
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Basta ya de malgastar el dinero en vitaminas y suplementos minerales
 Este es el demoledor título de un reciente editorial de la prestigiosa revista médica “Annals of Internal Medicine” que sirve de introducción a una serie de artículos científicos publicados sobre el posible papel de vitaminas y suplementos minerales en la prevención o progresión de diversas enfermedades crónicas y su efecto sobre la tasa de mortalidad.
Este es el demoledor título de un reciente editorial de la prestigiosa revista médica “Annals of Internal Medicine” que sirve de introducción a una serie de artículos científicos publicados sobre el posible papel de vitaminas y suplementos minerales en la prevención o progresión de diversas enfermedades crónicas y su efecto sobre la tasa de mortalidad.
View original post 1,498 more words
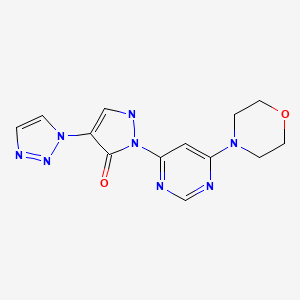
Iloprost (ciloprost)
– See more at: http://worlddrugtracker.blogspot.in/2014/01/iloprost-ciloprost-used-to-treat.html
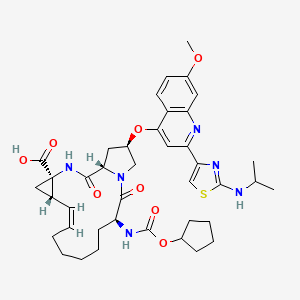
CILUPREVIR » All About Drugs
WANT TO KNOW ABOUT VIR SERIES CLICK
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
CILUPREVIR


DOI: 10.1002/9781118354520.ch10
Org Lett 2004, 6(17): 2901
http://pubs.acs.org/doi/full/10.1021/ol0489907
3. Efficient synthesis of (S)-2-(cyclopentyloxycarbonyl)-amino-8-nonenoic acid: Key buiding block for BILN 2061, an HCV NS3 protease inhibitor
Org Process Res Dev 2007, 11(1): 60
4. Chinese Journal of Chemistry, 2011 , vol. 29, 7 pg. 1489 – 1502
DOI: 10.1002/cjoc.201180270
http://onlinelibrary.wiley.com/doi/10.1002/cjoc.201180270/abstract;jsessionid=F5F4331F5A95D00728394A254C2B1AE7.f01t04
…………………………..
US 8222369
WO 2006071619
WO 2000059929
WO 2004092203
WO 2004039833
WO 2004037855
WO 2006036614
WO 2006033878
WO 2005042570
WO 2004093915
………………………………………………………………………..
https://www.google.co.in/patents/US8222369


…………………………………………………………………..
http://www.google.com/patents/WO2000059929A1
COMPD 822 IS CILUPREVIR IN TABLE 8
EXAMPLE 8 Synthesis of 4-hydroxy-7-methoxy-2[4(2-isopropylaminothiazolyl)] quinoline (8f ) Note: [ A variety of 2-alkylaminothiazolyl substituents were made using the same synthetic scheme where compound 8b was replaced by other alkyl thioureas.]


8b 8c 8d

A. The protocol used for the conversion of -anisidine to 8a was identical to that described in the literature: F.J. Brown et al. J. Med. Chem. 1989, 32 , 807-826. However, the purification procedure was modified to avoid purification by chromatography. The EtOAc phase containing the desired product was treated with a mixture of MgSO4, charcoal and 5% w/w (based on expected mass) silica gel. After filtration on celite, the product was triturated with ether. Compound 8a was obtained as a pale brown solid in >99% purity (as confirmed by HPLC).
B. A suspension of isopropyl thiourea (8b, 3.55 g, 30 mmol) and 3- bromopyruvic acid (8c, 5 g, 1 eq.) in dioxane (300 mL , 0.1 M) was heated to 80 °C.
Upon reaching 80 C the solution became clear and soon after the product precipitated as a white solid. After 2 hours of heating, the solution was cooled to RT and the white precipitate was filtered to obtain compound 8d in high purity (>98% purity as confirmed by NMR) and 94% yield (7.51 g). C. A mixture of the carboxylic acid 8d (4.85 g, 18.2 mmol) and the aniline derivative 8a (3 g, leq.) in pyridine (150 mL, 0.12 M) was cooled to -30 °C (upon cooling, the clear solution became partially a suspension). Phosphorus oxychloride (3.56 ml, 2.1 eq.) was then added slowly over a 5 min period. The reaction was stirred at -30 C for 1 h, the bath was removed and the reaction mixture was allowed to warm-up to RT. After 1.5 h the reaction mixture was poured into ice, the pH was adjusted to 11 with aqueous 3N NaOH, extracted with CH2C12, dried over anhydrous MgSO4, filtered and concentrated under vacuum. The beige solid was then purified by flash chromatography (45% EtOAc in hexane) to give compound 8e as a pale yellow solid in 73% yield (6.07 g). D. A solution of tBuOK (2.42 g, 21.6 mmol) in anhydrous tBuOH (40ml, 0.14 M, distilled from Mg metal) was heated to reflux. Compound 8e (1.8g, 5.4 mmol) was added portion-wise over 5 min and the dark red solution formed was stirred at reflux for an additional 20 min (completion of the reaction was monitored by HPLC). The mixture was cooled to RT and HCl was added (4 N in dioxane, 1.5 eq.). The mixture was then concentrated under vacuum, in order to assure that all of the
HCl and dioxane were removed, the product was re-dissolved twice in CH2C12 and dried under vacuum to finally obtain the HCl salt of compound 8f as a beige solid (1.62 g, 93% pure by HPLC). The product was then poured into a phosphate buffer
(IN NaH2PO4, pH=~4.5) and sonicated. The beige solid was filtered and dried under vacuum to give compound 8f (1.38 g, 81% yield) as a beige solid (91% pure by HPLC).
*H NMR (400 MHz, DMSO) δ 8.27 (s, IH), 8.12 (d, IH, J = 9.2 Hz), 7.97 (br.s, IH), 7.94 (s, IH), 7.43 (s, IH), 7.24 (dd, IH, J = 9.2, 2.2 Hz), 3.97 (m, IH), 3.94 (s, 3H), 1.24 (d, 2H, J = 6.4 Hz)
…………
METHYL ESTER
EXAMPLE 34c
Using the same procedure as described in example 34 but reacting bromoketone 34f with commercially available N-iso-propylthiourea gave # 822

Η NMR (400 MHz, DMSO-d6) δ 8.63 (s, IH), 8.33-8.23 (bs, IH), 8.21 (d, J = 9.2 Hz, IH), 8.04 (d, J = 8.3 Hz, IH), 7.86 (bs, IH), 7.77 (s, IH), 7.35-7.23 (m, 2H), 5.81 (bs, IH), 5.52 (dd, J = 8.5 Hz, IH), 5.27 (dd, J = 9.2 Hz, IH), 4.65 (d, J = 11.8 Hz, IH), 4.51 (dd, J = 7.6 Hz, IH), 4.37 (bs, IH), 4.15 (bs, IH), 4.07-3.98 (m, 2H), 3.97 (s, 3H), 3.88 (d, J = 8.9 Hz, IH), 2.60-2.53 (m, 2H), 2.47-2.37 (m, 2H), 2.19-2.10 (dd, J = 9.2 Hz, IH), 1.80-1.64 (m, 2H), 1.63-1.29 (m, 13H), 1.27 and 1.25 (2 x d, J – 6.5 Hz, 6H), 1.23-1.09 (m, 2H). MS; es+: 775.0 (M + H)+, es : 772.9 (M – H)\
CILUPREVIR IS FREE ACID OF ABOVE AND HAS ENTRY 822 TABLE 8
………
FREE AMINO COMPD
(Table 8)




A. To a solution of the macrocyclic intermediate 23b (13.05 g, 27.2 mmol, 1.0 eq.), Ph3P (14.28 g, 54.4 mmol, 2.0 eq) and 2-carboxymethoxy-4-hydroxy-7- methoxyquinoline (WO 00/09543 & WO 00/09558) (6.67 g, 28.6 mmol, 1.05 eq) in
THF (450 mL) at 0°C, DIAD (10.75 mL, 54.6 mmol, 2.0 eq) was added dropwise over a period of 15 min. The ice bath was then removed and the reaction mixture was stirred at RT for 3 h. After the complete conversion of starting material to products, the solvent was evaporated under vacuum, the remaining mixture diluted with
EtOAc, washed with saturated NaHCO3 (2x) and brine (lx), the organic layer was dried over anhydrous MgSO4, filtered and evaporated to dryness. Pure compound 34a was obtained after flash column chromatography; the column was eluted first with hexane/EtOAc (50:50), followed by CHCl3/EtOAc (95:5) to remove Ph3PO and
DIAD byproducts and elution of the impurities was monitored by TLC. Finally, the desired product 34a was eluted from the column with CHC13/ EtOAc (70:30).
Usually, the chromatography step had to be repeated 2-3 times before compound 34a could be isolated in high purity as a white solid with an overall yield of 68% (12.8 g, 99.5% pure by HPLC).
B. To a solution of the Boc-protected intermediate 34a (1.567g) in CH2C12 (15 mL), 4N HCl in dioxane (12 mL) was added and the reaction mixture was stirred at RT for 1 h. [In the event that a thick gel would form half way through the reaction period, an additional 10 mL CH2C12 was added.] Upon completion of the deprotection the solvents were evaporate to dryness to obtain a yellow solid and a paste like material. The mixture was redissolved in approximately 5% MeOH in
CH2C12 and re-evaporated to dryness under vacuum to obtain compound 34b as a yellow solid, which was used in the next step without any purification. C. To a solution of cyclopentanol (614 μL, 6.76 mmoL) in THF (15 mL), a solution of phosgene in toluene (1.93 M, 5.96 mL, 11.502 mmol) was added dropwise and the mixture was stirred at R.T. for 2 h to form the cyclopentyl chloroformate reagent (z). After that period, approximately half of the solvent was removed by evaporation under vacuum, the remaining light yellow solution was diluted by the addition of CH2C12 (5 mL) and concentrated to half of its original volume, in order to assure the removal of all excess phosgene. The above solution of the cyclopentyl chloroformate reagent was further diluted with THF (15 mL) and added to the amine-2HCl salt 34b. The mixture was cooled to 0 C in an ice bath, the pH was adjusted to -8.5-9 with the addition of Et3N (added dropwise) and the reaction mixture was stirred at 0 C for 1 h. After that period, the mixture was diluted with
EtOAc, washed with water (lx), saturated NaHCO3 (2x), H2O (2x) and brine (lx).
The organic layer was dried over anhydrous MgSO4, filtered and evaporated under vacuum to obtain a yellow-amber foam. Compound 34c was obtained as a white foam after purification by flash column chromatography (using a solvent gradient from 30% hexane to 20% hexane in EtOAc as the eluent) in 80% yield (1.27 g) and >93% purity. D. The dimethyl ester 34c (1.17g) was dissolved in a mixture of
THF/MeOH/H2O (20 mL, 2:1:1 ratio), and an aqueous solution of NaOH (1.8 mL,
IN, 1 eq.) was added. The reaction mixture was stirred at RT for 1 h before it was evaporated to dryness to obtain the sodium salt 34d as a white solid (-1.66 mmol). Compound 34d was used in the next step without purification.
E. The crude sodium salt 34d (1.66 mmoL) was dissolved in THF (17 mL), Et3N was added and the mixture was cooled to 0 C in an ice bath. Isobutylchloroformate
(322 μl, 2.5 mmol) was added dropwise and the mixture was stirred at 0 C for 75 min. After that period, diazomethane (15 mL) was added and stirring was continued at 0 C for 30 min and then at RT for an additional 1 h. Most of the solvent was evaporated to dryness under vacuum, the remaining mixture was diluted with EtOAc, washed with saturated NaHCO3 (2x), H2O (2x) and brine (lx), dried over anhydrous MgSO4, filtered and evaporated to dryness to obtain compound 34e as a light yellow foam (1.2g, -1.66 mmol). The diazoketone intermediate 34e was used in the next step without purification.
F. The diazoketone 34e (1.2g, 1.66 mmoL) dissolved in THF (17 mL) was cooled to 0 C in an ice bath. A solution of aqueous HBr (48%, 1.24 mL) was added dropwise and the reaction mixture was stirred at 0 C for 1 h. The mixture was then diluted with EtOAc, wash with saturated NaHCO3 (2x), H2O (2x) and brine (lx), the organic layer was dried over anhydrous MgSO4, filtered and evaporated to dryness to obtain the β-bromoketone intermediate 34f as a light yellow foam (-1.657 mmol).
G. To a solution of the bromoketone 34f (600 mg,0.779 mmol) in isopropanol (5 mL), thiourea (118 mg, 1.55 mmol) was added and the reaction mixture was placed in a pre-heated oil bath at 75 C where it was allowed to stir for 1 hr. The isopropanol was then removed under vacuum and the product dissolved in EtOAc
(100 mL). The solution was washed with saturated NaHCO3 and brine, the organic layer was dried over anhydrous Na2SO4, filtered and evaporated to afford the crude product 34g (522 mg) as a red-brown solid. This material was used in the final step without any further purification.
H. The crude methyl ester 34g (122 mg, 0.163 mmol) was dissolved in a solution of THF/MeOH/H2O (2:1:1 ratio, 4 mL) and saponified using LiOH»H2O (89 mg, 2.14 mmol). The hydrolysis reaction was carried out over a 12-15 h period at RT. The solvents were then removed under vacuum and the crude product purified by C18 reversed phase HPLC, using a solvent gradient from 10% CH3CN in H2O to 100%
CH3CN, to afford the HCV protease inhibitor #812 as a yellow solid (24 mg, 20% overall yield for the conversion of intermediate 34f to inhibitor #812).
*H NMR (400 MHz, DMSO-d6) δ 8.63 (s, IH), 8.26-8.15 (m, 2H), 7.79 (bs, IH), 7.72
(bs, IH), 7.50 (bs, 2H), 7.33-7.25 (m, 2H), 5.77 (bs, IH), 5.52 (dd, J = 8.3 Hz, IH), 5.27 (dd, J = 9.2 Hz, IH), 4.64 (d, J = 10.8 Hz, IH), 4.50 (dd, J = 8.3 Hz, IH), 4.39-4.31 (m, IH), 4.08-3.99 (m, 2H), 3.94 (s, 3H), 3.87 (d, J = 9.5 Hz, 2H), 2.65-2.53 (m, 2H), 2.46- 2.36 (m, 2H), 2.20-2.12 (dd, J = 8.6 Hz, IH), 1.80-1.64 (m, 2H), 1.63-1.06 (m, 14H). MS; es+: 733.2 (M + H)+, es“: 731.2 (M – H)\
………………..
http://www.google.com/patents/WO2006036614A2
(Z)-( 1S,4R, 14S, 18R)- 14-Cyclopentyloxycarbonylamino- 18-[2-(2- isopropylamino-thiazol-4-yl)-7-methoxy-quinolin-4-yloxy]-2,15-dioxo-3,16-diaza- tricyclo[14.3.0.0 ‘ ]nonadec-7-ene-4-carboxylic acid , whose chemical structure is as follows:

, provided for in Tsantrizos et al., U.S. Patent No. 6,608,027 Bl,
…………………………
https://www.google.co.in/patents/WO2005090383A2
ENTRY 218

…………………..
http://www.google.com/patents/WO2004039833A1

……………..
nmr
Synthesis of BILN 2061, an HCV NS3 protease inhibitor with proven antiviral effect in humans
Org Lett 2004, 6(17): 2901
http://pubs.acs.org/doi/full/10.1021/ol0489907
http://pubs.acs.org/doi/suppl/10.1021/ol0489907/suppl_file/ol0489907si20040715_032207.pdf procedure
http://pubs.acs.org/doi/suppl/10.1021/ol0489907/suppl_file/ol0489907si20040715_032254.pdf nmr spectra
BILN 2061:
Methyl ester 18 (2.69 g, 3.41 mmol) was dissolved in a mixture of THF
(40 mL), MeOH (20 mL) and water (20 mL) and added LiOH.H2O (1.14 g, 27.3 mmol).The resulting mixture was left to stir at RT for 15 h. The solvents were then removedunder reduced pressure and the crude product was redissolved with EtOAc and dilutedwith brine. The pH of the aqueous layer was adjusted to 6 with aqueous HCl (1N) and theaqueous phase was extracted with EtOAc (3x). The combined organic phase werewashed with water, brine, dried over MgSO4 and concentrated under reduced pressure toafford BILN 2061 as a yellow solid (2.63 g, 99% yield). HPLC(A) 99%, MS m/z (ES+)773 (M+H)+, (ES-) 775 (M-H)-;
1H NMR (DMSO-d6) δ 8.63 (s, 1H), 8.26-8.15 (m, 2H),
7.79 (bs, 1H), 7.72 (bs, 1H), 7.50 (bs, 2H), 7.33-7.25 (m, 2H), 5.77 (bs, 1H), 5.52 (dd, J=8.3 Hz, 1H), 5.27 (dd, J= 9.2 Hz, 1H), 4.64 (d, J= 10.8 Hz, 1H), 4.50 (dd, J= 8.3 Hz, 1H),4.39-4.31 (m, 1H), 4.08-3.99 (m, 2H), 3.94 (s, 3H), 3.87 (d, J= 9.5 Hz, 2H), 2.65-2.53(m, 2H), 2.46-2.36 (m, 2H), 2.20-2.12 (dd, J= 8.6 Hz, 1H), 1.80-1.64 (m, 2H), 1.63-1.06(m, 14H); HRMS calcd for C40H51N6O8S: 775.3489; found: 775.3476
…………………………
| WO2007019674A1 | Aug 3, 2006 | Feb 22, 2007 | Boehringer Ingelheim Int | Viral polymerase inhibitors |
| WO2010021717A2 * | Aug 20, 2009 | Feb 25, 2010 | Sequoia Pharmaceuticals, Inc. | Hcv protease inhibitors |
| WO2010080874A1 | Jan 7, 2010 | Jul 15, 2010 | Scynexis, Inc. | Cyclosporine derivative for use in the treatment of hcv and hiv infection |
| EP1455809A2 * | Dec 13, 2002 | Sep 15, 2004 | Bristol-Myers Squibb Co. | Inhibitors of hepatitis c virus |
| EP2364984A1 | Aug 28, 2006 | Sep 14, 2011 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine proteases |
| EP2366704A1 | Aug 28, 2006 | Sep 21, 2011 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine proteases |
| US7368452 | Jul 18, 2006 | May 6, 2008 | Enanta Pharmaceuticals, Inc. | Quinoxalinyl macrocyclic hepatitis C serine protease inhibitors |
| US7608590 | Jan 28, 2005 | Oct 27, 2009 | Medivir Ab | HCV NS-3 serine protease inhibitors |
| US7671032 | Jan 28, 2005 | Mar 2, 2010 | Medivir Ab | HCV NS-3 serine protease inhibitors |
| US7816348 | Jan 29, 2007 | Oct 19, 2010 | Boehringer Ingelheim International Gmbh | Viral polymerase inhibitors |
| US7897622 | Aug 10, 2007 | Mar 1, 2011 | Boehringer Ingelheim International Gmbh | Viral polymerase inhibitors |
| US8148399 | Jul 28, 2006 | Apr 3, 2012 | Tibotec Pharmaceuticals Ltd. | Macrocyclic inhibitors of hepatitis C virus |
| US8153800 | Aug 3, 2011 | Apr 10, 2012 | Tibotec Pharmaceuticals Ltd. | Macrocyclic inhibitors of hepatitis C virus |
| US8242140 | Jul 31, 2008 | Aug 14, 2012 | Boehringer Ingelheim International Gmbh | Viral polymerase inhibitors |
| US8349869 | Mar 6, 2012 | Jan 8, 2013 | Tibotec Pharmaceuticals Ltd. | Macrocylic inhibitors of hepatitis C virus |
| US8476257 | Dec 3, 2008 | Jul 2, 2013 | Boehringer Ingelheim International Gmbh | Viral polymerase inhibitors |
| US8541402 | May 3, 2012 | Sep 24, 2013 | Boehringer Ingelheim International Gmbh | Viral polymerase inhibitors |
| WO2000059929A1 * | Apr 3, 2000 | Oct 12, 2000 | Boehringer Ingelheim Ca Ltd | Macrocyclic peptides active against the hepatitis c virus |
| WO2003053349A2 * | Dec 13, 2002 | Jul 3, 2003 | Squibb Bristol Myers Co | Inhibitors of hepatitis c virus |
| WO2003064455A2 * | Jan 24, 2003 | Aug 7, 2003 | Boehringer Ingelheim Ca Ltd | Macrocyclic peptides active against the hepatitis c virus |
| WO2003066103A1 * | Feb 5, 2003 | Aug 14, 2003 | Boehringer Ingelheim Pharma | Pharmaceutical compositions for hepatitis c viral protease inhibitors |
………………………………………………………………………………………….


WANT TO KNOW ABOUT VIR SERIES CLICK
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
Orphan Drugs: GlaxoSmithKline’s Combo Melanoma Treatment
GlaxoSmithKline (GSK) receives FDA approval in May 2013, for two orphan drugs for the treatment of patients with advanced or unresectable melanoma :
Both drugs are approved as single agents, not as a combination treatment. Tafinlar, a BRAF inhibitor, is approved for melanoma whose tumors express the BRAF V600E gene mutation. Mekinist, a MEK inhibitor, is approved for melanoma whose tumors express the BRAF V600E or V600K gene mutations. Both drugs are approved with a companion genetic test, ThxID BRAF test, made by France’s bioMérieux, which will help determine if a patient’s melanoma cells have the V600E or V600K mutation in the BRAF gene. About 50% of Melanomas arising in the skin have a BRAF gene mutation.
GSK announces on January 8, that the FDA gives the first combination oral targeted therapy, Tafinlar + Mekinist, accelerated approval for the treatment of unresectable or metastatic melanoma, with BRAF V600E or V600K mutations…
View original post 153 more words
TIDEGLUSIB ..An NSAID and neuroprotective agent.


Tideglusib
M.Wt: 334.39
Formula: C19H14N2O2S
CAS No.: 865854-05-3
4-Benzyl-2-(naphthalen-1-yl)-1,2,4-thiadiazolidine-3,5-dione
Glycogen Synthase Kinase 3 beta (GSK-3beta; tau Protein Kinase I) Inhibitors
Treatment of Neurologic Drugs (Miscellaneous)
Alzheimer’s Dementia, Treatment ofCerebrovascular Diseases, NP031112; NP-031112, Nypta Zentylor
- NP 031112
- NP-12
- NP031112
- Tideglusib
- UNII-Q747Y6TT42
Noscira (Originator)
Tideglusib (NP-12, NP031112) is a potent, selective and irreversible[1] small molecule non-ATP-competitive GSK3 inhibitor that has been investigated as a potential treatment for Alzheimer’s disease and paralysis supranuclear palsy in Phase IIa[2] and IIb clinical trials.[3][4][5][6] The first clinical trial conducted with tideglusib to be published (in English, at least) was phase II and demonstrated that overall tideglusib was well tolerated, except for some moderate, asymptomatic, fully reversible increases in liver enzymes (≥2.5xULN; where ULN=Upper Limit of Normal).[4]
tideglusib
NP-031112 is an inhibitor of glycogen synthase kinase-3 beta (GSK-3beta) in early clinical development for the oral treatment of Alzheimer’s disease. The compound had been in phase II clinical trials for the treatment of progressive supranuclear palsy and for the treatment of Alzheimer’s disease; however the development was discontinued in 2011 and 2012 respectively, due to lack of efficacy.
The neuroprotective effects demonstrated in animal studies have also suggested its potential use in stroke and other brain disorders. It is being developed by Noscira (formerly known as NeuroPharma). In 2009, orphan drug designation was received in the E.U. and the U.S. for the treatment of progressive supranuclear palsy. In 2010, fast track designation was assigned in the U.S. by Noscira for this indication.
Fast Track status is granted to facilitate development and expedite the review of a drug for a serious or potentially fatal illness and to meet an unmet medical need
The Phase II trial for Progressive Supranuclear Palsy (PSP) commenced in December 2009 and is currently in progress
Belen Sopesen, CEO of Noscira: ‘Fast Track status is very positive for the company and is an incentive to continue advancing in the clinical development of Tideglusib (ZentylorTM) in Progressive Supranuclear Palsy’
Overexpression of GSK-3 leads to hyperphosphorylation of the tau protein, an anomaly which occurs in a number of neurodegenerative diseases known collectively as tauopathies, which include Alzheimer’s disease (AD), Progressive Supranuclear Palsy (PSP) and Pick disease. NP-12 is a GSK-3 inhibitor with oral bioavailability and great therapeutic potential as a disease-modifying treatment for Alzheimer’s.
NP-12 is currently undergoing clinical trials for Alzheimer’s disease in the EU. NP-12, the only GSK-3 inhibitor under clinical development for AD, has proven to be capable of acting on all of the histopathological lesions associated with the disease in experimental models: it reduces phosphorylation of the tau protein and hippocampal and entorhinal cortex neuron loss, improves spatial memory deficits and significantly reduces the accumulation of amyloid plaques in the brain. NP-12 also provides neuroprotection in vivo and has a potent anti-inflammatory effect in a range of animal models.
About Progressive Supranuclear Palsy
PSP is a neurodegenerative disease characterized by oculomotor disturbances, specifically difficulties in moving the eye vertically, falling down and Parkinsonian symptoms.
The disease affects an estimated 5-6.4 out of every 100,000 people.
There is currently no treatment capable of delaying or altering the progression of the illness.

TIDEGLUSIB
- Domínguez, JM; Fuertes, A; Orozco, L; del Monte-Millán, M; Delgado, E; Medina, M (January 2012). “Evidence for Irreversible Inhibition of Glycogen Synthase Kinase-3 by Tideglusib”. The Journal of Biological Chemistry 287 (2): 893–904.doi:10.1074/jbc.M111.306472. PMC 3256883. PMID 22102280.
- Teodoro Del Ser (2010). “Phase IIa clinical trial on Alzheimer’s disease with NP12, a GSK3 inhibitor”. Alzheimer’s & Dementia 6 (4): S147. doi:10.1016/j.jalz.2010.05.455.
- Eldar-Finkelman, H; Martinez, A (2011). “GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS”. Frontiers in Molecular Neuroscience 4: 32.doi:10.3389/fnmol.2011.00032. PMC 3204427. PMID 22065134.
- Del Ser, T; Steinwachs, KC; Gertz, HJ; Andrés, MV; Gómez-Carrillo, B; Medina, M; Vericat, JA; Redondo, P et al. (2013). “Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: A pilot study”. Journal of Alzheimer’s disease 33 (1): 205–15.doi:10.3233/JAD-2012-120805. PMID 22936007.
- “FDA Grants Fast Track Status to Tideglusib (ZentylorTM) for Progressive Supranuclear Palsy”. PR Newswire Europe Including UK Disclose. 10 September 2010. Retrieved 11 August 2013.
- Dominguez, JM; Fuertes, A; Orozco, L; Del Monte-Millan, M; Delgado, E; Medina, M (2011). “Evidence for Irreversible Inhibition of Glycogen Synthase Kinase-3 by Tideglusib”. Journal of Biological Chemistry 287 (2): 893–904.doi:10.1074/jbc.M111.306472. PMC 3256883. PMID 22102280.
- WO 2005097117
- WO 2006045581
- WO 2006084934
- WO 2008057933
- WO 2011151359
- Evidence for irreversible inhibition of glycogen synthase kinase-3β by tideglusib.
Domínguez JM, Fuertes A, Orozco L, del Monte-Millán M, Delgado E, Medina M.
J Biol Chem. 2012 Jan 6;287(2):893-904. doi: 10.1074/jbc.M111.306472. Epub 2011 Nov 18
13. MARTINEZ A ET AL.: “First Non-ATP Competitive Glycogen Synthase Kinase 3.beta. (GSK-3.beta.) Inhibitors: Thiadiazolidinones (TDZD) as Potential Drugs for the Treatment of Alzheimer’s Disease” JOURNAL OF MEDICINAL CHEMISTRY, vol. 45, no. 6, 2002, pages 1292-1299
|
4-18-2012
|
GSK-3 Inhibitors
|
|
|
5-13-2009
|
GSK-3 inhibitors
|
|
|
6-27-2008
|
Use Of Heterocyclic Compounds As Neurogenic Agents
|
CLINICAL TRIALS
http://clinicaltrials.gov/search/intervention=NP+031112
http://clinicaltrials.gov/show/NCT01350362
………….
http://www.google.com/patents/WO2005097117
For example, the following procedure can be used to produce 4-N-benzyl substituted thiadiazolidinones :

The general experimental procedure of Scheme 1 is described for example in Slomczynska,
U.; Barany, G., “Efficient Synthesis of l,2,4-Dithiazolidine-3,5-diones (Dithiasuccinoyl- amines) and observations on formation of l,2,4-Thiadiazolidine-3,5-dione by related
Chemistry”, J. Heterocyclic Chem., 1984, 21, 241-246.
For example, sulfuryl chloride is added dropwise with stirring, under nitrogen atmosphere, preferably at low temperature, preferably at about 5 °C, to a solution of benzyl isothiocyanate and the isocyanate indicated in each case, in a suitable solvent such as hexane, ether or THF. When the addition is finished, the mixture is left to react, for example by stirring for 20 hours at room temperature. After this time, the resulting product is isolated by conventional methods such as suction filtration or solvent evaporation and then, the purification is performed (e.g. by recristallization or silica gel column chromatography using the appropriate eluent). Other alternative procedures will be apparent to the person skilled in the art, such as the use of any other chlorinating agent instead of sulfuryl chloride, variations in the order of addition of the reactants and reaction conditions (solvents, temperature, etc).
Example 2
4-Benzyl-2-naphthalen-l-yl-[l,2,4]thiadiazolidine-3,5-dione (2)
Reagents: Benzyl-isothiocianate (13 mmol, 1.72 mL), 1-naphthyl-isocyanate (13 mmol, 1.9 mL) and SO2CI2 (13 mmol, 1.04 mL) in hexane (50 mL). Isolation: filtration of reaction mixture. Purification: recrystallization from EtOH. Yield: 3.8 g (87%), white needles. mp= 150 °C
1H-RMN (CDC13): 4.9 (s, 2H, CH2PI1); 7.3-7.9 (m, 12Η, arom.) 13C-RMN (CDCI3): 46.5 (CH2Ph); 128.3; 128.6; 129.0; 135.0 (C arom, Ph); 122.0; 125.3; 126.8; 127.2; 127.5; 128.5; 130.8; 134.4 (C arom, naphthyl); 152.2 (3-00); 165.9 (5- C=O).
Anal (C19H14N2O2S), C, H, N, S
Sulfuryl chloride is added dropwise with stirring, under nitrogen atmosphere, at 5 °C to a solution of benzyl isothiocyanate and the isocyanate indicated in each case, in hexane, ether or THF. When the addition is finished, the mixture is stirred for 20 hours at room temperature. After this time, the resulting product is isolated by suction filtration or by solvent evaporation and then, the purification is performed by recristallization or silica gel column chromatography using the appropriate eluent. More details can be found in Slomczynska, U.; Barany, G., “Efficient Synthesis of l,2,4-Dithiazolidine-3,5-diones (Dithiasuccinoyl-amines) and observations on formation of l,2,4-Thiadiazolidine-3,5-dione by related Chemistry”, J Heterocyclic Client., 1984, 21, 241-246.
…………
| WO2006045581A1 * | Oct 21, 2005 | May 4, 2006 | Neuropharma Sa | The use of 1, 2, 4-thiadiazolidine-3, 5-diones as ppar activators |
| WO2011151359A1 | Jun 1, 2011 | Dec 8, 2011 | Noscira, S.A. | Combined treatment with a cholinesterase inhibitor and a thiadiazolidinedione derivative |
| WO2013124413A1 | Feb 22, 2013 | Aug 29, 2013 | Noscira, S.A. | Thiadiazolidinediones as gsk-3 inhibitors |
| EP2177510A1 | Oct 17, 2008 | Apr 21, 2010 | Universität des Saarlandes | Allosteric protein kinase modulators |
| EP2527323A1 | May 24, 2011 | Nov 28, 2012 | Noscira, S.A. | Urea carbonyl disulfide derivatives and their therapeutic uses |
………..


THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family

MIDAZOLAM


MIDAZOLAM
8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine
59467-70-8 CAS NO OF FREE BASE
59467-94-6 MALEATE, Launched – 1982, Roche (Originator)
59467-96-8 (HCl)
A short-acting hypnotic-sedative drug with anxiolytic and amnestic properties. It is used in dentistry, cardiac surgery, endoscopic procedures, as preanesthetic medication, and as an adjunct to local anesthesia. The short duration and cardiorespiratory stability makes it useful in poor-risk, elderly, and cardiac patients. It is water-soluble at pH less than 4 and lipid-soluble at physiological pH.
Midazolam (/mɪˈdæzəlæm/, marketed in English-speaking countries and Mexico under the trade names Dormicum, Hypnovel, andVersed,) is a short-acting drug in the benzodiazepine class developed by Hoffmann-La Roche in the 1970s. The drug is used for treatment of acute seizures, moderate to severe insomnia, and for inducing sedation and amnesia before medical procedures. It possesses profoundly potentanxiolytic, amnestic, hypnotic, anticonvulsant, skeletal muscle relaxant, and sedative properties.[6][7][8] Midazolam has a fast recovery time and is the most commonly used benzodiazepine as a premedication for sedation; less commonly it is used for induction and maintenance of anesthesia.Flumazenil, a benzodiazepine antagonist drug, can be used to treat an overdose of midazolam, as well as to reverse sedation.[7] However, flumazenil can trigger seizures in mixed overdoses and in benzodiazepine-dependent individuals, so is not used in most cases.[9][10]
midazolam
Administration of midazolam by the intranasal or the buccal route (absorption via the gums and cheek) as an alternative to rectally administereddiazepam is becoming increasingly popular for the emergency treatment of seizures in children. Midazolam is also used for endoscopyprocedural sedation and sedation in intensive care. The anterograde amnesia property of midazolam is useful for premedication before surgery to inhibit unpleasant memories. Midazolam, like many other benzodiazepines, has a rapid onset of action, high effectiveness and low toxicity level. Drawbacks of midazolam include drug interactions, tolerance, and withdrawal syndrome, as well as adverse events including cognitive impairment and sedation. Paradoxical effects occasionally occur, most commonly in children and the elderly, particularly after intravenous administration. The drug has also recently been hastily introduced for use in executions in the USA in combination with other drugs.
Midazolam is a short-acting benzodiazepine in adults with an elimination half-life of one to four hours; however, in the elderly, as well as young children and adolescents, the elimination half-life is longer. Midazolam is metabolised into an active metabolite alpha1-hydroxymidazolam. Age related deficits, renal and liver status affect the pharmacokinetic factors of midazolam as well as its active metabolite. However, the active metabolite of midazolam is minor and contributes to only 10 percent of biological activity of midazolam. Midazolam is poorly absorbed orally with only 50 percent of the drug reaching the bloodstream. Midazolam is metabolised by cytochrome P450 (CYP) enzymes and by glucuronide conjugation. The therapeutic as well as adverse effects of midazolam are due to its effects on the GABAA receptors; midazolam does not activate GABAA receptors directly but, as with other benzodiazepines, it enhances the effect of the neurotransmitter GABA on the GABAA receptors (↑ frequency of Cl− channel opening) resulting in neural inhibition. Almost all of the properties can be explained by the actions of benzodiazepines on GABAA receptors. This results in the following pharmacological properties being produced: sedation, hypnotic, anxiolytic, anterograde amnesia, muscle relaxation and anti-convulsant.Midazolam maleate is a benzodiazepine that is commercialized by Astellas Pharma and Roche as an intravenous or intramuscular injection for the long-term sedation of mechanically ventilated patients under intensive care. The drug is also available in a tablet formulation, and is currently distributed in various markets, including Germany, Japan, Switzerland and the U.K. In March 2002, two lots of a syrup formulation were recalled in the U.S. due to the potential presence of a crystalline precipitate of an insoluble complex of midazolam and saccharin. Subsequently, the injection and syrup formulations of the product were both withdrawn from the U.S. market. In 2010, a Pediatric Use Marketing Authorization (PUMA) was filed for approval in the E.U. by ViroPharma for the treatment of prolonged, acute, convulsive seizures in infants, toddlers, children and adolescents, from 3 months to less than 18 years. In 2011, a positive opinion was assigned to the PUMA and final approval was assigned in June 2011. The product was launched in the U.S. in November 2011. This product was filed for approval in Japan in 2013 by Astellas Pharma for the conscious sedation in dentistry and dental surgery. In the same year the product was approved for this indication.
In terms of clinical development, a nasal formulation of the drug is in phase III clinical trials at Upsher-Smith for rescue treatment of seizures in patients on stable anti-epileptic drug regimens who require control of intermittent bouts of increased seizure activity (seizure clusters). The Hopitaux de Paris had been developing a sublingual tablet formulation of midazolam to be used in combination with morphine for the treatment of pain in children following bone fractures; however, no recent development has been reported for this indication. NovaDel Pharma had been developing the compound preclinically for the treatment of generalized anxiety, however no recent developments have been reported.
Midazolam achieves its therapeutic effect through interaction with the gamma-aminobutyric acid benzodiazepine (GABA-BZ) receptor complex. Subunit modulation of the GABA-BZ receptor chloride channel macromolecular complex is hypothesized to be responsible for some of the pharmacological properties of benzodiazepines, which include sedative, anxiolytic, muscle relaxant, and anticonvulsive effects in animal models. GABA acts at inhibitory synapses in the brain by binding to specific transmembrane receptors in the plasma membrane of both pre- and post-synaptic neurons, opening ion channels and bringing about a hyperpolarization via either chloride or potassium ion flow.
In 2008, fast track designation was assigned to midazolam maleate in the U.S. for the treatment of seizure disorders.
In 2009, Orphan Drug Designation was received in the U.S. by for the treatment of seizure disorders in patients who require control of intermittent bouts of increased seizure activity (e.g. acute repetitive seizures, seizure clusters). This designation was assigned in the U.S. for the treatment of nerve agent-induced seizures.
In 2010, midazolam maleate was licensed to Upsher-Smith by Ikano Therapeutics for the treatment of acute repetitive seizure in patients with epilepsy. However, in 2010, Ikano closed and dissolved its business. Previously, Ikano had transferred to Upsher-Smith ownership of it nasal midazolam maleate program.
Midazolam is among about 35 benzodiazepines which are currently used medically, and was synthesised in 1975 by Walser and Fryer at Hoffmann-LaRoche, Inc in the United States.Owing to its water solubility, it was found to be less likely to cause thrombophlebitis than similar drugs.The anticonvulsant properties of midazolam were studied in the late 1970s, but not until the 1990s did it emerge as an effective treatment for convulsive status epilepticus. As of 2010, it is the most commonly used benzodiazepine in anesthetic medicine. In acute medicine, midazolam has become more popular than other benzodiazepines, such as lorazepam and diazepam, because it is shorter lasting, is more potent, and causes less pain at the injection site.Midazolam is also becoming increasingly popular in veterinary medicine due to its water solubility.
Midazolam is a water-soluble benzodiazepine available as a sterile, nonpyrogenic parenteral dosage form for intravenous or intramuscular injection. Each mL contains midazolam hydrochloride equivalent to 1 mg or 5 mg midazolam compounded with 0.8% sodium chloride and 0.01% edetate disodium with 1% benzyl alcohol as preservative, and sodium hydroxide and/or hydrochloric acid for pH adjustment. pH 2.9-3.7.
Midazolam is a white to light yellow crystalline compound, insoluble in water. The hydrochloride salt of midazolam, which is formed in situ, is soluble in aqueous solutions. Chemically, midazolam HCl is 8-chloro-6-(2-fluorophenyl)-1-methyl-4H– imidazo[1,5-a] [1,4] benzodiazepine hydrochloride. Midazolam hydrochloride has the molecular formula C18H13ClFN3•HCl, a calculated molecular weight of 362.25 and the following structural formula:
 |
In the Netherlands, midazolam is a List II drug of the Opium Law. Midazolam is a Schedule IV drug under the Convention on Psychotropic Substances. In the United Kingdom, midazolam is a Class C controlled drug. In the United States, midazolam (DEA number 2884) is on the Schedule IV list of the Controlled Substances Act as a non-narcotic agent with low potential for abuse.
midaolam hydrochloride NDA 018654, 075154
REF
U.S. Pat. No. 4,280,957
U.S. Pat. No. 5,693,795
U.S. Pat. No. 6,512,114
Midazolam Maleate
Drugs Fut 1978, 3(11): 822
Bioorganic and Medicinal Chemistry, 2012 , vol. 20, 18 pg. 5658 – 5667
Journal of Heterocyclic Chemistry, 1983 , vol. 20, 3 pg. 551 – 558.. 32 maleate
WO 2001070744
WO 2001002402
WO 2012075286
US2011/275799 A1… no 5
Journal of Organic Chemistry, 1978 , vol. 43, p. 936,942, mp free base, nmr
| US4280957 | May 15, 1978 | Jul 28, 1981 | Hoffmann-La Roche Inc. | Imidazodiazepines and processes therefor |
| US6262260 * | Mar 23, 2000 | Jul 17, 2001 | Abbott Laboratories | Process for the preparation of midazolam |
| US6512114 | Jun 30, 1999 | Jan 28, 2003 | Abbott Laboratories | Process for the preparation of Midazolam |
……………………….
introduction
4H-imidazo[1,5-a][1,4]benzodiazepines or, more simply, imidazobenzodiazepines, are a class of benzodiazepines having the general formula (I),

wherein the 1,4-diazepine ring is fused with a 1,3-imidazole ring. The main compounds part of the 4H-imidazo[1,5-a][1,4]benzodiazepines are Midazolam of formula (IV):

an active ingredient currently commercially available as a hydrochloride salt under the name of Versed or Hypnovel for anaesthetic and sedative use and the maleate salt currently commercially available under the name Dormicum or Flormidal.
Other important compounds are Climazolam of formula (VII):

Imidazenil of formula (VIII):

1-Hydroxymidazolam of formula (IX):

and Desmethyl midazolam of formula (X):

all these being biologically active substances and having psychotropic and sedative action.
The synthesis of the Midazolam as described in U.S. Pat. No. 4,280,957 of Hoffmann-La Roche provides for the decarboxylation reaction of the 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine-3-carboxylic acid of formula (VI) according to the following scheme:

The process for preparing the intermediate (VI) via basic hydrolysis of the corresponding ester is described in such patent publication and it is well known in the art.
The thermal decarboxylation reaction in high boiling solvent such as mineral oil at 230° C. for 5 min results in a mixture of products of Midazolam of formula (IV) and of Isomidazolam of formula (IV-bis), a non-pharmacologically active isomer, at a 80:20 ratio. The two products are separated by chromatography.
At industrial level, the formation of the Isomidazolam isomer impurity requires a further isomerisation reaction performed on the mixture of the two compounds to convert the isomer into the active product. The reaction mixture obtained from the thermal decarboxylation is thus subjected to basic treatment under the action of KOH in EtOH followed by an acid treatment which thus provides a mixture of Midazolam-Isomidazolam at a 95:5 ratio. The final removal of the Isomidazolam impurity from the product occurs through crystallisation of the product from AcOEt and EtOH. In order to limit this isomerisation treatment, in the subsequent U.S. Pat. No. 5,693,795 of Hoffmann-La Roche dated 1999, there is described a process for performing the decarboxylation of the compound of formula (VI) in n-butanol in a continuous tubular reactor with a 4 minutes permanence period with a yield between 47-77%. However, the reaction, performed at high temperature and pressure (280° C., 100 bars) results in the formation of a considerable percentage of Isomidazolam (85:15 Midazolam/Isomidazolam ratio) which still requires the basic isomerisation step.
Lastly, in U.S. Pat. No. 6,512,114 of Abbott Laboratories there is described the decarboxylation of the compound of formula (VI) in mineral oil or in N,N-Dimethylacetamide (DMA) at 160-230° C. for at least 3 hours obtaining a 3/1 to 6/1 Midazolam/Isomidazolam ratio with a yield of isolated product equal to just 54%.
Though performed using dedicated apparatus and in extreme conditions, the prior art processes do not allow selectively performing the decarboxylation reaction of the intermediate (VI) to Midazolam thus requiring a further synthetic passage followed by crystallisation with ensuing reduction of the overall yield.
Midazolam (8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine) is represented by the following structural formula (I):

Midazolam is a central nervous system (CNS) depressant, used for short term treatment of insomnia. Like other benzodiazepines, midazolam binds to benzodiazepine receptors in the brain and spinal cord and is thus used as a short-acting hypnotic-sedative drug with anxiolytic and amnestic properties. It is currently used in dentistry, cardiac surgery, endoscopic procedures, as a preanesthetic medication, as an adjunct to local anesthesia and as a skeletal muscle relaxant. Depending on the pH value, midazolam can exist in solution as a closed ring form (I) as well as an open ring form (IA), which are in equilibrium, as shown in Scheme 1:

The amount of the open ring form (IA) is dependent upon the pH value of the solution. At a pH value of about 3, the content of the open ring form (IA) can be 40%, while at pH value of 7.5, the closed ring form (I) can be more than 90%.
Clinical studies have demonstrated that there are no significant differences in the clinical activity between midazolam hydrochloride and midazolam maleate, however the use of intravenous midazolam hydrochloride has been associated, in some cases, with respiratory depression and arrest.
U.S Pat. No. 4,280,957 (hereinafter the ‘957 patent) describes a synthetic process for preparing midazolam, which is depicted in Scheme 2 below. This process includes reacting 2-aminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-bezodiazepine (II) with acetic anhydride in dichloromethane to produce 2-acetamido-methyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-bezodiazepine (III). The latter is heated with polyphosphoric acid at 150° C. to produce 8-chloro-6-(2-fluorophenyl)-3a,4-dihydro-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine of formula (IV), which is purified by column chromatography. Compound IV is then mixed with toluene and manganese dioxide and heated to reflux to afford midazolam base, which is crystallized from ether to yield a product with mp of 152-154° C.

The ‘957 patent further describes an alternative process which includes reacting 2-aminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-bezodiazepine (II) (optionally as a dimaleate salt) with triethylorthoacetate in ethanol and in the presence of p-toluenesulfonic acid to afford 8-chloro-6-(2-fluorophenyl)-3a,4-dihydro-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine (IV). This product is dissolved in xylene and treated with activated manganese dioxide to afford the crude base, which is reacted in situ with maleic acid in ethanol and crystallized by addition of ether to produce the midazolam maleate having melting point of 148-151° C. The process is depicted in Scheme 3 below.

The preparation of midazolam maleate from the isolated midazolam base is also described in a further example of the ‘957 Patent, wherein a warm solution of midazolam base in ethanol is combined with a warm solution of maleic acid in ethanol. The mixture is diluted with ether and at least part of the solvents is evaporated using a steam bath to obtain crystalline midazolam maleate having melting point of 148-151° C. The yield and the purity of the obtained midazolam maleate are not disclosed.
U.S. Pat. No. 6,512,114 (hereinafter the ‘114 patent) describes another synthetic process for preparing midazolam, which is depicted in Scheme 4 below. According to this Process, the starting material 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine-3-carboxylic acid (V) is heated in mineral oil for 3 hours at 230° C. until it is decarboxylated, followed by treatment with potassium tert-butoxide, to afford midazolm (I), isomidazolam (VI) and a midazolam dimmer (VII). Midazolam base is obtained in 54.5% yield after two re-crystallizations from ethyl acetate and heptane; however, the purity of the product is not disclosed.

The preparation of midazolam by conventional routes is liable to produce impurities such as isomidazolam (VI) and a midazolam dimmer (VII), and possibly other impurities. There is, therefore, a need in the art for a midazolam purification process that will provide highly pure midazolam containing minimal amounts of impurities produced. The present invention provides such a process.
This example describes the preparation of midazolam base as taught in the ‘957 patent.
16 g (0.03 mol) of 2-aminomethyl-7-chloro-5-(2-fluorophenyl)-2,3-dihydro-1H-1,4-bezodiazepine dimaleate was dissolved in 200 ml of toluene and 10 ml of 25% ammonium hydroxide solution was added and mixing was maintained for an hour. Then, the phases were separated and the toluene phase was dried by azeotropic distillation using a Dean Stark apparatus. 7 ml (0.038 mol) of triethylorthoacetate was added and the solution was heated to reflux for 4 hours, after which time the solution was left to cool to ambient temperature. 25 ml of methyl tert-butyl ether was added and the mixture was cooled overnight to produce 8-chloro-6-(2-fluorophenyl)-3a,4-dihydro-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine, which was isolated by filtration. The product was mixed with 200 ml of toluene and dried by azeotropic distillation using a Dean Stark apparatus. Then, 30 g of manganese dioxide was added and the mixture was heated to reflux for two hours. The excess manganese dioxide was filtered off to afford a solution of midazolam base in toluene, which was evaporated to obtain a product having 97.9% purity and containing 0.44% of impurity VIII and 1.14% of impurity IX (according to HPLC).
…………………………
EXAMPLE 28
2-Aminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-benzodiazepine dimaleate
A suspension of 17 g (0.05 m) of 7-chloro-1,3-dihydro-5-(2-fluorophenyl)-2-nitromethylene-2H-1,4-benzodiazepine-4-oxide in 200 ml of tetrahydrofuran and 100 ml of methanol was hydrogenated in presence of 17 g of Raney nickel at an initial pressure of 155 psi for 24 hrs. The catalyst was removed by filtration and the filtrate was evaporated. The residue was dissolved in 50 ml of 2-propanol and warmed on the steambath. A warm solution of 17 g of maleic acid in 60 ml of ethanol was added and the salt was allowed to crystallize by cooling in the ice bath. The final product consisted of yellow crystals with mp 196
EXAMPLE 14
8-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine
Acetic anhydride, 7 ml., was added to a solution of 6.16 g. of crude 2-aminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-benzodiazepine in 200 ml. of methylene chloride. The solution was layered with 200 ml. of saturated aqueous sodium bicarbonate and the mixture was stirred for 20 minutes. The organic layer was separated, washed with sodium bicarbonate, dried over sodium sulfate and evaporated to leave 6.2 g. resinous 2-acetaminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-benzodiazepine. This material was heated with 40 g. of polyphosphoric acid at 150 water, made alkaline with ammonia and ice and extracted with methylene chloride. The extracts were dried and evaporated and the residue (5.7 g.) was chromatographed over 120 g. of silica gel using 20% methanol in methylene chloride. The clean fractions were combined and evaporated to yield resinous 8-chloro-3a,4-dihydro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[ 1,5-a][1,4]benzodiazepine. A mixture of this material with 500 ml. of toluene and 30 g. of manganese dioxide was heated to reflux for 11/2 hours. The manganese dioxide was separated by filtration over celite. The filtrate was evaporated and the residue was crystallized from ether to yield a product with m.p. 152 was recrystallized from methylene chloride/hexane
EXAMPLE 49
8-Chloro-6-(2-fluorophenyl)-1-methyl-6H-imidazo[1,5-a][1,4]benzodiazepine
Potassium t-butoxide, 0.625 g. (5.5 mmol), was added to a solution of 1.625 g. (5 mmol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine in 20 ml. of dimethylformamide cooled to -30 nitrogen for 10 min. at -30 ml. of glacial acetic acid and was then partitioned between aqueous bicarbonate and toluene/methylene chloride (3:1 v/v). The organic layer was separated, dried and evaporated. The residue was chromatographed over 60 g. of silica gel using 25% (v/v) methylene chloride in ethyl acetate. The less polar product was eluted first and was crystallized from ethylacetate/hexane to yield product with m.p. 180
EXAMPLE 50
8-Chloro-6-(2-fluorphenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine
Potassium t-butoxide, 0.125 g. (1.1 mmol) was added to a solution of 0.325 g. (1 mmol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-6H-imidazo[1,5-a][1,4]benzodiazepine in 20 ml. of dimethylformamide cooled to -30 -30 by addition of 0.2 ml. of glacial acetic acid and was partitioned between aqueous sodium bicarbonate and methylene chloridetoluene (1:3). The organic phase was washed with water, dried and evaporated. The residue was chromatographed over 20 g. of silica gel using ethyl acetate for elution. After elution of starting material, product was collected and crystallized from ether/hexane, m.p. 156
hyd and dihydrochloride
EXAMPLE 24
8-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine dihydrochloride
A solution of 0.32 g (1 mmol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine in 5 ml of ethanol was treated with excess ethanolic hydrogen chloride. The salt was crystallized by addition of 2-propanol and ether. The colorless crystals were collected, washed with ether and dried to leave a final product with mp 290
EXAMPLE 258-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine hydrochloride
A solution of 0.325 g (1 mmol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine in 3 ml of ethanol was combined with a suspension of 0.4 g (1 mmol) of the dihydrochloride of this compound in 5 ml of ethanol. After filtration, the solution was treated with ether and heated on the steambath for 5 min to crystallize. The crystals were collected, washed with ether and dried to leave the monohydrochloride with mp 295
maleate
EXAMPLE 22
8-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine maleate
A warm solution of 6.5 g (0.02 m) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine in 30 ml of ethanol was combined with a warm solution of 2.6 g (0.022 m) of maleic acid in 20 ml of ethanol. The mixture was diluted with 150 ml of ether and heated on the steam bath for 3 min. After cooling, the crystals were collected, washed with ether and dried in vacuo to yield a final product with mp 148
…
Synthesis
Midazolam, can be described according to scheme 4 indicated below:

 was prepared according to processes known in the art (e.g. U.S. Pat. No. 4,280,957) which comprise the basic hydrolysis of the corresponding ester.
was prepared according to processes known in the art (e.g. U.S. Pat. No. 4,280,957) which comprise the basic hydrolysis of the corresponding ester.For the reactions performed in the microreactor, the solutions containing the substrates to be decarboxylated were loaded into 5 and 10 mL gastight glass syringes (Hamilton, item n. 81527, 81627) mounted on syringe pumps (KD Scientifics, model KDS100). A pipe made of PTFE® (OD=1.58 mm, ID=0.8 mm, Supelco, item n. 58696-U) was used for making the reaction channel.A counterpressure valve sold by Swagelok (item n. SS-SS1-VH) was used for regulating the flow within the channel.Example 1Synthesis of the Compound of Formula (V)—Example of the Invention

50 g (0.135 mol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepin-3-carboxylic acid of formula (VI) and 250 mL of ethanol were loaded into a two-neck 500 mL flask, equipped with a magnetic stirrer. 40 mL of an aqueous solution of 1 M HCl are dripped in about 10 minutes. The open di-hydrochloride intermediate of formula (V) starts precipitating into the reaction environment already after 3 minutes from the beginning of the addition of the acid solution. The mixture is maintained stirred at RT for 3 hrs and then it is filtered on buckner washing the solid with ethanol. The moist product is dried in an oven under vacuum at 60° C. up to reaching a constant weight. A light yellow crystalline product is obtained (51.5 g, 83% yield). The crude product was used for the decarboxylation without further purifications.
ESI-MS [MeCN+0.1% HCOOH]: m/z 388 (V); 370 (VI).
1H-NMR (250 MHz, CD3OD): 2.52 (s, 3H); 4.27-4.41 (m, 2H); 7.22-8.1 (m, 7H). M.p.: 217° C.
Example 2
Synthesis of Midazolam of Formula (IV)—Performed in Batch—Example of the Invention

30 g (0.065 mol) of 5-(aminomethyl)-1-{(4-chloro-2-[(2-fluorophenyl)carbonyl]phenyl}-2-methyl-1H-imidazole-4-carboxylic acid dihydrochloride of formula (V) and 90 mL of NMP are loaded into a three-neck 250 mL flask, equipped with a magnetic stirrer and coolant. The mass is heated using an oil bath at T=195-203° C. for one hour. Thus, 1 mL of solution is collected for performing HPLC analysis. The reaction product is Midazolam having 82% titre (w/w) (determined via HPLC titre correcting it using the solvent) and it contains 1% of Isomidazolam. The product is extracted using Isopropyl acetate after raising the pH to 10 by adding aqueous Na2CO3.
Example 3
Synthesis of Midazolam of Formula (IV)—Performed in a Micro-Reactor—Example of the Invention

3.22 g (7 mmol) of 5-(aminomethyl)-1-{4-chloro-2-[(2-fluorophenyl)carbonyl]phenyl}-2-methyl-1H-imidazole-4-carboxylic acid dihydrochloride of formula (V) and 10 mL of NMP are loaded into a 10 mL flask equipped with a magnetic stirrer. In order to facilitate the complete solubilisation of the substrate, it is necessary to slightly heat the reaction mixture (about 40° C.) for a few minutes. The solution thus obtained is transferred into a 10 mL gastight glass syringe mounted on a KDS100 syringe pump (FIG. 1) and the flow is regulated at 1.0 mL/h so as to set a residence period of 30 minutes at 200° C. The reaction product is Midazolam having an 89% titre (w/w) (determined via HPLC titre correcting it using the solvent) and containing 3% (w/w) of Isomidazolam.
Example 4Synthesis of Midazolam of formula (IV)—Comparison of the InventionA table is reported which summarises the results of the decarboxylation of the compound of formula (V) and (V-bis) (for the latter see Examples 6 and 7) obtained according to some embodiments of the invention and those obtained by way of experiment through the decarboxylation of the intermediate of formula (VI) (process of the prior art) both performed in 3 volumes of NMP at 200° C., both in batch method (Example 4) and in continuous method with the microreactor (MR) made of PTFE of FIG. 1. (Examples 4-1, 4-2, 4-3).
| Example | substrate | Mode | Solv. | T° C. | t min. | Midazolam (p/p) | Isomidaz. (P/P) |
| 2 | (V) | Batch | NMP | 200 | 60 | 82 | 1 |
| 3 | (V) | MR | NMP | 200 | 30 | 89 | 3 |
| 7 | (V-bis) | Batch | NMP | 200 | 60 | 68 | 3 |
| 4 | (VI) | Batch | NMP | 200 | 60 | 78 | 18 |
| 4-1 | (VI) | MR | NMP | 200 | 38 | 81 | 17 |
| 4-2 | (VI) | MR | NMP | 200 | 20 | 77 | 18 |
| 4-3 | (VI) | MR | NMP | 200 | 15 | 58 | 22 |
| U.S. Pat. No. | (VI) | Tubular | n-BuOH | 290 | 4 | 85 * | 15 * |
| 5,693,795 | reactor | ||||||
| U.S. Pat. No. | (VI) | Batch | Olio | 230 | 180 | 75 * | 25 * |
| 6,512,114 | min. | 87.5 * | 12.5 * | ||||
| or DMA | |||||||
| * = Midazolam/Isomidazolam ratio only (other impurities not considered). | |||||||
The product of the comparative experiments 4, 4-1, 4-2, 4-3 and of the two USA patents should be subjected to a further isomerisation process to reduce the high amount of Isomidazolam so as to be able to obtain Midazolam free of Isomidazolam after further crystallization, which would not be required for the product obtained according to the invention (examples 2 and 3).

A 4-neck RBF was charged under nitrogen flow with: 10 g of Midazolam (IV) (prepared according to example 2) and 40 mL of Ethanol. The slurry was stirred until complete dissolution at 25/30° C. In an other flask was prepared the following solution: 3.72 g of maleic acid are dissolved in 15 mL of Ethanol. The slurry was stirred until complete dissolution at 25/30° C. The maleic acid solution is dropped in 30/40 minutes and keeping T=25/30° C. into the solution containing Midazolam. The slurry was cooled down at −15° C. in one hour and kept at that temperature for at least 2 hours. The slurry was then filtered and the cake was washed with 40 mL of cool Ethanol. The filter was discharged and the product was dried at 40° C. under vacuum for 2 hours and then at 60° C. for 8 hours. 12.8 g of Midazolam Maleate as white solid were collected (Molar yield=94.5%). m.p.=149-152° C. (by DSC).

A 4-neck RBF was charged under nitrogen flow with: 1 g of Midazolam (IV) (prepared according to example 2) and 15 mL of Ethanol. The slurry was stirred until complete dissolution at 25/30° C. 5 mL of a ethanolic solution of Hydrochloric acid 2N were slowly added. 20 mL of Isopropanol were added over 30 minutes at RT. The slurry was cooled down at −15° C. in one hour and kept at that temperature for at least 2 hours. The slurry was then filtered and the cake was washed with 10 mL of cool isopropanol. The filter was discharged and the product was dried at 40° C. under vacuum for 2 hours and then at 60° C. for 8 hours. Midazolam dihydrochloride as white solid was collected.
MIDAZOLAM HYDROCHLORIDE
Example 10
Preparation of 8-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine hydrochloride (Midazolam hydrochloride)

A 4-neck RBF was charged under nitrogen flow with: 1 g of Midazolam (IV) (prepared according to example 2) and 10 mL of Ethanol. The slurry was stirred until complete dissolution at 25/30° C. In an other flask was prepared the following suspension: 1.22 g of Midazolam dihydrochloride (prepared according to example 9) and 15 mL of Ethanol. The Midazolam ethanolic solution was added to the Midazolam dihydrochloride suspension. After filtration, the solution was treated with MTBE and heated at 60° C. until crystallization. After cooling to RT, the slurry was filtered, the cake washed with MTBE and the product was dried to provide Midazolam (mono)hydrochloride as a white solid.
…..


NEW DRUG APPROVALS
ONE TIME
$10.00
DAPAGLIFLOZIN…FDA approves AZ diabetes drug Farxiga

DAPAGLIFLOZIN, BMS-512148
The US Food and Drug Administration has finally approved AstraZeneca’s diabetes drug Farxiga but is insisting on six post-marketing studies, including a cardiovascular outcomes trial.
The approval was expected given that the agency’s Endocrinologic and Metabolic Drugs Advisory Committee voted 13-1 last month that the benefits of Farxiga (dapagliflozin), already marketed in Europe as Forxiga, outweigh identified risks. The FDA rejected the drug in January 2012 due to concerns about possible liver damage and the potential link with breast and bladder cancer.

Orphan Drugs: FDA PDUFA Dates January – February 2014
.
.
.
.
Investors and traders will be watching several orphan drug FDA PDUFA dates in January and February 2014.
FDA PDUFA Dates January – February 2014
| Generic Name | Trade Name | Sponsor | Indication | 2014 PDUFA Date |
| Tasimelteon | Hetlioz | Vanda Pharmaceuticals | Non-24 Hour Sleep/Wake Disorder in Blind Individuals Without Light Perception | 01/31 |
| Droxidopa * | Northera | Chelsea Therapeutics | Symptomatic Neurogenic Orthostatic Hypotension For Patients With Primary Autonomic Failure | 02/14 |
| Metreleptin | Bristol-Myers Squibb | Lipodystrophy | 02/27 | |
| Elosulfase Alfa ** | Vimizim | BioMarin Pharmaceutical | Mucopolysaccharidosis Type IV A (Morquio A Syndrome) | 02/28 |
| Ibrutinib | Imbruvica | Pharmacyclics | Chronic/Small-Cell Lymphocytic Leukemia | 2/28 |
.
* Resubmission
** Priority Review
Please note the links for the following 2 chart columns above :
1) “Generic Name” Column Link = Is a source for the FDA PDUFA Date
2) “Indication” Column Link = Is the FDA Orphan Drug Product Designation Database Link.
Please…
View original post 33 more words
