
Home » Posts tagged 'Prader-Willi syndrome'
Tag Archives: Prader-Willi syndrome
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
HM04 or H0900
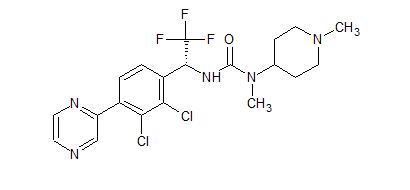
![3-[(1R)-1-(2,3-Dichloro-4-pyrazin-2-ylphenyl)-2,2,2-trifluoroethyl]-1-methyl-1-(1-methylpiperidin-4-yl)urea.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=91884613&t=l)
HM04 or H0900
Cas 1808913-24-7
(R)-3-(1-(2,3-dichloro-4-(pyrazin-2-yl)phenyl)-2,2,2-trifluoroethyl)-1-methyl-1-(1-methylpiperidin-4-yl) urea
The compound was disclosed in WO2015134839 . Helsinn under license from Novo Nordisk , is investigating ghrelin antagonists for treating obesity, Prader-Willi syndrome and other metabolic disorders; in May 2015, the program was listed as being in preclinical development
Helps reducing ghrelin signaling activity and treating disorder associated with an increase in ghrelin level (eg food abuse, alcohol addiction, and Prader-Willi syndrome).
Ghrelin, a growth hormone-releasing peptide produced by ghrelinergic cells in the gastrointestinal tract, is understood to function as a neuropeptide that regulates energy metabolism by stimulating appetite. The modulation, for example inhibition, of ghrelin signaling, through the ghrelin/growth hormone secretagogue receptor (GHS-Rla), is an attractive target for pharmacological treatment of disorders associated with high ghrelin level. Potential disorders for treatment using ghrelin modulators include food abuse (such as binge eating, obesity, hyperphagia (or uncontrollable appetite), post-dieting body weight rebound (including post-dieting hyperphagia), alcohol addiction, and genetic diseases associated with increased ghrelin level (e.g., Prader-Willi syndrome (PWS)).
PATENT
US 20150252021

PATENT
WO2015134839
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015134839
Example 1
nthesis of Intermediate lk
Intermediate k
Step 1:
To a solution of la (100 g, 0.62 mol) in DMF (1.2 L) was added N-bromosuccinimide (110 g, 0.62 mol) at 0 °C. The mixture was stirred at room temperature for 4 h, then water (800 mL) was added and the resulting mixture was extracted with EtOAc (3 x 500 mL). The combined organic layers were dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was triturated with petroleum ether to provide lb (133.7 g, 89% yield) as a brown solid. !H-NMR (CDC13, 300 MHz): δ= 7.30 (d, 1 H), 6.59 (d, 1 H), 4.22 (br, 2 H). LC-MS: 241 [M+l]+.
Step 2:
To a solution of lb (133.7 g, 0.55 mol) in dry CH2C12 (1.5 L) was added acetic anhydride (110 g, 0.62 mol) dropwise over a period of 20 minutes at room temperature. The mixture was stirred at room temperature overnight, then diluted with CH2C12 (300 mL) and washed with water (150 mL) and brine (200 mL). The organic layer was separated, dried over anhydrous Na2SC>4 and concentrated under reduced pressure. The residue was triturated with petroleum ether (300 mL) to provide compound lc (143.0 g, 91% yield) as a white solid. ¾-NMR (CDC13, 400 MHz): δ= 8.26 (d, 1 H), 7.63 (br, 1 H), 7.54 (d, 1 H), 2.26 (s, 3 H). LC-MS: 280 [M-l]“.
Step 3:
A mixture of compound lc (50.0 g, 0.18 mol), butyl vinyl ether (Id, 89.0 g, 0.89 mol), bis(l,3-diphenylphosphino)propane (DPPP, 22.0 g, 0.053 mol), TEA (100 mL, 0.71 mol) and Pd(OAc)2 (6.4 g, 0.027 mol) in DMSO (1.2 L) was heated at 130 °C under N2 overnight. After the reaction was completed, the mixture was cooled to 0 °C and 2N HC1 (480 mL) was added dropwise over a period of 30 minutes. Then, the mixture was extracted with EtOAc (3 x 100 mL). The combined organic layers were dried over anhydrous a2S04 and concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc: PE=1 : 10) to provide le (19.5 g, 45% yield) as a yellow solid. 1H-NMR (CDC13, 400 MHz): 3= 8.46 (d, 1 H), 7.82 (br, 1 H), 7.51 (d, 1 H), 2.63 (s, 3 H), 2.29 (s, 3 H). LC-MS: 244 [M-l]“.
Step 4:
To a solution of le (21.9 g, 89.4 mmol) in MeOH (350 mL) was added 2N NaOH solution (350 mL) at room temperature. The mixture was heated at 50 °C overnight, then cooled and concentrated under reduced pressure. The resulting solid was triturated with water (100 mL) for 30 min and filtered to provide If (18.0 g, 98% yield) as a brown solid. ¾-NMR (CDC13, 400 MHz): 3= 7.48 (d, 1 H), 6.68 (d, 1 H), 4.56 (br, 2 H), 2.62 (s, 3 H). LC-MS: 202[M-1]\
Step 5:
To a mixture of compound If (18.0 g, 89.2 mmol) and ice (360 g) in cone. HC1 (180 mL) was added a solution of NaN02 (9.2 g, 133.7 mmol) in water (20 mL) dropwise over a period of 30 minutes, and the resulting mixture stirred in an ice bath for 30 min. A solution of KI (74.0 g, 446 mmol) in water (360 mL) was added dropwise over 45 min at 0 °C. The mixture was stirred for 30 min and then extracted with EtOAc (3 x 100 mL). The combined organic layers were dried over anhydrous Na2SC>4 and concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc: PE=1 :40) to provide lg (23.9 g, 86% yield) as a yellow solid. 1H-NMR (CDC13, 400 MHz): 3= 7.6 (d, 1 H), 7.06 (d, 1 H), 2.62 (s, 3 H).
Step 6:
To a solution of lg (23.9 g, 76.1 mmol) in MeOH (100 mL)/THF (100 mL) was slowly added NaB¾ (2.9 g, 76.1 mmol) at 0 °C. The mixture was stirred at room temperature for 5 min, and then quenched with water (100 mL). The mixture was extracted with EtOAc (3 x 100 mL). The combined organic layers were dried over anhydrous Na2SC>4 and concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc: PE=1 : 10) to provide lh (22.4 g, 93% yield) as a white solid. 1H-NMR (CDC13, 400 MHz): 3= 7.81 (d, 1 H), 7.26 (d, 1 H), 5.23 (q, 1 H), 2.17 (br, 1 H), 1.47 (d, 3 H).
Step 7:
To a mixture of lh (22.4 g, 70.9 mmol), phthalimide (12.5 g, 85.0 mmol) and PPh3 (22.3 g, 85.0 mmol) in dry THF (450 mL) was added DIAD (21.5 g, 106.3 mmol) at room temperature under N2 protection. The mixture was stirred at room temperature overnight and then concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc: PE=1 : 15) to provide li (18.5 g, 58% yield) as a white solid. 1H-NMR (CDC13, 400 MHz): 3= 7.78-7.84 (m, 3 H), 7.70-7.73 (m, 2 H), 7.41-7.43 (d, 1 H), 5.76-5.81 (q, 1 H), 1.84 (d, 3 H).
Step 8:
A solution of li (7.2 g, 16.2 mmol) and hydrazine hydrate (98%, 4.0 g, 80.9 mmol) in MeOH (150 mL) was heated under reflux for 2 h, then cooled and concentrated under reduced pressure. The residue was diluted with water (100 mL) and extracted with CH2C12 (3 x 100 mL). The combined organic layers were dried over anhydrous Na2SC>4 and concentrated under reduced pressure to give lj (3.8 g, 75% yield) as a white solid. 1H-NMR (CDC13, 400 MHz): 3= 7.81 (d, 1 H), 7.25 (d, 1 H), 4.55 (q, 1 H), 1.36-1.38 (d, 3 H). LC-MS: 316 [M+l]+.
Step 9:
To a solution of lj (41. Og, 0.13 mol) in methyl tert-butyl ether (750 mL) was added slowly a solution of D-mandelic acid (7.8 g, 0.052 mol) in methyl tert-butyl ether (1 10 mL) at 45°C. The mixture was stirred at this temperature for 30 min then cooled and filtered. White solid obtained was partitioned between 5% NaOH solution (300 mL) and methyl tert-butyl ether (300 mL). The bi -phases were separated and the aqueous phase was extracted with methyl tert-butyl ether (300 mL). The combined organic layer was concentrated to provide Intermediate lk (12 g, 58.5% yield) as a white solid (ee%=98.0%, Chiralpak AD-H, 5 μπι, 4.6*250mm, mobile phase: Hex: EtOH : DEA=80 : 20 : 0.2), retention time = 6.408 min).
Example 2
Synthesis of Compoun
A suspension of N-methyl-4-piperidone 2a (13.3 g, 58.6 mmol), NH2Me (30% in MeOH, 100 mL) and Pd/C (0.66 g) in MeOH (200 mL) was heated at 60 °C under H2 atmosphere (50 psi) overnight, then cooled and filtered. The filtrate was concentrated under reduced pressure and the residue was dissolved in HC1 in dioxane (3N, 100 mL) and stirred for 30 min. The precipitate was filtered and washed with EtOAc (50 mL) to provide 2b (7.7g, 54% yield) as white powder. 1H-NMR (DMSO, 400 MHz): δ= 9.50 (br, 2 H), 3.48 (d, 2 H), 3.15-3.16 (m, 1 H), 2.96-3.01 (m, 2 H), 2.70 (s, 3 H), 2.51 (s, 3 H), 2.22-2.28 (m, 2 H), 1.94-2.02 (m, 2 H), LC-MS: 129 [M+l]+ .
Example 20
Synthesis of H0900
Step 1:
To a mixture of 16d (32 g, 120 mmol) in dry CH2CI2 (800 mL) was added Dess-Martin peroxide reagent (76 g, 180 mmol) portion- wise at 0 °C. The mixture was stirred at room temperature for 1 h, then diluted with DCM (800 mL), washed with aqueous NaHC03 solution (300 mL) and brine (300 mL). The organic phase was separated, dried over anhydrous Na2S04 and
concentrated under reduced pressure to afford crude 18a (31.4 g) which was used directly in the next step without further purification.
Step 2:
To a solution of 18a (12 g, 40 mmol) and 3b (22.2 g, 60 mmol) in DME (560 mL) were added Pd(PPh3)4 (9.25 g, 8 mmol) and Cul (1.52 g, 8 mmol) at room temperature. The mixture was stirred at 90 °C overnight, then concentrated under reduced pressure. The residue was purified with silica gel column chromatography (silica, EA : PE = 1 :5) to provide 18b (8.0 g, 79.3%) as a white solid. LC-MS: 253 [M+l]+.
Step 3:
To a solution of 18b (7 g, 27.7 mmol) and (¾)-tert-butylsulfinamide (7.27 g, 30.56 mmol) in dry THF (200 mL) was added Ti(i-OPr)4 (15.7 g, 55.4 mmol) dropwise at room temperature. The mixture was stirred at 80 °C overnight, and then cooled. Ethyl acetate (40 mL) was added, the resulting mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified with silica gel column chromatography (silica, EA:PE =1 :5) to provide 18c (6.8 g, 69%) as a yellow solid. 1H-NMR (CDC13, 400 MHz): 3= 9.10 (s, 1H), 8.97 (s, 1H), 8.72 (s, 1H), 8.64 (d, 1H),8.12 (d, 1H), 7.59 (d, 1H), 1.30 (s, 9H).LC-MS: 356 [M+l]+.
Step 4:
To a stirred solution of 18c (6.8 g, 19 mmol) and Tetrabutylammonium difluorotnphenylsilicate (15.8 g, 29 mmol) in dry THF (250 mL) was added a solution of TMSCF3 (11 g, 77 mmol) in anhydrous THF (50 mL) at -65 °C. The mixture was then stirred at -65 °C for 2 h, and at that point aqueous NH4CI solution (250 mL) was added. The mixture was diluted with ethyl acetate (250 mL), washed with brine (250 mL), dried over anhydrous Na2SC>4 and concentrated under reduced pressure. The residue was purified with silica gel column chromatography (silica, EA : PE=1 :2) to provide 18d (4.3 g, 52%) as a yellow solid. LC-MS: 426 [M+l]+.
Step 5:
To a stirred solution of 18d (4.3 g, 10.1 mmol) in MeOH (40 mL) was added a solution of HCl/MeOH (4N, 40 mL) at room temperature. The mixture was stirred for 1 h, then concentrated under reduced pressure. The residue was triturated with ethyl acetate (40 mL) to afford crude 18e (4.3g) which was directly in the next step without further purification. LC-MS: 322 [M+l]+.
Step 6:
To a solution of 18e (2.7 g, 7.1 mmol), 2b (3.4 g, 21.3 mmol) and TEA (80 mL) in DCM (220 mL) was added thiphosgene (3.15 g, 10.6 mmol) in DCM (40 mL) dropwise at 0 °C. The solution was warmed to ambient temperature and stirred for 1 h, then diluted with DCM ( 100 mL) and washed with aqueous Na2C03 solution (100 mL) and brine (100 mL). The organic layer was separated, dried over anhydrous Na2SC>4 and concentrated. The residue was purified with silica gel column chromatography (silica, DCM : CH3OH=10 : 1) to provide crude H0900 (2.13 g, ee%=92.5%) which was further purified through chiral separation to afford H0900 (1.6 g, 49% yield) as a white solid. (ee%=98.5%, Chiralpak IC 5um, 4.6*250mm, Phase: Hex: EtOH:
DEA=90: 10:0.2), retention tine =12.829 min. 1H-NMR (CDC13, 400 MHz): δ= 8.86 (d, 1H), 8.63 (dd, 1H), 8.55 (d, 1H), 7.47 (d, 1H), 7.40 (d, 1H), 6.28 (m, 1H), 5.18 (d, 1H), 4.12 (m, 1H), 2.88 (t, 2H), 2.77 (s, 3H), 2.22 (s, 3H), 2.05 (m, 2H), 2.48 (m, 2H), 1.52 (m, 2H), 1.73-1.49 (m, 4H). LC-MS: 476 [M+l]+.
PATENT
WO-2019118298
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019118298&tab=PCTDESCRIPTION&maxRec=1000
Novel crystalline fumarate salt forms of (R)-3-(1-(2,3-dichloro-4-(pyrazin-2-yl)phenyl)-2,2,2-trifluoroethyl)-1-methyl-1-(1-methylpiperidin-4-yl) urea (also referred to as HM04 or H0900; designated as Forms 1-4), process for their preparation and compositions comprising them are claimed.
PWS occurs in approximately 1 in 10,000 births and is associated with deletion or lack of expression of region 15ql 1.2 of the paternal chromosome 15.
Characteristics of PWS include short stature, low muscle tone, and hyperphagia. Growth hormone replacement is frequently used to treat growth deficiencies and hypotonia. However, treatment for the insatiable appetite is lacking and PWS children can mature into adults suffering from obesity and type 2 diabetes. Levels of ghrelin are generally elevated in PWS; however, the relationship with ghrelin signaling and food intake in PWS remains unclear. See Purtell L., et ah, In adults with Prader-Willi syndrome, elevated ghrelin levels are more consistent with hyperphagia than high PYY and GLP-l levels. Neuropeptides. 201 l;45(4):30l-7; Cummings D.E., et ah, Elevated plasma ghrelin levels in Prader Willi syndrome. Nature Medicine . 2002;8(7):643-4; DelParigi A., et ah, High circulating ghrelin: a potential cause for hyperphagia and obesity in Prader-Willi syndrome. The Journal of Clinical Endocrinology and Metabolism. 2002;87(l2):546l-4.
[005] Accordingly, it is desirable to find treatments that effectively inhibit GHSRla, that are tolerable to the patient, and that do not interfere with other functions of the growth hormones. GHSRla modulators, including inhibitors such as (R)-3-(l-(2,3-dichloro-4-(pyrazin-2-yl)phenyl)-2, 2, 2-trifluoroethyl)-l -methyl- l-(l-m ethylpiperidin-4-yl) urea (HM04, H0900) depicted below, are reported in LT.S. Patent No. 9,546,157.
Step 1 : Synthesis of compound 2A
[00106] 2,2,6,6-tetramethylpiperidine (7.20 kg, 51.1 mol, 3.0 eq.,
KF=0.30%) was added into a 100 L reactor equipped with a temperature probe and overhead stirrer and mixed at RT under nitrogen protection. THF (50 L) was added into the reactor and stirred. The vessel was purged with nitrogen three times and cooled to 0 °C. n-BuLi (20.4 L, 3.0 eq.; 2.5 M hexane solution) was added to the mixture dropwise while keeping the temperature at about 0 °C to about 5 °C for over one hour. The color of the solution turned yellow. The mixture was stirred at about 0 °C to about 5 °C for 30 minutes. The mixture was cooled to about -78 °C to about -70 °C to form Solution A.
[00107] Compound 1 (3.25 kg, 17.0 mol. 1.0 eq., KF=0.03%) was dissolved in 15 L of THF to form Solution B.
[00108] Solution B was added to solution A dropwise at a temperature of about -70 °C to about -78 °C over one hour and then stirred for 30 minutes to form solution C. Tri-isopropyl borate ((i-PrO)3B) (3.52 kg, 18.7 mol., 1.1 eq.) was added dropwise into solution C over 10 minutes. The reaction mixture was stirred at a
temperature of about -70 °C to about -78 °C for one hour. HC1 (40 L, 3M, 7.0 eq.) was added over 30 minutes to quench the reaction. A 10 degree rise in temperature was noted.
[00109] The resulting aqueous layer was separated and extracted with EtOAc (40 L). The aqueous layer was separated and extracted twice again with EtOAc (35 L, 30 L). The organic layers were combined resulting in about 160 L of liquid. The combined organic layer was washed twice with 50 L of a 1M aqueous HC1 solution saturated with NaCl. The organic layer was concentrated to about 5 L in a 50 L rotavapor at a temperature of about 50 °C to about 55 °C under 30-40 mmHg for about 8 hours.
[00110] The residual EtOAc was swapped with DME for 3 times (10 L x 3). The organic layer was concentrated in the 50 L rotavapor at a temperature of about 50 °C to about 55 °C under 30-40 mmHg for about 6 hours. Each time about 5 L of residual remained. DME (20 L) was added to the residual to obtain a deep brown solution of 14.2% compound 2A (3.55 kg in 25 kg of solution; 88.8% yield; 97.4% purity (AETC by HPLC, retention time = 1.6 minutes); 0.24% residual ethyl acetate). 1H-NMR (400 MHz, DMSO): 5=8.55 (s, 2H), 7.36 (d, 1H), 7.69 (d, 1H). A second batch of compound 2A was prepared by the same method to produce 3.29 kg (95.4% purity, 82.3% yield, 0.11% residual ethyl acetate).
[00111] Step 2: Synthesis of Compound 3A
C! , N
M
K2CO3 (I .O equiv)
2A OH
DME/H20 3:1 (20 vol), 50 e C 3A N
[00112] Compound 2 A (2.91 kg in 20.5 kg solution) was added into a 100
L reactor at room temperature under nitrogen. DME (45 mL), 2-chloropyrazin (1.42 kg,
12.4 mol., 1.0 eq.), and Pd(dppf)Cl2 (10% w/w, 291 g) were added sequentially, and each
mixed at room temperature under nitrogen. Nitrogen was bubbled into the mixture for 20
minutes and the resulting mixture was purged and filled with nitrogen (3 times). The
mixture was heated to 48-52°C over 60 minutes. K2CO3 (2.57 kg, 18.6 mol, 1.5 eq.) was
added to 22 L of water in another reactor at room temperature and then added dropwise to
the compound 2 A mixture over 10 minutes. The mixture was stirred at 48-52°C for 16
hours and then cooled to room temperature. This procedure was repeated twice and all
three batches were combined.
[00113] An aqueous solution of K2CO3 (1.0 kg) was dissolved in 22 L of
water and added to the combined mixture to adjust the pH to 9. TBME (50 L) was added
into the mixture and filtered (PET filter, 3-5 pm, 205g/m2) to remove about 50 g of
sticky, brown solid material (catalyst analog). The aqueous layer was twice separated and
extracted with TBME (40 L, 40L).
[00114] The aqueous layer was combined with the aqueous layer of a
fourth batch prepared according to the above method. The pH of the combined aqueous layers was adjusted to pH<3 with HC1 (2N, 48 L). The solid precipitated out slowly as
the mixture was stirred at room temperature for 1 hour. The mixture was filtered (PET
filter, 3-5 pm, 205g/m2) over 30 minutes to obtain 20 kg of wet product. ACN (40 L) was
added into a 100 L reactor equipped with an overhead stirrer at room temperature. The 20
kg of wet product was added into the reactor and the reaction mixture heated to reflux
and stirred at reflux for 4 hours. The reaction mixture was cooled to room temperature
over 3 hours (around 15 °C/hour) and filtered to obtain 8.5 kg of wet solid. The wet solid
was dried under vacuum (20-30 mmHg) at 50-55 °C for 15 hours to obtain compound 3 A
as a pale white solid (6.1 kg; 97.4% purity (AUC by HPLC, retention time = 3.7
minutes); 83.8% yield). 1H-NMR (400 MHz, DMSO): 5=7.67 (d, 1H), 7.82 (d, 1H), 8.75
(d, 1H), 8.82 (t, 1H), 8.98 (d, 1H), 13.89 (bs, 1H).
[00115] Step 3: Synthesis of compound 6A
3A 6A N
N
[00116] Compound 3 A (6.1 kg, 22.7 mol, 1.0 eq.) was added into a 100 L
reactor equipped with a temperature probe, overhead stirrer, and condenser. Methanol
(92 L) was added into the reactor at room temperature. The mixture was cooled to
0-10 °C and added with SOCk (5.4 kg, 45.3 mol, 2.0 eq.) dropwise at 0-10 °C over 30
minutes. The reaction mixture was heated to reflux (65 °C) and stirred at reflux for 15
hours. A suspension was formed. Most of the solvent and SOCk was removed under
vacuum distillation until about 30 L remained. The mixture was concentrated under
vacuum (30-40 mmHg) at 50-55 °C for about 6 hours. Water (10 L) was added to the residual at -5 to 15 °C. The pH was adjusted to 8-9 with an aqueous solution of K2CO3 (200 g, dissolved in 2L of water) at -5 to 15 °C. The resulting aqueous layer was extracted twice with isopropyl acetate (25 L, 25 L). The combination of organic layers (about 50 kg) was washed with 20 L of NaHCCb aqueous layer. The organic layer was separated and washed with 10 L of of an aqueous solution of NaHCCb. All the aqueous layers were combined (55.8 kg). The organic layer was filtered through a silica pad (30 cm) and the pad washed with extra isopropyl acetate until the compound 6 A was filtered from the silica gel (about 3 hours). The organic layer was concentrated to about 5 L. THF (10 L) was added to the residual and concentrated to about 5 L (3 times) under vacuum (30-40 mmHg) at 50-55 °C for about 3 hours. Another 10 L of THF was added to the residual concentrate, giving a concentrated solution of compound 6A (15.8 kg; 32.83%,
5.19 kg compound 6A in solution; 97.9% purity (AUC by HPLC, retention time = 8.5 min); 80.8% yield). 1H-NMR (400 MHz, DMSO): 5=3.98 (s, 3H), 7.54 (d, 1H), 7.78 (d, 1H), 8.63 (d, 1H), 8.72 (t, 1H), 8.94 (d, 1H).
[00117] Step 4: Synthesis of compound 6B
[00118] THF (26 L) was added into a 100 L reactor equipped with a temperature probe and overhead stirrer under nitrogen. DIBAL-H (26 kg, 46 mol, 5.0 eq.) was added and the system purged and filled with nitrogen three times. The mixture was cooled to -78 to -70 °C to form solution A. A room temperature solution of compound 6A (2.6 kg, 9.2 mol, 1.0 eq.) in 52 L of THF was added dropwise at -78 to -70 °C over 30 minutes under nitrogen. The mixture was warmed to -30 °C over about 5-6 hours. The reaction mixture was stirred at -40 to -30 °C for 30 minutes. The mixture was slowly added to 42 L of 2N HCL over 1 hour reaching a maximum temperature of 35 °C. The mixture was extracted with 26 L of isopropyl acetate. The organic layer was separated and washed with 30 L of brine. This procedure was repeated and both batches of organic layer were combined and concentrated from about 100 L to about 5-10 L under vacuum.
A solid slowly formed during concentration. The mixture was cooled to 5-15 °C and stirred for 1 hour. The mixture was filtered (30-50 pm) over 30 minutes. The solid was dried under vacuum at 50 °C for 6 hours to obtain compound 6B as a brown solid (2.1 kg; 97.5% purity (AUC by HPLC, retention time = 8.6 min); 45.7% yield). 1H-NMR (400 MHz, DMSO): d = 4.65 (d, 2H), 5.68 (t, 1H), 7.62 (d, 1H), 7.68 (d, 1H), 8.72 (d, 1H),
8.80 (t, 1H), 8.94 (d, 1H).
[00119] Step 5: Synthesis of compound 7
[00120] DMSO (10 L) was added to a 50 L flask equipped with a temperature probe and overhead stirrer under nitrogen at room temperature. Compound 6B (2.05 kg, 8.04 mol, 1.0 eq.) was added under nitrogen at room temperature. Et3N (8 L) was added under nitrogen at RT and the mixture was then cooled to 15-20 °C.
SCb. pyridine (5.1 kg, 32.08 mol, 4.0 eq.) was dissolved into 10 L of DMSO at 5-15 °C in a separate flask and added to the mixture dropwise over 3.5 hours at about 20 °C. The reaction mixture was transferred to 70 L of ice-water. The suspension mixture was stirred at 0-10 °C for 1 hour and filtered (PET, 3-5 pm, 205 g/m2) by centrifuge over 1.5 hours to obtain compound 7 as a brown solid. The solid was dissolved in 35 L of DCM at room temperature. The resulting DCM layer was washed with 5 L of brine. The organic layer was separated and concentrated under vacuum at 40-45 °C to dryness to obtain compound 7 as a brown solid (2.33 kg; 96.3% purity (AEiC by HPLC, retention time = 9.2 minutes); 93.5% yield). 1H-NMR (400 MHz, DMSO): d = 7.67 (d, 1H), 7.99 (d, 1H), 8.67 (d, 1H), 8.75 (s, 1H), 8.99 (d, 1H), 10.56 (s, 1H).
[00121] Step 6: Synthesis of compound 8
[00122] THF (23 L) was added to a 50 L flask equipped with a temperature probe and overhead stirrer under nitrogen at room temperature. Compound 7 (2.3 kg, 9.1 mol, 1.0 eq.) and (S)-2-methylpropane-2-sulfmamide (1.21 kg, 10 mol, 1.1 eq.) were added sequentially to the flask under nitrogen. Ti(OEt)4 (6.22 kg, 27.3 mol, 3.0 eq.) was added dropwise to the flask over 1 hour at 30-35 °C under nitrogen. The system was purged with nitrogen three times and then the mixture was stirred at room temperature for 2 hours. Isopropyl acetate (40 L) was added to the reaction mixture. The entire reaction mixture was then charged to 20 L of brine while stirring slowly at RT. A lot of solid was formed and no heat release was observed. The solid (about 18 kg) was filtered using centrifuge, and then the solid was slurried with 20 L of isopropyl acetate again for 20 minutes, and filtered again, resulting is slightly less solid (17.3 kg). The filtrates were then combined and washed with 20 L of brine. The organic layer was separated and concentrated in a rotavapor under vacuum (30-40 mmHg) at 40-50 °C for about 4 hours to remove the solvents and obtain a brown oil (compound 8). The oil was dissolved in DMF to obtain a black solution (7.36 kg; 40.1%; 3.0 kg compound 8 in solution; 92.1% purity (AUC by HPLC, retention time = 9.7 minutes); >100% yield). 1H-NMR (400 MHz, CDCb): d = 1.30 (s, 9H), 7.59 (d, 1H), 8.11 (d, 1H), 8.64 (s, 1H), 8.73 (m, 1H), 8.97 (s, 1H), 9.10 (s, 1H).
[00123] Step 7: Synthesis of compound 11
O
S
10 s C
8
11 N
[00124] DMF (26 L, 10 v/w) was added to a 50 L flask equipped with a temperature probe and overhead stirrer under nitrogen at 15 °C. Compound 8 (7.3 kg of
DMF solution, containing 2.9 kg, 8.1 mol, 1.0 eq.) and TBAA (2.44 kg, 8.1 mol, 1.0 eq.) were added sequentially to the flask under nitrogen. The mixture was cooled to 0-10 °C.
TMSCF3 (2.88 kg, 20.3 mol, 2.5 eq.) was then added to the flask over 60 min at 0-10 °C.
The reaction mixture was stirred at 0-5 °C under nitrogen protection for 3 hours.
Isopropyl acetate (60 L) was added to the mixture, followed by the addition of 45 L of
NaHCCb under stirring at 5-25 °C. The organic layer was separated, washed three times with NaHC03 (30 L x 3), and concentrated from 60 kg to 2.5 kg of brown oil. The oil product was dissolved in 20 L of TBME and filtered through a pad of silica gel (about 40 cm high, 30 cm diameter) over 2 hours to obtain 2.14 kg of compound 1 1 in TBME solution. The solution was concentrated at 45-50 °C to dryness to obtain compound 1 1 as a black oil (1.85 kg; 85.2% purity (AETC by HPLC, retention time = 9.1 minutes, 9.6 minutes for diastereoisomer); 53.6% yield). 1H-NMR (400 MHz, CDCh): d = 1.33 (s, 9H), 3.82-3.85 (d, 1H), 5.61-5.66 (m, 1H), 7.53-7.60 (m, 2H), 8.63-8.64 (d, 1H), 8.71-8.72 (m, 1H), 8.95 (s, 1H).
[00125] Step 8: Synthesis of compound 12 (free base)
[00126] Compound 1 1 (1.8 kg, 4.23 mol, 1.0 eq., crude) was added to a 50 L reactor equipped with a temperature probe and overhead stirrer under nitrogen at 25 °C. Anhydrous MeOH (18 L) was added to dissolve compound 1 1. Then MeOH/HCl (18 L, 1 N) was added dropwise at 25-30 °C over 10 minutes and the mixture was stirred at 25-30 °C for 1 hour. Water (15 L) was added to the reaction and the mixture concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 4 hours to remove the solvent. The pH of the mixture was adjusted to 10 with 5 L of K2CO3 solution. 20 L of EtOAc was then added to the mixture and the organic layer was separated and the aqueous layer extracted twice with EtOAc (15 L x 2). The organic layers were combined and washed with 10 L of brine. The combined organic layers contained 996 g of
compound 12 in 40 kg of EtOAc solution (84% purity (AUC by HPLC, retention time =
2.8 minutes). The organic layers were concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 3 hours to a 7.5 kg volume of compound 12 in EtOAc solution (83% purity (AETC by HPLC, retention time = 2.7 minutes).
[00127] In a separate 50 L reactor equipped with a temperature probe and overhead stirrer, D-CSA was added (930 g, 4.0 mol, 1.0 eq. to 1.26 kg compound 12) and stirred at room temperature under nitrogen. EtOAc (10 L) and then the EtOAc solution of compound 12 (1.26 kg, 3.9 mol, 1.0 eq.) were each sequentially added to the reactor. The mixture was stirred at room temperature for 1 hour and slowly became a suspension. The mixture was filtered by centrifuge and washed with EtOAc to produce 2.3 kg of compound 12 as an off-white solid (96.0% purity).
[00128] The solid product, 20 L of EtOAc, and 10 L of 10% aqueous K2CO3 were added sequentially to a 50 L flask and stirred at room temperature until no solid remained (pH = 9-10). The organic layer was separated and the aqueous layer extracted twice with EtOAc (10 L x 2). The organic layers were combined (about 32 kg) and washed with 10 L of brine. The organic layer contained 716 g of compound 12 in
31.8 kg of solution.
[00129] The organic layer was concentrated under vacuum at 45-50 °C to about 8 L. Activated carbon (200 g) was added to the organic layer and the mixture stirred at 60-70 °C for 1 hour, cooled to room temperature, and filtered using a Buchner funnel and filter paper (pore size: 30-50 pm) over 30 minutes to remove the activated carbon. The mixture was concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 3 hours to yield 710 g of compound 12 as a yellow solid (99.4% purity). [00130] D-CSA (410 g, 1.77 mol, 1.0 eq. to 680 g compound 12), 3.4 L iPrOH, and 68 mL of water were added sequentially to a 10 L reactor equipped with a temperature probe and overhead stirrer and stirred at room temperature under nitrogen. The mixture was heated to reflux (84 °C) to form solution A after 1 hour. Compound 12 (680 g) was dissolved in 3.4 L of iPrOH and added into solution A for one partition. A clear solution was formed and the temperature decreased to 65 °C. The mixture was stirred at 65 °C for about 15 minutes after which a solid appeared. The mixture was cooled to 10 °C over 2 hours, stirred at 10 °C for an additional 30 minutes, and filtered through a Buchner funnel and filter paper (pore size: 30-50 pm) over 30 minutes to collect the 1.1 kg of white solid.
[00131] EtOAc (10 L), 1.1 kg of white solid product, and 5 L of 10% K2CO3 were added sequentially to a 20 L flask and mixed for 5 minutes. The solid dissolved (pH = 9-10). The EtOAc layer was separated and the aqueous layer extracted twice with EtOAc (5 L each). The organic layers were combined (about 20 L), washed with 5 L of brine, and concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-55 °C for about 3 hours to remove most of the solution and until the residue weight reached 1 kg. Heptanes (1 L) was added to the mixture and stirred at room temperature for 30 minutes. The mixture was filtered using a Buchner funnel and filter paper (pore size: 30-50 pm) over 30 minutes to obtain 419 g of compound 12 base as a white solid (99.7% purity). The filtrate was concentrated to 135 g of compound 12 as a yellow solid (98.7% purity). 1H-NMR (400 MHz, CDCh): d = 1.85 (bs, 2H), 5.17 (m, 1H), 7.56 (d, 1H), 7.68 (d, 1H), 8.62 (d, 1H), 8.70-8.71 (m, 1H), 8.93 (s, 1H). Combined, the products resulted in a 40.7% yield of compound 12.
[00132] Step 9: Synthesis of compound 10
10A 10
[00133] Pd/C (40 g, 5% w/w) was added into a 10 L autoclave reactor at room temperature under nitrogen. THF (2 L), 2 L of methylamine (27%-30% alcoholic solution, 2.1 eq.), and 800 g of compound 10A (7 mol, 1.0 eq.) were sequentially added into the reactor. The system was purged with hydrogen three times. The mixture was stirred at hydrogen pressure (50 psi) at 70-75 °C overnight and was then filtered using a Biichner funnel and filter paper (pore size: 30-50 pm) over 10 minutes to remove the Pd/C. The filtrate was concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 3 hours to obtain 933 g of yellow oil. The mixture was distilled without a column at atmospheric pressure and the 140-170 °C portion was collected to obtain 763 g of compound 10 as a colorless oil (98.6% purity (AUC by HPLC, retention time = 4.8 minutes); 84.2% yield; 8000 ppm residual ethanol). A portion of the oil (563 g) was distilled using a 3 cm column at atmospheric pressure and the 140-170 °C portion was collected to obtain 510 g of compound 10 (75.8% yield; 134 ppm residual ethanol). 1H-NMR (400 MHz, CDCb): d = 0.82 (bs, 1H), 1.10-1.12 (q, 2H), 1.66 (d, 2H), 1.73-1.81 (t, 2H), 2.05 (s, 3H), 2.08-2.19 (m, 1H), 2.22 (s, 3H), 2.60 (d, 2H).
[00134] Step 10: Synthesis of HM04 fumarate salt
[00135] DCM (1L), 200 g CDI (1.23 mol, 2.0 eq.), and 35 g DABCO (0.31 mol, 0.5 eq.) were sequentially added into a 3 L reactor equipped with a temperature probe and overhead stirrer, and stirred at room temperature under nitrogen. The mixture was cooled to -10 to -5 °C. Compound 12 (200 g) was dissolved in 1 L of DCM and added into the mixture dropwise over 1 hour, followed by stirring for 16 hours at -10 to -5 °C. Compound 10 (159 g, 1.24 mol, 2.0 eq.) was added at -10 to 0 °C over 10 minutes. The mixture was then warmed to 0 to 5 °C and held for 2 hours. The mixture was concentrated under vacuum at 40-45 °C to about 1 L. HC1 (1 L of 1 N) was added to the residual and concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 2 hours to remove the DCM. Another 3 L of 1N HC1 was added to the residual and extracted three times with TBME (4 L, 2 L, 2 L). The aqueous layer was slowly adjusted to pH = 9-10 with 20% aqueous K2CO3 (about 1.5 L) and extracted with DCM (2 L x 3). The organic layers were combined (about 4 L) and washed three times with 0.25 N KH2PO4 (1.2 L x 3). The organic layer was washed with 2 L of brine to bring the pH to neutral and concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 2 hours to 450 g (335 mL). MTBE (1.5 L) was added to the residual and distilled until 500 mL of liquid was collected. This step was repeated four times with the addition of 500 mL of TBME and collection of 500 mL of distillate, with the exception that 330 mL of liquid was collected at the final distillation. About 1 to 1.2 L of residual remained in the flask. The residual was slowly cooled to room temperature and stirred at room temperature overnight. The mixture was filtered, washed twice with TBME (400 mL x 2), and dried to obtain 192 g of HM04 free base a light yellow solid (99.3% purity (AUC by HPLC, retention time = 11.0 minutes). The product on the wall was dissolved in DCM and concentrated under vacuum to obtain 22 g of HM04 free base as a brown sticky oil (97.6% purity). The filtrate was concentrated under vacuum to obtain 22.5 g of yellow solid (94.0% purity).
[00136] HM04 free base (187 g, 0.39 mol, 1.0 eq., 99.3% purity) and 1.9 L of ACN were sequentially added to a 3 L flask equipped with a temperature probe and overhead stirrer and stirred at 15 °C under nitrogen to obtain a light-yellow suspension. Fumaric acid (45.6 g, 0.39 mol, 1.0 eq.) was added to the flask and generated a white suspension after 1 minute. The reaction suspension was stirred overnight at room temperature, filtered (15-20 pm, ash<0.l5), washed twice with ACN (50 mL x 2), and dried under vacuum at 50 °C for 6 hours to obtain 207 g of HM04 fumarate salt as a light yellow solid (99.4% purity (AUC by HPLC, retention time = 11.1 minutes); 57.8% yield; 3100 ppm residual ACN). The filtrate was concentrated under vacuum to obtain 20.1 g of HM04 fumarate salt as a light yellow solid (97.3% purity).
[00137] A portion of the product (117 g) was further dried in a vacuum oven (20-40 mmHg) to lower the residual acetonitrile content. After drying at 60 °C for 6 hours, 15 hours, and 72 hours; and at 65 °C for 18 hours, the residual acetonitrile content was measured as 3100 ppm, 2570 ppm, 1300 ppm, and 256 ppm, respectively. After the drying process, 98 g of HM04 fumarate salt was isolated (99.4% purity (AUC by HPLC, retention time = 11.0 minutes); 1H-NMR (400 MHz, DMSO): d = 1.49-1.58 (m, 2H),
1.81-1.92 (m, 2H), 2.44-2.53 (m, 5H), 2.78 (s, 3H), 3.12 (m, 2H), 4.06-4.13 (m, 1H), 6.36-6.41 (m, 1H), 6.55 (s, 2H), 7.47 (d, 1H), 7.73 (d, 1H), 8.11 (d, 1H), 8.75 (d, 1H),
8.81-8.82 (m, 1H), 8.99 (d, 1H). The yield of 98g of HM04 fumarate salt isolated after drying the partial batch was extrapolated over the whole batch to calculate an
approximate yield of 48% for step 10.
[00138] XRPD analysis of HM04 fumarate salt products obtained after drying at 60 °C for 6 hours, 15 hours, and 72 hours; and at 65 °C for 18 hours was performed (see Figures 6-9, respectively). The XRPD profile showed that the HM04 fumarate salt product was consistent with Form 1.
Example 6. Streamlined Synthesis of HM04 Fumarate Salt Form 1
[00139] The overall yield of HM04 fumarate salt produced using Step 10 of Example 5 was calculated as approximately 48%. In order to increase the overall yield, a streamlined synthesis was investigated that eliminated the step of isolating HM04 free base. In particular, step 10 of the method of Example 5 shown in Figure 5 was changed. An overview of the streamlined synthesis beginning after step 9 of Example 5 is shown in Figure 10.
[00140] Streamlined HM04 Fumarate Salt Trial 1 : PCM (121.4 g). CPI (20.0 g, 123 mmol, 2 eq.) and DABCO (3.5 g, 31 mmol) were sequentially added into an inertized 1 L reactor. The mixture was cooled to -10 °C. Separately, a solution of DCM (132.5 g) and compound 12 (20.0 g, 62.1 mmol) were charged into a vessel and stirred until a solution was obtained. This solution was dropped into the 1 L reactor over 33 minutes by keeping the internal temperature at -10 to -5 °C. At the end of the addition, the vessel was rinsed with DCM (7.0 g), which was then added to the reaction mixture.
After stirring overnight (19 hours) and positive IPC, compound 10 (15.9 g, 124 mmol, 2 eq.) was added over 15 minutes and the vessel rinsed with DCM (9.0 g). After heating at 0 °C, 1 hour of stirring, positive IPC, and a further 1.5 hours of stirring, the mixture was heated at room temperature and charged with water (200.1 g). The aqueous layer was separated and the organic layer extracted twice with 1 N HC1 (201, 200 g). The combined aqueous layers containing the product were washed with TBME (148 g). After removal of the organic layer, the aqueous layer was charged with DCM (265.0 g) and 50% K2CO3 solution (about 240 ml) until reaching pH 9.61.
[00141] Meanwhile, a solution of KH2PO4 (8.2 g) in water (240 g) was prepared. The organic layer containing the product was charged with the KH2PO4 solution until reaching pH 7.12 (142.2 g). After separation of the aqueous layer, the organic layer was washed with water (200 g). After separation of the aqueous layer, the organic layer was evaporated at 50 °C. ACN (314.4 g) was added and the solvent distilled again at 70-75 °C under vacuum. ACN (235.8 g) was added and the solvent distilled again under vacuum. ACN (141.5 g) was added, the resulting solution polish filtered and the filter washed with ACN (16 g). After heating at 60 °C, fumaric acid (7.2 g, 62 mmol) was added to the solution, causing a white precipitate. After cooling to 20 °C over 1 hour, the suspension was filtered and washed twice with TBME (2 x 30 g). After drying on the filter with nitrogen flow, 70.7 g of wet raw product was obtained. This was slurried with TBME (177.0 g) for 1 hour, filtered, and washed with TBME (70 g). After drying on the filter under nitrogen flow, 33.0 g of wet product was obtained. Heating at 50 °C under vacuum afforded the dry product as a white powder of HM04 fumarate salt (21.1 g;
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9926337 | SUBSTITUTED ASYMMETRIC UREAS AND MEDICAL USES THEREOF | 2016-12-02 | |
| US9546157 | p-Substituted Asymmetric Ureas and Medical Uses Thereof | 2015-03-06 | 2015-09-10 |
////////////HM04, H0900, Helsinn, Novo Nordisk, PRECLINICAL, obesity, Prader-Willi syndrome, ghrelin
CN(C1CCN(C)CC1)C(=O)N[C@H](c3ccc(c2cnccn2)c(Cl)c3Cl)C(F)(F)F
Beloranib, 성분명 벨로라닙 ZGN-433….Zafgen’s Prader-Willi syndrome therapy receives orphan drug designation in Europe


Beloranib
CAS 251111-30-5 (beloranib),529511-79-3 (beloranib hemioxalate)
(E)-(3R,4S,5S,6R)-5-methoxy-4-((2R,3R)-2-methyl-3-(3-methylbut-2-en-1-yl)oxiran-2-yl)-1-oxaspiro[2.5]octan-6-yl 3-(4-(2-(dimethylamino)ethoxy)phenyl)acrylate
6-O-(4-dimethylaminoethoxy)cinnamoyl fumagillol
Mechanism of Action:methionine aminopeptidase 2 (MetAP2) inhibitor
Indication:Obesity US Patent : US6063812 Patent Exp Date: May 13, 2019
Originator: Chong Kun Dang (CKD) Pharma (종근당) Chong Kun Dang Pharm Corp
Developer: Zafgen Inc. (자프젠)Zafgen Corporation
Zafgen’s Prader-Willi syndrome therapy receives orphan drug designation in Europe The European Commission (EC) has granted orphan drug designation to US-based Zafgen for its beloranib for treating Prader-Willi syndrome. Beloranib is a potent inhibitor of Methionine aminopeptidase-2 that reduces hunger while stimulating the use of stored fat as an energy source (MetAP2). MetAP2 is an enzyme that modulates the activity of key cellular processes that control metabolism. http://www.pharmaceutical-technology.com/news/newszafgens-prader-willi-syndrome-therapy-receives-orphan-drug-designation-in-europe-4316842?WT.mc_id=DN_News
INTRODUCTION Beloranib is an experimental drug candidate for the treatment of obesity. It was discovered by CKD Pharmaceuticals and is currently being developed by Zafgen. Beloranib, an analog of the natural chemical compound fumagillin, is an inhibitor of the enzyme METAP2. It was originally designed as angiogenesis inhibitor for the treatment of cancer. However, once the potential anti-obesity effects of METAP2 inhibition became apparent, the clinical development began to focus on these effects and beloranib has shown positive results in preliminary clinical trials for this indication. At such low doses, says Thomas E. Hughes, president and chief executive officer of Zafgen, toxicity concerns tend to evaporate, in part because so little opportunity exists to inhibit off-target proteins.
Zafgen, a small pharmaceutical company in Cambridge, Mass., sees high selectivity and low toxicity with its covalent molecule for treating obesity, beloranib hemioxalate, also known as ZGN-433. “You’re passing a wave of the molecule through the body,” he says. “It hits the different tissues, silences the target enzyme where it finds it, and then it goes away.” Zafgen’s drug candidate inhibits an enzyme called methionine aminopeptidase 2 (MetAP2), which had been of interest in oncology circles until it turned out to be a poor target for treating cancer in mice. However, animals treated with a MetAP2 inhibitor lost weight. Zafgen pursued the enzyme as a target for obesity. Its drug candidate contains a spiroepoxide that bonds with a histidine in the protein’s active site.
ZGN-433 has undergone a Phase I clinical trial, in which obese volunteers lost up to 2 lb per week. It will enter Phase II trials within a year, Hughes says, funded by $33 million the company raised from investors. With dosing of up to 2 mg twice per week, ZGN-433 reaches a maximum concentration in the body of just a few nanomolar for several hours before the body quickly eliminates it, Hughes says. During that time, the drug is much more likely to interact with MetAP2 than with anything else. “You’re flying under the radar of a lot of concerns,” he says. “Drug-drug interactions are not an issue. There’s just not enough inhibitor to go around.
The same is true for off-target inhibition: The chance of off-target toxicity is largely gone.” Proponents of covalent inhibitors are quick to point out that dozens of such drugs are already on the market. They include aspirin, the world’s most widely used medicine; penicillin and related antibiotics; and recently developed blockbusters such as Plavix, Prevacid, and Nexium. The drugs treat a broad range of conditions, and many have minimal side effects, even when taken for years. By one count, of the marketed drugs that inhibit enzymes, more than one-third work by covalent modification (Biochemistry, DOI: 10.1021/bi050247e).
![]()
6-O-(4-dimethylaminoethoxy) cinnamoyl fumagillol hemioxalate
| Beloranib, ZGN-433, CKD-732 | |
|---|---|
 |
|
| Identifiers | |
| CAS number | 251111-30-5 |
| PubChem | 6918502 |
| ChemSpider | 26286923 |
| UNII | FI471K8BU6 |
| Jmol-3D images | Image 1 |
| Properties | |
| Molecular formula | C29H41NO6 |
| Molar mass | 499.64 g mol−1 |
| Except where noted otherwise, data are given for materials in their standard state (at 25 °C (77 °F), 100 kPa) | |
Beloranib (previously known as CKD-732; ZGN-433), a methionine aminopeptidase 2 (MetAP2) inhibitor originally designed as an anticancer agent, is being developed by Zafgen as a first-in-class obesity therapy. Beloranib, a twice-daily injection, is discovered by korean company Chong Kun Dang (CKD) Pharmaceuticals and was licensed to Cambridge, MA-based startup Zafgen, Inc. Zafgen holds exclusive worldwide rights (exclusive of Korea) for development and commercialization of beloranib. Beloranib, an analog of the antimicrobial agent fumagillin, is an inhibitor of the enzyme METAP2 involved in fatty acid production. It was originally designed as angiogenesis inhibitor for the treatment of cancer. However, once the potential anti-obesity effects of METAP2 inhibition became apparent, the clinical development began to focus on these effects.
Zafgen has chosen to develop beloranib not for the folks that need to shed a few pounds, but for severely obese people, and smaller groups of patients with rare and dangerous conditions. In January 2013, beloranib was granted orphan drug designation by the U.S. Food and Drug Administration to treat a rare genetic condition known as Prader-Willi Syndrome (PWS) that causes obesity through compulsive eating. Zafgen plans to seek the same designation for beloranib in craniopharyngioma (a rare benign brain tumor) related obesity as well. By going after these orphan indications, Zafgen can get onto the market quicker and cheaper than if it went straight for the larger obesity market. Zafgen recently completed two Phase 2a clinical trials evaluating beloranib’s ability to reduce body weight and to improve hyperphagia, one in PWS patients and one in severely obese patients. In its Phase 2a clinical trials, Zafgen observed reductions in body weight, body mass and body fat content in both patient populations and reductions in hyperphagia-related behaviors in PWS patients.
On June 19, 2014, Zafgen Inc. raised $96 million in its initial public offering (IPO) on the Nasdaq under the symbol “ZFGN” amid strong demand from investors. With its IPO cash, Zafgen plans to initiate its Phase 3 clinical program, consisting of two Phase 3 clinical trials, of beloranib in PWS patients, with the first Phase 3 trial to start in the second half of 2014, after finalizing the program design based on ongoing conversations with the FDA and certain European regulatory authorities. Zafgen is also planning a phase 2a trial in craniopharyngioma, and a Phase 2b trila in patients with severe obesity, all this year. The composition of matter patent (US6063812) on beloranib will each expire in May 2019. Zafgen owns two issued U.S. patents relating to beloranib polymorph compositions of matter that will expire in 2031 and two issued U.S. patents to methods of treating obesity that will expire in 2029.  Beloranib is an experimental drug candidate for the treatment of obesity. It was discovered by CKD Pharmaceuticals and is currently being developed by Zafgen.[1] Beloranib, an analog of the natural chemical compound fumagillin, is an inhibitor of the enzyme METAP2.[2] It was originally designed as angiogenesis inhibitor for the treatment of cancer.[3] However, once the potential anti-obesity effects of METAP2 inhibition became apparent, the clinical development began to focus on these effects and beloranib has shown positive results in preliminary clinical trials for this indication.[4][5]
Beloranib is an experimental drug candidate for the treatment of obesity. It was discovered by CKD Pharmaceuticals and is currently being developed by Zafgen.[1] Beloranib, an analog of the natural chemical compound fumagillin, is an inhibitor of the enzyme METAP2.[2] It was originally designed as angiogenesis inhibitor for the treatment of cancer.[3] However, once the potential anti-obesity effects of METAP2 inhibition became apparent, the clinical development began to focus on these effects and beloranib has shown positive results in preliminary clinical trials for this indication.[4][5]
………………………………..
http://www.google.com/patents/WO2005082349A1?cl=en
compound O-(4- dimethylaminoethoxycinnamoyl)fumagillol can be used in the form of a salt, e.g., acetate, lactate, benzoate, salicylate, mandelate, oxalate, methanesulfonate, or p- toluenesulfonate. Korean Patent No. 0357542 and its corresponding patents (U.S. Patent No. 6,063,812, Japanese Patent No. 3370985, and European Patent No. 1077964), filed by the present applicant, disclose fumagiUol derivatives, including the compounds used in the present invention. The composition of the present invention can be prepared in combination with pharmaceutically acceptable carriers commonly used in pharmaceutical formulations.
………………………..
http://www.google.com/patents/WO2012064838A1?cl=en
MetAP2 encodes a protein that functions at least in part by enzymatically removing the amino terminal methionine residue from certain newly translated proteins, such as, glyceraldehyde-3- phosphate dehydrogenase (Warder et al. (2008) J Proteome Res 7:4807). Increased expression of the MetAP2 gene has been historically associated with various forms of cancer. Molecules inhibiting the enzymatic activity of MetAP2 have been identified and have been explored for their utility in the treatment of various tumor types (Wang et al. (2003) Cancer Res 63:7861) and infectious diseases, such as, microsporidiosis, leishmaniasis, and malaria (Zhang et al. (2002) J. Biomed Sci. 9:34). Notably, inhibition of MetAP2 activity in obese and obese-diabetic animals leads to a reduction in body weight in part by increasing the oxidation of fat and in part by reducing the consumption of food (Rupnick et al. (2002) Proc Natl Acad Sci USA 99: 10730). [0003] 6-O-(4-Dimethylaminoethoxy)cinnamoyl fumagillol is a METAP2 inhibitor and is useful in the treatment of, e.g., obesity. 6-O-(4-Dimethylaminoethoxy)cinnamoyl fumagillol is characterized by formula I:

Example 1 [0060] Crystalline, Form A material of 6-O-(4-dimethylaminoethoxy)cinnamoyl fumagillol was prepared as follows: [0061] Approximately 423 mg of amorphous gum/oil-like 6-O-(4- dimethylaminoethoxy)cinnamoyl fumagillol free base compound was dissolved in ca. 6 mL of diisopropylether (IPE). The solution was allowed to stir for ca. 24 hours at ambient temperature (18-22°C) during which time solid precipitated. The resulting solid was isolated by filtration and dried under vacuum at ambient for ca. 4 hours (yield 35.8 %).
…………………..

………………….
http://www.google.com/patents/WO1999059986A1?cl=en
Example 14 : 0-(4-dimethylaminocinnamoyl)fumagillol 1) To a solution of 4-dimethylaminocinnamic acid (950 mg) in toluene (20 ml), dipyridyl disulfide (1.64 g) and triphenyl phosphine (1.97 g) were added, and the mixture was stirred for 12 hours. 2) The resultant solution of 1) was added to fumagillol (500 mg) at room temperature. Sodium hydride (142 mg) was added thereto, and the reaction mixture was stirred for 30 minutes. After adding saturated ammonium chloride solution (20 ml), the reaction mixture was extracted with ethyl acetate (100 ml). The organic layer was washed with brine and dried over anhydrous magnesium sulfate. After filtering, the solvent was distilled off under reduced pressure, and the residue was purified by column chromatography (eluent: ethyl acetate/ n-hexane = 1/2) to obtain yellow solid (470 mg). ‘H-NMR (CDCI3) δ : 7.60 (d, IH, J=15.8Hz), 7.41 (d, 2H, J=8.9Hz), 6.67 (d, 2H, J=8.9Hz), 6.27 (d, IH, J=15.8Hz), 5.71 (m, IH), 5.22 (bit, IH), 3.70 (dd, IH, J=2.8, 11.0Hz), 3.45 (s, 3H), 3.02 (s, 6H), 3.01 (d, IH, J=4.3Hz), 2.63 (t, IH, J=6.3Hz), 2.56 (d, IH, J=4.3Hz), 2.41 – 1.81 (m, 6H), 1.75 (s, 3H), 1.67 (s, 3H), 1.22 (s, 3H), 1.15 – 1.06 (m, IH)
………..
Organic Letters, 16(3), 792-795; 2014

An efficient, two-step construction of highly complex alkaloid-like compounds from the natural product fumagillol is described. This approach, which mimics a biosynthetic cyclase/oxidase sequence, allows for rapid and efficient structure elaboration of the basic fumagillol scaffold with a variety of readily available coupling partners. Mechanistic experiments leading to the discovery of an oxygen-directed oxidative Mannich reaction are also described.
References
- “News Release: Zafgen Secures $33 Million Series C Financing”. Zafgen, Inc. July 7, 2011.
- Chun, E; Han, CK; Yoon, JH; Sim, TB; Kim, YK; Lee, KY (2005). “Novel inhibitors targeted to methionine aminopeptidase 2 (MetAP2) strongly inhibit the growth of cancers in xenografted nude model”. International Journal of Cancer. Journal International Du Cancer 114 (1): 124–30. doi:10.1002/ijc.20687. PMID 15523682.
- Kim, EJ; Shin, WH (2005). “General pharmacology of CKD-732, a new anticancer agent: effects on central nervous, cardiovascular, and respiratory system”. Biological & Pharmaceutical Bulletin 28 (2): 217–23. doi:10.1248/bpb.28.217. PMID 15684472.
- “Zafgen Announces Positive Topline Phase 1b Data for ZGN-433 in Obesity”. MedNews. Drugs.com. 5 January 2011.
- “Fat-busting pill helps obese to shed two pounds a week – without changing their diets”. UK Daily Mail. 11 January 2011.
MORE REF Grenning, Alexander J. et al.Remodeling of Fumagillol: Discovery of an Oxygen-Directed Oxidative Mannich Reaction.Organic Letters, 16(3), 792-795; 2014
Hughes, T. E.; Kim, D. D.; Marjason, J.; Proietto, J.; Whitehead, J. P.; Vath, J. E. Ascending dose-controlled trial of beloranib, a novel obesity treatment for safety, tolerability, and weight loss in obese women. Obesity (2013), 21(9), 1782-1788.
Chung Il Hong, Jung Woo Kim, Sang Joon Lee, Soon Kil Ahn, Nam Song Choi, Ryung Kee Hong, Hyoung Sik Chun, Seung Kee Moon, Cheol Kyu Han. Angiogenesis inhibitors, antiarthritic agents and anticarcinogenic agents plus synthesis. US patent Number US6063812 A, Also published as CA2331873A1, CA2331873C, CN1301260A, CN100352810C, DE69903279D1, DE69903279T2, EP1077964A1,EP1077964B1,WO1999059986A1, Filing date: May 13, 1999.Original Assignee:Chong Kun Dang Corporation Crawford, Thomas; Reece, Hayley A.Preparation of crystalline forms of 6-O-(4-dimethylaminoethoxy)cinnamoylfumagillol.PCT Int. Appl. (2012), WO2012064838 A1, 20120518
Egorov, Maxim et al. Preparation of fumagillol derivatives useful for the treatment or prevention of bone tumors. PCT Int. Appl., WO2012130906, 04 Oct 2012
Stevenson, Cheri A.; Akullian, Laura C.; Petter, Russell C.; Kane, John J.; Hammond, Charles E.; Yin, Mao; Yurkovetskiy, Aleksandr.Preparation of biocompatible biodegradable fumagillin analog conjugates for the treatment of cancer. PCT Int. Appl. (2009), WO2009073445 A2, 20090611
Lee, Hong Woo et al.Design, synthesis, and antiangiogenic effects of a series of potent novel fumagillin analogues.Chemical & Pharmaceutical Bulletin, 55(7), 1024-1029; 2007
Lee, Hong Woo et al.Selective N-demethylation of tertiary aminofumagillols with selenium dioxide via a non-classical Polonovski type reaction.Heterocycles, 68(5), 915-932; 2006
|
References OTHERS |
1: Yin SQ, Wang JJ, Zhang CM, Liu ZP. The development of MetAP-2 inhibitors in cancer treatment. Curr Med Chem. 2012;19(7):1021-35. Review. PubMed PMID: 22229417.
2: Shin SJ, Ahn JB, Park KS, Lee YJ, Hong YS, Kim TW, Kim HR, Rha SY, Roh JK, Kim DH, Kim C, Chung HC. A Phase Ib pharmacokinetic study of the anti-angiogenic agent CKD-732 used in combination with capecitabine and oxaliplatin (XELOX) in metastatic colorectal cancer patients who progressed on irinotecan-based chemotherapy. Invest New Drugs. 2012 Apr;30(2):672-80. doi: 10.1007/s10637-010-9625-x. Epub 2010 Dec 29. PubMed PMID: 21188464.
3: Shin SJ, Jeung HC, Ahn JB, Rha SY, Roh JK, Park KS, Kim DH, Kim C, Chung HC. A phase I pharmacokinetic and pharmacodynamic study of CKD-732, an antiangiogenic agent, in patients with refractory solid cancer. Invest New Drugs. 2010 Oct;28(5):650-8. doi: 10.1007/s10637-009-9287-8. Epub 2009 Jul 8. PubMed PMID: 19585083.
4: Rhee Y, Park SY, Kim YM, Lee S, Lim SK. Angiogenesis inhibitor attenuates parathyroid hormone-induced anabolic effect. Biomed Pharmacother. 2009 Jan;63(1):63-8. doi: 10.1016/j.biopha.2007.10.013. Epub 2007 Nov 20. PubMed PMID: 18457934.
5: Kim YM, An JJ, Jin YJ, Rhee Y, Cha BS, Lee HC, Lim SK. Assessment of the anti-obesity effects of the TNP-470 analog, CKD-732. J Mol Endocrinol. 2007 Apr;38(4):455-65. PubMed PMID: 17446235.
6: Kim EJ, Shin WH. General pharmacology of CKD-732, a new anticancer agent: effects on central nervous, cardiovascular, and respiratory system. Biol Pharm Bull. 2005 Feb;28(2):217-23. PubMed PMID: 15684472.
7: Chun E, Han CK, Yoon JH, Sim TB, Kim YK, Lee KY. Novel inhibitors targeted to methionine aminopeptidase 2 (MetAP2) strongly inhibit the growth of cancers in xenografted nude model. Int J Cancer. 2005 Mar 10;114(1):124-30. PubMed PMID: 15523682.
8: Lee HS, Choi WK, Son HJ, Lee SS, Kim JK, Ahn SK, Hong CI, Min HK, Kim M, Myung SW. Absorption, distribution, metabolism, and excretion of CKD-732, a novel antiangiogenic fumagillin derivative, in rats, mice, and dogs. Arch Pharm Res. 2004 Feb;27(2):265-72. PubMed PMID: 15029870.
9: Kim JH, Lee SK, Ki MH, Choi WK, Ahn SK, Shin HJ, Hong CI. Development of parenteral formulation for a novel angiogenesis inhibitor, CKD-732 through complexation with hydroxypropyl-beta-cyclodextrin. Int J Pharm. 2004 Mar 19;272(1-2):79-89. PubMed PMID: 15019071.
10: Myung SW, Kim HY, Min HK, Kim DH, Kim M, Cho HW, Lee HS, Kim JK, Hong CI. The identification of in vitro metabolites of CKD-732 by liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 2002;16(21):2048-53. PubMed PMID: 12391579.
| WO2007072083A1 | Dec 22, 2006 | Jun 28, 2007 | Prosidion Ltd | Treatment of type 2 diabetes with a combination of dpiv inhibitor and metformin or thiazolidinedione |
| WO2011085201A1 * | Jan 7, 2011 | Jul 14, 2011 | Zafgen Corporation | Fumagillol type compounds and methods of making and using same |
| WO2011088055A2 * | Jan 11, 2011 | Jul 21, 2011 | Zafgen Corporation | Methods and compositions for treating cardiovascular disorders |
| WO2012064838A1 | Nov 9, 2011 | May 18, 2012 | Zafgen Corporation | Crystalline solids of a metap-2 inhibitor and methods of making and using same |
| WO2013169727A1 * | May 7, 2013 | Nov 14, 2013 | Zafgen, Inc. | Polymorphic salt of the oxalate salt of 6 – o – ( 4 – dimethylaminoethoxy) cinnarnoyl fumagillol and methods of making and using same |
| WO2013169857A1 * | May 8, 2013 | Nov 14, 2013 | Zafgen, Inc. | Treating hypothalamic obesity with metap2 inhibitors |
| EP2317845A1 * | Jul 17, 2009 | May 11, 2011 | Zafgen, Inc. | Methods of treating an overweight or obese subject |
| US8349891 | Aug 7, 2012 | Jan 8, 2013 | Zafgen, Inc. | Crystalline solids of a MetAP-2 inhibitor and methods of making and using same |
| US8367721 | Aug 7, 2012 | Feb 5, 2013 | Zafgen, Inc. | Methods of treating an overweight or obese subject |
| US8642650 | Dec 4, 2009 | Feb 4, 2014 | Zafgen, Inc. | Methods of treating an overweight or obese subject |
| US8735447 | Nov 16, 2012 | May 27, 2014 | Zafgen, Inc. | Crystalline solids of a MetAP-2 inhibitor and methods of making and using same |
| US20130018095 * | Jan 7, 2011 | Jan 17, 2013 | Vath James E | Fumigillol Type Compounds and Methods of Making and Using Same |
| WO2003027104A1 * | Jun 11, 2002 | Apr 3, 2003 | Byung-Ha Chang | Fumagillol derivatives and preparing method thereof |
| EP0682020A1 * | Aug 31, 1989 | Nov 15, 1995 | Takeda Chemical Industries, Ltd. | Fumagillol derivatives useful as angiogenesis inhibitors |
| US6040337 * | May 13, 1999 | Mar 21, 2000 | Chong Kun Dang Corporation | 5-demethoxyfumagillol derivatives and processes for preparing the same |
| US6063812 * | May 13, 1999 | May 16, 2000 | Chong Kun Dang Corporation | Angiogenesis inhibitors, antiarthritic agents and anticarcinogenic agents plus synthesis |
| WO1999059986A1 * | May 11, 1999 | Nov 25, 1999 | Soon Kil Ahn | Fumagillol derivatives and processes for preparing the same |
| WO2005082349A1 | Feb 25, 2005 | Sep 9, 2005 | Chong Kun Dang Pharm Corp | Composition for the treatment of obesity comprising fumagillol derivative |
| WO2010065883A2 | Dec 4, 2009 | Jun 10, 2010 | Zafgen Corporation | Method of treating an overweight or obese subject |
| KIM ET AL. JOURNAL OF MOLECULAR ENDOCRINOLOGY vol. 38, 2007, pages 455 – 465 | ||
| 2 | RUPNICK ET AL. PROC NATL ACAD SCI USA vol. 99, 2002, page 10730 | |
| 3 | WANG ET AL. CANCER RES vol. 63, 2003, page 7861 | |
| 4 | WARDER ET AL. J PROTEOME RES vol. 7, 2008, page 4807 | |
| 5 | * | YOO MEE KIM ET AL: “Assessment of the anti-obesity effects of the TNP-470 analog, CKD-732“, JOURNAL OF MOLECULAR ENDOCRINOLOGY, SOCIETY FOR ENDOCRINOLOGY, GB, vol. 38, no. 4, 1 April 2007 (2007-04-01), pages 455-465, XP002632891, ISSN: 0952-5041, DOI: 10.1677/JME.1.02165 |
| 6 | ZHANG ET AL. J. BIOMED SCI. vol. 9, 2002, page 34 |
………

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
web link

New Drug Approvals, ALL ABOUT DRUGS, WORLD DRUG TRACKER
MEDICINAL CHEM INTERNATIONAL, DRUG SYN INTERNATIONAL
SCALEUP OF DRUGS, ALL FOR DRUGS ON WEB,
MY CHINA, VIETNAM AND JAPAN BLOGS
ICELAND, RUSSIA, ARAB
BOBRDOBR, BLAND ICELAND, 100zakladok, adfty
GROUPS
you can post articles and will be administered by me on the google group which is very popular across the world
OPD GROUPSPACES, SCOOP OCI, organic-process-development GOOGLE, TVINX, MENDELEY WDT, SCIPEOPLE OPD, EPERNICUS OPD, SYNTHETIC ORGANIC CHEMISTRYLinkedIn group, DIIGO OPD, LINKEDIN OPD, WDT LINKEDIN, WDTI ZING


