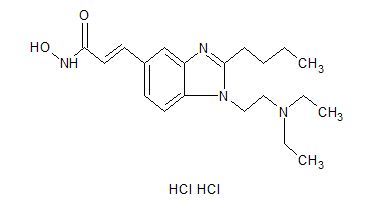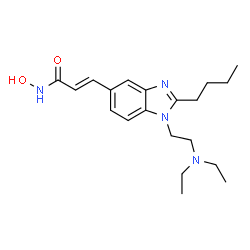
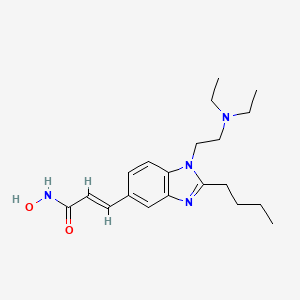

Pracinostat
- Molecular Formula C20H30N4O2
- Average mass 358.478 Da
2-Propenamide, 3-[2-butyl-1-[2-(diethylamino)ethyl]-1H-benzimidazol-5-yl]-N-hydroxy-, (2E)-
SB939
(2E)-3-{2-butyl-1-[2-(diethylamino)ethyl]-1,3-benzodiazol-5-yl}-N-hydroxyprop-2-enamide
N-hydroxy-1-[(4-methoxyphenyl)methyl]-1H-indole-6-carboxamide
929016-98-8 DI HCl salt, C20 H30 N4 O2 . 2 Cl H, 431.4
929016-96-6 (free base)
929016-97-7 (trifluoroacetate)
S*BIO (Originator)
Leukemia, acute myeloid, phase 3, helsinn
CAS 929016-98-8 DI HCl salt, C20 H30 N4 O2 . 2 Cl H, 431.4
E)-3-[2-Butyl-1-(2-diethylaminoethyl)-1H-benzimidazol-5-yl]-N-hydroxyacrylamide Dihydrochloride Salt
Pracinostat (SB939) is an orally bioavailable, small-molecule histone deacetylase (HDAC) inhibitor based on hydroxamic acid with potential anti-tumor activity characterized by favorable physicochemical, pharmaceutical, and pharmacokinetic properties.
WO-2017192451 describes Novel polymorphic crystalline forms of pracinostat (designated as Form 3) and their hydrates, processes for their preparation and compositions and combination comprising them are claimed. Also claimed is their use for inhibiting histone deacetylase and treating cancer, such as myelodysplastic syndrome (MDS), acute myeloid leukemia (AML), breast cancer, colon cancer, prostate cancer, pancreas cancer, leukemia, lymphoma, ovary cancer, melanoma and neuroblastoma.
See WO2014070948 , Helsinn , under sub-license from MEI Pharma (under license from S*Bio), is developing pracinostat, an oral HDAC inhibitor, for treating hematological tumors, including AML, MDS and myelofibrosis.
The oncolytic agent pracinostat hydrochloride is an antagonist of histone deacetylase 1 (HDAC1) and 2 (HDAC2) that was discovered by the Singapore-based company S*BIO. Helsinn obtained the exlusive development and commercialization rights in July 2016, and is conducting phase III clinical trials in combination with azacitidine in adults with newly diagnosed acute myeloid leukemia. Phase II trials are also under way for the treatment of previously untreated intermediate-2 or high risk myelodysplastic syndrome patients and for the treatment of primary or post essential thrombocythemia/polycythemia vera) in combination with ruxolitinib.In North America, S*BIO had been conducting phase II clinical trials of pracinostat hydrochloride in patients with solid tumors and for the treatment of myeloproliferative diseases and phase I clinical trials in patients with leukemia; however, recent progress reports are not available at present. The University of Queensland had been evaluating the compound in preclinical studies for malaria.

University of Queensland

MEI Pharma
The Canadian Cancer Society Research Institute (the research branch of the Canadian Cancer Society upon its integration with the National Cancer Institute of Canada to form the new Canadian Cancer Society) is conducting phase II clinical trials in Canada for the treatment of recurrent or metastatic prostate cancer.

Canadian Cancer Society Research Institute
In 2012, the product was licensed to MEI Pharma by S*BIO on a worldwide basis. In 2016, MEI Pharma and Helsinn entered into a licensing, development and commercialization agreement by which Helsinn obtained exclusive worldwide rights (including manufacturing and commercialization rights).

HELSINN
In 2014, the FDA assigned an orphan drug designation to MEI Pharma for the treatment of acute myeloid leukemia. In 2016, the product received breakthrough therapy designation in the U.S. in combination with azacitidine for the treatment of patients with newly diagnosed acute myeloid leukemia (AML) who are older than 75 years of age or unfit for intensive chemotherapy.
Pracinostat is an orally available, small-molecule histone deacetylase (HDAC) inhibitor with potential antineoplastic activity. Pracinostat inhibits HDACs, which may result in the accumulation of highly acetylated histones, followed by the induction of chromatin remodeling; the selective transcription of tumor suppressor genes; the tumor suppressor protein-mediated inhibition of tumor cell division; and, finally, the induction of tumor cell apoptosis. This agent may possess improved metabolic, pharmacokinetic and pharmacological properties compared to other HDAC inhibitors.
Pracinostat is a novel HDAC inhibitor with improved in vivo properties compared to other HDAC inhibitors currently in clinical trials, allowing oral dosing. Data demonstrate that Pracinostat is a potent and effective anti-tumor drug with potential as an oral therapy for a variety of human hematological and solid tumors
SYNTHESIS


Clinically tested HDAC inhibitors.
Activity
Pracinostat selectively inhibits HDAC class I,II,IV without class III and HDAC6 in class IV,[1] but has no effect on other Zn-binding enzymes, receptors, and ion channels. It accumulates in tumor cells and exerts a continuous inhibition to histone deacetylase,resulting in acetylated histones accumulation, chromatin remodeling, tumor suppressor genes transcription, and ultimately, apoptosis of tumor cells.[2]
Clinical medication
Clinical studies suggests that pracinostat has potential best pharmacokinetic properties when compared to other oral HDAC inhibitors.[3]In March 2014, pracinostat has granted Orphan Drug for acute myelocytic leukemia (AML) and for the treatment of T-cell lymphoma by the Food and Drug Administration.
Clinical Trials
PATENT
WO2005028447
Scheme I
Scheme II
 Scheme III
Scheme III
 Scheme IV
Scheme IV
 Scheme V
Scheme V
PAPER
Discovery of (2E)-3-{2-Butyl-1-[2-(diethylamino)ethyl]-1H-benzimidazol-5-yl}-N-hydroxyacrylamide (SB939), an Orally Active Histone Deacetylase Inhibitor with a Superior Preclinical Profile
Haishan Wang*†, Niefang Yu†, Dizhong Chen†, Ken Chi Lik Lee†, Pek Ling Lye†, Joyce Wei Wei Chang†, Weiping Deng†, Melvin Chi Yeh Ng†, Ting Lu†, Mui Ling Khoo†, Anders Poulsen†, Kanda Sangthongpitag‡, Xiaofeng Wu‡, Changyong Hu‡, Kee Chuan Goh‡, Xukun Wang‡, Lijuan Fang‡, Kay Lin Goh‡, Hwee Hoon Khng‡, Siok Kun Goh‡, Pauline Yeo§, Xin Liu§, Zahid Bonday‡, Jeanette M. Wood‡, Brian W. Dymock†, Ethirajulu Kantharaj§, and Eric T. Sun†
†Chemistry Discovery, ‡Biology Discovery, and §Pre-Clinical Development, S*BIO Pte Ltd., 1 Science Park Road, No. 05-09 The Capricorn, Singapore Science Park II, Singapore 117528, Singapore
J. Med. Chem., 2011, 54 (13), pp 4694–4720
DOI: 10.1021/jm2003552
Abstract

A series of 3-(1,2-disubstituted-1H-benzimidazol-5-yl)-N-hydroxyacrylamides (1) were designed and synthesized as HDAC inhibitors. Extensive SARs have been established for in vitro potency (HDAC1 enzyme and COLO 205 cellular IC50), liver microsomal stability (t1/2), cytochrome P450 inhibitory (3A4 IC50), and clogP, among others. These parameters were fine-tuned by carefully adjusting the substituents at positions 1 and 2 of the benzimidazole ring. After comprehensive in vitro and in vivo profiling of the selected compounds, SB939 (3) was identified as a preclinical development candidate. 3 is a potent pan-HDAC inhibitor with excellent druglike properties, is highly efficacious in in vivo tumor models (HCT-116, PC-3, A2780, MV4-11, Ramos), and has high and dose-proportional oral exposures and very good ADME, safety, and pharmaceutical properties. When orally dosed to tumor-bearing mice, 3 is enriched in tumor tissue which may contribute to its potent antitumor activity and prolonged duration of action. 3 is currently being tested in phase I and phase II clinical trials.
(E)-3-[2-Butyl-1-(2-diethylaminoethyl)-1H-benzimidazol-5-yl]-N-hydroxyacrylamide Dihydrochloride Salt (3)
The freebase of 3 was prepared according to procedure D. The hydroxamic acid moiety was identified by 1H–15N HSQC (DMSO-d6) with δN = 169.0 ppm (CONHOH). Other nitrogens in 3were identified by 1H–15N HMBC (DMSO-d6) with δN of 241.4 ppm for N3 of the benzimidazole ring, 152.3 ppm for N1, and 41.3 ppm for the diethylamino group (reference to nitromethane δN = 380.0 ppm in CDCl3). The dihydrochloride salt of 3 was prepared according to procedure D as white or off-white solid or powder in ∼60% yield from 9 in two steps. LC–MS m/z 359.2 ([M + H]+).
1H NMR (DMSO-d6) δ 11.79 (brs, 1H, NH or OH), 10.92 (very br s, 1H), 8.18 (d, J = 8.6 Hz, 1H), 7.97 (s, 1H), 7.79 (d, J = 8.6 Hz, 1H), 7.64 (d, J = 15.8 Hz, 1H), 6.65 (d, J = 15.8 Hz, 1H), 5.01 (t-like, J = 7.7 Hz, 2H), 3.48 (m, 2H), 3.30–3.19 (m, 6H), 1.87 (quintet, J = 7.8 Hz, 2H), 1.47 (sextet, J = 7.5 Hz, 2H), 1.29 (t, J = 7.2 Hz, 6H), 0.97 (t, J = 7.3 Hz, 3H);
13C NMR (DMSO-d6) δ 162.3, 156.0, 137.3 (CH), 132.8, 132.3, 132.0 (br, identified by HMBC), 124.7 (CH), 120.2 (CH), 113.1 (2 × CH), 48.2, 46.3, 39.0, 28.1, 25.0, 21.7, 13.6, 8.3.
Anal. (C20H30N4O2·2HCl·0.265H2O) C, H, N, Cl. Water content = 1.09% (Karl Fisher method). HRMS (ESI) m/z [M + H]+ calcd for C20H31N4O2, 359.2442; found, 359.2449.
PATENT
WO 2007030080
http://google.com/patents/WO2007030080A1?cl=en
SEE
WO 2008108741
WO 2014070948
Patent
WO-2017192451
References
- Jump up^ “In vitro enzyme activity of SB939 and SAHA”. 22 Aug 2014.
- Jump up^ “The oral HDAC inhibitor pracinostat (SB939) is efficacious and synergistic with the JAK2 inhibitor pacritinib (SB1518) in preclinical models of AML”. Blood Cancer Journal. doi:10.1038/bcj.2012.14.
- Jump up^ Veronica Novotny-Diermayr; et al. (March 9, 2010). “SB939, a Novel Potent and Orally Active Histone Deacetylase Inhibitor with High Tumor Exposure and Efficacy in Mouse Models of Colorectal Cancer”. Mol Cancer Ther. doi:10.1158/1535-7163.MCT-09-0689.
| Cited Patent |
Filing date |
Publication date |
Applicant |
Title |
| WO2005028447A1 * |
Sep 21, 2004 |
Mar 31, 2005 |
S*Bio Pte Ltd |
Benzimidazole derivates: preparation and pharmaceutical applications |
| US20050137234 * |
Dec 14, 2004 |
Jun 23, 2005 |
Syrrx, Inc. |
Histone deacetylase inhibitors |
| Citing Patent |
Filing date |
Publication date |
Applicant |
Title |
| WO2009084544A1 * |
Dec 24, 2008 |
Jul 9, 2009 |
Idemitsu Kosan Co., Ltd. |
Nitrogen-containing heterocyclic derivative and organic electroluminescent device using the same |
| WO2010043953A2 * |
Oct 14, 2009 |
Apr 22, 2010 |
Orchid Research Laboratories Ltd. |
Novel bridged cyclic compounds as histone deacetylase inhibitors |
| WO2010043953A3 * |
Oct 14, 2009 |
Mar 24, 2011 |
Orchid Research Laboratories Ltd. |
Novel bridged cyclic compounds as histone deacetylase inhibitors |
| WO2017030938A1 * |
Aug 12, 2016 |
Feb 23, 2017 |
Incyte Corporation |
Heterocyclic compounds and uses thereof |
| DE102007037579A1 |
Aug 9, 2007 |
Feb 19, 2009 |
Emc Microcollections Gmbh |
Neue Benzimidazol-2-yl-alkylamine und ihre Anwendung als mikrobizide Wirkstoffe |
| US8865912 |
Jan 27, 2014 |
Oct 21, 2014 |
Glaxosmithkline Llc |
Benzimidazole derivatives as PI3 kinase inhibitors |
| US9024029 |
Sep 3, 2013 |
May 5, 2015 |
Mei Pharma, Inc. |
Benzimidazole derivatives: preparation and pharmaceutical applications |
| US9062003 |
Sep 9, 2014 |
Jun 23, 2015 |
Glaxosmithkline Llc |
Benzimidazole derivatives as PI3 kinase inhibitors |
| US9156797 |
May 15, 2015 |
Oct 13, 2015 |
Glaxosmithkline Llc |
Benzimidazole derivatives as PI3 kinase inhibitors |
| US9402829 |
Feb 20, 2015 |
Aug 2, 2016 |
Mei Pharma, Inc. |
Benzimidazole derivatives: preparation and pharmaceutical applications |
| US9717713 |
Jun 10, 2016 |
Aug 1, 2017 |
Mei Pharma, Inc. |
Benzimidazole derivatives: preparation and pharmaceutical applications |
Pracinostat
|
| Names |
| IUPAC name
(E)-3-(2-Butyl-1-(2-(diethylamino)ethyl)-1H-benzo[d]imidazol-5-yl)-N-hydroxyacrylamide
|
| Other names
Pracinostat
|
| Identifiers |
|
|
|
|
|
|
| ChemSpider |
|
|
|
|
|
|
|
|
| Properties |
|
|
C20H30N4O2 |
| Molar mass |
358.49 g·mol−1 |
| Density |
1.1±0.1 g/cm3 |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
//////////////Pracinostat, PCI 34051, SB939, orphan drug designation, Leukemia, acute myeloid, phase 3, helsinn
CCCCC1=NC2=C(N1CCN(CC)CC)C=CC(=C2)C=CC(=O)NO
“NEW DRUG APPROVALS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....