
Home » Posts tagged 'tyrosine kinase inhibitor'
Tag Archives: tyrosine kinase inhibitor
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Deulorlatinib


Deulorlatinib
CAS 2131126-33-3
MFC21H162H3FN6O2, MW 409.4 g/mol

- (10R)-7-Amino-12-fluoro-10,15,16,17-tetrahydro-10,16-dimethyl-2-(methyl-d3)-15-oxo-2H-4,8-methenopyrazolo[4,3-h][2,5,11]benzoxadiazacyclotetradecine-3-carbonitrile
- 2H-4,8-Methenopyrazolo[4,3-h][2,5,11]benzoxadiazacyclotetradecine-3-carbonitrile, 7-amino-12-fluoro-10,15,16,17-tetrahydro-10,16-dimethyl-2-(methyl-d3)-15-oxo-, (10R)-
(10R)-7-amino-12-fluoro-2-(2H3)methyl-10,16-dimethyl15-oxo-10,15,16,17-tetrahydro-2H-8,4-
(metheno)pyrazolo[4,3-h][2,5,11]benzoxadiazacyclotetradecine-3-carbonitrile
tyrosine kinase inhibitor, antineoplastic, 7PW3UT8C9B, TGRX 326, TGRX-326
Deulorlatinib is an orally bioavailable inhibitor of the receptor tyrosine kinases anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1), with potential antineoplastic activity. Upon oral administration, deulorlatinib targets, binds to and inhibits the activity of ALK and ROS1, which leads to the disruption of ALK- and ROS1-mediated signaling and the inhibition of cell growth in ALK- and ROS1-expressing tumor cells. ALK belongs to the insulin receptor superfamily and plays an important role in nervous system development. ALK is not expressed in healthy adult human tissue but ALK dysregulation and gene rearrangements are associated with a variety of tumor cell types. ROS1, overexpressed in certain cancer cells, plays a key role in cell growth and survival of cancer cells.
- TGRX-326 Chinese Phase III for Advanced Non-small Cell Lung Cancer (NSCLC)CTID: NCT06082635Phase: Phase 3Status: Active, not recruitingDate: 2025-05-18
- TGRX-326 Pharmacokinetic Drug InteractionCTID: NCT06294561Phase: Phase 1Status: CompletedDate: 2024-06-27
- TGRX-326 Chinese Phase I for Advanced Non-small Cell Lung Cancer (NSCLC)CTID: NCT05441956Phase: Phase 1Status: Active, not recruitingDate: 2025-05-18
- TGRX-326 Chinese Phase II for Advanced Non-small Cell Lung Cancer (NSCLC)CTID: NCT05955391Phase: Phase 2Status: Active, not recruitingDate: 2025-05-18
SYN
WO 2017/148325 A1
syn
https://patentscope.wipo.int/search/en/detail.jsf?docId=US348430040&_cid=P11-MKG9AH-82468-1



Example 6: Synthesis of (10R)-7-amino-12-fluoro-2-(methyl-d3)-10,16-dimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]benzoxadiazacyclotetradecine-3-carbonitrile (the Compound of Formula (A))



| To a 250 mL three-necked flask equipped with magnetic stirring were added the compound of formula (J) (7.0 g, 42.2 mmol) and anhydrous dichloromethane (120 mL), and stirred until the solution became clear. The compound of formula (H) (8.77 g, 46.4 mmol) and then triethylamine (4.69 g, 46.4 mmol) were successively added. The mixture was stirred at room temperature under nitrogen atmosphere for 30 minutes to give a pale yellow clear solution for further use. |
| Alkylation of the Compound of Formula (E-a) with the Compound of Formula (F) to Form the Compound of Formula (D-a): |
| To another 250 mL three-necked flask equipped with magnetic stirring were added the compound of formula (E-a) (11.2 g, 59.3 mmol) and acetonitrile (200 mL), and cesium carbonate (25.7 g, 79.0 mmol) was added with stirring. The mixture was heated to 50° C. under nitrogen atmosphere, and stirred at this temperature for 30 min. The above-mentioned solution of the compound of formula (F) in acetonitrile was slowly added dropwise at 50° C. over 10 minutes. After the dropwise addition was completed, the mixture was reacted with stirring at this temperature for 2 hours. By TLC (DCM:MeOH=20:1) and HPLC monitoring, the reaction was completed. After cooling to room temperature, the reaction was quenched by adding water (200 mL). The reaction solution was diluted with ethyl acetate (300 mL), stirred for 5 minutes, and then filtered through Celite to remove insoluble solids. The filter cake was washed with ethyl acetate (50 mL). The organic layer was separated from the filtrate, and the aqueous phase was extracted with ethyl acetate (60 mL×2). The organic phases were combined, washed with a saturated aqueous solution of sodium carbonate (100 mL×3) and then saturated brine (60 mL), dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated to dryness under reduced pressure to give 17.5 g of a brown solid in a yield of 90.1% and a purity (HPLC) of >85% (ee>95%). LC-MS (APCI): m/z=390.1 (M+1) +. |
| Introduction of Boc Protecting Group into the Compound of Formula (D-a) to Form the Compound of Formula (C): |
| To a 250 mL single-necked flask equipped with magnetic stirring were added the compound of formula (D-a) (17.5 g, 35.8 mmol) and dichloromethane (200 mL), and stirred until the solution became clear. Triethylamine (14.5 g, 143.2 mmol) and then DMAP (850 mg, 7.2 mmol) were successively added. Boc2O (23.4 g, 107.4 mmol) was slowly added dropwise, and the mixture was reacted with stirring at room temperature under nitrogen atmosphere overnight. By TLC (DCM:MeOH=20:1) and HPLC monitoring, the reaction was completed. The reaction solution was evaporated under reduced pressure to remove the solvent, and the residue was purified by silica gel column chromatography (EA/PE=0-35%) to give 15.4 g of a white solid in a yield of 62.4% and a purity (HPLC) of >95% (ee>95%). LC-MS (APCI): m/z=590.1 (M+1−100) +. 1H NMR (300 MHz, CDCl 3) (δ/ppm): 8.06 (d, J=1.8 Hz, 1H), 7.53-7.48 (m, 1H), 7.24-7.20 (m, 2H), 7.04-6.98 (m, 1H), 6.81 (s, 1H), 5.66-5.59 (m, 1H), 4.89-4.69 (m, 2H), 2.97 (s, 3H), 1.58 (d, J=6.0 Hz, 3H), 1.47 (s, 18H). |
| Cyclization of the Compound of Formula (C) Using Palladium Catalyst to Form the Compound of Formula (B): |
| To a 500 mL single-necked flask equipped with magnetic stirring were added the compound of formula (C) (15.4 g, 22.3 mmol) and 2-methyl-2-butanol (300 mL), and stirred until the solution became clear. Potassium acetate (6.56 g, 66.9 mmol) was added. The system was evacuated with suction and purged with nitrogen gas three times. Palladium acetate (0.75 g, 3.35 mmol) and n-butylbis(1-adamantyl)phosphine (1.60 g, 4.46 mmol) were quickly added. The system was evacuated with suction and purged with nitrogen gas three times. The reaction solution was heated to 110° C. under nitrogen atmosphere, and reacted with stirring at this temperature overnight. By TLC (PE:EA=1:1) and HPLC monitoring, the reaction was completed. The reaction solution was cooled to room temperature, diluted with dichloromethane (300 mL), and filtered through Celite to remove insoluble solids. The filter cake was washed with dichloromethane (50 mL). The filtrates were combined, and concentrated to dryness under reduced pressure. To the residue was added acetonitrile (150 mL), and the mixture was heated to reflux for 1 hour. The oil bath was removed, and the mixture was allowed to slowly cool to room temperature. A large amount of a white solid precipitated out, and the precipitated solid was filtered. The filter cake was washed with acetonitrile (10 mL), and dried to give 8.2 g of a white solids in a yield of 60.4% and a purity (HPLC) of >99.5% (ee>99.9%). LC-MS (APCI): m/z=510.1 (M+1−100) +. 1H NMR (300 MHz, CDCl 3) (δ/ppm): 8.22 (d, J=1.8 Hz, 1H), 7.29-7.25 (m, 1H), 7.22-7.16 (m, 2H), 7.03-6.96 (m, 1H), 5.76-5.70 (m, 1H), 4.42 (q, J=14.1 Hz, 2H), 3.15 (s, 3H), 1.76 (d, J=6.0 Hz, 3H), 1.44 (s, 18H). |
| Removal of the Boc from the Compound of Formula (B) Using an Acid to Form the Compound of Formula (A): |

To a 250 mL single-necked flask equipped with magnetic stirring were added the compound of formula (B) (8.2 g, 13.5 mmol) and dichloromethane (100 mL), and stirred until the solution became clear. The mixture was cooled in an ice-water bath, and trifluoroacetic acid (20 mL) was slowly added dropwise. After the addition was completed, the ice bath was removed, and the mixture was reacted with stirring at room temperature for 2 hours. By TLC (DCM:MeOH=20:1) and HPLC monitoring, the reaction was completed. The reaction solution was evaporated under reduced pressure to remove the organic solvent. Dichloromethane (100 mL) and a saturated aqueous solution of sodium bicarbonate (60 mL) were added under cooling, and the mixture was stirred for 10 minutes. The organic phase was separated, and the aqueous layer was extracted with dichloromethane (50 mL×2). The organic phases were combined, washed successively with water (30 mL) and then saturated brine (500 mL), dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to give 5.1 g of an amorphous white solid in a yield of 92.6% and a purity (HPLC) of >99.5% (ee>99.9%). LC-MS (APCI): m/z=410.2 (M+1) +. 1H NMR (300 MHz, CDCl 3) (δ) ppm 7.79 (d, J=1.8 Hz, 1H), 7.31-7.27 (m, 1H), 7.23-7.19 (m, 1H), 7.06-6.97 (m, 1H), 6.87 (d, J=1.8 Hz, 1H), 5.75-5.70 (m, 1H), 5.09 (br s, 2H), 4.40 (q, J=14.1 Hz, 2H), 3.12 (s, 3H), 1.78 (d, J=6.6 Hz, 3H).
PAT
Preparation method for deuterated macrocyclic compound
Publication Number: US-2022024908-A1
Priority Date: 2018-11-28
ADVERTISEMENT
Advect Process Systems Ltd.
ADVECT PROCESS SYSTEMS CANADA LTD
51 Beechwood Rd., Cambridge, ON Canada N1S 3S1, Call Now +1 306 850 6737, Mail Now, ask@advectprocess.com



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
//////deulorlatinib, tyrosine kinase inhibitor, antineoplastic, 7PW3UT8C9B, TGRX 326, TGRX-326
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
NEXT
ADVERTISEMENT
Advect Process Systems Ltd.
ADVECT PROCESS SYSTEMS CANADA LTD
51 Beechwood Rd., Cambridge, ON Canada N1S 3S1
Call Now +1 306 850 6737, Mail Now, ask@advectprocess.com
Soquelitinib
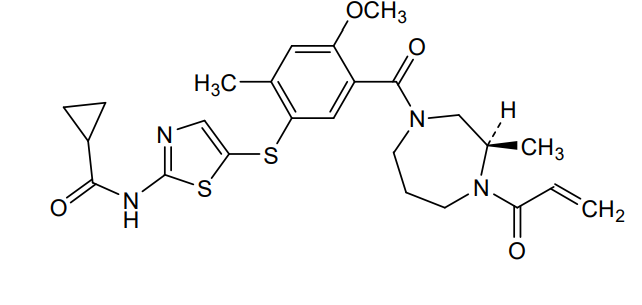
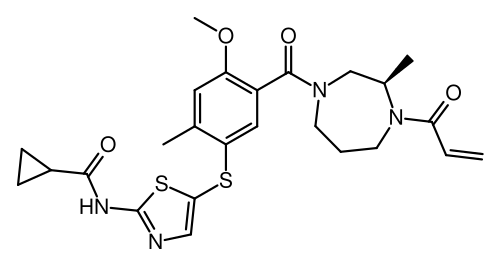
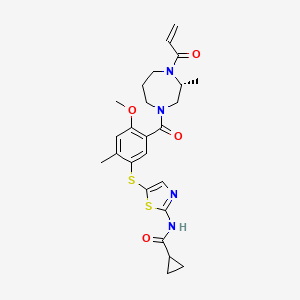
Soquelitinib
CAS 2226636-04-8
MF C25H30N4O4S2, 514.7 g/mol
N-[5-({4-methoxy-2-methyl-5-[(3R)-3-methyl-4-(prop-2-enoyl)-1,4-diazepane-1-carbonyl]phenyl}sulfanyl)-1,3-thiazol-2-yl]cyclopropane-1-carboxamide
tyrosine kinase inhibitor, antineoplastic, CPI818, CPI-000818, CPI596, CP I818, CPI 000818, CP I596, 6I5H17AN3I,
Soquelitinib (CPI-818) is an experimental drug which acts as a selective inhibitor of the enzyme interleukin-2-inducible T-cell kinase (ITK). It is in clinical trials for the treatment of T-cell lymphoma.[1][2]
Soquelitinib is an orally available, small-molecule, irreversible inhibitor of interleukin-2 inducible T-cell kinase (ITK) with potential immunomodulatory and antineoplastic activities. Upon oral administration, soquelitinib selectively and covalently binds to the cysteine residue at position 442 (CYS-442) of ITK, thereby disrupting ITK-mediated signal transduction, while sparing tyrosine-protein kinase TXK (resting lymphocyte kinase, RLK) activity. This may abrogate T-cell receptor (TCR) signaling through ITK and inhibit TCR-induced proliferation of malignant T-cells. Additionally, inhibiting ITK activation may prevent the upregulation of GATA-3, a transcription factor that drives T-helper 2 (Th2) cell differentiation and is overexpressed in certain T-cell lymphomas. Thus, selective inhibition of ITK may inhibit Th2 responses without affecting T-helper 1 (Th1)-dependent immunity. ITK, a member of the Tec family of non-receptor protein tyrosine kinases plays a significant role in the T-cell development, differentiation and production of pro-inflammatory cytokines.
- Safety, Tolerability, and Preliminary Efficacy of Soquelitinib in Participants With Moderate to Severe ADCTID: NCT06345404Phase: Phase 1Status: RecruitingDate: 2025-07-22
- Study of the ITK Inhibitor Soquelitinib to Reduce Lymphoproliferation and Improve Cytopenias in Autoimmune Lymphoproliferative Syndrome (ALPS)-FAS PatientsCTID: NCT06730126Phase: Phase 2Status: RecruitingDate: 2025-05-31
- Soquelitinib vs Standard of Care in Participants With Relapsed/Refractory Peripheral T-cell Lymphoma Not Otherwise Specified, Follicular Helper T-cell Lymphomas, or Systemic Anaplastic Large-cell LymphomaCTID: NCT06561048Phase: Phase 3Status: RecruitingDate: 2025-04-17
- A Dose Escalation Study Evaluating CPI-818 in Relapsed/Refractory T-Cell LymphomaCTID: NCT03952078Phase: Phase 1Status: Active, not recruitingDate: 2025-04-16
Syn
- US11008314,
- https://patentscope.wipo.int/search/en/detail.jsf?docId=US278926237&_cid=P10-MISM56-82578
- SIMILAR
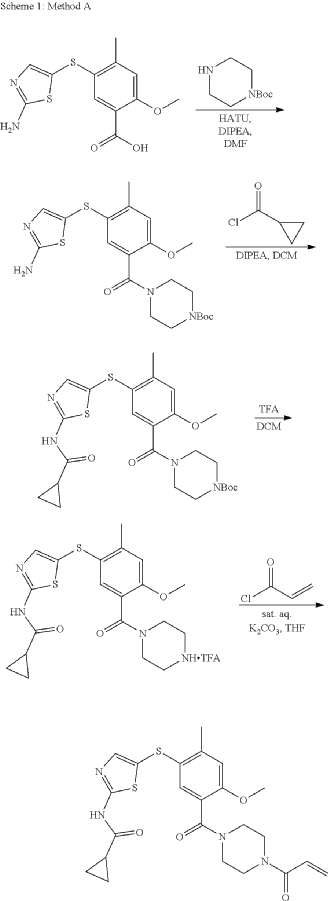

Syn
- WO2018089261 COMPD 44
- https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018089261&_cid=P10-MISM0C-78029-1
SYN
Embodiment B23. A method for an Th2/ITK-mediated disease in a patient in need thereof, the method comprising administering to the patient about 250 mg to about 1,000 mg per day of a compound of Formula (A) or a pharmaceutically acceptable salt thereof, wherein the compound of Formula (A) is:
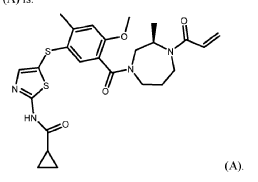
REF
https://www.nature.com/articles/s44386-024-00002-1
Pat
- Compounds and methods for modulating interleukin-2-inducible t-cell kinasePublication Number: US-2022363676-A1Priority Date: 2016-11-03
- Compounds and methods for modulating Interleukin-2-inducible T-cell kinasePublication Number: US-11897874-B2Priority Date: 2016-11-03Grant Date: 2024-02-13
- Itk inhibitors for increasing th1 cell activityPublication Number: WO-2023196278-A1Priority Date: 2022-04-05
- Compounds and methods for modulating interleukin-2-inducible t-cell kinasePublication Number: US-2019375743-A1Priority Date: 2016-11-03
- Compounds and methods for modulating interleukin-2-inducible t-cell kinasePublication Number: WO-2018089261-A2Priority Date: 2016-11-03
- Compounds and methods for modulating interleukin-2-inducible t-cell kinasePublication Number: US-11008314-B2Priority Date: 2016-11-03Grant Date: 2021-05-18
- Compounds and methods for modulating interleukin-2-inducible t-cell kinasePublication Number: EP-3534899-B1Priority Date: 2016-11-03Grant Date: 2022-06-01
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Khodadoust MS, Feldman TA, Yoon DH, Yannakou CK, Radeski D, Kim YH, et al. (November 2020). “Cpi-818, an oral interleukin-2-inducible T-cell kinase inhibitor, is well-tolerated and active in patients with T-cell lymphoma”. Blood. 136: 19–20. doi:10.1182/blood-2020-137782.
- Hsu LY, Rosenbaum JT, Verner E, Jones WB, Hill CM, Janc JW, et al. (December 2024). “Synthesis and characterization of soquelitinib a selective ITK inhibitor that modulates tumor immunity”. npj Drug Discovery. 1 (1) 2: 1–4. doi:10.1038/s44386-024-00002-1.
| Identifiers | |
|---|---|
| IUPAC name | |
| CAS Number | 2226636-04-8 |
| PubChem CID | 134517711 |
| DrugBank | DB18749 |
| ChemSpider | 129629996 |
| UNII | 6I5H17AN3I |
| KEGG | D12762 |
| Chemical and physical data | |
| Formula | C25H30N4O4S2 |
| Molar mass | 514.66 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////////////Soquelitinib, tyrosine kinase inhibitor, antineoplastic, CPI818, CPI-000818, CPI596, CP I818, CPI 000818, CP I596, 6I5H17AN3I,
Nefextinib
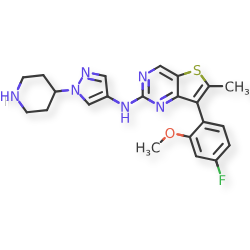
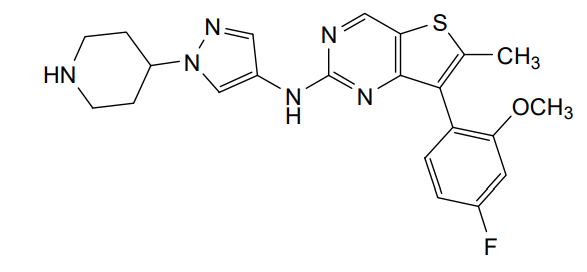
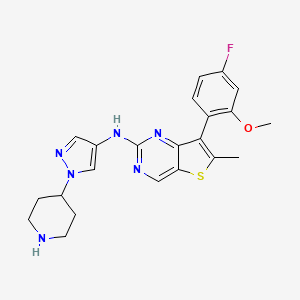
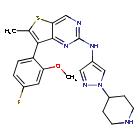
Nefextinib
CAS 2070931-57-4
MF C22H23FN6OS MW 438.52
7-(4-fluoro-2-methoxyphenyl)-6-methyl-N-[1-(piperidin4-yl)-1H-pyrazol-4-yl]thieno[3,2-d]pyrimidin-2-amine
7-(4-FLUORO-2-METHOXYPHENYL)-6-METHYL-N-(1-(PIPERIDIN-4-YL)-1H-PYRAZOL-4-YL) THIENO (3,2-D)PYRIMIDIN-2-AMINE
tyrosine kinase inhibitor, antineoplastic, DL772G3NN7, MAX-40279, MAX 40279
Nefextinib is an orally bioavailable inhibitor of the fibroblast growth factor receptor (FGFR) and FMS-like tyrosine kinase 3 (FLT3; CD135; STK1; FLK2), with potential antineoplastic activity. Upon oral administration, nefextinib binds to and inhibits both FGFR and FLT3, including FLT3 mutant forms, which results in the inhibition of FGFR/FLT3-mediated signal transduction pathways. This inhibits proliferation in FGFR/FLT3-overexpressing tumor cells. FGFR, a family of receptor tyrosine kinases, is upregulated in many tumor cell types. FLT3, a class III receptor tyrosine kinase (RTK), is overexpressed or mutated in most B-lineage neoplasms and in acute myeloid leukemias. They both play key roles in cellular proliferation and survival.
- A Phase 2 Study to Evaluate the Safety and Efficacy of Max-40279-01 in Patients With Advanced Gastric Cancer or Gastroesophageal Junction CancerCTID: NCT05395780Phase: Phase 2Status: Unknown statusDate: 2022-06-02
- MAX-40279 in Subjects With Acute Myelogenous Leukemia (AML)CTID: NCT03412292Phase: Phase 1Status: Unknown statusDate: 2022-01-19
- MAX-40279-01 in Patients With Advanced Solid TumorsCTID: NCT04183764Phase: Phase 1Status: Unknown statusDate: 2022-01-19
- Study of MAX-40279 in Patients With Relapsed or Refractory Acute Myelogenous Leukemia (AML)CTID: NCT04187495Phase: Phase 1Status: Unknown statusDate: 2022-01-19
- A Clincal Study of Max-40279-01 in Patients With Advanced Colorectal CancerCTID: NCT05130021Phase: Phase 2Status: Unknown statusDate: 2021-12-06
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017012559&_cid=P22-MHRG1L-67142-1
[0488]N-[7-(4-fluoro-2-methoxyphenyl)-6-methylthieno[3,2-d]pyrimidin-2-yl]-1-(piperidin-4-yl)-1H-pyrazol-4-amine (compound 31)
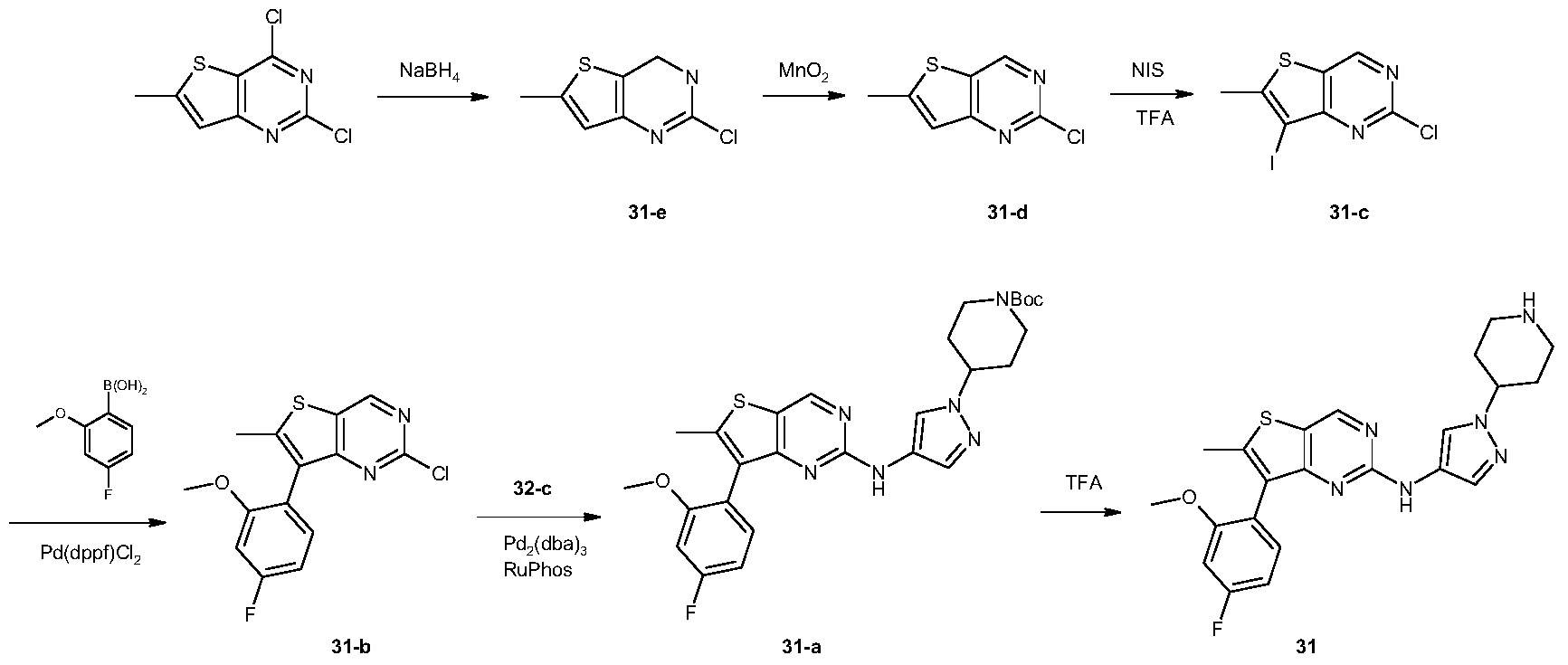
[0491]2,4-Dichloro-6-methylthiophene[3,2-d]pyrimidine (10 g, 45.6 mmol) was dissolved in tetrahydrofuran (100 mL) and ethanol (100 mL). The reaction mixture was cooled to 0 °C, and sodium borohydride (12.5 g, 198 mmol) was added in portions. The reaction mixture was brought to room temperature and stirred for 16 hours. It was then diluted with water (500 mL) and adjusted to pH 7 with 1 N hydrochloric acid solution. The aqueous phase was extracted with ethyl acetate (150 mL × 3). The organic phase was washed successively with water (100 mL × 3) and saturated brine (100 mL), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give a white solid 31-e (7.5 g, yield: 88%). This product required no further purification. LC-MS (ESI): m/z = 187 [M+H] + .
[0492]Synthesis of compound 31-d
[0493]Compound 31-e (7.5 g, 40 mmol) was dissolved in chloroform (300 mL) at 0 °C, and activated manganese dioxide (35 g, 400 mmol) was added. The reaction mixture was brought to room temperature and stirred for 16 hours. The reaction mixture was filtered through diatomaceous earth, and the filter cake was washed with chloroform (100 mL × 3). The combined filtrates were concentrated under reduced pressure to give a white solid 31-d (6.6 g, yield: 89%), which did not require further purification. LC-MS (ESI): m/z = 185 [M + H]+.
[0494]Synthesis of compound 31-c
[0495]Compound 31-d (3.1 g, 16.8 mmol) was dissolved in trifluoroacetic acid (30 mL) at 0 °C. N-iodosuccinimide (5.7 g, 25.3 mmol) was added in portions. The reaction mixture was brought to room temperature and stirred for 1 hour. The reaction was quenched with water (50 mL) and extracted with dichloromethane (50 mL × 3). The organic phase was washed successively with water (50 mL × 3) and saturated brine (50 mL), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give a white solid 31-c (4.9 g, yield: 94%). This product required no further purification. LC-MS (ESI): m/z = 311 [M + H] + .
[0496]Synthesis of compound 31-b
[0497]Compound 31-c (615 mg, 1.98 mmol), 2-methoxy-4-fluorophenylboronic acid (405 mg, 2.38 mmol), and sodium carbonate (630 mg, 5.94 mmol) were suspended in dioxane (5 mL) and water (5 mL). A [1,1′-bis(diphenylphosphine)ferrocene]palladium dichloride dichloromethane complex (163 mg, 0.2 mmol) was added. The mixture was purged three times with nitrogen and heated to 80 °C for 16 hours. After cooling to room temperature, the reaction solution was concentrated under reduced pressure. The residue was separated into layers by dichloromethane (50 mL) and water (50 mL). The organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated and purified by silica gel column chromatography (petroleum ether:dichloromethane = 1:1) to give a white solid 31-b (240 mg, yield: 39%). LC-MS (ESI): m/z = 309 [M+H] + .
[0498]Synthesis of compound 31-a
[0499]Compound 31-b (240 mg, 0.78 mmol) and compound 32-c (208 mg, 0.78 mmol) were dissolved in N,N-dimethylformamide (3 mL), and potassium carbonate (323 mg, 2.34 mmol), 2-dicyclohexylphosphine-2′,6′-diisopropoxy-1,1′-biphenyl (112 mg, 0.24 mmol), and tris(dibenzylacetone)palladium (134 mg, 0.24 mmol) were added. The reaction was carried out under nitrogen protection at 110 °C for 16 hours. After cooling to room temperature, the reaction mixture was separated into layers by dichloromethane (50 mL) and water (50 mL). The organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel thin-layer chromatography (petroleum ether: ethyl acetate = 1:1) to give a yellow viscous oil 31-a (190 mg, yield: 45%). LC-MS(ESI): m/z = 539[M+H] + .
[0500]Synthesis of Compound 31
[0501]31-a (190 mg, 0.35 mmol) was dissolved in dichloromethane (3 mL), and trifluoroacetic acid (3 mL) was added. The mixture was stirred at room temperature for 3 hours. The reaction solution was concentrated under reduced pressure, and the residue was separated into layers by ethyl acetate (50 mL) and 1N hydrochloric acid aqueous solution (50 mL). The aqueous phase was adjusted to pH = 10 with saturated potassium carbonate aqueous solution, and a solid precipitated. The solid was filtered, and the filter cake was washed with water (20 mL × 3). The solid was dried under vacuum to give a light yellow solid 31 (22 mg, yield: 14%). LC-MS (ESI): m/z = 439 [M+H] + .
[0502]
1H-NMR(400MHz,MeOD)δ:8.78(d,J=5Hz,1H),7.87(s,1H),7.48(s,1H),7.35(m,1H),7.05(dd,J=11Hz,J=2Hz,1H),6.91(m,1H),4.10(m,1H),3.79(s,3H),3.22(m,2H),2.77(m,2H),2.47(s,3H),2.03(m,2H),1.73(m,2H)ppm
PAT
- Condensed ring pyrimidine compound, intermediate, its preparation method, composition and applicationPublication Number: CN-106366093-BPriority Date: 2015-07-21Grant Date: 2020-08-18
- Condensation ring pyrimidine compounds, intermediates, methods for producing them, compositions and applicationsPublication Number: JP-6875372-B2Priority Date: 2015-07-21Grant Date: 2021-05-26
- Condensed ring pyrimidine compounds, intermediates, preparation methods, compositions and applications thereofPublication Number: KR-102591886-B1Priority Date: 2015-07-21Grant Date: 2023-10-20
- Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereofPublication Number: EP-3354653-B1Priority Date: 2015-07-21Grant Date: 2019-09-04
- Fused ring pyrimidine compounds, intermediates, production methods, compositions and applications thereofPublication Number: JP-2018520202-APriority Date: 2015-07-21
- Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereofPublication Number: US-10494378-B2Priority Date: 2015-07-21Grant Date: 2019-12-03
- Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereofPublication Number: US-2018208604-A1Priority Date: 2015-07-21
- Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereofPublication Number: WO-2017012559-A1Priority Date: 2015-07-21
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////nefextinib, tyrosine kinase inhibitor, antineoplastic, DL772G3NN7, MAX-40279, MAX 40279
Lunbotinib
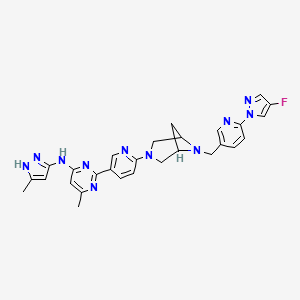
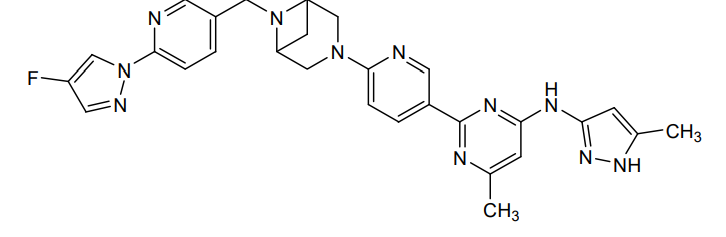
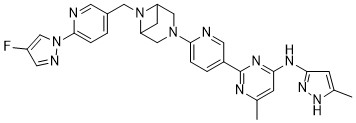
Lunbotinib
CAS 2479961-46-9
MF C28H28FN11 MW537.6 g/mol
2-[6-(6-{[6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl]methyl}-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl]-6-methyl-N-(5-methyl1H-pyrazol-3-yl)pyrimidin-4-amine
tyrosine kinase inhibitor, antineoplastic, KL3T9ZU6HQ
- 2-(6-(6-((6-(4-fluoropyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo(3.1.1)heptan-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine
- 2-[6-[6-[[6-(4-fluoropyrazol-1-yl)pyridin-3-yl]methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl]pyridin-3-yl]-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine
Lunbotinib is an orally bioavailable selective inhibitor of the proto-oncogene receptor tyrosine kinase rearranged during transfection (RET), with potential antineoplastic activity. Upon oral administration, lunbotinib selectively binds to various RET fusions and mutations, including solvent front resistance mutations, and inhibits the activity of RET. This results in an inhibition of cell growth of tumors that exhibit increased RET activity due to these fusions and mutations. RET overexpression, activating mutations, and fusions result in the upregulation and/or overactivation of RET tyrosine kinase activity in various cancer cell types. Dysregulated RET activity plays a key role in the development and progression of certain cancers. Lunbotinib is able to penetrate the blood-brain barrier (BBB) and may also be able to overcome resistance mechanisms to first generation selective RET inhibitors (SRIs).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020168939&_cid=P12-MHKH7H-14851-1
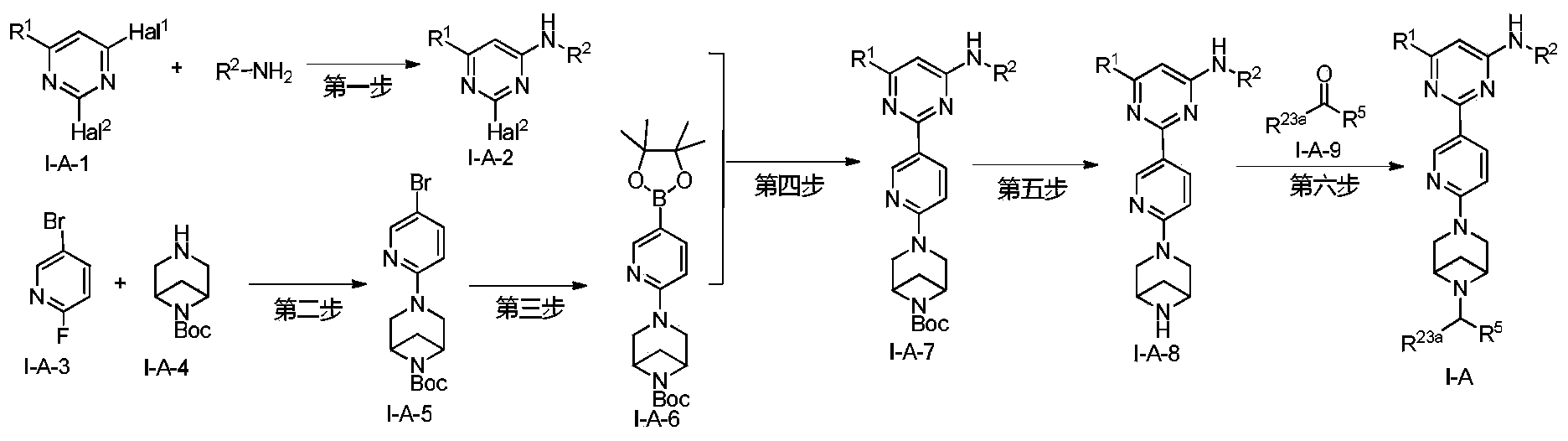
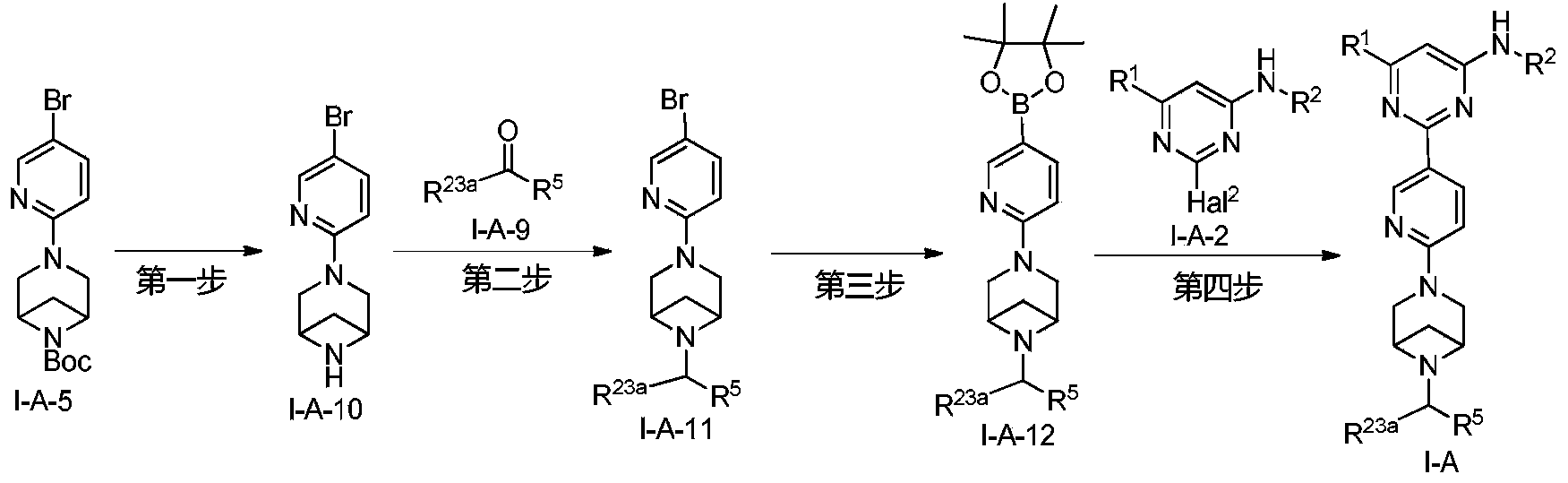
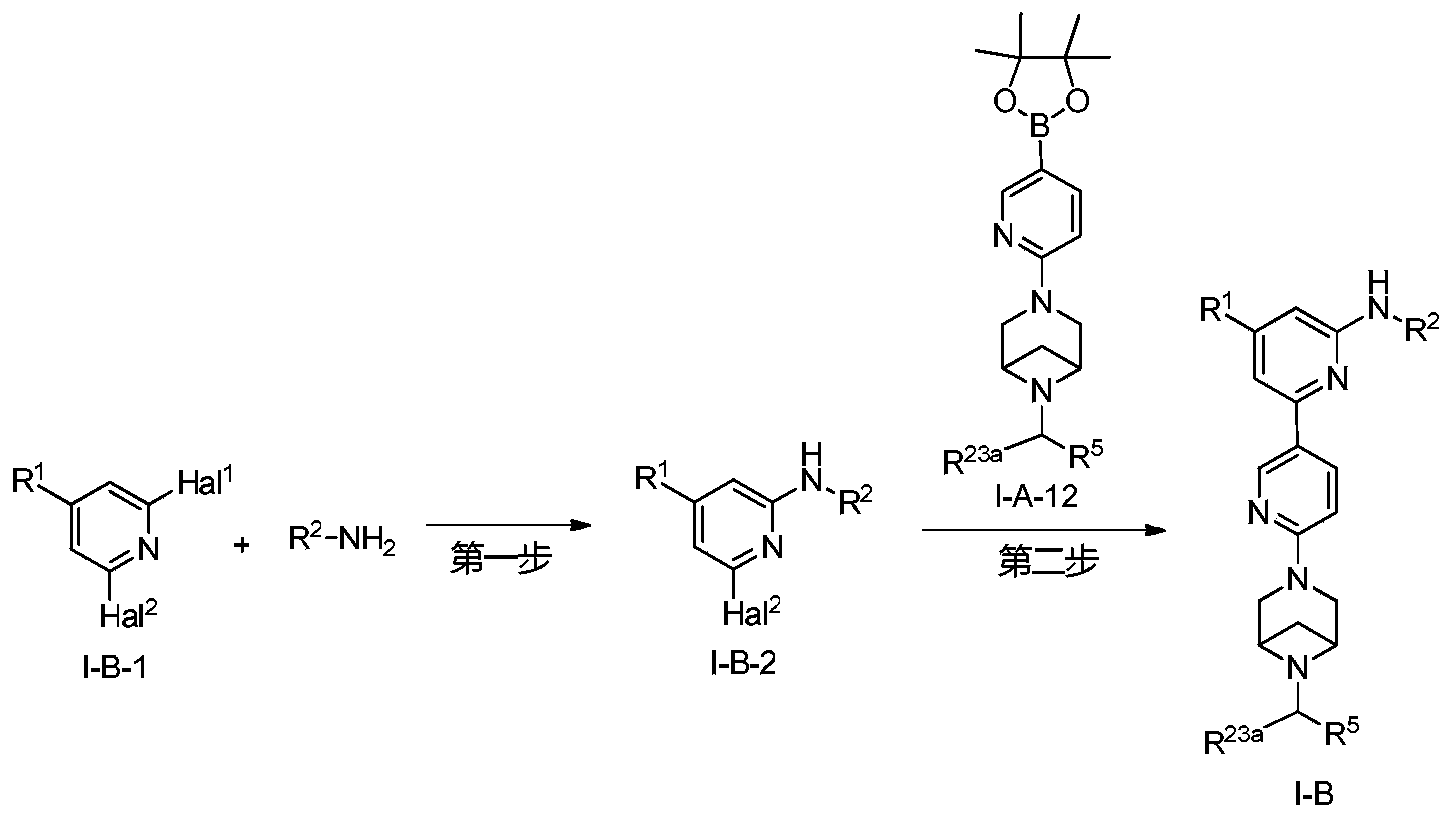
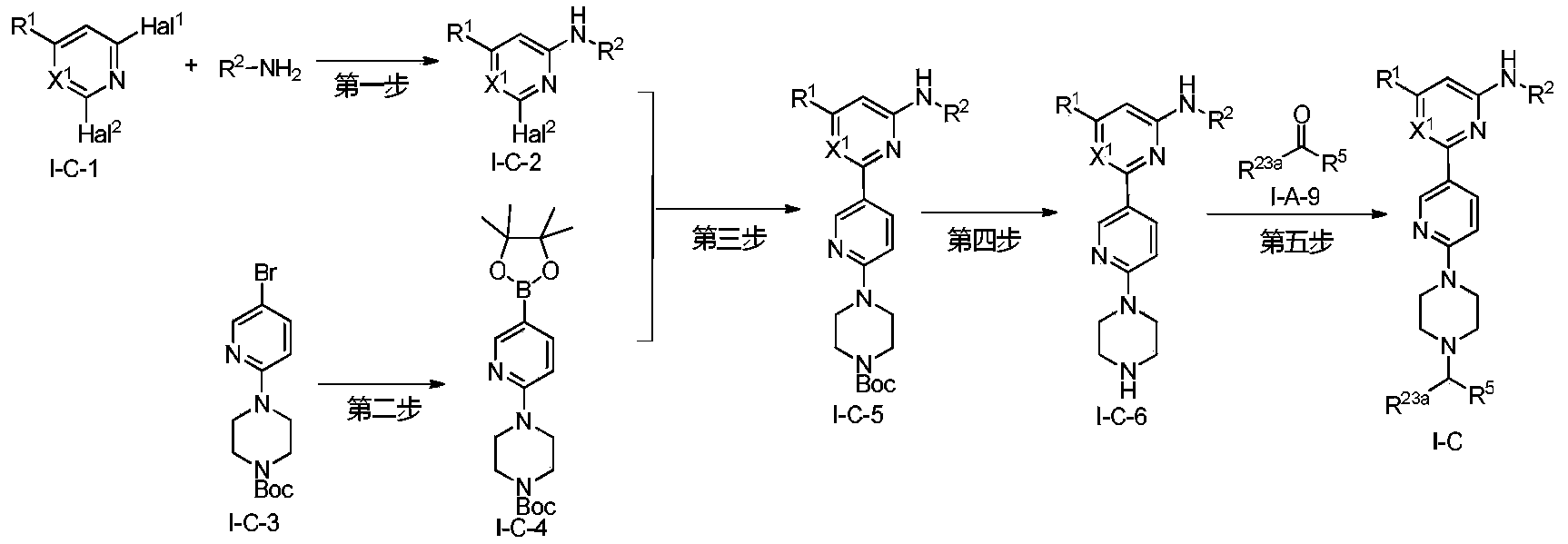
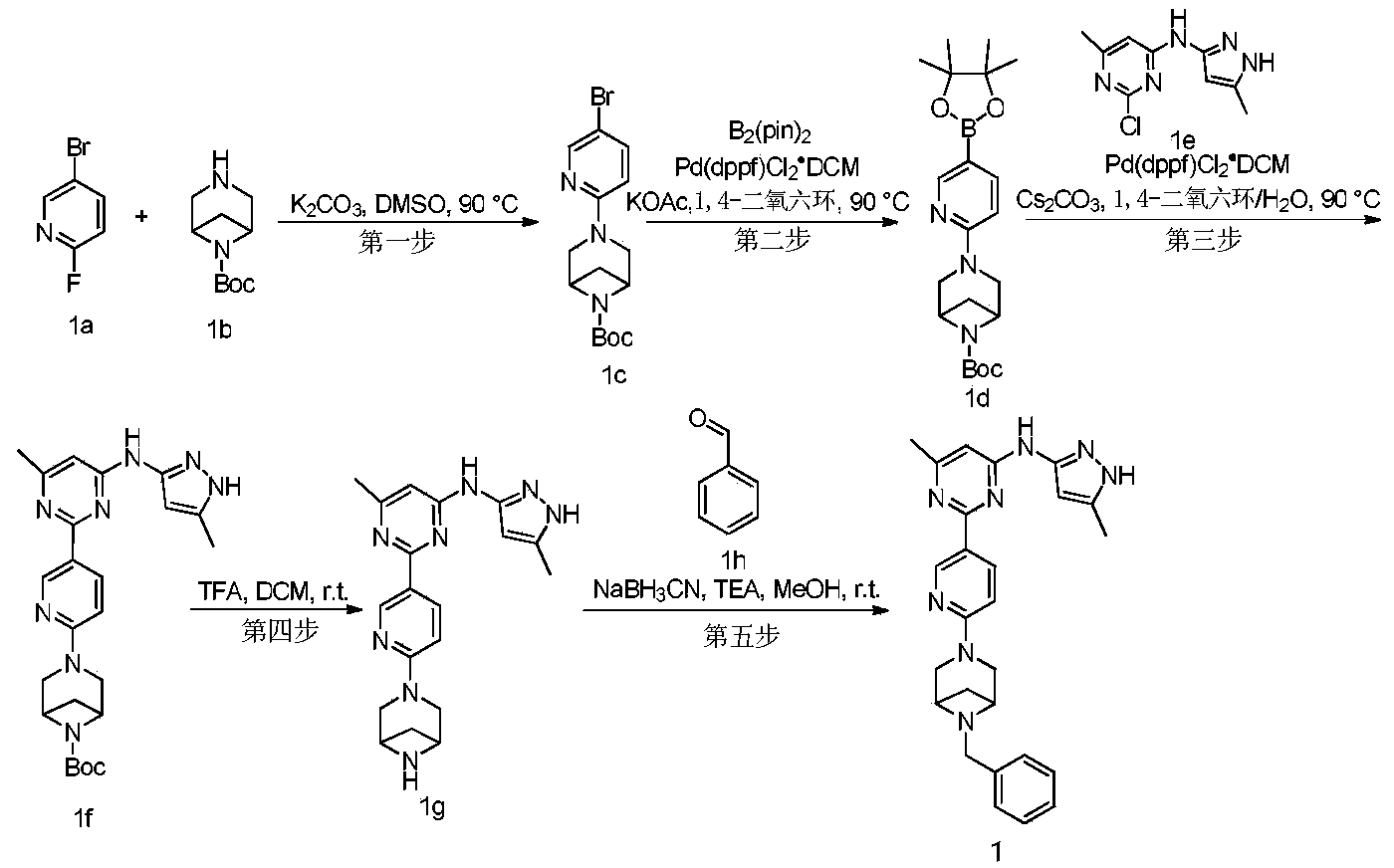
Example 6: 2-(6-(6-((6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptane-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (Compound 17)
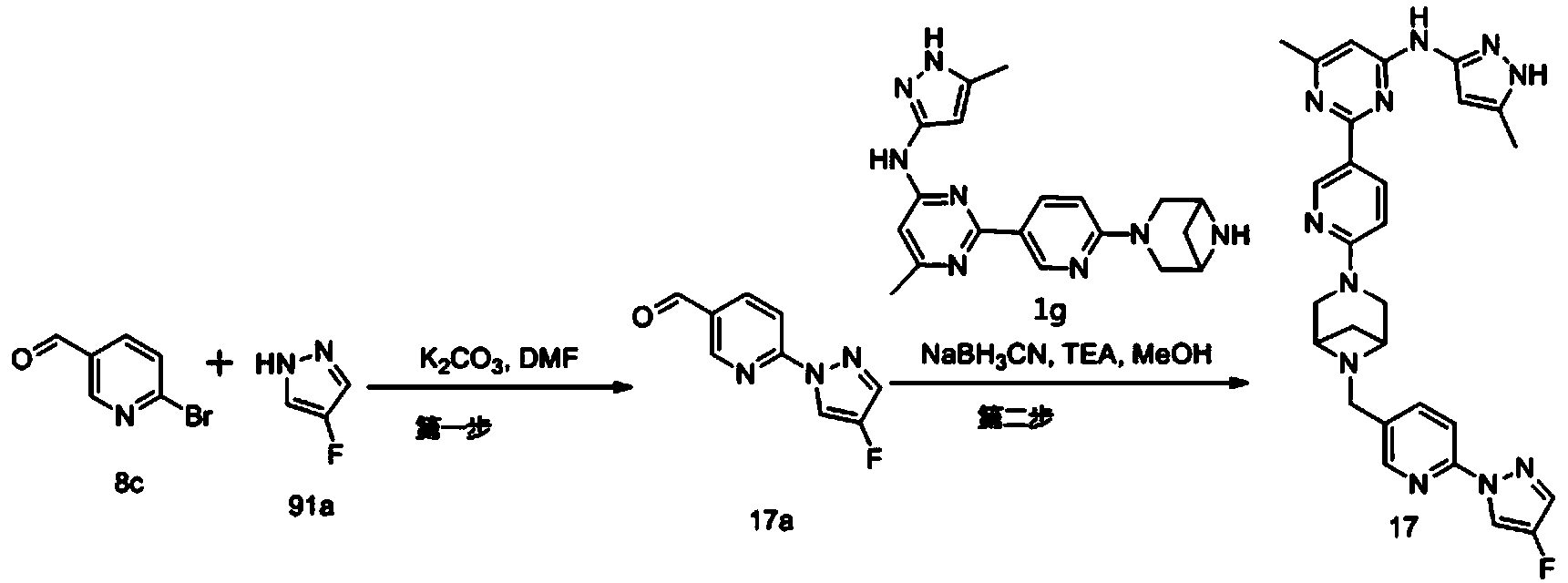
Step 1: Preparation of 6-(4-fluoro-1H-pyrazol-1-yl)nicotinaldehyde (compound 17a)
[0396]Compound 8c (2.0 g), 91a hydrochloride (1.58 g), and potassium carbonate (4.45 g) were sequentially added to DMF (15 mL), and the mixture was heated to 80 °C and stirred for 14 h. The reaction mixture was cooled to room temperature, diluted with water (100 mL), and extracted with DCM (50 mL x 2). The organic phases were combined, washed with water and saturated brine, dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure, and purified by silica gel column chromatography (PE:EA = 10:1) to give compound 17a (0.81 g). MS m/z (ESI): 192.1 [M+H]
[0397]Step 2: Preparation of 2-(6-(6-((6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptane-3-yl)pyridin-3-yl)-6-methyl-N-(5-methyl-1H-pyrazol-3-yl)pyrimidin-4-amine (compound 17)
[0398]1 g of trifluoroacetate (22.82 mg) and compound 17a (27.47 mg) were added to methanol (1.0 mL), followed by the sequential addition of triethylamine (4.45 mg) and sodium cyanoborohydride (13.86 mg), and the reaction was carried out at room temperature for 14 h. After the reaction was completed, the reaction solution was concentrated to dryness under reduced pressure and purified by Prep-HPLC to obtain compound 17 (7.0 mg). MS m/z (ESI): 538.3 [M+H]
[0399]
1H NMR(400MHz,DMSO-d 6)δ11.98(s,1H),9.66(s,1H),9.12(d,J=2.16Hz,1H),8.67(dd,J=4.54,0.64Hz,1H),8.43(dd,J=8.94,2.28Hz,1H),8.41(d,J=1.68,1H),7.98(dd,J=8.48Hz,2.12 1H),7.92(d,J=4.28,1H),7.87(d,J=8.4,1H),6.78(d,J=9.0Hz,2H),6.31(br,1H),3.78-3.71(m,4H),3.68-3.52(m,4H),2.59-2.52(m,1H),2.33(s,3H),2.25(s,3H),1.60(d,J=8.36Hz,1H).
PAT
- Heterocyclic compound, pharmaceutical composition comprising same, preparation method therefor, and use thereofPublication Number: US-2022144847-A1Priority Date: 2019-02-19
- Heterocyclic compounds, pharmaceutical compositions containing the same and preparation methods and uses thereofPublication Number: CN-113316578-BPriority Date: 2019-02-19Grant Date: 2023-10-31
- Heterocyclic compounds, pharmaceutical compositions containing the same and preparation methods and uses thereofPublication Number: CN-117263945-APriority Date: 2019-02-19
- Heterocyclic compounds, pharmaceutical compositions containing the same and preparation methods and uses thereofPublication Number: CN-117327078-APriority Date: 2019-02-19
- Heterocyclic compounds, pharmaceutical compositions containing same, methods for their preparation and usePublication Number: JP-7615056-B2Priority Date: 2019-02-19Grant Date: 2025-01-16
- Salt and crystal form of pyrimidine compound, and preparation methods thereforPublication Number: US-2023295174-A1Priority Date: 2020-07-28
- Heterocyclic compound, pharmaceutical composition comprising same, preparation method therefor, and use thereofPublication Number: WO-2020168939-A1Priority Date: 2019-02-19
- Heterocyclic compounds, pharmaceutical compositions containing the same, and preparation methods and uses thereofPublication Number: CN-113316578-APriority Date: 2019-02-19
- Heterocyclic compound, pharmaceutical composition comprising same, preparation method therefor, and use thereofPublication Number: EP-3929198-A1Priority Date: 2019-02-19
- Heterocyclic compounds, drug compositions containing them, methods of their manufacture and usePublication Number: JP-2022521859-APriority Date: 2019-02-19
- Use of heterocyclic compound for treating diseases related to ret genetic change and method thereforPublication Number: WO-2024240017-A1Priority Date: 2023-05-19
- Uses and methods of heterocyclic compounds for treating diseases associated with kinase resistance mutationsPublication Number: CN-116801882-APriority Date: 2021-03-24
- Use of heterocyclic compound in treating diseases related to kinase drug-resistant mutation and method thereforPublication Number: EP-4316490-A1Priority Date: 2021-03-24
- Salt and crystal form of pyrimidine compound, and preparation methods thereforPublication Number: EP-4190781-A1Priority Date: 2020-07-28
- Salts, crystal forms of pyrimidine compounds and methods for their preparationPublication Number: JP-2023535361-APriority Date: 2020-07-28
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////Lunbotinib, tyrosine kinase inhibitor, antineoplastic, KL3T9ZU6HQ
Momelotinib


Momelotinib
414.47, C23H22N6O2,
1056634-68-4
FDA 2023, Ojjaara,
| To treat intermediate or high-risk myelofibrosis in adults with anemia Drug Trials Snapshot |
N-(Cyanomethyl)-4-[2-(4-morpholin-4-ylanilino)pyrimidin-4-yl]benzamide
N-(Cyanomethyl)-4-[2-[4-(4-morpholinyl)phenylamino]pyrimidin-4-yl]benzamide
Jak2 tyrosine kinase inhibitor; Jak1 tyrosine kinase inhibitor
Inflammatory disease; Myelofibrosis; Myeloproliferative disorder; Pancreatic ductal adenocarcinoma; Polycythemia vera
CYT 387; CYT-387; momelotinib)
GS-0387
CYT387 sulfate saltCAS No: 1056636-06-6
 CYT387 Mesylate CAS No: 1056636-07-7
CYT387 Mesylate CAS No: 1056636-07-7
DI HCL SALT 1380317-28-1
Momelotinib, sold under the brand name Ojjaara among others, is an anticancer medication used for the treatment of myelofibrosis.[5] It is a Janus kinase inhibitor and it is taken by mouth.[5]
The most common adverse reactions include dizziness, fatigue, bacterial infection, hemorrhage, thrombocytopenia, diarrhea, and nausea.[8]
Momelotinib was approved for medical use in the United States in September 2023,[5][8][9] and in the European Union in January 2024.[6][10]
CYT387 is an ATP-competitive small molecule JAK1 / JAK2 inhibitor with IC50 of 11 and 18 nM for JAK1 and JAK2, respectively. CYT387 is useful for treatment of myeloproliferative disorders and anti-cancer.
CYT-387 is a potent, orally administered JAK1/JAK2/ Tyk2 inhibitor in phase III clinical studiest at Gilead for the treatment of post-polycythemia vera, for the treatment of primary myelofibrosis and for the treatment of post-essential thrombocythemia. Phase II studies are also ongoing, in combination with gemcitabine and nab-paclitaxel, in adults with untreated metastatic pancreatic ductal adenocarcinoma.
The compound possesses an excellent selectivity and safety profile. In 2010 and 2011, orphan drug designation was assigned by the FDA and the EMA, respectively, for the treatment of myelofibrosis. In 2011, orphan drug designation was assigned by the EMA for the treatment of post-essential thrombocythemia myelofibrosis and for the treatment of post-polycythemia vera myelofibrosis.
PAT
http://www.google.com.ar/patents/US8486941?cl=ja
N-(cyanomethyl)-4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzamide
| 3 |
 |
414.18 | 1H NMR (300 MHz, d6-DMSO): δ 9.47 (1 H, s), 9.32 (1 H, t, J = 5.5 Hz), 8.54 (1 H, d, J = 5.0 Hz), 8.27 (2 H, d, J = 8.7 Hz), 8.02 (2 H, d, J = 8.2 Hz), 7.67 (2 H, d, J = 9.1 Hz), 7.41 (1 H, d, J = 5.5 Hz), 6.93 (2 H, d, J = 9.1 Hz), 4.36 (2 H, d, J = 5.5 Hz), 3.75 (4 H, m), 3.05 (4 H, m). | m/z 415.3 [M + H]+ | N-(cyanomethyl)-4-(2-(4- morpholinophenylamino)pyrimidin- 4-yl)benzamide |
Example 1Synthesis of Compound 3
A mixture of 4-ethoxycarbonylphenyl boronic acid (23.11 g, 119 mmol), 2,4-dichloropyrimidine (16.90 g, 113 mmol), toluene (230 mL) and aqueous sodium carbonate (2 M, 56 mL) was stirred vigorously and nitrogen was bubbled through the suspension for 15 minutes. Tetrakis(triphenylphosphine)palladium[0] (2.61 g, 2.26 mmol) was added. Nitrogen was bubbled through for another 10 min., the mixture was heated to 100° C., then at 75° C. overnight. The mixture was cooled, diluted with ethyl acetate (200 mL), water (100 mL) was added and the layers were separated. The aqueous layer was extracted with ethyl acetate (100 ml) and the two organic extracts were combined. The organics were washed with brine, filtered through sodium sulfate, concentrated, and the resultant solid was triturated with methanol (100 mL) and filtered. The solids were washed with methanol (2×30 mL) and air dried. This material was dissolved in acetonitrile (150 mL) and dichloromethane (200 mL), stirred with MP.TMT Pd-scavenging resin (Agronaut part number 800471) (7.5 g) over 2 days. The solution was filtered, the solids were washed with dichloromethane (2×100 mL), and the filtrate concentrated to give ethyl 4-(2-chloropyrimidin-4-yl)benzoate as an off-white solid (17.73 g, 60%)—additional washing with dichloromethane yielded a further 1.38 g and 0.5 g of product. 1H NMR (300 MHz, d6-DMSO) δ 8.89 (1H, d, J=5.0 Hz); 8.32 (2H, d, J=8.7 Hz); 8.22 (1H, d, J=5.5 Hz); 8.12 (2H, d, J=8.7 Hz); 4.35 (2H, q, J=7.1 Hz); 1.34 (3H, t, J=7.1 Hz); LC-ESI-MS (method B): rt 7.3 min.; m/z 263.0/265.0 [M+H]+.
A mixture of ethyl 4-(2-chloropyrimidin-4-yl)benzoate (26.15 g, 99.7 mmol) and 4-morpholinoaniline (23.10 g, 129.6 mmol) was suspended in 1,4-dioxane (250 mL). p-Toluenesulfonic acid monohydrate (17.07 g, 89.73 mmol) was added. The mixture was heated at reflux for 40 h., cooled to ambient temperature, concentrated then the residue was partitioned between ethyl acetate and 1:1 saturated sodium bicarbonate/water (1 L total). The organic phase was washed with water (2×100 mL) and concentrated. The aqueous phase was extracted with dichloromethane (3×200 mL). The material which precipitated during this workup was collected by filtration and set aside. The liquid organics were combined, concentrated, triturated with methanol (200 mL) and filtered to yield additional yellow solid. The solids were combined, suspended in methanol (500 mL), allowed to stand overnight then sonicated and filtered. The solids were washed with methanol (2×50 mL) to give, after drying, ethyl 4-(2-(4-morphonlinophenylamino)pyrimidin-4-yl)benzoate (35.39 g, 88%). 1H NMR (300 MHz, d6-DMSO) δ 9.49 (1H, s); 8.54 (1H, d, J=5.0 Hz); 8.27 (2H, d, J=8.7 Hz); 8.10 (2H, d, J=8.7 Hz), 7.66 (2H, d, J=9.1 Hz); 7.38 (1H, d, J=5.0 Hz); 6.93 (2H, d, J=8.7 Hz); 4.35 (2H, q, J=6.9 Hz), 3.73 (4H, m); 3.04 (4H, m); 1.34 (3H, t, J=6.9 Hz); LC-ESI-MS (method B): rt 7.5 min.; m/z 404.1 [M+H].
A solution of ethyl 4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzoate (35.39 g, 87.6 mmol) in 3:1 methanol/tetrahydrofuran (350 mL) was treated with lithium hydroxide (4.41 g, 183.9 mmol) in water (90 mL). The mixture was heated at reflux for 2 h., cooled, concentrated and acidified with hydrochloric acid (2M, 92.5 mL, 185 mmol). The dark precipitate was filtered, washed with water, and dried under vacuum. The solid was ground to a powder with a mortar and pestle, triturated with methanol (500 mL) then filtered again to yield 4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzoic acid as a muddy solid. This material was washed with ether, air dried overnight, and ground to a fine powder with mortar and pestle. On the basis of mass recovery (34.49 g) the yield was assumed to be quantitative. 1H NMR (300 MHz, d6-DMSO) δ 9.47 (1H, s); 8.53 (1H, d, J=5.2 Hz); 8.24 (2H, d, J=8.5 Hz); 8.08 (2H, d, J=8.8 Hz), 7.66 (2H, d, J=9.1 Hz); 7.37 (1H, d, J=5.2 Hz); 6.93 (2H, d, J=9.1 Hz); 3.73 (4H, m); 3.04 (4H, m). LC-ESI-MS (method C): rt 7.3 min.; m/z 377.1 [M+H]+.
To a suspension of 4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzoic acid (theoretically 32.59 g, 86.6 mmol) in DMF (400 mL) was added triethylamine (72.4 mL, 519.6 mmol, 6 eq.) The mixture was sonicated to ensure dissolution. Aminoacetonitrile hydrochloride (16.02 g, 173.2 mmol) was added followed by N-hydroxybenzotriazole (anhydrous, 14.04 g, 103.8 mmol) and 1-ethyl-3-(dimethylaminopropyl)carbodiimide hydrochloride (19.92 g, 103.8 mmol). The suspension was stirred vigorously overnight. The solvent was evaporated under reduced pressure, the residue was diluted with 5% sodium bicarbonate (400 mL) and water (300 mL), giving a yellow solid, which was broken up and filtered. The solids were washed several times with 100 mL portions of water, triturated with hot methanol/dichloromethane (500 mL, 1:1), concentrated to a volume of approximately 300 mL), cooled and filtered. The solids were washed with cold methanol (3×100 mL), ether (200 mL) and hexane (200 mL) prior to drying to afford
Compound 3 (31.69 g, 88%). M.p. 238-243° C.
Microanalysis: Found C, 66.52; H, 5.41; N, 20.21. C23H26N6O10S2 requires C, 66.65; H, 5.35; N, 20.28%.
13C NMR (75.5 MHz, d6-DMSO) δ 166.04, 162.34, 160.26, 159.14, 146.14, 139.87, 134.44, 132.73, 127.80, 126.84, 120.29, 117.49, 115.50, 107.51, 66.06, 49.16, 27.68.
1H NMR GIVEN ABOVE
Example 6Salt Formation from Compound 3
Compound 3 (10.0 g) was suspended in methanol (1 L). Concentrated sulfuric acid (10.52 g, 90% w/w) was added dropwise to the stirring solution. A clear brown solution resulted and a solid lump formed. The solution was filtered quickly then allowed to continue stirring for 3 h (a second precipitate appeared within minutes). After this time the pale yellow precipitate was collected by filtration, washed with methanol (10 mL) then dried under vacuum overnight to afford 4-(4-(4-(4-(cyanomethylcarbamoyl)phenyl)pyrimidin-1-ium-2-ylamino)phenyl)morpholin-4-ium hydrogensulfate, as a pale yellow solid (10.20 g, 69%). m.p. 205° C. Microanalysis: Found C, 45.18; H, 4.36; N, 13.84; S, 10.24. C23H26N6O10S2 requires C, 45.24; H, 4.29; N, 13.76; S 10.50%. 1H NMR (300 MHz, d6-DMSO) δ 9.85 (br. s, 1H), 9.34 (t, J=5.4 Hz, 1H), 8.59 (d, J=5.2 Hz, 1H), 8.27 (d, J=8.5 Hz, 2H), 8.03 (d, J=8.5 Hz, 2H), 7.83 (d, J=8.4 Hz, 2H), 7.50 (d, J=5.2 Hz, 1H), 7.34 (br. s, 2H), 4.36 (d, J=5.4 Hz, 2H), 3.89 (br. s, 4H), 3.37 (br. s, 4H); 13C NMR (75.5 MHz, d6-DMSO) δ 166.07, 163.36, 159.20, 158.48, 140.19, 139.34, 136.45, 134.89, 128.00, 127.22, 121.13, 119.89, 117.59, 109.05, 64.02, 54.04, 27.82. LC-ESI-MS (method D): rt 10.0 min.; m/z 415.1 [M+H]+.
Compound 3 (0.25 g) was suspended in methanol (25 ml). Methane sulfonic acid (0.255 g) was added dropwise to the stirring solution and a clear brown solution resulted. The solution was allowed to stir for 3 h, after which the volume was reduced to 9 ml. The resultant precipitate was collected and dried under vacuum for 8 h to afford 4-(4-(4-(4-(cyanomethylcarbamoyl)phenyl)pyrimidin-1-ium-2-ylamino)phenyl)morpholin-4-ium methanesulfonate as a pale yellow solid (0.22 g). m.p. 208° C. 1H NMR (300 MHz, d6-DMSO) δ 9.83 (br. s, 1H), 9.35 (t, J=5.3 Hz, 1H), 8.59 (d, J=5.1 Hz, 1H), 8.28 (d, J=8.5 Hz, 2H), 8.04 (d, J=8.5 Hz, 2H), 7.83 (d, J=9.0 Hz, 2H), 7.50 (d, J=5.5 Hz, 1H), 7.31 (d, J=9.0 Hz, 2H), 4.36 (d, J=5.5 Hz, 2H), 3.88 (m, 4H), 3.35 (br. s, 4H), 2.36 (s, 6H); LC-ESI-MS (method D): rt 10.2 min.; m/z 415.3 [M+H]+.
Compound 3 (0.50 g) was suspended in methanol (45 ml). A freshly prepared solution of hydrochloric acid in methanol (2.6 ml, HCl conc. 40 mg/ml) was added dropwise to the stirring solution and a clear brown solution resulted. The solution was allowed to stir for 2 h, then the resultant precipitate was collected, washed with methanol (5 ml) and dried under vacuum for 8 h to afford 4-(4-(4-(4-(cyanomethylcarbamoyl)phenyl)pyrimidin-1-ium-2-ylamino)phenyl)morpholin-4-ium chloride a pale yellow solid (0.30 g). m.p. 210° C. 1H NMR (300 MHz, d6-DMSO) 1H NMR (300 MHz, DMSO) δ 9.92 (br. s, 1H), 9.42 (t, J=5.3, 1H), 8.62 (d, J=4.8, 1H), 8.29 (d, J=8.1, 2H), 8.06 (d, J=8.1, 2H), 7.89 (d, J=9.0, 2H), 7.53 (br. s, 3H), 4.36 (d, J=5.4, 2H), 3.82 (br. s, 4H), 3.43 (br. s, 4H)
LC-ESI-MS (method D): rt 10.3 min.; m/z 415.3 [M+H]+.
PAT
WO 2014114274
References on CYT387
. [1] A Pardanani et al CYT387, a Selective JAK1 / JAK2 inhibitor: in vitroassessment of kinase selectivity and preclinical s using Cell lines and Primary cells from polycythemia vera Patients Leukemia (2009) 23, 1441-1445
Abstract
Somatic mutations in Janus kinase 2 (JAK2), including JAK2V617F, result in dysregulated JAK-signal transducer and activator transcription (STAT) signaling, which is implicated in myeloproliferative neoplasm (MPN) pathogenesis. CYT387 is an ATP-competitive small molecule that potently inhibits JAK1 / JAK2 kinases ( IC (50) = 11 and 18 nM, respectively), with significantly less activity against other kinases, including JAK3 (IC (50) = 155 nM). CYT387 inhibits growth of Ba / F3-JAK2V617F and human erythroleukemia (HEL) cells ( IC (50) approximately 1500 nM) or Ba / F3-MPLW515L cells (IC (50) = 200 nM), but has considerably less activity against BCR-ABL harboring K562 cells (IC = 58 000 nM). Cell lines harboring mutated JAK2 alleles (CHRF-288-11 or Ba / F3-TEL-JAK2) were inhibited more potently than the corresponding pair harboring mutated JAK3 alleles (CMK or Ba / F3-TEL-JAK3), and STAT-5 phosphorylation was inhibited in HEL cells with an IC (50) = 400 nM. …
[2]. Tyner Jeffrey W. et al CYT387, a novel JAK2 inhibitor, induces Hematologic Responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms Blood June 24, 2010vol. no 115. 255232-5240
Abstract
Activating alleles of Janus kinase 2 (JAK2) SUCH as JAK2 (V617F) are Central to the pathogenesis of myeloproliferative neoplasms (MPN), suggesting Small molecule inhibitors targeting JAK2 That May be therapeutically Useful. IDENTIFIED We have an aminopyrimidine derivative ( CYT387), which inhibits JAK1, JAK2, and tyrosine kinase 2 (TYK2) at low nanomolar concentrations, with few additional targets. Between 0.5 and 1.5muM CYT387 caused growth suppression and apoptosis in JAK2-dependent hematopoietic cell lines, while nonhematopoietic cell lines were unaffected. In a murine MPN model, CYT387 normalized white cell counts, hematocrit, spleen size, and restored physiologic levels of inflammatory cytokines. Despite the hematologic responses and reduction of the JAK2 (V617F) allele burden, JAK2 (V617F) cells persisted and MPN recurred upon cessation of treatment, suggesting JAK2 inhibitors That May be Unable to Eliminate JAK2 (V617F) cells, Consistent with Preliminary results from Clinical Trials of JAK2 inhibitors in myelofibrosis. …
[3]. Sparidans RW, Durmus S, Xu N, Schinkel AH, Schellens JH, Beijnen JH.Liquid chromatography-tandem mass spectrometric assay for the JAK2 inhibitor CYT387 in plasma.J Chromatogr B Analyt Technol Biomed Life Sci 2012 May 1; 895-896:. 174-7 Epub 2012 Mar 23..
abstract
A quantitative bioanalytical Liquid Chromatography-Tandem Mass spectrometric (LC-MS / MS) assay for the JAK2 inhibitor CYT387 WAS Developed and validated. Plasma samples Were Treated using pre-Protein precipitation with acetonitrile containing cediranib as Internal Standard. The extract WAS Directly Injected into the chromatographic system after dilution with water. This system consisted of a sub-2 μm particle, trifunctional bonded octadecyl silica column with a gradient using 0.005% (v / v) of formic acid in a mixture of water and methanol. The eluate was transferred into the electrospray interface with positive ionization and the analyte was detected in the selected reaction monitoring mode of a triple quadrupole mass spectrometer. The assay was validated in a 0.25-1000 ng / ml calibration range. Within day precisions were 3.0-13.5%, BETWEEN Day Precisions 5.7% and 14.5%. Accuracies Were BETWEEN 96% and 113% for the Whole Calibration range. The Drug WAS stable under All Relevant Analytical Conditions. Finally, the assay successfully WAS Used to ASSESS Drug Levels in mice.
[4] . Monaghan KA, Khong T, Burns CJ, Spencer A.The novel JAK inhibitor CYT387 suppresses Multiple Signalling pathways, and induces apoptosis in Prevents Proliferation phenotypically Diverse myeloma cells.Leukemia 2011 Dec; 25 (12):. 1891-9.
Abstract
Janus kinases (JAKs) are involved in various signalling pathways exploited by malignant cells. In multiple myeloma (MM), the interleukin-6 / JAK / signal transducers and activators of transcription (IL-6 / JAK / STAT) pathway has been the focus of research for a number of years and IL-6 has an established role in MM drug resistance. JAKs therefore make a rational drug target for anti-MM therapy. CYT387 is a novel, orally bioavailable JAK1 / 2 inhibitor, which has recently been described. This preclinical evaluation of CYT387 for treatment of MM demonstrated that CYT387 was able to prevent IL-6-induced phosphorylation of STAT3 and greatly decrease IL-6- and insulin-like growth factor-1-induced phosphorylation of AKT and extracellular signal-regulated kinase in human myeloma cell lines (HMCL). CYT387 inhibited MM proliferation in a time- and dose-dependent manner in 6/8 HMCL, and this was not abrogated by the addition of exogenous IL-6 (3/3 HMCL). Cell cycling was inhibited with a G (2) / M accumulation of cells, and apoptosis was induced by CYT387 in all HMCL tested (3/3). CYT387 synergised in killing HMCL when used in combination with the conventional anti-MM therapies melphalan and bortezomib. Importantly, WAS Also apoptosis induced in Primary Patient MM cells (N = 6) with CYT387 as a single agent, and synergy WAS Seen Again when Combined with Conventional therapies.
[5]. Tyner JW, Bumm TG, Deininger J, Wood L, Aichberger KJ, Loriaux MM, Druker BJ, Burns CJ, Fantino E, Deininger MW.CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms.Blood 2010 Jun 24; 115 (25):. 5232- 40. Epub 2010 Apr 12.
Abstract
Activating alleles of Janus kinase 2 (JAK2) SUCH as JAK2 (V617F) are Central to the pathogenesis of myeloproliferative neoplasms (MPN), suggesting Small molecule inhibitors targeting JAK2 That May be therapeutically Useful. We have IDENTIFIED an aminopyrimidine derivative (CYT387), which inhibits JAK1, JAK2, and tyrosine kinase 2 (TYK2) at low nanomolar concentrations, with few additional targets. Between 0.5 and 1.5muM CYT387 caused growth suppression and apoptosis in JAK2-dependent hematopoietic cell lines, while nonhematopoietic cell lines were unaffected. In a murine MPN model, CYT387 normalized white cell counts, hematocrit, spleen size, and restored physiologic levels of inflammatory cytokines. Despite the hematologic responses and reduction of the JAK2 (V617F) allele burden, JAK2 (V617F) cells persisted and MPN recurred upon cessation of treatment, suggesting that JAK2 inhibitors may be unable to eliminate JAK2 (V617F) cells, consistent with preliminary results from clinical trials of JAK2 inhibitors in myelofibrosis. While the clinical benefit of JAK2 inhibitors may be substantial, not the least due to reduction of inflammatory cytokines and symptomatic improvement, our data add to increasing evidence that kinase inhibitor monotherapy of malignant disease is not curative, suggesting a need for drug combinations to optimally target the malignant cells.
JAKs are kinases which phosphorylate a group of proteins called Signal Transduction and Activators of Transcription or STATs. When phosphorylated, STATs dimerize, translocate to the nucleus and activate expression of genes which lead to, amongst other things, cellular proliferation.
The central role played by the JAK family of protein tyrosine kinases in the cytokine dependent regulation of both proliferation and end function of several important cell types indicates that agents capable of inhibiting the JAK kinases are useful in the prevention and chemotherapeutic treatment of disease states dependent on these enzymes. Potent and specific inhibitors of each of the currently known four JAK family members will provide a means of inhibiting the action of the cytokines that drive immunological and inflammatory diseases.
Myeloproliferative disorders (MPD) include, among others, polycythemia vera (PV), primary myelofibrosis, thrombocythemia, essential thrombocythemia (ET), idiopathic myelofibrosis (IMF), chronic myelogenous leukemia (CML), systemic mastocystosis (SM), chronic neutrophilic leukemia (CNL), myelodisplastic syndrome (MDS) and systemic mast cell disease (SMCD). JAK2 is a member of the JAK family of kinases in which a specific mutation (JAK2V617F) has been found in 99% of polycythemia vera (PV) patients and 50% of essential thrombocytopenia (ET) and idiopathic myelofibrosis (MF). This mutation is thought to activate JAK2, giving weight to the proposition that a JAK2 inhibitor will be useful in treating these types of diseases.
Asthma is a complex disorder characterized by local and systemic allergic inflammation and reversible airway obstruction. Asthma symptoms, especially shortness of breath, are a consequence to airway obstruction, and death is almost invariably due to asphyxiation. Airway Hyper Responsiveness (AHR), and mucus hyper secretion by goblet cells are two of the principle causes of airway obstruction in asthma patients. Intriguingly recent work in animal experimental models of asthma has underscored the importance of IL-13 as a key player in the pathology of asthma. Using a specific IL-13 blocker, it has been demonstrated that IL-13 acts independently of IL-4 and may be capable of inducing the entire allergic asthma phenotype, without the induction of IgE (i.e. in a non-atopic fashion). This and other models have pointed to an important second tier mechanism for elicitating the pathophysiology of asthma, that is not dependent on the production of IgE by resident B-cells or the presence of eonisophils. A direct induction of AHR by IL-13, represents an important process that is likely to be an excellent target for intervention by new therapies. A contemplated effect of a JAK2 inhibitor to the lungs would result in the suppression of the local release of IL-13 mediated IgE production, and therefore reduction in histaminine release by mast cells and eosinophils. This and other consequences of the absence of IL-13 indicate that many of the effects of asthma may be alleviated through administration of a JAK2 inhibitor to the lungs.
Chronic Obstructive Pulmonary Disease (COPD) is a term which refers to a large group of lung diseases which can interfere with normal breathing. Current clinical guidelines define COPD as a disease state characterized by airflow limitation which is not fully reversible. The airflow limitation is usually both progressive and associated with an abnormal inflammatory response of the lungs to noxious particles and gases, particularly cigarette smoke and pollution. Several studies have pointed to an association between increased production of IL-13 and COPD, lending support to the proposition that the potential alleviation of asthma symptoms by use of a JAK2 inhibitor, may also be achieved in COPD. COPD patients have a variety of symptoms including cough, shortness of breath, and excessive production of sputum. COPD includes several clinical respiratory syndromes including chronic bronchitis and emphysema.
Chronic bronchitis is a long standing inflammation of the bronchi which causes increased production of mucus and other changes. The patient’s symptoms are cough and expectoration of sputum. Chronic bronchitis can lead to more frequent and severe respiratory infections, narrowing and plugging of the bronchi, difficult breathing and disability.
Emphysema is a chronic lung disease which affects the alveoli and/or the ends of the smallest bronchi. The lung loses its elasticity and therefore these areas of the lungs become enlarged. These enlarged areas trap stale air and do not effectively exchange it with fresh air. This results in difficult breathing and may result in insufficient oxygen being delivered to the blood. The predominant symptom in patients with emphysema is shortness of breath.
Additionally, there is evidence of STAT activation in malignant tumors, among them lung, breast, colon, ovarian, prostate and liver cancer, as well as Hodgkins lymphoma, multiple myeloma and hepatocellular carcinoma. Chromosomal translocations involving JAK2 fusions to Tel, Bcr and PCM1 have been described in a number of hematopoietic malignancies including chronic myelogenous leukemia (CML), acute myelogenous leukemia (AML), chronic eosinophilic leukemia (CEL), myelodisplastic syndrome (MDS), myeloproliferative disease (MPD) and acute lymphocytic leukemia (ALL). This suggests treatment of hyperproliferative disorders such as cancers including multiple myeloma; prostate, breast and lung cancer; Hodgkin’s Lymphoma; CML; AML; CEL; MDS; ALL; B-cell Chronic Lymphocytic Leukemia; metastatic melanoma; glioma; and hepatoma, by JAK inhibitors is indicated.
Potent inhibitors of JAK2, in addition to the above, will also be useful in vascular disease such as hypertension, hypertrophy, cardiac ischemia, heart failure (including systolic heart failure and diastolic heart failure), migraine and related cerebrovascular disorders, stroke, Raynaud’s phenomenon, POEMS syndrome, Prinzmetal’s angina, vasculitides, such as Takayasu’s arteritis and Wegener’s granulomatosis, peripheral arterial disease, heart disease and pulmonary arterial hypertension.
Pulmonary arterial hypertension (PAH) is a pulmonary vascular disease affecting the pulmonary arterioles resulting in an elevation in pulmonary artery pressure and pulmonary vascular resistance but with normal or only mildly elevated left-sided filling pressures. PAH is caused by a constellation of diseases that affect the pulmonary vasculature. PAH can be caused by or associated with collagen vascular disorders such as systemic sclerosis (scleroderma), uncorrected congenital heart disease, liver disease, portal hypertension, HIV infection, Hepatitis C, certain toxins, splenectomy, hereditary hemorrhagic teleangiectasia, and primary genetic abnormalities. In particular, a mutation in the bone morphogenetic protein type 2 receptor (a TGF-b receptor) has been identified as a cause of familial primary pulmonary hypertension (PPH). It is estimated that 6% of cases of PPH are familial, and that the rest are “sporadic.” The incidence of PPH is estimated to be approximately 1 case per 1 million population. Secondary causes of PAH have a much higher incidence. The pathologic signature of PAH is the plexiform lesion of the lung which consists of obliterative endothelial cell proliferation and vascular smooth muscle cell hypertrophy in small precapillary pulmonary arterioles. PAH is a progressive disease associated with a high mortality. Patients with PAH may develop right ventricular (RV) failure. The extent of RV failure predicts outcome. The JAK/STAT pathway has recently been implicated in the pathophysiology of PAH. JAKs are kinases which phosphorylate a group of proteins called Signal Transduction and Activators of Transcription or STATs. When phosphorylated, STATs dimerize, translocate to the nucleus and activate expression of genes which lead to proliferation of endothelial cells and smooth muscle cells, and cause hypertrophy of cardiac myocytes. There are three different isoforms of JAK: JAK1, JAK2, and JAK3. Another protein with high homology to JAKs is designated Tyk2. An emerging body of data has shown that the phosphorylation of STAT3, a substrate for JAK2, is increased in animal models of PAH. In the rat monocrotaline model, there was increased phosphorylation of the promitogenic transcription factor STAT3. In this same study pulmonary arterial endothelial cells (PAECs) treated with monocrotaline developed hyperactivation of STAT3. A promitogenic agent or protein is an agent or protein that induces or contributes to the induction of cellular proliferation. Therefore, one effect of JAK2 inhibition would be to decrease proliferation of endothelial cells or other cells, such as smooth muscle cells. A contemplated effect of a JAK2 inhibitor would be to decrease the proliferation of endothelial cells or other cells which obstruct the pulmonary arteriolar lumen. By decreasing the obstructive proliferation of cells, a JAK2 inhibitor could be an effective treatment of PAH.
Additionally the use of JAK kinase inhibitors for the treatment of viral diseases and metabolic diseases is indicated.
Although the other members of the JAK family are expressed by essentially all tissues, JAK3 expression appears to be limited to hematopoetic cells. This is consistent with its essential role in signalling through the receptors for IL-2, IL4, IL-7, IL-9 and IL-15 by non-covalent association of JAK3 with the gamma chain common to these multichain receptors. Males with X-linked severe combined immunodeficiency (XSCID) have defects in the common cytokine receptor gamma chain (gamma c) gene that encodes a shared, essential component of the receptors of interleukin-2 (IL-2), IL-4, IL-7, IL-9, and IL-15. An XSCID syndrome in which patients with either mutated or severely reduced levels of JAK3 protein has been identified, suggesting that immunosuppression should result from blocking signalling through the JAK3 pathway. Gene Knock out studies in mice have suggested that JAK3 not only plays a critical role in B and T lymphocyte maturation, but that JAK3 is constitutively required to maintain T cell function. Taken together with the biochemical evidence for the involvement of JAK3 in signalling events downstream of the IL-2 and IL-4 receptor, these human and mouse mutation studies suggest that modulation of immune activity through the inhibition of JAK3 could prove useful in the treatment of T-cell and B-cell proliferative disorders such as transplant rejection and autoimmune diseases. Conversely undesired inhibition of JAK3 could have a devastating effect on the immune status of an individual treated with drug.
Although the inhibition of various types of protein kinases, targeting a range of disease states, is clearly beneficial, it has been to date demonstrated that the identification of a compound which is selective for a protein kinase of interest, and has good “drug like” properties such as high oral bioavailability, is a challenging goal. In addition, it is well established that the predictability of inhibition, or selectivity, in the development of kinase inhibitors is quite low, regardless of the level sequence similarity between the enzymes being targeted.
The challenges in developing therapeutically appropriate JAK2 inhibitors for use in treatment kinase associated diseases such as immunological and inflammatory diseases including organ transplants; hyperproliferative diseases including cancer and myeloproliferative diseases; viral diseases; metabolic diseases; and vascular diseases include designing a compound with appropriate specificity which also has good drug-likeliness.
There is therefore a continuing need to design and/or identify compounds which specifically inhibit the JAK family of kinases, and particularly compounds which may preferentially inhibit one of the JAK kinases relative to the other JAK kinases, particularly JAK2. There is a need for such compounds for the treatment of a range of diseases.

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Omjjara (GlaxoSmithKline Australia Pty Ltd)”. Therapeutic Goods Administration (TGA). 14 January 2025. Retrieved 20 January 2025.
- https://www.tga.gov.au/resources/artg/442230 [bare URL]
- “Notice: Multiple additions to the Prescription Drug List (PDL) [2024-12-20]”. Health Canada. 20 December 2024. Retrieved 21 December 2024.
- “Ojjaara product information”. Health Canada. 8 November 2024. Retrieved 27 December 2024.
- “Ojjaara- momelotinib tablet”. DailyMed. U.S. National Library of Medicine. 15 September 2023. Archived from the original on 30 November 2023. Retrieved 20 September 2023.
- “Omjjara EPAR”. European Medicines Agency. 5 August 2011. Retrieved 18 March 2024.
- “Omjjara Product information”. Union Register of medicinal products. 26 January 2024. Retrieved 18 March 2024.
- “FDA Roundup: September 19, 2023”. U.S. Food and Drug Administration (FDA) (Press release). 19 September 2023. Archived from the original on 21 September 2023. Retrieved 20 September 2023.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - “Novel Drug Approvals for 2023”. U.S. Food and Drug Administration (FDA). 15 September 2023. Archived from the original on 21 January 2023. Retrieved 20 September 2023. This article incorporates text from this source, which is in the public domain.
- “GSK’s Omjjara Authorized in EU for Treating Myelofibrosis With Anemia”. MarketWatch. Retrieved 30 January 2024.
- Pardanani A, Lasho T, Smith G, Burns CJ, Fantino E, Tefferi A (August 2009). “CYT387, a selective JAK1/JAK2 inhibitor: in vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients”. Leukemia. 23 (8): 1441–1445. doi:10.1038/leu.2009.50. PMID 19295546. S2CID 26947444.
- “Omjjara: Pending EC decision”. European Medicines Agency (EMA). 10 November 2023. Archived from the original on 29 November 2023. Retrieved 5 December 2023.
External links
- Clinical trial number NCT04173494 for “A Study of Momelotinib Versus Danazol in Symptomatic and Anemic Myelofibrosis Patients (MOMENTUM)” at ClinicalTrials.gov
- Clinical trial number NCT01969838 for “Momelotinib Versus Ruxolitinib in Subjects With Myelofibrosis (Simplify 1)” at ClinicalTrials.gov
 |
|
| Names | |
|---|---|
| Preferred IUPAC name
N-(Cyanomethyl)-4-{2-[4-(morpholin-4-yl)anilino]pyrimidin-4-yl}benzamide
|
|
Other names
|
|
| Identifiers | |
|
|
|
3D model (JSmol)
|
|
| ChEBI | |
| ChEMBL | |
| ChemSpider | |
| DrugBank |
|
| KEGG | |
|
PubChem CID
|
|
| UNII |
|
|
CompTox Dashboard (EPA)
|
|
| Properties | |
| C23H22N6O2 | |
| Molar mass | 414.469 g·mol−1 |
| Pharmacology | |
| L01EJ04 (WHO) | |
| By mouth | |
| Legal status | |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
| Clinical data | |
|---|---|
| Other names | Momelotinib hydrochloride hydrate (JAN JP), Momelotinib dihydrochloride (USAN US) |
| License data |
|
| Identifiers | |
| PDB ligand | |
| CompTox Dashboard (EPA) | |
//////////Momelotinib, APPROVALS 2023, FDA 2023, Ojjaara, high-risk myelofibrosis, anemia, APPROVALS 2024, EU 2024, EMA 2024
REF
European Journal of Medicinal Chemistry 265 (2024) 116124
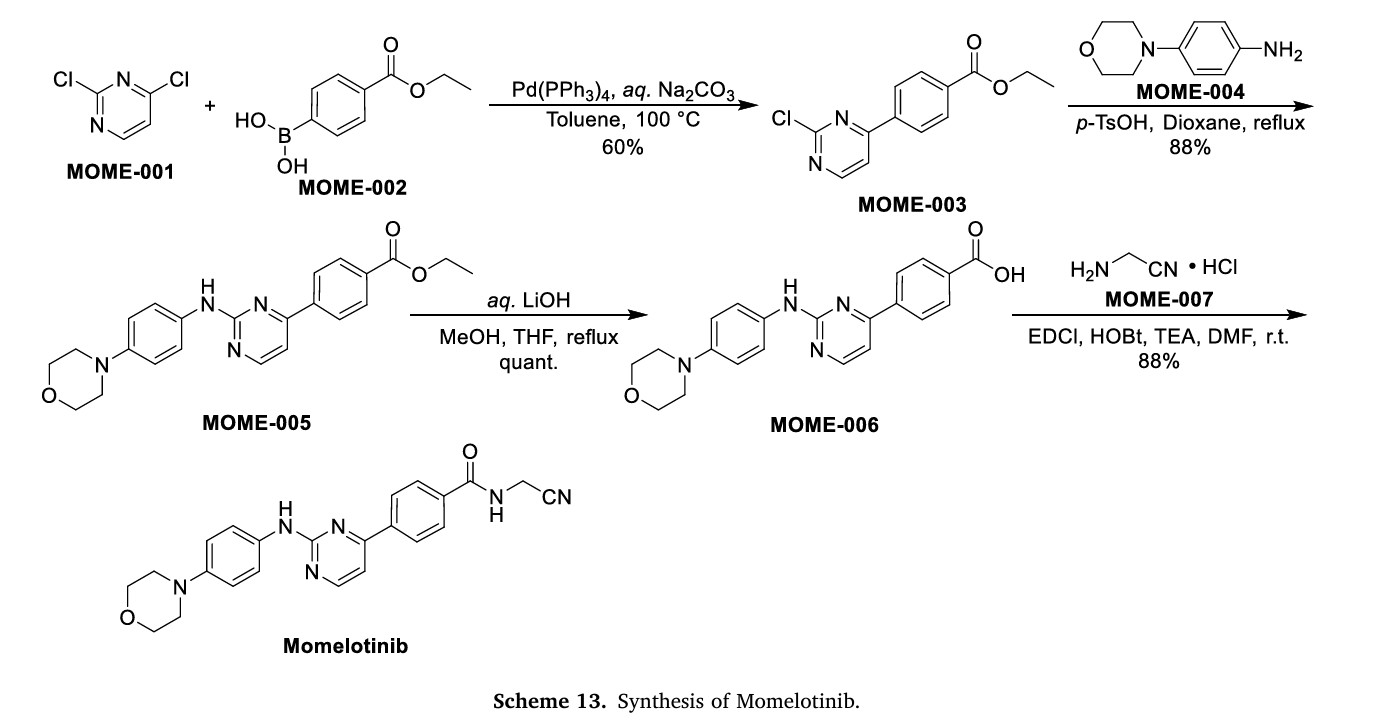
Scheme 13 illustrates the synthesis of Momelotinib Dihydrochloride [48]. The Pd(PPh3) 4-catalyzed Suzuki coupling reaction between 2,4-dichloropyrimidine (MOME-001) and boronic acid MOME-002
resulted in the formation of MOME-003. Subsequently, MOME-003 underwent a substitution reaction with aniline MOME-004 in the presence of p-toluenesulfonic acid (TsOH), yielding MOME-005.
MOME-005 was hydrolyzed by lithium hydroxide, leading to the formation of carboxylic acid MOME-006. MOME-006 underwent amidation with 2-aminoacetonitrile hydrochloride (MOME-007) to produce
Momelotinib.
[48] G.D. Smith, R. Fida, M.M. Kowalski, N-(cyanomethyl)-4-[2-[[4-(4-morpholinyl)
phenyl]amino]-4-pyrimidinyl]-benzamide [CYT387] or a Related Compound,
2012. WO2012071612A1.
.

{kind=link}
{kind=link}