hydroxide solution (15 g, 0.3761mol, 1.13 equiv, in 50 mL water) was added followed by methoxylamine hydrochloride (37.5 g x 81.59% Purity= 30.596 g, 0.366 mol, 1.1 equiv) at room temperature under stirring. The reaction mixture was stirred at room temperature for 1 to 2 h. The reaction was monitored by HPLC. The reaction mixture was concentrated at 40- 45°C under vacuum to get a thick residue. Water (250 mL) was added at room temperature and the reaction mixture was stirred for half an hour. The white solid was filtered, washed with water (50 mL), and dried at 40 to 45°C under reduced pressure. O-methyl 2- adamantanone oxime (57 g, 95 % yield) was obtained as a white solid.
(M++l) 180, 1HNMR (400 MHz, CDCl3 ): δ 1.98 – 1.79 (m, 12H), 2.53 (s, IH), 3.46 ( s, IH), 3.81 (s, 3H).
Example 2: Preparation of 4-(methoxycarbonvmethvPcvclohexanone A high pressure autoclave was charged with a mixture of methyl (4- hydroxyphenyl)acetate (50 g, 0.30 mol), palladium ( 5g) (10 %) on carbon (50 % wet) and O- xylene (250 mL). The reaction mixture was stirred under 110 to 115 psi of hydrogen pressure for 7 to 8 h at 1400C. The reaction was monitored by HPLC. The reaction mixture was then cooled to room temperature, and the catalyst was filtered off. Filtrate was concentrated under reduced pressure to get 4-(methoxycarbonylmethyl)cyclohexanone as light yellow to colorless oily liquid (48.7 g, 97.4 %).
(M++!) 171, ‘ HNMR (400 MHz, CDCl 3): δ 1.48 – 1.51 ( m, 2H), 2.1 1-2.07 (m, 2H), 2.4- 2.23 (m, 7H), 3.7 (s, 3H).
Example 3: Preparation of methyl (Is, 4s)-dispiro [cyclohexane-l, 3′-f 1,2,4] trioxolane-5′, 2″-tricvclor3.3.1.13–71decan1-4-ylacetate
A solution of O-methyl-2-adamantanone oxime (example 1) (11.06 g, 61.7 mmol, 1.5 equiv.) and 4-(methoxycarbonymethyl)cyclohexanone (example 2) (7.0 g, 41.1 mmol, 1 equiv.) in cyclohexane ( 200ml) and dichloromethane (40 mL) was treated with ozone (ozone was produced with an OREC ozone generator [0.6 L/min. O2, 60 V] passed through an empty gas washing bottle that was cooled to -780C). The solvent was removed after the reaction was complete. After removal of solvents, the crude product was purified by crystallization from 80% aqueous ethanol (200 mL) to afford the title compound as a colorless solid. Yield: 10.83 g, 78%, mp: 96-980C; 1HNMR (500 Hz3CDCl3): δ 1.20-1.33 (m, 2H), 1.61-2.09 (m, 5 21H), 2.22 (d, J = 6.8Hz, 2H), 3.67(s,3H).
Example 4: Preparation of (Is, 4s)-dispiro [cyclohexane-1, 3′-[l,2,4] trioxolane-5′, 2″- tricvclo [3.3.1.13‘7] decanl-4-ylacetic acid
Sodium hydroxide (3.86 g, 96.57 mmol, 3 equiv.) in water (80 mL) was added to a solution of methyl (\s, 4s)-dispiro [cyclohexane-1, 3′-[l,2,4] trioxolane-5′, 2″-tricyclo
10 [3.3.1.I3‘7] decan]-4-ylacetate (example 3) (10.83 g, 32.19 mmol, 1 equiv.) in 95% ethanol (150 mL). The mixture was stirred at 500C for about 4 h, cooled to O0C, and treated with IM hydrochloric acid (129ml, 4 equiv). The precipitate was collected by filtration, washed with 50 % aqueous ethanol (150 mL) and dried in vacuum at 40 0C to give the title compound as colorless solid. Yield: 9.952 g, 96%, mp: 146-1480C ( 95% ethanol), 1HNMR (500 Hz,
15 CDCl3): δ 1.19-1.41 (m,2H), 1.60-2.05 (m,21H), 2.27 (d, J=6.8 Hz,2H).
Example 5: Preparation of c?s-adamantane-2-spiro-3′-8′-[[[(2′-amino-2′-methyl propyl) amino] carbonyl] methyl]-! ‘, T , 4’-trioxaspiro [4.5] decane
Method A:
(Is, 4s)-dispiro[cyclohexane- 1 ,3 ‘-[ 1 ,2,4]trioxolane-5 ‘,2 ‘ ‘-tricyclo[3.3.1.13‘7]decan]-4-
.0 ylacetic acid (example 4) (5 g ,15.5mmol, 1 equiv) was mixed with triethylamine (2.5 g , 24.8 mmol, 1.6 equiv) in 100ml of dichloromethane. The reaction mixture was cooled to – 1O0C to 00C. Ethyl chloro formate (1.68 g, 17 mmol, 1.0 equiv) in 15 mL dichloromethane was charged to the above reaction mixture at – 100C to 00C. The reaction mixture was stirred at the same temperature for 10 to 30 minutes. The resulting mixed anhydride reaction mixture
15 was added dropwise to a previously prepared solution of l,2-diamino-2-methylpropane (1.64 g, 18.6 mmol, 1.2 equiv), in 100 mL dichloromethane at -100C to O0C. The temperature of reaction mixture was raised to room temperature. The reaction mixture was stirred at the same temperature till the reaction was complete. Reaction monitoring was done by thin layer chromatography using 5 to 10% methanol in dichloromethane. The reaction was complete
>0 within 2 h. Nitrogen atmosphere was maintained throughout the reaction. Water (50 mL) was charged, organic layer was separated and washed with 10% sodium bicarbonate solution (50 mL) and water (50 mL) at room temperature. The organic layer was dried over sodium sulphate and the solvent was removed at 25 to 4O0C under reduced pressure. Hexane (50ml) was added to obtain residue under stirring at room temperature. The mixture was filtered and washed with 5 mL of chilled hexane. The solid was dried under reduced pressure at room 5 temperature.
Yield: 5.2 g (85.4 %), (M++l) 393, 1HNMR (400 MHz, DMSO-J6 ): δ 0.929 ( s, 6H), 1.105 – 1.079 (m, 2H), 1.887-1.641 (m, 21H), 2.030-2.017 (d, 2H), 2.928 (d, 2H).
Method B:
(Is, 4s)-dispiro [cyclohexane-1, 3′-[l,2,4] trioxolane-5′, 2″-tricyclo [3.3.1.I3‘7]
10 decan]-4-ylacetic acid (example 4) (10 g, 31mmol, 1 equiv) was treated with isobutyl chloroformate (4.5 g, 33mmol, 1.1 equiv) in presence of organic base like triethyl amine (5 g, 49.6mmol, 1.6 equiv) at 00C to 7°C in 250ml of dichloromethane. The solution was stirred at O0C to 7°C for aboutlO to 30 minutes. To the above reaction mixture, previously prepared solution of l,2-diamino-2-methylpropane (3.27 g, 37 mmol, 1.2 equiv), in 50 mL of
15 dichloromethane was added at O0C to 7°C in one lot. The temperature of reaction mixture was raised to room temperature. The reaction mixture was stirred at the room temperature till reaction was over. Reaction monitoring was done by thin layer chromatography using 5 to 10% methanol in dichloromethane. Reaction was complete within 2 h. The reaction nitrogen atmosphere was maintained throughout the reaction. Water (250 mL) was charged, organic
20 layer was separated and washed with 10% sodium bicarbonate solution (200 mL) and water (100 mL) at room temperature and the solvent was removed at 25 to 4O0C under reduced pressure. Hexane (100ml) was added to the residue, under stirring, at room temperature. The mixture was filtered and washed with chilled hexane (10 mL). The resultant solid was dried under reduced pressure at room temperature. Yield: 10.63 g (87%), (M++l) 393, 1HNMR
>5 (400 MHz, DMSO-J6 ) :δ 0.928 ( s, 6H), 1.102 – 1.074 (m, 2H), 1.859-1.616 (m, 21H), 2.031- 2.013 (d, 2H), 2.94-2.925 (d, 2H). Method C:
(\s, 4s)-dispiro[cyclohexane-l,3′-[l,2,4]trioxolane-5′,2″-tricyclo[3.3.1.13>7]decan]-4- ylacetic acid (example 4) (5 g, 15.5mmol, 1 equiv) was treated with pivaloyl chloride (1.87 g, 15.5 mmol, 1 equiv) and triethylamine (2.5gm, 24.8mmol, 1.6 equiv) at -15°C to -100C in dichloromethane (125 mL). The solution was stirred at -150C to -100C for aboutlO to 30 minutes. It resulted in the formation of mixed anydride. To the above reaction mixture, previously prepared solution of 1 ,2-diamino-2-methylpropane (1.64 g, 18.6 mmol, 1.2 equiv) in 25 mL dichloromethane was added at -15°C to -100C. The temperature of reaction mixture was raised to room temperature. The reaction mixture was stirred at the room temperature till reaction was over. Reaction monitoring was done by thin layer chromatography using 5 to 10% methanol in dichloromethane. The reaction was complete within 2 h. Nitrogen atmosphere was maintained throughout the reaction. Water (125 mL) was charged, organic layer was separated and washed with 50 mL of 10% sodium bicarbonate solution and 125 mL of water, respectively at room temperature. Finally solvent was removed at 25 to 4O0C under reduced pressure. 50 mL of 5% Ethyl acetate – hexane solvent mixture was added to the residue under stirring at room temperature. The mixture was filtered and washed with 5 mL of chilled hexane. Solid was dried under reduced pressure at room temperature. Yield: 5.03 g (83 %), (M++l) 393, 1JINMR (400 MHz, OMSO-d6 ):δ 0.93 ( s, 6H), 1.113 – 1.069 (m, 2H), 1.861-1.644 (m, 21H), 2.033-2.015 (d, 2H), 2.948-2.933 (d, 2H).
Example 6: Preparation of c/s-adamantane-2-spiro-3′ -8 ‘-πT(2′-amino-2’ -methyl propyl) amino! carbonyl] methyli-l ‘, 2\ 4′-U-JoXaSpJrQ [4.51 decane maleate To a solution of c/s-adamantane-2-spiro-3′-8′-[[[(2′-amino-2’-methyl propyl) amino] carbonyl] methyl]-! ‘, 2′, 4’-trioxaspiro [4.5] decane (example 5) (60 g, 0.153 moles) in ethanol (150 mL) was added a solution of maleic acid (17.3 g, 0.15 moles, 0.98 equiv. in ethanol 90 mL) and the reaction mixture was stirred for about 1 h. To this clear solution, n- heptane (720 mL) was added at room temperature in 1 h and the reaction mixture was stirred for 3 h. It was then cooled to 0 to 100C and filtered. The cake was washed with n-heptane (60 mL) and dried under vacuum at 40-450C.
Yield: 67 g, 77.4%, mp: 1490C (decomp), (M++l) 393.5, 1HNMR (300 MHz, DMSO-^ ): δ 1.05-1.11 (2H,m), 1.18 (6H,s), 1.64-1.89 (21H,m), 2.07(2H,d), 3.21 (2H,d), 6.06 (2H,d), 7.797 (2H, bs), 8.07 (IH, t).



References
- Dong, Yuxiang; Wittlin, Sergio; Sriraghavan, Kamaraj; Chollet, Jacques; Charman, Susan A.; Charman, William N.; Scheurer, Christian; Urwyler, Heinrich et al. (2010). “The Structure−Activity Relationship of the Antimalarial Ozonide Arterolane (OZ277)”. Journal of Medicinal Chemistry 53 (1): 481–91. doi:10.1021/jm901473s. PMID 19924861.
- Blow to Ranbaxy drug research plans at LiveMint.com, Sep 21 2007
- Vennerstrom, Jonathan L.; Arbe-Barnes, Sarah; Brun, Reto; Charman, Susan A.; Chiu, Francis C. K.; Chollet, Jacques; Dong, Yuxiang; Dorn, Arnulf et al. (2004). “Identification of an antimalarial synthetic trioxolane drug development candidate”. Nature 430 (7002): 900–4.doi:10.1038/nature02779. PMID 15318224.
- In the Pipeline: “Ozonides As Drugs: What Will They Think Of Next?”, by Derek Lowe, November 23, 2009, at Corante.com
- Indian company starts Phase III trials of synthetic artemisinin, May 4 2009, at the WorldWide Antimalarial Resistance Network
- http://www.nature.com/nature/journal/v430/n7002/full/nature02779.html
|
5-27-2011
|
PROCESS FOR THE PREPARATION OF DISPIRO 1,2,4-TRIOXOLANE ANTIMALARIALS (OZ277)
|
|
|
2-13-2009
|
STABLE DOSAGE FORMS OF SPIRO AND DISPIRO 1,2,4-TRIOXOLANE ANTIMALARIALS
|
|
|
6-15-2005
|
Spiro and dispiro 1,2,4-trioxolane antimalarials
|
|
|
11-31-2004
|
Spiro and dispiro 1,2,4-trixolane antimalarials
|
ANTIMALARIALS




http://www.rsc.org/chemistryworld/2013/03/new-antimalarial-drug-class-resistance-elq-300-quinolone

http://www.nature.com/nrd/journal/v9/n11/full/nrd3301.html
Structure of NITD609; the 1R,3Sconfiguration is fundamental for its antimalarial activity
