
Home » Posts tagged 'sun pharma'
Tag Archives: sun pharma
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Sun Pharma and Merck & Co. Inc. Enter into Licensing Agreement for Tildrakizumab, MK 3222

Tildrakizumab (MK-3222)
| Company | Merck & Co. Inc. |
| Description | Anti-IL-23 antibody |
| Molecular Target | Interleukin-23 (IL-23) |
| Mechanism of Action | Antibody |
| Therapeutic Modality | Biologic: Antibody |
| Latest Stage of Development | Phase III |
| Standard Indication | Psoriasis |
| Indication Details | Treat moderate to severe chronic plaque psoriasis |
| Regulatory Designation | |
| Partner | Sun Pharmaceutical Industries Ltd. |
Tildrakizumab is a monoclonal antibody designed for the treatment of immunologically mediated inflammatory disorders.[1]
Tildrakizumab was designed to block interleukin-23, a cytokine that plays an important role in managing the immune system and autoimmune disease. Originally developed by Schering-Plough, this drug is now part of Merck‘s clinical program, following that company’s acquisition of Schering-Plough.
Sun Pharmaceutical acquired worldwide rights to tildrakizumab for use in all human indications from Merck in exchange for an upfront payment of U.S. $80 million. Upon product approval, Sun Pharmaceutical will be responsible for regulatory activities, including subsequent submissions, pharmacovigilance, post approval studies, manufacturing and commercialization of the approved product. [2]
As of March 2014, the drug was in phase III clinical trials for plaque psoriasis. The two trials will enroll a total of nearly 2000 patients, and preliminary results are expected in June, 2015. [3][4]

References
- 1 Statement On A Nonproprietary Name Adopted By The USAN Council – Tildrakizumab, American Medical Association.
- 2
- http://www.merck.com/licensing/our-partnership/sunpharma_partnership.html
- 3
- http://clinicaltrials.gov/ct2/show/NCT01729754?term=SCH-900222&phase=2&fund=2&rank=1
- 4
http://clinicaltrials.gov/ct2/show/NCT01722331?term=SCH-900222&phase=2&fund=2&rank=2
Sun Pharma and Merck & Co. Inc. Enter into Licensing Agreement for Tildrakizumab, MK 3222
WHITEHOUSE STATION, N.J., and MUMBAI, India, Wednesday, September 17, 2014 (BUSINESS WIRE) – Merck & Co., Inc., (NYSE:MRK), known as MSD outside the United States and Canada, and Sun Pharmaceutical Industries Ltd. (Reuters: SUN.BO, Bloomberg: SUNP IN, NSE: SUNPHARMA, BSE: 524715) through their respective subsidiaries, today announced an exclusive worldwide licensing agreement for Merck’s investigational therapeutic antibody candidate, tildrakizumab, (MK-3222), which is currently being evaluated in Phase 3 registration trials for the treatment of chronic plaque psoriasis, a skin ailment.
Under terms of the agreement, Sun Pharma will acquire worldwide rights to tildrakizumab for use in all human indications from Merck in exchange for an upfront payment of U.S. $80 million. Merck will continue all clinical development and regulatory activities, which will be funded by Sun Pharma. Upon product approval, Sun Pharma will be responsible for regulatory activities, including subsequent submissions, pharmacovigilance, post approval studies, manufacturing and commercialization of the approved product. Merck is eligible to receive undisclosed payments associated with regulatory (including product approval) and sales milestones, as well as tiered royalties ranging from mid-single digit through teen percentage rates on sales.
“Consistent with our previously announced global initiative to sharpen our commercial and R&D focus, including prioritizing our late stage pipeline candidates, we are pleased to enter into this agreement with Sun Pharma to help realize the potential of tildrakizumab for patients with chronic plaque psoriasis,” said Iain D. Dukes, Ph.D., senior vice president, Business Development and Licensing, Merck Research Laboratories.
“Sun Pharma is very pleased to enter into this collaboration with Merck, a recognized leader in the field of inflammatory/immunology therapies, for this late-stage candidate for chronic plaque psoriasis,” said Kirti Ganorkar, senior vice president, Business Development, Sun Pharma. “This collaboration is a part of our strategy towards building our pipeline of innovative dermatology products in a market with strong growth potential.”
The transaction is subject to customary closing conditions, including the requirements under the Hart Scott-Rodino Antitrust Improvements Act.
About Tildrakizumab
Tildrakizumab is an investigational humanized, anti-IL-23p19 monoclonal antibody that binds specifically to IL-23p19 and is therefore designed to selectively block the cytokine IL-23. Human genetics suggest that inhibiting IL-23 is effective for treating inflammatory conditions. In clinical studies for the treatment of chronic plaque psoriasis, tildrakizumab demonstrates efficacy in blocking inflammation by blocking IL-23. Other potential indications, which may be evaluated in future, include psoriatic arthritis and Crohn’s Disease.
Further details of the Phase 3 clinical trials can be found at: http://clinicaltrials.gov
About Merck
Today’s Merck is a global healthcare leader working to help the world be well. Merck is known as MSD outside the United States and Canada. Through our prescription medicines, vaccines, biologic therapies, and consumer care and animal health products, we work with customers and operate in more than 140 countries to deliver innovative health solutions. We also demonstrate our commitment to increasing access to healthcare through far-reaching policies, programs and partnerships. For more information, visit www.merck.com and connect with us on Twitter, Facebook and YouTube.
About Sun Pharma
Established in 1983, listed since 1994 and headquartered in India, Sun Pharmaceutical Industries Ltd. (Reuters: SUN.BO, Bloomberg: SUNP IN, NSE: SUNPHARMA, BSE: 524715) is an international specialty pharmaceutical company with over 75% sales from global markets. It manufactures and markets a large basket of pharmaceutical formulations as branded generics as well as generics in US, India and several other markets across the world. For the year ending March 2014, overall revenues were at US$2.7 billion, of which US contributed US$1.6 billion. In India, the company is a leader in niche therapy areas of psychiatry, neurology, cardiology, nephrology, gastroenterology, orthopedics and ophthalmology. The company has strong skills in product development, process chemistry, and manufacturing of complex dosage forms. More information about the company can be found at www.sunpharma.com.
| Monoclonal antibody | |
|---|---|
| Type | ? |
| Source | Humanized (from mouse) |
| Target | IL23 |
| Identifiers | |
| CAS Number | 1326244-10-3 |
| ATC code | none |
| ChemSpider | none |
| Chemical data | |
| Formula | C6426H9918N1698O2000S46 |
| Molar mass | 144.4 kg/mol |
///////Sun Pharma, Merck & Co. Inc, Licensing Agreement, Tildrakizumab, mk 3222
WO 2016024224, New Patent, Trelagliptin, SUN PHARMA


WO 2016024224, New Patent, Trelagliptin, SUN PHARMA
SUN PHARMACEUTICAL INDUSTRIES LIMITED [IN/IN]; Sun House, Plot No. 201 B/1 Western Express Highway Goregaon (E) Mumbai, Maharashtra 400 063 (IN)
BARMAN, Dhiren, Chandra; (IN).
NATH, Asok; (IN).
PRASAD, Mohan; (IN)
The present invention provides a process for the preparation of 4-fluoro-2- methylbenzonitrile of Formula (II), and its use for the preparation of trelagliptin or its salts. The present invention provides an efficient, simple, and commercially friendly process for the preparation of 4-fluoro-2-methylbenzonitrile, which is used as an intermediate for the preparation of trelagliptin or its salts. The present invention avoids the use of toxic and hazardous reagents, high boiling solvents, and bromo intermediates such as 2-bromo-5-fluorotoluene, which is lachrymatory in nature and thus difficult to handle at a commercial scale.
Trelagliptin is a dipeptidyl peptidase IV (DPP-IV) inhibitor, chemically designated as 2- [[6-[(3i?)-3 -aminopiperidin- 1 -yl] -3 -methyl -2,4-dioxopyrimidin- 1 -yljmethyl] -4-fluorobenzonitrile, represented by Formula I.

Formula I
Trelagliptin is administered as a succinate salt of Formula la, chemically designated as 2-[[6-[(3i?)-3-aminopiperidin-l-yl]-3-methyl-2,4-dioxopyrimidin-l-yl]methyl]-4-fluorobenzonitrile butanedioic acid (1 : 1).

Formula la
U.S. Patent Nos. 7,795,428, 8,288,539, and 8,222,411 provide a process for the preparation of 4-fluoro-2-methylbenzonitrile by reacting 2-bromo-5-fluorotoluene with copper (I) cyanide in N,N-dimethylformamide.
Chinese Patent No. CN 102964196 provides a process for the preparation of 4-fluoro-2-methylbenzonitrile by reacting 4-fluoro-2-methylbenzyl alcohol with cuprous iodide in the presence of 2,2′-bipyridine and 2,2,6,6-tetramethylpiperidine oxide (TEMPO) in an anhydrous ethanol.
Copper (I) cyanide is toxic to humans, and therefore its use in the manufacture of a drug substance is not advisable. In addition, 2-bromo-5-fluorotoluene is converted to 4-fluoro-2-methylbenzonitrile by refluxing in N,N-dimethylformamide at 152°C to 155°C for 24 hours. This leads to some charring, resulting in a tedious work-up process and low yield. Furthermore, the use of reagents like cuprous iodide, 2,2′-bipyridine, and 2,2,6,6-tetramethylpiperidine oxide (TEMPO) is hazardous and/or environmentally-unfriendly, and therefore their use in the manufacture of a drug substance is not desirable.
The present invention provides an efficient, simple, and commercially friendly process for the preparation of 4-fluoro-2-methylbenzonitrile, which is used as an intermediate for the preparation of trelagliptin or its salts. The present invention avoids the use of toxic and hazardous reagents, high boiling solvents, and bromo intermediates such as 2-bromo-5-fluorotoluene, which is lachrymatory in nature and thus difficult to handle at a commercial scale.
EXAMPLES
Example 1 : Preparation of 4-fluoro-2-methylbenzaldoxime
4-Fluoro-2-methylbenzaldehyde (1.38 g) was added to ethanol (10 mL) to obtain a solution. To this solution, hydroxylamine hydrochloride (2.76 g) and pyridine (1 mL) were added, and then the mixture was stirred at 20°C to 25 °C for 3 hours. The solvent was recovered up to maximum extent from the reaction mixture under reduced pressure to afford the title compound. Yield: 3.1 g
Example 2: Preparation of 4-fluoro-2-methylbenzaldoxime
4-Fluoro-2-methylbenzaldehyde (5 g) was added to ethanol (37 mL) to obtain a solution. To this solution, hydroxylamine hydrochloride (10 g) and N,N-diisopropylethylamine (3.6 mL) were added, and then the mixture was stirred at 20°C to 25 °C for 2 hours. The solvent was recovered up to maximum extent from the reaction mixture under reduced pressure to afford the title compound. Yield: 3.1 g
Example 3 : Preparation of 4-fluoro-2-methylbenzaldoxime
4-Fluoro-2-methylbenzaldehyde (10 g) was added to ethanol (40 mL) to obtain a solution. To this solution, hydroxylamine hydrochloride (20 g) and N,N-diisopropylethylamine (7.5 mL) were added, and then the mixture was stirred at 20°C to 25 °C for 4 hours. The solvent was recovered from the reaction mixture under reduced pressure to afford the title compound. Yield: 11.0 g
Example 4: Preparation of 4-fluoro-2-methylbenzaldoxime
4-Fluoro-2-methylbenzaldehyde (50 g) was added to ethanol (500 mL) to obtain a solution. To this solution, hydroxylamine hydrochloride (70 g) and N,N-diisopropylethylamine (36 mL) were added, and then the mixture was stirred at 20°C to 25 °C for 6 hours. The solvent was recovered from the reaction mixture under reduced pressure to afford the title compound. Yield: 51.0 g
Example 5 : Preparation of 4-fluoro-2-methylbenzaldoxime
4-Fluoro-2-methylbenzaldehyde (20 g) was added to ethanol (200 mL) to obtain a solution. To this solution, hydroxylamine hydrochloride (20 g) and N,N-diisopropylethylamine (18 mL) were added, and then the mixture was stirred at 20°C to 25 °C for 4 hours. The solvent was recovered from the reaction mixture under reduced pressure to obtain a residue. Deionized water (60 mL) was charged into the residue, and then the slurry was stirred at 0°C to 5°C for 1 hour. The solid obtained was filtered, then washed with deionized water (2 x 20 mL). The wet solid was dried in an air oven at 40°C to 45 °C for 4 hours to 5 hours. The crude product obtained was recrystallized in ethanol (50 mL) to afford the pure title compound. Yield: 21.0 g
Example 6: Preparation of 4-fluoro-2-methylbenzaldoxime
4-Fluoro-2-methyl benzaldehyde (50 g) was added to ethanol (500 mL) to obtain a solution. To this solution, hydroxylamine hydrochloride (50 g) and N,N-diisopropylethylamine (46.4 mL) were added, and then the mixture was stirred at 20°C to 25 °C for 4 hours. The solvent was recovered from the reaction mixture under reduced pressure to obtain a residue. Deionized water (150 mL) was charged to the residue, and then the slurry was stirred at 0°C to 5°C for 1 hour. The solid obtained was filtered, then washed with deionized water (2 x 50 mL). The wet solid was dried in an air oven at 40°C to 45 °C for 4 hours to 5 hours. The crude product obtained was recrystallized in ethanol (200 mL) to afford the pure title compound. Yield: 53.5 g
Example 7: Preparation of 4-fluoro-2-methylbenzonitrile
4-Fluoro-2-methylbenzaldoxime (3.1 g) and phosphorous pentoxide (1 g) were added to toluene (30 mL) to obtain a reaction mixture. The reaction mixture was refluxed at 110°C to 115°C for 24 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 25°C to 30°C. Deionized water (30 mL) was added to the mixture and then the layers were separated. The organic layer was concentrated under reduced pressure to afford the title compound. Yield: 1.1 g
Example 8: Preparation of 4-fluoro-2-methylbenzonitrile
4-Fluoro-2-methylbenzaldoxime (3 g) and phosphorous pentoxide (2 g) were added to toluene (30 mL) to obtain a reaction mixture. The reaction mixture was refluxed at 110°C to 115°C for 24 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 25°C to 30°C. Deionized water (30 mL) was added to the mixture and then the layers were separated. The organic layer was concentrated under reduced pressure to afford the title compound. Yield: 1.0 g
Example 9: Preparation of 4-fluoro-2-methylbenzonitrile
4-Fluoro-2-methylbenzaldoxime (5 g) and concentrated sulphuric acid (2 mL) were added to toluene (100 mL) to obtain a reaction mixture. The reaction mixture was refluxed at 110°C to 115°C for 5 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 25°C to 30°C. Deionized water (50 mL) was added to the mixture and then the layers were separated. The organic layer was concentrated under reduced pressure to afford the title compound. Yield: 3.24 g
Example 10: Preparation of 4-fluoro-2-methylbenzonitrile
4-Fluoro-2-methylbenzaldoxime (25 g) and concentrated sulphuric acid (35 g) were added to toluene (500 mL) to obtain a reaction mixture. The reaction mixture was refluxed at 110°C to 115°C for 6 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 25°C to 30°C. Deionized water (250 mL) was added to the mixture and then the layers were separated. The organic layer was concentrated under reduced pressure to afford the title compound. Yield: 20.5 g
Example 11 : Preparation of 4-fluoro-2-methylbenzonitrile
4-Fluoro-2-methyl benzaldoxime (5 g) and sodium bisulphate monohydrate (3.1 g) were added to toluene (50 mL) to obtain a reaction mixture. The reaction mixture was refluxed at 110°C to 115°C for 12 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 25°C to 30°C, then filtered, and then washed with toluene (10 mL). The filtrate was concentrated under reduced pressure to afford the title compound. Yield: 3.0 g
Example 12: Preparation of 4-fluoro-2-methylbenzonitrile
4-Fluoro-2-methyl benzaldoxime (50 g) and sodium bisulphate monohydrate (31.6 g) were added to toluene (500 mL) to obtain a reaction mixture. The reaction mixture was refluxed at 110°C to 115°C using a Dean-Stark apparatus for 12 hours. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to 25 °C to 30°C, then filtered, and then washed with toluene (100 mL). The filtrate was concentrated under reduced pressure to afford a crude product. The crude product obtained was recrystallized in a mixture of toluene (200 mL) and hexane (500 mL) to afford the title compound.
Yield: 38.0 g


Sun Pharma managing director Dilip Shanghvi.
/////////////WO 2016024224, New Patent, Trelagliptin, SUN PHARMA
WO 2016024289, NILOTINIB, New Patent by SUN PHARMA
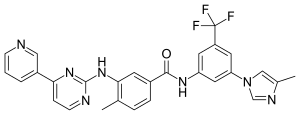
NILOTINIB

WO 2016024289, NILOTINIB, New Patent by SUN
SUN PHARMACEUTICAL INDUSTRIES LTD [IN/IN]; 17/B, Mahal Industrial Estate, Off Mahakali Caves Road, Andheri (east), Mumbai 400093 (IN)
THENNATI, Rajamannar; (IN).
KILARU, Srinivasu; (IN).
VALANCE SURENDRAKUMAR, Macwan; (IN).
SHRIPRAKASH DHAR, Dwivedi; (IN)
The present invention provides novel salts of nilotinib and polymorphs thereof. The acid addition salts of nilotinib with benzenesulfonic acid, butanedisulfonic acid, 1-5- naphthalenedisulfonic acid, naphthalene-1-sulfonic acid and 1-hydroxynaphthoic acid; hydrates and anhydrates thereof.
Nilotinib, 4-methyl-N-[3-(4-methyl-lH-imidazol-l-yl)-5-(trifluoromethyl)phenyl]-3-[[4-(3-pyridinyl)-2-pyrimidinyl] amino] -benzamide, having the following formula

is marketed under the name Tasigna® in US and Europe. Tasigna contains nilotinib monohydrate monohydrochloride salt and is available as capsules for the treatment of adult patients with newly diagnosed Philadelphia chromosome positive chronic myeloid leukemia (Ph+ CML) in chronic phase. Tasigna is also indicated for the treatment of chronic phase and accelerated phase Philadelphia chromosome positive chronic myelogenous leukemia (Ph+ CML) in adult patients resistant or intolerant to prior therapy that included imatinib.
Nilotinib is considered a low solubility/low permeability (class IV) compound in the Biopharmaceutics Classification System (BCS). Therefore, dissolution of nilotinib can potentially be rate limiting step for in-vivo absorption. It is soluble in acidic media; being practically insoluble in buffer solutions of pH 4.5 and higher.
WIPO publication 2014059518A1 discloses crystalline forms of nilotinib hydrochloride and methods of the preparation of various crystalline solvates of nilotinib hydrochloride including benzyl alcohol, acetic acid and propylene glycol.
WIPO publication 2011033307A1 discloses nilotinib dihydrochloride and its hydrates and method for their preparation.
WIPO publication 2011163222A1 discloses the preparation of nilotinib salts and crystalline forms thereof. The salts of nilotinib disclosed are hydrochloride, fumarate, 2-chloromandelate, succinate, adipate, L-tartrate, glutarate, p-toluenesulfonate, camphorsulfonate, glutamate, palmitate, quinate, citrate, maleate, acetate, L-malate, L-aspartate, formate, hydrobromide, oxalate and malonate.
WIPO publication number 2011086541A1 discloses a nilotinib monohydrochloride monohydrate salt and methods for preparing.
WIPO publication number 2010054056A2 describes several crystalline forms of nilotinib hydrochloride.
WIPO publication number 2007/015871A1 discloses the preparation of nilotinib salts and crystalline forms thereof. The salts are mixtures of nilotinib and one acid wherein the acids are selected from the group consisting of hydrochloric acid, phosphoric acid, sulfuric acid, sulfonic acid, methane sulfonic acid, ethane sulfonic acid, benzene sulfonic acid, p-toluene sul- fonic acid, citric acid, fumaric acid, gentisic acid, malonic acid, maleic acid, and tartaric acid.
WIPO publication number 2007015870A2 discloses several nilotinib salts including amorphous and crystalline forms of nilotinib free base, nilotinib HC1 and nilotinib sulfate along with their hydrate and solvates.
EXAMPLES:
Example 1: Preparation of nilotinib benzenesulfonate crystalline Form I
Nilotinib base (1 g) was suspended in water (20 ml). A solution of benzenesulfonic acid (0.4 g) in water (3ml) was added and the content was heated at 60 °C for 2-3 h. The mixture was cooled to 25-30 °C, filtered, washed with water (3 x 5 ml) and dried under vacuum for 2 h at 50-55 °C.
1H NMR (500 MHz, DMSO-d6) δ 2.40 (s,3H), 2.42 (s,3H), 7.35-7.37 (m,3H), 7.51-7.66 (m,5H),7.83 (d,lH), 7.96 (s,lH),8.08 (s,lH),8.30 (s,lH) 8.39 (s,lH),8.54 (d,lH), 8.61 (d,lH), 8.64 (s,lH), 8.75 (d,lH), 9.25 (s,lH), 9.34 (d,lH), 9.61 (s,lH), 10.84 (s,lH).
The salt provides an XRPD pattern substantially same as set forth in FIG. 1.
Example 2: Preparation of nilotinib butanedisulfonate (2: 1) crystalline Form II
Nilotinib base (100 g) was dissolved in 20 % water in THF solution (2000 ml) at 60-65 °C and insoluble matter was filtered. The filtrate was concentrated under vacuum below 60 °C. Filtered water (1000 ml) was added to the reaction mixture and it was heated at 50-55 °C, followed by addition of 1,4-butanedisulfonic acid -60% aqueous solution (28.6 ml) at same temperature. The content was stirred at 50-55 °C for 2-3h. Reaction mixture as cooled to 25-30 °C and product was filtered, washed with water (200 ml x 2) and dried in air oven at 50-55 °C (yield: 115 g).
Sun Pharma managing director Dilip Shanghvi.
Purity (by HPLC):99.76%
1H NMR (400 MHz,DMSO-d6) δ 1.63-1.66(m,2H), 2.40(d,3H),2.42(s,3H),2.43-2.47(m,2H), 7.51-7.62(m,3H),7.85(dd,lH),7.96(s,lH),8.08(s,lH),8.34(s,lH),8.38(d,lH),8.52-8.55(m,lH), 8.60-8.62 (m,2H), 8.75(d,lH), 9.25(S,1H),9.34(S,1H),9.59(S,1H),10.86(S,1H)
Water content: 7.95 %.
The salt has a XRPD pattern substantially same as set forth in FIG. 2.
Example 3: Preparation of nilotinib butanedisulfonate (2: 1) crystalline Form II
Nilotinib base (300 g) was suspended in methanol (3000 ml) and aqueous hydrochloric acid was added to get pH less than 2. Reaction contents were heated at reflux and was filtered and washed with methanol (100 ml). 5% (w/w) NaOH (1200 ml) solution was added at 40-45 °C within 15 min, reaction mixture was stirred for 2h. Product was filtered, washed with water
(300 ml x 3) and dried for lh. Wet material was suspended in water (3000 ml), heated at 50- 55 °C followed by addition of 1,4-butanedisulfonic acid -60% aqueous solution. The reaction mixture was stirred at 50-55°C for 2hrs. Product was filtered at room temperature, washed with water (500 ml x 2) and dried in air oven at 50-55 °C (yield: 293 g).
Purity (by HPLC): 99.88 %
1H NMR (400 MHz,DMSO-d6+TFA-dl) δ 1.75-1.78(m,2H), 2.36(d,3H),2.38(s,3H),2.69- 2.72(m,2H),7.45(d,lH),7.68(d,lH),7.83(s,lH),7.88(dd,lH),7.97(s,lH),8.16-8.19(m,lH), 8.35
(s,2H), 8.63(d,lH),8.68(d,lH),9.04(d,lH),9.21(d,lH),9.53(br s,lH),9.69(d,lH)10.80 (s,lH)
Water content: 6.44 %
Example 4: Preparation of nilotinib butanedisulfonate (2: 1) crystalline Form III
Nilotinib butanedisulfonate (210g) was dissolved in acetic acid water mixture (50:50) (2520 ml) at 75-80 °C and was filtered to remove insoluble matter and washed with acetic acid water mixture (50:50) (210 ml). Water (3150ml) was added to the filtrate and stirred first at room temperature and then at 0-5 °C. Product was filtered and washed with water. Material was dried in air oven at 70-75 °C. Dried material was leached with methanol (3438 ml) at reflux temperature, filtered and dried in air oven 70-75°C (yield: 152.6 g)
Purity (by HPLC): 99.89 %
1H NMR (400 MHz,DMSO-d6+TFA-dl) δ 1.73-1.77(m,2H), 2.40(s,6H),2.67-2.70(m,2H), 7.50 (d,lH), 7.70(d,lH), 7.88-7.92(m,2H), 8.07(s,lH),8.23 (dd,lH), 8.34(s,2H), 8.67 (d,lH), 8.72 (d,lH), 9.09(d,lH), 9.23 (s,lH), 9.54(d,lH), 9.74(d,lH), 10.86(s,lH).
Water content: 0.61 %
The salt provides an XRPD pattern substantially same as set forth in FIG. 3.
Example 5: Preparation of crystalline form of nilotinib butanedisulfonate (2: 1)
Crystalline Nilotinib butanedisulfonate (1 g) of Example 2 was suspended in methanol (20 ml) and was stirred at reflux for 60 min. The mixture was cooled to room temperature. Solid was filtered, washed with methanol (2 ml x 3) and dried in air oven at 70-75°C (yield: 0.8 g)
Example 6: Preparation of nilotinib butanedisulfonate (1: 1) crystalline Form IV
Nilotinib base (20 g) was suspended in methanol (800 ml) and 1,4-butanedisulfonic acid -60
% aqueous solution (6 ml) was added at 50-55 °C, and was filtered to remove insoluble matter. Filtrate was stirred at room temperature for 2-3 h. Product formed was filtered, washed with methanol (20 ml x 2) and dried the product in air oven at 70-75 °C (yield: 18.4 g).
Purity (by HPLC):99.86 %
1H NMR (400 MHz,DMSO-d6) δ 1.64-1.68(m,4H), 2.47-2.5 l(m,4H), 2.41(s,3H), 2.42(d,3H), 7.52(d,lH), 7.83-7.89(m,2H), 7.99(s,lH), 8.15(s,lH), 8.36 (d,lH), 8.39(s,lH), 8.65-8.66(m,2H), 8.79(d,lH), 8.89(br s,lH), 9.36(s,lH), 9.41(br s,lH), 9.74(d,lH), 10.91(s,lH).
The salt has XRPD pattern substantially same as set forth in FIG. 4.
Example 7: Preparation of nilotinib 1,5-napthalenedisulfonic acid salt (2: 1) crystalline Form V
Nilotinib base (1 g) was suspended in water (20 ml). A solution of 1,5-napthalenedisulfonic acid (0.4 g; 0.6 eq.) in water (5ml) was added and the content was heated at 50-55 °C for lh. The mixture was cooled to 25-30 °C, filtered and washed with water (10 ml). The product was dried in air oven at 50-55°C (yield: 1.2 g).
1H NMR (400 MHz,DMSO-d6) δ 2.39 (s,3H), 2.42 (s,3H), 7.45-7.61 (m,4H),7.84 (d,lH), 7.97(s,2H),8.08 (m,lH),8.31 (s,lH) 8.38 (s,lH),8.55 (d,lH), 8.63 (s,2H), 8.75 (s,lH), 8.92 (d,lH), 9.26 (s, 1H), 9.34 (s,lH),9.62 (s,lH), 10.85 (s,lH).
The salt has a XRPD pattern substantially same as set forth in FIG. 5.
Example 8: Preparation of nilotinib 1,5-napthalenedisulfonic acid salt (1: 1) crystalline Form VI
Nilotinib base (1 g) was suspended in water (20 ml). A solution of 1,5-napthalenedisulfonic acid (0.8 g; 1.2eq) in water (5 ml) was added and the content was heated at 50-55 °C for 1 h. The mixture was cooled to 25-30 °C, filtered, washed with water (10 ml) and dried in air oven at 50-55 °C (yield: 1.4g).
1H NMR(400 MHz,DMSO-d6) δ 2.40 (s,3H),2.41 (s,3H), 7.43-7.52 (m,3H),7.61 (d,lH), 7.85-7.99(m,5H),8.11 (s,lH),8.34 (s,2H), 8.64-8.67 (m,2H), 8.89-8.92 (m,4H),9.40(d,2H), 9.72 (s,lH), 10.87 (s,lH).
The salt has a XRPD pattern substantially same as set forth in FIG. 6.
Example 9: Preparation of nilotinib napthalene-1- sulfonic acid salt crystalline Form VII Nilotinib base (1 g) was suspended in water (10 ml) and heated to 50-55 °C. A solution of napthelene-1 -sulfonic acid and methanol (10 ml) was added to it and heated at 70-75 °C for 30 min. The mixture was cooled to 25-30 °C and stirred for 10 min. The product was filtered, washed with water (2 x 2 ml) and dried under vacuum for 1-2 h at 50-55 °C.
1H NMR (400 MHz,DMSO-d6) δ 2.41 (s,3H),2.42 (s,3H), 7.46-7.58 (m,5H), 7.70-8.00 (m,7H)8.11(s,lH)8.31(s,lH),8.37(s,lH),8.63-8.66 (m,3H), 8.81-8.89 (m,2H), 9.31 (s,lH), 9.37 (d,lH), 9.71 (d,lH), 10.86 (s,lH)
The salt has a XRPD pattern substantially same as set forth in FIG. 7.
Example 10: Preparation of nilotinib l-hydroxy-2-napthoic acid salt crystalline Form VIII Nilotinib base (1 g) was suspended in water (20 ml) and heated to 50-55 °C. l-Hydroxy-2-napthoic acid was added to it and the content was heated at 50-55 °C for 1 h. Methanol (5 ml) was added to the mixture and stirred for 30 min. The content was filtered, washed with water (2 x 2 ml) and dried under vacuum for 1 h at 50-55 °C.
1H NMR (400 MHz, DMSO-d6) δ 2.25 (s,3H), 2.41 (s,3H), 7.40-7.92 (m,l lH), 8.23-8.73 (m,8H), 9.24 (s,lH), 9.34(s,lH), 10.70 (s,lH).
The salt has a XRPD pattern substantially same as set forth in FIG. 8.
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-methyl-N-[3-(4-methyl-1H-imidazol-1-yl)- 5-(trifluoromethyl)phenyl]-3- [(4-pyridin-3-ylpyrimidin-2-yl) amino]benzamide
|
|
| Clinical data | |
| Trade names | Tasigna |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a608002 |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy category |
|
| Legal status | |
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 30%[1] |
| Protein binding | 98%[1] |
| Metabolism | Hepatic (mostly CYP3A4-mediated)[1] |
| Biological half-life | 15-17 hours[1] |
| Excretion | Faeces (93%)[1] |
| Identifiers | |
| CAS Number | 641571-10-0(base) |
| ATC code | L01XE08 |
| PubChem | CID 644241 |
| IUPHAR/BPS | 5697 |
| DrugBank | DB04868 |
| ChemSpider | 559260 |
| UNII | F41401512X |
| KEGG | D08953 |
| ChEBI | CHEBI:52172 |
| ChEMBL | CHEMBL255863 |
| PDB ligand ID | NIL (PDBe, RCSB PDB) |
| Chemical data | |
| Formula | C28H22F3N7O |
| Molar mass | 529.5245 g/mol |
//////////////WO 2016024289, WO-2016024289, NILOTINIB, New Patent, SUN
Cc1ccc(cc1Nc2nccc(n2)c3cccnc3)C(=O)Nc4cc(cc(c4)n5cc(nc5)C)C(F)(F)F
Sun Pharma, Merck & Co Inc ink pact for Tildrakizumab

Sep 17, 2014,
Under terms of the agreement, Sun Pharma will acquire worldwide rights to tildrakizumab for use in all human indications from Merck in exchange for an upfront payment of USD 80 million.
Pharma major Sun Pharmaceutical Industries today entered into a licensing agreement with Merck & Co Inc for investigational therapeutic antibody candidate, tildrakizumab to be used for treatment of plaque psoriasis. Under terms of the agreement, Sun Pharma will acquire worldwide rights to tildrakizumab for use in all human indications from Merck in exchange for an upfront payment of USD 80 million, the companies said in a joint statement. Tildrakizumab is being evaluated in Phase III registration trials for the treatment of chronic plaque psoriasis, a skin ailment. “Merck will continue all clinical development and regulatory activities, which will be funded by Sun Pharma. Upon product approval, Sun Pharma will be responsible for regulatory activities, including subsequent submissions, pharmacovigilance, post approval studies, manufacturing and commercialisation of the approved product,” it added.


Sun Pharma managing director Dilip Shanghvi.


| Monoclonal antibody | |
|---|---|
| Source | Humanized (from mouse) |
| Target | IL23 |
| Clinical data | |
| Legal status |
?
|
| Identifiers | |
| CAS number | 1326244-10-3 |
| ATC code | None |
| Chemical data | |
| Formula | C6426H9918N1698O2000S46 |
| Mol. mass | 144.4 kDa |
Tildrakizumab is a monoclonal antibody designed for the treatment of immunologically mediated inflammatory disorders.[1]
Tildrakizumab was designed to block interleukin-23, a cytokine that plays an important role in managing the immune system andautoimmune disease. Originally developed by Schering-Plough, this drug is now part of Merck‘s clinical program, following that company’s acquisition of Schering-Plough.
As of March 2014, the drug was in phase III clinical trials for plaque psoriasis. The two trials will enroll a total of nearly 2000 patients, and preliminary results are expected in June, 2015. [2][3]
References
FDA OKs Sun Pharma for Generic Januvia, Glumetza
Sun Pharma announces tentative USFDA approvals for generic Januvia® and generic Glumetza®
SUNPHARMA, BSE: 524715) announced that the US FDA has granted its subsidiary, two tentative approvals for its Abbreviated New Drug Applications (ANDA) for generic version of Januvia®, Sitagliptin Tablets and generic version of Glumetza®, Metformin HCl Extended-release tablets.
read more at pharmalive
http://www.pharmalive.com/fda-oks-sun-pharma-for-generic-januvia-glumetza
Sun Pharma Global FZE, First Generic Version of Cancer Drug Doxil (doxorubicin hydrochloride liposome injection) Approved
Sun Pharma Global FZE drug, approved by USFDA
DOXIL (doxorubicin HCl liposome injection) is doxorubicin hydrochloride (HCl) encapsulated in STEALTH® liposomes for intravenous administration.
Doxorubicin is an anthracycline topoisomerase inhibitor isolated from Streptomyces peucetius var. caesius.
Doxorubicin HCl, which is the established name for (8S,10S)-10-[(3-amino-2,3,6-trideoxyα- L-lyxo-hexopyranosyl)oxy]-8-glycolyl-7,8,9,10-tetrahydro-6,8,11-trihydroxy-1-methoxy5,12- naphthacenedione hydrochloride, has the following structure:
 |
The molecular formula of the drug is C27H29NO11•HCl; its molecular weight is 579.99.
DOXIL (doxorubicin hcl liposome injection) is provided as a sterile, translucent, red liposomal dispersion in 10-mL or 30-Ml glass, single use vials. Each vial contains 20 mg or 50 mg doxorubicin HCl at a concentration of 2 mg/mL and a pH of 6.5. The STEALTH® liposome carriers are composed of N-(carbonyl-methoxypolyethylene glycol 2000)-1,2-distearoyl-sn-glycero3- phosphoethanolamine sodium salt (MPEG-DSPE), 3.19 mg/mL; fully hydrogenated soy phosphatidylcholine (HSPC), 9.58 mg/mL; and cholesterol, 3.19 mg/mL. Each mL also contains ammonium sulfate, approximately 2 mg; histidine as a buffer; hydrochloric acid and/or sodium hydroxide for pH control; and sucrose to maintain isotonicity. Greater than 90% of the drug is encapsulated in the STEALTH® liposomes
MONDAY Feb. 4, 2013 — The first generic version of the cancer drug Doxil (doxorubicin hydrochloride liposome injection) has been approved by the U.S. Food and Drug Administration, which says the action should help relieve shortages of the brand-name medication.
Doxil is on the agency’s drug shortage list. The list empowers the FDA’s Office of Generic Drugs to grant priority review to generic equivalents, the agency said Monday in a news release.
Noting that generics were of the same quality and strength as the original drugs, the FDA said: “Generic manufacturing and packaging sites must pass the same quality standards as those of brand-name drugs.”
Generic Doxil will be produced by Sun Pharma Global FZE in 20 milligram and 50 milligram vials.

{kind=link}