
Home » Posts tagged 'SOLID TUMOUR'
Tag Archives: SOLID TUMOUR
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
CK-101
![N-[3-[2-[2,3-Difluoro-4-[4-(2-hydroxyethyl)piperazin-1-yl]anilino]quinazolin-8-yl]phenyl]prop-2-enamide.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=117909640&t=l)

CK-101, RX-518
CAS 1660963-42-7
N-[3-[2-[[2,3-Difluoro-4-[4-(2-hydroxyethyl)piperazin-1-yl]phenyl]amino]quinazolin-8-yl]phenyl]acrylamide
N-(3-(2-((2,3-Difluoro-4-(4-(2-hydroxyethyl)piperazin-1-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide
EGFR-IN-3
UNII-708TLB8J3Y
Suzhou NeuPharma (Originator)
Checkpoint Therapeutics
Non-Small Cell Lung Cancer Therapy
Solid Tumors Therapy
PHASE 2 Checkpoint Therapeutics, Cancer, lung (non-small cell) (NSCLC), solid tumour
RX518(CK-101) is an orally available third-generation and selective inhibitor of certain epidermal growth factor receptor (EGFR) activating mutations, including the resistance mutation T790M, and the L858R and exon 19 deletion (del 19) mutations, with potential antineoplastic activity.
In August 2019, Suzhou Neupharma and its licensee Checkpoint Therapeutics are developing CK-101 (phase II clinical trial), a novel third-generation, covalent, EGFR inhibitor, as a capsule formulation, for the treatment of cancers including NSCLC and other advanced solid tumors. In September 2017, the FDA granted Orphan Drug designation to this compound, for the treatment of EGFR mutation-positive NSCLC; in January 2018, the capsule was being developed as a class 1 chemical drug in China.
CK-101 (RX-518), a small-molecule inhibitor of epidermal growth factor receptor (EGFR), is in early clinical development at Checkpoint Therapeutics and Suzhou NeuPharma for the potential treatment of EGFR-mutated non-small cell lung cancer (NSCLC) and other advanced solid malignancies.
In 2015, Suzhou NeuPharma granted a global development and commercialization license to its EGFR inhibitor program, excluding certain Asian countries, to Coronado Biosciences (now Fortress Biotech). Subsequently, Coronado assigned the newly acquired program to its subsidiary Checkpoint Therapeutics.
In 2017, the product was granted orphan drug designation in the U.S. for the treatment of EGFR mutation-positive NSCLC.
There are at least 400 enzymes identified as protein kinases. These enzymes catalyze the phosphorylation of target protein substrates. The phosphorylation is usually a transfer reaction of a phosphate group from ATP to the protein substrate. The specific structure in the target substrate to which the phosphate is transferred is a tyrosine, serine or threonine residue. Since these amino acid residues are the target structures for the phosphoryl transfer, these protein kinase enzymes are commonly referred to as tyrosine kinases or serine/threonine kinases.
[0003] The phosphorylation reactions, and counteracting phosphatase reactions, at the tyrosine, serine and threonine residues are involved in countless cellular processes that underlie responses to diverse intracellular signals (typically mediated through cellular receptors), regulation of cellular functions, and activation or deactivation of cellular processes. A cascade of protein kinases often participate in intracellular signal transduction and are necessary for the realization of these cellular processes. Because of their ubiquity in these processes, the protein kinases can be found as an integral part of the plasma membrane or as cytoplasmic enzymes or localized in the nucleus, often as components of enzyme complexes. In many instances, these protein kinases are an essential element of enzyme and structural protein complexes that determine where and when a cellular process occurs within a cell.
[0004] The identification of effective small compounds which specifically inhibit signal transduction and cellular proliferation by modulating the activity of tyrosine and serine/threonine kinases to regulate and modulate abnormal or inappropriate cell proliferation, differentiation, or metabolism is therefore desirable. In particular, the identification of compounds that specifically inhibit the function of a kinase which is essential for processes leading to cancer would be beneficial.
[0005] While such compounds are often initially evaluated for their activity when dissolved in solution, solid state characteristics such as polymorphism are also important. Polymorphic forms of a drug substance, such as a kinase inhibitor, can have different physical properties, including melting point, apparent solubility, dissolution rate, optical and mechanical properties, vapor pressure, and density. These properties can have a direct effect on the ability to process or manufacture a drug substance and the drug product. Moreover, differences in these properties
can and often lead to different pharmacokinetics profiles for different polymorphic forms of a drug. Therefore, polymorphism is often an important factor under regulatory review of the ‘sameness’ of drug products from various manufacturers. For example, polymorphism has been evaluated in many multi-million dollar and even multi-billion dollar drugs, such as warfarin sodium, famotidine, and ranitidine. Polymorphism can affect the quality, safety, and/or efficacy of a drug product, such as a kinase inhibitor. Thus, there still remains a need for polymorphs of kinase inhibitors. The present disclosure addresses this need and provides related advantages as well.
PATENT
WO2015027222
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015027222


PATENT
WO-2019157225
Crystalline form II-VIII of the compound presumed to be CK-101 (first disclosed in WO2015027222 ), for treating a disorder mediated by epidermal growth factor receptor (EGFR) eg cancer.
SCHEME A
Scheme B
General Procedures
Example 1: Preparation of the compound of Formula I (N-(3-(2-((2,3-difluoro-4-(4-(2-hydroxyethyl)piperazin-l-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide)
[0253] To a solution of l,2,3-trifluoro-4-nitrobenzene (2.5 g, 14 mmol, 1.0 eq.) in DMF (20 mL) was added K2C03 (3.8 g, 28 mmol, 2.0 eq.) followed by 2-(piperazin-l-yl)ethanol (1.8 g, 14 mmol, 1.0 eq.) at 0 °C and the mixture was stirred at r.t. overnight. The mixture was poured into ice-water (200 mL), filtered and dried in vacuo to afford 2-(4-(2,3-difluoro-4-nitrophenyl)piperazin-l-yl)ethanol (2.7 g, 67.5%).
[0254] To a solution of 2-(4-(2,3-difluoro-4-nitrophenyl)piperazin-l-yl)ethanol (2.7 g, 9.0 mmol) in MeOH (30 mL) was added Pd/C (270 mg) and the resulting mixture was stirred at r.t.
overnight. The Pd/C was removed by filtration and the filtrate was concentrated to afford 2-(4-(4-amino-2,3-difluorophenyl)piperazin-l-yl)ethanol (2.39 g, 99% yield) as off-white solid.
[0255] To a solution of 8-bromo-2-chloroquinazoline (15.4 g, 63.6 mmol, 1 eq. ) and (3-aminophenyl)boronic acid (8.7 g, 63.6 mmol, 1 eq.) in dioxane/H20 (200 mL/20 mL) was added Na2C03 (13.5 g, 127.2 mmol, 2 eq.), followed by Pd(dppf)Cl2 (2.6 g, 3.2 mmol, 0.05 eq.) under N2, then the mixture was stirred at 80 °C for 12 h. Then the solution was cooled to r.t.,
concentrated and the residue was purified via column chromatography (PE/EA=3 :2, v/v) to afford 3-(2-chloroquinazolin-8-yl)aniline as yellow solid (8.7 g, 53.7% yield).
[0256] To a solution of 3-(2-chloroquinazolin-8-yl)aniline (8.7 g, 34 mmol, 1 eq.) in DCM ( 200 mL ) cooled in ice-bath was added TEA (9.5 mL, 68 mmol, 2 eq. ), followed by acryloyl chloride (4.1 mL, 51 mmol, 1.5 eq.) dropwise. The resulting mixture was stirred at r.t. for 1 h, then washed with brine, dried over anhydrous N2S04 concentrated and the residue was purified via column chromatography (PE/EA=l : 1, v:v) to afford N-(3-(2-chloroquinazolin-8-yl)phenyl)acryl amide as yellow solid(6.6 g, 65% yield).
[0257] To a suspension of 2-(4-(4-amino-2,3-difluorophenyl)piperazin-l-yl)ethanol (83 mg,
0.32 mmol, 1 eq.) and N-(3-(2-chloroquinazolin-8-yl)phenyl)acrylamide (100 mg, 0.32 mmol, 1 eq.) in n-BuOH (5 mL) was added TFA (68 mg, 0.64 mmol, 2 eq.) and the resulting mixture was stirred at 90 °C overnight. The mixture was concentrated, diluted with DCM (20 mL) , washed with Na2C03 solution (20 mL), dried over anhydrous Na2S04, concentrated and the residue was purified via column chromatography (MeOH/DCM=l/30, v:v) to afford N-(3-(2-((2,3-difluoro-4-(4-(2-hydroxyethyl)piperazin-l-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide as a yellow solid(l6.3 mg, 9.5% yield). LRMS (M+H+) m/z calculated 531.2, found 531.2. 1H NMR
(CD3OD, 400 MHz) d 9.21 (s, 1 H), 7.19-8.01 (m, 10 H), 8.90 (s, 1 H), 6.41-6.49 (m, 3 H), 5.86 (m, 1 H), 3.98-4.01 (m, 3 H), 3.70-3.76 (m, 3 H), 3.40-3.49 (m, 2 H), 3.37-3.39 (m, 4 H), 3.18 (m, 2H).
Example 2. Preparation of Form I of the compound of Formula I
[0258] Crude compound of Formula I (~30 g, 75% of weight based assay) was dissolved in ethyl acetate (3 L) at 55-65 °C under nitrogen. The resulting solution was filtered via silica gel pad and washed with ethyl acetate (3 L><2) at 55-65 °C. The filtrate was concentrated via vacuum at 30-40 °C to ~2.4 L. The mixture was heated up to 75-85 °C and maintained about 1 hour.
Then cooled down to 50-60 °C and maintained about 2 hours. The heat-cooling operation was repeated again and the mixture was then cooled down to 20-30 °C and stirred for 3 hours. The resulting mixture was filtered and washed with ethyl acetate (60 mL><2). The wet cake was dried via vacuum at 30-40 °C to get (about 16 g) of the purified Form I of the compound of Formula I.
Example 3. Preparation of Form III of the compound of Formula I
[0259] The compound of Formula I (2 g) was dissolved in EtOH (40 mL) at 75-85 °C under nitrogen. n-Heptane (40 mL) was added dropwise into reaction at 75-85 °C. The mixture was stirred at 75-85 °C for 1 hour. Then cooled down to 50-60 °C and maintained about 2 hours. The heat-cooling operation was repeated again and continued to cool the mixture down to 20-30 °C and stirred for 3 hours. The resulting mixture was filtered and washed with EtOH/n-Heptane (1/1, 5 mL><2). The wet cake was dried via vacuum at 30-40 °C to get the purified Form III of the compound of Formula I (1.7 g).
Example 4. Preparation of Form IV of the compound of Formula I The crude compound of Formula I (15 g) was dissolved in ethyl acetate (600 mL) at 75-85 °C under nitrogen and treated with anhydrous Na2S04, activated carbon, silica metal scavenger for 1 hour. The resulting mixture was filtered via neutral Al203 and washed with ethyl acetate (300 mL><2) at 75-85 °C. The filtrate was concentrated under vacuum at 30-40 °C and swapped with DCM (150 mL). n-Heptane (75 mL) was added into this DCM solution at 35-45 °C, and then the mixture was cooled down to 20-30 °C slowly. The resulting mixture was filtered and washed with DCM/n-Heptane (2/1, 10 mL><3). The wet cake was dried via vacuum at 35-40 °C to get the purified Form IV of the compound of Formula I (9.6 g).
Example 5. Preparation of Form V of the compound of Formula I
[0260] Polymorph Form III of the compound of Formula I was dried in oven at 80 °C for 2 days to obtain the polymorph Form V.
Example 6. Preparation of Form VI of the compound of Formula I
[0261] The compound of Formula I (1 g) was dissolved in IPA (20 mL) at 75-85 °C under nitrogen. n-Heptane (20 mL) was added dropwise into reaction at 75-85 °C. The mixture was stirred at 45-55 °C for 16 hours. Then heated up to 75-85 °C and maintained about 0.5 hour.
Then cooled down to 45-55 °C for 0.5 hour and continued to cool the mixture down to 20-30 °C and stirred for 3 hours. Filtered and washed with IPA/n-Heptane (1/1, 3 mL><2). The wet cake was dried via vacuum at 75-80 °C for 2 hours to get the purified Form VI of the compound of Formula I.
Example 7. Preparation of Form VIII of the compound of Formula I
[0262] The polymorph Form VI of the compound of Formula I was dried in oven at 80 °C for 2 days to obtain the polymorph Form VIII.
Example 8. X-ray powder diffraction (XRD)
[0263] X-ray powder diffraction (XRD) patterns were obtained on a Bruker D8 Advance. A CuK source (=1.54056 angstrom) operating minimally at 40 kV and 40 mA scans each sample between 4 and 40 degrees 2-theta. The step size is 0.05°C and scan speed is 0.5 second per step.
Example 9. Thermogravimetric Analyses (TGA)
[0264] Thermogravimetric analyses were carried out on a TA Instrument TGA unit (Model TGA 500). Samples were heated in platinum pans from ambient to 300 °C at 10 °C/min with a nitrogen purge of 60mL/min (sample purge) and 40mL/min (balance purge). The TGA temperature was calibrated with nickel standard, MP=354.4 °C. The weight calibration was performed with manufacturer-supplied standards and verified against sodium citrate dihydrate desolvation.
Example 10. Differential scanning calorimetry (DSC)
[0265] Differential scanning calorimetry analyses were carried out on a TA Instrument DSC unit (Model DSC 1000 or 2000). Samples were heated in non-hermetic aluminum pans from ambient to 300 °C at 10 °C/min with a nitrogen purge of 50mL/min. The DSC temperature was calibrated with indium standard, onset of l56-l58°C, enthalpy of 25-29J/g.
Example 11. Hygroscopicity (DVS)
[0266] The moisture sorption profile was generated at 25°C using a DVS Moisture Balance Flow System (Model Advantage) with the following conditions: sample size approximately 5 to 10 mg, drying 25°C for 60 minutes, adsorption range 0% to 95% RH, desorption range 95% to 0% RH, and step interval 5%. The equilibrium criterion was <0.01% weight change in 5 minutes for a maximum of 120 minutes.
Example 12: Microscopy
[0267] Microscopy was performed using a Leica DMLP polarized light microscope equipped with 2.5X, 10X and 20X objectives and a digital camera to capture images showing particle shape, size, and crystallinity. Crossed polars were used to show birefringence and crystal habit for the samples dispersed in immersion oil.
Example 13: HPLC
[0256] HPLCs were preformed using the following instrument and/or conditions.
///////////////CK-101 , CK 101 , CK101 , phase II , Suzhou Neupharma, Checkpoint Therapeutics , Orphan Drug designation, EGFR mutation-positive NSCLC, NSCLC, CANCER, SOLID TUMOUR, China, RX-518, AK543910
OCCN1CCN(CC1)c5ccc(Nc2nc3c(cccc3cn2)c4cccc(NC(=O)C=C)c4)c(F)c5F
Picropodophyllin
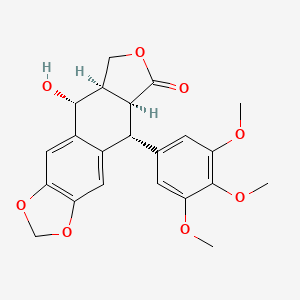
Picropodophyllin
Picropodophyllotoxin
CAS 477-47-4
AXL1717, NSC 36407, BRN 0099161
414.4 g/mol, C22H22O8
(5R,5aR,8aS,9R)-5-hydroxy-9-(3,4,5-trimethoxyphenyl)-5a,6,8a,9-tetrahydro-5H-[2]benzofuro[5,6-f][1,3]benzodioxol-8-one
Furo(3′,4′:6,7)naphtho(2,3-d)-1,3-dioxol-6(5aH)-one, 5,8,8a,9-tetrahydro-9-hydroxy-5-(3,4,5-trimethoxyphenyl)-, (5R-(5-alpha,5a-alpha,8a-alpha,9-alpha))-
5-19-10-00665 (Beilstein Handbook Reference)
Axelar is developing picropodophyllin, a small-molecule IGF-1 receptor antagonist for the treatment of cancer including NSCLC and malignant astrocytoma. In February 2019, a phase Ia study was planned to initiate for solid tumor in March 2019.
Picropodophyllin is a cyclolignan alkaloid found in the mayapple plant family (Podophyllum peltatum), and a small molecule inhibitor of the insulin-like growth factor 1 receptor (IGF1R) with potential antineoplastic activity. Picropodophyllin specifically inhibits the activity and downregulates the cellular expression of IGF1R without interfering with activities of other growth factor receptors, such as receptors for insulin, epidermal growth factor, platelet-derived growth factor, fibroblast growth factor and mast/stem cell growth factor (KIT). This agent shows potent activity in the suppression o f tumor cell proliferation and the induction of tumor cell apoptosis. IGF1R, a receptor tyrosine kinase overexpressed in a variety of human cancers, plays a critical role in the growth and survival of many types of cancer cells.
Picropodophyllotoxin is an organic heterotetracyclic compound that has a furonaphthodioxole skeleton bearing 3,4,5-trimethoxyphenyl and hydroxy substituents. It has a role as an antineoplastic agent, a tyrosine kinase inhibitor, an insulin-like growth factor receptor 1 antagonist and a plant metabolite. It is a lignan, a furonaphthodioxole and an organic heterotetracyclic compound.
Picropodophyllin has been investigated for the treatment of Non Small Cell Lung Cancer.
One of the largest challenges in pharmaceutical drug development is that drug compounds often are poorly soluble, or even insoluble, in aqeous media. Insufficient drug solubility means insufficient bioavailability, as well as poor plasma exposure of the drug when administered to humans and animals. Variability of plasma exposure in humans is yet a problem when developing drugs which are poorly soluble, or even insoluble, in aqeous media.
It is estimated that between 40% and 70 % of all new chemical entities identified in drug discovery programs, are insufficiently soluble in aqeous media (M. Lindenberg, S et al: European Journal of Pharmaceutics and Biopharmaceuticals, vol. 58, no.2, pp. 265-278, 2004). Scientists have investigated various ways of solving the problem with poor drug solubility in order to enhance bioavailability of poorly absorbed drugs, aiming at increasing their clinical efficacy when administered orally.
Technologies such as increase of the surface area and hence dissolution may sometimes solve solubility problems. Other techniques that may also solve bioavailability problems are addition of surfactants and polymers. However, each chemical compound has its own unique chemical and physical properties, and hence has its own unique challenges when being formulated into a pharmaceutical product that can exert its clinical efficacy.
Picropodophyllin is an insulin-like growth factor-1 receptor inhibitor fiGF-lR inhibitor) small-molecule compound belonging to the class of compounds denominated cyclolignans, having the chemical structure:
The patent applicant is presently entering clinical phase II development with its development compound picropodophyllin (AXL1717). However, picropodophyllin is poorly soluble in aqueous media. In a phase I clinical study performed by the applicant in 2012 (Ekman S et al; Acta Oncologica, 2016; 55: pp. 140-148), it was discovered that picropodophyllin, when administered as an oral suspension to lung cancer patients, resulted in unacceptable variability in drug exposure. A large variability in plasma exposure of the active drug picropodophyllin occurred not only within certain patients, but also between several patients.
Yet a problem with administering picropodophyllin as an aqeous solution, is that due to the poor solubility in aqueous media, it is difficult or even impossible to reach the required therapeutic doses.
The compound picropodophyllin is furthermore physically unstable, and transforms from amorphous picropodophyllin into crystalline picropodophyllin. Yet a stability problem with picropodophyllin is that it is chemically unstable in solution.

Product case, WO02102804
Patent
WO-2019130194
Novel amorphous forms of picropodophyllin , processes for their preparation and compositions comprising them are claimed. Also claims are their use for treating cancers, such as neurologic cancer, lung cancer, breast cancer, head and neck cancer, gastrointestinal cancer, genitourinary cancer, gynecologic cancer, hematologic cancer, musculoskeletal cancer, skin cancer, endocrine cancer, and eye cancers. , claiming picropodophyllin derivatives as modulators of insulin-like growth factor-1 receptor (IGF-1), useful for treating cancers, assigned to Axelar AB ,
CLIP

CLIP
https://pubs.rsc.org/en/content/articlelanding/2004/cc/b312245j/unauth#!divAbstract
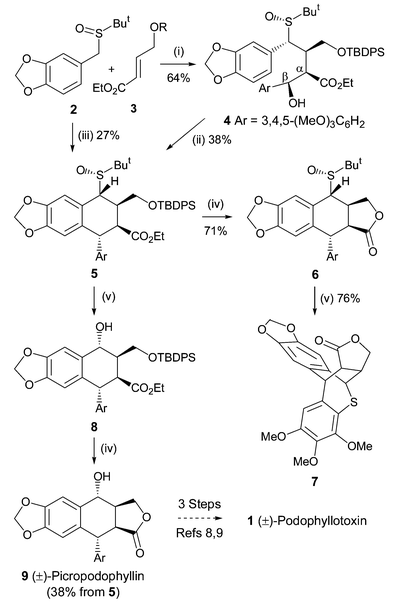
http://www.rsc.org/suppdata/cc/b3/b312245j/b312245j.pdf
dH(CDCl3; 300 MHz; Me4Si): 2.64-2.78 (1 H, m, 3-H), 3.23 (1 H, dd, J 4.4 and 8.2, 2-H), 3.81 (6 H, s, 2 x OMe), 3.85 (3 H, s, OMe), 4.09 (1 H, d, J 4.4, 1-H), 4.38–4.59 (3 H, m, 11-H2 and 4-H), 5.91 (1 H, d, J 1.5, OCH2O), 5.93 (1 H, d, J 1.5, OCH2O), 6.35 (1 H, s, 5-H/8-H), 6.46 (1 H, s, 2’-H and 6’-H) and 7.07 (1 H, s, 5-H/8-H).
CLIP
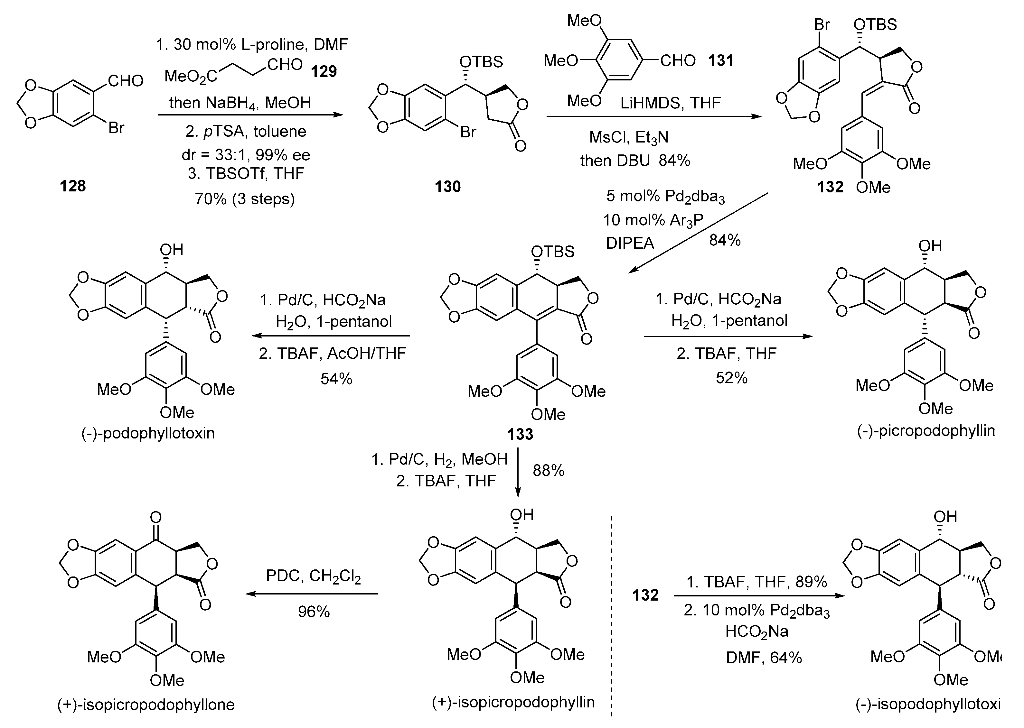
PAPER
Organic Letters (2018), 20(6), 1651-1654
https://pubs.acs.org/doi/abs/10.1021/acs.orglett.8b00408

A nickel-catalyzed reductive cascade approach to the efficient construction of diastereodivergent cores embedded in podophyllum lignans is developed for the first time. Their gram-scale access paved the way for unified syntheses of naturally occurring podophyllotoxin and other members.
Synthesis of (−)-Podophyllotoxin (1)
https://pubs.acs.org/doi/suppl/10.1021/acs.orglett.8b00408/suppl_file/ol8b00408_si_001.pdf
The residue was purified by flash column chromatography (petroleum ether/EtOAc = 4 : 1 → petroleum ether/EtOAc = 2 : 1) on silica gel to afford 1 (8.6 mg, 87% yield) as a white solid; Rf = 0.23 (petroleum ether/EtOAc = 1 : 1); [α]20 D = –115.00 (c = 1.00, CHCl3) [ref.13: [α]20 D = –101.7 (c = 0.55, EtOH)]; Mp. 167–168 °C; 1H NMR (400 MHz, CDCl3): δ = 7.11 (s, 1H), 6.51 (s, 1H), 6.37 (s, 2H), 5.98 (s, 1H), 5.96 (s, 1H), 4.77 (t, J = 8.4 Hz, 1H), 4.60 (t, J = 8.0 Hz, 1H), 4.59 (d, J = 4.4 Hz, 1H), 4.08 (dd, J = 9.6, 8.8 Hz, 1H), 3.81 (s, 3H), 3.75 (s, 6H), 2.84 (dd, J = 14.0, 4.4 Hz, 1H), 2.83−2.74 (m, 1H), 2.13 (d, J = 8.0 Hz, 1H, −OH) ppm; 13C NMR (100 MHz, CDCl3): δ = 174.6, 152.5 (2C), 147.7, 147.6, 137.1, 135.5, 133.3, 131.0, 109.7, 108.4 (2C), 106.3, 101.4, 72.6, 71.4, 60.7, 56.2 (2C), 45.2, 44.1, 40.6 ppm.

https://pubs.acs.org/doi/suppl/10.1021/acs.orglett.8b00408/suppl_file/ol8b00408_si_002.pdf


PAPER
Organic Letters (2017), 19(24), 6530-6533
https://pubs.acs.org/doi/abs/10.1021/acs.orglett.7b03236

he first catalytic enantioselective total synthesis of (−)-podophyllotoxin is accomplished by a challenging organocatalytic cross-aldol Heck cyclization and distal stereocontrolled transfer hydrogenation in five steps from three aldehydes. Reversal of selectivity in hydrogenation led to the syntheses of other stereoisomers from the common precursor.
https://pubs.acs.org/doi/suppl/10.1021/acs.orglett.7b03236/suppl_file/ol7b03236_si_001.pdf
(-)-Picropodophyllin 4. The lactone 5 (0.2 g, 0.38 mmol) was taken in 1-pentanol (5 mL) in a double neck RB flask at rt. Water (0.14 mL, 7.6 mmol) was added to above mixture and it was then degassed with argon followed by addition of Pd/C (0.04 g, 20% by wt.) and HCO2Na (0.78g, 11.4 mmol). The reaction mixture was heated at 40 °C for 12 h. On completion, the reaction mixture was diluted with EtOAc (200 mL), filtered through a celite pad and solvent was removed under vacuum. This crude mixture was dissolved in THF (3.8 mL), TBAF (1.9 mL, 1.9 mmol, 1M in THF) was added and stirred for 6 h at 27 °C. On completion, EtOAc (250 mL) was added, washed with water (100 mL), brine and dried over Na2SO4. After removal of solvent, the crude product was purified by column chromatography (hexanes-EtOAc, 3:2) to get the title compound as a white solid (0.082 g, 52%): Rf 0.32 (hexanes/EtOAc, 1:1); [α]25 D = -10.6 (c = 0.4, CHCl3) [lit. -10 (c = 0.3, CHCl3), -11 (c = 0.41, CHCl3)]3a,b;
Mp 214-216 °C; 1H NMR (600 MHz, CDCl3) δ 7.05 (s, 1H), 6.47 (s, 2H), 6.41 (s, 1H), 5.95 (d, J = 14.1 Hz, 2H), 4.5 (m, 2H), 4.44 (t, J = 8.0 Hz, 1H), 4.15 (d, J = 4.1 Hz, 1H), 3.86 (s, 3H), 3.83 (s, 6H), 3.24 (dd, J = 8.7, 5.0 Hz, 1H), 2.75 (m, 1H), 2.12 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 177.6, 153.7, 147.5, 147.1, 139.3, 137.4, 131.9, 130.6, 109.3, 105.9, 105.5, 101.2, 69.8, 69.6, 60.9, 56.3, 45.4, 44.1, 42.7; HRMS (ESI-TOF) m/z 437.1219 [(M+Na)+ ; calcd for C22H22O8Na+ : 437.1212].
PAPER
The Journal of organic chemistry (2000), 65(3), 847-60.
https://pubs.acs.org/doi/abs/10.1021/jo991582+

REF
Berichte der Deutschen Chemischen Gesellschaft [Abteilung] B: Abhandlungen (1932), 65B, 1846.
Justus Liebigs Annalen der Chemie (1932), 499, 59-76.
Justus Liebigs Annalen der Chemie (1932), 494, 126-42.
Journal of the American Chemical Society (1954), 76, 5890-1
Helvetica Chimica Acta (1954), 37, 190-202.
Journal of the American Chemical Society (1988), 110(23), 7854-8.
//////////////Picropodophyllin, AXL1717, NSC 36407, BRN 0099161, Picropodophyllotoxin, AXELAR, PHASE 1, CANCER, neurologic cancer, lung cancer, breast cancer, head and neck cancer, gastrointestinal cancer, genitourinary cancer, gynecologic cancer, hematologic cancer, musculoskeletal cancer, skin cancer, endocrine cancer, eye cancers, NSCLC, malignant astrocytoma, SOLID TUMOUR
COC1=CC(=CC(=C1OC)OC)C2C3C(COC3=O)C(C4=CC5=C(C=C24)OCO5)O
Podofilox, Podophyllotoxin, Wartec, Condyline, Condylox
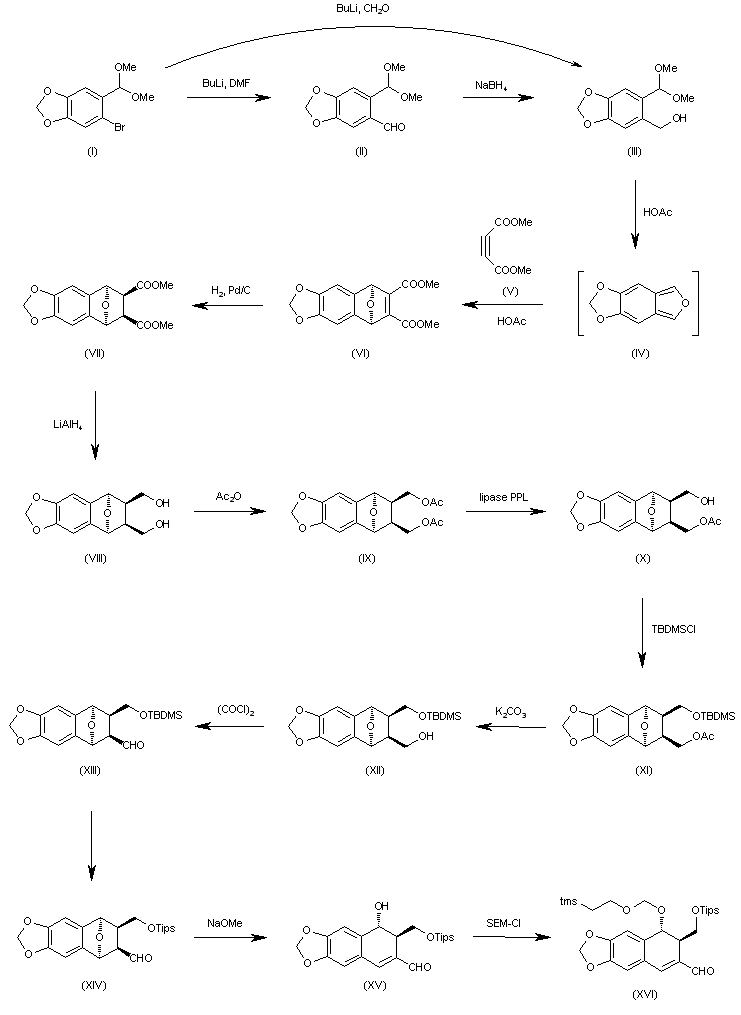
| J Org Chem 2000,65(3),847 |
The formylation of 6-bromo-1,3-benzodioxole-5-carbaldehyde dimethyl acetal (I) with BuLi and DMF gives the 6-formyl derivative (II), which is reduced with NaBH4 in ethanol to yield the corresponding carbinol (III). The cyclization of (III) with dimethyl acetylenedicarboxylate (V) in hot acetic acid (through the nonisolated intermediate (IV)) affords dimethyl 1,4-epoxy-6,7-(methylenedioxy)naphthalene-2,3-dicarboxylate (VI), which is hydrogenated with H2 over Pd/C in ethyl acetate to give the (1R*,2S*,3R*,4S*)-tetrahydro derivative (VII). The reduction of (VII) with LiAlH4 in refluxing ethyl ether affords the corresponding bis carbinol (VIII), which is treated with acetic anhydride to afford the diacetate (IX). The enzymatic monodeacetylation of (VIII) with PPL enzyme in DMSO/buffer gives (1R,2R,3S,4S)-2-(acetoxymethyl)-1,4-epoxy-3-(hydroxymethyl)-6,7-(methylenedioxy)-1,2,3,4-tetrahydronaphthalene (X), which is silylated with TBDMS-Cl and imidazole in DMF yielding the silyl ether (XI). The hydrolysis of the acetoxy group of (XI) with K2CO3 in methanol affords the carbinol (XII), which is oxidized with oxalyl chloride in dichloromethane affording the carbaldehyde (XIII). The exchange of the silyl protecting group of (XIII) (for stability problems) provided the triisopropylsilyl ether (XIV), which is treated with sodium methoxide in methanol to open the epoxide ring yielding the hydroxy aldehyde (XV). The protection of the hydroxy group of (XV) with 2-(trimethylsilyl)ethoxymethyl chloride and DIEA in dichloromethane provides the corresponding ether (XVI). The carbinol (III) can also be obtained directly from 6-bromo-1,3-benzodioxole-5-carbaldehyde dimethyl acetal (I) by reaction with formaldehyde and BuLi in THF.
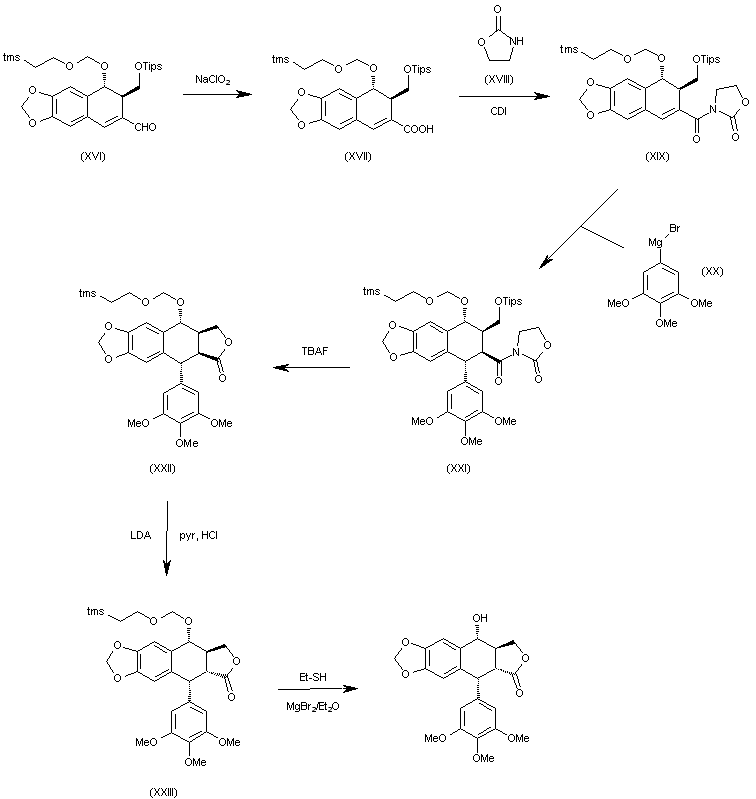
The oxidation of the aldehyde group of (XVI) with NaClO2 in tert-butanol affords the corresponding carboxylic acid (XVII), which is condensed with 2-oxazolidinone (XVIII) by means of carbonyldiimidazole (CDI) in THF to give the acyl imidazolide (XIX). The arylation of (XIX) with 3,4,5-trimethoxyphenylmagnesium bromide (XX) in THF yields the expected addition product (XXI), which is cyclized by means of TBAF in hot THF to afford the tetracyclic intermediate (XXII). Isomerization of the cis-lactone ring of (XXII) with LDA in THF affords intermediate (XXIII) with its lactone ring with the correct trans-conformation. Finally, this compound is deprotected with ethyl mercaptane and MgBr2 in ethyl ether to provide the target compound.
Synthesis 1992,719
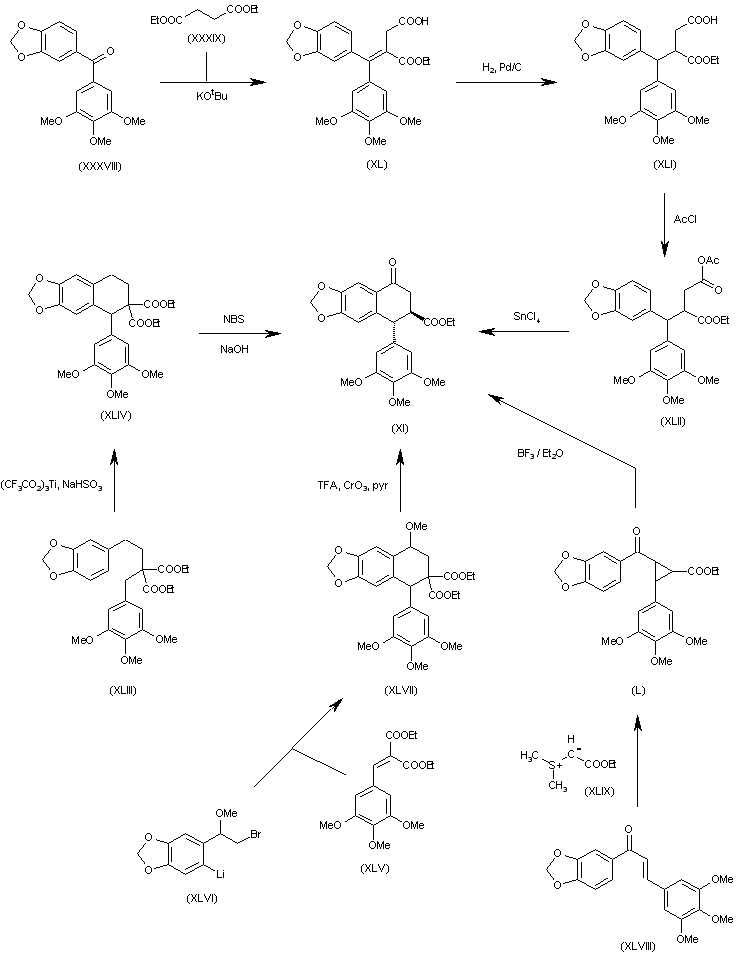
The intermediate trans-8-oxo-5-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetra-hydronaphtho[2,3-d][1,3]benzodioxole-6-carboxylic acid ethyl ester (XI) has been obtained by several different ways: (a) The condensation of benzophenone (XXXVIII) with diethyl malonate (XXXIX) by means of t-BuOK gives the alkylidenemalonate (XL), which is hydrogenated with H2 over Pd/C to the alkylmalonate hemiester (XLI). The reaction of (XLI) with acetyl chloride affords the mixed anhydride (XLII), which is finally cyclized to the target (XI) by means of SnCl4. (b) The cyclization of the malonic ester derivative (XLIII) by means of Ti(CF3–CO2)3 gives the 5-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydronaphtho [2,3-d][1,3]dioxole-6,6-dicarboxylic acid dimethyl ester (XLIV), which is finally oxidized and decarboxylated with NBS and NaOH in methanol to afford the target intermediate (XI). (c) The cyclization of the benzylidenemalonate (XLV) with the aryllithium derivative (XLVI) gives the 8-methoxy-5-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydronaphtho[2,3-d][1,3]dioxole-6,6-dicarboxylic acid dimethyl ester (XLVII), which is demethylated with TFA and oxidized with CrO3 and pyridine to the target compound (XI). (d) The cyclopropanation of the chalcone (XLVIII) with (ethoxycarbonyl) (dimethylsulfonium)methylide (XLIX) gives the cyclopropanecarboxylate (L), which is finally rearranged with BF3/Et2O to the target intermediate (IX).
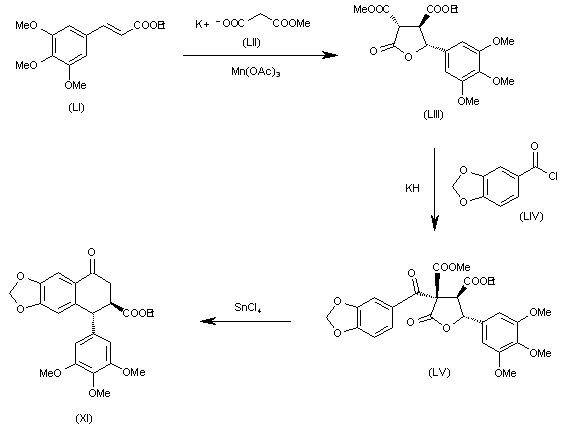
The cyclization of 3,4,5-trimethoxycinnamic acid ethyl ester (LI) with malonic acid ethyl ester potassium salt (LII) by means of Mn(OAc)3 gives the tetrahydrofuranone (LIII), which is acylated with 1,3-benzodioxol-5-ylcarbonyl chloride (LIV) yielding the tetrahydrofuranone (LV). Finally, this compound is rearranged and decarboxylated with SnCl4 to the target intermediate (XI).
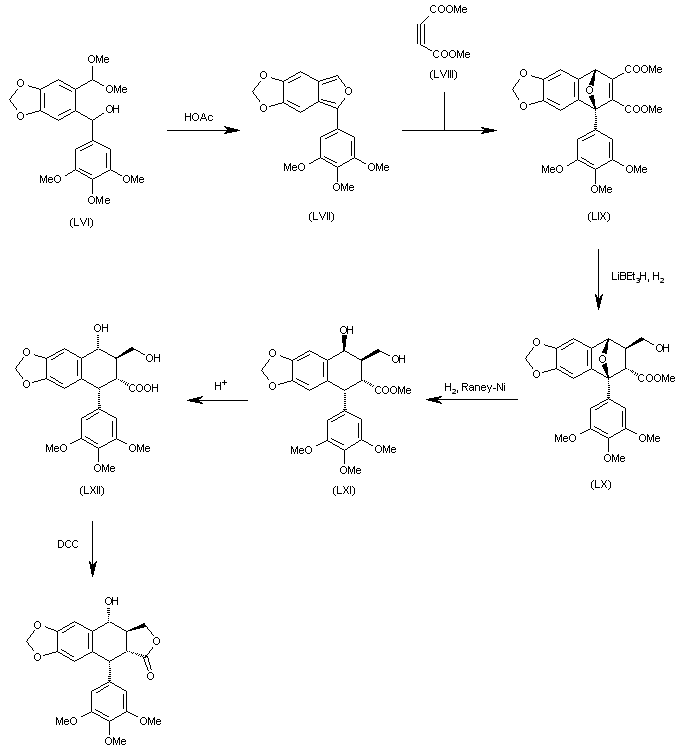
The cyclization of 6-[1-hydroxy-1-(3,4,5-trimethoxyphenyl)methyl]-1,3-benzodioxol-5-carbaldehyde dimethylacetal (LVI) by means of AcOH gives 5-(3,4,5-trimethoxyphenyl)-1,3-dioxolo[4,5-f]isobenzofuran (LVII), which is submitted to a Diels-Alder cyclization with acetylenedicarboxylic acid dimethyl ester (LVIII) yielding the epoxy derivative (LIX). The selective reduction of (LIX) with LiBEt3H and H2 affords the carbinol (LX), which is treated with H2 over RaNi in order to open the epoxide ring to give the diol (LXI) with the wrong configuration at the secondary OH group. The treatment of (LXI) with aqueous acid isomerizes the secondary OH group to (LXII) with the suitable configuration. Finally, this compound is cyclized with DCC to the desired target compound.
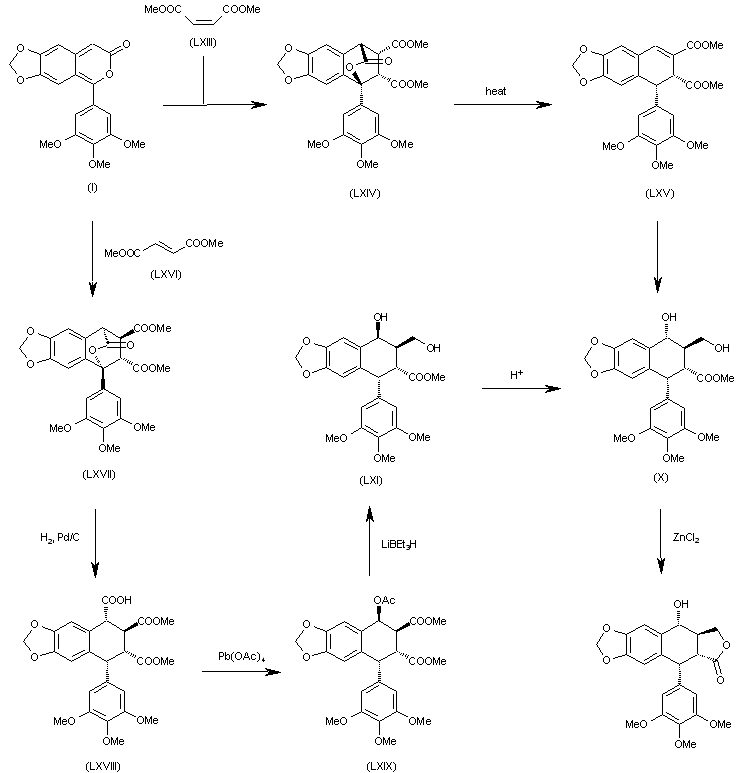
The Diels-Alder cyclization of 5-(3,4,5-trimethoxyphenyl)-7H-pyrano[3,4-f][1,3]benzodioxol-7-one (I) with dimethyl maleate (LXIII) gives the expected adduct (LXIV), which by thermal extrusion of CO2 yields the dihydronaphthodioxole (LXV). This compound is then converted to dihydroxycompound (X), which is finally cyclized by means of ZnCl2 to provide the target compound. The Diels-Alder cyclization of 5-(3,4,5-trimethoxyphenyl)-7H-pyrano[3,4-f][1,3]benzodioxol-7-one (I) with dimethyl fumarate (LXVI) gives the expected adduct (LXVII), which by hydrogenation with H2 over Pd/C yields the tricarboxylic acid derivative (LXVIII). The reaction of (LXVIII) with Pb(OAc)4 affords the acetoxy derivative (LXIX), which is selectively reduced with LiBEt3H providing the diol (LXI) with the wrong configuration at the secondary OH group. The treatment of (LXI) with aqueous acid isomerizes the secondary OH group to give the previously described (X) with the suitable configuration.
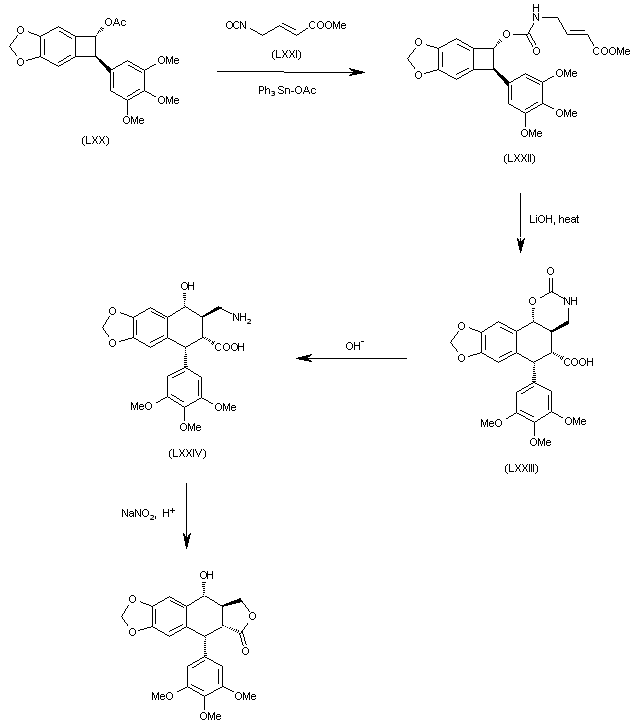
The reaction of benzocyclobutane derivative (LXX) with isocyanate (LXXI) by means of Ph3SnOAc gives the carbamate (LXXII), which is cyclized by a thermal treatment with LiOH yielding the tetracyclic carboxylic acid (LXXIII). The opening of the oxazinone ring of (LXXIII) in basic medium affords the tricyclic amino acid (LXXIV), which is finally cyclized to the target compound by reaction with sodium nitrite in acidic medium (pH = 4).
J Chem Soc Chem Commun 1993,1200
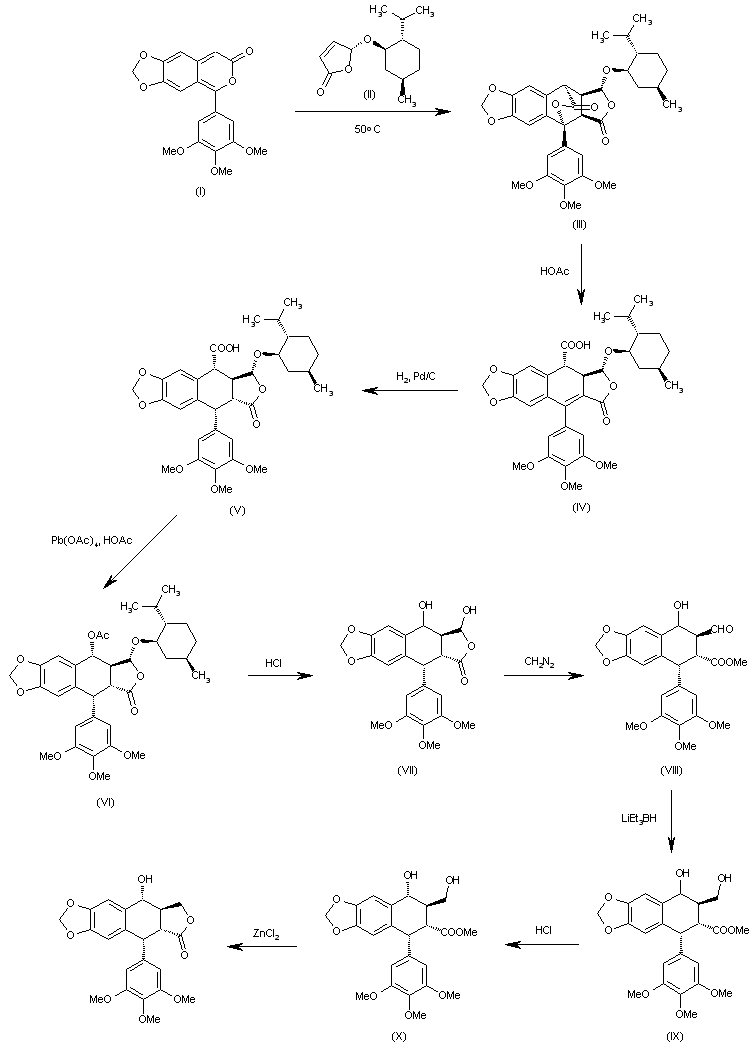
The Diels-Alder cyclization of 5-(3,4,5-trimethoxyphenyl)-7H-pyrano[3,4-f][1,3]benzodioxol-7-one (I) with the chiral dihydrofuranone (II) in hot acetonitrile gives the pentacyclic anhydride (III), which is opened with warm acetic acid yielding the carboxylic acid (IV). Hydrogenation of the benzylic double bond of (IV) with H2 over Pd/C affords (V), which is treated with lead tetraacetate and acetic acid in THF to give the acetoxy compound (VI). The hydrolysis of the acetoxy group and the menthol hemiacetal group with HCl in hot dioxane yields the diol (VII), which is treated with diazomethane in ether/methanol affording the aldehyde (VIII). The reduction of the aldehyde group of (VIII) with LiEt3BH in THF gives the diol (IX) as a diastereomeric mixture, which is treated with HCl in THF to afford the diol (X) with the right conformation. Finally, this compound is lactonized to the target compound with ZnCl2 in THF.
//////////
