
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Peramivir, a Flu Treatment

PERAMIVIR
CAS 330600-85-6
- Molecular FormulaC15H28N4O4
- Average mass328.407 Da
(1S,2S,3R,4R)-4-carbamimidamido-3-[(1S)-1-acetamido-2-ethylbutyl]-2-hydroxycyclopentane-1-carboxylic acid
- RWJ 270201
- RWJ-270201
- RWJ270201
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Peramivir hydrate | QW7Y7ZR15U | 1041434-82-5 | RFUCJKFZFXNIGB-ZBBHRWOZSA-N |
| Molecular Weight | 382.45 |
| Formula | C15H28N4O4 • 3H2O |
| CAS No. | 330600-85-6 (Peramivir); 1041434-82-5 (Peramivir Trihydrate); |
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Rapivab | Solution | 600 mg/60mL | Intravenous | BioCryst Pharmaceuticals, Inc. | 2014-12-20 | Not applicable |  |
|
| Rapivab | Solution | 600 mg/60mL | Intravenous | Seqirus USA Inc. | 2014-12-20 | Not applicable | |
|
| Rapivab | Solution | 10 mg | Intravenous | Biocryst Pharmaceuticals Inc | Not applicable | Not applicable |  |
- Drug Name:
- Peramivir Hydrate
- Research Code:
- S-021812
- Trade Name:
- Rapiacta® / Peramiflu®
- MOA:
- Neuraminidase inhibitor
- Indication:
- Influenza infection
- Status:
- Approved
- Company:
- BioCryst (Originator) , Shionogi
- Sales:
- $21.6 Million (Y2015);

$23.7 Million (Y2014);
$20 Million (Y2013);
$24.1 Million (Y2012);
$17.7 Million (Y2011); - ATC Code:
Approved Countries or Area
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-12-19 | Marketing approval | Rapivab | Influenza infection | Solution | 200 mg/20 ml | BioCryst | Standard |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2010-01-13 | Marketing approval | Rapiacta | Influenza infection | Injection, Solution | Eq. 150 mg/300 mg Peramivir | BioCryst, Shionogi |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2013-04-05 | Marketing approval | 力纬 | Influenza infection | Injection, Solution | 100ml | 广州南新制药 | 1.1 |
Shionogi , GC Pharma and Seqirus , under license from BioCryst Pharmaceuticals (which licensed the program from the University of Alabama ), have developed and launched an iv formulation of the influenza neuraminidase inhibitor peramivir.
Peramivir hydrate was approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on January 13, 2010, and approved by the U.S. Food and Drug Administration (FDA) on December 19, 2014. It was co-developed and co-marketed as Rapiacta® by BioCryst and Shionogi in Japan.
Peramivir hydrate is a neuraminidase inhibitor, acting as a transition-state analogue inhibitor of influenza neuraminidase and thereby preventing new viruses from emerging from infected cells. It is indicated for the treatment of influenza A and B viral infections.
Rapiacta® is available as injection solution for intravenous, containing 150 mg/300 mg Peramivir hydrate. The recommended dose is once a day by an intravenous drip infusion over a period of 15 minutes or longer.
Peramivir (trade name Rapivab) is an antiviral drug developed by BioCryst Pharmaceuticals for the treatment of influenza. Peramivir is a neuraminidase inhibitor, acting as a transition-state analogue inhibitor of influenza neuraminidase and thereby preventing new viruses from emerging from infected cells. It is approved for intravenous administration.[1]
In October 2009, the U.S. Food and Drug Administration (FDA) issued an emergency use authorization (EUA) for the use of peramivir based on safety data from phase I, phase II, and limited phase III trial data. The emergency use authorization for peramivir expired in June 2010.[2][3] On 19 December 2014, the FDA approved peramivir to treat influenza infection in adults.[1]
Peramivir has also been approved in Japan and South Korea and is available in Japan as Rapiacta and in South Korea as Peramiflu.[ As of 2015, it is the only intravenous option for treating swine flu.
Peramivir is an antiviral agent developed by Biocryst Pharmaceuticals to treat influenza A/B. The development of peramivir has been supported by the US Department of Health and Human Services as part of the government’s effort to prepare for a flu pandemic. Being an influenza virus neuraminidase inhibitor, peramivir works by preventing new viruses from emerging from infected cells. Due to the poor oral bioavailability, the oral formulation of the drug was previously abandoned by Johnson and Johnson Company. The injectable intravenous formulation of peramivir was approved by the FDA in September 2017 for the treatment of acute uncomplicated influenza to pediatric patients 2 years and older who have been symptomatic for no more than two days.
History
An intramuscular (IM) peramivir phase II study for seasonal influenza in 2008–2009 found no effect for the primary endpoint of improvement in the median time to alleviation of symptoms in subjects with confirmed, acute, uncomplicated influenza infection versus placebo.
In October 2009, it was reported that the experimental antiviral drug peramivir had been “life-saving” effective in intravenous treating 8 serious cases of swine flu.[4] On October 23, the U.S. Food and Drug Administration (FDA) issued an Emergency Use Authorization for peramivir, allowing the use of the drug in intravenous form for hospitalized patients only in cases where the other available methods of treatment are ineffective or unavailable;[5] for instance, if oseltamivir resistance develops and a person is unable to take zanamivir via the inhaled route. The U.S. government (department of Health and Human Services) gave BioCryst Pharmaceuticals more than $77 million to finish the Phase III clinical development of peramivir. In 2009 the department of Health and Human Services had already given about $180 million to the program.[6] Biocryst also donated 1200 courses of treatment to the US department of Health and Human Services.[7] The Emergency Use Authorization expired on June 23, 2010. In 2011 a phase III trial found the median durations of influenza symptoms were the same with 1 intravenous injection of peramivir against 5 days of oral oseltamivir for people with seasonal influenza virus infection.[8]
In 2012 BioCryst reported that it should halt enrollment on its study for intravenous peramivir in potentially life-threatened people after an interim analysis led trial monitors to conclude that it would be futile to continue and the trial should be terminated. The difference between peramivir and control group (oral oseltamivir) for the primary endpoint, clinical or virologic, was small.[9] In 2013 the Biomedical Advanced Research and Development Authority (BARDA/HHS) released new funding under the current $234.8 million contract to enable completion of a New Drug Application filing for intravenous (IV) peramivir.[10]
According to a research report published in June 2011, a new variant of swine flu had emerged in Asia with a genetic adaptation (a S247N neuraminidase mutation) giving some resistance to oseltamivir and zanamivir, but no significant reduction in sensitivity to peramivir.[11][12] But a H274Y virus mutation showed resistance to oseltamivir and peramivir, but not to zanamivir, and only in N1 neuraminidases.[13] Ultimately 3.2% (19/599) of A(H1N1)pdm09 viruses collected between 2009 and 2012 had highly reduced peramivir inhibition due to the H275Y NA mutation.[14]
BioCryst Pharmaceuticals submitted a new drug application (NDA) to the U.S. Food and Drug Administration (FDA) for intravenous peramivir in December 2013.[15] Peramivir (Rapivab) was approved for intravenous administration in December 2014.[1][16]

Reference:1. WO9933781A1 / US6562861B1.

Reference:1. CN102372657A.

Reference:1. CN102633686A.

Reference:1. J. Med. Chem. 2001, 44, 4379-4392.



PATENT
peramivir’s product case, WO9933781 ,
hold SPC protection in the EU states until December 2023, and contain one of the Orange Book (OB) listed patents, US6562861 , that was due to expire in the US in December 2023. In January 2021, the US FDA’s OB was seen to list patents describing peramivir that expire in 2027.
PATENT
WO-2021002689
Process for preparing an inhibitor of neuraminidase infection ie peramivir trihydrate crystal from the free form of the drug ie peramivir. Discloses the use of peramivir trihydrate in treating influenza infection. Represents the first PCT filing from CKD Bio Corp that focuses on peramivir; however, this case was first seen as a Korean national filing that is published in February 2020.
PATENT
https://patents.google.com/patent/WO2012145932A1/en
Peramivir has the chemical name of
(1^,2^,3^, 4R)-3-[(llS)-l-acetamido-2-ethyl-butyl] -4-(diaminomethylideneamino)- 2-hydroxy-cyclopentane-l-carboxylicacid, and has the following structure:

(I)
[0003] Peramivir is currently being developed as an antiviral drug, and in particular, for treatment of influenza. Acting as a neuraminidase inhibitor, peramivir can efficiently inhibit the replication of all type of influenza viruses. Peramivir can be administered via injection, and is known to be well-tolerated and cause only mild adverse effect.
[0004] Several processes relating to the preparation of peramivir are disclosed in CN1227466, CN1282316, and WO01/00577A1.
[0005] As shown in Scheme 1, CN 1227466 discloses a process comprising ring-opening of chiral 2-azabicyclo [2.2.1] hept-5-en-3-one, followed by
amino-protecting reaction, Diel- Alder conjugate cycloaddition, reduction, acetylation, guanidylation and finally hydrolyzation to yield peramivir. The major drawback of this process is the use of highly expensive starting material 1. In addition, this process is not suitable for scale-up.
Scheme 1

[0006] WO2009021404 discloses a method comprising reacting
N-Boc-protected chiral 2-azabicyclo [2.2.1] hept-5-en-3-one and
2-ethylbutylaldehyde as starting material to prepare peramivir as illustrated in Scheme 2.
Scheme 2

eram v r
EXAMPLES
Example 1
1 (liS’,4J/?)-methyl-4-(2,3-bis(ieri-butoxycarbonyl)guanidino)cyclopent-2-ene- carboxylate (13)

11 12 13 [0063] To a mixture of (lS^R^methyl 4-aminocyclopent-2-enecarboxylate tartaric acid salt 11 ( 7.29 g, 25 mmol) in dichloromethane (150 mL), was added Et3N (9 mL, 65 mmol) at 0 °C, and the resulting mixture was stirred for 15 min. To this, tert-butyl (lH-pyrazol-l-yl)methylenedicarbamate 12 was added. After addition, the reaction was monitored for completion by TLC ( PE: EtOAc=5 : 1 ) . The organic layers were washed with water and brine and dried over anhydrous Na2S04. The mixture was filtered and concentrated to give 13 as a white solid, which was used in the next step without further purification.
[0064] MS (M+l ) : 384.
[0065] ‘H NMR (400 MHz, CDC13) δ 11.49 (s, 1H), 8.53 (d, J= 8.4 Hz, 1H), 5.94 – 5.83 (m, 2H), 5.38 – 5.31 (m, 1H), 3.73 (s, 3H), 3.56 – 3.44 (m, 1H), 2.60 (dt, J= 14.0, 8.5 Hz, 1H), 1.94 (dt, J= 13.9, 4.7 Hz, 1H), 1.50 (d, J= 7.4 Hz, 18H) (See attached Chart 1)
Example 2
2 (3aJ/?,4J/?,6iS’,6aiS -methyl-4-(2,3-bis(ieri-butoxycarbonyl)guanidino)-3-(pentan -3-yl)-4,5,6,6a-tetrahydro-3aH-cyclopenta[d]isoxazole-6-carboxylate (5)

14 85% a) Preparation of 2-Ethyl-N-hydroxybutanimidoyl chloride (14):
[0066] Hydroxylamine hydrochloride (7.2g, 0.1 mol) was dissolved in water (7 mL). Toluene (27 mL) was added, followed by addition of 2-ethylbutylraldehyde (lOg, 0.1 mol). The two-phase mixture was stirred vigorously while cooling.
Sodium hydroxide solution (ca.30%, 14.6g, O. l lmol) was added slowly (addition is very exothermic) to maintain a temperature between 15-25 °C. The mixture was stirred for 60 min, then allowed to stand to separate the layers. The organic extract was washed with water and brine, dried over Na2S04, and directly used in the next step.
[0067] N-Chlorosuccinimide (NCS) (13.3g, 0.1 mol) was suspended in dimethylformamide (DMF) (17ml) and cooled to about 10 °C. The toluene solution prepared above (3 .15 mol) was added dropwise with sufficient cooling to maintain the reaction temperature betweenlO-25°C. After addition, the reaction was monitored by TLC until completion of the reaction. Water (100ml) was added slowly (slightly exothermic) while maintaining the temperature at 15-25 °C. The two-phase mixture was stirred for 15 min at 15-25 °C and the layers were separated. The water layer was extracted with toluene (10ml) and the organic layer washed with water (3 X 20ml) and brine, dried over Na2S04, and directly used in the next step. b)
Preparation of (3aJ/?,4J/?,6iS’,6aiS -methyl-4-(2,3-bis(ieri-butoxycarbonyl)-guan idine)-3 -(pentan-3 -yl)-4,5,6,6a-tetrahy dro-3 aH-cy clopenta [d] is oxazole-6- carboxylate (15)
[0068] 13 (from example 1, 9.2g, 0.024 mol) was dissolved in toluene (100 mL) and triethylamine (10. Og, 0.099 mol) and the reaction mixture was heated to 60-70 °C. 2- Ethyl-N-hydroxylbutanimidoyl chloride 14 (from example 2a, 14.8 g, 0.099 mol) in toluene (40 mL) was added to the above solution. A white solid
(triethylammonium chloride) was formed. After addition, the reaction was
monitored for completion by TLC ( PE: EtOAc=3 : 1. The reaction mixture was cooled to 20-25 °C, the precipitate was removed by filtration and the filter cake was washed with toluene (50 g). The organic filtrate was washed with water, brine, and dried over anhydrous Na2S04. The mixture was filtered and concentrated by rotary evaporation. The resulting residue was purified by silica gel flash column
chromatography using PE/EtOAc (30 : 1-4: 1, v/v) to give 15 as a white solid.
[0069] Yield : 10.0 g (85%).
[0070] MS (M+l ) :497.
[0071] ‘H NMR (400 MHz, CDC13) δ 11.29 (s, 1H), 8.55 (d, J= 6.4 Hz, 1H), 5.30 (dd, J= 9.1, 1.5 Hz, 1H), 4.53 (d, J= 4.8 Hz, 1H), 3.78 (s, 3H), 3.70 (d, J = 9.1 Hz, 1H), 3.25 (t, J= 5.4 Hz, 1H), 2.93 – 2.84 (m, 1H), 2.20 (dd, J= 7.6, 3.7 Hz, 2H), 1.87 – 1.60 (m, 4H), 1.49 (d, J= 5.0 Hz, 18H), 0.95 (t, J= 7.4 Hz, 3H), 0.87 (t, J= 7.5 Hz, 3H). (See attached Chart 2)
Example 3 3. (1^,2^,3^,4^)-ιη6ίΗγ1-3-(1-306ίαιηΐάο-2-6ίΗγ1 υίγ1)-4-(2,3- Ϊ8(ί^ί- butoxycarb -onyl) guanidino)-2-hydroxycyclopentanecarboxylate (16):

[0072] Compound 15 ( from example 2, 5.0 g, 10.08 mmol) and nickel chloride hexahydrate (2.5g, 10.5 mmol) were dissolved in methanol (40 mL). The green solution was cooled to -15 °C, while a suspension formed. Sodium borohydride
( 0.456 g, 12 mmol) was added to the reaction mixture at -10 to -5 °C (reaction is highly exothermic). A black suspension was formed along with gas evolution.
After complete addition of the sodium borohydride solution, the reaction mixture was stirred until TLC showed 15 was fully consumed. A solution of acetic
anhydride ( 15g, 0.13 mol) was added slowly and maintained the reaction temperature at 0-5 °C, the reaction mixture was stirred for 2-12 h at 0 °C (The black solution change into green solution ), The pH of the mixture was adjusted to ~9 by addition of 25% aq. ammonium hydroxide. The mixture was concentrated by rotary evaporator. The resulting residue was diluted with water (30 mL) and extracted with EtOAc (50 mL><3). The combined organic extracts were washed with water and brine and dried over anhydrous Na2S04. The mixture was filtered and concentrated by rotary evaporation. The residue was purified by flash chromatography using DCM/Methanol (100:0 to 100:2, v/v) to give 16 as a white solid. [0073] Yield: 3.8 g (71%).
[0074] MS (M+l ) : 543.
[0075] ‘H NMR (400 MHz, CDC13) δ 11.39 (s, 1H), 8.72 (d, J= 9.9 Hz, 1H), 8.59 (d, J= 8.5 Hz, 1H), 4.53 – 4.39 (m, 1H), 4.26 (d, J= 16.4 Hz, 2H), 3.96 (t, J = 10.2 Hz, 1H), 3.71 (s, 3H), 2.90 – 2.75 (m, 1H), 2.53 (dt, J= 13.6, 8.8 Hz, 1H), 2.10 (s, 3H), 2.03 (d, J= 6.3 Hz, 1H), 1.90 – 1.76 (m, 1H), 1.38 (dd, J= 73.9, 7.9 Hz, 18H), 1.25 (ddd, J= 15.2, 13.1, 7.3 Hz, 4H), 0.79 (t, J= 7.3 Hz, 3H), 0.75 (dd, J = 14.1, 6.9 Hz, 3H). (See attached Chart 3)
Example 4
4. (1^,2^,3^,4^)-3-(1-306ίαιηΐάο-2-6ίΗγ1 υίγ1)-4-(2,3- Ϊ8(ί^ί- butoxycarbonyl)gu -anidino)-2-hydroxycyclopentanecarboxylic acid (17)

[0076] To a mixture of compound 16 ( from example 3, 2.0 g, 3.69 mmol) in MeOH/THF (1 : 1, v/v, 12 mL), was added aq. NaOH (IN, 7 mL) at room
temperature. After completion of the reaction (monitored by TLC,
DCM:MeOH=10: l, the mixture was concentrated by rotary evaporation. The resulting solution was neutralized to pH 7 using ice-cold 1 N HCl aq. solution and quickly extracted with EtOAc twice. The combined organic extracts were washed with water, brine, and dried over anhydrous Na2S04. The mixture was filtered and the filtrate was concentrated by rotary evaporation. The resulting white foam was washed triturated by hexane, filtered, dried to give 17 as a white solid
[0077] Yield: 1.6 g (84%).
[0078] MS (M+l ) : 529 o
[0079] ‘H NMR (400 MHz, CDC13) δ 11.41 (s, 1H), 8.80 (d, J= 9.8 Hz, 1H), 8.62 (d, J= 8.3 Hz, 1H), 4.43 (dd, J= 23.3, 14.3 Hz, 2H), 4.00 (t, J= 9.8 Hz, 1H), 2.83 (s, 1H), 2.53 (dt, J= 16.9, 8.4 Hz, 1H), 2.14 (s, 3H), 1.91 (dd, J= 12.5, 6.0 Hz, 1H), 1.46 (dd, J = 30.1, 9.5 Hz, 18H), 1.47 – 1.14 (m, 6H), 0.97 – 0.69 (m, 6H). (See attached Chart 4)
Example 5
5. (1^,2^,3^,4^)-3-(1-306ίαιηΐάο-2-6ίΗγ1 υίγ1)-4^υαηΐ(1ΐηο-2- hydroxycyclopent -anecarboxylic acid (Peramivir I)

[0080] Compound 17 ( from example 4, 1.1 g, 2 mmol) was dissolved in aq. HC1 ( 6N, 6 mL, 36 mmol) at 0 °C. The mixture was stirred at room temperature overnight. The resulting solution was neutralized to pH 6-7 using ice-cold 1 N NaOH aq. solution. The mixture was concentrated to 1.5 ml by rotary evaporation. To this, methanol (20 mL) was added. The precipitate was filtered, and the filtrate was concentrated. The resulting white solid was recrystallized from
methanol/water (1 : 1, v/v) to give Peramivir I as a white solid.
[0081] Yield: 500 mg (73%).
[0082] MS (M+l ) : 329.
[0083] H NMR (400 MHz, D20) δ 4.21 (d, J= 10.6 Hz, 2H), 3.70 (dd, J= 14.6, 9.0 Hz, 1H), 2.57 (d, J= 4.8 Hz, 1H), 2.40 (dt, J= 17.7, 8.9 Hz, 1H), 2.14 – 2.01 (m, 1H), 1.81 (s, 3H), 1.75 – 1.58 (m, 1H), 1.31 (s, 3H), 0.78 (ddd, J= 21.6, 18.6, 6.8 Hz, 8H). (See attached Chart 5)
SYN

Method of synthesis
i. Lactan is dissolved I alcoholic solution, esterification by ring opening under HCl and acid catalysis.
ii. Compound formed is reacted with hydrochloride and tert-butyl dicarbonate.
iii. Reaction with oxammonium hydrochloride reactant salt in alcohol-carbonate system.
iv. Cycloaddition reaction takes place under base catalysis.
v. Hydrogenation in alcoholic solution to generate intermediate compound.
vi. Taking off BOC protect hydrochloride
vii. Two guanidine radicals are put in the reaction vessel, system made of alcohol, organic solvent, aqueous sodium hydroxide solution.
viii. Taking off BOC, product is obtained under organic acid.
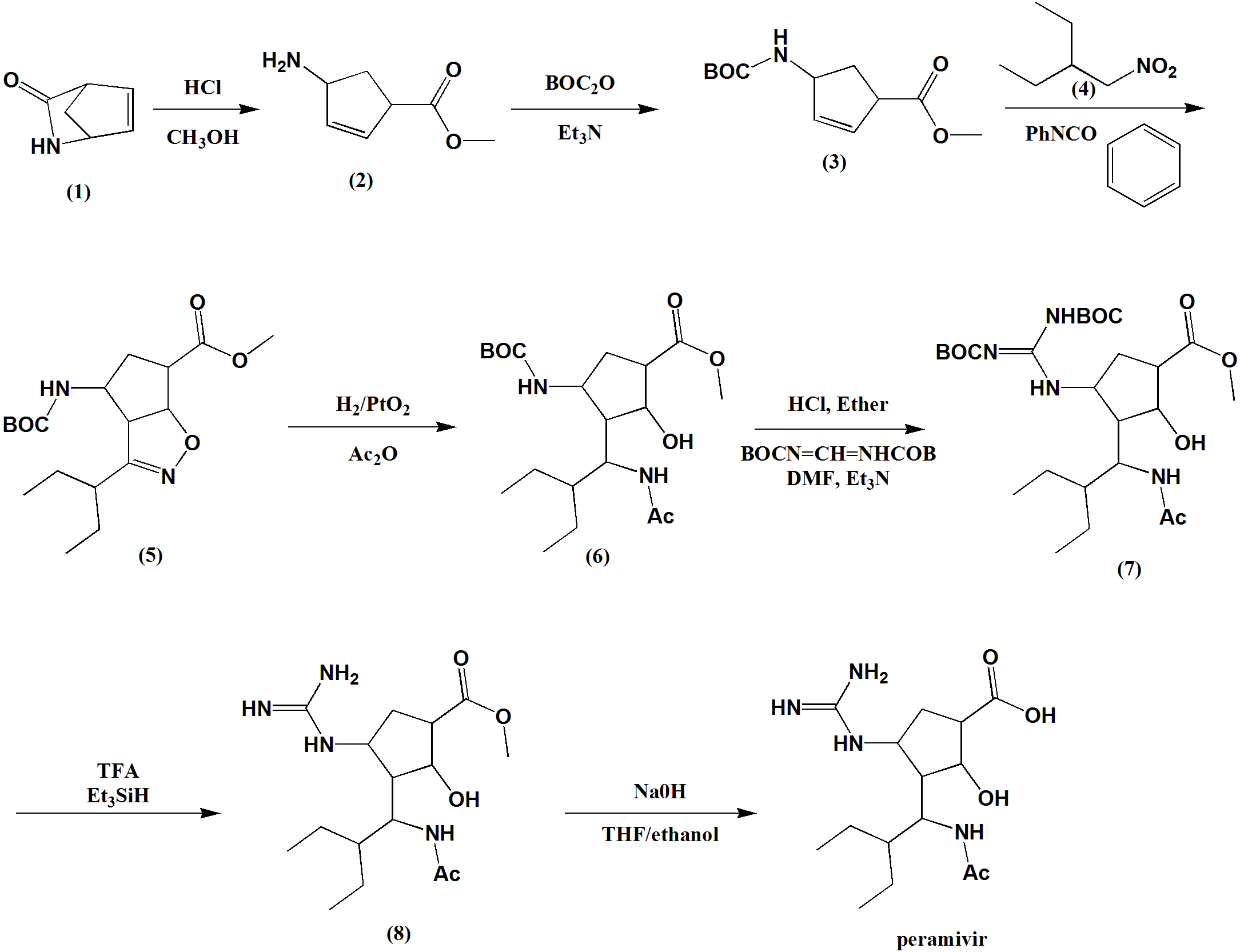
SYN
https://www.tandfonline.com/doi/abs/10.1080/00397911.2012.729279?journalCode=lsyc20
Abstract
An improved and convenient synthetic route for the synthesis of peramivir has been developed with a total 34% yield. The process was improved from previous methods in three key reaction steps including 1,3-dipolar cycloaddition, reductive ring cleavage of the isoxazoline, and incorporation of the peripheral guanidino group. First, an activated sodium hypochlorite (Cl% = 10%) was employed for the catalytic 1,3-dipolar cycloaddition, and 61–68% yields were obtained. Second, the NaBH4-NiCl2 was used as a new reducing reagent instead of the expensive catalyst PtO2. Most important, an innovative and environmentally friendly method of guanylation in the final step was developed using chloroformamidine hydrochloride as the amidino reagent, which avoided the use of the highlytoxic reagent of HgCl2 and made the process greener.
Supplemental materials are available for this article. Go to the publisher’s online edition of Synthetic Communications® to view the free supplemental file.
GRAPHICAL ABSTRACT


Peramivir – Synthetic Route 1

Synthetic Reference
Chand, Pooran; Kotian, Pravin L.; Dehghani, Ali; El-Kattan, Yahya; Lin, Tsu-Hsing; Hutchison, Tracy L.; Babu, Y. Sudhakar; Bantia, Shanta; Elliott, Arthur J.; Montgomery, John A. Systematic Structure-Based Design and Stereoselective Synthesis of Novel Multi-Substituted Cyclopentane Derivatives with Potent Anti-influenza Activity. Journal of Medicinal Chemistry. Volume 44. Issue 25. Pages 4379-4392. Journal. (2001).
Synthetic Reference
Jia, Fei; Hong, Juan; Sun, Ping-Hua; Chen, Jian-Xin; Chen, Wei-Min. Facile synthesis of the neuraminidase inhibitor peramivir. Synthetic Communications. Volume 43. Issue 19. Pages 2641-2647. Journal; Online Computer File. (2013).
Peramivir – Synthetic Route 3

Peramivir – Synthetic Route 4

Synthetic Reference
Li, Mingjie; Zhu, Quanming; Wang, Chunyan. A process for preparing peramivir. Assignee Shandong Luoxin Pharmacy Stock Co., Ltd., Peop. Rep. China. CN 103524383. (2014).
Synthetic Reference
Ge, Min; Li, Liang; Hu, Chunchen; Wang, Huaiqiu; Fu, Mingwei; Li, Lingchao. Synthesis of Peramivir via 3+2 ring closure. Assignee Acesys Pharmatech Ltd., Peop. Rep. China. CN 108997171. (2018).
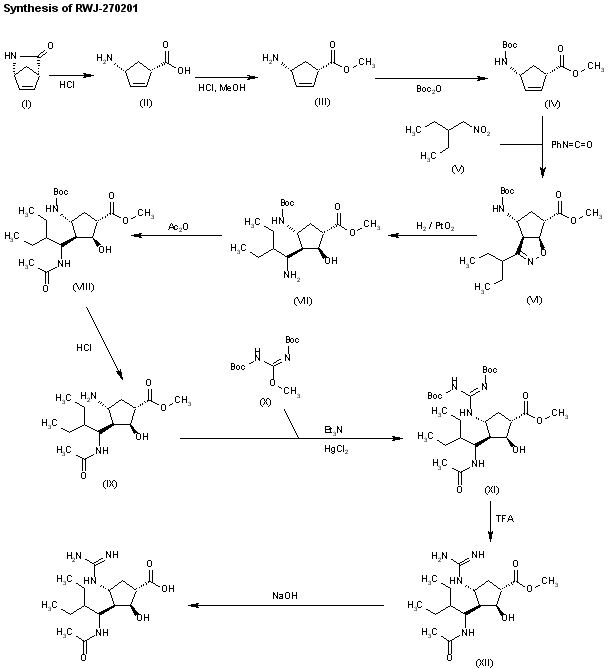
SYN 1
WO 9933781
Ring opening of cis-2-azabicyclo[2.2.1]hept-5-en-3-one (I) with HCl in refluxing methanol gives cis-4-amino-2-cyclopentene-1-carboxylic acid (II), which is esterified with HCl/methanol, yielding the methyl ester (III). The protection of (III) with tert-butoxycarbonyl anhydride and triethylamine in dichloromethane affords carbamate (IV), which is cyclized with 2-ethyl-1-nitrobutane (V) by means of phenyl isocyanate in refluxing benzene to give the cyclopenta-oxazoline (VI). Ring opening of (VI) by hydrogenation with H2 over PtO2 in HCl/methanol yields amine (VII), which is acylated with acetic anhydride and triethylamine in dichloromethane to afford acetamide (VIII). The deprotection of (VIII) with HCl in ethyl ether gives the cyclopentylamine (IX), which is condensed with N,N’-bis(tert-butoxycarbonyl)-O-methylisourea (X) by means of HgCl2 in DMF to yield the protected guanidino compound (XI). Deprotection of the guanidino group of (XI) with trifluoroacetic acid in dichloromethane affords the methyl ester (XII), which is finally hydrolyzed with NaOH in THF/methanol.
SYN 2
J Med Chem 2000,43(19),3482
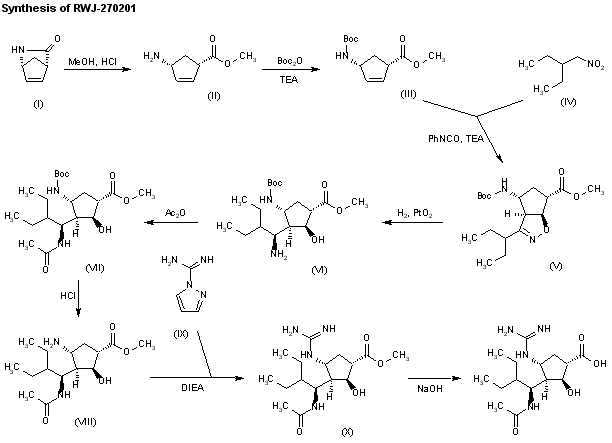
The stereospecific synthesis of BCX-1812(RWJ-270201) has been reported: Ring opening of (-)-2-azabicyclo[2.2.1]hept-5-en-3-one (I) with methanolic HCl gives the methyl ester (II), which is N-protected with Boc2O and TEA yielding the carbamate (III). Cyclization of (III) with 2-ethyl-1-nitrobutane (IV) by means of phenyl isocyanate and TEA affords the bicyclic compound (V) and other isomers. Compound (V) is isolated from the mixture and then hydrogenated with H2 over PtO2 in MeOH and a catalytic amount of HCl to provide amine (VI). Reaction of (VI) with acetic anhydride gives the acetamide (VII), which is Boc-deprotected with ethereal HCl yielding amine (VIII). The guanylation of (VIII) with pyrazolecarboxamidine hydrochloride (IX) and DIEA affords the guanidino derivative (X), which is finally hydrolyzed with NaOH to the desired (1S,2S,3R,4R,1’S)-diastereomer.
SYN 3
J Med Chem 2001,44(25),4379
The ring opening of (-)-2-azabicyclo [2,2,1]hept-5-en-3-one (I) with methanolic HCl gives the methyl ester (II), which is N-protected with Boc2O and TEA, yielding the carbamate (III). The cyclization of (III) with 2-ethyl-1-nitrobutane (IV) by means of phenyl isocyanate and TEA affords the bicyclic compound (V) along with other isomers that are separated by column chromatography. Compound (V) is hydrogenated with H2 over PtO2 in methanol to provide amine (VI), which is acylated with Ac2O, giving the acetamide (VII). N-Boc deprotection of (VII) by means of HCl in ethyl ether yields the amine (VIII), which is condensed with protected isothiourea (IX) by means of HgCl2 to afford the guanidine derivative (X). The hydrolysis of (X) with NaOH provides the carboxylic acid (XI), which is finally deprotected with trifluoroacetic acid to yield the target cyclopentanecarboxylic acid derivative.

References
- ^ Jump up to:a b c “Drug Approval Package: Rapivab (peramivir) Injection NDA #206426”. U.S. Food and Drug Administration (FDA). 16 January 2015. Retrieved 11 February 2020.
- ^ Thorlund K, Awad T, Boivin G, Thabane L (May 2011). “Systematic review of influenza resistance to the neuraminidase inhibitors”. BMC Infectious Diseases. 11 (1): 134. doi:10.1186/1471-2334-11-134. PMC 3123567. PMID 21592407.
- ^ “Peramivir authorized for Emergency use”. LifeHugger. 2009-12-04. Archived from the original on 2011-07-13. Retrieved 2009-12-04.
- ^ “Life-Saving H1N1 Drug Unavailable to Most”. CBS Evening News. Atlanta, GA, USA: CBS Interactive. 2009-10-19. Retrieved 2009-10-20.
- ^ “Emergency Use Authorization Granted For BioCryst’s Peramivir”. Reuters. 2009-10-24.
- ^ “Feds hand BioCryst $77M for anti-viral trial”. Fierce biotech. September 21, 2009.
- ^ “FDA Authorizes Emergency Use of Intravenous Antiviral Peramivir for 2009 H1N1 Influenza for Certain Patients, Settings”. Reuters. 2009-10-24.
- ^ Kohno S, Yen MY, Cheong HJ, Hirotsu N, Ishida T, Kadota J, et al. (November 2011). “Phase III randomized, double-blind study comparing single-dose intravenous peramivir with oral oseltamivir in patients with seasonal influenza virus infection”. Antimicrobial Agents and Chemotherapy. 55 (11): 5267–76. doi:10.1128/AAC.00360-11. PMC 3195028. PMID 21825298.
- ^ “BioCryst scraps $235M late-stage flu drug program backed by feds”. Fierce Biotech. November 8, 2012.
- ^ “BioCryst to File Peramivir NDA Supported by BARDA/HHS Funding”. Fierce Biotech. July 11, 2013.
- ^ Hurt, A.C. (9 June 2011). “Increased detection in Australia and Singapore of a novel influenza A(H1N1)2009 variant with reduced oseltamivir and zanamivir sensitivity due to a S247N neuraminidase mutation”. Eurosurveillance.
- ^ Hirschler, Ben (2011-06-10). “Swine flu starting to show resistance to drugs”. Reuters.
- ^ McKimm-Breschkin JL (January 2013). “Influenza neuraminidase inhibitors: antiviral action and mechanisms of resistance”. Influenza and Other Respiratory Viruses. 7 Suppl 1: 25–36. doi:10.1111/irv.12047. PMC 4942987. PMID 23279894.
- ^ Leang SK, Kwok S, Sullivan SG, Maurer-Stroh S, Kelso A, Barr IG, Hurt AC (March 2014). “Peramivir and laninamivir susceptibility of circulating influenza A and B viruses”. Influenza and Other Respiratory Viruses. 8 (2): 135–9. doi:10.1111/irv.12187. PMC 4186459. PMID 24734292.
- ^ “BioCryst Files Peramivir NDA for the Treatment of Influenza”(Press release). BioCryst Pharmaceuticals. 2013-12-20.
- ^ “Rapivab: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 11 February 2020.
External links
- “Peramivir”. Drug Information Portal. U.S. National Library of Medicine.
- “Peramivir: Requirements for Administration under EUA”. Lifehugger. Archived from the original on 2011-07-13.
- “CDC H1N1 Flu – Termination of the Emergency Use Authorization (EUA) of Medical Products and Devices”. U.S. Centers for Disease Control and Prevention (CDC).
- Clinical trial number NCT00957996 for “Safety Study of IV Peramivir in Hospitalized Subjects With Confirmed or Suspected Influenza” at ClinicalTrials.gov
- “A Novel Anti-viral Drug for Influenza, RAPIACTA Launch” (PDF). Shionogi & Co., Ltd. January 26, 2010. Archived from the original (PDF) on 2013-05-22.
 |
|
| Clinical data | |
|---|---|
| Trade names | Rapivab |
| AHFS/Drugs.com | Monograph |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Intravenous |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 100% (IV) |
| Elimination half-life | 7.7 to 20.8 hours (in patients with normal renal function) |
| Excretion | Kidney |
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII |
|
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C15H28N4O4 |
| Molar mass | 328.413 g·mol−1 |
| 3D model (JSmol) | |
| |
|
| Influenza (Flu) |
|---|
 |
////////////////PERAMIVIR, RWJ 270201, RWJ-270201, RWJ270201M, BCX 1812, RWJ 270201
/////////////////

PERAMIVIR
(1S,2S,3S,4R)-3-[(1S)-1-acetamido-2-ethyl-butyl]-4- (diaminomethylideneamino)-2-hydroxy-cyclopentane- 1-carboxylic acid
8th, APRIL 2013
In the wake of the growing bird flu outbreak, China’s SFDA will fast-track approvals of a flu treatment – not a vaccine – known as peramivir (peramivir sodium chloride injection). The drug is a neuraminidase inhibitor, which works by preventing the virus from moving from an infected cell to infect other cells. Guangzhou Nanxin Pharma has been approved to produce the medicine, and it expects to begin distributing the drug in 30 days. Other China pharmas have also filed for approval to make the drug or conduct human clinical trials
Peramivir is an experimental antiviral drug developed by BioCryst Pharmaceuticals for the treatment of influenza. It has been authorized for the emergency use of treatment of certain hospitalized patients with known or suspected 2009 H1N1 influenza.[1]
Peramivir is a neuraminidase inhibitor, acting as a transition-state analogue inhibitor of influenza neuraminidase and thereby preventing new viruses from emerging from infected cells.
The development of peramivir is supported by the US Department of Health and Human Services as part of the US government’s effort to prepare against the threat of an influenza pandemic.[2]
Peramivir is already available in Japan as RAPIACTA (R) and also available in South Korea as PERAMIFLU. Peramivir is currently the only intravenous option for treating swine flu. The drug is in Phase III studies in US.[3][4]
Use in treating Influenza A (H1N1) “Swine Flu”
In October 2009, it was reported that the experimental antiviral drug peramivir had been effective in treating serious cases of swine flu.[5] On October 23, the U.S. Food and Drug Administration (FDA) issued an Emergency Use Authorization for Peramivir, allowing the use of the drug in intravenous form for hospitalized patients only in cases where the other available methods of treatment are ineffective or unavailable;[6] for instance, if oseltamivir (Tamiflu) resistance develops and a person is unable to take Relenza via the inhaled route. The Emergency Use Authorization expired on June 23, 2010.
Biocryst also donated 1200 courses of treatment to the US department of Health and Human Services.[7]
According to a research report published in June 2011, a novel variant of swine flu has emerged in Asia with a genetic adaptation giving some resistance to Roche’s (ROG.VX) Tamiflu and GlaxoSmithKline’s (GSK.L) Relenza, the two mainstay drugs used to tackle the disease. There was no significant reduction in sensitivity to Peramivir. [8]
Initial treatment courses are for 5 to 10 days duration. Treatment beyond 10 days is permitted depending on clinical presentation such as critical illness (e.g., respiratory failure or intensive care unit admission), continued viral shedding or unresolved clinical influenza illness.[9]
- “Peramivir authorized for Emergency use”. LifeHugger. 2009-12-04. Retrieved 2009-12-04.
- “HHS Pursues Advance Development of New Influenza Antiviral Drug” (Press release). USDepartment of Health and Human Services. 2007-01-04. Retrieved 2007-05-25.
- “Evaluation of the Efficacy and Safety of Peramivir in Subjects With Uncomplicated Acute Influenza”.National Institutes of Health. 2007-03-16. Retrieved 2007-05-25.
- “Evaluation of the Efficacy and Safety of Peramivir in Adults With Acute Serious or Potentially Life-Threatening Influenza”. National Institutes of Health. 2007-03-28. Retrieved 2007-05-25.
- “Life-Saving H1N1 Drug Unavailable to Most”. CBS Evening News (Atlanta, GA, USA: CBS Interactive). 2009-10-19. Retrieved 2009-10-20.
- “Emergency Use Authorization Granted For BioCryst’s Peramivir”. Reuters. 2009-10-24.
- “FDA Authorizes Emergency Use of Intravenous Antiviral Peramivir for 2009 H1N1 Influenza for Certain Patients, Settings”. Reuters. 2009-10-24.
- Hirschler, Ben (2011-06-10). “Swine flu starting to show resistance to drugs”. Reuters.
- “Peramivir: Dosage and Administration”. LifeHugger. 2009-12-04. Retrieved 2009-12-04.
Gilead Submits NDA to FDA for Sofosbuvir for the Treatment of Hepatitis C

Sofosbuvir
Isopropyl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyl-tetrahydrofuran-2-yl]methoxy-phenoxy-phosphoryl]amino]propanoate

9 APRIL 2013
Gilead Sciences today announced that the company has submitted a New Drug Application (NDA) to the U.S. Food and Drug Administration (FDA) for approval of sofosbuvir, a once-daily oral nucleotide analogue for the treatment of chronic hepatitis C virus (HCV) infection. The data submitted in this NDA support the use of sofosbuvir and ribavirin (RBV) as an all-oral therapy for patients with genotype 2 and 3 HCV infection, and for sofosbuvir in combination with RBV and pegylated interferon (peg-IFN) for treatment-naïve patients with genotype 1, 4, 5 and 6 HCV infection.
Chronic HCV infection affects up to four million Americans, particularly individuals born between 1946 and 1964. The disease is the leading cause of liver cancer and liver transplantation in the United States. Treatment for HCV currently includes 24-48 weeks of therapy with peg-IFN, which has to be injected and is associated with significant side effects, leaving some patients unable to complete therapy. If approved, sofosbuvir would shorten HCV therapy to 12 to 16 weeks, and depending on the genotype, would either eliminate or reduce the duration of peg-IFN injections.
“Current therapies are not suitable for large numbers of patients with HCV infection, and are challenging to take and tolerate,” said John C. Martin, PhD, Chairman and Chief Executive Officer of Gilead Sciences. “Sofosbuvir’s antiviral potency, safety profile and once-daily administration have the potential to improve cure rates by simplifying and shortening therapy for patients with this disease.”
The sofosbuvir NDA is supported primarily by data from four phase 3 studies, NEUTRINO, FISSION, POSITRON and FUSION, in which 12 or 16 weeks of sofosbuvir-based therapy was found to be superior or non-inferior to currently available treatment options or historical controls, based on the proportion of patients who had a sustained virologic response (HCV undetectable) 12 weeks after completing therapy (SVR12). Patients who achieve SVR12 are considered cured of HCV.
Gilead plans to file for regulatory approval of sofosbuvir in other geographies, including the European Union, in the second quarter of 2013. The European Medicines Agency (EMA) has accepted Gilead’s request for accelerated assessment for sofosbuvir, a designation that is granted to new medicines of major public health interest. Accelerated assessment could shorten the EMA’s review time of sofosbuvir by two months. Granting of accelerated assessment does not guarantee a positive opinion from the CHMP or approval by the European Commission.
Sofosbuvir (formerly PSI-7977 or GS-7977) is an experimental drug candidate for the treatment of hepatitis C.[1] It was discovered at Pharmasset and then acquired for development by Gilead Sciences. It is currently in Phase III clinical trials.[2]
Sofosbuvir is a prodrug that is metabolized to the active antiviral agent 2′-deoxy-2′-α-fluoro-β-C-methyluridine-5′-monophosphate.[3]
Sofosbuvir is a nucleotide analogue inhibitor of the hepatitis C virus (HCV) polymerase.[4] The HCV polymerase or NS5B protein is a RNA-dependent RNA polymerase critical for the viral cycle.
Sofosbuvir is being studied in combination with pegylated interferon and ribavirin, with ribavirin alone, and with other direct-acting antiviral agents.[5] It has shown excellent clinical efficacy when used either with pegylated interferon/ribavirin or in interferon-free combinations. In particular, combinations of sofosbuvir with NS5A inhibitors, such as daclatasvir or GS-5885, have shown sustained virological response rates of up to 100% in people infected with HCV.[6]
- Sofia, M. J.; Bao, D.; Chang, W.; Du, J.; Nagarathnam, D.; Rachakonda, S.; Reddy, P. G.; Ross, B. S. et al. (2010). “Discovery of a β-d-2′-Deoxy-2′-α-fluoro-2′-β-C-methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis C Virus”. Journal of Medicinal Chemistry 53 (19): 7202–7218. doi:10.1021/jm100863x. PMID 20845908. edit
- “PSI-7977″. Gilead Sciences.
- Murakami, E.; Tolstykh, T.; Bao, H.; Niu, C.; Steuer, H. M. M.; Bao, D.; Chang, W.; Espiritu, C. et al. (2010). “Mechanism of Activation of PSI-7851 and Its Diastereoisomer PSI-7977″. Journal of Biological Chemistry 285 (45): 34337–34347. doi:10.1074/jbc.M110.161802. PMC 2966047. PMID 20801890. edit
- Alejandro Soza (November 11, 2012). “Sofosbuvir”. Hepaton.
- Tom Murphy (November 21, 2011). “Gilead Sciences to buy Pharmasset for $11 billion”. Bloomberg Businessweek.
- http://www.gilead.com/pr_1757156
- AASLD: PSI-7977 plus Ribavirin Can Cure Hepatitis C in 12 Weeks without Interferon. Highleyman, L. HIVandHepatitis.com. 8 November 2011.
- Nucleotide Polymerase Inhibitor Sofosbuvir plus Ribavirin for Hepatitis C. Gane, E et al. New England Journal of Medicine 368:3444. January 3, 2013.
- CROI 2013: Sofosbuvir + Ledipasvir + Ribavirin Combo for HCV Produces 100% Sustained Response. Highleyman, L. HIVandHepatitis.com. 4 March 2013.
Lundbeck has presented promising data on Brintellix, its recently-filed investigational antidepressant co-developed with Takeda.

vortioxetine
9 APRIL 2013
Lundbeck has presented promising data on Brintellix, its recently-filed investigational antidepressant co-developed with Takeda.
Vortioxetine (code name Lu AA21004) is an experimental drug currently under development by Lundbeck and Takeda for the treatment of major depressive disorder(MDD) and generalized anxiety disorder (GAD).Commercial name chosen is Brintellix.
Regulatory approval for the treatment of MDD for the European market has been filed in September 2012, for the United States in October 2012, and filing for Canada should follow. Filing for the Japanese market is expected in 2013
The Danish drugmaker announced results for the REVIVE study which compared Brintellix (vortioxetine) with Servier’s Valdoxan (agomelatine), Servier’s in adults with major depression (MDD) who changed antidepressant after an inadequate response to commonly-prescribed selective serotonin reuptake inhibitors (SSRIs) or serotonin–norepinephrine reuptake inhibitors (SNRIs). Lundbeck noted that as one of the newest antidepressants, agomelatine was chosen as a comparator because of its different mode of action from conventional SSRI/SNRI therapies.
Lundbeck noted that few randomised, double-blind trials looking at MDD patients who were unresponsive to first-line antidepressants have been conducted and “this is one of these few studies which also shows a significant difference between treatments.” On the primary efficacy endpoint for REVIVE, Brintellix was statistically significantly superior to agomelatine by 2.2 points on the Montgomery–Asberg Depression Rating Scale (MADRS), a ten-item questionnaire used to measure severity of the disorder.
Brintellix is under review on both sides of the Atlantic and is one of three new products, Lundbeck hopes to launch this year. The other two, which are already approved in some territories, are its once-monthly version of Abilify (aripiprazole) for schizophrenia and the alcohol dependence treatment Selincro (nalmefene); indeed, Lundbeck also presented new data on the later from three Phase III studies that “consistently show a significant reduction in alcohol consumption” in patients with high-risk drinking levels.
Bayer PAH drug Riociguat gets priority review at FDA

RIOCIQUAT
CAS NO 625115-55-1
Methyl N-[4,6-Diamino-2-[1-[(2-fluorophenyl)methyl]-1H-pyrazolo[3,4-b]pyridin-3-yl]-5-pyrimidinyl]-N-methyl-carbaminate
9 APRIL2013
Bayer has been boosted by the news that regulators in the USA are fast-tracking the German group’s investigational pulmonary arterial hypertension riociguat.
The US Food and Drug Administration has granted priority review to the New Drug Application for riociguat, which Bayer filed in February on both sides of the Atlantic for PAH and a related condition, inoperable chronic thromboembolic pulmonary hypertension (CTEPH). The FDA bestows a priority review on medicines that offer major advances in care or that provide a treatment where no adequate therapy exists. The agency aims to complete its assessment within eight months from the submission of the NDA, rather than the standard 12 months.
Riociguat (BAY 63-2521) is a novel drug that is currently in clinical development by Bayer. It is a stimulator of soluble guanylate cyclase (sGC). At the moment Phase III clinical trialsinvestigate the use of riociguat as a new approach to treat two forms of pulmonary hypertension (PH): chronic thromboembolic pulmonary hypertension (CTEPH) andpulmonary arterial hypertension (PAH). Riociguat constitutes the first drug of a novel class of sGC stimulators
The submissions are based on two Phase III studies and riociguat, the first member of a novel class of compounds called stimulators of soluble guanylate cyclase (sGC), met its primary endpoint in both trials, a change in exercise capacity after 12- or 16 weeks respectively. The drug was generally well tolerated, with a good safety profile.

If approved, riociguat would be going up against Actelion’s Tracleer (bosentan) and Gilead Sciences/GlaxoSmithKline’s Letairis/Volibris (ambrisentan). Actelion, which has dominated the PAH market, has already filed its follow-up to Tracleer, Opsumit (macitentan).
Links
FDA Approves Diclegis for Pregnant Women Experiencing Nausea and Vomiting

doxylamine

pyridoxine
8 april 2013
The U.S. Food and Drug Administration today approved Diclegis (doxylamine succinate and pyridoxine hydrochloride) to treat pregnant women experiencing nausea and vomiting. Diclegis is a delayed-release tablet intended for women who have not adequately responded to conservative management of nausea and vomiting during pregnancy, such as dietary and lifestyle modifications.
These modifications include eating several small meals instead of three large meals, eating bland foods that are low in fat and easy to digest and avoiding smells that can trigger nausea. “Many women experience nausea and vomiting during pregnancy, and sometimes these symptoms are not adequately managed through recommended changes in diet and lifestyle,” said Hylton V. Joffe, M.D., M.M.Sc., director of the Division of Reproductive and Urologic Products in the FDA’s Center for Drug Evaluation and Research.
“Diclegis is now the only FDA-approved treatment for nausea and vomiting due to pregnancy, providing a therapeutic option for pregnant women seeking relief from these symptoms.” Diclegis was studied in 261 women experiencing nausea and vomiting due to pregnancy. Study participants in the clinical trial were at least 18 years old and had been pregnant for at least 7 weeks and up to 14 weeks. Women were randomly assigned to receive two weeks of treatment with Diclegis or a placebo.
The study results showed that women taking Diclegis experienced greater improvement in nausea and vomiting than those taking placebo. Additionally, observational (epidemiological) studies have shown that the combination of active ingredients in Diclegis does not pose an increased risk of harm to the fetus. Diclegis is taken daily. Tablets must be taken whole on an empty stomach. The recommended starting dose is two tablets taken at bedtime.
If symptoms are not adequately controlled, the dose can be increased to a maximum recommended dose of four tablets daily (one in the morning, one mid-afternoon and two at bedtime). Nausea and vomiting due to pregnancy usually improve after the first trimester. Health care professionals should reassess their patients for continued need for Diclegis as pregnancy progresses. Drowsiness or sleepiness, which can be severe, is the most common side effect reported by women taking Diclegis.
Women should avoid using Diclegis when engaging in activities requiring mental alertness, such as driving or operating heavy machinery, until cleared to do so by their health care provider.
Diclegis is marketed by Duchesnay Inc., based in Blainville, Québec, Canada.
AURVEDA-Shatavari, wonder herb for women’s overall health and vitality .

Asparagus racemosus (Satavar, Shatavari, or Shatamull) is a species of asparagus common throughout Sri Lanka, India and the Himalayas. It grows one to two metres tall and prefers to take root in gravelly, rocky soils high up in piedmont plains, at 1,300–1,400 metres elevation).It was botanically described in 1799. Due to its multiple uses, the demand for Asparagus racemosus is constantly on the rise. Due to destructive harvesting, combined with habitat destruction, and deforestation, the plant is now considered ‘endangered’ in its natural habitat.
Asparagus racemosus (Shatavari) is recommended in Ayurvedic texts for the prevention and treatment of gastric ulcers, dyspepsia and as a galactogogue. A. racemosus has also been used successfully by some Ayurvedic practitioners for nervous disorders.
Shatawari has different names in the different Indian languages, such as Shatuli, Vrishya and other terms. In Nepal it is called Kurilo. The name Shatawari means “curer of a hundred diseases” (shat: “hundred”; vari: “curer”).
Shatavari is mentioned under six important rasayanas in ayurveda. Rasayanas are those plant drugs which promote general well being of an individual by increasing cellular vitality or resistance. This bitter sweet herb is especially used in Ayurveda to correct Pitta dosha imbalance.
Shatavari: This powerful herb strengthens the female organs, enhancing fertility and sexual vitality. Works on the reproductive, respiratory, circulatory and digestive systems. Excellent herb to cool Pitta and to bring moisture and lustre to the skin, while nourishing the cells and tissues. Shatavari soothes and protects the dry and inflamed membranes of the lungs, stomach, kidneys and sexual organs. Found in our Pitta spicebodhi herbal body bar and our spicebodhimama massage oil.
Asparagamine A, a polycyclic alkaloid with antitumor activity against a variety of cell lines was isolated from the dried roots and subsequently synthesized to allow for the construction of analogs.
Two new steroidal saponins, shatavaroside A and shatavaroside B together with a known saponin, filiasparoside C, were isolated from the roots of Asparagus racemosus.
Five steroidal saponins, shatavarins VI-X, together with five known saponins, shatavarin I (or asparoside B), shatavarin IV (or asparinin B), shatavarin V, immunoside and schidigerasaponin D5 (or asparanin A), have been isolated from the roots of Asparagus racemosus.
Isoflavone, 8-methoxy-5,6,4′-trihydroxyisoflavone 7-O-beta-D-glucopyranoside
Asparagus racemosus is an important medicinal plant of tropical and subtropical India. Its medicinal usage has been reported in the Indian and British Pharmacopoeias and in traditional systems of medicine such as Ayurveda, Unani and Siddha. It is mainly known for its phytoestrogenic properties. In Ayurveda, Asparagus racemosus has been described as a rasayana herb and has been used extensively as an adaptogen to increase the non-specific resistance of organisms against a variety of stresses. Besides use in the treatment of diarrhoea and dysentery, the plant also has antioxidant, immunostimulant, anti-dyspepsia and antitussive effects.”
The roots are used in Ayurvedic medicine, following a regimen of processing and drying. It is generally used as a uterine tonic, as agalactogogue (to improve breast milk), in hyperacidity, and as a general health tonic.
The reputed adaptogenic effects of Shatavari may be attributed to its concentrations of saponins,known as Shatavarins.
conclusion
Shatavari or Asparagus racemosus is a popular herb aptly called the “Female Health Formula”. Asparagus racemosus is the most commonly used Asparagus species in ayurveda and indigenous medicine in India. The plant is called shatawar in Hindi and in Sanskrit this plant is called shatavari which means ‘able to have one hundred husbands’. In Ayurveda this amazing herb is known as the “queen of herbs” because it promotes love and positive emotions and is very useful in strengthening the reproductive system of women. It is also a very important herb for women’s overall health and vitality
.
Pharmacyclics Announces Third Breakthrough Therapy Designation for Ibrutinib from the U.S. Food and Drug Administration

IBRUTINIB
1-[(3R)-3-[4-amino-3-(4-phenoxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]piperidin-1-yl]prop-2-en-1-one
SUNNYVALE, Calif., April 8, 2013
Pharmacyclics, Inc. announced today that the U.S. Food and Drug Administration (FDA) has granted an additional Breakthrough Therapy Designation for the investigational oral agent ibrutinib as monotherapy for the treatment of chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) patients with deletion of the short arm of chromosome 17 (deletion 17p). Patients harboring a deletion within chromosome 17 generally have poor response to chemoimmunotherapy and have limited treatment options. The presence of deletion 17p is one of the worst prognostic factors in patients with CLL.
In February 2013, FDA granted Breakthrough Therapy Designations for ibrutinib as a monotherapy for the treatment of patients with relapsed or refractory mantle cell lymphoma (MCL) and as a monotherapy for the treatment of patients with Waldenstrom’s macroglobulinemia (WM), both of which are also B-cell malignancies. Ibrutinib is jointly being developed by Pharmacyclics and Janssen for treatment of B-cell malignancies.
The Breakthrough Therapy Designation is intended to expedite the development and review of a potential new drug for serious or life-threatening diseases where “preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development.” The designation of a drug as a Breakthrough Therapy was enacted as part of the 2012 Food and Drug Administration Safety and Innovation Act. Pharmacyclics, together with Janssen, is working with the FDA to determine the implications of this Breakthrough Therapy Designation to the ongoing and planned development and the FDA filing requirements for the use of ibrutinib in CLL patients with deletion 17p.
The FDA Breakthrough Therapy Designation for ibrutinib in CLL patients with deletion 17p was based on data from pre-clinical and clinical studies where ibrutinib as a monotherapy was used to treat patients with this disease. Ibrutinib has the potential to improve the outcome in this serious and life-threatening disease which has a poor prognosis. In addition, Pharmacyclics and Janssen have recently initiated a Phase II study of ibrutinib in patients with CLL deletion 17p, RESONATE™ -17, which is a single-arm, open-label, multi-center trial using ibrutinib as a monotherapy in patients who have deletion 17p and who did not respond to or relapsed after at least one prior CLL treatment (a high unmet need population). The primary endpoint of the study will be overall response rate. This global study opened this year and Pharmacyclics plans to enroll 111 patients worldwide.
About Chronic Lymphocytic Leukemia
Chronic Lymphocytic Leukemia (CLL) is a slow-growing blood cancer that starts in the white blood cells (lymphocytes), most commonly from B-cells. CLL is the second most common adult leukemia. Approximately 16,000 patients in the US are diagnosed each year with CLL. The prevalence of CLL is approximately 113,000 in the US. The disease is a chronic disease of the elderly with an average survival of about 5 years. Patients commonly receive multiple lines of treatment over the course of their disease.
In CLL the genetic mutation 17p deletion occurs when the short arm of chromosome 17 is missing. Del 17p is associated with abnormalities of a key tumor suppressor gene, TP53, which results in poor response to chemoimmunotherapy and worse treatment outcomes. It occurs in about 7% of treatment naive CLL patients and is estimated to be approximately 20% to 40% of relapsed or refractory patients harboring the mutation.
About Ibrutinib
Ibrutinib , previously publicly known as PCI-32765, is an experimental drug candidate for the treatment of various types of cancer. It was first synthesized at Celera Genomics as a selective inhibitor of Bruton’s tyrosine kinase (Btk).It was later discovered to have anti-lymphoma properties in vivo by scientists at Pharmacyclics, Inc.Ibrutinib is currently under development by Pharmacyclics, Inc and Johnson & Johnson‘sJanssen Pharmaceutical division for chronic lymphocytic leukemia, mantle cell lymphoma,diffuse large B-cell lymphoma, and multiple myeloma. It also has potential effects against autoimmune arthritis.
Janssen Biotech, Inc. and Pharmacyclics entered a collaboration and license agreement in December 2011 to co-develop and co-commercialize ibrutinib. Ibrutinib was designed to specifically target and selectively inhibit an enzyme called Bruton’s tyrosine kinase (BTK). BTK is a key mediator of at least three critical B-cell pro-survival mechanisms occurring in parallel – regulation of apoptosis, adhesion, and cell migration and homing. Through these multiple signals, BTK regulation helps to direct malignant B-cells to lymphoid tissues, thus allowing access to a micro environment necessary for survival.
The effectiveness of ibrutinib alone or in combination with other treatments is being studied in several B-cell malignancies, including chronic lymphocytic leukemia/small lymphocytic lymphoma, mantle cell lymphoma, diffuse large B-cell lymphoma, follicular lymphoma, Waldenstrom’s macroglobulinemia and multiple myeloma. To date five Phase III trials have been initiated with ibrutinib and a total of 26 trials are currently registered on www.clinicaltrials.gov.
About Pharmacyclics
Pharmacyclics® is a clinical-stage biopharmaceutical company focused on developing and commercializing innovative small-molecule drugs for the treatment of cancer and immune mediated diseases. Our mission and goal is to build a viable biopharmaceutical company that designs, develops and commercializes novel therapies intended to improve quality of life, increase duration of life and resolve serious unmet medical healthcare needs; and to identify promising product candidates based on scientific development and administrational expertise, develop our products in a rapid, cost-efficient manner and pursue commercialization and/or development partners when and where appropriate.
Presently, Pharmacyclics has three product candidates in clinical development and several preclinical molecules in lead optimization. The Company is committed to high standards of ethics, scientific rigor, and operational efficiency as it moves each of these programs to viable commercialization.
The Company is headquartered in Sunnyvale, California and is listed on NASDAQ under the symbol PCYC. To learn more about how Pharmacyclics advances science to improve human healthcare visit at http://www.pharmacyclics.com.
FDA Extends Delcath’s Melblez Cancer System Review

Melphalan hydrochloride (trade name Alkeran) is a chemotherapy drug belonging to the class of nitrogen mustard alkylating agents.
An alkylating agent adds an alkyl group (CnH2n+1) to DNA. It attaches the alkyl group to the guanine base of DNA, at the number 7 nitrogen atom of the imidazole ring.
Otherwise known as L-Phenylalanine Mustard, or L-PAM, melphalan is a phenylalaninederivative of mechlorethamine.
AYURVEDA HERBS-HOLY BASIL (Ocimum sanctum)

HOLY BASIL (Ocimum sanctum): Also known as Tulsi, this plant is actually considered sacred by many people in India. As such, it can be found growing in temple gardens, where the rich fragrance opens respiratory passages and some say, help the spirit soar.
Tulsi is one of the most sacred plants of India. Basil opens the heart and mind, bestowing the energy of love and devotion. Basil strengthens the immune system, increasing prana or life force and improving the memory. A nerve tonic, improving absorption and strengthening the nerve tissue, also used externally for various skin conditions. This plant is found in most East Indian households as it absorbs positive ions, energizes negative ions, and liberates ozone from the suns rays. Found in our spicenlightenment Vata aromatherapy Candle and spicebodhi Vata body bar.
Holy Basil’s key compounds, including eugenol and caryophyllene, are similar to those found in oregano (Origanum vulgare) and it shares the anti-inflammatory, antipyretic, and analgesic actions typical of the oregano family (Padalia RC, Verma RS. Nat Prod Res. 2011;25(6):569-75. Godhwani S, et al. J Ethnopharmacol. 1987;21:153-63).
This plant is also native to West Africa. In Sierra Leone, it is called ‘Fever Plant.’ The various fixed oil compounds found in the plant have shown extensive antimicrobial and antifungal activity against a variety of pathogens including Escherichia coli and Candida albicans. In classical Ayurveda, Holy Basil was used as an anti-tussive, to clear “excess dampness in the lungs.” Recent human trials have validated this, the data showing that this herb can increase lung capacity as well as reduce labored breathing.
It has also been shown to significantly reduce several measures of stress in generalized anxiety disorder (GAD) patients.
Holy basil can be taken in capsule, tea and in liquid forms. It is dispensed in 600-700 mg doses, twice daily, before meals. Allow 2-4 weeks for optimal results.
New combination therapy cures patient with advanced ovarian cancer

A novel ovarian cancer treatment made from tumour cells has cured a woman in the US with an advanced form of the disease, scientists at the Perelman School of Medicine at the University of Pennsylvania have announced.
During a preliminary trial of the two-step immunotherapy, the patient achieved complete remission, while seven other women had no measurable disease at the end of the study.
The therapy includes a personalised immune cell vaccination made from the patients’ live tumour cells and adoptive T-cell therapy.
Both treatments are given in conjunction with Avastin (bevacizumab), a drug developed by Roche that controls the blood vessel growth that feeds tumours.
The second step in the study involved the isolation of immune cells, known as dendritic cells, from the patients’ blood through a process called apheresis, similar to the process used for blood donation.
Announcing its findings at the American Association of Cancer Research (AACR) Annual Meeting in Washington DC on Saturday, the research team reported that in the study of 31 patients, vaccination therapy alone showed a 61% clinical benefit, and the combination of both therapies benefited around 75% of participants.
Ovarian cancer is the fifth leading cause of cancer-related deaths among women in the US, taking the lives of 14,000 people each year.
Lead author of the study Lana Kandalaft said; “Given these grim outcomes, there is definitely a vast unmet need for the development of novel, alternate therapies.”
“This is the first time such a combination immunotherapy approach has been used for patients with ovarian cancer, and we believe the results are leading us toward a completely new way to treat this disease.”
The vaccine trial is still open to accrual to test new combinatorial strategies.
